 Pietro Guaraldi1Isabella Allegri2Alessandra Ariatti3Tommaso Baldini4
Pietro Guaraldi1Isabella Allegri2Alessandra Ariatti3Tommaso Baldini4 Andrea Barbieri5Federico Barocelli6Michela Bartolotti7Elena Biagini8,9Francesca Bianchi10Annamaria Borghi11Claudia Borghi12
Andrea Barbieri5Federico Barocelli6Michela Bartolotti7Elena Biagini8,9Francesca Bianchi10Annamaria Borghi11Claudia Borghi12 Giuseppe Boriani5
Giuseppe Boriani5 Ilaria Cani1Samuela Carigi13Luca Codeluppi14Marco Currò Dossi15Francesca Dalpozzo16
Ilaria Cani1Samuela Carigi13Luca Codeluppi14Marco Currò Dossi15Francesca Dalpozzo16 Roberto D’Angelo1Riccardo De Gennaro17Francesco Di Spigno18Elisa Gardini19Gianluca Lanati20
Roberto D’Angelo1Riccardo De Gennaro17Francesco Di Spigno18Elisa Gardini19Gianluca Lanati20 Chiara Leuzzi21Francesca Marzo13
Chiara Leuzzi21Francesca Marzo13 Marco Masullo1Gaia Mazzanti22Elisa Merli23Agnese Milandri24Dennis Monari12Enrica Perugini25Alberto Ponziani26,27Emanuela Postiglione15Marta Rasia28
Marco Masullo1Gaia Mazzanti22Elisa Merli23Agnese Milandri24Dennis Monari12Enrica Perugini25Alberto Ponziani26,27Emanuela Postiglione15Marta Rasia28 Rita Rinaldi1Davide Scancarello29
Rita Rinaldi1Davide Scancarello29 Kevin Serafini5
Kevin Serafini5 Matteo Serenelli30Maurizio Sguazzotti25Ernesto Siena31Anna Maria Simone32
Matteo Serenelli30Maurizio Sguazzotti25Ernesto Siena31Anna Maria Simone32 Chiara Terracciano33Chiara Valenti21
Chiara Terracciano33Chiara Valenti21 Giovanni Vitale26Maria Vitiello34
Giovanni Vitale26Maria Vitiello34 Simone Longhi8*
Simone Longhi8*- 1IRCCS Istituto delle Scienze Neurologiche di Bologna, Bologna, Italy
- 2Department of Neurology, University Hospital of Parma, Parma, Italy
- 3Department of Biomedical, Metabolic Neural Sciences, University of Modena, Modena, Italy
- 4Department of Neurology, Infermi Hospital, AUSL della Romagna, Rimini, Italy
- 5Cardiology Division, Department of Biomedical, Metabolic and Neural Sciences, University of Modena and Reggio Emilia, Policlinico di Modena, Modena, Italy
- 6 Cardiology Division, Parma University Hospital, Parma, Italy
- 7Cardiology Unit, Bufalini Hospital, AUSL della Romagna, Cesena, Italy
- 8Cardiology Unit, Department of Cardiac Thoracic and Vascular, IRCCS Azienda Ospedaliero-Universitaria di Bologna, Bologna, Italy
- 9European Reference Network for Rare, Low Prevalence and Complex Diseases of the Heart-ERN GUARD- Heart, Bologna, Italy
- 10Neurology Unit, Bufalini Hospital Cesena, AUSL della Romagna, Cesena, Italy
- 11IRCCS Istituto delle Scienze Neurologiche di Bologna, UOC Neurologia Rete Stroke Metropolitana Bologna, Bologna, Italy
- 12Cardiology Unit, Ramazzini Hospital, Carpi, Italy
- 13Cardiology Unit, Infermi Hospital, AUSL della Romagna, Rimini, Italy
- 14Neurology Unit, Department of Neuromotor and Rehabilitation, Azienda USL- IRCCS di Reggio Emilia, Reggio Emilia, Italy
- 15Neurology Unit, ‘Santa Maria delle Croci’ Hospital, AUSL della Romagna, Ravenna, Italy
- 16Neurology Unit, Ospedale di Lugo, AUSL della Romgana, Lugo, Italy
- 17Department of Neuroscience and Rehabilitation, Azienda Ospedaliero-Universitaria S, Anna, Ferrara, Italy
- 18Cardiology Unit of Emergency Department, Guglielmo da Saliceto Hospital, Piacenza, Italy
- 19Cardiology Unit, ‘Morgagni–Pierantoni’ Hospital, AUSL della Romagna, Forli, Italy
- 20Cardiology Unit, Castel San Giovanni Hospital, Castel San Giovanni, Italy
- 21Cardiology Unit, IRCCS Arcispedale S. Maria Nuova, Reggio Emilia, Italy
- 22Cardiology Unit, Bellaria Hospital, IRCCS Istituto delle Scienze Neurologiche di Bologna, Bologna, Italy
- 23Cardiology Unit, ‘Degli Infermi’ Hospital, AUSL della Romagna, Faenza, Italy
- 24Cardiology Unit, Bentivoglio Hospital, Bologna, Italy
- 25Cardiology Unit, Maggiore Hospital, Bologna, Italy
- 26Cardiology Unit, Santa Maria della Scaletta Hospital, Imola, Italy
- 27Department of Experimental, Diagnostic and Specialty Medicine, University of Bologna, Bologna, Italy
- 28Cardiology Unit, Ospedale di Fidenza, Fidenza, Italy
- 29Cardiology Unit, ‘Santa Maria delle Croci’ Hospital, AUSL della Romagna, Ravenna, Italy
- 30Cardiology Unit, Azienda Ospedaliero Universitaria di Ferrara, Ferrara, Italy
- 31Neurology Unit, Ospedale di Fidenza, Fidenza, Italy
- 32Neurology Unit, Ramazzini Hospital, AUSL Modena, Carpi, Italy
- 33Neurology Unit, 'Guglielmo da Saliceto' Hospital, Piacenza, Italy
- 34Neurology and Stroke Unit, Department of Neuroscience, Bufalini Hospital, Cesena, Italy
Transthyretin amyloidosis (ATTR) is a rare disease caused by the extracellular accumulation of misfolded transthyretin (TTR) amyloid fibrils. ATTR can be either hereditary (ATTRv) or acquired (wtATTR). ATTRv is caused by a mutation in the transthyretin gene (TTR) with an autosomal dominant pattern of inheritance. In ATTRv amyloidosis, some patients primarily exhibit symptoms of polyneuropathy others mainly or exclusively present with symptoms of cardiomyopathy. However, many patients present with a multisystemic involvement that includes sensory, motor, autonomic, and cardiac symptoms. Early diagnosis and detection of disease progression are emerging as a crucial need for ATTR amyloidosis in order to significantly impact survival, patients’ functions and quality of life. Currently, parameters to be monitored in ATTR patients in the real life might refer to some publicly available recommendations regarding the monitoring and assessment of disease progression in the real-world setting of patients with ATTRv. Nonetheless, a standardized disease monitoring protocol has not been established in Italy, posing a significant unmet need for a prompt and equal access to care. Therefore, in the Emilia-Romagna Region the “ATTR Working Group” has sought to tailor the recommendations to the Regional “real clinical setting” in order to optimize and standardize a monitoring protocol aimed at identifying disease progression. Patients’ and carriers’ access to uniform monitoring routes across the entire Region ensures optimal disease management and economic sustainability.
Introduction
Transthyretin amyloidosis (ATTR) is a rare disease caused by the extracellular accumulation of misfolded transthyretin (TTR) amyloid fibrils. This accumulation can affect multiple sites, including the nerves, heart and gastrointestinal tract. ATTR can be either hereditary (ATTRv) or acquired (wtATTR). ATTRv is caused by a mutation in the transthyretin gene (TTR) with an autosomal dominant pattern of inheritance. Wild-type transthyretin amyloidosis (wtATTR) is an age-related pathological condition due to TTR protein tetramer instability which leads to amyloid fibrils deposition mainly in the heart (1, 2). The prevalence of wtATTR in the elderly population is estimated to be around 10–25% in individuals over the age of 80. However, it is plausible that wtATTR amyloidosis is actually underdiagnosed (3).
A recent national survey revealed a 50% increase in diagnosed ATTRv cases in Italy over a 4-year period, with a shift towards p.Ile88Leu and p.Phe84Leu variants, and a rising prevalence of mixed phenotypes, underlining the importance of broad-based genotype–phenotype monitoring protocols (4). Also, given the non-negligible prevalence of hereditary forms even among elderly ATTR-CM patients (5.3% overall, 13% in women ≥70 years), systematic genetic testing is warranted in all cases irrespective of age (5).
In ATTRv amyloidosis, some patients primarily exhibit symptoms of polyneuropathy, traditionally referred to as familial amyloidotic polyneuropathy (FAP) or ATTR polyneuropathy (ATTR-PN); others mainly or exclusively present with symptoms of cardiomyopathy, traditionally known as familial amyloidotic cardiomyopathy (FAC) or more recently as ATTR cardiac amyloidosis (ATTR-CA). However, many patients present with a multisystemic involvement that includes sensory, motor, autonomic, and cardiac symptoms. Notably, incomplete penetrance has been observed, meaning that not all individuals carrying a pathogenic TTR mutation will develop clinical manifestations of the disease. This variability complicates both diagnosis and genetic counseling. However, once symptoms of ATTRv amyloidosis emerge, the disease often progresses rapidly, leading to severe disability and reduced life expectancy, thus, monitoring of carriers is mandatory (1).
Importantly, the genotype–phenotype association can widely vary depending on the TTR variant which is prognostic of disease severity and progression rate over time. Indeed, in the Emilia-Romagna Region (Italy) the most prevalent phenotype is caused by the variant p.Ile88Leu, which is characterized by early overt cardiac symptoms.
In order to provide an optimal and appropriate health care service to ATTR patients, a structured referral network was established in 2022, involving both tertiary and peripheral ATTR-CA cardiology and neurology centers. This network, referred to as the ‘ATTR Working Group,’ brings together cardiologists and neurologists from across the Emilia-Romagna Region (6). The Working Group members work both in Academic and peripheral centers and collaborate to achieve the following goals:
• To ensure that all patients in the Emilia-Romagna Region have equal access to the best possible diagnostic and therapeutic opportunities through standardized approaches to the management of ATTR patients;
• To address Regional epidemiology-related needs of patients affected by specific TTR variant;
• To facilitate and support scientific data dissemination.
Early diagnosis and detection of disease progression are emerging as a crucial need for ATTR amyloidosis in order to significantly impact survival, patients’ functions and quality of life (7–9). Currently, parameters to be monitored in ATTR patients in the real life might refer to some publicly available recommendations regarding the monitoring and assessment of disease progression in the real-world setting of patients with ATTRv (1, 7–9). Recent evidence suggests that early myocardial deformation abnormalities, especially reduced left and right ventricular global longitudinal strain, may be present in asymptomatic ATTRv mutation carriers despite preserved ejection fraction, and may represent early echocardiographic markers of myocardial infiltration (10). Nonetheless, a standardized disease monitoring protocol has not been established in Italy, posing a significant unmet need for a prompt and equal access to care.
Therefore, in the Emilia-Romagna Region the “ATTR Working Group” has sought to tailor the above mentioned recommendations to the Regional “real clinical setting” in order to optimize and standardize a monitoring protocol aimed at identifying disease progression. Patients’ and carriers’ access to uniform monitoring routes across the entire Region ensures optimal disease management and economic sustainability.
Moreover, ATTRv patients and TTR variant carriers’ data are being collected through the REDCap web platform, allowing the possibility to retrospectively assess the evolving ATTR management associated with the implementation of the Regional monitoring protocol within the referral chain.
In this article we aim to describe the ATTRv monitoring protocol that has been employed in the Emilia-Romagna Region since January 2023.
Methods
To agree on a Regional monitoring protocol specifically tailored to the clinical needs of ATTRv patients and carriers, a specific survey of the previously identified centers (Working Group) managing ATTRv patients (6) was administered. The survey collected the monitoring methods employed at each center taking into account both the cardiological and neurological aspects related to ATTR patients (Supplementary Appendix 1). Through subsequent in-person and remote meetings, the results of the surveys highlighted the capabilities of the involved centers. Simultaneously, a review of the current cardiology guidelines and neurology recommendations available for the ATTRv setting was conducted (1, 7–9).
Results
The Working Group identified the three most frequently observed clinical scenarios in routine medical practice and agreed on the following definitions:
1. Asymptomatic carriers: individuals who carry the TTR gene mutation in one allele in absence of any symptoms and signs of disease.
2. ATTR patients:
• Cardiological phenotype: patients presenting with cardiac symptoms and/or signs without neurological symptoms and/or signs.
• Neurological/mixed phenotype: patients presenting with exclusively neurological symptoms and/or signs, or with mixed clinical characteristics (i.e., both cardiac and neurological involvement).
Monitoring asymptomatic carriers
According to current scientific literature, monitoring of asymptomatic carriers should begin approximately 10 years before the predicted age of disease onset (PADO) (8). In our protocol, however, monitoring is scheduled to start 15 years before PADO in order to account for potential anticipation phenomena and to minimize the risk of missing early signs of disease onset.
Specifically, asymptomatic carriers should undergo baseline clinical and neurophysiological evaluations, followed by annual assessments starting 15 years before the predicted onset of the disease in their family cluster. This recommendation is based in part on findings from a recent study by Cisneros-Barroso et al., which demonstrated that the onset of ATTRv occurs, on average, 16 years earlier in offspring compared to their parents (11).
The follow-up program includes clinical and instrumental multidisciplinary assessments to be conducted annually, biennially, or every three years (Table 1). Should one or more monitoring tests yield abnormal results, it is recommended that the patient undergo evaluation at the designated tertiary center.
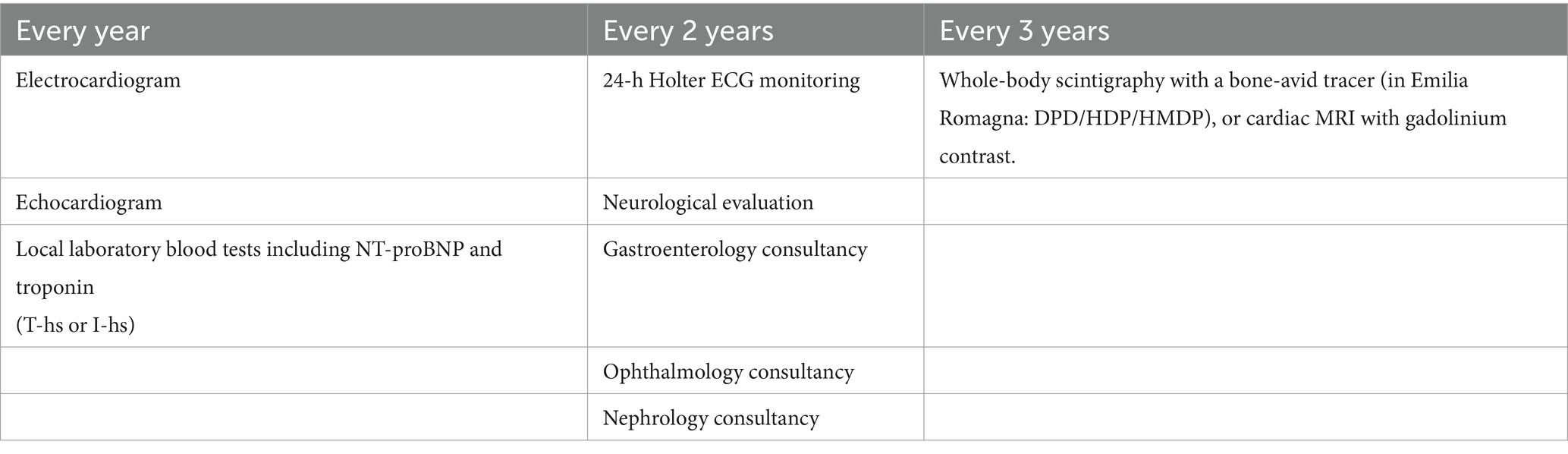
Table 1. Assessments type and timing of carrier monitoring.
Neurological monitoring of ATTR patients
From a neurological standpoint, the Working Group proposes a unified protocol of clinical and instrumental evaluations for both carriers and symptomatic patients, applied with different timing based on the patient category (Table 2).
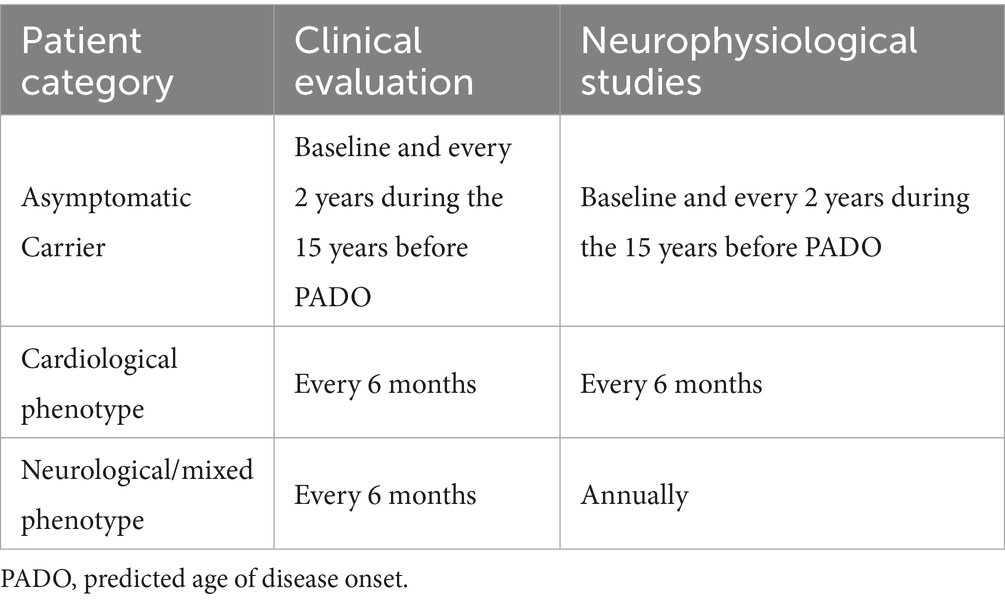
Table 2. Neurological follow-up across the three clinical scenarios of ATTRv.
Specifically, the working group recommends clinical evaluations every 6 months for all ATTR patients. Neurophysiological assessments should be performed annually in patients with a neurological or mixed phenotype, and every 6 months in patients with a predominantly cardiac phenotype.
This differentiated approach reflects the distinct prognostic trajectories of patients with cardiac amyloidosis (CA) and the variability in therapeutic options available for neurological versus cardiological involvement (8).
The neurological monitoring protocol includes: (1) clinical assessment, and (2) neurophysiological studies.
1. Clinical Assessment
Experts agree on performing a comprehensive series of clinical evaluations, which include a neurological physical examination and the use of validated self-administered scales and questionnaires, specifically:
• Complete neurological physical examination
• Measurement of weight and height for Body Mass Index (BMI, kg/m2) calculation
• Measurement of blood pressure (mmHg) and heart rate (bpm) in the supine position and after 3 min of standing (orthostasis)
• Application of scales selected for their relevance and feasibility in clinical practice:
FAP/PND (Familial Amyloidotic Polyneuropathy / PolyNeuropathy Disability): a straightforward scale used to classify patients based on ambulatory capability.
NIS (Neuropathy Impairment Score): this scale ranges from 0 to 244, with higher scores indicating more severe dysfunction. It is a composite score of clinical deficits (weakness, loss of reflexes, and sensory loss) derived from the assessment of muscle strength in 24 muscle groups, reflexes in 5 groups, and sensory function in all four limbs (11).
CADT (Compound Autonomic Dysfunction Test): a clinician-administered scale designed to assess autonomic symptoms and signs. The total score reflects the presence and severity of orthostatic hypotension, gastrointestinal, sphincteric, and sexual dysfunction symptoms.
R-ODS (Rasch-built Overall Disability Scale): a 24-item self-administered questionnaire that evaluates functional ability in daily activities. It is specific to patients with peripheral neuropathy and measures the condition’s impact of on everyday tasks. Scores on this scale help identify functional impairment, assessing an individual’s ability to function independently in daily life (7).
Norfolk QOL-DN (Norfolk Quality of Life-Diabetic Neuropathy): a 35-item self-administered questionnaire that assesses the quality of life in patients, with a scoring range from −4 to 136 (3). This tool is designed to evaluate symptoms associated with damage to different types of nerve fibers and is structured into five domains: physical functioning/large fiber neuropathy; activities of daily living; symptoms; small fiber neuropathy; and autonomic neuropathy (12, 23).
2. Neurophysiological Assessment
The Working Group proposes the same neurophysiological assessment protocol for all three patient groups, with timing variations based on the clinical scenario (as outlined in Table 1).
Neurophysiological tests will be performed according to a standardized methodology:
• With surface electrodes;
• Using the antidromic technique for sensory nerve conduction studies and ortodromic for motor nerves;
• Measuring amplitude of compoud motor action potential (CMAP) and sensory nerve action potential (SNAP) from the negative to the positive peak
• Unilaterally on the clinically most affected side, or randomly (right or left) in asymptomatic carriers for the Tibial, Peroneal, Radial, nerves; Bilaterally for the Median, Sural, Dorsal Sural nerves (the and Ulnar nerve is also examined bilaterally in presence of bilateral signs of distal slowing of the sensory and/or motor conduction of the Median nerve);
• Applying normative reference values derived from available scientific evidence (13–15) for each specific test.
Reference values for each parameter measured in the individual neurophysiological tests, along with detailed information on stimulation and recording techniques, are provided in Table 3
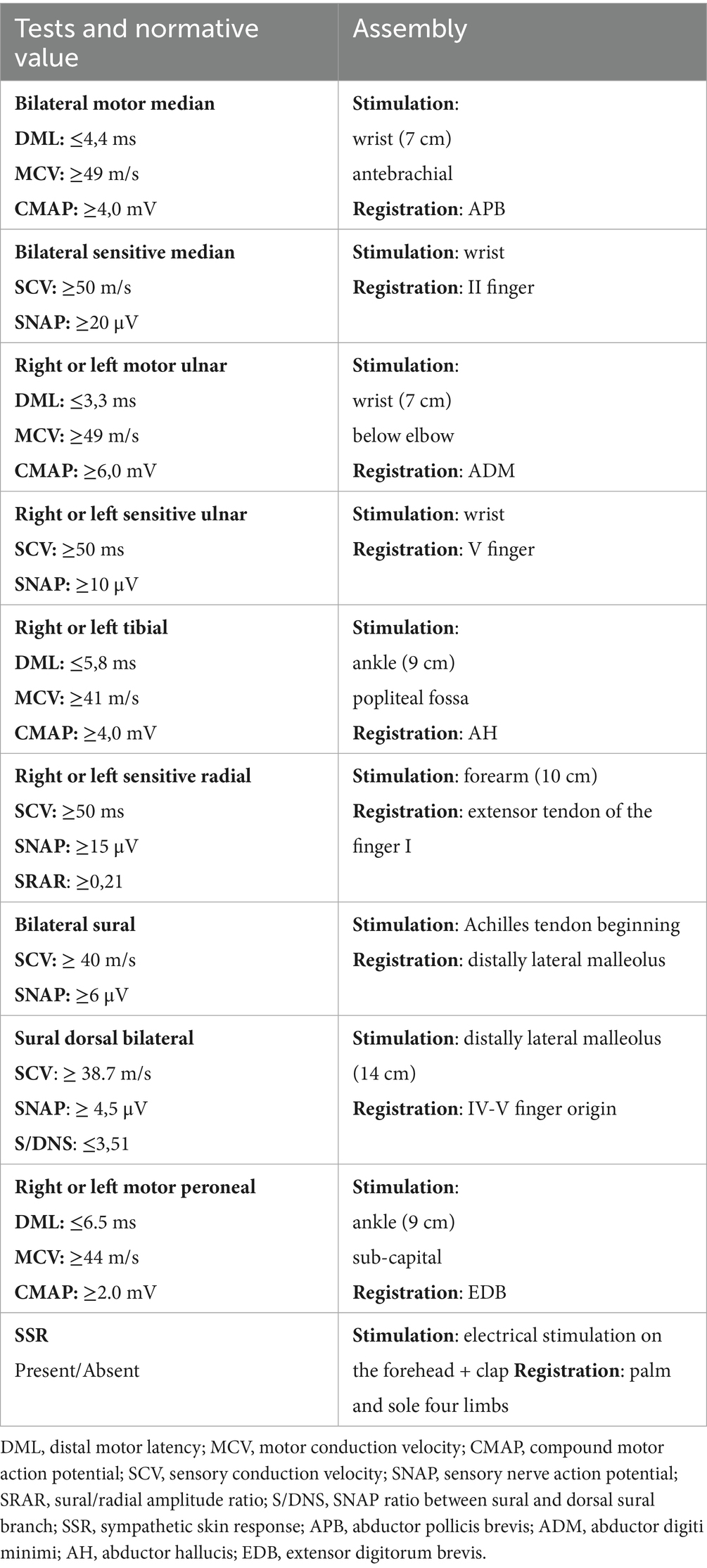
Table 3. Neurophysiological tests in the follow-up of patients with ATTR.
The most suitable neurophysiological scores applicable to real practice, derived from individual measurements, should be calculated as follows:
• Total NCS (Nerve Conduction Study): sum of sural SNAP, ulnar SAP, tibial CMAP, and peroneal CMAP (15).
• SNS (Sensory Neurophysiological Score): sum of sural SNAP and median SNAP (16).
• MNS (Motor Neurophysiological Score): sum of peroneal CMAP and median CMAP (16).
• Sural/DNS Ratio: ratio of sural SNAP to SNAP of the dorsal sural nerve branch (DNS) (14).
• SRAR (Sural/Radial Amplitude Ratio): ratio of sural SNAP to radial SNAP (13).
• SSRs (Sympathetic Skin Response score): sum of lower and upper limb SSRs on the same side (16).
Cardiological monitoring program for patients with ATTR-CA
For patients diagnosed with TTR-related cardiac amyloidosis, experts recommend cardiological evaluations every six months (i.e., cardiology visit, ECG, echocardiogram, and laboratory tests including NT-proBNP and local Troponin T-hs o I-hs), and annually (24-h Holter ECG).
In addition, it is recommended that the following assessments be performed every 6 months:
• Kansas City Cardiomyopathy Questionnaire (KCCQ)
• EuroQoL-5D (EQ-5D)
• 6-Minute Walk Test (6MWT)
Patients presenting with exclusively neurological involvement undergo the same evaluation protocol on a yearly basis.
The Working Group also agrees on the need for the following at the onset of specific symptoms and every 2 years afterwards:
• Nephrology consultation
• Gastroenterology consultation
• Ophthalmology consultation
Discussion
Implementation of a regionally shared protocol for the monitoring of ATTR amyloidosis represents a relevant advancement in standardizing care and addressing both diagnostic delays and heterogeneous disease management across healthcare settings. Compared with international recommendations (1, 7–9), the monitoring protocol established by the Emilia-Romagna ATTR Working Group incorporates core principles from existing guidelines, while tailoring them to the regional clinical reality, particularly regarding accessibility and the availability of resources in peripheral centers.
From a neurological standpoint, including of both clinical scales and neurophysiological studies within a structured timeline for asymptomatic carriers and symptomatic patients reflects an effort to balance sensitivity in early detection with feasibility in daily clinical practice. While the selected scales (NIS, FAP/PND, R-ODS, CADT, Norfolk QOL-DN) are consistent with international standards, the regional protocol emphasizes routine and scheduled use of such tools, which are often underutilized in real-world settings due to logistical constraints. Moreover, standardizing of nerve conduction techniques and adopting of composite scores (e.g., SNS, MNS, SRAR, Sural/DNS) aim to improve comparability across centers and over time, an essential step toward objective disease monitoring. These methodological refinements go beyond current recommendations by proposing specific parameters and thresholds, which may serve as a benchmark for other regional systems.
In the cardiology domain, the protocol remains aligned with ESC and AHA guidelines regarding frequency and type of assessments, including echocardiography, NT-proBNP, troponin levels, and quality of life measures such as KCCQ and EQ-5D. However, the regional approach underlines the importance of integrating functional tests (6MWT) and periodic imaging (in asymptomatic carriers whole-body scintigraphy or cardiac MRI) as part of routine monitoring, even without overt clinical progression. This reflects a proactive rather than reactive strategy, supported by evidence that early identification of subclinical changes can impact prognosis and therapeutic timing. Furthermore, while most centers adopt current cardiology staging systems, the protocol guides interdisciplinary consultations (e.g., nephrology, gastroenterology, ophthalmology), anticipating multisystemic involvement.
A key strength of this initiative lies in the formalization of a shared referral chain between tertiary and peripheral centers, allowing patients and asymptomatic carriers to access the same clinical tools regardless of geographic location. This organizational framework fosters equity of care, improves early detection, and enhances the regional capacity to manage a rare and complex condition. Continuous patient-level data collection through the REDCap-based registry provides the infrastructure for prospective outcome analyses. This will allow the identification of variability in care delivery, gaps in adherence to the protocol, and opportunities for improvement, consistent with the principles of implementation science (17). Future analyses will be essential to assess whether protocol adherence correlates with improved functional outcomes, delayed disease progression, or reduced healthcare utilization.
Some limitations must be acknowledged: although the survey captured variability across centers, some differences in resources and expertise may persist, potentially affecting the uniform application of monitoring tools. Secondly, while the protocol has been designed for generalizability, its applicability to other regions or healthcare systems may require adjustments.
Nonetheless, the Emilia-Romagna ATTR monitoring protocol demonstrates that it is possible to implement a structured, multidisciplinary, and scalable disease management model even within a heterogeneous regional context. Its success will depend not only on its initial design but also on continuous quality control, training of professionals, and alignment with evolving clinical evidence and therapeutic advances.
Emerging biomarkers and alternative monitoring strategies
While our regional protocol emphasizes standardized clinical, neurophysiological, and cardiological assessments, other innovative approaches are under investigation and may soon influence carrier monitoring strategies. Among these, cutaneous amyloid detection using minimally invasive skin punch biopsies has demonstrated promising diagnostic yield, even in pre-symptomatic stages, by identifying early amyloid deposition in peripheral small fibers (18, 19). This approach could provide an objective marker of disease onset in ATTRv carriers, potentially complementing or even anticipating changes detected by neurophysiological studies (20).
Another area of growing interest is the use of circulating biomarkers of axonal damage, particularly neurofilament light chain (NfL). Elevated plasma NfL levels have been associated with early axonal injury and correlate with disease severity in a variety of neurodegenerative and neuroinflammatory disorders (21). Recent studies in ATTRv carriers suggest that NfL may rise before overt clinical symptoms, providing a quantifiable, minimally invasive indicator of subclinical neurodegeneration (22).
Although neither cutaneous amyloid detection nor NfL assays are currently widely available in our region, their incorporation into future standardized protocols could further enhance early detection and risk stratification. By combining these emerging biomarkers with established clinical and instrumental tools, a multimodal monitoring approach may become feasible, ultimately improving personalized timing for therapeutic intervention.
Conclusion
The multidisciplinary and multicentric ATTR Working Group project represents a response to the diagnostic and therapeutic unmet needs in managing patients and asymptomatic carriers with ATTRv. Since 2022, it has involved all hospital facilities across the Emilia-Romagna region, enabling the identification and validation of a sustainable and standardized diagnostic and care pathway. The program is tailored to the specific needs of ATTRv patients, with particular emphasis on both clinical management (care and follow-up) and the practical and psychological dimensions, including quality of life (QoL).
The ATTRv monitoring protocol is continuously evolving, whereby analysis of patients’ data collected into a web-based registry, will enhance the clinical setting of ATTRv, promote the uptake of research findings into routine healthcare and inform Healthcare providers on patient care pathway efficiency and treatment outcomes.
Author contributions
PG: Writing – original draft, Writing – review & editing. IA: Methodology, Validation, Writing – review & editing. AA: Data curation, Methodology, Writing – review & editing. TB: Investigation, Project administration, Supervision, Writing – review & editing. ABa: Conceptualization, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing. FeB: Data curation, Resources, Validation, Writing – review & editing. MB: Methodology, Software, Validation, Writing – review & editing. EB: Formal analysis, Funding acquisition, Supervision, Writing – review & editing. FrB: Methodology, Validation, Visualization, Writing – review & editing. ABo: Data curation, Validation, Visualization, Writing – review & editing. CB: Data curation, Formal analysis, Visualization, Writing – review & editing. GB: Formal analysis, Investigation, Methodology, Visualization, Writing – review & editing. IC: Investigation, Methodology, Validation, Visualization, Writing – review & editing. SC: Investigation, Supervision, Validation, Visualization, Writing – review & editing. LC: Formal analysis, Supervision, Validation, Writing – review & editing. MC: Supervision, Validation, Visualization, Writing – review & editing. FD: Resources, Supervision, Validation, Writing – review & editing. RD'A: Validation, Visualization, Writing – review & editing. RG: Formal analysis, Supervision, Visualization, Writing – review & editing. FS: Data curation, Resources, Validation, Writing – review & editing. EG: Conceptualization, Supervision, Validation, Visualization, Writing – review & editing. GL: Supervision, Validation, Visualization, Writing – review & editing. CL: Conceptualization, Supervision, Validation, Visualization, Writing – review & editing. FM: Supervision, Validation, Visualization, Writing – review & editing. MM: Supervision, Validation, Visualization, Writing – review & editing. GM: Supervision, Validation, Visualization, Writing – review & editing. EM: Supervision, Validation, Visualization, Writing – review & editing. AM: Supervision, Validation, Visualization, Writing – review & editing. DM: Supervision, Validation, Visualization, Writing – review & editing. EnP: Project administration, Supervision, Validation, Visualization, Writing – review & editing. AP: Data curation, Supervision, Visualization, Writing – review & editing. EmP: Supervision, Validation, Visualization, Writing – review & editing. MR: Supervision, Validation, Visualization, Writing – review & editing. RR: Methodology, Supervision, Validation, Visualization, Writing – review & editing. DS: Supervision, Validation, Visualization, Writing – review & editing. KS: Supervision, Validation, Visualization, Writing – review & editing. MSe: Formal analysis, Supervision, Validation, Visualization, Writing – review & editing. MSg: Supervision, Validation, Visualization, Writing – review & editing. ES: Methodology, Supervision, Validation, Visualization, Writing – review & editing. AS: Supervision, Validation, Visualization, Writing – review & editing. CT: Supervision, Validation, Visualization, Writing – review & editing. CV: Supervision, Validation, Visualization, Writing – review & editing. GV: Methodology, Validation, Visualization, Writing – review & editing. MV: Supervision, Validation, Visualization, Writing – review & editing. SL: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The work reported in this publication was funded by the Italian Ministry of Health, RC-2025-2794599.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer LL declared a past co-authorship with the author IC to the handling editor.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2025.1666318/full#supplementary-material
References
1. Conceição, I, Coelho, T, Rapezzi, C, Parman, Y, Obici, L, Galán, L, et al. Assessment of patients with hereditary transthyretin amyloidosis - understanding the impact of management and disease progression. Amyloid. (2019) 26:103–11. doi: 10.1080/13506129.2019.1627312
2. Ton, VK, Mukherjee, M, and Judge, DP. Transthyretin cardiac amyloidosis: pathogenesis, treatments, and emerging role in heart failure with preserved ejection fraction. Clin Med Insights Cardiol. (2015) 8:39–44. doi: 10.4137/CMC.S15719
3. Kim, HM, Sohn, DW, and Paeng, JC. Prevalence of positive 99 mTc-DPD scintigraphy as an indicator of the prevalence of wild-type transthyretin amyloidosis in the elderly. Int Heart J. (2019) 60:643–7. doi: 10.1536/ihj.18-345
4. Fumagalli, C, Longhi, S, Aimo, A, Argirò, A, Barilaro, A, Biagini, E, et al. Change in prevalence of ATTR variants in Italy - results from a National Survey. Eur Heart J Qual Care Clin Outcomes. (2025). doi: 10.1093/ehjqcco/qcaf024
5. Maestro-Benedicto, A, Vela, P, de, F, Mora, N, Pomares, A, Gonzalez-Vioque, E, et al. Frequency of hereditary transthyretin amyloidosis among elderly patients with transthyretin cardiomyopathy. Eur J Heart Fail. (2022) 24:2367–73. doi: 10.1002/ejhf.2658
6. Longhi, S, Biagini, E, Guaraldi, P, Carigi, S, Dossi, MC, Bartolotti, M, et al. Temporal implementation of a regional referral pathway in transthyretin cardiac amyloidosis: Emilia-Romagna experience. J Cardiovasc Med (Hagerstown). (2024) 25:682–92. doi: 10.2459/JCM.0000000000001633
7. Adams, D, Algalarrondo, V, Polydefkis, M, Sarswat, N, Slama, MS, and Nativi-Nicolau, J. Expert opinion on monitoring symptomatic hereditary transthyretin-mediated amyloidosis and assessment of disease progression. Orphanet J Rare Dis. (2021) 16:411. doi: 10.1186/s13023-021-01960-9
8. Garcia-Pavia, P, Rapezzi, C, Adler, Y, Arad, M, Basso, C, Brucato, A, et al. Diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J. (2021) 42:1554–68. doi: 10.1093/eurheartj/ehab072
9. Condoluci, A, Théaudin, M, Schwotzer, R, Pazhenkottil, AP, Arosio, P, Averaimo, M, et al. Management of transthyretin amyloidosis. Swiss Med Wkly. (2021) 151:w30053. doi: 10.4414/smw.2021.w30053
10. Capeline, LS, Oishi, GSL, Mancuso, FJN, Squassante Capeline, L, Lobo Oishi, GS, Neves Mancuso, FJ, et al. Advanced echocardiographic assessment in transthyretin amyloidosis: early phenotype markers in mutation carriers. Eur Heart J Cardiovasc Imaging. (2025) 26:863–5. doi: 10.1093/ehjci/jeaf067
11. Cisneros-Barroso, E, González-Moreno, J, Rodríguez, A, Ripoll-Vera, T, Álvarez, J, Usón, M, et al. Anticipation on age at onset in kindreds with hereditary ATTRV30M amyloidosis from the Majorcan cluster. Amyloid. (2020) 27:254–8. doi: 10.1080/13506129.2020.1789580
12. Dyck, PJB, González-Duarte, A, Obici, L, Polydefkis, M, Wiesman, JF, Antonino, I, et al. Development of measures of polyneuropathy impairment in hATTR amyloidosis: from NIS to mNIS + 7. J Neurol Sci. (2019) 405:116424. doi: 10.1016/j.jns.2019.116424
13. Esper, GJ, Nardin, RA, Benatar, M, Sax, TW, Acosta, JA, and Raynor, EM. Sural and radial sensory responses in healthy adults: diagnostic implications for polyneuropathy. Muscle Nerve. (2005) 31:628–32. doi: 10.1002/mus.20313
14. Luigetti, M, Guglielmino, V, Romozzi, M, Romano, A, di Paolantonio, A, Bisogni, G, et al. Nerve conduction studies of dorsal sural nerve: normative data and its potential application in ATTRv pre-symptomatic subjects. Brain Sci. (2022) 12:1037. doi: 10.3390/brainsci12081037
16. Castro, J, Miranda, B, de Castro, I, and Conceição, I. Changes in nerve conduction studies predate clinical symptoms onset in early onset Val30Met hereditary ATTR amyloidosis. Eur J Neurol. (2022) 29:826–32. doi: 10.1111/ene.15176
17. Wensing, M. Implementation science in healthcare: introduction and perspective. Z Evid Fortbild Qual Gesundhwes. (2015) 109:97–102. doi: 10.1016/j.zefq.2015.02.014
18. Leonardi, L, Adam, C, Beaudonnet, G, Beauvais, D, Cauquil, C, Not, A, et al. Skin amyloid deposits and nerve fiber loss as markers of neuropathy onset and progression in hereditary transthyretin amyloidosis. Eur J Neurol. (2022) 29:1477–87. doi: 10.1111/ene.15268
19. Freeman, R, Gonzalez-Duarte, A, Barroso, F, Campagnolo, M, Rajan, S, Garcia, J, et al. Cutaneous amyloid is a biomarker in early ATTRv neuropathy and progresses across disease stages. Ann Clin Transl Neurol. (2022) 9:1370–83. doi: 10.1002/acn3.51636
20. Schulz, N, Beauvais, D, Cauquil, C, Labeyrie, C, Iliescu, I, Francou, B, et al. Intracutaneous amyloid deposition is associated with nerve conduction studies deterioration in presumed asymptomatic pathogenic variant TTR carriers. Eur J Neurol. (2025) 32:e70277. doi: 10.1111/ene.70277
21. Khalil, M, Teunissen, CE, Lehmann, S, Otto, M, Piehl, F, Ziemssen, T, et al. Neurofilaments as biomarkers in neurological disorders - towards clinical application. Nat Rev Neurol. (2024) 20:269–87. doi: 10.1038/s41582-024-00955-x
22. Romano, A, Primiano, G, Antonini, G, Ceccanti, M, Fenu, S, Forcina, F, et al. Serum neurofilament light chain: a promising early diagnostic biomarker for hereditary transthyretin amyloidosis? Eur J Neurol. (2024) 31:e16070. doi: 10.1111/ene.16070
Keywords: amyloid, transthyretin, multidisciplinary collaboration, standardized care, protocol, equity, equality, sustainability
Citation: Guaraldi P, Allegri I, Ariatti A, Baldini T, Barbieri A, Barocelli F, Bartolotti M, Biagini E, Bianchi F, Borghi A, Borghi C, Boriani G, Cani I, Carigi S, Codeluppi L, Currò Dossi M, Dalpozzo F, D’Angelo R, De Gennaro R, Di Spigno F, Gardini E, Lanati G, Leuzzi C, Marzo F, Masullo M, Mazzanti G, Merli E, Milandri A, Monari D, Perugini E, Ponziani A, Postiglione E, Rasia M, Rinaldi R, Scancarello D, Serafini K, Serenelli M, Sguazzotti M, Siena E, Simone AM, Terracciano C, Valenti C, Vitale G, Vitiello M and Longhi S (2025) Monitoring patients and asymptomatic carriers with hereditary transthyretin amyloidosis: regional protocol of Emilia-Romagna ATTR working group. Front. Neurol. 16:1666318. doi: 10.3389/fneur.2025.1666318
Edited by:
Giacomo Tini, Sapienza University of Rome, ItalyReviewed by:
Luca Leonardi, Sapienza University of Rome, ItalySteven Muller, University Medical Center Utrecht, Netherlands
Copyright © 2025 Guaraldi, Allegri, Ariatti, Baldini, Barbieri, Barocelli, Bartolotti, Biagini, Bianchi, Borghi, Borghi, Boriani, Cani, Carigi, Codeluppi, Currò Dossi, Dalpozzo, D’Angelo, De Gennaro, Di Spigno, Gardini, Lanati, Leuzzi, Marzo, Masullo, Mazzanti, Merli, Milandri, Monari, Perugini, Ponziani, Postiglione, Rasia, Rinaldi, Scancarello, Serafini, Serenelli, Sguazzotti, Siena, Simone, Terracciano, Valenti, Vitale, Vitiello and Longhi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Simone Longhi, bG9uZ2hpc2ltb25lQGdtYWlsLmNvbQ==