 Yi Luan
Yi Luan Hui Liu
Hui Liu Ying Luan3
Ying Luan3 Yang Yang
Yang Yang Jing Yang
Jing Yang Kai-Di Ren
Kai-Di Ren- 1Research Center for Clinical System Biology, Translational Medicine Center, the First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
- 2School of Laboratory Medicine, Xinxiang Medical University, Xinxiang, China
- 3Henan Key Laboratory of Precision Clinical Pharmacy, Zhengzhou University, Zhengzhou, China
- 4Department of Pharmacy, the First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
- 5Department of Physiology and Neurobiology, School of Basic Medical Sciences, Zhengzhou University, Zhengzhou, China
Atherosclerosis (AS) features include progressive hardening and reduced elasticity of arteries. AS is the leading cause of morbidity and mortality. An increasing amount of evidence showed that epigenetic modifications on genes serve are a main cause of several diseases, including AS. Histone deacetylases (HDACs) promote the deacetylation at lysine residues, thereby condensing the chromatin structures and further inhibiting the transcription of downstream genes. HDACs widely affect various physiological and pathological processes through transcriptional regulation or deacetylation of other non-histone proteins. In recent years, the role of HDACs in vascular systems has been revealed, and their effects on atherosclerosis have been widely reported. In this review, we discuss the members of HDACs in vascular systems, determine the diverse roles of HDACs in AS, and reveal the effects of HDAC inhibitors on AS progression. We provide new insights into the potential of HDAC inhibitors as drugs for AS treatment.
Introduction
As a critical potential pathology of cardiovascular disease (CVD), atherosclerosis (AS) is characterized by the progressive hardening and reduced elasticity of arteries; AS is a leading cause of morbidity and mortality (Aghamajidi et al., 2021). Its progression will ultimately lead to myocardial infraction, ischemic stroke, cerebrovascular incidents, and peripheral vascular disease, thereby increasing the risk of death (Yang et al., 2021). The most outstanding feature of AS is the plaque formation in the arteries. Vascular cell homeostasis low-density lipoprotein (LDL) oxidation, monocyte recruitment, macrophage-derived foam cell formation, and thrombus formation play important roles during AS progression (Luan et al., 2021). Among them, vascular homeostasis is one of the major AS risk factors (Rajendran et al., 2013). The maintenance of vascular homeostasis requires the joint participation of various vascular cells. Vascular cells are composed of endothelial cells (ECs) and smooth muscle cells (SMCs). The alteration of proliferation, migration, and apoptosis of ECs and SMCs is indispensable in AS (Dai et al., 2018). Endothelial dysfunction includes abnormal proliferation, migration, and apoptosis and contributes to enhanced endothelial permeability to lipoproteins, increased leucocyte migration and adhesion, and reduced nitric oxide production, thereby subsequently triggering fatty streak formation (Liu et al., 2007). The proliferation and migration of SMCs are critical in the formation of fatty steak, which induces advanced lesions and fibrous cap formation (Clarke et al., 2006). AS is very likely to form at certain areas of arteries, such as branching points and bends, because of the local disturbance of endothelial functions (Ravensbergen et al., 1998). In addition, lipid metabolism disorder also plays an important role in AS progression. As reported, LDL, especially that modified by oxidation, enzymatic processing, desialylation, and aggregation, is the main substance in the atherosclerotic lesions (Torzewski, 2021). These LDL modifications are prone to induce immune response; thus, the body forms highly atherogenic circulating LDL, which are involved in immune complexes (Zhang et al., 2018a). Other cell types are also involved in AS pathogenesis, such as macrophages and stem cells (Wang et al., 2015a; Takamura et al., 2017; Ruytinx et al., 2018; Kloc et al., 2020).
An increasing amount of evidence indicated that epigenetic modifications on genes are the main cause of many diseases, such as, cancer, and CVDs (Yang et al., 2021). Epigenetic modifications can modulate gene expression without altering gene sequences, thereby facilitating rapid and reversal of the regulation of targeted genes (Wee et al., 2014). Epigenetic modifications are composed of DNA and histone modifications. DNA modifications are inheritable, whereas histone modifications are not (Qin et al., 2021). Histone methylation and acetylation are the main forms of histone modification (Rajan et al., 2020). Acetylation changes the condensation of chromatin and has been considered as a therapeutic target. Histone acetylation is tightly controlled by histone acetyltransferases (HATs) and histone deacetylases (HDACs), which exert contradictory functions (Raman and Rai, 2018). Acetylation in histones and non-histones can be catalyzed by HATs and removed by HDACs. In most cases, HDACs repress gene expressions through interactions with histones and transcription factors (Gallinari et al., 2007).
The Classification of HDACs
HDACs promote the deacetylation at lysine residues and condense the chromatin structures, thereby further inhibiting the transcription of downstream genes (Walther et al., 2020). HDACs are composed of two families, namely, HDAC and sirtuin, which include 18 members (Dai et al., 2021). These members can be clearly classified into four groups, namely, Class I, II, III, and IV, based on the enzymatic activities, domain structures, functions, and sequence similarity (Figure 1) (Zhang et al., 2020). Class I HDACs (HDAC1/2/3/8) share high sequence similarity with yeast Rpd3 and are mostly located in the nucleus (Zaidi et al., 2020). Class II HDACs are subclassified into subclass IIa (HDAC4, 5, 7, and 9) and subclass IIb (HDAC6 and 10). Class IIa HDACs are specifically expressed in muscle and heart tissues; they translocate between cytoplasm and nucleus, interact with kinase proteins (calcium-independent protein kinases and the MAPK), and act as signal transducer (Ziegler et al., 2020). Class IIb members mostly localize in the cytoplasm and are distinguished from the class IIa members in terms of the tandem deacetylase domains (Yu et al., 2020). Class III is composed of the sirtuin family and contains seven members (SIRT1 to 7), which share high sequence similarity with the yeast protein silent information regulator 2 (Sir2) (Chen et al., 2020). These HDACs require NAD+ to catalyze the deacetylation reaction due to their conserved catalytic domain, whereas other HDACs rely on the binding of zinc molecule as an activator (Vaquero et al., 2007). SIRT1/2 shuttle between the nucleus and cytoplasm. SIRT6/7 are mostly in the nucleus, and SIRT3/4/5 are localized in the mitochondria (Figure 1) (Villalba and Alcaín, 2012). Class IV HDAC (HDAC11, the sole member in class IV) shares sequence homology with Class I and II members (Seto and Yoshida, 2014). HDAC11 modulates the protein stability of CDT1 and negatively modulates the expression of interleukin (IL)-10 and the activity of T cells, thereby indicating the potential role of HDAC11 in AS progression (Glozak and Seto, 2009). The classical HDAC family also consists of Class I, II, and IV besides Class III (Dai et al., 2021).
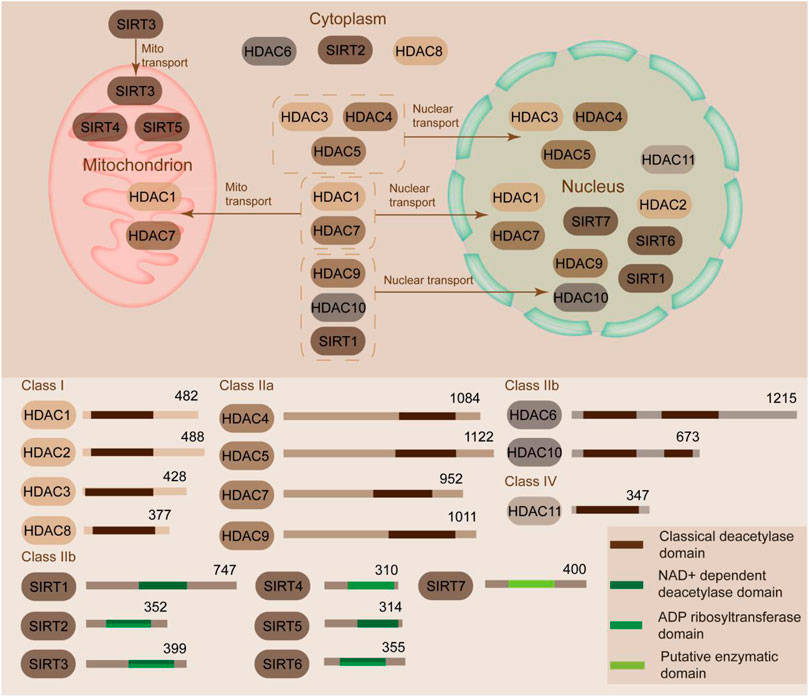
FIGURE 1. Classification and sublocation of HDACs. HDACs can be classified into Class I, II, III, and IV according to similarities. Class I HDACs (HDAC1/2/3/8) are mostly located in the nucleus. Class II HDACs are subclassified into subclass IIa (HDAC4, 5, 7 and 9) and subclass IIb (HDAC6 and 10). Class IIa HDACs translocate between cytoplasm and nucleus. Class IIb members mostly localize in the cytoplasm. Class III contains seven members (SIRT1 to 7). SIRT1/2 shuttle between the nucleus and cytoplasm. SIRT6/7 are mostly in the nucleus. SIRT3/4/5 are localized in the mitochondria. Class IV HDAC (HDAC11) is predominantly located in the nucleus.
Although HDACs are commonly recognized as enzymes that catalyze the removal of acetyl group from histones, present studies have identified many other non-histone substrates, such as NF-kB, E2F1, SP1, KLF2/4, and STAT1 (Villagra et al., 2010). Considering the diversity of HDAC substrates, they are also related to multiple cellular processes and several diseases, including AS (Ke et al., 2021). Notably, the sirtuin family is widely acknowledged because of its diverse roles in vascular functions. In this review, we discuss the role of HDACs in vascular function and AS process and the pharmacological effects of HDAC inhibitors (HDACi) on AS treatment.
HDACs in Modulating the Function of Endothelial Cells (ECs)
Endothelial cells (ECs) are a main type of cells in blood vessels that modulate vascular tone, blood coagulation, and mediate inflammatory reaction (Wautier and Wautier, 2021). HDACs are a major group of histone deacetylases that are extensively involved in endothelial cell function regulation (He et al., 2011). HDACs are critical in modulating the gene expressions involved in vascular homeostasis and vessel development as transcriptional cofactors (Figure 2) (Zhao et al., 2020). Among the HDACs, HDAC1/2/3 are reportedly involved in EC proliferation mediated by oscillatory shear stress, which increases the expression of cyclin A and decreases p21 (Bazou et al., 2016). HDAC2 overexpression suppresses vascular dysfunction induced by oxidized LDL (Pandey et al., 2014). HDAC3 mediates EC differentiation from embryonic stem cells and keeps endothelial integrity dependent on PI3K/Akt and TGFβ2 pathways (Wang et al., 2021). Moreover, the knockdown of HDAC3 is associated with the decrease of Nox4, a major source of reactive oxygen species (ROS) production in the vascular wall, thereby suggesting the critical role of HDAC3 in EC function (Fu et al., 2020).
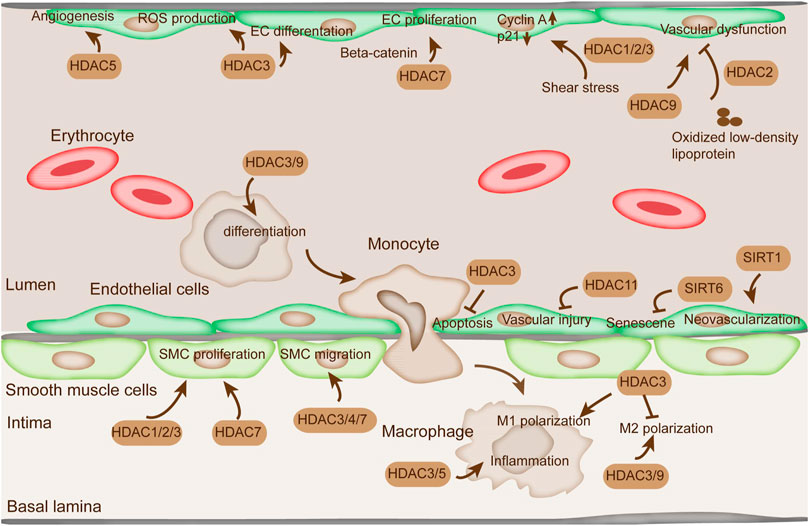
FIGURE 2. The function of HDACs in ECs, SMC, and macrophages. The alteration of proliferation, migration, and apoptosis of ECs and SMCs is indispensable in AS. HDAC1/2/3/7 regulate SMC proliferation. HDAC3/4/7 are involved in SMC migration. HDAC3/5 modulate inflammation in macrophage. HDAC3/9 regulate proinflammatory gene expression by modulating M2 macrophage polarization. HDAC11 plays a critical role in vascular injury. SIRT1 is directly related to EC senescence and apoptosis. SIRT6 protects EC from senescence. HDAC1/2/3 are reportedly involved in EC proliferation mediated by oscillatory shear stress. In addition, HDAC5 represses angiogenesis in ECs.
Class IIa HDACs are involved in vascular homeostasis response to signals in the modulation of gene expression (Clocchiatti et al., 2011). For instance, HDAC4/5 nuclear translocation is initiated by nitric oxide (NO) to modulate the activation of protein phosphatase 2A (PP2A) (Illi et al., 2008). Moreover, NO promotes the formation of a complex containing HDAC3, HDAC4, HDAC5, and an active PP2A (Illi et al., 2008). HDAC5 represses angiogenesis by mediating the expression profile of angiogenic genes in ECs (Shiva Shankar and Willems, 2014). Its overexpression weakens sprout formation, whereas its inhibition by the inhibitors manifests pro-angiogenic effect by inducing EC migration, sprouting, and tube formation. Previous studies indicated that HDAC7 regulated EC proliferation by modulating beta-catenin translocation. HDAC7 overexpression prevented nucleus beta-catenin translocation and inhibited EC proliferation (Margariti et al., 2010). HDAC7 knockdown promoted the nuclear translocation of beta-catenin and inhibited the levels of cyclin D1, cyclin E1, and E2F2, thereby causing EC hypertrophy. Moreover, HDAC7-mediated EC proliferation suppression can be partially ameliorated by VEGF through the induction of HDAC7 degradation via PLCg-IP3K signaling pathway (Chang et al., 2006). Another HDAC, HDAC9, affected EC dysfunction and permeability dysfunction in oxygen-glucose deprivation-induced ischemia in the cerebral hemisphere (Shi et al., 2016).
The sirtuin family is also involved in mediating the function of ECs (Matsushima and Sadoshima, 2015). SIRT1 is critical in modulating neovascularization by regulating angiogenic ability when stimulated by angiogenic cues (Botti et al., 2014). The deacetylase activity of SIRT1 on Foxo transcription factors repressed its transcription and restrained EC proliferation, migration, and neovessel formation (Gu et al., 2016). SIRT6 protected EC from senescence. SIRT6 depletion aggravated the percentage of senescent cells in HUVEC and aortic endothelial cells and suppressed the formation of tubule networks (Cardus et al., 2013). The sole member of class IV, HDAC11, restored the expression of angiogenic factor in response to carotid artery ligation in mice (Nunez-Alvarez and Suelves, 2021). HDAC11 depletion mitigated vascular injury in mice, suggesting its critical role in vascular injury (Nunez-Alvarez and Suelves, 2021).
The apoptosis of ECs also plays critical role in endothelial dysfunction during AS progression (Qin et al., 2017). In addition, recent studies implied that apoptosis in luminal EC probably induced the formation of thrombus on eroded plaques without rupture (Quillard et al., 2017). HDACs are believed to affect EC apoptosis. Knockdown of HDAC3 induced extensive membrane blebs and more Annexin V staining and reduced cell survival (Lee and Chiu, 2019). In addition, HDAC3 overexpression facilitated Akt phosphorylation and activated its kinase activity (Long et al., 2017). Therefore, HDAC3 is crucial in maintaining cell survival and prevents AS by activating Akt. In addition, SIRT1 is directly related to EC senescence and apoptosis (Liu et al., 2018). Hou et al. showed that SIRT1 prevented the externalization of early membrane apoptotic phosphatidylserine, and the DNA degradation was dependent on Akt1 and FoxO3a (Hou et al., 2011). Other studies demonstrated that SIRT1 mediated EC proliferation and senescence by modulating a serine/threonine kinase and tumor suppressor LKB1 (Zu et al., 2010). Inhibition of HDACs by valproic acid induced phosphorylation of extracellular signal-regulated kinase1/2 (ERK 1/2) and subsequently caused phosphorylation of Bcl-2 and EC apoptosis inhibition in response to serum starvation (Joanna et al., 2009).
HDAC Modulation in Smooth Muscle Cells (SMCs)
The proliferation of smooth muscle cells (SMCs) is necessary in the formation of neointima and arteriosclerosis (Daniel and Sedding, 2011). Following EC injury and activation, diverse growth factors (e.g., PDGF and TGF-beta) and cytokines (interferon-ɣ) were released and promote SMC proliferation, which aggravated the generation of advanced lesions during AS (Annoni et al., 1992). Interference in the level of HDAC1/2/3 attenuated SMC proliferation induced by mitogens (Figure 3) (Findeisen et al., 2011). The suppression in the activity of Classes I and II HDACs by apicidin inhibited proliferation in newborn pulmonary arterial SMCs and cell cycle arrest at G1 phase (Zhao et al., 2020). HDAC inhibition by butyrate abrogated Akt activation and subsequent downstream Akt targets, thereby promoting proliferation arrest (Zhao et al., 2020). Among SMCs, vascular smooth muscle cells (VSMCs) are crucial in regulating blood pressure and tissue repair (Jaminon et al., 2019). HDACs mediate the functions of VSMCs. SIRT1 could regulate VSMC proliferation and motility and induce cell cycle arrest at G1/S transition (Wang and Chen, 2020). Overexpression of the unspliced form of HDAC7 (HDAC7u) attenuated SMC proliferation by decreasing cyclin D1, whereas the spliced HDAC7 did not result in the same effect (Zhou et al., 2011a). HDAC7u showed binding with beta-catenin and inhibited its nuclear translocation; moreover, it mitigated beta-catenin activity (Zhou et al., 2011b). Knockdown of HDAC7 exacerbated neointimal formation in femoral artery wire injury animal model, indicating a potential therapeutic in AS (Margariti et al., 2009).
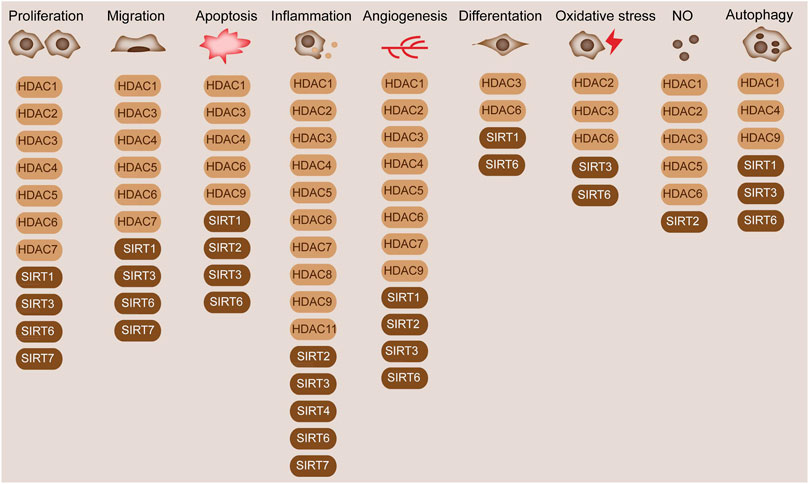
FIGURE 3. Diverse functions of HDACs in blood vessels. HDACs are widely involved in the cellular processes in blood vessels, such as cell proliferation, migration, and differentiation. HDAC1/2/3/4/5/6/7 and SIRT1/3/6/7 are involved in cell proliferation. HDAC1/3/4/5/6/7 and SIRT1/3/6/7 regulate cell migration. HDAC1/3/4/6/9 and SIRT1/2/3/6 mediate cell apoptosis. HDAC1/2/3/4/5/6/7/8/9/11 and SIRT2/3/4/6/7 modulate inflammation. HDAC1/2/3/4/5/6/7/9 and SIRT1/2/3/6 modulate angiogenesis. HDAC3/6 and SIRT1/6 modulate cell apoptosis. HDAC2/3/6 and SIRT3/6 are associated with oxidative stress. HDAC1/2/3/5/6 and SIRT2 are associated with NO production. HDAC1/4/9 and SIRT1/3/6 modulate cell autophagy.
In addition, VSMC migration is tightly related to vascular remodeling, which is likely to trigger AS progression (Lin et al., 2021). Mechanical cyclic strain reportedly suppressed the migration of SMCs along with the elevation of acetylated histone H3 and HDAC7 and decreased the level of HDAC3/4 (Figure 2) (Yan et al., 2009). Treatment with HDAC inhibitor tributyrin diminished VSMC migration and reduced the level of HDAC7 (Yan et al., 2009).
Also, the formation and the accumulation of lipid-loaded foam cells is the critical step in the pathophysiology of AS in the vascular wall (Linton et al., 2000). The uptake of modified lipoproteins in macrophage, such as oxidized LDL and native and modified LDL, drives macrophages to form foam cells (Linton et al., 2000). Recent studies also discovered that SMCs can also form foam cells. The inhibition of HDACs is involved in the formation of foam cells (Figure 3). MS‐275, which is known as a specific class I HDAC inhibitor, inhibits human foam cell formation (Jeanblanc et al., 2015).
The inversion of macrophage to foam cells plays a pro-atherosclerotic role in the AS process (Moore et al., 2013). The macrophage uptake of lipoproteins depends on scavenger receptors (SRs), including SR-A, CD36, and lectin-like oxidized LDL receptor-1 (LOX-1) (Chistiakov et al., 2016). The inhibition of SRs suppresses the ingestion of lipoproteins and hinders the AS process. Schaeffer et al. revealed that LOX-1 expression could be stimulated by pro-inflammatory cytokines in macrophage, which elevated the uptake of ox-LDL by nidus macrophages (Pirillo et al., 2013). The deacetylation of RelA/p65 by SIRT1 induced the suppression of NF-κB signaling pathway and LOX-1 in macrophage, diminished the uptake of oxLDL and foam cell formation, and subsequently reduced the risk of AS (Stein et al., 2010). Moreover, resveratrol decreased the uptake of oxLDL and protected from AS (Ou et al., 2006).
HDACs in Modulating the Function of Monocyte/Macrophage
Monocytes and macrophages are critical driving factors in the inflammatory disease process (Yang et al., 2014). They play an important role in the pathogenesis and the initiation of AS, because AS is lipid-driven and occurs with chronic inflammation (Naruszewicz, 1989). Monocytes and macrophages develop foam cells and pro-inflammatory phenotypes in response to oxidized LDLs (Adamson and Leitinger, 2011). Perturbation of this phenotype would bring beneficial outcomes in the management of the disease. An increasing amount of evidence pointed out that histone acetylation plays an important role in the modulation in monocytes and macrophages (Das Gupta et al., 2016). In particular, H3 acetylation plays an important role in macrophage phenotypic gene expression (Figure 2) (Zubair et al., 2021). Among the HDACs, HDAC3 acts as a key modulator in M1 macrophage polarization, thereby blocking M2 macrophage polarization (Figure 2) (Wang et al., 2015b). HDAC3 also inhibits NF-κB signaling by deacetylating NF-κB p65 subunit and inducing its association with the IκB-α (Rajendrasozhan et al., 2008). Moreover, HDAC3 is responsible for the modulation of lipopolysaccharide-induced M1 macrophage-associated inflammatory gene expression (Treuter et al., 2017). HDAC5 is also a modulator of inflammation in macrophages (Zhao et al., 2019).
Translocation of circulating monocytes to the artery wall is one of the prominent phenotypes in AS in endothelial dysfunction and lipoprotein retention (Manduteanu and Simionescu, 2012). Upon differentiation into macrophage, these cells play an important role in sustaining lipid homeostasis in the vessel wall and inflammatory mediator secretion. Lipoprotein uptake by macrophage in the initiation of plaque contributes to the formation of lipid-load macrophage foam cells, which are hallmarks of AS (Remmerie and Scott, 2018). These foam cells stick to the artery walls, resulting in adverse inflammatory response that induces the recruitment and activation of other immune cells. Thus, chronic inflammation is maintained, which stimulates the progression of plaque formation. The role of HDACs in this process should not be ignored.
Histone acetylation also plays an important role in the cholesterol metabolism of macrophages (Zubair et al., 2021). Alterations in SIRT1 and SIRT6 promote cholesterol efflux by activating ABCA-1 and ATP-binding cassette subfamily G member (ABCG-1), leading to reduced macrophage-derived foam cell formation (D'Onofrio et al., 2018). During AS, histone modification within the atherosclerotic plaque affects macrophage phenotype; acetylation level alteration on H3K9 and H3K27 has been detected from human advanced AS plaques (Zhu et al., 2021).
LDLR−/− mice, as a general AS mice model, were fed with atherogenic diet and exhibited elevated HDAC3 and HDAC9 expressions during monocyte differentiation to macrophages (Figure 3) (Davis and Gallagher, 2019). HDAC3/9 systemic or myeloid-specific depletion reduced AS by increasing M2 macrophage polarization and lowering proinflammation gene expression (Sanchez-Lopez et al., 2019). Genome-wide association studies depicted that HDAC9 genetic variants were associated with coronary artery disease and AS (Prestel et al., 2019). Systemic and bone marrow-specific depletion of HDAC9 brought about elevated lipid homeostatic genes, reduced inflammatory genes, and switched macrophage phenotype to the M2 state, thereby decreasing AS progression by increasing the acetylation of the promoter of ABCA-1 and ABCG-1 in macrophages (Cao et al., 2014). Simultaneously, HDAC3 depletion induced macrophage phenotype switch, increased anti-inflammatory cytokine secretion, and reduced pro-inflammatory cytokine, thereby suggesting the promotion effect of HDAC3 on AS. The above mentioned HDACs mediated the recruitment and differentiation of monocytes and modulated AS progression (Hoeksema et al., 2014). However, the underlying mechanism of HDAC-mediated monocyte differentiation remains unclear and needs future research.
The Functions of HDACs in the Pathogenesis of Atherosclerosis
AS is featured by the accumulated lipid and fatty streak lesion formation in the vessel, acting as the most prevalent disease in vasculature (Mundi et al., 2018). Notably, the recruitment of circulating monocytes to the arterial wall and lipoprotein retention is one of the earliest events of AS (Moore et al., 2013). The differentiation of monocytes to macrophages sustains the homeostasis of lipid in the vessel wall and secretion of inflammatory mediators, thereby functioning critically in the pathophysiology of AS (Rajendrasozhan et al., 2008). Macrophage lipid uptake in the initiation of plaque leads to the formation of lipid-loaded foam cells, which are hallmarks of AS (Rajendrasozhan et al., 2008). The presence of these foam cells at the artery wall stimulates disadvantageous immune response, which further leads to the recruitment and activation of other immune cells, maintains chronical immune response, and induces plaque progression (Wilson, 2010). Epigenetic modification in VSMCs (the dominant cell type in the arterial wall), such as acetylation, is reportedly associated with AS formation.
The effect of cholesterol metabolism on AS is obvious. The metabolic homeostasis of cholesterol includes cholesterol uptake, synthesis, and efflux and is critical in maintaining the homeostasis of blood vessel (Ghosh, 2011). Histone acetylation, which is affected by HDACs, is important in cholesterol metabolism. For instance, perturbations in SIRT1 and SIRT6 facilitate cholesterol efflux by activating ABCA-1 and ABCG-1, thereby leading to the reduced formation of macrophage-derived foam cells (Figure 4) (Ghosh, 2011). Apart from this, the alteration of histone acetylation by HDACs also affects macrophage phenotype in AS plaque. The alteration of acetylation on H3K9 and H3K27 is observed in advanced plaques compared with healthy samples (Lee et al., 2020). The levels of several HDAC, HDAC3, and HDAC9 could be stimulated in response to monocyte differentiation to macrophages in LDLR−/− mice kept on an atherogenic diet (Figure 4) (Rajendrasozhan et al., 2008). In addition, myeloid-specific deletion of HDAC9 and HDAC3 mitigates AS by promoting M2 macrophage polarization and reducing proinflammatory gene expression (Qiu et al., 2021). The mechanism of HDAC9 in promoting pro-inflammatory responses and augmenting the effects of atherosclerotic plaque vulnerability involves the binding of HDAC9 to IKKα and β, which contributes to their deacetylation and subsequent activation, thereby finally driving vascular inflammation (Van den Bossche et al., 2014).
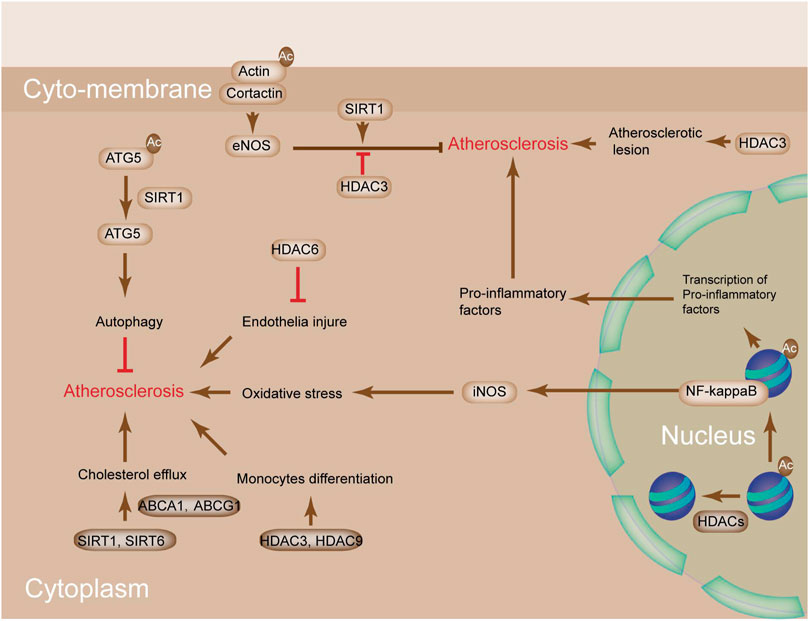
FIGURE 4. The potential mechanism of HDACs in atherosclerosis. SIRT1 and SIRT6 perturbation promotes cholesterol efflux by activating ABCA1 and ABCG1, leading to reduced macrophage-derived foam cell formation. Deacetylation of ATG5 by SIRT1 activates ATG5 to increase autophagy, which protects from AS. SIRT1-mediated deacetylation of cortactin promotes the translocation of cortactin to the cell periphery, whereas its interaction with cortical actin activates eNOS under shear stress conditions, which increases the bioavailability of nitric oxide (NO) and protects from AS. HDAC3/9 are stimulated in response to monocyte differentiation to macrophages in LDLR−/− mice kept on an atherogenic diet. HDAC6 is implicated in the prevention of endothelial injury and AS. NO bioavailability is increased by SIRT1-mediated deacetylation of eNOS and decreased by HDAC3-mediated deacetylation of eNOS. Acetylation of NF-κB subunits activates the expressions of Nos1, Nos2, and pro-inflammatory genes. High levels of NO and oxidative stress promote AS.
In addition to cholesterol metabolism, several biological processes are involved in the AS process, including the following: endothelial denudation, injury, activation, and shear stress; local platelets adherence; lipoprotein oxidation; lipoprotein aggregation; and inflammatory cytokine secretion and foam cell formation (Luchetti et al., 2017). The levels of Class I, IIa, IIb, and IV HDAC isoenzymes were dramatically augmented in human and mice AS tissues (Lian et al., 2020). Treatment with SAHA, a HDAC non-selective inhibitor, mitigated the level of atherosclerotic injury and reduced the production of ROS induced by NAPDH and pro-inflammation markers (Zhao et al., 2020). HDAC1 was suppressed in atherosclerotic lesions and aortic EC-treated with oxidized lipoproteins (Chen et al., 2020). Arginase 2 (Arg2) regulates the generation of endothelial nitric oxide, proliferation, fibrosis, and inflammation, making it a possible target for AS progression (Yang and Ming, 2014). Arg2 ablation by HDAC2 overexpression in human aortic ECs impaired EC activation induced by oxLDL (Pandey et al., 2014). Shear stress in the vicinity of disturbed flow could upregulate the expression of HDAC3. HDAC3 depletion aggravated atherosclerotic lesion in aortic isografts of ApoE-knockout mice, thereby indicating the protective role of HDAC3 in AS (Zhao et al., 2020).
HDACs are also involved in the modulation of cholesterol efflux (Cruz et al., 2021). The inhibition of HDACs upregulates the expression of cholesterol efflux genes ABCA1 and ABCG1. Perturbations in SIRT1 and SIRT6 facilitate cholesterol efflux by activating ABCA-1 and ABCG-1, leading to the reduced formation of macrophage-derived foam cells (Sosnowska et al., 2017). Deacetylation of autophagy protein 5 (ATG5) by SIRT1 activates ATG5 to increase autophagy, which protects from atherosclerosis (Jiang et al., 2016). Furthermore, HDAC3 remains the only HDAC that is upregulated in human atherosclerotic lesions and accompanied by inflammatory macrophages, thereby indicating that it could be a potential target in AS prevention. Macrophage-induced inflammatory response and SMC-elicited vascular remodeling are the two main pathophysiological features in AS (Deeb and Hajjar, 2016). CIITA, known as a major histocompatibility class II transactivator, is a key modulator in these processes. It modifies IFN-gamma-induced major histocompatibility class II activation and inhibits type I collagen (Wu et al., 2009). HDAC2 counteracts CIITA activation via the degradation of CIITA, which is dependent on deacetylation activity (Kong et al., 2009). HDAC6 activity was greatly induced in ApoE−/- mice fed on high-fat diet in spite of the unchanged protein level, thereby indicating that HDAC6 inhibition can prevent endothelial injury and AS (Xu et al., 2021). The phosphorylation of HDAC5 was induced in A10 vascular SMCs in response to IGF-1 (Truong et al., 2021). Interference of IGF-1 receptor tyrosine kinase and NAD(P)H oxidase mitigated IGF-1 induced HDAC5 phosphorylation, thereby suggesting that HDAC5 phosphorylation is related to NAD(P)H oxidase-induced ROS generation and vascular disorders (Zhao et al., 2020).
Inflammation is considered as a main inducer of AS. Histone H3 acetylation mediated by the type A KAT p300 transcription factor is a prerequisite for the expression of inflammatory genes in the VSMCs of rats (Sun et al., 2017). Furthermore, treatment with HDAC inhibitor trichostatin A promotes the expression of inflammatory cytokine tumor necrosis factor (TNF) and aggravates the development of neointima lesions in AS-susceptible LDLR−/- mice (Okamoto et al., 2006). Histone acetylation is important in AS progression, and the inflammatory status is modulated by HDAC activity (Cosío et al., 2004). Vascular injury decreases the expression of VSMC differentiation marker genes and transforms it into a more proliferative phenotype, thereby increasing the probability of AS (Morris et al., 2019). Promoting the differentiation of VSMCs by histone H4 acetylation can restore injury in response to vascular stress by assisting the combination of serum response factor (SRF) and myocardin with CArG elements (Xiao et al., 2012). This process can be reversed by Kruppel-like factor 4 (KLF4), which promotes the deacetylation of histone H4 by recruiting HDAC2 (McDonald et al., 2006). SIRT1 exerts its anti-atherosclerotic effect by deacetylating autophagy protein 5, thereby impeding the oxLDL-induced cytotoxicity. The inhibition of SIRT1 facilitated the development of atherosclerotic plaque formation in ApoE−/− mice (Yuan et al., 2020). Other studies also showed that SIRT1 might prevent atherosclerosis by modulating eNOS activation. In human umbilical vein endothelial cells (HUVECs), cortactin phosphorylation by AMPK, deacetylation by SIRT1, and eNOS deacetylation by SIRT1 had atheroprotective effects (Figure 4) (Shentu et al., 2016), whereas eNOS deacetylation by HDAC3 promoted AS. Different from the reduced level of eNOS in the atherosclerosis state, neuronal NO synthase (nNOS) levels were induced in neointimal and media VSMCs (Nakata et al., 2007). Inducible NO synthase (iNOS) was also induced in atherosclerotic situations and associated with oxidative stress and inflammation (Förstermann and Sessa, 2012). Nuclear factor-κB (NF-κB) is responsible for the induction of nNOS in VSMC and iNOS and pro-inflammatory genes in ECs. The transcriptional factor NF-κB is modulated by lysine acetylation. SIRT1 deacetylation on p65 destroys the interaction of p300 and NF-κB, thereby reducing NF-κB transcriptional activity (Figure 4) (Li et al., 2021). SIRT1 is modulated by iNOS levels through increase in the production of NO, activity of NF-κB, and expression of pro-inflammatory genes (Lee et al., 2009). These studies indicated that HDACs offered protected against AS.
The diverse functions of HDACs in AS have been mentioned above (Table 1). However, the role of HDACs in AS often leads to conflicting results. The overexpression of HDAC3, HDAC5, and HDAC7 displayed a pro-atherosclerotic feature (Zhang et al., 2018b). HDAC3 depletion induced the increase in the expressions of IL-4-activated genes and activated anti-inflammatory phenotype due to the reprogramming-like effect on macrophage (Kuznetsova et al., 2020). Also, the HDAC9 ablation in mice repressed the expression of inflammatory-related genes and cytokine secretion in macrophage in response to LPS (Liu et al., 2021). However, HDACs seemed to exhibit atherosclerosis protection in animal models (Halili et al., 2010). HDAC3 depletion in ECs facilitated the increase in neointimal formation, thereby implicating the beneficial role of HDAC3 in AS. Similarly, HDAC inhibitor treatment, such as that of Trichostatin A, by intraperitoneal injection resulted in augmented plaque size and macrophage infiltration in plaques in LDLR−/- mice (Manea et al., 2020). These findings suggested the complicated effects of HDACs on AS and indicated that HDACs in a specific cell type affect different phenotypes in atherogenic progression.
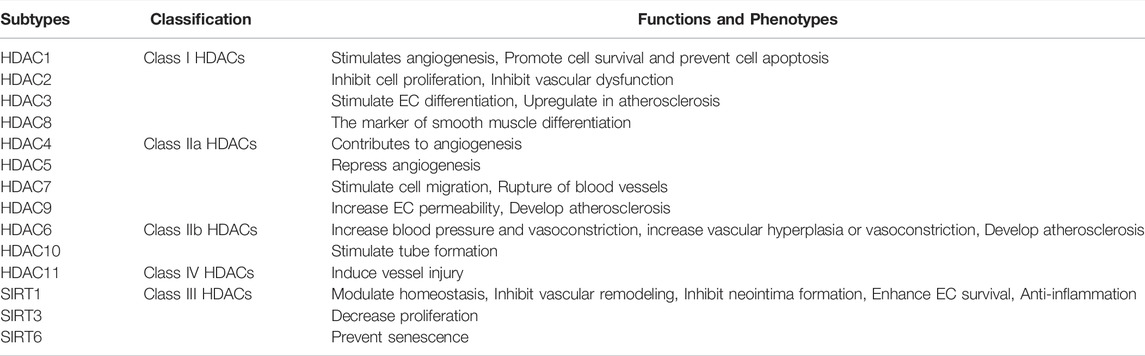
TABLE 1. Roles of HDACs in blood vessels and the pathology of AS.
Despite the diverse effects of HDACs in AS pathogenesis (Table 1), some issues need to be discussed. One question to address is whether the effects of HDACs on tissues or cell types are specific. Another problem is whether or not the effects of HDACs on the migration of SMCs and ECs are the same. To answer these questions, more experiments are required in future.
Promising Therapeutic Targets in AS
HDACs are involved in broad biological processes. Thus, multiple HDAC inhibitors are developed to target the catalytic domain in HDACs (Table 2). The inhibitors can be classified into four groups, namely, hydroxamic acids, short chain fatty acids, cyclic tetrapeptides, and benzamides, based on their structural diversity (Table 2) (Li and Seto, 2016).
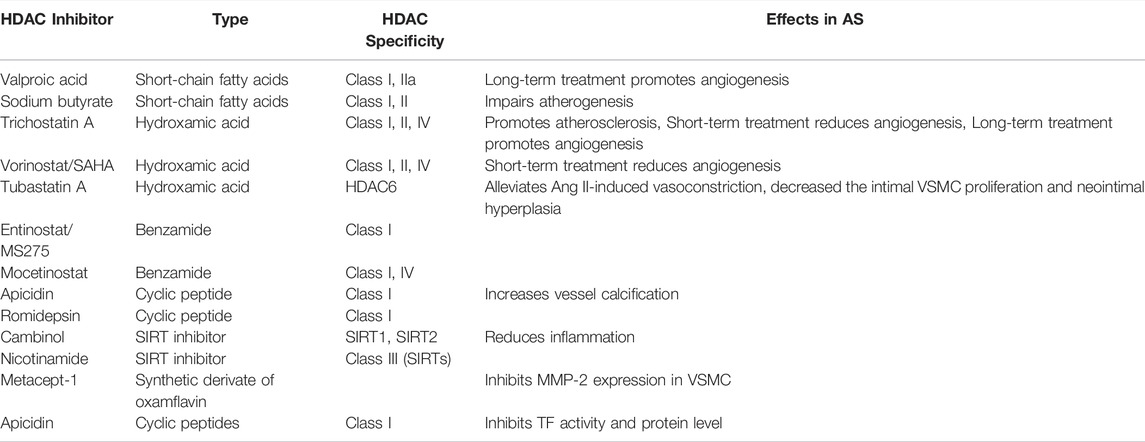
TABLE 2. Effects of HDACis in AS.
Inhibitors of HDACs generally cause growth arrest, cellular differentiation, and apoptosis. Thus, they are used for cancer treatment (Li and Seto, 2016). The HDAC non-selective inhibitors, SAHA/vorinostat and Romidepsin (Istodax, FK228), which are characterized by a relatively low IC50 for HDAC1/2, have been approved for the clinical treatment of cutaneous T cell lymphoma by FDA in the United States (Bertino and Otterson, 2011). Romidepsin facilitates the acetylation of non-histone substrates involved in the transcription of VCAM-1 in ECs, which mediates the AS process (Tambaro et al., 2010). In addition, Romidepsin treatment in Apoe−/- mice protected from diet-induced atherosclerotic lesion accumulation (Nicorescu et al., 2019). Given the elevated level of HDAC1/2 in advanced AS and the clinical availability of Romidepsin, the use and mechanism of this specific inhibitor in AS deserve further investigation. A potent HDAC inhibitor, suberoylanilide hydroxamic acid, exhibits anti-inflammatory properties by attenuating the LPS-induced expression of NF-κB-regulated cytokines (Zhao et al., 2015). Sodium valproate, a class I HDAC inhibitor, reportedly facilitated the phenotype switch of macrophage, thereby delaying AS progression (Chen et al., 2014). Pharmacological inhibition of HDAC1/2/3 by scriptaid prevented smooth muscle cell proliferation and neointima formation (Findeisen et al., 2011).
Commonly used HDAC inhibitors in experimental animal models and clinical trials are the Class I and II HDACs. These consist of the natural products butyrate and Trichostatin A (TSA). Although the inhibition of HDACs in macrophage brought about beneficial effects to AS, the broad usage of these inhibitors is limited by the observation that TSA unexpectedly promoted the progression of plaque expansion in an AS mouse model (Choi et al., 2005). This result might be due to the negative effects of TSA on other cell types in AS, such as ECs and SMCs. Therefore, the inhibition of HDACs in macrophage and monocytes is beneficial in the AS state. Findeisen et al. reported that scriptaid, a non-selective HDAC inhibitor, protected from neointimal thickening both in vitro and in vivo in a mouse model (Findeisen et al., 2011). Also, they demonstrated that scriptaid had no obvious toxicity at the dosage used.
Treatment with TMP195, a selective inhibitor of Class IIa HDAC, suppressed critical inflammatory pathways and mitigated atherogenesis in advanced stage AS, thereby offering a novel therapeutic strategy for reducing the consequence of vascular inflammation (Asare et al., 2020). Another Class II HDAC selective inhibitor, MC1568, rescued serum-dependent histone acetylation in NO-induced HUVECs (Illi et al., 2008). HDAC3-specific inhibitor RGFP966 suppressed endothelial-to-mesenchymal transition by modulating inflammatory response in AS (Chen et al., 2021).
Outlook
AS is a common pathological basis of cardiovascular and cerebrovascular diseases and seriously endangers human health. With the further understanding of the pathogenesis of AS, an increasing amount of evidence suggested the important role of HDACs in AS. The effects of HDACs on atherosclerosis are complex and multifaceted. In ECs, SMCs, and even macrophages, HDACs play different roles in regulating cell proliferation, migration, apoptosis, differentiation, inflammation, and oxidative stress (Zhou et al., 2011a; Grimaldi et al., 2015). It is difficult to characterize the overall effect of HDACs on AS. A growing number of recent studies suggested that HDACs can be used as potential therapeutic targets for AS (Neele et al., 2020; Wong et al., 2021). Inhibitors of HDACs can improve the symptoms of AS by precisely inhibiting the deacetylase activity of HDACs (Morrell et al., 2013). The development of inhibitors of HDACs is one of the current research hot spots. The current HDACi market, which includes existing drugs, is expected to expand into other indications, such as cardiovascular and cerebrovascular diseases (e.g., AS) (Bagchi and Weeks, 2019).
It is well known that the majority of existing or clinically available HDAC inhibitors are generic (Bondarev et al., 2021). The development of selective HDACi could reduce the side effects of other target activities, such as the potential generic toxicity of HDAC6 (Yang et al., 2017). However, as there are many subtypes of HDACs, and there are many similarities in the active domain and catalytic site among the subtypes, the development of HDAC inhibitors with high subtype selectivity is a breakthrough point that can be reached in the future and that will likely face great challenges in practical research. Clinical verification is needed to test the efficacy. In addition, the development of dual-target HDAC inhibitors is one of the current research directions (Jin et al., 2021). While remaining active against HDAC, they also act on one or more targets related to AS, which is worth studying in the future. Dual-target HDAC may be expanded and improved in terms of indications and efficacy to some extent, but problems (e.g., high toxicity of dual-target HDAC inhibition) exist (Peng et al., 2020; Kuznetsoff et al., 2021). These issues need to be addressed in future research works. HDACi has been extensively studied in recent years and has been regarded to have a potential therapeutic effect on many diseases, including neurodegenerative diseases, autoimmune diseases, acute graft-versus-host disease, and so on (Vojinovic et al., 2011; Ghiboub et al., 2021). Its potential therapeutic effect on AS is also worth exploring through further research. Other issues that need to be addressed are the targeting and specificity of HDACi. Researchers need to find out how to accurately make HDACi act on the AS region without changing the function of other normal cells. In future studies, more efficient and accurate HDAC inhibitors need to be developed, so AS to improve the therapeutic effect of related inhibitors on AS. With further research, more functions of HDACs in the pathogenesis of AS will be revealed, which will help us find a better plan to fight this disease.
Author Contributions
YiL, HL, YinL, K‐DR, and YY conceptualized and wrote the manuscript and created Figures. YY and JY contributed to the writing of the manuscript. YiL, YinL, and K‐DR reviewed and modified the manuscript. All authors approved the final version of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (Nos. 31900502; 81901590), Key scientific Research project of Henan Universities (No. 21A310027), and the Henan Medical Science and Technology Joint Building Program (No. LHGJ20190236).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adamson, S., and Leitinger, N. (2011). Phenotypic Modulation of Macrophages in Response to Plaque Lipids. Curr. Opin. Lipidol. 22 (5), 335–342. doi:10.1097/MOL.0b013e32834a97e4
Aghamajidi, A., Gorgani, M., Shahba, F., Shafaghat, Z., and Mojtabavi, N. (2021). The Potential Targets in Immunotherapy of Atherosclerosis. Int. Rev. Immunol. [Epub ahead of print], 1–18. doi:10.1080/08830185.2021.1988591
Annoni, G., Weiner, F. R., and Zern, M. A. (1992). Increased Transforming Growth Factor-Beta 1 Gene Expression in Human Liver Disease. J. Hepatol. 14 (2-3), 259–264. doi:10.1016/0168-8278(92)90168-o
Asare, Y., Campbell-James, T. A., Bokov, Y., Yu, L. L., Prestel, M., El Bounkari, O., et al. (2020). Histone Deacetylase 9 Activates IKK to Regulate Atherosclerotic Plaque Vulnerability. Circ. Res. 127 (6), 811–823. doi:10.1161/CIRCRESAHA.120.316743
Bagchi, R. A., and Weeks, K. L. (2019). Histone Deacetylases in Cardiovascular and Metabolic Diseases. J. Mol. Cel Cardiol 130, 151–159. doi:10.1016/j.yjmcc.2019.04.003
Bazou, D., Ng, M. R., Song, J. W., Chin, S. M., Maimon, N., and Munn, L. L. (2016). Flow-induced HDAC1 Phosphorylation and Nuclear export in Angiogenic Sprouting. Sci. Rep. 6, 34046. doi:10.1038/srep34046
Bertino, E. M., and Otterson, G. A. (2011). Romidepsin: a Novel Histone Deacetylase Inhibitor for Cancer. Expert Opin. Investig. Drugs 20 (8), 1151–1158. doi:10.1517/13543784.2011.594437
Bondarev, A. D., Attwood, M. M., Jonsson, J., Chubarev, V. N., Tarasov, V. V., and Schiöth, H. B. (2021). Recent Developments of HDAC Inhibitors: Emerging Indications and Novel Molecules. Br. J. Clin. Pharmacol. 87, 4577–4597. doi:10.1111/bcp.14889
Botti, C., Chiara, B., Caiafa, I., Ilaria, C., Coppola, A., Antonietta, C., et al. (2014). SIRT1 Inhibition Affects Angiogenic Properties of Human MSCs. Biomed. Res. Int. 2014, 783459. doi:10.1155/2014/783459
Cao, Q., Rong, S., Repa, J. J., St Clair, R., Parks, J. S., and Mishra, N. (2014). Histone Deacetylase 9 Represses Cholesterol Efflux and Alternatively Activated Macrophages in Atherosclerosis Development. Arterioscler Thromb. Vasc. Biol. 34 (9), 1871–1879. doi:10.1161/ATVBAHA.114.303393
Cardus, A., Uryga, A. K., Walters, G., and Erusalimsky, J. D. (2013). SIRT6 Protects Human Endothelial Cells from DNA Damage, Telomere Dysfunction, and Senescence. Cardiovasc. Res. 97 (3), 571–579. doi:10.1093/cvr/cvs352
Chang, S., Young, B. D., Li, S., Qi, X., Richardson, J. A., and Olson, E. N. (2006). Histone Deacetylase 7 Maintains Vascular Integrity by Repressing Matrix Metalloproteinase 10. Cell 126 (2), 321–334. doi:10.1016/j.cell.2006.05.040
Chen, L., Shang, C., Wang, B., Wang, G., Jin, Z., Yao, F., et al. (2021). HDAC3 Inhibitor Suppresses Endothelial-To-Mesenchymal Transition via Modulating Inflammatory Response in Atherosclerosis. Biochem. Pharmacol. 192, 114716. doi:10.1016/j.bcp.2021.114716
Chen, S., Wu, H., Klebe, D., Hong, Y., and Zhang, J. (2014). Valproic Acid: a New Candidate of Therapeutic Application for the Acute central Nervous System Injuries. Neurochem. Res. 39 (9), 1621–1633. doi:10.1007/s11064-014-1241-2
Chen, X., He, Y., Fu, W., Sahebkar, A., Tan, Y., Xu, S., et al. (2020). Histone Deacetylases (HDACs) and Atherosclerosis: A Mechanistic and Pharmacological Review. Front Cel Dev Biol 8, 581015. doi:10.3389/fcell.2020.581015
Chistiakov, D. A., Bobryshev, Y. V., and Orekhov, A. N. (2016). Macrophage-mediated Cholesterol Handling in Atherosclerosis. J. Cel Mol Med 20 (1), 17–28. doi:10.1111/jcmm.12689
Choi, J. H., Nam, K. H., Kim, J., Baek, M. W., Park, J. E., Park, H. Y., et al. (2005). Trichostatin A Exacerbates Atherosclerosis in Low Density Lipoprotein Receptor-Deficient Mice. Arterioscler Thromb. Vasc. Biol. 25 (11), 2404–2409. doi:10.1161/01.ATV.0000184758.07257.88
Clarke, M. C., Figg, N., Maguire, J. J., Davenport, A. P., Goddard, M., Littlewood, T. D., et al. (2006). Apoptosis of Vascular Smooth Muscle Cells Induces Features of Plaque Vulnerability in Atherosclerosis. Nat. Med. 12 (9), 1075–1080. doi:10.1038/nm1459
Clocchiatti, A., Florean, C., and Brancolini, C. (2011). Class IIa HDACs: from Important Roles in Differentiation to Possible Implications in Tumourigenesis. J. Cel Mol Med 15 (9), 1833–1846. doi:10.1111/j.1582-4934.2011.01321.x
Cosío, B. G., Mann, B., Ito, K., Jazrawi, E., Barnes, P. J., Chung, K. F., et al. (2004). Histone Acetylase and Deacetylase Activity in Alveolar Macrophages and Blood Mononocytes in Asthma. Am. J. Respir. Crit. Care Med. 170 (2), 141–147. doi:10.1164/rccm.200305-659OC
Cruz, D. L., Pipalia, N., Mao, S., Gadi, D., Liu, G., Grigalunas, M., et al. (2021). Inhibition of Histone Deacetylases 1, 2, and 3 Enhances Clearance of Cholesterol Accumulation in Niemann-Pick C1 Fibroblasts. ACS Pharmacol. Transl Sci. 4 (3), 1136–1148. doi:10.1021/acsptsci.1c00033
D'Onofrio, N., Servillo, L., and Balestrieri, M. L. (2018). SIRT1 and SIRT6 Signaling Pathways in Cardiovascular Disease Protection. Antioxid. Redox Signal. 28 (8), 711–732. doi:10.1089/ars.2017.7178
Dai, Y., Wei, T., Shen, Z., Bei, Y., Lin, H., and Dai, H. (2021). Classical HDACs in the Regulation of Neuroinflammation. Neurochem. Int. 150, 105182. doi:10.1016/j.neuint.2021.105182
Dai, Z., Zhu, M. M., Peng, Y., Jin, H., Machireddy, N., Qian, Z., et al. (2018). Endothelial and Smooth Muscle Cell Interaction via FoxM1 Signaling Mediates Vascular Remodeling and Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 198 (6), 788–802. doi:10.1164/rccm.201709-1835OC
Daniel, J. M., and Sedding, D. G. (2011). Circulating Smooth Muscle Progenitor Cells in Arterial Remodeling. J. Mol. Cel Cardiol 50 (2), 273–279. doi:10.1016/j.yjmcc.2010.10.030
Das Gupta, K., Shakespear, M. R., Iyer, A., Fairlie, D. P., and Sweet, M. J. (2016). Histone Deacetylases in Monocyte/macrophage Development, Activation and Metabolism: Refining HDAC Targets for Inflammatory and Infectious Diseases. Clin. Transl Immunol. 5 (1), e62. doi:10.1038/cti.2015.46
Davis, F. M., and Gallagher, K. A. (2019). Epigenetic Mechanisms in Monocytes/Macrophages Regulate Inflammation in Cardiometabolic and Vascular Disease. Arterioscler Thromb. Vasc. Biol. 39 (4), 623–634. doi:10.1161/ATVBAHA.118.312135
Deeb, R. S., and Hajjar, D. P. (2016). Repair Mechanisms in Oxidant-Driven Chronic Inflammatory Disease. Am. J. Pathol. 186 (7), 1736–1749. doi:10.1016/j.ajpath.2016.03.001
Findeisen, H. M., Gizard, F., Zhao, Y., Qing, H., Heywood, E. B., Jones, K. L., et al. (2011). Epigenetic Regulation of Vascular Smooth Muscle Cell Proliferation and Neointima Formation by Histone Deacetylase Inhibition. Arterioscler Thromb. Vasc. Biol. 31 (4), 851–860. doi:10.1161/ATVBAHA.110.221952
Förstermann, U., and Sessa, W. C. (2012). Nitric Oxide Synthases: Regulation and Function. Eur. Heart J. 33 (7), 837a–837d. doi:10.1093/eurheartj/ehr304
Fu, Y., Wang, Y., Gao, X., Li, H., and Yuan, Y. (2020). Dynamic Expression of HDAC3 in Db/db Mouse RGCs and its Relationship with Apoptosis and Autophagy. J. Diabetes Res. 2020, 6086780. doi:10.1155/2020/6086780
Gallinari, P., Di Marco, S., Jones, P., Pallaoro, M., and Steinkühler, C. (2007). HDACs, Histone Deacetylation and Gene Transcription: from Molecular Biology to Cancer Therapeutics. Cell Res 17 (3), 195–211. doi:10.1038/sj.cr.7310149
Ghiboub, M., Elfiky, A. M. I., de Winther, M. P. J., Harker, N. R., Tough, D. F., and de Jonge, W. J. (2021). Selective Targeting of Epigenetic Readers and Histone Deacetylases in Autoimmune and Inflammatory Diseases: Recent Advances and Future Perspectives. J. Pers Med. 11 (5). doi:10.3390/jpm11050336
Ghosh, S. (2011). Macrophage Cholesterol Homeostasis and Metabolic Diseases: Critical Role of Cholesteryl Ester Mobilization. Expert Rev. Cardiovasc. Ther. 9 (3), 329–340. doi:10.1586/erc.11.16
Glozak, M. A., and Seto, E. (2009). Acetylation/deacetylation Modulates the Stability of DNA Replication Licensing Factor Cdt1. J. Biol. Chem. 284 (17), 11446–11453. doi:10.1074/jbc.M809394200
Grimaldi, V., Vietri, M. T., Schiano, C., Picascia, A., De Pascale, M. R., Fiorito, C., et al. (2015). Epigenetic Reprogramming in Atherosclerosis. Curr. Atheroscler. Rep. 17 (2), 476. doi:10.1007/s11883-014-0476-3
Gu, X., Han, D., Chen, W., Zhang, L., Lin, Q., Gao, J., et al. (2016). SIRT1-mediated FoxOs Pathways Protect against Apoptosis by Promoting Autophagy in Osteoblast-like MC3T3-E1 Cells Exposed to Sodium Fluoride. Oncotarget 7 (40), 65218–65230. doi:10.18632/oncotarget.11573
Halili, M. A., Andrews, M. R., Labzin, L. I., Schroder, K., Matthias, G., Cao, C., et al. (2010). Differential Effects of Selective HDAC Inhibitors on Macrophage Inflammatory Responses to the Toll-like Receptor 4 Agonist LPS. J. Leukoc. Biol. 87 (6), 1103–1114. doi:10.1189/jlb.0509363
He, H. M., Li, A., Zhang, S. W., and Duan, M. L. (2011). The Effects of a Histone Deacetylase (HDAC) Inhibitor on Endotoxin-Induced Endothelial Cell Injury. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue 23 (10), 602–604.
Hoeksema, M. A., Gijbels, M. J., Van den Bossche, J., van der Velden, S., Sijm, A., Neele, A. E., et al. (2014). Targeting Macrophage Histone Deacetylase 3 Stabilizes Atherosclerotic Lesions. EMBO Mol. Med. 6 (9), 1124–1132. doi:10.15252/emmm.201404170
Hou, J., Wang, S., Shang, Y. C., Chong, Z. Z., and Maiese, K. (2011). Erythropoietin Employs Cell Longevity Pathways of SIRT1 to foster Endothelial Vascular Integrity during Oxidant Stress. Curr. Neurovasc Res. 8 (3), 220–235. doi:10.2174/156720211796558069
Illi, B., Dello Russo, C., Colussi, C., Rosati, J., Pallaoro, M., Spallotta, F., et al. (2008). Nitric Oxide Modulates Chromatin Folding in Human Endothelial Cells via Protein Phosphatase 2A Activation and Class II Histone Deacetylases Nuclear Shuttling. Circ. Res. 102 (1), 51–58. doi:10.1161/CIRCRESAHA.107.157305
Jaminon, A., Reesink, K., Kroon, A., and Schurgers, L. (2019). The Role of Vascular Smooth Muscle Cells in Arterial Remodeling: Focus on Calcification-Related Processes. Int. J. Mol. Sci. 20 (22). doi:10.3390/ijms20225694
Jeanblanc, J., Lemoine, S., Jeanblanc, V., Alaux-Cantin, S., and Naassila, M. (2015). The Class I-specific HDAC Inhibitor MS-275 Decreases Motivation to Consume Alcohol and Relapse in Heavy Drinking Rats. Int. J. Neuropsychopharmacol. 18 (9). doi:10.1093/ijnp/pyv029
Jiang, Q., Hao, R., Wang, W., Gao, H., and Wang, C. (2016). SIRT1/Atg5/autophagy Are Involved in the Antiatherosclerosis Effects of Ursolic Acid. Mol. Cel Biochem 420 (1-2), 171–184. doi:10.1007/s11010-016-2787-x
Jin, W., et al. (2021). Dual-target Inhibitors of Bromodomain-Containing Protein 4 (BRD4) in Cancer Therapy: Current Situation and Future Directions. Drug Discov Today.
Joanna, F., van Grunsven, L. A., Mathieu, V., Sarah, S., Sarah, D., Karin, V., et al. (2009). Histone Deacetylase Inhibition and the Regulation of Cell Growth with Particular Reference to Liver Pathobiology. J. Cel Mol Med 13 (9B), 2990–3005. doi:10.1111/j.1582-4934.2009.00831.x
Ke, X., Lin, Z., Ye, Z., Leng, M., Chen, B., Jiang, C., et al. (2021). Histone Deacetylases in the Pathogenesis of Diabetic Cardiomyopathy. Front. Endocrinol. (Lausanne) 12, 679655. doi:10.3389/fendo.2021.679655
Kloc, M., Uosef, A., Kubiak, J. Z., and Ghobrial, R. M. (2020). Role of Macrophages and RhoA Pathway in Atherosclerosis. Int. J. Mol. Sci. 22 (1). doi:10.3390/ijms22010216
Kong, X., Fang, M., Li, P., Fang, F., and Xu, Y. (2009). HDAC2 Deacetylates Class II Transactivator and Suppresses its Activity in Macrophages and Smooth Muscle Cells. J. Mol. Cel Cardiol 46 (3), 292–299. doi:10.1016/j.yjmcc.2008.10.023
Kuznetsoff, J. N., Owens, D. A., Lopez, A., Rodriguez, D. A., Chee, N. T., Kurtenbach, S., et al. (2021). Dual Screen for Efficacy and Toxicity Identifies HDAC Inhibitor with Distinctive Activity Spectrum for BAP1-Mutant Uveal Melanoma. Mol. Cancer Res. 19 (2), 215–222. doi:10.1158/1541-7786.mcr-20-0434
Kuznetsova, T., Prange, K. H. M., Glass, C. K., and de Winther, M. P. J. (2020). Transcriptional and Epigenetic Regulation of Macrophages in Atherosclerosis. Nat. Rev. Cardiol. 17 (4), 216–228. doi:10.1038/s41569-019-0265-3
Lee, D. Y., and Chiu, J. J. (2019). Atherosclerosis and Flow: Roles of Epigenetic Modulation in Vascular Endothelium. J. Biomed. Sci. 26 (1), 56. doi:10.1186/s12929-019-0551-8
Lee, H. T., Oh, S., Ro, D. H., Yoo, H., and Kwon, Y. W. (2020). The Key Role of DNA Methylation and Histone Acetylation in Epigenetics of Atherosclerosis. J. Lipid Atheroscler. 9 (3), 419–434. doi:10.12997/jla.2020.9.3.419
Lee, J. H., Song, M. Y., Song, E. K., Kim, E. K., Moon, W. S., Han, M. K., et al. (2009). Overexpression of SIRT1 Protects Pancreatic Beta-Cells against Cytokine Toxicity by Suppressing the Nuclear Factor-kappaB Signaling Pathway. Diabetes 58 (2), 344–351. doi:10.2337/db07-1795
Li, D., Yang, Y., Wang, S., He, X., Liu, M., Bai, B., et al. (2021). Role of Acetylation in Doxorubicin-Induced Cardiotoxicity. Redox Biol. 46, 102089. doi:10.1016/j.redox.2021.102089
Li, Y., and Seto, E. (2016). HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb Perspect. Med. 6 (10). doi:10.1101/cshperspect.a026831
Lian, B., Pei, Y. C., Jiang, Y. Z., Xue, M. Z., Li, D. Q., Li, X. G., et al. (2020). Truncated HDAC9 Identified by Integrated Genome-wide Screen as the Key Modulator for Paclitaxel Resistance in Triple-Negative Breast Cancer. Theranostics 10 (24), 11092–11109. doi:10.7150/thno.44997
Lin, H. H., Hsieh, M. C., Wang, C. P., Yu, P. R., Lee, M. S., and Chen, J. H. (2021). Anti-Atherosclerotic Effect of Gossypetin on Abnormal Vascular Smooth Muscle Cell Proliferation and Migration. Antioxidants (Basel) 10 (9). doi:10.3390/antiox10091357
Linton, M. R. F., et al. (2000). “The Role of Lipids and Lipoproteins in Atherosclerosis,” in Endotext (South Dartmouth (MA: MDText.com, Inc.).
Liu, H., Yuan, L., Xu, S., and Wang, K. (2007). Endothelial Cell and Macrophage Regulation of Vascular Smooth Muscle Cell Calcification Modulated by Cholestane-3beta, 5alpha, 6beta-Triol. Cell Biol Int 31 (9), 900–907. doi:10.1016/j.cellbi.2007.02.009
Liu, T., Ma, X., Ouyang, T., Chen, H., Lin, J., Liu, J., et al. (2018). SIRT1 Reverses Senescence via Enhancing Autophagy and Attenuates Oxidative Stress-Induced Apoptosis through Promoting P53 Degradation. Int. J. Biol. Macromol 117, 225–234. doi:10.1016/j.ijbiomac.2018.05.174
Liu, Y., Du, M., and Lin, H. Y. (2021). Histone Deacetylase 9 Deficiency Exaggerates Uterine M2 Macrophage Polarization. J. Cel Mol Med 25 (16), 7690–7708. doi:10.1111/jcmm.16616
Long, J., Fang, W. Y., Chang, L., Gao, W. H., Shen, Y., Jia, M. Y., et al. (2017). Targeting HDAC3, a New Partner Protein of AKT in the Reversal of Chemoresistance in Acute Myeloid Leukemia via DNA Damage Response. Leukemia 31 (12), 2761–2770. doi:10.1038/leu.2017.130
Luan, Y., Luan, Y., Yuan, R. X., Feng, Q., Chen, X., and Yang, Y. (2021). Structure and Function of Mitochondria-Associated Endoplasmic Reticulum Membranes (MAMs) and Their Role in Cardiovascular Diseases. Oxid Med. Cel Longev 2021, 4578809. doi:10.1155/2021/4578809
Luchetti, F., Crinelli, R., Cesarini, E., Canonico, B., Guidi, L., Zerbinati, C., et al. (2017). Endothelial Cells, Endoplasmic Reticulum Stress and Oxysterols. Redox Biol. 13, 581–587. doi:10.1016/j.redox.2017.07.014
Manduteanu, I., and Simionescu, M. (2012). Inflammation in Atherosclerosis: a Cause or a Result of Vascular Disorders? J. Cel Mol Med 16 (9), 1978–1990. doi:10.1111/j.1582-4934.2012.01552.x
Manea, S. A., Vlad, M. L., Fenyo, I. M., Lazar, A. G., Raicu, M., Muresian, H., et al. (2020). Pharmacological Inhibition of Histone Deacetylase Reduces NADPH Oxidase Expression, Oxidative Stress and the Progression of Atherosclerotic Lesions in Hypercholesterolemic Apolipoprotein E-Deficient Mice; Potential Implications for Human Atherosclerosis. Redox Biol. 28, 101338. doi:10.1016/j.redox.2019.101338
Margariti, A., Xiao, Q., Zampetaki, A., Zhang, Z., Li, H., Martin, D., et al. (2009). Splicing of HDAC7 Modulates the SRF-Myocardin Complex during Stem-Cell Differentiation towards Smooth Muscle Cells. J. Cel Sci 122 (Pt 4), 460–470. doi:10.1242/jcs.034850
Margariti, A., Zampetaki, A., Xiao, Q., Zhou, B., Karamariti, E., Martin, D., et al. (2010). Histone Deacetylase 7 Controls Endothelial Cell Growth through Modulation of Beta-Catenin. Circ. Res. 106 (7), 1202–1211. doi:10.1161/CIRCRESAHA.109.213165
Matsushima, S., and Sadoshima, J. (2015). The Role of Sirtuins in Cardiac Disease. Am. J. Physiol. Heart Circ. Physiol. 309 (9), H1375–H1389. doi:10.1152/ajpheart.00053.2015
McDonald, O. G., Wamhoff, B. R., Hoofnagle, M. H., and Owens, G. K. (2006). Control of SRF Binding to CArG Box Chromatin Regulates Smooth Muscle Gene Expression In Vivo. J. Clin. Invest. 116 (1), 36–48. doi:10.1172/JCI26505
Moore, K. J., Sheedy, F. J., and Fisher, E. A. (2013). Macrophages in Atherosclerosis: a Dynamic Balance. Nat. Rev. Immunol. 13 (10), 709–721. doi:10.1038/nri3520
Morrell, N. W., Archer, S. L., Defelice, A., Evans, S., Fiszman, M., Martin, T., et al. (2013). Anticipated Classes of New Medications and Molecular Targets for Pulmonary Arterial Hypertension. Pulm. Circ. 3 (1), 226–244. doi:10.4103/2045-8932.109940
Morris, H. E., Neves, K. B., Montezano, A. C., MacLean, M. R., and Touyz, R. M. (2019). Notch3 Signalling and Vascular Remodelling in Pulmonary Arterial Hypertension. Clin. Sci. (Lond) 133 (24), 2481–2498. doi:10.1042/CS20190835
Mundi, S., Massaro, M., Scoditti, E., Carluccio, M. A., van Hinsbergh, V. W. M., Iruela-Arispe, M. L., et al. (2018). Endothelial Permeability, LDL Deposition, and Cardiovascular Risk Factors-A Review. Cardiovasc. Res. 114 (1), 35–52. doi:10.1093/cvr/cvx226
Nakata, S., Tsutsui, M., Shimokawa, H., Yamashita, T., Tanimoto, A., Tasaki, H., et al. (2007). Statin Treatment Upregulates Vascular Neuronal Nitric Oxide Synthase through Akt/NF-kappaB Pathway. Arterioscler Thromb. Vasc. Biol. 27 (1), 92–98. doi:10.1161/01.ATV.0000251615.61858.33
Naruszewicz, M. (1989). Macrophages in the Pathogenesis of Atherosclerosis. Advances in Science and Personal Observations. Kardiol Pol. 32 (1), 27–34.
Neele, A. E., Willemsen, L., Chen, H. J., Dzobo, K. E., and de Winther, M. P. J. (2020). Targeting Epigenetics as Atherosclerosis Treatment: an Updated View. Curr. Opin. Lipidol. 31 (6), 324–330. doi:10.1097/MOL.0000000000000711
Nicorescu, I., Dallinga, G. M., de Winther, M. P. J., Stroes, E. S. G., and Bahjat, M. (2019). Potential Epigenetic Therapeutics for Atherosclerosis Treatment. Atherosclerosis 281, 189–197. doi:10.1016/j.atherosclerosis.2018.10.006
Nunez-Alvarez, Y., and Suelves, M. (2021). HDAC11: a Multifaceted Histone Deacetylase with Proficient Fatty Deacylase Activity and its Roles in Physiological Processes. FEBS J. [Epub ahead of print]. doi:10.1111/febs.15895
Okamoto, H., Fujioka, Y., Takahashi, A., Takahashi, T., Taniguchi, T., Ishikawa, Y., et al. (2006). Trichostatin A, an Inhibitor of Histone Deacetylase, Inhibits Smooth Muscle Cell Proliferation via Induction of p21(WAF1). J. Atheroscler. Thromb. 13 (4), 183–191. doi:10.5551/jat.13.183
Ou, H. C., Chou, F. P., Sheen, H. M., Lin, T. M., Yang, C. H., and Huey-Herng Sheu, W. (2006). Resveratrol, a Polyphenolic Compound in Red Wine, Protects against Oxidized LDL-Induced Cytotoxicity in Endothelial Cells. Clin. Chim. Acta 364 (1-2), 196–204. doi:10.1016/j.cccn.2005.06.018
Pandey, D., Sikka, G., Bergman, Y., Kim, J. H., Ryoo, S., Romer, L., et al. (2014). Transcriptional Regulation of Endothelial Arginase 2 by Histone Deacetylase 2. Arterioscler Thromb. Vasc. Biol. 34 (7), 1556–1566. doi:10.1161/ATVBAHA.114.303685
Peng, X., Sun, Z., Kuang, P., and Chen, J. (2020). Recent Progress on HDAC Inhibitors with Dual Targeting Capabilities for Cancer Treatment. Eur. J. Med. Chem. 208, 112831. doi:10.1016/j.ejmech.2020.112831
Pirillo, A., Norata, G. D., and Catapano, A. L. (2013). LOX-1, OxLDL, and Atherosclerosis. Mediators Inflamm. 2013, 152786. doi:10.1155/2013/152786
Prestel, M., Prell-Schicker, C., Webb, T., Malik, R., Lindner, B., Ziesch, N., et al. (2019). The Atherosclerosis Risk Variant Rs2107595 Mediates Allele-specific Transcriptional Regulation of HDAC9 via E2F3 and Rb1. Stroke 50 (10), 2651–2660. doi:10.1161/STROKEAHA.119.026112
Qin, B., Shu, Y., Xiao, L., Lu, T., Lin, Y., Yang, H., et al. (2017). MicroRNA-150 Targets ELK1 and Modulates the Apoptosis Induced by Ox-LDL in Endothelial Cells. Mol. Cel Biochem 429 (1-2), 45–58. doi:10.1007/s11010-016-2935-3
Qin, J., Guo, N., Tong, J., and Wang, Z. (2021). Function of Histone Methylation and Acetylation Modifiers in Cardiac Hypertrophy. J. Mol. Cel Cardiol 159, 120–129. doi:10.1016/j.yjmcc.2021.06.011
Qiu, M., Xu, E., and Zhan, L. (2021). Epigenetic Regulations of Microglia/Macrophage Polarization in Ischemic Stroke. Front. Mol. Neurosci. 14, 697416. doi:10.3389/fnmol.2021.697416
Quillard, T., Franck, G., Mawson, T., Folco, E., and Libby, P. (2017). Mechanisms of Erosion of Atherosclerotic Plaques. Curr. Opin. Lipidol. 28 (5), 434–441. doi:10.1097/MOL.0000000000000440
Rajan, P. K., Udoh, U. A., Sanabria, J. D., Banerjee, M., Smith, G., Schade, M. S., et al. (2020). The Role of Histone Acetylation-/Methylation-Mediated Apoptotic Gene Regulation in Hepatocellular Carcinoma. Int. J. Mol. Sci. 21 (23). doi:10.3390/ijms21238894
Rajendran, P., Rengarajan, T., Thangavel, J., Nishigaki, Y., Sakthisekaran, D., Sethi, G., et al. (2013). The Vascular Endothelium and Human Diseases. Int. J. Biol. Sci. 9 (10), 1057–1069. doi:10.7150/ijbs.7502
Rajendrasozhan, S., Yang, S. R., Edirisinghe, I., Yao, H., Adenuga, D., and Rahman, I. (2008). Deacetylases and NF-kappaB in Redox Regulation of Cigarette Smoke-Induced Lung Inflammation: Epigenetics in Pathogenesis of COPD. Antioxid. Redox Signal. 10 (4), 799–811. doi:10.1089/ars.2007.1938
Raman, A. T., and Rai, K. (2018). Loss of Histone Acetylation and H3K4 Methylation Promotes Melanocytic Malignant Transformation. Mol. Cel Oncol 5 (3), e1359229. doi:10.1080/23723556.2017.1359229
Ravensbergen, J., Ravensbergen, J. W., Krijger, J. K., Hillen, B., and Hoogstraten, H. W. (1998). Localizing Role of Hemodynamics in Atherosclerosis in Several Human Vertebrobasilar junction Geometries. Arterioscler Thromb. Vasc. Biol. 18 (5), 708–716. doi:10.1161/01.atv.18.5.708
Remmerie, A., and Scott, C. L. (2018). Macrophages and Lipid Metabolism. Cell Immunol 330, 27–42. doi:10.1016/j.cellimm.2018.01.020
Ruytinx, P., Proost, P., Van Damme, J., and Struyf, S. (2018). Chemokine-Induced Macrophage Polarization in Inflammatory Conditions. Front. Immunol. 9, 1930. doi:10.3389/fimmu.2018.01930
Sanchez-Lopez, E., Zhong, Z., Stubelius, A., Sweeney, S. R., Booshehri, L. M., Antonucci, L., et al. (2019). Choline Uptake and Metabolism Modulate Macrophage IL-1β and IL-18 Production. Cell Metab 29 (6), 1350–e7. doi:10.1016/j.cmet.2019.03.011
Seto, E., and Yoshida, M. (2014). Erasers of Histone Acetylation: the Histone Deacetylase Enzymes. Cold Spring Harb Perspect. Biol. 6 (4), a018713. doi:10.1101/cshperspect.a018713
Shentu, T. P., He, M., Sun, X., Zhang, J., Zhang, F., Gongol, B., et al. (2016). AMP-activated Protein Kinase and Sirtuin 1 Coregulation of Cortactin Contributes to Endothelial Function. Arterioscler Thromb. Vasc. Biol. 36 (12), 2358–2368. doi:10.1161/ATVBAHA.116.307871
Shi, W., Wei, X., Wang, Z., Han, H., Fu, Y., Liu, J., et al. (2016). HDAC9 Exacerbates Endothelial Injury in Cerebral Ischaemia/reperfusion Injury. J. Cel Mol Med 20 (6), 1139–1149. doi:10.1111/jcmm.12803
Shiva Shankar, T. V., and Willems, L. (2014). Epigenetic Modulators Mitigate Angiogenesis through a Complex Transcriptomic Network. Vascul Pharmacol. 60 (2), 57–66. doi:10.1016/j.vph.2014.01.003
Sosnowska, B., Mazidi, M., Penson, P., Gluba-Brzózka, A., Rysz, J., and Banach, M. (2017). The Sirtuin Family Members SIRT1, SIRT3 and SIRT6: Their Role in Vascular Biology and Atherogenesis. Atherosclerosis 265, 275–282. doi:10.1016/j.atherosclerosis.2017.08.027
Stein, S., Lohmann, C., Schäfer, N., Hofmann, J., Rohrer, L., Besler, C., et al. (2010). SIRT1 Decreases Lox-1-Mediated Foam Cell Formation in Atherogenesis. Eur. Heart J. 31 (18), 2301–2309. doi:10.1093/eurheartj/ehq107
Sun, H. J., Ren, X. S., Xiong, X. Q., Chen, Y. Z., Zhao, M. X., Wang, J. J., et al. (2017). NLRP3 Inflammasome Activation Contributes to VSMC Phenotypic Transformation and Proliferation in Hypertension. Cell Death Dis 8 (10), e3074. doi:10.1038/cddis.2017.470
Takamura, T. A., Tsuchiya, T., Oda, M., Watanabe, M., Saito, R., Sato-Ishida, R., et al. (2017). Circulating Malondialdehyde-Modified Low-Density Lipoprotein (MDA-LDL) as a Novel Predictor of Clinical Outcome after Endovascular Therapy in Patients with Peripheral Artery Disease (PAD). Atherosclerosis 263, 192–197. doi:10.1016/j.atherosclerosis.2017.06.029
Tambaro, F. P., Dell'aversana, C., Carafa, V., Nebbioso, A., Radic, B., Ferrara, F., et al. (2010). Histone Deacetylase Inhibitors: Clinical Implications for Hematological Malignancies. Clin. Epigenetics 1 (1-2), 25–44. doi:10.1007/s13148-010-0006-2
Torzewski, M. (2021). The Initial Human Atherosclerotic Lesion and Lipoprotein Modification-A Deep Connection. Int. J. Mol. Sci. 22 (21). doi:10.3390/ijms222111488
Treuter, E., Fan, R., Huang, Z., Jakobsson, T., and Venteclef, N. (2017). Transcriptional Repression in Macrophages-Basic Mechanisms and Alterations in Metabolic Inflammatory Diseases. FEBS Lett. 591 (19), 2959–2977. doi:10.1002/1873-3468.12850
Truong, V., Jain, A., Anand-Srivastava, M. B., and Srivastava, A. K. (2021). Angiotensin II-Induced Histone Deacetylase 5 Phosphorylation, Nuclear export, and Egr-1 Expression Are Mediated by Akt Pathway in A10 Vascular Smooth Muscle Cells. Am. J. Physiol. Heart Circ. Physiol. 320 (4), H1543–H1554. doi:10.1152/ajpheart.00683.2020
Van den Bossche, J., Neele, A. E., Hoeksema, M. A., de Heij, F., Boshuizen, M. C., van der Velden, S., et al. (2014). Inhibiting Epigenetic Enzymes to Improve Atherogenic Macrophage Functions. Biochem. Biophys. Res. Commun. 455 (3-4), 396–402. doi:10.1016/j.bbrc.2014.11.029
Vaquero, A., Sternglanz, R., and Reinberg, D. (2007). NAD+-dependent Deacetylation of H4 Lysine 16 by Class III HDACs. Oncogene 26 (37), 5505–5520. doi:10.1038/sj.onc.1210617
Villagra, A., Sotomayor, E. M., and Seto, E. (2010). Histone Deacetylases and the Immunological Network: Implications in Cancer and Inflammation. Oncogene 29 (2), 157–173. doi:10.1038/onc.2009.334
Villalba, J. M., and Alcaín, F. J. (2012). Sirtuin Activators and Inhibitors. Biofactors 38 (5), 349–359. doi:10.1002/biof.1032
Vojinovic, J., Damjanov, N., D'Urzo, C., Furlan, A., Susic, G., Pasic, S., et al. (2011). Safety and Efficacy of an Oral Histone Deacetylase Inhibitor in Systemic-Onset Juvenile Idiopathic Arthritis. Arthritis Rheum. 63 (5), 1452–1458. doi:10.1002/art.30238
Walther, M., Schrahn, S., Krauss, V., Lein, S., Kessler, J., Jenuwein, T., et al. (2020). Heterochromatin Formation in Drosophila Requires Genome-wide Histone Deacetylation in Cleavage Chromatin before Mid-blastula Transition in Early Embryogenesis. Chromosoma 129 (1), 83–98. doi:10.1007/s00412-020-00732-x
Wang, F., and Chen, H. Z. (2020). Histone Deacetylase SIRT1, Smooth Muscle Cell Function, and Vascular Diseases. Front. Pharmacol. 11, 537519. doi:10.3389/fphar.2020.537519
Wang, J., Xu, X., Li, P., Zhang, B., and Zhang, J. (2021). HDAC3 Protects against Atherosclerosis through Inhibition of Inflammation via the microRNA-19b/PPARγ/NF-Κb axis. Atherosclerosis 323, 1–12. doi:10.1016/j.atherosclerosis.2021.02.013
Wang, X., Cao, K., Sun, X., Chen, Y., Duan, Z., Sun, L., et al. (2015). Macrophages in Spinal Cord Injury: Phenotypic and Functional Change from Exposure to Myelin Debris. Glia 63 (4), 635–651. doi:10.1002/glia.22774
Wang, Z. X., Wang, C. Q., Li, X. Y., Feng, G. K., Zhu, H. L., Ding, Y., et al. (2015). Mesenchymal Stem Cells Alleviate Atherosclerosis by Elevating Number and Function of CD4(+)CD25 (+)FOXP3 (+) Regulatory T-Cells and Inhibiting Macrophage Foam Cell Formation. Mol. Cel Biochem 400 (1-2), 163–172. doi:10.1007/s11010-014-2272-3
Wautier, J. L., and Wautier, M. P. (2021). Endothelial Cell Participation in Inflammatory Reaction. Int. J. Mol. Sci. 22 (12). doi:10.3390/ijms22126341
Wee, S., Dhanak, D., Li, H., Armstrong, S. A., Copeland, R. A., Sims, R., et al. (2014). Targeting Epigenetic Regulators for Cancer Therapy. Ann. N. Y Acad. Sci. 1309, 30–36. doi:10.1111/nyas.12356
Wilson, H. M. (2010). Macrophages Heterogeneity in Atherosclerosis - Implications for Therapy. J. Cel Mol Med 14 (8), 2055–2065. doi:10.1111/j.1582-4934.2010.01121.x
Wong, L. M., Phoon, L. Q., and Wei, L. K. (2021). Epigenetics Modifications in Large-Artery Atherosclerosis: A Systematic Review. J. Stroke Cerebrovasc. Dis. 30 (12), 106033. doi:10.1016/j.jstrokecerebrovasdis.2021.106033
Wu, X., Kong, X., Luchsinger, L., Smith, B. D., and Xu, Y. (2009). Regulating the Activity of Class II Transactivator by Posttranslational Modifications: Exploring the Possibilities. Mol. Cel Biol 29 (21), 5639–5644. doi:10.1128/MCB.00661-09
Xiao, Q., Pepe, A. E., Wang, G., Luo, Z., Zhang, L., Zeng, L., et al. (2012). Nrf3-Pla2g7 Interaction Plays an Essential Role in Smooth Muscle Differentiation from Stem Cells. Arterioscler Thromb. Vasc. Biol. 32 (3), 730–744. doi:10.1161/ATVBAHA.111.243188
Xu, S., Chen, H., Ni, H., and Dai, Q. (2021). Targeting HDAC6 Attenuates Nicotine-Induced Macrophage Pyroptosis via NF-κB/NLRP3 Pathway. Atherosclerosis 317, 1–9. doi:10.1016/j.atherosclerosis.2020.11.021
Yan, Z. Q., Yao, Q. P., Zhang, M. L., Qi, Y. X., Guo, Z. Y., Shen, B. R., et al. (2009). Histone Deacetylases Modulate Vascular Smooth Muscle Cell Migration Induced by Cyclic Mechanical Strain. J. Biomech. 42 (7), 945–948. doi:10.1016/j.jbiomech.2009.01.012
Yang, J., Zhang, L., Yu, C., Yang, X. F., and Wang, H. (2014). Monocyte and Macrophage Differentiation: Circulation Inflammatory Monocyte as Biomarker for Inflammatory Diseases. Biomark Res. 2 (1), 1. doi:10.1186/2050-7771-2-1
Yang, S. S., Zhang, R., Wang, G., and Zhang, Y. F. (2017). The Development Prospection of HDAC Inhibitors as a Potential Therapeutic Direction in Alzheimer's Disease. Transl Neurodegener 6, 19. doi:10.1186/s40035-017-0089-1
Yang, Y., Luan, Y., Yuan, R. X., and Luan, Y. (2021). Histone Methylation Related Therapeutic Challenge in Cardiovascular Diseases. Front. Cardiovasc. Med. 8, 710053. doi:10.3389/fcvm.2021.710053
Yang, Z., and Ming, X. F. (2014). Functions of Arginase Isoforms in Macrophage Inflammatory Responses: Impact on Cardiovascular Diseases and Metabolic Disorders. Front. Immunol. 5, 533. doi:10.3389/fimmu.2014.00533
Yu, W., Liu, J., Yu, Y., Zhang, V., Clausen, D., Kelly, J., et al. (2020). Discovery of Ethyl Ketone-Based HDACs 1, 2, and 3 Selective Inhibitors for HIV Latency Reactivation. Bioorg. Med. Chem. Lett. 30 (13), 127197. doi:10.1016/j.bmcl.2020.127197
Yuan, P., Hu, Q., He, X., Long, Y., Song, X., Wu, F., et al. (2020). Laminar Flow Inhibits the Hippo/YAP Pathway via Autophagy and SIRT1-Mediated Deacetylation against Atherosclerosis. Cel Death Dis 11 (2), 141. doi:10.1038/s41419-020-2343-1
Zaidi, S. A. H., Guzman, W., Singh, S., Mehrotra, S., and Husain, S. (2020). Changes in Class I and IIb HDACs by δ-Opioid in Chronic Rat Glaucoma Model. Invest. Ophthalmol. Vis. Sci. 61 (14), 4. doi:10.1167/iovs.61.14.4
Zhang, D., Hu, X., Li, J., Hoogstra-Berends, F., Zhuang, Q., Esteban, M. A., et al. (2018). Converse Role of Class I and Class IIa HDACs in the Progression of Atrial Fibrillation. J. Mol. Cel Cardiol 125, 39–49. doi:10.1016/j.yjmcc.2018.09.010
Zhang, H., Ji, L., Yang, Y., Zhang, X., Gang, Y., and Bai, L. (2020). The Role of HDACs and HDACi in Cartilage and Osteoarthritis. Front. Cel Dev Biol 8, 560117. doi:10.3389/fcell.2020.560117
Zhang, L., Cheng, H., Yue, Y., Li, S., Zhang, D., and He, R. (2018). H19 Knockdown Suppresses Proliferation and Induces Apoptosis by Regulating miR-148b/WNT/β-catenin in Ox-LDL -stimulated Vascular Smooth Muscle Cells. J. Biomed. Sci. 25 (1), 11. doi:10.1186/s12929-018-0418-4
Zhao, T. C., Wang, Z., and Zhao, T. Y. (2020). The Important Role of Histone Deacetylases in Modulating Vascular Physiology and Arteriosclerosis. Atherosclerosis 303, 36–42. doi:10.1016/j.atherosclerosis.2020.04.020
Zhao, Y., Ma, G., and Yang, X. (2019). HDAC5 Promotes Mycoplasma Pneumoniae-Induced Inflammation in Macrophages through NF-Κb Activation. Life Sci. 221, 13–19. doi:10.1016/j.lfs.2019.02.004
Zhao, Y., Zhou, P., Liu, B., Bambakidis, T., Mazitschek, R., Alam, H. B., et al. (2015). Protective Effect of Suberoylanilide Hydroxamic Acid against Lipopolysaccharide-Induced Liver Damage in Rodents. J. Surg. Res. 194 (2), 544–550. doi:10.1016/j.jss.2014.10.056
Zhou, B., Margariti, A., Zeng, L., Habi, O., Xiao, Q., Martin, D., et al. (2011). Splicing of Histone Deacetylase 7 Modulates Smooth Muscle Cell Proliferation and Neointima Formation through Nuclear β-catenin Translocation. Arterioscler Thromb. Vasc. Biol. 31 (11), 2676–2684. doi:10.1161/ATVBAHA.111.230888
Zhou, B., Margariti, A., Zeng, L., and Xu, Q. (2011). Role of Histone Deacetylases in Vascular Cell Homeostasis and Arteriosclerosis. Cardiovasc. Res. 90 (3), 413–420. doi:10.1093/cvr/cvr003
Zhu, X., Chen, Z., Shen, W., Huang, G., Sedivy, J. M., Wang, H., et al. (2021). Inflammation, Epigenetics, and Metabolism Converge to Cell Senescence and Ageing: the Regulation and Intervention. Signal. Transduct Target. Ther. 6 (1), 245. doi:10.1038/s41392-021-00646-9
Ziegler, N., Raichur, S., Brunner, B., Hemmann, U., Stolte, M., Schwahn, U., et al. (2020). Liver-Specific Knockdown of Class IIa HDACs Has Limited Efficacy on Glucose Metabolism but Entails Severe Organ Side Effects in Mice. Front. Endocrinol. (Lausanne) 11, 598. doi:10.3389/fendo.2020.00598
Zu, Y., Liu, L., Lee, M. Y., Xu, C., Liang, Y., Man, R. Y., et al. (2010). SIRT1 Promotes Proliferation and Prevents Senescence through Targeting LKB1 in Primary Porcine Aortic Endothelial Cells. Circ. Res. 106 (8), 1384–1393. doi:10.1161/CIRCRESAHA.109.215483
Keywords: atherosclerosis (AS), histone deacetylases (HDACs), vascular systems, HDAC inhibitors, deacetylation
Citation: Luan Y, Liu H, Luan Y, Yang Y, Yang J and Ren K-D (2022) New Insight in HDACs: Potential Therapeutic Targets for the Treatment of Atherosclerosis. Front. Pharmacol. 13:863677. doi: 10.3389/fphar.2022.863677
Received: 27 January 2022; Accepted: 21 March 2022;
Published: 21 April 2022.
Edited by:
Prasanth Puthanveetil, Midwestern University, United StatesReviewed by:
Yanni Xu, Chinese Academy of Medical Sciences, ChinaVeronika Borutinskaite, Vilnius University, Lithuania
Copyright © 2022 Luan, Liu, Luan, Yang, Yang and Ren. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yang Yang, yangyangbio@163.com; Jing Yang, jingyang_0101@163.com; Kai-Di Ren, renkd006@163.com
†These authors have contributed equally to this work