 Omar Shennib
Omar Shennib Olivia Raines1
Olivia Raines1 Megan E. Williams
Megan E. Williams- 1Department of Neurobiology, School of Medicine, University of Utah, Salt Lake City, UT, United States
- 2Division of Pediatric Neurology, Department of Pediatrics, School of Medicine, University of Utah, Salt Lake City, UT, United States
The synaptic adhesion molecule KIRREL3 regulates synapse development in mice and is implicated in human neurological disorders, including autism spectrum disorder, intellectual disability, and Jacobsen syndrome (chromosome 11q deletion syndrome). However, its status as a definitive human disease gene remains unresolved, likely due to the rarity of KIRREL3-related disorders and significant gaps in understanding its molecular mechanisms. Current knowledge is further fragmented across disparate clinical and basic research reports, often buried in supplemental data. This review synthesizes existing evidence to enable clinicians and scientists to better evaluate KIRREL3 variants as potentially disease causing. We review its conserved role in mediating neuron-to-neuron interactions during axon targeting and synapse formation in mice and how disruptions to these interactions could contribute to neurological pathology in humans. We also discuss how disease-associated variants alter KIRREL3 function. Our analysis underscores the need for integrated studies spanning basic and clinical investigation to validate KIRREL3’s disease association and advance future interventions for KIRREL3-related disorders.
Introduction
KIRREL3 is a member of the small IRM (Irregular cell recognition module) gene family and the only one repeatedly linked to neurological disorders (1–5). Like all IRM proteins, KIRREL3 is a single-pass transmembrane protein (1, 6). IRM proteins are best characterized in non-neural tissues, where they play critical roles in kidney slit diaphragm formation and muscle cell fusion (6–9). These proteins also contribute to neurodevelopmental processes in model organisms, including Drosophila eye formation and C. elegans synaptic connectivity (6, 7, 10–29). Because this important foundational work on invertebrate IRM proteins was reviewed elsewhere (30) and our goal is to translate Kirrel3 knowledge to human neurological diseases, we focus this review on Kirrel3 in brain function and dysfunction of mammals from mice to humans.
Over the past two decades, human genetic studies suggested associations between KIRREL3 (OMIM: 607761) and brain conditions such as autism spectrum disorder, intellectual disability, Jacobsen syndrome, and developmental delay (4, 31–37). However, the role of KIRREL3 as a disease-associated gene remains unresolved in the clinic even though there is supportive evidence of its variant pathogenicity in the scientific literature. Many different types of genetic variants in the KIRREL3 gene, including missense mutations, truncating variants, splice-site alterations, and copy number variants, are reported in the public databases gnomAD, UCSC, VariCarta, and ClinVar (38–41). While some of these variants are classified as pathogenic or likely pathogenic, many others remain classified as variants of uncertain significance (VUS) due to insufficient evidence (41). This uncertainty stems from several factors, including small sample sizes in initial studies, limited functional validation, lack of inheritance information, and inconsistent phenotypic presentations among individuals with KIRREL3 variants. Furthermore, the lack of large-scale, systematic analyses on KIRREL3-related disorders contributes to the unresolved role of KIRREL3 in disease. This leaves clinicians and researchers without clear guidelines for interpretation.
The goal of this work is to provide a side-by-side analysis of what is currently known about KIRREL3 protein function in the mammalian brain, as well as what is known about Kirrel3 gene variants and their associations with neurological diseases. Furthermore, recent analyses also found increases KIRREL3 gene expression to glioma cancer (32, 33). Misexpression of cell adhesion molecules is common in metastatic cancers, however, we will focus on neurological disorders in this review (42). This review will contribute to the understanding of KIRREL3 and serve as a model for resolving gene-disease associations surrounding other genes with similarly unclear disease associations.
KIRREL3 gene and isoforms expression
The human KIRREL3 gene (Kin of IRRE-like 3), also previously known as NEPH2 (43), is located on chromosome 11q24.2 (hg38 chr11:126,423,358-127,000,770), covering 577.413 Kb, with 21 exons (Figure 1A) (39, 44, 45). Its longest and most abundant transcript encodes a single-pass transmembrane protein that is 778 amino acids long with five extracellular immunoglobulin domains and an intracellular region ending in a PDZ binding domain (Figure 1B) (44, 46, 47). Targeted long-read mRNA sequencing revealed the full exon structure and alternative splicing pattern of Kirrel3 in the mouse brain (45). In mice, 19 alternatively spliced isoforms were identified including those with different intracellular domains and secreted forms. The study also analyzed publicly available long-read sequencing data from human brain tissue and identified 11 isoforms in the human brain (45). Although, this number is likely an underestimate because Kirrel3 transcripts are rare and a targeted approach was not used, the patterns were similar to mice in that human KIRREL3 isoforms also had different intracellular domains and secreted forms (45). Interestingly, cross-species comparisons of Kirrel3 transcripts revealed that humans and great apes uniquely possess an additional exon encoding a distinct 30 amino acid insertion in the intracellular domain, suggesting evolutionary specialization (45). However, future work is needed to directly test if different KIRREL3 isoforms, including the hominid-specific exon, have distinct functions in the brain.
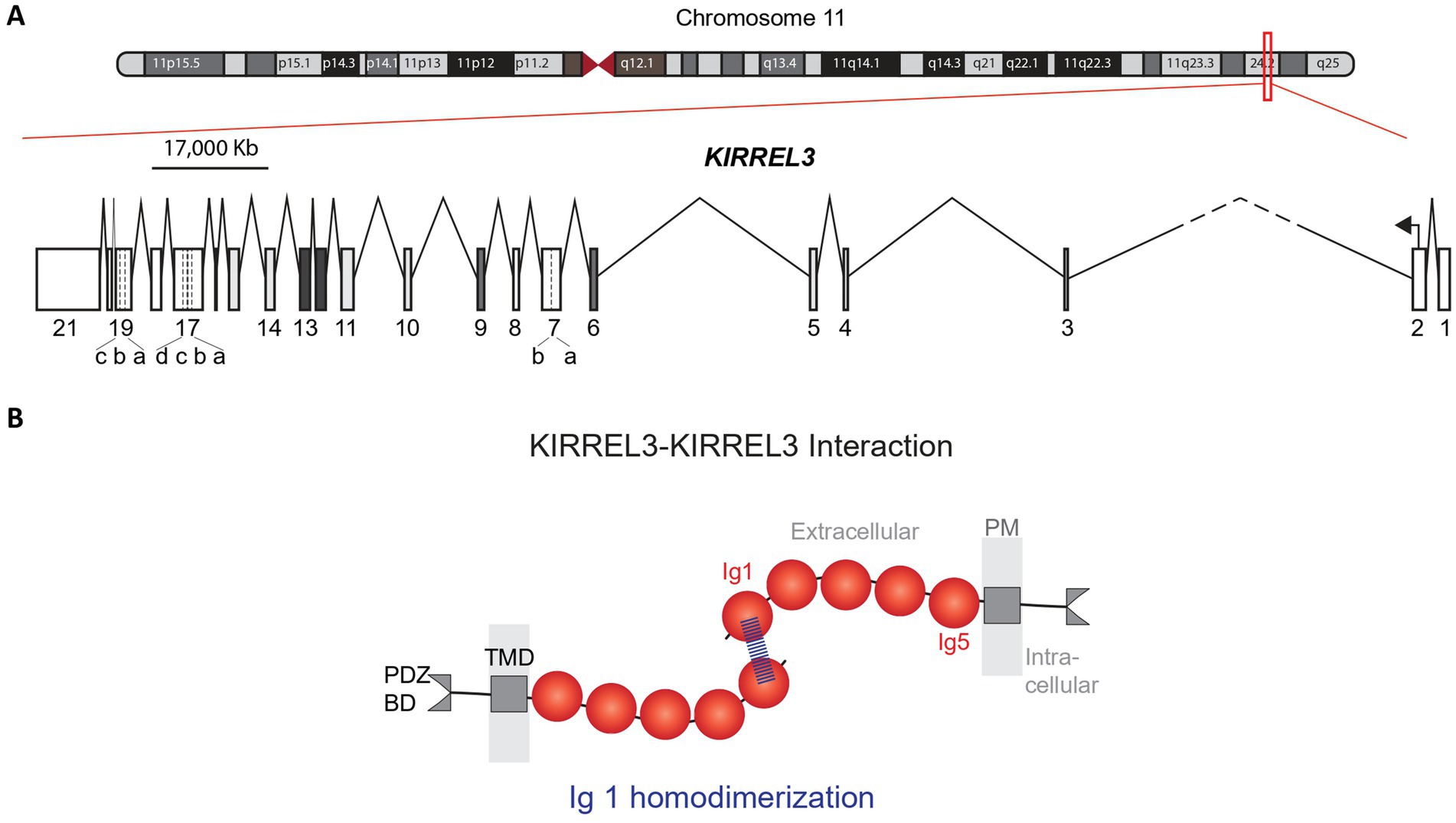
Figure 1. Human KIRREL3 gene and protein structures. (A) Genetic map of chromosome 11. Red box covers the KIRREL3 gene (577.413 Kb) on the q arm of chromosome 11. KIRREL3 schematic gene structure shows 21 exons (boxes) and introns (lines). Exon 7, 17, and 19 are reported in unique isoforms and have 5′ extensions and 3′ extensions, indicated by dotted lines. Intron 2–3 is cut short (dashes) to fit in the scaled illustration. (B) KIRREL3 protein structure and protein transhomophilic interaction. Binding structural assay shows the protein binding structure is through the IgD1 via hydrogen bonding at Q128 amino acid position. IgD, immunoglobulin domain; TMD, transmembrane domain; BD, binding domain; PM, plasma membrane.
The KIRREL3 mRNA is expressed in multiple tissues, including the kidney, skeletal muscle, and brain (16, 47, 48). To refine its expression profile using recent datasets, we analyzed RNA-seq and single-nucleus RNA sequencing available on GTEx, Human Protein Atlas, BioGPS, and Allen Brain Atlas (46, 49–51). We found that both human and mouse Kirrel3 mRNA have the highest expression in the brain with enrichment in the cerebral cortex, thalamus, cerebellum, amygdala, hippocampus, and olfactory bulb (Figures 2A,C). Within the brain, Kirrel3 transcription is enriched in three major cell populations: excitatory neurons (amygdala and hippocampal dentate gyrus (DG)), inhibitory neurons (LAMP5-LHX6, Chandelier, and MGE-derived interneurons), and oligodendrocytes (Figures 2B,D). The conserved expression pattern of Kirrel3 in both human and mouse brain cell types, and the high conservation in the gene (94.7%) and protein (98%) sequences, supports the use of mouse models to elucidate its cellular function (52–55).
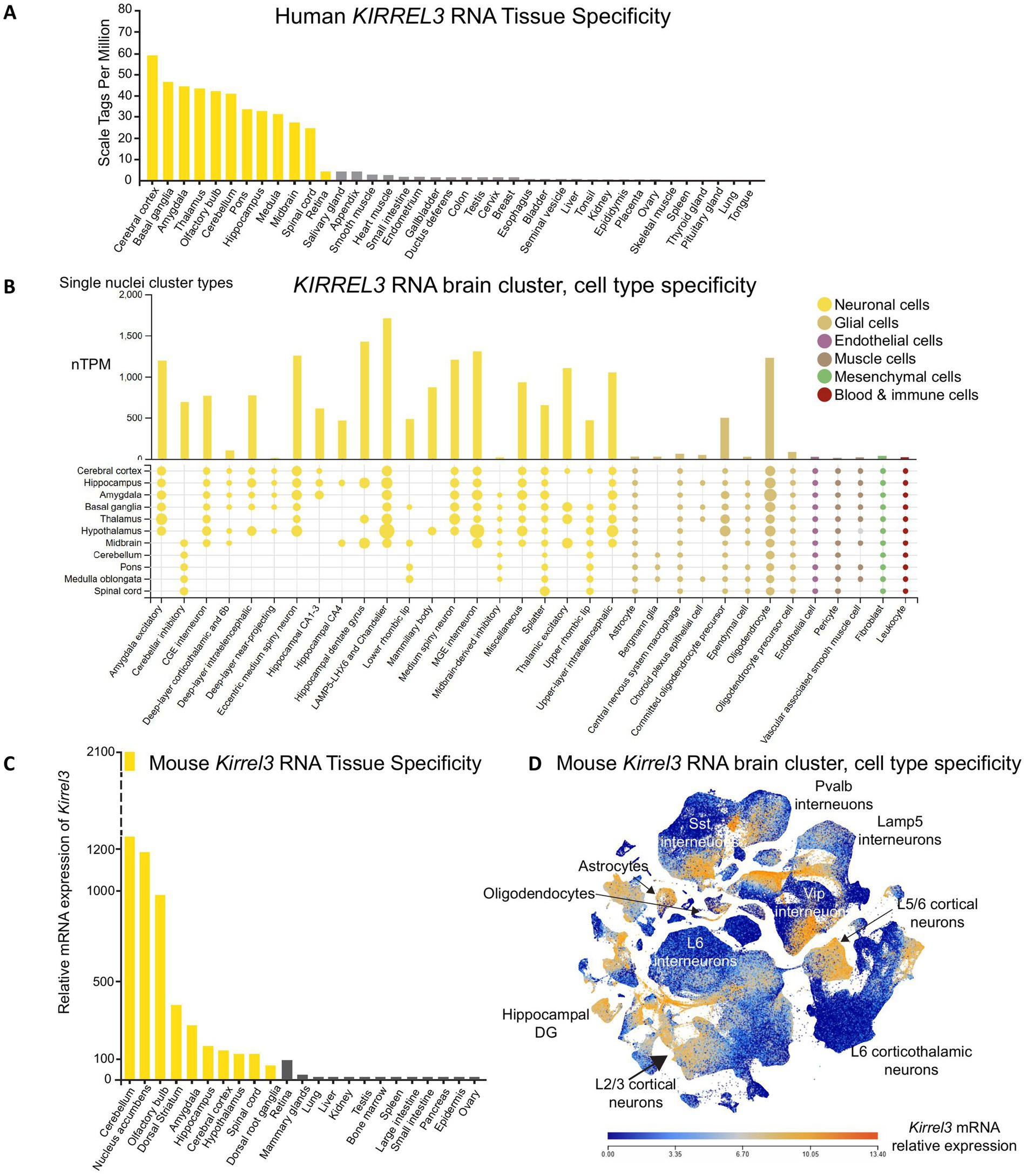
Figure 2. KIRREL3 expression in human and mouse. (A) RNA sequencing from adult human tissues show KIRREL3 mRNA expression levels higher in brain regions (yellow) than anywhere else. (B) Single nuclei sequencing from human brain shows that KIRREL3 copies are enriched in excitatory and inhibitory neurons, as well as gliodendrocytes. Y-axis shows transcript expression in normalized transcript per million (nTPM). Human transcript expression levels are based on transcriptomics data from two sources: HPA and GTEX. (C) Mouse Kirrel3 mRNA expression is similar to human with high expression in brain regions (yellow). (D) Scatter blot of mouse Kirrel3 transcript expression shows expression in specific cell types throughout the brain. Mouse transcriptomics data are from two sources: BioGFP and Allen Brain Atlas.
KIRREL3 behavioral studies in mouse
Several mouse models were developed to investigate Kirrel3’s role in neurological disorders, including a global knockout, a conditional knockout, and a point mutant knock-in mouse. Kirrel3 knockout mice were used to investigate behavioral phenotypes relevant to neurological disorders and compared to wildtype controls (56–59). Kirrel3 knockout mice exhibit subtle deficits in social recognition and novel object memory, which may be consistent with core features of autism spectrum disorder and intellectual disability in humans (56, 58). Moreover, a recent study revealed that selective activation of Kirrel3-expressing GABAergic neurons in the hippocampus impairs contextual memory discrimination, directly linking Kirrel3-expressing GABA neurons to cognitive processes (59).
Beyond memory functions, Kirrel3 knockout mice show distinct behavioral alterations across multiple neural systems. While basic olfactory function remains intact, knockout mice show reduced male–male aggression (56, 57, 60). Motor assessments yield complex findings, with knockout mice displaying both hyperactivity in open field tests and enhanced coordination on rotarod tasks (56–58, 61). Auditory system evaluation reveals a selective deficit in processing high-intensity sounds (110 dB), while baseline hearing remains unaffected (57). Though molecular mechanisms were investigated in hippocampal and olfactory circuits (See Molecular Mechanisms Section), significant gaps remain in understanding Kirrel3’s precise function in other brain regions. The lack of mechanistic studies in cerebellar and auditory circuits limits our interpretation of the observed behavioral phenotypes. Collectively, Kirrel3 knockout behavioral studies indicate that Kirrel3 subtly influences distinct brain functions in mice, including cognition, olfaction, motor control, and auditory processing. These functions are clinically relevant, as they are commonly disrupted in autism and intellectual disability (62–64). However, a caveat of mouse behavioral studies is that they were done in adult mice and genetic compensation can occur over time. More behavioral assays in juvenile mice are needed to directly assess the developing brain and ameliorate this caveat. Future research using conditional knockout models would clarify Kirrel3’s region-specific effects on mouse brain function. Additionally, although automated tracking methods are propelling the complexity of mouse behavioral research, mice inherently lack the subtle and complex behaviors of humans and direct translation from mouse to human should be interpreted with caution.
Molecular mechanisms of KIRREL3 in mouse
In general, Kirrel3 knockout mice exhibit normal brain and body size throughout development compared to wildtype controls (56–58). Macroscopic-level findings show that Kirrel3 manipulation does not alter gross neuroanatomy but instead affects ultrastructures (52, 56–58, 60, 65, 66). This is similar to autism where there is no clear or consistent pathology at a macroscopic structural level, but it is largely theorized that deficits are at a synaptic and circuit level (63, 67). Thus, we next review mechanistic studies of Kirrel3 in the mammalian brain.
Kirrel3 promotes hippocampal synapse formation
Kirrel3 is expressed selectively in two hippocampal cell populations: excitatory dentate gyrus (DG) neurons and a subset of inhibitory GABAergic neurons (47, 52, 58). Kirrel3 is important for the formation of specialized excitatory synapses onto GABAergic neurons in mice called mossy fiber filopodia synapses (Figures 3A,B) (52, 66). Kirrel3 knockout mice have a significant reduction in the formation of mossy fiber filopodia synapses (66). Because these synapses normally excite GABAergic neurons that, in turn, inhibit CA3 neurons, Kirrel3 knockout mice lack this inhibition and have a corresponding increase in CA3 neuron activity (Figure 3C) (52). Thus, a reduction in excitatory synapses actually increases net brain activity (because they specifically form on inhibitory neurons) and highlights why understanding the precise synapses regulated by neurodevelopmental risk genes is important for developing effective treatment strategies. Unlike disorders involving general excitatory synapse loss, where therapies might aim to boost neuronal activity, KIRREL3-related dysfunction may instead require interventions that decrease neuronal activity.
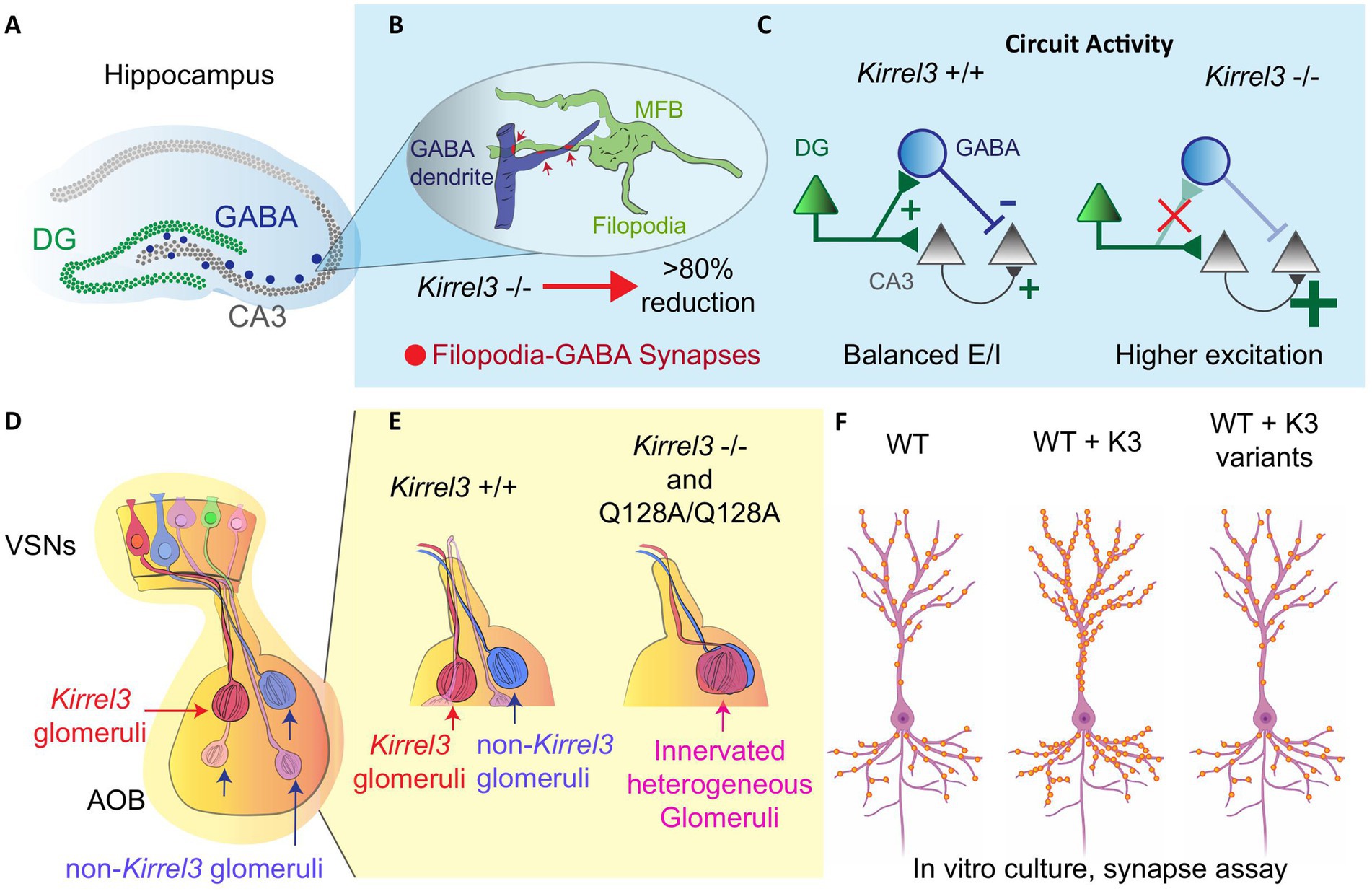
Figure 3. Kirrel3 functional studies in rodents. (A) A Hippocampal diagram and its relevant regions (DG, CA3 and GABA neurons). (B) Structure of mossy fiber botouns (MFB) and filopodia-GABA synapses in the CA3 region. In the Kirrel3 KO mouse, these synapses are reduced significantly. (C) CA3 neurons have higher excitation/inhibition ratio (E/I) in knockout mice. This is most likely due to the loss of feed-forward inhibition from the DG filopodia-GABA synapse reduction. (D) An olfactory system diagram shows vomeronasal sensory neurons (VSN) projecting axons to form synaptic Kirrel3 homogeneous glomeruli in the accessory olfactory bulb (AOB). (E) Kirrel3 KO and point mutant (Q128A) mice show overall a reduction in glomeruli number compared to wild-type (WT) mouse. (F) In vitro hippocampal culture, over expressing Kirrel3 (K3) patient variants shows similar synapse number to the no overexpression condition in the WT. Only over-expression of Kirrel3 wild-type copy increases the synapse number. This indicates that the Kirrel3 patient variants alter Kirrel3’s synaptic function.
A subsequent study did not directly study KIRREL3 protein function but instead studied the role of the GABA neurons that express Kirrel3 in learning, memory, and circuit activity (59). When these Kirrel3-expressing GABA neurons were activated with chemogenetics, they impaired memory and powerfully suppressed CA3 activity (59), confirming their capacity for circuit inhibition. However, it remains untested whether such activation could rescue the hyperactivity phenotype in knockout mice, a crucial next step for therapeutic development. These results are particularly significant given the established role of GABAergic dysfunction in autism spectrum disorders (68). Importantly, Kirrel3’s expression pattern extends far beyond the hippocampus, and is expressed by specific cell types in many brain regions (Figure 2) (46, 49, 51). Comparative studies across brain regions are needed to test whether Kirrel3’s hippocampal mechanisms generalize to other areas.
Kirrel3 axon coalescence role in the olfactory system
Within the olfactory system, Kirrel3 is expressed in discrete subsets of vomeronasal sensory neurons that converge to form homogeneous glomeruli in the accessory olfactory bulb (Figure 3D) (48, 60, 69, 70). This restricted expression implicates Kirrel3 in helping to establish part of the glomerular map essential for odor and pheromonal processing. Kirrel3 knockout mice display defects in posterior accessory olfactory glomeruli, which appear disorganized and significantly reduced in number compared to wildtype controls (Figure 3E) (48, 60). KIRREL3 is known to mediate synapse formation and cell adhesion through homophilic binding in trans where the KIRREL3 extracellular domain from one cell binds the KIRREL3 extracellular domain on another cell synapses (2, 6, 48, 52, 65, 71, 72). This was shown to occur in vivo through study of a p.Q128A point mutant that breaks the KIRREL3 homophilic trans-cellular binding (71). The mutant mouse phenocopied the glomeruli disruption in knockout mice and microscopic investigation showed that these glomeruli are innervated and heterogeneous (Figure 3E) (71). This indicates that KIRREL3-KIRREL3 trans-cellular binding is necessary for its function in the mouse brain. Olfactory deficits in KIRREL3-related disorders remain understudied, and this wiring mechanism might have broad relevance. The accessory olfactory bulb shares developmental pathways with social brain circuits, and disrupted pheromone processing could contribute to the social behavior impairments seen in patients.
KIRREL3 variants in human neurological disorders
KIRREL3-related disorders have broad phenotypic spectrum and variable symptom severity. Affected individuals present with neurodevelopmental conditions, including autism, intellectual disability, developmental delay, ADHD, or bipolar disorder, often accompanied by dysmorphic facial features, e.g., coarse facies, large ears, and flat nasal bridge (Table 1). Moreover, symptom severity ranges from mild to profound. We explore specific variants and severity correlations in a later section (SNV in clinical cases and Table 2). The link between KIRREL3 and neurological disorders was first discovered through two foundational studies. Grossfeld et al. (73) first identified deletions encompassing KIRREL3 in patients with 11q terminal deletion syndrome. Subsequently, Bhalla et al. (31) made two key discoveries. First, they identified deletion variants in KIRREL3, including a balanced translocation. This chromosomal translocation disrupted both KIRREL3 and CDH15 in a severely affected female patient with an IQ of 16. Second, they reported the first pathogenic single nucleotide variants in KIRREL3. These included three missense mutations, p.R40W, p.R336Q, p.V731F, found in unrelated intellectual disability patients (31, 74). These three variants showed rare allele frequencies (0.0000016–0.000075) suggesting negative selection, and subsequent functional studies confirmed their impairment of synapse formation in vitro (2).
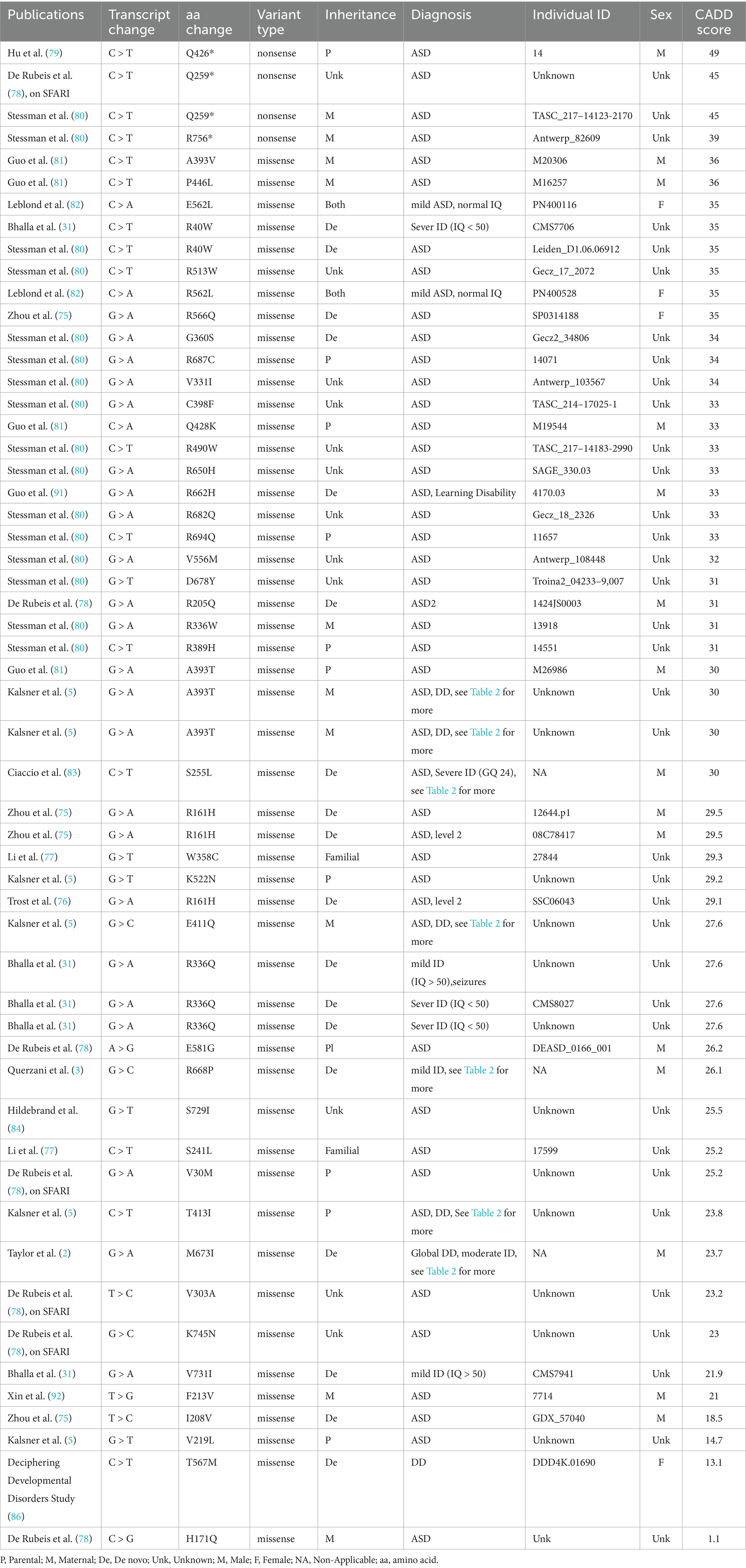
Table 1. Manual curation of well-reported exonic variants.
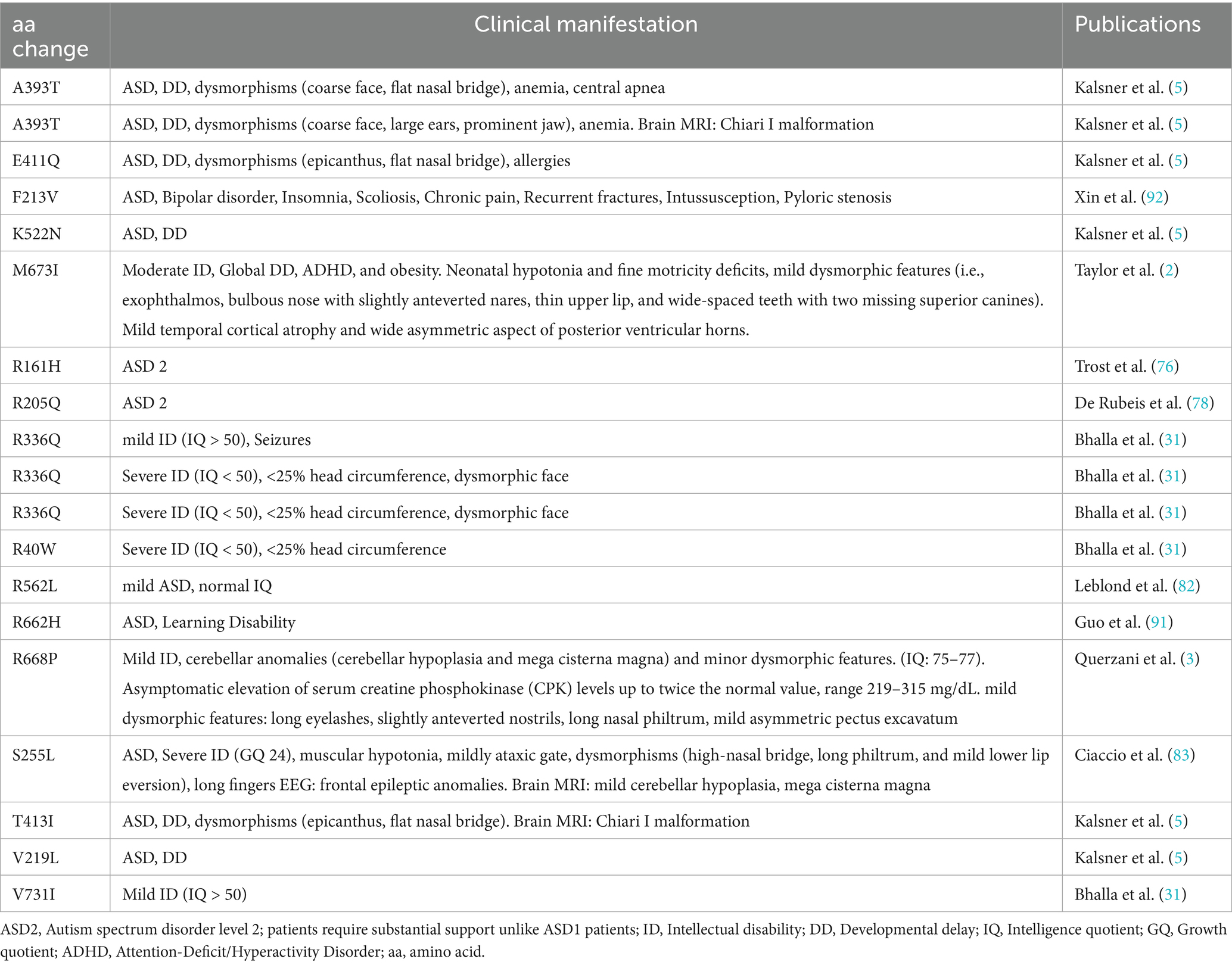
Table 2. KIRREL3 variants reported with detailed clinical manifestation.
Testing the function of human variants in rodent neurons
KIRREL3 missense variants identified in patients with autism and intellectual disability were functionally tested in a series of experiments using cultured cells and mouse neurons (2). Exogenous expression of wildtype Kirrel3 traffics to the cell membrane, accumulates at synapses, causes cells to adhere to one another, and induces extra synapses in cultured mouse hippocampal excitatory neurons (2). Six variants were tested and trafficked correctly (R40W, R161H, R205Q, R336Q, M673I, and V731F); however, five out of the six variants, excluding M673I, had a reduced ability to induce synapses in cultured hippocampal neurons (Figure 3F) (2). Two variants that were not synaptogenic, p.R205Q and p.V731F, were unable to mediate trans-cellular adhesion, explaining their inability to mediate synapse formation (2). However, p.R40W, p.R161H and p.R336Q variants were not synaptogenic but could mediate cell adhesion normally (2). Together, this indicates that KIRREL3 trans-cellular binding is necessary but not sufficient for its synaptic function. This provides strong support that KIRREL3 is more than just synaptic glue and is also an active molecule in synapse formation. To our knowledge, this is the only study to date to investigate the function of KIRREL3 missense variants found in patients with neurodevelopmental disorders. The fact that most disease-associated variants (5/6) have a clear functional deficit in cell and neuron culture assays strongly suggests that they may contribute to pathogenicity in humans.
SNVs in clinical cases
To gain a better picture of KIRREL3 SNV types from a clinical database, we analyzed 116 clinically validated KIRREL3 variants from ClinVar (41). Human KIRREL3 gene and transcript (NM_032531.4) references are annotated by the NCBI Homo sapiens Annotation Release 109.20200815; however, for the newly discovered exons, we used the most recent published data (45). For allele and variant frequencies, we used gnomAD Joint Variant Frequencies 4.0 v2 (74). We calculated CADD score, if not reported already, for variants in Table 1 using https://cadd.gs.washington.edu/snv.
This analysis reveals two main points. First, variants are distributed across the entire protein (Figures 4A,B), with no obvious clustering toward specific domains. Second, most variants (56%) remain classified as variants of uncertain significance (VUS), with only 2% currently designated as pathogenic on ClinVar. Even functionally tested variants like p.R40W and p.V731F remain classified as VUS, despite evidence that they impair synapse formation in mice (2). This conservative approach suggests current guidelines may underestimate KIRREL3’s clinical importance.
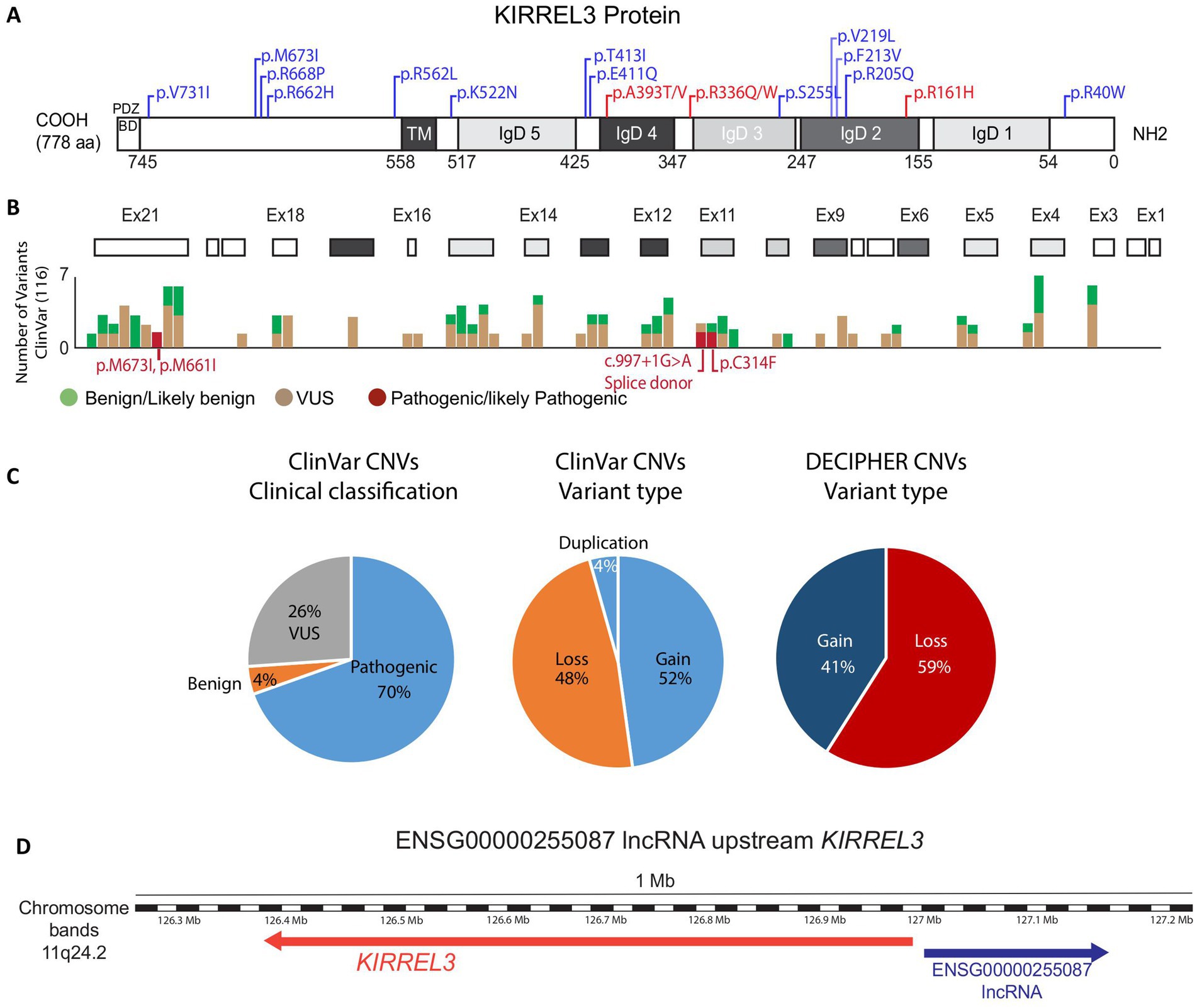
Figure 4. KIRREL3 clinical variants. (A) KIRREL3 protein domains as predicted by SMART and UniProtKB/TrEMBL. Numbers in the bottom are amino acid (aa) number order. Domains are grey shaded to match their corresponding exons in (B). Well-reported variants with detailed clinical manifestations are indicated on the protein domains, see Table 2. Variants with 1 or 2 occurrences are blue, variants with ≥3 occurrence are red. (B) Exons (boxes) are aligned with clinical variants reported on ClinVar. (C) Statistical counts on clinical variants types reported in ClinVar and DECIPHER. (D) Genetic map of KIRREL3 and Kirrel3-IncRNA. IncRNA’s associated mutation was reported in an ASD patient as a potential regulator of the gene. See Methods for details. IgD, immunoglobulin domain; TM, transmembrane; BD, binding domain; VUS, variant of uncertain significance.
We also searched for genetic evidence to establish KIRREL3 as a risk factor for neurological disorders in large-scale studies on autism spectrum disorder and intellectual disability (FDR < 0.1) (2, 3, 5, 31, 75–84). From these studies, we curated 55 exonic SNVs that are well-phenotyped, and the connections are supported by computational predictions showing that many exonic missense variants have high pathogenicity scores (CADD ≥30), indicating strong likelihood of deleterious effects (Table 1) (85). Furthermore, we found that there is a predominance of de novo mutations, with equal maternal and paternal inheritance (75, 76, 80, 81, 86). Interestingly, the extracellular region has three recurrent pathogenic variants, each observed in ≥3 unrelated cases and associated with severe phenotypes, p.R161H, p.R336Q/W, p.A393T/V (Figure 4A in red, Table 2). Table 2 highlights the variants with reported detailed clinical manifestations. Interestingly, both extracellular variants, p.R161H and p.R336Q, disrupt synapse induction in mice (2). The 19 intracellular variants remain poorly characterized beyond the PDZ-binding domain’s putative synaptic scaffolding role (2, 10, 83). The recurrence of pathogenic extracellular variants exhibiting similar severe phenotypes suggests their clinical significance, warranting prioritization in genetic testing. Intracellular variants remain less understood but may contribute to variable expressivity. Future work should address the intracellular domain’s unexplored contributions to KIRREL3-related pathogenesis through structural biology and proteomic approaches. The combination of domain-wide variant distribution, functional evidence, and genetic associations supports KIRREL3 as a clinically significant risk gene in neurological disorders. Improved variant interpretation methods are needed to fully capture the variants’ pathogenic potential.
CNVs in clinical cases
KIRREL3 deletions are a consistent feature of Jacobsen syndrome (chromosome 11q deletion syndrome), which typically results from large (7–20 Mb) terminal deletions at 11q23.3-qter that encompass KIRREL3 (11q24.2) (34, 36, 87). Jacobsen syndrome is a rare genetic disorder characterized by developmental delays, craniofacial dysmorphisms, and variable congenital anomalies. The syndrome’s characteristic intellectual disability appears specifically linked to loss of KIRREL3 gene. This is because two unusual cases with microdeletions (∼700 kb) sparing KIRREL3 presented without intellectual disability (88). This genotype–phenotype correlation strongly suggests KIRREL3 loss is the key contributor to cognitive impairment in Jacobsen syndrome.
We also analyzed other publicly available CNVs in ClinVar and DECIPHER in the same way we analyzed the SNVs in the previous section. KIRREL3 CNVs show significantly high pathogenicity rates, with nearly equal proportions of duplications and deletions (gain and loss) (Figure 4C). Furthermore, the gene’s exceptionally high probability of loss-of-function intolerance (pLI = 0.98), suggests haploinsufficiency as a key disease mechanism in humans (74–76, 80). However, critical gaps remain in correlating these genetic findings with detailed clinical outcomes, as most databases lack comprehensive phenotypic information.
Beyond CNVs that disrupt KIRREL3’s protein-coding sequence, regulatory mechanisms may also contribute to KIRREL3-related neurological disorders. A study of chromosomal abnormalities in genetically undiagnosed neurological cases identified a patient with ADHD and spatial coordination deficits carrying de novo balanced breakpoints at 11q24.2 (89, 90). While no pathogenic coding variants were detected, the breakpoints disrupted ENSG00000255087, a long non-coding RNA (lncRNA) located upstream of KIRREL3 (Figure 4D). Subsequent analysis demonstrated co-occurring KIRREL3 mRNA downregulation in this patient. This suggests that either the lncRNA regulates KIRREL3 gene expression or this region contains an enhancer regulatory element for KIRREL3 gene. These findings highlight the importance of investigating nearby regulatory element variants where non-coding disruptions can phenocopy coding mutations through transcriptional dysregulation.
Future perspectives
Moving forward, research on KIRREL3 should address several critical gaps to fully understand its role in neural circuit development and neurological disorders. While studies revealed essential Kirrel3 functions in hippocampal and olfactory systems, conditional knockout approaches targeting specific neuronal populations will be necessary to dissect its roles in cortical, cerebellar, and other neural circuits. The existence of multiple Kirrel3 isoforms, particularly the distinct secreted and transmembrane forms, presents another crucial area for exploration. Detailed characterization of isoform-specific localization, binding partners, and functional differences could explain the phenotypic variability observed in patients and provide mechanistic insights into how different mutations lead to diverse clinical outcomes.
There are currently no published studies on human iPSC-derived neurons from patients with KIRREL3 mutations. However, human cellular models will be essential for translating findings from animal studies to clinical applications. Patient-derived iPSC neurons with defined KIRREL3 variants offer a powerful platform to validate mechanisms observed in mice while capturing human-specific aspects of neural development. On the clinical front, establishing standardized phenotyping and reporting protocols across clinic centers will be critical for capturing the full spectrum of KIRREL3-related manifestations and correlating them with specific genetic changes.
These future perspectives require collaboration between neurobiologists studying synaptic development, geneticists characterizing variants, and clinicians caring for affected individuals. In conclusion, KIRREL3 emerges as a model for understanding how synaptic adhesion molecules contribute to neurological disorders. Its complex isoform regulation, region-specific circuit functions, and diverse mutational mechanisms illustrate the intricate relationship between molecular neurobiology and clinical presentation.
Author contributions
OS: Formal analysis, Visualization, Writing – original draft, Methodology, Validation, Project administration, Data curation, Investigation, Supervision, Software, Conceptualization, Writing – review & editing. OR: Conceptualization, Writing – review & editing, Investigation. ASK: Writing – review & editing, Validation. MEW: Supervision, Conceptualization, Validation, Project administration, Writing – review & editing, Investigation, Funding acquisition, Writing – original draft, Visualization, Resources.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the R01 MH125943 (MEW) R01 MH134515 (MEW), and Graduate Research Fellowship from University of Utah (OS), and the Pediatric Epilepsy Research Foundation (ASK).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that Gen AI was used in the creation of this manuscript. AI was only used to help edit the manuscript in catching grammatical errors. The content edited using the Generative AI has been checked for factual accuracy and plagiarism.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Fischbach, KF, Linneweber, GA, Felix Malte Andlauer, T, Hertenstein, A, Bonengel, B, and Chaudhary, K. The irre cell recognition module (IRM) proteins. J Neurogenet. (2009) 23:48–67. doi: 10.1080/01677060802471668
2. Taylor, MR, Martin, EA, Sinnen, B, Trilokekar, R, Ranza, E, Antonarakis, SE, et al. Kirrel3-mediated synapse formation is attenuated by disease-associated missense variants. J Neurosci. (2020) 40:5376–88. doi: 10.1523/JNEUROSCI.3058-19.2020
3. Querzani, A, Sirchia, F, Rustioni, G, Rossi, A, Orsini, A, Marseglia, GL, et al. KIRREL3-related disorders: a case report confirming the radiological features and expanding the clinical spectrum to a less severe phenotype. Ital J Pediatr. (2023) 49:99. doi: 10.1186/s13052-023-01488-7
4. Guerin, A, Stavropoulos, DJ, Diab, Y, Chénier, S, Christensen, H, Kahr, WHA, et al. Interstitial deletion of 11q-implicating the KIRREL3 gene in the neurocognitive delay associated with Jacobsen syndrome. Am J Med Genet A. (2012) 158a:2551–6. doi: 10.1002/ajmg.a.35621
5. Kalsner, L, Twachtman-Bassett, J, Tokarski, K, Stanley, C, Dumont-Mathieu, T, Cotney, J, et al. Genetic testing including targeted gene panel in a diverse clinical population of children with autism spectrum disorder: findings and implications. Mol Genet Genomic Med. (2018) 6:171–85. doi: 10.1002/mgg3.354
6. Neumann-Haefelin, E, Kramer-Zucker, A, Slanchev, K, Hartleben, B, Noutsou, F, Martin, K, et al. A model organism approach: defining the role of Neph proteins as regulators of neuron and kidney morphogenesis. Hum Mol Genet. (2010) 19:2347–59. doi: 10.1093/hmg/ddq108
7. Durcan, PJ, Al-Shanti, N, and Stewart, CE. Identification and characterization of novel Kirrel isoform during myogenesis. Physiol Rep. (2013) 1:e00044. doi: 10.1002/phy2.44
8. Donoviel, DB, Freed, DD, Vogel, H, Potter, DG, Hawkins, E, Barrish, JP, et al. Proteinuria and perinatal lethality in mice lacking NEPH1, a novel protein with homology to NEPHRIN. Mol Cell Biol. (2001) 21:4829–36. doi: 10.1128/MCB.21.14.4829-4836.2001
9. George, B, and Holzman, LB. Signaling from the podocyte intercellular junction to the actin cytoskeleton. Semin Nephrol. (2012) 32:307–18. doi: 10.1016/j.semnephrol.2012.06.002
10. Liu, YF, Sowell, SM, Luo, Y, Chaubey, A, Cameron, RS, Kim, HG, et al. Autism and intellectual disability-associated KIRREL3 interacts with neuronal proteins MAP1B and MYO16 with potential roles in neurodevelopment. PLoS One. (2015) 10:e0123106. doi: 10.1371/journal.pone.0123106
11. Chao, DL, and Shen, K. Functional dissection of SYG-1 and SYG-2, cell adhesion molecules required for selective synaptogenesis in C. elegans. Mol Cell Neurosci. (2008) 39:248–57. doi: 10.1016/j.mcn.2008.07.001
12. Wanner, N, Noutsou, F, Baumeister, R, Walz, G, Huber, TB, and Neumann-Haefelin, E. Functional and spatial analysis of C. elegans SYG-1 and SYG-2, orthologs of the Neph/nephrin cell adhesion module directing selective synaptogenesis. PLoS One. (2011) 6:e23598. doi: 10.1371/journal.pone.0023598
13. Shen, K, and Bargmann, CI. The immunoglobulin superfamily protein SYG-1 determines the location of specific synapses in C. elegans. Cell. (2003) 112:619–30. doi: 10.1016/S0092-8674(03)00113-2
14. Srinivas, BP, Woo, J, Leong, WY, and Roy, S. A conserved molecular pathway mediates myoblast fusion in insects and vertebrates. Nat Genet. (2007) 39:781–6. doi: 10.1038/ng2055
15. Tamir-Livne, Y, Mubariki, R, and Bengal, E. Adhesion molecule Kirrel3/Neph2 is required for the elongated shape of myocytes during skeletal muscle differentiation. Int J Dev Biol. (2017) 61:337–45. doi: 10.1387/ijdb.170005eb
16. Durcan, PJ, Conradie, JD, Van deVyver, M, and Myburgh, KH. Identification of novel Kirrel3 gene splice variants in adult human skeletal muscle. BMC Physiol. (2014) 14:11. doi: 10.1186/s12899-014-0011-3
17. Ristola, M, and Lehtonen, S. Functions of the podocyte proteins nephrin and Neph3 and the transcriptional regulation of their genes. Clin Sci (Lond). (2014) 126:315–28. doi: 10.1042/CS20130258
18. Yang, B, Zhang, X, Zhou, H, Zhang, X, Yang, W, Lu, J, et al. Preliminary study on the role and mechanism of KIRREL3 in the development of esophageal squamous cell carcinoma. Pathol Res Pract. (2022) 237:154025. doi: 10.1016/j.prp.2022.154025
19. Lüthy, K, Ahrens, B, Rawal, S, Lu, Z, Tarnogorska, D, Meinertzhagen, IA, et al. The irre cell recognition module (IRM) protein Kirre is required to form the reciprocal synaptic network of L4 neurons in the Drosophila lamina. J Neurogenet. (2014) 28:291–301. doi: 10.3109/01677063.2014.883390
20. Zhuang, S, Shao, H, Guo, F, Trimble, R, Pearce, E, and Abmayr, SM. Sns and Kirre, the Drosophila orthologs of Nephrin and Neph1, direct adhesion, fusion and formation of a slit diaphragm-like structure in insect nephrocytes. Development. (2009) 136:2335–44. doi: 10.1242/dev.031609
21. Tucker, DK, Adams, CS, Prasad, G, and Ackley, BD. The immunoglobulin superfamily members syg-2 and syg-1 regulate neurite development in C. elegans. J Dev Biol. (2022) 10:1003. doi: 10.3390/jdb10010003
22. Strünkelnberg, M, Bonengel, B, Moda, LM, Hertenstein, A, de Couet, HG, Ramos, RG, et al. Rst and its paralogue kirre act redundantly during embryonic muscle development in Drosophila. Development. (2001) 128:4229–39. doi: 10.1242/dev.128.21.4229
23. Shen, K, Fetter, RD, and Bargmann, CI. Synaptic specificity is generated by the synaptic guidepost protein SYG-2 and its receptor, SYG-1. Cell. (2004) 116:869–81. doi: 10.1016/S0092-8674(04)00251-X
24. Özkan, E, Chia, PH, Wang, RR, Goriatcheva, N, Borek, D, Otwinowski, Z, et al. Extracellular architecture of the SYG-1/SYG-2 adhesion complex instructs synaptogenesis. Cell. (2014) 156:482–94. doi: 10.1016/j.cell.2014.01.004
25. Machado, MC, Octacilio-Silva, S, Costa, MS, and Ramos, RG. Rst transcriptional activity influences kirre mRNA concentration in the Drosophila pupal retina during the final steps of ommatidial patterning. PLoS One. (2011) 6:e22536. doi: 10.1371/journal.pone.0022536
26. Chia, PH, Chen, B, Li, P, Rosen, MK, and Shen, K. Local F-actin network links synapse formation and axon branching. Cell. (2014) 156:208–20. doi: 10.1016/j.cell.2013.12.009
27. Bao, S, Fischbach, KF, Corbin, V, and Cagan, RL. Preferential adhesion maintains separation of ommatidia in the Drosophila eye. Dev Biol. (2010) 344:948–56. doi: 10.1016/j.ydbio.2010.06.013
28. Helmstädter, M, Lüthy, K, Gödel, M, Simons, M, Ashish,, Nihalani, D, et al. Functional study of mammalian Neph proteins in Drosophila melanogaster. PLoS One. (2012) 7:e40300. doi: 10.1371/journal.pone.0040300
29. Haralalka, S, Shelton, C, Cartwright, HN, Guo, F, Trimble, R, Kumar, RP, et al. Live imaging provides new insights on dynamic F-actin filopodia and differential endocytosis during myoblast fusion in Drosophila. PLoS One. (2014) 9:e114126. doi: 10.1371/journal.pone.0114126
30. Helmstädter, M, Höhne, M, and Huber, TB. A brief overview on IRM function across evolution. J Neurogenet. (2014) 28:264–9. doi: 10.3109/01677063.2014.918976
31. Bhalla, K, Luo, Y, Buchan, T, Beachem, MA, Guzauskas, GF, Ladd, S, et al. Alterations in CDH15 and KIRREL3 in patients with mild to severe intellectual disability. Am J Hum Genet. (2008) 83:703–13. doi: 10.1016/j.ajhg.2008.10.020
32. Kotian, N, Troike, KM, Curran, KN, Lathia, JD, and McDonald, JA. A drosophila RNAi screen reveals conserved glioblastoma-related adhesion genes that regulate collective cell migration. G3. (2021) 12:356. doi: 10.1093/g3journal/jkab356
33. Yi, GZ, Zhang, HY, Que, TS, Qu, SQ, Li, ZY, Qi, ST, et al. Identification of the clinical and genetic characteristics of gliomas with gene fusions by integrated genomic and transcriptomic analysis. Eur J Med Res. (2025) 30:49. doi: 10.1186/s40001-025-02306-y
34. Serra, G, Memo, L, Antona, V, Corsello, G, Favero, V, Lago, P, et al. Jacobsen syndrome and neonatal bleeding: report on two unrelated patients. Ital J Pediatr. (2021) 47:147. doi: 10.1186/s13052-021-01108-2
35. Linares Chávez, EP, Toral López, J, Valdés Miranda, JM, González Huerta, LM, Perez Cabrera, A, del Refugio Rivera Vega, M, et al. Jacobsen syndrome: surgical complications due to unsuspected diagnosis, the importance of molecular studies in patients with Craniosynostosis. Mol Syndromol. (2016) 6:229–35. doi: 10.1159/000442477
36. Anzick, S, Thurm, A, Burkett, S, Velez, D, Cho, E, Chlebowski, C, et al. Chromoanasynthesis as a cause of Jacobsen syndrome. Am J Med Genet A. (2020) 182:2533–9. doi: 10.1002/ajmg.a.61824
37. Baccarin, M, Picinelli, C, Tomaiuolo, P, Castronovo, P, Costa, A, Verdecchia, M, et al. Appropriateness of array-CGH in the ADHD clinics: a comparative study. Genes Brain Behav. (2020) 19:e12651. doi: 10.1111/gbb.12651
38. Collins, RL, Brand, H, Karczewski, KJ, Zhao, X, Alföldi, J, Francioli, LC, et al. A structural variation reference for medical and population genetics. Nature. (2020) 581:444–51. doi: 10.1038/s41586-020-2287-8
39. Perez, G, Barber, GP, Benet-Pages, A, Casper, J, Clawson, H, Diekhans, M, et al. The UCSC genome browser database: 2025 update. Nucleic Acids Res. (2025) 53:D1243–d1249. doi: 10.1093/nar/gkae974
40. Belmadani, M, Jacobson, M, Holmes, N, Phan, M, Nguyen, T, Pavlidis, P, et al. VariCarta: a comprehensive database of harmonized genomic variants found in autism Spectrum disorder sequencing studies. Autism Res. (2019) 12:1728–36. doi: 10.1002/aur.2236
41. Landrum, MJ, Lee, JM, Benson, M, Brown, GR, Chao, C, Chitipiralla, S, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. (2018) 46:D1062–d1067. doi: 10.1093/nar/gkx1153
42. Makrilia, N, Kollias, A, Manolopoulos, L, and Syrigos, K. Cell adhesion molecules: role and clinical significance in cancer. Cancer Investig. (2009) 27:1023–37. doi: 10.3109/07357900902769749
43. Sellin, L, Huber, TB, Gerke, P, Quack, I, Pavenstädt, H, and Walz, G. NEPH1 defines a novel family of podocin interacting proteins. FASEB J 17:115–7. doi: 10.1096/fj.02-0242fje
44. Dyer, SC, Austine-Orimoloye, O, Azov, AG, Barba, M, Barnes, I, Barrera-Enriquez, VP, et al. Ensembl 2025. Nucleic Acids Res. (2025) 53:D948–d957. doi: 10.1093/nar/gkae1071
45. Traenkner, D, et al. Modular splicing is linked to evolution in the synapse-specificity molecule Kirrel3. eNeuro. (2023) 10:ENEURO.0253-0223.2023. doi: 10.1523/ENEURO.0253-23.2023
46. Siletti, K, Hodge, R, Mossi Albiach, A, Lee, KW, Ding, SL, Hu, L, et al. Transcriptomic diversity of cell types across the adult human brain. Science. (2023) 382:eadd7046. doi: 10.1126/science.add7046
47. Tamura, S, Morikawa, Y, Hisaoka, T, Ueno, H, Kitamura, T, and Senba, E. Expression of mKirre, a mammalian homolog of Drosophila kirre, in the developing and adult mouse brain. Neuroscience. (2005) 133:615–24. doi: 10.1016/j.neuroscience.2005.03.030
48. Serizawa, S, Miyamichi, K, Takeuchi, H, Yamagishi, Y, Suzuki, M, and Sakano, H. A neuronal identity code for the odorant receptor-specific and activity-dependent axon sorting. Cell. (2006) 127:1057–69. doi: 10.1016/j.cell.2006.10.031
49. Yao, Z, van Velthoven, CTJ, Nguyen, TN, Goldy, J, Sedeno-Cortes, AE, Baftizadeh, F, et al. A taxonomy of transcriptomic cell types across the isocortex and hippocampal formation. Cell. (2021) 184:3222–3241.e26. doi: 10.1016/j.cell.2021.04.021
50. Wu, C, Jin, X, Tsueng, G, Afrasiabi, C, and Su, AI. BioGPS: building your own mash-up of gene annotations and expression profiles. Nucleic Acids Res. (2016) 44:D313–6. doi: 10.1093/nar/gkv1104
51. Karlsson, M, Zhang, C, Méar, L, Zhong, W, Digre, A, Katona, B, et al. A single-cell type transcriptomics map of human tissues. Sci Adv. (2021) 7:169. doi: 10.1126/sciadv.abh2169
52. Martin, EA, Muralidhar, S, Wang, Z, Cervantes, DC, Basu, R, Taylor, MR, et al. Correction: the intellectual disability gene Kirrel3 regulates target-specific mossy fiber synapse development in the hippocampus. eLife. (2016) 5:18706. doi: 10.7554/eLife.18706
53. Breschi, A, Gingeras, TR, and Guigó, R. Comparative transcriptomics in human and mouse. Nat Rev Genet. (2017) 18:425–40. doi: 10.1038/nrg.2017.19
54. Monaco, G, van Dam, S, Casal Novo Ribeiro, JL, Larbi, A, and de Magalhães, JP. A comparison of human and mouse gene co-expression networks reveals conservation and divergence at the tissue, pathway and disease levels. BMC Evol Biol. (2015) 15:259. doi: 10.1186/s12862-015-0534-7
55. Su, AI, Cooke, MP, Ching, KA, Hakak, Y, Walker, JR, Wiltshire, T, et al. Large-scale analysis of the human and mouse transcriptomes. Proc Natl Acad Sci USA. (2002) 99:4465–70. doi: 10.1073/pnas.012025199
56. Hisaoka, T, Komori, T, Kitamura, T, and Morikawa, Y. Abnormal behaviours relevant to neurodevelopmental disorders in Kirrel3-knockout mice. Sci Rep. (2018) 8:1408. doi: 10.1038/s41598-018-19844-7
57. Völker, LA, Maar, BA, Pulido Guevara, BA, Bilkei-Gorzo, A, Zimmer, A, Brönneke, H, et al. Neph2/Kirrel3 regulates sensory input, motor coordination, and home-cage activity in rodents. Genes Brain Behav. (2018) 17:e12516. doi: 10.1111/gbb.12516
58. Choi, SY, Han, K, Cutforth, T, Chung, W, Park, H, Lee, D, et al. Mice lacking the synaptic adhesion molecule Neph2/Kirrel3 display moderate hyperactivity and defective novel object preference. Front Cell Neurosci. (2015) 9:283. doi: 10.3389/fncel.2015.00283
59. Tuñon-Ortiz, A, Tränkner, D, Peterson, CM, Shennib, O, Ye, F, Shi, J, et al. Inhibitory neurons marked by the connectivity molecule Kirrel3 regulate memory precision. J Neurosci. (2025):e1760242025. doi: 10.1523/JNEUROSCI.1760-24.2025
60. Prince, JE, Brignall, AC, Cutforth, T, Shen, K, and Cloutier, JF. Kirrel3 is required for the coalescence of vomeronasal sensory neuron axons into glomeruli and for male-male aggression. Development. (2013) 140:2398–408. doi: 10.1242/dev.087262
61. Völker, LA, Petry, M, Abdelsabour-Khalaf, M, Schweizer, H, Yusuf, F, Busch, T, et al. Comparative analysis of Neph gene expression in mouse and chicken development. Histochem Cell Biol. (2012) 137:355–66. doi: 10.1007/s00418-011-0903-2
62. Danesh, AA, Howery, S, Aazh, H, Kaf, W, and Eshraghi, AA. Hyperacusis in autism Spectrum disorders. Audiol Res. (2021) 11:547–56. doi: 10.3390/audiolres11040049
63. Amaral, DG, Schumann, CM, and Nordahl, CW. Neuroanatomy of autism. Trends Neurosci. (2008) 31:137–45. doi: 10.1016/j.tins.2007.12.005
64. Hazen, EP, Stornelli, JL, O'Rourke, JA, Koesterer, K, and McDougle, CJ. Sensory symptoms in autism spectrum disorders. Harv Rev Psychiatry. (2014) 22:112–24. doi: 10.1097/01.HRP.0000445143.08773.58
65. Roh, JD, Choi, SY, Cho, YS, Choi, TY, Park, JS, Cutforth, T, et al. Increased excitatory synaptic transmission of dentate granule neurons in mice lacking PSD-95-interacting adhesion molecule Neph2/Kirrel3 during the early postnatal period. Front Mol Neurosci. (2017) 10:81. doi: 10.3389/fnmol.2017.00081
66. Martin, EA, Woodruff, D, Rawson, RL, and Williams, ME. Examining hippocampal mossy fiber synapses by 3D electron microscopy in wildtype and Kirrel3 knockout mice. eNeuro. (2017) 4:88. doi: 10.1523/ENEURO.0088-17.2017
67. Bourgeron, T. From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat Rev Neurosci. (2015) 16:551–63. doi: 10.1038/nrn3992
68. Zhao, H, Mao, X, Zhu, C, Zou, X, Peng, F, Yang, W, et al. GABAergic system dysfunction in autism spectrum disorders. Front Cell Dev Biol. (2021) 9:781327. doi: 10.3389/fcell.2021.781327
69. Morikawa, Y, Komori, T, Hisaoka, T, Ueno, H, Kitamura, T, and Senba, E. Expression of mKirre in the developing sensory pathways: its close apposition to nephrin-expressing cells. Neuroscience. (2007) 150:880–6. doi: 10.1016/j.neuroscience.2007.10.013
70. Imai, T, and Sakano, H. Axon-axon interactions in neuronal circuit assembly: lessons from olfactory map formation. Eur J Neurosci. (2011) 34:1647–54. doi: 10.1111/j.1460-9568.2011.07817.x
71. Wang, J, Vaddadi, N, Pak, JS, Park, Y, Quilez, S, Roman, CA, et al. Molecular and structural basis of olfactory sensory neuron axon coalescence by Kirrel receptors. Cell Rep. (2021) 37:109940. doi: 10.1016/j.celrep.2021.109940
72. Brusés, JL. Identification of gene transcripts expressed by postsynaptic neurons during synapse formation encoding cell surface proteins with presumptive synaptogenic activity. Synapse. (2010) 64:47–60. doi: 10.1002/syn.20702
73. Grossfeld, PD, Mattina, T, Lai, Z, Favier, R, Jones, KL, Cotter, F, et al. The 11q terminal deletion disorder: a prospective study of 110 cases. Am J Med Genet A. (2004) 129a:51–61. doi: 10.1002/ajmg.a.30090
74. Karczewski, KJ, Francioli, LC, Tiao, G, Cummings, BB, Alföldi, J, Wang, Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. (2020) 581:434–43. doi: 10.1038/s41586-020-2308-7
75. Zhou, X, Feliciano, P, Shu, C, Wang, T, Astrovskaya, I, Hall, JB, et al. Integrating de novo and inherited variants in 42,607 autism cases identifies mutations in new moderate-risk genes. Nat Genet. (2022) 54:1305–19. doi: 10.1038/s41588-022-01148-2
76. Trost, B, Thiruvahindrapuram, B, Chan, AJS, Engchuan, W, Higginbotham, EJ, Howe, JL, et al. Genomic architecture of autism from comprehensive whole-genome sequence annotation. Cell. (2022) 185:4409–4427.e18. doi: 10.1016/j.cell.2022.10.009
77. Li, J, Wang, L, Guo, H, Shi, L, Zhang, K, Tang, M, et al. Targeted sequencing and functional analysis reveal brain-size-related genes and their networks in autism spectrum disorders. Mol Psychiatry. (2017) 22:1282–90. doi: 10.1038/mp.2017.140
78. De Rubeis, S, He, X, Goldberg, AP, Poultney, CS, Samocha, K, Cicek, AE, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. (2014) 515:209–15. doi: 10.1038/nature13772
79. Hu, C, Wang, Y, Li, C, Mei, L, Zhou, B, Li, D, et al. Targeted sequencing and clinical strategies in children with autism spectrum disorder: a cohort study. Front Genet. (2023) 14:1083779. doi: 10.3389/fgene.2023.1083779
80. Stessman, HA, Xiong, B, Coe, BP, Wang, T, Hoekzema, K, Fenckova, M, et al. Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat Genet. (2017) 49:515–26. doi: 10.1038/ng.3792
81. Guo, H, Wang, T, Wu, H, Long, M, Coe, BP, Li, H, et al. Inherited and multiple de novo mutations in autism/developmental delay risk genes suggest a multifactorial model. Mol Autism. (2018) 9:64. doi: 10.1186/s13229-018-0247-z
82. Leblond, CS, Cliquet, F, Carton, C, Huguet, G, Mathieu, A, Kergrohen, T, et al. Both rare and common genetic variants contribute to autism in the Faroe Islands. NPJ Genom Med. (2019) 4:1. doi: 10.1038/s41525-018-0075-2
83. Ciaccio, C, Leonardi, E, Polli, R, Murgia, A, D'Arrigo, S, Granocchio, E, et al. A missense De Novo variant in the CASK-interactor KIRREL3 gene leading to neurodevelopmental disorder with mild cerebellar hypoplasia. Neuropediatrics. (2021) 52:484–8. doi: 10.1055/s-0041-1725964
84. Hildebrand, MS, Jackson, VE, Scerri, TS, van Reyk, O, Coleman, M, Braden, RO, et al. Severe childhood speech disorder: gene discovery highlights transcriptional dysregulation. Neurology. (2020) 94:e2148–67. doi: 10.1212/WNL.0000000000009441
85. Kircher, M, Witten, DM, Jain, P, O'Roak, BJ, Cooper, GM, and Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. (2014) 46:310–5. doi: 10.1038/ng.2892
86. Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature. (2017) 542:433–8. doi: 10.1038/nature21062
87. Mattina, T, Perrotta, CS, and Grossfeld, P. Jacobsen syndrome. Orphanet J Rare Dis. (2009) 4:9. doi: 10.1186/1750-1172-4-9
88. Conrad, S, Demurger, F, Moradkhani, K, Pichon, O, le Caignec, C, Pascal, C, et al. 11q24.2q24.3 microdeletion in two families presenting features of Jacobsen syndrome, without intellectual disability: role of FLI1, ETS1, and SENCR long noncoding RNA. Am J Med Genet A. (2019) 179:993–1000. doi: 10.1002/ajmg.a.61113
89. Talkowski, ME, Rosenfeld, JA, Blumenthal, I, Pillalamarri, V, Chiang, C, Heilbut, A, et al. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell. (2012) 149:525–37. doi: 10.1016/j.cell.2012.03.028
90. Andersen, RE, Alkuraya, IF, Ajeesh, A, Sakamoto, T, Mena, EL, Amr, SS, et al. Chromosomal structural rearrangements implicate long non-coding RNAs in rare germline disorders. Hum Genet. (2024) 143:921–38. doi: 10.1007/s00439-024-02693-y
91. Guo, H, Duyzend, MH, Coe, BP, Baker, C, Hoekzema, K, Gerdts, J, et al. Genome sequencing identifies multiple deleterious variants in autism patients with more severe phenotypes. Genet Med. (2019) 21:1611–1620. doi: 10.1038/s41436-018-0380-2
Keywords: gene disease association, genotype–phenotype correlation, clinical variant, neurodevelopmental disorders, autism, intellectual disability, cell adhesion molecule, Kirrel3
Citation: Shennib O, Raines O, Karamian AS and Williams ME (2025) Evaluation of the synapse adhesion molecule Kirrel3 in neurological disease. Front. Neurol. 16:1662931. doi: 10.3389/fneur.2025.1662931
Edited by:
Minerva Carrasquillo, Mayo Clinic Florida, United StatesReviewed by:
Ramachandran Prakasam, Washington University in St. Louis, United StatesLe Wang, University of California, San Diego, United States
Copyright © 2025 Shennib, Raines, Karamian and Williams. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Omar Shennib, b21hci5zaGVubmliQG5ldXJvLnV0YWguZWR1; Megan E. Williams, bWVnYW4ud2lsbGlhbXNAbmV1cm8udXRhaC5lZHU=