Abstract
Objective (background):
The aim of this study is to analyze the current research status and future prospects of the field of developmental and epileptic encephalopathy (DEE) and gene through bibliometric methods. It aims to explore the trends and potential developments in this field.
Methods:
A systematic search of the DEE and gene literature from 2001 to 2025.2 was conducted using the Web of Science core collection database. Supplementary PubMed searches for this field’s clinical research trends ensured verified data comprehensiveness and methodological rigor. Quantitative analysis of co-authorship networks was performed using VOSviewer and CiteSpace tools.
Results:
A total of 1,022 articles related to the field of DEE and gene were included in this study, authored by 8,355 researchers affiliated with 7,238 institutions across 315 countries. United States emerged as the leading research countries in this field, with the National Institute of Health and Medical Research. Professor Ingrid Scheffer had the highest number of publications in this field, and the journal Epilepsy had the highest citation count. The research hotspots in this field revolved around epilepsy, mutations, epileptic encephalopathy, de novo mutations, and seizure.
Conclusion:
The research on DEE and gene is currently experiencing rapid growth. The field is expanding, and the research is becoming more in-depth.
1 Introduction
Developmental and epileptic encephalopathies (DEEs) are a group of heterogeneous epilepsy syndromes defined by developmental impairment, which is closely linked to both the underlying pathological etiology and the persistent epileptic process itself. Clinically, they impose a heavy burden on pediatric populations: 1 in 340 children under 16 have concurrent epilepsy and developmental impairment, while the prevalence of DEEs specifically reaches 1 in 590 children (1). This high incidence highlights the urgency of exploring DEE etiologies and advancing research to improve clinical management—particularly given that genetic mutations account for a large proportion of cases, with 112 DEE-associated genes identified to date (2), including GABRA1 (3), TANC2 (4), CSMD1 (5), and GABRB3 (6), alongside other origins like structural brain abnormalities and metabolic disorders (7). The International League Against Epilepsy (ILAE) has guided the standardized evolution of the DEE concept. In 2010, it proposed “epileptic encephalopathy” and defined “developmental encephalopathy” as age-worsening developmental delay/disability caused by underlying factors (8). A key finding—that cognitive impairment and epilepsy from the same gene are independent (some patients show pre-seizure delay or ongoing decline even with controlled seizures)—led to the formal introduction of “DEEs” in 2017: diseases characterized by drug-resistant epilepsy, abnormal EEG, and impaired brain development, encompassing both etiology-driven developmental encephalopathy and epileptic activity-related neurocognitive issues (9). In 2022, the ILAE merged DEE-related syndromes and classified them by onset age, further standardizing research and clinical practice (10).
While DEEs and gene research have grown substantially, these knowledge structure, research trends, and collaborative networks remain unexamined via systematic bibliometrics. This study addresses this gap by analyzing DEE and gene-related literature from 2001 to February 2025 using VOSviewer and CiteSpace. By mapping collaborations, identifying hotspots, and tracing evolutionary trajectories, we aim to clarify future research directions for the field.
2 Research methods and data sources
2.1 Data sources
The relevant articles on gene and DEEs were retrieved from the Web of Science database. Additionally, a literature search was performed through PubMed to supplement the trends of clinical research in this field, so as to ensure data comprehensiveness and methodological rigor. The search formula is “(TS = (epileptic encephalopathy)) OR TS = (developmental encephalopathy) OR TS = (developmental and epileptic encephalopathy) OR TS = (Epileptic Syndrome with Cognitive Impairment)) OR TS = (Epilepsy-Associated Encephalopathy)) OR TS = (Seizure-Induced Encephalopathy)) AND TS = (gene)) AND TS = (genetic).” This study covers all types of articles from 2001 to Feb 2025. After screening and removing duplicates, a total of 1,022 publications in the field were obtained (Figure 1). These records were written by 8,355 authors from 7,238 institutions in 315 countries and cited 25,625 references from 261 different journals.
Figure 1
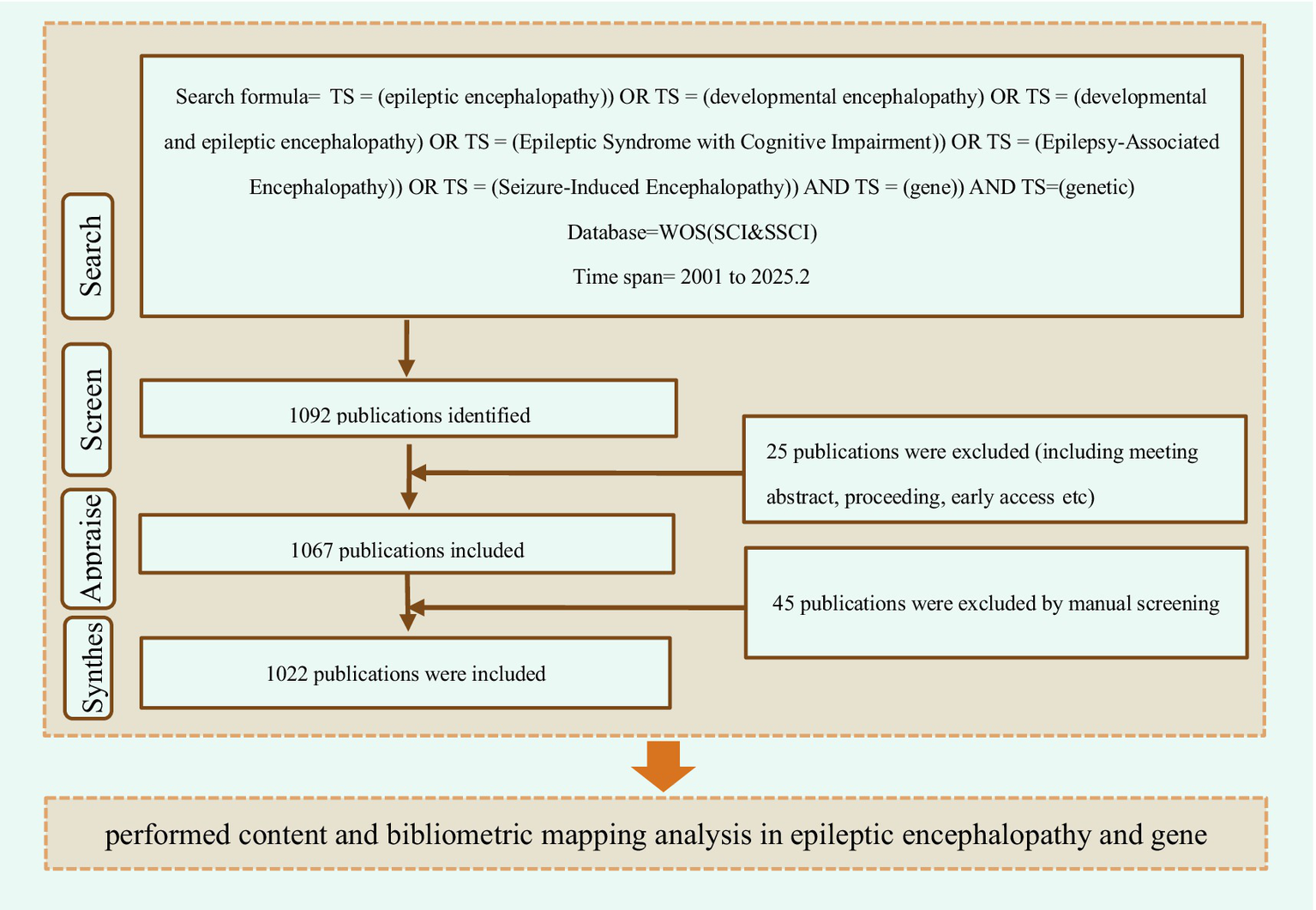
Inclusion and analysis flow chart.
2.2 Research methods
VOSviewer 1.6.19 and CiteSpace 6.3.10 were utilized to analyze the relevant literature from the Web of Science database in this study. First, a thesaurus was constructed by VOSviewer, and synonyms were merged in the literature (11). Subsequently, the data downloaded from the Web of Science was processed using CiteSpace and VOSviewer to create a new project to visualize analysis (12, 13). The parameter setting of CiteSpace was Method (LLR), Time Splicing (2001–2025), Text Processing (all term source), and Node types (choose the node we need). The parameter setting of the VOSviewer was chosen type of data, data source, and analysis and counting method. Finally, bibliometric maps were constructed by the CiteSpace and VOSviewer software, including scientific productions, authors, countries, institutions, co-cited journals, co-cited references, and keywords.
3 Results
3.1 Temporal distribution map of publications
The overall trend of research publications on gene and DEEs from 2001 to the present is shown in Figure 2. As the figure shows, in 2010, the International League Against Epilepsy (ILAE) first proposed the concept of epileptic encephalopathy, and the literature publication has established an annually rapidly increasing trend, reaching a peak in 2024. This indicates that the field of epileptic encephalopathy and gene received increasing attention from scholars and became a prominent research hotspot in recent years.
Figure 2
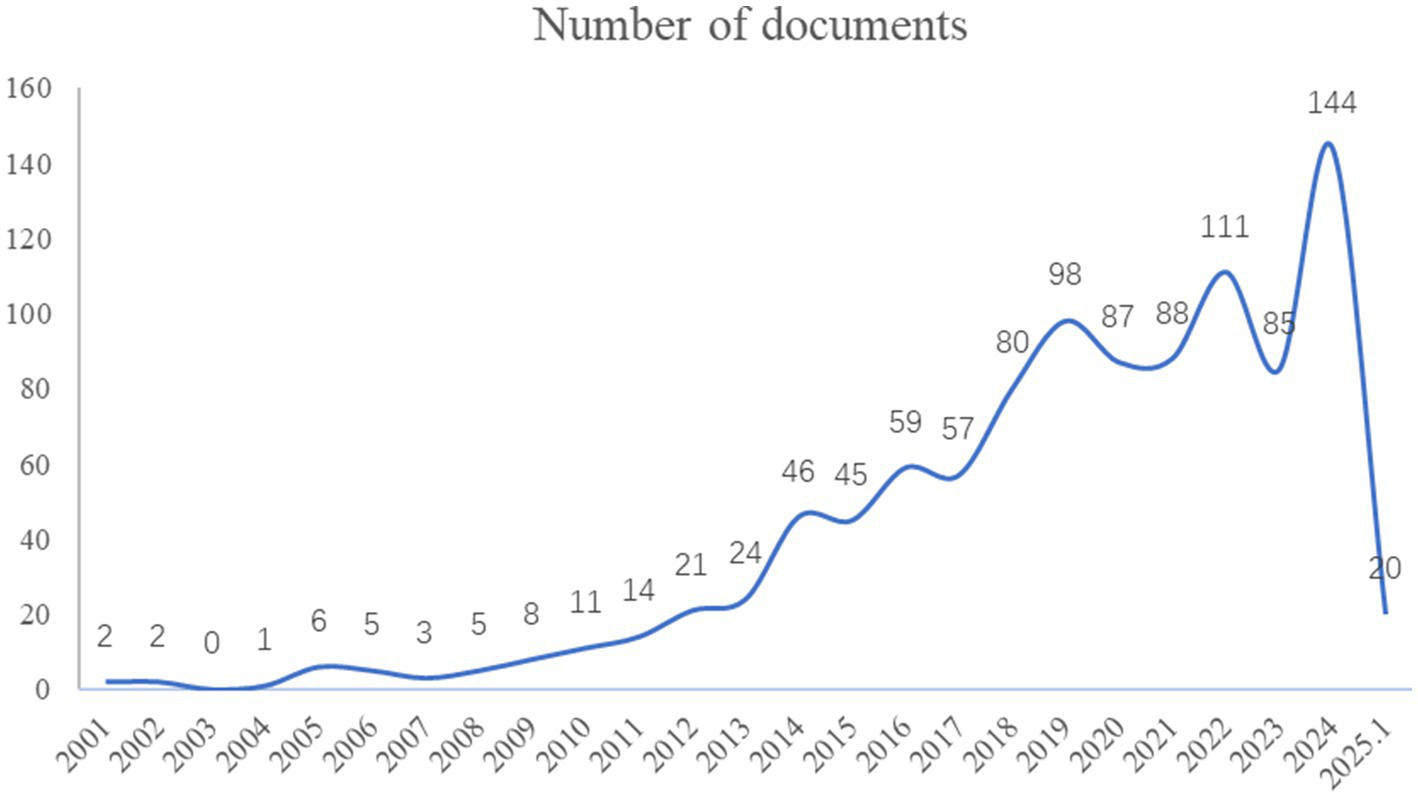
The annual analysis of research publications related to epileptic encephalopathy and gene.
3.2 Analysis of authors
Bibliometric analysis of the authors can help us identify the representative scholars in the field (13). The publications were authored by 8,355 researchers in the analysis. The top five authors in terms of publications are presented in Table 1. Professor Ingrid Scheffer, from The University of Melbourne, Australia, was the highest productive author in the field. It can be seen from Figure 3 that Professor Ingrid Scheffer has close connections with each author. The result indicates that these authors have a significant influence in the field of epileptic encephalopathy and gene.
Table 1
| Author | Number of publications | Citation count | TLS |
|---|---|---|---|
| Ingrid Scheffer | 34 | 2,490 | 585 |
| Guerrini Renzo | 23 | 1,060 | 310 |
| Rikke Steensbjerre Møller | 22 | 615 | 298 |
| Nicola Specchio | 18 | 1,058 | 267 |
| Samuel F. Berkovic | 18 | 719 | 267 |
Top five authors in the field of epileptic encephalopathy and gene based on publication frequency.
Figure 3
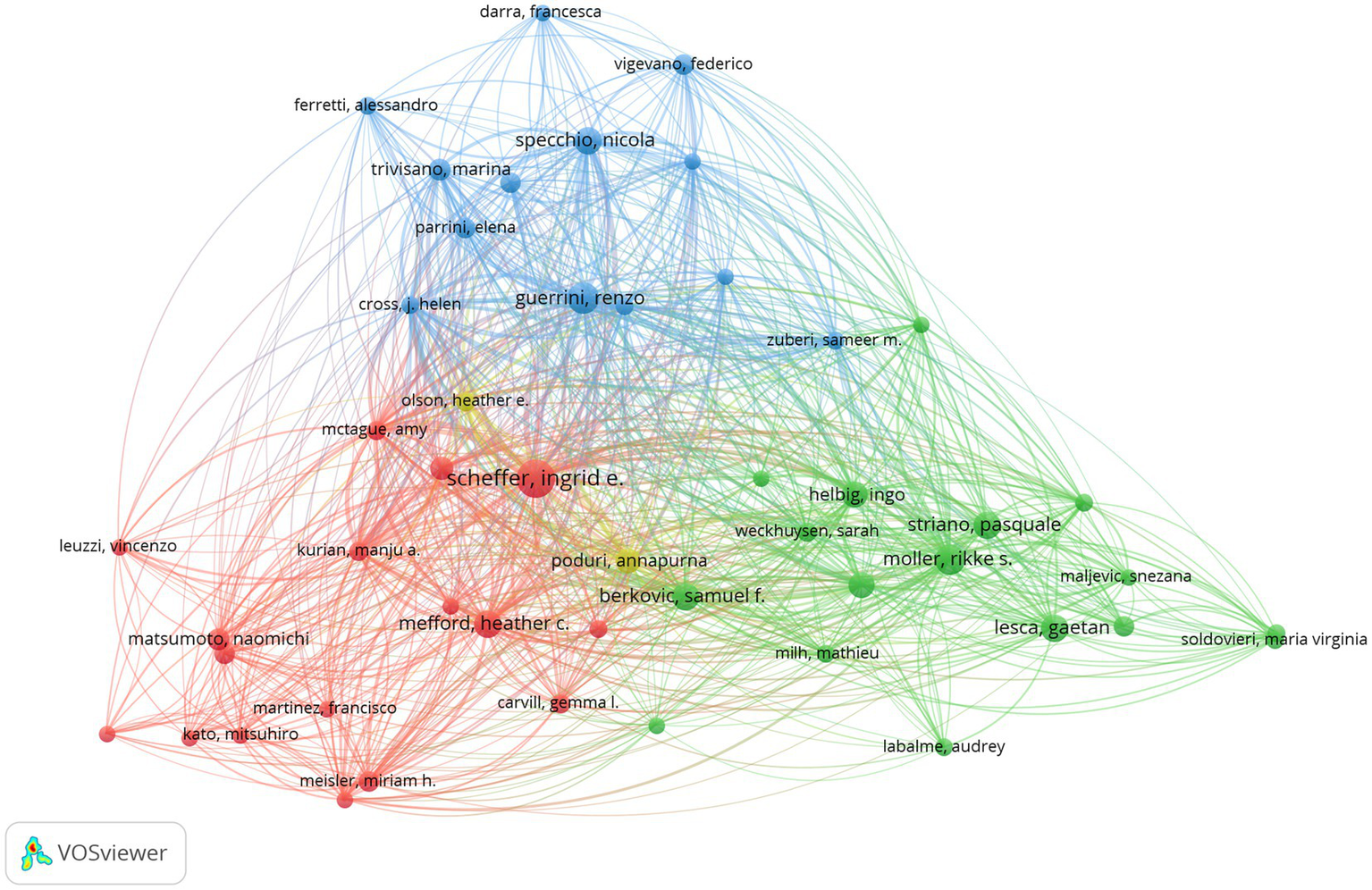
Author analysis of research on epileptic encephalopathy and gene, where circles represent authors, and lines denote their collaborations.
3.3 Analysis of countries
In this study, the contributions of 315 countries in the field of epileptic encephalopathy and gene were analyzed. The top five countries published 901 articles, accounting for 88.16% of the total literature in this field. To gain a clearer understanding of each country’s contributions, we used VOS viewer to filter countries with five or more publications to conduct visualized analysis (Figure 4). At the same time, the top ten countries in terms of publications are listed in Table 2. The publications among countries were imbalanced. The United States (306, Centrality = 0.02), Italy (192, Centrality = 0.01), and China (153, Centrality = 0.00) had the highest number of publications, surpassing other countries by a considerable margin. This indicates that a majority of the research articles in this field originate from a few countries.
Figure 4
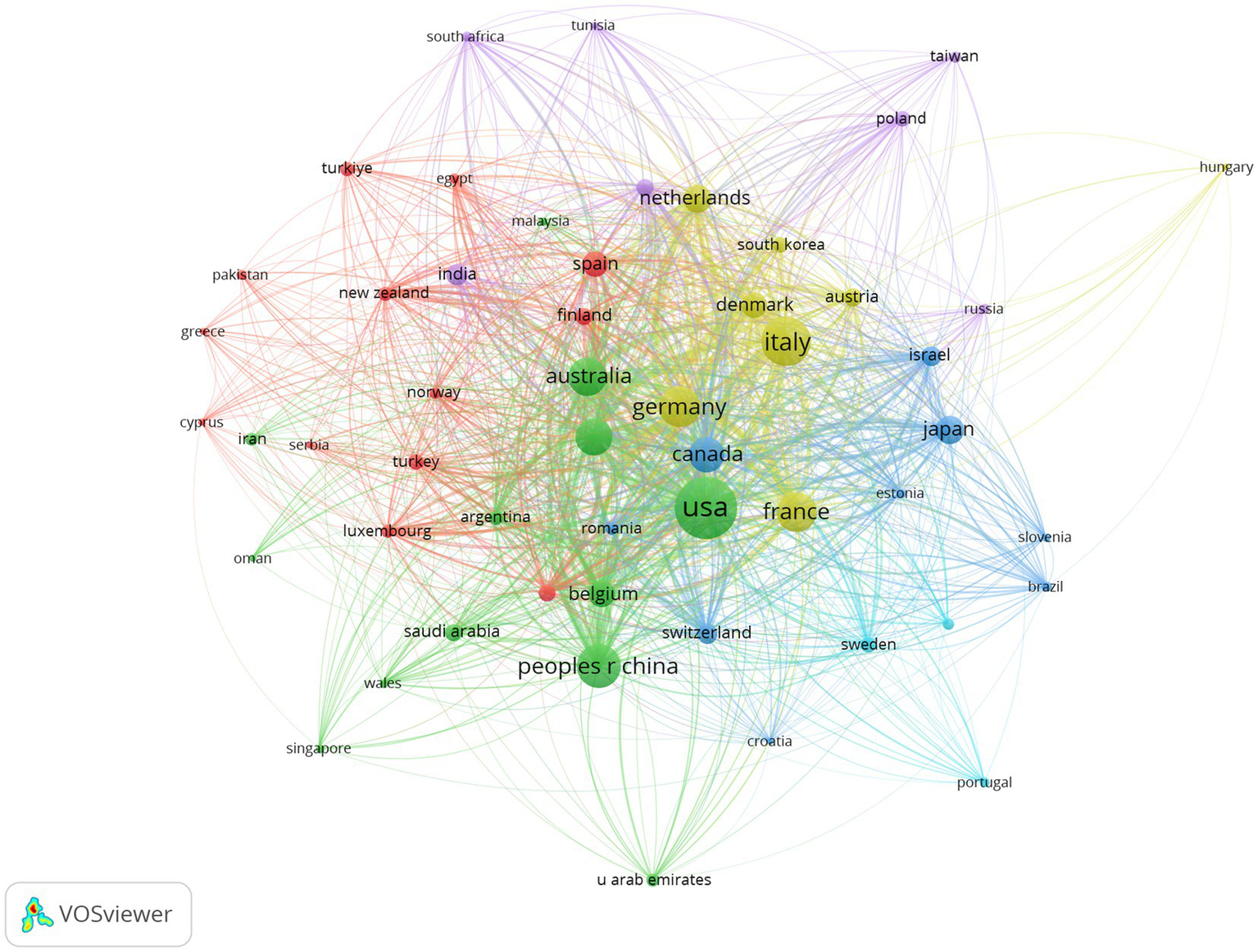
Network visualization of national collaborations in developmental and epileptic encephalopathies and gene research, where circles represent countries, and lines denote their collaborations.
Table 2
| Rank | Country | Publication count | Centrality | Rank | Institution | Publication count |
|---|---|---|---|---|---|---|
| 1 | United States | 306 | 0.02 | 1 | National Institute of Health and Medical Research | 88 |
| 2 | Italy | 192 | 0.01 | 2 | The University of Melbourne | 83 |
| 3 | China | 153 | 0.00 | 3 | Florey Institute of Neuroscience and Mental Health | 71 |
| 4 | Germany | 138 | 0.06 | 4 | The University of London | 70 |
| 5 | France | 122 | 0.04 | 5 | Harvard University | 65 |
| 6 | Australia | 115 | 0.06 | 6 | The University College London | 64 |
| 7 | England | 108 | 0.03 | 7 | Assistance Publique – Hopitaux de Paris | 60 |
| 8 | Canada | 98 | 0.09 | 8 | Harvard Medical School | 57 |
| 9 | Netherlands | 68 | 0.02 | 9 | Royal Children’s Hospital of Melbourne | 56 |
| 10 | Japan | 63 | 0.05 | 10 | Centre national de la recherche scientifique | 55 |
Top 10 countries and institutions in developmental and epileptic encephalopathies and gene relationship research publications.
3.4 Analysis of institutions
Since 2001, 7,238 institutions have published literature in the field. Table 2 presents the top ten most productive institutions. The institutions with the most published were the National Institute of Health and Medical Research (n = 88), followed by The University of Melbourne (n = 83), and Florey Institute of Neuroscience and Mental Health (n = 71). As can be seen in Table 2 and Figure 5, the number of papers of the top institutions accounts for a relatively small proportion of total publications, which indicates that the formation of the preferential attachment effect has not yet emerged.
Figure 5
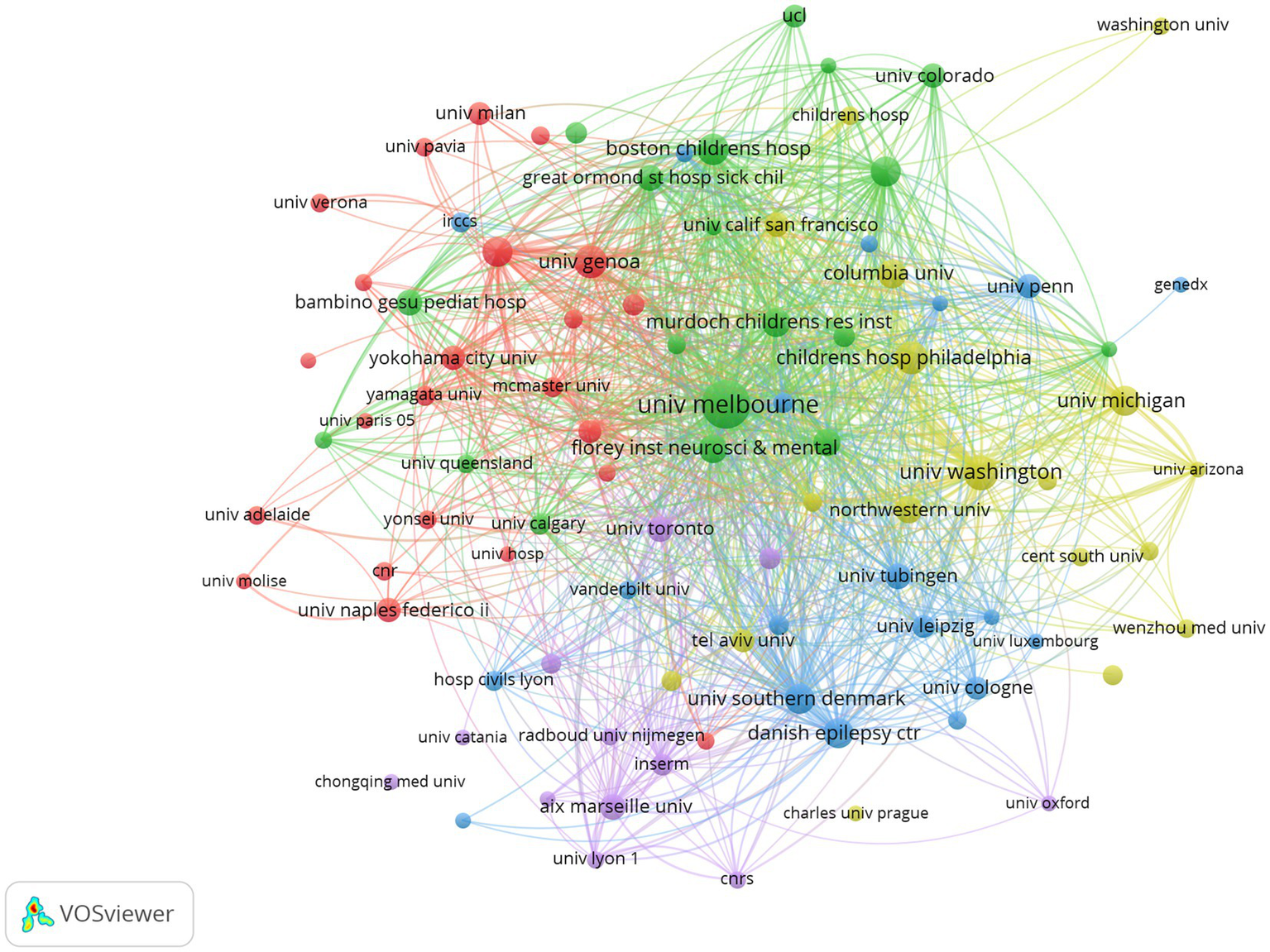
Collaboration network of institutions in developmental and epileptic encephalopathies and gene research, where circles represent institutions, and lines denote their collaborations.
3.5 Analysis of co-citation journals
Co-citation is the frequency with that two articles are cited together. The top 10 co-citation journals are shown in Table 3, with half of them belonging to Q1. The journal with the highest co-citation is Epilepsy (70, Q2, IF = 2.30), followed by Seizure-european journal of epilepsy (36, Q3, IF = 2.70) and The American Journal of Human Genetics (26, Q1, IF = 8.18). These findings show that the co-citation journals in the field of epileptic encephalopathy and gene have high quality and impact. The top 36 journals, being co-cited by at least 7 times, were used for the analysis through the VOSviewer tool. (Figure 6).
Table 3
| Rank | Cited journals | Co-citation frequency | JCR | IF |
|---|---|---|---|---|
| 1 | Epilepsy | 70 | Q2 | 2.30 |
| 2 | Seizure-european journal of epilepsy | 36 | Q3 | 2.70 |
| 3 | The American Journal of Human Genetics | 26 | Q1 | 8.18 |
| 4 | Frontiers in Neurology | 26 | Q3 | 2.7 |
| 5 | European journal of paediatric neurology | 25 | Q1 | 4.8 |
| 6 | Brain & development | 25 | Q1 | 3.8 |
| 7 | Brain | 24 | Q1 | 10.78 |
| 8 | Epileptic disorders | 23 | Q3 | 1.9 |
| 9 | Molecular Genetics & Genomic Medicine | 22 | Q4 | 1.5 |
| 10 | International journal of molecular sciences | 21 | Q1 | 4.9 |
Top 10 co-citation journals in developmental and epileptic encephalopathies and gene research.
Figure 6
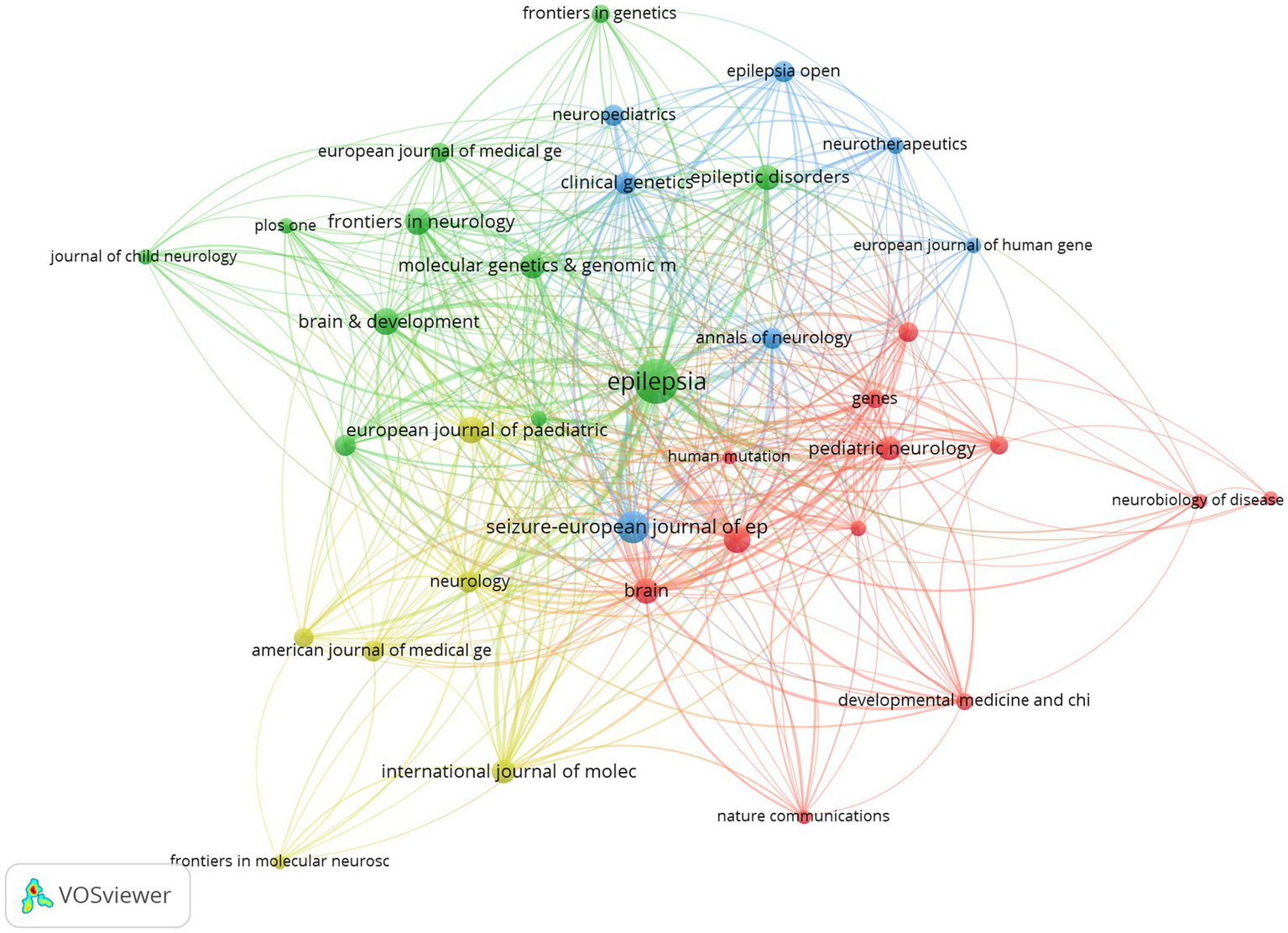
The co-citation network of journals in epileptic encephalopathy and gene research, where circles represent journals, and lines denote their collaborations.
3.6 Analysis of co-citation references
Since 2001, there are a total of 25,625 co-citation references taken into analysis in the field. The top five co-citation references were listed in the Table 4. In this article, “De novo mutations in epileptic encephalopathies” was written by Andrew S. Allen, and had the highest number of citations (n = 111). He highlighted the strong correlations between de novo mutations in the genes GABRB3 and ALG13 and epileptic encephalopathies, such as infantile spasms and Lennox–Gastaut syndrome, and demonstrated an enrichment of these mutations within particular gene sets, notably those regulated by the fragile X protein. And the top 10 co-citation references with the strongest burst strength were displayed in Figure 7, and the burst strength values ranged from 15.59 to 33.58. Among them, the reference with the highest burst strength (33.58) came from “ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology.”
Table 4
| Reference | Co-citation count | Authors |
|---|---|---|
| De novo mutations in epileptic encephalopathies | 111 | Andrew S Allen |
| Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1 | 111 | Gemma L Carvill |
| KCNQ2 encephalopathy: emerging phenotype of a neonatal epileptic encephalopathy | 77 | Sarah Weckhuysen |
| De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy | 72 | Hirotomo Saitsu |
| Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009 | 71 | Anne T Berg |
Co-citation references.
Figure 7
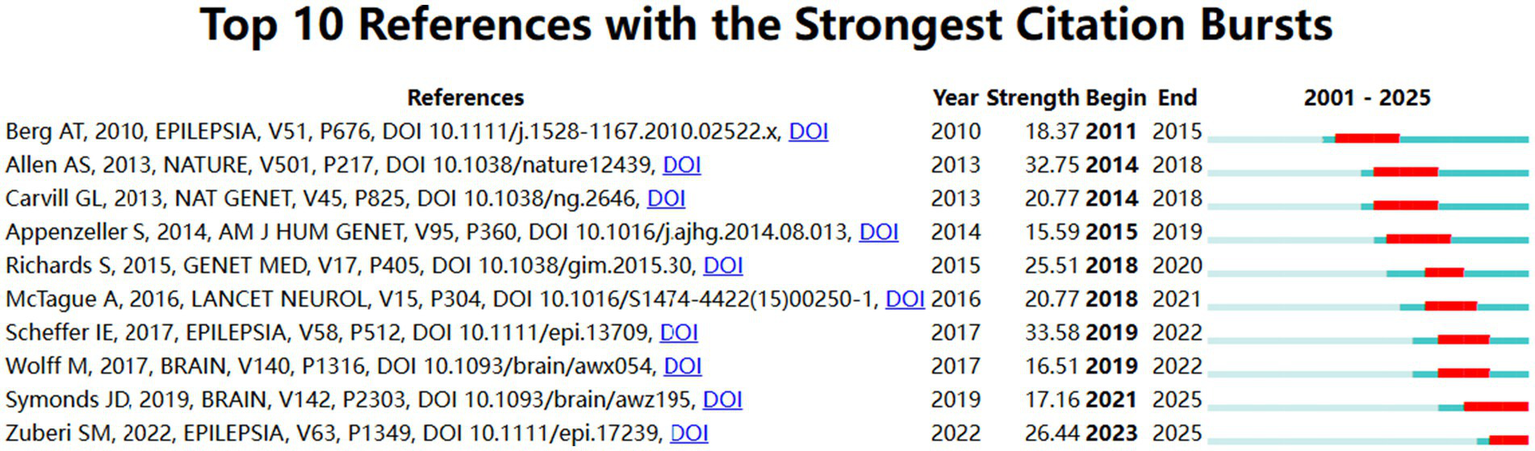
Visual analysis of literature emergence in developmental and epileptic encephalopathies and gene research. Burst strength and time duration of the top 10 references with the strongest citation bursts.
3.7 Analysis of keyword
High-frequency keywords in articles reflect the hot spots of a research field (14). Table 5 lists the top 10 most frequently occurring keywords in the field. It can be seen that high-frequency keywords such as epilepsy, mutations, epileptic encephalopathy, de novo mutations, and seizure constitute the representative academic terminology in the field, which show the hotspots in the field. Furthermore, we selected 154 keywords that appeared with a frequency greater than or equal to 10 for visual analysis (Figure 8).
Table 5
| Keywords | Frequency | TLS | Keywords | Frequency | TLS |
|---|---|---|---|---|---|
| epilepsy | 294 | 1,529 | Intellectual disability | 131 | 695 |
| mutations | 224 | 1,025 | encephalopathy | 121 | 627 |
| epileptic encephalopathy | 206 | 962 | gene | 115 | 480 |
| de novo mutations | 184 | 1,084 | variants | 114 | 676 |
| seizure | 152 | 884 | Dravet syndrome | 112 | 729 |
Top 10 most frequently occurring high-frequency keywords in developmental and epileptic encephalopathies and gene research.
Figure 8
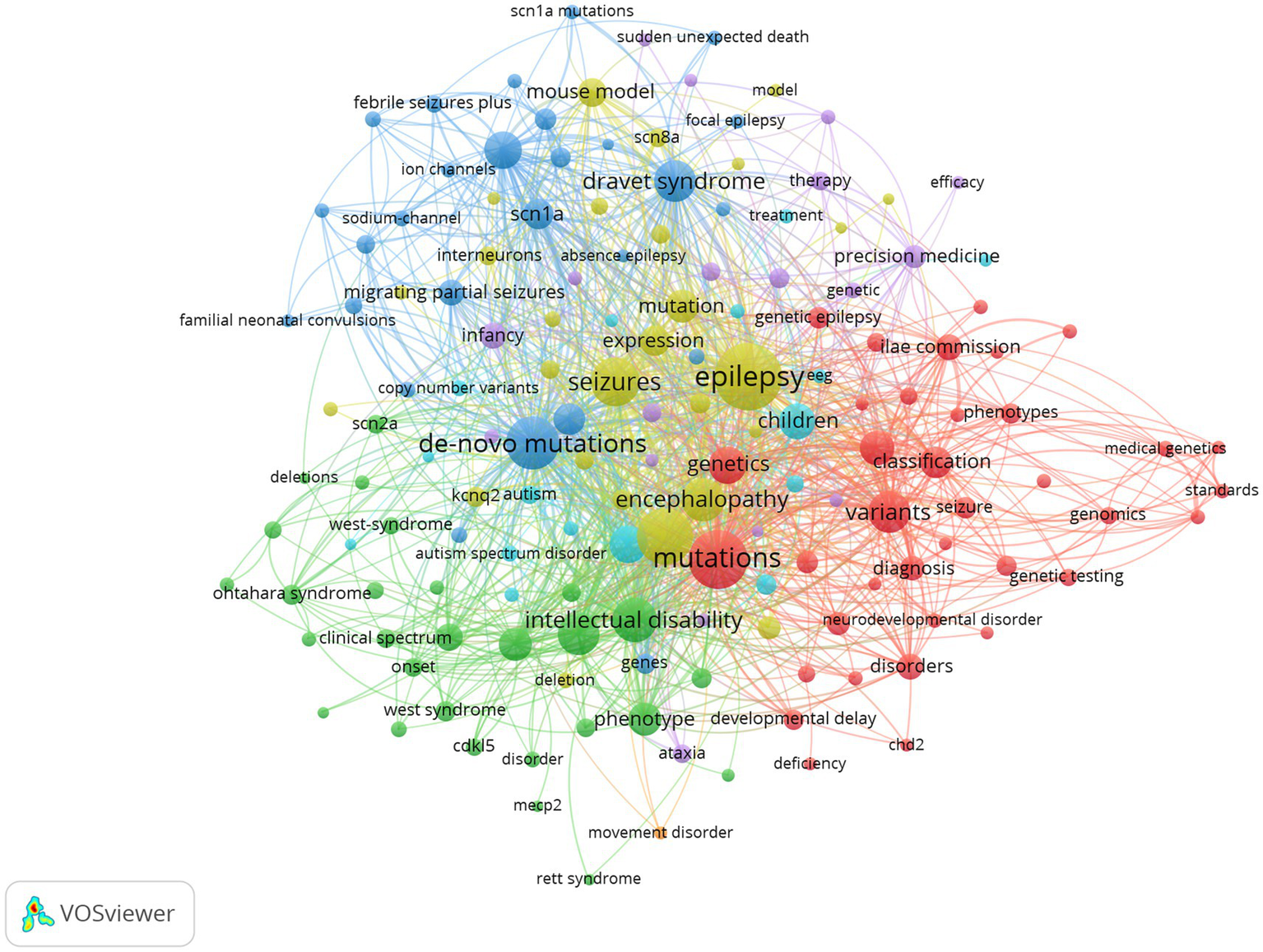
Keyword visualization map of developmental and epileptic encephalopathies and gene, where circles represent keywords, and lines denote their collaborations.
4 Discussion
As the research on the developmental and epileptic encephalopathies has deepened, scholars have increasingly focused on the relationship between developmental and epileptic encephalopathies and gene. In recent years, the number of studies in this field has grown significantly. This study quantitatively analyzed scientific outputs, countries, institutions, journals, authors, references, and keywords. Such an analysis is likely to assist in discerning the latest advancements, evolutionary trajectories, cutting - edge research hotspots, and future research trends in this area.
The temporal distribution of publications reveals a clear upward trajectory, with a marked acceleration after 2010 and a peak in 2024. This trend is closely aligned with the conceptual evolution of DEEs led by the International League Against Epilepsy (ILAE): the 2010 proposal of “epileptic encephalopathy” and “developmental encephalopathy” clarified the core pathological features of the disease, laying the foundation for targeted research The 2017 formal definition of “DEE” further integrated these two concepts, emphasizing the heterogeneity of drug-resistant epilepsy and EEG abnormalities, which standardized research objectives and promoted interdisciplinary collaboration. The 2022 ILAE classification of DEE-related syndromes by onset age further refined clinical and research frameworks, driving a surge in studies focusing on age-specific phenotypes and etiologies.
Analysis of countries and institutions reveals an uneven yet collaborative global research landscape in the field of DEE: the top five countries account for 88.16% of all publications, with the United States taking the lead—this dominance stems from strong funding support (e.g., from the U.S. National Institutes of Health), advanced technical platforms (such as gene editing and animal model development), and interdisciplinary teams integrating neurology, genetics, and molecular biology. Italy and Australia also stand out as key contributors, driven by robust clinical research networks focused on pediatric epileptic encephalopathies, exemplified by institutions like the University of Melbourne and the Florey Institute of Neuroscience and Mental Health. At the institutional level, the National Institute of Health and Medical Research ranks first in publication count, reflecting the role of public research institutions in coordinating large-scale genetic cohort studies. However, the “preferential attachment effect” has not yet fully materialized, as the top 10 institutions contribute only a small proportion of total publications—this creates opportunities for emerging institutions, especially those in low- and middle-income countries, to engage in global collaboration, a need made urgent given the global burden of DEEs (affecting 1 in 590 children under 16 years of age).
The identification of core authors further clarifies the intellectual backbone of the field. Professor Ingrid E. Scheffer led with 34 publications and 2,490 citations, reflecting her pivotal role in bridging clinical and genetic research—including contributions to DEE classification and the identification of SCN1A variants in Dravet syndrome, a prototypical DEE (15). Her close collaborations with other top authors have facilitated the validation of genotype–phenotype correlations, such as the link between SCN1A haploinsufficiency and GABAergic interneuron dysfunction (16, 17). Other key authors, such as Samuel F. Berkovic and Nicola Specchio, have focused on translational research—including the development of preclinical models for DEEs and the evaluation of non-pharmacological treatments (18–20). The collaborative networks among these authors highlight the importance of cross-institutional and cross-national partnerships in resolving the complexity of DEE genetics.
Genetic factors are the primary cause of DEE, mainly driven by monogenic variants (with de novo variants predominating) and supplemented by inherited patterns like autosomal dominant/recessive and X-linked inheritance. Pathogenic genes disrupt key neurodevelopmental processes, leading to imbalanced neuronal excitability and neurodevelopmental disorders. Major mechanisms include: (1) Ion transport abnormalities: variants in genes like SCN1A (LOF causes Dravet syndrome, GOF leads to EIDEE), KCNQ2, and CACNA1A impair transmembrane ion flux, triggering excessive neuronal excitability; (2) Synaptic transmission defects: variants in genes such as SYNGAP1, STXBP1, and newly identified TANC2 disrupt neuronal signal communication (e.g., TANC2 variants destabilize synapses and increase seizure susceptibility in Drosophila); (3) Signal transduction disorders (e.g., DEPDC5 in the mTOR pathway); and (4) Epigenetic dysregulation (e.g., EP400 for chromatin remodeling, MECP2 for transcriptional activation) (4, 15, 21, 22). These gene-mediated abnormalities collectively manifest as DEE phenotypes like epilepsy and intellectual disability.
4.1 Strengths and limitations
This study conducted a bibliometric analysis of research literature on developmental and epileptic encephalopathies and gene in the WOS database. However, the comprehensiveness and authority of the analysis results may be affected due to limitations in the database and language types. To enhance the reliability of the analysis, we plan to expand to other relevant databases and further investigate the research on developmental and epileptic encephalopathies in its interdisciplinary fields to improve our analysis results.
5 Conclusion
This study conducted a quantitative and visual analysis in the field of DEE and gene. With the help of CiteSpace and VOSviewer, we have a deeper understanding of the latest progress, evolution paths, frontier research hot spots, and future research trends in the field. And the field of DEE and gene research has received extensive attention from countries and institutions, especially the United States. Professor Ingrid Scheffer had the highest number of publications in this field, and the journal Epilepsy had the highest citation count. The research hotspots in this field revolved around epilepsy, mutations, epileptic encephalopathy, de novo mutations, and seizure. Overall, our results will help researchers understand the research trend, and provide clues for future research directions in the field.
Statements
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
Author contributions
W-hL: Methodology, Data curation, Conceptualization, Investigation, Formal analysis, Writing – original draft. S-qZ: Formal analysis, Investigation, Funding acquisition, Writing – original draft. YD: Project administration, Investigation, Data curation, Formal analysis, Writing – original draft. YZ: Writing – review & editing, Software, Investigation, Resources, Project administration. HM: Writing – review & editing.
Funding
The author(s) declared that financial support was received for this work and/or its publication. This study was supported by the Medical Science and Technology Research of Guangdong Province (No. A2023136) and the Science and Technology Projects in Guangzhou (No. 2024A03J0816).
Acknowledgments
The help of related authors and clinicians participating in this work is greatly appreciated.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that Gen AI was used in the creation of this manuscript. The authors would like to express our gratitude to the generative AI technology (Doubao) for it assistance in refining the language and enhancing the clarity of this paper.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Correction note
A correction has been made to this article. Details can be found at: 10.3389/fneur.2026.1781415.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1.
Poke G Stanley J Scheffer IE Sadleir LG . Epidemiology of developmental and epileptic encephalopathy and of intellectual disability and epilepsy in children. Neurology. (2023) 100:e1363–75. doi: 10.1212/WNL.0000000000206758,
2.
Zhang MW Liang XY Wang J Gao LD Liao HJ He YH et al . Epilepsy-associated genes: an update. Seizure. (2024) 116:4–13. doi: 10.1016/j.seizure.2023.09.021,
3.
Liu WH Luo S Zhang DM Lin ZS Lan S Li X et al . De novo GABRA1 variants in childhood epilepsies and the molecular subregional effects. Front Mol Neurosci. (2023) 16:1321090. doi: 10.3389/fnmol.2023.1321090,
4.
Luo S Zhang W‐J Jiang M Ren R‐N Liu L Li Y‐L et al . De novo TANC2 variants caused developmental and epileptic encephalopathy and epilepsy. Epilepsia. (2025) 66:2365–78. doi: 10.1111/epi.18358,
5.
Zhang W Luo S Jiang M Chen Y Ren R Wu Y et al . CSMD1 as a causative gene of developmental and epileptic encephalopathy and generalized epilepsies. Genes Dis. (2025) 12:101473. doi: 10.1016/j.gendis.2024.101473,
6.
Yang Y Zeng Q Cheng M Niu X Xiangwei W Gong P et al . GABRB3-related epilepsy: novel variants, clinical features and therapeutic implications. J Neurol. (2022) 269:2649–65. doi: 10.1007/s00415-021-10834-w,
7.
Medyanik AD Anisimova PE Kustova AO Tarabykin VS Kondakova EV . Developmental and epileptic encephalopathy: pathogenesis of intellectual disability beyond Channelopathies. Biomolecules. (2025) 15:133. doi: 10.3390/biom15010133,
8.
Kwan P Arzimanoglou A Berg AT Brodie MJ Allen Hauser W Mathern G et al . Definition of drug resistant epilepsy: consensus proposal by the ad hoc task force of the ILAE commission on therapeutic strategies. Epilepsia. (2010) 51:1069–77. doi: 10.1111/j.1528-1167.2009.02397.x,
9.
Scheffer IE Berkovic S Capovilla G Connolly MB French J Guilhoto L et al . ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. (2017) 58:512–21. doi: 10.1111/epi.13709,
10.
Zuberi SM Wirrell E Yozawitz E Wilmshurst JM Specchio N Riney K et al . ILAE classification and definition of epilepsy syndromes with onset in neonates and infants: position statement by the ILAE task force on nosology and definitions. Epilepsia. (2022) 63:1349–97. doi: 10.1111/epi.17239,
11.
van Eck NJ Waltman L . Software survey: VOSviewer, a computer program for bibliometric mapping. Scientometrics. (2010) 84:523–38. doi: 10.1007/s11192-009-0146-3,
12.
Wu H Cheng K Guo Q Yang W Tong L Wang Y et al . Mapping knowledge structure and themes trends of osteoporosis in rheumatoid arthritis: a bibliometric analysis. Front Med. (2021) 8:787228. doi: 10.3389/fmed.2021.787228,
13.
Liu W Li H Lin S . Research hotspots and frontiers of alcohol and epilepsy: a bibliometric analysis. Neuropsychopharmacol Rep. (2024) 44:342–55. doi: 10.1002/npr2.12421,
14.
Yuan J Liu Y Zhang T Zheng C Ding X Zhu C et al . Traditional Chinese medicine for breast cancer treatment: a bibliometric and visualization analysis. Pharm Biol. (2024) 62:499–512. doi: 10.1080/13880209.2024.2359105,
15.
Scheffer IE Zuberi S Mefford HC Guerrini R McTague A . Developmental and epileptic encephalopathies. Nat Rev Dis Primers. (2024) 10:61. doi: 10.1038/s41572-024-00546-6,
16.
Scheffer IE Zhang YH Jansen FE Dibbens L . Dravet syndrome or genetic (generalized) epilepsy with febrile seizures plus?Brain Dev. (2009) 31:394–400. doi: 10.1016/j.braindev.2009.01.001,
17.
Mulley JC Scheffer IE Petrou S Berkovic SF . Channelopathies as a genetic cause of epilepsy. Curr Opin Neurol. (2003) 16:171–6. doi: 10.1097/00019052-200304000-00009
18.
Marini C Scheffer IE Nabbout R Mei D Cox K Dibbens LM et al . SCN1A duplications and deletions detected in Dravet syndrome: implications for molecular diagnosis. Epilepsia. (2009) 50:1670–8. doi: 10.1111/j.1528-1167.2009.02013.x,
19.
Carvill GL McMahon J Schneider A Zemel M Myers CT Saykally J et al . Mutations in the GABA transporter SLC6A1 cause epilepsy with myoclonic-atonic seizures. Am J Hum Genet. (2015) 96:808–15. doi: 10.1016/j.ajhg.2015.02.016,
20.
Tan C Shard C Ranieri E Hynes K Pham DH Leach D et al . Mutations of protocadherin 19 in female epilepsy (PCDH19-FE) lead to allopregnanolone deficiency. Hum Mol Genet. (2015) 24:5250–9. doi: 10.1093/hmg/ddv245,
21.
Luo S Wang PY Liu WH Liang XY Gu YJ Ye YC et al . Age at onset of genetic disease and genetic dependent stage: evidence from cases with SCN1A variants. Brain. (2025) 148:e56–60. doi: 10.1093/brain/awaf064,
22.
Luo S Wang PY Zhou P Zhang WJ Gu YJ Liang XY et al . Variants in EP400, encoding a chromatin remodeler, cause epilepsy with neurodevelopmental disorders. Am J Hum Genet. (2025) 112:87–105. doi: 10.1016/j.ajhg.2024.11.010,
Summary
Keywords
developmental and epileptic encephalopathies, gene, bibliometric analysis, Web of Science, VOSviewer, CiteSpace
Citation
Liu W-h, Zhang S-q, Ding Y, Zhao Y and Meng H (2025) Exploring the landscape of developmental and epileptic encephalopathies and gene research global dynamics via bibliometric study 2001–2025. Front. Neurol. 16:1733441. doi: 10.3389/fneur.2025.1733441
Received
27 October 2025
Revised
17 November 2025
Accepted
26 November 2025
Published
16 December 2025
Corrected
20 January 2026
Volume
16 - 2025
Edited by
Magdeldin Elgizouli, University of Zurich, Switzerland
Reviewed by
Sifen Xie, Guangdong 999 Brain Hospital, China
Chen Yujie, Zhuzhou Central Hospital, China
Updates
Copyright
© 2025 Liu, Zhang, Ding, Zhao and Meng.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Heng Meng, memphisheng@163.com; Ying Zhao, 396302462@qq.com
†These authors have contributed equally to this work and share first authorship
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.