 Eugene Lin
Eugene Lin Po-Hsiu Kuo
Po-Hsiu Kuo Yu-Li Liu
Yu-Li Liu Younger W.-Y. Yu5
Younger W.-Y. Yu5 Albert C. Yang
Albert C. Yang Shih-Jen Tsai
Shih-Jen Tsai- 1Department of Electrical Engineering, University of Washington, Seattle, WA, United States
- 2Graduate Institute of Biomedical Sciences, China Medical University, Taichung, Taiwan
- 3Department of Public Health, Institute of Epidemiology and Preventive Medicine, National Taiwan University, Taipei, Taiwan
- 4Center for Neuropsychiatric Research, National Health Research Institutes, Miaoli County, Taiwan
- 5Yu's Psychiatric Clinic, Kaohsiung, Taiwan
- 6Department of Psychiatry, Taipei Veterans General Hospital, Taipei, Taiwan
- 7Division of Psychiatry, National Yang-Ming University, Taipei, Taiwan
- 8Division of Interdisciplinary Medicine and Biotechnology, Beth Israel Deaconess Medical Center/Harvard Medical School, Boston, MA, United States
- 9Institute of Brain Science, National Yang-Ming University, Taipei, Taiwan
In the wake of recent advances in scientific research, personalized medicine using deep learning techniques represents a new paradigm. In this work, our goal was to establish deep learning models which distinguish responders from non-responders, and also to predict possible antidepressant treatment outcomes in major depressive disorder (MDD). To uncover relationships between the responsiveness of antidepressant treatment and biomarkers, we developed a deep learning prediction approach resulting from the analysis of genetic and clinical factors such as single nucleotide polymorphisms (SNPs), age, sex, baseline Hamilton Rating Scale for Depression score, depressive episodes, marital status, and suicide attempt status of MDD patients. The cohort consisted of 455 patients who were treated with selective serotonin reuptake inhibitors (treatment-response rate = 61.0%; remission rate = 33.0%). By using the SNP dataset that was original to a genome-wide association study, we selected 10 SNPs (including ABCA13 rs4917029, BNIP3 rs9419139, CACNA1E rs704329, EXOC4 rs6978272, GRIN2B rs7954376, LHFPL3 rs4352778, NELL1 rs2139423, NUAK1 rs2956406, PREX1 rs4810894, and SLIT3 rs139863958) which were associated with antidepressant treatment response. Furthermore, we pinpointed 10 SNPs (including ARNTL rs11022778, CAMK1D rs2724812, GABRB3 rs12904459, GRM8 rs35864549, NAALADL2 rs9878985, NCALD rs483986, PLA2G4A rs12046378, PROK2 rs73103153, RBFOX1 rs17134927, and ZNF536 rs77554113) in relation to remission. Then, we employed multilayer feedforward neural networks (MFNNs) containing 1–3 hidden layers and compared MFNN models with logistic regression models. Our analysis results revealed that the MFNN model with 2 hidden layers (area under the receiver operating characteristic curve (AUC) = 0.8228 ± 0.0571; sensitivity = 0.7546 ± 0.0619; specificity = 0.6922 ± 0.0765) performed maximally among predictive models to infer the complex relationship between antidepressant treatment response and biomarkers. In addition, the MFNN model with 3 hidden layers (AUC = 0.8060 ± 0.0722; sensitivity = 0.7732 ± 0.0583; specificity = 0.6623 ± 0.0853) achieved best among predictive models to predict remission. Our study indicates that the deep MFNN framework may provide a suitable method to establish a tool for distinguishing treatment responders from non-responders prior to antidepressant therapy.
Introduction
Personalized medicine, an emerging paradigm of medicine, is developing into the cornerstone of healthcare practice in terms of medical decisions and treatments tailored to the individual patient (1, 2). More precisely, patients are partitioned into subgroups by genetic and clinical characteristics, thereby medications could be tailored to specific patients with comparable genetic and clinical biomarkers (3). More broadly, personalized medicine promises to offer accurate diagnostic and therapeutic approaches in a patient-specific manner during all stages of patient care, including prevention, diagnosis, prognosis, treatment, and follow-up (4). The usage of genetic and clinical biomarkers has highlighted a key role in personalized medicine in the field of chronic diseases such as psychiatric or mental disorders (5, 6). Although the integration of personalized medicine into clinical decision making is still emerging, symbolic progress has recently been made by using genetic and clinical information to facilitate better predictions of patients' responses to targeted therapy (7). For instance, several genome-wide association studies (GWAS) have been carried out to pinpoint susceptible genetic loci influencing antidepressant treatment response as an entity (5, 6, 8). Moreover, accumulating evidence implicates that carefully chosen single nucleotide polymorphisms (SNPs) could be utilized as genetic biomarkers to infer clinical treatment outcomes and adverse drug reactions in patients with major depressive disorder (MDD) treated with antidepressants (5, 6, 8).
Recent advances in deep learning have demonstrated its power to learn and recognize complex non-linear hierarchical patterns based on large-scale empirical data (9). In general, the objective of deep learning is to facilitate an algorithm to learn a hierarchical representation of the data via multiple layers of abstraction such as multi-layer feedforward neural networks (MFNNs) (10). Due to new techniques such as the deployment of General-Purpose Computing on Graphics Processing Units, deep learning has carried out state-of-the-art performances on a wide variety of applications such as molecular biology (11). In the generic terms, the workflow for a deep learning algorithm is comprised of three portions including the model building from example inputs, evaluation and tuning of the model, and then the model production in prediction-making (12). In other words, a deep learning algorithm for classification applications such as medical diagnosis in personalized medicine is a procedure for choosing the best hypothesis from a set of alternatives that fit a set of observations (13).
The use of personalized medicine in terms of predicting antidepressant treatment response is still in its infancy. Scant human studies have investigated methods to build prediction models for estimating antidepressant treatment response. A study by Kautzky et al. suggested that a random forest prediction model for treatment outcome correctly identified 25% of responders by using 3 SNPs (including BDNF rs6265, PPP3CC rs7430, and HTR2A rs6313) and a clinical variable (that is, melancholia) (14). The following study by Patel et al. reported that an alternating decision tree model estimated treatment response with 89% accuracy by using structural imaging, age and mini-mental status examination scores (15). Moreover, another study by Chekroud et al. implicated that a machine learning model predicted clinical remission by using 25 variables with 59% accuracy (16). Iniesta et al. also demonstrated that regularized regression models based on clinical and demographical characteristics can predict response with clinically meaningful accuracy (17). Finally, a recent study by Maciukiewicz et al. showed that a support vector machine model forecasted treatment response with 52% accuracy by using SNPs (18).
In light of the aforementioned considerations, we hypothesized that it could be feasible and effective to use deep learning to build predictive models of antidepressant treatment outcome. To the best of our knowledge, no previous studies have been performed to evaluate predictive models for drug efficacy of antidepressants by using deep learning techniques. First, we explored for susceptibility loci by conducting a GWAS study with antidepressant treatment response per se in a hypothesis-free manner. Then, we combined genetic and clinical variables to optimize prediction of antidepressant treatment outcome using deep MFNN models. We selected the deep MFNN models because these models can be commonly utilized to solve complex applications in classification and predictive modeling and these models possess the benefits of fault tolerance, non-linearity, integrality, and real-time operations (19, 20).
Materials and Methods
Study Population
The study cohort is comprised of 455 patients, 268 patients from NHRI (The National Health Research Institutes) and 187 patients from TVGH (Taipei Veterans General Hospital), who were diagnosed with MDD in two central institutes in Taiwan. Subjects were part of the International SSRI Pharmacogenomics Consortium (ISPC) project encompassing 7 member sites from 5 countries (21). The diagnosis was assessed by board-certified psychiatrists who interviewed outpatients and obtained medical records. The inclusion criteria were that (1) subjects were Taiwanese with a minimum baseline score of 14 on the 21-item Hamilton Rating Scale for Depression (HRSD), and (2) either subjects were first-episode cases or had withdrawal of antidepressants for more than 2 weeks prior to entry into the study. Exclusion criteria were additional current DSM-IV Axis I diagnoses (including substance abuse, generalized anxiety disorders, panic disorders, or obsessive compulsive disorders), personality disorders, pregnancy, recent suicide attempt, and major medical and/or neurological disorders. These patients were treated with selective serotonin reuptake inhibitors (SSRIs), which include escitalopram (38.5%), paroxetine (38.5%), fluoxetine (18.3%) and citalopram (4.8%). Patients were assessed repeatedly at baseline and week 2, 4, and 8 using the 21-item HRSD.
Experiments were conducted in accordance with the Declaration of Helsinki and approved by the Institutional Review Board of Taipei Veterans General Hospital (VGHIRB No.: 2014-06-001B). Written informed consent was obtained from all participants ensuring adequate understanding of the study.
Measurement
Measurements of treatment response were obtained for participants as follows (22). First, we measured the sum score of 21-item HRSD at the 8th week of antidepressant treatment and recoded the results as “non-remitted” if the sum score was greater than 7 and as “remitted” otherwise. Second, we measured the percentage change of HRSD (that is, %ΔHRSD) and recoded the results as “non-response” if the percentage change was greater than −50% and as “response” otherwise.
Genotyping Data and Quality Controls
For all participants, SNP genotyping was carried out using Illumina HumanOmniExpressExome BeadChips in the International SSRI Pharmacogenomics Consortium. A total of 455 subjects were genotyped with 951,123 SNPs.
First, we performed quality control procedures with each individual, including kinship, sample quality, and population stratification (23). Then, plate-wise genotyping biases were checked. Samples with plate pass rate greater than 97% were retained for the subsequent analyses. We removed a total of 18 samples (11 from NHRI and 7 from TVGH) during this step. Secondly, the inbreeding coefficient and identity by state (IBS) were examined, and thereby we eliminated samples with strong kinship. In total, 9 subjects (4 from NHRI and 5 from TVGH) were removed due to the similarity measures far away from the clustering (that is, outliers in terms of the IBS distance). Thirdly, a multidimensional scaling analysis method was utilized with the genome-wide IBS pairwise distance to eliminate outliers. Our results showed that none was away from the clustering on the scatter plot. Finally, 7 patients treated with sertraline (SSRI) and venlafaxine (serotonin and norepinephrine reuptake inhibitor) were excluded. As a result, 421 MDD patients were retained for the subsequent analyses.
In addition, we performed quality control procedures as follows for SNP exclusion from further analyses (24). We removed SNPs which failed the Hardy-Weinberg tests with a P-value less than 0.0001, genotype missing rate greater than 5%, minor allele frequency (MAF) smaller than 0.05, or bad calling ones in clustering (for example, heterozygous genotypes were falsely called as homozygous, or homozygous genotypes were incorrectly called as heterozygous). After performing the aforementioned quality control procedures, a total of 647,030 SNPs in the samples were retained for the subsequent imputation analysis. The genotyping call rate was 99.9% for all subjects.
Imputation
Imputation was carried out using IMPUTE2 v3 (25), with haplotype reference panels (https://mathgen.stats.ox.ac.uk/impute/data_download_1000G_phase1_integrated_SHAPEIT2.html) released in March/April 2012 from the 1000 Genomes Project on the basis of HapMap build 37. Only imputed SNPs with high genotype information contents (that is, IMPUTE information score > 0.5) were used in the subsequent association analyses. In total, 30,040,257 SNPs were imputed with high confidence for each individual in the samples. Then, we removed markers which failed the Hardy-Weinberg tests with a p-value less than 0.0001, genotype missing rate greater than 5%, MAF smaller than 0.05, or bad calling ones in clustering. As a result, a total of 4,241,701 SNPs were retained for the subsequent analyses.
Statistical Analysis
The Student's t-test was conducted to measure the difference in the means of two continuous variables. We performed the chi-square test for categorical data. In order to evaluate the relation of the investigated SNPs with antidepressant treatment outcome, we conducted a logistic regression analysis to evaluate the odds ratios (ORs) and their 95% confidence intervals (CIs), adjusting for covariates including age, sex, and site (26). Multiple testing was adjusted by the Bonferroni correction. The criterion for significance was set at P < 0.05 for all tests. Data are presented as the mean ± standard deviation.
To investigate SNP-SNP interactions, we leveraged the generalized multifactor dimensionality reduction (GMDR) method (27). We tested two-way interactions using 10-fold cross-validation. The GMDR software provides some output parameters, including the testing accuracy and empirical P-values, to assess each selected interaction. Moreover, we provided age, sex, and site as covariates for SNP-SNP interaction models in our interaction analyses. Permutation testing obtains empirical P-values of prediction accuracy as a benchmark based on 1,000 shuffles.
Predictive Model Algorithms
In this study, we employed a key deep learning technique, MFNNs. Logistic regression analysis, the standard method for clinical classification (10), was employed to compare with the MFNN models. The Waikato Environment for Knowledge Analysis (WEKA) software (10) was utilized to perform these predictive models.
An MFNN model consists of one input layer, one or multiple hidden layers and one output layer. The MFNN model is one category of artificial neural network algorithms where networks between entities construct no directed cycles (28). In other words, a loop or cycle does not occur in the network because the data only relays in an onward direction from the input neuron panel, through various panels of the hidden neuron portions (if any), and then to the output neuron panel.
Moreover, the principal operation of the MFNN model is subdivided into the learning and retrieving stages in terms of an algorithmic point of view (19). In the learning stage of the MFNN model, the back-propagation algorithm (29) is leveraged for the learning strategy. Additionally, in the retrieving stage, the MFNN model repeats via all the panels to carry out the retrieval process at the output panel in keeping with the inputs of test patterns. On the other hand, from a structural point of view, the MFNN model is an iterative and spatial neural network that possesses various panels of hidden neuron portions among the input and output neuron panels (19).
Here, to train the MFNN models, WEKA's parameters were chosen as follows: the momentum = 0.01, the learning rate = 0.001, the batch size = 100, and the number of epochs = 500.
Evaluation of the Predictive Performance
The receiver operating characteristic (ROC) methodology was employed and the area under the ROC curve (AUC) was calculated to evaluate the performance of predictive models (30–32). The AUC of a predictive model can be viewed as the probability that the predictive model will rank a randomly selected positive sample higher than a randomly selected negative one (32). Because AUC is a better performance metric than accuracy, most researchers have adopted AUC for estimating predictive capability of predictive models nowadays (32). The better the prediction model, the higher the AUC (30, 32). In the present work, we utilized AUC to evaluate the performance of various prediction models on a dataset. Additionally, sensitivity (i.e., the proportion of correctly predicted responders of all tested responders) and specificity (i.e., the proportion of correctly predicted non-responders of all the tested non-responders) were measured.
Moreover, the repeated 10-fold cross-validation method was employed to investigate the generalization of the predictive algorithms generated by the aforementioned models (30, 33). Firstly, we randomly split the whole dataset into 10 separate segments. Secondly, in order to evaluate the predictive performance, we trained the predictive model using nine-tenths of the data and tested the predictive model with the remaining tenth of data. Next, we repeated the previous step nine more times by leaving out distinct nine-tenths of the data as training data and a distinct tenth of data as testing data. Finally, we reported the average estimation over all runs by processing the aforementioned regular 10-fold cross-validation for 10 times with distinct batches of data. We estimated the performance of all predictive models using repeated 10-fold cross-validation testing.
Results
A total of 421 MDD patients (mean age of 43.7 years and 28.7% of males) were retained for the following analyses after we removed the subjects with missing or incomplete data. Table 1 describes the demographic and clinical characteristics of the study population, including 257 antidepressant treatment responders and 164 treatment non-responders. The treatment-response rate in our cohort was 61.0%. Additionally, the remission rate was 33.0%. Six clinical biomarkers were used in the subsequent deep learning analyses, including age at time of consent, sex, marital status (or in a long-term relationship), the number of depressive episodes until time of study enrollment, 21-item HRSD at baseline, and the status of whether the patients had previously attempted suicide. There was a significant difference in the number of depressive episodes (P = 0.039) and 21-item HRSD at baseline (P = 0.013) between the treatment response and non-response subjects (Table 1). Furthermore, there was a significant difference in 21-item HRSD at baseline (P = 0.022) between the remitted and non-remitted subjects (Table 1).
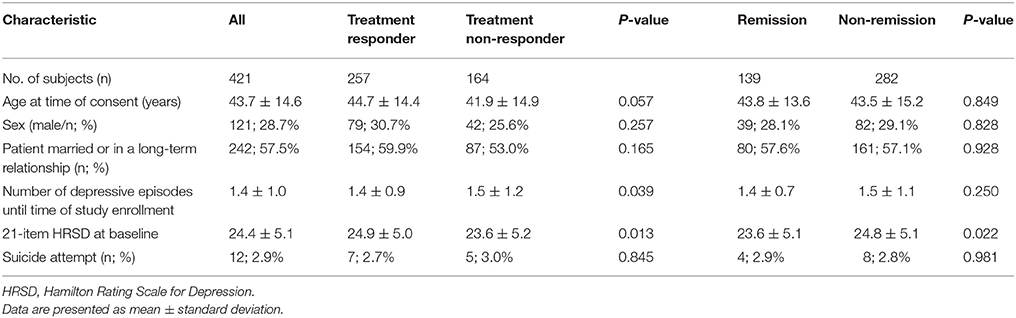
Table 1. Demographic and clinical characteristics of study subjects.
First, we investigated the association between antidepressant treatment response and 4,241,701 SNPs assessed in a GWAS study. None of these SNPs reached the genome-wide significance level (P < 1.2 × 10−8) after Bonferroni correction for the multiple comparisons. For further investigation in the subsequent deep learning analyses, we identified 10 key SNPs showing an evidence of association with antidepressant treatment responders per se with the criterion of a significant level of P < 7.5 × 10−5 (Table 2). The top-rated SNPs encompass rs4917029 adjacent to the ATP binding cassette subfamily A member 13 (ABCA13) gene, rs9419139 adjacent to the BCL2 interacting protein 3 (BNIP3) gene, rs704329 in the calcium voltage-gated channel subunit alpha1 E (CACNA1E) gene, rs6978272 in the exocyst complex component 4 (EXOC4) gene, rs7954376 adjacent to the glutamate ionotropic receptor NMDA type subunit 2B (GRIN2B) gene, rs4352778 in the LHFPL tetraspan subfamily member 3 (LHFPL3) gene, rs2139423 in the neural EGFL like 1 (NELL1) gene, rs2956406 in the NUAK family kinase 1 (NUAK1) gene, rs4810894 adjacent to the phosphatidylinositol-3,4,5-trisphosphate dependent Rac exchange factor 1 (PREX1) gene, and rs139863958 adjacent to the slit guidance ligand 3 (SLIT3) gene. For example, as demonstrated in Table 2 for the rs4810894 SNP of the PREX1 gene, there was an indication of an association with antidepressant treatment response after adjustment of covariates such as age, sex, and site for the dominant model (OR = 2.70; 95% CI = 1.78–4.10; P = 3.20 × 10−6). Similarly, there was an indication of an association with antidepressant treatment response among the subjects after adjustment of covariates for the dominant model for the rs7954376 SNP of the GRIN2B gene (OR = 0.29; 95% CI = 0.17–0.49; P = 3.96 × 10−6).
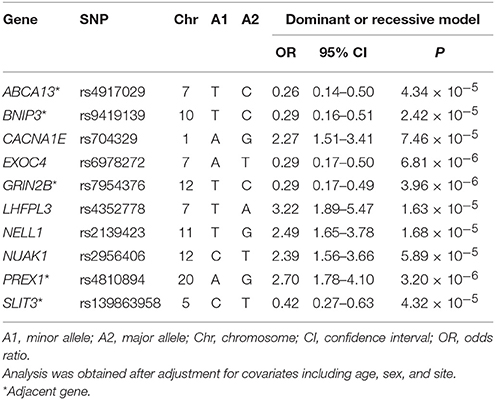
Table 2. Odds ratio analysis with odds ratios after adjustment for covariates between treatment response and the top 10 SNPs.
In addition, we pinpointed 10 key SNPs showing an evidence of association with remission per se with the criterion of a significant level of P < 9.9 × 10−5 (Table 3) for further investigation in the subsequent deep learning analyses. The top-rated SNPs encompass rs11022778 in the aryl hydrocarbon receptor nuclear translocator like (ARNTL) gene, rs2724812 in the calcium/calmodulin dependent protein kinase ID (CAMK1D) gene, rs12904459 adjacent to the gamma-aminobutyric acid type A receptor beta3 subunit (GABRB3) gene, rs35864549 adjacent to the glutamate metabotropic receptor 8 (GRM8) gene, rs9878985 in the N-acetylated alpha-linked acidic dipeptidase like 2 (NAALADL2) gene, rs483986 in the neurocalcin delta (NCALD) gene, rs12046378 adjacent to the phospholipase A2 group IVA (PLA2G4A) gene, rs73103153 adjacent to the prokineticin 2 (PROK2) gene, rs17134927 in the RNA binding fox-1 homolog 1 (RBFOX1) gene, and rs77554113 adjacent to the zinc finger protein 536 (ZNF536) gene. For instance, as shown in Table 3 for the rs77554113 SNP of the ZNF536 gene, there was an indication of an association with remission after adjustment of covariates such as age, sex, and site for the dominant model (OR = 4.81; 95% CI = 2.54–9.13; P = 1.49 × 10−6). Similarly, there was an indication of an association with remission among the subjects after adjustment of covariates for the dominant model for the rs12904459 SNP of the GABRB3 gene (OR = 2.53; 95% CI = 1.64–3.90; P = 2.58 × 10−5).
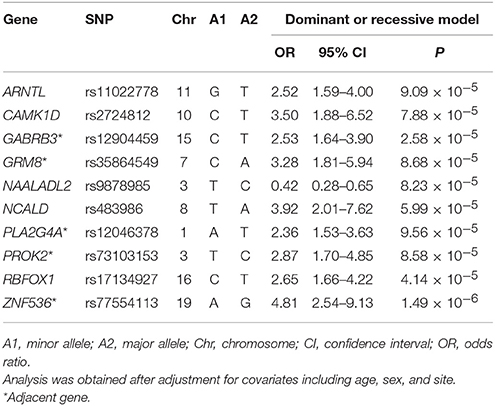
Table 3. Odds ratio analysis with odds ratios after adjustment for covariates between remission and the top 10 SNPs.
Furthermore, we integrated the aforementioned 6 clinical biomarkers with the top 10 SNPs of treatment response to build the predictive models for antidepressant treatment response by using the deep MFNN framework. Table 4 summarizes the results of repeated 10-fold cross-validation experiments by deep MFNN models and logistic regression using 16 biomarkers including the aforementioned 10 SNPs of treatment response and 6 clinical biomarkers. For deep MFNN models, we performed a series of different architectures containing 1, 2, and 3 hidden layers. Figure 1 shows an example of architecture of the MFNN model with 3 hidden layers. To measure the performance of prediction models, we used the ROC methodology and calculated the AUC, sensitivity, and specificity for these four predictive models using 16 biomarkers. As indicated in Table 4, the average values of AUC for the deep MFNN prediction models of 1, 2, and 3 hidden layers were 0.8211 ± 0.0571, 0.8228 ± 0.0571, and 0.8220 ± 0.0570, respectively. Of all the MFNN prediction models, the deep MFNN model with 2 hidden layers gave better performance than the other two models in terms of AUC. Among all four predictive models, the deep MFNN model with 2 hidden layers performed best, outperforming the logistic regression model (AUC = 0.8168 ± 0.0553) in terms of AUC. For comparison, we also built the predictive models for antidepressant treatment response using only 6 clinical biomarkers. The models with 16 biomarkers performed better than the ones with only 6 clinical biomarkers (Table 4).
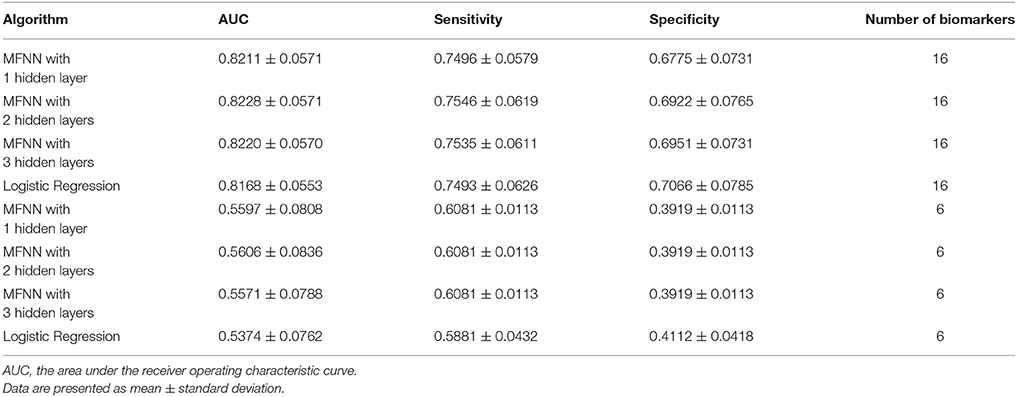
Table 4. The results of repeated 10-fold cross-validation experiments for predicting treatment response using multilayer feedforward neural networks (MFNNs) and logistic regression with 16 biomarkers and 6 clinical biomarkers only.
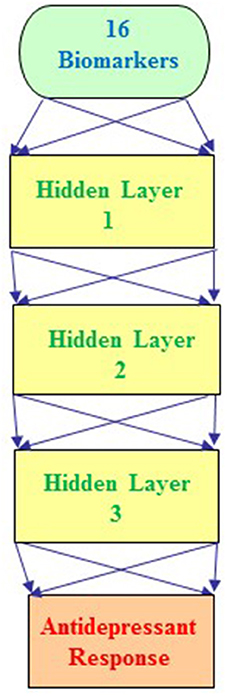
Figure 1. An example architecture of a multilayer feedforward neural network (MFNN) model with 3 hidden layers. The MFNN model contains 16 units in the input layer corresponding to 16 biomarkers (including 10 SNPs and 6 clinical predictors). The MFNN model is configured with 2 units in the output layer corresponding to antidepressant treatment outcome (that is, antidepressant treatment responders and non-responders).
Moreover, we combined the aforementioned 6 clinical biomarkers with the top 10 SNPs of remission to construct the predictive models for remission by using the deep MFNN framework. Table 5 summarizes the results of repeated 10-fold cross-validation experiments by deep MFNN models and logistic regression using 16 biomarkers including the aforementioned 10 SNPs of remission and 6 clinical biomarkers. For deep MFNN models, we achieved a series of different architectures containing 1, 2, and 3 hidden layers. As indicated in Table 5, the average values of AUC for the deep MFNN prediction models of 1, 2, and 3 hidden layers were 0.8042 ± 0.0729, 0.8047 ± 0.0727, and 0.8060 ± 0.0722, respectively. Of all the MFNN prediction models, the deep MFNN model with 3 hidden layers gave better performance than the other two models in terms of AUC. Among all four predictive models, the deep MFNN model with 3 hidden layers performed best, outperforming the logistic regression model (AUC = 0.7985 ± 0.0772) in terms of AUC. For comparison, we also built the predictive models for remission using only 6 clinical biomarkers. The models with 16 biomarkers performed better than the ones with only 6 clinical biomarkers (Table 5).
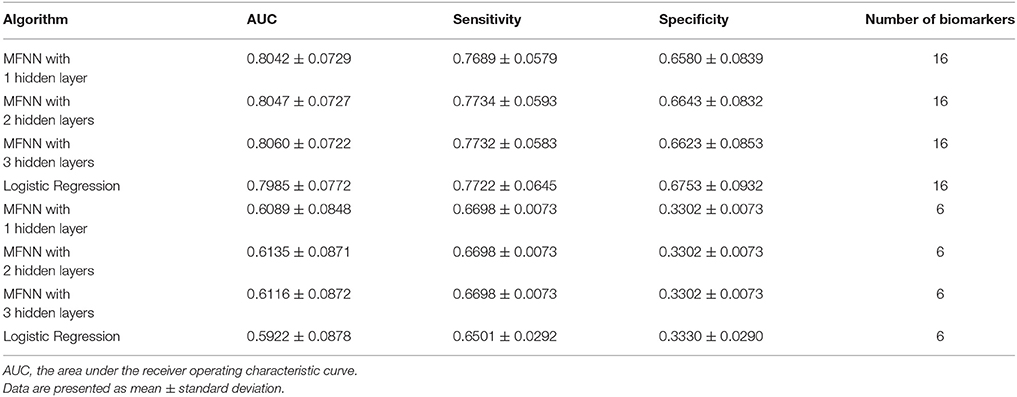
Table 5. The results of repeated 10-fold cross-validation experiments for predicting remission using multilayer feedforward neural networks (MFNNs) and logistic regression with 16 biomarkers and 6 clinical biomarkers only.
Finally, the GMDR analysis was used to assess SNP-SNP interactions between the top 10 SNPs in antidepressant treatment response including age, sex, and site as covariates. Table 6 summarizes the results obtained from GMDR analysis for SNP-SNP interaction models with covariate adjustment. As shown in Table 6, there was a significant SNP-SNP interaction involving BNIP3 rs9419139 and PREX1 rs4810894 (P < 0.001) in influencing antidepressant treatment response. We also assessed SNP-SNP interactions between the top 10 SNPs in remission including age, sex, and site as covariates. As shown in Table 6, there was a significant SNP-SNP interaction involving ARNTL rs11022778 and ZNF536 rs77554113 (P < 0.001) in influencing remission.
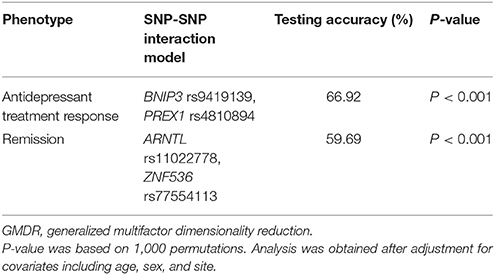
Table 6. SNP-SNP interaction models identified by the GMDR method with adjustment for age, sex, and site.
Discussion
Our analysis is the first study to date to leverage deep learning for building predictive models of antidepressant treatment outcome among Taiwanese MDD individuals with the GWAS data. In this study, we pinpointed that the deep MFNN model with 2 hidden layers outperformed the logistic regression model as well as other predictive models in terms of AUC for distinguishing antidepressant treatment responders and non-responders in MDD. We also found that the deep MFNN model with three hidden layers exceeded the logistic regression model as well as other predictive models in terms of AUC for predicting remission in MDD. Additionally, we identified 26 predictive variables of antidepressants, including 6 clinical biomarkers, 10 SNPs for treatment response, and 10 SNPs for remission. The 6 clinical biomarkers encompass the patient's age at time of consent, sex, marital status (or in a long-term relationship), the number of depressive episodes until time of study enrollment, 21-item HRSD at baseline, and the status of suicide attempts. By using the GWAS data, we further tracked down the top 10 SNPs showing an evidence of association with antidepressant treatment response as well as remission, respectively. Moreover, we combined the 6 clinical predictors with the 10 genetic variants to establish the predictive models of antidepressant treatment outcome as well as remission by using the deep MFNN framework. Our data also indicated that our deep MFNN models may provide a suitable approach to create predictive models for forecasting antidepressant treatment response as well as remission with clinically meaningful accuracy. Therefore, our deep MFNN approach is a proof of concept of a deep learning predictive tool for antidepressant efficacy prior to antidepressant treatment.
In the present study, we found that 10 potential SNPs (including ABCA13 rs4917029, BNIP3 rs9419139, CACNA1E rs704329, EXOC4 rs6978272, GRIN2B rs7954376, LHFPL3 rs4352778, NELL1 rs2139423, NUAK1 rs2956406, PREX1 rs4810894, and SLIT3 rs139863958) may play an important role in the modulation of antidepressant treatment response in a Taiwanese population by using a GWAS study. However, to our knowledge, no studies have been implicated these SNPs for drug efficacy of antidepressants. The functional relevance of the effects of these SNPs on antidepressant treatment response remains to be elucidated. The BNIP3 gene is located on chromosome 10q26.3 and encodes a mitochondrial protein which is implicated in several functions linked to antidepressive effects as well as the action of antidepressants (34). The ABCA13, PREX1, and SLIT3 genes have been suggested to link with MDD (35–37). The CACNA1E gene has been shown to be associated with bipolar disorder (38). The GRIN2B and EXOC4 genes have been found to be in relation to schizophrenia (39, 40). The LHFPL3, NELL1, and NUAK1 gene might be connected to the synapse function, mental development delay, and neuropsychiatric disorders, respectively (41–43).
Moreover, we identified 10 potential SNPs (including ARNTL rs11022778, CAMK1D rs2724812, GABRB3 rs12904459, GRM8 rs35864549, NAALADL2 rs9878985, NCALD rs483986, PLA2G4A rs12046378, PROK2 rs73103153, RBFOX1 rs17134927, and ZNF536 rs77554113) to be associated with remission in a Taiwanese population by using a GWAS study. The ARNTL gene, located on chromosome 11p15.3, is one of the clock genes, representing the hallmark of circadian rhythms that have been shown to influence the behavioral effects of psychoactive drugs (44). ARNTL rs11022778 SNP has also been suggested to be involved to in the susceptibility of mood disorders (both MDD and bipolar disorders) and suicide attempts (45, 46). The GRM8, PLA2G4A, and PROK2 genes were found to be associated with MDD (47–49). The NCALD and ZNF536 genes have been shown to be linked with bipolar disorder (50, 51). The CAMK1D, GABRB3, and RBFOX1 genes have been suggested to contribute to schizophrenia physiopathology (52–54). The NAALADL2 gene has been implicated to cause autism spectrum disorder, a neurodevelopmental disorder (55).
On another note, our deep MFNN framework utilized 6 clinical biomarkers encompassing demographics data (such as age, sex, marital status), total scores on baseline severity measures (such as 21-item HRSD at baseline), recurrent episodes (that is, the number of depressive episodes until time of study enrollment), and the status of suicide attempts. These 6 clinical biomarkers have been previously tested as predictors for predicting possible antidepressant treatment outcome in MDD in other studies (17, 56). However, to the best of our knowledge, no prior studies have been evaluated for drug efficacy of antidepressants by combining these clinical predictors with the SNP information. There are more potential clinical biomarkers such as body mass index, MDD subtypes, the age of onset of MDD, social support, previous antidepressant treatments, and early life stressful events that could possibly influence antidepressants treatment response (17, 56). However, our study did not comprise these clinical biomarkers due to lack of data.
Remarkably, another intriguing finding was that we further inferred the epistatic effects between BNIP3 rs9419139 and PREX1 rs4810894 in influencing antidepressant treatment response as well as the epistatic effects between ARNTL rs11022778 and ZNF536 rs77554113 in influencing remission by using the GMDR approach. To our knowledge, no other study has been conducted to weigh SNP-SNP interactions between these SNPs. The ARNTL and BNIP3 gene have been implicated to influence psychoactive drugs and antidepressants (34, 44). The PREX1 and ZNF536 genes have been shown to be associated with MDD and bipolar disorder, respectively (37, 50). The biological mechanisms of these SNP-SNP interactions on antidepressant treatment outcome remain to be elucidated.
This study has both limitations and strengths. The major weakness is that the novel findings related to the selected SNPs were not validated by other independent cohorts and the MFNN models were not verified by an independent cohort or dataset. Both the sensitivity and specificity of the model were also not adequate enough to predict treatment response in clinical practice. In addition, our study was carried out using four different SSRI antidepressants and some patients had concomitant use of alprazolam, lorazepam or clonazepam for insomnia. Moreover, the present study was underpowered because of limited sample size, and few replications of the effects and uncertain biological mechanisms were found for some selected SNPs on antidepressant treatment outcome. Therefore, our findings warrant much more research to evaluate whether the observations are reproducible in various ethnic populations (57). The use of SNPs in this study serves as a strategy for building the MFNN models, and these selected SNPs will need to be replaced with significant SNPs from much larger and more rigorous studies in the future. Future clinical trials are needed to facilitate a comprehensive assessment of the predictive models for antidepressant treatment outcome with different ethnic populations (57). By contrast, the main strength of our work is that we utilized rigorously phenotyped antidepressant response in MDD patients to assess influential SNPs among the investigated genes.
Conclusions
In conclusion, we carried out deep learning predictive models for assessing antidepressant treatment outcome in Taiwanese subjects. Our findings demonstrate that our deep MFNN framework may provide a plausible way to build predictive models for estimating antidepressant treatment response with clinically meaningful accuracy. Thus, we could anticipate that the results of our studies could be generalized for genomics studies of personalized medicine in predicting treatment response for human disorders and could be employed to establish molecular diagnostic and prognostic tools over the next few years. It is essential to evolve further understandings into the role of these deep learning predictive models investigated in this study by using independent replication studies with larger sample sizes.
Ethics Statement
This study was approved by the Institutional Review Board of Taipei Veterans General Hospital (VGHIRB No.: 2014-06-001B) and complies with the Declaration of Helsinki. Informed written consent was obtained from all participants.
Author Contributions
Study conception and design: EL, P-HK, and S-JT. Acquisition of data: P-HK, Y-LL, YY, and AY. Analysis and interpretation of data: EL and S-JT. Draft manuscript: EL. All authors read and approved the final manuscript.
Funding
This work was supported by Taipei Veterans General Hospital, Taiwan (Grant V105D17-002-MY2-2) and by the Ministry of Science and Technology, Taiwan (Grant MOST 107-2634-F-075-002; MOST 107-2634-F-002-019).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Translational Resource Center for Genomic Medicine of National Research Program for Biopharmaceuticals for their data service. Genotyping of MDD subjects was funded by the RIKEN Center for Integrative Medical Science, Yokohama, Japan. We thank Emily Ting for English editing.
References
1. Katsanis SH, Javitt G, Hudson K. Public health. A case study of personalized medicine. Science (2008) 320:53–4. doi: 10.1126/science.1156604
2. Snyderman R. Personalized health care: from theory to practice. Biotechnol J. (2012) 7:973–9. doi: 10.1002/biot.201100297
3. Sadee W, Dai Z. Pharmacogenetics/genomics and personalized medicine. Hum Mol Genet. (2005) 14 Spec No. 2:R207–14. doi: 10.1093/hmg/ddi261
4. Lane HY, Tsai GE, Lin E. Assessing gene-gene interactions in pharmacogenomics. Mol Diagn Ther. (2012) 16:15–27. doi: 10.2165/11597270-000000000-00000
5. Lin E, Chen PS. Pharmacogenomics with antidepressants in the STAR*D study. Pharmacogenomics (2008) 9:935–46. doi: 10.2217/14622416.9.7.935
6. Lin E, Lane HY. Genome-wide association studies in pharmacogenomics of antidepressants. Pharmacogenomics (2015) 16:555–66. doi: 10.2217/pgs.15.5
7. Hamburg MA, Collins FS. The path to personalized medicine. N Engl J Med. (2010) 363:301–4. doi: 10.1056/NEJMp1006304
8. Lin E, Tsai SJ. Genome-wide microarray analysis of gene expression profiling in major depression and antidepressant therapy. Prog Neuropsychopharmacol Biol Psychiatry (2016) 64:334–40. doi: 10.1016/j.pnpbp.2015.02.008
9. Bengio Y, Courville A, Vincent P. Representation learning: a review and new perspectives. IEEE Trans Pattern Anal Mach Intell. (2013) 35:1798–828. doi: 10.1109/TPAMI.2013.50
10. Witten IHFE. Data Mining: Practical Machine Learning Tools and Techniques. Francisco, CA: Morgan Kaufmann Publishers (2005).
11. Chen Y, Li Y, Narayan R, Subramanian A, Xie X. Gene expression inference with deep learning. Bioinformatics (2016) 32:1832–9. doi: 10.1093/bioinformatics/btw074
12. Pham T, Tran T, Phung D, Venkatesh S. Predicting healthcare trajectories from medical records: a deep learning approach. J Biomed Inform. (2017) 69:218–29. doi: 10.1016/j.jbi.2017.04.001
13. Lin E, Lane HY. Machine learning and systems genomics approaches for multi-omics data. Biomark Res (2017) 5:2. doi: 10.1186/s40364-017-0082-y
14. Kautzky A, Baldinger P, Souery D, Montgomery S, Mendlewicz J, Zohar J, et al. The combined effect of genetic polymorphisms and clinical parameters on treatment outcome in treatment-resistant depression. Eur Neuropsychopharmacol. (2015) 25:441–53. doi: 10.1016/j.euroneuro.2015.01.001
15. Patel MJ, Andreescu C, Price JC, Edelman KL, Reynolds CF III, Aizenstein HJ. Machine learning approaches for integrating clinical and imaging features in late-life depression classification and response prediction. Int J Geriatr Psychiatry (2015) 30:1056–67. doi: 10.1002/gps.4262
16. Chekroud AM, Zotti RJ, Shehzad Z, Gueorguieva R, Johnson MK, Trivedi MH, et al. Cross-trial prediction of treatment outcome in depression: a machine learning approach. Lancet Psychiatry (2016) 3:243–50. doi: 10.1016/S2215-0366(15)00471-X
17. Iniesta R, Malki K, Maier W, Rietschel M, Mors O, Hauser J, et al. Combining clinical variables to optimize prediction of antidepressant treatment outcomes. J Psychiatr Res. (2016) 78:94–102. doi: 10.1016/j.jpsychires.2016.03.016
18. Maciukiewicz M, Marshe VS, Hauschild AC, Foster JA, Rotzinger S, Kennedy JL, et al. GWAS-based machine learning approach to predict duloxetine response in major depressive disorder. J Psychiatr Res. (2018) 99:62–8. doi: 10.1016/j.jpsychires.2017.12.009
20. Lin E, Hwang Y, Wang SC, Gu ZJ, Chen EY. An artificial neural network approach to the drug efficacy of interferon treatments. Pharmacogenomics (2006) 7:1017–24. doi: 10.2217/14622416.7.7.1017
21. Biernacka JM, Sangkuhl K, Jenkins G, Whaley RM, Barman P, Batzler A, et al. The International SSRI Pharmacogenomics Consortium (ISPC): a genome-wide association study of antidepressant treatment response. Transl Psychiatry (2015) 5:e553. doi: 10.1038/tp.2015.47
22. Lin KM, Tsou HH, Tsai IJ, Hsiao MC, Hsiao CF, Liu CY, et al. CYP1A2 genetic polymorphisms are associated with treatment response to the antidepressant paroxetine. Pharmacogenomics (2010) 11:1535–43. doi: 10.2217/pgs.10.128
23. Lin E, Kuo PH, Liu YL, Yang AC, Tsai SJ. Transforming growth factor-beta signaling pathway-associated genes SMAD2 and TGFBR2 are implicated in metabolic syndrome in a Taiwanese population. Sci Rep. (2017) 7:13589. doi: 10.1038/s41598-017-14025-4
24. Lin E, Tsai SJ, Kuo PH, Liu YL, Yang AC, Kao CF, et al. The rs1277306 variant of the REST gene confers susceptibility to cognitive aging in an elderly taiwanese population. Dement Geriatr Cogn Disord. (2017) 43:119–27. doi: 10.1159/000455833
25. Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. (2009) 5:e1000529. doi: 10.1371/journal.pgen.1000529
26. Lin E, Kuo PH, Liu YL, Yang AC, Kao CF, Tsai SJ. Effects of circadian clock genes and health-related behavior on metabolic syndrome in a Taiwanese population: Evidence from association and interaction analysis. PLoS ONE (2017) 12:e0173861. doi: 10.1371/journal.pone.0173861
27. Lou XY, Chen GB, Yan L, Ma JZ, Zhu J, Elston RC, et al. A generalized combinatorial approach for detecting gene-by-gene and gene-by-environment interactions with application to nicotine dependence. Am J Hum Genet. (2007) 80:1125–37. doi: 10.1086/518312
29. Rumelhart DE, Hinton GE, William RJ. Learning internal representation by error propagation. In: Parallel Distributed Processing: Explorations. The Micro-Structure of Cognition. 1. Cambridge, MA: MIT Press (1996). p. 318–62.
30. Huang LC, Hsu SY, Lin E. A comparison of classification methods for predicting Chronic Fatigue Syndrome based on genetic data. J Transl Med. (2009) 7:81. doi: 10.1186/1479-5876-7-81
31. Lin E, Hwang Y. A support vector machine approach to assess drug efficacy of interferon-alpha and ribavirin combination therapy. Mol Diagn Ther. (2008) 12:219–23. doi: 10.1007/BF03256287
32. Linden A. Measuring diagnostic and predictive accuracy in disease management: an introduction to receiver operating characteristic (ROC) analysis. J Eval Clin Pract. (2006) 12:132–9. doi: 10.1111/j.1365-2753.2005.00598.x
33. Lin E, Hsu SY. A Bayesian approach to gene-gene and gene-environment interactions in chronic fatigue syndrome. Pharmacogenomics (2009) 10:35–42. doi: 10.2217/14622416.10.1.35
34. Tohda M, Hayashi H, Sukma M, Tanaka K. BNIP-3: a novel candidate for an intrinsic depression-related factor found in NG108-15 cells treated with Hochu-ekki-to, a traditional oriental medicine, or typical antidepressants. Neurosci Res. (2008) 62:1–8. doi: 10.1016/j.neures.2008.05.007
35. Chen J, Khan RAW, Wang M, He K, Wang Q, Li Z, et al. Association between the variability of the ABCA13 gene and the risk of major depressive disorder and schizophrenia in the Han Chinese population. World J Biol Psychiatry (2017) 18:550–6. doi: 10.1080/15622975.2016.1245442
36. Glessner JT, Wang K, Sleiman PM, Zhang H, Kim CE, Flory JH, et al. Duplication of the SLIT3 locus on 5q35.1 predisposes to major depressive disorder. PLoS ONE (2010) 5:e15463. doi: 10.1371/journal.pone.0015463
37. Li J, Chai A, Wang L, Ma Y, Wu Z, Yu H, et al. Synaptic P-Rex1 signaling regulates hippocampal long-term depression and autism-like social behavior. Proc Natl Acad Sci USA. (2015) 112:E6964–72. doi: 10.1073/pnas.1512913112
38. Jan WC, Yang SY, Chuang LC, Lu RB, Lu MK, Sun HS, et al. Exploring the associations between genetic variants in genes encoding for subunits of calcium channel and subtypes of bipolar disorder. J Affect Disord. (2014) 157:80–6. doi: 10.1016/j.jad.2013.12.044
39. Guo Z, Niu W, Bi Y, Zhang R, Ren D, Hu J, et al. A study of single nucleotide polymorphisms of GRIN2B in schizophrenia from Chinese Han population. Neurosci Lett. (2016) 630:132–5. doi: 10.1016/j.neulet.2016.07.038
40. Merico D, Zarrei M, Costain G, Ogura L, Alipanahi B, Gazzellone MJ, et al. Whole-genome sequencing suggests schizophrenia risk mechanisms in humans with 22q11.2 deletion syndrome. G3 (Bethesda) (2015) 5:2453–61. doi: 10.1534/g3.115.021345
41. Alemany S, Ribases M, Vilor-Tejedor N, Bustamante M, Sanchez-Mora C, Bosch R, et al. New suggestive genetic loci and biological pathways for attention function in adult attention-deficit/hyperactivity disorder. Am J Med Genet B Neuropsychiatr Genet. (2015) 168:459–70. doi: 10.1002/ajmg.b.32341
42. Dateki S, Watanabe S, Kinoshita F, Yoshiura KI, Moriuchi H. Identification of 11p14.1-p15.3 deletion probably associated with short stature, relative macrocephaly, and delayed closure of the fontanelles. Am J Med Genet A. (2017) 173:217–20. doi: 10.1002/ajmg.a.37978
43. Davenport EC, Pendolino V, Kontou G, McGee TP, Sheehan DF, Lopez-Domenech G, et al. An essential role for the tetraspanin LHFPL4 in the cell-type-specific targeting and clustering of synaptic GABAA receptors. Cell Rep. (2017) 21:70–83. doi: 10.1016/j.celrep.2017.09.025
44. Manev H, Uz T. Clock genes: influencing and being influenced by psychoactive drugs. Trends Pharmacol Sci. (2006) 27:186–9. doi: 10.1016/j.tips.2006.02.003
45. Dmitrzak-Weglarz MP, Pawlak JM, Maciukiewicz M, Moczko J, Wilkosc M, Leszczynska-Rodziewicz A, et al. Clock gene variants differentiate mood disorders. Mol Biol Rep. (2015) 42:277–88. doi: 10.1007/s11033-014-3770-9
46. Pawlak J, Dmitrzak-Weglarz M, Maciukiewicz M, Wilkosc M, Leszczynska-Rodziewicz A, Zaremba D, et al. Suicidal behavior in the context of disrupted rhythmicity in bipolar disorder–data from an association study of suicide attempts with clock genes. Psychiatry Res. (2015) 226:517–20. doi: 10.1016/j.psychres.2015.01.010
47. Pae CU, Yu HS, Kim JJ, Lee CU, Lee SJ, Lee KU, et al. BanI polymorphism of the cytosolic phospholipase A2 gene and mood disorders in the Korean population. Neuropsychobiology (2004) 49:185–8. doi: 10.1159/000077364
48. Spijker S, Van Zanten JS, De Jong S, Penninx BW, van Dyck R, Zitman FG, et al. Stimulated gene expression profiles as a blood marker of major depressive disorder. Biol Psychiatry (2010) 68:179–86. doi: 10.1016/j.biopsych.2010.03.017
49. Terracciano A, Tanaka T, Sutin AR, Sanna S, Deiana B, Lai S, et al. Genome-wide association scan of trait depression. Biol Psychiatry (2010) 68:811–7. doi: 10.1016/j.biopsych.2010.06.030
50. Winham SJ, Cuellar-Barboza AB, McElroy SL, Oliveros A, Crow S, Colby CL, et al. Bipolar disorder with comorbid binge eating history: a genome-wide association study implicates APOB. J Affect Disord. (2014) 165:151–8. doi: 10.1016/j.jad.2014.04.026
51. Xu W, Cohen-Woods S, Chen Q, Noor A, Knight J, Hosang G, et al. Genome-wide association study of bipolar disorder in Canadian and UK populations corroborates disease loci including SYNE1 and CSMD1. BMC Med Genet. (2014) 15:2. doi: 10.1186/1471-2350-15-2
52. Gadelha A, Coleman J, Breen G, Mazzoti DR, Yonamine CM, Pellegrino R, et al. Genome-wide investigation of schizophrenia associated plasma Ndel1 enzyme activity. Schizophr Res. (2016) 172:60–7. doi: 10.1016/j.schres.2016.01.043
53. Li J, Yoshikawa A, Brennan MD, Ramsey TL, Meltzer HY. Genetic predictors of antipsychotic response to lurasidone identified in a genome wide association study and by schizophrenia risk genes. Schizophr Res. (2018) 192:194–204. doi: 10.1016/j.schres.2017.04.009
54. Sun J, Jayathilake K, Zhao Z, Meltzer HY. Investigating association of four gene regions (GABRB3, MAOB, PAH, and SLC6A4) with five symptoms in schizophrenia. Psychiatry Res. (2012) 198:202–6. doi: 10.1016/j.psychres.2011.12.035
55. Kuo PH, Chuang LC, Su MH, Chen CH, Chen CH, Wu JY, et al. Genome-wide association study for autism spectrum disorder in taiwanese han population. PLoS ONE (2015) 10:e0138695. doi: 10.1371/journal.pone.0138695
56. Perlis RH. A clinical risk stratification tool for predicting treatment resistance in major depressive disorder. Biol Psychiatry (2013) 74:7–14. doi: 10.1016/j.biopsych.2012.12.007
Keywords: antidepressant, deep learning, genome-wide association studies, major depressive disorder, multilayer feedforward neural networks, personalized medicine, single nucleotide polymorphisms
Citation: Lin E, Kuo P-H, Liu Y-L, Yu YW-Y, Yang AC and Tsai S-J (2018) A Deep Learning Approach for Predicting Antidepressant Response in Major Depression Using Clinical and Genetic Biomarkers. Front. Psychiatry 9:290. doi: 10.3389/fpsyt.2018.00290
Received: 26 February 2018; Accepted: 12 June 2018;
Published: 06 July 2018.
Edited by:
Chad A. Bousman, University of Calgary, CanadaReviewed by:
Eva E. Redei, Northwestern University, United StatesJin Li, Chinese Academy of Sciences, China
Yun-Ai Su, Peking University Institute of Mental Health, China
Copyright © 2018 Lin, Kuo, Liu, Yu, Yang and Tsai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eugene Lin, lines@uw.edu
Shih-Jen Tsai, tsai610913@gmail.com