 Collin J. Byrne
Collin J. Byrne Sandhya Khurana
Sandhya Khurana Aseem Kumar3,4
Aseem Kumar3,4 T. C. Tai
T. C. Tai- 1Department of Biology, Laurentian University, Sudbury, ON, Canada
- 2Medical Sciences Division, Northern Ontario School of Medicine, Sudbury, ON, Canada
- 3Department of Chemistry and Biochemistry, Laurentian University, Sudbury, ON, Canada
- 4Biomolecular Sciences Program, Laurentian University, Sudbury, ON, Canada
The immune system is increasingly recognized for its role in the genesis and progression of hypertension. The adrenal gland is a major site that coordinates the stress response via the hypothalamic-pituitary-adrenal axis and the sympathetic-adrenal system. Catecholamines released from the adrenal medulla function in the neuro-hormonal regulation of blood pressure and have a well-established link to hypertension. The immune system has an active role in the progression of hypertension and cytokines are powerful modulators of adrenal cell function. Adrenal medullary cells integrate neural, hormonal, and immune signals. Changes in adrenal cytokines during the progression of hypertension may promote blood pressure elevation by influencing catecholamine biosynthesis. This review highlights the potential interactions of cytokine signaling networks with those of catecholamine biosynthesis within the adrenal, and discusses the role of cytokines in the coordination of blood pressure regulation and the stress response.
Introduction
Hypertension—The Roles of Catecholamines and Inflammation
Hypertension
Approximately one in five Canadian adults live with hypertension; globally about 40% of adults over the age of 25 are hypertensive (1, 2). Recent guidelines have defined stage 1 hypertension as mean systolic blood pressure (SBP) ≥ 130 mm Hg or mean diastolic blood pressure (DBP) ≥ 80 mm Hg, and stage 2 hypertension as SBP ≥ 140 or DBP ≥ 90 mm Hg (3). Elevated blood pressure (BP) can cause changes in arterial structure that can increase risk of stroke, heart disease, kidney failure, and other diseases (4). Hypertension is a leading cause of renal failure, second only to diabetes (5). Globally, high BP is attributed to 54.5% of deaths caused by ischemic heart disease, 58.3% of deaths caused by hemorrhagic stroke and 50% of deaths caused by ischemic stroke (6). The significant health risks associated with hypertension have made it the world's leading risk factor for death, estimated as the cause of 13.5% of premature deaths (7). A majority of adults in both developing and developed countries have BP that is higher than is optimal and the risk of death from high BP is particularly great in lower income countries (4, 8). Globally, the cost attributed to BP above optimal levels, including both prehypertension and hypertension, is estimated at US$370 billion, roughly 10% of money spent on healthcare (9). Most recent estimates affirm that the treatment of hypertension is one of the most cost-effective approaches available for increasing quality-adjusted life-years and decreasing preventable deaths (10).
Hypertension can be classified into the categories of essential (or primary) and non-essential (or secondary) hypertension. Hypertension is diagnosed as essential when there is no discernable underlying cause. Essential hypertension is often attributed to a combination of genetic and environmental factors. Non-essential hypertension is directly linked to a pre-existing medical condition such as sleep apnoea, kidney damage, or diseases that consist of abnormal hormone biosynthesis (11, 12). Only a small minority (5–10%) of hypertension diagnoses are classified as non-essential, leaving the remaining majority (90–95%) of diagnosis to be classified as essential hypertension (13).
Like asthma, obesity, diabetes, and a multitude of other pathophysiological conditions, essential hypertension is a multigenic disease that is highly influenced by environmental factors (14). Multigenic traits involve multiple genes and do not have a single recognizable pattern of inheritance, as do single-locus Mendelian traits. Only a small proportion of cases of hypertension are directly caused by individual alleles, which show distinct inheritance patterns within families.
Recent estimates of hypertension awareness and control have demonstrated increased proportions of people who are aware of their condition, who receive treatment, and who have controlled their BP with medication (8). However, despite increased awareness and an abundance of available interventions, hypertension remains prevalent worldwide (6).
Regulation of Blood Pressure
There is a panoply of treatments available for reducing BP and combating hypertension. This is, in part, due to the numerous physiological parameters that influence BP and that are accessible targets for treatment. Blood pressure is the product of cardiac output and total systemic vascular resistance. These variables are dependent on parameters such as blood volume, extracellular fluid volume, arterial and venous compliance, and resistance to venous return (see Figure 1). Changes in the structure and function of kidneys, blood vessels, and the heart are regulated by neuroendocrine feedback mechanisms and serve to control blood volume and vascular resistance (16). Antihypertensive therapies must necessarily act upon the physiological mechanisms that control cardiac output and vascular resistance to effectively regulate BP.
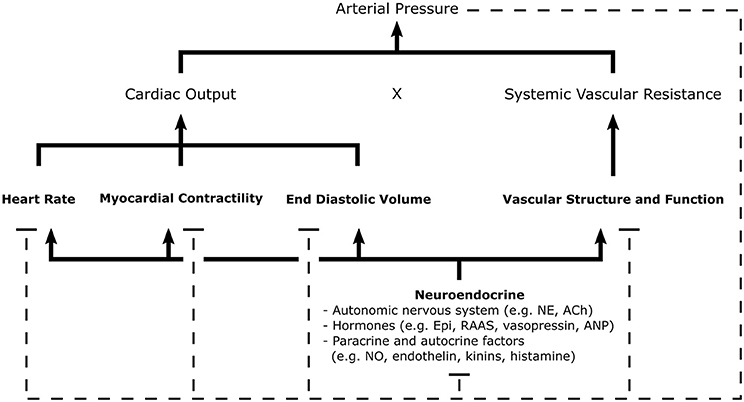
Figure 1. Schematic of the general mechanisms for blood pressure regulation. Arterial pressure is the product of cardiac output and systemic vascular resistance, parameters regulated by neuroendocrine signals which control cardiac, renal, and vascular function. Negative feedback pathways, depicted by dashed lines, are central to the maintenance homeostasis. Various sensors of arterial pressure mediate feedback by modulating sympathetic and parasympathetic tone; thereby, influencing many elements of cardiovascular function. The kidneys play a major role in the regulation of blood pressure through the RAAS, controlling pressure-natriuresis and pressure diuresis-mechanisms which determine fluid volume. Autocrine and paracrine mechanisms allow individual tissues to autoregulate vascular tone and blood flow through local release of vasoactive substances. Ach, Acetylcholine; ANP, Atrial Natriuretic Peptide; Epi, Epinephrine; NE, Norepinephrine; NO, Nitric Oxide; RAAS, Renin-Angiotensin-Aldosterone System [Concept derived from Cowley (15)].
The drugs available for treating hypertension fall into three general categories: diuretics (including thiazides, loop diuretics, aldosterone blockers, and potassium sparers), adrenergic inhibitors (including peripheral inhibitors, β-blockers, central α2-agonists, α1-blockers, and combined α- β- blockers), and vasodilators (including direct vasodilators, angiotensin-converting enzyme [ACE] inhibitors, calcium channel blockers, and angiotensin [Ang] II receptor blockers) (10). Further, recent efforts have been made to develop surgical interventions for treating hypertension when other approaches prove inadequate. These include renal sympathetic denervation and the implantation of devices which electrically stimulate activation of the carotid baroreflex (17, 18). Changes in lifestyle, when possible, are also effective measures for treating hypertension and are recommended in current treatment guidelines by expert committees (19–22). Some of these lifestyle changes include the consumption of a healthy diet, engaging in regular physical activity, minimization of alcohol consumption, maintenance of healthy body weight (BMI 18.5–24.9 kg/m2), maintenance of a moderate waist circumference (<102 cm for men, <88 cm for women), moderate sodium intake (<2,000 mg/day), and living in a smoke-free environment. Treatment recommendations for hypertension generally include a combination of antihypertensive drugs and appropriate lifestyle modifications, together with careful self-monitoring (3, 21).
Hypertension and Catecholamine Biosynthesis
Adrenergic inhibitors are useful pharmacologic agents for treating hypertension. Adrenergic signaling via α- and β-adrenergic receptors can influence cardiac output and peripheral resistance. The catecholamines (CAs) dopamine (DA), norepinephrine (NE), and epinephrine (Epi) are involved in the regulation of the cardiovascular system. These CAs are produced by neuroendocrine cells and have dual hormone and neurotransmitter functions.
The biosynthesis of CAs begins with the hydroxylation of tyrosine by the enzyme tyrosine hydroxylase (TH), producing L-3,4-dihydroxyphenylalanine (L-DOPA) (23). Next, L-DOPA is decarboxylated by the enzyme L-aromatic amino acid decarboxylase (AADC), converting it to DA (24). DA is then hydroxylated to produce NE; a reaction catalyzed by dopamine β-hydroxylase (DBH) (25). This sequence of reactions, which convert tyrosine to NE, is common in postganglionic neurons of the sympathetic nervous system and in specific brain regions such as the locus coeruleus. In adrenal chromaffin cells, and a small number of brain neurons, one additional biosynthetic step occurs, consisting of the methylation of NE by phenylethanolamine N-methyltransferase (PNMT) to produce Epi (26). Epi is the major secretory product of the adrenal gland, which is also the most abundant source of secreted Epi in the body (27, 28). CAs in the adrenal medulla are sequestered in CA storage vesicles of chromaffin cells. When stimulated, chromaffin cells release CAs from their vesicles through Ca+2-mediated exocytosis (29, 30).
Once released into circulation, CAs can interact with numerous adrenergic receptor types expressed in a variety of tissues. All CA receptors are G protein-coupled receptors (31). There are multiple forms of DA receptor, and they can be categorized in at least five (D1−5) different subtypes. Adrenergic receptor subtypes include α1-, α2-, β1-, β2-, and β3- adrenergic receptors, some of which can be divided into further subtypes. Adrenergic receptors are activated by the CAs Epi and NE, with each receptor having a distinct affinity for each ligand. Through these receptors, CAs can signal to numerous tissues throughout the body to produce a wide and coordinated physiological response.
The distribution and function of DA receptors suggests that DA may decrease BP by synergistically enhancing vasodilation, inhibiting synaptic NE release, decreasing circulating CAs, inhibiting aldosterone secretion and inhibiting sodium reabsorption in the kidney (32, 33). The α-adrenoceptors are important for the maintenance of vascular tone and promotion of smooth muscle contraction in other parts of the body. Sympathetic stimulation of α1-adrenoceptors is a major mechanism for sympathetic-mediated vasoconstriction (34). β-adrenergic receptors are expressed in airway smooth muscle, epithelium, endothelium, immunocytes, and myocardium (35). In cardiac tissue, although all three types are present, β1-adrenergic receptors are the major β-adrenoceptor type expressed. β1- and β2-adrenoceptor-mediated actions in the heart include positive inotropic (increased contractility), chronotropic (increased heart rate), dromotropic (increased conductivity), and bathmotropic (increased threshold of excitation) effects (36). β3-adrenoceptors require higher concentrations of CAs for activation than other β-adrenoceptors, and β3-adenoceptor signaling is suggested to counteract effects of β1- and β2-adrenoceptor activation, thus mediating a protective feedback loop to prevent adrenergic overstimulation.
Elevated plasma levels of Epi and NE have been reported in animal models of hypertension as well as in patients with essential hypertension (37–41). CA biosynthesis is dependent upon and correlated with the activity of the CA biosynthetic enzymes (40). Control of CA biosynthesis occurs primarily through the enzymes TH, DBH, and PNMT. AADC activity is typically high and is not rate limiting under normal physiological conditions (42, 43). In adrenal chromaffin cells, the expression of AADC is regulated to a lesser degree than the other CA biosynthetic enzymes (44–46). Regulation of TH, DBH, and PNMT is achieved through both transcriptional and post-transcriptional mechanisms (47–50). Transcript levels of TH and PNMT, and activities of TH and DBH are enhanced in the adrenal medullas of genetically hypertensive rat models (51–54). Further, PNMT is one of the putative gene loci linked to hypertension in genetic studies (55–57). Prolonged elevation of plasma CA levels can contribute to cardiac dysfunction by the over activation of vascular smooth muscle cells, resulting in ischemia and functional hypoxia; and oxidative damage (through the formation of oxidized CAs and oxygen free radicals), resulting in ultrastructural changes and cellular damage in cardiomyocytes (36). Oxidative damage may also lead to immune activation that contributes to the further progression of cardiovascular dysfunction (vide infra). Taken together, CAs play a role in hypertension, and the molecular mechanisms that regulate the CA biosynthetic enzymes and their activities are of interest for understanding the progression of hypertension and cardiovascular disease.
Hypertension and Inflammation
The role of inflammation in the genesis of hypertension and accompanying organ damage is well established (16). Inflammation is one of the most important factors contributing to cardiovascular risk; and it is a major contributor to the formation, progression and destabilization of atherosclerotic lesions (58–60). The link between immune and cardiovascular function is apparent in major immune diseases including rheumatic diseases, HIV, and psoriasis. Cardiovascular pathologies are the leading cause of premature mortality in patients with autoimmune rheumatic diseases (61). Individuals with HIV infection have higher risk of cardiovascular disease (CVD), arterial stiffness, systolic, and pulse pressures than matching uninfected individuals (62). A recent meta-analysis of observational studies concluded that psoriasis, a chronic inflammatory skin condition, is associated with increased prevalence and incidence of hypertension and that odds of hypertension increase with severity of psoriasis (63). Inflammation is an essential component of many diseases, and the connections between innate and adaptive immunity, hypertension, and CVD add support to the role of the immune system in cardiovascular pathology (64).
A prospective cohort study of 20,525 women concluded that high plasma levels of the inflammatory biomarker C-reactive protein are predictive for the development of hypertension (65). Several studies have supported immune involvement in the elevated BP of spontaneously hypertensive rats (SHR). Purcell and Gattone (66) found that young SHRs have an elevated rate of nerve growth into the thymus, a primary lymphoid organ important in T-cell development, compared to their normotensive Wistar Kyoto (WKY) counterparts. Others have found that treatment of SHR with antithymocyte serum or with the immunosuppressant cyclophosphamide lowers BP (67, 68). Later studies established the role of the adaptive immune response in hypertension after finding that mice with a genetic deletion in recombinase-activating protein (RAG-1−/−), which lack T- and B-lymphocytes, experience blunted hypertension in response to both Ang II and deoxycorticosterone acetate (DOCA)-salt; adoptive transfer of T-cells restored the elevation in BP (69). This study also identified the role of the cytokine TNF-α in BP elevation when mice treated with Ang II responded with both increased BP and increased synthesis of TNF-α from T-cells; anti-TNF-α therapy with etanercept (a TNF-α inhibitor) blunted Ang II-mediated elevations in BP (69). Taken together, these studies suggest the potential for enhanced neural activation of T-cells in hypertension as well as a functional importance of cytokine signaling in BP regulation. Also supporting the role of cytokines in hypertension, multiple studies have identified altered profiles of pro- and anti-inflammatory cytokines and cytokine production capacity in humans when comparing hypertensive or pre-hypertensive patients to control subjects (70–73). Although results are sometimes conflicting, these studies implicate cytokines such as IL-1, IL-4, IL-6, IL-7, IL-13, TNF-α, and CCL2 in human hypertension. Other cytokines not analyzed or not changed in circulating concentration may also be important in hypertension because of the potential for local regulatory effects at important centers for BP regulation.
Introduction of exogenous cytokine has been reported to induce changes in BP. The cytokines IL-2 and IL-10 predominantly decrease BP whereas the cytokine IL-6 predominantly increases BP (74–79). Other cytokines, such as TNF-α and IL-1, appear to have more intricate effects in relation to BP regulation. Regulation of BP by cytokines may be mediated by signaling to neural control centers or by direct actions on peripheral tissues. In a screening experiment for changes in chemokine expression in DOCA/salt-induced hypertensive mice, transcript of the chemokine CCL2 and its receptor CCR2 were increased in aortas after the onset of hypertension. Treatment of mice with the CCR2 antagonist, INCB3344, substantially reduced DOCA/salt-induced infiltration of macrophages in aortic wall and DOCA/salt-induced elevations in BP (80). IL-1 can modulate BP by influencing activity of neurohormonal control centers in the brain (81–84). IL-1 can also modify vascular reactivity to NE (85). Human cancer patients who receive high doses of IL-2 demonstrate hemodynamic changes which suggest decreased peripheral resistance but increased cardiac output, with an overall reduction in mean arterial pressure (86, 87). Similar hemodynamic changes and hypotension are observed in experimental animals (88, 89). Interestingly, IL-2 can decrease circulating Epi in mice, and reversal of IL-2-induced hypotension is associated with a rise in circulating CAs (89). Like IL-2, experimentally-induced elevations in IL-10 levels are associated with decreases in BP in animal models (82, 90, 91). IL-2 and other cytokines may reduce CA-mediated vasoconstriction by enhancing superoxide generation, a molecule that can interact with CAs and inhibit their signaling function (92–94). In contrast to many other cytokines, IL-6 appears to increase BP. In a large cohort study of healthy men, after controlling for age and other cardiac risk factors, baseline plasma IL-6 levels were positively associated with increased systolic and diastolic BP, pulse pressure, and mean arterial pressure (95). In animal experiments, IL-6 is shown to contribute to Ang II-induced, but not salt-sensitive hypertension (75, 76, 96, 97). Pro-hypertensive effects of IL-6 may be mediated, in part, by vasoconstrictive effects of the cytokine that could result in increased peripheral resistance (85, 98). The major proinflammatory cytokine TNF-α can induce hypotension; however, inhibition of TNF with etanercept can prevent elevations in BP caused by Ang II infusion, high-fructose feeding, and chronic inflammation in animal models (69, 99–102). TNF-α inhibition does not influence BP in salt-dependent hypertension (103). TNF-α can directly modulate arterial tone, and TNF-mediated signaling is involved in cardiac remodeling (85, 98). Prenatal exposure to elevated levels of cytokines may also contribute to hypertension during adulthood (104). This may involve morphological changes in tissues, as suggested by findings of increased adrenergic innervation of the thymus in humans provided with IFN-α therapy (105). The BP-regulatory effects of cytokines are complex, affecting multiple physiological systems in combination with hormones, neurotransmitters, and other signaling molecules. Future investigations into the integration of cytokine signaling with the other BP regulatory systems will help to illuminate the events responsible for BP dysregulation in disease.
In describing a model for the interaction of inflammation and hypertension, Harrison et al. (106) hypothesized that modest elevations in BP (to values of SBP ~135–140 mm Hg), largely caused by activity of the CNS, trigger immune changes that lead to hypertension (see Figure 2) (106). In this model, initial elevations in BP are responsible for neoantigen formation from oxidation and altered mechanical forces in vasculature. Neoantigens then induce inflammatory responses in the kidneys, blood vessels and other tissues, where lymphocyte infiltration and expression of inflammatory mediators such as CCL2, IL-1, IL-6, IL-17, and TNF-α is increased (69, 107–109). These cytokines and inflammatory mediators work in concert with CAs and other BP-elevating hormones leading to vascular and renal dysfunction and initiating a more severe hypertensive state (106). This feed-forward model described by Harrison et al. (106) was foretelling of findings by Kirabo et al. (110), whose work outlined a mechanism for hypertension based on an autoimmune-like reaction. In this mechanism, initial increases in BP lead to oxidative stress and lipid peroxidation which results in neoantigen formation, initiation of T-cell proliferation and cytokine biosynthesis, and further increases in BP. Recent studies also demonstrate that immune cell function can be altered by high salt microenvironments in the skin and skeletal muscle, with salt acting as a proinflammatory stimulus for the development of hypertension (111, 112). With the support of these and other findings, a new paradigm is being established that implicates inflammation in the elevation of BP and progression of hypertension.
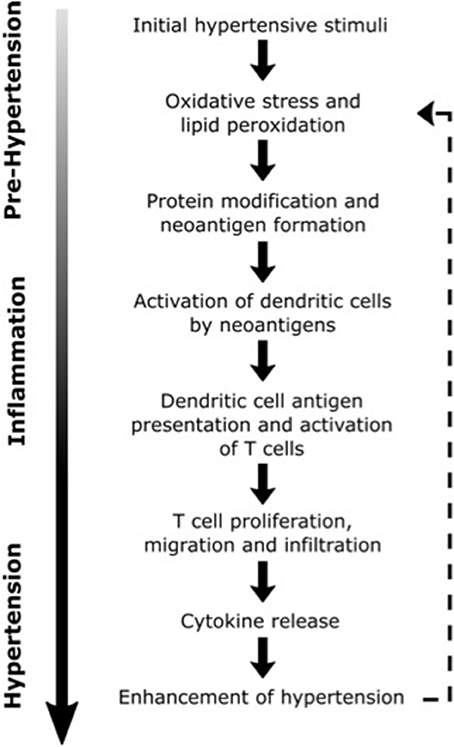
Figure 2. Possible inflammatory processes contributing to the progression of hypertension. Dashed line illustrates the positive feedback loop that may lead to further elevations in blood pressure [Concept derived from Harrison (106)].
The Major Mechanisms of Adrenal Medullary Regulation
The adrenal gland is a key organ involved in the physiological adaptation to stress. The “fight-or-flight” response, first described by Cannon in the early twentieth century, is characterized by increased BP, increased heart rate, increased cardiac output, and changes in vascular and respiratory smooth muscle tone (113, 114). The two major hormones secreted into circulation that facilitate the physiological stress response include cortisol and Epi, both being primarily products of the adrenal cortex and medulla, respectively (28). There are two major effector circuits that are activated when the CNS perceives or anticipates a stress. They include the hypothalamic-pituitary-adrenal (HPA) axis, which stimulates the adrenal medulla through a hormonal mechanism, and the sympathetic-adrenal (SA) axis, which stimulates the adrenal medulla through a neural mechanism (115). These axes are in many ways physically distinct but they also have overlapping CNS components and physiological functions. Initiation of the physiological stress response, involving either HPA or SA axis, is primarily derived from structures of the limbic system. Termination of the stress response, caused by hormonal and neural feedback, also involves many of these same limbic structures. Integration of hormonal and neural signaling cascades allows the HPA and SA axis to function cooperatively while also tailoring individual responses to the specific initiating stimuli (116).
Hypothalamic-Pituitary-Adrenal Axis
The HPA axis consists of the paraventricular nucleus (PVN) of the hypothalamus, the anterior pituitary gland and the adrenal gland (117). The HPA axis is activated when afferent neurons from multiple brain regions stimulate hypophysiotrophic neurons of the paraventricular nucleus, inducing them to release corticotropin-releasing hormone (CRH), and vasopressin (see Figure 3). CRH and vasopressin then travel through hypophysial portal vessels to the anterior pituitary. The axons of CRH neurons present in the PVN project to the median eminence through the lateral retrochiasmatic area. CRH released from the outer layer of the median eminence binds to receptors on pituitary corticotropes and promotes the secretion of adrenocorticotrophic hormone (ACTH) into systemic circulation (118, 119). In the presence of CRH, vasopressin has a synergistic effect, enhancing secretion of ACTH into circulation. ACTH then travels to parenchymal cells of the adrenocortical zona fasciculata, where it binds to plasma membrane receptors and initiates a rapid increase in the biosynthesis and secretion of glucocorticoids (GCs). Once in systemic circulation, GCs bind to ubiquitously expressed intracellular glucocorticoid receptors (GRs) to induce physiological adaptations to the initial stressor. An intra-adrenal portal vascular system allows the exposure of adrenal medullary cells to especially high concentrations of GCs released from the adrenal cortex (120). GCs produce their cellular effects primarily by regulating transcription. Endogenous cortisol (corticosterone in rodents) is a lipid-soluble steroid hormone that binds to the cytoplasmic GR. Prior to ligand binding, GR is located in the cytoplasm as a multiprotein complex (121). HSP90, one of the proteins in this complex, maintains the cytoplasmic retention of GR until binding of a ligand to GR causes dissociation from the complex and translocation into the nucleus. GR then homodimerizes and binds to glucocorticoid response elements (GRE) in the promoter regions of target genes directly, or interacts with other transcription factor proteins causing transactivation or transrepression (121). An alternate manner in which GCs can initiate transcription or signaling events is via membrane receptor activation, which, when coupled with G-proteins can induce downstream signaling cascades. This alternate mechanism of signaling activation triggered by GCs is exerted in situations wherein rapid functional changes associated with GC mediated signaling activation are critical (122).
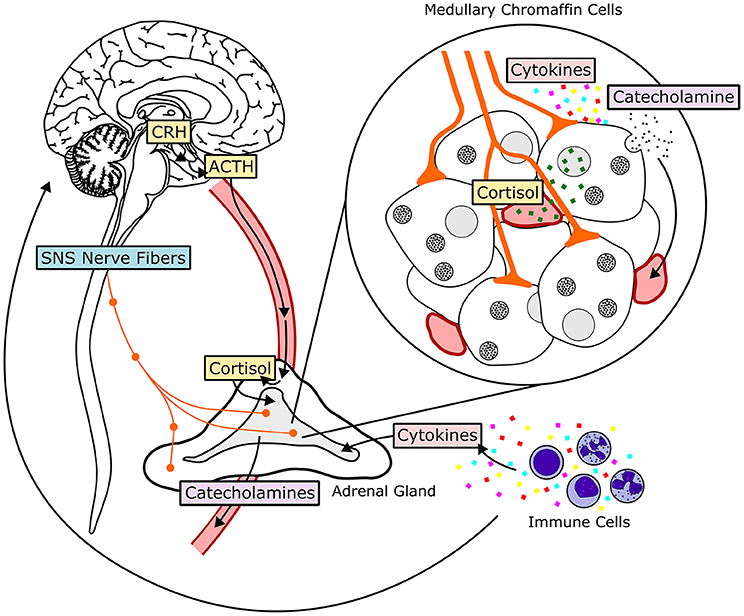
Figure 3. Hormonal and neural mechanisms regulating adrenal medullary chromaffin cells. The HPA-axis, comprised of the hormones corticotropin-releasing hormone (CRH), adrenocorticotropic hormone (ACTH), and cortisol, is shown in yellow. The SA-axis, comprised of afferent preganglionic sympathetic nervous system (SNS) fibers, is shown in blue. Green squares represent glucocorticoid (cortisol) produced in the adrenal cortex and traveling to the adrenal medulla through vasculature. Acetylcholine, pituitary adenylate cyclase-activating peptide, and other neurotransmitters are released from synaptic terminals. Cytokines, transported to the adrenal medulla or produced locally, influence adrenal chromaffin cell function and response to HPA- and SA-axis activation. Both glucocorticoids and sympathetic input stimulate release of catecholamines, primarily epinephrine, from chromaffin cells by exocytosis. Epinephrine then enters systemic circulation and travels to target tissues throughout the body.
GCs are important regulators of homeostasis during basal conditions and during stress. They are critical for regulating cardiovascular, immune, metabolic, developmental, and reproductive processes (117). For example, the strong influence of GCs on immune function has allowed them to become some of the most commonly used compounds for therapeutic treatment of inflammatory, autoimmune, and lymphoproliferative disorders. In human peripheral blood mononuclear cells, ~20% of genes are regulated either positively or negatively by GCs (123). The effects of GCs on cardiovascular regulation are also important. Cortisol is a regulator of BP in humans and can lead to hypertension when in excess (124). Chronically elevated GC, either endogenous (as seen in cases of adrenal hyperplasia or dysplasia or elevated ACTH expression), or by exogenous means (when administered for immunosuppressant therapy), can cause a clinical disorder known as Cushing syndrome. It is estimated that 80% of patients with endogenous elevated GCs, and 20% of corticosteroid therapy patients with Cushing syndrome present with elevated BP (125, 126).
One way that GCs can influence BP is by influencing CA biosynthesis and secretion. GCs directly increase the release of CAs by sympathetic nerves and adrenal medullary cells (127, 128). Early evidence of GC control over adrenal CA biosynthesis was demonstrated in experiments performed by Wurtman and Axelrod (129), who reported that ablation of the pituitary gland decreases PNMT activity, which can be restored by addition of ACTH or GC (129). Later, in vitro and in vivo experiments confirmed that GCs are responsible for increasing PNMT mRNA expression (128, 130, 131), increasing the amount of functional intronless mRNA splice variant (49), increasing PNMT activity (49), and enhancing PNMT protein stability via regulation of the co-substrate S-adenosyl-methionine (132–134) in adrenal chromaffin cells. Although, Greene and Tischler (135), previously thought that PC12 cells do not synthesize PNMT or epinephrine, and are primarily noradrenergic, later studies by Kim et al. (136) and Byrd et al. (137) showed that these cells do indeed express low levels of PNMT and Epi, and the expression of both is significantly increased in the presence of the synthetic GC, dexamethasone (135–137).
Studies in rat pheochromocytoma cells show that, in addition to PNMT, GCs regulate the other CA biosynthetic enzymes to produce parallel increases in their transcript level and activity (136, 138–141). Similar observations pertaining to regulation of enzyme transcript levels have also been made with primary cultures of bovine adrenal medullary cells; however, unlike rat pheochromocytoma cells, in bovine chromaffin cell primary cultures DBH transcript does not appear to be regulated by GC (50, 142).
Thus, GCs can increase transcript of TH, DBH, and PNMT. The site critical for GC responsiveness of the rat TH gene is located at about −5.7 kb and it closely resembles the activator protein 1 (AP-1) binding site (143). This finding is consistent with earlier observations that the proximal promoter region (−773 to +27 bp) is not sufficient for GC regulation of the TH gene (138, 139). Another functional GRE has been identified at ~2.4 kb upstream in the mouse TH promoter (144). Several putative GREs have been identified in the first 1 kb of the upstream rat DBH gene, with corresponding sequences in the human DBH promoter (140). Although long exposure with GCs can increase transcript levels of DBH in PC12 cells, functionality of putative GREs in the DBH promoter has not yet been proven (140). GCs are also important regulators of PNMT transcription (131). Three functional GREs have been identified in the proximal 1 kb rat PNMT promoter, and activation at these sites can be synergistically regulated by the transcription factors early growth response 1 (Egr1) and activator protein 2 (AP-2) (145–147).
Sympathetic-Adrenal Axis
Working alongside the HPA-axis, the SA-axis, consisting of the direct innervation of adrenal medullary chromaffin cells by the sympathetic nervous system, also signals the adrenal medulla to synthesize and secrete Epi (148). Stress signals, primarily originating from limbic structures, are transmitted to preganglionic sympathetic neurons in the intermediolateral cell column of the thoracolumbar spinal cord which project, via the splanchnic nerve, to chromaffin cells of the adrenal medulla (116). The cortex is also innervated by the splanchnic nerve and neurotransmitters such as acetylcholine (ACh), released at the adrenocortical junction, regulate steroid biosynthesis and can influence vasculature to regulate adrenal perfusion (149–152). The neural stimulus is delivered to each chromaffin cell by several synaptic boutons and compelling evidence now suggests that gap junctions also help to propagate electrochemical signals between neighboring adrenocortical cells (153, 154). A combination of neurotransmitters and neuropeptides such as neuropeptide Y (NPY), acetylcholine (ACh), and vasoactive intestinal peptide (VIP) are released from sympathetic nerve terminals and bind to plasma membrane receptors on chromaffin cells. These substances stimulate the release of large amounts of stored CAs from chromaffin cell vesicles via Ca+2-mediated alteration of action potential and exocytosis; the frequency of these action potentials is dependent on the concentration of ACh (155–160). ACh directed CA secretion can also be mediated in the presence of K+ and Na+ induced membrane depolarization (161, 162). Additionally, the adrenal cortex receives input from medullary ganglion cells that synthesize NE, NPY, and VIP, amongst other biomolecules; this paracrine interaction can also influence steroidogenesis (149). The extrinsic innervation of the adrenal gland, and intrinsic neural networks within it allow for an integrated signaling and fine tuning of adrenal function (150, 163). Due to the direct innervations of adrenal chromaffin cells, the SA effector circuit has a short latency in comparison to excitation via the HPA axis, which is generally longer lasting and slower to respond (164). Stimulation of chromaffin cell activity by the SA axis may contribute to hypertension through either an increase in sympathetic nerve firing or an unusually high sensitivity of chromaffin cells to sympathetic stimulation (165–168).
Synaptic transmission at the SA synapse is mediated by the small molecule transmitter acetylcholine (ACh) and by neuroactive peptides. The frequency of action potential firing at sympathetic nerve terminals influences the types of neurotransmitters released from the presynaptic nerve at the SA synapse. Stress is associated with high frequency splanchnic nerve firing, whereas basal sympathetic tone is characterized by lower frequency firing (169). In the preganglionic sympathetic nerves at the SA synapse, small synaptic vesicles (SSVs) contain ACh and large dense core vesicles (LDCVs) contain neuropeptides such as pituitary adenylate cyclase-activating peptide (PACAP). During high frequency firing both LDCVs and SSVs are released from the presynaptic nerve terminals. During basal conditions, only SSVs are released (170). Both PACAP and ACh are integral at the SA synapse for promoting CA biosynthesis and secretion (115).
ACh is perhaps the best characterized molecule for synaptic transmission from the splanchnic nerve to the adrenal medulla. ACh binds to both nicotinic and muscarinic plasma membrane receptors on chromaffin cells (mAChRs and nAChRs, respectively). nAChRs are also classified as muscle or neuronal nAChRs depending on their site of expression. Although both AChR types can promote CA release, the reliance on stimulation via mAChRs or nAChRs is species dependent. For example in bovine adrenals, nAChRs are primarily responsible for cholinergic transmission and CA release, in chickens, the mAChRs are the key players, whereas in other species it could be both (142, 171–173). Structurally, neuronal nAChRs are heterodimeric proteins made of 5 subunits, two α, and three β subunits, combinations of which can give rise to numerous receptor subtypes, with the α3β4 being the most pertinent nAChR for CA secretion (172). The nAChRs are ligand gated cation channels mediating quick excitation responses, while mAChRs are metabotropic receptors coupled with G-proteins resulting in slower neuronal signaling (172, 173). There are 5 isoforms of the mAChRs, M1-M5, which are species specific and are coupled with different Gα proteins and signaling pathways (173).
Activation of nAChRs increases TH mRNA in chromaffin cells in a protein kinase A (PKA)-dependent manner (142, 174, 175). Cholinergic stimulation of chromaffin cells also induces PNMT promoter-driven luciferase activity through a PKA-dependent mechanism (176). Additionally, in vitro and in vivo evidence supports the role of mAChRs in activating PNMT expression, via induction of the transcription factor Egr1 (177, 178).
Evolutionarily conserved, PACAP belongs to the vasoactive intestinal protein (VIP) family of peptides. PACAP is primarily released from LDCVs during high frequency neuronal firing, and is important for generating sustained increases in CA synthesis and secretion by chromaffin cells (179, 180). The PACAP precursor is processed into two bioactive forms, namely PACAP38 and PACAP27. The PAC1 receptor (PAC1R), which belongs to the subclass B1 GPCR, is selective for PACAP38/PACAP27, while VPAC1 and VPAC2 have affinities for both PACAP and VIP (181). PAC1R signals through Gαs, which regulates adenylyl cyclase (182). Binding of PACAP to PAC1R can signal through the conventional cyclic adenosine monophosphate (cAMP)-PKA pathway and at least two other insulated, cAMP-sensitive signaling pathways involving the signal transduction proteins exchange protein directly activated by cAMP (Epac) and the extracellular signal regulated kinases (ERK) 1 and 2 (183–185). The PAC1R can also stimulate Gαq, which activates a phospholipase C (PLC)-protein kinase C (PKC) pathway (186).
PACAP is now recognized as a critical peptide for signaling at the “splanchnicoadrenomedullary” junction under situations of stress (187, 188). PACAP is capable of upregulating chromaffin cell expression of TH, DBH, and PNMT transcripts (189–191). In studies using PACAP−/− mice, the biosynthesis of TH and PNMT transcripts was significantly reduced in animals exposed to restraint stress, possibly due to blunted Egr1 and cFos; under sustained stress, reduction in CRH mRNA in the PVN and circulating corticosterone was observed indicating that PACAP is critical in the stress response (190, 192). In addition to regulation of CA biosynthesis, other studies have demonstrated the importance of PACAP in regulating CA secretion from adrenal cells, and its role in nerve firing (180, 184, 193, 194). Taken together, these studies suggest a central role for PACAP in HPA axis function during stress. Consistent with this role, disrupted PACAP signaling has been correlated with anxiety, depression, behavioral and cognitive changes, and other psychopathologies (195–198).
As mentioned above, signaling via cAMP is an important molecular mechanism induced by both ACh and PACAP, and is involved in the regulation of CA biosynthetic enzymes in adrenal chromaffin cells. In primary cultured bovine adrenomedullary chromaffin cells, cAMP signaling produces synchronized increases in both transcript and activity levels of TH, DBH, and PNMT (27). Similar activation of the CA biosynthetic enzymes by cAMP signaling occurs in rat chromaffin cells (138–140, 176, 199). It should be noted that in both rat and bovine models, the induction of PNMT by cAMP is relatively small compared to the induction of TH and DBH. Signaling by cAMP activates PKA and can lead to tissue-specific induction of other signaling pathways. For example, in PC12 cells, PACAP activates PKA signaling as well as signaling through the mitogen-activated protein kinases (MAPKs) p38 and ERK1/2 via a PKA dependent mechanism (200, 201). Signaling by ERK1/2, downstream of PKA activation, may contribute to PNMT transcriptional activation (191).
The promoters of TH, DBH, and PNMT all contain motifs that can bind many common transcription factors. The transcription factors specificity protein 1 (Sp1), AP-2, and Egr1 all have functional consensus sequences in the promoters of TH, DBH, and PNMT rat genes. AP-1 and cAMP-responsive element (CRE) motifs are also present within the rat TH and DBH promoters (155, 199). Induction of Egr1 in rat chromaffin cells occurs through a cAMP/PKA signaling mechanism (202, 203). In rat chromaffin cells, Egr1 regulates transcription of TH, DBH, and PNMT (199, 204, 205). Splanchnic nerve activation, the cholinergic agonists nicotine and muscarine, and the neuropeptide PACAP are all inducers of Egr1 (176, 177). Transcriptional activation of TH and DBH also occur through cAMP induction of transcription factor binding to AP-1 and CRE motifs (206–209). Other transcription factors potentially involved in the activation of CA biosynthetic enzymes by neural stimuli include Sp1 and AP-2 (176, 191). Taken together, these studies suggest that the neuronal regulation of chromaffin cells involves a number of transcription factors which can act individually, or cooperatively to regulate expression of the enzymes responsible for CA biosynthesis.
Chronic Stress and Hypertension
The coordinated functionality of both HPA and SA axes is essential for BP homeostasis. The functional balance between these axes is critical in the biosynthesis of CAs and in the maintenance of BP (116, 210). Exposure to chronic stress, however, leads to sustained elevated blood levels of CAs that can have adverse affects on health and have been correlated with hypertension (211–213). Studies of panic disorder, and chronic mental stress have documented co-morbidity of these conditions with hypertension, and reported elevated circulating cortisol and Epi, and an increased expression of PNMT in the sympathetic nerve fibers of affected patients (214, 215). Chronic stress results in SA overdrive thereby altering adrenergic output; this consequently elevates BP causing physiological and metabolic changes (216, 217). Repeated exposure to stress elevates Epi, and is correlated with hypertension (41, 218, 219). The extent of Epi activation is dependent on the type of stressor and duration of stress exposure (155, 220). Psychological stress has been correlated with elevated cortisol and Epi, with consequences on metabolism and physiological functions; this has been reported in people who experience chronic work stress, and consequently develop metabolic syndrome (221, 222). Indeed, stressors such as mood disorders, low social support, socioeconomic background etc. are factors that can influence cardiovascular health; effective strategies to manage cardiovascular health should, if possible, encompass interventions that target these psychosocial stressors (223, 224). Individuals with anxiety, especially as seen in those who experience post-traumatic stress disorder, are susceptible to developing hypertension, and have an elevated risk for other cardiovascular pathologies, possibly due to over activity of the HPA (225–227). Additionally, other factors such as physical fitness, diet, smoking, age, sex, and obesity can influence the onset and/or severity of hypertension (3, 228–230).
Cytokine-Mediated Regulation of Catecholamine Biosynthesis
Investigations into the potential role of cytokines in regulating CA biosynthesis by the adrenal gland were, in part, inspired by insights gained from studying depression (231). Depression can be induced by alterations in NE and other neurotransmitter levels, and sympathetic hyperactivity is a well characterized attribute of the condition (232). It has also been reported that a large proportion of patients receiving IFN-α therapy for treatment of cancer or infectious disease develop a behavioral syndrome that is very similar to major depression (232). This finding led to questions about the influence of cytokines on neurotransmitter synthesis, and the role of cytokines in regulating neural activity. Interestingly, depression is now associated both with elevations in plasma levels of proinflammatory cytokines and increased risk of hypertension, cardiovascular morbidity, and mortality (233–235). Although the causal relationships are not yet resolved, possible influences of inflammatory mediators such as cytokines on catecholaminergic cell function are now being investigated for their contribution to hypertension and CVD.
In humans, treatment with IFN-α increases circulating levels of NE and Epi (236, 237). Both intravenous and intracerebroventricular administration of IL-1 to rats has been reported to increase plasma levels of NE and Epi, along with increased renal sympathetic nerve activity, SBP, and heart rate (238, 239). Central administration of IL-1 to rats has also been reported to increase ACTH secretion (240). These findings suggest that IL-1 can activate SA and HPA axes by direct stimulation of regulatory centers within the brain. In humans, peripheral administration of IL-6 increases plasma cortisol and NE but does not affect plasma Epi levels (241–244). Studies have suggested that peripherally, but not centrally administered, TNF-α elevates plasma CA levels in rats (245, 246). Increased expression of IL-10 in the brain can inhibit elevations in plasma NE resulting from myocardial infarction in rats (247). Numerous cytokines, including IFNs, IL-1, IL-2, IL-6, and TNF-α induce changes in brain CA synthesis or metabolism. Often, excitatory or inhibitory effects of cytokines in the brain are regionally dependent. Many of these same cytokines also modulate CA levels in the hypothalamus and influence function of the HPA axis (248, 249). For example, central and peripheral administrations of IFN-α both alter levels of DA and NE in specific regions of the brain (250–252). The patterns of altered CA levels differ depending on the location, central or peripheral, of IFN-α administration. This suggests that direct and indirect sensing of cytokines by the brain induce unique responses in CA synthesis by neural tissues. Numerous studies report similar regulatory effects for other cytokines in relation to brain CA synthesis. In peripheral tissues, the effects of centrally or peripherally administered cytokines on CA levels and CA turnover is tissue-specific, suggesting that cytokines can influence sympathetic activity both directly and indirectly, and that modulation of sympathetic nerve activity is specific rather than global (253–259).
Cytokines have also been reported to regulate CA biosynthetic enzymes in vivo. In vivo studies using rats demonstrate that the cytokines IFN-α, IL-1β, and TNF-α regulate the CA biosynthetic enzyme TH in catecholaminergic cells of the brain and carotid body (251, 260–262). Interestingly, centrally administered cytokines can regulate CA biosynthetic enzymes in the adrenal medulla as well, likely through indirect mechanisms involving neural activation at the level of the CNS and downstream effects mediated by the HPA or SA axes (263). For a more complete presentation of cytokine effects in the brain, HPA and SA axis, several reviews are available (248, 249, 264, 265). In sites of CA biosynthesis outside the brain, the influence of cytokine signaling is only beginning to be understood.
Cytokine Expression by Adrenal Chromaffin Cells
Adrenal cytokines can originate either systemically or locally; both situations have possible importance to cytokine-mediated regulation of adrenal function during hypertension. Numerous studies have identified unique profiles of circulating and tissue-expressed cytokines in hypertensive animal and human subjects (73, 80, 82, 95). Even during normal physiological conditions, cytokines are expressed at detectable levels by adrenal medullary tissue. Cytokines are expressed at varying levels throughout the adrenal gland (266, 267). The highest levels of expression are most commonly observed in the cortex or steroid-producing cells within the medulla, although expression of cytokines by chromaffin cells themselves has also been demonstrated in a number of studies (see Table 1). In humans, as in many species, the adrenal medulla is contiguous with the adrenal cortex, meaning that chromaffin cells are in direct contact with steroidogenic cells (303). Chromaffin cells are also receptive to many cytokines that are produced locally in the adrenal gland (see Table 1). Receptiveness to cytokines is demonstrated either by expression of cytokine receptors or by response of isolated chromaffin cells to cytokines. In instances where the cytokine and its receptor are co-expressed, or when a locally produced cytokine can elicit a response in chromaffin cells, there is a possibility of autocrine or paracrine signaling that may influence endocrine function of the adrenal medulla (317).
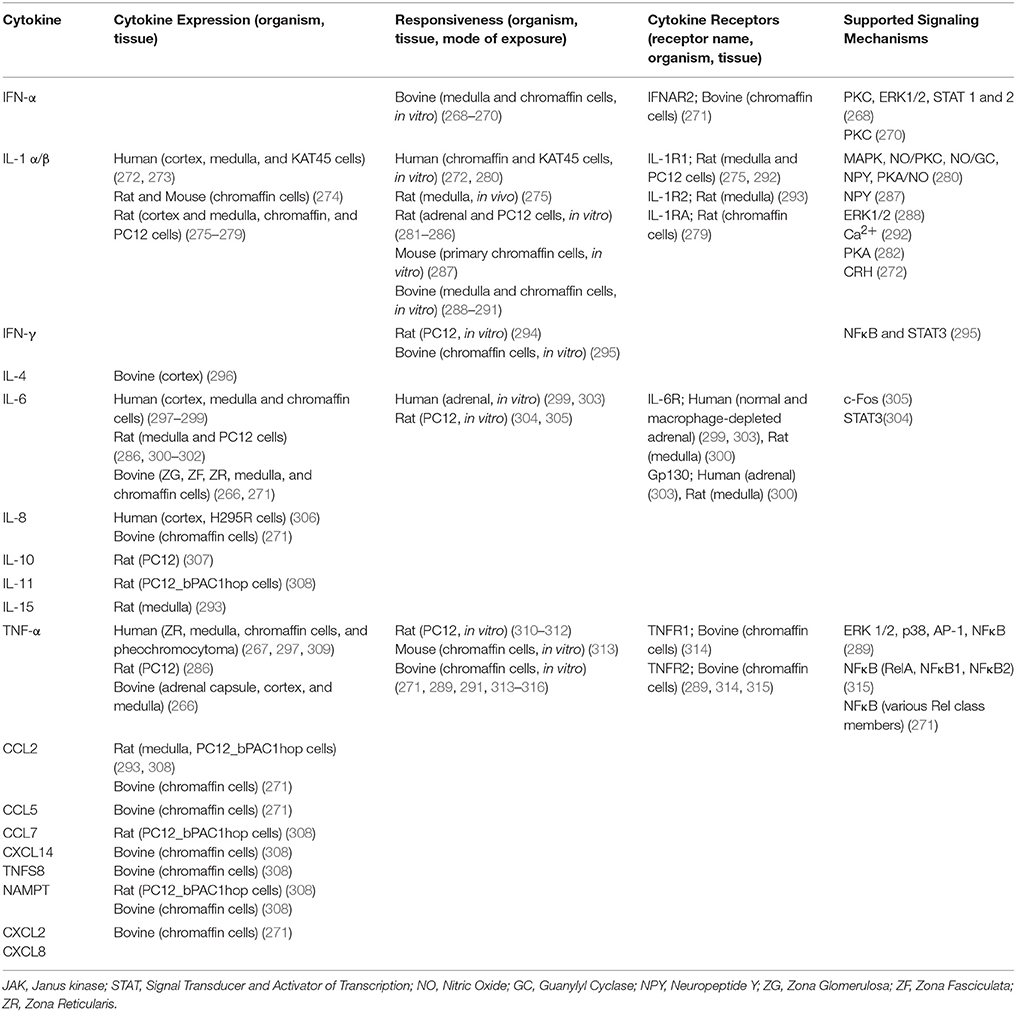
Table 1. Cytokine expression, responsiveness, and signaling observed in adrenal gland.
Cytokine Signaling in Chromaffin Cells
Wherever they may originate, there is now strong evidence that cytokines profoundly influence the adrenal medulla by inducing changes in secretion, intracellular signaling, gene transcription, and translation (318). The cytokines most studied for their influence on adrenal chromaffin cell function include IFN-α, IL-1β, IL-6, and TNF-α. These cytokines have likely received particular attention because they are prominent mediators of the systemic acute phase inflammatory response.
IFN-α is a type I interferon and signals via the IFN-α receptor (IFNAR) complex, which includes IFNAR1 and IFNAR2 subunits. Transcript expression of IFNAR2 has been reported to increase in response to TNF-α treatment of bovine adrenal chromaffin cells (271). In many cells, binding of ligand to IFNAR induces activation of janus kinase (JAK)/signal transducer and activator of transcription (STAT) signaling, with the phosphorylation of STAT1 and STAT2, which dimerize to form two different transcriptional activator complexes (a STAT1 homodimer and STAT1-STAT2-IRF9 heterotrimer). IFN-α can also activate other members of the STAT family (319). Treatment of bovine chromaffin cells with IFN-α induces phosphorylation, increased expression, and nuclear translocation of STAT1 and STAT2 (268). Further, IFN-α induces an increase in STAT3 phosphorylation but only increases nuclear STAT3 in a small proportion of cells (268). IFN-α also induces ERK1/2 signaling downstream of PKC activation in chromaffin cells (268, 320). Similar to IL-1, IFN-α inhibits ACh-stimulated CA secretion from chromaffin cells (269). IFN-α also suppresses NE uptake by cultured bovine chromaffin cells (270). IFN-α induces PKC- and ERK1/2-dependent phosphorylation of TH at the serine (Ser)-31 site (no change in phosphorylation at Ser-19 or -40), a post-translational modification that is linked to increased TH protein stability and activity (268, 321, 322). ERK1/2 activation has also been reported to contribute to histamine and Ang II-induced increases in TH Ser-31 phosphorylation in bovine adrenal chromaffin cells (323, 324). Similar mechanisms of post-translational regulation of TH by ERK1/2 in adrenal chromaffin cells may be utilized by other ERK1/2-activating cytokines.
IL-1β increases protein levels of the CA biosynthetic enzyme TH and, like IFN-α, induces phosphorylation of TH, in this case at the Ser-40 site which decreases inhibitory feedback of CAs on TH activity (280, 325). Induction of TH phosphorylation by either IL-1β or IFN-α is transient (lasting <30 min) (268, 280). Long-term (24 h) incubation with IL-1β does increase total TH protein, while incubation with IFN-α has not yet been demonstrated to change TH protein level (268, 280). IL-1β-induced phosphorylation of TH at other Ser sites and the involvement of ERK1/2 signaling in IL-1β-induced TH regulation have not been investigated. IL-1 receptors IL-1R1 and IL-1R2 are both expressed by rat adrenal medullary cells (275, 293, 292). IL-1R1 is responsible for transmembrane signaling and IL-1R2 is a decoy receptor that acts as an endogenous inhibitor, like IL-1RA, to IL-1 signaling (326). IL-1 exists in two forms, IL-1α and IL-1β. Although they are structurally very different, both IL-1α and IL-1β bind to the IL-1Rs and the neurochemical effects of both forms are similar (248). The similarity in effects of IL-1α and IL-1β is observed in adrenal chromaffin cells as well (280–282). Stimulation of chromaffin cells with IL-1 can induce PKA, ERK1/2, nitric oxide (NO)/PKC, and NO/guanylyl cyclase intracellular signaling mechanisms (280–282). Some IL-1-induced effects in chromaffin cells rely on intermediate autocrine signaling by factors such as NPY and CRH. IL-1 induction of NPY is responsible for downstream activation of PKA/NO, as well as ERK1/2, PKC and guanylyl cyclase pathways (280). IL-1-induced CRH expression can trigger a signaling loop, where CRH stimulates chromaffin cells to produce more IL-1β (272, 283). Exposure to IL-1 can also cause increased expression of IL-1R1 in PC12 cells (292). An autocrine signaling loop utilizing IL-1 is supported in vivo. Intravenous injection of IL-1β has been reported to increase IL-1β and IL-1R1 mRNA levels in the medulla of rats (275).
IL-1 alone has been reported to stimulate CA release from cultures of primary adrenal chromaffin cells and from pheochromocytomas (272, 280, 282, 284, 290). A significant portion of IL-1 induction of CA secretion relies on intermediate autocrine signaling by NPY (280, 287). In contrast to basal application of IL-1, when combined with ACh, IL-1 has an inhibitory effect on chromaffin cell CA release (288). IL-1 may function in the homeostatic control of CAs, where in the absence of stimulation by other sources, IL-1 enhances CA secretion, but when other activators are present IL-1 dampens their effects on CA secretion. An auto-regulatory mechanism utilizing IL-1 is supported in vivo, as administration of cholinergic agonists increases IL-1 mRNA and decreases IL-1 protein stores in rat adrenals, suggesting enhanced IL-1 secretion in response to cholinergic stimulation (274, 276). Interestingly, it has been reported that medullary expression of IL-1R2 is increased by immobilization stress (293). If IL-1 is involved in homeostatic control of CAs, increased expression of the decoy receptor IL-1R2 may be a stress-specific response, whereby IL-1R2 prevents IL-1 from dampening CA release in response to a psychological stressor. Differential regulation of CA biosynthetic enzymes has been reported in response to long-term exposure to cold or immobilization stress (220, 327, 328).
IL-6 binds to a receptor complex consisting of IL-6 receptor (IL-6R) and glycoprotein (gp) 130 components. IL-6 binds to IL-6R, which can exist in either soluble or membrane-bound forms. The receptor-ligand complex then couples to the gp130 signal transducing component, promoting dimerization of gp130, facilitating downstream signaling (329). JAK/STAT3 and MAPK/ERK signaling are common IL-6 activated pathways and are induced in neurons exposed to IL-6 (330). IL-6-induced STAT3 signaling has been reported in PC12 cells, and both STAT3 and ERK1/2 signaling mechanisms are supported by preliminary investigations using bovine chromaffin cells (304, 318, 331). The signal transducing component of the IL-6R complex is shared with other IL-6 family cytokines, which, like IL-6, bind to ligand-specific receptors which then complex with a gp130. Therefore, it is unsurprising that adrenal chromaffin cells are also responsive to other IL-6 family cytokines (300). In immune cells, IL-6 activates AP-1 via a Ras-dependent MAPK signaling mechanism (332). Activation of this transcription factor may also occur in chromaffin cells, as IL-6 has been reported to induce increases in c-fos transcript in PC12 cells (305). In PC12 cells, high concentrations of IL-6 were found to have an inhibitory effect the basal CA-producing function, decreasing DA and NE release as well as TH protein (304).
TNF-α binds to the plasma membrane situated receptors TNF receptor (TNFR) 1 and TNFR2. Binding of ligand to these receptors can initiate signaling cascades that utilize numerous protein kinases and lead to the activation of two major transcription factors, AP-1 and NFκB (289, 333). Bovine chromaffin cells have been found to express TNFR1 and TNFR2; however, TNFR1 appears to be the more consistently expressed of the two (289, 314, 315). A detailed investigation by Ait-Ali et al. (289) found that TNF-α signaling in bovine chromaffin cells relies on ERK1/2 and p38 signal transduction mechanisms. Further, this study determined that activation of the transcription factor AP-1 occurs downstream of ERK1/2 activation, also finding that TNF-α induces NFκB transcription factor activity in an ERK1/2-independent manner. In chromaffin cells, TNF-α-induced NFκB signaling appears to involve primarily the p65 subunit, and possibly p52 and p50 subunits. Thus, transcriptional regulation likely occurs primarily via the p65 homodimer (315). The inhibitor of NFκB, PDTC, blocks the enhancement of the transcript levels of some genes by TNF-α (315). Later experiments demonstrated that a variety of Rel family transcription factors are likely involved in the transcriptional regulatory effects of TNF-α (271). TNF-α has been reported to induce and to modulate neuropeptide transcript and protein in bovine adrenal chromaffin cells (289, 291, 313). The effects of TNF-α on CA synthesis have not been thoroughly investigated. A study of cytokine modulation of hypoxic response found that TNF-α can inhibit hypoxic induction of TH transcript (310). An oligonucleotide microarray analysis of TNF-α-induced changes in bovine chromaffin cell transcriptome identified upregulation of PNMT transcript after long (48 h), but not short (6 h) exposures to the cytokine (315).
In addition to direct signaling mechanisms of cytokines (see Figure 4), evidence is now emerging that cytokines can induce long-term changes in chromaffin cells through the activation of autocrine signaling loops. It is a well-established phenomenon that in immune cells, cytokines favor their own synthesis and that of other cytokines, resulting in the formation of autocrine signaling cascades (334). Two long established examples are IL-1 and TNF-α, which can stimulate their own production, along with numerous other cytokines and inflammatory mediators (335–338). These autocrine signaling loops can be self-regulating by stimulating the production of anti-inflammatory molecules such as IL-10 (339, 340). By inducing the production of autocrine signaling molecules, cytokines may initiate long-term signaling programs in the adrenal medulla (266, 291, 313). For example, in primary cultures of bovine adrenal chromaffin cells, treatment with IL-1α has been reported to induce synthesis of the cytokines IL-6 and TNF-α, as well as the neuropeptides VIP, NPY, and Met-Enkephalin (266, 291). Intermediate autocrine signaling by NPY is critical for CA regulatory effects of IL-1 in chromaffin cells (280, 287).
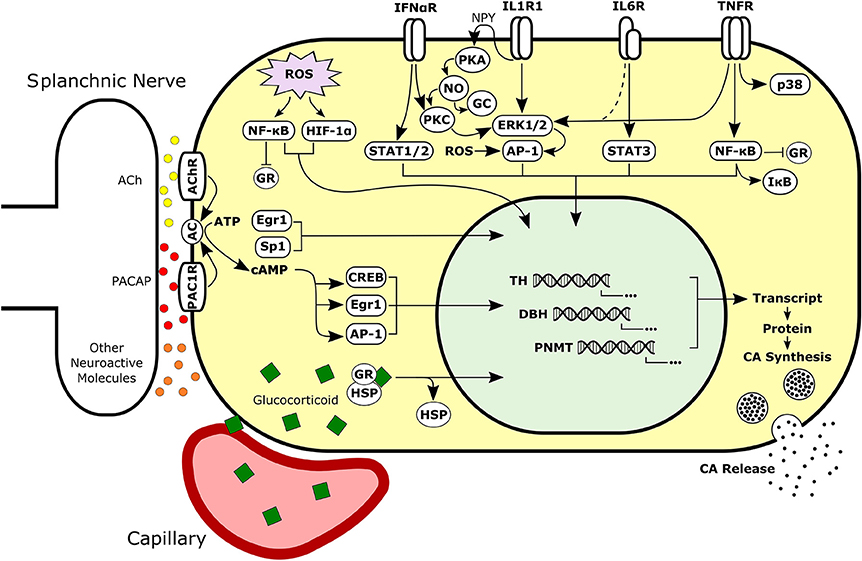
Figure 4. Simplified schematic of neural, hormonal, redox, and immune signaling pathways activated in adrenal chromaffin cells. Intracellular signals may be integrated to regulate synthesis and secretion of CAs by chromaffin cells during normal or pathological conditions. Chromaffin cells are responsive to extracellular signaling molecules in the adrenal such as glucocorticoid, cytokines, and neurotransmitters. Sympathetic neurons release neuroactive molecules including both small molecule neurotransmitters (e.g., ACh and ATP) and peptide neurotransmitters (e.g., PACAP and substance P). Dashed line represents signaling mechanism supported by unpublished findings. Ach, Acetylcholine; PACAP, Pituitary Adenylate Cyclase-Activating Peptide; AC, Adenylyl Cyclase; CA, Catecholamine; GR, Glucocorticoid Receptor; HSP, Heat Shock Protein; NPY, Neuropeptide Y; PKA, Protein Kinase A; NO, Nitric Oxide; PKC, Protein Kinase C; GC, Guanylyl Cyclase.
Whether the responses of medullary cells to cytokines primarily functions for protective action against inflammatory stimuli or if cytokines are a normal part of the diverse informational molecules that constantly regulate chromaffin cell homeostatic function, local changes in cytokine signaling within the medulla have the potential to exacerbate dysfunctional CA synthesis. The regulation of adrenal function by cytokines and the importance of immune mechanisms in contributing to the progression of hypertension and CVD are summarized above. The bi-directional relationship of the immune and neuro-endocrine systems conceivably provides fitness advantages to organisms and may be a physiologically important part of maintaining health, dysfunction of which may result in pathology. The “neuro-immune circuit” has helped to explain perplexing phenomena such as the co-morbidity of neuropsychiatric symptoms and inflammatory disease (341). Similarly, integration of immune and adrenal functions provides an explanation for the etiology of inflammation-related hypertension and may help to elucidate mechanisms of essential hypertension.
Cytokine Modulation of Glucocorticoid Signaling in Chromaffin Cells
GCs and transmitters released at splanchnic-adrenal medullary synapses are important informational molecules which control Epi biosynthesis during normal and stress conditions [see (115) and references therein]. Chromaffin cells must coordinate intracellular signaling pathways induced by these and other informational molecules in order to produce appropriate responses under diverse physiological conditions. Cytokines produced either systemically or locally may be significant modulators of adrenal CA biosynthesis by altering chromaffin cell response to GCs and neurotransmitters. How, and to what extent, chromaffin cells simultaneously process information from immune and stress circuits is not well-understood.
A number of cytokines, including IFN-α, IL-1, IL-2, and TNF-α, have been reported to have inhibitory effects either on GC-induced GR nuclear translocation, GR-GRE binding, or GR-mediated gene transcription in diverse cell types (342, 343). In mouse hippocampal HT22 cells, inhibition of GR transcriptional activity by IFN-α is dependent on JAK/STAT signaling pathway. STAT5 (not STAT1 or STAT2) appears to be the major mediator of IFN-α inhibitory effects on GR function in HT22 cells. Co-immunoprecipitation revealed that phosphorylated STAT5 binds to GR within the nucleus, and IFN-α-induced repression of GR function does not rely on inhibition of GR protein expression or nuclear translocation (344). In bovine chromaffin cells, IFN-α does not induce phosphorylation of STAT5; instead, IFN-α primarily activates STAT1 and STAT2 (268). A trimeric complex consisting of STAT1, STAT2, and interferon regulatory factor (IRF) 9, induced by IFN-α/β receptor activation, is directly regulated by the nuclear coactivator of GR, glucocorticoid receptor-interacting protein (GRIP) 1 in murine macrophages (345). Occupancy of target promoters by the STAT1-STAT2-IRF9 complex, pre-initiation complex assembly, and type I IFN stimulated gene expression were reported to be repressed by dexamethasone (Dex)-induced depletion of GRIP. Inhibition of IFN-induced gene expression by Dex appears to be caused by sequestration of GRIP1 from its functional activity as a coactivator for the STAT1-STAT2-IRF9 complex by GR. Conceivably, as long as GRIP1 protein levels are sufficiently low in adrenal chromaffin cells, this competitive mechanism could allow for the converse effect, whereby induction of the STAT1-STAT2-IRF9 complex by IFN-α leads to an inhibition of GR transcriptional effects through the repression of GRIP1 coactivation of GR.
In mouse fibroblast cells, inhibition of GR-GRE binding and GR-mediated promoter activation by IL-1 has been reported to depend on p38 signaling (346). The mechanism of p38 regulation of GR appears to involve direct phosphorylation of GR at the Ser-211 site (human) which modulates recruitment of GR coactivators to GRE-containing promoters (346, 347). So far, p38 signaling has not been demonstrated as a significant contributor in IL-1 regulation of chromaffin cell function (288). Activation of p38 signaling pathway could possibly be a mechanism for inhibition of GR function by other cytokines, such as TNF-α, which have been reported to induce p38 signaling in chromaffin cells (289).
IL-2 inhibition of GR is dependent on signal transduction by p38 (reported in murine HT-2 T-helper cell line) and by JAK3 (reported in human PBMCs), both contributing to inhibition of GR nuclear translocation (348, 349). In a murine T-lymphocyte cell line (CTLL-2), IL-2 represses GR transactivation through direct protein-protein interactions of IL-2-induced STAT5 with GR following translocation to the nucleus (350).
In human keratinocyte-derived HaCaT cells, TNF-α inhibits GC-induced transcriptional activation and GR nuclear translocation through a MEK-1/ERK-dependent mechanism (351). In human epithelial-derived HeLa cells, the inhibitory effect of MAPK signaling on GR function is demonstrated by inhibition of transcriptional activation of GR-responsive luciferase constructs in the presence of constitutively activated p38 or JNK (352). In human colon carcinoma-derived HCT116 cells, TNF-α can suppress GR transactivation by induction of FLICE-associated huge protein, which competes with GR to bind the nuclear coactivator GRIP1 (353). Additionally, mutual repression of GR and NFκB function may result from transrepression (also known as tethering), caused by direct protein-protein interactions within the nucleus, or from competition for the cofactors CBP and steroid receptor coactivator-1 (354–356).
IL-6 enhances GR function through a STAT3-dependent mechanism in rat hepatoma H4IIE cells. Like STAT5, STAT3 forms complexes with GR in the nucleus; however, unlike STAT5, formation of a STAT3-GR complex appears to enhance promoter activation, both of IL-6 responsive elements and of GREs (357).
IL-10 is another cytokine reported to have synergistic rather than inhibitory effects on GR function. In a study using human leukocytes (U937 cells and whole blood cell cultures), treatment with the anti-inflammatory cytokine IL-10 had opposite effects to TNF-α. Inhibition of IL-6 secretion in whole-blood cell cultures and promotion of IL-1R antagonist secretion by human monocyte-derived U937 cells are two effects of Dex treatment. TNF-α supresses, whereas IL-10 intensifies these Dex-induced activities (358). The mechanism of IL-10 and GR signaling crosstalk is not yet understood; however, the effect may be achieved through an increase in GR expression (358). Other possible mechanisms include IL-10-induced activation of STAT3, or IL-10-induced inhibition of NFκB, decreasing the transrepression effect (355, 359, 360).
The mechanisms of cytokine-induced GR regulation involve intermediate signaling molecules such as MAPKs and transcription factors including STAT1, STAT2, STAT3, STAT5, NFκB, and AP-1. In chromaffin cells, the most commonly reported signaling pathway by cytokines is that of the MAPK transduction molecules, particularly ERK1/2 (318). The MAPKs JNK and ERK can inhibit GR-mediated transcriptional activation by direct and indirect phosphorylation of GR at Ser-246 in rats (Ser-226 in humans), which may prevent signaling by promoting nuclear export of the transcription factor (361, 362). ERK1/2 signaling can also lead to AP-1 transcription factor activity. In chromaffin cells, both IL-6 and TNF-α have been reported to induce activation of AP-1 subunits (289, 305). Treatment of bovine chromaffin cells with TNF-α increases transcriptional activation of AP-1 responsive promoter elements and increases binding of fos and jun proteins to TPA-responsive element (TRE) and CRE sequences through an ERK1/2-dependent mechanism (289). In humans, GC resistance is positively correlated with expression of c-fos in peripheral blood mononuclear cells in vivo (363). The interaction between GR and AP-1 is mutually repressive, involving tethering at the GR-DNA binding domain; thus, inhibition of GR function may occur after GR translocation to the nucleus (364–367). Activation of AP-1 signaling may be a component of chromaffin cell regulation by other ERK1/2-inducing cytokines, such as IFN-α and IL-1, and leading to insensitivity of chromaffin cells to GCs.
Cytokine Modulation of Neurotransmitter and cAMP Signaling in Chromaffin Cells
Signal integration of neurotransmitters such as ACh and PACAP with some cytokines has been demonstrated in chromaffin cells. IL-1 inhibits ACh-induced CA release via reducing Ca2+ influx in bovine chromaffin cells; this is triggered by ERK1/2 signaling pathways (288). Similarly, IFN-α has been reported to inhibit ACh-induced CA secretion and Ca2+ influx in bovine chromaffin cells (269). Chromaffin cell response to the neuropeptide PACAP is also modified by cytokine exposure. Combined treatment with PACAP and TNF-α synergistically upregulates VIP and galanin expression in bovine chromaffin cells (313).
The signaling pathways of cAMP and cytokines may be integrated through interactions at MAPK, JAK-STAT, and NFκB. A number of studies report effects of cAMP/PKA signaling on the function of these other signaling molecules. In a human T-lymphocyte cell line (Jurkat cells) cAMP increases ERK1/2 activity by inhibiting function of haematopoietic protein tyrosine phosphatase, a negative regulator of ERK1/2 and p38 (368). In experiments using mouse (70Z/3) and human (Jurkat) lymphocyte cell lines, PKA was reported to contribute to the activation of NFκB by phosphorylating p65 following dissociation from IκB (369). In human PBMCs, cAMP has an inhibitory effect on IL-6-induced STAT1 and STAT3 binding to DNA and prevents IL-6 induction of an IL-6-responsive gene harboring a STAT1 and STAT3 binding sequence (370). Compared to cAMP regulatory effects on cytokine signaling pathways, the effects of cytokines on cAMP signaling are less well-understood. This may in part be because cAMP activation occurs early in intracellular signaling cascades and because the cellular output of cAMP activation is highly tissue-dependent (371, 372). In chromaffin cells, transcription factors involved in the response to cAMP/PKA signaling include Egr1, AP-1, and CREB (202, 203, 209). Signaling via cAMP in chromaffin cells may also lead to activation of the MAPKs p38 and ERK1/2 (200, 201). In bovine chromaffin cells, the cytokines IL-1 and TNF-α have been reported to enhance induction of VIP and substance P expression by forskolin (Forsk), an activator of adenylyl cyclase (291). The mechanisms of cytokine modulation of cAMP signaling in chromaffin cells have not been defined.
Free Radicals, Cytokines, and Hypertension
Oxidative stress and reactive oxygen species (ROS) are active contributors to the pathogenesis of hypertension and CVD. For example, ROS-mediated signaling and activation of CA biosynthesis have been associated with elevated BP in patients who suffer from obstructive sleep apnoea and as such experience intermittent hypoxia (IH) (373, 374). Adrenal derived cells exposed to IH or sustained hypoxia show ROS-mediated elevation in CA secretion, upregulation in TH, DBH and PNMT, driven by HIF-1α, Egr1, and Sp1 (375–384). Furthermore, numerous studies have highlighted the interrelationship between the immune response and oxidative stress in organs such as the heart, brain, or the kidney, ultimately leading to vessel and organ damage, and hypertension (64, 385, 386). Oxidative stress can be a major contributor of immune activation in hypertension via an autoimmune-like reaction driven by isoketals (products of ROS-mediated fatty acid oxidation) that serve as auto- antigens (106). Like HIF-1α, the transcription factor NFκβ (a transcription factor with complicated roles in inflammatory signaling pathways) is oxygen-sensitive, and release of mitochondrial H2O2 has been demonstrated to activate it (387, 388). Activation of NFκβ, Nrf2 or other proinflammatory transcription factors via ROS can modulate the biosynthesis of proinflammatory cytokines, and other molecules like VEGF, which can affect vascular damage associated with hypertension. Moreover, studies report a correlation of BP with circulating cytokines, and oxidative stress parameters; proinflammatory cytokines can lead to more ROS generation perpetuating the effects on the hypertensive state (389). For example, treatment with AngII inhibitors lowered pro-inflammatory cytokines and reduced parameters of oxidative stress in hypertensives, while dietary antioxidant intervention leads to lowered inflammatory markers such as CRP and IL-6, and improvement in BP (69, 390–392).
Concluding Remarks
Numerous cytokines regulate expression of enzymes responsible for biogenesis of CAs, the major secretory product of chromaffin cells and important regulators of BP homeostasis. Constitutively expressed cytokines may have an important function in homeostatic control of CA biosynthesis and may modify CA biosynthesis during inflammation. Further, adrenal regulation by cytokines could be an important innate mechanism for preventing the progression of hypertension, by dampening CA biosynthesis with the development of inflammation. Additionally, the inhibition of GC-induced adrenal medullary activation by cytokines may be part of an autoregulatory loop to prevent medullary over-stimulation particularly when inflammation induces a compensatory increase in GC secretion (an important endogenous anti-inflammatory molecule) (393). Increased concentration of GCs in the adrenal medulla, in the absence of such an inhibitory mechanism, would result in increased CA release (394). Thus, immune changes that coincide with hypertension could signal an adaptive inhibition of CA biosynthesis, preventing adrenal medullary over-activation through cytokine-mediated antagonism of GC-induced chromaffin cell activation. Both effects may be protective mechanisms against the development of hypertension; disturbance of such mechanisms, either by changes in local adrenal cytokine concentrations or by disruption of chromaffin cell sensitivity to cytokines, could be contributing factors to the progression of hypertension. Future investigations to determine changes in local cytokine concentrations in the adrenal medulla during prehypertension and overt hypertension will provide better insight into the relevance of cytokine-chromaffin cell signaling in this disease. Moreover, in addition to their effects in the adrenal, many cytokines also modulate CA levels in the hypothalamus and influence function of the HPA axis, and conceivably the neuro-endocrine circuit (248, 249).
The microenvironment of the adrenal gland is a viable locale for cross talk between endocrine pathways and immune response networks (395). Intermediary signaling molecules like ROS could be involved in integration of the signaling networks. Overall, the molecular mechanisms involved in integrating hormonal, neural, immune, and possibly redox inputs to the adrenal medulla remain to be elucidated. The patterns of inter-adrenal cytokine regulatory effects on CA enzyme expression could provide insight into potential converging points of interaction between these pathways (Figure 4). These networks involve direct or indirect, bidirectional interactions at the cellular and/or molecular levels of CAs, GCs, cytokines, and ROS. Understanding these unique interactions will help to improve our current understanding of adrenal functioning and HPA regulation in hypertension.
Author Contributions
CB, SK, AK, and TT contributed conception and structure and focus of the review manuscript; CB and SK compiled published reports and manuscripts relevant to the review and provided summaries and wrote the first draft of the manuscript; AK and TT provided critical reviews and edits of manuscript versions. All authors contributed to manuscript revision, read and approved the submitted version.
Funding
TT was funded by grants from the Canadian Institutes of Health Research (OPG/119463), Natural Sciences and Engineering Council of Canada (RGPIN/312776) and the NOSMFA Research Development Fund.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to acknowledge that this manuscript includes content from Collin Byrne's Master's thesis, published online by Laurentian University, Sudbury, Ontario (396).
Abbreviations
AADC, Aromatic amino acid decarboxylase; ACh, Acetylcholine; ACTH, Adrenocorticotropic hormone; Ang, Angiotensin; AP-1, Activator protein 1; AP-2, Activator protein 2; BP, Blood Pressure; CA, Catecholamine; CBP, CREB-binding protein; CNS, Central nervous system; CRE, cAMP response element; CRH, Corticotropin-releasing hormone; CVD, Cardiovascular disease; DA, Dopamine; DBH, Dopamine β-hydroxylase; DBP, Diastolic blood pressure; DOCA, Deoxycorticosterone acetate; Egr1, Early growth response 1; Epac, Exchange protein directly activated by cAMP; Epi, Epinephrine; ERK, Extracellular signal regulated kinases; GC, Glucocorticoid; GRE, Glucocorticoid response element; GRIP, Glucocorticoid receptor-interacting protein; HPA, Hypothalamic-pituitary-adrenal; IFNAR, IFN-α receptor; IL-1R, IL-1 receptor; IL-6R, IL-6 receptor; IRF, Interferon regulatory factor; JAK, Janus kinase; LDCV, Large dense core vesicle; L-DOPA, L-3,4-dihydroxyphenylalanine; MAPK, Mitogen-activated protein kinase; NE, Norepinephrine; NO, Nitric oxide; PACAP, Pituitary adenylate cyclase-activating peptide; PKA, Protein kinase A; PKC, Protein kinase C; PLC, Phospholipase C; PNMT, Phenylethanolamine N-methyltransferase; RAAS, Renin-angiotensin-aldosterone system; SA, Sympathetic-adrenal; SBP, Systolic blood pressure; Ser, Serine; SHR, Spontaneously hypertensive rat; SNS, Sympathetic nervous system; Sp1, Specificity protein 1; SSV, Small synaptic vesicle; STAT, Signal transducer and activator of transcription; TH, Tyrosine hydroxylase; TNFR, TNF receptor; TRE, TPA-responsive element.
References
1. Wilkins K, Campbell N, Joffres M, McAlister F, Nichol M, Quach S, et al. Blood Pressure in Canadian Adults. Health Report, Statistics Canada (2010). p. 37–46.
2. Weaver CG, Clement FM, Campbell NRC, James MT, Klarenbach SW, Hemmelgarn BR, et al. Healthcare costs attributable to hypertension: canadian population-based cohort study. Hypertension (2015) 66:502–8. doi: 10.1161/HYPERTENSIONAHA.115.05702
3. Whelton PK, Carey RM, Aronow WS, Ovbiagele B, Casey DE, Smith SC, et al. High blood pressure clinical practice guideline: executive summary 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the prevention, detection, evaluation, and management of high blood pressure in adults executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension (2017) 71:1269–324. doi: 10.1161/HYP.0000000000000066
4. World Health Organization. Global Health Risks: Mortality and Burden of Disease Attributable to Selected Major Risks. Geneva: World Health Organization (2009).
5. Rosamond W, Flegal K, Furie K, Go A, Greenlund K, Haase N, et al. Heart disease and stroke statistics - 2008 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation (2008) 117:e25–146. doi: 10.1161/CIRCULATIONAHA.107.187998
6. Forouzanfar MH, Liu P, Roth GA, Ng M, Biryukov S, Marczak L, et al. Global burden of hypertension and systolic blood pressure of at least 110 to 115mmHg, 1990-2015. JAMA (2017) 317:165–82. doi: 10.1001/jama.2016.19043
7. Lawes CMM, Vander Hoorn S, Rodgers A. Global burden of blood-pressure-related disease, 2001. Lancet (2008) 371:1513–8. doi: 10.1016/S0140-6736(08)60655-8
8. Wilkins K, Campbell N, Joffres M, McAlister F, Nichol M, Quach S, et al. Blood Pressure in Canadian Adults. Health Reports, Statistics Canada (2010). p. 37–46.
9. Gaziano TA, Bitton A, Anand S, Weinstein MC. The global cost of nonoptimal blood pressure. J Hypertens. (2009) 27:1472–7. doi: 10.1097/HJH.0b013e32832a9ba3
10. Kaplan NM, Victor RG. Cost-effectiveness of treating hypertension. In: Goolsby J, Vosburgh, A, editors, Kaplan's Clinical Hypertension: Eleventh Edition. Philadelphia, PA: Lippincott Williams & Wilkins (2014). p. 162–163.
11. Pedrosa RP, Drager LF, Gonzaga CC, Sousa MG, De Paula LKG, Amaro ACS, et al. Obstructive sleep apnea: the most common secondary cause of hypertension associated with resistant hypertension. Hypertension (2011) 58:811–7. doi: 10.1161/HYPERTENSIONAHA.111.179788
12. Leung AA, Daskalopoulou SS, Dasgupta K, McBrien K, Butalia S, Zarnke KB, et al. Hypertension Canada's 2017 guidelines for diagnosis, risk assessment, prevention, and treatment of hypertension in adults. Can J Cardiol. (2017) 33:557–76. doi: 10.1016/j.cjca.2017.03.005
13. Rimoldi SF, Scherrer U, Messerli FH. Secondary arterial hypertension: when, who, and how to screen? Eur Heart J. (2014) 35:1245–54. doi: 10.1093/eurheartj/eht534
14. Padmanabhan S, Caulfield M, Dominiczak AF. Genetic and molecular aspects of hypertension. Circ Res. (2015) 116:937–59. doi: 10.1161/CIRCRESAHA.116.303647
15. Cowley AW. The genetic dissection of essential hypertension. Nat Rev Genet. (2006) 7:829–40. doi: 10.1038/nrg1967
16. Coffman TM. Under pressure: the search for the essential mechanisms of hypertension. Nat Med. (2011) 17:1402–9. doi: 10.1038/nm.2541
17. Krum H, Schlaich MP, Sobotka PA, Böhm M, Mahfoud F, Rocha-Singh K, et al. Percutaneous renal denervation in patients with treatment-resistant hypertension: final 3-year report of the Symplicity HTN-1 study. Lancet (2014) 383:622–9. doi: 10.1016/S0140-6736(13)62192-3
18. Lohmeier TE, Iliescu R. Chronic lowering of blood pressure by carotid baroreflex activation: mechanisms and potential for hypertension therapy. Hypertension (2011) 57:880–6. doi: 10.1161/HYPERTENSIONAHA.108.119859
19. Krause T, Lovibond K, Caulfield M, McCormack T, Williams B. Management of hypertension: summary of NICE guidance. BMJ (2011) 343:d4891. doi: 10.1136/bmj.d4891
20. Mancia G, Fagard R, Narkiewicz K, Redon J, Zanchetti A, Böhm M, et al. 2013 ESH/ESC guidelines for the management of arterial hypertension: the Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). J Hypertens. (2013) 31:1281–357. doi: 10.1097/01.hjh.0000431740.32696.cc
21. Hypertension Canada. CHEP Recommendations for the Management of Hypertension. Markham, ON: Hypertension Canada (2015).
22. James PA, Oparil S, Carter BL, Cushman WC, Dennison-Himmelfarb C, Handler J, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA (2014) 311:507–20. doi: 10.1001/jama.2013.284427
23. Nagatsu T, Levitt M, Udenfriend S. Tyrosine hydroxylase. The initial step in norepinephrine biosynthesis. J Biol Chem. (1964) 239:2910–7.
24. Christenson JG, Dairman W, Udenfriend S. On the identity of DOPA decarboxylase and 5-hydroxytryptophan decarboxylase (immunological titration-aromatic L-amino acid decarboxylase-serotonin-dopamine-norepinephrine). Proc Natl Acad Sci USA. (1972) 69:343–7.
25. Weinshilboum R, Axelrod J. Serum dopamine-beta-hydroxylase activity. Circ Res. (1971) 28:307–15.
26. Axelrod J. Purification and properties of phenylethanolamine-N-methyl transferase. J Biol Chem. (1962) 237:1657–60.
27. Hwang O, Kim ML, Lee JD. Differential induction of gene expression of catecholamine biosynthetic enzymes and preferential increase in norepinephrine by forskolin. Biochem Pharmacol. (1994) 48:1927–34.
28. Wong DL, Tank AW. Stress-induced catecholaminergic function: transcriptional and post-transcriptional control. Stress (2007) 10:121–30. doi: 10.1080/10253890701393529
29. Douglas WW. Stimulus-secretion coupling: the concept and clues from chromaffin and other cells. Br J Pharmacol. (1968) 34:451–74.
30. Smith U, Smith DS, Winkler H, Ryan JW. Exocytosis in the adrenal medulla demonstrated by freeze-etching. Science (1973) 179:79–82.
31. Caron MG, Lefkowitz RJ. Catecholamine receptors: structure, function, and regulation. In: C. Wayne Bardin, editor. Recent Prog Horm Res. 48th Edn. (San Diego, CA: Academic Press, Inc.) (1993). p. 277–90.
32. Missale C, Nash SR, Robinson SW, Jaber M, Caron MG. Dopamine receptors: from structure to function. Physiol Rev. (1998) 78:189–225.
33. Carey RM. Renal dopamine system: paracrine regulator of sodium homeostasis and blood pressure. Hypertension (2001) 38:297–302. doi: 10.1161/hy0901.096422
34. Guimarães S, Moura D. Vascular adrenoceptors: an update. Pharmacol. Rev. (2001) 53:319–56. Available online at: http://pharmrev.aspetjournals.org/content/53/2/319/tab-article-info
35. Johnson M. Molecular mechanisms of beta(2)-adrenergic receptor function, response, and regulation. J Allergy Clin Immunol. (2006) 117:18–24; quiz: 25. doi: 10.1016/j.jaci.2005.11.012
36. Adameova A, Abdellatif Y, Dhalla NS. Role of the excessive amounts of circulating catecholamines and glucocorticoids in stress-induced heart disease. Can J Physiol Pharmacol. (2009) 87:493–514. doi: 10.1139/y09-042
37. Bühler F, Amann F, Bolli P. Elevated adrenaline and increased -adrenoceptor-mediated vasoconstriction in essential hypertension. J Cardiovasc Pharmacol. (1982) 4:134–8.
38. Borkowski KR, Quinn P. Adrenaline and the development of genetic hypertension. J Hypertens Suppl. (1984) 2:S81–3.
39. Jablonskis LT, Howe PR. Elevated plasma adrenaline in spontaneously hypertensive rats. Blood Pressure (1994) 3:106–11.
41. Goldstein DS. Plasma catecholamines and essential hypertension. An analytical review. Hypertension (1983) 5:86–99.
42. Berry MD, Juorio AV, Li XM, Boulton AA. Aromatic L-amino acid decarboxylase: a neglected and misunderstood enzyme. Neurochem Res. (1996) 21:1075–87.
43. Bowsher RR, Henry DP. Aromatic L-amino acid decarboxylase. In: Boulton AA, Baker GB, Yu PH, editors. Neuromethods Neurotransmitter Enzymes, Vol. 5 (Totowa, NJ: Humana Press) (1986). p. 33–78.
44. Kvetnansky R, Weise VK, Kopin IJ. Elevation of adrenal tyrosine hydroxylase and phenylethanolamine-N-methyl transferase by repeated immobilization of rats. Endocrinology (1970) 87:744–9.
45. Kvetnansky R, Gewirtz GP, Weise VK, Kopin IJ. Catecholamine-synthesizing enzymes in the rat adrenal gland during exposure to cold. Am J Physiol. (1971) 220:928–31.
46. Thoenen H. Comparison between the effect of neuronal activity and nerve growth factor on the enzymes involved in the synthesis of norepinephrine. Pharmacol Rev. (1972) 24:255–67.
47. Tai TC, Wong DL. Protein kinase A and protein kinase C signaling pathway interaction in phenylethanolamine N-methyltransferase gene regulation. J Neurochem. (2003) 85:816–29. doi: 10.1046/j.1471-4159.2003.01728.x
48. Tekin I, Roskoski R, Carkaci-Salli N, Vrana KE. Complex molecular regulation of tyrosine hydroxylase. J Neural Transm. (2014) 121:1451–81. doi: 10.1007/s00702-014-1238-7
49. Unsworth BR, Hayman GT, Carroll A, Lelkes PI. Tissue-specific alternative mRNA splicing of phenylethanolamine N-methyltransferase (PNMT) during development by intron retention. Int J Dev Neurosci. (1999) 17:45–55.
50. Hwang O, Joh TH. Effects of cAMP, glucocorticoids, and calcium on dopamine beta-hydroxylase gene expression in bovine chromaffin cells. J Mol Neurosci. (1993) 4:173–83.
51. Reja V, Goodchild AK, Pilowsky PM. Catecholamine-related gene expression correlates with blood pressures in SHR. Hypertension (2002) 40:342–7. doi: 10.1161/01.HYP.0000027684.06638.63
52. Nguyen P, Peltsch H, de Wit J, Crispo J, Ubriaco G, Eibl J, et al. Regulation of the phenylethanolamine N-methyltransferase gene in the adrenal gland of the spontaneous hypertensive rat. Neurosci Lett. (2009) 461:280–4. doi: 10.1016/j.neulet.2009.06.022
53. Nagatsu T, Kato T, Numata(sudo) Y, Ikuta K, Sano M. Norepinephrine-synthesizing enzymes in brain, adrenals and peripheral sympathetic nerves of spontaneously hypertensive rats. Jpn J Pharmacol. (1977) 27:531–5.
54. Grobecker H, Saavedra JM, Weise VK. Biosynthetic enzyme activities and catecholamines in adrenal glands of genetic and experimental hypertensive rats. Circ Res. (1982) 50:742–6.
55. Kaneda N, Ichinose H, Kobayashi K, Oka K, Kishi F, Nakazawa A, et al. Molecular cloning of cDNA and chromosomal assignment of the gene for human phenylethanolamine N-methyltransferase, the enzyme for epinephrine biosynthesis. J Biol Chem. (1988) 263:7672–7.
56. Koike G, Jacob HJ, Krieger JE, Szpirer C, Hoehe MR, Horiuchi M, et al. Investigation of the phenylethanolamine N-methyltransferase gene as a candidate gene for hypertension. Hypertension (1995) 26:595–601. Available online at: http://pharmrev.aspetjournals.org/content/52/4/595/tab-article-info
57. Hoehe MR, Plaetke R, Otterud B, Stauffer D, Holik J, Byerley WF, et al. Genetic linkage of the human gene for phenylethanol-amine N-methyltransferase (PNMT), the adrenaline-synthesizing enzyme, to DNA markers on chromosome 17q21-q22. Hum Mol Genet. (1992) 1:175–8.
58. Hollan I, Meroni PL, Ahearn JM, Cohen Tervaert JW, Curran S, Goodyear CS, et al. Cardiovascular disease in autoimmune rheumatic diseases. Autoimmun Rev. (2013) 12:1004–15. doi: 10.1016/j.autrev.2013.03.013
59. Libby P. Inflammation and cardiovascular disease mechanisms. Am J Clin Nutr. (2006) 83:456S–60S. doi: 10.1093/ajcn/83.2.456S
60. Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. (2006) 6:508–19. doi: 10.1038/nri1882
61. Zinger H, Sherer Y, Shoenfeld Y. Atherosclerosis in autoimmune rheumatic diseases-mechanisms and clinical findings. Clin Rev Allergy Immunol. (2009) 37:20–8. doi: 10.1007/s12016-008-8094-x
62. Schillaci G, Maggi P, Madeddu G, Pucci G, Mazzotta E, Penco G, et al. Symmetric ambulatory arterial stiffness index and 24-h pulse pressure in HIV infection: results of a nationwide cross-sectional study. J Hypertens. (2013) 31:560–7. doi: 10.1097/HJH.0b013e32835ca949
63. Armstrong AW, Harskamp CT, Armstrong EJ. The association between psoriasis and hypertension: a systematic review and meta-analysis of observational studies. J Hypertens. (2013) 31:433–42. discussion: 442–3. doi: 10.1097/HJH.0b013e32835bcce1
64. Norlander AE, Madhur MS, Harrison DG. The immunology of hypertension. J Exp Med. (2018) 215:21–33. doi: 10.1084/jem.20171773
65. Sesso HD, Buring JE, Rifai N, Blake GJ, Gaziano JM, Ridker PM. C-reactive protein and the risk of developing hypertension. JAMA (2003) 290:2945–51. doi: 10.1001/jama.290.22.2945
66. Purcell ES, Gattone VH. Immune system of the spontaneously hypertensive rat. I. Sympathetic innervation. Exp Neurol. (1992) 117:44–50.
67. Dzielak DJ. Immune mechanisms in experimental and essential hypertension. Am J Physiol. (1991) 260:R459–67.
68. Bendich A, Belisle EH, Strausser HR. Immune system modulation and its effect on the blood pressure of the spontaneously hypertensive male and female rat. Biochem Biophys Res Commun. (1981) 99:600–7.
69. Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, et al. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. (2007) 204:2449–60. doi: 10.1084/jem.20070657
70. Peeters AC, Netea MG, Janssen MC, Kullberg BJ, Van der Meer JW, Thien T. Pro-inflammatory cytokines in patients with essential hypertension. Eur J Clin Invest. (2001) 31:31–6. doi: 10.1046/j.1365-2362.2001.00743.x
71. Stumpf C, Auer C, Yilmaz A, Lewczuk P, Klinghammer L, Schneider M, et al. Serum levels of the Th1 chemoattractant interferon-gamma-inducible protein (IP) 10 are elevated in patients with essential hypertension. Hypertens Res. (2011) 34:484–8. doi: 10.1038/hr.2010.258
72. Stumpf C, John S, Jukic J, Yilmaz A, Raaz D, Schmieder RE, et al. Enhanced levels of platelet P-selectin and circulating cytokines in young patients with mild arterial hypertension. J Hypertens. (2005) 23:995–1000. doi: 10.1097/01.hjh.0000166840.63312.12
73. Chrysohoou C, Pitsavos C, Panagiotakos DB, Skoumas J, Stefanadis C. Association between prehypertension status and inflammatory markers related to atherosclerotic disease: the ATTICA Study. Am J Hypertens. (2004) 17:568–73. doi: 10.1016/j.amjhyper.2004.03.675
74. Granger JP. An emerging role for inflammatory cytokines in hypertension. Am J Physiol. (2006) 290:H923–4. doi: 10.1152/ajpheart.01278.2005
75. Lee DL, Sturgis LC, Labazi H, Osborne JB, Fleming C, Pollock JS, et al. Angiotensin II hypertension is attenuated in interleukin-6 knockout mice. Am J Physiol Heart Circ Physiol. (2006) 290:935–40. doi: 10.1152/ajpheart.00708.2005
76. Brands MW, Banes-Berceli AKL, Inscho EW, Al-Azawi H, Allen AJ, Labazi H. Interleukin 6 knockout prevents angiotensin II hypertension: role of renal vasoconstriction and janus kinase 2/signal transducer and activator of transcription 3 activation. Hypertension (2010) 56:879–84. doi: 10.1161/HYPERTENSIONAHA.110.158071
77. Lima VV, Zemse SM, Chiao CW, Bomfim GF, Tostes RC, Clinton Webb R, et al. Interleukin-10 limits increased blood pressure and vascular RhoA/Rho-kinase signaling in angiotensin II-infused mice. Life Sci. (2016) 145:137–43. doi: 10.1016/j.lfs.2015.12.009
78. Ishimitsu T, Uehara Y, Numabe A, Tsukada H, Ogawa Y, Yagi S. Antihypertensive effect of interleukin-2 in salt-sensitive Dahl rats. Hypertension (1994) 23:68–73.
80. Chan CT, Moore JP, Budzyn K, Guida E, Diep H, Vinh A, et al. Reversal of vascular macrophage accumulation and hypertension by a CCR2 antagonist in deoxycorticosterone/salt-treated mice. Hypertension (2012) 60:1207–12. doi: 10.1161/HYPERTENSIONAHA.112.201251
81. McCann SM, Kimura M, Karanth S, Yu WH, Mastronardi CA, Rettori V. The mechanism of action of cytokines to control the release of hypothalamic and pituitary hormones in infection. Ann NY Acad Sci. (2006) 917:4–18. doi: 10.1111/j.1749-6632.2000.tb05368.x
82. Shi P, Diez-Freire C, Jun JY, Qi Y, Katovich MJ, Li Q, et al. Brain microglial cytokines in neurogenic hypertension. Hypertension (2010) 56:297–303. doi: 10.1161/HYPERTENSIONAHA.110.150409
83. Weidenfeld J, Abramsky O, Ovadia H. Evidence for the involvement of the central adrenergic system in interleukin 1-induced adrenocortical response. Neuropharmacology (1989) 28:1411–4.
84. Wilson BC, Fulop K, Summerlee AJ. Changing effect of i.c.v. IL-1 beta on vasopressin release in anaesthetized, female rats at different stages of lactation: role of prostaglandins and noradrenaline. J Neuroendocrinol. (1996) 8:915–20.
85. Baudry N, Rasetti C, Vicaut E. Differences between cytokine effects in the microcirculation of the rat. Am J Physiol. (1996) 271:H1186–92.
86. Gaynor ER, Vitek L, Sticklin L, Creekmore SP, Ferraro ME, Thomas JX, et al. The hemodynamic effects of treatment with interleukin-2 and lymphokine-activated killer cells. Ann Intern Med. (1988) 109:953–8.
87. Quan W, Ramirez M, Taylor C, Quan F, Vinogradov M, Walker P. Administration of high-dose continuous infusion interleukin-2 to patients age 70 or over. Cancer Biother Radiopharm. (2005) 20:11–5. doi: 10.1089/cbr.2005.20.11
88. Zeilender S, Davis D, Fairman RP, Glauser FL. Inotropic and vasoactive drug treatment of interleukin 2 induced hypotension in sheep. Cancer Res. (1989) 49:4423–6.
89. Samlowski WE, Petersen R, Cuzzocrea S, Macarthur H, Burton D, McGregor JR, et al. A nonpeptidyl mimic of superoxide dismutase, M40403, inhibits dose-limiting hypotension associated with interleukin-2 and increases its antitumor effects. Nat Med. (2003) 9:750–5. doi: 10.1038/nm874
90. Tinsley JH, South S, Chiasson VL, Mitchell BM. Interleukin-10 reduces inflammation, endothelial dysfunction, and blood pressure in hypertensive pregnant rats. Am J Physiol Regul Integr Compar Physiol. (2010) 298:R713–9. doi: 10.1152/ajpregu.00712.2009
91. Viel EC, Lemarié C a, Benkirane K, Paradis P, Schiffrin EL. Immune regulation and vascular inflammation in genetic hypertension. Am J Physiol Heart Circ Physiol. (2010) 298:938–44. doi: 10.1152/ajpheart.00707.2009
92. Faggioni R, Allavena P, Cantoni L, Carelli M, Demitri MT, Delgado R, et al. Mechanisms of interleukin-2-induced hydrothoraxy in mice: protective effect of endotoxin tolerance and dexamethasone and possible role of reactive oxygen intermediates. J Immunother Emphas Tumor Immunol. (1994) 15:194–201.
93. Suzuki K, Furui H, Kaneko M, Takagi K, Satake T. Priming effect of recombinant human interleukin-2 and recombinant human interferon-gamma on human neutrophil superoxide production. Arzneimittel-Forschung (1990) 40:1176–9.
94. Macarthur H, Westfall TC, Riley DP, Misko TP, Salvemini D. Inactivation of catecholamines by superoxide gives new insights on the pathogenesis of septic shock. Proc Natl Acad Sci USA. (2000) 97:9753–8. doi: 10.1073/pnas.97.17.9753
95. Chae CU, Lee RT, Rifai N, Ridker PM. Blood pressure and inflammation in apparently healthy men. Hypertension (2001) 38:399–403. doi: 10.1161/01.HYP.38.3.399
96. Schrader LI, Kinzenbaw DA, Johnson AW, Faraci FM, Didion SP. IL-6 deficiency protects against angiotensin II induced endothelial dysfunction and hypertrophy. Arterioscler Thromb Vasc Biol. (2007) 27:2576–81. doi: 10.1161/ATVBAHA.107.153080
97. Sturgis LC, Cannon JG, Schreihofer DA, Brands MW. The role of aldosterone in mediating the dependence of angiotensin hypertension on IL-6. Am J Physiol Regul Integr Compar Physiol. (2009) 297:1742–8. doi: 10.1152/ajpregu.90995.2008
98. Iversen PO, Nicolaysen A, Kvernebo K, Benestad HB, Nicolaysen G. Human cytokines modulate arterial vascular tone via endothelial receptors. Pflügers Archiv. (1999) 439:93–100.
99. Feinberg B, Kurzrock R, Talpaz M, Blick M, Saks S, Gutterman JU. A phase I trial of intravenously-administered recombinant tumor necrosis factor-alpha in cancer patients. J Clin Oncol. (1988) 6:1328–34.
100. Saks S, Rosenblum M. Recombinant human TNF-alpha: preclinical studies and results from early clinical trials. Immunol Ser. (1992) 56:567–87.
101. Venegas-Pont M, Manigrasso MB, Grifoni SC, LaMarca BB, Maric C, Racusen LC, et al. Tumor necrosis factor-alpha antagonist etanercept decreases blood pressure and protects the kidney in a mouse model of systemic lupus erythematosus. Hypertension (2010) 56:643–9. doi: 10.1161/HYPERTENSIONAHA.110.157685
102. Tran LT, MacLeod KM, McNeill JH. Chronic etanercept treatment prevents the development of hypertension in fructose-fed rats. Mol Cell Biochem. (2009) 330:219–28. doi: 10.1007/s11010-009-0136-z
103. Elmarakby AA, Quigley JE, Imig JD, Pollock JS, Pollock DM. TNF-alpha inhibition reduces renal injury in DOCA-salt hypertensive rats. Am J Physiol Regul Integr Compar Physiol. (2008) 294:R76–83. doi: 10.1152/ajpregu.00466.2007
104. Samuelsson AM, Alexanderson C, Mölne J, Haraldsson B, Hansell P, Holmäng A. Prenatal exposure to interleukin-6 results in hypertension and alterations in the renin-angiotensin system of the rat. J Physiol. (2006) 575:855–67. doi: 10.1113/jphysiol.2006.111260
105. Cavallotti D, Artico M, Iannetti G, Cavallotti C. Occurrence of adrenergic nerve fibers in human thymus during immune response. Neurochem Int. (2002) 40:211–21. doi: 10.1016/S0197-0186(01)00074-2
106. Harrison DG, Guzik TJ, Lob HE, Madhur MS, Marvar PJ, Thabet SR, et al. Inflammation, immunity, and hypertension. Hypertension (2011) 57:132–40. doi: 10.1161/HYPERTENSIONAHA.110.163576
107. Blasi ER, Rocha R, Rudolph AE, Blomme EAG, Polly ML, McMahon EG. Aldosterone/salt induces renal inflammation and fibrosis in hypertensive rats. Kidney Int. (2003) 63:1791–800. doi: 10.1046/j.1523-1755.2003.00929.x
108. Crowley SD, Frey CW, Gould SK, Griffiths R, Ruiz P, Burchette JL, et al. Stimulation of lymphocyte responses by angiotensin II promotes kidney injury in hypertension. Am J Physiol Renal Physiol. (2008) 295:F515–24. doi: 10.1152/ajprenal.00527.2007
109. Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, et al. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension (2010) 55:500–7. doi: 10.1161/HYPERTENSIONAHA.109.145094
110. Kirabo A, Fontana V, de Faria APC, Loperena R, Galindo CL, Wu J, et al. DC isoketal-modified proteins activate T cells and promote hypertension. J Clin Invest. (2014) 124:4642–56. doi: 10.1172/JCI74084
111. Foss JD, Kirabo A, Harrison DG. Do high-salt microenvironments drive hypertensive inflammation? Am J Physiol Regul Integr Comp Physiol. (2017) 312:R1–4. doi: 10.1152/ajpregu.00414.2016
112. Kirabo A. A new paradigm of sodium regulation in inflammation and hypertension. Am J Physiol Regul Integr Comp Physiol. (2017) 313:R706–710. doi: 10.1152/ajpregu.00250.2017
113. Curtis BM, O'Keefe JH. Autonomic tone as a cardiovascular risk factor: the dangers of chronic fight or flight. Mayo Clin Proc Mayo Clin. (2002) 77:45–54. doi: 10.4065/77.1.45
114. Cannon W, La Paz D de. Emotional stimulation of adrenal secretion. Am J Physiol. (1911) 28:64–70.
115. Wong DL. Epinephrine biosynthesis: hormonal and neural control during stress. Cell Mol Neurobiol. (2006) 26:891–900. doi: 10.1007/s10571-006-9056-6
116. Ulrich-Lai YM, Herman JP. Neural regulation of endocrine and autonomic stress responses. Nat Rev Neurosci. (2009) 10:397–409. doi: 10.1038/nrn2647
117. Smith SM, Vale WW. The role of the hypothalamic-pituitary-adrenal axis in neuroendocrine responses to stress. Dialogues Clin Neurosci. (2006) 8:383–95. Available online at: https://www.dialogues-cns.org/wp-content/uploads/issues/08/DialoguesClinNeurosci-8-383.pdf
118. Palkovits M. Neuropeptides in the hypothalamo-hypophyseal system: lateral retrochiasmatic area as a common gate for neuronal fibers towards the median eminence. Peptides (1984) 5(Suppl. 1):35–9.
119. Romanov RA, Alpár A, Hokfelt T, Harkany T. Molecular diversity of corticotropin-releasing hormone mRNA-containing neurons in the hypothalamus. J Endocrinol. (2017) 232:R161–72. doi: 10.1530/JOE-16-0256
120. Wurtman RJ. Stress and the adrenocortical control of epinephrine synthesis. Metabolism (2002) 51:11–4. doi: 10.1053/meta.2002.33185
121. Nicolaides NC, Galata Z, Kino T, Chrousos GP, Charmandari E. The human glucocorticoid receptor: molecular basis of biologic function. Steroids (2010) 75:1–12. doi: 10.1016/j.steroids.2009.09.002
122. Tasker JG, Di S, Malcher-Lopes R. Minireview: rapid glucocorticoid signaling via membrane-associated receptors. Endocrinology (2006) 147:5549–56. doi: 10.1210/en.2006-0981
123. Galon J, Franchimont D, Hiroi N, Frey G, Boettner A, Ehrhart-Bornstein M, et al. Gene profiling reveals unknown enhancing and suppressive actions of glucocorticoids on immune cells. FASEB J. (2002) 16:61–71. doi: 10.1096/fj.01-0245com
124. Kelly JJ, Mangos G, Williamson PM, Whitworth JA. Cortisol and hypertension. Clin Exp Pharmacol Physiol Suppl. (1998) 25:S51–6.
125. Cicala MV, Mantero F. Hypertension in Cushing's syndrome: from pathogenesis to treatment. Neuroendocrinology (2010) 92:44–9. doi: 10.1159/000314315
126. Peppa M, Krania M, Raptis SA. Hypertension and other morbidities with Cushing's syndrome associated with corticosteroids: a review. Integr Blood Pressure Control (2011) 4:7–16. doi: 10.2147/IBPC.S9486
127. Pilipović I, Radojević K, Perišić M, Leposavić G. Glucocorticoid-catecholamine interplay within the composite thymopoietic regulatory network. Ann NY Acad Sci. (2012) 1261:34–41. doi: 10.1111/j.1749-6632.2012.06623.x
128. Sharara-Chami RI, Joachim M, Pacak K, Majzoub JA. Glucocorticoid treatment–effect on adrenal medullary catecholamine production. Shock (2010) 33:213–7. doi: 10.1097/SHK.0b013e3181af0633
129. Wurtman RJ, Axelrod J. Adrenaline synthesis: control by the pituitary gland and adrenal glucocorticoids. Science (1965) 150:1464–5. doi: 10.1126/science.150.3702.1464
130. Evinger MJ, Towle AC, Park DH, Lee P, Joh TH. Glucocorticoids stimulate transcription of the rat phenylethanolamine N-methyltransferase (PNMT) gene in vivo and in vitro. Cell Mol Neurobiol. (1992) 12:193–215. doi: 10.1007/BF00712926
131. Wong DL, Lesage A, Siddall B, Funder JW. Glucocorticoid regulation of phenylethanolamine N-methyltransferase in vivo. FASEB J. (1992) 6:3310–5.
132. Berenbeim DM, Wong DL, Masover SJ, Ciaranello RD. Regulation of synthesis and degradation of rat adrenal phenylethanolamine N-methyltransferase. III. Stabilization of PNMT against thermal and tryptic degradation by S-adenosylmethionine. Mol Pharmacol. (1979) 16:482–90.
133. Wong DL, Hayashi RJ, Ciaranello RD. Regulation of biogenic amine methyltransferases by glucocorticoids via S-adenosylmethionine and its metabolizing enzymes, methionine adenosyltransferase and S-adenosylhomocysteine hydrolase. Brain Res. (1985) 330:209–16.
134. Wong DL, Zager EL, Ciaranello RD. Effects of hypophysectomy and dexamethasone administration on central and peripheral S-adenosylmethionine levels. J Neurosci. (1982) 2:758–64.
135. Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci USA. (1976) 73:2424–8.
136. Kim KT, Park DH, Joh TH. Parallel up-regulation of catecholamine biosynthetic enzymes by dexamethasone in PC12 cells. J Neurochem. (1993) 60:946–51.
137. Byrd JC, Hadjiconstantinou M, Cavalla D. Epinephrine synthesis in the PC12 pheochromocytoma cell line. Eur J Pharmacol. (1986) 127:139–42.
138. Lewis EJ, Harrington CA, Chikaraishi DM. Transcriptional regulation of the tyrosine hydroxylase gene by glucocorticoid and cyclic AMP. Proc Natl Acad Sci USA. (1987) 84:3550–4.
139. Lewis EJ, Tank AW, Weiner N, Chikaraishi DM. Regulation of tyrosine hydroxylase mRNA by glucocorticoid and cyclic AMP in a rat pheochromocytoma cell line. Isolation of a cDNA clone for tyrosine hydroxylase mRNA. J Biol Chem. (1983) 258:14632–7.
140. McMahon A, Sabban EL. Regulation of expression of dopamine beta-hydroxylase in PC12 cells by glucocorticoids and cyclic AMP analogues. J Neurochem. (1992) 59:2040–7.
141. Tischler AS, Perlman RL, Morse GM, Sheard BE. Glucocorticoids increase catecholamine synthesis and storage in PC12 pheochromocytoma cell cultures. J Neurochem. (1983) 40:364–70.
142. Stachowiak MK, Hong JS, Viveros OH. Coordinate and differential regulation of phenylethanolamine N-methyltransferase, tyrosine hydroxylase and proenkephalin mRNAs by neural and hormonal mechanisms in cultured bovine adrenal medullary cells. Brain Res. (1990) 510:277–88.
143. Rani CSS, Elango N, Wang SS, Kobayashi K, Strong R. Identification of an activator protein-1-like sequence as the glucocorticoid response element in the rat tyrosine hydroxylase gene. Mol Pharmacol. (2009) 75:589–98. doi: 10.1124/mol.108.051219
144. Hagerty T, Fernandez E, Lynch K, Wang SS, Morgan WW, Strong R. Interaction of a glucocorticoid-responsive element with regulatory sequences in the promoter region of the mouse tyrosine hydroxylase gene. J Neurochem. (2001) 78:1379–88. doi: 10.1046/j.1471-4159.2001.00521.x
145. Tai TC, Claycomb R, Her S, Bloom AK, Wong DL. Glucocorticoid responsiveness of the rat phenylethanolamine N-methyltransferase gene. Mol Pharmacol. (2002) 61:1385–92. doi: 10.1124/mol.61.6.1385
146. Ross ME, Evinger MJ, Hyman SE, Carroll JM, Mucke L, Comb M, et al. Identification of a functional glucocorticoid response element in the phenylethanolamine N-methyltransferase promoter using fusion genes introduced into chromaffin cells in primary culture. J Neurosci. (1990) 10:520–30.
147. Wong DL, Siddall BJ, Ebert SN, Bell RA, Her S. Phenylethanolamine N-methyltransferase gene expression: synergistic activation by Egr-1, AP-2 and the glucocorticoid receptor. Brain Res Mol Brain Res. (1998) 61:154–61.
148. Axelrod J, Reisine TD. Stress hormones: their interaction and regulation. Science (1984) 224:452–9.
149. Ehrhart-Bornstein M, Hinson JOYP, Bornstein SR, Scherbaum WA, Vinson GP. Intraadrenal interactions in the regulation of adrenocortical steroidogenesis. Endocr Rev. (2017) 19:101–43. doi: 10.1210/edrv.19.2.0326
150. Coupland RE, Parker TL, Kesse WK, Mohamed AA. The innervation of the adrenal gland. III. Vagal innervation. J Anat. (1989) 163:173–81.
151. Charlton BG, Nkomazana OF, McGadey J, Neal DE. A preliminary study of acetylcholinesterase-positive innervation in the human adrenal cortex. J Anat. (1991) 176:99–104.
152. Engeland WC. Functional innervation of the adrenal cortex by the splanchnic nerve. Hormone Metab Res. (1998) 30:311–4.
153. Colomer C, Martin AO, Desarménien MG, Guérineau NC. Gap junction-mediated intercellular communication in the adrenal medulla: an additional ingredient of stimulus-secretion coupling regulation. Biochim Biophys Acta (2012) 1818:1937–51. doi: 10.1016/j.bbamem.2011.07.034
154. Desarménien MG, Jourdan C, Toutain B, Vessières E, Hormuzdi SG, Guérineau NC. Gap junction signalling is a stress-regulated component of adrenal neuroendocrine stimulus-secretion coupling in vivo. Nat. Commun. (2013) 4:2938. doi: 10.1038/ncomms3938
155. Kvetnansky R, Sabban EL, Palkovits M. Catecholaminergic systems in stress: structural and molecular genetic approaches. Physiol Rev. (2009) 89:535–606. doi: 10.1152/physrev.00042.2006
156. Brandt BL, Hagiwara S, Kidokoro Y, Miyazaki S. Action potentials in the rat chromaffin cell and effects of acetylcholine. J Physiol. (1976) 263:417–39.
157. Wightman RM, Jankowski JA, Kennedy RT, Kawagoe KT, Schroeder TJ, Leszczyszyn DJ, et al. Temporally resolved catecholamine spikes correspond to single vesicle release from individual chromaffin cells. Proc Natl Acad Sci USA. (1991) 88:10754–8.
158. Gandía L, López MG, Villarroya M, Gilabert JA, Cárdenas A, García AG, et al. Blocking effects of otilonium on Ca2+ channels and secretion in rat chromaffin cells. Eur J Pharmacol (1996) 298:199–205.
159. Alamo L, García AG, Borges R. Electrically-evoked catecholamine release from cat adrenals. Role of cholinergic receptors. Biochem Pharmacol. (1991) 42:973–8.
160. Kidokoro Y, Ritchie AK, Hagiwara S. Effect of tetrodotoxin on adrenaline secretion in the perfused rat adrenal medulla. Nature (1979) 278:63–5.
161. Kidokoro* Y, Miyazaki S, Ozawa S. Acetylcholine-induced membrane depolarization and potential fluctuations in the rat adrenal chromaffin cell. J Physiol. 1982:203–20.
162. Kidokoro Y, Ritchie AK. Chromaffin cell action potentials and their possible role in adrenaline secretion from rat adrenal medulla. J Physiol. (1980) 307:199–216.
164. Droste SK, de Groote L, Atkinson HC, Lightman SL, Reul JMHM, Linthorst ACE. Corticosterone levels in the brain show a distinct ultradian rhythm but a delayed response to forced swim stress. Endocrinology (2008) 149:3244–53. doi: 10.1210/en.2008-0103
165. Schlaich MP, Lambert E, Kaye DM, Krozowski Z, Campbell DJ, Lambert G, et al. Sympathetic augmentation in hypertension: role of nerve firing, norepinephrine reuptake, and Angiotensin neuromodulation. Hypertension (2004) 43:169–75. doi: 10.1161/01.HYP.0000103160.35395.9E
166. Anderson EA, Sinkey CA, Lawton WJ, Mark AL. Elevated sympathetic nerve activity in borderline hypertensive humans. Evidence from direct intraneural recordings. Hypertension (1989) 14:177–83.
167. Segura-Chama P, López-Bistrain P, Pérez-Armendáriz EM, Jiménez-Pérez N, Millán-Aldaco D, Hernández-Cruz A. Enhanced Ca(2+)-induced Ca (2+) release from intracellular stores contributes to catecholamine hypersecretion in adrenal chromaffin cells from spontaneously hypertensive rats. Pflugers Arch. (2015) 467:2307–23. doi: 10.1007/s00424-015-1702-8
168. Fhaner MJ, Galligan JJ, Swain GM. Increased catecholamine secretion from single adrenal chromaffin cells in DOCA-salt hypertension is associated with potassium channel dysfunction. ACS Chem Neurosci. (2013) 4:1404–13.
169. Klevans LR, Gebber GL. Comparison of differential secretion of adrenal catecholamines by splanchnic nerve stimulation and cholinergic agents. J Pharmacol Exp Therap. (1970) 172:69–76.
170. Hökfelt T, Bartfai T, Bloom F. Neuropeptides: opportunities for drug discovery. Lancet Neurol. (2003) 2:463–72. doi: 10.1016/S1474-4422(03)00482-4
171. Evinger MJ, Ernsberger P, Regunathan S, Joh TH, Reis DJ. A single transmitter regulates gene expression through two separate mechanisms: cholinergic regulation of phenylethanolamine N-methyltransferase mRNA via nicotinic and muscarinic pathways. J Neurosci. (1994) 14:2106–16.
172. Criado M. Acetylcholine nicotinic receptor subtypes in chromaffin cells. Pflügers Arch. (2018) 470:13–20. doi: 10.1007/s00424-017-2050-7
173. Inoue M. Muscarinic receptors in adrenal chromaffin cells : physiological role and regulation of ion channels. Pflugers Arch. (2018) 470:29–38. doi: 10.1007/s00424-017-2047-2
174. Gueorguiev VD, Zeman RJ, Hiremagalur B, Menezes A, Sabban EL. Differing temporal roles of Ca2+ and cAMP in nicotine-elicited elevation of tyrosine hydroxylase mRNA. Am J Physiol. (1999) 276:54–65.
175. Hiremagalur B, Nankova B, Nitahara J, Zeman R, Sabban EL. Nicotine increases expression of tyrosine hydroxylase gene. Involvement of protein kinase A-mediated pathway. J Biol Chem. (1993) 268:23704–11.
176. Wong DL, Anderson LJ, Tai TC. Cholinergic and peptidergic regulation of phenylethanolamine N-methyltransferase gene expression. Ann NY Acad Sci. (2002) 971:19–26. doi: 10.1111/j.1749-6632.2002.tb04428.x
177. Morita K, Bell R, Siddall B, Wong D. Neural stimulation of Egr-1 messenger RNA expression in rat adrenal gland: possible relation to phenylethanolamine N-methyltransferase gene regulation. J Pharmacol Exp Ther. (1996) 279:379–85.
178. Morita K, Wong DL. Role of Egr-1 in cholinergic stimulation of phenylethanolamine N-methyltransferase promoter. J Neurochem. (1996) 67:1344–51.
179. Wakade AR. Noncholinergic transmitter(s) maintains secretion of catecholamines from rat adrenal medulla for several hours of continuous stimulation of splanchnic neurons. J Neurochem. (1988) 50:1302–8.
180. Taupenot L, Mahata M, Mahata SK, O'Connor DT. Time-dependent effects of the neuropeptide PACAP on catecholamine secretion: stimulation and desensitization. Hypertension (1999) 34:1152–62.
181. May V, Parsons RL. G Protein-coupled receptor endosomal signaling and regulation of neuronal excitability and stress responses: signaling options and lessons from the PAC1 receptor. J Cell Physiol. (2017) 232:698–706. doi: 10.1002/jcp.25615
182. Martin B, Lopez de Maturana R, Brenneman R, Walent T, Mattson MP, Maudsley S. Class II G protein-coupled receptors and their ligands in neuronal function and protection. Neuromolecular Medicine (2005) 7:3–36. doi: 10.1385/NMM:7:1-2:003
183. Gerdin MJ, Eiden LE. Regulation of PC12 cell differentiation by cAMP signaling to ERK independent of PKA: do all the connections add up? Sci STKE (2007) 2007:pe15. doi: 10.1126/stke.3822007pe15
184. Kuri BA, Chan S-A, Smith CB. PACAP regulates immediate catecholamine release from adrenal chromaffin cells in an activity-dependent manner through a protein kinase C-dependent pathway. J Neurochem. (2009) 110:1214–25. doi: 10.1111/j.1471-4159.2009.06206.x
185. Emery AC, Eiden LE. Signaling through the neuropeptide GPCR PAC1 induces neuritogenesis via a single linear cAMP- and ERK-dependent pathway using a novel cAMP sensor. FASEB J. (2012) 26:3199–211. doi: 10.1096/fj.11-203042
186. Yang K, Lei G, Jackson MF, Macdonald JF. The involvement of PACAP/VIP system in the synaptic transmission in the hippocampus. J Mol Neurosci. (2010) 42:319–26. doi: 10.1007/s12031-010-9372-7
187. Smith CB, Eiden LE. Is PACAP the major neurotransmitter for stress transduction at the adrenomedullary synapse? J Mol Neurosci. (2012) 48:403–12. doi: 10.1007/s12031-012-9749-x
188. Eiden LE, Emery AC, Zhang L, Smith CB. PACAP signaling in stress: insights from the chromaffin cell. Pflugers Arch. (2017) 470:79–88. doi: 10.1007/s00424-017-2062-3
189. Tönshoff C, Hemmick L, Evinger MJ. Pituitary adenylate cyclase activating polypeptide (PACAP) regulates expression of catecholamine biosynthetic enzyme genes in bovine adrenal chromaffin cells. J Mol Neurosci. (1997) 9:127–40.
190. Stroth N, Eiden LE. Stress hormone synthesis in mouse hypothalamus and adrenal gland triggered by restraint is dependent on pituitary adenylate cyclase-activating polypeptide signaling. Neuroscience (2010) 165:1025–30. doi: 10.1016/j.neuroscience.2009.11.023
191. Tai TC, Wong-Faull DC, Claycomb R, Aborn JL, Wong DL. PACAP-regulated phenylethanolamine N-methyltransferase gene expression. J Neurochem. (2010) 115:1195–205. doi: 10.1111/j.1471-4159.2010.07005.x
192. Stroth N, Liu Y, Aguilera G, Eiden LE. Pituitary adenylate cyclase-activating polypeptide controls stimulus-transcription coupling in the hypothalamic-pituitary-adrenal axis to mediate sustained hormone secretion during stress. J Neuroendocrinol. (2011) 23:944–55. doi: 10.1111/j.1365-2826.2011.02202.x
193. Fukushima Y, Hikichi H, Mizukami K, Nagayama T, Yoshida M, Suzuki-Kusaba M, et al. Role of endogenous PACAP in catecholamine secretion from the rat adrenal gland. Am J Physiol Regul Integr Comp Physiol. (2001) 281:R1562–1567. doi: 10.1152/ajpregu.2001.281.5.R1562
194. Stroth N, Kuri BA, Mustafa T, Chan SA, Smith CB, Eiden LE. PACAP controls adrenomedullary catecholamine secretion and expression of catecholamine biosynthetic enzymes at high splanchnic nerve firing rates characteristic of stress transduction in male mice. Endocrinology (2013) 154:330–9. doi: 10.1210/en.2012-1829
195. Hattori S, Takao K, Tanda K, Toyama K, Shintani N, Baba A, et al. Comprehensive behavioral analysis of pituitary adenylate cyclase-activating polypeptide (PACAP) knockout mice. Front Behav Neurosci. (2012) 6:1–18. doi: 10.3389/fnbeh.2012.00058
196. Hammack SE, May V, Hall JD. Pituitary adenylate cyclase activating polypeptide in stress- related disorders: data convergence from animal and human studies. Biol Psychiatry (2016) 78:167–77. doi: 10.1016/j.biopsych.2014.12.003
197. Gaszner B, Kormos V, Kozicz T, Hashimoto H, Reglodi D, Helyes Z. The behavioral phenotype of pituitary adenylate-cyclase activating polypeptide-deficient mice in anxiety and depression tests is accompanied by blunted c-Fos expression in the bed nucleus of the stria terminalis, central projecting Edinger-Westphal nucleu. Neuroscience (2012) 202:283–99. doi: 10.1016/j.neuroscience.2011.11.046
198. Kormos V, Gaszner B. Role of neuropeptides in anxiety, stress, and depression: from animals to humans. Neuropeptides (2013) 47:401–19. doi: 10.1016/j.npep.2013.10.014
199. Cheng S-Y, Serova LI, Glazkova D, Sabban EL. Regulation of rat dopamine beta-hydroxylase gene transcription by early growth response gene 1 (Egr1). Brain Res. (2008) 1193:1–11. doi: 10.1016/j.brainres.2007.11.055
200. Hansen TO, Rehfeld JF, Nielsen FC. Cyclic AMP-induced neuronal differentiation via activation of p38 mitogen-activated protein kinase. J Neurochem. (2000) 75:1870–7. doi: 10.1046/j.1471-4159.2000.0751870.x
201. Vaudry D, Falluel-Morel A, Bourgault S, Basille M, Burel D, Wurtz O, et al. Pituitary adenylate cyclase-activating polypeptide and its receptors: 20 years after the discovery. Pharmacol Rev. (2009) 61:283–357. doi: 10.1124/pr.109.001370
202. Tai TC, Morita K, Wong DL. Role of Egr-1 in cAMP-dependent protein kinase regulation of the phenylethanolamine N-methyltransferase gene. J Neurochem. (2001) 76:1851–9. doi: 10.1046/j.1471-4159.2001.00189.x
203. Ginty DD, Glowacka D, Bader DS, Hidaka H, Wagner JA. Induction of immediate early genes by Ca2+ influx requires cAMP-dependent protein kinase in PC12 cells. J Biol Chem. (1991) 266:17454–8.
204. Papanikolaou NA, Sabban EL. Ability of Egr1 to activate tyrosine hydroxylase transcription in PC12 cells. Cross-talk with AP-1 factors. J Biol Chem. (2000) 275:26683–9. doi: 10.1074/jbc.M000049200
205. Ebert SN, Balt SL, Hunter JP, Gashler A, Sukhatme V, Wong DL. Egr-1 activation of rat adrenal phenylethanolamine N-methyltransferase gene. J Biol Chem. (1994) 269:20885–98.
206. Kim KS, Lee MK, Carroll J, Joh TH. Both the basal and inducible transcription of the tyrosine hydroxylase gene are dependent upon a cAMP response element. J Biol Chem. (1993) 268:15689–95.
207. Lim J, Yang C, Hong SJ, Kim KS. Regulation of tyrosine hydroxylase gene transcription by the cAMP-signaling pathway: involvement of multiple transcription factors. Mol Cell Biochem. (2000) 212:51–60. doi: 10.1023/A:1007148719497
208. Stachowiak MK, Goc A, Hong JS, Kaplan BB, Stachowiak EK. Neural and hormonal regulation of the tyrosine hydroxylase gene in adrenal medullary cells: participation of c-fos and AP1 factors. Mol Cell Biochem. (1990) 1:202–13. doi: 10.1016/1044-7431(90)90003-M
209. Swanson DJ, Zellmer E, Lewis EJ. AP1 Proteins mediate the cAMP response of the dopamine beta-hydroxylase gene. J Biol Chem. (1998) 273:24065–74. doi: 10.1074/jbc.273.37.24065
210. Currie G, Freel EM, Perry CG, Dominiczak AF. Disorders of blood pressure regulation-role of catecholamine biosynthesis, release, and metabolism. Curr Hypertens Rep. (2012) 14:38–45. doi: 10.1007/s11906-011-0239-2
211. Sparrenberger F, Cichelero FT, Ascoli AM, Fonseca FP, Weiss G, Berwanger O, et al. Does psychosocial stress cause hypertension? A systematic review of observational studies. J Hum Hypertens. (2009) 23:12–9. doi: 10.1038/jhh.2008.74
212. Gasperin D, Netuveli G, Dias-da-Costa JS, Pattussi MP. Effect of psychological stress on blood pressure increase: a meta-analysis of cohort studies. Cadernos de Saúde Pública (2009) 25:715–26. doi: 10.1590/S0102-311X2009000400002
213. Liu M-Y, Li N, Li WA, Khan H. Association between psychosocial stress and hypertension: a systematic review and meta-analysis. Neurol Res. (2017) 39:573–80. doi: 10.1080/01616412.2017.1317904
214. Esler M, Schwarz R, Alvarenga M. Mental stress is a cause of cardiovascular diseases: from scepticism to certainty. Stress Health (2008) 24:175–80. doi: 10.1002/smi.1198
215. Esler M, Eikelis N, Schlaich M, Lambert G, Alvarenga M, Dawood T, et al. Chronic mental stress is a cause of essential hypertension: Presence of biological markers of stress. Clin Exp Pharmacol Physiol. (2008) 35:498–502. doi: 10.1111/j.1440-1681.2008.04904.x
216. Grassi G, Seravalle G, Quarti-Trevano F. The “neuroadrenergic hypothesis” in hypertension: current evidence. Exp Physiol. (2010) 95:581–6. doi: 10.1113/expphysiol.2009.047381
217. Grassi G, Quarti-Trevano F, Seravalle G, Dell'Oro R. Sympathetic overdrive and cardiovascular risk in the metabolic syndrome. Hypertens Res Off J Jpn Soc Hypertens. (2006) 17:473–81. doi: 10.1291/hypres.29.839
218. Wong DL, Tai TC, Wong-Faull DC, Claycomb R, Meloni EG, Myers KM, et al. Epinephrine: a short- and long-term regulator of stress and development of illness : a potential new role for epinephrine in stress. Cell Mol Neurobiol. (2012) 32:737–48. doi: 10.1007/s10571-011-9768-0
219. Rumantir MS, Jennings GL, Lambert GW, Kaye DM, Seals DR, Esler MD. The “adrenaline hypothesis” of hypertension revisited: evidence for adrenaline release from the heart of patients with essential hypertension. J Hypertens. (2000) 18:717–23. Available online at: https://insights.ovid.com/pubmed?pmid=10872556
220. Kvetnansky R. Stressor specificity and effect of prior experience on catecholamine biosynthetic enzyme phenylethanolamine N-methyltransferase. Ann NY Acad Sci. (2004) 1032:117–29. doi: 10.1196/annals.1314.009
221. Chandola T, Brunner E, Marmot M. Chronic stress at work and the metabolic syndrome: prospective study. Br Med J. (2006) 332:521–4. doi: 10.1136/bmj.38693.435301.80
222. Lambert EA, Lambert GW. Stress and its role in sympathetic nervous system activation in hypertension and the metabolic syndrome. Curr Hypertens Rep. (2011) 13:244–8. doi: 10.1007/s11906-011-0186-y
223. Rozanski A, Blumenthal JA, Kaplan J. Impact of psychological factors on the pathogenesis of cardiovascular disease and implications for therapy. Circulation (1999) 99:2192–217. doi: 10.1161/01.CIR.99.16.2192
224. Rozanski A, Blumenthal JA, Davidson KW, Saab PG, Kubzansky L. The epidemiology, pathophysiology, and management of psychosocial risk factors in cardiac practice: the emerging field of behavioral cardiology. J Am Coll Cardiol. (2005) 45:637–51. doi: 10.1016/j.jacc.2004.12.005
225. Edmondson D, von Känel R. Post-traumatic stress disorder and cardiovascular disease. Lancet Psychiatry (2017) 4:320–9. doi: 10.1016/S2215-0366(16)30377-7
226. Coughlin SS. Post-traumatic Stress Disorder and Cardiovascular Disease. Open Cardiovasc Med J (2011) 5:164–70. doi: 10.2174/1874192401105010164
227. Mehta D, Binder EB. Gene × environment vulnerability factors for PTSD: the HPA-axis. Neuropharmacology (2012) 62:654–62. doi: 10.1016/j.neuropharm.2011.03.009
228. Eckel RH, Jakicic JM, Ard JD, De Jesus JM, Houston Miller N, Hubbard VS, et al. 2013 AHA/ACC guideline on lifestyle management to reduce cardiovascular risk: a report of the American College of cardiology/American Heart Association task force on practice guidelines. Circulation (2014) 129(25 Suppl. 2):S76–99. doi: 10.1161/01.cir.0000437740.48606.d1
229. Sandberg K, Ji H. Sex differences in primary hypertension. Biol Sex Differ. (2012) 3:7. doi: 10.1186/2042-6410-3-7
230. Carretero OA, Oparil S. Essential hypertension. Part I: definition and etiology. Circulation (2000) 101:329–35. doi: 10.1161/01.CIR.101.3.329
231. Miller AH, Raison CL. The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol. (2016) 16:22–34. doi: 10.1038/nri.2015.5
232. Raison CL, Capuron L, Miller AH. Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol. (2006) 27:24–31. doi: 10.1016/j.it.2005.11.006
233. Serrano CV, Setani KT, Sakamoto E, Andrei AM, Fraguas R. Association between depression and development of coronary artery disease: pathophysiologic and diagnostic implications. Vasc Health Risk Manag. (2011) 7:159–64. doi: 10.2147/VHRM.S10783
234. Meng L, Chen D, Yang Y, Zheng Y, Hui R. Depression increases the risk of hypertension incidence: a meta-analysis of prospective cohort studies. J Hypertens. (2012) 30:842–51. doi: 10.1097/HJH.0b013e32835080b7
235. Dowlati Y, Herrmann N, Swardfager W, Liu H, Sham L, Reim EK, et al. A meta-analysis of cytokines in major depression. Biol Psychiatry (2010) 67:446–57. doi: 10.1016/j.biopsych.2009.09.033
236. Pende A, Musso NR, Vergassola C, Puppo F, Ioverno A, Criscuolo D, et al. Neuroendocrine effects of interferon alpha 2-a in healthy human subjects. J Biol Regul Homeostat Agents (1990) 4:67–72.
237. Corssmit EP, Heijligenberg R, Endert E, Ackermans MT, Sauerwein HP, Romijn JA. Endocrine and metabolic effects of interferon-alpha in humans. J Clin Endocrinol Metab. (1996) 81:3265–9.
238. Kannan H, Tanaka Y, Kunitake T, Ueta Y, Hayashida Y, Yamashita H. Activation of sympathetic outflow by recombinant human interleukin-1 beta in conscious rats. Am J Physiol. (1996) 270:R479–85. doi: 10.1152/ajpregu.1996.270.2.R479
239. Rivier C, Vale W, Brown M. In the rat, interleukin-1 alpha and -beta stimulate adrenocorticotropin and catecholamine release. Endocrinology (1989) 125:3096–102. doi: 10.1210/endo-125-6-3096
240. Hashimoto K, Hirasawa R, Makino S. Comparison of the effects of intra-third ventricular administration of interleukin-1 or platelet activating factor on ACTH secretion and the sympathetic-adrenomedullary system in conscious rats. Acta Med Okayama (1993) 47:1–6.
241. Stouthard JM, Romijn JA, Van der Poll T, Endert E, Klein S, Bakker PJ, et al. Endocrinologic and metabolic effects of interleukin-6 in humans. Am J Physiol. (1995) 268:813–9. doi: 10.1152/ajpendo.1995.268.5.E813
242. Torpy DJ, Papanicolaou DA, Lotsikas AJ, Wilder RL, Chrousos GP, Pillemer SR. Responses of the sympathetic nervous system and the hypothalamic-pituitary-adrenal axis to interleukin-6: a pilot study in fibromyalgia. Arthritis and Rheum. (2000) 43:872–80. doi: 10.1002/1529-0131(200004)43:4<872::AID-ANR19>3.0.CO;2-T
243. Steensberg A, Fischer CP, Keller C, Møller K, Pedersen BK. IL-6 enhances plasma IL-1ra, IL-10, and cortisol in humans. Am J Physiol Endocrinol Metab. (2003) 285:E433–7. doi: 10.1152/ajpendo.00074.2003
244. van Hall G, Steensberg A, Sacchetti M, Fischer C, Keller C, Schjerling P, et al. Interleukin-6 stimulates lipolysis and fat oxidation in humans. J Clin Endocrinol Metab. (2003) 88:3005–10. doi: 10.1210/jc.2002-021687
245. Darling G, Goldstein DS, Stull R, Gorschboth CM, Norton JA. Tumor necrosis factor: immune endocrine interaction. Surgery (1989) 106:1155–60.
246. De Laurentiis A, Pisera D, Caruso C, Candolfi M, Mohn C, Rettori V, et al. Lipopolysaccharide- and tumor necrosis factor-alpha-induced changes in prolactin secretion and dopaminergic activity in the hypothalamic-pituitary axis. Neuroimmunomodulation (2002) 10:30–9. doi: 10.1159/000064412
247. Yu Y, Zhang ZH, Wei SG, Chu Y, Weiss RM, Heistad DD, et al. Central gene transfer of interleukin-10 reduces hypothalamic inflammation and evidence of heart failure in rats after myocardial infarction. Circ Res. (2007) 101:304–12. doi: 10.1161/CIRCRESAHA.107.148940
248. Dunn AJ. Effects of cytokines and infections on brain neurochemistry. Clin Neurosci Res. (2006) 6:52–68. doi: 10.1016/j.cnr.2006.04.002
249. Haddad JJ, Saadé NE, Safieh-Garabedian B. Cytokines and neuro-immune-endocrine interactions: a role for the hypothalamic-pituitary-adrenal revolving axis. J Neuroimmunol. (2002) 133:1–19. doi: 10.1016/S0165-5728(02)00357-0
250. Kitagami T, Yamada K, Miura H, Hashimoto R, Nabeshima T, Ohta T. Mechanism of systemically injected interferon-alpha impeding monoamine biosynthesis in rats: role of nitric oxide as a signal crossing the blood-brain barrier. Brain Res. (2003) 978:104–14. doi: 10.1016/S0006-8993(03)02776-8
251. Kumai T, Tateishi T, Tanaka M, Watanabe M, Shimizu H, Kobayashi S. Effect of interferon-alpha on tyrosine hydroxylase and catecholamine levels in the brain of rats. Life Sci. (2000) 67:663–9. doi: 10.1016/S0024-3205(00)00660-3
252. Kamata M, Higuchi H, Yoshimoto M, Yoshida K, Shimizu T. Effect of single intracerebroventricular injection of alpha-interferon on monoamine concentrations in the rat brain. Eur Neuropsychopharmacol. (2000) 10:129–32. doi: 10.1016/S0924-977X(99)00067-X
253. Terao A, Oikawa M, Saito M. Tissue-specific increase in norepinephrine turnover by central interleukin-1, but not by interleukin-6, in rats. Am J Physiol. (1994) 266:400–4. doi: 10.1152/ajpregu.1994.266.2.R400
254. Vriend CY, Zuo L, Dyck DG, Nance DM, Greenberg AH. Central administration of interleukin-1 beta increases norepinephrine turnover in the spleen. Brain Res Bull. (1993) 31:39–42. doi: 10.1016/0361-9230(93)90008-Y
255. Saito M, Akiyoshi M, Shimizu Y. Possible role of the sympathetic nervous system in responses to interleukin-1. Brain Res Bull. 27:305–8. doi: 10.1016/0361-9230(91)90116-2
256. Akiyoshi M, Shimizu Y, Saito M. Interleukin-1 increases norepinephrine turnover in the spleen and lung in rats. Biochem Biophys Res Commun. (1990) 173:1266–70. doi: 10.1016/S0006-291X(05)80923-4
257. Hurst S, Collins SM. Interleukin-1 beta modulation of norepinephrine release from rat myenteric nerves. Am J Physiol. (1993) 264:30–5. doi: 10.1152/ajpgi.1993.264.1.G30
258. Bognar IT, Albrecht SA, Farasaty M, Schmitt E, Seidel G, Fuder H. Effects of human recombinant interleukins on stimulation-evoked noradrenaline overflow from the rat perfused spleen. Naunyn-Schmiedeberg's Arch Pharmacol. (1994) 349:497–502.
259. Sterin-Borda L, Perez Leiros C, Borda ES, de Bracco MM. Effects of IL-2 on the myocardium. Participation of the sympathetic system. J Mol Cell Cardiol. (1996) 28:2457–65. doi: 10.1006/jmcc.1996.0238
260. Kang YM, He RL, Yang LM, Qin DN, Guggilam A, Elks C, et al. Brain tumour necrosis factor-alpha modulates neurotransmitters in hypothalamic paraventricular nucleus in heart failure. Cardiovasc Res. (2009) 83:737–46. doi: 10.1093/cvr/cvp160
261. Sirivelu MP, MohanKumar PS, MohanKumar SMJ. Differential effects of systemic interleukin-1β on gene expression in brainstem noradrenergic nuclei. Life Sci. (2012) 90:77–81. doi: 10.1016/j.lfs.2011.10.006
262. Zhang XJ, Wang X, Xiong LZ, Fan J, Duan XL, Wang BR. Up-regulation of IL-1 receptor type I and tyrosine hydroxylase in the rat carotid body following intraperitoneal injection of IL-1beta. Histochem Cell Biol. (2007) 128:533–40. doi: 10.1007/s00418-007-0346-y
263. Sim YB, Park SH, Kang YJ, Jung JS, Ryu OH, Choi MG, et al. Interleukin-1β (IL-1β) increases pain behavior and the blood glucose level: possible involvement of sympathetic nervous system. Pharmacol Biochem Behav. (2012) 102:170–6. doi: 10.1016/j.pbb.2012.04.006
264. Elenkov IJ, Wilder RL, Chrousos GP, Vizi ES. The sympathetic nerve - an integrative interface between two supersystems: the brain and the immune system. Pharmacol Rev. (2000) 52:595–638. Available online at: http://pharmrev.aspetjournals.org/content/52/4/595/tab-article-info
265. Turnbull AV, Rivier CL. Regulation of the hypothalamic-pituitary-adrenal axis by cytokines: actions and mechanisms of action. Physiol Rev. (1999) 79:1–71. doi: 10.1152/physrev.1999.79.1.1
266. Call GB, Husein OF, McIlmoil CJ, Adams A, Heckmann RA, Judd AM. Bovine adrenal cells secrete interleukin-6 and tumor necrosis factor in vitro. Gen Compar Endocrinol. (2000) 118:249–61. doi: 10.1006/gcen.2000.7458
267. González-Hernández JA, Ehrhart-Bornstein M, Späth-Schwalbe E, Scherbaum WA, Bornstein SR. Human adrenal cells express tumor necrosis factor-alpha messenger ribonucleic acid: evidence for paracrine control of adrenal function. J Clin Endocrinol Metab. (1996) 81:807–13.
268. Douglas SA, Bunn SJ. Interferon-alpha signalling in bovine adrenal chromaffin cells: involvement of signal-transducer and activator of transcription 1 and 2, extracellular signal-regulated protein kinases 1/2 and serine 31 phosphorylation of tyrosine hydroxylase. J Neuroendocrinol. (2009) 21:200–7. doi: 10.1111/j.1365-2826.2009.01821.x
269. Tachikawa E, Kondo Y, Takahashi M, Kashimoto T, Yanagihara N, Toyohira Y, et al. Interferon-alpha reduces catecholamine secretion from bovine adrenal chromaffin cells stimulated by acetylcholine. Naunyn-Schmiedeberg's Archives of Pharmacol. (1997) 356:699–705.
270. Toyohira Y, Yanagihara N, Minami K, Ueno S, Uezono Y, Tachikawa E, et al. Down-regulation of the noradrenaline transporter by interferon-alpha in cultured bovine adrenal medullary cells. J Neurochem. (1998) 70:1441–7.
271. Samal B, Ait-Ali D, Bunn SJ, Mustafa T, Eiden LE. Discrete signal transduction pathway utilization by a neuropeptide (PACAP) and a cytokine (TNF-alpha) first messenger in chromaffin cells, inferred from coupled transcriptome-promoter analysis of regulated gene cohorts. Peptides (2013) 45:48–60. doi: 10.1016/j.peptides.2013.03.020
272. Venihaki M, Gravanis A, Margioris AN. KAT45 human pheochromocytoma cell line. A new model for the in vitro study of neuro-immuno-hormonal interactions. Ann NY Acad Sci. (1998) 840:425–33. doi: 10.1111/j.1749-6632.1998.tb09581.x
273. González-Hernández JA, Bornstein SR, Ehrhart-Bornstein M, Gschwend JE, Gwosdow A, Jirikowski G, et al. IL-1 is expressed in human adrenal gland in vivo. Possible role in a local immune-adrenal axis. Clin Exp Immunol. (1995) 99:137–41. doi: 10.1111/j.1365-2249.1995.tb03484.x
274. Schultzberg M, Andersson C, Undén A, Troye-Blomberg M, Svenson SB, Bartfai T. Interleukin-1 in adrenal chromaffin cells. Neuroscience (1989) 30:805–10. doi: 10.1016/0306-4522(89)90171-1
275. Engström L, Rosén K, Angel A, Fyrberg A, Mackerlova L, Konsman JP, et al. Systemic immune challenge activates an intrinsically regulated local inflammatory circuit in the adrenal gland. Endocrinology (2007) 149:1436–50. doi: 10.1210/en.2007-1456
276. Andersson C, Svenson SB, Van Deventer S, Cerami A, Bartfai T. Interleukin-1 alpha expression is inducible by cholinergic stimulation in the rat adrenal gland. Neuroscience (1992) 47:481–5. doi: 10.1016/0306-4522(92)90262-Z
277. Nobel CS, Schultzberg M. Induction of interleukin-1 beta mRNA and enkephalin mRNA in the rat adrenal gland by lipopolysaccharides studied by in situ hybridization histochemistry. Neuroimmunomodulation (1995) 2:61–73. doi: 10.1159/000096873
278. Alheim K, Andersson C, Tingsborg S, Ziolkowska M, Schultzberg M, Bartfai T. Interleukin 1 expression is inducible by nerve growth factor in PC12 pheochromocytoma cells. Proc Natl Acad Sci USA. (1991) 88:9302–6. doi: 10.1073/pnas.88.20.9302
279. Schultzberg M, Tingsborg S, Nobel S, Lundkvist J, Svenson S, Simoncsits A, et al. Interleukin-1 receptor antagonist protein and mRNA in the rat adrenal gland. J Interfer Cytokine Res. (1995) 15:721–9. doi: 10.1089/jir.1995.15.721
280. Rosmaninho-Salgado J, Araújo IM, Alvaro AR, Mendes AF, Ferreira L, Grouzmann E, et al. Regulation of catecholamine release and tyrosine hydroxylase in human adrenal chromaffin cells by interleukin-1beta: role of neuropeptide Y and nitric oxide. J Neurochem. (2009) 109:911–22. doi: 10.1111/j.1471-4159.2009.06023.x
281. Gwosdow AR. Mechanisms of interleukin-1-induced hormone secretion from the rat adrenal gland. Endocrine Res. (1995) 21:25–37. doi: 10.3109/07435809509030418
282. Joseph A, Kumar A, O'Connell NA, Agarwal RK, Gwosdow AR. Interleukin-1 alpha stimulates dopamine release by activating type II protein kinase A in PC-12 cells. Am J Physiol. (1995) 269:1083–8.
283. Venihaki M, Gravanis a, Margioris AN. Comparative study between normal rat chromaffin and PC12 rat pheochromocytoma cells: production and effects of corticotropin-releasing hormone. Endocrinology (1997) 138:698–704. doi: 10.1210/endo.138.2.4916
284. Gwosdow AR, O'Connell NA, Spencer JA, Kumar MS, Agarwal RK, Bode HH, et al. Interleukin-1-induced corticosterone release occurs by an adrenergic mechanism from rat adrenal gland. Am J Physiol. (1992) 263:461–6.
285. Li XM, Juorio A V, Boulton AA. Induction of aromatic L-amino acid decarboxylase mRNA by interleukin-1 beta and prostaglandin E2 in PC12 cells. Neurochem Res. (1994) 19:591–5. doi: 10.1007/BF00971335
286. Liu MT, Huang HM, Jeng KC, Ou SC, Kuo JS. Induction of cytokine genes and IL-1alpha by chemical hypoxia in PC12 cells. Life Sci. (2000) 67:2147–57. doi: 10.1016/S0024-3205(00)00811-0
287. Rosmaninho-Salgado J, Alvaro AR, Grouzmann E, Duarte EP, Cavadas C. Neuropeptide Y regulates catecholamine release evoked by interleukin-1beta in mouse chromaffin cells. Peptides (2007) 28:310–4. doi: 10.1016/j.peptides.2006.11.015
288. Morita K, Miyasako T, Kitayama S, Dohi T. Interleukin-1 inhibits voltage-dependent P/Q-type Ca2+ channel associated with the inhibition of the rise of intracellular free Ca2+ concentration and catecholamine release in adrenal chromaffin cells. Biochim Biophys Acta (2004) 1673:160–9. doi: 10.1016/j.bbagen.2004.04.012
289. Ait-Ali D, Turquier V, Grumolato L, Yon L, Jourdain M, Alexandre D, et al. The proinflammatory cytokines tumor necrosis factor-α and interleukin-1 stimulate neuropeptide gene transcription and secretion in adrenochromaffin cells via activation of extracellularly regulated kinase 1/2 and p38 protein kinases, and activator protein-1 transcription factors. Mol Endocrinol. (2004) 18:1721–39. doi: 10.1210/me.2003-0129
290. Yanagihara N, Minami K, Shirakawa F, Uezono Y, Kobayashi H, Eto S, et al. Stimulatory effect of IL-1 beta on catecholamine secretion from cultured bovine adrenal medullary cells. Biochem Biophys Res Commun. (1994) 198:81–7. doi: 10.1006/bbrc.1994.1012
291. Eskay RL, Eiden LE. Interleukin-1 alpha and tumor necrosis factor-alpha differentially regulate enkephalin, vasoactive intestinal polypeptide, neurotensin, and substance P biosynthesis in chromaffin cells. Endocrinology (1992) 130:2252–8.
292. Shu HF, Wang BR, Bi H, Pei JM, Wang X, Fan J, et al. PC12 cells express IL-1 receptor type I and response to IL-1beta stimulation. Respir Physiol Neurobiol. (2007) 157:187–95. doi: 10.1016/j.resp.2007.01.010
293. Liu X, Serova L, Kvetnanský R, Sabban EL. Identifying the stress transcriptome in the adrenal medulla following acute and repeated immobilization. Ann NY Acad Sci. (2008) 1148:1–28. doi: 10.1196/annals.1410.082
294. Cho SG, Yi SY, Yoo YS. IFNgamma and TNFalpha synergistically induce neurite outgrowth on PC12 cells. Neurosci Lett. (2005) 378:49–54. doi: 10.1016/j.neulet.2004.12.073
295. Pérez-Rodríguez R, Roncero C, Oliván AM, González MP, Oset-Gasque MJ. Signaling mechanisms of interferon gamma induced apoptosis in chromaffin cells: involvement of nNOS, iNOS, and NFkappaB. J Neurochem. (2009) 108:1083–96. doi: 10.1111/j.1471-4159.2008.05862.x
296. Woods AM, Judd AM. Interleukin-4 increases cortisol release and decreases adrenal androgen release from bovine adrenal cells. Domest Anim Endocrinol. (2008) 34:372–82. doi: 10.1016/j.domaniend.2007.10.004
297. Kontogeorgos G, Scheithauer BW, Kovacs K, Horvath E, Melmed S. Growth factors and cytokines in paragangliomas and pheochromocytomas, with special reference to sustentacular cells. Endocrine Pathol. (2002) 13:197–206. doi: 10.1385/EP:13:3:197
298. Kontogeorgos G, Messini I, Kyrodimou E, Giannikaki L, Zois E, Koukoulis G, et al. Immunohistochemical localization of interleukin-6 in peripheral human endocrine glands. Endocrine (2002) 17:135–40. doi: 10.1385/ENDO:17:2:135
299. Päth G, Bornstein SR, Ehrhart-bornstein M, Scherbaum WA. Interleukin-6 and the interleukin-6 receptor in the human adrenal gland: expression and effects on steroidogenesis. J Clin Endocrinol Metab. (1997) 82:2343–9.
300. Gadient RA, Lachmund A, Unsicker K, Otten U. Expression of interleukin-6 (IL-6) and IL-6 receptor mRNAs in rat adrenal medulla. Neurosci Lett. (1995) 194:17–20. doi: 10.1016/0304-3940(95)11708-5
301. Möller JC, Krüttgen A, Burmester R, Weis J, Oertel WH, Shooter EM. Release of interleukin-6 via the regulated secretory pathway in PC12 cells. Neurosci Lett. (2006) 400:75–9. doi: 10.1016/j.neulet.2006.02.018
302. Sallmann S, Jüttler E, Prinz S, Petersen N, Knopf U, Weiser T, et al. Induction of interleukin-6 by depolarization of neurons. J Neurosci. (2000) 20:8637–42. doi: 10.1523/JNEUROSCI.20-23-08637.2000
303. Willenberg HS, Päth G, Vögeli T a, Scherbaum WA, Bornstein SR. Role of interleukin-6 in stress response in normal and tumorous adrenal cells and during chronic inflammation. Ann NY Acad Sci. (2002) 966:304–14. doi: 10.1111/j.1749-6632.2002.tb04230.x
304. Li R, Hou J, Xu Q, Liu Q-J, Shen Y-J, Rodin G, et al. High level interleukin-6 in the medium of human pancreatic cancer cell culture suppresses production of neurotransmitters by PC12 cell line. Metab Brain Dis. (2012) 27:91–100. doi: 10.1007/s11011-011-9270-x
305. Satoh T, Nakamura S, Taga T, Matsuda T, Hirano T, Kishimoto T, et al. Induction of neuronal differentiation in PC12 cells by B-cell stimulatory factor 2/interleukin 6. Mol Cell Biol. (1988) 8:3546–9. doi: 10.1128/MCB.8.8.3546
306. Romero DG, Vergara GR, Zhu Z, Covington GS, Plonczynski MW, Yanes LL, et al. Interleukin-8 synthesis, regulation, and steroidogenic role in H295R human adrenocortical cells. Endocrinology (2006) 147:891–8. doi: 10.1210/en.2005-0951
307. Virmani A, Koverech A, Ali SF, Binienda ZK. Acetyl-L-Carnitine modulates TP53 and IL10 gene expression induced by 3-NPA evoked toxicity in PC12 cells. Curr Neuropharmacol. (2011) 9:195–9. doi: 10.2174/157015911795017182
308. Ait-Ali D, Samal B, Mustafa T, Eiden LE. Neuropeptides, growth factors, and cytokines: a cohort of informational molecules whose expression is up-regulated by the stress-associated slow transmitter PACAP in chromaffin cells. Cell Mol Neurobiol. (2010) 30:1441–9. doi: 10.1007/s10571-010-9620-y
309. Tong AL, Zeng ZP, Yang D, Li HZ, Li M, Chen S, et al. Expression of transforming growth factor alpha, tumor necrosis factor alpha, and vascular endothelial growth factor of human pheochromocytoma tissues. Zhongguo Yi Xue Ke Xue Yuan Xue Bao (2004) 26:426–31.
310. Goryo K, Torii S, Yasumoto K-I, Sogawa K. Tumour necrosis factor-α suppresses the hypoxic response by NF-κB-dependent induction of inhibitory PAS domain protein in PC12 cells. J Biochem. (2011) 150:311–8. doi: 10.1093/jb/mvr061
311. Trincavelli ML, Falleni A, Chelli B, Tuscano D, Costa B, Gremigni V, et al. A(2A) adenosine receptor ligands and proinflammatory cytokines induce PC 12 cell death through apoptosis. Biochem Pharmacology. (2003) 66:1953–62. doi: 10.1016/j.bcp.2003.07.006
312. Soeda S, Ochiai T, Paopong L, Tanaka H, Shoyama Y, Shimeno H. Crocin suppresses tumor necrosis factor-alpha-induced cell death of neuronally differentiated PC-12 cells. Life Sci. (2001) 69:2887–98. doi: 10.1016/S0024-3205(01)01357-1
313. Ait-Ali D, Stroth N, Sen JM, Eiden LE. PACAP-cytokine interactions govern adrenal neuropeptide biosynthesis after systemic administration of LPS. Neuropharmacology (2010) 58:208–14. doi: 10.1016/j.neuropharm.2009.07.034
314. Tamura R, Nemoto T, Maruta T, Onizuka S, Yanagita T, Wada A, et al. Up-regulation of NaV1.7 sodium channels expression by tumor necrosis factor-α in cultured bovine adrenal chromaffin cells and rat dorsal root ganglion neurons. Anesth Analg (2014) 118:318–24. doi: 10.1213/ANE.0000000000000085
315. Ait-Ali D, Turquier V, Tanguy Y, Thouënnon E, Ghzili H, Mounien L, et al. Tumor necrosis factor (TNF)-alpha persistently activates nuclear factor-kappaB signaling through the type 2 TNF receptor in chromaffin cells: implications for long-term regulation of neuropeptide gene expression in inflammation. Endocrinology (2008) 149:2840–52. doi: 10.1210/en.2007-1192
316. Xie J, Wang Y, Kolls J, Malinski T, Nelson S, Summer W, et al. Tumor necrosis factor alpha inhibits contractions to sympathetic nerve stimulation by a nitric oxide-dependent mechanism. Proc Soc Exp Biol Med Soc Exp Biol Med. (1993) 203:446–53.
317. Douglas SA, Sreenivasan D, Carman FH, Bunn SJ. Cytokine interactions with adrenal medullary chromaffin cells. Cell Mol Neurobiol. (2010) 30:1467–75. doi: 10.1007/s10571-010-9593-x
318. Bunn SJ, Ait-Ali D, Eiden LE. Immune-neuroendocrine integration at the adrenal gland: cytokine control of the adrenomedullary transcriptome. J Mol Neurosci. (2012) 48:413–9. doi: 10.1007/s12031-012-9745-1
319. Caraglia M, Marra M, Pelaia G, Maselli R, Caputi M, Marsico SA, et al. Alpha-interferon and its effects on signal transduction pathways. J Cell Physiol. (2005) 202:323–35. doi: 10.1002/jcp.20137
320. Toyohira Y, Yanagihara N, Minami K, Ueno S, Uezono Y, Tachikawa E, et al. Down-regulation of the noradrenaline transporter by interferon-α in cultured bovine adrenal medullary cells. J Neurochem. (2002) 70:1441–7. doi: 10.1046/j.1471-4159.1998.70041441.x
321. Moy LY, Tsai LH. Cyclin-dependent kinase 5 phosphorylates serine 31 of tyrosine hydroxylase and regulates its stability. J Biol Chem. (2004) 279:54487–93. doi: 10.1074/jbc.M406636200
322. Sutherland C, Alterio J, Campbell DG, Le Bourdellès B, Mallet J, Haavik J, et al. Phosphorylation and activation of human tyrosine hydroxylase in vitro by mitogen-activated protein (MAP) kinase and MAP-kinase-activated kinases 1 and 2. Eur J Biochem. (1993) 217:715–22.
323. Cammarota M, Bevilaqua LRM, Rostas JAP, Dunkley PR. Histamine activates tyrosine hydroxylase in bovine adrenal chromaffin cells through a pathway that involves ERK1/2 but not p38 or JNK. J Neurochem. (2003) 84:453–8. doi: 10.1046/j.1471-4159.2003.01517.x
324. Bobrovskaya L, Odell A, Leal RB, Dunkley PR. Tyrosine hydroxylase phosphorylation in bovine adrenal chromaffin cells: the role of MAPKs after angiotensin II stimulation. J Neurochem. (2001) 78:490–8. doi: 10.1046/j.1471-4159.2001.00445.x
325. Daubner SC, Lauriano C, Haycock JW, Fitzpatrick PF. Site-directed mutagenesis of serine 40 of rat tyrosine hydroxylase. Effects of dopamine and cAMP-dependent phosphorylation on enzyme activity. J Biol Chem. (1992) 267:12639–46.
326. Peters VA, Joesting JJ, Freund GG. IL-1 receptor 2 (IL-1R2) and its role in immune regulation. Brain Behav Immun. (2013) 32:1–8. doi: 10.1016/j.bbi.2012.11.006
327. Kvetnansky R, Jelokova J, Rusnak M, Dronjak S, Serova L, Nankova B, et al. Novel stressors exaggerate tyrosine hydroxylase gene expression in the adrenal medulla of rats exposed to long-term cold stress. In: McCarty R, Aguilera G, Sabban EL KR, editors. Stress Neural, Endocr. Mol. Stud. New York, NY: Taylor and Francis. (2002), p. 121–8.
328. Kvetnansky R, Rusnak M, Dronjak S, Krizanova O, Sabban EL. Effect of novel stressors on tyrosine hydroxylase gene expression in the adrenal medulla of repeatedly immobilized rats. Neurochem Res. (2003) 28:625–30. doi: 10.1023/A:1022846012174
329. Kamimura D, Ishihara K, Hirano T. IL-6 signal transduction and its physiological roles: the signal orchestration model. Rev Physiol Biochem Pharmacol. (2003) 149:1–38. doi: 10.1007/s10254-003-0012-2
330. Fang XX, Jiang XL, Han XH, Peng YP, Qiu YH. Neuroprotection of interleukin-6 against NMDA-induced neurotoxicity is mediated by JAK/STAT3, MAPK/ERK, and PI3K/AKT signaling pathways. Cell Mol Neurobiol. (2013) 33:241–51. doi: 10.1007/s10571-012-9891-6
332. Jones SA, Horiuchi S, Topley N, Yamamoto N, Fuller GM. The soluble interleukin 6 receptor: mechanisms of production and implications in disease. FASEB J. (2001) 15:43–58. doi: 10.1096/fj.99-1003rev
333. Baud V, Karin M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. (2001) 11:372–7. doi: 10.1016/S0962-8924(01)02064-5
334. Cavaillon JM, Adib-Conquy M, Fitting C, Adrie C, Payen D. Cytokine cascade in sepsis. Scand J Infect Dis. (2003) 35:535–44. doi: 10.1080/00365540310015935
335. Sparacio SM, Zhang Y, Vilcek J, Benveniste EN. Cytokine regulation of interleukin-6 gene expression in astrocytes involves activation of an NF-kappa B-like nuclear protein. J Neuroimmunol. (1992) 39:231–42. doi: 10.1016/0165-5728(92)90257-L
336. Dinarello CA, Ikejima T, Warner SJ, Orencole SF, Lonnemann G, Cannon JG, et al. Interleukin 1 induces interleukin 1. I. Induction of circulating interleukin 1 in rabbits in vivo and in human mononuclear cells in vitro. J Immunol. (1987) 139:1902–10.
337. Descoteaux A, Matlashewski G. Regulation of tumor necrosis factor gene expression and protein synthesis in murine macrophages treated with recombinant tumor necrosis factor. J Immunol. (1990) 145:846–53.
338. DeForge LE, Kenney JS, Jones ML, Warren JS, Remick DG. Biphasic production of IL-8 in lipopolysaccharide (LPS)-stimulated human whole blood. Separation of LPS- and cytokine-stimulated components using anti-tumor necrosis factor and anti-IL-1 antibodies. J Immunol. (1992) 148:2133–41.
339. de Waal Malefyt R, Figdor CG, Huijbens R, Mohan-Peterson S, Bennett B, Culpepper J, et al. Effects of IL-13 on phenotype, cytokine production, and cytotoxic function of human monocytes. Comparison with IL-4 and modulation by IFN-gamma or IL-10. J Immunol. (1993) 151:6370–81.
340. Wanidworanun C, Strober W. Predominant role of tumor necrosis factor-alpha in human monocyte IL-10 synthesis. J Immunol. (1993) 151:6853–61.
341. Irwin MR, Cole SW. Reciprocal regulation of the neural and innate immune systems. Nat Rev Immunol. (2011) 11:625–32. doi: 10.1038/nri3042
342. Smoak KA, Cidlowski JA. Mechanisms of glucocorticoid receptor signaling during inflammation. Mech Ageing Dev. (2004) 125:697–706. doi: 10.1016/j.mad.2004.06.010
343. Pace TWW, Miller AH. Cytokines and glucocorticoid receptor signaling. Relevance to major depression. Ann NY Acad Sci. (2009) 1179:86–105. doi: 10.1111/j.1749-6632.2009.04984.x
344. Hu F, Pace TWW, Miller AH. Interferon-alpha inhibits glucocorticoid receptor-mediated gene transcription via STAT5 activation in mouse HT22 cells. Brain Behav Immun. (2009) 23:455–63. doi: 10.1016/j.bbi.2009.01.001
345. Flammer JR, Dobrovolna J, Kennedy MA, Chinenov Y, Glass CK, Ivashkiv LB, et al. The type I interferon signaling pathway is a target for glucocorticoid inhibition. Mol Cell Biol. (2010) 30:4564–74. doi: 10.1128/MCB.00146-10
346. Miller AL, Webb MS, Copik AJ, Wang Y, Johnson BH, Kumar R, et al. p38 Mitogen-activated protein kinase (MAPK) is a key mediator in glucocorticoid-induced apoptosis of lymphoid cells: correlation between p38 MAPK activation and site-specific phosphorylation of the human glucocorticoid receptor at serine 211. Mol Endocrinol. (2005) 19:1569–83. doi: 10.1210/me.2004-0528
347. Kino T, Ichijo T, Amin ND, Kesavapany S, Wang Y, Kim N, et al. Cyclin-dependent kinase 5 differentially regulates the transcriptional activity of the glucocorticoid receptor through phosphorylation: clinical implications for the nervous system response to glucocorticoids and stress. Mol Endocrinol. (2007) 21:1552–68. doi: 10.1210/me.2006-0345
348. Goleva E, Kisich KO, Leung DYM. A role for STAT5 in the pathogenesis of IL-2-induced glucocorticoid resistance. J Immunol. (2002) 169:5934–40. doi: 10.4049/jimmunol.169.10.5934
349. Irusen E, Matthews JG, Takahashi A, Barnes PJ, Chung KF, Adcock IM. p38 Mitogen-activated protein kinase-induced glucocorticoid receptor phosphorylation reduces its activity: role in steroid-insensitive asthma. J Allergy Clin Immunol. (2002) 109:649–57. doi: 10.1067/mai.2002.122465
350. Biola A, Lefebvre P, Perrin-Wolff M, Sturm M, Bertoglio J, Pallardy M. Interleukin-2 inhibits glucocorticoid receptor transcriptional activity through a mechanism involving STAT5 (signal transducer and activator of transcription 5) but not AP-1. Mol Endocrinol. (2001) 15:1062–76. doi: 10.1210/mend.15.7.0657
351. Onda K, Nagashima M, Kawakubo Y, Inoue S, Hirano T, Oka K. Mitogen-activated protein kinase kinase 1/extracellular signal-regulated kinase (MEK-1/ERK) inhibitors sensitize reduced glucocorticoid response mediated by TNFalpha in human epidermal keratinocytes (HaCaT). Biochem Biophys Res Commun. (2006) 351:266–72. doi: 10.1016/j.bbrc.2006.10.032
352. Szatmáry Z, Garabedian MJ, Vilcek J. Inhibition of glucocorticoid receptor-mediated transcriptional activation by p38 mitogen-activated protein (MAP) kinase. J Biol Chem. (2004) 279:43708–15. doi: 10.1074/jbc.M406568200
353. Kino T, Chrousos GP. Tumor necrosis factor alpha receptor- and Fas-associated FLASH inhibit transcriptional activity of the glucocorticoid receptor by binding to and interfering with its interaction with p160 type nuclear receptor coactivators. J Biol Chem. (2003) 278:3023–9. doi: 10.1074/jbc.M209234200
354. Sheppard KA, Phelps KM, Williams AJ, Thanos D, Glass CK, Rosenfeld MG, et al. Nuclear integration of glucocorticoid receptor and nuclear factor-kappaB signaling by CREB-binding protein and steroid receptor coactivator-1. J Biol Chem. (1998) 273:29291–4. doi: 10.1074/jbc.273.45.29291
355. Ray A. Prefontaine KE. Physical association and functional antagonism between the p65 subunit of transcription factor NF-kappa B and the glucocorticoid receptor. Proc Natl Acad Sci USA. (1994) 91:752–6. doi: 10.1073/pnas.91.2.752
356. Caldenhoven E, Liden J, Wissink S, Van de Stolpe A, Raaijmakers J, Koenderman L, et al. Negative cross-talk between RelA and the glucocorticoid receptor: a possible mechanism for the antiinflammatory action of glucocorticoids. Mol Endocrinol. (1995) 9:401–12.
357. Zhang Z, Jones S, Hagood JS, Fuentes NL, Fuller GM. STAT3 acts as a co-activator of glucocorticoid receptor signaling. J Biol Chem. (1997) 272:30607–10. doi: 10.1074/jbc.272.49.30607
358. Franchimont D, Martens H, Hagelstein MT, Louis E, Dewe W, Chrousos GP, et al. Tumor necrosis factor alpha decreases, and interleukin-10 increases, the sensitivity of human monocytes to dexamethasone: potential regulation of the glucocorticoid receptor. J Clin Endocrinol Metab. (1999) 84:2834–9.
359. Wang P, Wu P, Siegel MI, Egan RW, Billah MM. Interleukin (IL)-10 inhibits nuclear factor kappa B (NF kappa B) activation in human monocytes. IL-10 and IL-4 suppress cytokine synthesis by different mechanisms. J Biol Chem. (1995) 270:9558–63. doi: 10.1074/jbc.270.16.9558
360. Williams L, Bradley L, Smith A, Foxwell B. Signal transducer and activator of transcription 3 is the dominant mediator of the anti-inflammatory effects of IL-10 in human macrophages. J Immunol. (2004) 172:567–76. doi: 10.4049/jimmunol.172.1.567
361. Rogatsky I, Logan SK, Garabedian MJ. Antagonism of glucocorticoid receptor transcriptional activation by the c-Jun N-terminal kinase. Proc Natl Acad Sci USA. (1998) 95:2050–5. doi: 10.1073/pnas.95.5.2050
362. Galliher-Beckley AJ, Cidlowski JA. Emerging roles of glucocorticoid receptor phosphorylation in modulating glucocorticoid hormone action in health and disease. IUBMB Life (2009) 61:979–86. doi: 10.1002/iub.245
363. Takahashi E, Onda K, Hirano T, Oka K, Maruoka N, Tsuyuguchi M, et al. Expression of c-fos, rather than c-jun or glucocorticoid-receptor mRNA, correlates with decreased glucocorticoid response of peripheral blood mononuclear cells in asthma. International Immunopharmacology (2002) 2:1419–27. doi: 10.1016/S1567-5769(02)00083-8
364. Rogatsky I, Luecke HF, Leitman DC, Yamamoto KR. Alternate surfaces of transcriptional coregulator GRIP1 function in different glucocorticoid receptor activation and repression contexts. Proc Natl Acad Sci USA. (2002) 99:16701–6. doi: 10.1073/pnas.262671599
365. Schüle R, Rangarajan P, Kliewer S, Ransone LJ, Bolado J, Yang N, et al. Functional antagonism between oncoprotein c-Jun and the glucocorticoid receptor. Cell (1990) 62:1217–26. doi: 10.1016/0092-8674(90)90397-W
366. Heck S, Kullmann M, Gast A, Ponta H, Rahmsdorf HJ, Herrlich P, et al. A distinct modulating domain in glucocorticoid receptor monomers in the repression of activity of the transcription factor AP-1. EMBO J. (1994) 13:4087–95.
367. Kassel O, Schneider S, Heilbock C, Litfin M, Göttlicher M, Herrlich P. A nuclear isoform of the focal adhesion LIM-domain protein Trip6 integrates activating and repressing signals at AP-1- and NF-kappaB-regulated promoters. Genes Dev. (2004) 18:2518–28. doi: 10.1101/gad.322404
368. Saxena M, Williams S, Taskén K, Mustelin T. Crosstalk between cAMP-dependent kinase and MAP kinase through a protein tyrosine phosphatase. Nat Cell Biol. (1999) 1:305–11. doi: 10.1038/13024
369. Zhong H, SuYang H, Erdjument-Bromage H, Tempst P, Ghosh S. The transcriptional activity of NF-kappaB is regulated by the IkappaB-associated PKAc subunit through a cyclic AMP-independent mechanism. Cell (1997) 89:413–24. doi: 10.1016/S0092-8674(00)80222-6
370. Sengupta TK, Schmitt EM, Ivashkiv LB. Inhibition of cytokines and JAK-STAT activation by distinct signaling pathways. Proc Natl Acad Sci USA. (1996) 93:9499–504. doi: 10.1073/pnas.93.18.9499
371. Taskén K, Aandahl EM. Localized effects of cAMP mediated by distinct routes of protein kinase A. Physiol Rev. (2004) 84:137–67. doi: 10.1152/physrev.00021.2003
372. Zhang X, Odom DT, Koo SH, Conkright MD, Canettieri G, Best J, et al. Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc Natl Acad Sci USA. (2005) 102:4459–64. doi: 10.1073/pnas.0501076102
373. Foster GE, Poulin MJ, Hanly PJ. Intermittent hypoxia and vascular function: implications for obstructive sleep apnoea. Exp Physiol. (2007) 92:51–65. doi: 10.1113/expphysiol.2006.035204
374. Fletcher EC. Invited review: physiological consequences of intermittent hypoxia: systemic blood pressure. J Appl Physiol. (2001) 90:1600–5. doi: 10.1152/jappl.2001.90.4.1600
375. Mochizuki-Oda N, Takeuchi Y, Matsumura K, Oosawa Y, Watanabe Y. Hypoxia-induced catecholamine release and intracellular Ca2+ increase via suppression of K+ channels in cultured rat adrenal chromaffin cells. J Neurochem. (1997) 69:377–87. doi: 10.1046/j.1471-4159.1997.69010377.x
376. Thompson RJ, Jackson A, Nurse CA. Developmental loss of hypoxic chemosensitivity in rat adrenomedullary chromaffin cells. J Physiol. (1997) 498(Pt 2):503–10.
377. Kuri BA, Khan SA, Chan SA, Prabhakar NR, Smith CB. Increased secretory capacity of mouse adrenal chromaffin cells by chronic intermittent hypoxia: involvement of protein kinase C. J Physiol. (2007) 584:313–9. doi: 10.1113/jphysiol.2007.140624
378. Souvannakitti D, Nanduri J, Yuan G, Kumar GK, Fox AP, Prabhakar NR. NADPH oxidase-dependent regulation of T-type Ca2+ channels and ryanodine receptors mediate the augmented exocytosis of catecholamines from intermittent hypoxia-treated neonatal rat chromaffin cells. J Neurosci. (2010) 30:10763–72. doi: 10.1523/JNEUROSCI.2307-10.2010
379. Kumar GK, Rai V, Sharma SD, Ramakrishnan DP, Peng YJ, Souvannakitti D, et al. Chronic intermittent hypoxia induces hypoxia-evoked catecholamine efflux in adult rat adrenal medulla via oxidative stress. J Physiol. (2006) 575:229–39. doi: 10.1113/jphysiol.2006.112524
380. Kim DK, Natarajan N, Prabhakar NR, Kumar GK. Facilitation of dopamine and acetylcholine release by intermittent hypoxia in PC12 cells: involvement of calcium and reactive oxygen species. J Appl Physiol. (2004) 96:1206–15; discussion: 1196. doi: 10.1152/japplphysiol.00879.2003
381. Yuan G, Adhikary G, McCormick AA, Holcroft JJ, Kumar GK, Prabhakar NR. Role of oxidative stress in intermittent hypoxia-induced immediate early gene activation in rat PC12 cells. J Physiol. (2004) 557:773–83. doi: 10.1113/jphysiol.2003.058503
382. Czyzyk-Krzeska MF, Furnari BA, Lawson EE, Millhorn DE. Hypoxia increases rate of transcription and stability of tyrosine hydroxylase mRNA in pheochromocytoma (PC12) cells. J Biol Chem. (1994) 269:760–4. Available online at: http://www.jbc.org/content/269/1/760.long
383. Khurana S, Peng S, McDonald E, Yates W, Venkataraman K, Tai TC. Phenylethanolamine N-methyltransferase Gene Expression in PC12 cells exposed to Intermittent Hypoxia. Neurosci Lett. (2018) 666:169–74. doi: 10.1016/j.neulet.2017.12.056
384. Tai TC, Wong-Faull DC, Claycomb R, Wong DL. Hypoxic stress-induced changes in adrenergic function: role of HIF1 alpha. J Neurochem. (2009) 109:513–24. doi: 10.1111/j.1471-4159.2009.05978.x
385. Vaziri ND, Rodríguez-Iturbe B. Mechanisms of Disease: oxidative stress and inflammation in the pathogenesis of hypertension. Nat Clin Pract Nephrol. (2006) 2:582. doi: 10.1038/ncpneph0283
386. Nosalski R, McGinnigle E, Siedlinski M, Guzik TJ. Novel immune mechanisms in hypertension and cardiovascular risk. Curr Cardiovasc Risk Rep. (2017) 11:12. doi: 10.1007/s12170-017-0537-6
387. Chandel NS, Trzyna WC, McClintock DS, Schumacker PT. Role of oxidants in NF-kappa B activation and TNF-alpha gene transcription induced by hypoxia and endotoxin. J Immunol. (2000) 165:1013–21. doi: 10.4049/jimmunol.165.2.1013
388. Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. (2009) 1:a001651. doi: 10.1101/cshperspect.a001651
389. Elmarakby AA, Sullivan JC. Relationship between oxidative stress and inflammatory cytokines in diabetic nephropathy. Cardiovasc Ther. (2012) 30:49–59. doi: 10.1111/j.1755-5922.2010.00218.x
390. Manabe S, Okura T, Watanabe S, Fukuoka T, Higaki J. Effects of angiotensin II receptor blockade with valsartan on pro-inflammatory cytokines in patients with essential hypertension. J Cardiovasc Pharmacol. (2005) 46:735–9. doi: 10.1097/01.fjc.0000185783.00391.60
391. Dandona P, Kumar V, Aljada A, Ghanim H, Syed T, Hofmayer D, et al. Angiotensin II receptor blocker valsartan suppresses reactive oxygen species generation in leukocytes, nuclear factor-kappa B, in mononuclear cells of normal subjects: evidence of an antiinflammatory action. J Clin Endocrinol Metab. (2003) 88:4496–501. doi: 10.1210/jc.2002-021836
392. Lopez-Garcia E, MB S, TT F, JB M, Rifai N, JE M, et al. Major dietary patterns are related to plasma concentrations of markers of inflammation and endothelial dysfunction. Am J Clin Nutr. (2004) 80:1029–35. doi: 10.1093/ajcn/80.4.1029
393. McEwen BS, Biron CA, Brunson KW, Bulloch K, Chambers WH, Dhabhar FS, et al. The role of adrenocorticoids as modulators of immune function in health and disease: neural, endocrine and immune interactions. Brain Res Brain Res Rev. (1997) 23:79–133. doi: 10.1016/S0165-0173(96)00012-4
394. Wurtman RJ, Axelrod J. Control of enzymatic synthesis of adrenaline in the adrenal medulla by adrenal cortical steroids. J Biol Chem. (1966) 241:2301–5.
Keywords: hypertension, adrenal medulla, catecholamine, immune, cytokine, glucocorticoid, epinephrine
Citation: Byrne CJ, Khurana S, Kumar A and Tai TC (2018) Inflammatory Signaling in Hypertension: Regulation of Adrenal Catecholamine Biosynthesis. Front. Endocrinol. 9:343. doi: 10.3389/fendo.2018.00343
Received: 22 March 2018; Accepted: 07 June 2018;
Published: 28 June 2018.
Edited by:
James A. Carr, Texas Tech University, United StatesReviewed by:
Ricardo Borges, Universidad de La Laguna, SpainBalazs Gaszner, University of Pécs, Hungary
Copyright © 2018 Byrne, Khurana, Kumar and Tai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: T. C. Tai, dGMudGFpQG5vc20uY2E=