 Julia Kolter1,2
Julia Kolter1,2 Philipp Henneke1,3*
Philipp Henneke1,3*
- 1Center for Chronic Immunodeficiency (CCI), Medical Center – University of Freiburg, Faculty of Medicine, University of Freiburg, Freiburg, Germany
- 2Faculty of Biology, University of Freiburg, Freiburg, Germany
- 3Center for Pediatrics and Adolescent Medicine, Medical Center – University of Freiburg, Freiburg, Germany
The pathogenesis of neonatal late-onset sepsis (LOD), which manifests between the third day and the third month of life, remains poorly understood. Group B Streptococcus (GBS) is the most important cause of LOD in infants without underlying diseases or prematurity and the third most frequent cause of meningitis in the Western world. On the other hand, GBS is a common intestinal colonizer in infants. Accordingly, despite its adaption to the human lower gastrointestinal tract, GBS has retained its potential virulence and its transition from a commensal to a dangerous pathogen is unpredictable in the individual. Several cellular innate immune mechanisms, in particular Toll-like receptors, the inflammasome and the cGAS pathway, are engaged by GBS effectors like nucleic acids. These are likely to impact on the GBS-specific host resistance. Given the long evolution of streptococci as a normal constituent of the human microbiota, the emergence of GBS as the dominant neonatal sepsis cause just about 50 years ago is remarkable. It appears that intensive usage of tetracycline starting in the 1940s has been a selection advantage for the currently dominant GBS clones with superior adhesive and invasive properties. The historical replacement of Group A by Group B streptococci as a leading neonatal pathogen and the higher frequency of other β-hemolytic streptococci in areas with low GBS prevalence suggests the existence of a confined streptococcal niche, where locally competing streptococcal species are subject to environmental and immunological selection pressure. Thus, it seems pivotal to resolve neonatal innate immunity at mucous surfaces and its impact on microbiome composition and quality, i.e., genetic heterogeneity and metabolism, at the microanatomical level. Then, designer pro- and prebiotics, such as attenuated strains of GBS, and oligonucleotide priming of mucosal immunity may unfold their potential and facilitate adaptation of potentially hazardous streptococci as part of a beneficial local microbiome, which is stabilized by mucocutaneous innate immunity.
Introduction
Neonatal sepsis occurs as two distinct clinical entities either in the first 72 h of life as early-onset disease (EOD), resulting from in utero or intrapartum infection, or during the following 3 months as late-onset sepsis (LOD). In both cases, the Gram-positive, β-hemolytic Group B Streptococcus (GBS) is one of the most prevalent bacterial species in blood and cerebrospinal fluid. As a consequence, pregnant women undergo routine or targeted screening for GBS in the last third of pregnancy in many Western European countries and the USA. In case of positive testing, women receive preventive intrapartum antibiotics during delivery (1). Since approximately 20–30% of all pregnant women are colonized, this prevention strategy affects an estimated 1 million women every year in the US alone. In other countries such as the Netherlands, a risk-based approach has been adopted, i.e., antibiotics are only administered in case of additional risk factors such as premature labor, intrapartum fever, bacteriuria, prolonged membrane rupture or previous children with GBS disease.
Before the use of antibiotic prophylaxis, the GBS sepsis incidence exceeded 1 in 1,000 children with high case fatality rates (2, 3). The role of GBS in neonatal sepsis may be due to (i) it being one of the most prevalent colonizers of the birth canal and thus among the first bacteria to get into contact with the newborn (4, 5), (ii) GBS carrying highly invasive properties, and (iii) a particular neonatal immunopathology induced by GBS. In EOD, the size and deposition site, e.g., the lung, of the GBS inoculum may be decisive factors. However, it is unresolved why GBS establishes as a harmless mucocutaneous colonizer in approximately 10% of infants in the first weeks of life, and overcomes epithelial barriers and cellular innate immunity only in less than one in thousand infants to cause LOD. In other words, it remains a puzzle which specific factors at the level of mucosal immunity and the local microbiome allow GBS to leave its colonizing niche, thus facilitating invasion in the individual child.
At the beginning of life, the developmental lines of the microbiota and of the local cellular innate immunity have to run with substantial interdependence. Both areas are subject to factors in cis and in trans, i.e., specific bacteria are influenced by the microbiota and by host immunity, and host cells are modulated by other host and microbial cells (6). In order to guarantee long-term ecologic stability, adaptation on either side of the host–microbe interface is required, both at the population level and in the individual cell. The putative contribution of variations in specific innate immune genes to neonatal sepsis has recently been discussed (7). The authors suggested that affected children may suffer from yet to be identified minor primary immunodeficiency. This is a tempting hypothesis, given the enormous gain in knowledge on single gene alterations leading to susceptibility to a narrow spectrum of microorganisms. On the other hand, there is no indication for inheritance of a specific neonatal sepsis risk. Moreover, LOD typically remains the only “suspicious” episode in the individual infection biography. Finally, preterm birth is a well-recognized risk factor of GBS sepsis. In preterm infants, several factors impact on the individual codevelopment of microbiota and immunity, in particular cesarean section and formula feeding, which modify the microbiome (8, 9), and antibiotic usage, which affects both the microbiome and myeloid cell development (10, 11).
The hypothesis underlying this review holds that aberrations in the codevelopment of microbiota and host immunity, rather than genetic variations in immune genes alone, shape the individual risk for neonatal GBS sepsis, in particular LOD.
GBS: Colonization and Virulence Factors
Neonatal GBS sepsis is a global problem with an overall incidence of around 0.5/1,000 live births. In contrast to the situation in Europe, American and African countries, GBS are reported to be a rare cause of neonatal colonization and sepsis in Southeast Asia (12, 13). However, the epidemiology in developing countries often suffers from constraints related to early deaths outside hospitals and low microbiological sensitivity of detection methods (13). In many, but not all Western European and North American countries, intrapartum antibiotic prophylaxis (IAP) has been associated with a decreased incidence of EOD while LOD rates remained unchanged (14–16). Notably, a substantial proportion of mothers whose infants developed EOD were tested negative before birth (1). It is unclear whether this phenomenon is due to false-negative test results or very recent GBS acquisition. Although, as outlined above, incidence and fatality rates are significantly higher in preterm than term infants (16–18), most cases occur in term infants (1) without clinical or laboratory evidence for immunodeficiency. LOD alone has an incidence of about 0.3–0.4 per 1,000 children and can develop randomly within the first 3 months after birth (19). It manifests more frequently as meningitis than EOD (17, 20). Conceptionally, these observations indicate that EOD and LOD originate from distinct biological processes or disturbances thereof.
Group B streptococcus is classified into 10 serotypes based on chemical structure and conformation of capsular polysaccharides. Serotyping relies on latex agglutination or multiplex PCR (21). In the past 30 years about 50% of the reported neonatal GBS sepsis cases worldwide were caused by serotype III strains (13). This indicates a considerable genetic homogeneity and stability in the pathogenic potential of GBS despite antibiotic selection pressure. Notably, Islam et al. did not detect any colonization by GBS of serotype III in their cohort of more than 600 infants in Bangladesh, while 6% of all infants were colonized by other serotypes, predominantly VII and Ia (22). It is very plausible yet uncertain that low circulation of highly invasive GBS strains underlies the low incidence of invasive neonatal GBS in several Asian countries (13).
In addition to the serotypes, GBS can be further classified by multilocus sequence typing, with more than 700 identified types (ST). The majority of human isolates belong to six clonal complexes (23, 24). EOD is significantly associated with serotype Ia strain ST-23 and closely related ST-24 as well as the ST-17 strain of serotype III (25, 26). LOD on the other hand is largely caused by ST-17 (20, 25). Moreover, ST-17 causes most cases of meningitis in EOD and LOD (27). In EOD, the distribution of invasive strains mainly corresponds to those colonizing the mothers (26). However, ST-17 shows an elevated disease-to-colonization ratio in EOD and LOD, i.e., it causes more cases of invasive disease than expected from its colonization rate of pregnant women (28–30). These observations, together with the characteristic expression of several virulence factors, have led to the term of a “hypervirulent” strain. Two of these factors, the hypervirulent GBS adhesin HvgA (27) and the serine-rich repeat glycoprotein Srr2 (31), are surface-anchored proteins which allow for adherence to epithelial cells and host plasma proteins. ST-17 strains also often carry the 2b pilus variant which contributes to invasion in mouse models (32, 33).
Most GBS strains produce surface-associated β-hemolysin which can damage membranes and promote barrier penetration (34). β-Hemolysin was found to be identical to the orange to red pigment of GBS, an ornithine rhamnolipid called granadaene (35). Both factors rely on the cyl operon which is controlled by the CovR/S two-component system. Strains mutated in CovR/S show hyperhemolysis and increased virulence (34, 35). For further detailed descriptions about GBS virulence factors, we refer to recent reviews (36, 37).
Routes of Infection
In EOD, GBS is usually transmitted from the colonized maternal vaginal tract during birth to the infant. Aspiration of contaminated fluids allows for bacterial entry via the respiratory tract in many cases, resulting in sepsis or pneumonia during the first days of life (38). The route of infection in LOD is less well understood. The gastrointestinal tract is considered to be a natural reservoir for sepsis pathogens in neonates (39, 40). GBS shares this niche with Escherichia coli, the second typical organism in neonatal sepsis. Yet, the point of time when GBS establishes colonization is highly variable. 50–70% of colonized mothers transfer GBS to their offspring during delivery (41, 42) and 50% of infants which later developed LOD were colonized with GBS at birth (43). It remains unknown how many of these infants were stably colonized between the first contact with GBS and the disease onset. Unfortunately, large-scale and longitudinal colonization data of mother-infant pairs before and after disease onset, which would allow resolving this LOD puzzle, are not available. In a case series, Carl et al. found that 7 out of 11 children with LOD by GBS, E. coli or Serratia marcescens produced at least one stool with the matching organism before bloodstream infection (39). However, only two infants with GBS sepsis contributed to this study and they showed a GBS positive stool only briefly before sepsis, indicating recent colonization or overgrowth in the gastrointestinal tract. Another longitudinal case study on LOD also found that GBS occurred in the stool 2 days before sepsis onset (44). In contrast, it has been shown for other infections, e.g., enterococcal or staphylococcal bloodstream infections, that children often have a pathogen-dominated gut flora before disease onset (44, 45). Thus, it is conceivable that GBS exposure constitutes a particular LOD risk to infants who failed to firmly establish GBS colonization after birth (46). However, it seems important to note that stool samples do not always adequately mirror the actual intestinal community (47).
Meningitis caused by serotype III strains is often linked to high-level bacteremia. Factors that enable serotype III strains to survive in the blood stream, i.e., escape of adaptive and innate immune mechanisms, such as antibody or complement-mediated phagocytosis may be responsible for this effect (48). While the route of infection has not been resolved with certainty in infants, several studies showed bacterial dissemination to the blood and CNS after intraperitoneal (49), subcutaneous (50, 51) and intragastral (27, 52) inoculation of GBS serotype III in neonatal mice and rats. ST-17 is also specifically found in cases of GBS meningitis after 3 months of age (53), indicating that this clonal complex has an increased capability of overcoming colonization site barriers and blood borne immunity and of invading the CNS.
The Neonatal Microbiome
The microbiome, defined as the microbial flora inhabiting the human body, constitutes an important factor in individual health and development. The composition of the microbiome is complex, distinct between individuals and subject to environmental changes and adaptation to host factors. Each body site contains a unique microbial community. Even within one niche such as the skin the composition varies depending on the exact location, i.e., the back skin shows a different microbial signature than the foot pad or the axillary vault (54). It seems self-evident that exposure to bacteria in the birth canal impacts on the colonizing flora in the infant. However, the fetus may be less sterile than thought, i.e., that the microbiome might develop already in utero. 16S rDNA sequencing of amniotic fluid, placenta samples and meconium revealed prenatal presence of bacteria with a predominance of Escherichia spp. (9, 55, 56). Of note, the Streptococcus genus was also detected in these samples, yet at very low abundance (56). Intrauterine colonization data have to be interpreted with some caution, since microbial viability is usually not confirmed and the risk of contamination is high in many of the investigated samples (57). Accordingly, the contribution of colonization in utero to microbiome development is still unclear, whereas that of colonization after rupture of fetal membranes is beyond doubt. As an example, vaginal delivery and cesarean section result in different bacterial communities on skin, nares, and gingiva (9). Yet, the impact of the delivery mode on the expansion and functional diversification after the first 6 weeks of life is surprisingly modest (9, 58). Instead, the infant’s microbiome follows a rather predictable successive colonization pattern and reaches a stable state resembling the adult microbiome already at 1–3 years of age (59–61). Oxygen abundance in the neonatal gut facilitates the colonization by facultative anaerobes, e.g., Lactobacillus and Streptococcus followed by Enterobacteriaceae. After oxygen is consumed and anaerobic conditions are established, obligate anaerobic species, e.g., Bifidobacterium, Bacteroides, and Clostridium spp. populate the intestine (62, 63). Administration of antibiotics, on the other hand, heavily affects the postnatal microbiome (8, 64, 65). Postnatal exposure to antibiotics alters the gut microbiome in the first 2–3 years of life by delaying microbiome development and altering phylogenetic diversity, e.g., affecting early colonization with Lactospiraceae spp. (8, 65). In addition, antibiotics reduce the stability of the microbiota composition as indicated by an increased variation between consecutive samples as compared to controls (65). Notably, very preterm infants with a gestational age of <33 weeks, who in many cases receive antibiotics within 24 h of birth, showed a 10-fold reduced bacterial diversity in comparison to term infants (66).
GBS as Part of the Human Microbiome
Streptococcus is, together with Lactobacillus, Staphylococcus, and Propionibacterium, one of the most commonly found bacterial genera in the neonatal intestine and oral cavity (9). Streptococcal species account for up to 10% of total bacteria in fecal samples during the first months of life (67–69). In pregnant women, GBS colonization is found in up to 30% of rectovaginal samples (28, 70, 71) and stable colonization with the same clone for several years has been demonstrated (4, 70). Spread from the gastrointestinal tract to the genital tract is considered to be a probable colonization sequence for GBS (4). Since strains might be lost or reacquired in relatively short time periods (72, 73), GBS screening is recommended relatively late in pregnancy, i.e., between gestational weeks 35 and 37 (74).
Colonization by GBS is not exclusively confined to humans. Instead, GBS was first described in the 1880s as a cause of mastitis in goats and cows and it is a frequent commensal in seals and fish (75, 76). Although rare, invasive GBS disease can be a zoonotic disease as outbreaks in adults have been linked to raw fish consumption (77). Moreover, the hypervirulent ST-17 strain, which emerged 40 years ago, shares greater genetic similarity with bovine than with many human strains, indicating that it originated from a bovine lineage. Therefore, GBS may—under very specific conditions—cross species barriers (28, 78). However, since virulent strains in humans are distinct from those causing disease in animals (26, 75), person-to-person transmission plays the primary role in human GBS dissemination. Data on GBS spread are largely confined to mother-infant pairs. In contrast, the contribution of fecal-oral transmission by other family members than the mother to GBS colonization of the infant remains unclear. While strains are largely shared between sexual partners (79, 80), cohabitation appears to play a minor role in transmission (81).
Intrapartum antibiotic prophylaxis during delivery may transiently increase the GBS colonization risk of the infant yet probably does not affect the relative abundance of Streptococcus spp. in the stool beyond the first few weeks of life (72). While a number of studies longitudinally analyzed the development of the microbiome after birth on the level of phylum, class or order, studies on species or even genus level, e.g., with a specific focus on Group A Streptococcus (GAS) or GBS are rare and do not allow for robust statements on this level of resolution. Infants which were tested negative for GBS after IAP administration frequently acquire maternal GBS strains at later time points (82). Breast milk is hence a probable source of GBS in LOD. Several LOD case studies detected GBS in breast milk (46, 83). However, it is often unclear whether GBS in breast milk results from maternal colonization or infant oropharyngeal contamination. Mutated strains from infants which have been detected in the maternal breast milk (84) support the latter hypothesis. On the other hand, positive cultures of breast milk correspond to heavy colonization of the newborn (82), which is in turn a risk factor for LOD, especially in the case of mastitis (18). Bacterial expansion in breast milk and subsequent uptake by the infant may favor heavy colonization and LOD recurrences. Finally, nosocomial GBS transmission can occur in the case of children with invasive devices (82), indicating again that LOD can be a smear infection in some cases.
Competing Microbes: GBS Needs to Find Its (Neonatal) Niche
Although GBS is the most prevalent streptococcal strain in neonatal sepsis, other streptococci, notably Groups A, D, and G, are isolated from blood cultures of newborns as well (22, 85, 86). Indeed, the connection of GBS and neonatal sepsis was only found in the 1960s and its predominance was established in the 1970s (24, 78). Prior to that, GAS and Streptococcus pneumoniae accounted for most neonatal sepsis cases (3, 87). As in other ecological niches, competition for nutrition and space occurs between bacterial species on colonized human body sites (88). Indeed, examples of mutual exclusion are found in the genus Streptococcus, e.g., in the case of Streptococcus mutans, the predominating cause of caries. The presence of other streptococcal species in the oral cavity, namely Streptococcus sanguinis and Streptococcus oligofermentans, is inversely correlated with the abundance of S. mutans which has been linked to the production of hydrogen peroxide in vitro (89, 90). Another example is the observation that Corynebacterium and Dolosigranulum in the upper respiratory tract are protective against colonization with Streptococcus pneumonia, which causes otitis media in infants after colonization of the airways (91). More importantly in the context of this review, growth of GBS is inhibited by Streptococcus salivarius both in vitro and in a vaginal colonization mouse model (92). Competitive growth was also shown for Bifidobacterium and GBS in vitro (93) and lactobacilli inhibited growth (94) and attachment of GBS to vaginal epithelial cells (95). In addition, Lactobacillus reuteri reduced vaginal colonization in a mouse model (96) and—importantly—as a probiotic in a placebo-controlled trial in pregnant women (97). These findings are in line with a very recent randomized, double-blind, placebo-controlled trial from Indian, where Lactobacillus plantarum plus fructooligosaccharide protected newborns from sepsis (98). In general, however, the presence of GBS appears not to be linked to an abnormal microbiome or a reduction of the predominant Lactobacillus genus in the vaginal tract of the mother (99–101). Interestingly, a small study found significant taxonomic differences in stools of 6-month infants, when mothers were GBS carriers, as compared to non-carriers (102). Yet, robust epidemiological evidence for a correlation of neonatal colonization with GBS and that of other specific intestinal commensals such as other streptococcal species is not existent.
Next to streptococci, staphylococci cause bacteremia and sepsis in newborns. Indeed, coagulase-negative staphylococci are the most common cause of nosocomial sepsis in newborns, yet do not play a role in healthy term infants. The generally more virulent S. aureus is isolated in variable frequency from neonatal blood cultures, but it is rarely found in cerebrospinal fluid (86). Furthermore, in view of the omnipresence of S. aureus as a colonizer in up to 50% of neonates, infants of this age group are not specifically susceptible to staphylococcal infections, unless they are subject to medical interventions such as indwelling catheters or surgery (85, 103). Hence, the contact with GBS and potentially other (beta-hemolytic) streptococci and the establishment of coexistence with these bacteria appears to impose a greater risk to the infant compared to other genii.
The Impact of Antibiotic Pressure and Resistance on LOD
The majority of GBS strains isolated from humans are resistant to the antibiotic tetracycline. Indeed, the insertion of tetracycline resistance (TcR) elements, i.e., the ribosomal protection proteins Tet(M) and Tet(O), in few GBS clones led to their selection and expansion after the onset of extensive tetracycline usage since 1948 (24). These clones have since replaced a prior diverse GBS population, concurrent with the rise of GBS as major cause of neonatal sepsis. Notably, TcR elements are the most widely spread resistance genes in the human gut microbiota (104). Moreover, a subset of GBS strains, especially ST-1, carry genes which confer general resistance to macrolids and lincosamides, i.e., the methylases erm(B) and erm(TR) (24). Resistance rates to clindamycin (lincosamid) and erythromycin (macrolide) range up to 30 and 50%, respectively (30, 71, 105, 106). A rise of resistance to fluoroquinolones has been described in serotype V strains (105, 107). In addition, GBS with reduced penicillin susceptibility due to mutations in the penicillin-binding proteins are isolated with increasing frequencies in Japan (108, 109) and were also reported to occur spontaneously in an American patient after prolonged penicillin treatment (110). In this context, it seems likely that the frequent use of antibiotics other than tetracyclines may also lead to selection of hypervirulent strains. In the Netherlands, the incidence of EOD caused by ST-17 has significantly increased after implementation of a risk-based approach of antibiotic prophylaxis (15). ST-17 strains are also significantly more prevalent in women with IAP as compared to other strains (72). Thus, a relatively short course of intrapartum antibiotics, usually penicillin and ampicillin, may allow for seeding and expansion of hypervirulent GBS strains, which may not affect the majority of infants but propagate LOD development in few colonized individuals.
In addition, the capsular serotypes of GBS are not fixed but subject to frequent exchange by conjugative transfer between strains, explaining for the diversity of serotypes within clonal complexes. Lately, serotype IV has emerged as a causative agent of adult GBS disease in the US (106, 111). This seems important, as serotype IV is not included in the latest efforts in vaccine development to capsular antigens of GBS. Sequencing has revealed that a predominating serotype IV strain acquired large genomic fragments by horizontal gene transfer from the hypervirulent ST-17 and ST-23 strains (112). Additionally, ST-17 strains with capsular switching to serotype IV have been identified in several countries (29, 113, 114). Since maternal antibodies can impact on colonization with the antibody-specific GBS strains in mothers and early infants (115–117), it remains an open question whether targeting certain serotypes may eventually select for strains which have acquired novel capsule genes and allow for their expansion.
Interestingly, single-nucleotide polymorphisms (SNPs) in virulence-associated genes were detected in neonatal invasive GBS strains in comparison to the respective colonizing strains from the mothers, possibly contributing to the transition from a maternal commensal to a neonatal pathogen (84). This suggests that mutations are positively selected for in the neonatal environment. Moreover, mutations in the virulence regulator CovR/S leading to hyperhemolytic activity were found in invasive isolates of women in preterm labor (35). The acquisition of antibiotic resistance, serotype switching and SNPs can therefore lead to microevolution in the individual newborn, which may explain the pathogenicity of GBS in only a very small number of infants.
The Role of Antibiotics and Dysbiosis in the Development of GBS Sepsis
The microbiota may have beneficial but also detrimental, acute, and chronic effects on infant health. Dysbiosis may predispose the neonatal intestine to inflammation (63) and facilitate the expansion of otherwise infrequent pathobionts (118, 119). Dysbiosis with lower bacterial diversity and decreased density of Propionibacterium spp. was found to precede the onset of necrotizing enterocolitis (NEC) (120, 121). Moreover, lactate-producing bacilli such as staphylococci and streptococci were reduced after birth in infants with NEC (68). Even though the increased prevalence of opportunistic pathogens such as uropathogenic E. coli (122) and Clostridium perfringens (68) has been linked to NEC, a common bacterial signature has not been found (121, 123). In addition, it is often unclear whether dysbiosis and the development of organ pathology are causally linked or whether they both depend on upstream disturbances, which may be diverse. Mai et al. found signs of dysbiosis in preterm infants already 2 weeks before onset of sepsis (124). Dysbiosis meant a delayed colonization with Proteobacteria and decreased density of Bifidobacteria spp. This observation receives support by the finding that Bifidobacterium spp. in the gut are protective for LOD (44), although the data on this issue are not fully consistent between studies (40). During sepsis, anaerobic Bacteroides and Bifidobacterium spp. were found to be decreased and aerobic Enterobacteria to be increased in affected infants as compared to non-septic twin controls (125). In view of these observations, a reduced intestinal Bifidobacterium density in infants whose mothers received IAP constitutes an important warning sign for the most careful usage of antibiotics in this sensitive period (93). In support of this notion, the risk for LOD caused by various pathogens including GBS in preterm infants is threefold higher after prolonged empirical antibiotic treatment (126). Antibiotics can affect the composition of the microbiome in many ways, including the depletion of competitive microbes, a delay in immune cell maturation (see below) and dysbiosis, all of which widen the niche for pathogenic bacteria.
Cellular Innate Immunity and Resistance to GBS
Group B streptococcus is also recognized as an important health threat in immunocompromised adults, i.e., the elderly and patients with diabetes mellitus or HIV infections. Notably, the most common manifestations are skin/soft tissue infections and bacteremia (127–129), indicating that in these patients barrier immunity is important for the normal containment of GBS, similar to the situation in infants. The immaturity of the neonatal immune system in comparison to that of the adult was reviewed in detail elsewhere (130–132) and we will therefore focus on selected GBS-related aspects.
Neonatal rodents show exquisite sensitivity for GBS. Neonatal rats succumb to doses as low as 10 CFU intraperitoneally, while adult rats require approximately 6-log higher inoculums for a similar mortality rate (49) even if their body weight is taken into account (50). Neonatal mice, which normally die after i.p. infection within 48 h, were protected by transfer of specific antiserum to the pregnant dam before delivery (133). This experimental data is in line with the protective role of maternal GBS antibodies in the protection from GBS EOD, which is the basis for the development of a maternal vaccine (36, 134). In contrast, the role of maternal antibodies in the prevention LOD development is less clear. Recently, it has been inferred that high antibody levels also prevent GBS colonization (42, 116, 117). Women with high serotype-specific titers had a significantly lower risk of rectovaginal colonization with the respective GBS strains (42). However, GBS antibody levels do not inversely correlate with the sepsis risk per se. Thus, it remains puzzling why only very few of the GBS exposed and/or colonized infants with low antibody levels develop LOD.
In the innate arm of the immune system, the family of Toll-like receptors (TLRs) is essential for the defense against invasive streptococcal infections. Children with genetic deficiency in MyD88, an essential adaptor for all TLRs but TLR3, or IRAK4, a kinase downstream of MyD88, have an approximately 50% risk of dying from invasive bacterial infections in the first 8 years of life. In most cases, streptococci are the causative organisms (135, 136). Furthermore, roughly one third of the affected children suffer from a sepsis episode in the first 3 months of life. Thus, the risk for early and late neonatal sepsis is approximately 1,000-fold higher in these infants than in newborn infants overall. It seems noteworthy that most isolates are either pneumococci or GAS, whereas only few cases of late neonatal sepsis and meningitis caused by GBS have been reported so far (135, 137). Whether this predominance of other streptococcal species is due to an altered microbiome in MyD88- and IRAK4-deficient individuals has not been explored so far. In mice with MyD88 deficiency, a gross deviation in microbiome composition cannot be observed (138, 139), although a generally increased risk for the invasion and dissemination of intestinal commensals was observed (140). Moreover, MyD88-deficient neonatal mice have not been studied in this context. The already exceptional susceptibility of neonatal mice for local GBS infections, with a 100,000-fold decreased LD90 (cfu/g bw) in 2-day-old mice as compared to adult mice, is further significantly increased in MyD88 deficiency (141, 142).
Within the MyD88-dependent TLR family, TLR2 activation by GBS lipoproteins (143, 144) and endosomal TLR-activation by single-stranded RNA are equally important. TLR13 is a common receptor of 16S rRNA from Gram-positive bacteria including GBS in mice (10, 145), whereas TLR8 is the incomplete analog in humans (146–148). TLR recognition by myeloid cells is highly site-specific, i.e., RNA sensing and TLR13 are crucial for recognition of GBS by resident mouse macrophages but not circulating blood monocytes (142). Interestingly, recognition of GBS and Gram-positive bacteria appears to rely more on endosomal TLRs than recognition of Gram-negative bacteria (149). This seems intriguing in the context of human neonatal mononuclear cells, which are particularly responsive to TLR8 ligands (150). Accordingly, recognition of bacterial RNA by TLRs is not only particularly important at the beginning of life, but may result in distinct immune activation patterns induced by Streptococcaceae and Enterobacteriaceae. It remains an appealing yet unproven hypothesis that TLR8-dependent immunopathology contributes to myeloid cell-mediated disturbance of mucocutaneous barrier integrity. In addition, TLR8 and 13 do not hold exclusive roles in the recognition of GBS RNA or nucleic acids in general. First, the NLRP3 inflammasome mediates GBS-induced formation of IL-1β and IL-18 in macrophages via recognition of ssRNA (151, 152). NLRP3 activation requires the induction of potassium efflux by a rhamnolipid of GBS, which also mediates cytolysis (35). Proper inflammasome activation is essential for the neonatal resistance against GBS (151). Next, GBS DNA engages the cytosolic signaling of cGAS and STING which leads to interferon (IFN)-β production and contributes to GBS immunity (153, 154). In addition, conventional dendritic cells secret type I IFNs in response to endosomal GBS RNA interacting with TLR7 (155). GBS may subvert nucleotide sensing via expression of ectonucleotidases (154, 156) (Figure 1). Similarly, the GBS hyaluronidase HylB blocks cellular activation by degrading host hyaluronic acid into fragments which bind and inhibit TLR2 (157). HylB was shown to promote vaginal colonization and ascending infections in mice (157, 158). How these enzymes impact on the sensing of colonizing GBS and of competing bacteria in neonates is currently unclear. It furthermore remains to be determined how the relatively increased TRIF-dependent pathway in neonates impacts on barrier defense against GBS (159). Any effect can be assumed to be indirect, since TRIF is redundant in GBS-mediated activation of phagocytes, although a role as a signaling intermediate in other (immune) cells cannot be excluded (149, 160).
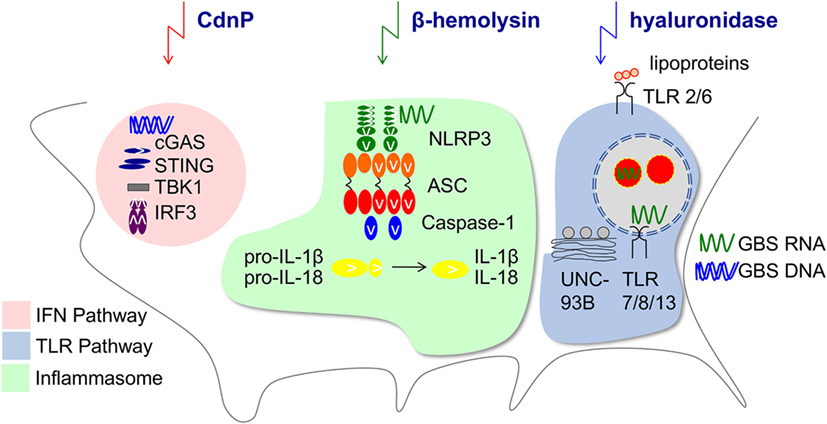
Figure 1. Innate immune pathways manipulated by Group B Streptococcus. Depicted is the impact of GBS on type I interferons (IFN) (153, 155), Toll-like receptor (TLR) (10, 149), and inflammasome (151) pathways by secreted bacterial factors. The ectonucleotidase CdnP hydrolyzes bacterial cyclic dinucleotides which otherwise activate STING and IFN-β production (154). Hemolysin contributes as second signal to the NLRP3 inflammasome activation (152). The GBS hyaluronidase can degrade pro-inflammatory hyaluronan polymers during tissue injury which normally bind to TLR2 and the resulting fragments block TLR2 signaling in the host (157).
Understanding the distinct TLR, inflammasome and cGAS engagement in the monocyte-macrophage lineage by GBS is of utmost importance, since macrophages are the dominant resident immune cells at mucocutaneous barriers, i.e., the dermis and the gut. They are crucially involved in barrier maintenance (161, 162), both by executing direct antimicrobial actions and by cytokine and chemokine dependent recruitment and activation of other immune cells. Development of the neonatal macrophage compartment is particularly well understood in the neonatal intestine, where the population of embryonic macrophages is replaced by monocyte-derived macrophages starting at weaning (163). It is tempting to speculate that macrophage maturation in the lamina propria directly impacts on the macrophage-driven recognition and elimination of invading GBS. Another TLR-based mechanism promoting susceptibility to GBS is the increased production of anti-inflammatory cytokines. Enhanced IL-10 concentrations in serum and cord blood are correlated with mortality in septic infants (164). Moreover, IL-10 has a major impact on intestinal barrier immunity, both in humans and mice. Yet, whereas too little IL-10 leads to spontaneous inflammation and colitis, increased IL-10 production impairs neutrophil recruitment into infected organs and thus decreases GBS clearance (164, 165). How increased IL-10 formation impacts on keeping GBS in a colonization—as opposed to an invasion—state is currently not known.
Impact of the Microbiome on the Developing Immunity
Numerous studies were initiated to understand the impact of the colonizing flora on the function of intestinal cells in general and the immune system in general. Research is usually based on germ-free mice and antibiotic treatments in order to understand the consequences of a reduction or absence of microorganisms. Evidence for immunological consequences of alterations in the microbiome was even found in cells very distant to the gastrointestinal tract such as brain microglia (166). In a highly interesting mouse study, exposure of the pregnant dam to antibiotics not only led to neutropenia in newborn mice, but subsequently increased the susceptibility to Gram-negative sepsis (10). A reduction in Gammaproteobacteria may mediate these effects, since their effector LPS induces granulocyte colony-stimulating factor production and consequently granulopoiesis. Recently, Josefsdottir et al. suggested that the microbiota is the cause of neutropenia and general depletion of hematopoietic stem cells across multiple lineages in antibiotic-treated mice (11). The phenotype could be partially rescued by fecal transfer. This experimental data is in line with the observation that administration of ceftalorine and β-lactam antibiotics can lead to neutropenia in patients (167, 168). Consequently, antibiotics appear to indirectly impact on the maturation of the immune response (169) and the resistance against neonatal sepsis pathogens. An overall smaller granulocyte pool in neonates (132) may further propagate the negative effects of antibiotics. Therefore, it seems that the immaturity of neonatal blood cells, including phagocytes and adaptive immune cells, might restrict the ability to fight off pathogens. Hence, in the stochastic event of pathogen invasion through the muco-cutaneous barrier, which may be potently responded to by the adult immune system, neonatal immunity may be overwhelmed, resulting in bacterial spread and sepsis (Figure 2). It remains incompletely understood whether the protection in the adult usually involves the resident immune cells at mucocutaneous sites, e.g., the lamina propria in the gut or the dermis in the skin, or whether circulating leukocytes are necessary for efficient barrier defense.
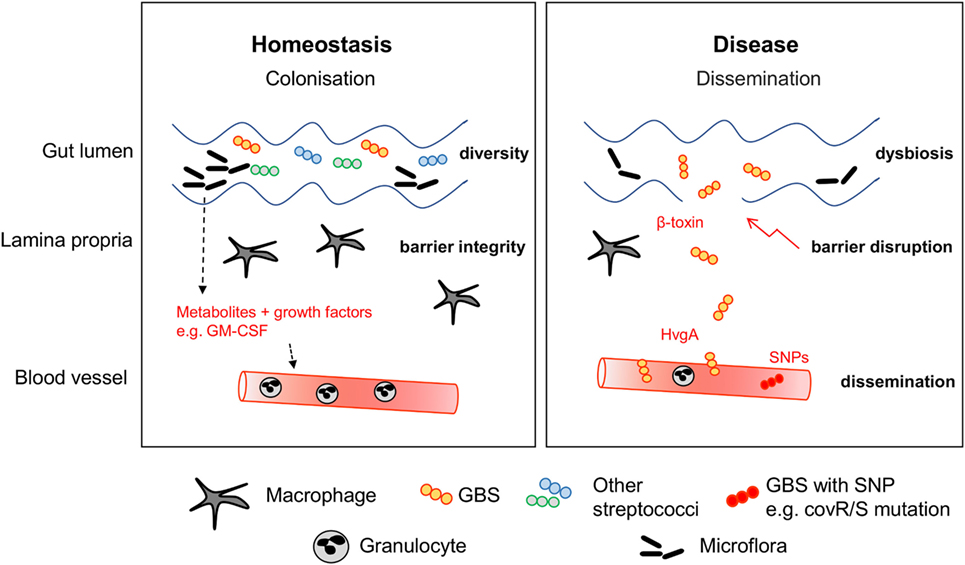
Figure 2. Stabilization of the mucocutaneous niche. During homeostasis, GBS colonizes the intestine of healthy infants. Macrophages and other immune cells guarantee barrier integrity by surveillance. Other commensal bacteria including streptococcal species form the niche. Disease can be preceded by multiple factors leading to dysbiosis, expansion of GBS and barrier disruption. Expression of virulence factors such as HvgA and β-toxin facilitate adhesion to epithelial cells and barrier disruption. Dissemination is often concurrent with mutations of the CovR/S virulence repressor.
Conclusion
The challenge to understand and ultimately prevent neonatal GBS sepsis comprises (i) the control of GBS transmission during and immediately after birth leading to EOD and (ii) the subsequent control of GBS as a mucocutaneous colonizer, when failure results in LOD. Whereas high maternal antibody titers, as induced by GBS vaccines, and IAP are established strategies to prevent EOD, similar strategies with proven efficacy for LOD reduction are missing. Based on experimental and observational evidence, it seems worth considering—and thus requires careful studies—whether antibiotic pressure during primary colonization of the intestine facilitates dysbiosis on the strain level and transient immunodeficiency in the individual child. Furthermore, capsular polysaccharide based vaccines may select for serotype-switched virulent strains as observed with ST-17 and allow for the expansion of other β-hemolytic streptococci than GBS.
The vast recent gain in knowledge on the coevolution of microbiome and cellular barrier defense make the design of novel approaches for neonatal sepsis prevention conceivable, although much preclinical work remains to be done first. Examples are designer probiotics, containing—among others—strains which occupy the streptococcal niche without risk of invasion. Immunomodulators that accelerate the maturation of the phagocyte population resident at mucocutaneous sites may be another strategy that holds potential. Yet, the variable conditions and demands at the beginning of life, e.g., that of very preterm infants or those requiring antibiotic therapy early on, make one-fits-all solutions to the neonatal sepsis conundrum unlikely and rather ask for individualized approaches.
Author Contributions
JK and PH wrote and edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
PH is supported by grants from the German Federal Ministry of Education and Research (Grant 01EO0803) and the German Research Foundation (CRC/TRR167).
References
1. Van Dyke MK, Phares CR, Lynfield R, Thomas AR, Arnold KE, Craig AS, et al. Evaluation of universal antenatal screening for group B Streptococcus. N Engl J Med (2009) 360:2626–36. doi:10.1056/NEJMoa0806820
2. Baltimore RS. Pathogenesis of neonatal group B streptococcal infections. Yale J Biol Med (1982) 55:291–5.
3. Bizzarro MJ, Raskind C, Baltimore RS, Gallagher PG. Seventy-five years of neonatal sepsis at Yale: 1928-2003. Pediatrics (2005) 116:595–602. doi:10.1542/peds.2005-0552
4. Dillon HC Jr, Gray E, Pass MA, Gray BM. Anorectal and vaginal carriage of group B streptococci during pregnancy. J Infect Dis (1982) 145:794–9. doi:10.1093/infdis/145.6.794
5. Bayo M, Berlanga M, Agut M. Vaginal microbiota in healthy pregnant women and prenatal screening of group B streptococci (GBS). Int Microbiol (2002) 5:87–90. doi:10.1007/s10123-002-0064-1
6. Thaiss CA, Zmora N, Levy M, Elinav E. The microbiome and innate immunity. Nature (2016) 535:65–74. doi:10.1038/nature18847
7. Borghesi A, Stronati M, Fellay J. Neonatal group B streptococcal disease in otherwise healthy infants: failure of specific neonatal immune responses. Front Immunol (2017) 8:215. doi:10.3389/fimmu.2017.00215
8. Bokulich NA, Chung J, Battaglia T, Henderson N, Jay M, Li H, et al. Antibiotics, birth mode, and diet shape microbiome maturation during early life. Sci Transl Med (2016) 8:343ra382. doi:10.1126/scitranslmed.aad7121
9. Chu DM, Ma J, Prince AL, Antony KM, Seferovic MD, Aagaard KM. Maturation of the infant microbiome community structure and function across multiple body sites and in relation to mode of delivery. Nat Med (2017) 23:314–26. doi:10.1038/nm.4272
10. Deshmukh HS, Liu Y, Menkiti OR, Mei J, Dai N, O’leary CE, et al. The microbiota regulates neutrophil homeostasis and host resistance to Escherichia coli K1 sepsis in neonatal mice. Nat Med (2014) 20(5):524–30. doi:10.1038/nm.3542
11. Josefsdottir K, Baldridge M, Kadmon C, King K. Antibiotics impair murine hematopoiesis by depleting the intestinal microbiota. Blood (2017) 129:729–39. doi:10.1182/blood-2016-03-708594
12. Darmstadt GL, Saha SK, Choi Y, El Arifeen S, Ahmed NU, Bari S, et al. Population-based incidence and etiology of community-acquired neonatal bacteremia in Mirzapur, Bangladesh: an observational study. J Infect Dis (2009) 200:906–15. doi:10.1086/605473
13. Edmond KM, Kortsalioudaki C, Scott S, Schrag SJ, Zaidi AKM, Cousens S, et al. Group B streptococcal disease in infants aged younger than 3 months: systematic review and meta-analysis. Lancet (2012) 379:547–56. doi:10.1016/S0140-6736(11)61651-6
14. Cohen-Wolkowiez M, Moran C, Benjamin DK, Cotten CM, Clark RH, Benjamin DK Jr, et al. Early and late onset sepsis in late preterm infants. Pediatr Infect Dis J (2009) 28:1052–6. doi:10.1097/INF.0b013e3181acf6bd
15. Bekker V, Bijlsma MW, Van De Beek D, Kuijpers TW, Van Der Ende A. Incidence of invasive group B streptococcal disease and pathogen genotype distribution in newborn babies in the Netherlands over 25 years: a nationwide surveillance study. Lancet Infect Dis (2014) 14:1083–9. doi:10.1016/S1473-3099(14)70919-3
16. Langley G, Schaffner W, Farley MM, Lynfield R, Bennett NM, Reingold A, et al. Twenty years of active bacterial core surveillance. Emerg Infect Dis (2015) 21:1520–8. doi:10.3201/eid2109.141333
17. Jordan HT, Farley MM, Craig A, Mohle-Boetani J, Harrison LH, Petit S, et al. Revisiting the need for vaccine prevention of late-onset neonatal group B streptococcal disease: a multistate, population-based analysis. Pediatr Infect Dis J (2008) 27:1057–64. doi:10.1097/INF.0b013e318180b3b9
18. Berardi A, Rossi C, Lugli L, Creti R, Bacchi Reggiani ML, Lanari M, et al. Group B Streptococcus late-onset disease: 2003-2010. Pediatrics (2013) 131:e361–8. doi:10.1542/peds.2012-1231
19. Madhi SA, Dangor Z, Heath PT, Schrag S, Izu A, Sobanjo-Ter Meulen A, et al. Considerations for a phase-III trial to evaluate a group B Streptococcus polysaccharide-protein conjugate vaccine in pregnant women for the prevention of early- and late-onset invasive disease in young-infants. Vaccine (2013) 31(Suppl 4):D52–7. doi:10.1016/j.vaccine.2013.02.029
20. Poyart C, Reglier-Poupet H, Tazi A, Billoet A, Dmytruk N, Bidet P, et al. Invasive group B streptococcal infections in infants, France. Emerg Infect Dis (2008) 14:1647–9. doi:10.3201/eid1410.080185
21. Poyart C, Tazi A, Reglier-Poupet H, Billoet A, Tavares N, Raymond J, et al. Multiplex PCR assay for rapid and accurate capsular typing of group B streptococci. J Clin Microbiol (2007) 45:1985–8. doi:10.1128/JCM.00159-07
22. Islam MS, Saha SK, Islam M, Modak JK, Shah R, Talukder RR, et al. Prevalence, serotype distribution and mortality risk associated with group B Streptococcus colonization of newborns in rural Bangladesh. Pediatr Infect Dis J (2016) 35:1309–12. doi:10.1097/INF.0000000000001306
23. Jones N, Bohnsack JF, Takahashi S, Oliver KA, Chan MS, Kunst F, et al. Multilocus sequence typing system for group B Streptococcus. J Clin Microbiol (2003) 41:2530–6. doi:10.1128/JCM.41.6.2530-2536.2003
24. Da Cunha V, Davies MR, Douarre PE, Rosinski-Chupin I, Margarit I, Spinali S, et al. Streptococcus agalactiae clones infecting humans were selected and fixed through the extensive use of tetracycline. Nat Commun (2014) 5:4544. doi:10.1038/ncomms5544
25. Martins ER, Pessanha MA, Ramirez M, Melo-Cristino J; Portuguese Group for the Study of Streptococcal Infections. Analysis of group B streptococcal isolates from infants and pregnant women in Portugal revealing two lineages with enhanced invasiveness. J Clin Microbiol (2007) 45:3224–9. doi:10.1128/JCM.01182-07
26. Bohnsack JF, Whiting A, Gottschalk M, Dunn DM, Weiss R, Azimi PH, et al. Population structure of invasive and colonizing strains of Streptococcus agalactiae from neonates of six U.S. Academic Centers from 1995 to 1999. J Clin Microbiol (2008) 46:1285–91. doi:10.1128/JCM.02105-07
27. Tazi A, Disson O, Bellais S, Bouaboud A, Dmytruk N, Dramsi S, et al. The surface protein HvgA mediates group B Streptococcus hypervirulence and meningeal tropism in neonates. J Exp Med (2010) 207:2313–22. doi:10.1084/jem.20092594
28. Jones N, Oliver KA, Barry J, Harding RM, Bisharat N, Spratt BG, et al. Enhanced invasiveness of bovine-derived neonatal sequence type 17 group B Streptococcus is independent of capsular serotype. Clin Infect Dis (2006) 42:915–24. doi:10.1086/500324
29. Seale AC, Koech AC, Sheppard AE, Barsosio HC, Langat J, Anyango E, et al. Maternal colonization with Streptococcus agalactiae and associated stillbirth and neonatal disease in coastal Kenya. Nat Microbiol (2016) 1:16067. doi:10.1038/nmicrobiol.2016.67
30. Martins ER, Pedroso-Roussado C, Melo-Cristino J, Ramirez M; Portuguese Group for the Study of Streptococcal Infections. Streptococcus agalactiae causing neonatal infections in Portugal (2005-2015): diversification and emergence of a CC17/PI-2b multidrug resistant sublineage. Front Microbiol (2017) 8:499. doi:10.3389/fmicb.2017.00499
31. Six A, Bellais S, Bouaboud A, Fouet A, Gabriel C, Tazi A, et al. Srr2, a multifaceted adhesin expressed by ST-17 hypervirulent group B Streptococcus involved in binding to both fibrinogen and plasminogen. Mol Microbiol (2015) 97(6):1209–22. doi:10.1111/mmi.13097
32. Lazzarin M, Mu R, Fabbrini M, Ghezzo C, Rinaudo CD, Doran KS, et al. Contribution of pilus type 2b to invasive disease caused by a Streptococcus agalactiae ST-17 strain. BMC Microbiol (2017) 17:148. doi:10.1186/s12866-017-1057-8
33. Perichon B, Szili N, Du Merle L, Rosinski-Chupin I, Gominet M, Bellais S, et al. Regulation of PI-2b pilus expression in hypervirulent Streptococcus agalactiae ST-17 BM110. PLoS One (2017) 12:e0169840. doi:10.1371/journal.pone.0169840
34. Rosa-Fraile M, Dramsi S, Spellerberg B. Group B streptococcal haemolysin and pigment, a tale of twins. FEMS Microbiol Rev (2014) 38(5):932–46. doi:10.1111/1574-6976.12071
35. Whidbey C, Harrell MI, Burnside K, Ngo L, Becraft AK, Iyer LM, et al. A hemolytic pigment of group B Streptococcus allows bacterial penetration of human placenta. J Exp Med (2013) 210:1265–81. doi:10.1084/jem.20122753
36. Kobayashi M, Vekemans J, Baker CJ, Ratner AJ, Le Doare K, Schrag SJ. Group B Streptococcus vaccine development: present status and future considerations, with emphasis on perspectives for low and middle income countries. F1000Res (2016) 5:2355. doi:10.12688/f1000research.9363.1
37. Vornhagen J, Adams Waldorf KM, Rajagopal L. Perinatal group B streptococcal infections: virulence factors, immunity, and prevention strategies. Trends Microbiol (2017) 25(11):919–31. doi:10.1016/j.tim.2017.05.013
38. Doran KS, Nizet V. Molecular pathogenesis of neonatal group B streptococcal infection: no longer in its infancy. Mol Microbiol (2004) 54:23–31. doi:10.1111/j.1365-2958.2004.04266.x
39. Carl MA, Ndao IM, Springman AC, Manning SD, Johnson JR, Johnston BD, et al. Sepsis from the gut: the enteric habitat of bacteria that cause late-onset neonatal bloodstream infections. Clin Infect Dis (2014) 58:1211–8. doi:10.1093/cid/ciu084
40. Taft DH, Ambalavanan N, Schibler KR, Yu Z, Newburg DS, Deshmukh H, et al. Center variation in intestinal microbiota prior to late-onset sepsis in preterm infants. PLoS One (2015) 10:e0130604. doi:10.1371/journal.pone.0130604
41. Melin P. Neonatal group B streptococcal disease: from pathogenesis to preventive strategies. Clin Microbiol Infect (2011) 17:1294–303. doi:10.1111/j.1469-0691.2011.03576.x
42. Le Doare K, Faal A, Jaiteh M, Sarfo F, Taylor S, Warburton F, et al. Association between functional antibody against group B Streptococcus and maternal and infant colonization in a Gambian cohort. Vaccine (2017) 35:2970–8. doi:10.1016/j.vaccine.2017.04.013
43. Dillon HC Jr, Khare S, Gray BM. Group B streptococcal carriage and disease: a 6-year prospective study. J Pediatr (1987) 110:31–6. doi:10.1016/S0022-3476(87)80283-4
44. Stewart CJ, Embleton ND, Marrs ECL, Smith DP, Fofanova T, Nelson A, et al. Longitudinal development of the gut microbiome and metabolome in preterm neonates with late onset sepsis and healthy controls. Microbiome (2017) 5:75. doi:10.1186/s40168-017-0295-1
45. Shaw AG, Sim K, Randell P, Cox MJ, Mcclure ZE, Li M-S, et al. Late-onset bloodstream infection and perturbed maturation of the gastrointestinal microbiota in premature Infants. PLoS One (2015) 10:e0132923. doi:10.1371/journal.pone.0132923
46. Elling R, Hufnagel M, De Zoysa A, Lander F, Zumstein K, Krueger M, et al. Synchronous recurrence of group B streptococcal late-onset sepsis in twins. Pediatrics (2014) 133:e1388–91. doi:10.1542/peds.2013-0426
47. Gevers D, Kugathasan S, Denson LA, Vazquez-Baeza Y, Van Treuren W, Ren B, et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe (2014) 15:382–92. doi:10.1016/j.chom.2014.02.005
48. van Sorge NM, Doran KS. Defense at the border: the blood-brain barrier versus bacterial foreigners. Future Microbiol (2012) 7:383–94. doi:10.2217/fmb.12.1
49. Ferrieri P, Burke B, Nelson J. Production of bacteremia and meningitis in infant rats with group B streptococcal serotypes. Infect Immun (1980) 27:1023–32.
50. Zeligs BJ, Armstrong CD, Walser JB, Bellanti JA. Age-dependent susceptibility of neonatal rats to group B streptococcal type III infection: correlation of severity of infection and response of myeloid pools. Infect Immun (1982) 37:255–63.
51. Mancuso G, Tomasello F, Migliardo M, Delfino D, Cochran J, Cook JA, et al. Beneficial effects of interleukin-6 in neonatal mouse models of group B streptococcal disease. Infect Immun (1994) 62:4997–5002.
52. Kim KS, Dunn K, Mcgeary SA, Stiehm ER. Efficacy of orally administered immune serum globulin against type III group B streptococcal colonization and systemic disease in an infant rat model. Pediatr Res (1984) 18:1329–31. doi:10.1203/00006450-198412000-00022
53. Florindo C, Gomes JP, Rato MG, Bernardino L, Spellerberg B, Santos-Sanches I, et al. Molecular epidemiology of group B streptococcal meningitis in children beyond the neonatal period from Angola. J Med Microbiol (2011) 60:1276–80. doi:10.1099/jmm.0.031674-0
54. Grice EA, Segre JA. The skin microbiome. Nat Rev Microbiol (2011) 9:244–53. doi:10.1038/nrmicro2537
55. Aagaard K, Ma J, Antony KM, Ganu R, Petrosino J, Versalovic J. The placenta harbors a unique microbiome. Sci Transl Med (2014) 6:237ra265. doi:10.1126/scitranslmed.3008599
56. Collado MC, Rautava S, Aakko J, Isolauri E, Salminen S. Human gut colonisation may be initiated in utero by distinct microbial communities in the placenta and amniotic fluid. Sci Rep (2016) 6:23129. doi:10.1038/srep23129
57. Lauder AP, Roche AM, Sherrill-Mix S, Bailey A, Laughlin AL, Bittinger K, et al. Comparison of placenta samples with contamination controls does not provide evidence for a distinct placenta microbiota. Microbiome (2016) 4:29. doi:10.1186/s40168-016-0172-3
58. La Rosa PS, Warner BB, Zhou Y, Weinstock GM, Sodergren E, Hall-Moore CM, et al. Patterned progression of bacterial populations in the premature infant gut. Proc Natl Acad Sci U S A (2014) 111:12522–7. doi:10.1073/pnas.1409497111
59. Palmer C, Bik EM, Digiulio DB, Relman DA, Brown PO. Development of the human infant intestinal microbiota. PLoS Biol (2007) 5:e177. doi:10.1371/journal.pbio.0050177
60. Koenig JE, Spor A, Scalfone N, Fricker AD, Stombaugh J, Knight R, et al. Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci U S A (2011) 108(Suppl 1):4578–85. doi:10.1073/pnas.1000081107
61. Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature (2012) 489:220–30. doi:10.1038/nature11550
62. Adlerberth I, Wold AE. Establishment of the gut microbiota in Western infants. Acta Paediatr (2009) 98:229–38. doi:10.1111/j.1651-2227.2008.01060.x
63. Tourneur E, Chassin C. Neonatal immune adaptation of the gut and its role during infections. Clin Dev Immunol (2013) 2013:270301. doi:10.1155/2013/270301
64. Penders J, Thijs C, Vink C, Stelma FF, Snijders B, Kummeling I, et al. Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics (2006) 118:511–21. doi:10.1542/peds.2005-2824
65. Yassour M, Vatanen T, Siljander H, Hamalainen AM, Harkonen T, Ryhanen SJ, et al. Natural history of the infant gut microbiome and impact of antibiotic treatment on bacterial strain diversity and stability. Sci Transl Med (2016) 8:343ra381. doi:10.1126/scitranslmed.aad0917
66. Gibson MK, Wang B, Ahmadi S, Burnham CA, Tarr PI, Warner BB, et al. Developmental dynamics of the preterm infant gut microbiota and antibiotic resistome. Nat Microbiol (2016) 1:16024. doi:10.1038/nmicrobiol.2016.24
67. Arboleya S, Sanchez B, Milani C, Duranti S, Solis G, Fernandez N, et al. Intestinal microbiota development in preterm neonates and effect of perinatal antibiotics. J Pediatr (2015) 166:538–44. doi:10.1016/j.jpeds.2014.09.041
68. Heida FH, Van Zoonen A, Hulscher JBF, Te Kiefte BJC, Wessels R, Kooi EMW, et al. A necrotizing enterocolitis-associated gut microbiota is present in the meconium: results of a prospective study. Clin Infect Dis (2016) 62:863–70. doi:10.1093/cid/ciw016
69. Mazzola G, Murphy K, Ross RP, Di Gioia D, Biavati B, Corvaglia LT, et al. Early gut microbiota perturbations following intrapartum antibiotic prophylaxis to prevent group B streptococcal disease. PLoS One (2016) 11:e0157527. doi:10.1371/journal.pone.0157527
70. Hansen SM, Uldbjerg N, Kilian M, Sorensen UBS. Dynamics of Streptococcus agalactiae colonization in women during and after pregnancy and in their infants. J Clin Microbiol (2004) 42:83–9. doi:10.1128/JCM.42.1.83-89.2004
71. Stoll BJ, Hansen NI, Sanchez PJ, Faix RG, Poindexter BB, Van Meurs KP, et al. Early onset neonatal sepsis: the burden of group B streptococcal and E. coli disease continues. Pediatrics (2011) 127:817–26. doi:10.1542/peds.2010-2217
72. Manning SD, Lewis MA, Springman AC, Lehotzky E, Whittam TS, Davies HD. Genotypic diversity and serotype distribution of group B Streptococcus isolated from women before and after delivery. Clin Infect Dis (2008) 46:1829–37. doi:10.1086/588296
73. Kwatra G, Adrian PV, Shiri T, Buchmann EJ, Cutland CL, Madhi SA. Serotype-specific acquisition and loss of group B Streptococcus recto-vaginal colonization in late pregnancy. PLoS One (2014) 9:e98778. doi:10.1371/journal.pone.0098778
74. Verani JR, Mcgee L, Schrag SJ. Prevention of perinatal group B streptococcal disease – revised guidelines from CDC, 2010. MMWR Recomm Rep (2010) 59:1–36.
75. Manning SD, Springman AC, Million AD, Milton NR, Mcnamara SE, Somsel PA, et al. Association of group B Streptococcus colonization and bovine exposure: a prospective cross-sectional cohort study. PLoS One (2010) 5:e8795. doi:10.1371/journal.pone.0008795
76. Delannoy CM, Crumlish M, Fontaine MC, Pollock J, Foster G, Dagleish MP, et al. Human Streptococcus agalactiae strains in aquatic mammals and fish. BMC Microbiol (2013) 13:41. doi:10.1186/1471-2180-13-41
77. Rajendram P, Mar Kyaw W, Leo YS, Ho H, Chen WK, Lin R, et al. Group B Streptococcus sequence type 283 disease linked to consumption of raw fish, Singapore. Emerg Infect Dis (2016) 22:1974–7. doi:10.3201/eid2211.160252
78. Bisharat N, Crook DW, Leigh J, Harding RM, Ward PN, Coffey TJ, et al. Hyperinvasive neonatal group B Streptococcus Has arisen from a bovine ancestor. J Clin Microbiol (2004) 42:2161–7. doi:10.1128/JCM.42.5.2161-2167.2004
79. Manning SD, Tallman P, Baker CJ, Gillespie B, Marrs CF, Foxman B. Determinants of co-colonization with group B Streptococcus among heterosexual college couples. Epidemiology (2002) 13:533–9. doi:10.1097/00001648-200209000-00008
80. Meyn LA, Moore DM, Hillier SL, Krohn MA. Association of sexual activity with colonization and vaginal acquisition of group B Streptococcus in nonpregnant women. Am J Epidemiol (2002) 155:949–57. doi:10.1093/aje/155.10.949
81. Manning SD, Neighbors K, Tallman PA, Gillespie B, Marrs CF, Borchardt SM, et al. Prevalence of group B Streptococcus colonization and potential for transmission by casual contact in healthy young men and women. Clin Infect Dis (2004) 39:380–8. doi:10.1086/422321
82. Berardi A, Rossi C, Creti R, China M, Gherardi G, Venturelli C, et al. Group B streptococcal colonization in 160 mother-baby pairs: a prospective cohort study. J Pediatrics (2013) 163:1099.e–104.e. doi:10.1016/j.jpeds.2013.05.064
83. Filleron A, Lombard F, Jacquot A, Jumas-Bilak E, Rodiere M, Cambonie G, et al. Group B streptococci in milk and late neonatal infections: an analysis of cases in the literature. Arch Dis Child Fetal Neonatal Ed (2014) 99:F41–7. doi:10.1136/archdischild-2013-304362
84. Almeida A, Villain A, Joubrel C, Touak G, Sauvage E, Rosinski-Chupin I, et al. Whole-genome comparison uncovers genomic mutations between group B streptococci sampled from infected newborns and their mothers. J Bacteriol (2015) 197:3354–66. doi:10.1128/JB.00429-15
85. Berkley JA, Lowe BS, Mwangi I, Williams T, Bauni E, Mwarumba S, et al. Bacteremia among children admitted to a rural hospital in Kenya. N Engl J Med (2005) 352:39–47. doi:10.1056/NEJMoa040275
86. Talbert AW, Mwaniki M, Mwarumba S, Newton CR, Berkley JA. Invasive bacterial infections in neonates and young infants born outside hospital admitted to a rural hospital in Kenya. Pediatr Infect Dis J (2010) 29:945–9. doi:10.1097/INF.0b013e3181dfca8c
87. Shah BA, Padbury JF. Neonatal sepsis: an old problem with new insights. Virulence (2014) 5:170–8. doi:10.4161/viru.26906
88. Hibbing ME, Fuqua C, Parsek MR, Peterson SB. Bacterial competition: surviving and thriving in the microbial jungle. Nat Rev Microbiol (2010) 8:15–25. doi:10.1038/nrmicro2259
89. Kreth J, Merritt J, Shi W, Qi F. Competition and coexistence between Streptococcus mutans and Streptococcus sanguinis in the dental biofilm. J Bacteriol (2005) 187:7193–203. doi:10.1128/JB.187.21.7193-7203.2005
90. Tong H, Chen W, Merritt J, Qi F, Shi W, Dong X. Streptococcus oligofermentans inhibits Streptococcus mutans through conversion of lactic acid into inhibitory H2O2: a possible counteroffensive strategy for interspecies competition. Mol Microbiol (2007) 63:872–80. doi:10.1111/j.1365-2958.2006.05546.x
91. Laufer AS, Metlay JP, Gent JF, Fennie KP, Kong Y, Pettigrew MM. Microbial communities of the upper respiratory tract and otitis media in children. MBio (2011) 2:e245–210. doi:10.1128/mBio.00245-10
92. Patras KA, Wescombe PA, Rosler B, Hale JD, Tagg JR, Doran KS. Streptococcus salivarius K12 limits group B Streptococcus vaginal colonization. Infect Immun (2015) 83:3438–44. doi:10.1128/IAI.00409-15
93. Aloisio I, Mazzola G, Corvaglia LT, Tonti G, Faldella G, Biavati B, et al. Influence of intrapartum antibiotic prophylaxis against group B Streptococcus on the early newborn gut composition and evaluation of the anti-Streptococcus activity of Bifidobacterium strains. Appl Microbiol Biotechnol (2014) 98:6051–60. doi:10.1007/s00253-014-5712-9
94. Ruiz FO, Gerbaldo G, Garcia MJ, Giordano W, Pascual L, Barberis IL. Synergistic effect between two bacteriocin-like inhibitory substances produced by lactobacilli strains with inhibitory activity for Streptococcus agalactiae. Curr Microbiol (2012) 64:349–56. doi:10.1007/s00284-011-0077-0
95. Zarate G, Nader-Macias ME. Influence of probiotic vaginal lactobacilli on in vitro adhesion of urogenital pathogens to vaginal epithelial cells. Lett Appl Microbiol (2006) 43:174–80. doi:10.1111/j.1472-765X.2006.01934.x
96. De Gregorio PR, Juarez Tomas MS, Leccese Terraf MC, Nader-Macias ME. Preventive effect of Lactobacillus reuteri CRL1324 on Group B Streptococcus vaginal colonization in an experimental mouse model. J Appl Microbiol (2015) 118:1034–47. doi:10.1111/jam.12739
97. Ho M, Chang YY, Chang WC, Lin HC, Wang MH, Lin WC, et al. Oral Lactobacillus rhamnosus GR-1 and Lactobacillus reuteri RC-14 to reduce group B Streptococcus colonization in pregnant women: a randomized controlled trial. Taiwan J Obstet Gynecol (2016) 55:515–8. doi:10.1016/j.tjog.2016.06.003
98. Panigrahi P, Parida S, Nanda NC, Satpathy R, Pradhan L, Chandel DS, et al. A randomized synbiotic trial to prevent sepsis among infants in rural India. Nature (2017) 548:407–12. doi:10.1038/nature23480
99. Brzychczy-Wloch M, Pabian W, Majewska E, Zuk MG, Kielbik J, Gosiewski T, et al. Dynamics of colonization with group B streptococci in relation to normal flora in women during subsequent trimesters of pregnancy. New Microbiol (2014) 37:307–19.
100. Rick AM, Aguilar A, Cortes R, Gordillo R, Melgar M, Samayoa-Reyes G, et al. Group B streptococci colonization in pregnant guatemalan women: prevalence, risk factors, and vaginal microbiome. Open Forum Infect Dis (2017) 4:ofx020. doi:10.1093/ofid/ofx020
101. Rosen GH, Randis TM, Desai PV, Sapra KJ, Ma B, Gajer P, et al. Group B Streptococcus and the vaginal microbiota. J Infect Dis (2017) 216(6):744–51. doi:10.1093/infdis/jix395
102. Cassidy-Bushrow AE, Sitarik A, Levin AM, Lynch SV, Havstad S, Ownby DR, et al. Maternal group B Streptococcus and the infant gut microbiota. J Dev Orig Health Dis (2016) 7:45–53. doi:10.1017/S2040174415001361
103. Lowy FD. Staphylococcus aureus infections. N Engl J Med (1998) 339:520–32. doi:10.1056/NEJM199808203390806
104. Hu Y, Yang X, Qin J, Lu N, Cheng G, Wu N, et al. Metagenome-wide analysis of antibiotic resistance genes in a large cohort of human gut microbiota. Nat Commun (2013) 4:2151. doi:10.1038/ncomms3151
105. Hays C, Louis M, Plainvert C, Dmytruk N, Touak G, Trieu-Cuot P, et al. Changing epidemiology of group B Streptococcus susceptibility to fluoroquinolones and aminoglycosides in France. Antimicrob Agents Chemother (2016) 60(12):7424–30.
106. Teatero S, Ramoutar E, Mcgeer A, Li A, Melano RG, Wasserscheid J, et al. Clonal complex 17 group B Streptococcus strains causing invasive disease in neonates and adults originate from the same genetic pool. Sci Rep (2016) 6:20047. doi:10.1038/srep20047
107. Kimura K, Nishiyama Y, Shimizu S, Wachino J, Matsui M, Suzuki S, et al. Screening for group B streptococci with reduced penicillin susceptibility in clinical isolates obtained between 1977 and 2005. Jpn J Infect Dis (2013) 66:222–5. doi:10.7883/yoken.66.222
108. Kimura K, Suzuki S, Wachino J, Kurokawa H, Yamane K, Shibata N, et al. First molecular characterization of group B streptococci with reduced penicillin susceptibility. Antimicrob Agents Chemother (2008) 52:2890–7. doi:10.1128/AAC.00185-08
109. Seki T, Kimura K, Reid ME, Miyazaki A, Banno H, Jin W, et al. High isolation rate of MDR group B streptococci with reduced penicillin susceptibility in Japan. J Antimicrob Chemother (2015) 70:2725–8. doi:10.1093/jac/dkv203
110. Longtin J, Vermeiren C, Shahinas D, Tamber GS, Mcgeer A, Low DE, et al. Novel mutations in a patient isolate of Streptococcus agalactiae with reduced penicillin susceptibility emerging after long-term oral suppressive therapy. Antimicrob Agents Chemother (2011) 55:2983–5. doi:10.1128/AAC.01243-10
111. Teatero S, Mcgeer A, Li A, Gomes J, Seah C, Demczuk W, et al. Population structure and antimicrobial resistance of invasive serotype IV group B Streptococcus, Toronto, Ontario, Canada. Emerg Infect Dis (2015) 21:585–91. doi:10.3201/eid2014.140759
112. Campisi E, Rinaudo CD, Donati C, Barucco M, Torricelli G, Edwards MS, et al. Serotype IV Streptococcus agalactiae ST-452 has arisen from large genomic recombination events between CC23 and the hypervirulent CC17 lineages. Sci Rep (2016) 6:29799. doi:10.1038/srep29799
113. Bellais S, Six A, Fouet A, Longo M, Dmytruk N, Glaser P, et al. Capsular switching in group B Streptococcus CC17 hypervirulent clone: a future challenge for polysaccharide vaccine development. J Infect Dis (2012) 206:1745–52. doi:10.1093/infdis/jis605
114. Meehan M, Cunney R, Cafferkey M. Molecular epidemiology of group B streptococci in Ireland reveals a diverse population with evidence of capsular switching. Eur J Clin Microbiol Infect Dis (2014) 33:1155–62. doi:10.1007/s10096-014-2055-5
115. Le Doare K, Kampmann B. Breast milk and Group B streptococcal infection: vector of transmission or vehicle for protection? Vaccine (2014) 32:3128–32. doi:10.1016/j.vaccine.2014.04.020
116. Kwatra G, Adrian PV, Shiri T, Buchmann EJ, Cutland CL, Madhi SA. Natural acquired humoral immunity against serotype-specific group B Streptococcus rectovaginal colonization acquisition in pregnant women. Clin Microbiol Infect (2015) 21(568):e513–21. doi:10.1016/j.cmi.2015.01.030
117. Baker JA, Lewis EL, Byland LM, Bonakdar M, Randis TM, Ratner AJ. Mucosal vaccination promotes clearance of Streptococcus agalactiae vaginal colonization. Vaccine (2017) 35:1273–80. doi:10.1016/j.vaccine.2017.01.029
118. Ayres JS, Trinidad NJ, Vance RE. Lethal inflammasome activation by a multidrug-resistant pathobiont upon antibiotic disruption of the microbiota. Nat Med (2012) 18:799–806. doi:10.1038/nm.2729
119. Stecher B, Maier L, Hardt WD. ‘Blooming’ in the gut: how dysbiosis might contribute to pathogen evolution. Nat Rev Microbiol (2013) 11:277–84. doi:10.1038/nrmicro2989
120. Morrow AL, Lagomarcino AJ, Schibler KR, Taft DH, Yu Z, Wang B, et al. Early microbial and metabolomic signatures predict later onset of necrotizing enterocolitis in preterm infants. Microbiome (2013) 1:13. doi:10.1186/2049-2618-1-13
121. Stewart CJ, Embleton ND, Marrs EC, Smith DP, Nelson A, Abdulkadir B, et al. Temporal bacterial and metabolic development of the preterm gut reveals specific signatures in health and disease. Microbiome (2016) 4:67. doi:10.1186/s40168-016-0216-8
122. Ward DV, Scholz M, Zolfo M, Taft DH, Schibler KR, Tett A, et al. Metagenomic sequencing with strain-level resolution implicates uropathogenic E. coli in necrotizing enterocolitis and mortality in preterm infants. Cell Rep (2016) 14:2912–24. doi:10.1016/j.celrep.2016.03.015
123. Pammi M, Cope J, Tarr PI, Warner BB, Morrow AL, Mai V, et al. Intestinal dysbiosis in preterm infants preceding necrotizing enterocolitis: a systematic review and meta-analysis. Microbiome (2017) 5:31. doi:10.1186/s40168-017-0248-8
124. Mai V, Torrazza RM, Ukhanova M, Wang X, Sun Y, Li N, et al. Distortions in development of intestinal microbiota associated with late onset sepsis in preterm infants. PLoS One (2013) 8:e52876. doi:10.1371/journal.pone.0052876
125. Cernada M, Bauerl C, Serna E, Collado MC, Martinez GP, Vento M. Sepsis in preterm infants causes alterations in mucosal gene expression and microbiota profiles compared to non-septic twins. Sci Rep (2016) 6:25497. doi:10.1038/srep25497
126. Kuppala VS, Meinzen-Derr J, Morrow AL, Schibler KR. Prolonged initial empirical antibiotic treatment is associated with adverse outcomes in premature infants. J Pediatr (2011) 159:720–5. doi:10.1016/j.jpeds.2011.05.033
127. Farley MM, Harvey RC, Stull T, Smith JD, Schuchat A, Wenger JD, et al. A population-based assessment of invasive disease due to group B Streptococcus in nonpregnant adults. N Engl J Med (1993) 328:1807–11. doi:10.1056/NEJM199306243282503
128. Skoff TH, Farley MM, Petit S, Craig AS, Schaffner W, Gershman K, et al. Increasing burden of invasive group B streptococcal disease in nonpregnant adults, 1990-2007. Clin Infect Dis (2009) 49:85–92. doi:10.1086/599369
129. Edwards MS, Rench MA, Rinaudo CD, Fabbrini M, Tuscano G, Buffi G, et al. Immune responses to invasive group B streptococcal disease in adults. Emerg Infect Dis (2016) 22:1877–83. doi:10.3201/eid2211.160914
130. Kenzel S, Henneke P. The innate immune system and its relevance to neonatal sepsis. Curr Opin Infect Dis (2006) 19:264–70. doi:10.1097/01.qco.0000224821.27482.bd
131. Levy O. Innate immunity of the newborn: basic mechanisms and clinical correlates. Nat Rev Immunol (2007) 7:379–90. doi:10.1038/nri2075
132. Basha S, Surendran N, Pichichero M. Immune responses in neonates. Expert Rev Clin Immunol (2014) 10:1171–84. doi:10.1586/1744666X.2014.942288
133. Rodewald AK, Onderdonk AB, Warren HB, Kasper DL. Neonatal mouse model of group B streptococcal infection. J Infect Dis (1992) 166:635–9. doi:10.1093/infdis/166.3.635
134. Heath PT. Status of vaccine research and development of vaccines for GBS. Vaccine (2016) 34:2876–9. doi:10.1016/j.vaccine.2015.12.072
135. von Bernuth H, Picard C, Jin Z, Pankla R, Xiao H, Ku CL, et al. Pyogenic bacterial infections in humans with MyD88 deficiency. Science (2008) 321:691–6. doi:10.1126/science.1158298
136. Picard C, Von Bernuth H, Ghandil P, Chrabieh M, Levy O, Arkwright PD, et al. Clinical features and outcome of patients with IRAK-4 and MyD88 deficiency. Medicine (Baltimore) (2010) 89:403–25. doi:10.1097/MD.0b013e3181fd8ec3
137. Krause JC, Ghandil P, Chrabieh M, Casanova JL, Picard C, Puel A, et al. Very late-onset group B Streptococcus meningitis, sepsis, and systemic shigellosis due to interleukin-1 receptor-associated kinase-4 deficiency. Clin Infect Dis (2009) 49:1393–6. doi:10.1086/630206
138. Vaishnava S, Yamamoto M, Severson KM, Ruhn KA, Yu X, Koren O, et al. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science (2011) 334:255–8. doi:10.1126/science.1209791
139. Ubeda C, Lipuma L, Gobourne A, Viale A, Leiner I, Equinda M, et al. Familial transmission rather than defective innate immunity shapes the distinct intestinal microbiota of TLR-deficient mice. J Exp Med (2012) 209:1445–56. doi:10.1084/jem.20120504
140. Slack E, Hapfelmeier S, Stecher B, Velykoredko Y, Stoel M, Lawson MA, et al. Innate and adaptive immunity cooperate flexibly to maintain host-microbiota mutualism. Science (2009) 325:617–20. doi:10.1126/science.1172747
141. Mancuso G, Midiri A, Beninati C, Biondo C, Galbo R, Akira S, et al. Dual role of TLR2 and myeloid differentiation factor 88 in a mouse model of invasive group B streptococcal disease. J Immunol (2004) 172:6324–9. doi:10.4049/jimmunol.172.10.6324
142. Kolter J, Feuerstein R, Spoeri E, Gharun K, Elling R, Trieu-Cuot P, et al. Streptococci engage TLR13 on myeloid cells in a site-specific fashion. J Immunol (2016) 196:2733–41. doi:10.4049/jimmunol.1501014
143. Henneke P, Takeuchi O, Malley R, Lien E, Ingalls RR, Freeman MW, et al. Cellular activation, phagocytosis, and bactericidal activity against group B Streptococcus involve parallel myeloid differentiation factor 88-dependent and independent signaling pathways. J Immunol (2002) 169:3970–7. doi:10.4049/jimmunol.169.7.3970
144. Henneke P, Dramsi S, Mancuso G, Chraibi K, Pellegrini E, Theilacker C, et al. Lipoproteins are critical TLR2 activating toxins in group B streptococcal sepsis. J Immunol (2008) 180:6149–58. doi:10.4049/jimmunol.180.9.6149
145. Oldenburg M, Kruger A, Ferstl R, Kaufmann A, Nees G, Sigmund A, et al. TLR13 recognizes bacterial 23S rRNA devoid of erythromycin resistance-forming modification. Science (2012) 337:1111–5. doi:10.1126/science.1220363
146. Bergstrom B, Aune MH, Awuh JA, Kojen JF, Blix KJ, Ryan L, et al. TLR8 senses Staphylococcus aureus RNA in human primary monocytes and macrophages and induces IFN-beta production via a TAK1-IKKbeta-IRF5 signaling pathway. J Immunol (2015) 195:1100–11. doi:10.4049/jimmunol.1403176
147. Kruger A, Oldenburg M, Chebrolu C, Beisser D, Kolter J, Sigmund AM, et al. Human TLR8 senses UR/URR motifs in bacterial and mitochondrial RNA. EMBO Rep (2015) 16:1656–63. doi:10.15252/embr.201540861
148. Ehrnström B, Beckwith KS, Yurchenko M, Moen SH, Kojen JF, Lentini G, et al. Toll-like receptor 8 is a major sensor of group B Streptococcus but not Escherichia coli in human primary monocytes and macrophages. Front Immunol (2017) 8:1243. doi:10.3389/fimmu.2017.01243
149. Deshmukh SD, Kremer B, Freudenberg M, Bauer S, Golenbock DT, Henneke P. Macrophages recognize streptococci through bacterial single-stranded RNA. EMBO Rep (2011) 12:71–6. doi:10.1038/embor.2010.189
150. Corbett NP, Blimkie D, Ho KC, Cai B, Sutherland DP, Kallos A, et al. Ontogeny of Toll-like receptor mediated cytokine responses of human blood mononuclear cells. PLoS One (2010) 5:e15041. doi:10.1371/journal.pone.0015041
151. Costa A, Gupta R, Signorino G, Malara A, Cardile F, Biondo C, et al. Activation of the NLRP3 inflammasome by group B streptococci. J Immunol (2012) 188:1953–60. doi:10.4049/jimmunol.1102543
152. Gupta R, Ghosh S, Monks B, Deoliveira R, Tzeng T, Kalantari P, et al. RNA and beta-hemolysin of group B Streptococcus induce IL-1beta by activating NLRP3 inflammasomes in mouse macrophages. J Biol Chem (2014) 289(20):13701–5. doi:10.1074/jbc.C114.548982
153. Charrel-Dennis M, Latz E, Halmen KA, Trieu-Cuot P, Fitzgerald KA, Kasper DL, et al. TLR-independent type I interferon induction in response to an extracellular bacterial pathogen via intracellular recognition of its DNA. Cell Host Microbe (2008) 4:543–54. doi:10.1016/j.chom.2008.11.002
154. Andrade WA, Firon A, Schmidt T, Hornung V, Fitzgerald KA, Kurt-Jones EA, et al. Group B Streptococcus degrades cyclic-di-AMP to modulate STING-dependent type I interferon production. Cell Host Microbe (2016) 20:49–59. doi:10.1016/j.chom.2016.06.003
155. Mancuso G, Gambuzza M, Midiri A, Biondo C, Papasergi S, Akira S, et al. Bacterial recognition by TLR7 in the lysosomes of conventional dendritic cells. Nat Immunol (2009) 10:587–94. doi:10.1038/ni.1733
156. Firon A, Dinis M, Raynal B, Poyart C, Trieu-Cuot P, Kaminski PA. Extracellular nucleotide catabolism by the group B Streptococcus ectonucleotidase NudP increases bacterial survival in blood. J Biol Chem (2014) 289:5479–89. doi:10.1074/jbc.M113.545632
157. Kolar SL, Kyme P, Tseng CW, Soliman A, Kaplan A, Liang J, et al. Group B Streptococcus evades host immunity by degrading hyaluronan. Cell Host Microbe (2015) 18:694–704. doi:10.1016/j.chom.2015.11.001
158. Vornhagen J, Quach P, Boldenow E, Merillat S, Whidbey C, Ngo LY, et al. Bacterial hyaluronidase promotes ascending GBS infection and preterm birth. MBio (2016) 7:e781–716. doi:10.1128/mBio.00781-16
159. Ulas T, Pirr S, Fehlhaber B, Bickes MS, Loof TG, Vogl T, et al. S100-alarmin-induced innate immune programming protects newborn infants from sepsis. Nat Immunol (2017) 18:622–32. doi:10.1038/ni1017-1173b
160. Kenzel S, Santos-Sierra S, Deshmukh SD, Moeller I, Ergin B, Fitzgerald KA, et al. Role of p38 and early growth response factor 1 in the macrophage response to group B Streptococcus. Infect Immun (2009) 77:2474–81. doi:10.1128/IAI.01343-08
161. Hoshi N, Schenten D, Nish SA, Walther Z, Gagliani N, Flavell RA, et al. MyD88 signalling in colonic mononuclear phagocytes drives colitis in IL-10-deficient mice. Nat Commun (2012) 3:1120. doi:10.1038/ncomms2113
162. Feuerstein R, Seidl M, Prinz M, Henneke P. MyD88 in macrophages is critical for abscess resolution in staphylococcal skin infection. J Immunol (2015) 194:2735–45. doi:10.4049/jimmunol.1402566
163. Bain CC, Bravo-Blas A, Scott CL, Gomez Perdiguero E, Geissmann F, Henri S, et al. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat Immunol (2014) 15:929–37. doi:10.1038/ni.2967
164. Madureira P, Andrade EB, Gama B, Oliveira L, Moreira S, Ribeiro A, et al. Inhibition of IL-10 production by maternal antibodies against group B Streptococcus GAPDH confers immunity to offspring by favoring neutrophil recruitment. PLoS Pathog (2011) 7:e1002363. doi:10.1371/journal.ppat.1002363
165. Andrade EB, Alves J, Madureira P, Oliveira L, Ribeiro A, Cordeiro-Da-Silva A, et al. TLR2-induced IL-10 production impairs neutrophil recruitment to infected tissues during neonatal bacterial sepsis. J Immunol (2013) 191:4759–68. doi:10.4049/jimmunol.1301752
166. Erny D, Hrabe De Angelis AL, Jaitin D, Wieghofer P, Staszewski O, David E, et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci (2015) 18:965–77. doi:10.1038/nn.4030
167. Olaison L, Belin L, Hogevik H, Alestig K. Incidence of beta-lactam-induced delayed hypersensitivity and neutropenia during treatment of infective endocarditis. Arch Intern Med (1999) 159:607–15. doi:10.1001/archinte.159.6.607
168. Furtek KJ, Kubiak DW, Barra M, Varughese CA, Ashbaugh CD, Koo S. High incidence of neutropenia in patients with prolonged ceftaroline exposure. J Antimicrob Chemother (2016) 71:2010–3. doi:10.1093/jac/dkw062
Keywords: S. agalactiae, Group B Streptococcus, cellular innate immunity, microbiome, colonization, sepsis
Citation: Kolter J and Henneke P (2017) Codevelopment of Microbiota and Innate Immunity and the Risk for Group B Streptococcal Disease. Front. Immunol. 8:1497. doi: 10.3389/fimmu.2017.01497
Received: 01 September 2017; Accepted: 24 October 2017;
Published: 10 November 2017
Edited by:
Kirsty Le Doare, Imperial College London, United KingdomReviewed by:
Paola Massari, Tufts University School of Medicine, United StatesPietro Speziale, University of Pavia, Italy
Copyright: © 2017 Kolter and Henneke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Philipp Henneke, cGhpbGlwcC5oZW5uZWtlQHVuaWtsaW5pay1mcmVpYnVyZy5kZQ==