 Mariana Fuertes1
Mariana Fuertes1 Julieta Tkatch2Josefina Rosmino2Leandro Nieto1
Julieta Tkatch2Josefina Rosmino2Leandro Nieto1 Mirtha Adriana Guitelman2
Mirtha Adriana Guitelman2 Eduardo Arzt1,3*
Eduardo Arzt1,3*
- 1Instituto de Investigación en Biomedicina de Buenos Aires (IBioBA) – CONICET – Partner Institute of the Max Planck Society, Buenos Aires, Argentina
- 2División Endocrinología, Hospital General de Agudos “Carlos G. Durand”, Buenos Aires, Argentina
- 3Departamento de Fisiología y Biología Molecular y Celular, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, Buenos Aires, Argentina
Cushing’s disease (CD) is an endocrine disorder originated by a corticotroph tumor. It is linked with high mortality and morbidity due to chronic hypercortisolism. Treatment goals are to control cortisol excess and achieve long-term remission, therefore, reducing both complications and patient’s mortality. First-line of treatment for CD is pituitary’s surgery. However, 30% of patients who undergo surgery experience recurrence in long-term follow-up. Persistent or recurrent CD demands second-line treatments, such as pituitary radiotherapy, adrenal surgery, and/or pharmacological therapy. The latter plays a key role in cortisol excess control. Its targets are inhibition of adrenocorticotropic hormone (ACTH) production, inhibition of adrenal steroidogenesis, or antagonism of cortisol action at its peripheral receptor. Retinoic acid (RA) is a metabolic product of vitamin A (retinol) and has been studied for its antiproliferative effects on corticotroph tumor cells. It has been shown that this drug regulates the expression of pro-opiomelanocortin (POMC), ACTH secretion, and tumor growth in corticotroph tumor mouse cell lines and in the nude mice experimental model, via inhibition of POMC transcription. It has been shown to result in tumor reduction, normalization of cortisol levels and clinical improvement in dogs treated with RA for 6 months. The orphan nuclear receptor COUP-TFI is expressed in normal corticotroph cells, but not in corticotroph tumoral cells, and inhibits RA pathways. A first clinical human study demonstrated clinical and biochemical effectiveness in 5/7 patients treated with RA for a period of up to 12 months. In a recent second clinical trial, 25% of 16 patients achieved eucortisolemia, and all achieved a cortisol reduction after 6- to 12-month treatment. The goal of this review is to discuss in the context of the available and future pharmacological treatments of CD, RA mechanisms of action on corticotroph tumor cells, and future perspectives, focusing on potential clinical implementation.
Introduction
Cushing’s disease (CD) is a severe endocrine disorder associated with increased morbidity and mortality, caused by an adrenocorticotropic hormone (ACTH) secreting pituitary adenoma (1). It is the most common form of endogenous Cushing’s syndrome (CS) (2), and it is the consequence of excessive secretion of cortisol from the adrenal glands. ACTH pituitary adenomas are microadenoma in more than 90% of cases (3).
The clinical phenotype involves features related to metabolic syndrome (diabetes mellitus, central obesity, hypertension, and dyslipidemia), muscle weakness, easy bruising, hirsutism, and cognitive disorders. The main cause of death is represented by cardiovascular disease (4–6).
First-line of treatment for CD is transsphenoidal surgery, aimed to remove the pituitary adenoma. However, success rates are variable (reported as 65–90%) depending on the surgeon’s expertise (5, 7). Pituitary radiotherapy and bilateral adrenalectomy are usually selected as second-line therapies when hypercortisolism persists; however, they are associated with long-term complications (6, 8).
Pharmacological treatment is recommended to control cortisol excess when surgery is contraindicated or non-curative, or while waiting for radiotherapy to come into effect (9, 10).
In this review, we will summarize the current state of art of pharmacological treatment of CD. We will focus on retinoic acid (RA), its molecular mechanism of action in different models of CD, its use, and future perspectives.
Pharmacological Therapy of CD
There are three pathophysiological mechanisms that are targeted by different treatments: central inhibition of ACTH secretion, adrenal-directed inhibition of steroidogenesis, and glucocorticoid receptor (GR) blockade (11) (Table 1).
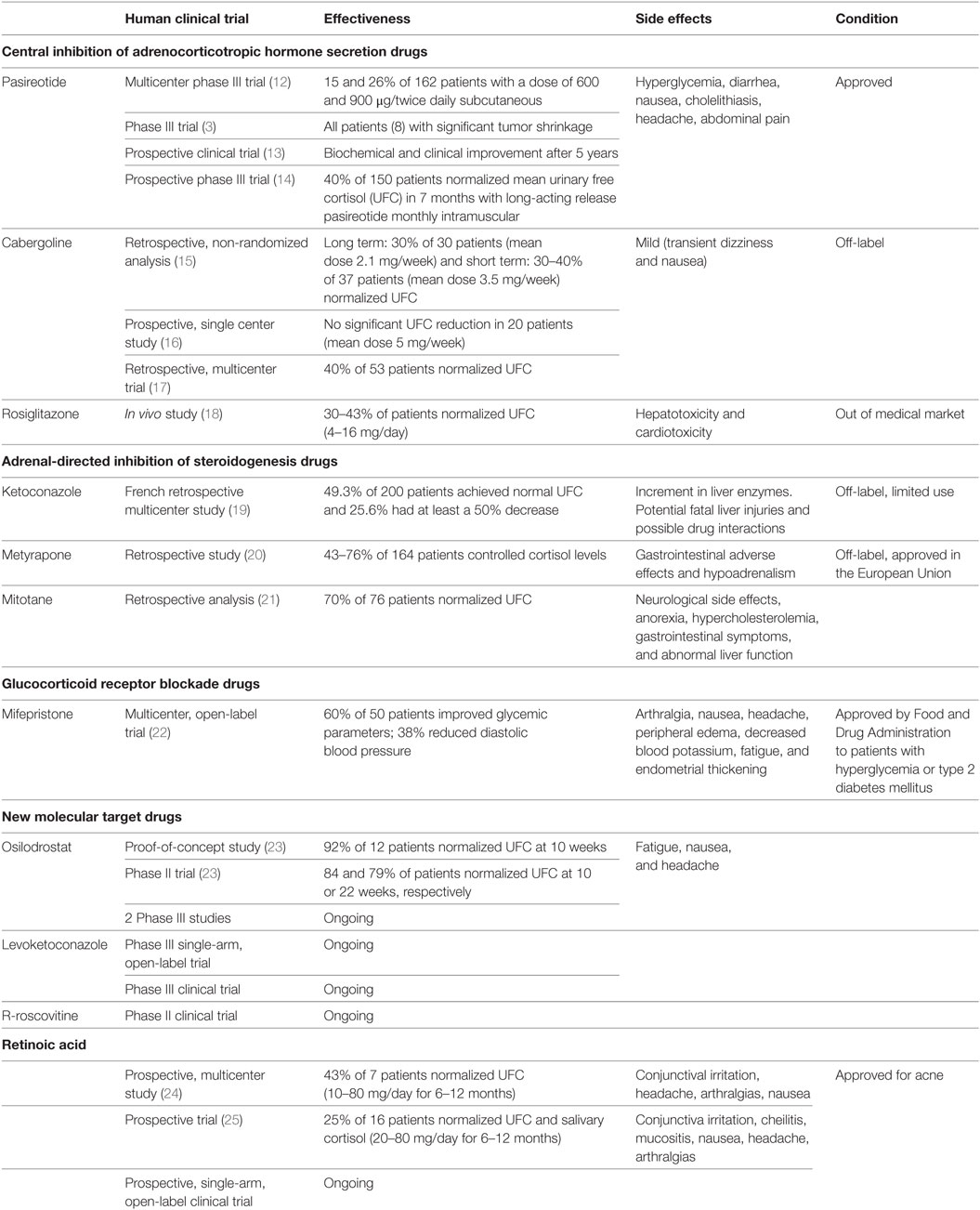
Table 1. Summary of pharmacological therapy and clinical trials of CD.
We will also discuss new molecular targets and other medical treatment perspectives. The availability of drugs with different mechanisms of action expand the therapeutic spectrum and allow drug combinations, as will be pointed, thus permitting cortisol levels normalization with the lowest rate of adverse effects.
Central Inhibition of ACTH Secretion
Corticotroph adenomas frequently express both dopamine receptors (DRs) and somatostatin transmembrane receptor (SSTR).
Somatostatin Receptor Ligands (SRLs)
Pasireotide (SOM230) is a novel somatostatin analog that binds to multiple SSTR with high affinity and has a 40-fold higher affinity for SSTR5 than octreotide. SSTR5 is the predominant SSTR expressed in corticotroph tumors (7, 26). Octreotide and lanreotide have high affinity for SSTR2 and moderate affinity for SSTR5. The expression of SSTR2 is reduced by hypercortisolism and SRLs acting on SSTR2 are not effective in CD patients (27, 28). SSTR5 expression may be less susceptible to the suppressive effect of cortisol (29–31).
Pasireotide is currently the only approved treatment for CD directed toward the pituitary gland. It was approved in 2012 in US and Europe for the treatment of adult CD patients who did not achieve remission after surgery or when it has been contraindicated (7, 28, 32). This drug has been shown to suppress both ACTH secretion and cell proliferation (11). A multicenter phase III study reported an effectiveness of this somatostatin analog in the subcutaneous formulation in 15 and 26% of a total of 162 patients with a dose of 600 and 900 µg/twice daily (12).
Hyperglycemia is the most common adverse effect occurring in 73% of the patients (12). Pasireotide-induced hyperglycemia is mediated by a reduction of insulin and incretin hormones secretion. Blood glucose levels must be monitored during pasireotide administration, mainly in patients with previous diabetes mellitus or impaired fasting blood glucose (12, 26). Diarrhea, nausea, cholelithiasis, headache, and abdominal pain are also common adverse events (31, 33). A subsequent phase III trial, on eight patients treated in a single center with pasireotide for 6 months, also indicated significant tumor shrinkage (3).
In a prospective, multicenter trial, pasireotide was given alone or in combination with cabergoline (CAB) with or without ketoconazole (KCN) (30). Additional medical therapy in combination with pasireotide may be of benefit for those patients in whom normalization of cortisol levels are not achieved (12, 30).
In a recent prospective clinical trial, a subset of patients with CD treated with pasireotide showed biochemical and clinical improvement after 5 years (13). This study suggests that pasireotide is an effective drug for the long-term treatment of patients with CD, especially when surgery fails or is contraindicated (13).
In the first prospective phase III trial of monthly intramuscular long-acting release pasireotide (LAR), pasireotide normalized mean urinary free cortisol (UFC) in about 40% of 150 patients with CD at month 7 and showed a similar safety profile than twice-daily subcutaneous pasireotide (14). LAR formulation and monthly administration is an efficient option in patients with CD persistent or recurrent, although has not yet been approved (14).
DR Agonists
Dopamine receptors are widely expressed in normal neuroendocrine tissues and pituitary adenomas, around 80% corticotroph tumors express dopamine receptor 2 (D2) (34). The DR family consists of five receptor subtypes that include D1-like (D1 and 4) and D2-like (D2, 3, and 4) receptors (6). In corticotroph adenomas, the suppressive action of the dopamine agonist CAB on ACTH secretion in CD patients has a positive correlation with the expression of D2 (34).
In a retrospective, non-randomized analysis of 30 CD patients treated for long term with CAB (mean dose 2.1 mg/week), nine patients (30%) showed a complete response after a mean of 37 months. Two patients did not respond after 2 and 5 years, respectively, but one patient transiently renormalized UFC after increase of CAB dosage. Four initial partial responders did not sustain a long-term response. Adverse effects were mild and included transient dizziness and nausea (15).
A response rate of approximately 30–40% at a high mean dose of 3.5 mg/week (0.5–7 mg/week) has been reported in CD patients (11, 15, 16, 35). Recent studies combining CAB and KCN showed that up to 79% of patients achieved normal levels of UFC (35). Unfortunately, since neither drug has been tested in controlled clinical trials, both are used as off-label therapy for the treatment of CD.
In a prospective single center study (16), 20 patients with CD were treated with CAB (median dose 5 mg/week) during a 6-week period. Prolactin levels were suppressed in all patients (demonstrating treatment compliance). Even though it had been previously suggested that higher doses of CAB improve hypercortisolism, no significant UFC reductions were observed in this trial, thus demonstrating a limited value of the use of CAB for the treatment of CD.
In a retrospective multicenter trial, 53 patients with CD received CAB as mono therapy and 9 patients received CAB complementary to other therapies. UFC normalization was observed in 21 of 53 patients who received CAB as monotherapy during a 12-month period (responding patients); 4 patients were considered partial responders and 28 patients did not modify or raise UFC levels. Among the responding patients, five developed corticoid insufficiency. CAB was withdrawn after 12 months in 28% of the responding patients, due to intolerance or escape. Hypercortisolism control was achieved in 23% of the overall patients, for 32.5 months. No significant differences were observed between baseline UFC levels, but the responding group received lower doses of CAB in comparison to non-responding patients (1.5 vs. 3.5 mg/week). Adequate response and tolerance to CAB was achieved in 20–25% of CD patients and basal UFC levels did not adequately predict the treatment response (17).
Peroxisome Proliferator-Activated Receptor (PPAR) Gamma Ligands
Peroxisome proliferator-activated receptor gamma ligands have shown antiproliferative and apoptotic activity in corticotropina murine models. In vivo studies using rosiglitazone from 4 to 16 mg/day in CD patients demonstrated UFC normalization between 30 and 43% of treated patients. Due to its hepatotoxicity and cardiotoxicity, this drug was taken off the medical market (18, 36, 37).
Adrenal-Directed Inhibition of Steroidogenesis
Adrenal steroidogenesis inhibitors are used as a cornerstone of medical treatment of CD (38). KCN, mitotane, etomidate, metyrapone, aminoglutethimide, and trilostane are drugs that inhibit adrenocortical steroidogenesis. Adrenal steroidogenesis inhibitors are generally used to control hypercortisolism in the short term. These drugs have the disadvantage of not acting directly on the pituitary tumor.
Ketoconazole
Ketoconazole, an antifungal substance, is a synthetic derivative of imidazole that diminishes adrenal steroidogenesis (400–1,200 mg). It is one of the most commonly used off-label drug in the treatment of CS (39). KCN blocks key cytochrome P450 (CYP) enzymes that participate in adrenal cortex steroidogenesis. In CS patients, it significantly reduces UFC with a 30–80% efficacy (28, 39).
In a French retrospective multicenter study, in 200 patients with active CD, KCN was administrated as monotherapy. In this study, 49.3% of patients achieved normal UFC, 25.6% had at least a 50% decrease, and in 25.4% UFC levels were unchanged. About half of the patients who received KCN before surgery (40 patients, median final dose of 600 mg/day) exhibited an improvement in hypokalemia, hypertension, and diabetes. Only 13.5 and 2.5% of patients shown mild and major increase in liver enzymes, respectively, that disappeared after drug withdrawal (19).
In 2013, the Food and Drug Administration (FDA) limited its use because of potential fatal liver injuries and possible drug interactions (40). However, the European Agency EMA had recommended a permission of a marketing authorization for KCN (HRA Pharma) in the treatment of CS.
Metyrapone
Metyrapone shows potent and selective inhibition of CYP11B1. Reductions in aldosterone and UFC were observed following metyrapone treatment. Gastrointestinal adverse effects and hypoadrenalism have been described. Metyrapone is employed clinically as an off label drug for CS treatment (20, 41).
In a retrospective study with 164 CS patients treated with metyrapone monotherapy, 43–76% of them managed to control cortisol levels. No escape was reported (20). In the European Union, it is approved for the treatment of CS (38).
Mitotane (OPDD)
Mitotane, a synthetic derivative of the pesticide dichlorodiphenyltrichloroethane (Lysodren®), is mainly used for its adrenolytic action in adrenocortical cancer. It has also a slow, but potent, inhibitory action on steroid biosynthesis when used at lower doses. Adrenal insufficiency (AI) is frequently observed, and replacement therapy with hydrocortisone is needed (11). After discontinuation of the treatment, around 70% of patients achieve normal UFC and about 10% may have sustained remission (11, 21).
A number of potential side effects have been associated with mitotane, including neurological side effects, anorexia, hypercholesterolemia, gastrointestinal symptoms, and abnormal liver function (38).
Etomidate
Etomidate blocks multiple steroidogenic enzymes including side-chain cleavage complex, aldosterone synthase, 17-hydroxylase, 11 beta-hydroxylase (CYP11B1), and 17–20 lyase enzymes (38).
Intravenous etomidate is often used in patients with severe hypercortisolism who cannot take oral medication (42). This drug is indicated in patients who are not immediate surgical candidates and for severe manifestations of CS such as hypertension, severe sepsis, or psychotic crises. Its use requires close monitoring at intensive care settings, for central nervous system depression. Close serum cortisol monitoring is indispensable to avoid AI. Etomidate use may be life-saving when all other treatments have failed (43).
Aminoglutethimide
Aminoglutethimide inhibits the conversion of cholesterol to pregnenolone (CYP11A1). Although cortisol decrease is gradual, some patients require replacement therapy with hydrocortisone. This drug has side effects that limit its use, including lethargy, sedation, dizziness, blurred vision, gastrointestinal discomfort, headache, myalgia, and skin rash (44). Aminoglutethimide is no longer used in patients with CD, due to the association with many unfavorable side effects (45).
Trilostane
Trilostane blocks steroidogenesis. This drug was effective for the treatment of CS in dogs. It did not show the same effectiveness in humans with CD (46). Trilostane is associated with major adverse effects including diarrhea, nausea, headache, asthenia, abdominal pain, paresthesias, and escape phenomenon. This drug is not used anymore for the treatment of CD, because of its limited efficacy, compared with other drugs (39).
GR Blockade
Glucocorticoid receptor blockade can be used in selected patients, but these drugs are associated with pharmacological interactions and adverse events related to their mechanism of action such as arthralgia, nausea, headache, peripheral edema, decreased blood potassium, endometrial thickening, and therefore require frequent monitoring. Moreover, they lack a reliable biomarker to monitor their efficacy; therefore, its use is limited.
Mifepristone
Mifepristone is a high-affinity non-selective GR antagonist that has greater affinity for the GR: 10 times compared with cortisol and 4 times compared with dexamethasone. It does not bind to the mineralocorticoid receptor (47). Its antagonism results in rapidly controlled systemic effects of cortisol excess in CS patients (48). Because the negative feedback is lost, leading to an increase in ACTH and subsequent cortisol levels, these parameters are not useful for monitoring the efficacy of the treatment or disease activity (47).
In a multicenter, open-label trial, mifepristone (at dose 300–1,200 mg daily) was given orally to 50 patients with CD associated with type 2 diabetes or hypertension, during a 6-month period. This drug showed an improvement of glycemic parameters (60% of patients) and diastolic blood pressure reduction (38% of patients) (ClinicalTrials.gov NCT00569582). The most common side effects are arthralgia, nausea, headache, peripheral edema, decreased blood potassium, fatigue, and endometrial thickening (22). The FDA approved mifepristone in 2012 and the specific indication is hyperglycemia or type 2 diabetes mellitus due to CS (22).
New Molecular Targets for CD in Clinical Trials
Osilodrostat (LCI 699)
Osilodrostat is an oral inhibitor of CYP11B1 that inhibits deoxycortisol hydroxylation to produce cortisol, the final step of its synthesis. The mechanism of action of this drug is similar to metyrapone (28).
In a 10-week, proof-of-concept study, this drug showed efficacy to normalize UFC in 11 of 12 patients with CD. It produces a rise in ACTH, 11-deoxycortisol, and 11-deoxycorticosterone that declined after discontinuation. Osilodrostat was generally well tolerated. Fatigue, nausea, and headache were the most frequent adverse events reported (23).
In a 22-week phase II trial in CD patients, 84 and 79% reached UFC normalization at 10 and 22 weeks, respectively (49). Plasma aldosterone and cortisol levels decreased in both studies, while an increase in the levels of their precursors was observed (23, 49).
Osilodrostat use as alternative treatment for CD is promising and two phase III studies are currently ongoing (50).
Levoketoconazole (COR-003)
Levoketoconazole is a drug in investigation for CS that acts similar to its enantiomer, KCN, but it is hypothesized to provide better efficacy and safety (50).
In vitro and in vivo studies showed that levoketoconazole might cause diarrhea, fatigue, dizziness, and abdominal pain. It might not be as damaging to the liver as KCN. Levoketoconazole could be used to treat endogenous CS of any cause in adult patients who are unwilling to have surgery, before surgery, or in whom surgery has failed, or while waiting for the effects of radiotherapy to occur. A phase III single-arm, open-label trial is currently ongoing to determine efficacy, safety, tolerability, and pharmacokinetics in CS patients (51). A phase III clinical trial to assess the safety and efficacy of levoketoconazole in the treatment of endogenous CS is being carried out (ClinicalTrials.gov Identifier: NCT03277690).
R-Roscovitine
R-roscovitine is a cyclin-dependent kinase (CDK) and cyclin E inhibitor that may be a potential therapeutic option for CD patients. R-roscovitine was first tested in a mouse model showing reduction of serum corticosterone and ACTH levels. R-roscovitine suppressed in vitro and in vivo ACTH expression, induced senescence and cell cycle exit in corticotroph tumor cells by overexpression of p27, p21, and p57, and downregulated cyclin E expression (52, 53). A phase II clinical trial in CD patients is ongoing.
Other Medical Prospective Treatments
Epidermal Growth Factor Receptor (EGFR)
Epidermal growth factor receptor family is considered a putative therapeutic target for CD. In murine studies, gefitinib (an inhibitor of EGFR tyrosine kinase activity) reduced tumor size and corticosterone levels, and reversed hypercortisolemia signs. EGFR signaling inhibition may be a novel strategy to treat CD, but further studies are needed (54, 55). A clinical trial is ongoing (ClinicalTrials.gov Identifier: NCT02484755).
Selective, Peptide Melanocortin 2 Receptor (MC2R) Antagonists
Adrenocorticotropic hormone acts on MC2R at the adrenal cortex and produces chronically increased circulating cortisol. A decrease in cortisol levels and adrenocortical androgen secretion (preserving mineralocorticoid hormone secretion) may be expected when the MC2R activation by ACTH is blocked. MC2R could therefore be a new target for the treatment of CD (56).
Chimeric Compounds
Chimeric compounds that bind to both D2R and SSTRs may act in synergy, with greater potency than each individual compound, to control ACTH release and tumor growth. SST/DA chimeras have the ability to interact with both receptors and present enhanced potency and efficacy than individual SSTR or DR agonists. Currently, second-generation chimeric compounds are under development (57). A recent study showed that the chimeric compound sst2/sst5/D2 (BIM-23A760) that acts through different molecular mechanisms, such as reduction of calcium concentration and inhibition of hormonal secretion on pituitary adenoma cells, represents a promising therapeutic tool (58).
Temozolomide Monotherapy and in Combination
Temozolomide is an oral alkylating chemotherapy agent, which is usually used for the treatment of high-grade astrocytoma, glioblastoma, and melanoma. This drug has shown promise as monotherapy and in combination with pasireotide, as a treatment for aggressive pituitary adenomas and carcinomas (7, 59).
Temozolomide causes deoxyribonucleic acid (DNA) damage because it depletes the DNA repair enzyme, O6-methylguanine-DNA-methyl transferase (MGMT), which results in tumor cell apoptosis. MGMT expression levels emerge as inversely related to therapeutic response to temozolomide. It has been used as a predictor of temozolomide effectiveness (60, 61). For aggressive corticotroph adenomas refractory to surgery, radiotherapy, or other medical treatment, temozolomide may represent an option (7).
Doxazosin
The selective alpha (1)-blocker doxazosin has shown inhibition of pituitary tumor cell proliferation in murine and human models and a decrease in plasma ACTH levels. There is not enough experience with this drug and further studies are needed (62).
MicroRNAs
MicroRNAs are small non-coding RNA molecules that regulate gene expression at the post-transcriptional level. MicroRNAs control different processes such us cell differentiation, cell growth, and apoptosis. Tumoral corticotroph cell proliferation may be limited through microRNAs genetic manipulation. It could therefore be a new treatment for CD, but future studies are necessary (63).
Monoclonal Antibodies
Bevacizumab, a monoclonal antibody, blocks the expression of vascular endothelial growth factor receptor 2. This receptor is overexpressed in aggressive pituitary tumors. Bevacizumab could be a novel target in the treatment of CD, decreasing cell proliferation in aggressive and fast-growing corticotroph tumors (64). Antiangiogenic therapy may be promising for the treatment of aggressive corticotroph tumors.
Rapamicin
The mTOR complex, a serine–threonine kinase, plays a key role in the cell growth, regulation, and proliferation. Rapamicin inhibits mTOR pathway and is a potential drug for the treatment of CD (65).
Testicular Orphan Nuclear Receptor 4 (TR4)
Overexpression of TR4 promotes corticotroph cell proliferation, ACTH secretion, and pro-opiomelanocortin (POMC) transcription. It could be another potential target to achieve control of hypercortisolism in patients with CD (66).
Retinoic Acid
Retinoic acid is a product of the metabolism of vitamin A/retinol, whose most used derivative is its cis isomer called Isotretinoin.
For many years, retinoids have been applied to therapeutically treat skin diseases, mainly acne. As a consequence of increasing understanding of their molecular action, their uses were diversified, involving their application in oncology and non-cancer-related applications (premalignant conditions and diseases such as chronic obstructive pulmonary, Alzheimer, and Parkinson) (67). Some groups have reported antitumorigenic effects for retinoids in several malignant diseases: epithelial tumors (68), cutaneous malignant diseases (69, 70), Kaposi’s sarcoma (71, 72), squamous cell carcinomas (73, 74), cutaneous T-cell lymphoma (75, 76), neuroblastoma (77), lung cancer (78), acute promyelocytic leukemia (79, 80), and others. In addition, immune stimulating effects of RA have been described (81).
There is evidence that retinoids participate in pituitary development (82) and adult pituitary gland has retinoid receptors, specific genes with retinoic acid response element (RARE) sequence and enzymes of the metabolism of retinoids (83–86).
Mechanism of Action of RA
The classical mechanism of RA action involves a nuclear hormone receptor-mediated activation of the transcriptional machinery on target genes, as well as activation of kinase cascades. This ability to activate multiple transcriptional and non-genomic programs gives RA enormous therapeutic potential.
Once in the target tissue, RA is delivered to its nuclear receptor through intracellular binding proteins such as cellular retinol-binding protein type II and fatty acid-binding protein 5 (87). RA interacts with two classes of receptors, RA receptor (RAR) (88) or retinoid X receptor (RXR) (89), each of which has three isoforms (α, β, and γ) (Figure 1). Both receptors belong to the steroid hormone/nuclear receptor superfamily along with estrogen, thyroid hormone, vitamin D, and orphan receptors (90). RARs and RXRs heterodimers act as RA ligands, but RXRs can heterodimerize with other nuclear receptors [vitamin D receptor, PPAR, liver X receptor, thyroid receptor, pregnane X receptor, RAR-related orphan receptor, farnesol X receptor (FXR), chicken ovoalbumin upstream promoter transcription factor (COUP-TF), nerve growth factor inducible B, Nur77-related receptor 1 (NURR1), nuclear orphan receptor 1, V-erbA-related protein 2, and constitutively active receptor (CAR)] (91) that act as rexinoid ligands. These nuclear receptors can activate transcription through the RA response elements RAREs in the target genes or assemble into co-repressor complexes for other gene pathways (92). The large number of possible complexes brings a very high degree of plasticity (93, 94). Moreover, RAR/RXRs contain several phosphorylation sites modified by kinases such as protein kinase A, mitogen- and stress-activated protein kinase 1, CDKs, and mitogen-activated protein kinases, which further makes the response more complex. An additional level of complexity is the fact that the magnitude and duration of the response is regulated through RARs and RXRs degradation by the proteasome.
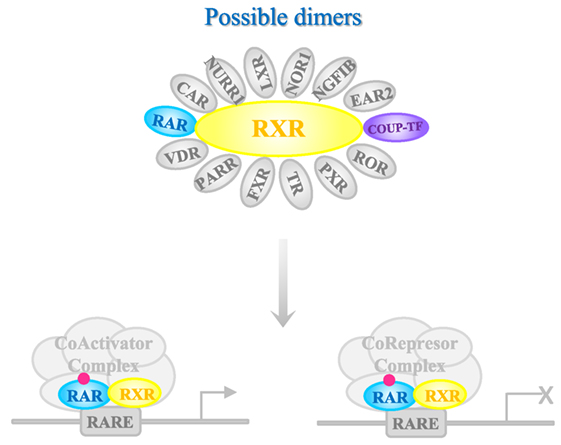
Figure 1. Mechanism of action of RA receptors (RARs). Retinoic acid (RA) nuclear receptor RARs and retinoid X receptors (RXRs) heterodimerize in the presence of the ligand that binds to RAR. The complex interacts with deoxyribonucleic acid containing specific RA responding element sequences [retinoic acid response element (RARE)], to mediate gene transcription. Chicken ovoalbumin upstream promoter transcription factor (COUP-TF) [as others nuclear receptors such as known ligand receptors vitamin D receptor (VDR), peroxisome proliferator-activated receptor (PPAR), liver X receptor (LXR), thyroid receptor, pregnane X receptor (PXR), RAR-related orphan receptor (ROR), and farnesol X receptor (FXR), or orphan receptors BGFIB, Nur77-related receptor 1 (NURR1), nuclear orphan receptor 1 (NOR-1), V-erbA-related protein 2 (EAR-2), and CAR] is able to dimerize with RXR. The complex with COUP-TF inhibits the effect of RA. Depending on the co-factors that are recognized by RAR-RXR complex, transcription of the target gene is activated or inhibited.
Chicken ovalbumin upstream promoter transcription factors belong to the superfamily of the steroid/thyroid hormone/vitamin receptors (95). COUP-TFs are supposed to have an important evolutionary and functional role given the high conservation of their amino acid sequence between species (96). COUP-TFs are orphan receptors because no endogenous ligands have been identified so far. COUP-TFI is more highly expressed in neuronal tissues of the central and peripheral nervous systems, whereas COUP-TFII is mainly present in developing organs (97–100).
Chicken ovoalbumin upstream promoter transcription factors are negative regulators of RA signaling (101–103). It has been reported that during development the signaling of COUP-TFs and retinoids have a functional crosstalk evidenced by the antagonistic action of COUP-TF on retinoid target genes and the fact that RA can modulate COUP-TFs expression (104, 105). In fact, COUP factors are considered as part of the RA signaling pathway (104–108). Besides, COUP-TFI, RARα, and RARβ knockout mice share common phenotypes (109–111).
RA in Corticotroph and Other Cellular Models
In AtT-20 pituitary ACTH-secreting tumor cells, an experimental cellular model of corticotroph adenomas, RA inhibits POMC transcription and stimulation of POMC by corticotropin-releasing hormone (CRH), being these effects highly specific because RA does not affect key factors of the corticotroph function such as cAMP response element-binding, GR, and paired-like homeodomain X 1 (112). In contrast to the action of RA showing POMC transcriptional inhibition, a synthetic agonist that acts only on RARα, Am80, has been described to increase POMC mRNA expression, CRH-induced ACTH secretion, and POMC promoter activity in AtT-20 corticotroph cells (113).
As part of its antitumorigenic action, RA inhibits ACTH and corticosterone-secreting cells proliferation. These antiproliferative effects involve transcription factors such as activator protein 1 and Nur77/Nurr1 (112). RA induces bone morphogenetic protein 4 (BMP-4), which participates in its antiproliferative effects (114) and has been also shown to be involved in the action of somatostatin analogs on corticotrophs (115). In GH3 and AtT-20 cells, RA-induced expression of BMP-4 involves chromatin remodeling (116). On the other hand, RA induces apoptosis/cell death in ACTH-secreting cells by inducing caspase-3 activity (112).
Furthermore, RA inhibits ACTH production in primary culture of human corticotrophins (112). In accordance, RA reduces ACTH endogenous production in tumor cells although it has no effect on non-tumoral pituitary cells (112) (Figure 2). In primary cultures from 10 human adrenal specimens, RA blunted ACTH receptor transcription (117).
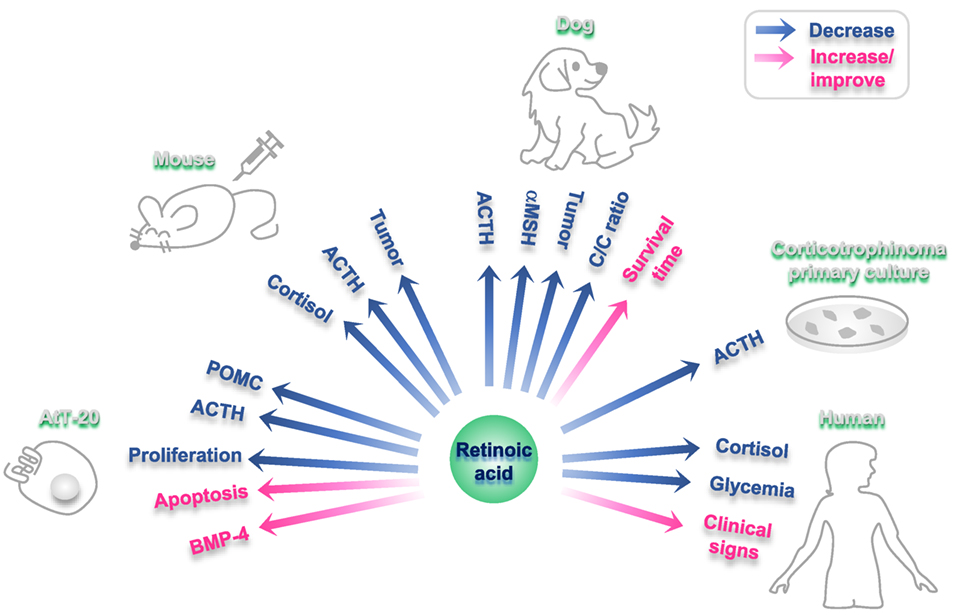
Figure 2. Effects of retinoic acid (RA) on different experimental models of Cushing disease. Blue arrows: hormones/factors/read-outs than decrease with RA administration; fuchsia arrows: hormones/factors/read-outs than increase/improve with RA administration.
RA in Rodent and Canine Models
In RA-treated AtT-20 xenographed nude mice, an in vivo model of corticotropina, RA reduces tumor mass and reverses endocrine alterations (112). RA also decreases plasma ACTH and cortisol, together with a reduction in adrenal hyperplasia and skin atrophy in these rodents (112).
Cushing’s disease is a common disorder in dogs (118). A dose of 2 mg/kg body weight/day of RA results effective and safe in this model (119). This treatment with RA reduces plasma ACTH and melanocyte-stimulating hormone alpha and diminishes the cortisol/creatinine urine ratio, leading to normalization of the gonadal axis and the estrous cycle (119). After a period of 180 days of treatment, RA reduced pituitary adenoma size, while survival time and clinical signs (food intake, skin appearance, and hair recovery) improved (119). RA action has not adverse events neither hepatotoxicity in dogs (119) (Figure 2).
Besides, in a poodle dog with Nelson’s syndrome with iatrogenic hypoadrenocorticism induced by mitotane, RA reduced plasma ACTH and macroadenoma size after 10 months of treatment (2 mg/kg/day) (120).
RA in Humans
In the first study in seven patients with CD, in five patients UFC levels decreased markedly after RA administration for 6–12 months (doses of 10–80 mg/day) and three of them reached normalization (24). The levels of plasma ACTH did not show a modification at the end of the treatment in responsive patients, while other signs showed an improvement (blood pressure, glycemia, and sings of hypercortisolism). Adverse events were mild and transient, including conjunctive irritation, headache, arthralgia, and nausea. Three patients complained of mild arthralgias, another three of xerophthalmia, two of abdominal pain, and one patient reported headaches. There were no reports of hepatic or renal dysfunction. One of the responding patient maintained normal UFC levels after isotretinoin withdrawal. This would be explained by a prolonged inhibitory effect of RA on corticotroph tumor cells (24).
In a recently prospective trial, isotretinoin was given orally for 6–12 months to 16 patients with persistent or recurrent CD in a dose ranging from 20 to 80 mg/day. Before drug administration and every month, clinical/biochemical data were registered. An initial response with normalization of the UFC and salivary cortisol was observed in six patients (37.5%). Subsequently, two patients escaped or relapsed. The group of responding patients had significantly lower mean age and lower mean basal levels of UFC, nocturnal salivary cortisol, and ACTH, in comparison with non-responders patients. Only patients who had UFC levels <2.5-fold ULN obtained UFC normalization. In a non-responding patient treated with a full-tolerated dose of CAB (3 mg/week), UFC levels were normalized after the addition of isotretinoin 50 mg/day. This combination allowed the reduction of CAB dose to 2 mg/day and hence the optimization of the tolerance profile. At the end of the study, four patients (25%) achieved the normalization of UFC levels. In the rest of the patients, UFC levels experienced up to 52.1% reduction. Isotretinoin side effects were experienced by seven patients (conjunctiva irritation, cheilitis, mucositis, nausea, headache, and arthralgias), though they were mostly transient. The authors of this study conclude that RA could be useful in patients with mild hypercortisolism (25).
The FDA approved RA for acne treatment in 1982. The usual dose for the treatment of acne vulgaris is 0.5–1 mg/kg/day. In patients with CD, in previous clinical trials, the initial dose was 20 mg/day and the maximum dose was 80 mg/day.
The described mild adverse effects for acne treatment include skin and annexes disorders, xerophthalmia, dry skin, conjunctiva irritation, dermatitis, pruritus, rash, hair fragility, and alopecia. Musculoskeletal system disorders: pain in muscle and joints, arthritis. Sensory disorders: photophobia, lenticular cataract, and keratitis. Gastrointestinal system disorders: nausea, intestinal inflammatory diseases such as colitis, ileitis, and hemorrhages. Laboratory findings: transaminases elevation (reversible and transient), increase in serum triglycerides and cholesterol, hyperuricemia. Although a causal relationship has not been established, high fasting blood sugar levels have been reported, and new cases of diabetes have been diagnosed during therapy. Serious adverse effects included psychiatric disorders, rash, metabolic disorders, increased intracranial pressure, renal, and abdominal disturbances. RA is contraindicated in patients with renal or hepatic insufficiency.
Retinoic acid is highly teratogenic; therefore, it is contraindicated not only in pregnant women but also in women of childbearing age. The latter group could only use RA in combination with two contraception methods (121).
In the two studies on patients with CD, RA has shown good response pattern in patients with mild hypercortisolism. It was generally well tolerated with transient and mild adverse effects (arthritis, xerophthalmia, nausea, and headache) (24, 25). This would represent an advantage, over other drugs such as pasireotide (worsening diabetes symptoms), KCN (hepatotoxicity), and mifepristone (arthralgia, peripheral edema, decreased blood potassium, fatigue, and endometrial thickening).
Future Perspectives
In primary culture of human corticotrophins, normal ACTH-secreting cells express COUP-TFI, but tumoral cells do not (112). Corticotroph normal cells do not respond to RA treatment and this may be attributed to the fact that they express the inhibitory factor COUP-TFI. In fact, when tumoral cells responding to RA are transfected with a COUP-TFI expression vector, the response to RA is abolished (112). In 34 specimens of human pituitary adenomas, 29 expressed COUP-TFI but only 5 of them presented expression of COUP (122). This interesting difference between normal/tumoral COUP-TFI expression makes this protein a promising biomarker of a possible response of the cells to RA action. In lung cancer, the antiproliferative action of RA depends on Nur77/COUP-TFI balance (123). Currently, there are no long-term studies involving RA. An ongoing controlled clinical trial in which both, the long-term efficacy of RA and the correlation with the expression of COUP-TFI are tested, would contribute to clarify the future perspective to implement RA as a potential drug for CD treatment.
Conclusion
Cushing’s disease presents high morbidity and mortality in the absence of proper treatment.
Although transsphenoidal surgery is the primary option, some patients are not candidates to surgery, or do not achieve remission after long-term control.
New knowledge about the mechanisms involved in the pathophysiology of CD led to the discovery of new targets for developing drugs that seek to reduce the secretion of ACTH. Targeting specific molecules, and searching biomarkers which predict response to different drugs, is the path to accomplish effective treatment. RA represents a new alternative to medical treatment in CD, but the true efficacy should be assessed in long-term controlled studies.
Author Contributions
MF, JR, MG, and EA conducted literature review and wrote the manuscript. EA and MG conceived the concept and idea of this article. JT and LN contributed to review, discussion, and conception of the work. MG and EA have critically read, revised, and edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The authors acknowledge the financial supports from Max Planck Society from Germany; University of Buenos Aires (UBA) from Argentina; the Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) from Argentina; the Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT) from Argentina; and Fondo para la Convergencia Estructural de Mercosur (FOCEM). JR is recipient of a fellowship for specializing in clinical research from ANPCyT.
Abbreviations
α-MSH, melanocyte-stimulating hormone alpha; ACTH, adrenocorticotropic hormone; AI, adrenal insufficiency; AP-1, activator protein 1; BMP-4, bone morphogenetic protein 4; CAB, carbergoline; CAR, constitutively active receptor; CD, Cushing’s disease; CDK, cyclin-dependent kinase; COR-003, levoketoconazole; COUP-TF, chicken ovoalbumin upstream promoter transcription factor; CRBPII, cellular retinol-binding protein type II; CREB, cAMP response element-binding; CRH, corticotropin-releasing hormone; CS, Cushing’s syndrome; CYP, cytochrome P450; CYP11B1, 11 beta-hydroxylase; D2, dopamine receptor 2; DNA, deoxyribonucleic acid; DR, dopamine receptor; EAR-2, V-erbA-related protein 2; FABP5, fatty acid-binding protein 5; FDA, Food and Drug Administration; FXR, farnesol X receptor; GR, glucocorticoid receptor; KCN, ketoconazole; LCI699, osilodrostat; LXR, liver X receptor; MAPK, mitogen-activated protein kinase; MC2R, melanocortin 2 receptor; MGMT, O6-methylguanine-DNA-methyl transferase; MSK1, mitogen- and stress-activated protein kinase 1; NGFIB, nerve growth factor inducible B; NOR1, nuclear orphan receptor 1; Nur77, nerve growth factor IB; NURR1, Nur77-related receptor 1; OPDD, mitotane; PKA, protein kinase A; POMC, pro-opiomelanocortin; PPAR, peroxisome proliferator-activated receptor; PTX1, paired-like homeodomain X 1; PXR, pregnane X receptor; RA, retinoic acid; RAR, retinoic acid receptor; RARE, retinoic acid response element; ROR, RAR-related orphan receptor; RXR, retinoid X receptor; SOM230, pasireotide; SRL, somatostatin receptor ligand; SSTR, somatostatin transmembrane receptor; TR, thyroid receptor; UFC, urinary free cortisol; VDR, vitamin D receptor.
References
1. Cannavo S, Messina E, Albani A, Ferrau F, Barresi V, Priola S, et al. Clinical management of critically ill patients with Cushing’s disease due to ACTH-secreting pituitary macroadenomas: effectiveness of presurgical treatment with pasireotide. Endocrine (2016) 52(3):481–7. doi:10.1007/s12020-015-0601-2
2. Lamos EM, Munir KM. Cushing disease: highlighting the importance of early diagnosis for both de novo and recurrent disease in light of evolving treatment patterns. Endocr Pract (2014) 20(9):945–55. doi:10.4158/EP14068.RA
3. Simeoli C, Auriemma RS, Tortora F, De Leo M, Iacuaniello D, Cozzolino A, et al. The treatment with pasireotide in Cushing’s disease: effects of long-term treatment on tumor mass in the experience of a single center. Endocrine (2015) 50(3):725–40. doi:10.1007/s12020-015-0557-2
4. Newell-Price J, Bertagna X, Grossman AB, Nieman LK. Cushing’s syndrome. Lancet (2006) 367(9522):1605–17. doi:10.1016/S0140-6736(06)68699-6
5. Steffensen C, Bak AM, Rubeck KZ, Jorgensen JO. Epidemiology of Cushing’s syndrome. Neuroendocrinology (2010) 92(Suppl 1):1–5. doi:10.1159/000314297
6. Feelders RA, Hofland LJ. Medical treatment of Cushing’s disease. J Clin Endocrinol Metab (2013) 98(2):425–38. doi:10.1210/jc.2012-3126
7. Fleseriu M, Petersenn S. New avenues in the medical treatment of Cushing’s disease: corticotroph tumor targeted therapy. J Neurooncol (2013) 114(1):1–11. doi:10.1007/s11060-013-1151-1
8. Becker G, Kocher M, Kortmann RD, Paulsen F, Jeremic B, Muller RP, et al. Radiation therapy in the multimodal treatment approach of pituitary adenoma. Strahlenther Onkol (2002) 178(4):173–86. doi:10.1007/s00066-002-0826-x
9. Tritos NA, Biller BM. Advances in medical therapies for Cushing’s syndrome. Discov Med (2012) 13(69):171–9.
10. van der Pas R, de Herder WW, Hofland LJ, Feelders RA. New developments in the medical treatment of Cushing’s syndrome. Endocr Relat Cancer (2012) 19(6):R205–23. doi:10.1530/ERC-12-0191
11. Langlois F, McCartney S, Fleseriu M. Recent progress in the medical therapy of pituitary tumors. Endocrinol Metab (Seoul) (2017) 32(2):162–70. doi:10.3803/EnM.2017.32.2.162
12. Colao A, Petersenn S, Newell-Price J, Findling JW, Gu F, Maldonado M, et al. A 12-month phase 3 study of pasireotide in Cushing’s disease. N Engl J Med (2012) 366(10):914–24. doi:10.1056/NEJMoa1105743
13. Petersenn S, Salgado LR, Schopohl J, Portocarrero-Ortiz L, Arnaldi G, Lacroix A, et al. Long-term treatment of Cushing’s disease with pasireotide: 5-year results from an open-label extension study of a phase III trial. Endocrine (2017) 57(1):156–65. doi:10.1007/s12020-017-1316-3
14. Lacroix A, Gu F, Gallardo W, Pivonello R, Yu Y, Witek P, et al. Efficacy and safety of once-monthly pasireotide in Cushing’s disease: a 12 month clinical trial. Lancet Diabetes Endocrinol (2018) 6(1):17–26. doi:10.1016/S2213-8587(17)30326-1
15. Godbout A, Manavela M, Danilowicz K, Beauregard H, Bruno OD, Lacroix A. Cabergoline monotherapy in the long-term treatment of Cushing’s disease. Eur J Endocrinol (2010) 163(5):709–16. doi:10.1530/EJE-10-0382
16. Burman P, Eden-Engstrom B, Ekman B, Karlsson FA, Schwarcz E, Wahlberg J. Limited value of cabergoline in Cushing’s disease: a prospective study of a 6-week treatment in 20 patients. Eur J Endocrinol (2016) 174(1):17–24. doi:10.1530/EJE-15-0807
17. Ferriere A, Cortet C, Chanson P, Delemer B, Caron P, Chabre O, et al. Cabergoline for Cushing’s disease: a large retrospective multicenter study. Eur J Endocrinol (2017) 176(3):305–14. doi:10.1530/EJE-16-0662
18. Pecori Giraldi F, Scaroni C, Arvat E, Martin M, Giordano R, Albiger N, et al. Effect of protracted treatment with rosiglitazone, a PPARgamma agonist, in patients with Cushing’s disease. Clin Endocrinol (Oxf) (2006) 64(2):219–24. doi:10.1111/j.1365-2265.2006.02452.x
19. Castinetti F, Guignat L, Giraud P, Muller M, Kamenicky P, Drui D, et al. Ketoconazole in Cushing’s disease: is it worth a try? J Clin Endocrinol Metab (2014) 99(5):1623–30. doi:10.1210/jc.2013-3628
20. Daniel E, Aylwin S, Mustafa O, Ball S, Munir A, Boelaert K, et al. Effectiveness of metyrapone in treating Cushing’s syndrome: a retrospective multicenter study in 195 patients. J Clin Endocrinol Metab (2015) 100(11):4146–54. doi:10.1210/jc.2015-2616
21. Baudry C, Coste J, Bou Khalil R, Silvera S, Guignat L, Guibourdenche J, et al. Efficiency and tolerance of mitotane in Cushing’s disease in 76 patients from a single center. Eur J Endocrinol (2012) 167(4):473–81. doi:10.1530/EJE-12-0358
22. Fleseriu M, Biller BM, Findling JW, Molitch ME, Schteingart DE, Gross C, et al. Mifepristone, a glucocorticoid receptor antagonist, produces clinical and metabolic benefits in patients with Cushing’s syndrome. J Clin Endocrinol Metab (2012) 97(6):2039–49. doi:10.1210/jc.2011-3350
23. Bertagna X, Pivonello R, Fleseriu M, Zhang Y, Robinson P, Taylor A, et al. LCI699, a potent 11beta-hydroxylase inhibitor, normalizes urinary cortisol in patients with Cushing’s disease: results from a multicenter, proof-of-concept study. J Clin Endocrinol Metab (2014) 99(4):1375–83. doi:10.1210/jc.2013-2117
24. Pecori Giraldi F, Ambrogio AG, Andrioli M, Sanguin F, Karamouzis I, Corsello SM, et al. Potential role for retinoic acid in patients with Cushing’s disease. J Clin Endocrinol Metab (2012) 97(10):3577–83. doi:10.1210/jc.2012-2328
25. Vilar L, Albuquerque JL, Lyra R, Trovao Diniz E, Rangel Filho F, Gadelha P, et al. The role of isotretinoin therapy for Cushing’s disease: results of a prospective study. Int J Endocrinol (2016) 2016:8173182. doi:10.1155/2016/8173182
26. Boscaro M, Ludlam WH, Atkinson B, Glusman JE, Petersenn S, Reincke M, et al. Treatment of pituitary-dependent Cushing’s disease with the multireceptor ligand somatostatin analog pasireotide (SOM230): a multicenter, phase II trial. J Clin Endocrinol Metab (2009) 94(1):115–22. doi:10.1210/jc.2008-1008
27. Stalla GK, Brockmeier SJ, Renner U, Newton C, Buchfelder M, Stalla J, et al. Octreotide exerts different effects in vivo and in vitro in Cushing’s disease. Eur J Endocrinol (1994) 130(2):125–31. doi:10.1530/eje.0.1300125
28. Cuevas-Ramos D, Lim DST, Fleseriu M. Update on medical treatment for Cushing’s disease. Clin Diabetes Endocrinol (2016) 2:16. doi:10.1186/s40842-016-0033-9
29. de Bruin C, Feelders RA, Waaijers AM, van Koetsveld PM, Sprij-Mooij DM, Lamberts SW, et al. Differential regulation of human dopamine D2 and somatostatin receptor subtype expression by glucocorticoids in vitro. J Mol Endocrinol (2009) 42(1):47–56. doi:10.1677/JME-08-0110
30. Feelders RA, de Bruin C, Pereira AM, Romijn JA, Netea-Maier RT, Hermus AR, et al. Pasireotide alone or with cabergoline and ketoconazole in Cushing’s disease. N Engl J Med (2010) 362(19):1846–8. doi:10.1056/NEJMc1000094
31. Ceccato F, Scaroni C, Boscaro M. Clinical use of pasireotide for Cushing’s disease in adults. Ther Clin Risk Manag (2015) 11:425–34. doi:10.2147/TCRM.S37314
32. Fleseriu M, Petersenn S. Medical management of Cushing’s disease: what is the future? Pituitary (2012) 15(3):330–41. doi:10.1007/s11102-012-0397-5
33. Colao A, De Block C, Gaztambide MS, Kumar S, Seufert J, Casanueva FF. Managing hyperglycemia in patients with Cushing’s disease treated with pasireotide: medical expert recommendations. Pituitary (2014) 17(2):180–6. doi:10.1007/s11102-013-0483-3
34. Pivonello R, Ferone D, de Herder WW, Kros JM, De Caro ML, Arvigo M, et al. Dopamine receptor expression and function in corticotroph pituitary tumors. J Clin Endocrinol Metab (2004) 89(5):2452–62. doi:10.1210/jc.2003-031746
35. Barbot M, Albiger N, Ceccato F, Zilio M, Frigo AC, Denaro L, et al. Combination therapy for Cushing’s disease: effectiveness of two schedules of treatment: should we start with cabergoline or ketoconazole? Pituitary (2014) 17(2):109–17. doi:10.1007/s11102-013-0475-3
36. Heaney AP, Fernando M, Yong WH, Melmed S. Functional PPAR-gamma receptor is a novel therapeutic target for ACTH-secreting pituitary adenomas. Nat Med (2002) 8(11):1281–7. doi:10.1038/nm784
37. Stone JC, Furuya-Kanamori L, Barendregt JJ, Doi SA. Was there really any evidence that rosiglitazone increased the risk of myocardial infarction or death from cardiovascular causes? Pharmacoepidemiol Drug Saf (2015) 24(3):223–7. doi:10.1002/pds.3736
38. Daniel E, Newell-Price JD. Therapy of endocrine disease: steroidogenesis enzyme inhibitors in Cushing’s syndrome. Eur J Endocrinol (2015) 172(6):R263–80. doi:10.1530/EJE-14-1014
39. Pivonello R, De Leo M, Cozzolino A, Colao A. The treatment of Cushing’s disease. Endocr Rev (2015) 36(4):385–486. doi:10.1210/er.2013-1048
40. Fleseriu M, Castinetti F. Updates on the role of adrenal steroidogenesis inhibitors in Cushing’s syndrome: a focus on novel therapies. Pituitary (2016) 19(6):643–53. doi:10.1007/s11102-016-0742-1
41. Rigel DF, Fu F, Beil M, Hu CW, Liang G, Jeng AY. Pharmacodynamic and pharmacokinetic characterization of the aldosterone synthase inhibitor FAD286 in two rodent models of hyperaldosteronism: comparison with the 11beta-hydroxylase inhibitor metyrapone. J Pharmacol Exp Ther (2010) 334(1):232–43. doi:10.1124/jpet.110.167148
42. Nieman LK, Biller BM, Findling JW, Murad MH, Newell-Price J, Savage MO, et al. Treatment of Cushing’s syndrome: an endocrine society clinical practice guideline. J Clin Endocrinol Metab (2015) 100(8):2807–31. doi:10.1210/jc.2015-1818
43. Preda VA, Sen J, Karavitaki N, Grossman AB. Etomidate in the management of hypercortisolaemia in Cushing’s syndrome: a review. Eur J Endocrinol (2012) 167(2):137–43. doi:10.1530/EJE-12-0274
44. Bertagna X, Guignat L, Groussin L, Bertherat J. Cushing’s disease. Best Pract Res Clin Endocrinol Metab (2009) 23(5):607–23. doi:10.1016/j.beem.2009.06.001
45. Feelders RA, Hofland LJ, de Herder WW. Medical treatment of Cushing’s syndrome: adrenal-blocking drugs and ketaconazole. Neuroendocrinology (2010) 92(Suppl 1):111–5. doi:10.1159/000314292
46. Igaz P, Tombol Z, Szabo PM, Liko I, Racz K. Steroid biosynthesis inhibitors in the therapy of hypercortisolism: theory and practice. Curr Med Chem (2008) 15(26):2734–47. doi:10.2174/092986708786242921
47. Bourgeois S, Pfahl M, Baulieu EE. DNA binding properties of glucocorticosteroid receptors bound to the steroid antagonist RU-486. EMBO J (1984) 3(4):751–5.
48. Johanssen S, Allolio B. Mifepristone (RU 486) in Cushing’s syndrome. Eur J Endocrinol (2007) 157(5):561–9. doi:10.1530/EJE-07-0458
49. Fleseriu M, Pivonello R, Young J, Hamrahian AH, Molitch ME, Shimizu C, et al. Osilodrostat, a potent oral 11beta-hydroxylase inhibitor: 22-week, prospective, phase II study in Cushing’s disease. Pituitary (2016) 19(2):138–48. doi:10.1007/s11102-015-0692-z
50. Guelho D, Grossman AB. Emerging drugs for Cushing’s disease. Expert Opin Emerg Drugs (2015) 20(3):463–78. doi:10.1517/14728214.2015.1047762
51. Salvatori R, DelConte A, Geer EB, Koziol T, Jorkasky D. An open-label study to assess the safety and efficacy of levoketoconazole (COR-003) in the treatment of endogenous Cushing’s syndrome. Endocrine Society’s 97th Annual Meeting and Expo. San Diego (2015).
52. Liu NA, Jiang H, Ben-Shlomo A, Wawrowsky K, Fan XM, Lin S, et al. Targeting zebrafish and murine pituitary corticotroph tumors with a cyclin-dependent kinase (CDK) inhibitor. Proc Natl Acad Sci U S A (2011) 108(20):8414–9. doi:10.1073/pnas.1018091108
53. Liu NA, Araki T, Cuevas-Ramos D, Hong J, Ben-Shlomo A, Tone Y, et al. Cyclin E-mediated human proopiomelanocortin regulation as a therapeutic target for Cushing disease. J Clin Endocrinol Metab (2015) 100(7):2557–64. doi:10.1210/jc.2015-1606
54. Fukuoka H, Cooper O, Ben-Shlomo A, Mamelak A, Ren SG, Bruyette D, et al. EGFR as a therapeutic target for human, canine, and mouse ACTH-secreting pituitary adenomas. J Clin Invest (2011) 121(12):4712–21. doi:10.1172/JCI60417
55. Ben-Shlomo A, Cooper O. Role of tyrosine kinase inhibitors in the treatment of pituitary tumours: from bench to bedside. Curr Opin Endocrinol Diabetes Obes (2017) 24(4):301–5. doi:10.1097/MED.0000000000000344
56. Halem HA, Ufret M, Jewett I, Mattei A, Bastille A, Beech J, et al. In vivo suppression of corticosterone in rodent models of Cushing’s disease with a selective, peptide MC2 receptor antagonist. Endocrine Society’s 98th Annual Meeting and Expo. Boston (2016).
57. Culler MD. Somatostatin-dopamine chimeras: a novel approach to treatment of neuroendocrine tumors. Horm Metab Res (2011) 43(12):854–7. doi:10.1055/s-0031-1287769
58. Ibanez-Costa A, Lopez-Sanchez LM, Gahete MD, Rivero-Cortes E, Vazquez-Borrego MC, Galvez MA, et al. BIM-23A760 influences key functional endpoints in pituitary adenomas and normal pituitaries: molecular mechanisms underlying the differential response in adenomas. Sci Rep (2017) 7:42002. doi:10.1038/srep42002
59. Bode H, Seiz M, Lammert A, Brockmann MA, Back W, Hammes HP, et al. SOM230 (pasireotide) and temozolomide achieve sustained control of tumour progression and ACTH secretion in pituitary carcinoma with widespread metastases. Exp Clin Endocrinol Diabetes (2010) 118(10):760–3. doi:10.1055/s-0030-1253419
60. Dillard TH, Gultekin SH, Delashaw JB Jr, Yedinak CG, Neuwelt EA, Fleseriu M. Temozolomide for corticotroph pituitary adenomas refractory to standard therapy. Pituitary (2011) 14(1):80–91. doi:10.1007/s11102-010-0264-1
61. Liu JK, Patel J, Eloy JA. The role of temozolomide in the treatment of aggressive pituitary tumors. J Clin Neurosci (2015) 22(6):923–9. doi:10.1016/j.jocn.2014.12.007
62. Fernando MA, Heaney AP. Alpha1-adrenergic receptor antagonists: novel therapy for pituitary adenomas. Mol Endocrinol (2005) 19(12):3085–96. doi:10.1210/me.2004-0471
63. Qian ZR, Asa SL, Siomi H, Siomi MC, Yoshimoto K, Yamada S, et al. Overexpression of HMGA2 relates to reduction of the let-7 and its relationship to clinicopathological features in pituitary adenomas. Mod Pathol (2009) 22(3):431–41. doi:10.1038/modpathol.2008.202
64. Ortiz LD, Syro LV, Scheithauer BW, Ersen A, Uribe H, Fadul CE, et al. Anti-VEGF therapy in pituitary carcinoma. Pituitary (2012) 15(3):445–9. doi:10.1007/s11102-011-0346-8
65. Ferone D, Pivonello C, Vitale G, Zatelli MC, Colao A, Pivonello R. Molecular basis of pharmacological therapy in Cushing’s disease. Endocrine (2014) 46(2):181–98. doi:10.1007/s12020-013-0098-5
66. Du L, Bergsneider M, Mirsadraei L, Young SH, Jonker JW, Downes M, et al. Evidence for orphan nuclear receptor TR4 in the etiology of Cushing disease. Proc Natl Acad Sci U S A (2013) 110(21):8555–60. doi:10.1073/pnas.1306182110
67. Labeur M, Paez-Pereda M, Arzt E, Stalla GK. Potential of retinoic acid derivatives for the treatment of corticotroph pituitary adenomas. Rev Endocr Metab Disord (2009) 10(2):103–9. doi:10.1007/s11154-008-9080-6
68. Hyams MN, Gallaher PD. Vitamin A therapy in the treatment of vulvar leucoplakia. Am J Obstet Gynecol (1950) 59(6):1346–54. doi:10.1016/0002-9378(50)90302-4
69. Zhang C, Duvic M. Retinoids: therapeutic applications and mechanisms of action in cutaneous T-cell lymphoma. Dermatol Ther (2003) 16(4):322–30. doi:10.1111/j.1396-0296.2003.01644.x
70. Weinstock MA, Bingham SF, Digiovanna JJ, Rizzo AE, Marcolivio K, Hall R, et al. Tretinoin and the prevention of keratinocyte carcinoma (basal and squamous cell carcinoma of the skin): a veterans affairs randomized chemoprevention trial. J Invest Dermatol (2012) 132(6):1583–90. doi:10.1038/jid.2011.483
71. Bodsworth NJ, Bloch M, Bower M, Donnell D, Yocum R, International Panretin Gel KS Study Group. Phase III vehicle-controlled, multi-centered study of topical alitretinoin gel 0.1% in cutaneous AIDS-related Kaposi’s sarcoma. Am J Clin Dermatol (2001) 2(2):77–87. doi:10.2165/00128071-200102020-00004
72. Miles SA, Dezube BJ, Lee JY, Krown SE, Fletcher MA, Saville MW, et al. Antitumor activity of oral 9-cis-retinoic acid in HIV-associated Kaposi’s sarcoma. AIDS (2002) 16(3):421–9. doi:10.1097/00002030-200202150-00014
73. Recchia F, Lalli A, Lombardo M, De Filippis S, Saggio G, Fabbri F, et al. Ifosfamide, cisplatin, and 13-cis retinoic acid for patients with advanced or recurrent squamous cell carcinoma of the head and neck: a phase I-II study. Cancer (2001) 92(4):814–21. doi:10.1002/1097-0142(20010815)92:4<814::AID-CNCR1387>3.0.CO;2-8
74. Atzpodien J, Kirchner H, Jonas U, Bergmann L, Schott H, Heynemann H, et al. Interleukin-2- and interferon alfa-2a-based immunochemotherapy in advanced renal cell carcinoma: a prospectively randomized trial of the German Cooperative Renal Carcinoma Chemoimmunotherapy group (DGCIN). J Clin Oncol (2004) 22(7):1188–94. doi:10.1200/JCO.2004.06.155
75. Straus DJ, Duvic M, Kuzel T, Horwitz S, Demierre MF, Myskowski P, et al. Results of a phase II trial of oral bexarotene (Targretin) combined with interferon alfa-2b (Intron-A) for patients with cutaneous T-cell lymphoma. Cancer (2007) 109(9):1799–803. doi:10.1002/cncr.22596
76. Quereux G, Saint-Jean M, Peuvrel L, Brocard A, Knol AC, Dreno B. Bexarotene in cutaneous T-cell lymphoma: third retrospective study of long-term cohort and review of the literature. Expert Opin Pharmacother (2013) 14(13):1711–21. doi:10.1517/14656566.2013.810718
77. Matthay KK, Reynolds CP, Seeger RC, Shimada H, Adkins ES, Haas-Kogan D, et al. Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: a Children’s Oncology Group study. J Clin Oncol (2009) 27(7):1007–13. doi:10.1200/JCO.2007.13.8925
78. Arrieta O, Gonzalez-De la Rosa CH, Arechaga-Ocampo E, Villanueva-Rodriguez G, Ceron-Lizarraga TL, Martinez-Barrera L, et al. Randomized phase II trial of all-trans-retinoic acid with chemotherapy based on paclitaxel and cisplatin as first-line treatment in patients with advanced non-small-cell lung cancer. J Clin Oncol (2010) 28(21):3463–71. doi:10.1200/JCO.2009.26.6452
79. Douer D, Zickl LN, Schiffer CA, Appelbaum FR, Feusner JH, Shepherd L, et al. All-trans retinoic acid and late relapses in acute promyelocytic leukemia: very long-term follow-up of the North American Intergroup study I0129. Leuk Res (2013) 37(7):795–801. doi:10.1016/j.leukres.2013.03.001
80. Lo-Coco F, Avvisati G, Vignetti M, Thiede C, Orlando SM, Iacobelli S, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med (2013) 369(2):111–21. doi:10.1056/NEJMoa1300874
82. Rhodes SJ, Chen R, DiMattia GE, Scully KM, Kalla KA, Lin SC, et al. A tissue-specific enhancer confers Pit-1-dependent morphogen inducibility and autoregulation on the pit-1 gene. Genes Dev (1993) 7(6):913–32. doi:10.1101/gad.7.6.913
83. Sanno N, Sugawara A, Teramoto A, Abe Y, Yen PM, Chin WW, et al. Immunohistochemical expression of retinoid X receptor isoforms in human pituitaries and pituitary adenomas. Neuroendocrinology (1997) 65(4):299–306. doi:10.1159/000127188
84. Drouin J, Maira M, Philips A. Novel mechanism of action for Nur77 and antagonism by glucocorticoids: a convergent mechanism for CRH activation and glucocorticoid repression of POMC gene transcription. J Steroid Biochem Mol Biol (1998) 65(1–6):59–63. doi:10.1016/S0960-0760(97)00180-5
85. Brown NS, Smart A, Sharma V, Brinkmeier ML, Greenlee L, Camper SA, et al. Thyroid hormone resistance and increased metabolic rate in the RXR-gamma-deficient mouse. J Clin Invest (2000) 106(1):73–9. doi:10.1172/JCI9422
86. Brossaud J, Pallet V, Corcuff JB. Vitamin A, endocrine tissues and hormones: interplay and interactions. Endocr Connect (2017) 6(7):R121–30. doi:10.1530/EC-17-0101
87. Napoli JL. Cellular retinoid binding-proteins, CRBP, CRABP, FABP5: effects on retinoid metabolism, function and related diseases. Pharmacol Ther (2017) 173:19–33. doi:10.1016/j.pharmthera.2017.01.004
88. Germain P, Chambon P, Eichele G, Evans RM, Lazar MA, Leid M, et al. International union of pharmacology. LX. Retinoic acid receptors. Pharmacol Rev (2006) 58(4):712–25. doi:10.1124/pr.58.4.7
89. Heyman RA, Mangelsdorf DJ, Dyck JA, Stein RB, Eichele G, Evans RM, et al. 9-cis retinoic acid is a high affinity ligand for the retinoid X receptor. Cell (1992) 68(2):397–406. doi:10.1016/0092-8674(92)90479-V
90. Whitfield GK, Jurutka PW, Haussler CA, Haussler MR. Steroid hormone receptors: evolution, ligands, and molecular basis of biologic function. J Cell Biochem Suppl (1999) 32–33:110–22. doi:10.1002/(SICI)1097-4644(1999)75:32+<110::AID-JCB14>3.0.CO;2-T
91. Williams GR, Franklyn JA. Physiology of the steroid-thyroid hormone nuclear receptor superfamily. Baillieres Clin Endocrinol Metab (1994) 8(2):241–66. doi:10.1016/S0950-351X(05)80251-4
92. Wei LN. Cellular retinoic acid binding proteins: genomic and non-genomic functions and their regulation. Subcell Biochem (2016) 81:163–78. doi:10.1007/978-94-024-0945-1_6
93. Liu Z, Hu Q, Rosenfeld MG. Complexity of the RAR-mediated transcriptional regulatory programs. Subcell Biochem (2014) 70:203–25. doi:10.1007/978-94-017-9050-5_10
94. Piskunov A, Al Tanoury Z, Rochette-Egly C. Nuclear and extra-nuclear effects of retinoid acid receptors: how they are interconnected. Subcell Biochem (2014) 70:103–27. doi:10.1007/978-94-017-9050-5_6
95. Wang LH, Tsai SY, Cook RG, Beattie WG, Tsai MJ, O’Malley BW. COUP transcription factor is a member of the steroid receptor superfamily. Nature (1989) 340(6229):163–6. doi:10.1038/340163a0
96. Tsai SY, Tsai MJ. Chick ovalbumin upstream promoter-transcription factors (COUP-TFs): coming of age. Endocr Rev (1997) 18(2):229–40. doi:10.1210/er.18.2.229
97. Qiu Y, Cooney AJ, Kuratani S, DeMayo FJ, Tsai SY, Tsai MJ. Spatiotemporal expression patterns of chicken ovalbumin upstream promoter-transcription factors in the developing mouse central nervous system: evidence for a role in segmental patterning of the diencephalon. Proc Natl Acad Sci U S A (1994) 91(10):4451–5. doi:10.1073/pnas.91.10.4451
98. Pereira FA, Qiu Y, Tsai MJ, Tsai SY. Chicken ovalbumin upstream promoter transcription factor (COUP-TF): expression during mouse embryogenesis. J Steroid Biochem Mol Biol (1995) 53(1–6):503–8. doi:10.1016/0960-0760(95)00097-J
99. Qin J, Suh JM, Kim BJ, Yu CT, Tanaka T, Kodama T, et al. The expression pattern of nuclear receptors during cerebellar development. Dev Dyn (2007) 236(3):810–20. doi:10.1002/dvdy.21060
100. Ip BK, Wappler I, Peters H, Lindsay S, Clowry GJ, Bayatti N. Investigating gradients of gene expression involved in early human cortical development. J Anat (2010) 217(4):300–11. doi:10.1111/j.1469-7580.2010.01259.x
101. Kliewer SA, Umesono K, Heyman RA, Mangelsdorf DJ, Dyck JA, Evans RM. Retinoid X receptor-COUP-TF interactions modulate retinoic acid signaling. Proc Natl Acad Sci U S A (1992) 89(4):1448–52. doi:10.1073/pnas.89.4.1448
102. Tran P, Zhang XK, Salbert G, Hermann T, Lehmann JM, Pfahl M. COUP orphan receptors are negative regulators of retinoic acid response pathways. Mol Cell Biol (1992) 12(10):4666–76. doi:10.1128/MCB.12.10.4666
103. Park JI, Tsai SY, Tsai MJ. Molecular mechanism of chicken ovalbumin upstream promoter-transcription factor (COUP-TF) actions. Keio J Med (2003) 52(3):174–81. doi:10.2302/kjm.52.174
104. Fjose A, Weber U, Mlodzik M. A novel vertebrate svp-related nuclear receptor is expressed as a step gradient in developing rhombomeres and is affected by retinoic acid. Mech Dev (1995) 52(2–3):233–46. doi:10.1016/0925-4773(95)00404-O
105. Brubaker K, McMillan M, Neuman T, Nornes HO. All-trans retinoic acid affects the expression of orphan receptors COUP-TF I and COUP-TF II in the developing neural tube. Brain Res Dev Brain Res (1996) 93(1–2):198–202. doi:10.1016/0165-3806(96)00007-7
106. Jonk LJ, de Jonge ME, Pals CE, Wissink S, Vervaart JM, Schoorlemmer J, et al. Cloning and expression during development of three murine members of the COUP family of nuclear orphan receptors. Mech Dev (1994) 47(1):81–97. doi:10.1016/0925-4773(94)90098-1
107. van der Wees J, Matharu PJ, de Roos K, Destree OH, Godsave SF, Durston AJ, et al. Developmental expression and differential regulation by retinoic acid of Xenopus COUP-TF-A and COUP-TF-B. Mech Dev (1996) 54(2):173–84. doi:10.1016/0925-4773(95)00471-8
108. Zhuang Y, Gudas LJ. Overexpression of COUP-TF1 in murine embryonic stem cells reduces retinoic acid-associated growth arrest and increases extraembryonic endoderm gene expression. Differentiation (2008) 76(7):760–71. doi:10.1111/j.1432-0436.2007.00258.x
109. Luo J, Sucov HM, Bader JA, Evans RM, Giguere V. Compound mutants for retinoic acid receptor (RAR) beta and RAR alpha 1 reveal developmental functions for multiple RAR beta isoforms. Mech Dev (1996) 55(1):33–44. doi:10.1016/0925-4773(95)00488-2
110. Qiu Y, Pereira FA, DeMayo FJ, Lydon JP, Tsai SY, Tsai MJ. Null mutation of mCOUP-TFI results in defects in morphogenesis of the glossopharyngeal ganglion, axonal projection, and arborization. Genes Dev (1997) 11(15):1925–37. doi:10.1101/gad.11.15.1925
111. Alfano C, Magrinelli E, Harb K, Studer M. The nuclear receptors COUP-TF: a long-lasting experience in forebrain assembly. Cell Mol Life Sci (2014) 71(1):43–62. doi:10.1007/s00018-013-1320-6
112. Paez-Pereda M, Kovalovsky D, Hopfner U, Theodoropoulou M, Pagotto U, Uhl E, et al. Retinoic acid prevents experimental Cushing syndrome. J Clin Invest (2001) 108(8):1123–31. doi:10.1172/JCI11098
113. Uruno A, Saito-Hakoda A, Yokoyama A, Kogure N, Matsuda K, Parvin R, et al. Retinoic acid receptor-alpha up-regulates proopiomelanocortin gene expression in AtT20 corticotroph cells. Endocr J (2014) 61(11):1105–14. doi:10.1507/endocrj.EJ14-0115
114. Giacomini D, Paez-Pereda M, Theodoropoulou M, Labeur M, Refojo D, Gerez J, et al. Bone morphogenetic protein-4 inhibits corticotroph tumor cells: involvement in the retinoic acid inhibitory action. Endocrinology (2006) 147(1):247–56. doi:10.1210/en.2005-0958
115. Tsukamoto N, Otsuka F, Miyoshi T, Yamanaka R, Inagaki K, Yamashita M, et al. Effects of bone morphogenetic protein (BMP) on adrenocorticotropin production by pituitary corticotrope cells: involvement of up-regulation of BMP receptor signaling by somatostatin analogs. Endocrinology (2010) 151(3):1129–41. doi:10.1210/en.2009-1102
116. Yacqub-Usman K, Duong CV, Clayton RN, Farrell WE. Preincubation of pituitary tumor cells with the epidrugs zebularine and trichostatin A are permissive for retinoic acid-augmented expression of the BMP-4 and D2R genes. Endocrinology (2013) 154(5):1711–21. doi:10.1210/en.2013-1061
117. Sesta A, Cassarino MF, Tapella L, Castelli L, Cavagnini F, Pecori Giraldi F. Effect of retinoic acid on human adrenal corticosteroid synthesis. Life Sci (2016) 151:277–80. doi:10.1016/j.lfs.2016.03.023
118. Ling GV, Stabenfeldt GH, Comer KM, Gribble DH, Schechter RD. Canine hyperadrenocorticism: pretreatment clinical and laboratory evaluation of 117 cases. J Am Vet Med Assoc (1979) 174(11):1211–5.
119. Castillo V, Giacomini D, Paez-Pereda M, Stalla J, Labeur M, Theodoropoulou M, et al. Retinoic acid as a novel medical therapy for Cushing’s disease in dogs. Endocrinology (2006) 147(9):4438–44. doi:10.1210/en.2006-0414
120. De Marco V, Pinto EM, Mendoca BB. Nelson’s syndrome in a poodle dog treated with retinoic acid. World Small Animal Veterinary Association World Congress 2009. São Paulo, Brazil (2009). 34 p.
121. Layton A. The use of isotretinoin in acne. Dermatoendocrinol (2009) 1(3):162–9. doi:10.4161/derm.1.3.9364
122. Bush ZM, Lopes MB, Hussaini IM, Jane JA Jr, Laws ER Jr, Vance ML. Immunohistochemistry of COUP-TFI: an adjuvant diagnostic tool for the identification of corticotroph microadenomas. Pituitary (2010) 13(1):1–7. doi:10.1007/s11102-009-0189-8
Keywords: Cushing disease, pharmacological treatment, adrenocorticotropic hormone, retinoic acid, chicken ovoalbumin upstream promoter transcription factor
Citation: Fuertes M, Tkatch J, Rosmino J, Nieto L, Guitelman MA and Arzt E (2018) New Insights in Cushing Disease Treatment With Focus on a Derivative of Vitamin A. Front. Endocrinol. 9:262. doi: 10.3389/fendo.2018.00262
Received: 16 February 2018; Accepted: 07 May 2018;
Published: 24 May 2018
Edited by:
Odelia Cooper, Cedars-Sinai Medical Center, United StatesReviewed by:
Eliza B. Geer, Memorial Sloan Kettering Cancer Center, United StatesLeila Warszawski, Instituto Estadual de Diabetes e Endocrinologia Luiz Capriglione, Brazil
Copyright: © 2018 Fuertes, Tkatch, Rosmino, Nieto, Guitelman and Arzt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eduardo Arzt, ZWFyenRAaWJpb2JhLW1wc3AtY29uaWNldC5nb3YuYXI=