 Mengyao Guo
Mengyao Guo Linyuan Qin
Linyuan Qin Hanlin Cai
Hanlin Cai Ruihan Wang
Ruihan Wang Caimei Luo
Caimei Luo Feng Yang
Feng Yang Shiyu Feng
Shiyu Feng Hui Gao
Hui Gao Qin Chen*
Qin Chen*- Department of Neurology, West China Hospital of Sichuan University, Chengdu, China
Familial frontotemporal dementia (FTD) is a genetically heterogeneous disease with various clinical manifestations, making it difficult to diagnose. There are three main gene mutations in familial FTD: repeat expansion in chromosome 9 open reading frame 72 (C9orf72), microtubule-associated protein tau (MAPT), and progranulin (GRN). These mutations can produce corresponding changes in fluid biomarkers years before symptoms appear. Therefore, biomarkers play a vital role in the diagnosis and treatment of familial FTD. In this review, we highlight fluid biomarkers in the blood and cerebrospinal fluid (CSF) that contribute to the clinical diagnosis of familial FTD, the study of disease pathophysiological mechanisms, and possibly be used as outcome endpoints in future clinical trials.
1 Introduction
Frontotemporal dementia (FTD) is a common form of early-onset dementia that predominantly impacts the frontal and temporal lobes, exhibiting diverse clinical and pathological characteristics (1, 2). It is characterized by significant personality, behavioral changes, and cognitive impairment (3). Approximately 10–30% of FTD are hereditary, exhibiting a distinct autosomal dominant inheritance pattern (4). An autosomal dominant inheritance pattern has been reported in 10–25% of families with FTD (5, 6). For clarity, we define familial FTD as cases with an identifiable autosomal dominant mutation. In contrast, sporadic FTD refers to phenotypically similar but genetically unconfirmed cases without a clear family history. The most common genetic mutations that cause familial FTD include repeat expansions in chromosome 9 open reading frame 72 (C9orf72), microtubule-associated protein tau (MAPT), and progranulin (GRN) (7). Less common genetic reasons include mutations in TBK1, TARDBP, VCP, FUS, CHMP2B, SQSTM1, and UBQLN2 (8). The pathological proteins generated by different genes display considerable heterogeneity, and the clinical manifestations arising from identical gene mutations and harmful protein deposits vary significantly depending on their deposition locations, posing a considerable challenge for clinical diagnosis (4, 9).
C9orf72 is the most common genetic cause of familial FTD (10). FTD associated with C9orf72 was caused by the amplification of the GGGGCC hexanucleotide repeat in the non-coding region of the gene. The length of this pathogenic repeat sequence may vary from 30 to several 1,000, whereas healthy individuals often possess fewer than 30 repetitions (11). The repeat sequences in C9orf72 can be transcribed into abnormal RNA transcripts. The RNA transcripts can be subsequently translated into dipeptide repeat proteins (DPRs), such as poly (GA), poly(GR), poly(PR), poly(PA), and poly(GP), which have toxic effects on neurons (12). C9orf72-associated FTD is predominantly linked to TDP-43 type B pathology, characterized by cytoplasmic inclusions of TDP-43 protein in neurons and glial cells, especially in the frontal and temporal lobes (13).
The gene encoding the progranulin (GRN) was also found on chromosome 17 (14). This mutation produces an aberrantly shortened progranulin mRNA transcript, resulting in diminished quantities of full-length functional progranulin proteins. The haploinsufficiency of progranulin disrupts normal lysosomal and neuronal functioning, thereby contributing to the pathophysiology of FTD-TDP (15).
The MAPT gene is located on chromosome 17q21.3, contains 16 exons, spans approximately 150 kb, encodes tau protein, and significantly influences neuronal integrity. Mutations in MAPT result in abnormal aggregation of tau proteins, ultimately leading to degeneration of glutamatergic neurons (16). The most prevalent MAPT mutations were P301L, V337M, R406W, and N27. The P301 mutation decreases tau affinity for microtubules while enhancing its aggregation and phosphorylation, thereby increasing the pathological accumulation of tau protein and resulting in neurodegeneration (17, 18).
Although the clinical manifestations and pathological characteristics can be linked, their association is typically limited (19). Therefore, sensitive biomarkers for familial FTD are necessary due to the heterogeneity of the disorder. Fluid biomarkers are molecules or chemicals present in physiological fluids that can fluctuate and indicate the presence of a disease (Table 1).
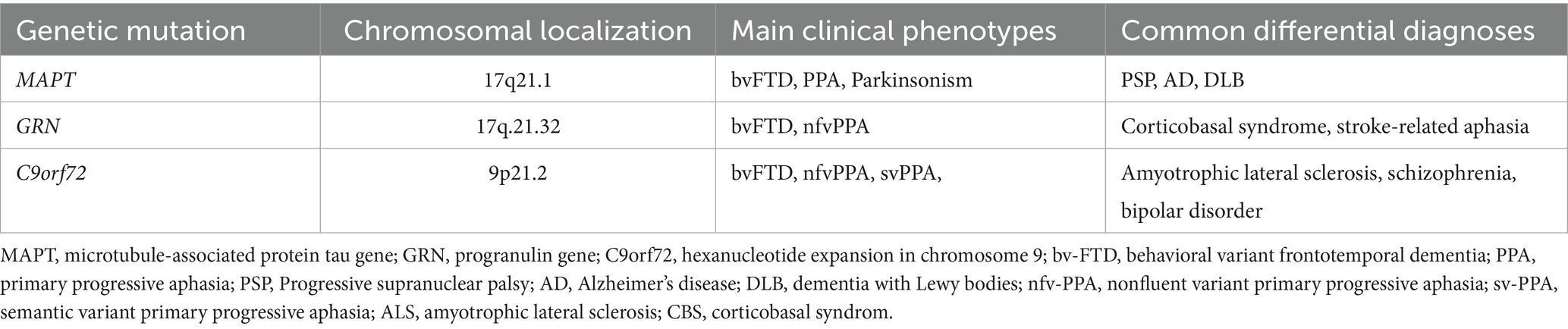
Table 1. Clinical phenotypes and differential diagnoses of familial FTD subtypes.
Research has shown that biomarkers in the blood and cerebrospinal fluid (CSF) have potential for investigating FTD. However, sporadic FTD accounts for more than 70% of all clinical cases, and its complicated etiology includes environmental factors, somatic mutations, genetic mosaicism, and challenging biomarker research (20). In contrast, familial FTD offers a more genetically defined framework, facilitating mechanistic investigation and reliable biomarker confirmation. Additionally, developing findings indicate that over 60% of sporadic FTD individuals exhibit overlapping endosomal-lysosomal biomarker profiles with familial subtypes, suggesting shared downstream pathogenic pathways (21). Thus, the familial FTD biomarker framework also serves as a potential reference for the classification, stratification, and therapeutic targeting of sporadic FTD (22).
Biomarkers can be classified into several functional categories. Diagnostic biomarkers help confirm the presence of disease, while predictive biomarkers identify individuals at risk of developing symptoms. Prognostic biomarkers provide information on disease progression, and monitoring biomarkers track treatment response or disease severity over time. Different biomarkers may serve distinct roles depending on genetic subtype, disease stage, and clinical presentation.
Overall, identifying fluid biomarkers of familial FTD is crucial for tracking disease development, anticipating treatment outcomes, and investigating potential pathophysiological alterations associated with the condition. In this review, we describe the most recent developments in fluid biomarkers associated with familial FTD.
2 Methods
We conducted an electronic search of the MEDLINE, PubMed, and Embase databases using a combination of several keywords. The following search terms were used as keywords to identify all relevant studies: (“FTD” OR “FTD” OR “frontotemporal dementia” OR “lobar degeneration” OR “frontotemporal lobar degeneration”) AND (“microtubule associated protein tau” OR “MAPT” OR “Progranulin” OR “GRN” OR “Progranulin” OR “GRN”) AND (“biomarker”). Related studies that contained these keywords from the references were also searched for potentially qualified studies. Studies were excluded if they were (1) reviews without original data; (2) unrelated to familial FTD; or (3) not written in English. We also excluded studies that did not differentiate between genetic subtypes of FTD.
3 Discussion
3.1 Biomarkers in the blood
Current research on familial FTD in the blood mainly involves three environments: serum, plasma, and small extracellular vesicles (sEVs). We reviewed and summarized these biomarkers in the blood (Table 2).
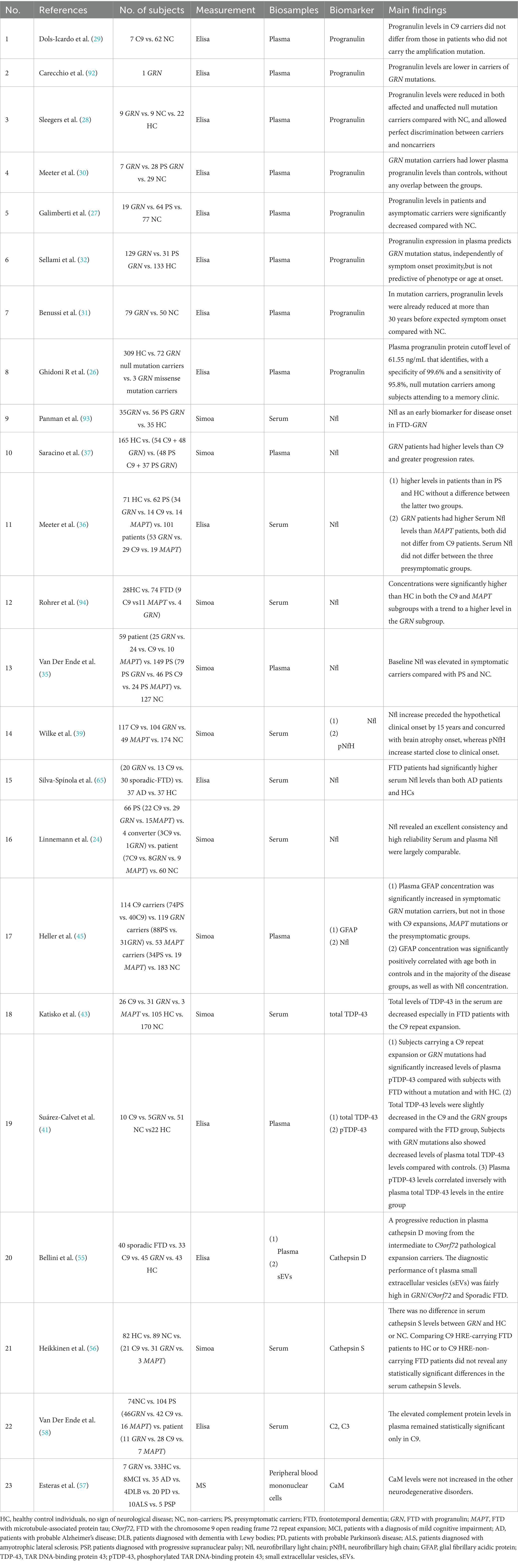
Table 2. Biomarkers in the blood.
3.1.1 Progranulin
Progranulin serves as a significant biomarker for detecting GRN mutations in FTD (23). Many studies have shown that a considerable reduction in progranulin levels is typical of individuals with GRN mutations and is not linked to other forms of familial FTD (14, 15, 24–27). Progranulin levels assessed by enzyme-linked immunosorbent assay (ELISA) accurately distinguished between GRN mutation carriers and healthy individuals, demonstrating a specificity of 99.6% and a sensitivity of 95.8% (28). Dols-Icardo et al. further demonstrated that progranulin levels remained unaffected by the C9orf72 mutation, indicating that progranulin serves as a specific biomarker for GRN-related FTD (29). Meeter et al. found that progranulin levels in the blood of a considerable cohort of presymptomatic GRN mutation carriers were significantly lower than those of age-matched healthy controls. This suggests that progranulin can be employed not only in symptomatic individuals but also in identifying mutation carriers before symptom manifestation, thus functioning as an effective early diagnostic tool (30). Benussi et al. also found that progranulin, as a “status” biomarker for GRN mutations, may serve as an early warning indicator prior to symptom manifestation, although exhibits minimal correlation with the rate of illness development (31). Sellami et al. suggested that this detection method is economically viable and appropriate for screening, allowing for patient selection without the need for costly genetic testing (32). In summary, measurement of progranulin in the blood can identify carriers of GRN mutations and can be used as an early diagnosis and cost-effective screening method. However, the presence of progranulin indicates only a possible gene mutation and has no correlation with the degree of neurodegeneration in the brain.
3.1.2 Neurofilament light chain protein
The neurofilament light chain (Nfl) is a subunit of neurofilaments (Nfs), which are cylindrical proteins located in the cytoplasm of neurons (33). Nfl is found in dendrites, neuronal bodies, and axons, contributing to the structural stability of neurons (34). Although Nfl is not a disease-specific biomarker, its elevated levels have been consistently observed in a range of neurodegenerative disorders, including FTD. Van Der Ende et al. found that serum Nfl levels were significantly elevated in symptomatic carriers relative to presymptomatic carriers and non-carriers, indicating clinical progression and highlights the potential value of serum Nfl as a candidate selection tool for disease progression (35). In GRN-associated FTD, serum Nfl levels are elevated two–three times 2–4 years prior to the onset of clinical symptoms, enabling dynamic monitoring of neuronal axonal damage and disease progression through regular blood tests (36). Saracino et al. further discovered that the concentration of Nfl in individuals with the GRN mutation was markedly elevated compared to those with the symptomatic C9orf72 mutation, indicating that Nfl may represent varying rates of pathological disease progression (37). Moreover, while Nfl levels are elevated in MAPT mutation carriers compared to healthy controls, the magnitude of elevation is typically lower than that observed in GRN and C9orf72 carriers, which is consistent with the relatively slower disease progression associated with MAPT-related pathology (38). Wilke et al. indicated that Nfl levels progressively increased over the 15 years preceding symptom manifestation, implying its potential as an early prognostic marker for familial FTD development (39). Linnemann et al. observed that the robust constancy of Nfl across multicenter investigations renders it a suitable biomarker for clinical trials, particularly for assessing disease progression and treatment response (24). Overall, Nfl levels in the blood indicate the underlying clinical burden of familial FTD and demonstrate the potential for differentiating genetic subtypes, particularly with high sensitivity and predictive value in GRN mutation carriers. Its levels progressively elevate years prior to the onset of symptoms and thus could be used to predict early diagnosis.
3.1.3 TAR DNA binding protein 43
TAR DNA-binding protein 43 (TDP-43) is an RNA-binding protein that induces aberrant protein aggregation in the cytoplasm during pathological conditions (40). The TDP-43 protein assay typically has two forms: (1) total TDP-43 levels and (2) phosphorylated TDP-43 (pTDP-43), which exhibit distinct alterations across several genetic subtypes of FTD. Suarez-Calvet et al. found that pTDP-43 levels increased in C9orf72 and GRN mutation carriers, whereas total TDP-43 levels decreased, indicating an inverse relationship between pTDP-43 and total TDP-43 (41). Changes in TDP-43 levels may indicate abnormalities in protein metabolism and disease mechanisms, particularly in processes related to protein aggregation and neurodegeneration. Under pathological conditions, TDP-43 undergoes post-translational modifications, primarily phosphorylation, resulting in cytoplasmic mislocalization and aggregation. Unlike total TDP-43, which includes both functional and diseased forms, pTDP-43 is disease-specific and serves as a hallmark of TDP-43 proteinopathies (42). Katisko et al. further observed that total TDP-43 levels were markedly diminished in individuals with the C9orf72 mutation, indicating that TDP-43 may possess diagnostic significance in C9orf72-associated FTD (43). Thus, TDP-43 and its phosphorylated form may function as biomarkers for the future diagnosis of familial FTD, especially in individuals carrying C9orf72 and GRN mutations.
3.1.4 Glial fibrillary acidic protein
Glial fibrillary acidic protein (GFAP) is considered a marker of astrocyte activation and may significantly contribute to the pathophysiology of GRN-associated FTD (44–46). GRN mutations result in diminished quantities of functional progranulin protein, thereby compromising lysosomal function and exacerbating neuro-inflammation. This failure in astrocytes induces a reactive state marked by increased cytokine release and modified homeostatic support, thereby facilitating disease development (47).
Activated astrocytes release inflammatory mediators that compromise neuronal connections and disrupt lysosomal function, hence exacerbating neurodegeneration (48, 49). Heller et al. showed that GFAP levels are significantly elevated in GRN mutations, particularly prior to symptom onset, implying that GFAP could serve as a valuable early biomarker for identifying the risk in these patients (45). More importantly, GFAP levels were found to be positively correlated with neurofilament light chain (Nfl) levels, which indicates the potential for employing both as dynamic surveillance indicators, which could improve the accuracy of disease classification (50) To summary, the GRN mutation is strongly associated with an intensified inflammatory response, and the increase in GFAP may indicate pathogenic activation of astrocytes in the early stages of the disease. Consequently, GFAP may serve as a possible biomarker for the early detection of GRN-associated FTD, and could elucidate the neuroinflammatory mechanisms underlying the disease. Nevertheless, existing research on GFAP expression in other genetic subgroups of FTD is limited. This gap highlights the need for further research.
3.1.5 Lysosomal proteases
Recent investigations on lysosomal proteins in familial FTD have predominantly concentrated on cathepsin D and S. Cathepsin D, an aspartic protease found in lysosomes that participates in proteolytic metabolism and regulates the digestion of hormones and antigens. It has been shown to be correlated with neurodegenerative alterations (51, 52). Animal models carrying GRN and C9orf72 mutations have shown a marked reduction in plasma Cathepsin D activity, suggesting a role in disease pathogenesis (53, 54). Consistent with these findings, human studies have reported a notable decrease in Cathepsin D levels in individuals with GRN and C9orf72 mutations along with a progressive decline in cathepsin D plasma levels from C9orf72 intermediate expansion carriers to C9orf72 pathological expansion carriers. Pathogenic expansions are characterized by hexanucleotide repeats exceeding 30 G4C2 repetitions, whereas intermediate expansions are defined as consisting of 12–30 hexanucleotide repeats. This suggests a dose-dependent influence of C9orf72 expansion on cathepsin D plasma levels (55). The decrease in Cathepsin D did not occur after the onset of symptoms but was already apparent during the asymptomatic phase. Thus, cathepsin D may serve as a presymptomatic biomarker. Furthermore, the diagnostic efficacy of cathepsin D concentration in extracellular vesicles as a criterion for distinguishing FTD patients from healthy controls was notably high (AUC = 0.85), exhibiting a sensitivity of 75.4% and a specificity of 76.7%. This indicates that extracellular vesicle-associated Cathepsin D may serve as a diagnostic biomarker, especially for identifying patients with pathogenic GRN or C9orf72 mutations.
Although Cathepsin D is an aspartic protease localized within lysosomes, Cathepsin S is a cysteine protease that is predominantly expressed by microglia and is involved in antigen presentation and immune regulation. In contrast to Cathepsin D, Heikkinen et al. found that serum Cathepsin S levels were not significantly different between patients with familial FTD and healthy controls. No significant differences were observed among FTD subtypes, including GRN and C9orf72 mutant carriers, MAPT mutation carriers, or sporadic cases, indicating that Cathepsin S is not a reliable biomarker for differentiating clinical, genetic, or pathological groupings (56).
Overall, Cathepsin D showed a notable decrease in GRN and C9orf72 carriers associated with disease progression and elevated copy number in C9orf72, thus signifying mutant gene carriers and pathological conditions, thereby functioning as a potential biomarker for screening and early detection of familial FTD. Further investigation of additional lysosomal protein types may uncover novel biologically significant biomarkers of familial FTD.
3.1.6 Other biomarkers in the blood
Calmodulin (CaM) in peripheral cells used as a potential biomarker to investigate the association between familial FTD and other degenerative disorders. Elevated CaM levels have been specifically observed in peripheral blood mononuclear cells in patients with AD but not in those with other neurodegenerative disorders. Consequently, CaM could aid in differential diagnosis in the differential diagnosis of familial FTD, providing complementary value to established core AD biomarkers, such as phosphorylated tau species, MTBR-tau isoforms, and Aβ42/40 ratio (57).
The complement system plays an important role in neuroinflammation and synaptic clearance and its activation may facilitate neurodegeneration. Van Der Ende et al. identified markedly increased concentrations of the complement proteins C2 and C3 in the plasma of C9orf72 mutant carriers, indicating a potential association between complement system overactivation and illness development (58). As these alterations manifest before symptom onset, testing for C2 and C3 may facilitate the early identification of illness risk in carriers of the C9orf72 mutation. Increased levels of complement proteins in various neurological illnesses suggest a generalized overexpression of the complement system rather than gene-or disease-specific upregulation (59–61). Thus, although complement proteins may assist in identifying a higher disease risk in C9orf72 carriers, their diagnostic specificity across other FTD genotypes remains unclear. Future research comparing complement levels in genetically related subtypes of FTD may further clarify potential gene-specific effects.
3.2 Biomarkers in the CSF
CSF biomarkers can accurately indicate disease progression with minimal interference. We reviewed and summarized the CSF biomarkers of familial FTD (Table 3).
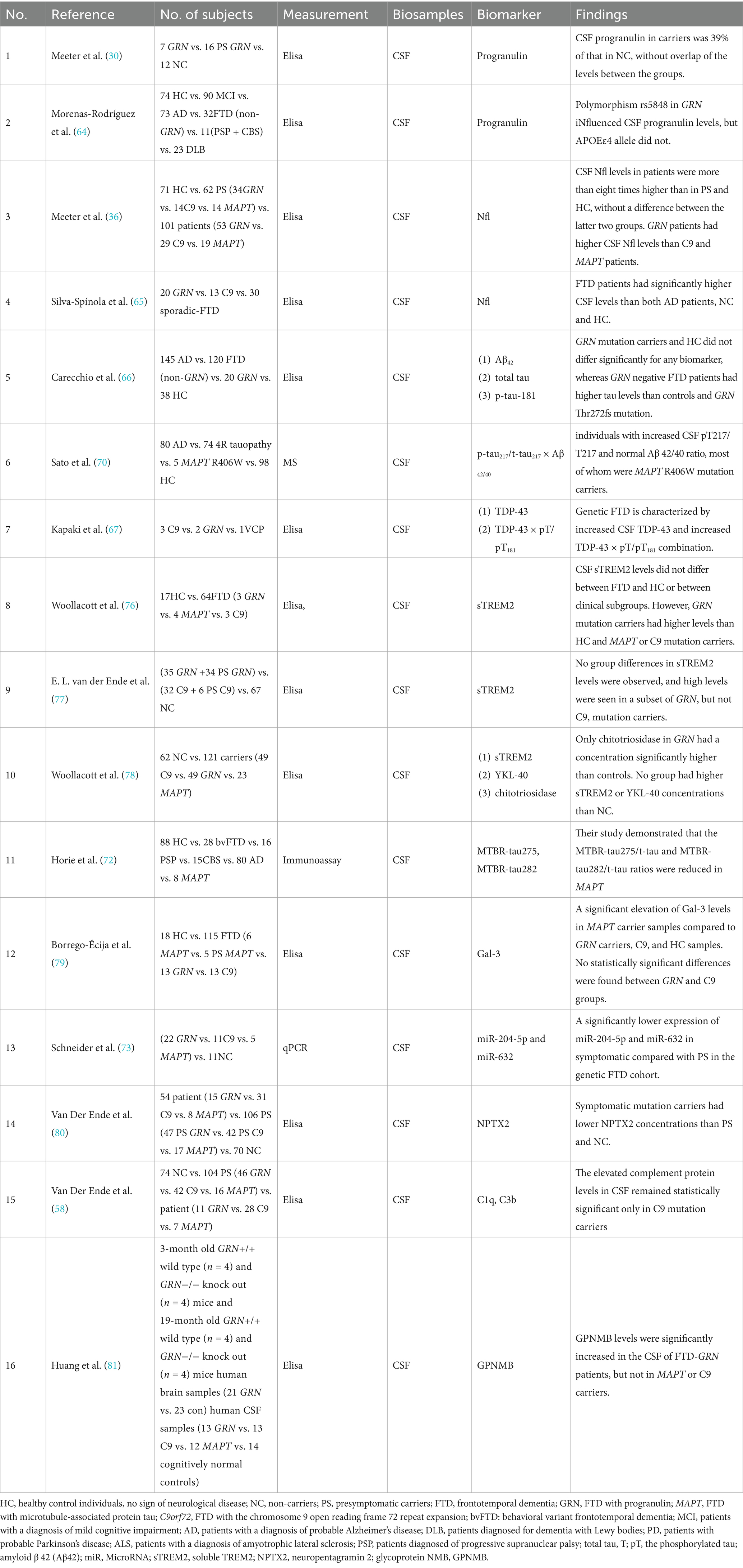
Table 3. Biomarkers in the CSF.
3.2.1 Progranulin
Progranulin has been examined in the CSF less extensively than in the blood, and the correlation between CSF and blood levels is relatively weak (62, 63). Unlike the blood test, which has a clear cut-off value, the evaluation of progranulin in CSF studies is uncertain. Research conducted by Meeter et al. indicated that individuals with GRN mutations exhibited markedly diminished progranulin levels, even prior to the onset of symptoms (30). Morenas-Rodríguez et al. found that CSF progranulin levels were strictly regulated and possessed no diagnostic utility except in cases of primary neurodegenerative dementia associated with GRN mutations (64). Thus, progranulin in CSF may serve as a specific biomarker for GRN mutations. However, precise cut-off value thresholds for potential clinical use necessitate further clarification.
3.2.2 Neurofilament light chain (Nfl)
Consistent with findings in blood, CSF Nfl levels also exhibit a progressive increase during the presymptomatic phase and correlate with brain atrophy and clinical decline (65). Importantly, CSF and serum Nfl levels are strongly correlated (r = 0.87, p < 0.001), supporting the use of less invasive serum testing for longitudinal monitoring. Unlike blood-based measurements, CSF Nfl may offer slightly higher sensitivity in distinguishing symptomatic from presymptomatic carriers, particularly in early-stage MAPT or GRN associated cases (36). Nfl studies in both blood and CSF have shown similar results, suggesting that although non-specific, Nfl could be used as a predictive biomarker of disease progression.
3.2.3 Amyloid and tau-related biomarkers
Amyloid beta (Aβ) and tau proteins are characteristic markers of Alzheimer’s (66, 67). Aβ PET imaging showed a low positivity rate in individuals diagnosed with FTD, particularly in cohorts with autopsy-confirmed diagnoses. Reimand et al. found that only 11.1% of patients with FTD showed Aβ PET positivity, primarily linked to concurrent AD rather than isolated FTD pathology (68). This supports the use of AD-associated biomarkers, including Aβ-PET or CSF Aβ42, as exclusionary tools in the context of FTD. Elevated levels of total tau (t-tau) and phosphorylated tau (p-tau), along with decreased Aβ42 in patients with AD, could be used as biomarkers to differentiate AD from FTD (69). The ratio of p-tau217/t-tau217 to Aβ 42/40 in CSF demonstrated efficacy as a composite biomarker to identify tau lesions in carriers of the MAPT R406W mutation, and could effectively distinguish them from cognitively normal individuals and those with other tauopathies (70). TDP-43 × p-tau/p-tau18 has also demonstrated diagnostic significance in familial FTD, particularly in association with the pathology of TDP-43. Kapaki et al. found that CSF TDP-43 levels, especially when analyzed alongside tau-based ratios (TDP-43 × t-tau/p-tau), were increased in genetic FTD cases with GRN and C9orf72 mutations, highlighting their diagnostic relevance for TDP-43-driven subtypes (67). The microtubule-binding region (MTBR) of tau represents the central area of tau aggregates within the brain along with truncated C-terminal tau fragments found in the CSF (71). Horie et al. demonstrated that the MTBR-tau275/t-tau and MTBR-tau282/t-tau ratios were significantly reduced in MAPT-associated FTD cases compared with cognitively normal controls and patients with AD (72). This reduction likely reflects distinct aggregation patterns and epitope accessibility of tau filaments in primary tauopathies versus AD, underscoring the diagnostic specificity of MTBR-based tau measurements. Given the substantial overlap in biomarker expression across neurodegenerative diseases and the genotypic heterogeneity inherent in FTD, diagnosis relies more on multimodal biomarker panels and personalized interpretations than on a single biomarker.
3.2.4 MicroRNA
MicroRNAs (miRNAs) within exosomes possess diagnostic potential for genetic FTD, and are gaining interest. Schneider et al. revealed a considerable reduction in the expression of miR-204-5p and miR-632 in symptomatic FTD, particularly among carriers of GRN mutations (73). In GRN-related FTD, downregulation of miR-204-5p and miR-632 leads to overexpression of the pro-apoptotic target HRK gene, which contributes to neuronal death in the frontal and temporal lobes (74, 75). However, because of the unique pathogenic mechanisms of different genotypes (such as C9orf72 and MAPT), the expression of the aforementioned miRNAs did not show significant changes; thus, detection efficacy was limited. These findings suggest that miR may be an early detection method for the diagnosis of familial FTD; however, further evidence is needed.
3.2.5 Soluble TREM2
Soluble TREM2 (sTREM2) serves as a biomarker for neuroinflammation and microglial activation. Woollacott et al. reported elevated sTREM2 levels in the CSF of symptomatic GRN mutation carriers, implying that sTREM2 may serve as a biomarker for neuronal damage in familial frontotemporal dementia (76). Conversely, Van der Ende et al. demonstrated no significant disparities in CSF sTREM2 levels between presymptomatic and symptomatic carriers of GRN or C9orf72 mutations and noncarriers (77). Owing to the limited and contradictory evidence, it is currently difficult to determine the potential importance of this biomarker.
3.2.6 Other biomarkers in the CSF
It was found that levels of chitotriosidase and Galectin-3 (Gal-3) were elevated in MAPT mutation carriers, potentially serving as markers of neuroinflammation and glial cell activation (78). Borrego-Écija et al. further showed that Gal-3 is significantly upregulated in MAPT-associated FTD, indicating its potential as a subtype-specific biomarker linked to neuroinflammation and glial cell activation (79). Van Der Ende et al. observed elevated levels of complement proteins in both CSF and plasma of genetic FTD cases through the GENFI study, further supporting the hypothesis that immune dysregulation is significant in disease pathology. These findings indicated that these proteins could function as biomarkers for MAPT-associated FTD subtypes. The observed heterogeneity among the various genetic forms of FTD, as noted by Van Der Ende et al., highlights the need for additional research to clarify the specific functions of these proteins in disease onset and progression.
Another potential biomarker is the decreased level of neuropentagramin 2 (NPTX2) in individuals with familial FTD, suggesting that NPTX2 may serve as a novel synaptic-derived biomarker of disease progression (80).
In animal experiments, glycoprotein non-metastatic melanoma protein B (GPNMB) levels were significantly elevated in the cerebrospinal fluid of FTD-GRN mice, whereas no such increase was observed in MAPT or C9orf72 mice. GPNMB may serve as a specific biomarker for GRN-associated FTD, facilitating monitoring of disease onset, progression, and response to treatment. Additionally, GPNMB expression was increased in brain tissue from human GRN-associated FTD samples, consistent with the results from GRN-deficient mouse models (81).
In C9orf72-associated FTD, five dipeptide repeats (DPR) are generated by this gene. However, only poly (GP) levels are quantifiable and may serve as diagnostic markers for patient screening, such as blood tests (82, 83). Poly (GP) levels may be used for early disease detection, stratification of mutation carriers, and evaluation of therapeutic efficacy in clinical trials.
Despite these promising findings, most candidate biomarkers remain at an exploratory stage. Extensive longitudinal and multicenter studies across diverse populations are required to confirm their diagnostic, prognostic, and therapeutic monitoring utility in familial FTD.
3.3 Other biomarkers
In addition to the blood and CSF, there is a paucity of investigations on other fluid biomarkers for familial FTD. In a single longitudinal cohort investigation, salivary lactoferrin, which serves as a biomarker to differentiate AD from FTD, exhibited over 87% sensitivity and 91% specificity. Saliva is readily accessible, and sampling is straightforward and noninvasive. However, the quality of a specimen is affected by various factors that may hinder its development (84).
3.4 Measurement technique
Tremendous breakthroughs have been made in the assessment of fluid biomarkers of familial FTD. The predominant markers under investigation are proteins, and the three most frequently employed methods are ELISA, single-molecule array (Simoa), and mass spectrometry (MS). Elisa, while widely used, exhibits notable limitations. Its sensitivity is constrained, particularly in detecting markers during the early stages of the disease or at low concentrations, which may remain undetected due to insufficient levels. Additionally, its dynamic range is restricted, potentially limiting its ability to capture subtle changes in biomarker expression (85–87). The Simoa technique, which is widely utilized today, is an ultrasensitive method for detecting protein biomarkers through single-molecule counting. The detection limit of this technology can achieve levels on the order of FML (fg/mL) (88). Simoa demonstrates the capability to identify extremely low concentrations of neural markers in comparison to Elisa, which is especially significant for the early diagnosis of familial FTD. Simoa instruments are costly, kits are relatively expensive, and their application scope is somewhat restricted. MS can be used to identify and measure biomarkers linked to familial frontotemporal dementia through analysis of proteins, metabolites, and peptides (89). This method is appropriate for the concurrent detection of multiple related biomarkers in familial FTD, identifying markers present in very low quantities, and facilitating the observation of minor changes in the early stages of the disease (90). However, MS imposes stringent criteria for sample extraction and pretreatment, resulting in a relatively complex operational process (91). Quantitative polymerase chain reaction (qPCR) has emerged as a complementary tool, particularly for the detection of nucleic acid–based biomarkers. The new techniques improve the sensitivity and specificity of the test, but they also require stricter procedures for processing specimens, which must be carried out in a licensed laboratory to ensure the accuracy of the test and its potential use in clinical diagnosis (Table 4).
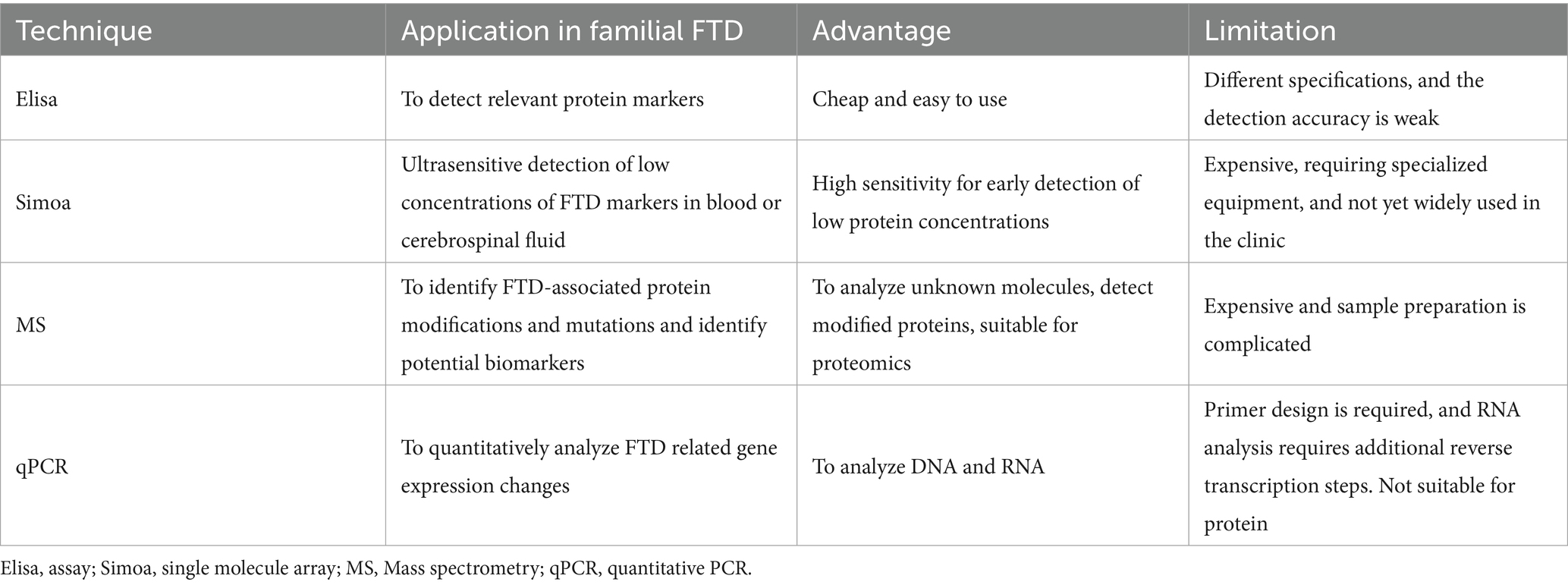
Table 4. Technique in familial FTD.
3.5 Challenges and limitations
Despite an increasing number of studies on fluid biomarkers of familial FTD, significant challenges and limitations remain. A significant number of the examined research are exploratory and utilize very small sample sizes, hence constraining statistical power and the generalizability. Diagnostic performance metrics are often unavailable, making it difficult to evaluate and compare biomarker efficacy in a clinically meaningful way.
The presence of significant clinicopathological variation among several genetic subgroups of familial FTD introduces further complexity. Biomarker expression may differ not only between genes but also among mutation carriers within the same family, and this heterogeneity is not generally acknowledged in the literature. Furthermore, the overlapping biomarker profiles among many neurodegenerative disorders, such as Alzheimer’s disease, amyotrophic lateral sclerosis, and atypical parkinsonian syndromes, may result in misinterpretation and diminish specificity.
A further limitation results from the prevalence of cross-sectional studies. In the absence of longitudinal data, evaluating the temporal dynamics of biomarker changes during the disease progression, particularly in the presymptomatic phase, is challenging. Variations in study design, inclusion criteria, comparison groups, and the utilization of diverse test platforms create further variability and confound inter-study comparisons.
Several biomarkers are not exclusive to familial FTD. Nfl and GFAP levels are elevated in various neurodegenerative diseases, including AD, to similar extents. Despite their variations across diseases, clinical cut-offs to differentiate between neurodegenerative diseases have not yet been established. The absence of specificity can result in diagnostic ambiguity, particularly in early or atypical cases.
In conclusion, fluid biomarkers present a significant potential for enhancing the diagnosis and monitoring of disease progression in familial FTD. However, their clinical application is currently constrained by challenges related to specificity, heterogeneity, sample availability, technological limitations, and economic viability. Addressing these limitations will require larger, multicenter, genotype-stratified longitudinal studies using harmonized protocols and standardized biomarker panels.
4 Conclusion
This review summarizes the recent fluid biomarker findings in familial frontotemporal dementia with respect to common genetic mutations. Several studies have been conducted in recent years on the fluid markers associated with familial FTD. In this review, we highlight fluid biomarkers in the blood and CSF that contribute to clinical diagnosis, disease progression surveillance, and pathophysiological mechanisms of familial FTD. Compared to expensive genetic tests, convenient and cost-effective fluid biomarkers are promising for use as screening tools and provide important information for disease prognosis. Future research should prioritize large-scale, multicenter longitudinal studies to validate candidate biomarkers across genetically stratified FTD cohorts. Moreover, integrating fluid biomarkers with advanced neuroimaging and multi-omics profiling could enhance diagnostic precision and therapeutic monitoring. The development of standardized, cost-effective, and scalable biomarker assays remains essential for clinical translation.
Author contributions
MG: Writing – original draft, Writing – review & editing. LQ: Data curation, Formal analysis, Resources, Writing – review & editing. HC: Data curation, Resources, Writing – review & editing. RW: Data curation, Resources, Writing – review & editing. CL: Writing – review & editing, Resources. FY: Data curation, Resources, Writing – review & editing. SF: Data curation, Resources, Writing – review & editing. HG: Conceptualization, Data curation, Resources, Supervision, Visualization, Writing – review & editing. QC: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was funded by the National Natural Science Foundation of China (No. U23A20422), STI2030-Major Projects Youth Scientist Program (No. 2022ZD0213600), and the National Natural Science Foundation of China (No. 82201608).
Acknowledgments
We would like to thank all the authors of the original studies included in this review for their contributions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Vieira, RT, Caixeta, L, Machado, S, Silva, AC, Nardi, AE, Arias-Carrión, O, et al. Epidemiology of early-onset dementia: a review of the literature. Clin Pract Epidemiol Ment Health. (2013) 9:88–95. doi: 10.2174/1745017901309010088
2. Shafiei, G, Bazinet, V, Dadar, M, Manera, AL, Collins, DL, Dagher, A, et al. Network structure and transcriptomic vulnerability shape atrophy in frontotemporal dementia. Brain J Neurol. (2023) 146:321–36. doi: 10.1093/brain/awac069
3. Rosen, HJ, Allison, SC, Schauer, GF, Gorno-Tempini, ML, Weiner, MW, and Miller, BL. Neuroanatomical correlates of behavioural disorders in dementia. Brain J Neurol. (2005) 128:2612–25. doi: 10.1093/brain/awh628
4. Bang, J, Spina, S, and Miller, BL. Frontotemporal dementia. Lancet. (2015) 386:1672–82. doi: 10.1016/S0140-6736(15)00461-4
5. Rosso, SM, Donker Kaat, L, Baks, T, Joosse, M, de Koning, I, Pijnenburg, Y, et al. Frontotemporal dementia in the Netherlands: patient characteristics and prevalence estimates from a population-based study. Brain J Neurol. (2003) 126:2016–22. doi: 10.1093/brain/awg204
6. Koçoğlu, C, Van Broeckhoven, C, and van der Zee, J. How network-based approaches can complement gene identification studies in frontotemporal dementia. Trends Genet. (2022) 38:944–55. doi: 10.1016/j.tig.2022.05.005
7. Rademakers, R, Neumann, M, and Mackenzie, IR. Advances in understanding the molecular basis of frontotemporal dementia. Nat Rev Neurol. (2012) 8:423–34. doi: 10.1038/nrneurol.2012.117
8. Sieben, A, van Langenhove, T, Engelborghs, S, Martin, JJ, Boon, P, Cras, P, et al. The genetics and neuropathology of frontotemporal lobar degeneration. Acta Neuropathol. (2012) 124:353–72. doi: 10.1007/s00401-012-1029-x
9. Das, J, Bhui, U, Chowdhary, S, Sarkar, S, Ghoshal, IK, Nayak, S, et al. Biomarkers unveiling neurodegeneration: keys to progression and therapeutic insights. Indian J Pharm Educ Res. (2025) 59:s1–s15. doi: 10.5530/ijper.20250179
10. Renton, AE, Majounie, E, Waite, A, Simón-Sánchez, J, Rollinson, S, Gibbs, JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. (2011) 72:257–68. doi: 10.1016/j.neuron.2011.09.010
11. Ash, PEA, Bieniek, KF, Gendron, TF, Caulfield, T, Lin, WL, DeJesus-Hernandez, M, et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. (2013) 77:639–46. doi: 10.1016/j.neuron.2013.02.004
12. Mori, K, Weng, S-M, Arzberger, T, May, S, Rentzsch, K, Kremmer, E, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. (2013) 339:1335–8. doi: 10.1126/science.1232927
13. Thumbadoo, KM, Dieriks, BV, Murray, HC, Swanson, MEV, Yoo, JH, Mehrabi, NF, et al. Hippocampal aggregation signatures of pathogenic UBQLN2 in amyotrophic lateral sclerosis and frontotemporal dementia. Brain. (2024) 147:3547–61. doi: 10.1093/brain/awae140
14. Baker, M, Mackenzie, IR, Pickering-Brown, SM, Gass, J, Rademakers, R, Lindholm, C, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. (2006) 442:916–9. doi: 10.1038/nature05016
15. Liu, C-J, and Bosch, X. Progranulin: a growth factor, a novel TNFR ligand and a drug target. Pharmacol Ther. (2012) 133:124–32. doi: 10.1016/j.pharmthera.2011.10.003
16. Bunker, JM, Kamath, K, Wilson, L, Jordan, MA, and Feinstein, SC. FTDP-17 mutations compromise the ability of tau to regulate microtubule dynamics in cells. J Biol Chem. (2006) 281:11856–63. doi: 10.1074/jbc.M509420200
17. Hutton, M, Lendon, CL, Rizzu, P, Baker, M, Froelich, S, Houlden, H, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. (1998) 393:702–5. doi: 10.1038/31508
18. Goedert, M, Eisenberg, DS, and Crowther, RA. Propagation of tau aggregates and neurodegeneration. Annu Rev Neurosci. (2017) 40:189–210. doi: 10.1146/annurev-neuro-072116-031153
19. Snowden, JS, Adams, J, Harris, J, Thompson, JC, Rollinson, S, Richardson, A, et al. Distinct clinical and pathological phenotypes in frontotemporal dementia associated with MAPT, PGRN and C9orf72 mutations. Amyotroph Lat Scl Frontotemporal Degener. (2015) 16:497–505. doi: 10.3109/21678421.2015.1074700
20. Liampas, I, Kyriakoulopoulou, P, Karakoida, V, Kavvoura, PA, Sgantzos, M, Bogdanos, DP, et al. Blood-based biomarkers in frontotemporal dementia: A narrative review. Int J Mol Sci. (2024) 25:11838. doi: 10.3390/ijms252111838
21. Del Campo, M, Zetterberg, H, Gandy, S, Onyike, CU, Oliveira, F, Udeh-Momoh, C, et al. New developments of biofluid-based biomarkers for routine diagnosis and disease trajectories in frontotemporal dementia. Alzheimers Dement. (2022) 18:2292–307. doi: 10.1002/alz.12643
22. Sogorb-Esteve, A, Weiner, S, Simrén, J, Swift, IJ, Bocchetta, M, Todd, EG, et al. Proteomic analysis reveals distinct cerebrospinal fluid signatures across genetic frontotemporal dementia subtypes. Sci Transl Med. (2025) 17:eadm9654. doi: 10.1126/scitranslmed.adm9654
23. Ghidoni, R, Benussi, L, Glionna, M, Franzoni, M, and Binetti, G. Low plasma progranulin levels predict progranulin mutations in frontotemporal lobar degeneration. Neurology. (2008) 71:1235–9. doi: 10.1212/01.wnl.0000325058.10218.fc
24. Linnemann, C, Wilke, C, Mengel, D, Zetterberg, H, Heller, C, Kuhle, J, et al. NfL reliability across laboratories, stage-dependent diagnostic performance and matrix comparability in genetic FTD: a large GENFI study. J Neurol Neurosurg Psychiatry. (2024) 95:822–8. doi: 10.1136/jnnp-2023-332464.
25. Finch, N, Baker, M, Crook, R, Swanson, K, Kuntz, K, Surtees, R, et al. Plasma progranulin levels predict progranulin mutation status in frontotemporal dementia patients and asymptomatic family members. Brain J Neurol. (2009) 132:583–91. doi: 10.1093/brain/awn352
26. Ghidoni, R, Stoppani, E, Rossi, G, Piccoli, E, Albertini, V, Paterlini, A, et al. Optimal plasma progranulin cutoff value for predicting null progranulin mutations in neurodegenerative diseases: a multicenter Italian study. Neurodegener Dis. (2012) 9:121–7. doi: 10.1159/000333132
27. Galimberti, D, Fumagalli, GG, Fenoglio, C, Cioffi, SMG, Arighi, A, Serpente, M, et al. Progranulin plasma levels predict the presence of GRN mutations in asymptomatic subjects and do not correlate with brain atrophy: results from the GENFI study. Neurobiol Aging. (2018) 62:245.e9–245.e12. doi: 10.1016/j.neurobiolaging.2017.10.016
28. Sleegers, K, Brouwers, N, van Damme, P, Engelborghs, S, Gijselinck, I, van der Zee, J, et al. Serum biomarker for progranulin-associated frontotemporal lobar degeneration. Ann Neurol. (2009) 65:603–9. doi: 10.1002/ana.21621
29. Dols-Icardo, O, Suárez-Calvet, M, Hernández, I, Amer, G, Antón-Aguirre, S, Alcolea, D, et al. Expansion mutation in C9ORF72 does not influence plasma progranulin levels in frontotemporal dementia. Neurobiol Aging. (2012) 33:1851.e17–9. doi: 10.1016/j.neurobiolaging.2012.03.005
30. Meeter, LHH, Patzke, H, Loewen, G, Dopper, EGP, Pijnenburg, YAL, van Minkelen, R, et al. Progranulin levels in plasma and cerebrospinal fluid in Granulin mutation carriers. Dement Geriatr Cogn Disord Extra. (2016) 6:330–40. doi: 10.1159/000447738
31. Benussi, A, Gazzina, S, Premi, E, Cosseddu, M, Archetti, S, Dell’Era, V, et al. Clinical and biomarker changes in presymptomatic genetic frontotemporal dementia. Neurobiol Aging. (2019) 76:133–40. doi: 10.1016/j.neurobiolaging.2018.12.018
32. Sellami, L, Rucheton, B, Ben Younes, I, Camuzat, A, Saracino, D, Rinaldi, D, et al. Plasma progranulin levels for frontotemporal dementia in clinical practice: a 10-year French experience. Neurobiol Aging. (2020) 91:167.e1–9. doi: 10.1016/j.neurobiolaging.2020.02.014
33. Disanto, G, Barro, C, Benkert, P, Naegelin, Y, Schädelin, S, Giardiello, A, et al. Serum Neurofilament light: A biomarker of neuronal damage in multiple sclerosis. Ann Neurol. (2017) 81:857–70. doi: 10.1002/ana.24954
34. Olsson, B, Portelius, E, Cullen, NC, Sandelius, Å, Zetterberg, H, Andreasson, U, et al. Association of Cerebrospinal Fluid Neurofilament Light Protein Levels with Cognition in patients with dementia, motor neuron disease, and movement disorders. JAMA Neurol. (2019) 76:318–25. doi: 10.1001/jamaneurol.2018.3746
35. Van Der Ende, EL, Meeter, LH, Poos, JM, Panman, JL, Jiskoot, LC, Dopper, EGP, et al. Serum neurofilament light chain in genetic frontotemporal dementia: a longitudinal, multicentre cohort study. Lancet Neurol. (2019) 18:1103–11. doi: 10.1016/S1474-4422(19)30354-0
36. Meeter, LH, Dopper, EG, Jiskoot, LC, Sanchez-Valle, R, Graff, C, Benussi, L, et al. Neurofilament light chain: a biomarker for genetic frontotemporal dementia. Ann Clin Transl Neurol. (2016) 3:623–36. doi: 10.1002/acn3.325
37. Saracino, D, Dorgham, K, Camuzat, A, Rinaldi, D, Rametti-Lacroux, A, Houot, M, et al. Plasma NfL levels and longitudinal change rates in C9orf72 and GRN-associated diseases: from tailored references to clinical applications. J Neurol Neurosurg Psychiatry. (2021) 92:1278–88. doi: 10.1136/jnnp-2021-326914
38. Gendron, TF, Heckman, MG, White, LJ, Veire, AM, Pedraza, O, Burch, AR, et al. Comprehensive cross-sectional and longitudinal analyses of plasma neurofilament light across FTD spectrum disorders. Cell Rep Med. (2022) 3:100607. doi: 10.1016/j.xcrm.2022.100607
39. Wilke, C, Reich, S, van Swieten, JC, Borroni, B, Sanchez-Valle, R, Moreno, F, et al. Stratifying the Presymptomatic phase of genetic frontotemporal dementia by serum NfL and pNfH: a longitudinal multicentre study. Ann Neurol. (2022) 91:33–47. doi: 10.1002/ana.26265
40. Mackenzie, IR, Rademakers, R, and Neumann, M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. The lancet. Neurology. (2010) 9:995–1007. doi: 10.1016/S1474-4422(10)70195-2
41. Suárez-Calvet, M, Dols-Icardo, O, Lladó, A, Sánchez-Valle, R, Hernández, I, Amer, G, et al. Plasma phosphorylated TDP-43 levels are elevated in patients with frontotemporal dementia carrying a C9orf72 repeat expansion or a GRN mutation. J Neurol Neurosurg Psychiatry. (2014) 85:684–91. doi: 10.1136/jnnp-2013-305972
42. Walker, AK, Spiller, KJ, Ge, G, Zheng, A, Xu, Y, Zhou, M, et al. Functional recovery in new mouse models of ALS/FTLD after clearance of pathological cytoplasmic TDP-43. Acta Neuropathol. (2015) 130:643–60. doi: 10.1007/s00401-015-1460-x
43. Katisko, K, Huber, N, Kokkola, T, Hartikainen, P, Krüger, J, Heikkinen, AL, et al. Serum total TDP-43 levels are decreased in frontotemporal dementia patients with C9orf72 repeat expansion or concomitant motoneuron disease phenotype. Alzheimer's Res Ther. (2022) 14:151. doi: 10.1186/s13195-022-01091-8
44. Colangelo, AM, Alberghina, L, and Papa, M. Astrogliosis as a therapeutic target for neurodegenerative diseases. Neurosci Lett. (2014) 565:59–64. doi: 10.1016/j.neulet.2014.01.014
45. Heller, C, Foiani, MS, Moore, K, Convery, R, Bocchetta, M, Neason, M, et al. Plasma glial fibrillary acidic protein is raised in progranulin-associated frontotemporal dementia. J Neurol Neurosurg Psychiatry. (2020) 91:263–70. doi: 10.1136/jnnp-2019-321954
46. Sheth, U, Öijerstedt, L, Heckman, MG, White, LJ, Heuer, HW, Lario Lago, A, et al. Comprehensive cross-sectional and longitudinal comparisons of plasma glial fibrillary acidic protein and neurofilament light across FTD spectrum disorders. Mol Neurodegener. (2025) 20:30. doi: 10.1186/s13024-025-00821-4
47. Simon, MJ, Logan, T, DeVos, SL, and di Paolo, G. Lysosomal functions of progranulin and implications for treatment of frontotemporal dementia. Trends Cell Biol. (2023) 33:324–39. doi: 10.1016/j.tcb.2022.09.006
48. Zhu, N, Santos-Santos, M, Illán-Gala, I, Montal, V, Estellés, T, Barroeta, I, et al. Plasma glial fibrillary acidic protein and neurofilament light chain for the diagnostic and prognostic evaluation of frontotemporal dementia. Transl Neurodegener. (2021) 10:50. doi: 10.1186/s40035-021-00275-w
49. Marsan, E, Velmeshev, D, Ramsey, A, Patel, RK, Zhang, J, Koontz, M, et al. Astroglial toxicity promotes synaptic degeneration in the thalamocortical circuit in frontotemporal dementia with GRN mutations. J Clin Invest. (2023) 133:e164919. doi: 10.1172/JCI164919
50. Baiardi, S, Quadalti, C, Mammana, A, Dellavalle, S, Zenesini, C, Sambati, L, et al. Diagnostic value of plasma p-tau181, NfL, and GFAP in a clinical setting cohort of prevalent neurodegenerative dementias. Alzheimer's Res Ther. (2022) 14:153. doi: 10.1186/s13195-022-01093-6
51. Conner, GE, and Richo, G. Isolation and characterization of a stable activation intermediate of the lysosomal aspartyl protease cathepsin D. Biochemistry. (1992) 31:1142–7. doi: 10.1021/bi00119a024
52. Yao, R-Q, Ren, C, Xia, ZF, and Yao, YM. Organelle-specific autophagy in inflammatory diseases: a potential therapeutic target underlying the quality control of multiple organelles. Autophagy. (2021) 17:385–401. doi: 10.1080/15548627.2020.1725377
53. Shao, Q, Yang, M, Liang, C, Ma, L, Zhang, W, Jiang, Z, et al. C9orf72 and smcr8 mutant mice reveal MTORC1 activation due to impaired lysosomal degradation and exocytosis. Autophagy. (2020) 16:1635–50. doi: 10.1080/15548627.2019.1703353
54. Arrant, AE, Onyilo, VC, Unger, DE, and Roberson, ED. Progranulin gene therapy improves lysosomal dysfunction and microglial pathology associated with frontotemporal dementia and neuronal ceroid Lipofuscinosis. J Neurosci. (2018) 38:2341–58. doi: 10.1523/JNEUROSCI.3081-17.2018
55. Bellini, S, Saraceno, C, Benussi, L, Geviti, A, Longobardi, A, Nicsanu, R, et al. Plasma small extracellular vesicle Cathepsin D dysregulation in GRN/C9orf72 and sporadic frontotemporal lobar degeneration. Int J Mol Sci. (2022) 23:10693. doi: 10.3390/ijms231810693
56. Heikkinen, S, Huber, N, Katisko, K, Kokkola, T, Hartikainen, P, Krüger, J, et al. Serum Cathepsin S levels do not show alterations in different clinical, neuropathological, or genetic subtypes of frontotemporal dementia patients nor in comparison to healthy control individuals. J Alzheimer's Dis. (2023) 93:395–401. doi: 10.3233/JAD-221060
57. Esteras, N, Alquézar, C, de la Encarnación, A, Villarejo, A, Bermejo-Pareja, F, and Martín-Requero, Á. Calmodulin levels in blood cells as a potential biomarker of Alzheimer's disease. Alzheimer's Res Ther. (2013) 5:55. doi: 10.1186/alzrt219
58. Van Der Ende, EL, Heller, C, Sogorb-Esteve, A, Swift, IJ, McFall, D, Peakman, G, et al. Elevated CSF and plasma complement proteins in genetic frontotemporal dementia: results from the GENFI study. J Neuroinflammation. (2022) 19:217. doi: 10.1186/s12974-022-02573-0
59. Morgan, AR, Touchard, S, Leckey, C, O'Hagan, C, and Nevado-Holgado, AJNIMA Consortium, et al. Inflammatory biomarkers in Alzheimer's disease plasma. Alzheimers Dement. (2019) 15:776–87. doi: 10.1016/j.jalz.2019.03.007
60. Goetzl, EJ, Schwartz, JB, Abner, EL, Jicha, GA, and Kapogiannis, D. High complement levels in astrocyte-derived exosomes of Alzheimer disease. Ann Neurol. (2018) 83:544–52. doi: 10.1002/ana.25172
61. Daborg, J, Andreasson, U, Pekna, M, Lautner, R, Hanse, E, Minthon, L, et al. Cerebrospinal fluid levels of complement proteins C3, C4 and CR1 in Alzheimer's disease. J Neural Transm. (2012) 119:789–97. doi: 10.1007/s00702-012-0797-8
62. Wilke, C, Gillardon, F, Deuschle, C, Dubois, E, Hobert, M, Müller vom Hagen, J, et al. Serum levels of Progranulin do not reflect cerebrospinal fluid levels in neurodegenerative disease. Curr Alzheimer Res. (2016) 13:654–62. doi: 10.2174/1567205013666160314151247
63. Nicholson, AM, Finch, NCA, Thomas, CS, Wojtas, A, Rutherford, NJ, Mielke, MM, et al. Progranulin protein levels are differently regulated in plasma and CSF. Neurology. (2014) 82:1871–8. doi: 10.1212/WNL.0000000000000445
64. Morenas-Rodríguez, E, Cervera-Carles, L, Vilaplana, E, Alcolea, D, Carmona-Iragui, M, Dols-Icardo, O, et al. Progranulin protein levels in cerebrospinal fluid in primary neurodegenerative dementias. J Alzheimer's Dis. (2016) 50:539–46. doi: 10.3233/JAD-150746
65. Silva-Spínola, A, Lima, M, Leitão, MJ, Durães, J, Tábuas-Pereira, M, Almeida, MR, et al. Serum neurofilament light chain as a surrogate of cognitive decline in sporadic and familial frontotemporal dementia. Eur J Neurol. (2022) 29:36–46. doi: 10.1111/ene.15058
66. Carecchio, M, Fenoglio, C, Cortini, F, Comi, C, Benussi, L, Ghidoni, R, et al. Cerebrospinal fluid biomarkers in Progranulin mutations carriers. J Alzheimer's Dis. (2011) 27:781–90. doi: 10.3233/JAD-2011-111046
67. Kapaki, E, Boufidou, F, Bourbouli, M, Pyrgelis, ES, Constantinides, VC, Anastassopoulou, C, et al. Cerebrospinal fluid biomarker profile in TDP-43-related genetic frontotemporal dementia. J Pers Med. (2022) 12:1747. doi: 10.3390/jpm12101747
68. Reimand, J, Boon, BDC, Collij, LE, Teunissen, CE, Rozemuller, AJM, van Berckel, BNM, et al. Amyloid-β PET and CSF in an autopsy-confirmed cohort. Ann Clin Transl Neurol. (2020) 7:2150–60. doi: 10.1002/acn3.51195
69. Scheltens, P, de Strooper, B, Kivipelto, M, Holstege, H, Chételat, G, Teunissen, CE, et al. Alzheimer's disease. Lancet. (2021) 397:1577–90. doi: 10.1016/S0140-6736(20)32205-4
70. Sato, C, Mallipeddi, N, Ghoshal, N, Wright, BA, Day, GS, Davis, AA, et al. MAPT R406W increases tau T217 phosphorylation in absence of amyloid pathology. Ann Clin Transl Neurol. (2021) 8:1817–30. doi: 10.1002/acn3.51435
71. Horie, K, Barthélemy, NR, Sato, C, and Bateman, RJ. CSF tau microtubule binding region identifies tau tangle and clinical stages of Alzheimer's disease. Brain J Neurol. (2021) 144:515–27. doi: 10.1093/brain/awaa373
72. Horie, K, Barthélemy, NR, Spina, S, VandeVrede, L, He, Y, Paterson, RW, et al. CSF tau microtubule-binding region identifies pathological changes in primary tauopathies. Nat Med. (2022) 28:2547–54. doi: 10.1038/s41591-022-02075-9
73. Schneider, R, McKeever, P, Kim, T, Graff, C, van Swieten, J, Karydas, A, et al. Downregulation of exosomal miR-204-5p and miR-632 as a biomarker for FTD: a GENFI study. J Neurol Neurosurg Psychiatry. (2018) 89:851–8. doi: 10.1136/jnnp-2017-317492
74. Rosen, EY, Wexler, EM, Versano, R, Coppola, G, Gao, F, Winden, KD, et al. Functional genomic analyses identify pathways dysregulated by progranulin deficiency, implicating Wnt signaling. Neuron. (2011) 71:1030–42. doi: 10.1016/j.neuron.2011.07.021
75. Alquézar, C, de la Encarnación, A, Moreno, F, de Munain, AL, and Martín-Requero, Á. Progranulin deficiency induces overactivation of WNT5A expression via TNF-α/NF-κB pathway in peripheral cells from frontotemporal dementia-linked granulin mutation carriers. J Psychiatry Neurosci. (2016) 41:225–39. doi: 10.1503/jpn.150131
76. Woollacott, IOC, Nicholas, JM, Heslegrave, A, Heller, C, Foiani, MS, Dick, KM, et al. Cerebrospinal fluid soluble TREM2 levels in frontotemporal dementia differ by genetic and pathological subgrou. Alzheimer's Res Ther. (2018) 10:79. doi: 10.1186/s13195-018-0405-8
77. van der Ende, EL, Morenas-Rodriguez, E, McMillan, C, Grossman, M, Irwin, D, Sanchez-Valle, R, et al. CSF sTREM2 is elevated in a subset in GRN-related frontotemporal dementia. Neurobiol Aging. (2021) 103:158.e1–5. doi: 10.1016/j.neurobiolaging.2021.02.024
78. Woollacott, IOC, Swift, IJ, Sogorb-Esteve, A, Heller, C, Knowles, K, Bouzigues, A, et al. CSF glial markers are elevated in a subset of patients with genetic frontotemporal dementia. Ann Clin Transl Neurol. (2022) 9:1764–77. doi: 10.1002/acn3.51672
79. Borrego-Écija, S, Pérez-Millan, A, Antonell, A, Fort-Aznar, L, Kaya-Tilki, E, León-Halcón, A, et al. Galectin-3 is upregulated in frontotemporal dementia patients with subtype specificity. Alzheimers Dement. (2023) 20:1515–26. doi: 10.1002/alz.13536
80. Van Der Ende, EL, Xiao, M, Xu, D, Poos, JM, Panman, JL, Jiskoot, LC, et al. Neuronal pentraxin 2: a synapse-derived CSF biomarker in genetic frontotemporal dementia. J Neurol Neurosurg Psychiatry. (2020) 91:612–21. doi: 10.1136/jnnp-2019-322493
81. Huang, M, Modeste, E, Dammer, E, Merino, P, Taylor, G, Duong, DM, et al. Network analysis of the progranulin-deficient mouse brain proteome reveals pathogenic mechanisms shared in human frontotemporal dementia caused by GRN mutations. Acta Neuropathol Commun. (2020) 8:163. doi: 10.1186/s40478-020-01037-x
82. Lehmer, C, Oeckl, P, Weishaupt, JH, Volk, AE, Diehl-Schmid, J, Schroeter, ML, et al. Poly-GP in cerebrospinal fluid links C9orf72-associated dipeptide repeat expression to the asymptomatic phase of ALS/FTD. EMBO Mol Med. (2017) 9:859–68. doi: 10.15252/emmm.201607486
83. Swift, IJ, Sogorb-Esteve, A, Heller, C, Synofzik, M, Otto, M, Graff, C, et al. Fluid biomarkers in frontotemporal dementia: past, present and future. J Neurol Neurosurg Psychiatry. (2021) 92:204–15. doi: 10.1136/jnnp-2020-323520
84. González-Sánchez, M, Bartolome, F, Antequera, D, Puertas-Martín, V, González, P, Gómez-Grande, A, et al. Decreased salivary lactoferrin levels are specific to Alzheimer's disease. EBioMedicine. (2020) 57:102834. doi: 10.1016/j.ebiom.2020.102834
85. Schuster, J, and Funke, SA. Methods for the specific detection and quantitation of amyloid-β oligomers in cerebrospinal fluid. J Alzheimers Dis. (2016) 53:53–67. doi: 10.3233/JAD-151029
86. Solje, E, Benussi, A, Buratti, E, Remes, AM, Haapasalo, A, and Borroni, B. State-of-the-art methods and emerging fluid biomarkers in the diagnostics of dementia-A short review and diagnostic algorithm. Diagnostics. (2021) 11:788. doi: 10.3390/diagnostics11050788.
87. Del Campo, M, Jongbloed, W, Twaalfhoven, HAM, Veerhuis, R, Blankenstein, MA, Teunissen, CE, et al. Facilitating the validation of novel protein biomarkers for dementia: an optimal workflow for the development of Sandwich immunoassays. Front Neurol. (2015) 6:202. doi: 10.3389/fneur.2015.00202
88. Rissin, DM, Kan, CW, Campbell, TG, Howes, SC, Fournier, DR, Song, L, et al. Single-molecule enzyme-linked immunosorbent assay detects serum proteins at subfemtomolar concentrations. Nat Biotechnol. (2010) 28:595–9. doi: 10.1038/nbt.1641
89. Katzeff, JS, Bright, F, Phan, K, Kril, JJ, Ittner, LM, Kassiou, M, et al. Biomarker discovery and development for frontotemporal dementia and amyotrophic lateral sclerosis. Brain J Neurol. (2022) 145:1598–609. doi: 10.1093/brain/awac077
90. Mol, MO, Miedema, SSM, van Swieten, JC, van Rooij, JGJ, and Dopper, EGP. Molecular pathways involved in frontotemporal lobar degeneration with TDP-43 Proteinopathy: what can we learn from proteomics? Int J Mol Sci. (2021) 22:10298. doi: 10.3390/ijms221910298
91. Agrawal, I, Tripathi, P, and Biswas, S. Mass spectrometry based protein biomarkers and drug target discovery and clinical diagnosis in age-related progressing neurodegenerative diseases. Drug Metab Rev. (2022) 54:22–36. doi: 10.1080/03602532.2022.2029475
92. Carecchio, M, Fenoglio, C, de Riz, M, Guidi, I, Comi, C, Cortini, F, et al. Progranulin plasma levels as potential biomarker for the identification of GRN deletion carriers. A case with atypical onset as clinical amnestic mild cognitive impairment converted to Alzheimer's disease. J Neurol Sci. (2009) 287:291–3. doi: 10.1016/j.jns.2009.07.011
93. Panman, JL, Venkatraghavan, V, van der Ende, E, Steketee, RME, Jiskoot, LC, Poos, JM, et al. Modelling the cascade of biomarker changes in GRN-related frontotemporal dementia. J Neurol Neurosurg Psychiatry. (2021) 92:494–501. doi: 10.1136/jnnp-2020-323541
Keywords: fluid biomarker, familial frontotemporal dementia, MAPT, GRN, C9orf72
Citation: Guo M, Qin L, Cai H, Wang R, Luo C, Yang F, Feng S, Gao H and Chen Q (2025) Fluid biomarkers in familial frontotemporal dementia: progress and prospects. Front. Neurol. 16:1663609. doi: 10.3389/fneur.2025.1663609
Edited by:
Nobuyuki Kobayashi, Mainrain Brain Inc., JapanReviewed by:
Nahid Olfati, University of California, San Diego, United StatesJoy Das, Lovely Professional University, India
Copyright © 2025 Guo, Qin, Cai, Wang, Luo, Yang, Feng, Gao and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qin Chen, Y2hlbi5xaW5Ac2N1LmVkdS5jbg==; Hui Gao, Z2FvaHVpXzEwMDhAcXEuY29t