Abstract
The neurodevelopmental disorder autism spectrum disorder (ASD) affects 0.5%–1% of the global population and is marked by ongoing difficulties in social communication and cognitive function. Interestingly, ASD has been reported to share a genetic origin with epilepsy, a condition marked by recurrent, unprovoked seizures. Both ASD and epilepsy are caused by multifactorial and multigenetic origin. Whereas the number of genes linked to ASD etiology are growing, the genetic basis of epilepsy is more diverging leading to distinct epileptic syndromes. Despite decades of discussion, a comprehensive understanding of the genetic interplay between these disorders remains elusive. Our article focuses on investigating the shared genetic basis of abnormalities in synaptic proteins, highlighting the presynaptic compartment, which is less explored compared to the postsynaptic elements. We identify those biological processes linked to the presynaptic compartment, such as presynaptic assembly, ATP metabolism, various aspects of the synaptic vesicle cycle, are commonly affected across conditions, as evidenced by the shared genetics. Hence, this study offers initial insights into presynaptic signaling, and further research could aid in developing improved therapeutic strategies by targeting these presynaptic processes.
1 Introduction
Autism spectrum disorder (ASD) is a neurodevelopmental disorder that typically manifest at birth, persisting throughout an individual’s life causing behavioral, cognitive, and social challenges. Epilepsy, on the other hand, can occur throughout the life span of an individual due to various other neurological compromises including stroke, tumors or other pathologies. Notably, epilepsy is a common comorbid condition in individuals with ASD. Although both environmental and genetic factors contribute to the co-occurrence of ASD and epilepsy (1), genetic factors play a predominant role in the development of these disorders. Recent studies highlight that the co-occurrence of ASD and epilepsy is largely driven by disruptions in fundamental neurodevelopmental pathways. Shared genetic mutations affecting ion channels, synaptic proteins, and transcription factors contribute to these disruptions, leading to altered neural connectivity and excitability that underlie both autistic behaviors and epileptic seizures (2). Approximately 10%–20% of individuals with ASD share genetic factors that overlap with epilepsy (3). Interestingly, about 30% of individuals diagnosed with epilepsy also meet certain diagnostic criteria for ASD (4, 5). Notably, epilepsy may potentially contribute to the development of ASD, or conversely, the abnormal brain circuitry underlying ASD could predispose individuals to epileptic seizures.
The genetic causes of ASD and epilepsy involve dysregulation of synaptic functions due to mutations in genes such as SYN1 (synapsin-1), SCN2A, and SCN8A (sodium voltage-gated channel alpha subunit 2 and 8), KCNQ2 and KCNQ5 (potassium voltage-gated channel subfamily Q member 2 and 5), SHANK3 (glutamate receptor signaling protein SH3 and multiple ankyrin repeat domains 3), GABRG2 or GABRG3 (gamma-aminobutyric acid type A receptor gamma subunits 2 and 3). These genes are typically linked to synaptic compartments, and span across the pre- and post-synapse (6–9). However, despite its critical roles in neurotransmitter maintenance and release, neural circuit development, and activity regulation, our understanding of the presynaptic compartment in relation to neuropathology remains elusive. Although many genes are shared between ASD and epilepsy, the effects of specific regulatory mutations—such as loss- or gain-of-function variants—on disease onset and severity remain poorly understood. This highlights the need for closer examination of the functional consequences of these variants.
Current knowledge of proteins localized to the presynaptic active zone, such as RIM (Rab3A-interacting molecule), RIM-BP (RIM-binding protein), BSN (Bassoon), PCLO (Piccolo), PPFIA1 (Liprin-α), is limited despite their crucial roles. These active zone-specific scaffolding molecules have been associated with various conditions including ASD, intellectual disability, epilepsy, or schizophrenia (10–16). Their implications in these diseases underscore their significant impact on synaptic transmission and circuit development.
In this study, we investigate the genetic associations between ASD and epilepsy, specifically exploring the signaling pathways mediated by presynaptic genes. Our objective is to shed light on potential alterations in presynaptic and overall synaptic functions, thereby characterizing the presynaptic compartment as a target for novel therapeutic drug interventions. By conducting a systematic literature review and employing subsequent synaptic enrichment analysis using the SynGO database, we identified common genes associated with both ASD and epilepsy, highlighting a significant subset of synaptic genes. Beyond cataloging shared genes, our analysis specifically focuses on the nature of identified variants (loss- versus gain-of-function) and their mechanistic impact on synaptic processes. This approach provides a more comprehensive understanding of the genetic and functional interplay underlying ASD and epilepsy comorbidity. Moreover, characterizing variants as loss- or gain-of-function will help identifying promising candidates for precision therapies targeting synaptic dysfunctions.
2 Methods
A list of genes linked to both ASD and epilepsy was compiled through an extensive literature search on google scholar and PubMed using the keywords “comorbidity of ASD and epilepsy”; “ASD in epilepsy”; “Epilepsy percentage in ASD”; “genetics of ASD and epilepsy” and from the databases for ASD (SFARI: https://gene.sfari.org/database/human-gene/), epilepsy (EpilepsyGene: http://www.wzgenomics.cn/EpilepsyGene/index.php; epiGAD: https://www.epigad.org/index.html; CarpeDG: http://carpedb.ua.edu/search.cfm). Common genes implicated in both diseases were compiled, and a list of associated synaptic genes was identified using the synaptic gene ontologies (SynGO) database (17). Using the domain ‘Cellular Components’ (location), genes localized to the presynaptic region were identified. Their involvement in various processes was further identified and focused on by using the domain ‘Biological Process’ (Supplementary Figure 1).
3 Results and discussion
In our SynGO analysis, we identified 49 synaptic genes out of 125 common genes (Supplementary Table 1). Among these, 16 genes are exclusively associated with presynaptic localization and function, while 19 genes are linked to postsynaptic roles. Employing SynGO enrichment analysis, we further identified several synaptic genes based on the localization and biological processes that are common to both ASD and epileptic phenotypes (Supplementary Figure 2A and Supplementary Table 2). Additionally, 14 genes are shared between the pre- and post-synapse (Supplementary Figure 2A). Based on this analysis, the presynaptic genes are specifically localized to various cellular components (Supplementary Figure 2B).
The list of identified common ASD-epilepsy genes localized at the presynaptic compartment are involved in various synaptic processes, including synaptic assembly, regulation of presynaptic processes, synaptic signaling, and metabolism (Figures 1A,B). Mutations in these associated genes or resulting protein dysfunctions have been shown to impact these processes during the progression of ASD and epilepsy.
Figure 1

(A) Sunburst image depicts the gene enrichment analyses for common synaptic genes associated with ASD and epilepsy, categorized by biological processes. (B) Summary of the SynGO gene ontology database, categorizing gene products based on their biological processes and the functional processes that they are linked to. Key process within the presynapse, such as the synaptic vesicle cycle and regulation of membrane potential, show significant enrichment. Additionally, processes related to synapse organization indicating the disruptions in overall synaptic function in both ASD and epilepsy, primarily originating from the presynaptic compartment.
There are nearly 40% of genes associated with both ASD and epilepsy are synaptic genes, as identified through a gene ontology study using SynGO and the presynaptic function is as crucial as postsynaptic function in disease pathogenesis. While much attention has been devoted to understanding the postsynaptic receptor signaling in disease progression and drug development, knowledge about the presynaptic compartment remains limited. Our analysis underscores the significant enrichment of various processes within the presynaptic compartment. Disruption of these processes could have profound impact on overall synaptic function (Figure 2B), highlighting the critical need to investigate presynaptic mechanisms for a comprehensive understanding of disorders.
Figure 2
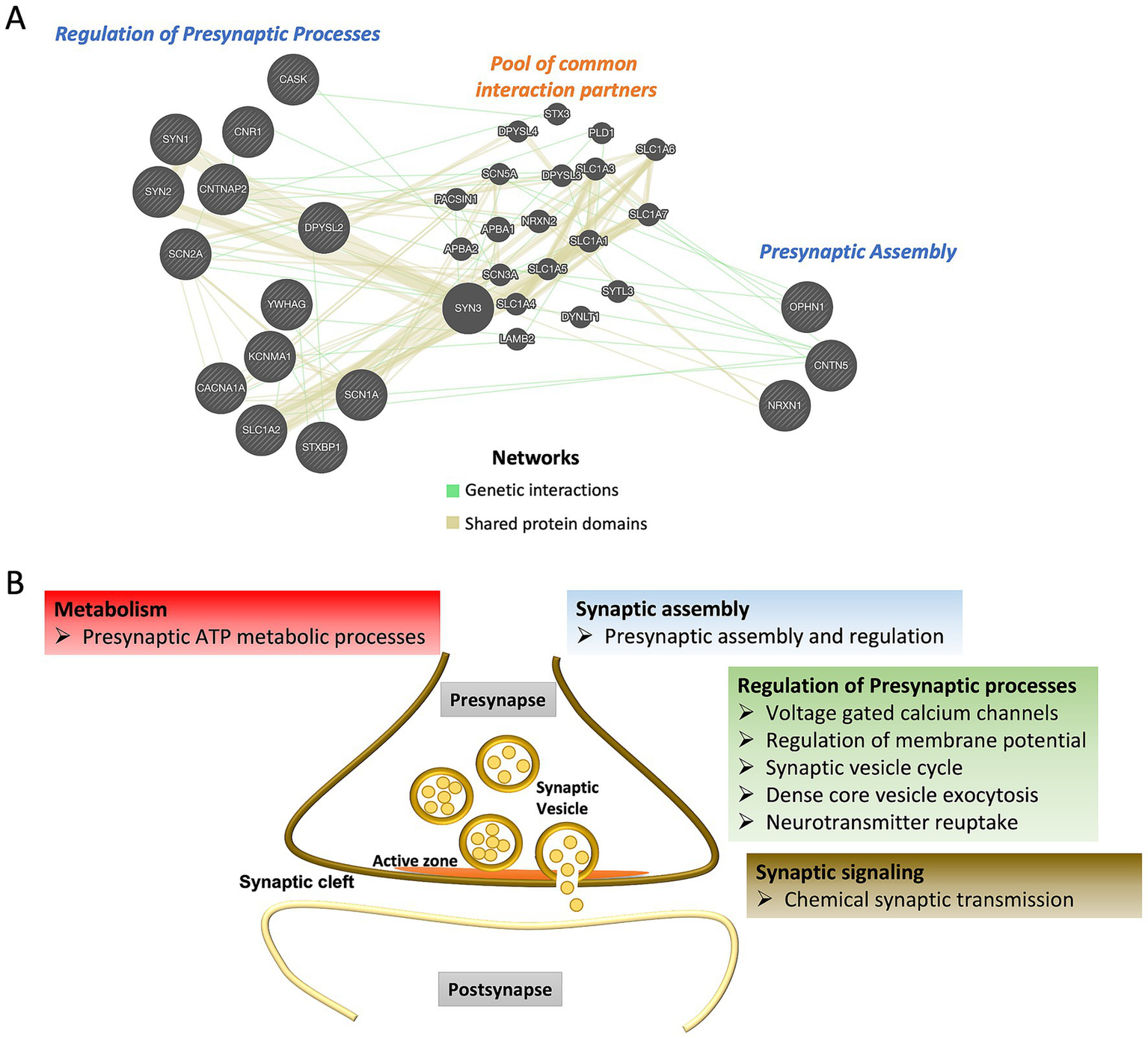
(A) Interactions among the shared ASD and epilepsy presynaptic genes regulating different biological functions. GeneMANIA database was used to identify interaction partners. (B) Schematic representation of presynaptic processes linked to common genes for ASD and epilepsy.
The Presynaptic Assembly involves three major steps: neuronal contact formation, synaptic precursor transport, and the cessation of transport processes at the contact sites. The CNTN5 gene encodes the protein Contactin-5, a member of the immunoglobulin superfamily of cell adhesion molecules critical for nervous system development, particularly in axonal contact formation. CNTN5 is primarily expressed postnatally in the central nervous system, including the cerebral cortex (auditory cortex), thalamus, and caudate putamen (18). Gene mutations or copy number variation (CNVs) in CNTN5 have been linked to ASD and epilepsy (19, 20). Loss of CNTN5 leads to synaptic dysfunction, resulting in heightened neuronal excitability (21).
OPHN1 encodes Oligophrenin-1, a Rho-GTPase-activating protein (RhoGAP) expressed ubiquitously in the developing brain. Oligophrenin-1 functions as extracellular growth and guiding signal mediators important for the linking of these signals originated from the cell-surface adhesion molecules to the intracellular signal transduction pathways. These pathways are crucial for neuronal morphogenesis, and cytoskeletal dynamics by orienting the actin molecules at axonal growth cones (22, 23). Deletion or mutations in OPHN1 are associated with nonspecific X-linked intellectual disability, ASD, intellectual disability, epilepsy, enlargement of ventricles in the brain, ataxia, and cerebellar hypoplasia (24). Loss of OPHN1 function results in impaired maturation of dendritic spines (25).
The NRXN1 gene encodes Neurexin 1, a presynaptically localized membrane protein involved in the formation of Ca2+-dependent surface receptor complexes. Neurexins form complexes with neuroligins, facilitating efficient synaptic contact formation and neurotransmission by linking calcium (Ca2+) channels to synaptic vesicles for exocytosis (26). The expression of different neurexins occurs during early cortical plate formation before extensive synaptogenesis takes place, with age-dependent increase in the expression of Neurexin1 (27). Mutations in the human NRXN1 gene have been implicated in several neuropathological conditions, including ASD, schizophrenia, autosomal recessive intellectual disability, Pitt-Hopkins-like syndrome, attention-deficit hyperactivity disorder (ADHD), and epilepsy (28, 29). Loss-of-function mutations in the NRXN1 gene disrupt protein–protein interactions, leading to synaptic dysfunctions, whereas gain-of-function mutations promote increased excitatory synaptogenesis and neuronal excitability, potentially via enhanced calcium signaling (30–32).
The Regulation of Presynaptic Processes, such as maintaining Ca2+ levels, ion channel activity to balance the membrane potential, and the synaptic vesicle cycle (encompassing exocytosis and neurotransmitter reuptake), is mediated by several proteins at the presynaptic terminal. The list of identified presynaptic genes from our analysis falls under the processes mentioned above that occur at the presynaptic compartment. CACNA1A encodes the α1A pore-forming subunit of the voltage-gated P/Q-type calcium channel (Cav2.1), which mediates its function at the presynaptic terminal (33). These channels are widely expressed throughout the central nervous system and are particularly abundant in brain regions such as the cerebellum, especially in Purkinje and granule cells (34). The Cav2.1 channel facilitates synaptic vesicle exocytosis through Ca2+-influx, thus playing a crucial role in neurotransmission. Haploinsufficiency or de novo mutations in the CACNA1A gene can lead to the development of epileptic encephalopathy, ASD, and schizophrenia (35).
The CNR1 gene encodes the type 1 cannabinoid receptor (CB1), which is part of the endocannabinoid system and is the receptor for the most widely used yet controversial psychoactive drug, cannabis. CNR1 expression is higher during the fetal stage compared to the postnatal stage in various brain areas, such as the prefrontal cortex, hippocampus, and caudate. The CB1 receptor, a G-protein-coupled receptor, is expressed presynaptically on neuronal terminals in brain regions including the hippocampus, amygdala, hypothalamus, midbrain, frontal cortex, and cerebellum, where it regulates the gamma-aminobutyric acid (GABA)ergic and glutamatergic transmission (36, 37). Genetic variations in the CNR1 gene are associated with neurological disorders, including ASD (38).
The Potassium Calcium-Activated Channel Subfamily M Alpha 1, encoded by KCNMA1 gene and commonly referred to as the Big K + (BK) channel exhibits exceptionally high conductance (>100 pS). These channels are predominantly expressed in the brain and muscle tissues and are classified within the voltage-gated K + channel family. BK channels are recognized for their ability to respond to changes in voltage, thereby regulating excitability through mediating potassium efflux, alongside intracellular calcium levels, making them pivotal in regulating neuronal and muscular function. Dysfunction or loss of BK channel can result from mutations or single nucleotide polymorphisms (SNPs) in the KCNMA1 gene. Such genetic alterations have been implicated in various disorders including autism, intellectual disability, epilepsy, hypertension, asthma (39, 40).
SCN1A and SCN2A encode the alpha subunit of the voltage-gated sodium channels Nav1.1 Nav1.2 (41). Both channels are expressed in the central nervous system and function as transmembrane protein complexes composed of glycosylated alpha subunits that form ion-conducting pores. Together, they play a crucial role in sodium exchange, as well as action potential generation and propagation among neurons, thus regulating excitability. Nav1.1 and Nav1.2 are widely distributed across the cerebral cortex, hippocampal CA3 and CA2 regions, dentate gyrus, thalamus, substantia nigra, putamen and cerebellum (42). SCN1A and SCN2A are considered risk genes for ASD due to their proximity to autism susceptibility loci on chromosomes (43). Additionally, mutations in these genes are associated with various forms of seizures, such as generalized epilepsy with febrile seizures plus or myoclonic epilepsy (44). Loss of SCN1A impairs inhibitory neuron excitability, leading to Dravet syndrome and ASD-like features, whereas gain-of-function mutations contribute to early-onset epilepsy and familial hemiplegic migraine type 3 (FHM3) (45). Similarly, gain-of-function variants in SCN2A are associated with early-infantile epilepsies (seizure onset before 3 months of age), while loss-of-function variants result in late-onset epilepsies and ASD/ID (46).
Synapsin family proteins, such as Synapsin1 encoded by SYN1 and Synapsin2 encoded by SYN2, are phosphoproteins that bind to synaptic vesicles (SVs). They are essential for neurotransmitter release and synaptic plasticity by participating in various steps of the SV cycle, including SV tethering, docking, fusion. These proteins also play an important role in synaptogenesis and have been implicated to be involved in key aspects of neuronal development, axonogenesis, and synaptic maintenance (47). As SYN1 and SYN2 are X-linked genes, mutations in these genes are associated to X-linked neurodevelopmental disorders, primarily affecting males with clinical presentation of epilepsy, learning disabilities, etc. Additionally, genetic variants in SYN1 and SYN2 are linked to ASD traits and X-linked intellectual disability across various ethnic backgrounds (47, 48). Mutations in SYN1 impair neurotransmitter release, neurite outgrowth, and synaptic vesicle pool trafficking (47, 49). Similarly, the loss-of-function mutations in SYN2 produce phenotypes nearly identical to those observed with SYN1 variants (48).
Calcium/Calmodulin Dependent Serine Protein Kinase (CASK) is a protein-coding gene belonging to the MAGUK (membrane-associated guanylate kinase) family of proteins and is ubiquitously expressed in the developing brain. At the presynaptic compartment, CASK regulates SV exocytosis, interacts with NRXN1, and contributes to maintaining the excitatory/inhibitory (E/I) balance by modulating ionotropic receptor trafficking (50). Located on the X-chromosome, loss of CASK is associated with X-linked intellectual disability, ASD and epilepsy (13). Recent studies have shown that loss-of-function mutations in CASK result in distinct phenotypes, including impaired neuronal outgrowth during development and reduced excitability during adulthood (51).
The STXBP1 gene encodes syntaxin-binding protein 1 (also known as MUNC18-1), which plays a role in neurotransmitter release by participating in SV cycle steps such as docking, priming and fusion through interactions with SNARE proteins (52). De novo heterozygous mutations in STXBP1 lead to severe forms of epileptic encephalopathies, including Ohtahara syndrome or Dravet syndrome (53). Mutations in the STXBP1 gene have been linked to intellectual disability and other neurodevelopmental conditions, such as ASD (54). While loss of STXBP1 leads to presynaptic dysfunction, neurodegeneration, and hyperexcitability (55, 56), gain-of-function mutations enhance synaptic functions (57).
The SLC1A2 gene encodes Solute Carrier Family1 Member2 (EAAT2), a member of the solute transporter protein family. SLC1A2 is responsible for clearing glutamate from the extracellular space between synapses and facilitates its reuptake to maintain excitatory neurotransmission. EAAT2 is the predominant glutamate transporters in the brain, accounting for over 95% of total glutamate uptake activity (58). Mutations in the SLC1A2 gene are primarily associated with epileptic encephalopathy, with some reports also linking them to ASD and intellectual disability (59). Mutations in SLC1A2 cause glutamate dysregulation, disrupted Ca2+ storage in the endoplasmic reticulum, and reduced EAAT2 expression and glutamate transport (60). Mild gain-of-function variants of SLC1A2 lead to modest increases in anion currents (61).
The CNTNAP2 gene, primarily active during the brain development, encodes the single-pass transmembrane protein contactin-associated protein-like 2 (CASPR2) protein. As a member of cell adhesion molecules, such as the neurexin superfamily, CASPR2 is crucial for synapse formation, neurite outgrowth and myelination through its interaction with contactin-1. The expression of CNTNAP2 is restricted to specific regions of the brain, including the cortex, striatum, and thalamus, thereby participating in the regulation of higher cognitive functions. Loss-of-function mutations in CNTNAP2 disrupt excitatory neuron development, reduce neurite branching and neuronal complexity, and impair cortical connectivity, contributing to intellectual disability, ASD, epilepsy, schizophrenia, and depression (62–65).
Dihydropyrimidinase-protein 2, also known as Collapsin response mediator protein-2, is encoded by the DPYSL2 gene and is crucial for neuronal development, cell migration and axonal growth and guidance, thus contributing to neuronal polarity. Dihydropyrimidinase-protein 2 is also involved in synaptic transmission, calcium homeostasis, neurotransmitter release, cytoskeletal dynamics and vesicle trafficking (66). Polymorphisms or mutations in DPYSL2 are associated with schizophrenia, intellectual disability, and epilepsy (67, 68). Loss of DPYSL2 leads to defects in axonal pruning and corpus callosal axon guidance (69).
The YWHAG gene encodes the adapter protein 14–3-3 protein gamma, a member of the 14–3-3 protein family, which is ubiquitously expressed in brain. 14–3-3 proteins bind to various other proteins containing phosphoserine sites and are involved in neuronal migration by mediating signal transduction. Through interactions with presynaptic active zone proteins, 14–3-3 regulates presynaptic remodeling during synaptic plasticity and long-term potentiation (70). De novo missense mutations in YWHAG are linked to epileptic encephalopathies, ASD and intellectual disability (71, 72).
Interactions among these genes (also known as epistasis) or the end products-proteins is responsible for physiological functions as well drive the complexity of disease pathology. List of identified shared presynaptic genes display interactions among and in between the biological processes arguing for a crosstalk among different functional aspects and synergistic approach in mediating the crucial synaptic functions (Figure 2A and Supplementary Table 3).
In conclusion, our analysis highlights the critical role of presynaptic signaling, which can be disrupted by mutations in genes commonly associated to both ASD and epilepsy. While the relationship between these two disorders has been described for decades, substantial evidence for a shared mechanistic basis underlying their core symptoms and for the efficacy of therapeutic intervention remains limited. Emerging data suggest that dysfunction of presynaptic genes is a key contributor to disease progression in both conditions. So far, a handful of studies have highlighted that targeting specific presynaptic components, such as receptors regulating neurotransmitter release or kinases essential for axonal transport, may offer promising avenues for pharmacological interventions (73, 74). Nevertheless, a more in-depth investigation into presynaptic signaling pathways and mechanisms mediating various presynaptic processes and assembly (Figure 2B) could provide additional targets for novel therapeutics. Future interventions should carefully consider the functional consequences of diverse gene mutations, including gain- and loss-of-function variants, to enable precision therapeutics for these comorbidities.
Statements
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
MS: Data curation, Formal analysis, Investigation, Methodology, Software, Visualization, Writing – original draft, Writing – review & editing. SP: Data curation, Formal analysis, Investigation, Methodology, Software, Visualization, Writing – original draft, Writing – review & editing. PD: Data curation, Formal analysis, Investigation, Visualization, Writing – review & editing. SM: Formal analysis, Methodology, Software, Writing – review & editing. BG: Formal analysis, Methodology, Visualization, Writing – review & editing. KS: Data curation, Formal analysis, Software, Writing – review & editing. JCK: Methodology, Visualization, Writing – original draft, Writing – review & editing. AA: Conceptualization, Investigation, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
We acknowledge the Department of Biotechnology, School of Bioengineering for the support and infrastructure. Further, we acknowledge the supported by School of Arts and Sciences, Sai University to AA. JCK is supported by the Deutsche Forschungsgemeinschaft (DFG, German Science Foundation—516641042).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2025.1677134/full#supplementary-material
References
1.
Lukmanji S Manji SA Kadhim S Sauro KM Wirrell EC Kwon CS et al . The co-occurrence of epilepsy and autism: a systematic review. Epilepsy Behav. (2019) 98:238–48. doi: 10.1016/j.yebeh.2019.07.037
2.
Specchio N di Micco V Aronica E Auvin S Balestrini S Brunklaus A et al . The epilepsy–autism phenotype associated with developmental and epileptic encephalopathies: new mechanism-based therapeutic options. Epilepsia. (2025) 66:970–87. doi: 10.1111/epi.18209
3.
Sgadò P Dunleavy M Genovesi S Provenzano G Bozzi Y . The role of GABAergic system in neurodevelopmental disorders: a focus on autism and epilepsy. Int J Physiol Pathophysiol Pharmacol. (2011) 3:223–35.
4.
Besag FMC Vasey MJ . Seizures and epilepsy in autism spectrum disorder. Psychiatr Clin North Am. (2021) 44:51–68. doi: 10.1016/j.psc.2020.11.005
5.
El Achkar CM Spence SJ . Clinical characteristics of children and young adults with co-occurring autism spectrum disorder and epilepsy. Epilepsy Behav. (2015) 47:183–90. doi: 10.1016/j.yebeh.2014.12.022
6.
Stessman HAF Xiong B Coe BP Wang T Hoekzema K Fenckova M et al . Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat Genet. (2017) 49:515–26. doi: 10.1038/ng.3792
7.
Wang J Lin ZJ Liu L Xu HQ Shi YW Yi YH et al . Epilepsy-associated genes. Seizure. (2017) 44:11–20. doi: 10.1016/j.seizure.2016.11.030
8.
Wang P Zhao D Lachman HM Zheng D . Enriched expression of genes associated with autism spectrum disorders in human inhibitory neurons. Transl Psychiatry. (2018) 8:13–3. doi: 10.1038/s41398-017-0058-6
9.
Zhang M-W Liang XY Wang J Gao LD Liao HJ He YH et al . Epilepsy-associated genes: an update. Seizure. (2024) 116:4–13. doi: 10.1016/j.seizure.2023.09.021
10.
Blondiaux A Jia S Annamneedi A Çalışkan G Nebel J Montenegro-Venegas C et al . Linking epileptic phenotypes and neural extracellular matrix remodeling signatures in mouse models of epilepsy. Neurobiol Dis. (2023) 188:106324. doi: 10.1016/j.nbd.2023.106324
11.
Chen C-H Huang Y-S Liao D-L Huang C-Y Lin C-H Fang T-H . Identification of rare mutations of two presynaptic cytomatrix genes BSN and PCLO in schizophrenia and bipolar disorder. JPM. (2021) 11:1057. doi: 10.3390/jpm11111057
12.
Froukh TJ . Next generation sequencing and genome-wide genotyping identify the genetic causes of intellectual disability in ten consanguineous families from Jordan. Tohoku J Exp Med. (2017) 243:297–309. doi: 10.1620/tjem.243.297
13.
Iossifov I O’Roak BJ Sanders SJ Ronemus M Krumm N Levy D et al . The contribution of de novo coding mutations to autism spectrum disorder. Nature. (2014) 515:216–21. doi: 10.1038/nature13908
14.
Kumar RA Sudi J Babatz TD Brune CW Oswald D Yen M et al . A de novo 1p34.2 microdeletion identifies the synaptic vesicle gene RIMS3 as a novel candidate for autism. J Med Genet. (2010) 47:81–90. doi: 10.1136/jmg.2008.065821
15.
Skotte L Fadista J Bybjerg-Grauholm J Appadurai V Hildebrand MS Hansen TF et al . Genome-wide association study of febrile seizures implicates fever response and neuronal excitability genes. Brain. (2022) 145:555–68. doi: 10.1093/brain/awab260
16.
Yeo XY Lim YT Chae WR Park C Park H Jung S . Alterations of presynaptic proteins in autism spectrum disorder. Front Mol Neurosci. (2022) 15:1062878. doi: 10.3389/fnmol.2022.1062878
17.
Koopmans F van Nierop P Andres-Alonso M Byrnes A Cijsouw T Coba MP et al . SynGO: an evidence-based, expert-curated knowledge base for the synapse. Neuron. (2019) 103:217–234.e4. doi: 10.1016/j.neuron.2019.05.002
18.
Kleijer KTE Zuko A Shimoda Y Watanabe K Burbach JPH . Contactin-5 expression during development and wiring of the thalamocortical system. Neuroscience. (2015) 310:106–13. doi: 10.1016/j.neuroscience.2015.09.039
19.
Epi4K Consortium . De novo mutations in epileptic encephalopathies. Nature. (2013) 501:217–21. doi: 10.1038/nature12439
20.
Mitani T Isikay S Gezdirici A Gulec EY Punetha J Fatih JM et al . High prevalence of multilocus pathogenic variation in neurodevelopmental disorders in the Turkish population. Am J Hum Genet. (2021) 108:1981–2005. doi: 10.1016/j.ajhg.2021.08.009
21.
Deneault E Faheem M White SH Rodrigues DC Sun S Wei W et al . CNTN5−/+or EHMT2−/+human iPSC-derived neurons from individuals with autism develop hyperactive neuronal networks. eLife. (2019) 8:e40092. doi: 10.7554/eLife.40092
22.
Santos-Rebouças CB Belet S Guedes de Almeida L Ribeiro MG Medina-Acosta E Bahia PRV et al . A novel in-frame deletion affecting the BAR domain of OPHN1 in a family with intellectual disability and hippocampal alterations. Eur J Hum Genet. (2014) 22:644–51. doi: 10.1038/ejhg.2013.216
23.
Brouns MR Matheson SF Settleman J . P190 RhoGAP is the principal Src substrate in brain and regulates axon outgrowth, guidance and fasciculation. Nat Cell Biol. (2001) 3:361–7. doi: 10.1038/35070042
24.
Aspromonte MC Bellini M Gasparini A Carraro M Bettella E Polli R et al . Characterization of intellectual disability and autism comorbidity through gene panel sequencing. Hum Mutat. (2019) 40:1346–63. doi: 10.1002/humu.23822
25.
Khelfaoui M Denis C van Galen E de Bock F Schmitt A Houbron C et al . Loss of X-linked mental retardation gene Oligophrenin1 in mice impairs spatial memory and leads to ventricular enlargement and dendritic spine immaturity. J Neurosci. (2007) 27:9439–50. doi: 10.1523/JNEUROSCI.2029-07.2007
26.
Missler M Zhang W Rohlmann A Kattenstroth G Hammer RE Gottmann K et al . Alpha-neurexins couple Ca2+ channels to synaptic vesicle exocytosis. Nature. (2003) 423:939–48. doi: 10.1038/nature01755
27.
Harkin LF Lindsay SJ Xu Y Alzu’bi A Ferrara A Gullon EA et al . Neurexins 1-3 each have a distinct pattern of expression in the early developing human cerebral cortex. Cerebral Cortex. (2017) 27:216–32. doi: 10.1093/cercor/bhw394
28.
Al Shehhi M Forman EB Fitzgerald JE V MI Krawczyk J Shen S . NRXN1 deletion syndrome; phenotypic and penetrance data from 34 families. Eur J Med Genet. (2019) 62:204–9. doi: 10.1016/j.ejmg.2018.07.015
29.
Schaaf CP Boone PM Sampath S Williams C Bader PI Mueller JM et al . Phenotypic spectrum and genotype-phenotype correlations of NRXN1 exon deletions. Eur J Hum Genet. (2012) 20:1240–7. doi: 10.1038/ejhg.2012.95
30.
Ishizuka K Yoshida T Kawabata T Imai A Mori H Kimura H et al . Functional characterization of rare NRXN1 variants identified in autism spectrum disorders and schizophrenia. J Neurodevelop Disord. (2020) 12:25. doi: 10.1186/s11689-020-09325-2
31.
Avazzadeh S McDonagh K Reilly J Wang Y Boomkamp SD McInerney V et al . Increased Ca2+ signaling in NRXN1α+/− neurons derived from ASD induced pluripotent stem cells. Mol Autism. (2019) 10:52. doi: 10.1186/s13229-019-0303-3
32.
Avazzadeh S Quinlan LR Reilly J McDonagh K Jalali A Wang Y et al . NRXN1α+/− is associated with increased excitability in ASD iPSC-derived neurons. BMC Neurosci. (2021) 22:56. doi: 10.1186/s12868-021-00661-0
33.
Fox PM Malepati S Manaster L Rossignol E Noebels JL . Developing a pathway to clinical trials for CACNA1A-related epilepsies: a patient organization perspective. Ther Adv Rare Dis. (2024) 5:26330040241245725. doi: 10.1177/26330040241245725
34.
Rajakulendran S Kaski D Hanna MG . Neuronal P/Q-type calcium channel dysfunction in inherited disorders of the CNS. Nat Rev Neurol. (2012) 8:86–96. doi: 10.1038/nrneurol.2011.228
35.
Damaj L Lupien-Meilleur A Lortie A Riou É Ospina LH Gagnon L et al . CACNA1A haploinsufficiency causes cognitive impairment, autism and epileptic encephalopathy with mild cerebellar symptoms. Eur J Hum Genet. (2015) 23:1505–12. doi: 10.1038/ejhg.2015.21
36.
Domenici MR Azad SC Marsicano G Schierloh A Wotjak CT Dodt HU et al . Cannabinoid receptor type 1 located on presynaptic terminals of principal neurons in the forebrain controls glutamatergic synaptic transmission. J Neurosci. (2006) 26:5794–9. doi: 10.1523/JNEUROSCI.0372-06.2006
37.
Katona I Sperlágh B Sı́k A Käfalvi A Vizi ES Mackie K et al . Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J Neurosci. (1999) 19:4544–58. doi: 10.1523/JNEUROSCI.19-11-04544.1999
38.
Smith DR Stanley CM Foss T Boles RG McKernan K . Rare genetic variants in the endocannabinoid system genes CNR1 and DAGLA are associated with neurological phenotypes in humans. PLoS One. (2017) 12:e0187926. doi: 10.1371/journal.pone.0187926
39.
Liang L Li X Moutton S Schrier Vergano SA Cogné B Saint-Martin A et al . De novo loss-of-function KCNMA1 variants are associated with a new multiple malformation syndrome and a broad spectrum of developmental and neurological phenotypes. Hum Mol Genet. (2019) 28:2937–51. doi: 10.1093/hmg/ddz117
40.
Yao Y Qu D Jing X Jia Y Zhong Q Zhuo L et al . Molecular mechanisms of epileptic encephalopathy caused by KCNMA1 loss-of-function mutations. Front Pharmacol. (2021) 12:775328. doi: 10.3389/fphar.2021.775328
41.
Liang L Fazel Darbandi S Pochareddy S Gulden FO Gilson MC Sheppard BK et al . Developmental dynamics of voltage-gated sodium channel isoform expression in the human and mouse brain. Genome Med. (2021) 13:135. doi: 10.1186/s13073-021-00949-0
42.
Goodwin G McMahon SB . The physiological function of different voltage-gated sodium channels in pain. Nat Rev Neurosci. (2021) 22:263–74. doi: 10.1038/s41583-021-00444-w
43.
Escayg A Heils A MacDonald BT Haug K Sander T Meisler MH . A novel SCN1A mutation associated with generalized epilepsy with febrile seizures plus--and prevalence of variants in patients with epilepsy. Am J Hum Genet. (2001) 68:866–73. doi: 10.1086/319524
44.
Claes L Del-Favero J Ceulemans B Lagae L Van Broeckhoven C De Jonghe P . De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet. (2001) 68:1327–32. doi: 10.1086/320609
45.
Brunklaus A Brünger T Feng T Fons C Lehikoinen A Panagiotakaki E et al . The gain of function SCN1A disorder spectrum: novel epilepsy phenotypes and therapeutic implications. Brain. (2022) 145:3816–31. doi: 10.1093/brain/awac210
46.
Berecki G Howell KB Heighway J Olivier N Rodda J Overmars I et al . Functional correlates of clinical phenotype and severity in recurrent SCN2A variants. Commun Biol. (2022) 5:515. doi: 10.1038/s42003-022-03454-1
47.
Fassio A Patry L Congia S Onofri F Piton A Gauthier J et al . SYN1 loss-of-function mutations in autism and partial epilepsy cause impaired synaptic function. Hum Mol Genet. (2011) 20:2297–307. doi: 10.1093/hmg/ddr122
48.
Corradi A Fadda M Piton A Patry L Marte A Rossi P et al . SYN2 is an autism predisposing gene: loss-of-function mutations alter synaptic vesicle cycling and axon outgrowth. Hum Mol Genet. (2014) 23:90–103. doi: 10.1093/hmg/ddt401
49.
Giannandrea M Guarnieri FC Gehring NH Monzani E Benfenati F Kulozik AE et al . Nonsense-mediated mRNA decay and loss-of-function of the protein underlie the X-linked epilepsy associated with the W356× mutation in Synapsin I. PLoS One. (2013) 8:e67724. doi: 10.1371/journal.pone.0067724
50.
Mori T Kasem EA Suzuki-Kouyama E Cao X Li X Kurihara T et al . Deficiency of calcium/calmodulin-dependent serine protein kinase disrupts the excitatory-inhibitory balance of synapses by down-regulating GluN2B. Mol Psychiatry. (2019) 24:1079–92. doi: 10.1038/s41380-018-0338-4
51.
McSweeney D Gabriel R Jin K Pang ZP Aronow B Pak C . CASK loss of function differentially regulates neuronal maturation and synaptic function in human induced cortical excitatory neurons. iScience. (2022) 25:105187. doi: 10.1016/j.isci.2022.105187
52.
Rizo J Xu J . The synaptic vesicle release machinery. Annu Rev Biophys. (2015) 44:339–67. doi: 10.1146/annurev-biophys-060414-034057
53.
Carvill GL Weckhuysen S McMahon JM Hartmann C Møller RS Hjalgrim H et al . GABRA1 and STXBP1: novel genetic causes of Dravet syndrome. Neurology. (2014) 82:1245–53. doi: 10.1212/WNL.0000000000000291
54.
Campbell IM Yatsenko SA Hixson P Reimschisel T Thomas M Wilson W et al . Novel 9q34.11 gene deletions encompassing combinations of four mendelian disease genes: STXBP1, SPTAN1, ENG, and TOR1A. Genet Med. (2012) 14:868–76. doi: 10.1038/gim.2012.65
55.
Kovačević J Maroteaux G Schut D Loos M Dubey M Pitsch J et al . Protein instability, haploinsufficiency, and cortical hyper-excitability underlie STXBP1 encephalopathy. Brain. (2018) 141:1350–74. doi: 10.1093/brain/awy046
56.
The DDD Study O’Brien S Ng-Cordell E Astle DE Scerif G Baker K . STXBP1-associated neurodevelopmental disorder: a comparative study of behavioural characteristics. J Neurodevelop Disord. (2019) 11:17. doi: 10.1186/s11689-019-9278-9
57.
Lammertse HCA van Berkel AA Iacomino M Toonen RF Striano P Gambardella A et al . Homozygous STXBP1 variant causes encephalopathy and gain-of-function in synaptic transmission. Brain. (2020) 143:441–51. doi: 10.1093/brain/awz391
58.
Otis TS Kavanaugh MP . Isolation of current components and partial reaction cycles in the glial glutamate transporter EAAT2. J Neurosci. (2000) 20:2749–57. doi: 10.1523/JNEUROSCI.20-08-02749.2000
59.
Mir A Almudhry M Alghamdi F Albaradie R Ibrahim M Aldurayhim F et al . SLC gene mutations and pediatric neurological disorders: diverse clinical phenotypes in a Saudi Arabian population. Hum Genet. (2022) 141:81–99. doi: 10.1007/s00439-021-02404-x
60.
Qu Q Zhang W Wang J Mai D Ren S Qu S et al . Functional investigation of SLC1A2 variants associated with epilepsy. Cell Death Dis. (2022) 13:1063. doi: 10.1038/s41419-022-05457-6
61.
Kovermann P Bayat A Fenger CD Leeuwen L Borovikov A Sharkov A et al . The severity of SLC1A2-associated neurodevelopmental disorders correlates with transporter dysfunction. EBioMedicine. (2025) 114:105648. doi: 10.1016/j.ebiom.2025.105648
62.
Lu P Wang F Zhou S Huang X Sun H Zhang YW et al . A novel CNTNAP2 mutation results in abnormal neuronal E/I balance. Front Neurol. (2021) 12:712773. doi: 10.3389/fneur.2021.712773
63.
Toma C Pierce KD Shaw AD Heath A Mitchell PB Schofield PR et al . Comprehensive cross-disorder analyses of CNTNAP2 suggest it is unlikely to be a primary risk gene for psychiatric disorders. PLoS Genet. (2018) 14:e1007535. doi: 10.1371/journal.pgen.1007535
64.
St George-Hyslop F Haneklaus M Kivisild T Livesey FJ . Loss of CNTNAP2 alters human cortical excitatory neuron differentiation and neural network development. Biol Psychiatry. (2023) 94:780–91. doi: 10.1016/j.biopsych.2023.03.014
65.
Liska A Bertero A Gomolka R Sabbioni M Galbusera A Barsotti N et al . Homozygous loss of autism-risk gene CNTNAP2 results in reduced local and long-range prefrontal functional connectivity. Cereb Cortex. (2018) 28:1141–53. doi: 10.1093/cercor/bhx022
66.
Brittain JM Piekarz AD Wang Y Kondo T Cummins TR Khanna R . An atypical role for collapsin response mediator protein 2 (CRMP-2) in neurotransmitter release via interaction with presynaptic voltage-gated calcium channels. J Biol Chem. (2009) 284:31375–90. doi: 10.1074/jbc.M109.009951
67.
Suzuki H Li S Tokutomi T Takeuchi C Takahashi M Yamada M et al . De novo non-synonymous DPYSL2 (CRMP2) variants in two patients with intellectual disabilities and documentation of functional relevance through zebrafish rescue and cellular transfection experiments. Hum Mol Genet. (2022) 31:4173–82. doi: 10.1093/hmg/ddac166
68.
Tabarés-Seisdedos R Rubenstein JLR . Chromosome 8p as a potential hub for developmental neuropsychiatric disorders: implications for schizophrenia, autism and cancer. Mol Psychiatry. (2009) 14:563–89. doi: 10.1038/mp.2009.2
69.
Desprez F Ung DC Vourc’h P Jeanne M Laumonnier F . Contribution of the dihydropyrimidinase-like proteins family in synaptic physiology and in neurodevelopmental disorders. Front Neurosci. (2023) 17:1154446. doi: 10.3389/fnins.2023.1154446
70.
Schröder MS Stellmacher A Romorini S Marini C Montenegro-Venegas C Altrock WD et al . Regulation of presynaptic anchoring of the scaffold protein bassoon by phosphorylation-dependent interaction with 14-3-3 adaptor proteins. PLoS One. (2013) 8:e58814. doi: 10.1371/journal.pone.0058814
71.
Fusco C Micale L Augello B Teresa Pellico M Menghini D Alfieri P et al . Smaller and larger deletions of the Williams Beuren syndrome region implicate genes involved in mild facial phenotype, epilepsy and autistic traits. Eur J Hum Genet. (2014) 22:64–70. doi: 10.1038/ejhg.2013.101
72.
Kanani F Titheradge H Cooper N Elmslie F Lees MM Juusola J et al . Expanding the genotype–phenotype correlation of de novo heterozygous missense variants in YWHAG as a cause of developmental and epileptic encephalopathy. Am J Med Genet A. (2020) 182:713–20. doi: 10.1002/ajmg.a.61483
73.
Berth SH Lloyd TE . Disruption of axonal transport in neurodegeneration. J Clin Invest. (2023) 133:e168554. doi: 10.1172/JCI168554
74.
Olivero G Grilli M Marchi M Pittaluga A . Metamodulation of presynaptic NMDA receptors: new perspectives for pharmacological interventions. Neuropharmacology. (2023) 234:109570. doi: 10.1016/j.neuropharm.2023.109570
Summary
Keywords
ASD, epilepsy, presynaptic genes, comorbidity, abnormal synaptic plasticity, synaptic organization
Citation
Sharma M, Pamidi SC, Divi PK, Mohapatra S, George B, Sneha KP, Kreutzmann JC and Annamneedi A (2025) Genetic crosstalk of autism spectrum disorders and epilepsy: an insight into the presynapse. Front. Neurol. 16:1677134. doi: 10.3389/fneur.2025.1677134
Received
31 July 2025
Accepted
16 October 2025
Published
03 November 2025
Volume
16 - 2025
Edited by
Atsushi Ishii, University of Arizona, United States
Reviewed by
Dominique Debanne, Unité de Neurobiologie des canaux Ioniques et de la Synapse (UNIS), France
Updates
Copyright
© 2025 Sharma, Pamidi, Divi, Mohapatra, George, Sneha, Kreutzmann and Annamneedi.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anil Annamneedi, anil.a@saiuniversity.edu.in
†These authors have contributed equally to this work and share first authorship
‡ORCID: Anil Annamneedi, https://orcid.org/0000-0002-6743-8627
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.