 Qiming Tan1†
Qiming Tan1† Seun E. Akindehin2†
Seun E. Akindehin2† Camila E. Orsso3Richelle C. Waldner1
Camila E. Orsso3Richelle C. Waldner1 Richard D. DiMarchi4
Richard D. DiMarchi4 Timo D. Müller2*†
Timo D. Müller2*† Andrea M. Haqq1,3*†
Andrea M. Haqq1,3*†- 1Department of Pediatrics, University of Alberta, Edmonton, AB, Canada
- 2Institute for Diabetes and Obesity, Helmholtz Diabetes Center at Helmholtz Zentrum München, Germany and German Center for Diabetes Research (DZD), Munich, Germany
- 3Department of Agricultural Food & Nutritional Science, University of Alberta, Edmonton, AB, Canada
- 4Department of Chemistry, Indiana University, Bloomington, IN, United States
The incretin hormone glucagon-like peptide-1 (GLP-1) has received enormous attention during the past three decades as a therapeutic target for the treatment of obesity and type 2 diabetes. Continuous improvement of the pharmacokinetic profile of GLP-1R agonists, starting from native hormone with a half-life of ~2–3 min to the development of twice daily, daily and even once-weekly drugs highlight the pharmaceutical evolution of GLP-1-based medicines. In contrast to GLP-1, the incretin hormone glucose-dependent insulinotropic polypeptide (GIP) received little attention as a pharmacological target, because of conflicting observations that argue activation or inhibition of the GIP receptor (GIPR) provides beneficial effects on systemic metabolism. Interest in GIPR agonism for the treatment of obesity and diabetes was recently propelled by the clinical success of unimolecular dual-agonists targeting the receptors for GIP and GLP-1, with reported significantly improved body weight and glucose control in patients with obesity and type II diabetes. Here we review the biology and pharmacology of GLP-1 and GIP and discuss recent advances in incretin-based pharmacotherapies.
1 Introduction
Obesity, diagnosed as a body mass index (BMI) ≥ 30 kg/m2, is a progressive, chronic disease that has grown to pandemic prevalence over the past decades (1). Obesity substantially increases the risk of type-2 diabetes (T2D), cardiometabolic diseases, osteoarthritis, neurological and mental disorders as well as several forms of cancer, resulting in premature disability and demise (Figure 1) (2, 3). Depending on the severity of the disease and the age at diagnosis, long-term health complications may last a lifetime and worsen the therapeutic outcome for multiple associated chronic diseases (2). Unsurprisingly, obesity leads to excess medical costs and imposes a large economic burden on individuals, families, health care systems, and societies (2, 4).
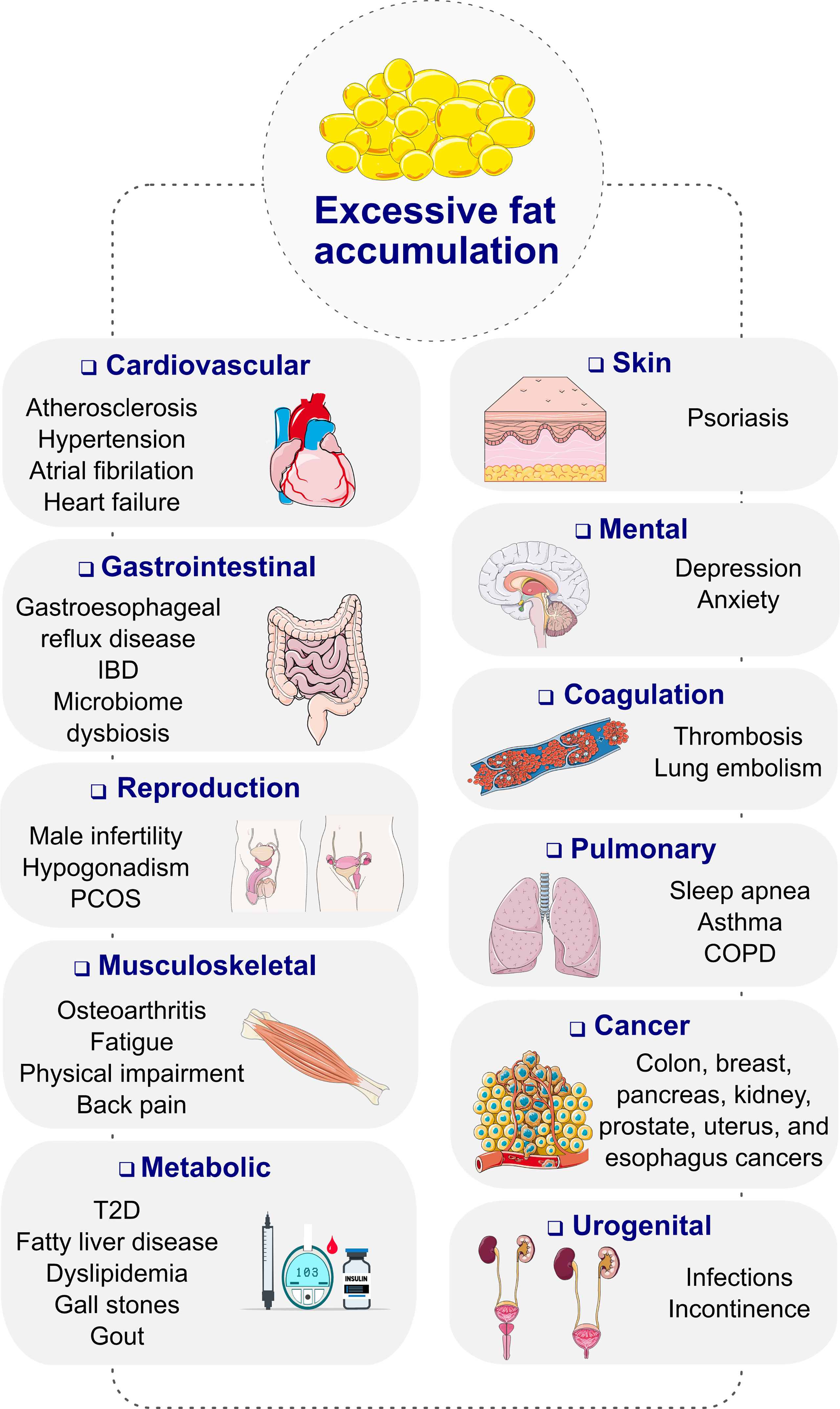
Figure 1 Complications of obesity. COPD, chronic obstructive pulmonary disease; IBD, inflammatory bowel disease; PCOS, polycystic ovary syndrome; T2D, type 2 diabetes. Images retrieved from smart.servier.com.
While traditionally recognized primarily as a disease of the elderly, T2D is currently one of the most frequently diagnosed preventable chronic diseases in middle age, as well as children and adolescents (5, 6). Excess body fat along with age constitute the two most important risk factors for the premature development of T2D. Some studies report that >80% of youth and ~50% of adults with T2D are overweight or obese at point of diagnosis (6, 7). Early onset T2D relative to late-onset disease is associated with a more rapid deterioration of β-cell function, emphasizing the importance for early diagnosis and treatment initiation (8). Obesity-related mechanisms that are potentially linked to the severity of the disease include adipocyte lipid spillover, ectopic fat accumulation and tissue inflammation (9). Sizeable weight loss not only improves glucometabolic health, it may also reduce the risk for obesity-linked co-morbidities, increase life expectancy, and improve quality of life (10–12). Therapies aiming to decrease body weight are consequently a valuable strategy to delay the onset and decrease the risk of T2D, as well as managing established disease (13).
Lifestyle modifications, such as balanced nutrition, calorie restriction and physical exercise, remain the cornerstone of any weight loss intervention. However, lifestyle changes alone are insufficiently efficacious and sustainable as a stand-alone therapy, possibly because physiological adaptations conspire to promote weight regain following diet-induced weight loss (14). Genetic and environmental factors may further undermined treatment efficacy (15). Polygenetic gene variants, each accounting for only a small difference in body weight, may sum up to sizably affect body mass and may hinder susceptibility of an individual to respond to a weight loss intervention (16, 17). There are also less frequent variants with larger effects leading to early onset of severe obesity in humans. Several syndromic and monogenic disorders of obesity that have been identified include Prader–Willi syndrome, Bardet–Biedl syndrome, and loss-of-function mutations in the genes encoding for pro-opiomelanocortin (POMC), leptin, leptin receptor (LEPR) or the melanocortin-4 receptor (MC4R) (17).
Pharmacotherapy as an adjunct to lifestyle adjustments is often used to enhance weight loss efficacy (18). However, a key obstacle in the development of anti-obesity medication is that rodent studies proved largely incapable to predict cardiovascular safety in humans (13, 19, 20). Also, the heterogeneity of patient cohorts, with many individuals being of advanced age and at high risk for development of cardiovascular diseases (CVD), represents an obstacle that is not easy to address with pharmacotherapy (19). Consistent with this, a series of previously employed anti-obesity medications were withdrawn soon after approval due to unforeseen adverse effects on the cardiovascular system (19–21). Furthermore, when given at tolerable doses, pharmacotherapy rarely decreases body weight >10%. Notable exceptions are semaglutide 2.4 mg (Wegovy® Novo Nordisk, Copenhagen, Denmark), a long-acting agonist at the glucagon-like peptide-1 receptor (GLP-1R) (22), and the experimental drug candidate tirzepatide, a dual-agonist at the receptors for GLP-1 and the glucose-dependent insulinotropic polypeptide (GIP) (23). Each of these peptides decrease body weight with a favorable safety profile in the majority of patients by >10% (24–28). While the clinical success of these drugs sets the stage for a new era in anti-obesity medication, there remains considerable controversy as to how GIP regulates metabolism and whether GIP receptor agonism or antagonism is a preferred treatment for obesity and T2D. In this manuscript, we provide an overview of the mechanistic biology and in vivo pharmacology of GLP-1 and GIP. We summarize recent clinical results with molecules that target each receptor and discuss recurrent questions related to their mode-of-action.
2 Glucagon-Like Peptide-1 (GLP-1)
2.1 The Physiology of GLP-1
GLP-1 is encoded by proglucagon, a 158 amino acid precursor protein, that is predominantly expressed in the gut, pancreas, and distinct neuronal populations of the hindbrain (29). In the brain and the intestine, proglucagon is cleaved by the action of the prohormone convertase 1/3 (PC1/3) into GLP-1, GLP-2, glicentin, glicentin-related polypeptide (GRPP), and oxyntomodulin (OXM) (29–31). In the pancreatic α-cells, proglucagon is cleaved by PC2 into glucagon, GRPP and the major proglucagon fragment (MPGF). In the intestine, GLP-1 is secreted from enteroendocrine L-cells located in the gut epithelium. The density of the L-cells is low in the duodenum and jejunum and it is high in the ileum and colon (29). Nutrients stimulating the secretion of GLP-1 in the intestine include monosaccharides such as glucose, galactose, fructose (32–34), fatty acids (35, 36), as well as proteins (37) and amino acids, particularly glutamine and glycine (38, 39). The relevance of endocrine factors to promote GLP-1 secretion seem to vary across species and may include acetylcholine, insulin, ghrelin, GIP, and gastrin-releasing peptide (29). Circulating levels of total GLP-1 are low during fasting (~5 pmol/l) and rapidly rise up to 40 pmol/l shortly after a meal (40). Consistent with the ability of GLP-1 to accelerate glucose-stimulation of insulin secretion (GSIS), the meal-induced rise in plasma GLP-1 is paralleled by enhanced insulin immunoreactivity (40).
GLP-1 promotes its biological action through binding to the GLP-1 receptor (GLP-1R), a 7 transmembrane G protein-coupled receptor of the class B family (41). GLP-1R signals primarily via the Gαs pathway, and hence accelerates intracellular levels of cAMP (42). GLP-1R can also recruit, and induce signaling, via the Gαq and β-arrestin pathways and knockdown of β-arrestin-1 in rat insulinoma (INS1) cells decreases the ability of GLP-1 to promote GSIS (43). Immunohistochemical studies in tissues from humans and non-human primates show widespread distribution of GLP-1R in the brain and in the periphery (44). These data largely align with studies in rodents in which the abundance of the GLP-1R transcript was assessed using mice that express green fluorescent protein (GFP) under control of the GLP-1R promoter (45). Consistent with the ability of GLP-1R agonists to decrease homeostatic and hedonic food intake (29, 29), expression GLP-1R is found in the rodent hypothalamus (ARC, VMH, DMH, PVH, LH), hindbrain (AP, NTS, ventrolateral medulla) and telencephalon (amygdala, olfactory bulb, preoptic area, nucleus accumbens) (45, 46). In the pancreas, GLP-1R is solidly expressed in the β- and δ-cells but is only found in a small portion of α-cells (45). No expression of GLP-1R is found in the liver and the thyroid gland (44).
Albeit best known for its glycemic effects, GLP-1 is a pleiotropic hormone with a series of metabolic effects beyond the regulation of glucose metabolism (Figure 2A). Apart from its ability to act on the pancreas to enhance GSIS and to inhibit the secretion of glucagon, GLP-1 decreases body weight by decreasing homeostatic and hedonic food intake (29). GLP-1R agonism further inhibits gastric emptying (47); has cardio- and neuroprotective effects (48); lowers inflammation and apoptosis (49–53); stimulates β-cell proliferation in rodents (54); and exerts positive effects on learning, memory, and reward behavior (55, 56). Endogenous GLP-1 is mainly produced as GLP-1(7-36NH2), with a low proportion produced as GLP-1(7-37) and an even a lower portion as GLP-1(1-37) or GLP-1(1-36NH2) (29, 57). Native GLP-1 has a half-life of just ~2–3 min (58–60), which results mainly from rapid in vivo proteolysis by the dipeptidylpeptidase-4 (DPP-4) and fast renal elimination. DPP-4 cleaves GLP-1(7-36NH2) and GLP-1(7-37) at the second N-terminal amino acid (Ala8) position, leading to metabolically metabolites GLP-1(9-36NH2) and GLP-1(9-37) of much reduced potency (61). Despite species-related differences, GLP-1 is also subject to degradation by the neutral endopeptidase (NEP) 24.11, which cleaves GLP-1 at its central and C-terminal positions Asp15, Ser18, Tyr19, Glu27, Phe28 and Trp31 (61, 62). The relevance of NEP 24.11 to cleave GLP-1 varies among species and while it contributes substantially to GLP-1 degradation in mice and pigs (63, 64), it’s relevance in humans has long been questioned. Nonetheless, more recent data show that NEP 24.11 also plays a physiological relevant role for degradation of GLP-1 in humans (65).
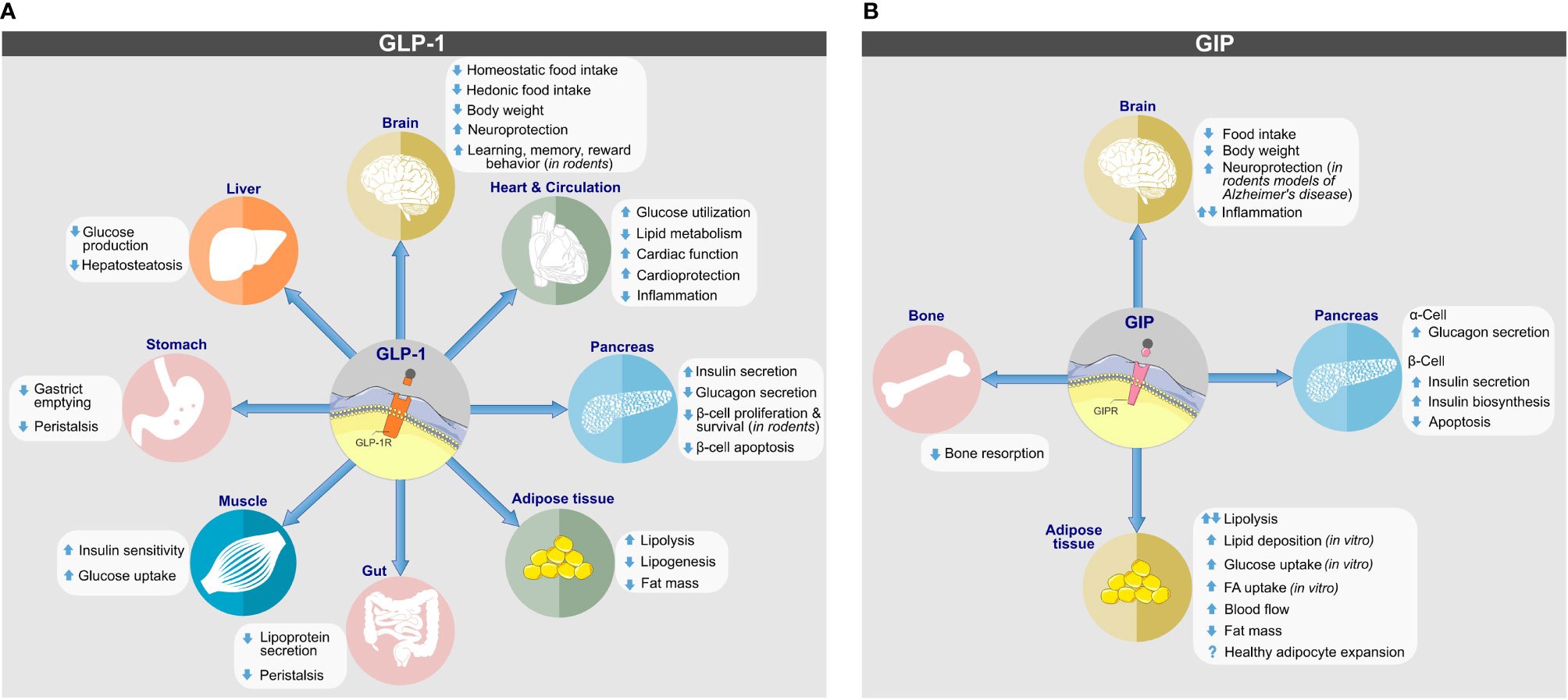
Figure 2 Biological actions of (A) GLP-1 and (B) GIP on target tissues. Direct and indirect effects are depicted. Images retrieved from smart.servier.com.
Consequential due to its short half-life, native GLP-1 has only limited pharmacological potential and only ~10–15% of active GLP-1 is estimated to reach the general circulation (59, 66–68). Nonetheless, emphasizing its therapeutic implication, 6-wk continuous infusion of GLP-1(7-36NH2) at a rate of 4.8 pmol-1 kg-1 min-1 improved glycemic control and insulin sensitivity in patients with T2D (69). Despite the lack of singular attribution to enhanced GLP-1 action, similar results have been demonstrated following administration of a DPP-4 inhibitor (70–73).
Notably, as comprehensively reviewed elsewhere (20, 29, 74), GLP-1 improves glycemic control via several complementary mechanisms. In the pancreas, GLP-1 directly acts on the β-cells to promote GSIS via the action of PKA and Epac2 (29). Both pathways are equally important for the insulinotropic effect of GLP-1 and ensure that GLP-1 primarily enhances insulin secretion under conditions of hyperglycemia (29). Other than stimulating the secretion of insulin in a glucose-dependent manner, GLP-1 also promotes the production of insulin via activation of Pdx1, which binds to the insulin promoter and activates its expression (29). GLP-1 also lowers blood glucose by inhibiting the release of glucagon and thus inhibiting hepatic glucose production (29). Clamp studies in patients with T2D indicate that the insulinotropic and glucagonostatic effects of GLP-1 contribute equally to decreasing blood glucose (75). While GLP-1 directly acts on the β-cells to stimulate insulin secretion, GLP-1 inhibition of glucagon secretion seems to be indirectly triggered via paracrine effects in the islets. Accordingly, GLP-1 stimulates the secretion of insulin, zinc, GABA, amylin, and somatostatin, all of which inhibit glucagon secretion (30). Supporting this notion of a paracrine effect is that the GLP-1 receptor is only expressed in a small subset of α-cells (45). However, GLP-1 does not only decrease blood glucose via its direct effects on the islets, it also inhibits gastric emptying and thereby slows glucose entry into the circulation (47, 76–78). Emphasizing the importance of GLP-1-mediated inhibition on gastric emptying for the regulation of blood glucose, antagonizing GLP-1’s effect on gastric emptying by co-infusion of GLP-1 with erythromycin during a liquid meal diminished GLP-1’s ability to decrease post-prandial hyperglycemia in patients with T2D (79).
In summary, GLP-1 improves fasting blood glucose through its direct action on the pancreatic islets and decreases postprandial hyperglycemia through inhibition of gastric emptying, and thus reduced glucose entry into circulation (74). These differences in glucose regulation translate into important pharmacological differences. Short-acting GLP-1 mimetics (e.g. exenatide BID, lixisenatide) are self-administered prior to a meal and, in conjunction with their short half-life of 2–3 h, display substantial fluctuations in circulation, with highest levels during the prandial and early post-prandial state, and lowest levels in the fasting periods between meals. Due to their relatively high plasma levels at the time of meal intake, the short-acting GLP-1 mimetics relative to the long-acting GLP-1R agonists display a higher tendency to affect GI motility and to decrease post-prandial hyperglycemia (74). On the contrary, long-acting GLP-1R agonists are less prone to affect GI motility and primarily decrease fasting blood glucose levels via direct action in the pancreas (74). With stable plasma concentrations and lower efficacy on GI motility, the long-acting GLP-1 mimetics are less prone to incur adverse gastrointestinal side effects (80) and generally have a higher potential to decrease body weight (74).
2.2 Pharmacological Advances in GLP-1R Agonism
A variety of structurally and chemically refined GLP-1R analogs have received extensive attention and have been implemented in clinical use for the treatment of T2D and obesity (Figure 3) (29). Clinical success in the treatment of obesity has been established for liraglutide 3 mg (Saxenda®, Novo Nordisk, Denmark) (81) and more recently for semaglutide 2.4 mg (Wegovy®, Novo Nordisk, Denmark) (13, 28). Also, molecules with simultaneous activity at the receptor for GIP (82–85) or glucagon (86, 87) have shown promising results for this application. The continuous improvement of the pharmacokinetic profile of GLP-1R agonism, starting from a native hormone with a half-life of ~2–3 min to the development of twice daily (exenatide BID), daily (liraglutide, lixisenatide) and even weekly (exenatide ER, albiglutide, dulaglutide, semaglutide) formulations, highlight the pharmaceutical advancement in this arena. Additionally, the recent development of an orally available preparation of semaglutide (Rybelsus®, Novo Nordisk), and the recruitment of GLP-1 into unimolecular pharmacology with GIP, glucagon (and others) (20, 29, 88) exemplifies how GLP-1-based drug development and innovation advanced over recent years. It emphasizes how structural refinements and biochemical modifications can extend the application of GLP-1R agonists from the treatment of T2D to obesity (13).
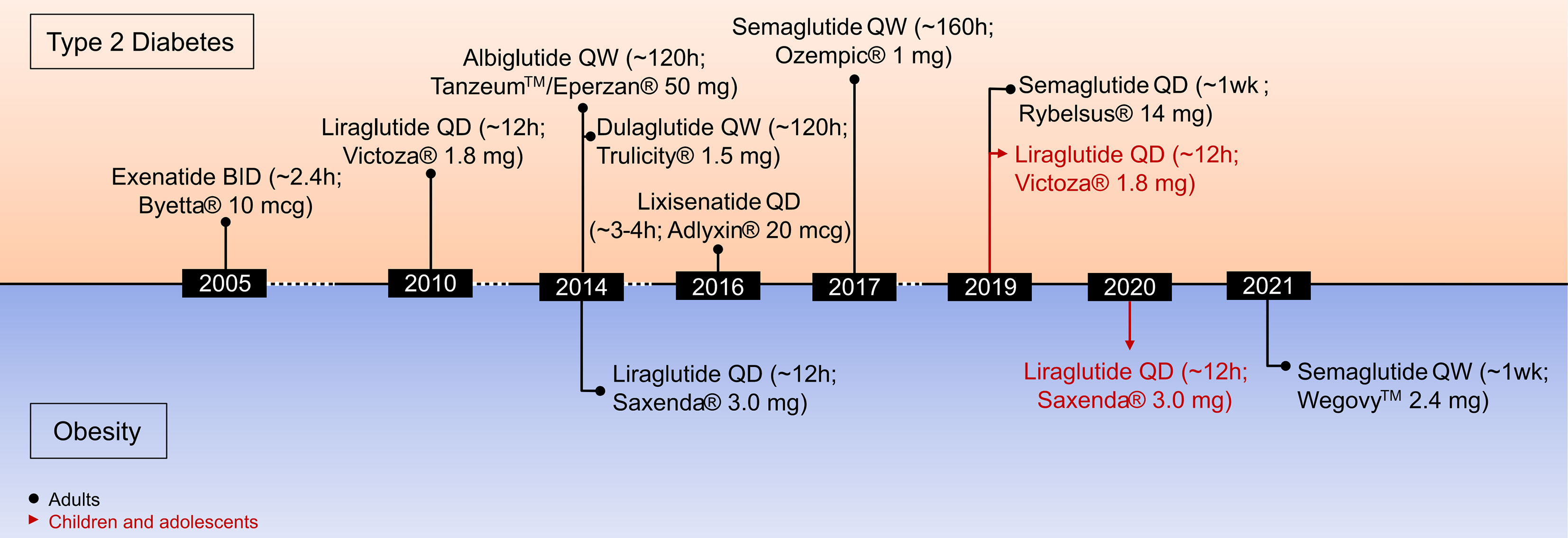
Figure 3 Timeline of drug approvals by the U.S. Food and Drug Administration.
2.3 Methods to Extend GLP-1’s Half-Life
The short action profile of native GLP-1 imposes a major challenge towards its successful clinical utilization. To overcome this limitation, various strategies have been applied to extend the half-life of GLP-1 and to accelerate its in vivo action and potency (Figure 4). The efficacy of a drug in a biological system is influenced by factors that include the stability of the active molecule and its rate of diffusion into and elimination from circulation. Methods to improve drug efficacy included structural and chemical modifications targeted to increase molecular stability and activity, to improve biodistribution/bioavailability, and to delay renal elimination (89).
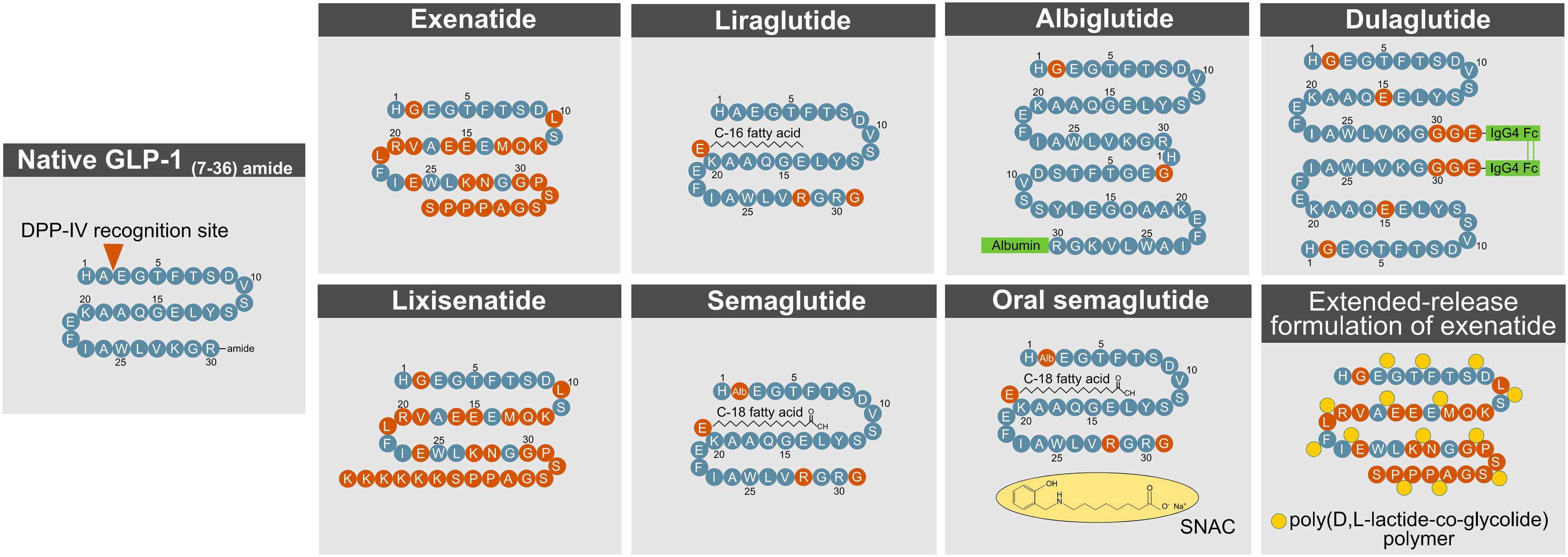
Figure 4 Methods to enhance GLP- 1R action. DPP- 4, dipeptidylpeptidase-4; GLP- 1, glucagon-like peptide-1.
2.3.1 Protection From DPP-IV Cleavage
A commonly used procedure to improve the half-life of active GLP-1 is a modification at the second N-terminal amino acid position (Ala8) to protect from degradation by DPP-4. Such structural modification has been applied to exenatide, lixisenatide, semaglutide, dulaglutide, and albiglutide.
Exenatide and lixisenatide are synthetic peptides that fully resemble the sequence of exendin-4, a GLP-1 paralog that is naturally found in the saliva of the gila monster (heloderma suspectum). Both exenatide and lixisenatide contain the full sequence of exendin-4, but lixisenatide is extended on the C-terminus to possess six additional lysine residues (20). Exendin-4 has a glycine at the second N-terminal position (Gly8), which prevents the molecule from being fully recognized by DPP-4. Relative to native GLP-1, exendin-4 (and thus exenatide and lixisenatide) is further differentiated with amino acid substitutions in the middle segment of the sequence, which render the drug less susceptible to degradation by NEP 24.11 (62). The C-terminus of exendin-4 relative to native GLP-1 is extended by nine amino acids, which enhances its secondary structure and improves chemical stability (90). Despite being protected from DPP-4 cleavage, exendin-4 still undergoes rapid renal elimination, which in humans leads to a half-life of ~2.5 h (exenatide) and ~3–4 h (lixisenatide), respectively (29). Consistent with this notion, plasma clearance of exendin-4 is reduced 4.4-fold in nephrectomized rats (91) and 3.4-fold in patients with end-stage renal disease (92). With only 53% sequence homology to native GLP-1, exendin-4 has the limitation that ~40% of patients treated with exenatide (93–95), and ~60% of patients treated with lixisenatide (96), develop antibodies against the molecule, but this does not seem to negatively affect glucose handling or the prevalence of adverse effects. While exenatide, lixisenatide, dulaglutide, and albiglutide possess a glycine at their second N-terminal amino acid position, semaglutide bears an aminoisobutyric acid (AIB) at this residue to protect from DPP-4 inactivation.
2.3.2 Covalent Binding to Albumin
Albiglutide (GlaxoSmithKline, London, UK) is a sixty amino acid tandem of two DPP-4 protected GLP-1 molecules that are covalently fused to human albumin (20). The chemical conjugation to albumin enhances GLP-1’s proteolytic stability and simultaneously delays renal elimination, which manifests as a half-life of ~120 h in humans (97). The long half-life of albumin, and potentially that of albumin-fused drugs, is derived from albumin’s ability to escape intracellular degradation by binding to the neonatal Fc receptor (FcRn), in a pH-dependent fashion (98–100). Upon endocytosis by the endothelial cells, albumin binds to the Fc receptor (FcRn) during endosomal acidification, which sorts the intracellular trafficking of the albumin away from degradative lysosomes and back to the plasma membrane where it can once again reenter the general circulation (98–100). Underlining the relevance of this intracellular mechanism, circulating levels of albumin are decreased by 40–50% in FcRn-deficient mice (99, 100). Furthermore, as the molecular size of a drug greatly affects its renal elimination (89, 101), larger molecules exhibit an inherently greater circulatory half-life due to physical hindrance in glomerular filtration (89). Thus, the fusion and binding of GLP-1 to albumin, or the linkage of several GLP-1 molecules with an Fc fragment, naturally promote a delay in renal clearance due to the increased size of the molecule.
2.3.3 Acylation With a Fatty Acid
The strategic fusion of GLP-1 to a fatty acid has been applied for liraglutide (Novo Nordisk, Denmark) and semaglutide (Novo Nordisk, Denmark). Liraglutide is linked to palmitic acid (C16:0) via a gamma glutamic acid spacer at the lysine residue at position 26 (102). The fatty acid promotes formation of a self-associated multimolecular complex, which protracts the diffusion of the molecule from the site of injection. A delay in renal clearance of the drug is achieved by the reversible non-covalent binding of the fatty acid moiety to human serum albumin (HSA). Although liraglutide carries the native GLP-1 alanine residue at the second N-terminal position (Ala8), the fatty acid-mediated albumin binding promotes self-oligomerization and thereby improves the proteolytic stability of the molecule to further decrease the drug’s susceptibility to DPP-4 cleavage. This chemical modification manifests in a half-life of ~12 h in humans (103). Semaglutide is structurally identical to liraglutide with the exception that the Ala8 residue is substituted with AIB to further protect from DPP-IV recognition, and that the palmitic acid (C16:0) is exchanged with a dicarboxylic-stearic acid (C18:0). Impressively, these modifications manifest a half-life of ~160 h upon subcutaneous injection in humans (104). Of note, both liraglutide and semaglutide show great sequence homology (>95%) to native GLP-1, which decreases the likelihood of immunogenicity.
2.3.4 Pegylation
Polyethylene glycol (PEG) is a synthetic water-soluble inert polymer with the potential to enhance a drug’s half-life by slowing down its rate of renal clearance. Pegylation of a drug can also enhance its aqueous solubility, protect against in vivo proteolysis, and enhance toxicological safety and potentially even its non-immunogenic resilience (105). The linkage between the PEG and a conjugated molecule/peptide can either be stable or degradable, with the latter often being used for prodrugs. Overall, pegylation does not seem to affect the folding and stability of the conjugated protein as assessed by circular dichroism, ultraviolet absorption, or NMR spectroscopy (106–110). This observation seems particularly important since misfolded proteins might lose in vivo potency and be subject to enhanced proteolysis and potential immunogenicity. Various FDA approved pegylated proteins are currently in therapeutic use. The first FDA approved pegylated drug was Adagen® (Enzon Pharmaceuticals, new Jersey, USA) which was approved by the FDA in 1990 for adenosine deaminase deficiency associated with severe combined immunodeficiency disease (SCID). The PEG modification can, in rare cases, lead to vacuolation or the generation of antibodies against the PEG. As it pertains to drugs to control body weight and/or glycemia, preclinically tested pegylated anorectics include leptin (111), FGF21 (112–117), GIP (118, 119), GLP-1R agonists (120–122), NPY2 receptor agonists (123) and unimolecular dual-agonists targeting the receptors for GLP-1 and glucagon (124, 125) or GLP-1 and GIP (126).
2.3.5 Fc Fusion
Linkage of two GLP-1 molecules via the Fc fragment of a monoclonal antibody (IgG4) has been applied for dulaglutide. The sixty amino acid molecule comprises two Gly8-modified DPP-4 protected GLP-1 molecules in which the C-termini are fused at a Gly36 residue to IgG4 Fc fragments. Notably, while dulaglutide carries a glycine at position 8 to protect from DPP-4 cleavage, Fc fusion to native GLP-1 is sufficient to reduce DPP-4 degradation by 4-5-fold relative to native GLP-1 (127). The Arg36 residue of native GLP-1 was substituted with Gly36 to serve as an anchor for the IgG4 Fc fragments and to decrease the possibility of T-cell epitope interaction (128). Furthermore, the native GLP-1 Gly22 residue is exchanged with glutamic acid, which stabilizes the secondary structure and enhances potency (128). The rationale of extending the half-life of GLP-1 via fusion to an IgG4 Fc fragment is similar to its fusion to albumin. As with albumin, the Fc complex binds upon endocytosis to the Fc receptor (FcRn) in the acidic endosomal compartments with the consequence that the FcRn-bound Fc complex is recycled back to the plasma membrane and secreted back into general circulation (101, 129–131). The Fc fragment, and the two GLP-1 motifs fused via the Fc fragment, also further increase the size of the molecule, and thereby naturally decrease its renal elimination (89, 101).
2.3.6 Sustained-Release Formulations
Bydureon® (AstraZeneca, Wilmington, USA) is an extended-release (ER) formulation of exenatide. The drug is self-applied on a weekly basis independent of meal patterns. The extended release is achieved through incorporation of exenatide (exendin-4) into 0.06 mm-diameter biodegradable microspheres, which comprise a 50:50 poly(D,L-lactide-co-glycolide) (PLG) polymer along with sucrose (132). Exenatide ER contains encapsulated exenatide at a concentration of 5 mg per 100 mg of microspheres (132). In the human body, the PLG polymers slowly degrade through the non-catalyzed hydrolysis of the ester linkages into lactic acid and glycolic acid, which are finally eliminated as carbon dioxide and water (132).
During the protracted degradation of PLG, exendin-4 is sustainably released into the general circulation for a period of several (~7) weeks, yielding therapeutic levels after two weeks and a steady state after 6–7 weeks (132). The release of exenatide into the general circulation occurs in three stages. During the initial phase, the freshly injected microspheres hydrate and immediately release loose and cell-surface-bound exenatide (< 1%) into the circulation (132). In the second phase, the polymer hydrolyzes into smaller fragments and, upon reaching a size of ~20 kDa, promotes the constant diffusion of exenatide into the circulation. In the erosion phase, the PLG polymers fully hydrolyze and eventually release all remaining exenatide (132). Reflecting this 3-phase diffusion, a first peak of exenatide release (1–2% of the total area under the plasma concentration curve) is observed in the first 48 h after the injection, followed by two peaks after approximately two and seven weeks, respectively (132).
The prolonged rise to achieve steady state plasma concentrations seems to have beneficial effects on tolerability since the frequency of nausea and vomiting is reduced in patients treated with exenatide ER relative to treatment with exenatide BID (80, 133). Exenatide ER is also superior over exenatide BID in preventing episodes of hyperglycemia, as indicated by a greater decrease in HbA1c (80). Weight loss, however, does not seem to be overtly different between patients treated with exenatide ER and exenatide BID (80). The continuous steady-state plasma concentration of exenatide (which is structurally identical in both formulations) nonetheless seems to be associated with a greater immunogenic liability for exenatide ER. In line with this notion, anti-exenatide antibodies are detected in ~70% of patients treated with exenatide ER relative to >40% of patients treated with exenatide BID (80).
2.3.7 Oral Semaglutide
A general limitation to oral administration of GLP-1 mimetics is poor absorption via the GI-tract and quick degradation by proteolytic enzymes and the acidic environment of the stomach. For this reason, peptide GLP-1R agonists are not available as oral preparations and rather have to be subcutaneously self-injected by the patient. Difficulties and discomfort with self-injected medication is a factor that negatively affects patient compliance and quality of life (134–136). A milestone in GLP-1-based pharmacotherapies is the recent development of an oral available formulation of semaglutide (Rybelsus®, Novo Nordisk, Copenhagen, Denmark). In Rybelsus®, semaglutide is co-formulated with sodium N-[8-(2-hydroxybenzoyl)aminocaprylate] (SNAC), which shields the molecule from enzymatic and acidic degradation and accelerates its site-directed release and absorption in the stomach (137). As demonstrated in humans, the SNAC-linked semaglutide tablet undergoes surface erosion in the stomach with the result that the non-covalent linkage between semaglutide and SNAC dissolves and releases free semaglutide into the circulation (137). Another possibility to orally engage GLP-1R activity is through administration of smaller molecule mimetics. Two conventional small molecule agonist with agonism at GLP-1R have recently been disclosed and are in clinical evaluation for the oral treatment of diabetes (138–140).
2.4 Recent Clinical Advances of GLP-1R Agonists for the Treatment of Obesity and Diabetes
Liraglutide 3 mg (Saxenda®, Novo Nordisk, Copenhagen, Denmark) was approved in 2014 for the treatment of obesity in adults and in 2020 for the treatment of obesity in children aged 12 and older (Figure 3) (141). Between 50 and 70% of the patients treated with liraglutide 3mg achieve a body weight reduction > 5% while between 6 and 35% achieve weight loss > 10% (142). Of appreciable note, in contrast to a vast majority of previously employed anti-obesity medications (13, 20), GLP-1R agonism improves cardiovascular (CV) health in patients with T2D (48, 143). Consistent with this, a recent meta-analysis assessing CV outcome of different GLP-1R agonists across eight CV outcome trials comprising 60,080 patients with T2D. It demonstrated that GLP-1R agonists reduce the risk of a major adverse cardiovascular event (MACE) by 14%, death by 13%, non-fatal stroke by 12%, and broad composite kidney outcome by 17% (144).
In June 2021, the FDA approved semaglutide 2.4 mg (Wegovy®) in adjunct to lifestyle programs for the treatment of obesity in adults (145). In non-diabetic individuals overweight or obese, 68 weeks of treatment with semaglutide (2.4 mg QW), decreased body weight by 14.9% relative to 2.4% in placebo treated controls (28). Impressively, 86.4% of patients treated with semaglutide showed weight loss >5% (vs. 31.5% in placebo controls), while 69.1% (vs. 12.0% in placebo controls) lost >10%, 50.5% (vs. 4.9% in placebo controls) >15% and 32% (vs. 1.7% in placebo controls) >20% (28). Similar to the observation in patients with T2D (48, 143), participants with obesity receiving semaglutide 2.4 mg had greater improvement of markers indicative of cardiometabolic health (28). Like with other GLP-1R agonists, adverse effects of semaglutide are predominantly of gastrointestinal nature, with the most common being nausea, diarrhea, vomiting, and constipation (28). Notably, while a series of clinical studies confirms the ability of semaglutide 2.4 mg to lower body weight >10% in most patients, the magnitude of weight loss is considerably lower in diabetic relative to non-diabetic patients with obesity (28, 146–148).
3 Glucose-Dependent Insulinotropic Polypeptide (GIP)
3.1 The Physiology of GIP
GIP is derived from posttranslational cleavage of proGIP, a 153 amino acid preprohormone that is expressed in the enteroendocrine K-cells of the upper intestine, the pancreatic α-cells and potentially the CNS (149). The majority of circulating GIP refers to GIP(1-42), which gets cleaved from proGIP in the intestine by the action of PC1/3, but also a shorter form, GIP(1-30NH2), is produced in the intestine and the pancreatic α-cells by cleavage of proGIP by PC2 (149). Both forms are of equal insulinotropic potency in mice (150) and both carry an alanine at their second N-terminal residue, which render the molecules susceptible for DPP-4 degradation (151). Similar to GLP-1, GIP undergoes rapid renal elimination and consistent with this, intact GIP is quickly cleared from the circulation with a half-life of four minutes and without major difference between healthy subjects and individuals with T2D (151). Primarily secreted in response to the ingestion of fat, GIP promotes its biological action through binding to the GIP receptor (GIPR), a class B GPCR that, similar to GLP-1R and belonging to the glucagon receptor family (149). In mice, Gipr is ubiquitously expressed in the endocrine pancreas with comparable mRNA quantities in the α- β- and δ-cells (152). GIPR is further expressed in adipocytes (153), myeloid cells (154), the endothelium of the heart and blood vessels, the pituitary and the inner layers of the adrenal cortex (155). In the brain, Gipr is found in the cerebral cortex, hippocampus, olfactory bulb as well as in the hypothalamus and the hindbrain (155–157).
3.2 GIP Regulation of Lipid Metabolism
GIP is best known for its ability to act on the pancreatic islets where depending on blood glucose concentration it stimulates the secretion of insulin or glucagon (Figure 2B) (158). The insulinotropic effect of GIP is diminished in patients with type-2 diabetes (159) but is restored upon near normalization of glycemia upon 4-week administration of insulin (160).
Beyond its glycemic effects, GIP decreases bone resorption (161), has neuroprotective effects in animal models of Alzheimer’s disease (162), and regulates lipid metabolism (163). As comprehensively reviewed previously (149, 163–165) GIP regulation of energy and lipid metabolism is partially conflicting, since activation and inhibition of GIPR signaling can both decrease body weight and fat mass in rodents (149, 164).
As demonstrated in vitro in cultured adipocytes (166, 167) and in vivo in Zucker rats (167), GIP enhances the activity of lipoprotein lipase (LPL), which promotes adipose tissue lipid disposal by enhancing hydrolytic cleavage of circulating triglycerides (TAG) into fatty acids (FA) and monoacylglycerol, which gets re-esterified and stored in adipose tissue. Consistent with this, GIP promotes clearance of circulating TAGs in dogs (168) and enhances adipose tissue TAG accumulation in rats (169) and humans (170). Under conditions of hyperglycemia and hyperinsulinemia, GIP increases adipose tissue blood flow and TAG clearance in healthy lean subjects (171). It further enhances FA synthesis in adipose tissue explants (172) and potentiates insulin-stimulated uptake of FA into the adipocytes (173). Collectively, GIP can act on the adipose tissue to increase adipose tissue blood flow, to enhance LPL-induced TAG clearance from the circulation and to stimulate adipocyte lipid storage. Recently, it was hypothesized that GIP may also facilitate healthy white adipose tissue expansion and thereby protect from adipocyte lipid spill-over and ectopic accumulation of lipids (163). This hypothesis is anchored on the observations that GIP is a target of PPARγ, a master regulator of adipogenesis (174), the expression of GIP increases during adipocyte differentiation (175, 176), and that knockdown of GIPR impairs adipocyte development (175).
3.3 Regulation of Energy Metabolism by GIPR Agonism and Antagonism
Consistent with the proposed role of GIP in lipid storage, mice with global loss of Gipr are lean and exhibit decreased body weight gain when fed a high-fat diet (HFD) (177). Body weight is also decreased in Gipr deficient ob/ob mice relative to ob/ob controls (177) and is decreased in mice with deletion of Gipr in the adipose tissue (178) and the CNS (179). However, the body weight difference between wt mice and CNS- or adipose-specific Gipr ko mice is only mild in comparison to the global loss of Gipr (177–179), indicating that lack of GIPR signaling in these tissues does not fully explain the phenotype of the global Gipr ko mice. No difference in body weight is seen between wt mice and mice that lack Gipr in the brown adipose tissue (BAT) (180) or the pancreatic β-cells (181).
The observation that global loss of Gipr protects from diet-induced obesity (177) and that GIP can promote adipocyte lipid storage (163) has inspired the development of GIPR antagonists for the treatment of obesity (182). Among the most prominent GIPR antagonists are natural (183) and biochemically modified (fatty-acylated) (184) forms of N-terminally truncated GIP as well as GIPR neutralizing antibodies (181, 185, 186). When administered peripherally, particularly antibody-based GIPR antagonists demonstrate some potential to prevent the development of HFD-induced obesity in rodents (181, 186), but GIPR antagonists show only modest, if any ability to decrease body weight once obesity is already established (181, 183–186). However, one study demonstrated that central administration of an antibody-based GIPR antagonist remarkably decreased body weight in DIO mice, potentially through mechanisms that include restoration of leptin sensitivity (185). Enhanced leptin action can however not be the sole mechanism by which centrally applied GIPR antagonists decrease body weight, since loss of GIPR in leptin deficient ob/ob mice still decreases body weight relative to ob/ob controls (177).
While GIPR antagonism has only little effects to decrease body weight in DIO mice (181, 183–186), its combination with GLP-1R agonism decreases body weight beyond what is possible with either monotherapy alone (181). Interestingly, this observation is surprisingly similar to long-acting (acylated) GIPR agonists when co-injected with fatty-acyl GLP-1 (126). When given at a dose of 3 nmol/kg/day, fatty-acyl GIP fails to affect body weight in DIO mice, but when given as an adjunct to fatty-acyl GLP-1 (3 nmol/kg/day), weight loss in the co-therapy is synergistically greater than treatment with GIP or GLP-1 alone (126). The mechanism of how GIPR and GLP-1R agonism synergizes to enhance body weight loss are not known, but a series of experimental results indicates that GIP acts on the GIP receptor in the brain to decrease body weight via inhibition of food intake (156, 179, 187). Consistent with this, GIPR is expressed in the hypothalamus (156) and the hindbrain (157, 188) and DREADD-mediated activation of GIPR neurons/cells in the hypothalamus decreases food intake in rodents (156). Central and peripheral administration of a long-acting (fatty-acylated) GIP increases cFOS neuronal activation in key feeding areas of the hypothalamus (179) and several long-acting GIPR agonists have been shown to decreases body weight and food intake in DIO mice without affecting energy expenditure (179, 184). How pharmacological activation and inhibition of GIPR both improve energy and lipid metabolism is subject of intense scientific investigation. Relevant hypotheses include that GIPR agonists may desensitize GIP receptor signaling (189), or that GIPR agonists and antagonists improve metabolism through independent mechanisms (187). Arguing against a role of GIPR agonists as functional antagonists is the observation that single central administration of acylated GIP is sufficient to rapidly lower food intake within hours after its administration (179) and that even DREADD-mediated (non-GIPR ligand-induced) activation of GIPR neurons decreases food intake in mice (156). Also, expression of GIPR is not decreased upon chronic treatment of DIO mice with acyl-GIP in either the hypothalamus or the adipose tissue (179) and dual-agonists targeting the receptors for GLP-1 and GIP do not show enhanced internalization of the GIP receptor (42, 190).
The ability of GIPR agonists to decrease body weight and food intake vanishes in global Gipr ko mice but is preserved in GLP-1R ko mice (179, 184), indicating that GIPR agonists lower body weight and food intake independent of GLP-1R signaling via the GIP receptor. When administered centrally (icv), fatty-acyl GIP decreases body weight and food intake in HFD-fed wildtype mice but not in mice with CNS deletion of Gipr (179). When administered peripherally, fatty-acyl GIP fails to affect food intake in CNS Gipr ko mice but shows partially preserved ability to decrease body weight (179). These data indicate that fatty-acyl GIP acts in the brain to decrease body weight via inhibition of food intake but also decreases body weight independent of GIPR in the CNS (179). Although GIPR agonists do not require GLP-1R to decrease body weight (179, 184), GIPR agonism was recently shown to attenuate the emetic effects of GLP-1R agonism in mice, rats and musk shrews (188). Given the role of the caudal hindbrain in emetic/aversive behavior (191) and in mediating GLP-1 effects on food intake (56, 192, 193), these data collectively suggest that GIP potentially acts on the hindbrain-hypothalamus axis to decrease food intake and to improve tolerability of GLP-1R agonism.
3.4 GIPR/GLP-1R Dual-Agonists
The observation that weight loss in DIO mice is enhanced by co-therapy with GIPR and GLP-1R agonists (126) has resulted in the development of unimolecular GLP-1R/GIPR dual-agonist peptides (82, 126). The first of such dual-agonist, MAR709, shows nearly balanced activity at both target receptors and improves body weight and glycemia with greater potency relative to pharmacokinetically-matched GLP-1 in rodents with diet- and genetically induced obesity (126). In a 12-wk phase II study, MAR709 (a.k.a. NNC0090-2746) reduced body weight and blood glucose in patients with T2D, but the drug candidate at the single tested dose was not superior to dose titrated treatment with liraglutide (83). In 2020, Novo Nordisk announced discontinuation of MAR709 due to success of semaglutide 2.4 mg in clinical trials and in favor of other potentially more effective drugs in their clinical pipeline (13).
Another GIPR/GLP-1R co-agonist, Tirzepatide (a.k.a. LY3298176) has been developed by Eli Lilly (82). The molecule is based on the human GIP sequence, in which GLP-1R residues were introduced to yield a fivefold greater potency at the human GIPR relative to GLP-1R (82). Fatty-acylation of the lysine 20 residue with a C20 diacid allows covalent binding to albumin, which results in a half-life of ~160 hrs in humans (82). When given at equimolar concentrations, tirzepatide shows greater weight loss but equal improvement in glucose metabolism relative to treatment with semaglutide in DIO mice (82). Interestingly, when given at a daily dose of 10 nmol/kg, tirzepatide improves glucose metabolism but fails to affect body weight in GLP-1R ko mice (194). These data are seemingly in contrast to previous reports showing preserved ability of long-acting GIPR agonists to decrease body weight in GLP-1R ko mice (179, 184). Importantly, human GIP is only a weak and partial agonist at the mouse relative to the human GIP receptor (195), and all currently available data on the pharmacokinetics of tirzepatide are based on human GIPR (82). It warrants clarification whether tirzepatide shows preserved potency at the mouse GIPR.
In recent phase III clinical trials, tirzepatide showed profound ability to improve body weight and glycemia in patients with obesity and T2D. Depending on the dose (5, 10, or 15 mg QW), 40-52 weeks of treatment with tirzepatide, decreased HbA1c between -1.87 and – 2.59%, with 81 – 97% of patients achieving a HbA1c <7%, and 23 – 62% of patients achieving HbA1c <5.7% (24–27, 196). Serum levels of fasting glucose decreased between 43.6 – 67.9 mg/dl while body weight decreased relative baseline between 6.2 – 12.9% (24–27, 196). After 40 weeks of treatment, 47 – 57% of patients treated with tirzepatide 15mg QW decreased body weight >10%, relative to 1% in patients treated with placebo, while 27 – 32% of patients decreased body weight >15% (0% in placebo controls) (25, 27, 196). Treatment with tirzepatide was at all tested doses (5, 10, 15 mg QW) superior to treatment with semaglutide 1mg QW to decrease HbA1c, fasting levels of blood glucose and body weight (25). It warrants to be determined how tirzepatide compares to the recently approved semaglutide 2.4 mg. Interestingly, while tirzepatide shows comparable ability to decrease body weight in obese type-2 diabetic vs non-diabetic individuals (24–27), Semaglutide 2.4 mg is considerably less efficacious in obese diabetic relative to obese non-diabetic individuals (28, 146–148).
4 Outlook
The results of completed tirzepatide trials are quite exciting. However, some effort is still required to ensure such therapeutic advance will be applicable to all populations for whom it is intended. Racial and ethnic minorities carry a disproportionate burden of obesity and T2D in the general population, but their enrolment in the completed tirzepatide trials was lower than expected, which raises concerns regarding the generalizability of these trials. Female participants were reasonably represented in the completed trials, but no assessment of biological sex differences in drug efficacy and safety profiles was made (24–27, 196). T2D is increasingly diagnosed in children, adolescents, and young adults; there is a need for more efficacious, reliable therapies approved for younger patients. The heterogeneous and multifactorial etiology of T2D and obesity combined with the physiologic complexity of human metabolic system contributes to wide variations in clinical phenotypes and individual response to treatment. Adequate demographic representation across races and ethnic groups, geographic areas, sexes and genders, and age groups may lead to more robust and complete data that broaden the understanding of inter-individual response variability, which may in turn help expand the population of patients responsive to new medications such as tirzepatide.
It is well recognized that T2D and obesity treatments may be best suited for precision medicine approaches rather than a “one size fits all” paradigm. While considerable effort continues to be invested into developing more pharmacotherapies for T2D and diabetes, it is of paramount importance that use of the currently available medications is optimized—that is, to provide the right treatment to the right patient, at the right time. The completed trials of tirzepatide demonstrate meaningful improvement of glycemic and weight control with this drug. However, as with any medical therapy, there are large inter-individual differences in response among participants, including the possibility of less than 5% weight loss to 20% or greater weight loss (24–27, 196), suggesting that individualization and precision treatment might be necessary to achieve greater disease modification. Thus, the challenge for scientists and physicians is to identify and validate biomarkers for drug selection. In future trials, accurate and timely detection of individual response to investigational drugs and reporting individual response data will be helpful in differentiating treatment responders from non-responders. The overall goal is to utilize specific genetic, clinical, and biochemical characteristics of individual patients to tailor treatment to achieve optimal outcomes. Moreover, from a clinical perspective, it will be equally important to decipher mechanisms by which GIP acts on the brain to decrease body weight and to determine which brain areas are involved. Much further work is also needed to establish how GIPR antagonism decreases body weight (centrally or peripherally mediated) and how GIPR and GLP-1R agonism synergizes to decrease body weight.
Author Contributions
QT and SA co-wrote sections and edited the manuscript. CO made the figures and edited the manuscript. RW edited the manuscript. RD edited and revised the manuscript and co-wrote sections. TM conceptualized and wrote the manuscript. AH co-conceptualized the manuscript, co-wrote sections, and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
QT received research funding from the International Helmholtz Research School for Diabetes and the Alberta Diabetes Institute. SA received research funding from the Helmholtz Association (Helmholtz Diabetes School). TM received research funding from the German Research Foundation (TRR152 and TRR296). AH received research funding from the Canadian Institutes of Health Research and Weston Family Foundation (Pro00069477 and Pro00100067).
Conflict of Interest
AH is an investigator on clinical trials for Rhythm Pharmaceuticals, Inc., and Levo Therapeutics, has received grant funding from the W. Garfield Weston Foundation, and has served as a speaker for Rhythm Pharmaceuticals, Inc. TM receives research funding by Novo Nordisk, but these funds are unrelated the here described work. TM further received speaking fees from Eli Lilly, Novo Nordisk, Mercodia, AstraZeneca, Berlin Chemie, and Sanofi Aventis. RD is a co-inventor on intellectual property owned by Indiana University and licensed to Novo Nordisk. He was recently employed by Novo Nordisk and previously Lilly Research Laboratories.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a shared affiliation with several of the authors SA and TM.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Afshin A, Forouzanfar MH, Reitsma MB, Sur P, Estep K, Lee A, et al. Health Effects of Overweight and Obesity in 195 Countries Over 25 Years. N Engl J Med (2017) 377(1):13–27. doi: 10.1056/NEJMoa1614362
2. Reilly JJ, Kelly J. Long-Term Impact of Overweight and Obesity in Childhood and Adolescence on Morbidity and Premature Mortality in Adulthood: Systematic Review. Int J Obes (Lond) (2011) 35(7):891–8. doi: 10.1038/ijo.2010.222
3. Bray GA, Kim KK, Wilding JPH, Federation WO. Obesity: A Chronic Relapsing Progressive Disease Process. A Position Statement of the World Obesity Federation. Obes Rev (2017) 18(7):715–23. doi: 10.1111/obr.12551
4. Tremmel M, Gerdtham UG, Nilsson PM, Saha S. Economic Burden of Obesity: A Systematic Literature Review. Int J Environ Res Public Health (2017) 14(4):435. doi: 10.3390/ijerph14040435
5. Chen L, Magliano DJ, Zimmet PZ. The Worldwide Epidemiology of Type 2 Diabetes Mellitus–Present and Future Perspectives. Nat Rev Endocrinol (2011) 8(4):228–36. doi: 10.1038/nrendo.2011.183
6. Candler TP, Mahmoud O, Lynn RM, Majbar AA, Barrett TG, Shield JPH. Continuing Rise of Type 2 Diabetes Incidence in Children and Young People in the UK. Diabetes Med (2018) 35(6):737–44. doi: 10.1111/dme.13609
7. Lascar N, Brown J, Pattison H, Barnett AH, Bailey CJ, Bellary S. Type 2 Diabetes in Adolescents and Young Adults. Lancet Diabetes Endocrinol (2018) 6(1):69–80. doi: 10.1016/S2213-8587(17)30186-9
8. Magliano DJ, Sacre JW, Harding JL, Gregg EW, Zimmet PZ, Shaw JE. Young-Onset Type 2 Diabetes Mellitus - Implications for Morbidity and Mortality. Nat Rev Endocrinol (2020) 16(6):321–31. doi: 10.1038/s41574-020-0334-z
9. Goossens GH, Blaak EE. Adipose Tissue Dysfunction and Impaired Metabolic Health in Human Obesity: A Matter of Oxygen? Front Endocrinol (Lausanne) (2015) 6:55. doi: 10.3389/fendo.2015.00055
10. Rueda-Clausen CF, Ogunleye AA, Sharma AM. Health Benefits of Long-Term Weight-Loss Maintenance. Annu Rev Nutr (2015) 35:475–516. doi: 10.1146/annurev-nutr-071714-034434
11. Wing RR, Lang W, Wadden TA, Safford M, Knowler WC, Bertoni AG, et al. Benefits of Modest Weight Loss in Improving Cardiovascular Risk Factors in Overweight and Obese Individuals With Type 2 Diabetes. Diabetes Care (2011) 34(7):1481–6. doi: 10.2337/dc10-2415
12. Douketis JD, Macie C, Thabane L, Williamson DF. Systematic Review of Long-Term Weight Loss Studies in Obese Adults: Clinical Significance and Applicability to Clinical Practice. Int J Obes (Lond) (2005) 29(10):1153–67. doi: 10.1038/sj.ijo.0802982
13. Muller TD, Bluher M, Tschop MH, DiMarchi RD. Anti-Obesity Drug Discovery: Advances and Challenges. Nat Rev Drug Discovery (2021) 1–23. doi: 10.1038/s41573-021-00337-8
14. Heiman ML, Greenway FL. A Healthy Gastrointestinal Microbiome is Dependent on Dietary Diversity. Mol Metab (2016) 5(5):317–20. doi: 10.1016/j.molmet.2016.02.005
15. Qasim A, Turcotte M, de Souza RJ, Samaan MC, Champredon D, Dushoff J, et al. On the Origin of Obesity: Identifying the Biological, Environmental and Cultural Drivers of Genetic Risk Among Human Populations. Obes Rev (2018) 19(2):121–49. doi: 10.1111/obr.12625
16. Pigeyre M, Yazdi FT, Kaur Y, Meyre D. Recent Progress in Genetics, Epigenetics and Metagenomics Unveils the Pathophysiology of Human Obesity. Clin Sci (Lond) (2016) 130(12):943–86. doi: 10.1042/CS20160136
17. Kaur Y, de Souza RJ, Gibson WT, Meyre D. A Systematic Review of Genetic Syndromes With Obesity. Obes Rev (2017) 18(6):603–34. doi: 10.1111/obr.12531
18. Babiker A, Al Dubayee M. Anti-Diabetic Medications: How to Make a Choice? Sudan J Paediatr (2017) 17(2):11–20. doi: 10.24911/SJP.2017.2.12
19. Onakpoya IJ, Heneghan CJ, Aronson JK. Post-Marketing Withdrawal of Anti-Obesity Medicinal Products Because of Adverse Drug Reactions: A Systematic Review. BMC Med (2016) 14(1):191. doi: 10.1186/s12916-016-0735-y
20. Muller TD, Clemmensen C, Finan B, DiMarchi RD, Tschop MH. Anti-Obesity Therapy: From Rainbow Pills to Polyagonists. Pharmacol Rev (2018) 70(4):712–46. doi: 10.1124/pr.117.014803
21. Comerma-Steffensen S, Grann M, Andersen CU, Rungby J, Simonsen U. Cardiovascular Effects of Current and Future Anti-Obesity Drugs. Curr Vasc Pharmacol (2014) 12(3):493–504. doi: 10.2174/1570161112666140423223529
22. Singh G, Krauthamer M, Bjalme-Evans M. Wegovy (Semaglutide): A New Weight Loss Drug for Chronic Weight Management. J Investig Med (2021) 70:5–13. doi: 10.1136/jim-2021-001952
23. Min T, Bain SC. The Role of Tirzepatide, Dual GIP and GLP-1 Receptor Agonist, in the Management of Type 2 Diabetes: The SURPASS Clinical Trials. Diabetes Ther (2021) 12(1):143–57. doi: 10.1007/s13300-020-00981-0
24. Del Prato S, Kahn SE, Pavo I, Weerakkody GJ, Yang Z, Doupis J, et al. Tirzepatide Versus Insulin Glargine in Type 2 Diabetes and Increased Cardiovascular Risk (SURPASS-4): A Randomised, Open-Label, Parallel-Group, Multicentre, Phase 3 Trial. Lancet (2021) 398:1811–24. doi: 10.1016/S0140-6736(21)02188-7.
25. Frias JP, Davies MJ, Rosenstock J, Perez Manghi FC, Fernandez Lando L, Bergman BK, et al. Tirzepatide Versus Semaglutide Once Weekly in Patients With Type 2 Diabetes. N Engl J Med (2021) 385(6):503–15. doi: 10.1056/NEJMoa2107519
26. Ludvik B, Giorgino F, Jodar E, Frias JP, Fernandez Lando L, Brown K, et al. Once-Weekly Tirzepatide Versus Once-Daily Insulin Degludec as Add-on to Metformin With or Without SGLT2 Inhibitors in Patients With Type 2 Diabetes (SURPASS-3): A Randomised, Open-Label, Parallel-Group, Phase 3 Trial. Lancet (2021) 398(10300):583–98. doi: 10.1016/S0140-6736(21)01443-4
27. Rosenstock J, Wysham C, Frias JP, Kaneko S, Lee CJ, Fernandez Lando L, et al. Efficacy and Safety of a Novel Dual GIP and GLP-1 Receptor Agonist Tirzepatide in Patients With Type 2 Diabetes (SURPASS-1): A Double-Blind, Randomised, Phase 3 Trial. Lancet (2021) 398(10295):143–55. doi: 10.1016/S0140-6736(21)01324-6
28. Wilding JPH, Batterham RL, Calanna S, Davies M, Van Gaal LF, Lingvay I, et al. Once-Weekly Semaglutide in Adults With Overweight or Obesity. N Engl J Med (2021) 384(11):989. doi: 10.1056/NEJMoa2032183
29. Müller TD, Finan B, Bloom SR, D’Alessio D, Drucker DJ, Flatt PR, et al. Glucagon-Like Peptide 1 (GLP-1). Mol Metab (2019) 30:72–130. doi: 10.1016/j.molmet.2019.09.010
30. Muller TD, Finan B, Clemmensen C, DiMarchi RD, Tschop MH. The New Biology and Pharmacology of Glucagon. Physiol Rev (2017) 97(2):721–66. doi: 10.1152/physrev.00025.2016
31. Zeigerer A, Sekar R, Kleinert M, Nason S, Habegger KM, Muller TD. Glucagon’s Metabolic Action in Health and Disease. Compr Physiol (2021) 11(2):1759–83. doi: 10.1002/cphy.c200013
32. Herrmann C, Goke R, Richter G, Fehmann HC, Arnold R, Goke B. Glucagon-Like Peptide-1 and Glucose-Dependent Insulin-Releasing Polypeptide Plasma Levels in Response to Nutrients. Digestion (1995) 56(2):117–26. doi: 10.1159/000201231
33. Kuhre RE, Gribble FM, Hartmann B, Reimann F, Windelov JA, Rehfeld JF, et al. Fructose Stimulates GLP-1 But Not GIP Secretion in Mice, Rats, and Humans. Am J Physiol Gastrointest Liver Physiol (2014) 306(7):G622–30. doi: 10.1152/ajpgi.00372.2013
34. Steinert RE, Frey F, Topfer A, Drewe J, Beglinger C. Effects of Carbohydrate Sugars and Artificial Sweeteners on Appetite and the Secretion of Gastrointestinal Satiety Peptides. Br J Nutr (2011) 105(9):1320–8. doi: 10.1017/S000711451000512X
35. Hirasawa A, Tsumaya K, Awaji T, Katsuma S, Adachi T, Yamada M, et al. Free Fatty Acids Regulate Gut Incretin Glucagon-Like Peptide-1 Secretion Through GPR120. Nat Med (2005) 11(1):90–4. doi: 10.1038/nm1168
36. Thomsen C, Rasmussen O, Lousen T, Holst JJ, Fenselau S, Schrezenmeir J, et al. Differential Effects of Saturated and Monounsaturated Fatty Acids on Postprandial Lipemia and Incretin Responses in Healthy Subjects. Am J Clin Nutr (1999) 69(6):1135–43. doi: 10.1093/ajcn/69.6.1135
37. Calbet JA, Holst JJ. Gastric Emptying, Gastric Secretion and Enterogastrone Response After Administration of Milk Proteins or Their Peptide Hydrolysates in Humans. Eur J Nutr (2004) 43(3):127–39. doi: 10.1007/s00394-004-0448-4
38. Gameiro A, Reimann F, Habib AM, O’Malley D, Williams L, Simpson AK, et al. The Neurotransmitters Glycine and GABA Stimulate Glucagon-Like Peptide-1 Release From the GLUTag Cell Line. J Physiol (2005) 569(Pt 3):761–72. doi: 10.1113/jphysiol.2005.098962
39. Reimann F, Williams L, da Silva Xavier G, Rutter GA, Gribble FM. Glutamine Potently Stimulates Glucagon-Like Peptide-1 Secretion From GLUTag Cells. Diabetologia (2004) 47(9):1592–601. doi: 10.1007/s00125-004-1498-0
40. Orskov C, Wettergren A, Holst JJ. Secretion of the Incretin Hormones Glucagon-Like Peptide-1 and Gastric Inhibitory Polypeptide Correlates With Insulin Secretion in Normal Man Throughout the Day. Scand J Gastroenterol (1996) 31(7):665–70. doi: 10.3109/00365529609009147
41. Mayo KE, Miller LJ, Bataille D, Dalle S, Goke B, Thorens B, et al. International Union of Pharmacology. XXXV. The Glucagon Receptor Family. Pharmacol Rev (2003) 55(1):167–94. doi: 10.1124/pr.55.1.6
42. Novikoff A, O’Brien SL, Bernecker M, Grandl G, Kleinert M, Knerr PJ, et al. Spatiotemporal GLP-1 and GIP Receptor Signaling and Trafficking/Recycling Dynamics Induced by Selected Receptor Mono- and Dual-Agonists. Mol Metab (2021) 49:101181. doi: 10.1016/j.molmet.2021.101181
43. Sonoda N, Imamura T, Yoshizaki T, Babendure JL, Lu JC, Olefsky JM. Beta-Arrestin-1 Mediates Glucagon-Like Peptide-1 Signaling to Insulin Secretion in Cultured Pancreatic Beta Cells. Proc Natl Acad Sci USA (2008) 105(18):6614–9. doi: 10.1073/pnas.0710402105
44. Pyke C, Heller RS, Kirk RK, Orskov C, Reedtz-Runge S, Kaastrup P, et al. GLP-1 Receptor Localization in Monkey and Human Tissue: Novel Distribution Revealed With Extensively Validated Monoclonal Antibody. Endocrinology (2014) 155(4):1280–90. doi: 10.1210/en.2013-1934
45. Richards P, Parker HE, Adriaenssens AE, Hodgson JM, Cork SC, Trapp S, et al. Identification and Characterization of GLP-1 Receptor-Expressing Cells Using a New Transgenic Mouse Model. Diabetes (2014) 63(4):1224–33. doi: 10.2337/db13-1440
46. Jensen CB, Pyke C, Rasch MG, Dahl AB, Knudsen LB, Secher A. Characterization of the Glucagonlike Peptide-1 Receptor in Male Mouse Brain Using a Novel Antibody and In Situ Hybridization. Endocrinology (2018) 159(2):665–75. doi: 10.1210/en.2017-00812
47. Willms B, Werner J, Holst JJ, Orskov C, Creutzfeldt W, Nauck MA. Gastric Emptying, Glucose Responses, and Insulin Secretion After a Liquid Test Meal: Effects of Exogenous Glucagon-Like Peptide-1 (GLP-1)-(7-36) Amide in Type 2 (Noninsulin-Dependent) Diabetic Patients. J Clin Endocrinol Metab (1996) 81(1):327–32. doi: 10.1210/jcem.81.1.8550773
48. Marso SP, Daniels GH, Brown-Frandsen K, Kristensen P, Mann JF, Nauck MA, et al. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N Engl J Med (2016) 375(4):311–22. doi: 10.1056/NEJMoa1603827
49. Kapodistria K, Tsilibary EP, Kotsopoulou E, Moustardas P, Kitsiou P. Liraglutide, a Human Glucagon-Like Peptide-1 Analogue, Stimulates AKT-Dependent Survival Signalling and Inhibits Pancreatic Beta-Cell Apoptosis. J Cell Mol Med (2018) 22(6):2970–80. doi: 10.1111/jcmm.13259
50. Li Y, Hansotia T, Yusta B, Ris F, Halban PA, Drucker DJ. Glucagon-Like Peptide-1 Receptor Signaling Modulates Beta Cell Apoptosis. J Biol Chem (2003) 278(1):471–8. doi: 10.1074/jbc.M209423200
51. Sun L, Dai Y, Wang C, Chu Y, Su X, Yang J, et al. Novel Pentapeptide GLP-1 (32-36) Amide Inhibits Beta-Cell Apoptosis In Vitro and Improves Glucose Disposal in Streptozotocin-Induced Diabetic Mice. Chem Biol Drug Des (2015) 86(6):1482–90. doi: 10.1111/cbdd.12615
52. Khalilnezhad A, Taskiran D. Protective Effects of Glucagon-Like Peptide-1 (GLP-1) Analogue Exenatide Against Glucose and Fructose-Induced Neurotoxicity. Int J Neurosci (2018) 129:1–24. doi: 10.1080/00207454.2018.1543671
53. Liao P, Yang D, Liu D, Zheng Y. GLP-1 and Ghrelin Attenuate High Glucose/High Lipid-Induced Apoptosis and Senescence of Human Microvascular Endothelial Cells. Cell Physiol Biochem (2017) 44(5):1842–55. doi: 10.1159/000485820
54. Xu G, Stoffers DA, Habener JF, Bonner-Weir S. Exendin-4 Stimulates Both Beta-Cell Replication and Neogenesis, Resulting in Increased Beta-Cell Mass and Improved Glucose Tolerance in Diabetic Rats. Diabetes (1999) 48(12):2270–6. doi: 10.2337/diabetes.48.12.2270
55. During MJ, Cao L, Zuzga DS, Francis JS, Fitzsimons HL, Jiao X, et al. Glucagon-Like Peptide-1 Receptor is Involved in Learning and Neuroprotection. Nat Med (2003) 9(9):1173–9. doi: 10.1038/nm919
56. Alhadeff AL, Grill HJ. Hindbrain Nucleus Tractus Solitarius Glucagon-Like Peptide-1 Receptor Signaling Reduces Appetitive and Motivational Aspects of Feeding. Am J Physiol Regul Integr Comp Physiol (2014) 307(4):R465–70. doi: 10.1152/ajpregu.00179.2014
57. Orskov C, Rabenhoj L, Wettergren A, Kofod H, Holst JJ. Tissue and Plasma Concentrations of Amidated and Glycine-Extended Glucagon-Like Peptide I in Humans. Diabetes (1994) 43(4):535–9. doi: 10.2337/diab.43.4.535
58. Hui H, Farilla L, Merkel P, Perfetti R. The Short Half-Life of Glucagon-Like Peptide-1 in Plasma Does Not Reflect its Long-Lasting Beneficial Effects. Eur J Endocrinol (2002) 146(6):863–9. doi: 10.1530/eje.0.1460863
59. Deacon CF, Pridal L, Klarskov L, Olesen M, Holst JJ. Glucagon-Like Peptide 1 Undergoes Differential Tissue-Specific Metabolism in the Anesthetized Pig. Am J Physiol (1996) 271(3 Pt 1):E458–64. doi: 10.1152/ajpendo.1996.271.3.E458
60. Meier JJ, Nauck MA, Kranz D, Holst JJ, Deacon CF, Gaeckler D, et al. Secretion, Degradation, and Elimination of Glucagon-Like Peptide 1 and Gastric Inhibitory Polypeptide in Patients With Chronic Renal Insufficiency and Healthy Control Subjects. Diabetes (2004) 53(3):654–62. doi: 10.2337/diabetes.53.3.654
61. Deacon CF. Circulation and Degradation of GIP and GLP-1. Horm Metab Res (2004) 36(11-12):761–5. doi: 10.1055/s-2004-826160
62. Hupe-Sodmann K, McGregor GP, Bridenbaugh R, Goke R, Goke B, Thole H, et al. Characterisation of the Processing by Human Neutral Endopeptidase 24.11 of GLP-1(7-36) Amide and Comparison of the Substrate Specificity of the Enzyme for Other Glucagon-Like Peptides. Regul Pept (1995) 58(3):149–56. doi: 10.1016/0167-0115(95)00063-h
63. Plamboeck A, Holst JJ, Carr RD, Deacon CF. Neutral Endopeptidase 24.11 and Dipeptidyl Peptidase IV Are Both Mediators of the Degradation of Glucagon-Like Peptide 1 in the Anaesthetised Pig. Diabetologia (2005) 48(9):1882–90. doi: 10.1007/0-306-47920-6_36
64. Windelov JA, Wewer Albrechtsen NJ, Kuhre RE, Jepsen SL, Hornburg D, Pedersen J, et al. Why is it So Difficult to Measure Glucagon-Like Peptide-1 in a Mouse? Diabetologia (2017) 60(10):2066–75. doi: 10.1007/s00125-017-4347-7
65. Wewer Albrechtsen NJ, Mark PD, Terzic D, Hansen LH, Andersen UO, Hartmann B, et al. Sacubitril/valsartan Augments Postprandial Plasma Concentrations of Active GLP-1 When Combined With Sitagliptin in Men. J Clin Endocrinol Metab (2019) 104:3868–76. doi: 10.1210/jc.2019-00515
66. Holst JJ. The Physiology of Glucagon-Like Peptide 1. Physiol Rev (2007) 87(4):1409–39. doi: 10.1152/physrev.00034.2006
67. Hansen L, Deacon CF, Orskov C, Holst JJ. Glucagon-Like Peptide-1-(7-36)Amide is Transformed to Glucagon-Like Peptide-1-(9-36)Amide by Dipeptidyl Peptidase IV in the Capillaries Supplying the L Cells of the Porcine Intestine. Endocrinology (1999) 140(11):5356–63. doi: 10.1210/endo.140.11.7143
68. Hansen L, Hartmann B, Bisgaard T, Mineo H, Jorgensen PN, Holst JJ. Somatostatin Restrains the Secretion of Glucagon-Like Peptide-1 and -2 From Isolated Perfused Porcine Ileum. Am J Physiol Endocrinol Metab (2000) 278(6):E1010–8. doi: 10.1152/ajpendo.2000.278.6.E1010
69. Zander M, Madsbad S, Madsen JL, Holst JJ. Effect of 6-Week Course of Glucagon-Like Peptide 1 on Glycaemic Control, Insulin Sensitivity, and Beta-Cell Function in Type 2 Diabetes: A Parallel-Group Study. Lancet (2002) 359(9309):824–30. doi: 10.1016/S0140-6736(02)07952-7
70. Ahren B, Simonsson E, Larsson H, Landin-Olsson M, Torgeirsson H, Jansson PA, et al. Inhibition of Dipeptidyl Peptidase IV Improves Metabolic Control Over a 4-Week Study Period in Type 2 Diabetes. Diabetes Care (2002) 25(5):869–75. doi: 10.2337/diacare.25.5.869
71. Bergman AJ, Stevens C, Zhou Y, Yi B, Laethem M, De Smet M, et al. Pharmacokinetic and Pharmacodynamic Properties of Multiple Oral Doses of Sitagliptin, a Dipeptidyl Peptidase-IV Inhibitor: A Double-Blind, Randomized, Placebo-Controlled Study in Healthy Male Volunteers. Clin Ther (2006) 28(1):55–72. doi: 10.1016/j.clinthera.2006.01.015
72. D’Alessio DA, Denney AM, Hermiller LM, Prigeon RL, Martin JM, Tharp WG, et al. Treatment With the Dipeptidyl Peptidase-4 Inhibitor Vildagliptin Improves Fasting Islet-Cell Function in Subjects With Type 2 Diabetes. J Clin Endocrinol Metab (2009) 94(1):81–8. doi: 10.1210/jc.2008-1135
73. He YL, Serra D, Wang Y, Campestrini J, Riviere GJ, Deacon CF, et al. Pharmacokinetics and Pharmacodynamics of Vildagliptin in Patients With Type 2 Diabetes Mellitus. Clin Pharmacokinet (2007) 46(7):577–88. doi: 10.2165/00003088-200746070-00003
74. Meier JJ. GLP-1 Receptor Agonists for Individualized Treatment of Type 2 Diabetes Mellitus. Nat Rev Endocrinol (2012) 8(12):728–42. doi: 10.1038/nrendo.2012.140
75. Hare KJ, Vilsboll T, Asmar M, Deacon CF, Knop FK, Holst JJ. The Glucagonostatic and Insulinotropic Effects of Glucagon-Like Peptide 1 Contribute Equally to its Glucose-Lowering Action. Diabetes (2010) 59(7):1765–70. doi: 10.2337/db09-1414
76. Linnebjerg H, Park S, Kothare PA, Trautmann ME, Mace K, Fineman M, et al. Effect of Exenatide on Gastric Emptying and Relationship to Postprandial Glycemia in Type 2 Diabetes. Regul Pept (2008) 151(1-3):123–9. doi: 10.1016/j.regpep.2008.07.003
77. Little TJ, Pilichiewicz AN, Russo A, Phillips L, Jones KL, Nauck MA, et al. Effects of Intravenous Glucagon-Like Peptide-1 on Gastric Emptying and Intragastric Distribution in Healthy Subjects: Relationships With Postprandial Glycemic and Insulinemic Responses. J Clin Endocrinol Metab (2006) 91(5):1916–23. doi: 10.1210/jc.2005-2220
78. Wettergren A, Schjoldager B, Mortensen PE, Myhre J, Christiansen J, Holst JJ. Truncated GLP-1 (Proglucagon 78-107-Amide) Inhibits Gastric and Pancreatic Functions in Man. Dig Dis Sci (1993) 38(4):665–73. doi: 10.1007/BF01316798
79. Meier JJ, Kemmeries G, Holst JJ, Nauck MA. Erythromycin Antagonizes the Deceleration of Gastric Emptying by Glucagon-Like Peptide 1 and Unmasks its Insulinotropic Effect in Healthy Subjects. Diabetes (2005) 54(7):2212–8. doi: 10.2337/diabetes.54.7.2212
80. Drucker DJ, Buse JB, Taylor K, Kendall DM, Trautmann M, Zhuang D, et al. Exenatide Once Weekly Versus Twice Daily for the Treatment of Type 2 Diabetes: A Randomised, Open-Label, Non-Inferiority Study. Lancet (2008) 372(9645):1240–50. doi: 10.1016/S0140-6736(08)61206-4
81. Pi-Sunyer X, Astrup A, Fujioka K, Greenway F, Halpern A, Krempf M, et al. A Randomized, Controlled Trial of 3.0 Mg of Liraglutide in Weight Management. N Engl J Med (2015) 373(1):11–22. doi: 10.1056/NEJMoa1411892
82. Coskun T, Sloop KW, Loghin C, Alsina-Fernandez J, Urva S, Bokvist KB, et al. LY3298176, a Novel Dual GIP and GLP-1 Receptor Agonist for the Treatment of Type 2 Diabetes Mellitus: From Discovery to Clinical Proof of Concept. Mol Metab (2018) 18:3–14. doi: 10.1016/j.molmet.2018.09.009
83. Frias JP, Bastyr EJ 3rd, Vignati L, Tschop MH, Schmitt C, Owen K, et al. The Sustained Effects of a Dual GIP/GLP-1 Receptor Agonist, NNC0090-2746, in Patients With Type 2 Diabetes. Cell Metab (2017) 26(2):343–52.e2. doi: 10.1016/j.cmet.2017.07.011
84. Frias JP, Nauck MA, Van J, Benson C, Bray R, Cui X, et al. Efficacy and Tolerability of Tirzepatide, a Dual GIP and GLP-1 Receptor Agonist in Patients With Type 2 Diabetes: A 12-Week, Randomised, Double-Blind, Placebo-Controlled Study to Evaluate Different Dose-Escalation Regimens. Diabetes Obes Metab (2020) 22:938–46. doi: 10.1111/dom.13979
85. Frias JP, Nauck MA, Van J, Kutner ME, Cui X, Benson C, et al. Efficacy and Safety of LY3298176, a Novel Dual GIP and GLP-1 Receptor Agonist, in Patients With Type 2 Diabetes: A Randomised, Placebo-Controlled and Active Comparator-Controlled Phase 2 Trial. Lancet (2018) 392(10160):2180–93. doi: 10.1016/S0140-6736(18)32260-8
86. Ambery P, Parker VE, Stumvoll M, Posch MG, Heise T, Plum-Moerschel L, et al. MEDI0382, a GLP-1 and Glucagon Receptor Dual Agonist, in Obese or Overweight Patients With Type 2 Diabetes: A Randomised, Controlled, Double-Blind, Ascending Dose and Phase 2a Study. Lancet (2018) 391(10140):2607–18. doi: 10.1016/S0140-6736(18)30726-8
87. Ambery PD, Klammt S, Posch MG, Petrone M, Pu W, Rondinone C, et al. MEDI0382, a GLP-1/Glucagon Receptor Dual Agonist, Meets Safety and Tolerability Endpoints in a Single-Dose, Healthy-Subject, Randomized, Phase 1 Study. Br J Clin Pharmacol (2018) 84(10):2325–35. doi: 10.1111/bcp.13688
88. Tschop MH, Finan B, Clemmensen C, Gelfanov V, Perez-Tilve D, Muller TD, et al. Unimolecular Polypharmacy for Treatment of Diabetes and Obesity. Cell Metab (2016) 24(1):51–62. doi: 10.1016/j.cmet.2016.06.021
89. Lien S, Lowman HB. Therapeutic Peptides. Trends Biotechnol (2003) 21(12):556–62. doi: 10.1016/j.tibtech.2003.10.005
90. Neidigh JW, Fesinmeyer RM, Prickett KS, Andersen NH. Exendin-4 and Glucagon-Like-Peptide-1: NMR Structural Comparisons in the Solution and Micelle-Associated States. Biochemistry (2001) 40(44):13188–200. doi: 10.1021/bi010902s
91. Parkes D, Jodka C, Smith P, Nayak S, Rinehart L, Gingerich R, et al. Pharmacokinetic Actions of Exendin-4 in the Rat: Comparison With Glucagon-Like Peptide-1. Drug Dev Res (2001) 53:260–7. doi: 10.1002/ddr.1195
92. BYETTA. Full Prescribing Information. (2005). FDA file retrieved from https://www.accessdata.fda.gov/drugsatfda_docs/label/2005/021773lbl.pdf Issued April 2005.
93. Buse JB, Henry RR, Han J, Kim DD, Fineman MS, Baron AD, et al. Effects of Exenatide (Exendin-4) on Glycemic Control Over 30 Weeks in Sulfonylurea-Treated Patients With Type 2 Diabetes. Diabetes Care (2004) 27(11):2628–35. doi: 10.2337/diacare.27.11.2628
94. DeFronzo RA, Ratner RE, Han J, Kim DD, Fineman MS, Baron AD. Effects of Exenatide (Exendin-4) on Glycemic Control and Weight Over 30 Weeks in Metformin-Treated Patients With Type 2 Diabetes. Diabetes Care (2005) 28(5):1092–100. doi: 10.2337/diacare.28.5.1092
95. Kendall DM, Riddle MC, Rosenstock J, Zhuang D, Kim DD, Fineman MS, et al. Effects of Exenatide (Exendin-4) on Glycemic Control Over 30 Weeks in Patients With Type 2 Diabetes Treated With Metformin and a Sulfonylurea. Diabetes Care (2005) 28(5):1083–91. doi: 10.2337/diacare.28.5.1083
96. Fonseca VA, Alvarado-Ruiz R, Raccah D, Boka G, Miossec P, Gerich JE, et al. Efficacy and Safety of the Once-Daily GLP-1 Receptor Agonist Lixisenatide in Monotherapy: A Randomized, Double-Blind, Placebo-Controlled Trial in Patients With Type 2 Diabetes (GetGoal-Mono). Diabetes Care (2012) 35(6):1225–31. doi: 10.2337/dc11-1935
97. Bush MA, Matthews JE, De Boever EH, Dobbins RL, Hodge RJ, Walker SE, et al. Safety, Tolerability, Pharmacodynamics and Pharmacokinetics of Albiglutide, a Long-Acting Glucagon-Like Peptide-1 Mimetic, in Healthy Subjects. Diabetes Obes Metab (2009) 11(5):498–505. doi: 10.1111/j.1463-1326.2008.00992.x
98. Andersen JT, Dalhus B, Cameron J, Daba MB, Plumridge A, Evans L, et al. Structure-Based Mutagenesis Reveals the Albumin-Binding Site of the Neonatal Fc Receptor. Nat Commun (2012) 3:610. doi: 10.1038/ncomms1607
99. Chaudhury C, Mehnaz S, Robinson JM, Hayton WL, Pearl DK, Roopenian DC, et al. The Major Histocompatibility Complex-Related Fc Receptor for IgG (FcRn) Binds Albumin and Prolongs its Lifespan. J Exp Med (2003) 197(3):315–22. doi: 10.1084/jem.20021829
100. Kim J, Bronson CL, Hayton WL, Radmacher MD, Roopenian DC, Robinson JM, et al. Albumin Turnover: FcRn-Mediated Recycling Saves as Much Albumin From Degradation as the Liver Produces. Am J Physiol Gastrointest Liver Physiol (2006) 290(2):G352–60. doi: 10.1152/ajpgi.00286.2005
101. Cavaco M, Castanho M, Neves V. Peptibodies: An Elegant Solution for a Long-Standing Problem. Biopolymers (2017) 110:e23095. doi: 10.1002/bip.23095
102. Knudsen LB, Lau J. The Discovery and Development of Liraglutide and Semaglutide. Front Endocrinol (Lausanne) (2019) 10:155. doi: 10.3389/fendo.2019.00155
103. Agerso H, Jensen LB, Elbrond B, Rolan P, Zdravkovic M. The Pharmacokinetics, Pharmacodynamics, Safety and Tolerability of NN2211, a New Long-Acting GLP-1 Derivative, in Healthy Men. Diabetologia (2002) 45(2):195–202. doi: 10.1007/s00125-001-0719-z
104. Gotfredsen CF, Molck AM, Thorup I, Nyborg NC, Salanti Z, Knudsen LB, et al. The Human GLP-1 Analogs Liraglutide and Semaglutide: Absence of Histopathological Effects on the Pancreas in Nonhuman Primates. Diabetes (2014) 63(7):2486–97. doi: 10.2337/db13-1087
105. Turecek PL, Bossard MJ, Schoetens F, Ivens IA. PEGylation of Biopharmaceuticals: A Review of Chemistry and Nonclinical Safety Information of Approved Drugs. J Pharm Sci (2016) 105(2):460–75. doi: 10.1016/j.xphs.2015.11.015
106. Digilio G, Barbero L, Bracco C, Corpillo D, Esposito P, Piquet G, et al. NMR Structure of Two Novel Polyethylene Glycol Conjugates of the Human Growth Hormone-Releasing Factor, hGRF(1-29)-Nh2. J Am Chem Soc (2003) 125(12):3458–70. doi: 10.1021/ja021264j
107. Hinds KD, Kim SW. Effects of PEG Conjugation on Insulin Properties. Adv Drug Deliv Rev (2002) 54(4):505–30. doi: 10.1016/S0169-409X(02)00025-X
108. Meng F, Manjula BN, Smith PK, Acharya SA. PEGylation of Human Serum Albumin: Reaction of PEG-Phenyl-Isothiocyanate With Protein. Bioconjug Chem (2008) 19(7):1352–60. doi: 10.1021/bc7003878
109. Pasut G, Veronese FM. State of the Art in PEGylation: The Great Versatility Achieved After Forty Years of Research. J Control Release (2012) 161(2):461–72. doi: 10.1016/j.jconrel.2011.10.037
110. Veronese FM, Sartore L, Schiavon O, Caliceti P. A Comparative Study of Enzymatic, Structural, and Pharmacokinetic Properties of Superoxide Dismutase Isolated From Two Sources and Modified by Monomethoxypolyethylene Glycol Using Different Methods of Coupling. Ann N Y Acad Sci (1990) 613:468–74. doi: 10.1111/j.1749-6632.1990.tb18202.x
111. Muller TD, Sullivan LM, Habegger K, Yi CX, Kabra D, Grant E, et al. Restoration of Leptin Responsiveness in Diet-Induced Obese Mice Using an Optimized Leptin Analog in Combination With Exendin-4 or FGF21. J Pept Sci (2012) 18(6):383–93. doi: 10.1002/psc.2408
112. Camacho RC, Zafian PT, Achanfuo-Yeboah J, Manibusan A, Berger JP. Pegylated Fgf21 Rapidly Normalizes Insulin-Stimulated Glucose Utilization in Diet-Induced Insulin Resistant Mice. Eur J Pharmacol (2013) 715(1-3):41–5. doi: 10.1016/j.ejphar.2013.06.023
113. Huang Z, Wang H, Lu M, Sun C, Wu X, Tan Y, et al. A Better Anti-Diabetic Recombinant Human Fibroblast Growth Factor 21 (Rhfgf21) Modified With Polyethylene Glycol. PloS One (2011) 6(6):e20669. doi: 10.1371/journal.pone.0020669
114. Mu J, Pinkstaff J, Li Z, Skidmore L, Li N, Myler H, et al. FGF21 Analogs of Sustained Action Enabled by Orthogonal Biosynthesis Demonstrate Enhanced Antidiabetic Pharmacology in Rodents. Diabetes (2012) 61(2):505–12. doi: 10.2337/db11-0838
115. Song L, Zhu Y, Wang H, Belov AA, Niu J, Shi L, et al. A Solid-Phase PEGylation Strategy for Protein Therapeutics Using a Potent FGF21 Analog. Biomaterials (2014) 35(19):5206–15. doi: 10.1016/j.biomaterials.2014.03.023
116. Xu P, Ye X, Zhang Y, Yuan Q, Liu M, Wu Q, et al. Long-Acting Hypoglycemic Effects of PEGylated FGF21 and Insulin Glargine in Mice With Type 1 Diabetes. J Diabetes Complications (2015) 29(1):5–12. doi: 10.1016/j.jdiacomp.2014.10.001
117. Zhao L, Wang H, Xie J, Chen Z, Li X, Niu J. Potent Long-Acting Rhfgf21 Analog for Treatment of Diabetic Nephropathy in Db/Db and DIO Mice. BMC Biotechnol (2017) 17(1):58. doi: 10.1186/s12896-017-0368-z
118. Salhanick AI, Clairmont KB, Buckholz TM, Pellegrino CM, Ha S, Lumb KJ. Contribution of Site-Specific PEGylation to the Dipeptidyl Peptidase IV Stability of Glucose-Dependent Insulinotropic Polypeptide. Bioorg Med Chem Lett (2005) 15(18):4114–7. doi: 10.1016/j.bmcl.2005.06.002
119. Yanagimachi T, Fujita Y, Takeda Y, Honjo J, Atageldiyeva KK, Takiyama Y, et al. Pancreatic Glucose-Dependent Insulinotropic Polypeptide (GIP) (1-30) Expression is Upregulated in Diabetes and PEGylated GIP(1-30) can Suppress the Progression of Low-Dose-STZ-Induced Hyperglycaemia in Mice. Diabetologia (2016) 59(3):533–41. doi: 10.1007/s00125-015-3842-y
120. Han J, Fei Y, Zhou F, Chen X, Zhang Y, Liu L, et al. Xenopus-Derived Glucagon-Like Peptide-1 and Polyethylene-Glycosylated Glucagon-Like Peptide-1 Receptor Agonists: Long-Acting Hypoglycaemic and Insulinotropic Activities With Potential Therapeutic Utilities. Br J Pharmacol (2018) 175(3):544–57. doi: 10.1111/bph.14107
121. Tang D, Tian H, Wu J, Cheng J, Luo C, Sai W, et al. C-Terminal Site-Specific PEGylated Exendin-4 Analog: A Long-Acting Glucagon Like Peptide-1 Receptor Agonist, on Glycemic Control and Beta Cell Function in Diabetic Db/Db Mice. J Pharmacol Sci (2018) 138(1):23–30. doi: 10.1016/j.jphs.2018.08.009
122. Thi Nguyen NT, Jung S, Lee SH, Bae ON, Lee EK. Mono-PEGylates of Exenatide in Branched and Dimeric Structures can Improve In Vivo Stability and Hypoglycemic Bioactivity. J Biotechnol (2019) 306:89–96. doi: 10.1016/j.jbiotec.2019.09.016
123. DeCarr LB, Buckholz TM, Milardo LF, Mays MR, Ortiz A, Lumb KJ. A Long-Acting Selective Neuropeptide Y2 Receptor PEGylated Peptide Agonist Reduces Food Intake in Mice. Bioorg Med Chem Lett (2007) 17(7):1916–9. doi: 10.1016/j.bmcl.2007.01.045
124. Clemmensen C, Chabenne J, Finan B, Sullivan L, Fischer K, Kuchler D, et al. GLP-1/Glucagon Coagonism Restores Leptin Responsiveness in Obese Mice Chronically Maintained on an Obesogenic Diet. Diabetes (2014) 63(4):1422–7. doi: 10.2337/db13-1609
125. Day JW, Ottaway N, Patterson JT, Gelfanov V, Smiley D, Gidda J, et al. A New Glucagon and GLP-1 Co-Agonist Eliminates Obesity in Rodents. Nat Chem Biol (2009) 5(10):749–57. doi: 10.1038/nchembio.209
126. Finan B, Ma T, Ottaway N, Müller TD, Habegger KM, Heppner KM, et al. Unimolecular Dual Incretins Maximize Metabolic Benefits in Rodents, Monkeys, and Humans. Sci Transl Med (2013) 5(209):209ra151. doi: 10.1126/scitranslmed.3007218
127. Kim DM, Chu SH, Kim S, Park YW, Kim SS. Fc Fusion to Glucagon-Like Peptide-1 Inhibits Degradation by Human DPP-IV, Increasing its Half-Life in Serum and Inducing a Potent Activity for Human GLP-1 Receptor Activation. BMB Rep (2009) 42(4):212–6. doi: 10.5483/BMBRep.2009.42.4.212
128. Glaesner W, Vick AM, Millican R, Ellis B, Tschang SH, Tian Y, et al. Engineering and Characterization of the Long-Acting Glucagon-Like Peptide-1 Analogue LY2189265, an Fc Fusion Protein. Diabetes Metab Res Rev (2010) 26(4):287–96. doi: 10.1002/dmrr.1080
129. Ishii-Watabe A, Suzuki T, Tada M, Kawanishi T, Yamaguchi T, Kawasaki N. [FcRn, a Critical Regulator of Antibody Pharmacokinetics]. Nihon Yakurigaku Zasshi (2010) 136(5):280–4. doi: 10.1254/fpj.136.280
130. Suzuki T, Ishii-Watabe A, Tada M, Kobayashi T, Kanayasu-Toyoda T, Kawanishi T, et al. Importance of Neonatal FcR in Regulating the Serum Half-Life of Therapeutic Proteins Containing the Fc Domain of Human IgG1: A Comparative Study of the Affinity of Monoclonal Antibodies and Fc-Fusion Proteins to Human Neonatal FcR. J Immunol (2010) 184(4):1968–76. doi: 10.4049/jimmunol.0903296
131. Tzaban S, Massol RH, Yen E, Hamman W, Frank SR, Lapierre LA, et al. The Recycling and Transcytotic Pathways for IgG Transport by FcRn Are Distinct and Display an Inherent Polarity. J Cell Biol (2009) 185(4):673–84. doi: 10.1083/jcb.200809122
132. DeYoung MB, MacConell L, Sarin V, Trautmann M, Herbert P. Encapsulation of Exenatide in Poly-(D,L-Lactide-Co-Glycolide) Microspheres Produced an Investigational Long-Acting Once-Weekly Formulation for Type 2 Diabetes. Diabetes Technol Ther (2011) 13(11):1145–54. doi: 10.1089/dia.2011.0050
133. Bergenstal RM, Wysham C, Macconell L, Malloy J, Walsh B, Yan P, et al. Efficacy and Safety of Exenatide Once Weekly Versus Sitagliptin or Pioglitazone as an Adjunct to Metformin for Treatment of Type 2 Diabetes (DURATION-2): A Randomised Trial. Lancet (2010) 376(9739):431–9. doi: 10.1016/S0140-6736(10)60590-9
134. Cooke CE, Lee HY, Tong YP, Haines ST. Persistence With Injectable Antidiabetic Agents in Members With Type 2 Diabetes in a Commercial Managed Care Organization. Curr Med Res Opin (2010) 26(1):231–8. doi: 10.1185/03007990903421994
135. Guerci B, Chanan N, Kaur S, Jasso-Mosqueda JG, Lew E. Lack of Treatment Persistence and Treatment Nonadherence as Barriers to Glycaemic Control in Patients With Type 2 Diabetes. Diabetes Ther (2019) 10(2):437–49. doi: 10.1007/s13300-019-0590-x
136. Spain CV, Wright JJ, Hahn RM, Wivel A, Martin AA. Self-Reported Barriers to Adherence and Persistence to Treatment With Injectable Medications for Type 2 Diabetes. Clin Ther (2016) 38(7):1653–64 e1. doi: 10.1016/j.clinthera.2016.05.009
137. Buckley ST, Baekdal TA, Vegge A, Maarbjerg SJ, Pyke C, Ahnfelt-Ronne J, et al. Transcellular Stomach Absorption of a Derivatized Glucagon-Like Peptide-1 Receptor Agonist. Sci Transl Med (2018) 10(467):eaar7047. doi: 10.1126/scitranslmed.aar7047
139. Aspnes GE, Bagley SW, Cutro JM, Dowling MS, Edmonds D, Flanagan ME, et al. GLP-1 Agonists and Uses Thereof. New York, NY, USA: Pfizer Inc. 2018.
140. Kawai T, Tanino F, Fukazawa M, Ogawa K, Nagao S, Yoshino H, et al. OWL833, an Orally Active Nonpeptide GLP-1 Receptor Agonist, Improves Glucose Tolerance by Increasing Insulin Secretion and Reduces Food Intake of Cynomolgus Monkeys. Diabetes (2018) 67(Supplement 1):1118–P. doi: 10.2337/db18-1118-P
141. FDA. FDA Approves Weight Management Drug for Patients Aged 12 and Older. 2020. FDA website in the section of News & Events for Human Drugs. FDA approves weight management drug for patients aged 12 and older (issued December 04, 2020) retrieved from https://www.fda.gov/drugs/news-events-human-drugs/fda-approves-weight-management-drug-patients-aged-12-and-older.
142. Mehta A, Marso SP, Neeland IJ. Liraglutide for Weight Management: A Critical Review of the Evidence. Obes Sci Pract (2017) 3(1):3–14. doi: 10.1002/osp4.84
143. Marso SP, Bain SC, Consoli A, Eliaschewitz FG, Jodar E, Leiter LA, et al. Semaglutide and Cardiovascular Outcomes in Patients With Type 2 Diabetes. N Engl J Med (2016) 375(19):1834–44. doi: 10.1056/NEJMoa1607141
144. Giugliano D, Scappaticcio L, Longo M, Caruso P, Maiorino MI, Bellastella G, et al. GLP-1 Receptor Agonists and Cardiorenal Outcomes in Type 2 Diabetes: An Updated Meta-Analysis of Eight CVOTs. Cardiovasc Diabetol (2021) 20(1):189. doi: 10.1186/s12933-021-01366-8
145. FDA. FDA Approves New Drug Treatment for Chronic Weight Management, First Since 2014. (2021). FDA website in the section of News & Events for Human Drugs. FDA approves weight management drug for patients aged 12 and older (issued December 04, 2020) retrieved from https://www.fda.gov/drugs/news-events-human-drugs/fda-approves-weight-management-drug-patients-aged-12-and-older.
146. Davies M, Faerch L, Jeppesen OK, Pakseresht A, Pedersen SD, Perreault L, et al. Semaglutide 2.4 Mg Once a Week in Adults With Overweight or Obesity, and Type 2 Diabetes (STEP 2): A Randomised, Double-Blind, Double-Dummy, Placebo-Controlled, Phase 3 Trial. Lancet (2021) 397(10278):971–84. doi: 10.1016/S0140-6736(21)00213-0
147. Rubino D, Abrahamsson N, Davies M, Hesse D, Greenway FL, Jensen C, et al. Effect of Continued Weekly Subcutaneous Semaglutide vs Placebo on Weight Loss Maintenance in Adults With Overweight or Obesity: The STEP 4 Randomized Clinical Trial. JAMA (2021) 325(14):1414–25. doi: 10.1001/jama.2021.3224
148. Wadden TA, Bailey TS, Billings LK, Davies M, Frias JP, Koroleva A, et al. Effect of Subcutaneous Semaglutide vs Placebo as an Adjunct to Intensive Behavioral Therapy on Body Weight in Adults With Overweight or Obesity: The STEP 3 Randomized Clinical Trial. JAMA (2021) 325(14):1403–13. doi: 10.1001/jama.2021.1831
149. Finan B, Muller TD, Clemmensen C, Perez-Tilve D, DiMarchi RD, Tschop MH. Reappraisal of GIP Pharmacology for Metabolic Diseases. Trends Mol Med (2016) 22(5):359–76. doi: 10.1016/j.molmed.2016.03.005
150. Gault VA, Porter DW, Irwin N, Flatt PR. Comparison of Sub-Chronic Metabolic Effects of Stable Forms of Naturally Occurring GIP(1-30) and GIP(1-42) in High-Fat Fed Mice. J Endocrinol (2011) 208(3):265–71. doi: 10.1530/JOE-10-0419
151. Vilsboll T, Agerso H, Lauritsen T, Deacon CF, Aaboe K, Madsbad S, et al. The Elimination Rates of Intact GIP as Well as its Primary Metabolite, GIP 3-42, Are Similar in Type 2 Diabetic Patients and Healthy Subjects. Regul Pept (2006) 137(3):168–72. doi: 10.1016/j.regpep.2006.07.007
152. DiGruccio MR, Mawla AM, Donaldson CJ, Noguchi GM, Vaughan J, Cowing-Zitron C, et al. Comprehensive Alpha, Beta and Delta Cell Transcriptomes Reveal That Ghrelin Selectively Activates Delta Cells and Promotes Somatostatin Release From Pancreatic Islets. Mol Metab (2016) 5(7):449–58. doi: 10.1016/j.molmet.2016.04.007
153. Yip RG, Boylan MO, Kieffer TJ, Wolfe MM. Functional GIP Receptors Are Present on Adipocytes. Endocrinology (1998) 139(9):4004–7. doi: 10.1210/endo.139.9.6288
154. Mantelmacher FD, Zvibel I, Cohen K, Epshtein A, Pasmanik-Chor M, Vogl T, et al. GIP Regulates Inflammation and Body Weight by Restraining Myeloid-Cell-Derived S100A8/A9. Nat Metab (2019) 1(1):58–69. doi: 10.1038/s42255-018-0001-z
155. Metzger VL, Baker RJ, Schingoethe DJ. Responses of Rumen Microflora to High-Concentrate Low-Roughage Diets Containing Whey Products. J Dairy Sci (1976) 59(10):1769–75. doi: 10.3168/jds.S0022-0302(76)84436-0
156. Adriaenssens AE, Biggs EK, Darwish T, Tadross J, Sukthankar T, Girish M, et al. Glucose-Dependent Insulinotropic Polypeptide Receptor-Expressing Cells in the Hypothalamus Regulate Food Intake. Cell Metab (2019) 30(5):987–96 e6. doi: 10.1016/j.cmet.2019.07.013
157. Dowsett GKC, Lam BYH, Tadross JA, Cimino I, Rimmington D, Coll AP, et al. A Survey of the Mouse Hindbrain in the Fed and Fasted States Using Single-Nucleus RNA Sequencing. Mol Metab (2021) 53:101240. doi: 10.1016/j.molmet.2021.101240
158. Christensen M, Vedtofte L, Holst JJ, Vilsboll T, Knop FK. Glucose-Dependent Insulinotropic Polypeptide: A Bifunctional Glucose-Dependent Regulator of Glucagon and Insulin Secretion in Humans. Diabetes (2011) 60(12):3103–9. doi: 10.2337/db11-0979
159. Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W. Preserved Incretin Activity of Glucagon-Like Peptide 1 [7-36 Amide] But Not of Synthetic Human Gastric Inhibitory Polypeptide in Patients With Type-2 Diabetes Mellitus. J Clin Invest (1993) 91(1):301–7. doi: 10.1172/JCI116186
160. Hojberg PV, Vilsboll T, Rabol R, Knop FK, Bache M, Krarup T, et al. Four Weeks of Near-Normalisation of Blood Glucose Improves the Insulin Response to Glucagon-Like Peptide-1 and Glucose-Dependent Insulinotropic Polypeptide in Patients With Type 2 Diabetes. Diabetologia (2009) 52(2):199–207. doi: 10.1007/s00125-008-1195-5
161. Stensen S, Gasbjerg LS, Helsted MM, Hartmann B, Christensen MB, Knop FK. GIP and the Gut-Bone Axis - Physiological, Pathophysiological and Potential Therapeutic Implications. Peptides (2020) 125:170197. doi: 10.1016/j.peptides.2019.170197
162. Zhang ZQ, Hölscher C. GIP has Neuroprotective Effects in Alzheimer and Parkinson’s Disease Models. Peptides (2020) 125:170184. doi: 10.1016/j.peptides.2019.170184
163. Samms RJ, Coghlan MP, Sloop KW. How May GIP Enhance the Therapeutic Efficacy of GLP-1? Trends Endocrinol Metab (2020) 31(6):410–21. doi: 10.1016/j.tem.2020.02.006
164. Campbell JE. Targeting the GIPR for Obesity: To Agonize or Antagonize? Potential Mechanisms Mol Metab (2021) 46:101139. doi: 10.1016/j.molmet.2020.101139
165. Campbell JE, Drucker DJ. Pharmacology, Physiology, and Mechanisms of Incretin Hormone Action. Cell Metab (2013) 17(6):819–37. doi: 10.1016/j.cmet.2013.04.008
166. Eckel RH, Fujimoto WY, Brunzell JD. Gastric Inhibitory Polypeptide Enhanced Lipoprotein Lipase Activity in Cultured Preadipocytes. Diabetes (1979) 28(12):1141–2. doi: 10.2337/diab.28.12.1141
167. Kim SJ, Nian C, McIntosh CH. GIP Increases Human Adipocyte LPL Expression Through CREB and TORC2-Mediated Trans-Activation of the LPL Gene. J Lipid Res (2010) 51(11):3145–57. doi: 10.1194/jlr.M006841
168. Wasada T, McCorkle K, Harris V, Kawai K, Howard B, Unger RH. Effect of Gastric Inhibitory Polypeptide on Plasma Levels of Chylomicron Triglycerides in Dogs. J Clin Invest (1981) 68(4):1106–7. doi: 10.1172/JCI110335
169. Kim SJ, Nian C, McIntosh CH. Activation of Lipoprotein Lipase by Glucose-Dependent Insulinotropic Polypeptide in Adipocytes. A Role for a Protein Kinase B, LKB1, and AMP-Activated Protein Kinase Cascade. J Biol Chem (2007) 282(12):8557–67. doi: 10.1074/jbc.M609088200
170. Thondam SK, Daousi C, Wilding JP, Holst JJ, Ameen GI, Yang C, et al. Glucose-Dependent Insulinotropic Polypeptide Promotes Lipid Deposition in Subcutaneous Adipocytes in Obese Type 2 Diabetes Patients: A Maladaptive Response. Am J Physiol Endocrinol Metab (2017) 312(3):E224–wfi 233. doi: 10.1152/ajpendo.00347.2016
171. Asmar M, Simonsen L, Madsbad S, Stallknecht B, Holst JJ, Bulow J. Glucose-Dependent Insulinotropic Polypeptide may Enhance Fatty Acid Re-Esterification in Subcutaneous Abdominal Adipose Tissue in Lean Humans. Diabetes (2010) 59(9):2160–3. doi: 10.2337/db10-0098
172. Oben J, Morgan L, Fletcher J, Marks V. Effect of the Entero-Pancreatic Hormones, Gastric Inhibitory Polypeptide and Glucagon-Like Polypeptide-1(7-36) Amide, on Fatty Acid Synthesis in Explants of Rat Adipose Tissue. J Endocrinol (1991) 130(2):267–72. doi: 10.1677/joe.0.1300267
173. Beck B, Max JP. Gastric Inhibitory Polypeptide Enhancement of the Insulin Effect on Fatty Acid Incorporation Into Adipose Tissue in the Rat. Regul Pept (1983) 7(1):3–8. doi: 10.1016/0167-0115(83)90276-8
174. Kim SJ, Nian C, McIntosh CH. Adipocyte Expression of the Glucose-Dependent Insulinotropic Polypeptide Receptor Involves Gene Regulation by PPARgamma and Histone Acetylation. J Lipid Res (2011) 52(4):759–70. doi: 10.1194/jlr.M012203
175. Song DH, Getty-Kaushik L, Tseng E, Simon J, Corkey BE, Wolfe MM. Glucose-Dependent Insulinotropic Polypeptide Enhances Adipocyte Development and Glucose Uptake in Part Through Akt Activation. Gastroenterology (2007) 133(6):1796–805. doi: 10.1053/j.gastro.2007.09.005
176. Weaver RE, Donnelly D, Wabitsch M, Grant PJ, Balmforth AJ. Functional Expression of Glucose-Dependent Insulinotropic Polypeptide Receptors is Coupled to Differentiation in a Human Adipocyte Model. Int J Obes (Lond) (2008) 32(11):1705–11. doi: 10.1038/ijo.2008.148
177. Miyawaki K, Yamada Y, Ban N, Ihara Y, Tsukiyama K, Zhou H, et al. Inhibition of Gastric Inhibitory Polypeptide Signaling Prevents Obesity. Nat Med (2002) 8(7):738–42. doi: 10.1038/nm727
178. Joo E, Harada N, Yamane S, Fukushima T, Taura D, Iwasaki K, et al. Inhibition of Gastric Inhibitory Polypeptide Receptor Signaling in Adipose Tissue Reduces Insulin Resistance and Hepatic Steatosis in High-Fat Diet-Fed Mice. Diabetes (2017) 66(4):868–79. doi: 10.2337/db16-0758
179. Zhang Q, Delessa CT, Augustin R, Bakhti M, Collden G, Drucker DJ, et al. The Glucose-Dependent Insulinotropic Polypeptide (GIP) Regulates Body Weight and Food Intake via CNS-GIPR Signaling. Cell Metab (2021) 33(4):833–44 e5. doi: 10.1016/j.cmet.2021.01.015
180. Beaudry JL, Kaur KD, Varin EM, Baggio LL, Cao X, Mulvihill EE, et al. Physiological Roles of the GIP Receptor in Murine Brown Adipose Tissue. Mol Metab (2019) 28:14–25. doi: 10.1016/j.molmet.2019.08.006
181. Killion EA, Wang J, Yie J, Shi SD, Bates D, Min X, et al. Anti-Obesity Effects of GIPR Antagonists Alone and in Combination With GLP-1R Agonists in Preclinical Models. Sci Transl Med (2018) 10(472). doi: 10.1126/scitranslmed.aat3392
182. Holst JJ, Rosenkilde MM. Recent Advances of GIP and Future Horizons. Peptides (2020) 125:170230. doi: 10.1016/j.peptides.2019.170230
183. Baldassano S, Gasbjerg LS, Kizilkaya HS, Rosenkilde MM, Holst JJ, Hartmann B. Increased Body Weight and Fat Mass After Subchronic GIP Receptor Antagonist, But Not GLP-2 Receptor Antagonist, Administration in Rats. Front Endocrinol (Lausanne) (2019) 10:492. doi: 10.3389/fendo.2019.00492
184. Mroz PA, Finan B, Gelfanov V, Yang B, Tschop MH, DiMarchi RD, et al. Optimized GIP Analogs Promote Body Weight Lowering in Mice Through GIPR Agonism Not Antagonism. Mol Metab (2019) 20:51–62. doi: 10.1016/j.molmet.2018.12.001
185. Kaneko K, Fu Y, Lin HY, Cordonier EL, Mo Q, Gao Y, et al. Gut-Derived GIP Activates Central Rap1 to Impair Neural Leptin Sensitivity During Overnutrition. J Clin Invest (2019) 129(9):3786–91. doi: 10.1172/JCI126107
186. Svendsen B, Capozzi ME, Nui J, Hannou SA, Finan B, Naylor J, et al. Pharmacological Antagonism of the Incretin System Protects Against Diet-Induced Obesity. Mol Metab (2020) 32:44–55. doi: 10.1016/j.molmet.2019.11.018
187. Adriaenssens AE, Gribble FM, Reimann F. The Glucose-Dependent Insulinotropic Polypeptide Signaling Axis in the Central Nervous System. Peptides (2020) 125:170194. doi: 10.1016/j.peptides.2019.170194
188. Borner T, Geisler CE, Fortin SM, Cosgrove R, Alsina-Fernandez J, Dogra M, et al. GIP Receptor Agonism Attenuates GLP-1 Receptor Agonist-Induced Nausea and Emesis in Preclinical Models. Diabetes (2021) 70(11):2545–53. doi: 10.2337/db21-0459
189. Holst JJ, Rosenkilde MM. GIP as a Therapeutic Target in Diabetes and Obesity: Insight From Incretin Co-Agonists. J Clin Endocrinol Metab (2020) 105(8):e2710–6. doi: 10.1210/clinem/dgaa327
190. Willard FS, Douros JD, Gabe MB, Showalter AD, Wainscott DB, Suter TM, et al. Tirzepatide is an Imbalanced and Biased Dual GIP and GLP-1 Receptor Agonist. JCI Insight (2020) 5(17):140532. doi: 10.1172/jci.insight.140532
191. Hornby PJ. Central Neurocircuitry Associated With Emesis. Am J Med (2001) 111 Suppl 8A:106S–12S. doi: 10.1016/S0002-9343(01)00849-X
192. Hayes MR, Skibicka KP, Grill HJ. Caudal Brainstem Processing is Sufficient for Behavioral, Sympathetic, and Parasympathetic Responses Driven by Peripheral and Hindbrain Glucagon-Like-Peptide-1 Receptor Stimulation. Endocrinology (2008) 149(8):4059–68. doi: 10.1210/en.2007-1743
193. Hayes MR, Leichner TM, Zhao S, Lee GS, Chowansky A, Zimmer D, et al. Intracellular Signals Mediating the Food Intake-Suppressive Effects of Hindbrain Glucagon-Like Peptide-1 Receptor Activation. Cell Metab (2011) 13(3):320–30. doi: 10.1016/j.cmet.2011.02.001
194. Samms RJ, Christe ME, Collins KA, Pirro V, Droz BA, Holland AK, et al. GIPR Agonism Mediates Weight-Independent Insulin Sensitization by Tirzepatide in Obese Mice. J Clin Invest (2021) 131(12):146353. doi: 10.1172/JCI146353
195. Sparre-Ulrich AH, Hansen LS, Svendsen B, Christensen M, Knop FK, Hartmann B, et al. Species-Specific Action of (Pro3)GIP - a Full Agonist at Human GIP Receptors, But a Partial Agonist and Competitive Antagonist at Rat and Mouse GIP Receptors. Br J Pharmacol (2016) 173(1):27–38. doi: 10.1111/bph.13323
Keywords: incretin, GLP-1, GIP, obesity, diabetes, drug
Citation: Tan Q, Akindehin SE, Orsso CE, Waldner RC, DiMarchi RD, Müller TD and Haqq AM (2022) Recent Advances in Incretin-Based Pharmacotherapies for the Treatment of Obesity and Diabetes. Front. Endocrinol. 13:838410. doi: 10.3389/fendo.2022.838410
Received: 17 December 2021; Accepted: 07 February 2022;
Published: 01 March 2022.
Edited by:
Susanna Hofmann, Helmholtz Association of German Research Centres (HZ), GermanyReviewed by:
Victor Alan Gault, Ulster University, United KingdomNicolai Jacob Wewer Albrechtsen, Rigshospitalet, Denmark
Copyright © 2022 Tan, Akindehin, Orsso, Waldner, DiMarchi, Müller and Haqq. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Timo D. Müller, dGltby5tdWVsbGVyQGhlbG1ob2x0ei1tdWVuY2hlbi5kZQ==; Andrea M. Haqq, aGFxcUB1YWxiZXJ0YS5jYQ==
†These authors have contributed equally to this work