 Zoltan Spolarics
Zoltan Spolarics Geber Peña1
Geber Peña1- 1Department of Surgery, Rutgers-New Jersey Medical School, Newark, NJ, United States
- 2Department of Pathology and Laboratory Medicine, Rutgers-New Jersey Medical School, Newark, NJ, United States
Females have a longer lifespan and better general health than males. Considerable number of studies also demonstrated that, after trauma and sepsis, females present better outcomes as compared to males indicating sex-related differences in the innate immune response. The current notion is that differences in the immuno-modulatory effects of sex hormones are the underlying causative mechanism. However, the field remains controversial and the exclusive role of sex hormones has been challenged. Here, we propose that polymorphic X-linked immune competent genes, which are abundant in the population are important players in sex-based immuno-modulation and play a key role in causing sex-related outcome differences following trauma or sepsis. We describe the differences in X chromosome (ChrX) regulation between males and females and its consequences in the context of common X-linked polymorphisms at the individual as well as population level. We also discuss the potential pathophysiological and immune-modulatory aspects of ChrX cellular mosaicism, which is unique to females and how this may contribute to sex-biased immune-modulation. The potential confounding effects of ChrX skewing of cell progenitors at the bone marrow is also presented together with aspects of acute trauma-induced de novo ChrX skewing at the periphery. In support of the hypothesis, novel observations indicating ChrX skewing in a female trauma cohort as well as case studies depicting the temporal relationship between trauma-induced cellular skewing and the clinical course are also described. Finally, we list and discuss a selected set of polymorphic X-linked genes, which are frequent in the population and have key regulatory or metabolic functions in the innate immune response and, therefore, are primary candidates for mediating sex-biased immune responses. We conclude that sex-related differences in a variety of disease processes including the innate inflammatory response to injury and infection may be related to the abundance of X-linked polymorphic immune-competent genes, differences in ChrX regulation, and inheritance patterns between the sexes and the presence of X-linked cellular mosaicism, which is unique to females.
Introduction
Females have better general health and a longer lifespan than males (1, 2). Sex-related disparities in the immune response show a mixed pattern. Whereas autoimmune diseases are predominant in females (3–6), a large number of studies demonstrated that following trauma and sepsis females present improved clinical course and better outcomes as compared to males (7–22). However, the field remains controversial as some investigations showed no female benefit or a few studies even suggested a female disadvantage following infection (23–25). These controversies may be related to the fact that most human studies were designed to simply compare males versus females or after stratification by sex hormone status without considering genetic contributions (26–28). Animal studies using genetically homogenous inbred strains are more convincing in showing a female benefit but the aspect of genetic polymorphism as it exists in humans is lacking in these models.
Whereas ample evidence exists to support the important immuno-modulatory role of sex hormones (7–13, 26–32), sex-based outcome differences associated with the innate immune response are unlikely to be solely dependent on their effects. For example, observations on young prepubertal children indicated that boys have higher mortality rate than girls after burns (33). Infection-induced mortality rate was also higher in newborn boys than girls (34, 35). Male newborns displayed more frequent viral and bacterial respiratory infections and were more prone to sepsis and meningitis than girls (35). Additionally, sex differences in disease processes including sepsis were shown to manifest between postmenopausal women and older men (36, 37). Even animal models using castrated or ovariectomized animals or sex hormone replacements found that sex-related differences were only partially diminished between males and females (38–40). Finally, studies testing the beneficial effects of sex hormone treatment after injury or sepsis employed pharmacological doses of estrogen. Therefore steroid-like effects or secondary changes in the ACTH axis may have contributed to the specific hormonal effects (41–46). These observations indicate that factors other than sex hormones likely contribute to sex-dimorphic outcomes (20, 21, 35, 47, 48). Whereas studies have started to address the role of X chromosome (ChrX) in autoimmune diseases (3–6), the role of sex chromosomes in contributing to sex-related immunomodulation during the innate immune response has not been thoroughly examined.
We previously proposed that common non-pathological X-linked genetic polymorphisms may be important in modulating sex-based differences in innate immunity and neglecting the genetic aspect may be part of the reasons of current controversies in this field (16, 48). The proposed ChrX hypothesis has been tested and confirmed in animal proof-of-concept studies (49–51) and recently published human investigations also support the concept (52). The aim of the current article is to present the hypothesis in a new light and in the context of recent clinical observations. We present how the inherently polymorphic ChrX and uniquely female ChrX mosaicism may contribute to immuno-modulation at the individual subject as well as human population level. Furthermore, we identify and discuss selected polymorphic X-linked candidate genes, which we believe are likely players in sex-based immunomodulation via cellular mosaicism and sex-linked inheritance patterns. New experimental findings will also be shown. Aspects of autoimmune diseases, sex hormone effects, and the potential contribution of Y chromosome genes will be discussed only to the extent that it is related to the proposed concept.
ChrX-Mosaicism and Cellular Variability
Females inherit two naturally polymorphic parental X chromosomes (ChrXs) whereas males inherit a single ChrX passed on from the mother. Therefore, females carry polymorphic X-linked alleles from both parents while males carry only the maternal variants (53, 54). To compensate for the potential double-dosed gene expression in females as compared to males, cells undergo random ChrX inactivation during early female embryonic development. The process involves methylation of one of the ChrXs rendering it inactive for gene expression (55–57). Although the cells retain and copy the inactive DNA during cell divisions, the expression-block is retained for the entire lifespan of the cell. These epigenetic changes and the random selection of ChrX inactivation within a cell result in females showing cellular mosaicism for the expression of X-linked polymorphic proteins or differences in gene regulation due to allelic variations between mosaic cells. The ratio of circulating blood cells with the active respective parental ChrXs, on average, approximates to one-to-one in younger healthy individuals (56–58). In blood, mosaic cell populations are dispersed homogenously, however, because organs start developing from cellular islands during embryogenesis, the distribution of X-linked mosaicism is patch-like and inhomogeneous in solid organs and tissues (59). The likely consequence of X-linked mosaicism is that the innate genetic polymorphisms of the ChrXs will result in distinct variations in mosaic cellular subsets resulting in phenotypes with different regulatory potentials or functional responsiveness within a female subject (48). This is in contrast with males who lack X-linked cell mosaicism and in whom polymorphisms from the maternal ChrX are the only drivers of X-linked cellular variability (Figure 1). Based on these considerations, we propose that increased cellular variability through ChrX mosaicism in otherwise healthy females may be an advantageous condition during the innate immune response by providing improved functional flexibility to dynamically changing pathophysiological conditions. These differences in cellular phenotypes may be a contributing factor to sex-based outcome differences and immune-modulation. The concept of improved cellular adaptability in females is supported by the fact that ChrX skewing is frequently observed in healthy females or in severe X-linked genetic defects and it is defined as increased numbers of one of the mosaic subpopulations relative to the other (60–62).
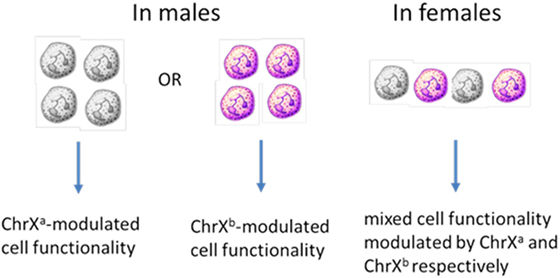
Figure 1. ChrX-linked cellular mosaicism for the expression of variant proteins results in phenotype diversity in females but increased functional polarity in males.
During inflammation, de novo, acute and functional ChrX cellular skewing may occur resulting from different degrees of cell trafficking, recruitment to injury or infection sites, or dissimilar rates in necrosis, apoptosis, and cell proliferation driven by X-linked allelic variations (Figure 2). This de novo X-linked cellular skewing is expected to be temporary and reversible. When the inflammatory response dissipates and infection or injury resolves, the original cell ratio is expected to become rebalanced. The exception is when the inflammatory response results in irreversibly skewed cell ratios in bone marrow progenitors, which is an unlikely scenario. Our previous observations from mice studies using X-linked knockout and mosaic models for CYBB (gp91phox) and interleukin-1 receptor-associated kinase 1 (IRAK1) support this hypothesis (49–51). These studies showed similar mosaic cell ratios between BM and blood in healthy animals. However, endotoxemia or sepsis resulted in ChrX skewing in circulating blood and immune-competent organs. These investigations also revealed that animals with ChrX-linked mosaicism showed improved outcome as compared to WT or deficient animals but the mechanisms of protection were different in the gp91phox- and IRAK1-deficient models (49–51).
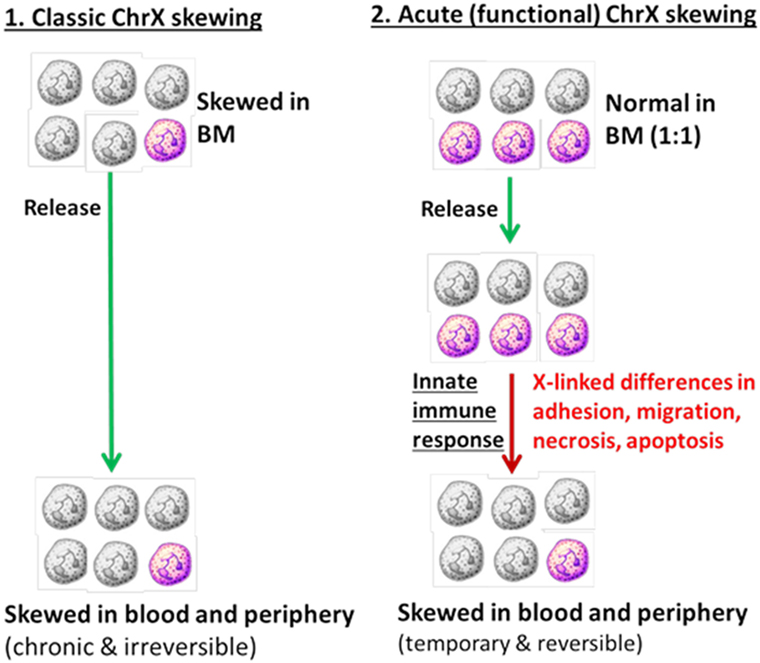
Figure 2. Cellular skewing driven by primary changes in bone marrow progenitors (Part 1) versus selection of mosaic subpopulations at the periphery driven by X-linked allelic variants (Part 2).
Observations from these animal studies also suggested that the presence of different mosaic cell populations may provide the conditions for intercellular communication between mosaic cells. An interplay among mosaic cellular variants in females could “buffer” the inflammatory response by downregulating hyper-active cell populations during excessive inflammation or in contrast, may compensate for immuno-paralysis, thereby improving the clinical course. Evidently, this potentially beneficial “buffering” mechanism is lacking in single-ChrX males.
However, it needs mentioning that ChrX skewing may also be disadvantageous in some cases. For example, gross and spontaneous ChrX skewing of BM progenitors in healthy females may precondition immune cells for a “biased” cell response to injury or infection driven by polymorphisms of one of the ChrXs. Moreover, extreme ChrX skewing in a female manifests a male-like phenotype because cells expressing one of the two ChrXs dominate in the circulation. This possibility is realistic as spontaneous ChrX cellular skewing in the bone marrow and blood is common in otherwise healthy females especially at older age (63–65).
Impacts of Common X-Linked Polymorphisms at the Population Level
The potential effects of X-linked polymorphisms in the context of sex-based outcome differences are expected to be significant due to several factors (66). First, because of the X-linked inheritance pattern, it is evident that all clinically important X-linked polymorphisms have a greater impact on the male than female population in contrast with polymorphic alleles linked to the autosomal chromosome, which show no sex bias (48, 53). For example, a 20% allele frequency of an X-linked variant allele in a randomly mating population or a population sample showing Hardy–Weinberg equilibrium will result in genotype frequencies in females as 4% affected (homozygous), 32% mosaic, and 64% normal. In males of the same population, the genotype distribution will be 20% affected (hemizygous) and 80% normal, which indicates that males are affected five times more than females in this example. In addition, a large segment of the female population presents cellular mosaicism for expressing the WT and risk allele, respectively (48). Females who have mosaicism for the expression of the WT and risk alleles are likely to present their own phenotypes, which are different from females carrying cells homozygous either for the variant or WT alleles as demonstrated in our mice studies (49–51). These facts indicate that part of the female health advantages over males could be related to, or at least impacted by, sex-biased distributions of common X-linked polymorphic alleles together with the presence of uniquely female X-linked cellular mosaicism (48).
Second, X-linked cellular variability becomes even more complex after considering the possibilities produced by gametal ChrX recombination in mothers followed by subsequent random ChrX inactivation in female embryos. Sex chromosome recombination is different from that of autosomal chromosomes because recombination between two ChrXs can take place only in females. In males, recombination between the sex chromosomes is limited to the small pseudo-autosomal regions at the tips of the Y and X chromosomes (59). For example, consider two X-linked alleles producing functionally related or interacting protein A and B and their allelic variants a and b in a population. If these two related allelic mutations reside in different recombination blocks, then recombination followed by ChrX inactivation theoretically will result in 10 different mosaic cell pairs with associated phenotypes in females: AB + AB, AB + Ab, AB + aB, AB + ab, Ab + Ab, Ab + ab, Ab + aB, aB + aB, aB + ab, ab + ab. However, in males, the potential genotypic combinations are limited to produce only four cellular phenotypes in the absence of cellular mosaicism (AB, Ab, aB, ab). Assuming more interrelated but independently recombined X-linked variants, the male–female difference in the possible numbers of mosaic phenotypes increases exponentially reaching almost a 100-fold difference at four related alleles (Figure 3). Most of these combinations are not likely to produce distinct phenotypes in females and the X-linked pattern distorts the population frequency of these genotype–phenotype combinations; however, the fact remains unquestionable that the potential for a markedly increased variability to manifest in females as compared to males is there. Furthermore, studies have also indicated that up to 15% of genes may escape permanent silencing on the inactive ChrX (55, 59), which may also add to increased cellular diversity in females compared to males. These facts not only indicate inherent difficulties in predicting phenotype-genotype relationships in the context of sex but also complicate the evaluation of genetic association studies testing X-linked genes (48, 53).
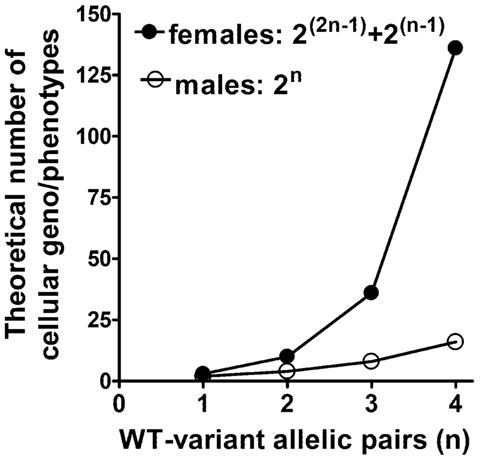
Figure 3. Possible number of different cellular phenotypes in female ChrX mosaicisms versus single-ChrX carrying males. The number of cellular phenotypes are shown as the function of the number of functionally interrelated polymorphic gene variant pairs which reside in different recombination regions of the ChrX.
The facts and considerations presented so far indicate that X-linked polymorphisms including known risk alleles together with other common allelic variations with immune-modulatory potentials are likely contributors to sex-based differences in the innate immune response and post-injury recovery. The unique distribution and different regulation of ChrXs between the sexes present a more polarized cellular machinery in males as compared to females at the individual level (Figure 4, top). The duality of cellular machinery in females should blunt polarity resulting in buffered responses either by simply “diluting” polarized cells or through cell-cell interactions and consequent positive and negative feedback mechanisms (Figure 4A). At the population level, these X-linked differences are expected to result in an increased representation of males at the extreme boundaries of the response distribution curve (Figure 4B).
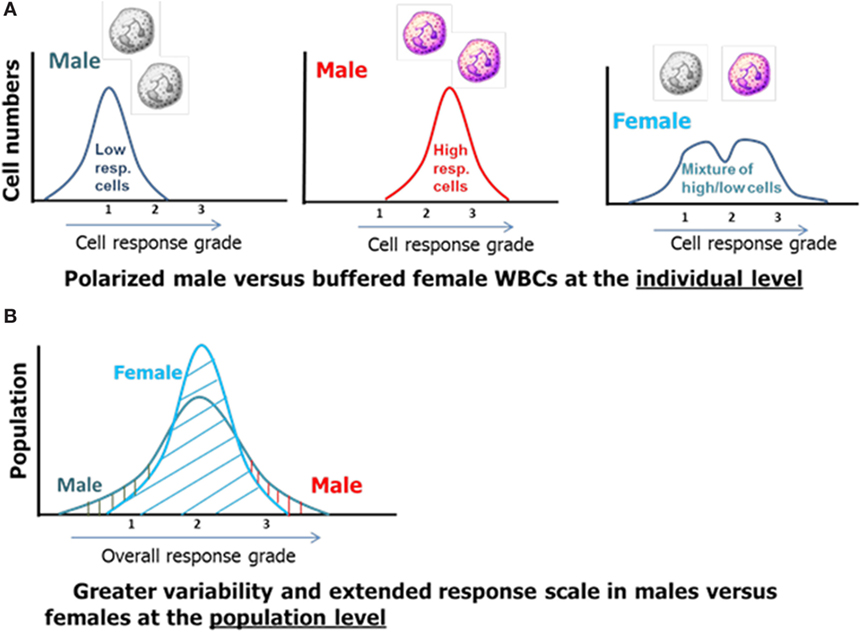
Figure 4. The effect of X-linked cellular mosaicism in females and its absence in males at the individual (A) and population level (B).
In summary, in any clinical cohorts of mixed sex, males hemizygous for the risk allele will always be more represented than homozygous females and a large segment of the female subject will show mosaicism for the alleles with their own unique phenotypes. Subjects who show mosaicism for the expression of the variant and WT alleles may also be further subdivided by the presence or absence of spontaneous or pathology-induced ChrX skewing. In those individuals who present ChrX skewing, it also needs to be tested whether the “direction” of ChrX skewing manifests cellular subset ratios moving toward or against variant allele-expressing populations. This is important because without knowing the “direction” of ChrX skewing, it is impossible to conclude whether the mosaic subjects are rendered quasi WT or quasi variant phenotype, thereby being protected or prone to pathology. These facts and reflections indicate that genetic association studies investigating cohorts of mixed sex should consider aspects of X-linked polymorphisms, cellular mosaicism, and the status and direction of chronic or acute ChrX skewing in the analyses. This will help in identifying female subgroups within a cohort with distinct genotype–phenotype relationships and associated pathology and thus could help in resolving continuing controversies in the field.
Testing the Hypothesis in Humans
As discussed earlier, observations from animal studies using X-linked genetic knockouts combined with X-linked cellular mosaicisms supported the proposed concept at the proof-of-principle level (49–51). However, the proposed hypothesis is based on the notion that naturally occurring X-linked risk alleles in combination with other common polymorphisms with immune-modulatory potentials drive sex-related disparities in the innate immune response in humans. Therefore, only human clinical studies can adequately test these questions. To achieve this goal, we postulate that investigating trauma patients may present advantages over other clinical conditions of infection, which developed secondary to immunocompromised status or chronic diseases. The reason is that, in trauma patients, the pathology is initiated suddenly at the time of injury, which is followed by a temporal cascade of events manifesting a sterile non-specific inflammatory response. Those patients who survive their initial injuries frequently present secondary clinical complications, which may manifest in infection leading to sepsis and organ dysfunction and culminates in late post-injury mortality in some patients. Additionally, a large proportion of trauma victims are relatively young or middle-aged, which lessens analytical difficulties related to comorbidities and age. Thus, the well-defined starting point at the time of injury followed by a cascading pathology with chances for secondary complications in a trauma cohort provides a clinical situation, which could manifest acute and functional ChrX skewing.
Our recently published study conducted on 39 female trauma patients with moderate to severe injuries (52) indicated that at admission, two-third of the patients displayed X-linked mosaic cell ratios between 1 and 3. About a third of the patients presented ratios at the 3–7 range and three patients displayed markedly skewed mosaic cell ratios in the 8–30 range. Testing serial blood samples during the clinical course showed additional changes in cell-ratios ranging between 20 and 900% over initial. Changes in mosaic cell ratios during the injury course correlated with the severity of trauma need for ventilator support and development of pneumonia (52).
We also conducted a follow-up study testing the same question on a subsequent independent trauma cohort with a sample size of 60 (Figure 5). These new observations confirmed previous findings indicating that about a third of the patients presents trauma-induced de novo ChrX skewing at a degree greater than 30% over initial during the hospital course (Figure 5). The most marked changes in post-injury ChrX skewing seem to occur within the first 3–6 days after injury. In recovering patients, toward the end of the hospital stay, skewed cell ratios begin to return to initial values of ChrX ratios similar to that found at admission (Figure 5).
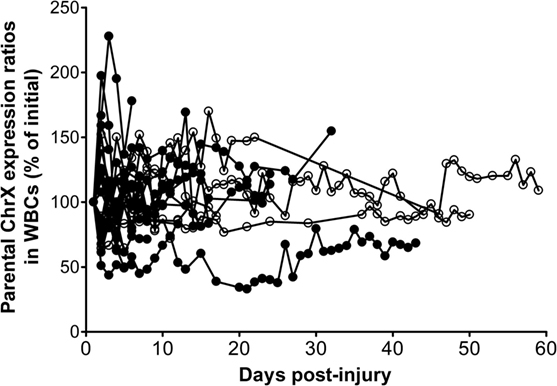
Figure 5. Alterations in ChrX ratios in white blood cell (WBC) during the trauma course. ChrX ratios are expressed as percent of initial measured in the first blood sample drawn at admission. ChrX active/inactive-ratios in circulating WBCs were determined using the variable length polymorphism at the HUMARA locus as we described in detail earlier (52).
To gain more insights into how ChrX skewing may relate to the fine clinical course, we also present actual white blood cell (WBC) numbers expressing respective parental ChrXs from three patients from this cohort to present architypes for typical patterns.
Figure 6A indicates a patient with similar initial WBC numbers expressing respective parental ChrXs measured at admission. There is a transient and reversible increase in X-linked WBC selection peaking at day-3 post injury returning to initial values at day 4. Just after the diagnosis of clinical sepsis on day 4 (arrow), ChrX skewing becomes evident again and lasts for the remaining of the hospital course with only small additional variations.
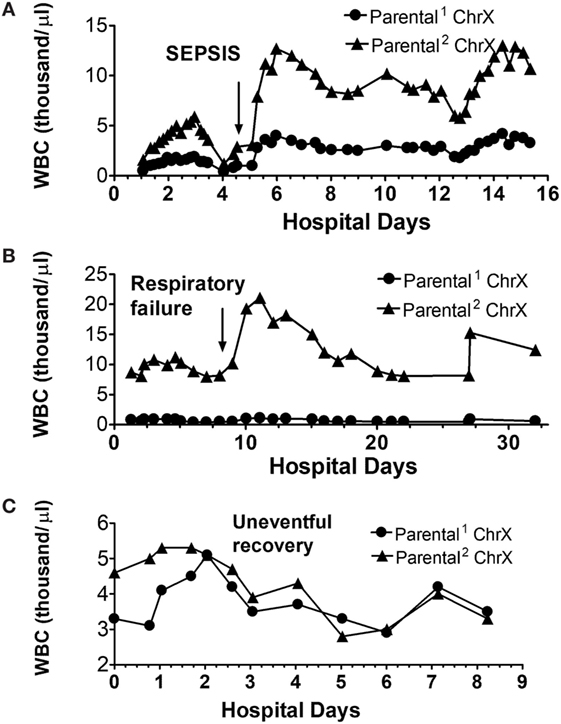
Figure 6. Alterations in circulating white blood cell (WBC) numbers expressing respective parental ChrXs during complicated (A,B) and uncomplicated (C) clinical courses. ChrX active/inactive-ratios in circulating WBCs from serial samples were determined using the variable length polymorphism at the HUMARA locus as we described in detail earlier (52). From the ChrX ratios combined with the accompanying circulating WBC counts, number of cells expressing respective parental ChrXs were calculated. Arrows indicate the time of onset of sepsis or respiratory failure.
The second patient (Figure 6B) already showed marked ChrX skewing in WBCs at admission, which was followed by an additional increase of the dominant mosaic subset peaking at day 5 and returning to original levels by day 7. After the diagnosis of respiratory failure, a marked increase in WBC number occurred, but it was restricted to the already dominant mosaic subpopulation of WBCs.
Finally, Figure 6C depicts a patient with a non-complicated “uneventful” hospital course indicating normalization of WBC ChrX skewing by day-2 followed by small variations in WBC numbers without ChrX skewing during the remaining course.
The observed pattern of these case studies is consistent with the proposed hypothesis and supports the notion that X-linked cell selections occur during the inflammatory response to trauma and most likely also manifests after other inflammatory and infectious conditions. Trauma-induced ChrX skewing seems to be in a temporal relationship with the onset and progression of a clinical complication and associated pathophysiological challenges. It however, remains to be tested whether ChrX skewing represents a clinical advantage following trauma as the phenomenon may simply be a manifestation of X-linked functional differences among mosaic subsets without changing clinical outcome. At this point, it cannot be concluded that the outcome advantage of females after trauma is directly associated with increased ChrX skewing. Nevertheless, the potential manifestation of X-linked, variable, buffered, and adaptive cellular responses in females, and the fact that these X-linked mechanism is lacking in males, is likely to benefit most of the females during an innate inflammatory response under a variety of clinical conditions.
X-Linked Genes of Immuno-Modulation
Past and recent advances in whole genome sequencing, new generation expression profiling, and identifying allelic variations opened revolutionary new possibilities in elucidating genetic components of disease susceptibility and outcome. New allelic variants are identified with increasing speed and the information is readily available from a variety of databases. These robust data sets offer great possibilities for data mining across a variety of genetic backgrounds and pathological conditions. Although the quality of the information in these databases can be inconsistent, for example, some of the genes are studied in great details, others neglected. Therefore, using “educated guesses” in identifying relevant X-linked candidate genes in seeking insights to genotype–phenotype relationships remains a valid and productive approach. In this section, we highlight polymorphic X-linked genes that are widespread in the population and were shown to have immuno-modulatory effects or clinical phenotypes. We propose that allelic polymorphic forms of these genes either alone or in combination are most likely to be responsible or at least contribute to sex-related differences in the immune response and immuno-modulation.
ChrX is medium-sized among chromosomes and carries about 1,200 genes (67). ChrX is relatively rich in highly polymorphic immune competent genes including mediators of the toll-like receptor (TLR)-mediated signaling pathway, TLR7, TLR8, IRAK1, NEMO (IKKγ), NKRF, NKAP, Bruton’s kinase (BTK). Other X-linked proteins with immune competent functions include CD40 ligand (CD40L), FOXP3, IL13R, BMX, IL2RG, and TMP1. In addition, ChrX codes for enzymes involved in redox regulation and apoptosis, NOX1, NOX2, glucose-6-phosphate dehydrogenase (G6PD), XIAP, and AIF (more details in Table 1).
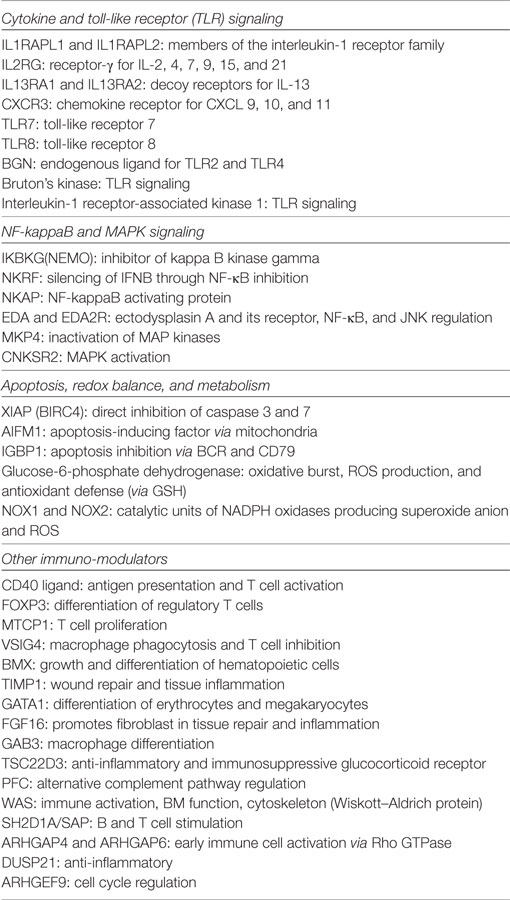
Table 1. X-linked genes with major roles in the inflammatory response.
Interleukin-1 receptor-associated kinase 1 haplotype is one of the primary candidate gene contributing to sex-related outcome differences following trauma and sepsis (68–70). IRAK1 is a member of the intracellular TLR/IL1R/MyD88 adapter complex (71–75). Upon TLR or IL-1β stimuli, IRAK1 becomes phosphorylated and mediates the recruitment and rearrangement of other regulatory proteins, which subsequently leads to IKKγ (NEMO), NFκB, and MAPKs activation resulting in cell activation and cytokine production (71–76). IRAK1 activation is pro-inflammatory (76–78). Animal studies indicate that inhibition or lack of IRAK1 expression is beneficial in sepsis, burn or endotoxemia (79–84).
The human IRAK1 haplotype is composed of seven SNPs five intronic and two exonic. The two exonic polymorphisms results in amino acid (aa) changes at 196 (phe > ser) and 532 (ser > leu). The change at aa 532 seems to be the critical mutation as it persistently increases kinase activity of IRAK1 and augments cell activation (70, 85) in accordance with similar observation from animal studies (79–84).
The clinical impact of IRAK1 variants is well established, as independent studies on septic European as well as Asian cohorts (46, 69, 70) indicated doubled incidence of septic shock, pneumonia, and mortality as compared to WT individuals. In these sepsis studies (69, 70), IRAK1-mosaic females were merged with the WT group, therefore, the effects of mosaicism remained elusive. A later study conducted on trauma patients (68) indicated markedly worsened late post-injury mortality rate of individuals with this haplotype as compared to WT, whereas late mortality rate of “heterozygous” mosaic females was at midway between WT and variant patients. However, the number of homozygous variant and “heterozygous” mosaic female subjects was small in the cohort and aspects or ChrX mosaicism or accompanying ChrX skewing were not examined (68).
The population frequency of the IRAK1 haplotype is 20–40% in whites and Hispanics (86–88). The frequency distribution is reversed in the Asian population, where the variant haplotype may reach 70% prevalence (22, 46, 88, 89). It is important to mention that although most individuals of African descent do not carry the full IRAK1 haplotype, the allelic variation causing the clinically important amino acid change is present at 30–40% frequency. Thus, the X-linked inheritance pattern, high population frequency, and abundance of cellular mosaicism for WT and variant IRAK1 make it likely that the IRAK1 haplotype contributes to sex-biased immuno-modulation across all races and ethnicities.
The variant IRAK1 has also been shown to be associated with increased susceptibility to systemic lupus erythematosus (SLE) (86, 88) an autoimmune disease affecting predominantly females (4, 90, 91). Intriguingly, in the rare cases when SLE develops in males, the clinical course is worsened and mortality is greater than that of observed in females (90, 91). This apparent discrepancy, however, is consistent with the proposed ChrX hypothesis. Namely, cellular mosaicism for X-linked risk alleles including IRAK1 combined with ChrX reactivation (47, 92, 93) could account for the increased susceptibility of females to SLE but, at the same time, ChrX mosaicism may buffer the SLE-associated inflammatory response and alleviate organ dysfunction. In contrast, the lack of ChrX mosaicism in males lessens the chance to the initial onset of SLE but when develops the associated inflammation is more robust due to an “un-buffered” response resulting in more severe organ dysfunction and increased mortality (90, 91). This interconnected relationship of X-linked disease susceptibility, the presence or absence of ChrX mosaicism and associated “immuno-buffering,” or lack of, further signify the complexities of sex-based immuno-modulation.
Among the identified TLRs, TLR7 and TLR8 are X-linked. Clinically important allelic variants have been described impacting susceptibility to tuberculosis, altered macrophage function, allergic and autoimmune conditions including SLE (94, 95) and complications of viral infections (95–101). The population frequency of the minor alleles ranges between 15 and 40% in the European and African population. Interestingly, in the East Asian population, the variant TLR7 and TLR8 alleles are more abundant than the ancestral WT similarly to that observed for IRAK1 population distributions. TLR7 plays an important role in the recognition and subsequent signaling of single-stranded RNA from viruses including influenza, measles, and dengue (102–104). The clinical course of these virus infections was reported to be worse in women than men (24, 25). Polymorphisms of TLR7 and TLR8 have been shown to impact the clinical course of measles (105), but it remains to be tested how ChrX mosaicism for TLR variants impacts outcome after these virus infections.
Nuclear factor kappa B (NFκB) activation is achieved by the phosphorylation of the NFκB-inhibitory proteins (IκB) through the actions of IκB-kinases (IKKα and β). IκB-kinases are in complexes with the X-linked NFκB essential modulator, IKKγ (NEMO) (106–111). Upon a variety of stimuli, including TLR, DAMP, and PAMP activation and subsequent signaling, NEMO becomes activated (112, 113) and interacts with IκB-kinases. Complete lack of NEMO activity is lethal in males, whereas heterozygous mosaic females present an immune-compromised clinical condition (incontinentia pigmenti) (114, 115). The reported SNPs on exons, introns, and flexing gene regions are increasing. Some of these new mutations are associated with complex immune pathologies. Thus, polymorphic forms of NEMO together with allelic variants of other X-linked members of the NFκB regulatory cascade (Table 1) are likely candidates for sex-related immuno modulation.
Bruton’s kinase also participates in TLR-mediated NFκB and p38 MAPK activation affecting TNFα, IL-1β, and IL-12 production as well as iNOS expression and Ca2+ release (116, 117). Severe BTK mutations causes X-liked agammaglobulinemia (XLA) in humans and X-linked immune deficiency in mice (Xid) (116, 117). Several polymorphic alleles causing different degree of XLA phenotypes have been reported (118–120).
FOXP3 is a key regulator of T cell activation and CD40L a transmembrane signaling molecule involved in platelet, endothelial, and immune cell activation. FOXP3 is highly polymorphic and clinically relevant allelic variants have been reported to be present in the approximate range of 10–35% in the population and allele frequencies show marked differences among various ethnic groups (121). The variants have been shown to be associated with autoimmune syndromes, allergic conditions, and cancer (122–127).
X-linked G6PD is the rate limiting enzyme of the pentose phosphate shunt and supplies reduced NADPH as the electron donor for superoxide production in phagocytes. Through the actions of peroxidases and glutathione reductase, G6PD also plays a critical role in supporting antioxidant pathways (128). G6PD is highly polymorphic. The allele frequencies of the common African and Mediterranean polymorphic forms that results in an 80–90% decrease in the enzyme activity of red blood cells may reach 10–25% in populations living in or migrated from sub-Saharan Africa or other malaria endemic regions (129, 130). Whereas the common African Mediterranean polymorphisms affect primarily red blood cell functions due to decreased antioxidant capacity, polymorphic forms have been shown to modulate cytokine production (131, 132) and worsen the clinical course in sepsis and endotoxemia (129–131). A null mutation of G6PD causes chronic granulomatous disease (CGD)-like syndromes and is incompatible with life in hemizygous males (129, 130). Studies on severely injured African American male trauma patients indicated that the common African G6PD variants were associated with increased incidence of infection and sepsis, alterations in monocyte activation, and worsened secondary anemia as compared to individuals with the WT allele (133). The impact of G6PD polymorphic variants on female trauma patients has not been tested, but the X-linked distribution and mosaic expression in females could impact the trauma course contributing to sex-biased outcomes.
Another likely X-linked candidate gene is CYBB (gp91phox, NOX2) encoding for the catalytic subunits of NADPH oxidase, which is responsible for superoxide production by phagocytes during the oxidative burst. Mutations in this gene are the most frequent causes of CGD (134). CGD patients present recurrent bacterial and fungal infections due to inefficient killing of pathogens by phagocytes. Null mutation of this protein is usually fatal in hemizygous males due to recurrent infections and uncontrollable sepsis. Clinical symptoms are related to the type of mutations, which are rather heterogeneous and newly described mutations continue to emerge (135–138). Animal studies suggested that mice with WBC mosaicism for NOX2-WT and NOX2-KO have a dual advantage. In these mosaic animals, efficient bacterial killing was maintained whereas oxidative stress-mediated organ damage was diminished, thereby resulting in improved survival of mosaic mice as compared to homozygous WT or full NOX2 deficiency (50).
All the remaining immune-competent and apoptotic genes listed in Table 1 were reported to have dozens or some of them hundreds of exonic and intronic allelic variants as well as base changes in flanking regulatory regions. Reliable population frequency data are lacking for most of the genes listed and the associated clinical phenotypes are available only to those few mutations, which causes severe pathological phenotype. Nonetheless, conceptually, these polymorphic variants could contribute to increased X-linked cellular functional variability and immuno-modulation. Evidently, there could be many more X-linked polymorphisms impacting genes encoding for regulatory proteins, receptors, and enzymes of various metabolic functions, which may contribute either directly or indirectly to increased cellular variability via ChrX mosaicism and associated immuno-modulation.
Finally, it is worth mentioning that ChrX is one of the richest among chromosomes in encoding miRNAs many of which having immuno-regulatory functions (35, 139). Some of these miRNAs are polymorphic and were shown to impact T cell responses or their presence was associated with autoimmune conditions including Crohn’s disease and rheumatoid arthritis (35, 139, 140). Because miRNAs have multiple genetic targets within a cell, mosaic distribution of polymorphic miRNAs in females may also participate in producing unique X-linked phenotypes. Recent studies showed sex-biased roles of X-linked miRNAs in rheumatoid arthritis (140) consistent with this notion.
Conclusion
We provided a set of facts, arguments, and supporting observations, which indicate that sex-related differences during the inflammatory response may be related to X-linked polymorphic genes and the associated increase in cellular variability in females. The fact that several key regulatory and metabolic proteins participating in signal transduction, apoptosis, carbohydrate metabolism, superoxide production and xenobiotic, and antioxidant defenses are encoded on the X chromosome and their polymorphic variants are widespread in the population strongly supports this hypothesis.
Inflammation is often considered to be a double-edged sword because it is required for the control and elimination of invading microorganisms; however, an overwhelming inflammatory response may lead to organ dysfunction or septic shock and death. In males, the cells of innate immunity carry one allelic compilation of the gene pool that may be adequate, hypo- or hyperresponsive under a pathological condition. In females, the two sets of innate immune cells differing in X-linked parental alleles with different regulatory and activation capacity could represent a more balanced and more adaptive system, which better adjusts to dynamically changing pathophysiology during the inflammatory response.
These differences in cellular functional variability between males and females and the male-biased X-linked inheritance pattern are likely contributors to sex hormone effects in causing sex-associated outcome differences in the population. Pursuing further this concept will be valuable because it can reveal novel aspects of cellular defense mechanisms, which could be utilized in developing improved therapeutic routines. Furthermore, testing for X-linked risk alleles together with the determination of ChrX skewing in females will help to stratify female subgroups or identify outliers within a cohort. The unique regulation of ChrX should be calculated into study designs and considered at the analytical stages of clinical studies investigating mixed cohorts especially with a focus on infection and immunity.
Ethics Statement
This study was carried out in accordance with the recommendations of the Institutional Review Board of Rutgers-New Jersey Medical School with written informed consent from subjects in accordance with the Declaration of Helsinki. The protocol was approved by the Institutional Review Board of Rutgers-New Jersey Medical School.
Author Contributions
ZS: primary contributor of developing the concept, planning and organizing experiments, analyzing data, and formulating the manuscript. GP, YQ, RD, and DL: participated in conceptual aspects, planning and analyzing experiments, and formulation of manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by National Institute of Health (NIH), National Institute of General Medical Sciences, Grant RO1 GM118602.
References
1. Oksuzyan A, Juel K, Vaupel JW, Christensen K. Men: good health and high mortality. Sex differences in health and aging. Aging Clin Exp Res (2008) 20:91–102. doi:10.1007/BF03324754
2. Ostan R, Monti D, Gueresi P, Bussolotto M, Franceschi C, Baggio G. Gender, aging and longevity in humans: an update of an intriguing/neglected scenario paving the way to a gender-specific medicine. Clin Sci (Lond) (2016) 130:1711–25. doi:10.1042/CS20160004
3. Klein SL, Flanagan KL. Sex differences in immune responses. Nat Rev Immunol (2016) 16:626–38. doi:10.1038/nri.2016.90
4. Rubtsova K, Marrack P, Rubtsov AV. Sexual dimorphism in autoimmunity. J Clin Invest (2015) 125:2187–93. doi:10.1172/JCI78082
5. Hewagama A, Richardson B. The genetics and epigenetics of autoimmune diseases. J Autoimmun (2009) 33:3–11. doi:10.1016/j.jaut.2009.03.007
6. Invernizzi P, Pasini S, Selmi C, Gershwin ME, Podda M. Female predominance and X chromosome defects in autoimmune diseases. J Autoimmun (2009) 33:12–6. doi:10.1016/j.jaut.2009.03.005
7. Angele MK, Schwacha MG, Ayala A, Chaudry IH. Effect of gender and sex hormones on immune responses following shock. Shock (2000) 14:81–90. doi:10.1097/00024382-200014020-00001
8. Kovacs EJ, Messingham KA, Gregory MS. Estrogen regulation of immune responses after injury. Mol Cell Endocrinol (2002) 193:129–35. doi:10.1016/S0303-7207(02)00106-5
9. Choudhry MA, Bland KI, Chaudry IH. Gender and susceptibility to sepsis following trauma. Endocr Metab Immune Disord Drug Targets (2006) 6:127–35. doi:10.2174/187153006777442422
10. Offner PJ, Moore EE, Biffl WL. Male gender is a risk factor for major infections after surgery. Arch Surg (1999) 134:935–8. doi:10.1001/archsurg.134.9.935
11. Wichmann MW, Inthorn D, Andress HJ, Schildberg FW. Incidence and mortality of severe sepsis in surgical intensive care patients: the influence of patient gender on disease process and outcome. Intensive Care Med (2000) 26:167–72. doi:10.1007/s001340050041
12. Schroder J, Kahlke V, Book M, Stuber F. Gender differences in sepsis: genetically determined? Shock (2000) 14:307–10. doi:10.1097/00024382-200014030-00011
13. Sperry JL, Friese RS, Frankel HL, West MA, Cuschieri J, Moore EE, et al. Male gender is associated with excessive IL-6 expression following severe injury. J Trauma (2008) 64:572–8. doi:10.1097/TA.0b013e3181650fdf
14. Deitch EA, Livingston DH, Lavery RF, Monaghan SF, Bongu A, Machiedo GW. Hormonally active women tolerate shock-trauma better than do men: a prospective study of over 4000 trauma patients. Ann Surg (2007) 246:447–53. doi:10.1097/SLA.0b013e318148566
15. Guidry CA, Swenson BR, Davies SW, Dossett LA, Popovsky KA, Bonatti H, et al. Sex- and diagnosis-dependent differences in mortality and admission cytokine levels among patients admitted for intensive care. Crit Care Med (2014) 42:1110–20. doi:10.1097/CCM.0000000000000139
16. Spolarics Z. In gender-based outcomes, sex hormones may be important but it is in the genes. Crit Care Med (2014) 42:1294–5. doi:10.1097/CCM.0000000000000268
17. Kawasaki T, Chaudry IH. The effects of estrogen on various organs: therapeutic approach for sepsis, trauma, and reperfusion injury. Part 2: liver, intestine, spleen, and kidney. J Anesth (2012) 26:892–9. doi:10.1007/s00540-012-1426-2
18. Mostafa G, Huynh T, Sing RF, Miles WS, Norton HJ, Thomason MH. Gender-related outcomes in trauma. J Trauma (2002) 53(3):430–4. doi:10.1097/01.TA.0000022347.85881.A8
19. Haider AH, Crompton JG, Chang DC, Efron DT, Haut ER, Handly N, et al. Evidence of hormonal basis for improved survival among females with trauma-associated shock: an analysis of the National Trauma Data Bank. J Trauma (2010) 69:537–40. doi:10.1097/TA.0b013e3181efc67b
20. Migeon BR. The role of X inactivation and cellular mosaicism in women’s health and sex-specific diseases. JAMA (2006) 295:1428–33. doi:10.1001/jama.295.12.1428
21. Migeon BR. Why females are mosaics, X-chromosome inactivation, and sex differences in disease. Gend Med (2007) 4:97–105. doi:10.1016/S1550-8579(07)80024-6
22. Sperry JL, Vodovotz Y, Ferrell RE, Namas R, Chai YM, Feng QM, et al. Racial disparities and sex-based outcomes differences after severe injury. J Am Coll Surg (2012) 214:973–80. doi:10.1016/j.jamcollsurg.2012.02.020
23. Papathanassoglou E, Middleton N, Benbenishty J, Williams G, Christofi MD, Hegadoren K. Systematic review of gender-dependent outcomes in sepsis. Nurs Crit Care (2017) 22:284–92. doi:10.1111/nicc.12280
24. Klein SL, Marriott I, Fish EN. Sex-based differences in immune function and responses to vaccination. Trans R Soc Trop Med Hyg (2015) 109:9–15. doi:10.1093/trstmh/tru167
25. Klein SL, Hodgson A, Robinson DP. Mechanisms of sex disparities in influenza pathogenesis. J Leukoc Biol (2012) 92:67–73. doi:10.1189/jlb.0811427
26. Maggio M, Ceda GP, Lauretani F, Bandinelli S, Metter EJ, Artoni A, et al. Estradiol and inflammatory markers in older men. J Clin Endocrinol Metab (2009) 94(2):518–22. doi:10.1210/jc.2008-0940
27. Weitzmann MN, Pacifici R. Estrogen regulation of immune cell bone interactions. Ann N Y Acad Sci (2006) 1068:256–74. doi:10.1196/annals.1346.030
28. Liu PY, Death AK, Handelsman DJ. Androgens and cardiovascular disease. Endocr Rev (2003) 24:313–40. doi:10.1210/er.2003-0005
29. Chen CC, Parker CR Jr. Adrenal androgens and the immune system. Semin Reprod Med (2004) 22:369–77. doi:10.1055/s-2004-861553
30. Aube H, Milan C, Blettery B. Risk factors for septic shock in the early management of bacteremia. Am J Med (1992) 93:283–8. doi:10.1016/0002-9343(92)90234-3
31. Barrow RE, Herndon DN. Thermal burns, gender, and survival. Lancet (1988) 2:1076–7. doi:10.1016/S0140-6736(88)90093-1
32. Angus DC, Wax RS. Epidemiology of sepsis: an update. Crit Care Med (2001) 29:S109–16. doi:10.1097/00003246-200107001-00035
33. Barrow RE, Herndon DN. Incidence of mortality in boys and girls after severe thermal burns. Surg Gynecol Obstet (1990) 170:295–8.
34. Wells JC. Natural selection and sex differences in morbidity and mortality in early life. J Theor Biol (2000) 202:65–76. doi:10.1006/jtbi.1999.1044
35. Libert C, Dejager L, Pinheiro I. The X chromosome in immune functions: when a chromosome makes the difference. Nat Rev Immunol (2010) 10:594–604. doi:10.1038/nri2815
36. Adrie C, Azoulay E, Francais A, Clec’h C, Darques L, Schwebel C, et al. Influence of gender on the outcome of severe sepsis: a reappraisal. Chest (2007) 132:1786–93. doi:10.1378/chest.07-0420
37. Crimmins EM, Hayward MD, Saito Y. Differentials in active life expectancy in the older population of the United States. J Gerontol B Psychol Sci Soc Sci (1996) 51:S111–20. doi:10.1093/geronb/51B.3.S111
38. Abdelfattah KR, Gatson JW, Maass DL, Wolf SE, Minei JP, Wigginton JG. 17beta-Estradiol reappropriates mass lost to the hypermetabolic state in thermally injured rats. J Surg Res (2013) 181:136–41. doi:10.1016/j.jss.2012.06.010
39. Angele MK, Pratschke S, Hubbard WJ, Chaudry IH. Gender differences in sepsis: cardiovascular and immunological aspects. Virulence (2014) 5:12–9. doi:10.4161/viru.26982
40. Sperry JL, Nathens AB, Frankel HL, Vanek SL, Moore EE, Maier RV, et al. Characterization of the gender dimorphism after injury and hemorrhagic shock: are hormonal differences responsible? Crit Care Med (2008) 36:1838–45. doi:10.1097/CCM.0b013e3181760c14
41. Redei E, Li L, Halasz I, McGivern RF, Aird F. Fast glucocorticoid feedback inhibition of ACTH secretion in the ovariectomized rat: effect of chronic estrogen and progesterone. Neuroendocrinology (1994) 60:113–23. doi:10.1159/000126741
42. Sharma A, Aoun P, Wigham J, Weist S, Veldhuis JD. Gender determines ACTH recovery from hypercortisolemia in healthy older humans. Metabolism (2013) 62:1819–29. doi:10.1016/j.metabol.2013.08.014
43. Plechner AJ. Cortisol abnormality as a cause of elevated estrogen and immune destabilization: insights for human medicine from a veterinary perspective. Med Hypotheses (2004) 62:575–81. doi:10.1016/j.mehy.2003.12.005
44. Sharma AN, Aoun P, Wigham JR, Weist SM, Veldhuis JD. Estradiol, but not testosterone, heightens cortisol-mediated negative feedback on pulsatile ACTH secretion and ACTH approximate entropy in unstressed older men and women. Am J Physiol Regul Integr Comp Physiol (2014) 306:R627–35. doi:10.1152/ajpregu.00551.2013
45. Adams TE, Sakurai H, Adams BM. Effect of stress-like concentrations of cortisol on estradiol-dependent expression of gonadotropin-releasing hormone receptor in orchidectomized sheep. Biol Reprod (1999) 60:164–8. doi:10.1095/biolreprod60.1.164
46. Fang Y, Zhang L, Zhou GQ, Wang ZF, Zeng ZS, Luo ZY, et al. Functional polymorphism in exon 5 and variant haplotype of the interleukin-1 receptor-associated kinase 1 gene are associated with susceptibility to and severity of sepsis in the Chinese population. Chin Med J (Engl) (2011) 124:2248–53.
47. Forsdyke DR. X chromosome reactivation perturbs intracellular self/not-self discrimination. Immunol Cell Biol (2009) 87:525–8. doi:10.1038/icb.2009.39
48. Spolarics Z. The X-files of inflammation: cellular mosaicism of X-linked polymorphic genes and the female advantage in the host response to injury and infection. Shock (2007) 27:597–604. doi:10.1097/SHK.0b013e31802e40bd
49. Chandra R, Federici S, Hasko G, Deitch EA, Spolarics Z. Female X-chromosome mosaicism for gp91phox expression diversifies leukocyte responses during endotoxemia. Crit Care Med (2010) 38:2003–10. doi:10.1097/CCM.0b013e3181eb9ed6
50. Chandra R, Federici S, Nemeth ZH, Horvath B, Pacher P, Hasko G, et al. Female X-chromosome mosaicism for NOX2 deficiency presents unique inflammatory phenotype and improves outcome in polymicrobial sepsis. J Immunol (2011) 186:6465–73. doi:10.4049/jimmunol.1100205
51. Chandra R, Federici S, Nemeth ZH, Csoka B, Thomas JA, Donnelly R, et al. Cellular mosaicism for X-linked polymorphisms and IRAK1 expression presents a distinct phenotype and improves survival following sepsis. J Leukoc Biol (2014) 95:497–507. doi:10.1189/jlb.0713397
52. Pena G, Michalski C, Donnelly RJ, Qin Y, Sifri ZC, Mosenthal AC, et al. Trauma-induced acute X chromosome skewing in white blood cells represents an immuno-modulatory mechanism unique to females and a likely contributor to sex-based outcome differences. Shock (2017) 47:402–8. doi:10.1097/SHK.0000000000000764
53. Schaffner SF. The X chromosome in population genetics. Nat Rev Genet (2004) 5:43–51. doi:10.1038/nrg1247
54. Sabeti PC, Schaffner SF, Fry B, Lohmueller J, Varilly P, Shamovsky O, et al. Positive natural selection in the human lineage. Science (2006) 312:1614–20. doi:10.1126/science.1124309
55. Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature (2005) 434:400–4. doi:10.1038/nature03479
56. Goto T, Monk M. Regulation of X-chromosome inactivation in development in mice and humans. Microbiol Mol Biol Rev (1998) 62:362–78.
57. Heard E, Disteche CM. Dosage compensation in mammals: fine-tuning the expression of the X chromosome. Genes Dev (2006) 20:1848–67. doi:10.1101/gad.1422906
58. Latham KE. X chromosome imprinting and inactivation in preimplantation mammalian embryos. Trends Genet (2005) 21:120–7. doi:10.1016/j.tig.2004.12.003
59. Gunter C. Genome biology: she moves in mysterious ways. Nature (2005) 434:279–80. doi:10.1038/434279a
60. Abkowitz JL, Taboada M, Shelton GH, Catlin SN, Guttorp P, Kiklevich JV. An X chromosome gene regulates hematopoietic stem cell kinetics. Proc Natl Acad Sci U S A (1998) 95:3862–6. doi:10.1073/pnas.95.7.3862
61. Hatakeyama C, Anderson CL, Beever CL, Penaherrera MS, Brown CJ, Robinson WP. The dynamics of X-inactivation skewing as women age. Clin Genet (2004) 66:327–32. doi:10.1111/j.1399-0004.2004.00310.x
62. Brown CJ. Skewed X-chromosome inactivation: cause or consequence? J Natl Cancer Inst (1999) 91:304–5. doi:10.1093/jnci/91.4.304
63. Busque L, Paquette Y, Provost S, Roy DC, Levine RL, Mollica L, et al. Skewing of X-inactivation ratios in blood cells of aging women is confirmed by independent methodologies. Blood (2009) 113:3472–4. doi:10.1182/blood-2008-12-195677
64. Minks J, Robinson WP, Brown CJ. A skewed view of X chromosome inactivation. J Clin Invest (2008) 118:20–3. doi:10.1172/JCI34470
65. van Dijk JP, Heuver L, Stevens-Linders E, Jansen JH, Mensink EJ, Raymakers RA, et al. Acquired skewing of lyonization remains stable for a prolonged period in healthy blood donors. Leukemia (2002) 16:362–7. doi:10.1038/sj.leu.2402379
66. Clayton DG. Sex chromosomes and genetic association studies. Genome Med (2009) 1:110. doi:10.1186/gm110
67. Ross MT, Grafham DV, Coffey AJ, Scherer S, McLay K, Muzny D, et al. The DNA sequence of the human X chromosome. Nature (2005) 434:325–37. doi:10.1038/nature03440
68. Sperry JL, Zolin S, Zuckerbraun BS, Vodovotz Y, Namas R, Neal MD, et al. X chromosome-linked IRAK-1 polymorphism is a strong predictor of multiple organ failure and mortality postinjury. Ann Surg (2014) 260:698–703. doi:10.1097/SLA.0000000000000918
69. Toubiana J, Courtine E, Pene F, Viallon V, Asfar P, Daubin C, et al. IRAK1 functional genetic variant affects severity of septic shock. Crit Care Med (2010) 38:2287–94. doi:10.1097/CCM.0b013e3181f9f9c7
70. Arcaroli J, Silva E, Maloney JP, He Q, Svetkauskaite D, Murphy JR, et al. Variant IRAK-1 haplotype is associated with increased nuclear factor-kappaB activation and worse outcomes in sepsis. Am J Respir Crit Care Med (2006) 173:1335–41. doi:10.1164/rccm.200603-341OC
71. O’Neill LA. How toll-like receptors signal: what we know and what we don’t know. Curr Opin Immunol (2006) 18:3–9. doi:10.1016/j.coi.2005.11.012
72. Lye E, Mirtsos C, Suzuki N, Suzuki S, Yeh WC. The role of interleukin 1 receptor-associated kinase-4 (IRAK-4) kinase activity in IRAK-4-mediated signaling. J Biol Chem (2004) 279:40653–8. doi:10.1074/jbc.M402666200
73. Janssens S, Beyaert R. Functional diversity and regulation of different interleukin-1 receptor-associated kinase (IRAK) family members. Mol Cell (2003) 11:293–302. doi:10.1016/S1097-2765(03)00053-4
74. Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev (2004) 18:2195–224. doi:10.1101/gad.1228704
75. Gottipati S, Rao NL, Fung-Leung WP. IRAK1: a critical signaling mediator of innate immunity. Cell Signal (2008) 20:269–76. doi:10.1016/j.cellsig.2007.08.009
76. Huang Y, Li T, Sane DC, Li L. IRAK1 serves as a novel regulator essential for lipopolysaccharide-induced interleukin-10 gene expression. J Biol Chem (2004) 279:51697–703. doi:10.1074/jbc.M410369200
77. Thomas JA, Allen JL, Tsen M, Dubnicoff T, Danao J, Liao XC, et al. Impaired cytokine signaling in mice lacking the IL-1 receptor-associated kinase. J Immunol (1999) 163:978–84.
78. Maitra U, Deng H, Glaros T, Baker B, Capelluto DG, Li Z, et al. Molecular mechanisms responsible for the selective and low-grade induction of proinflammatory mediators in murine macrophages by lipopolysaccharide. J Immunol (2012) 189:1014–23. doi:10.4049/jimmunol.1200857
79. Chandra R, Federici S, Bishwas T, Nemeth ZH, Deitch EA, Thomas JA, et al. IRAK1-dependent signaling mediates mortality in polymicrobial sepsis. Inflammation (2013) 36:1503–12. doi:10.1007/s10753-013-9692-1
80. Verdrengh M, Thomas JA, Hultgren OH. IL-1 receptor-associated kinase 1 mediates protection against Staphylococcus aureus infection. Microbes Infect (2004) 6:1268–72. doi:10.1016/j.micinf.2004.08.009
81. Deng C, Radu C, Diab A, Tsen MF, Hussain R, Cowdery JS, et al. IL-1 receptor-associated kinase 1 regulates susceptibility to organ-specific autoimmunity. J Immunol (2003) 170:2833–42. doi:10.4049/jimmunol.170.6.2833
82. Swantek JL, Tsen MF, Cobb MH, Thomas JA. IL-1 receptor-associated kinase modulates host responsiveness to endotoxin. J Immunol (2000) 164:4301–6. doi:10.4049/jimmunol.164.8.4301
83. Thomas JA, Tsen MF, White DJ, Horton JW. IRAK contributes to burn-triggered myocardial contractile dysfunction. Am J Physiol Heart Circ Physiol (2002) 283:H829–36. doi:10.1152/ajpheart.00416.2001
84. Thomas JA, Haudek SB, Koroglu T, Tsen MF, Bryant DD, White DJ, et al. IRAK1 deletion disrupts cardiac Toll/IL-1 signaling and protects against contractile dysfunction. Am J Physiol Heart Circ Physiol (2003) 285:H597–606. doi:10.1152/ajpheart.0655.2001
85. Liu G, Tsuruta Y, Gao Z, Park YJ, Abraham E. Variant IL-1 receptor-associated kinase-1 mediates increased NF-kappa B activity. J Immunol (2007) 179:4125–34. doi:10.4049/jimmunol.179.6.4125
86. Jacob CO, Zhu J, Armstrong DL, Yan M, Han J, Zhou XJ, et al. Identification of IRAK1 as a risk gene with critical role in the pathogenesis of systemic lupus erythematosus. Proc Natl Acad Sci U S A (2009) 106:6256–61. doi:10.1073/pnas.0901181106
87. Sanchez E, Comeau ME, Freedman BI, Kelly JA, Kaufman KM, Langefeld CD, et al. Identification of novel genetic susceptibility loci in African American lupus patients in a candidate gene association study. Arthritis Rheum (2011) 63:3493–501. doi:10.1002/art.30563
88. Kaufman KM, Zhao J, Kelly JA, Hughes T, Adler A, Sanchez E, et al. Fine mapping of Xq28: both MECP2 and IRAK1 contribute to risk for systemic lupus erythematosus in multiple ancestral groups. Ann Rheum Dis (2013) 72:437–44. doi:10.1136/annrheumdis-2012-201851
89. Ishida R, Emi M, Ezura Y, Iwasaki H, Yoshida H, Suzuki T, et al. Association of a haplotype (196Phe/532Ser) in the interleukin-1-receptor-associated kinase (IRAK1) gene with low radial bone mineral density in two independent populations. J Bone Miner Res (2003) 18:419–23. doi:10.1359/jbmr.2003.18.3.419
90. Margery-Muir AA, Bundell C, Nelson D, Groth DM, Wetherall JD. Gender balance in patients with systemic lupus erythematosus. Autoimmun Rev (2017) 16:258–68. doi:10.1016/j.autrev.2017.01.007
91. Yacoub Wasef SZ. Gender differences in systemic lupus erythematosus. Gend Med (2004) 1:12–7. doi:10.1016/S1550-8579(04)80006-8
92. Long H, Yin H, Wang L, Gershwin ME, Lu Q. The critical role of epigenetics in systemic lupus erythematosus and autoimmunity. J Autoimmun (2016) 74:118–38. doi:10.1016/j.jaut.2016.06.020
93. Wang J, Syrett CM, Kramer MC, Basu A, Atchison ML, Anguera MC. Unusual maintenance of X chromosome inactivation predisposes female lymphocytes for increased expression from the inactive X. Proc Natl Acad Sci U S A (2016) 113:E2029–38. doi:10.1073/pnas.1520113113
94. Guo Y, Chai Q, Zhao Y, Li P, Qiao J, Huang J. Increased activation of toll-like receptors-7 and -8 of peripheral blood mononuclear cells and upregulated serum cytokines in patients with pediatric systemic lupus erythematosus. Int J Clin Exp Med (2015) 8:20472–80.
95. Lee YH, Choi SJ, Ji JD, Song GG. Association between toll-like receptor polymorphisms and systemic lupus erythematosus: a meta-analysis update. Lupus (2016) 25:593–601. doi:10.1177/0961203315622823
96. Gantier MP, Irving AT, Kaparakis-Liaskos M, Xu D, Evans VA, Cameron PU, et al. Genetic modulation of TLR8 response following bacterial phagocytosis. Hum Mutat (2010) 31:1069–79. doi:10.1002/humu.21321
97. Nilsson D, Andiappan AK, Hallden C, De YW, Sall T, Tim CF, et al. Toll-like receptor gene polymorphisms are associated with allergic rhinitis: a case control study. BMC Med Genet (2012) 13:66. doi:10.1186/1471-2350-13-66
98. Mackelprang RD, Bigham AW, Celum C, de BG, Beima-Sofie K, John-Stewart G, et al. Toll-like receptor polymorphism associations with HIV-1 outcomes among sub-Saharan Africans. J Infect Dis (2014) 209:1623–7. doi:10.1093/infdis/jit807
99. Xiao W, Liu Z, Lin J, Li J, Wu K, Ma Y, et al. Association of toll-like receptor 7 and 8 gene polymorphisms with Graves’ disease in Chinese Cantonese population. Tissue Antigens (2015) 85:29–34. doi:10.1111/tan.12479
100. Zhang Q, Qian FH, Yin XW, Cao Q, Bai JL, Du Q, et al. Associations of toll-like receptor 7 and 8 polymorphisms with asthma and asthma-related phenotypes in a Chinese Han population. Iran J Allergy Asthma Immunol (2015) 14:569–80.
101. Tormanen S, Korppi M, Terasjarvi J, Vuononvirta J, Koponen P, Helminen M, et al. Polymorphism in the gene encoding toll-like receptor 10 may be associated with asthma after bronchiolitis. Sci Rep (2017) 7:2956. doi:10.1038/s41598-017-03429-x
102. Wang JP, Liu P, Latz E, Golenbock DT, Finberg RW, Libraty DH. Flavivirus activation of plasmacytoid dendritic cells delineates key elements of TLR7 signaling beyond endosomal recognition. J Immunol (2006) 177:7114–21. doi:10.4049/jimmunol.177.10.7114
103. Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, et al. Recognition of single-stranded RNA viruses by toll-like receptor 7. Proc Natl Acad Sci U S A (2004) 101:5598–603. doi:10.1073/pnas.0400937101
104. Whitehorn J, Simmons CP. The pathogenesis of dengue. Vaccine (2011) 29:7221–8. doi:10.1016/j.vaccine.2011.07.022
105. Clifford HD, Yerkovich ST, Khoo SK, Zhang G, Upham J, Le Souef PN, et al. Toll-like receptor 7 and 8 polymorphisms: associations with functional effects and cellular and antibody responses to measles virus and vaccine. Immunogenetics (2012) 64:219–28. doi:10.1007/s00251-011-0574-0
106. Tato CM, Hunter CA. Host-pathogen interactions: subversion and utilization of the NF-kappa B pathway during infection. Infect Immun (2002) 70:3311–7. doi:10.1128/IAI.70.7.3311-3317.2002
107. Bubici C, Papa S, Pham CG, Zazzeroni F, Franzoso G. NF-kappaB and JNK: an intricate affair. Cell Cycle (2004) 3:1524–9. doi:10.4161/cc.3.12.1321
108. Wu CJ, Conze DB, Li T, Srinivasula SM, Ashwell JD. NEMO is a sensor of Lys 63-linked polyubiquitination and functions in NF-kappaB activation. Nat Cell Biol (2006) 8:398–406. doi:10.1038/ncb1384
109. Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell (2006) 22:245–57. doi:10.1016/j.molcel.2006.03.026
110. Burns KA, Martinon F. Inflammatory diseases: is ubiquitinated NEMO at the hub? Curr Biol (2004) 14:R1040–2. doi:10.1016/j.cub.2004.11.040
111. Bartek J, Lukas J. Cell biology. The stress of finding NEMO. Science (2006) 311:1110–1. doi:10.1126/science.1124540
112. Wuerzberger-Davis SM, Nakamura Y, Seufzer BJ, Miyamoto S. NF-kappaB activation by combinations of NEMO SUMOylation and ATM activation stresses in the absence of DNA damage. Oncogene (2006) 26(5):641–51. doi:10.1038/sj.onc.1209815
113. Mabb AM, Wuerzberger-Davis SM, Miyamoto S. PIASy mediates NEMO sumoylation and NF-kappaB activation in response to genotoxic stress. Nat Cell Biol (2006) 8(9):986–93. doi:10.1038/ncb1458
114. Smahi A, Courtois G, Vabres P, Yamaoka S, Heuertz S, Munnich A, et al. Genomic rearrangement in NEMO impairs NF-kappaB activation and is a cause of incontinentia pigmenti. The International Incontinentia Pigmenti (IP) Consortium. Nature (2000) 405:466–72. doi:10.1038/35013114
115. Nelson DL. NEMO, NFkappaB signaling and incontinentia pigmenti. Curr Opin Genet Dev (2006) 16:282–8. doi:10.1016/j.gde.2006.04.013
116. Gray P, Dunne A, Brikos C, Jefferies CA, Doyle SL, O’Neill LA. MyD88 adapter-like (Mal) is phosphorylated by Bruton’s tyrosine kinase during TLR2 and TLR4 signal transduction. J Biol Chem (2006) 281:10489–95. doi:10.1074/jbc.M508892200
117. Jefferies CA, O’Neill LA. Bruton’s tyrosine kinase (Btk)-the critical tyrosine kinase in LPS signalling? Immunol Lett (2004) 92:15–22. doi:10.1016/j.imlet.2003.11.017
118. Vihinen M, Kwan SP, Lester T, Ochs HD, Resnick I, Valiaho J, et al. Mutations of the human BTK gene coding for bruton tyrosine kinase in X-linked agammaglobulinemia. Hum Mutat (1999) 13:280–5. doi:10.1002/(SICI)1098-1004(1999)13:4<280::AID-HUMU3>3.0.CO;2-L
119. Lindvall JM, Blomberg KE, Valiaho J, Vargas L, Heinonen JE, Berglof A, et al. Bruton’s tyrosine kinase: cell biology, sequence conservation, mutation spectrum, siRNA modifications, and expression profiling. Immunol Rev (2005) 203:200–15. doi:10.1111/j.0105-2896.2005.00225.x
120. Lopez-Granados E, Perez DD, Ferreira CA, Fontan CG, Garcia Rodriguez MC. A genotype-phenotype correlation study in a group of 54 patients with X-linked agammaglobulinemia. J Allergy Clin Immunol (2005) 116:690–7. doi:10.1016/j.jaci.2005.04.043
121. Oda JM, Hirata BK, Guembarovski RL, Watanabe MA. Genetic polymorphism in FOXP3 gene: imbalance in regulatory T-cell role and development of human diseases. J Genet (2013) 92:163–71. doi:10.1007/s12041-013-0213-7
122. He YQ, Bo Q, Yong W, Qiu ZX, Li YL, Li WM. FoxP3 genetic variants and risk of non-small cell lung cancer in the Chinese Han population. Gene (2013) 531:422–5. doi:10.1016/j.gene.2013.08.066
123. Jiang LL, Ruan LW. Association between FOXP3 promoter polymorphisms and cancer risk: a meta-analysis. Oncol Lett (2014) 8:2795–9. doi:10.3892/ol.2014.2585
124. Piao Z, Kim HJ, Choi JY, Hong CR, Lee JW, Kang HJ, et al. Effect of FOXP3 polymorphism on the clinical outcomes after allogeneic hematopoietic stem cell transplantation in pediatric acute leukemia patients. Int Immunopharmacol (2016) 31:132–9. doi:10.1016/j.intimp.2015.12.022
125. Zhang DG, Xia XP, Wu H, Lin XQ, Jiang LJ, Ding R, et al. [Association of ulcerative colitis with fork head/winged helix transcription factor-3 gene polymorphisms in Chinese patients]. Zhonghua Nei Ke Za Zhi (2017) 56:188–93. doi:10.3760/cma.j.issn.0578-1426.2017.03.008
126. In JW, Lee N, Roh EY, Shin S, Park KU, Song EY. Association of aplastic anemia and FoxP3 gene polymorphisms in Koreans. Hematology (2017) 22:149–54. doi:10.1080/10245332.2016.1238645
127. Koukouikila-Koussounda F, Ntoumi F, Ndounga M, Tong HV, Abena AA, Velavan TP. Genetic evidence of regulatory gene variants of the STAT6, IL10R and FOXP3 locus as a susceptibility factor in uncomplicated malaria and parasitaemia in Congolese children. Malar J (2013) 12:9. doi:10.1186/1475-2875-12-9
128. Spolarics Z. Endotoxemia, pentose cycle, and the oxidant/antioxidant balance in the hepatic sinusoid. J Leukoc Biol (1998) 63:534–41.
129. Luzzatto L, Mehta A. Glucose 6 phosphate dehydrogenase deficiency. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. New York, NY: McGrawHill (1995). p. 3367–98.
130. Beutler E. G6PD: population genetics and clinical manifestations. Blood Rev (1996) 10:45–52. doi:10.1016/S0268-960X(96)90019-3
131. Mordmuller B, Turrini F, Long H, Kremsner PG, Arese P. Neutrophils and monocytes from subjects with the Mediterranean G6PD variant: effect of Plasmodium falciparum hemozoin on G6PD activity, oxidative burst and cytokine production. Eur Cytokine Netw (1998) 9:239–45.
132. Wilmanski J, Siddiqi M, Deitch EA, Spolarics Z. Augmented IL-10 production and redox-dependent signaling pathways in glucose-6-phosphate dehydrogenase-deficient mouse peritoneal macrophages. J Leukoc Biol (2005) 78:85–94. doi:10.1189/jlb.0105010
133. Spolarics Z, Siddiqi M, Siegel JH, Garcia ZC, Stein DS, Ong H, et al. Increased incidence of sepsis and altered monocyte functions in severely injured type A- glucose-6-phosphate dehydrogenase-deficient African American trauma patients. Crit Care Med (2001) 29:728–36. doi:10.1097/00003246-200104000-00005
134. Koker MY, Sanal O, de Boer M, Tezcan I, Metin A, Tan C, et al. Skewing of X-chromosome inactivation in three generations of carriers with X-linked chronic granulomatous disease within one family. Eur J Clin Invest (2006) 36:257–64. doi:10.1111/j.1365-2362.2006.01619.x
135. Stasia MJ, Bordigoni P, Floret D, Brion JP, Bost-Bru C, Michel G, et al. Characterization of six novel mutations in the CYBB gene leading to different sub-types of X-linked chronic granulomatous disease. Hum Genet (2005) 116:72–82. doi:10.1007/s00439-004-1208-5
136. Wolach B, Ash S, Gavrieli R, Stark B, Yaniv I, Roos D. Acute lymphoblastic leukemia in a patient with chronic granulomatous disease and a novel mutation in CYBB: first report. Am J Hematol (2005) 80:50–4. doi:10.1002/ajh.20424
137. Rump A, Rosen-Wolff A, Gahr M, Seidenberg J, Roos C, Walter L, et al. A splice-supporting intronic mutation in the last bp position of a cryptic exon within intron 6 of the CYBB gene induces its incorporation into the mRNA causing chronic granulomatous disease (CGD). Gene (2006) 371:174–81. doi:10.1016/j.gene.2005.11.036
138. von Goessel H, Hossle JP, Seger R, Gungor T. Characterization of 17 new cases of X-linked chronic granulomatous disease with seven novel mutations in the CYBB gene. Exp Hematol (2006) 34:528–35. doi:10.1016/j.exphem.2006.01.005
139. Pinheiro I, Dejager L, Libert C. X-chromosome-located microRNAs in immunity: might they explain male/female differences? The X chromosome-genomic context may affect X-located miRNAs and downstream signaling, thereby contributing to the enhanced immune response of females. Bioessays (2011) 33:791–802. doi:10.1002/bies.201100047
Keywords: sexual dimorphism, X chromosome inactivation, cellular mosaicism, X chromosome skewing, infection, injury, sepsis
Citation: Spolarics Z, Peña G, Qin Y, Donnelly RJ and Livingston DH (2017) Inherent X-Linked Genetic Variability and Cellular Mosaicism Unique to Females Contribute to Sex-Related Differences in the Innate Immune Response. Front. Immunol. 8:1455. doi: 10.3389/fimmu.2017.01455
Received: 12 September 2017; Accepted: 18 October 2017;
Published: 13 November 2017
Edited by:
Georges Jacques Casimir, Free University of Brussels, BelgiumReviewed by:
Hiroyuki Oshiumi, Kumamoto University, JapanJames R. Whiteford, Queen Mary University of London, United Kingdom
Rodrigo Tinoco Figueiredo, Universidade Federal do Rio de Janeiro, Brazil
Copyright: © 2017 Spolarics, Peña, Qin, Donnelly and Livingston. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zoltan Spolarics, c3BvbGFyaWNAbmptcy5ydXRnZXJzLmVkdQ==