 Christophe Pedros
Christophe Pedros Amnon Altman
Amnon Altman Kok-Fai Kong
Kok-Fai Kong- Division of Cell Biology, La Jolla Institute for Allergy and Immunology, La Jolla, CA, United States
Follicular helper T (TFH) cells represent a highly specialized CD4+ T cell subpopulation that supports the generation of germinal centers (GC) and provides B cells with critical signals promoting antibody class switching, generation of high affinity antibodies, and memory formation. TFH cells are characterized by the expression of the chemokine receptor CXCR5, the transcription factor Bcl-6, costimulatory molecules ICOS, and PD-1, and the production of cytokine IL-21. The acquisition of a TFH phenotype is a complex and multistep process that involves signals received through engagement of the TCR along with a multitude of costimulatory molecules and cytokines receptors. Members of the Tumor necrosis factor Receptor Associated Factors (TRAF) represent one of the major classes of signaling mediators involved in the differentiation and functions of TFH cells. TRAF molecules are the canonical adaptor molecules that physically interact with members of the Tumor Necrosis Factor Receptor Superfamily (TNFRSF) and actively modulate their downstream signaling cascades through their adaptor function and/or E3 ubiquitin ligase activity. OX-40, GITR, and 4-1BB are the TRAF-dependent TNFRSF members that have been implicated in the differentiation and functions of TFH cells. On the other hand, emerging data demonstrate that TRAF proteins also participate in signaling from the TCR and CD28, which deliver critical signals leading to the differentiation of TFH cells. More intriguingly, we recently showed that the cytoplasmic tail of ICOS contains a conserved TANK-binding kinase 1 (TBK1)-binding motif that is shared with TBK1-binding TRAF proteins. The presence of this TRAF-mimicking signaling module downstream of ICOS is required to mediate the maturation step during TFH differentiation. In addition, JAK-STAT pathways emanating from IL-2, IL-6, IL-21, and IL-27 cytokine receptors affect TFH development, and crosstalk between TRAF-mediated pathways and the JAK-STAT pathways can contribute to generate integrated signals required to drive and sustain TFH differentiation. In this review, we will introduce the molecular interactions and the major signaling pathways controlling the differentiation of TFH cells. In each case, we will highlight the contributions of TRAF proteins to these signaling pathways. Finally, we will discuss the role of individual TRAF proteins in the regulation of T cell-dependent humoral responses.
Introduction
Production of high-affinity immunoglobulins (Ig) by B cells represents an essential component of protective immunity against pathogens. Antibodies (Abs) function through various mechanisms including specific binding and neutralization of pathogens or toxins, activation of the classical complement pathway, opsonization of pathogens through phagocytosis by innate immune cells, and induction of antibody-dependent cell cytotoxicity (1). The initial activation of naïve B cell leads to the production of secreted IgM and cell surface-bound IgD. After activation, B cells undergo class-switch and acquire the capacity to produce Abs belonging to the IgA, IgE, or IgG sub-classes, depending on environmental cues. These Ig subclasses, which differ in their heavy chains, function through different mechanisms and provide adaptability in response to the diverse forms of foreign antigens. Activated B cells can also undergo somatic hypermutations in the complementarity determining regions of the antigen-binding fragment (Fab), leading to the generation and selection of Ab-forming B cells expressing high-affinity Ig (1). B cells which lose affinity for their target or acquire autoreactivity during this process are eliminated. These B cell maturation events occur in specialized zones of the secondary lymphoid organs, dubbed the germinal centers (GC). GC B cells can differentiate into long-lived plasma cells, providing long lasting memory, and protection. The initial activation of a naïve B cell is T cell-independent, but the maturation events that lead to the generation of high affinity and long lasting protective Ab responses is critically dependent on help signals delivered by a specific CD4+ T cell population, known as follicular helper T (TFH) cells. TFH cells are characterized by the expression of the transcription factor Bcl6, the chemokine receptor CXCR5, ICOS and PD-1. They provide B cells with essential maturation signals, promote GC formation and reactions, and govern the development of high-affinity Abs (2–4). Expression of the costimulatory molecule CD40L by TFH cells plays a critical role in B cell activation and maturation, and the production of IL-21 and other cytokines by GC TFH cells influence B cell proliferation, survival and isotype switch.
Deficiency of TFH cells, such as in humans suffering from the X-linked lymphoproliferative disease (XLP) or in Bcl6fl/fl Cd4Cre mice, results in disruption of GC responses, impaired Ab production, and defective memory formation following immunization or infection (5, 6). In humans, several genetic mutations that affect TFH cell differentiation or function have been associated with primary immunodeficiencies characterized by failure to develop protective antibody responses such as the XLP, hyper-IgM syndrome, and common variable immunodeficiency (CVID) [reviewed in (7)]. On the other hand, dysregulated TFH responses, and uncontrolled GC reactions can lead to the production of autoantibodies implicated in the pathogenesis of several autoimmune diseases including systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), and multiple sclerosis (MS) [reviewed in (7)]. Dysregulated TFH responses can also contribute to allergic responses (8), favor the development of B cell malignancies such as follicular lymphomas (9, 10), and even give rise to several subsets of T cell lymphomas such as angioimmunoblastic T-cell lymphoma, follicular T cell lymphoma, and nodal peripheral lymphoma with TFH phenotype (11, 12). Among the mechanisms that dampen GC reactions and Ab responses, follicular regulatory T (TFR) cells represent a highly specialized subpopulation of Foxp3+ regulatory T cells (Tregs) that co-express Bcl6 and CXCR5. TFR cells have the ability inhibit TFH and B cell responses occurring in the GC [reviewed in (13)]. In Bcl6fl/fl Foxp3Cre mice, TFR deficiency leads to the development of late onset spontaneous autoimmune diseases and enhanced susceptibility to Ab-mediated autoimmunity (14). The involvement of TFR cells in the pathogenesis of human autoimmune diseases remains speculative, but alteration of the TFR:TFH ratio is observed in the blood of patients suffering from several autoimmune diseases [reviewed in (15)].
In light of the key contributions of TFH cells to immune responses, strategies aimed at promoting TFH responses have the potential to improve protective Ab responses against pathogens and vaccines efficacy. On the other hand, inhibiting TFH development or function could be of use for the treatment of immune-mediated diseases or malignancies where increased TFH and GC activity contribute to the disease development or severity such as myasthenia gravis, autoimmune thyroid disease, SLE or RA. Understanding the mechanisms and intracellular signaling pathways that control TFH differentiation and functions is therefore of paramount importance.
In this review, we will first chronicle the spatiotemporal cellular interactions during the multistage TFH differentiation process. Then, we will review the molecular interactions and the intracellular signaling pathways of the T cell receptor (TCR), costimulatory molecules of the immunoglobulin superfamily (IgSF), and tumor necrosis factor receptor superfamily (TNFRSF), and cytokine signaling that play major roles in the differentiation, maintenance, and functions of TFH cells. In each case, we will discuss the known contribution of the tumor necrosis factor receptor associated factors (TRAF) in these signaling pathways. Members of the TRAF family of proteins (TRAF1–6) have been initially identified for their modulation of signaling cascades downstream of TNFRSF members through their adaptor function and/or E3 ubiquitin ligase activity (16). The TRAF-dependent TNFRSF OX-40 (17, 18), GITR (19), and 4-1BB (20, 21) are implicated in the differentiation and functions of TFH cells. TRAF proteins can also participate in signaling from the TCR and the costimulatory receptor CD28 (22–28), which deliver critical signals leading to the differentiation of TFH cells. Engagement of the CD28-related costimulatory receptor ICOS is critical for TFH differentiation (29–31). ICOS plays an important role in TFH differentiation by recruiting phosphatidylinositol 3-kinase (PI3K) (31). Interestingly, ICOS does not recruit TRAFs directly but its cytoplasmic tail contains a binding motif for the TRAF family member-associated NF-κB activator (TANK)-binding kinase 1 (TBK1). This TBK1-binding motif is also present in TRAF2, 3 and 5, the TRAF proteins known to bind TBK1 (32). The presence of this motif in ICOS and the expression of TBK1 are required for the late step of TFH differentiation (32). Furthermore, TRAF proteins can also interfere with the JAK-STAT pathways that are activated downstream of the IL-2, IL-7, IL-6, IL-21, and IL-27 cytokine receptors (33–36) and might therefore affect TFH development by modulating cytokine signaling.
Following discussion of the surface receptors regulating TFH development, we will summarize the TRAF-dependent canonical and non-canonical NF-κB pathways that lead to the differentiation and functions of T-dependent Ab responses. Finally, we will focus on the role of individual TRAF proteins in the regulation of T cell-dependent humoral responses, and discuss their potential contributions at the mechanistic level based on their involvement in the multiple signaling pathways that affect humoral responses.
Cellular Interactions in TFH Differentiation
Differentiation of TFH cells is a complex multistep process. It involves sequential interactions between CD4+ T cells and professional antigen-presenting cells (APC), namely, dendritic cells (DCs), and B cells. Using traceable immunization and pathogen infection models, the TFH differentiation process can be divided into three spatiotemporal phases: (1) Initiation of TFH differentiation by DC priming of naïve CD4+ T cells in the T cell zone of the secondary lymphoid organs; (2) TFH maturation induced by interactions with cognate B cells at the T-B border; and (3) the functional/maintenance phase, within the GC [reviewed in (37, 38)]. The antigen-specific interactions between developing TFH and B cells provide a bidirectional communication that is critical for the maturation of both adaptive immune cells.
During the first few days (days 1–3) following immunization or viral infection, DCs, which are activated at the inflammatory site, enter secondary lymphoid organs and present the engulfed foreign peptides to naïve T cells at the interfollicular and paracortical T cell zones (39). Naïve T cells recognizing the peptide-MHC complex are activated and primed, leading to the induction of the transcription factor Bcl6 (40, 41). Bcl6, the master regulator of TFH cells, is a transcriptional repressor that antagonizes the expression of other lineage-specific transcription factors (42) and microRNAs (43). Bcl6 represses CCR7, the chemokine receptor for the chemokine CCL19 and CCL21 predominantly expressed in the T cell zone, and indirectly promotes the expression of the chemokine receptor CXCR5, the receptor for CXCL13 produced within the B cell zones. As a result of this shift in surface chemokine receptors, these Bcl6+CXCR5+ pre-TFH cells are no longer retained in the T cell zones, but are attracted along the CXCL13 chemokine gradient toward the T-B border (44). Several costimulatory molecules, such as ICOS, OX40, and CD40L, are also upregulated at the priming stage, regulating the migration, differentiation, and commitment to the TFH cell fate.
During the following few days (day 4–6), the second step of TFH differentiation begins at the T-B border, where pre-TFH cells seek out and interact with cognate B cells. Successful interactions with B cells provide pre-TFH cells with critical signals that ensure the continuation of TFH differentiation programming. During this stage, the expression of Bcl6 and CXCR5 continues to rise, promoting the migration of TFH cells deeper into the B cell follicles, and acquisition of the capacity to help B cells (45). In turn, B cells receive reciprocal signals from differentiating TFH cells, promoting their maturation and entry into the B cell follicles. Only stable T-B conjugates further migrate into the GC (46).
The third phase (day 7 and beyond following immunization or infection) occurs within the GC. Fully differentiated TFH cells localized in the B cell follicles, dubbed GC TFH cells, are characterized by the highest expression of CXCR5 and Bcl6 as well as high expression of PD-1 (44). Through their high expression of CD40L and production of the cytokines IL-4 and IL-21, GC TFH cells control GC B cell proliferation and survival, and drive affinity maturation and the generation of memory B cells. GC TFH can express IL-21 or IL-4 alone or in combination. IL-21-producing TFH cells are efficient in promoting somatic hypermutation, whereas IL-4-producing GC TFH have higher CD40L expression and are able to induce isotype switching and plasma cell differentiation (47). GC TFH cells require continuous antigenic stimulation for their maintenance. In the presence of further antigenic stimulation, long-lived memory TFH cells can persist and rapidly recall the TFH program upon reactivation (48).
The aforementioned cellular interactions between T cells and APCs influence TFH differentiation through a variety of signals delivered through engagement of the TCR, costimulatory molecules, and cytokine receptors. We will first discuss the molecules at play, and then review the implication of TRAF proteins in their signaling pathways.
Molecular Interactions in TFH Differentiation and Functions
Upon sequential interactions with DCs and B cells, the TFH differentiation program is initiated and maintained through integration of multiple signals received from the TCR, costimulatory and coinhibitory receptors, and cytokine receptors. In this part, we will review the role of these signals in TFH differentiation and the contribution of TRAFs in the signaling pathways that they trigger (summarized in Figure 1).
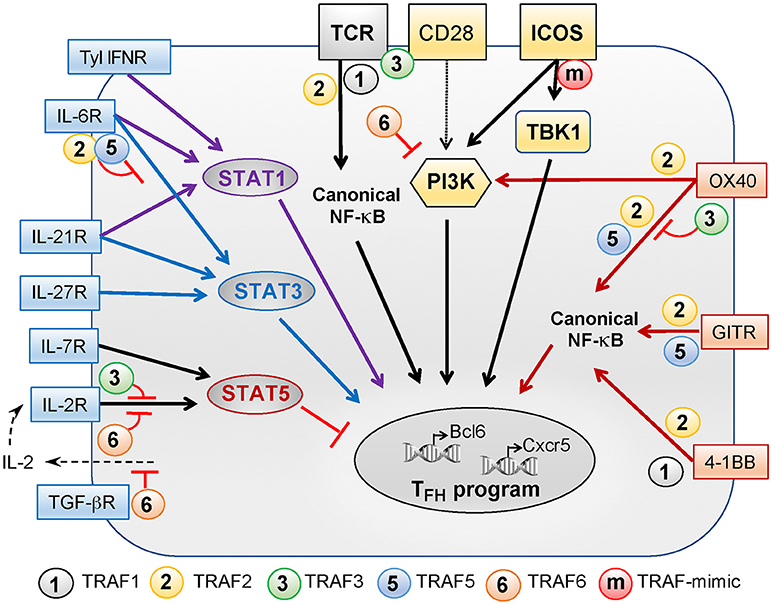
Figure 1. Role of TRAFs in TCR-, costimulatory receptor-, and cytokine receptor-associated signaling pathways controlling TFH differentiation. TFH differentiation depends on a variety of signals received through the TCR, costimulatory receptors of the Ig superfamily (yellow boxes), costimulatory proteins of the TNF receptor superfamily (orange boxes), and cytokine receptors (blue boxes). TRAF1, −2, −3, and −6 regulate TCR signals but TRAF3 activation depends on additional CD28 signaling. TRAF2 contributes to activation of the canonical NF-κB pathway that is critical for TFH differentiation. CD28 and ICOS plays a key role in TFH differentiation and can both activate PI3K. PI3K activation by ICOS is critical for TFH differentiation as compared to CD28-induced PI3K signals. Recently, we revealed that the TANK-binding kinase TBK1 functions downstream of ICOS to promote TFH differentiation. TRAFs are not recruited to ICOS but the presence of a TRAF-like motif in the intracellular tail of ICOS bypasses the need for TRAFs to recruit TBK1. The TNFR family members OX40, GITR, and 4-1BB signal through TRAFs and also contribute to TFH differentiation. TRAF2 is involved in recruitment and activation of PI3K by OX40. TRAF2 and−5 promote NF-κB pathway activation downstream OX40 and GITR while TRAF1 and−2 associate with 4-1BB to promote this pathway. TRAF3 plays a regulatory role in OX40 signaling. Signals through type 1 IFN, IL-6, and IL-21 receptors converge through STAT1 activation, positively regulating TFH differentiation. Signaling through IL-6, IL-21, and IL-27 receptors activates STAT3 to promote TFH differentiation. TRAF2 and−5 can both inhibit IL-6 mediated activation of STAT3. STAT5, a negative regulator of TFH differentiation, can be activated by signals through the IL-2 and IL-7 receptors. TRAF3 and−6 both negatively regulate IL-2R-induced signaling. Finally, TRAF6 is involved in the suppression of IL-2 production in the presence of TGF-β and, thus, could indirectly promote early TFH differentiation by limiting signals received through the IL-2R.
TCR Signaling
Engagement of the TCR is the initial and central event that triggers naïve T cell activation and differentiation. Together with other factors, including engagement of costimulatory or inhibitory receptors and cytokine signaling, the strength and duration of TCR signals impact the outcome of T cell activation and differentiation.
Using TCR-transgenic T cells with varying binding affinities to a pigeon cytochrome C peptide, it was revealed that T cells with a high-affinity TCR preferentially develop into CXCR5+ TFH (49). Concomitantly, a knock-in mouse strain expressing a mutated, non-signaling CD3ζ chain showed a selective defect in the generation of TFH cells (50). However, a high-affinity TCR does not appear to be an absolute prerequisite for TFH differentiation as TFH cells can also be generated after priming with intermediate and low affinity antigens (51). In the latter cases, B cells appear to play a key role in driving the differentiation of TFH cells with low TCR affinity (51). Additionally, experiments with different doses of antigen reveal that, for a given TCR affinity, increasing the amount of antigen available (45, 52) or a second peptide immunization that prolong antigen presentation (53) favors TFH differentiation. In contrast, another group demonstrated that the differentiation of TFH cells is reduced upon immunization with high doses of strong agonist peptide, as compared to lower doses (54). Differences in the inflammatory environment generated by the different antigen delivery systems might therefore influence the strength of TCR signals in favoring or antagonizing TFH differentiation. Taken together, our current understanding is that strong and sustained TCR–DC interactions promote TFH differentiation. Indeed, intravital imaging analysis reveals that sustained T-DC interactions promote TFH differentiation (52, 55).
TRAF1, 2, 3, or 6 can positively or negatively modulate signaling downstream of the TCR-CD3 complex (Figure 1). For example, Traf1−/− CD8+ T cells exhibit increased levels of active p52 after anti-CD3 stimulation, indicating that TRAF1 restrains the activation of the non-canonical NF-κB pathway in the absence of costimulation (23). As a result, Traf1−/− T cells hyperproliferate in response to stimulation with anti-CD3 Ab (22, 23). In contrast, Traf2−/− CD4+ T cells show reduced proliferation and activation after in vitro anti-CD3 stimulation (24). TRAF2 plays a positive role in the regulation of NF-κB signaling as Traf2−/−Tnf −/− T cells display a constitutively active non-canonical NF-κB pathway (56). In the absence of TRAF3, T cells exhibit reduced proliferation and cytokine production following costimulation with anti-CD3/CD28 Abs, reflecting an impaired activation of TCR signaling molecules Zap70, LAT, Erk, and PLCγ1 (25). Furthermore, TRAF3 has been shown to sequester the membrane localization of the kinase Csk and the phosphatase PTPN22, two known inhibitors of the TCR signaling, thereby reducing the threshold of T cell activation (26). On the other hand, Traf6−/− T cells hyperproliferate in vitro in response to stimulation with anti-CD3 Ab alone, bypassing the requirement for costimulation. Interestingly, the NF-κB pathway is independent of TRAF6. Instead, Traf6−/− T cells exhibit constitutive activation of phosphatidyl-inositol 3-kinase (PI3K), demonstrating that TRAF6 negatively regulates PI3K signaling following TCR engagement (27). In addition, TRAF6 can also be recruited to the T cell immunological synapse through the adaptor molecule LAT, promoting its ubiquitination and phosphorylation and positively regulating the activation of the calcium-sensing transcription factor, nuclear factor of activated T cells (NFAT) (28). Hence, it is becoming increasingly apparent that TRAF1, 2, 3, and 6 can influence the quality and intensity of TCR signaling through various mechanisms. However, it remains to be determined whether this TRAF-dependent modulation of TCR signaling is necessary and/or sufficient to significantly impact the differentiation of TFH cells.
Costimulatory Signaling
CD28 Signaling
Activated DCs present pathogen-derived peptide antigens associated with MHC class II molecules and upregulate the costimulatory ligands CD80 and 86, which interact with the costimulatory receptor CD28 on T cells. Interestingly, there is a selective preference for CD86 over CD80 to induce the formation of TFH cells (57, 58). This reflects the fact that CD86 is a higher affinity ligand of CD28 (59). As a result, the CD86-CD28 interaction is less likely to be attenuated by the competing CD86-CTLA-4 interaction, and, therefore, could deliver a more sustained stimulatory signal than CD80.
Signals elicited through CD28 are essential for the activation of naïve T cells and their development into all effector T cell subsets. The differentiation of TFH cells is no exception to this rule. The importance of CD28 for T-dependent Ab responses has been demonstrated using two different genetic models. First, Cd28−/− mice are deficient in GC formation and exhibit a delay in serum IgG titers following immunization with the hapten nitrophenol (NP) conjugated to chicken γ-globulin (NP-CGG) (60). Lack of CD28 costimulation in Cd28−/− T cells intrinsically inhibits the upregulation of the TFH master transcription factor Bcl-6 and, thus, all subsequent TFH differentiation steps are abrogated (61). Second, using a transgenic mouse strain ectopically expressing the soluble CD28 competitor, CTLA4–IgG fusion protein that blocks the interaction between CD28 and CD80/86, the T cell-dependent GC responses and antigen-specific TFH cells are dramatically attenuated (62, 63). However, this defect can be compensated by the coinjection of the NP-CGG antigen and an agonistic anti-CD28 Ab (63), because the latter bypasses the inhibitory effect of CTLA4-Ig. On the contrary, GC reactions in CTLA4-Ig mice are not restored when the agonistic anti-CD28 Ab is administered 10 days after immunization (64). Similarly, blocking CD28 by injection of CTLA4-Ig in wt mice 6–7 days post-immunization does not negatively impact TFH differentiation (61). Altogether, these results suggest that CD28 plays a key role during early T cell priming but not during the later phase of TFH maturation or maintenance in the GC. Consistent with this notion, the absence of CD80 specifically on DCs abolishes TFH differentiation whereas the absence of CD80 expression on B cells does not (65).
The signaling events that mediate CD28 function have been extensively studied, and signaling molecules that bind to specific motifs within the cytoplasmic tail of CD28 have been identified. The proximal tyrosine motif (YMNM) binds and activates the p85α subunit of PI3K as well as other adaptor proteins, including Grb2 and GADS. The distal proline-rich motif (PYAP) binds and activates Src family kinases and, indirectly, protein kinase C-θ (PKCθ) (66, 67). Using knock-in mouse strains expressing CD28 with mutations in either the proximal tyrosine motif or the distal proline-rich motif, it was demonstrated that the formation of GC and isotype switching are dependent on the PYAP motif, whereas the PI3K-binding YMNM sequence is dispensable (68). These results imply that CD28-mediated Lck and PKCθ signaling are critical for TFH differentiation. However, PI3K signaling mediated by CD28 is less important than PI3K signaling emanating from ICOS (see below).
ICOS Signaling
In humans, ICOS deficiency results in severe impairment of germinal center formation and inability to mount antibody responses against infection or vaccination (69, 70). Since its initial characterization (71), it has been established that ICOS is a major driver of T-dependent Ab responses and GC reactions. Icos−/− mice have defective GCs, impaired humoral response to antigens, and lack immunological memory (72–74). Similarly, ICOS ligand (ICOSL) deficiency or blockade of ICOS-ICOSL interaction using an anti-ICOSL Ab strongly reduces TFH development (29, 30). However, the temporal requirement for ICOS signals during the complex TFH differentiation process appears to vary depending on the experimental model. In an acute infection model, ICOS is required for the early CXCR5+Bcl6+ TFH differentiation of antigen-specific T cells as early as 3 days following infection with lymphocytic choriomeningitis virus (LCMV) (32, 75). Consistent with ICOS signaling during the early DC-T cell engagement favoring TFH differentiation through Bcl6 induction, ICOSL expression on CD8α− DCs favors the initiation of CXCR5+Bcl6+ TFH differentiation (76). In stark contrast, the early expression of Bcl6 by ovalbumin-specific OT-II CD4+ T cells is not affected by ICOS deficiency 3 days following NP-OVA immunization (61). Similarly, Icos−/− mice show intact TFH differentiation for as long as 6 days following infection with the non-lethal strain of malaria, Plasmodium chabaudi (77), indicating that early TFH differentiation can occur in an ICOS-independent manner in some models.
In addition to the priming stage, the ICOS-ICOSL interaction between TFH and B cells is also required for the maturation of developing TFH cells. Administration of an anti-ICOSL blocking Ab drastically curtails the TFH cell population in various infection models (61, 75, 77). Similarly, the expression of ICOSL by B cells is required for the generation of TFH cells (78). Additionally, ICOS is required for close contacts between T and B cells in the GC, promoting the expression of CD40L at the T cell surface and delivery of contact-dependent help to B cells (79).
ICOS-mediated activation of PI3K
PI3K signaling has been implicated as an important mediator downstream of several T cell molecules (TCR, CD28, CTLA-4, and ICOS). PI3K is a heterodimer consisting of a p110 catalytic subunit (of either the α, β, γ, or δ isoform) and a regulatory subunit, which can be p85α, p55α, p50α, p85β, or p55γ. The relevance of the ICOS-mediated PI3K signaling in the differentiation of TFH cells has been elegantly demonstrated using a knock-in mouse strain expressing an ICOS mutant incapable of binding PI3K (ICOS-YF). Similar to Icos−/− mice, ICOS-YF knock-in mice fail to generate TFH cells and GC reactions (31). The phenotype of ICOS-YF mice is in stark contrast to the CD28-YF mice, which are capable of mounting T cell-dependent Ab responses (68). This is consistent with the fact that ICOS delivers a more potent PI3K signaling than CD28 in T cells (80).
Although the PI3K-binding motifs of CD28 and ICOS differ by a single amino acid, i.e., YMNM in CD28 and YMFM in ICOS, the resulting difference in hydrophobicity property of these motifs confers a significant alteration in T cell signaling (81). ICOS triggering not only promotes the physical interaction between ICOS and the PI3K regulatory subunits p85α and p50α in activated T cells, but also promotes their recruitment to CD28 “in trans,” in the absence of CD28 ligation (80). Because p50α is the most potent isoform in regulating the kinase activity of PI3K (82, 83), ICOS ligation induces a higher PI3K activity as compared with CD28 ligation and delivers a more potent costimulatory signal favorable for the differentiation of TFH cells.
To understand the role of PI3K in the generation of humoral responses, several complementary approaches have been used. First, Ab responses, including isotype switching, GC formation, and GC B cells, are severely impaired in p110δ−/− mice following hapten-induced T-dependent and T-independent challenges (84). Second, using a mouse strain expressing a catalytically inactive form of p110δ (p110δD910A), but intact (active) p110α, and p110β isoforms, the abrogation of p110δ lipid kinase activity alone was sufficient to result in a near complete absence of GC and a profound reduction of serum IgG titers following immunization with T-dependent or T-independent antigens (85). However, these initial observations are confounded by the combined functional defects in T and B cell compartments. Third, a T cell-specific deletion of the p110δ catalytic subunit in p110δfl/fl Cd4Cre mice results in a nearly absence of CXCR5+PD-1+ GC TFH cells, and a significant reduction of GC B cells, GC reactions, and Ab affinity maturation following immunization with NP conjugated to keyhole limpet hemocyanin (KLH) (86). These findings reveal the non-redundant and T cell-intrinsic role of p110δ in TFH cell development. Fourth, p110δfl/fl Ox40Cre mice show similar defects in humoral responses following immunization (86). Since OX40 is expressed following TCR- and CD28-mediated T cell activation, ablation of p110δ at this later time point indicates that this catalytic subunit is crucial for T-dependent Ab responses after the initial activation of naïve T cells (86). Fifth, the magnitude and output from GC reactions are unperturbed in immunized p110δfl/fl Cd19Cre mice (86), implying that p110δ is dispensable in B cells, and/or that other PI3K catalytic subunits may contribute in a redundant manner to the GC reactions. Sixth, combined deletion of genes that encode four PI3K regulatory isoforms normally expressed in T cells (p85α, p55α, p50α, p85β) results in a drastic deficiency in T cell help to B cells in vivo. These mice display a significant reduction in GC numbers and size, as well as the production of class-switched Abs following immunization (87). Taken together, these data indicate that the ICOS-mediated PI3K pathway is crucial for T-dependent Ab responses.
Importance of PI3K-independent ICOS signaling
The PI3K-binding YMFM motif is a crucial feature of ICOS signaling in mediating the differentiation and functions of TFH cells. However, the knock-in ICOS-YF mouse strain, in which the association between ICOS and PI3K is selectively lost, is not a true phenocopy of Icos−/− mice (31). For example, in a model of respiratory infection with Chlamydia muridarum, ICOS-YF mice develop a much milder disease as compared to Icos−/− mice, albeit they are still not fully protected (88). Th17 responses negatively correlate with disease severity and are strongly reduced in Icos−/− mice but partially retained in ICOS-YF mice. Similarly, the severity of graft-vs.-host disease in ICOS-YF mice is intermediate between wild-type (wt) and Icos−/− mice, in a model of MHC-mismatched bone marrow transplantation (89). Interestingly, in this model, CD8+ T cells from ICOS-YF mice induce a disease indistinguishable from that induced by wt CD8+ T cells, whereas ICOS-YF and Icos−/− CD4+ T cells behave similarly. In vitro, ligation of ICOS induces T cell activation, calcium flux and proliferation of CD8± T cells in a PI3K-independent manner (89). Similarly, the PI3K-independent role of ICOS in activating calcium flux was demonstrated in CD4± T cells (31). Altogether, these data strongly evince the presence of important PI3K-independent pathway(s) downstream of ICOS.
TRAF-mimicking ICOS signaling
The aforementioned studies also pose a conundrum because other than its PI3K-binding motif, the cytoplasmic tail of ICOS lacks canonical motifs for protein-protein interactions. To resolve this issue, we looked for potential evolutionarily conserved sequence(s) in the cytoplasmic tail of ICOS (32). Remarkably, in addition to the YMFM motif, we found two additional highly conserved motifs in the intracellular domain of ICOS. They are the IProx motif (SSSVHDPNGE) and a more distal motif (AVNTAKK). Using an unbiased proteomics approach, TANK-binding kinase 1 (TBK1), a non-canonical member of the inhibitor of transcription factor NF-κB kinase (IKK) family, was unexpectedly found to interact with the serine-rich IProx motif. Mutation of this specific motif abrogated TBK1 binding to ICOS, but did not affect ICOS ability to recruit PI3K (32).
Similar to the mutation of the PI3K-binding motif (ICOS-YF), Icos−/− CD4+ T cells reconstituted with a mutated IProx motif (mIProx) displayed impaired CXCR5+PD-1+ GC TFH differentiation, GC formation, and IgG responses. Moreover, TBK1 knockdown in T cells resulted in defective humoral responses in response to acute LCMV infection (32). Although Icos−/− CD4+ T cells reconstituted with an ICOS-YF mutant fail to generate nascent CXCR5+ Bcl6+ TFH cells, this initial step of TFH development was not compromised in T cells expressing mIProx (32). Consistently, TBK1 was also dispensable for the development of nascent TFH cells, indicating that signals mediated by TBK1 binding to the ICOS IProx motif 'license' nascent TFH cells to enter the GC phase of TFH cell development. In agreement with our findings, it has recently been demonstrated that therapeutic inhibition of TBK1 reduced the number of GC TFH and their expression of Bcl6, caused a reduction in GC size, diminished the anti-collagen Ab levels and alleviated the progression of established collagen induced arthritis (90).
In contrast to the interaction between ICOS and PI3K, which can be induced by anti-CD3 or anti-ICOS stimulation alone, TBK1 coimmunoprecipitates with ICOS only when T cells are stimulated with a combination of anti-CD3 plus anti-ICOS Abs (32). These combined stimuli are physiologically provided by the strong cognate interaction that occurs in vivo between T cells and APCs. Hence, the requirement for activation of ICOS-TBK1 signaling is more stringent than that for the ICOS-PI3K pathway.
We further found that despite the known ability of TRAF2, 3, and 5 to physically interact with TBK1 (91–93), these TRAFs proteins were not corecruited with TBK1 to ICOS upon stimulation (32). Unexpectedly, the serine-rich IProx motif in ICOS turned out to be highly homologous with a region of TRAF2 and TRAF3 known as the “serine tongs,” which consists of the sequence SSSxxxPxGD/E (where 'S' is serine, “x” is any amino acid, “P” is proline, “G” is glycine and “D/E” indicates aspartic acid or glutamic acid). Substitution of this region in TRAF2 and TRAF3 with a string of alanines abolished their ability to bind TBK1. Thus, this sequence, which is also present in a similar form in the cytoplasmic region of ICOS, represents a previously unknown consensus TBK1-binding motif. The presence of this motif in ICOS therefore allows it to directly recruit TBK1, obviating the need for TRAF proteins as intermediary partners for TBK1 activation (Figure 1).
ICOS-dependent calcium signaling
The ability of ICOS to potentiate TCR-induced calcium flux is conserved in Icos−/− CD4± T cells expressing an ICOS mutant where most of the cytoplasmic tail is truncated, including the PI3K and TBK1 binding motifs (94), demonstrating that the ICOS-triggering calcium flux is independent of PI3K and TBK1. Interestingly, a short membrane anchoring sequence consisting of the sequence KKKY (where “K” is lysine and “Y” is tryptophan) is present in this mutant. Mutation of the KKKY motif in full-length ICOS dampens the calcium response in T cells, showing that this motif is both necessary and sufficient for calcium flux (94). This motif in ICOS is likely to positively regulate TFH responses since ICOS engagement and calcium flux promote CD40L surface expression (79), a critical requirement for TFH to provide B cells with contact dependent help signals.
OX40 Signaling
CD28 costimulation induces the expression of OX40, a TNFRSF member, on T cells (63). OX40 stimulation is involved in upregulation of Cxcr5 mRNAs (95), and higher expression of OX40 has been reported on TFH cells (49). However, the degree to which OX40 influences the development of TFH cells and Ab responses is highly context-dependent. Ox40−/− mice are able to mount effective humoral responses against acute LCMV, vesicular stomatitis virus (VSV), and influenza A virus infections, suggesting a non-essential role for OX40 in TFH development (96). Similarly, the absence of OX40 does not affect the expression of CXCR5 on antigen-specific CD4+ T cells and the development of IgG1 responses after infection with the rodent roundworm Heligmosomoides polygyrus (97). In stark contrast, OX40 is required to mount an efficient TFH and humoral response against chronic infection with the Clone 13 strain of LCMV. Ox40−/− mice are not able to control viral replication (17). In a Vaccinia virus infection model, Ox40−/− mice also exhibit a dramatically reduced TFH differentiation and Ab response. Blocking experiments using an anti-OX40L Ab showed that the OX40-OX40L interaction is required for both TFH generation and maintenance in this model (18). Variations in the expression of OX40 by CXCR5+ CD4 T cells in different mouse strains might account for the differential impact of OX40L Ab treatment on TFH generation and GC responses (29).
OX40 signaling can induce the expression of multiple TFH molecules, including CXCR5 and IL-21, by human T cells, and likely contributes to the pathogenic role of TFH cells in SLE (98). Mechanistically, TRAF2, 3, and 5 are recruited to the cytoplasmic tail of OX40 [Figure 1; (99, 100)]. However, TRAF2 plays a more important role in OX40 signaling by promoting the recruitment of PI3K, AKT, PKCθ, and IKKα, β, and γ, which trigger the mTOR and the canonical NF-κB pathways (99–101).
GITR Signaling
GITR expression is induced late during the maturation phase of GC TFH cells (102). The number of TFH cells is not affected in Gitr−/− mice during the first week of chronic infection with LCMV Clone 13. This is consistent with findings that the initial production of LCMV-specific IgG is not affected. However, Ab titers do not increase in Gitr−/− mice beyond the first week of infection. This defect is associated with an increase in the proportion of splenic Foxp3+ CXCR5+PD-1+ TFR cells, and a reduction in Foxp3− CXCR5+PD-1+ TFH cell numbers, suggesting that GITR plays a role in regulating the ratio between TFR and TFH cells. This GITR-mediated function is T-cell intrinsic because in mixed bone marrow chimera experiments, the TFH cell population is diminished in Gitr−/− CD4+ T cells, as compared to the wt CD4+ TFH cells following chronic LCMV infection (102). Consistent with a role for GITR signals in promoting humoral responses, administration of a recombinant GITR ligand protein enhanced the frequency of CXCR5+ICOS+ TFH cells and the expression of Bcl6 and IL-21 in a model of collagen-induced arthritis (19). Conversely, blocking GITR signals using a GITR-Fc fusion protein reduced the frequency of TFH cells, IgG production, and disease severity (19).
As a member of the TNFRSF, several TRAF molecules interact with the cytoplasmic tail of GITR. In CD8+ T cells, TRAF2 and 5 are involved in activation of the canonical NF-κB pathway triggered by GITR stimulation [Figure 1; (103)]. Additionally, the GITR-TRAF5 axis is known to activate the MAP kinase signaling pathways because Traf5−/− CD4+ T cells are defective in the activation of p38 and ERK kinases (104). On the other hand, TRAF3 has been demonstrated to inhibit the activation of the non-canonical NF-κB pathway triggered by GITR engagement (105). However, it is unclear which of these TRAF molecule(s) in the GITR signaling pathway plays a more prominent role in the maintenance of TFH cells.
CD40L Signaling
CD40L is rapidly upregulated upon TCR and costimulatory receptor ligation, and/or cytokine signaling (106). CD40L expressed on T cells is the ligand for the TNFRSF member CD40 expressed on B cells. CD40-CD40L signaling is essential for the development of T cell-dependent humoral responses. Cd40−/− or Cd40l−/− mice are severely defective in their ability to generate GC or develop IgG responses (107, 108). Similarly, individuals deficient for CD40 or CD40L suffer from hyper-IgM syndrome characterized by elevated IgM level, disrupted GC formation and reduced IgG, IgA and IgE levels (7). The requirement for CD40L signals received by T cells appears to be dispensable for early TFH differentiation during the DC priming phase (75), consistent with the fact that CD40L is highly expressed after the priming phase. Similarly, CD40 expression by DC is dispensable for an efficient TFH and IgG response, whereas CD40 expression on B cells is absolutely required for the generation of GC and TFH development (65). Interestingly, the intrinsic role of CD40L signaling in T cells does not appear to be critical for TFH differentiation as wt and Cd40l−/− antigen-specific T cells expand and differentiate into TFH cells to a comparable extent in a cotransfer experiment (65).
Very little is known about CD40L signaling in T cells. However, engagement of B cell-expressed CD40 by CD40L directly or indirectly recruits TRAF1, −2, −3, −5, and −6 to its cytoplasmic domain [reviewed in (109)]. The persistent TRAF-dependent CD40 signaling in B cells, delivered by CD40L expressed by T cells is considered to be one of the most potent signals in mediating different aspects of B cell biology, including differentiation, survival, proliferation, expression of costimulatory molecules, and cytokines, maturation of GC B cells, isotype switching, somatic hypermutation, and formation of long-lived plasma cells and memory B cells.
4-1BB Signaling
4-1BB (CD137 or TNFRSF9) is highly expressed on human TFH cells (110). However, 4-1bb−/− mice show no impairment of IgG production following VSV infection (111). Similarly, the absence of 4-1BB ligand in 4-1bbl−/− mice does not affect the T cell-dependent Ab responses (112). These data imply that 4-1BB and its ligand might be dispensable for the generation of T cell-dependent humoral responses in rodents. However, in vivo treatment with an agonistic anti-4-1BB Ab inhibits T cell-dependent Ab responses in various mouse models (20, 21, 113). In vivo administration of an agonistic anti-4-1BB Ab at the time of priming strongly reduces the development of Ab responses to T cell-dependent antigens (20). Additionally, treatment with an agonistic anti-4-1BB Ab suppresses the ongoing CD4+ T cell-dependent autoantibody production in the NZB × NZW mouse model of SLE (21). Therefore, excessive 4-1BB signals during both the initiation of Ab responses and their maintenance could negatively modulate TFH differentiation and/or functions. The exact mechanisms by which these agonistic Abs influence T cell-dependent humoral responses warrants careful interpretation because the expression of 4-1BB is not restricted to T cells. For instance, one study suggested that anti-4-1BB Ab treatment blocks GC formation by downregulating the follicular dendritic cell (FDC) network (114), a specialized subset of follicle-residing cells that support the GC reaction.
Cytoplasmic TRAF1 and TRAF2 are recruited to 4-1BB upon stimulation [Figure 1; (100, 115)]. TRAF1 is required for the activation of the classical NF-κB pathway following 4-1BB engagement (23). Following stimulation with an agonistic anti-4-1BB Ab, 4-1BB is internalized to an endosomal compartment. TRAF2, and its K63 polyubiquitination activity, colocalizes with 4-1BB in endosomes. The TRAF2-associated E3 ubiquitin ligase activity and K63 polyubiquitination are required for the 4-1BB-mediated activation of the classical NF-κB pathway (116). Additionally, TRAF2 mediates the p38 MAP kinase pathway downstream of 4-1BB as T cells expressing a dominant negative form of TRAF2 lose the ability to signal via the p38 cascade (117).
Coinhibitory Signaling
CTLA-4 Signaling
The inhibitory receptor CTLA-4 is constitutively expressed on Tregs and highly expressed on TFR cells (118). CTLA-4 plays a key role in the suppressive functions of Tregs (119) and Ctla4−/− mice develop systemic immune dysregulation, including increased TFH and GC B cell responses (58). Short-term blockade with anti-CTLA-4 Ab or Treg-specific deletion of CTLA-4 increases TFH and GC B cell responses in vivo (58, 120, 121) and reduces the ability of TFR cells to inhibit B cell activation in vitro upon coculture with TFH cells (120). CTLA-4 expressed by Tregs/TFR cells therefore has a major influence on TFH responses. Similar to the findings in mice, heterozygous, deleterious mutations in the human CTLA4 gene manifest an immune dysregulation disorder, characterized by lymphocytic infiltration of multiple non-lymphoid organs. These individuals exhibit increased frequency of circulating CXCR5±PD1± TFH cells, which is normalized in response to treatment with CTLA4-Ig therapy (122).
CTLA-4 is also expressed by TFH cells, although at a lower level than in TFR cells (120). The cell-intrinsic role of CTLA-4 in TFH differentiation and functions is far less defined than its cell-extrinsic role through Tregs and TFR cells. One study reported that late deletion of CTLA-4 from in vivo differentiated TFH cells using an inducible Cre/lox system increased their ability to induce isotype class switching and IgG production upon coculture with B cells (120). TFH-expressed CTLA-4 might therefore function to limit the B cell-stimulating activity of TFH cells in a cell-intrinsic manner. Its contribution to TFH differentiation during interactions between nascent TFH cells and B cells is currently unknown.
CTLA-4 delivers its negative signaling via multiple mechanisms. At the cell surface, CTLA-4 competes with CD28 for access to the CD80/86 ligands. Through a process called trogocytosis, CTLA-4 removes CD80/86 ligands from the surface of APCs, further limiting the availability of these ligands for CD28 (123). Intracellularly, the tyrosine-phosphorylated cytoplasmic domain of CTLA-4 can interact with the phosphatases SHP-2 and PP2A (124, 125). Altogether, these CTLA-4-mediated cell-extrinsic and cell-intrinsic mechanisms dampen signaling downstream of the TCR and CD28. As such, it is conceivable that TRAF molecules, which can modulate TCR and CD28 signaling (see above), may potentially influence the CTLA-4-mediated regulatory pathway to modulate T cell signaling during an immune response. On the other hand, in Foxp3+ Tregs, CTLA-4 recruits the kinase PKCη to potentiate its suppressive functions in vitro and in vivo (126, 127). The CTLA-4-PKCη complex promotes the activation of the canonical NF-κB pathway in Tregs, representing a unique positive signaling event (126). It remains, however, to be determined whether and how the CTLA-4-PKCη axis regulates the activity of the TFR subset. Although two other members of the novel PKC family, PKCδ and PKCε, have been shown to promote TRAF2 phosphorylation, IKK, and NF-κB activation in response to TNFα (128), it is unknown whether TRAFs are involved in the CTLA4-PKCη signal transduction pathway.
PD-1 Signaling
GC TFH cells express high levels of PD-1, consistent with this immunomodulatory protein being upregulated following chronic TCR stimulation, such as in the case of persistent interaction between T and B cells, which occurs during TFH differentiation. The PD-1 ligands, PD-L1, and PD-L2, are also highly expressed by GC B cells (129). PD-1 inhibits T cell activation by suppressing CD28 costimulatory signaling (130). In the absence of PD-1, early TFH differentiation is not affected, but the GC TFH cell population is enriched at later time points (129). Similar studies investigating the role of PD-1-PD-L1 interaction in TFH responses consistently report an expansion of TFH cells in Pdl1−/− mice and PD-1-deficient (Pdcd1−/−) mice, respectively, following protein immunization and viral infection (131–133). These findings reveal that PD-1 signaling can limit the proliferation of TFH cells (134).
Surprisingly, the absence of PD-1 signals leads to a reduction of B cell responses in some studies, despite an expansion of the TFH cell population (129, 131, 133). In one study, the increased TFH cell numbers observed in Pdcd1−/− mice is associated with a reduced synthesis of Il4 and Il21 mRNA by these cells (129), potentially explaining the reduced GC B cell responses. The discrepancy between the increased TFH cell numbers and the reduced B cell responses could also result, in part, from the contributions of PD-1 to the TFR cell population. In one study, Pdcd1−/− mice have elevated numbers of TFR cells that display enhanced suppressive activity following immunization with NP-OVA (135). The contribution of this suppressive population has not been assessed in other studies. It is possible that PD-1 affects the ratio between TFH and TFR cells differently in various models.
The ability of PD-1 to inhibit T cell activation depends on the recruitment of phosphatases SHP-1 and SHP-2 to the cytoplasmic domain of PD-1 (136, 137). More importantly, CD28 costimulatory signaling is distinctively sensitive to the PD-1-associated phosphatase activity (130). The recruitment of p85α and the phosphorylation of the CD28-associated kinases, Lck and PKCθ, are attenuated by the PD-1-SHP complex (130, 136). Interestingly, TRAF6 interacts with SHP-1, and this molecular complex restrains the phosphorylation of the p85α subunit of PI3K and the activation of the canonical NF-κB pathway (138), suggesting that TRAF6 might interfere with the co-inhibitory signaling of PD-1.
BTLA Signaling
BTLA, the ligand of the TNFRSF member HVEM, is highly expressed on TFH cells. Btla−/− mice have elevated level of IgG in response to the T-cell dependent NP-KLH antigen (139). Moreover, Btla−/− mice produce autoantibodies spontaneously (140), indicating that BTLA acts as a negative regulator of the humoral response. Upon immunization, TFH generation is not affected in Btla−/− mice, but the number of GC B cells is elevated (141). BTLA acts in a T cell-intrinsic fashion as Btla−/− CD4+ T cells activated in vitro in presence of IL-6 increase the production of IL-21, and promote IgG2a and IgG2b Ab responses upon in vivo transfer.
Similar to PD-1, BTLA relies on dual tyrosine phosphorylation motifs in its cytoplasmic tail to recruit SHP-1 and SHP-2 in T cells (142). Because TRAF6 interacts with SHP-1 (138), it is conceivable that TRAF6 might affect BTLA signaling.
Cytokines and STAT Signaling
In addition to the TCR and costimulatory receptors, interactions of autocrine or paracrine cytokines with their cognate receptors provide essential signals that regulate the differentiation and function of TFH cells. The signal transducer and activator of transcription (STAT) proteins are critical integrators of cytokine signals. Multiple STAT molecules can be activated simultaneously by one or more cytokines (143). The differentiation of TFH cells is positively or negatively modulated by STAT3-dependent cytokines (IL-6, IL-21, IL-27) and STAT5-dependent cytokines (IL-2 and IL-7), respectively. Interestingly, several TRAF molecules are involved in these cytokine/STAT signaling cascades.
IL-6
IL-6 is a pleiotropic cytokine that plays a major role in inflammation. Several studies have independently demonstrated the importance of IL-6 in TFH generation (78, 144, 145). Indeed, Il6−/− mice display reduced GC formation and humoral responses (146). An initial spike of IL-6 production is detected on days 1–3 in both acute and chronic LCMV infection models (145). At the T cell priming stage, conventional DC secrete large amounts of IL-6 upon activation. IL-6 can transiently induce the expression of the transcription factor Bcl6 and cytokine IL-21 (42, 78), creating a positive feedback loop for enforcing the TFH cell fate. Hence, the early programming of TFH cells is abated in the absence of IL-6 (144). However, during chronic infection with LCMV Clone 13, a second wave of IL-6 expression is observed 3 weeks post-infection. FDC are responsible for the production of IL-6 at this late phase of viral infection (145). Administration of an IL-6-neutralizing Ab or IL-6R-blocking Ab 20 days after infection reduces Bcl6 expression, TFH and GC B cells (145). Interference with IL-6 functions also impairs the host's ability to clear the virus, indicating a late, but critical, role of IL-6 in maintaining an intact humoral response. However, other studies demonstrate that the differentiation of TFH cells is not compromised in Il6−/− mice or upon IL-6 neutralization (147–149), indicating that other signals, including IL-21, may compensate for the absence of IL-6 (see below).
Interestingly, the impact of T cell-specific deletion of IL-6Rα is less profound than the systemic deletion of IL-6 (144, 149, 150). The proportion of CXCR5+PD-1+ TFH cells is moderately reduced in Il6rafl/fl Cd4Cre mice following antigen immunization. Antigen-specific TFH cells generated in vivo in the absence of IL-6Rα show reduced expression of Bcl-6 and IL-21 (150). However, there is a significant reduction in the fraction of GC B cells and plasma cells, implying that IL-6Rα signaling is indispensable for TFH cell functions. At the molecular level, the IL-6 receptor is composed of IL-6Rα and the glycoprotein 130 (gp130), a signal transducer common to IL-6 receptor family members. TRAF2 and TRAF5 constitutively associate with gp130 (35, 36). This interaction suppresses the recruitment of STAT3 to the IL-6R complex, because they compete for the same binding site on gp130 (35, 36). Therefore, TRAF2 and TRAF5 are negative regulators of the IL-6R signaling pathway that could potentially limit the induction and functions of TFH cells (Figure 1).
IL-21
IL-21 is a member of the common γ-chain family of cytokines produced by activated T and B cells. Its cognate receptor, IL-21R, is also highly expressed on TFH cells (151) and GC B cells (152). Interestingly, lack of IL-21 or IL-21R does not affect the initial differentiation and expansion of TFH cells (153, 154). However, the contraction of CXCR5+ PD-1+ GC TFH cells occurs at a faster rate in Il21−/− or Il21r−/− mice after the first week of antigen challenge (153, 154). Although T cells are found in the GCs, these T cells are not able to support GC reactions in Il21−/− or Il21r−/− mice, leading to diminished levels of GC B cells, plasma cells, and serum IgG. Taken together, these data suggest that the IL-21-IL-21R axis is required for the TFH cell persistence and functions.
Despite supportive evidence, the T-cell intrinsic role of IL-21 in the generation of humoral responses is hotly contested. Studies using Il21−/− and Il21r−/− mice show that the in vivo generation of CXCR5+PD-1+ TFH cells in these mice is as robust as in wt mice following NP-KLH (152) or NP-CGG (147) immunization, or infection with LCMV (148) or Influenza (149). Several additional studies provide potential insights into this discrepancy: First, while the loss of either IL-6 or IL-21 alone has only a marginal effect on TFH development and GC formation in response to acute viral infection, the simultaneous loss of both cytokines in Il6−/−Il21−/− mice (149) or the neutralization of IL-6 in Il21−/− mice (148), significantly blunts the antiviral Ab responses. These results indicate that IL-6 and IL-21 can act redundantly or complementarily to promote TFH development. This is mechanistically conceivable because IL-6 and IL-21 signal predominantly through the same intracellular signal transducer, STAT3 (see below). Second, even in the presence of an intact TFH cell population, Il21−/− and Il21r−/− mice are severely defective in mounting Ab responses. Mixed bone marrow chimera experiments revealed that IL-21 acts directly on B cells (152). In the absence of IL-21, the proliferation of GC B cells is significantly curtailed at the later stage of viral infection. However, as mentioned earlier, TFH and B cells in GCs are mutually dependent on each other. The absence of either IL-21 or IL-21R on either T or B cells could lead to similar defects. Therefore, the impaired T cell-dependent humoral responses in intact Il21−/− and Il21r−/− mice do not reveal whether IL-21 acts in an autocrine fashion or, alternatively, whether T and/or B cells respond to IL-21 in a paracrine fashion. Transfer of wt or Il21r−/− T cells and B cells into irradiated recipient mice shows that the presence of IL-21R on both T and B cells is required for the optimal production of high-affinity Abs in response to LCMV infection (154). Nonetheless, owing to the essential and yet complicated roles of IL-21 in T-dependent humoral responses, a “cleaner” experimental setup, based on the inducible Cre/lox system, would be required to rigorously dissect the spatiotemporal functions of IL-21 and/or IL-21R in T and B cells.
TRAF5 acts a negative regulator of IL-21 production. Traf5−/− CD4+ T cells secrete significantly elevated amount of IL-21 upon CD3 plus CD28 costimulation in the presence of IL-6 and TGF-β (35). This effect is dependent on the presence of IL-6 as the binding of TRAF5 to the IL-6R complex restricts the activation of STAT3 (Figure 1).
IL-27
IL-27 is a member of the IL-6/IL-12 family of cytokines, which binds to a heterodimeric receptor consisting of IL-27Rα and gp130 subunits. Il27ra−/− mice display defective development of CXCR5+PD-1+ GC TFH cells (155). Stimulation with recombinant IL-27 in vitro enhances ICOS expression and IL-21 production from naïve CD4+ T cells (155, 156). Additionally, IL-27 is required to promote the maturation of GC B cells (157). In vivo, IL-27 promotes T-dependent Ab responses through a combination of T- and B cell-intrinsic mechanisms (157). Because IL-6 and IL-27 share the gp130 subunit, it is possible that TRAF2 and TRAF5 could similarly modulate the signaling events downstream of IL-27R, and hence, alter the differentiation and functions of TFH cells.
Type I IFN
The dependence on STAT1 for the early stage of TFH differentiation (144) implies a role for type I IFNs (IFNα/β) in this process, because STAT1 is the key transcription regulator downstream of the type I IFN signaling pathway. IFNα/β are ubiquitous cytokines produced by innate immune cells during the early phase of viral infection. An early report demonstrates that exogenous administration of IFNα/β strongly promotes the production of IgG in a dose-dependent manner following antigen immunization (158). Conversely, in the absence of the IFNα/β receptor (IFNAR) subunit IFNAR1, the differentiation of TFH cells, migration of TFH cells into the GC, and B cell responses are impaired following immunization (159–161). Mechanistically, IFNα/β signaling in DC induces the production of IL-6, which in turn promotes TFH differentiation in vivo (159). In vitro treatment of CD4 T cells with IFNα/β induces the expression of Bcl6, CXCR5 and PD-1, but not the production of IL-21, suggesting that T cell-intrinsic IFNα/β signaling can positively contribute to the TFH differentiation. In agreement, in mixed bone marrow chimera experiments, the TFH differentiation of Ifnar−/− T cells is compromised, compared to wt T cells in the same recipients, demonstrating a T cell-intrinsic role of IFNα/β in the TFH differentiation following immunization (160, 161). Paradoxically, in the context of experimental Plasmodium infection, the differentiation of TFH cells, GC B cells, and Ab responses are significantly enhanced in Ifnar1−/− mice or upon anti-IFNAR1 Ab neutralization (162, 163), implying a negative role of IFNα/β signaling in parasitic infections. The contrasting roles of IFNα/β in the differentiation and functions of TFH cells might reflect the differential requirement of IFNα/β and its signaling in viral vs. parasitic infections.
Upon stimulation with IFNβ, TRAF2 coimmunoprecipitates with the IFNAR1 subunit of the IFN receptor complex (164). Analysis of Traf2−/− mouse embryonic fibroblasts (MEF) shows that the formation of the p52-p65 complex in the non-canonical NF-κB signaling pathway is absent upon stimulation with IFNβ. However, the IFN-induced activation of the canonical NF-κB pathway, and the phosphorylation of STAT1, STAT2, and STAT3 are indistinguishable between WT and Traf2−/− MEFs. Therefore, in lieu of TRAF2, other TRAF molecules might regulate these latter signaling cascades in response to IFNα stimulation (see below).
STAT1 and STAT3
As described above, IL-6,−21, and−27 promote the differentiation, persistence and functions of TFH cells in a T cell-intrinsic manner. A common feature among these TFH-inducing cytokines is their signaling via the transcription factors STAT1 and STAT3. Not surprisingly, STAT1 or STAT3 deficiency affects the generation of T-dependent B cell memory and high affinity Ab-secreting cells. The lack of STAT3 leads to profound defects in the acquisition of B cell help functions. T-cell specific deletion of STAT3 significantly impairs the number of CXCR5+ TFH cells, GC B cells, and IgG levels in mice following challenge with antigen plus adjuvant or LCMV infection (78, 165, 166). In humans, individuals with missense mutations or short deletions of STAT3 suffer from Hyper-IgE syndrome, a primary immunodeficiency characterized by heightened susceptibility to Staphylococcus aureus and Candida albicans. T cells from these STAT3 mutated individuals fail to upregulate IL-21 and provide help to B cells upon in vitro culture and a reduction of circulating CXCR5± CD4± T cells is observed in patients suffering from hyper IgE syndrome resulting from STAT3 mutations (167).
In addition, type I interferon also mediates its signaling through STAT1. Knockdown of STAT1 in mouse T cells results in defective generation of early CXCR5+Bcl6+ TFH cells 2 days after infection. This defect is more pronounced when both STAT1 and STAT3 are absent, suggesting a redundant role of these transcriptional regulators as downstream mediators of IL-6, −21, −27, and type I interferon (144).
The crosstalk between TRAF proteins and STATs has only been studied in recent years. As aforementioned, gp130, which mediates signaling downstream of IL-6 and IL-21, interacts with TRAF2 and TRAF5. Traf5−/− CD4+ T cells exhibit an elevated phosphorylation of JAK1 kinase upon stimulation with IL-6, suggesting that the recruitment of these TRAFs to the IL-6R and IL-21R complexes limits the phosphorylation of JAK1 in T cells (168).
In addition, TRAF6 associates with, and mediates the ubiquitination of STAT3 in fibroblasts (169). This interaction represses the transcriptional activity of STAT3 and downregulates the expression of STAT3-regulated genes upon stimulation with IFNα (169), suggesting that TRAF6 acts as a negative signaling mediator of STAT3 downstream of Type I interferon signaling. TRAF3 inhibits STAT3 activation downstream of IL-6R signaling in B cells (170). Moreover, TRAF3 is required for the association of the phosphatase PTPN22 with JAK1, which in turn inhibits STAT3 phosphorylation (170). TRAF3 and−6 might negatively regulate STAT3 activity in T cells via similar mechanisms.
IL-12
IL-12 is well known for its key role in inducing Th1 differentiation in both mouse and humans. However, in rodents, stimulation of naïve mouse CD4 T cells in vitro in the presence of IL-12 induces the expression of both the TFH transcription factor Bcl6 and the Th1 transcription factor T-bet (171). IL-21±, IFNγ±, and IL-21± IFNγ± cells are simultaneously present in the in vitro culture. However, the percentage of IL-21 producing cells declines rapidly over time in favor of IFNγ± cells, coinciding with reduction of Bcl6 expression in favor of T-bet (171).
IL-12 has been shown to mediate the differentiation of human TFH cells. Activated DC can induce naïve human CD4 T cells to produce IL-21 in an IL-12 dependent manner (172), and conversely, naïve human T cells primed with IL-12 can induce B cells to produce Ig in vitro (172). In vitro stimulation of naïve human CD4 T cells in the presence of IL-12 also induces the expression of CXCR5, Bcl6, and ICOS (172–174). The role of IL-12 signals for TFH generation is also important for in vivo responses as individuals deficient in the IL12-receptor subunit IL-12Rβ1 display less circulating CXCR5± CD4 T cells, altered GC responses, and reduced numbers of memory B cells (174). Induction of IL-21 and Bcl6 by IL-12 depends on the transcription factor STAT4 (171, 172). GC TFH in human tonsils show high levels of activated STAT4, suggesting that they could be actively receiving IL-12 signals in vivo (174). Concomitantly, the generation of TFH and GC B cells is impaired in Stat4−/− mice 4 days following immunization, but not at later stages (171). To date, no TRAF activity has been identified in the IL-12R or STAT4 signaling.
IL-2
IL-2 acts primarily on T cells via the IL-2R, consisting of the α, β, and the common γ subunits. The high-affinity IL-2Rα, CD25, is differentially expressed in TFH and non-TFH cells. CD25 is downregulated in Bcl6+CXCR5+ TFH cells, whereas CD25+ T cells express the transcription factor Blimp1, which is antagonistic to Bcl6 (75). These findings support the notion that TFH cells do not require IL-2 signaling for their differentiation and functions. In fact, the expression of Bcl6 is elevated under limiting IL-2 conditions. The accumulated Bcl6 proteins in turn bind to DNA and repress its direct target Prdm1 (which encodes the transcriptional repressor Blimp-1) (175). Reduction of IL-2 signaling results in increased TFH cell differentiation during the early DC priming phase in Il2ra+/−− mice (176) or upon anti-IL-2 Ab-mediated neutralization. Correspondingly, treatment with recombinant IL-2 impairs TFH differentiation and suppresses GC B cell responses (177).
TRAF3 and 6 are both negative regulators of IL-2 signaling (Figure 1). TRAF3 is recruited to the IL-2 receptor complex and promotes the recruitment of the phosphatase TCPTP. Traf3fl/fl Cd4Cre T cells show enhanced phosphorylation of Jak1, Jak3, and STAT5 upon IL-2 stimulation (33). Additionally, TRAF6 coimmunoprecipitates with IL-2Rβ in 293T cells coexpressing those two proteins, and preactivated Traf6−/− CD4+ T cells display enhanced phosphorylation and activation of Jak1 and Erk in response to IL-2 (34). TRAF3 and TRAF6 could therefore contribute to the control of TFH differentiation by modulating IL-2 signals.
IL-7
IL-7, a member of the IL-2 cytokine family, is important for T and B cell survival, proliferation and development. Similar to CD25, IL-7Rα expression is strongly downregulated during TFH differentiation, as early as 3 days following LCMV infection (178). IL-7Rα is then progressively reexpressed and GC TFH cells express high IL-7Rα levels (178), consistent with a role for IL-7 in the long-term survival of memory T cells. The early downregulation of IL-7Rα suggests a negative role for IL-7 signals in the differentiation of TFH cells. Indeed, Bcl6 represses IL-7R (179) and, reciprocally, TFH exposure to IL-7 represses the expression of the key TFH genes, Bcl6 and Cxcr5 (180). Consistent with these findings, administration of anti-IL7Rα Ab enhances TFH development and GC reactions, whereas transgenic expression of IL-7Rα by CD4+ T cells reduces their TFH differentiation (179). Intriguingly, one study showed the opposite, i.e., positive role of IL-7 on TFH cells. The administration of exogenous Fc-fused IL-7 significantly increases both CD4+ and CD8+ T cell responses induced by a DNA vaccine (181). The enhancement of CD4+ T cell responses was accompanied by the expansion of TFH cells, GC B cells, and GC reactions (181). The enhanced development of TFH cells in this experimental model is not dramatically affected by IL-6 and IL-21 neutralization, suggesting an independent role of IL-7 in TFH differentiation. No TRAF activity has been associated with IL-7R.
STAT5
IL-2 and IL-7, which negatively regulate TFH generation, signal through STAT5. STAT5 acts as a transcriptional repressor for the expression of Bcl6 (182). Inhibition of Bcl6 expression correlates with the enhanced binding of STAT5 to the Bcl6 promoter region in Th1 cells stimulated with IL-2 in vitro (175). Similarly, in IL-7 stimulated cells, there is an increase of STAT5 binding to the Bcl6 gene promoter, leading to a reduction in Bcl6 expression (180). Accordingly, T cell-specific deletion of STAT5 increases TFH cell development, GC B cell numbers, and Ab levels following immunization (183). On the other hand, the presence of a constitutively active STAT5 mutant in antigen-specific T cells blocks the differentiation of TFH cells following LCMV infection (176).
TRAF3 and 6 are recruited to the IL-2R and negatively regulate its signaling activity (33, 34). After IL-2 stimulation, activation of STAT5 is enhanced in Traf3fl/fl Cd4Cre T cells, suggesting that TRAF3 acts as a negative regulator of STAT5 (33).
TGF-β
In the human immune system, TGF-β alone is insufficient to induce expression of the TFH cell phenotype (184). In vitro TGF-β stimulation in combination with IL-12 or IL-23 optimally promotes the expression of TFH-associated molecules Bcl6, CXCR5, ICOS and IL-21, and antagonizes Blimp1 expression, in naïve human CD4+ T cells (184). Elevated phosphorylation of Smad2, a downstream effector of TGF-β signaling, is found in T cells localized close to the GC in tonsils, suggesting that TGF-β signaling is likely to participate in human TFH differentiation (184). However, the requirement of TGF-β for TFH differentiation appears to be species-specific as in vitro stimulation of murine T cells with TGF-β inhibits the induction of Bcl6, IL-21 and ICOS (42, 184, 185). In contrast, experiments using adoptive transfer of antigen-specific Tgfbr2−/− T cells revealed that T cell-intrinsic TGF-β signaling is required for the differentiation of CXCR5+PD-1+ TFH cells and the generation of GC B cell and Ab responses in vivo following LCMV infection (186). TGF-β suppresses the expression of CD25. The absence of IL-2 signaling, in turn, is beneficial for the early induction of TFH cells.
In vitro stimulation of Traf6fl/fl Cd4cre murine T cells in the presence of TGF-β shows enhanced and sustained Smad2 and Smad3 phosphorylation. This sustained TGF-β signaling results in lower Il2 mRNA and protein levels (187). Therefore, TRAF6 acts as a negative regulator of Smad-mediated TGF-β signaling in T cells, and thus, may influence the differentiation and functions of TFH cells (Figure 1).
TRAF-Mediated Canonical and Non-Canonical NF-κB Signaling in TFH Cell Differentiation
TRAF family members are critical signal transducers that relay signals between stimulus-sensing surface receptors and transcription regulators, ultimately leading to a change in gene expression. Many studies using different cell types and stimuli reveal that TRAF family members are involved in the activation of the transcription factors of the NF-κB family. NF-κB can be activated via two major pathways: the canonical and non-canonical signaling pathways [reviewed in (188, 189)]. Briefly, the canonical NF-κB pathway is controlled by TAK1 kinase activation that leads to the ubiquitination and proteasomal degradation of IκB family members, resulting in the release and nuclear translocation of the NF-κB1/p50–RelA/p65 and NF-κB1/p50–c-Rel dimers. On the other hand, activation of the non-canonical NF-κB pathway depends on the NF-κB-inducing kinase NIK. NIK can phosphorylate and activate IKKα, which in turn promotes p100 processing to generate NF-κB2/p52 and allow its nuclear translocation together with RelB. In the absence of activating signals, constitutive ubiquitination and degradation of NIK ensures the repression of the non-canonical NF-κB pathway. Herein, we will focus on the role of TRAF proteins in the canonical and non-canonical NF-κB signaling pathways (summarized in Figure 2) and discuss how TRAF-mediated NF-κB signaling can contribute to TFH differentiation and T-dependent humoral responses. Readers are advised to refer to other chapters in this volume to gain a broader perspective of TRAF-mediated canonical and non-canonical NF-κB pathways in the immune system.
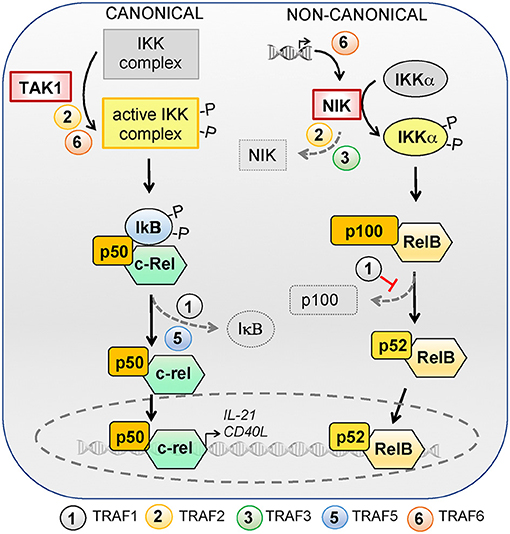
Figure 2. Role of TRAFs in the canonical and non-canonical NF-κB signaling pathways. NF-κB can be activated via the canonical or non-canonical signaling pathways. The canonical pathway is controlled by TAK1 kinase activation, which activates the IKK complex and leads to ubiquitylation and proteasomal degradation of IκB family members, resulting in the release and nuclear translocation of NF-kB1/p50–RelA/p65 and NF-κB1/p50–c-Rel dimers. RelA/p65 is dispensable for TFH differentiation but c-Rel regulates the expression of IL-21 and CD40L and is required for TFH differentiation. TRAF2 and−6 favor IKK complex activation by TAK1 and TRAF1 and−5 are required for optimal IκB degradation. The activation of the non-canonical NF-κB pathway depends on the NF-κB-inducing kinase NIK. NIK can phosphorylate and activate IKKα, which in turn promotes p100 processing to generate NF-κB2/p52 and allows nuclear translocation of NF-κB2/p52 and RelB. In the absence of activating signals, constitutive ubiquitylation and degradation of NIK ensures repression of the non-canonical NF-κB pathway. NIK deficiency in T cells does not impact TFH differentiation. TRAFs regulate the non-canonical NF-κB pathway by modulating NIK expression levels: TRAF6 is involved in transcriptional regulation of Nik whereas TRAF2 and−3 contribute to its degradation. TRAF1 restrains the non-canonical NF-κB pathway activation by inhibiting p100 processing.
TRAF-Mediated Canonical NF-κB Signaling in TFH Cells
Several studies demonstrate the T cell-intrinsic requirement for canonical NF-κB signaling in TFH differentiation. First, genetic ablation of the transcriptional subunit, NF-κB1/p50, in OT-II CD4+ T cells selectively impairs the upregulation of CXCR5 following immunization, leading to a severe defect in the generation of CXCR5+PD1+ GC TFH cells and GC B cell responses (190). Second, because the NF-κB1/p50 subunit dimerizes with RelA/p65 or c-Rel, Rel−/− mice (deficient for c-Rel) display defects in T cell-dependent humoral immunity (191). Subsequently, it was demonstrated that the mRNA and protein levels of IL-21 are reduced in Rel−/− T cells, indicating that c-Rel positively regulates the expression of IL-21 in T cells (192). Moreover, the expression of c-Rel is regulated by a microRNA, miR-155 (193). T cell-specific ablation of miR-155 promotes the degradation of c-Rel, which impedes the upregulation of CD40L in mir155−/− T cells, and severely impairs TFH differentiation and B cell Ab responses in vivo (193). This defect can be restored by over-expression of c-Rel in mir155−/− T cells, pointing to a T cell-intrinsic role for c-Rel in the control of TFH differentiation. Interestingly, the other NF-κB1/p50 partner, RelA/p65, is dispensable for TFH differentiation (194).
TRAF1, 2, 5, and 6 can positively regulate the activation of the canonical NF-κB pathway (Figure 2). Traf1−/− T cells show reduced IκB degradation upon stimulation with anti-4-1BB Ab (23). TRAF2 knockdown impairs the canonical NF-κB activation induced by anti-CD3/CD28 stimulation in Jurkat T cells (195), by anti-OX40 stimulation in T cell hybridomas (101), or by anti-4-1BB in HEK293T fibroblasts (116). Preactivated Traf5−/− T cells also show reduced canonical NF-κB activation upon treatment with anti-GITR Ab (104). In addition, knockdown of TRAF6 hinders the activation of the canonical NF-κB pathway in Jurkat T cells stimulated with anti-CD3/CD28 Ab (195), and the degradation of IκB is delayed in Traf6−/− T cells (27). Hence, TRAF1, 2, 5, and 6 can contribute to TFH differentiation by positively regulating the canonical NF-κB pathway.
TRAF-Mediated Non-Canonical NF-κB Signaling in TFH Cells
The role of non-canonical NF-κB signaling in T cell-dependent Ab responses has been extensively studied in Nik−/− mice, which display an impaired development of CXCR5+ PD-1+ GC TFH cells (196). However, TFH differentiation was not affected when NIK deficiency was restricted to T cells using an adoptive transfer model, implying that the role of NIK in TFH differentiation is not T cell-intrinsic (196). Instead, the expression of NIK in B cells is required for the optimal expression of ICOSL, and, thus, the promotion of TFH differentiation. These findings suggest that the non-canonical NF-κB signaling pathway in B cells, but not in T cells, is required for humoral responses. Although the deletion of either RelB or NF-κB2/p52 does not affect B cell responses, genetic ablation of both RelB and NF-κB2/p52 in GC B cells dramatically impedes GC reactions (197). Therefore, the non-canonical NF-κB signaling is more important in B cells, which in turn could affect the differentiation and maintenance of GC TFH cells.
TRAF2 and TRAF3 play a negative role in the control of the non-canonical NF-κB pathway (Figure 2). The absence of either TRAF2 or TRAF3 results in the constitutive activation of this pathway in T cells (25, 198). TRAF2 and TRAF3 form a complex with cIAP1 and cIAP2, which are E3 ubiquitin ligases responsible for NIK ubiquitination and degradation. In the absence of TRAF2 or TRAF3, the cIAP-TRAF complex is disrupted, allowing an increase of NIK protein level and aberrant activation of the non-canonical NF-κB pathway (199–201). In a similar fashion, TRAF1 has also been found to restrain the non-canonical NF-κB pathway in response to stimulation with anti-CD3 Ab (23). On the other hand, TRAF6 acts as a positive regulator by inducting the expression of NIK, resulting in activation of the non-canonical NF-κB pathway in the presence of OX40 ligation (202).
As T cell-specific Nik deficiency did not affect TFH differentiation (196), modulation of the non-canonical NF-κB pathway by TRAF2, 3, and 6 is not likely to directly impact on TFH differentiation. However, because the non-canonical NF-κB pathway is important for B cell maturation, which in turn is required to maintain TFH cells, TRAF1, 2, 3, and 6 might contribute to the overall T-dependent and T-independent Ab responses.
T Cell- and B Cell-Intrinsic Roles of TRAFs in Humoral Responses
In this part, we will review the contribution of individual TRAF proteins to the development of T cell-dependent humoral responses and discuss whether each TRAF member influences humoral responses through T cell-intrinsic or B cell-intrinsic pathways. In humans, single nucleotide polymorphisms of several members of the TRAF family are associated with the development of SLE and RA (16), two autoimmune disorders with excessive TFH responses and GC reactions (7). However, the mechanisms by which TRAFs contribute to disease susceptibility or development are unknown. In this part we will infer the potential contributions of each TRAF family member to the differentiation of TFH cells at the mechanistic level, in light of the known role of TRAFs in the signaling pathways controlling TFH differentiation reviewed in the preceding sections.
TRAF1
Traf1−/− mice display normal T cell and B cell lymphocyte development (22). Increased T cell proliferation of Traf1−/− T cells is observed in response to anti-CD3 Ab (22, 23) or antigen stimulation (203). Traf1−/− CD4+ T cells express higher levels of the Th2 cytokines IL-4, IL-5, and IL-13 upon in vitro stimulation. Accordingly, transfer of OVA-stimulated Traf1−/− CD4+ T cells into naïve wt recipients trigger an enhanced asthmatic response following aerosol inhalation with ovalbumin as compared to the transfer of OVA-stimulated wt CD4+ T cells (203). TRAF1 has been reported to associate with CD40 (204). However, the proliferation of Traf1−/− B cells is not affected upon in vitro stimulation with anti-IgM or anti-CD40 Abs, or in vivo challenge with T cell-independent antigens (22). Traf1−/− mice display normal IgG1, IgG2a, and IgE anti-ovalbumin responses, suggestive of an intact B cell isotype switching and T cell help (22). These data suggest that TRAF1 is dispensable for the development of T cell-dependent humoral responses.
TRAF2
Traf2−/− mice are embryonic lethal as a result of excessive TNFα production. Simultaneous deletion of the TNFα-TNFR1 axis results in partial rescue of the Traf2−/−Tnf−/− or Traf2−/−Tnfr1−/− animals (205). Traf2−/−Tnf−/− mice display normal IgM levels in response to VSV infection, but the IgG responses are abrogated. B cells from Traf2−/−Tnfr1−/− mice fail to proliferate and activate NF-κB in response to in vitro anti-CD40 stimulation (205). These data are consistent with the fact that TRAF2 interacts with CD40 (204), and that this interaction is essential for isotype switching (206). However, the fact that these models lack TRAF2 and TNFα-TNFR1 signaling confounds the interpretation regarding the actual role of TRAF2 in the development of T cell-dependent Ab responses.
To better define the roles of TRAF2 in B cells, Traf2fl/fl Mx1Cre and Traf2fl/fl Cd19Cre mice were generated, resulting in B cell-specific TRAF2 deletion (198, 207). Unexpectedly, B cell specific TRAF2 deficiency resulted in increased B cell numbers in the secondary lymphoid organs. Traf2−/− B cells display enhanced survival, increased cell size and constitutive activation of the non-canonical NF-κB pathway. Nevertheless, the CD40-mediated activation of the canonical NF-κB pathway and B cell proliferation are impaired in the absence of TRAF2, implying a positive regulatory role of TRAF2 in CD40 signaling. Similar to the Traf2fl/fl Mx1Cre and Traf2fl/fl Cd19Cre mice, mice expressing a TRAF2 dominant negative transgene (TRAF2-DN) devoid of the N-terminal RING and zinc finger domains display splenomegaly and lymphadenopathy (208). Surprisingly, the canonical NF-κB pathway is unperturbed in B cells isolated from TRAF2-DN mice in response to CD40L or TNFα stimulation. Instead, the activation of the JNK pathway is dependent on TRAF2 following CD40L or TNFα stimulation (208). The functional discrepancy between Traf2−/− and TRAF2-DN-expressing B cells could be explained by the conservation of TRAF2-mediated protein-protein interactions in TRAF2-DN-expressing B cells.
The contribution of TRAF2 to T cell functions and its implication in the regulation of Ab responses is under-explored. T cells from TRAF2-DN (208, 209) and Traf2fl/fl LckCre mice (24) show defective in vitro T cell proliferation in response to anti-TCR stimulation or allogenic APCs. Traf2−/− T cells show a propensity to skew into the Th2 lineage upon in vitro polarization and their Th17 differentiation is impaired (24). This is associated with reduced JNK and canonical NF-κB pathway activation following stimulation with TNFα. To date, a T cell-intrinsic role for TRAF2 in TFH differentiation has not been reported. However, because TRAF2 is involved in recruitment of PI3K to OX40 (210) and activation of the classical NF-κB pathway downstream of OX40 (101) and GITR (103), two key molecules promoting TFH differentiation, TRAF2 could potentially play a positive regulatory role in the differentiation of TFH cells. On the other hand, TRAF2 has been shown to restrain IL-6 signaling, suggestive of a negative role in TFH differentiation [Figures 1, 2; (36)]. Therefore, the overall role of TRAF2 in T-dependent Ab responses awaits further exploration.
TRAF3
Traf3−/− mice die within 10 days of birth. To assess the contribution of TRAF3 to T cell-dependent Ab responses, fetal liver cells were used to reconstitute the hematopoietic system of sublethally irradiated recipients (211). Traf3−/− fetal liver cells could reconstitute the T cell, B cell, granulocytic, and erythroid lineages, and reconstituted recipients survived longer than 6 months. Using this chimeric system, recipient mice reconstituted with Traf3−/− cells failed to produce antigen-specific IgG in response to T cell-dependent antigens. However, the proliferation of Traf3−/− B cells in response to stimuli such as anti-IgM Ab and CD40L was normal. Because the T cell recall response after in vivo immunization is dramatically reduced in the absence of TRAF3, it was concluded that TRAF3 is required for T cell help (211).
To circumvent the early postnatal lethality of Traf3−/− mice, Traf3fl/fl Cd19Cre mice with B cell-specific TRAF3 deletion were generated (198, 212). These mice exhibited splenomegaly and lymphoadenopathy, with a concomitant elevation of follicular B cells, spontaneous GC formation, hyperimmunoglobulinemia, T cell-independent Ab responses, and exacerbated autoimmune manifestations (212). At the molecular level, Traf3−/− B cells exhibit constitutive activation of the non-canonical NF-κB pathway, supporting the survival of B cells. In spite of all these B cell defects, the development of GC B cells in Traf3fl/fl Cd19Cre mice following immunization remained intact (213). Paradoxically, B cell-specific overexpression of TRAF3 in a transgenic mouse strain induced excessive systemic inflammation, autoimmunity, and hyperimmunoglobulinemia at an old age (214). These transgenic mice are hyperresponsive to T-dependent and T-independent antigen challenges, despite the fact that the over-expression of TRAF3 does not alter the CD40-mediated NF-κB and MAP kinase pathways in B cells. Altogether, these results indicate an important, yet complicated, role of TRAF3 in regulating B cell homeostasis.
To understand the roles of TRAF3 in T cell biology, Traf3fl/fl Cd4Cre mice were generated. These Traf3fl/fl CD4Cre mice are born at the expected Mendelian ratio, and they survive and breed normally (25). Following immunization with a T-dependent antigen, the antigen-specific IgG1 Abs are nearly absent in these mice, indicative of a T cell-intrinsic role of TRAF3 (25). In a Listeria monocytogenes infection model, Traf3fl/fl Cd4Cre mice are much more sensitive to bacterial challenge, displaying a higher bacterial load and lower numbers of IFNγ-producing T cells in the liver, demonstrating that Traf3−/− T cells are compromised (25). Mechanistically, TRAF3 is recruited to the TCR-CD28 complex and it participates in the activation of proximal TCR signaling (Figure 1). In its absence, proliferation and cytokine production are impaired in stimulated CD4+ and CD8+ T cells.
Interestingly, Treg-selective TRAF3 ablation in Traf3fl/fl Foxp3Cre mice leads to a marked reduction in TFR cell induction following immunization, resulting in increased expression of Bcl6, Cxcr5 and the cytokines genes Il-4, Il-10, Il-17, and Ifng by TFH cells, coupled with sustained GC reactions and production of high-affinity IgG Abs (215). The expression of ICOS is reduced in Traf3−/− TFR cells because of the inactivation of TRAF3-dependent ERK and AP-1 signaling pathways (215). However, whether TRAF3 influences TFH differentiation in a cell-intrinsic way remains to be determined. Interestingly, alternative splicing of TRAF3 can generates a TRAF3 isoform that mediates activation of the non-canonical NF-κB pathway and the production of CXCL13 by T cells (216). Although the relevance of TRAF3-controlled CXCL13 production in vivo in T cell-dependent Ab responses remains to be elucidated, these data suggest a positive role for TRAF3 in GC formation by favoring TFH cells migration into the GC. TRAF3 might also positively influence TFH differentiation by enhancing TCR- and CD28-induced signaling (25, 26) and restraining IL-2R signals [Figure 1; (33)]. Conversely, TRAF3 can inhibit IL-6R signaling in B cells (170) and negatively regulate OX40-induced NF-κB signaling in HEK293T cells (217). It is currently unknown whether a similar TRAF3-mediated regulation of IL-6R and NF-κB signaling occurs in primary T cells.
TRAF4
Traf4−/− mice show normal T and B cell differentiation. The T cell-dependent IgG response to OVA immunization is unaffected in Traf4−/− mice (218). Although the TFH cell population has not been investigated in this study, this finding suggests that TRAF4 is dispensable for the differentiation and functions of TFH cells. No other studies to date have demonstrated a role for TRAF4 in primary T cell functions.
TRAF5
Traf5−/− mice show unaltered development of T and B cell lineages (219, 220). Traf5−/− mice produce similar titers of IgG1 Ab than wt controls following antigen immunization, but there is a slight reduction in Ab affinity maturation (219). Traf5−/− T cells produce increased amounts of IL-4 and IL-5 in response to OX40 stimulation in vitro, and develop a more severe Th2-driven allergic lung inflammation following antigen immunization and airway challenge (220). In this model, Traf5−/− mice produce enhanced levels of OVA-specific IgE. Altogether, these data suggest that the T cell-dependent class switching and production of Ab in vivo are not dramatically affected by TRAF5 deficiency.
Notably, Traf5−/− T cells display impaired GITR signaling, with decreased canonical NF-κB, Erk, and p38 activation, and exhibit reduced proliferation and IL-2 production upon stimulation in presence of anti-GITR Ab (104). TRAF5 constitutively associates with the gp130 subunit of the IL-6R and negatively regulates IL-6R signaling by suppressing the recruitment of STAT3 to the IL-6R complex (35, 36). The role of TRAF5 in the development and maintenance of TFH responses in vivo remains to be investigated. Because GITR signaling (19), activation of the canonical NF-κB pathway (190, 191), and IL-6 signaling (78, 144, 145) are all important for TFH development, one could predict that TRAF5 is also involved in modulation of TFH differentiation by integrating these signals (Figures 1, 2).
TRAF6
Traf6−/− mice die prematurely within 17–19 days after birth, displaying severe osteopetrosis, splenomegaly, thymic atrophy, and defects in lymph node organogenesis (221, 222). B cells isolated from these mice fail to proliferate in response to anti-CD40 stimulation, indicating that TRAF6 is a mediator of CD40 signaling (222). These ex vivo data are consistent with an in vitro study showing that a B cell line expressing a CD40 mutant incapable of binding TRAF6 fails to secrete IL-6 and Ig following anti-CD40 stimulation (223). Similarly, in transgenic mice expressing a CD40 mutant incapable of TRAF6 recruitment, the generation of plasma cells, IgG production, and affinity maturation are severely compromised upon antigen challenge (224). B cells from Traf6fl/fl Cd19Cre mice display defects in proliferation, IL-6 production and phosphorylation of p38 MAP kinase upon stimulation with anti-CD40 Ab (225). In vivo antigen challenge also reveals the requirement of TRAF6 in T-dependent production of IgG, generation of long-lived plasma cells, isotype switching, and affinity maturation (225).
In T cells, TRAF6 acts as a negative rheostat. Traf6fl/fl LckCre mice develop a systemic inflammatory disease with increased production of Th2 cytokines, significant expansion of the B cell compartment and elevated serum Ab titers, including anti double-stranded DNA Abs (27). Traf6−/− T cells are hyperproliferative and display constitutively active PI3K-AKT signaling. These hyperreactive T cells are refractory to Treg-mediated suppression (27). Additionally, Traf6−/− T cells exhibit enhanced Th17 differentiation in vivo and in vitro in the presence of TGF-β (187). This effect was due to increased responsiveness of Traf6−/− T cells to TGF-β as TRAF6 impedes the production of IL-2 (187), which is a known inhibitor of Th17 differentiation (226). TRAF6 can bind to IL-2Rβ and inhibit Jak1 activation induced by IL-2 (34). Modulation of PI3K, TGF-β, and IL-2 signaling pathways by TRAF6 could directly affect TFH differentiation (Figure 1).
Summary
The differentiation of TFH cells is a complex process controlled by the integration of multiple signals. Many studies support the conclusion that TRAF proteins are important modulators of T-dependent and T-independent humoral responses. TRAFs act through a variety of mechanisms: modulation of TCR signals and integration of costimulatory and cytokine signals. As detailed above, TRAF2, 3, 5, and 6 are the most relevant ones involved in many TFH-inducing and TFH-antagonistic signaling pathways. However, the exact mechanisms of how each of these TRAF family members contributes to TFH differentiation remain elusive. Precise elucidation of the relevant mechanisms has been challenging for several reasons. First, TRAFs have dual functions as E3 ubiquitin ligases as well as molecular adaptors for protein-protein interactions; second, TRAFs are ubiquitously expressed in innate and adaptive immune cells as well as in non-immune cells; and finally, TRAFs are involved in a variety of signaling pathways that reinforce and/or neutralize each other. All these factors confound the interpretation of results derived from systemic deletion of TRAF proteins in vivo.
Future studies addressing TRAF-related mechanisms will be facilitated by the modern genome editing tools that simplify the generation of knock-in or cell type-specific knockout mice. For example, the E3 ligase activity of TRAF proteins could be specifically attenuated using the CRISPR-Cas system to differentiate the enzymatic vs. adaptor function of these proteins in various signaling pathways. Other genomic technologies such as single-cell RNA-Seq and CyTOF could be incorporated into various experiments to simultaneously profile the gene and protein expression alterations of different immune and non-immune cell populations. These approaches will provide a broader perspective of the role of TRAFs in different cell types during an immune response.
Studies on the role of TRAFs in TFH differentiation and B cell responses are of therapeutic interest as modulation of TFH differentiation has the potential to either reduce pathological Ab production in autoimmune and inflammatory diseases, or favor the development of long-lasting and high affinity humoral responses in the context of vaccination or treatment of infectious diseases.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This is publication number #3228 from the La Jolla Institute for Allergy and Immunology. CP is a Cancer Research Institute Irvington Fellow supported by the Cancer Research Institute.
References
1. Schroeder HW, Cavacini L. Structure and function of immunoglobulins. J Allergy Clin Immun. (2010) 125:S41–52. doi: 10.1016/j.jaci.2009.09.046
2. Breitfeld D, Ohl L, Kremmer E, Ellwart J, Sallusto F, Lipp M, et al. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J Exp Med. (2000) 192:1545–52. doi: 10.1084/jem.192.11.1545
3. Schaerli P, Willimann K, Lang AB, Lipp M, Loetscher P, Moser B. CXC chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. J Exp Med. (2000) 192:1553–62. doi: 10.1084/jem.192.11.1553
4. Kim CH, Rott JS, Clark-Lewis I, Campbell DJ, Wu L, Butcher EC. Subspecialization of CXCR5+ T cells: B helper activity is focused in a germinal center-localized subset of CXCR5+ T cells. J Exp Med. (2001) 193:1373–81. doi: 10.1084/jem.193.12.1373
5. Kaji T, Ishige A, Hikida M, Taka J, Hijikata A, Kubo M, et al. Distinct cellular pathways select germline-encoded and somatically mutated antibodies into immunological memory. J Exp Med. (2012) 209:2079–97. doi: 10.1084/jem.20120127
6. Hollister K, Kusam S, Wu H, Clegg N, Mondal A, Sawant DV, et al. Insights into the role of Bcl6 in follicular Th cells using a new conditional mutant mouse model. J Immunol. (2013) 191:3705–11. doi: 10.4049/jimmunol.1300378
7. Ma CS, Deenick EK. Human T follicular helper (Tfh) cells and disease. Immunol Cell Biol. (2014) 92:64–71. doi: 10.1038/icb.2013.55
8. Ballesteros-Tato A, Randall TD, Lund FE, Spolski R, Leonard WJ, Leon B. T follicular helper cell plasticity shapes pathogenic T helper 2 cell-mediated immunity to inhaled house dust mite. Immunity (2016) 44:259–73. doi: 10.1016/j.immuni.2015.11.017
9. Ame-Thomas P, Le Priol J, Yssel H, Caron G, Pangault C, Jean R, et al. Characterization of intratumoral follicular helper T cells in follicular lymphoma: role in the survival of malignant B cells. Leukemia (2012) 26:1053–63. doi: 10.1038/leu.2011.301
10. Rawal S, Chu FL, Zhang M, Park HJ, Nattamai D, Kannan S, et al. Cross talk between follicular Th cells and tumor cells in human follicular lymphoma promotes immune evasion in the tumor microenvironment. J Immunol. (2013) 190:6681–93. doi: 10.4049/jimmunol.1201363
11. Gaulard P, de Leval L. Follicular helper T cells: implications in neoplastic hematopathology. Semin Diagn Pathol. (2011) 28:202–13. doi: 10.1053/j.semdp.2011.03.003
12. Fujisawa M, Chiba S, Sakata-Yanagimoto M. Recent progress in the understanding of angioimmunoblastic T-cell lymphoma. J Clin Exp Hematop. (2017) 57:109–19. doi: 10.3960/jslrt.17019
13. Sage PT, Sharpe AH. T follicular regulatory cells. Immunol Rev. (2016) 271:246–59. doi: 10.1111/imr.12411
14. Fu W, Liu X, Lin X, Feng H, Sun L, Li S, et al. Deficiency in T follicular regulatory cells promotes autoimmunity. J Exp Med. (2018) 215:815–25. doi: 10.1084/jem.20170901
15. Gong Y, Tong J, Wang S. Are follicular regulatory T cells involved in autoimmune diseases? Front Immunol. (2017) 8:1790. doi: 10.3389/fimmu.2017.01790
17. Boettler T, Moeckel F, Cheng Y, Heeg M, Salek-Ardakani S, Crotty S, et al. OX40 facilitates control of a persistent virus infection. PLoS Pathog. (2012)8:e1002913. doi: 10.1371/journal.ppat.1002913
18. Tahiliani V, Hutchinson TE, Abboud G, Croft M, Salek-Ardakani S. OX40 cooperates with ICOS to amplify follicular Th cell development and germinal center reactions during infection. J Immunol. (2017) 198:218–28. doi: 10.4049/jimmunol.1601356
19. Ma J, Feng D, Wei Y, Tian J, Tang X, Rui K, et al. Blockade of glucocorticoid-induced tumor necrosis factor-receptor-related protein signaling ameliorates murine collagen-induced arthritis by modulating follicular helper T cells. Am J Pathol. (2016) 186:1559–67. doi: 10.1016/j.ajpath.2016.02.010
20. Mittler RS, Bailey TS, Klussman K, Trailsmith MD, Hoffmann MK. Anti-4-1BB monoclonal antibodies abrogate T cell-dependent humoral immune responses in vivo through the induction of helper T cell anergy. J Exp Med. (1999) 190:1535–40. doi: 10.1084/jem.190.10.1535
21. Foell J, Strahotin S, O'Neil SP, McCausland MM, Suwyn C, Haber M, et al. CD137 costimulatory T cell receptor engagement reverses acute disease in lupus-prone NZB x NZW F1 mice. J Clin Invest. (2003) 111:1505–18. doi: 10.1172/JCI200317662
22. Tsitsikov EN, Laouini D, Dunn IF, Sannikova TY, Davidson L, Alt FW, et al. TRAF1 is a negative regulator of TNF signaling. Enhanced TNF signaling in TRAF1-deficient mice. Immunity (2001) 15:647–57. doi: 10.1016/S1074-7613(01)00207-2
23. McPherson AJ, Snell LM, Mak TW, Watts TH. Opposing roles for TRAF1 in the alternative versus classical NF-kappaB pathway in T cells. J Biol Chem. (2012) 287:23010–9. doi: 10.1074/jbc.M112.350538
24. Villanueva JE, Walters SN, Saito M, Malle EK, Zammit NW, Watson KA, et al. Targeted deletion of Traf2 allows immunosuppression-free islet allograft survival in mice. Diabetologia (2017) 60:679–89. doi: 10.1007/s00125-016-4198-7
25. Xie P, Kraus ZJ, Stunz LL, Liu Y, Bishop GA. TNF receptor-associated factor 3 is required for T cell-mediated immunity and TCR/CD28 signaling. J Immunol. (2011) 186:143–55. doi: 10.4049/jimmunol.1000290
26. Wallis AM, Wallace EC, Hostager BS, Yi ZJ, Houtman CD, Bishop GA. TRAF3 enhances TCR signaling by regulating the inhibitors Csk and PTPN22. Sci Rep. (2017) 7:2081. doi: 10.1038/s41598-017-02280-4
27. King CG, Kobayashi T, Cejas PJ, Kim T, Yoon K, Kim GK, et al. TRAF6 is a T cell-intrinsic negative regulator required for the maintenance of immune homeostasis. Nat Med. (2006) 12:1088–92. doi: 10.1038/nm1449
28. Xie JJ, Liang JQ, Diao LH, Altman A, Li Y. TNFR-associated factor 6 regulates TCR signaling via interaction with and modification of LAT adapter. J Immunol. (2013) 190:4027–36. doi: 10.4049/jimmunol.1202742
29. Akiba H, Takeda K, Kojima Y, Usui Y, Harada N, Yamazaki T, et al. The role of ICOS in the CXCR5+ follicular B helper T cell maintenance in vivo. J Immunol. (2005) 175:2340–8. doi: 10.4049/jimmunol.175.4.2340
30. Hu YL, Metz DP, Chung J, Siu G, Zhang M. B7RP-1 blockade ameliorates autoimmunity through regulation of follicular helper T cells. J Immunol. (2009) 182:1421–8. doi: 10.4049/jimmunol.182.3.1421
31. Gigoux M, Shang J, Pak Y, Xu M, Choe J, Mak TW, et al. Inducible costimulator promotes helper T-cell differentiation through phosphoinositide 3-kinase. Proc Nat Acad Sci USA. (2009) 106:20371–6. doi: 10.1073/pnas.0911573106
32. Pedros C, Zhang Y, Hu JK, Choi YS, Canonigo-Balancio AJ, Yates JR III, et al. A TRAF-like motif of the inducible costimulator ICOS controls development of germinal center TFH cells via the kinase TBK1. Nat Immunol. (2016) 17:825–33. doi: 10.1038/ni.3463
33. Yi Z, Lin WW, Stunz LL, Bishop GA. The adaptor TRAF3 restrains the lineage determination of thymic regulatory T cells by modulating signaling via the receptor for IL-2. Nat Immunol. (2014) 15:866–74. doi: 10.1038/ni.2944
34. Motegi H, Shimo Y, Akiyama T, Inoue J-I. TRAF6 negatively regulates the Jak1-Erk pathway in interleukin-2 signaling. Genes Cells (2011) 16:179–89. doi: 10.1111/j.1365-2443.2010.01474.x
35. Nagashima H, Okuyama Y, Asao A, Kawabe T, Yamaki S, Nakano H, et al. The adaptor TRAF5 limits the differentiation of inflammatory CD4+ T cells by antagonizing signaling via the receptor for IL-6. Nat Immunol. (2014) 15:449–56. doi: 10.1038/ni.2863
36. Nagashima H, Okuyama Y, Hayashi T, Ishii N, So T. TNFR-associated factors 2 and 5 differentially regulate the instructive IL-6 receptor signaling required for Th17 development. J Immunol. (2016) 196:4082–9. doi: 10.4049/jimmunol.1501610
37. Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol. (2011) 29:621–63. doi: 10.1146/annurev-immunol-031210-101400
38. Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity (2014) 41:529–42. doi: 10.1016/j.immuni.2014.10.004
39. Kerfoot SM, Yaari G, Patel JR, Johnson KL, Gonzalez DG, Kleinstein SH, et al. Germinal center B cell and T follicular helper cell development initiates in the interfollicular zone. Immunity (2011) 34:947–60. doi: 10.1016/j.immuni.2011.03.024
40. Baumjohann D, Okada T, Ansel KM. Cutting edge: distinct waves of BCL6 expression during T follicular helper cell development. J Immunol. (2011) 187:2089–92. doi: 10.4049/jimmunol.1101393
41. Kitano M, Moriyama S, Ando Y, Hikida M, Mori Y, Kurosaki T, et al. Bcl6 protein expression shapes pre-germinal center B cell dynamics and follicular helper T cell heterogeneity. Immunity (2011) 34:961–72. doi: 10.1016/j.immuni.2011.03.025
42. Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, et al. Bcl6 mediates the development of T follicular helper cells. Science (2009) 325:1001–5. doi: 10.1126/science.1176676
43. Yu D, Rao S, Tsai LM, Lee SK, He Y, Sutcliffe EL, et al. The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity (2009) 31:457–68. doi: 10.1016/j.immuni.2009.07.002
44. Haynes NM, Allen CDC, Lesley R, Ansel KM, Killeen N, Cyster JG. Role of CXCR5 and CCR7 in follicular Th cell positioning and appearance of a programmed cell death gene-1high germinal center-associated subpopulation. J Immunol. (2007) 179:5099–108. doi: 10.4049/jimmunol.179.8.5099
45. Baumjohann D, Preite S, Reboldi A, Ronchi F, Ansel KM, Lanzavecchia A, et al. Persistent antigen and germinal center B cells sustain T follicular helper cell responses and phenotype. Immunity (2013) 38:596–605. doi: 10.1016/j.immuni.2012.11.020
46. Qi H, Cannons JL, Klauschen F, Schwartzberg PL, Germain RN. SAP-controlled T-B cell interactions underlie germinal centre formation. Nature (2008) 455:764–9. doi: 10.1038/nature07345
47. Weinstein JS, Herman EI, Lainez B, Licona-Limon P, Esplugues E, Flavell R, et al. TFH cells progressively differentiate to regulate the germinal center response. Nat Immunol. (2016) 17:1197–205. doi: 10.1038/ni.3554
48. Hale JS, Ahmed R. Memory T follicular helper CD4 T cells. Front Immunol. (2015) 6:16. doi: 10.3389/fimmu.2015.00016
49. Fazilleau N, McHeyzer-Williams LJ, Rosen H, McHeyzer-Williams MG. The function of follicular helper T cells is regulated by the strength of T cell antigen receptor binding. Nat Immunol. (2009) 10:375–84. doi: 10.1038/ni.1704
50. Hwang S, Palin AC, Li L, Song KD, Lee J, Love PE. (2015). TCR ITAM multiplicity is required for the generation of follicular helper T-cells. Nat Commun. 6:6982. doi: 10.1038/ncomms7982
51. Keck S, Schmaler M, Ganter S, Wyss L, Oberle S, Huseby ES, et al. Antigen affinity and antigen dose exert distinct influences on CD4 T-cell differentiation. Proc Nat Acad Sci USA. (2014) 111:14852–7. doi: 10.1073/pnas.1403271111
52. Tubo NJ, Pagan AJ, Taylor JJ, Nelson RW, Linehan JL, Ertelt JM, et al. Single naive CD4+ T cells from a diverse repertoire produce different effector cell types during infection. Cell (2013) 153:785–96. doi: 10.1016/j.cell.2013.04.007
53. Deenick EK, Chan A, Ma CS, Gatto D, Schwartzberg PL, Brink R, et al. Follicular helper T cell differentiation requires continuous antigen presentation that is independent of unique B cell signaling. Immunity (2010) 33:241–53. doi: 10.1016/j.immuni.2010.07.015
54. Krishnamoorthy V, Kannanganat S, Maienschein-Cline M, Cook SL, Chen JJ, Bahroos N, et al. The IRF4 gene regulatory module functions as a read-write integrator to dynamically coordinate t helper cell fate. Immunity (2017) 47:481–97.e7. doi: 10.1016/j.immuni.2017.09.001
55. Benson RA, MacLeod MKL, Hale BG, Patakas A, Garside P, Brewer JM. Antigen presentation kinetics control T cell/dendritic cell interactions and follicular helper T cell generation in vivo. Elife (2015) 4:e06994. doi: 10.7554/eLife.06994
56. Lin WJ, Su YW, Lu YC, Hao Z, Chio IIC, Chen NJ, et al. Crucial role for TNF receptor-associated factor 2 (TRAF2) in regulating NFkappaB2 signaling that contributes to autoimmunity. Proc Nat Acad Sci USA. (2011) 108:18354–9. doi: 10.1073/pnas.1109427108
57. Salek-Ardakani S, Choi YS, Rafii-El-Idrissi Benhnia M, Flynn R, Arens R, Shoenberger S. B cell-specific expression of B7-2 is required for follicular Th cell function in response to vaccinia virus. J Immunol. (2011) 186:5294–303. doi: 10.4049/jimmunol.1100406
58. Wang CJ, Heuts F, Ovcinnikovs V, Wardzinski L, Bowers C, Schmidt EM, et al. CTLA-4 controls follicular helper T-cell differentiation by regulating the strength of CD28 engagement. Proc Nat Acad Sci USA. (2015) 112:524–9. doi: 10.1073/pnas.1414576112
59. Collins AV, Brodie DW, Gilbert RJ, Iaboni A, Manso-Sancho R, Walse B, et al. The interaction properties of costimulatory molecules revisited. Immunity (2002) 17:201–10. doi: 10.1016/S1074-7613(02)00362-X
60. Ferguson SE, Han S, Kelsoe G, Thompson CB. CD28 is required for germinal center formation. J Immunol. (1996) 156:4576–81.
61. Weber JP, Fuhrmann F, Feist RK, Lahmann A, Al Baz MS, Gentz LJ, et al. ICOS maintains the T follicular helper cell phenotype by down-regulating Kruppel-like factor 2. J Exp Med. (2015) 212:217–33. doi: 10.1084/jem.20141432
62. Lane P, Burdet C, Hubele S, Scheidegger D, Muller U, McConnell F, et al. B cell function in mice transgenic for mCTLA4-H gamma 1: lack of germinal centers correlated with poor affinity maturation and class switching despite normal priming of CD4+ T cells. J Exp Med. (1994) 179:819–30. doi: 10.1084/jem.179.3.819
63. Walker LS, Gulbranson-Judge A, Flynn S, Brocker T, Raykundalia C, Goodall M, et al. Compromised OX40 function in CD28-deficient mice is linked with failure to develop CXC chemokine receptor 5-positive CD4 cells and germinal centers. J Exp Med. (1999) 190:1115–22. doi: 10.1084/jem.190.8.1115
64. Walker LSK, Wiggett HE, Gaspal FMC, Raykundalia CR, Goodall MD, Toellner K-M, et al. Established T cell-driven germinal center B cell proliferation is independent of CD28 signaling but is tightly regulated through CTLA-4. J Immunol. (2003) 170:91–8. doi: 10.4049/jimmunol.170.1.91
65. Watanabe M, Fujihara C, Radtke AJ, Chiang YJ, Bhatia S, Germain RN, et al. Co-stimulatory function in primary germinal center responses: CD40 and B7 are required on distinct antigen-presenting cells. J Exp Med. (2017) 214:2795–810. doi: 10.1084/jem.20161955
66. Kong KF, Yokosuka T, Canonigo-Balancio AJ, Isakov N, Saito T, Altman A. A motif in the V3 domain of the kinase PKC-theta determines its localization in the immunological synapse and functions in T cells via association with CD28. Nat Immunol. (2011) 12:1105–12. doi: 10.1038/ni.2120
67. Altman A, Kong KF. Protein kinase C enzymes in the hematopoietic and immune systems. Annu Rev Immunol. (2016) 34:511–38. doi: 10.1146/annurev-immunol-041015-055347
68. Dodson LF, Boomer JS, Deppong CM, Shah DD, Sim J, Bricker TL, et al. Targeted knock-in mice expressing mutations of CD28 reveal an essential pathway for costimulation. Mol Cell Biol. (2009) 29:3710–21. doi: 10.1128/MCB.01869-08
69. Grimbacher B, Hutloff A, Schlesier M, Glocker E, Warnatz K, Drager R, et al. Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nat Immunol. (2003) 4:261–8. doi: 10.1038/ni902
70. Yong PF, Salzer U, Grimbacher B. The role of costimulation in antibody deficiencies: ICOS and common variable immunodeficiency. Immunol Rev. (2009) 229:101–13. doi: 10.1111/j.1600-065X.2009.00764.x
71. Hutloff A, Dittrich AM, Beier KC, Eljaschewitsch B, Kraft R. Anagnostopoulos, I., and Kroczek, R. A., ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature (1999) 397:263–6. doi: 10.1038/16717
72. Dong C, Juedes AE, Temann UA, Shresta S, Allison JP, Ruddle NH, et al. ICOS co-stimulatory receptor is essential for T-cell activation and function. Nature (2001) 409:97–101. doi: 10.1038/35051100
73. Tafuri A, Shahinian A, Bladt F, Yoshinaga SK, Jordana M, Wakeham A, et al. ICOS is essential for effective T-helper-cell responses. Nature (2001) 409:105–9. doi: 10.1038/35051113
74. McAdam AJ, Greenwald RJ, Levin MA, Chernova T, Malenkovich N, Ling V, et al. ICOS is critical for CD40-mediated antibody class switching. Nature (2001) 409:102–5. doi: 10.1038/35051107
75. Choi YS, Kageyama R, Eto D, Escobar TC, Johnston RJ, Monticelli L, et al. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity (2011) 34:932–46. doi: 10.1016/j.immuni.2011.03.023
76. Shin C, Han J-A, Koh H, Choi B, Cho Y, Jeong H, et al. CD8alpha− dendritic cells induce antigen-specific T follicular helper cells generating efficient humoral immune responses. Cell Rep. (2015) 11:1929–40. doi: 10.1016/j.celrep.2015.05.042
77. Wikenheiser DJ, Ghosh D, Kennedy B, Stumhofer JS. The costimulatory molecule ICOS regulates host Th1 and follicular Th cell differentiation in response to Plasmodium chabaudi chabaudi AS infection. J Immunol. (2016) 196:778–91. doi: 10.4049/jimmunol.1403206
78. Nurieva RI, Chung Y, Hwang D, Yang XO, Kang HS, Ma L, et al. Generation of T follicular helper cells is mediated by interleukin-21 but independent of T helper 1, 2, or 17 cell lineages. Immunity (2008) 29:138–49. doi: 10.1016/j.immuni.2008.05.009
79. Liu D, Xu H, Shih C, Wan Z, Ma X, Ma W, et al. T-B-cell entanglement and ICOSL-driven feed-forward regulation of germinal centre reaction. Nature (2015) 517:214–8. doi: 10.1038/nature13803
80. Fos C, Salles A, Lang V, Carrette F, Audebert S, Pastor S, et al. ICOS ligation recruits the p50alpha PI3K regulatory subunit to the immunological synapse. J Immunol. (2008) 181:1969–77. doi: 10.4049/jimmunol.181.3.1969
81. Watanabe R, Harada Y, Takeda K, Takahashi J, Ohnuki K, Ogawa S, et al. Grb2 and Gads exhibit different interactions with CD28 and play distinct roles in CD28-mediated costimulation. J Immunol. (2006) 177:1085–91. doi: 10.4049/jimmunol.177.2.1085
82. Inukai K, Funaki M, Ogihara T, Katagiri H, Kanda A, Anai M, et al. p85alpha gene generates three isoforms of regulatory subunit for phosphatidylinositol 3-kinase (PI 3-Kinase), p50alpha, p55alpha, and p85alpha, with different PI 3-kinase activity elevating responses to insulin. J Biol Chem. (1997) 272:7873–82. doi: 10.1074/jbc.272.12.7873
83. Inukai I, Funaki M, Anai M, Ogihara T, Katagiri H, Fukushima Y, et al. Five isoforms of the phosphatidylinositol 3-kinase regulatory subunit exhibit different associations with receptor tyrosine kinases and their tyrosine phosphorylations. Febs Lett. (2001) 490:32–8. doi: 10.1016/S0014-5793(01)02132-9
84. Clayton E, Bardi G, Bell SE, Chantry D, Downes CP, Gray A, et al. A crucial role for the p110delta subunit of phosphatidylinositol 3-kinase in B cell development and activation. J Exp Med. (2002) 196:753–63. doi: 10.1084/jem.20020805
85. Okkenhaug K, Bilancio A, Farjot G, Priddle H, Sancho S, Peskett E, et al. Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science (2002) 297:1031–4. doi: 10.1126/science.1073560
86. Rolf J, Bell SE, Kovesdi D, Janas ML, Soond DR, Webb LMC, et al. Phosphoinositide 3-kinase activity in T cells regulates the magnitude of the germinal center reaction. J Immunol. (2010) 185:4042–52. doi: 10.4049/jimmunol.1001730
87. Deane JA, Kharas MG, Oak JS, Stiles LN, Luo J, Moore TI, et al. T-cell function is partially maintained in the absence of class IA phosphoinositide 3-kinase signaling. Blood (2007) 109:2894–902.
88. Gao XL, Gigoux M, Yang J, Leconte J, Yang X, Suh WK. Anti-chlamydial Th17 responses are controlled by the inducible costimulator partially through phosphoinositide 3-kinase signaling. PLoS ONE (2012) 7–12:e52657. doi: 10.1371/journal.pone.0052657
89. Li J, Heinrichs J, Leconte J, Haarberg K, Semple K, Liu C, et al. Phosphatidylinositol 3-kinase-independent signaling pathways contribute to ICOS-mediated T cell costimulation in acute graft-versus-host disease in mice. J Immunol. (2013) 191:200–7. doi: 10.4049/jimmunol.1203485
90. Louis C, Ngo D, D'Silva DB, Hansen J, Phillipson L, Jousset H, et al. Therapeutic effects of a TBK1 kinase inhibitor in germinal center-driven, autoantibody-mediated inflammatory arthritis. Arthritis Rheumatol. (2018). doi: 10.1002/art.40670
91. Pomerantz JL, Baltimore D. NF-kappaB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J. (1999) 18:6694–704. doi: 10.1093/emboj/18.23.6694
92. Oganesyan G, Saha SK, Guo B, He JQ, Shahangian A, Zarnegar B, et al. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature (2006) 439:208–11. doi: 10.1038/nature04374
93. Fang R, Jiang QF, Zhou X, Wang CG, Guan YK, Tao JL, et al. MAVS activates TBK1 and IKK epsilon through TRAFs in NEMO dependent and independent manner. PLoS Pathog. (2017) 13:e1006720. doi: 10.1371/journal.ppat.1006720
94. Leconte J, Bagherzadeh Yazdchi S, Panneton V, Suh WK. Inducible costimulator (ICOS) potentiates TCR-induced calcium flux by augmenting PLCgamma1 activation and actin remodeling. Mol Immunol. (2016) 79:38–46. doi: 10.1016/j.molimm.2016.09.022
95. Flynn S, Toellner KM, Raykundalia C, Goodall M, Lane P. CD4 T cell cytokine differentiation: the B cell activation molecule, OX40 ligand, instructs CD4 T cells to express interleukin 4 and upregulates expression of the chemokine receptor, Blr-1. J Exp Med. (1998) 188:297–304. doi: 10.1084/jem.188.2.297
96. Kopf M, Ruedl C, Schmitz N, Gallimore A, Lefrang K, Ecabert B, et al. OX40-deficient mice are defective in Th cell proliferation but are competent in generating B cell and CTL Responses after virus infection. Immunity (1999) 11:699–708. doi: 10.1016/S1074-7613(00)80144-2
97. Ekkens MJ, Liu Z, Liu Q, Whitmire J, Xiao S, Foster A, et al. The role of OX40 ligand interactions in the development of the Th2 response to the gastrointestinal nematode parasite Heligmosomoides polygyrus. J Immunol. (2003) 170:384–93. doi: 10.4049/jimmunol.170.1.384
98. Jacquemin C, Schmitt N, Contin-Bordes C, Liu Y, Narayanan P, Seneschal J, et al. OX40 ligand contributes to human lupus pathogenesis by promoting T follicular helper response. Immunity (2015) 42:1159–70. doi: 10.1016/j.immuni.2015.05.012
99. Kawamata S, Hori T, Imura A, Takaori-Kondo A, Uchiyama T. Activation of OX40 signal transduction pathways leads to tumor necrosis factor receptor-associated factor (TRAF) 2- and TRAF5-mediated NF-kappaB activation. J Biol Chem. (1998) 273:5808–14. doi: 10.1074/jbc.273.10.5808
100. Arch RH, Thompson CB. 4-1BB and Ox40 are members of a tumor necrosis factor (TNF)-nerve growth factor receptor subfamily that bind TNF receptor-associated factors and activate nuclear factor kappaB. Mol Cell Biol. (1998) 18:558–65. doi: 10.1128/MCB.18.1.558
101. So T, Soroosh P, Eun SY, Altman A, Croft M. Antigen-independent signalosome of CARMA1, PKCtheta, and TNF receptor-associated factor 2 (TRAF2) determines NF-kappaB signaling in T cells. Proc Nat Acad Sci USA. (2011) 108:2903–8. doi: 10.1073/pnas.1008765108
102. Clouthier DL, Zhou AC, Wortzman ME, Luft O, Levy GA, Watts TH. GITR intrinsically sustains early type 1 and late follicular helper CD4 T cell accumulation to control a chronic viral infection. PLoS Pathog. (2015) 11:e1004517. doi: 10.1371/journal.ppat.1004517
103. Snell LM, McPherson AJ, Lin GHY, Sakaguchi S, Pandolfi PP, Riccardi C, et al. CD8 T cell-intrinsic GITR is required for T cell clonal expansion and mouse survival following severe influenza infection. J Immunol. (2010) 185:7223–34. doi: 10.4049/jimmunol.1001912
104. Esparza EM, Lindsten T, Stockhausen JM, Arch RH. Tumor necrosis factor receptor (TNFR)-associated factor 5 is a critical intermediate of costimulatory signaling pathways triggered by glucocorticoid-induced TNFR in T cells. J Biol Chem. (2006) 281:8559–64. doi: 10.1074/jbc.M512915200
105. Hauer J, Puschner S, Ramakrishnan P, Simon U, Bongers M, Federle C, et al. TNF receptor (TNFR)-associated factor (TRAF) 3 serves as an inhibitor of TRAF2/5-mediated activation of the noncanonical NF-kappaB pathway by TRAF-binding TNFRs. Proc Nat Acad Sci USA. (2005) 102:2874–9. doi: 10.1073/pnas.0500187102
106. de Boer M, Kasran A, Kwekkeboom J, Walter H, Vandenberghe P, Ceuppens JL. Ligation of B7 with CD28/CTLA-4 on T cells results in CD40 ligand expression, interleukin-4 secretion and efficient help for antibody production by B cells. Eur J Immunol. (1993) 23:3120–5. doi: 10.1002/eji.1830231212
107. van Essen D, Kikutani H, Gray D. CD40 ligand-transduced co-stimulation of T cells in the development of helper function. Nature (1995) 378:620–3. doi: 10.1038/378620a0
108. Grewal IS, Xu J, Flavell RA. Impairment of antigen-specific T-cell priming in mice lacking CD40 ligand. Nature (1995) 378:617–20. doi: 10.1038/378617a0
109. Bishop GA, Moore CR, Xie P, Stunz LL, Kraus ZJ. TRAF proteins in CD40 signaling. Adv Exp Med Biol. (2007) 597:131–51. doi: 10.1007/978-0-387-70630-6_11
110. Alfaro C, Echeveste JI, Rodriguez-Ruiz ME, Solorzano JL, Perez-Gracia JL, Idoate MA, et al. Functional expression of CD137 (4-1BB) on T helper follicular cells. Oncoimmunology (2015) 4:e1054597. doi: 10.1080/2162402X.2015.1054597
111. Kwon BS, Hurtado JC, Lee ZH, Kwack KB, Seo SK, Choi BK, et al. Immune responses in 4-1BB (CD137)-deficient mice. J Immunol. (2002) 168:5483–90. doi: 10.4049/jimmunol.168.11.5483
112. DeBenedette MA, Wen T, Bachmann MF, Ohashi PS, Barber BH, Stocking KL, et al. Analysis of 4-1BB ligand (4-1BBL)-deficient mice and of mice lacking both 4-1BBL and CD28 reveals a role for 4-1BBL in skin allograft rejection and in the cytotoxic T cell response to influenza virus. J Immunol. (1999) 163:4833–41.
113. Hong HJ, Lee JW, Park SS, Kang YJ, Chang SY, Kim KM, et al. A humanized anti−4-1BB monoclonal antibody suppresses antigen-induced humoral immune response in nonhuman primates. J Immunother. (2000) 23:613–21. doi: 10.1097/00002371-200011000-00002
114. Sun Y, Blink SE, Chen JH, Fu YX. (2005). Regulation of follicular dendritic cell networks by activated T cells: the role of CD137 signaling. J Immunol. 175:884–90. doi: 10.4049/jimmunol.175.2.884
115. Saoulli K, Lee SY, Cannons JL, Yeh WC, Santana A, Goldstein MD, et al. CD28-independent, TRAF2-dependent costimulation of resting T cells by 4-1BB ligand. J Exp Med. (1998) 187:1849–62. doi: 10.1084/jem.187.11.1849
116. Martinez-Forero I, Azpilikueta A, Bolanos-Mateo E, Nistal-Villan E, Palazon A, Teijeira A, et al. T cell costimulation with anti-CD137 monoclonal antibodies is mediated by K63-polyubiquitin-dependent signals from endosomes. J Immunol. (2013) 190:6694–706. doi: 10.4049/jimmunol.1203010
117. Cannons JL, Choi Y, Watts TH. Role of TNF receptor-associated factor 2 and p38 mitogen-activated protein kinase activation during 4-1BB-dependent immune response. J Immunol. (2000) 165:6193–204. doi: 10.4049/jimmunol.165.11.6193
118. Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med. (2011) 17:975–82. doi: 10.1038/nm.2425
119. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science (2008) 322:271–5. doi: 10.1126/science.1160062
120. Sage PT, Paterson AM, Lovitch SB, Sharpe AH. The coinhibitory receptor CTLA-4 controls B cell responses by modulating T follicular helper, T follicular regulatory, and T regulatory cells. Immunity (2014) 41:1026–39. doi: 10.1016/j.immuni.2014.12.005
121. Wing JB, Ise W, Kurosaki T, Sakaguchi S. Regulatory T cells control antigen-specific expansion of Tfh cell number and humoral immune responses via the coreceptor CTLA-4. Immunity (2014) 41:1013–25. doi: 10.1016/j.immuni.2014.12.006
122. Alroqi FJ, Charbonnier LM, Baris S, Kiykim A, Chou J, Platt CD, et al. (2018). Exaggerated follicular helper T-cell responses in patients with LRBA deficiency caused by failure of CTLA4-mediated regulation. J Allergy Clin Immun. 141:1050–9.e10. doi: 10.1016/j.jaci.2017.05.022
123. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science (2011) 332:600–3. doi: 10.1126/science.1202947
124. Marengere LE, Waterhouse P, Duncan GS, Mittrucker HW, Feng GS, Mak TW. Regulation of T cell receptor signaling by tyrosine phosphatase SYP association with CTLA-4. Science (1996) 272:1170–3. doi: 10.1126/science.272.5265.1170
125. Chuang E, Fisher TS, Morgan RW, Robbins MD, Duerr JM, Vander Heiden MG, et al. The CD28 and CTLA-4 receptors associate with the serine/threonine phosphatase PP2A. Immunity (2000) 13:313–22. doi: 10.1016/S1074-7613(00)00031-5
126. Kong KF, Fu G, Zhang Y, Yokosuka T, Casas J, Canonigo-Balancio AJ, et al. Protein kinase C-eta controls CTLA-4-mediated regulatory T cell function. Nat Immunol. (2014) 15:465–72. doi: 10.1038/ni.2866
127. Pedros C, Canonigo-Balancio AJ, Kong KF, Altman A. Requirement of Treg-intrinsic CTLA4/PKCη signaling pathway for suppressing tumor immunity. JCI Insight (2017) 2:e95692. doi: 10.1172/jci.insight.95692
128. Li S, Wang L, Dorf ME. PKC phosphorylation of TRAF2 mediates IKKalpha/beta recruitment and K63-linked polyubiquitination. Mol Cell (2009) 33:30–42. doi: 10.1016/j.molcel.2008.11.023
129. Good-Jacobson KL, Szumilas CG, Chen L, Sharpe AH, Tomayko MM, Shlomchik MJ. PD-1 regulates germinal center B cell survival and the formation and affinity of long-lived plasma cells. Nat Immunol. (2010) 11:535–42. doi: 10.1038/ni.1877
130. Hui EF, Cheung J, Zhu J, Su XL, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science (2017) 355:1428–33. doi: 10.1126/science.aaf1292
131. Hamel KM, Cao Y, Wang Y, Rodeghero R, Kobezda T, Chen L, et al. B7-H1 expression on non-B and non-T cells promotes distinct effects on T- and B-cell responses in autoimmune arthritis. Eur J Immunol. (2010) 40:3117–27. doi: 10.1002/eji.201040690
132. Hams E, McCarron MJ, Amu S, Yagita H, Azuma M, Chen L, et al. Blockade of B7-H1 (programmed death ligand 1) enhances humoral immunity by positively regulating the generation of T follicular helper cells. J Immunol. (2011) 186:5648–55. doi: 10.4049/jimmunol.1003161
133. Kawamoto S, Tran TH, Maruya M, Suzuki K, Doi Y, Tsutsui Y, et al. The inhibitory receptor PD-1 regulates IgA selection and bacterial composition in the gut. Science (2012) 336:485–9. doi: 10.1126/science.1217718
134. Cubas RA, Mudd JC, Savoye AL, Perreau M, van Grevenynghe J, Metcalf T, et al. Inadequate T follicular cell help impairs B cell immunity during HIV infection. Nat Med. (2013) 19:494–9. doi: 10.1038/nm.3109
135. Sage PT, Francisco LM, Carman CV, Sharpe AH. The receptor PD-1 controls follicular regulatory T cells in the lymph nodes and blood. Nat Immunol. (2013) 14:152–61. doi: 10.1038/ni.2496
136. Sheppard KA, Fitz LJ, Lee JM, Benander C, George JA, Wooters J, et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. Febs Lett. (2004) 574:37–41. doi: 10.1016/j.febslet.2004.07.083
137. Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol. (2004) 173:945–54. doi: 10.4049/jimmunol.173.2.945
138. Zhang ZY, Jimi E, Bothwell ALM. Receptor activator of NF-kappa B ligand stimulates recruitment of SHP-1 to the complex containing TNFR-associated factor 6 that regulates osteoclastogenesis. J Immunol. (2003) 171:3620–6. doi: 10.4049/jimmunol.171.7.3620
139. Watanabe N, Gavrieli M, Sedy JR, Yang JF, Fallarino F, Loftin SK, et al. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat Immunol. (2003) 4:670–9. doi: 10.1038/ni944
140. Oya Y, Watanabe N, Owada T, Oki M, Hirose K, Suto A, et al. Development of autoimmune hepatitis-like disease and production of autoantibodies to nuclear antigens in mice lacking B and T lymphocyte attenuator. Arthritis Rheum. (2008) 58:2498–510. doi: 10.1002/art.23674
141. Kashiwakuma D, Suto A, Hiramatsu Y, Ikeda K, Takatori H, Suzuki K, et al. B and T lymphocyte attenuator suppresses IL-21 production from follicular Th cells and subsequent humoral immune responses. J Immunol. (2010) 185:2730–6. doi: 10.4049/jimmunol.0903839
142. Gavrieli M, Watanabe N, Loftin SK, Murphy TL, Murphy KM. Characterization of phosphotyrosine binding motifs in the cytoplasmic domain of B and T lymphocyte attenuator required for association with protein tyrosine phosphatases SHP-1 and SHP-2. Biochem Biophys Res Commun. (2003) 312:1236–43. doi: 10.1016/j.bbrc.2003.11.070
143. Villarino AV, Kanno Y, O'Shea JJ. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat Immunol. (2017) 18:374–84. doi: 10.1038/ni.3691
144. Choi YS, Eto D, Yang JA, Lao C, Crotty S. Cutting edge: STAT1 is required for IL-6-mediated Bcl6 induction for early follicular helper cell differentiation. J Immunol. (2013) 190:3049–53. doi: 10.4049/jimmunol.1203032
145. Harker JA, Lewis GM, Mack L, Zuniga EI. Late interleukin-6 escalates T follicular helper cell responses and controls a chronic viral infection. Science (2011) 334:825–9. doi: 10.1126/science.1208421
146. Kopf M, Herren S, Wiles MV, Pepys MB, Kosco-Vilbois MH. Interleukin 6 influences germinal center development and antibody production via a contribution of C3 complement component. J Exp Med. (1998) 188:1895–906. doi: 10.1084/jem.188.10.1895
147. Poholek AC, Hansen K, Hernandez SG, Eto D, Chandele A, Weinstein JS, et al. In vivo regulation of Bcl6 and T follicular helper cell development. J Immunol. (2010) 185:313–26. doi: 10.4049/jimmunol.0904023
148. Eto D, Lao C, DiToro D, Barnett B, Escobar TC, Kageyama R, et al. IL-21 and IL-6 are critical for different aspects of B cell immunity and redundantly induce optimal follicular helper CD4 T cell (Tfh) differentiation. PLoS ONE (2011) 6:e17739. doi: 10.1371/journal.pone.0017739
149. Karnowski A, Chevrier S, Belz GT, Mount A, Emslie D, D'Costa K, et al. B and T cells collaborate in antiviral responses via IL-6, IL-21, and transcriptional activator and coactivator, Oct2 and OBF-1. J Exp Med. (2012) 209:2049–64. doi: 10.1084/jem.20111504
150. Nish SA, Schenten D, Wunderlich FT, Pope SD, Gao Y, Hoshi N, et al. T cell-intrinsic role of IL-6 signaling in primary and memory responses. Elife (2014) 3:e01949. doi: 10.7554/eLife.01949
151. Vogelzang A, McGuire HM, Yu D, Sprent J, Mackay CR, King C. A fundamental role for interleukin-21 in the generation of T follicular helper cells. Immunity (2008) 29:127–37. doi: 10.1016/j.immuni.2008.06.001
152. Zotos D, Coquet JM, Zhang Y, Light A, D'Costa K, Kallies A., et al. IL-21 regulates germinal center B cell differentiation and proliferation through a B cell-intrinsic mechanism. J Exp Med. (2010) 207:365–78. doi: 10.1084/jem.20091777
153. Linterman MA, Beaton L, Yu D, Ramiscal RR, Srivastava M, Hogan JJ, et al. IL-21 acts directly on B cells to regulate Bcl-6 expression and germinal center responses. J Exp Med. (2010) 207:353–63. doi: 10.1084/jem.20091738
154. Rasheed MA, Latner DR, Aubert RD, Gourley T, Spolski R, Davis CW, et al. Interleukin-21 is a critical cytokine for the generation of virus-specific long-lived plasma cells. J Virol. (2013) 87:7737–46. doi: 10.1128/JVI.00063-13
155. Batten M, Ramamoorthi N, Kljavin NM, Ma CS, Cox JH, Dengler HS, et al. IL-27 supports germinal center function by enhancing IL-21 production and the function of T follicular helper cells. J Exp Med. (2010) 207:2895–906. doi: 10.1084/jem.20100064
156. Pot C, Jin H, Awasthi A, Liu SM, Lai C-Y, Madan R, et al. Cutting edge: IL-27 induces the transcription factor c-Maf, cytokine IL-21, and the costimulatory receptor ICOS that coordinately act together to promote differentiation of IL-10-producing Tr1 cells. J Immunol. (2009) 183:797–801. doi: 10.4049/jimmunol.0901233
157. Vijayan D, Mohd Redzwan N, Avery DT, Wirasinha RC, Brink R, Walters G, et al. IL-27 directly enhances germinal center B cell activity and potentiates lupus in sanroque mice. J Immunol. (2016) 197:3008–17. doi: 10.4049/jimmunol.1600652
158. Le Bon A, Schiavoni G, D'Agostino G, Gresser I, Belardelli F, Tough DF. Type i interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity (2001) 14:461–70. doi: 10.1016/S1074-7613(01)00126-1
159. Cucak H, Yrlid U, Reizis B, Kalinke U, Johansson-Lindbom B. Type I interferon signaling in dendritic cells stimulates the development of lymph-node-resident T follicular helper cells. Immunity (2009) 31:491–501. doi: 10.1016/j.immuni.2009.07.005
160. Riteau N, Radtke AJ, Shenderov K, Mittereder L, Oland SD, Hieny S, et al. Water-in-oil-only adjuvants selectively promote T follicular helper cell polarization through a type I IFN and IL-6-dependent pathway. J Immunol. (2016) 197:3884–93. doi: 10.4049/jimmunol.1600883
161. Loetsch C, Warren J, Laskowski A, Vazquez-Lombardi R, Jandl C, Langley DB, et al. Cytosolic recognition of RNA drives the immune response to heterologous erythrocytes. Cell Rep. (2017) 21:1624–38. doi: 10.1016/j.celrep.2017.10.044
162. Sebina I, James KR, Soon MS, Fogg LG, Best SE, Labastida Rivera F, et al. IFNAR1-Signalling obstructs ICOS-mediated humoral immunity during non-lethal blood-stage plasmodium infection. PLoS Pathog. (2016) 12:e1005999. doi: 10.1371/journal.ppat.1005999
163. Zander RA, Guthmiller JJ, Graham AC, Pope RL, Burke BE, Carr DJ, et al. Type I interferons induce T regulatory 1 responses and restrict humoral immunity during experimental malaria. PLoS Pathog. (2016) 12:e1005945. doi: 10.1371/journal.ppat.1005945
164. Yang CH, Murti A, Pfeffer SR, Fan M, Du Z, Pfeffer LM. The role of TRAF2 binding to the type I interferon receptor in alternative NF kappaB activation and antiviral response. J Biol Chem. (2008) 283:14309–16. doi: 10.1074/jbc.M708895200
165. Eddahri F, Denanglaire S, Bureau F, Spolski R, Leonard WJ, Leo O, et al. Interleukin-6/STAT3 signaling regulates the ability of naive T cells to acquire B-cell help capacities. Blood (2009) 113:2426–33. doi: 10.1182/blood-2008-04-154682
166. Ray JP, Marshall HD, Laidlaw BJ, Staron MM, Kaech SM, Craft J. Transcription factor STAT3 and type I interferons are corepressive insulators for differentiation of follicular helper and T helper 1 cells. Immunity (2014) 40:367–77. doi: 10.1016/j.immuni.2014.02.005
167. Ma CS, Avery DT, Chan A, Batten M, Bustamante J, Boisson-Dupuis S, et al. Functional STAT3 deficiency compromises the generation of human T follicular helper cells. Blood (2012) 119:3997–4008. doi: 10.1182/blood-2011-11-392985
168. Kimura M, Nagashima H, Okuyama Y, Ishii N, So T. TRAF2 and TRAF5 associated with the signal transducing receptor gp130 limit IL-6-driven transphosphorylation of JAK1 through the inhibition of proximal JAK-JAK interaction. Int Immunol. (2018) 30:291–9. doi: 10.1093/intimm/dxy029
169. Wei J, Yuan Y, Jin C, Chen H, Leng L, He F, et al. The ubiquitin ligase TRAF6 negatively regulates the JAK-STAT signaling pathway by binding to STAT3 and mediating its ubiquitination. PLoS ONE (2012) 7:e49567. doi: 10.1371/journal.pone.0049567
170. Lin WW, Yi Z, Stunz LL, Maine CJ, Sherman LA, Bishop GA. The adaptor protein TRAF3 inhibits interleukin-6 receptor signaling in B cells to limit plasma cell development. Sci Signal. (2015) 8:ra88. doi: 10.1126/scisignal.aaa5157
171. Nakayamada S, Kanno Y, Takahashi H, Jankovic D, Lu KT, Johnson TA, et al. Early Th1 cell differentiation is marked by a Tfh cell-like transition. Immunity (2011) 35:919–31. doi: 10.1016/j.immuni.2011.11.012
172. Schmitt N, Morita R, Bourdery L, Bentebibel SE, Zurawski SM, Banchereau J, et al. Human dendritic cells induce the differentiation of interleukin-21-producing T follicular helper-like cells through interleukin-12. Immunity (2009) 31:158–69. doi: 10.1016/j.immuni.2009.04.016
173. Ma CS, Suryani S, Avery DT, Chan A, Nanan R, Santner-Nanan B, et al. Early commitment of naive human CD4+ T cells to the T follicular helper (T(FH)) cell lineage is induced by IL-12. Immunol Cell Biol. (2009) 87:590–600. doi: 10.1038/icb.2009.64
174. Schmitt N, Bustamante J, Bourdery L, Bentebibel SE, Boisson-Dupuis S, Hamlin F, et al. IL-12 receptor beta1 deficiency alters in vivo T follicular helper cell response in humans. Blood (2013) 121:3375–85. doi: 10.1182/blood-2012-08-448902
175. Oestreich KJ, Mohn SE, Weinmann AS. Molecular mechanisms that control the expression and activity of Bcl-6 in TH1 cells to regulate flexibility with a TFH-like gene profile. Nat Immunol. (2012) 13:405–11. doi: 10.1038/ni.2242
176. Johnston RJ, Choi YS, Diamond JA, Yang JA, Crotty S. STAT5 is a potent negative regulator of TFH cell differentiation. J Exp Med. (2012) 209:243–50. doi: 10.1084/jem.20111174
177. Ballesteros-Tato A, Leon B, Graf BA, Moquin A, Adams PS, Lund FE, et al. Interleukin-2 inhibits germinal center formation by limiting T follicular helper cell differentiation. Immunity (2012) 36:847–56. doi: 10.1016/j.immuni.2012.02.012
178. Choi YS, Yang JA, Yusuf I, Johnston RJ, Greenbaum J, Peters B, et al. Bcl6 expressing follicular helper CD4 T cells are fate committed early and have the capacity to form memory. J Immunol. (2013) 190:4014–26. doi: 10.4049/jimmunol.1202963
179. Liu X, Lu H, Chen T, Nallaparaju KC, Yan X, Tanaka S, et al. Genome-wide analysis identifies Bcl6-controlled regulatory networks during T follicular helper cell differentiation. Cell Rep. (2016) 14:1735–47. doi: 10.1016/j.celrep.2016.01.038
180. McDonald PW, Read KA, Baker CE, Anderson AE, Powell MD, Ballesteros-Tato A, et al. IL-7 signalling represses Bcl-6 and the TFH gene program. Nat Commun. (2016) 7:10285. doi: 10.1038/ncomms10285
181. Seo YB, Im SJ, Namkoong H, Kim SW, Choi YW, Kang MC, et al. Crucial roles of interleukin-7 in the development of T follicular helper cells and in the induction of humoral immunity. J Virol. (2014) 88:8998–9009. doi: 10.1128/JVI.00534-14
182. Walker SR, Nelson EA, Frank DA. STAT5 represses BCL6 expression by binding to a regulatory region frequently mutated in lymphomas. Oncogene (2007) 26:224–33. doi: 10.1038/sj.onc.1209775
183. Nurieva RI, Podd A, Chen Y, Alekseev AM, Yu M, Qi X, et al. STAT5 protein negatively regulates T follicular helper (Tfh) cell generation and function. J Biol Chem. (2012) 287:11234–9. doi: 10.1074/jbc.M111.324046
184. Schmitt N, Liu Y, Bentebibel SE, Munagala I, Bourdery L, Venuprasad K, et al. The cytokine TGF-beta co-opts signaling via STAT3-STAT4 to promote the differentiation of human TFH cells. Nat Immunol. (2014) 15:856–65. doi: 10.1038/ni.2947
185. Suto A, Kashiwakuma D, Kagami S-I, Hirose K, Watanabe N, Yokote K, et al. Development and characterization of IL-21-producing CD4+ T cells. J Exp Med. (2008) 205:1369–79. doi: 10.1084/jem.20072057
186. Marshall HD, Ray JP, Laidlaw BJ, Zhang N, Gawande D, Staron MM, et al. The transforming growth factor beta signaling pathway is critical for the formation of CD4 T follicular helper cells and isotype-switched antibody responses in the lung mucosa. Elife (2015) 4:e04851. doi: 10.7554/eLife.04851
187. Cejas PJ, Walsh MC, Pearce EL, Han D, Harms GM, Artis D, et al. TRAF6 inhibits Th17 differentiation and TGF-beta-mediated suppression of IL-2. Blood (2010) 115:4750–7. doi: 10.1182/blood-2009-09-242768
188. Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. (2009) 27:693–733. doi: 10.1146/annurev.immunol.021908.132641
189. Sun SC. The non-canonical NF-kappaB pathway in immunity and inflammation. Nat Rev Immunol. (2017) 17:545–58. doi: 10.1038/nri.2017.52
190. Serre K, Mohr E, Benezech C, Bird R, Khan M, Caamano JH, et al. Selective effects of NF-kappaB1 deficiency in CD4+ T cells on Th2 and TFh induction by alum-precipitated protein vaccines. Eur J Immunol. (2011) 41:1573–82. doi: 10.1002/eji.201041126
191. Kontgen F, Grumont RJ, Strasser A, Metcalf D, Li R, Tarlinton D, et al. Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev. (1995) 9:1965–77. doi: 10.1101/gad.9.16.1965
192. Chen G, Hardy K, Bunting K, Daley S, Ma L, Shannon MF. Regulation of the IL-21 gene by the NF-kappaB transcription factor c-Rel. J Immunol. (2010) 185:2350–9. doi: 10.4049/jimmunol.1000317
193. Liu WH, Kang SG, Huang Z, Wu C-J, Jin HY, Maine CJ, et al. A miR-155-Peli1-c-Rel pathway controls the generation and function of T follicular helper cells. J Exp Med. (2016) 213:1901–19. doi: 10.1084/jem.20160204
194. Vasanthakumar A, Liao Y, Teh P, Pascutti MF, Oja AE, Garnham AL, et al. The TNF Receptor superfamily-NF-kappaB axis is critical to maintain effector regulatory T cells in lymphoid and non-lymphoid tissues. Cell Rep. (2017) 20:2906–20. doi: 10.1016/j.celrep.2017.08.068
195. Sun L, Deng L, Ea CK, Xia ZP, Chen ZJ. The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol Cell (2004) 14:289–301. doi: 10.1016/S1097-2765(04)00236-9
196. Hu H, Wu X, Jin W, Chang M, Cheng X, Sun S-C. Noncanonical NF-κB regulates inducible costimulator (ICOS) ligand expression and T follicular helper cell development. Proc Nat Acad Sci USA. (2011) 108:12827–32. doi: 10.1073/pnas.1105774108
197. De Silva NS, Anderson MM, Carette A, Silva K, Heise N, Bhagat G, et al. Transcription factors of the alternative NF-kappaB pathway are required for germinal center B-cell development. Proc Nat Acad Sci USA. (2016) 113:9063–8. doi: 10.1073/pnas.1602728113
198. Gardam S, Sierro F, Basten A, Mackay F, Brink R. TRAF2 and TRAF3 signal adapters act cooperatively to control the maturation and survival signals delivered to B cells by the BAFF receptor. Immunity (2008) 28:391–401. doi: 10.1016/j.immuni.2008.01.009
199. Liao G, Zhang M, Harhaj EW, Sun SC. Regulation of the NF-kappaB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J Biol Chem. (2004) 279:26243–50. doi: 10.1074/jbc.M403286200
200. Vallabhapurapu S, Matsuzawa A, Zhang W, Tseng PH, Keats JJ, Wang H, et al. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-kappaB signaling. Nat Immunol. (2008) 9:1364–70. doi: 10.1038/ni.1678
201. Zarnegar BJ, Wang Y, Mahoney DJ, Dempsey PW, Cheung HH, He J, et al. Noncanonical NF-kappaB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat Immunol. (2008) 9:1371–8. doi: 10.1038/ni.1676
202. Xiao X, Balasubramanian S, Liu W, Chu X, Wang H, Taparowsky EJ, et al. OX40 signaling favors the induction of T(H)9 cells and airway inflammation. Nat Immunol. (2012) 13:981–90. doi: 10.1038/ni.2390
203. Bryce PJ, Oyoshi MK, Kawamoto S, Oettgen HC, Tsitsikov EN. TRAF1 regulates Th2 differentiation, allergic inflammation and nuclear localization of the Th2 transcription factor, NIP45. Int Immunol. (2006) 18:101–11. doi: 10.1093/intimm/dxh354
204. Pullen SS, Miller HG, Everdeen DS, Dang TTA, Crute JJ, Kehry MR. CD40-Tumor necrosis factor receptor-associated factor (TRAF) interactions: regulation of CD40 signaling through multiple TRAF binding sites and TRAF hetero-oligomerization. Biochemistry (1998) 37:11836–45. doi: 10.1021/bi981067q
205. Nguyen LT, Duncan GS, Mirtsos C, Ng M, Speiser DE, Shahinian A, et al. TRAF2 deficiency results in hyperactivity of certain TNFR1 signals and impairment of CD40-mediated responses. Immunity (1999) 11:379–89. doi: 10.1016/S1074-7613(00)80113-2
206. Jabara HH, Weng Y, Sannikova T, Geha RS. TRAF2 and TRAF3 independently mediate Ig class switching driven by CD40. Int Immunol. (2009) 21:477–88. doi: 10.1093/intimm/dxp013
207. Grech AP, Amesbury M, Chan T, Gardam S, Basten A, Brink R. TRAF2 differentially regulates the canonical and noncanonical pathways of NF-kappaB activation in mature B cells. Immunity (2004) 21:629–42. doi: 10.1016/j.immuni.2004.09.011
208. Lee SY, Reichlin A, Santana A, Sokol KA, Nussenzweig MC, Choi Y. TRAF2 is essential for JNK but not NF-kappaB activation and regulates lymphocyte proliferation and survival. Immunity (1997) 7:703–13. doi: 10.1016/S1074-7613(00)80390-8
209. Cannons JL, Bertram EM, Watts TH. Cutting edge: profound defect in T cell responses in TNF receptor-associated factor 2 dominant negative mice. J Immunol. (2002) 169:2828–31. doi: 10.4049/jimmunol.169.6.2828
210. So T, Choi H, Croft M. OX40 complexes with phosphoinositide 3-kinase and protein kinase B (PKB) to augment TCR-dependent PKB signaling. J Immunol. (2011) 186:3547–55. doi: 10.4049/jimmunol.1003156
211. Xu Y, Cheng G, Baltimore D. Targeted disruption of TRAF3 leads to postnatal lethality and defective T-dependent immune responses. Immunity (1996) 5:407–15. doi: 10.1016/S1074-7613(00)80497-5
212. Xie P, Stunz LL, Larison KD, Yang B, Bishop GA. Tumor necrosis factor receptor-associated factor 3 is a critical regulator of B cell homeostasis in secondary lymphoid organs. Immunity (2007) 27:253–67. doi: 10.1016/j.immuni.2007.07.012
213. Gardam S, Turner VM, Anderton H, Limaye S, Basten A, Koentgen F, et al. Deletion of cIAP1 and cIAP2 in murine B lymphocytes constitutively activates cell survival pathways and inactivates the germinal center response. Blood (2011) 117:4041–51. doi: 10.1182/blood-2010-10-312793
214. Zapata JM, Llobet D, Krajewska M, Lefebvre S, Kress CL, Reed JC. Lymphocyte-specific TRAF3 transgenic mice have enhanced humoral responses and develop plasmacytosis, autoimmunity, inflammation, and cancer. Blood (2009) 113:4595–603. doi: 10.1182/blood-2008-07-165456
215. Chang JH, Hu H, Jin J, Puebla-Osorio N, Xiao Y, Gilbert BE, et al. TRAF3 regulates the effector function of regulatory T cells and humoral immune responses. J Exp Med. (2014) 211:137–51. doi: 10.1084/jem.20131019
216. Michel M, Wilhelmi I, Schultz AS, Preussner M, Heyd F. Activation-induced tumor necrosis factor receptor-associated factor 3 (Traf3) alternative splicing controls the noncanonical nuclear factor kappaB pathway and chemokine expression in human T cells. J Biol Chem. (2014) 289:13651–60. doi: 10.1074/jbc.M113.526269
217. Takaori-Kondo A, Hori T, Fukunaga K, Morita R, Kawamata S, Uchiyama T. Both amino- and carboxyl-terminal domains of TRAF3 negatively regulate NF-kappaB activation induced by OX40 signaling. Biochem Biophys Res Commun. (2000) 272:856–63. doi: 10.1006/bbrc.2000.2860
218. Cherfils-Vicini J, Vingert B, Varin A, Tartour E, Fridman W-H, Sautès-Fridman C, et al. (2008). Characterization of immune functions in TRAF4-deficient mice. Immunology 124:562–74. doi: 10.1111/j.1365-2567.2008.02810.x
219. Nakano H, Sakon S, Koseki H, Takemori T, Tada K, Matsumoto M, et al. Targeted disruption of Traf5 gene causes defects in CD40- and CD27-mediated lymphocyte activation. Proc Nat Acad Sci USA. (1999) 96:9803–8. doi: 10.1073/pnas.96.17.9803
220. So T, Salek-Ardakani S, Nakano H, Ware CF, Croft M. TNF receptor-associated factor 5 limits the induction of Th2 immune responses. J Immunol. (2004) 172:4292–7. doi: 10.4049/jimmunol.172.7.4292
221. Naito A, Azuma S, Tanaka S, Miyazaki T, Takaki S, Takatsu K, et al. Severe osteopetrosis, defective interleukin-1 signalling and lymph node organogenesis in TRAF6-deficient mice. Genes Cells (1999) 4:353–62. doi: 10.1046/j.1365-2443.1999.00265.x
222. Lomaga MA, Yeh WC, Sarosi I, Duncan GS, Furlonger C, Ho A, et al. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. (1999) 13:1015–24. doi: 10.1101/gad.13.8.1015
223. Jalukar SV, Hostager BS, Bishop GA. Characterization of the roles of TNF receptor-associated factor 6 in CD40-mediated B lymphocyte effector functions. J Immunol. (2000) 164:623–30. doi: 10.4049/jimmunol.164.2.623
224. Ahonen C, Manning E, Erickson LD, O'Connor B, Lind EF, Pullen SS, et al. The CD40-TRAF6 axis controls affinity maturation and the generation of long-lived plasma cells. Nat Immunol. (2002) 3:451–6. doi: 10.1038/ni792
225. Kobayashi T, Kim TS, Jacob A, Walsh MC, Kadono Y, Fuentes-Panana E, et al. TRAF6 is required for generation of the B-1a B cell compartment as well as T cell-dependent and -independent humoral immune responses. PLoS ONE (2009) 4:e4736. doi: 10.1371/journal.pone.0004736
Keywords: TRAF, follicular helper T cell, antibody response, TCR signaling, costimulation signaling, cytokine signaling, NF-κB
Citation: Pedros C, Altman A and Kong KF (2018) Role of TRAFs in Signaling Pathways Controlling T Follicular Helper Cell Differentiation and T Cell-Dependent Antibody Responses. Front. Immunol. 9:2412. doi: 10.3389/fimmu.2018.02412
Received: 04 September 2018; Accepted: 28 September 2018;
Published: 22 October 2018.
Edited by:
Tania H. Watts, University of Toronto, CanadaReviewed by:
Andreas Hutloff, Deutsches Rheuma-Forschungszentrum (DRFZ), GermanyWoong-Kyung Suh, Institut de Recherches Cliniques de Montreal (IRCM), Canada
Copyright © 2018 Pedros, Altman and Kong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christophe Pedros, Y3BlZHJvc0Bsamkub3Jn
Kok-Fai Kong, a29rZmFpa29uZ0BnbWFpbC5jb20=