 Judith Wienke
Judith Wienke Claire T. Deakin2,3,4
Claire T. Deakin2,3,4 Lucy R. Wedderburn
Lucy R. Wedderburn Femke van Wijk
Femke van Wijk- 1Laboratory of Translational Immunology, University Medical Center Utrecht, Utrecht, Netherlands
- 2UCL Great Ormond Street Institute of Child Health, University College London, London, United Kingdom
- 3NHR Biomedical Research Center at Great Ormond Hospital, London, United Kingdom
- 4Arthritis Research UK Center for Adolescent Rheumatology, UCL, UCLH and GOSH, London, United Kingdom
- 5Pediatric Rheumatology and Immunology, University Medical Center Utrecht, Utrecht, Netherlands
Juvenile Dermatomyositis (JDM) is a systemic immune-mediated disease of childhood, characterized by muscle weakness, and a typical skin rash. Other organ systems and tissues such as the lungs, heart, and intestines can be involved, but may be under-evaluated. The inflammatory process in JDM is characterized by an interferon signature and infiltration of immune cells such as T cells and plasmacytoid dendritic cells into the affected tissues. Vasculopathy due to loss and dysfunction of endothelial cells as a result of the inflammation is thought to underlie the symptoms in most organs and tissues. JDM is a heterogeneous disease, and several disease phenotypes, each with a varying combination of affected tissues and organs, are linked to the presence of myositis autoantibodies. These autoantibodies have therefore been extensively studied as biomarkers for the disease phenotype and its associated prognosis. Next to identifying the JDM phenotype, monitoring of disease activity and disease-inflicted damage not only in muscle and skin, but also in other organs and tissues, is an important part of clinical follow-up, as these are key determinants for the long-term outcomes of patients. Various monitoring tools are currently available, among which clinical assessment, histopathological investigation of muscle and skin biopsies, and laboratory testing of blood for specific biomarkers. These investigations also give novel insights into the underlying immunological processes that drive inflammation in JDM and suggest a strong link between the interferon signature and vasculopathy. New tools are being developed in the quest for minimally invasive, but sensitive and specific diagnostic methods that correlate well with clinical symptoms or reflect local, low-grade inflammation. In this review we will discuss the types of (extra)muscular tissue inflammation in JDM and their relation to vasculopathic changes, critically assess the available diagnostic methods including myositis autoantibodies and newly identified biomarkers, and reflect on the immunopathogenic implications of identified markers.
Introduction
Juvenile Dermatomyositis (JDM) is a systemic immune-mediated disease of childhood. It is the most common idiopathic inflammatory myopathy in children, with an incidence of 2–4/million/year (1). Although the exact etiology is still elusive, both genetic and environmental factors are thought to play a role in the development of the disease (2–5). JDM is characterized by inflammation of skeletal muscles and skin, leading to muscle weakness and a typical skin rash of the face and hands (heliotrope rash and Gottron's papules, respectively), which are also used as classification criteria (6, 7). Next to the muscle and skin, other organs can be affected. Vital organ involvement, especially of the lungs, is still the major cause of death in JDM patients (8, 9). Although rare, cardiac involvement and microangiopathy of the intestine, brain and kidneys have been described (10). Thus, rather than being confined to specific tissues, JDM is a truly systemic disease, which can affect multiple organ systems.
Before the introduction of corticosteroids as a treatment option, mortality and morbidity among JDM patients were high, and long-term outcomes were not the primary focus. Since then, mortality rates have dropped from over 30% to 2–3% (11). With increasing survival, long-term outcomes become an important concern of patients and physicians, as patients' quality of life and societal participation depend on it. Long-term outcomes are likely dependent on various factors such as disease severity and activity, response to treatment and medication side effects which together determine the cumulative organ and tissue damage.
Especially low-grade inflammation and extramuscular manifestations of the disease are difficult to investigate in routine clinical care and may therefore be overlooked. Unrecognized, local inflammation leading to tissue damage and subsequent organ dysfunction may have serious consequences for short-term and long-term outcomes. So far, reliable assessment of disease activity and the type and extent of tissue involvement has been rather challenging. Current clinical tools for assessment of disease activity require active collaboration of patients, which can be difficult for young, unwell children. Detecting low-grade inflammation or differentiating clinically between various causes of muscle impairment is even more challenging. Hence, there is a great need for minimally invasive, objective and reliable diagnostic tools for the assessment and monitoring of (low-grade) disease activity and related organ involvement. Optimally, such tools could guide clinical decision making, facilitate individually tailored treatment regimens, and reduce the risk of over- and under-treatment.
In this review we will discuss the types of (extra)muscular tissue involvement that have been described in JDM and their relation to vasculopathic changes, critically assess the available diagnostic and monitoring tools and reflect on the immunopathogenic implications of identified markers.
Signs of Systemic Disease Activity in JDM Based on affected Tissues and Organs
JDM patients can present with a spectrum of symptoms. Most, but not all patients, have the classic combination of muscle involvement and typical skin rashes. Approximately 1–5% of JDM patients present with amyopathic JDM, but it was estimated that 26% of these patients will eventually progress to classical JDM, which can occur up to years after onset (12). This indicates that the phenotype can evolve over the course of the disease, possibly also dependent on treatment. True amyopathic JDM however is very rare and mild muscle involvement may be present but missed (13). Amyopathic JDM generally has a relatively mild disease course with fewer systemic manifestations, less required immunosuppressive treatment and a good prognosis (12, 14, 15).
(Sub)Cutaneous and Other Extramuscular Symptoms
Cutaneous symptoms can range from the pathognomonic heliotrope rash and Gottron's papules, to photosensitive rashes such as malar and truncal erythema, and severe complications such as skin ulceration and dystrophic calcinosis. Calcinosis occurs in 12–47% of patients and can occur in the skin and in subcutaneous, myofascial, or muscle tissue. Most often it is a long-term complication and its presence has been associated with delayed diagnosis and more severe disease with poorer functional outcomes. Effective treatment of calcinosis is still challenging, but aggressive high-dose immunosuppression or, in very severe cases, autologous stem cell transplantation have been shown to be able to reverse calcinosis, suggesting that chronic (low-grade) inflammation may be accountable for calcifications (16–20). Cutaneous and oral ulceration affects up to 30% of patients and is thought to result from occlusive endartheropathy of the small vessels (10, 21). Lipodystrophy affects 8–14% of JDM patients and is often associated with hormonal and metabolic changes (10, 22–24). We suspect that patients with lipodystrophy may therefore have an increased risk of cardiovascular events in the long-term. Limb edema and arthritis are also common, occurring in 11–32 and 23–58% of patients, respectively (10).
Next to the skin and musculoskeletal system, other organ systems can be involved, of which the lung is the most frequently affected. Up to 75% of children with JDM develop respiratory involvement, which may result from a complication of respiratory muscle weakness or immunosuppressive therapy, or from interstitial lung disease (ILD) (25, 26). ILD occurs in 8–19% of juvenile myositis patients and has been described as the major cause of death in JDM (27–30). Cardiac involvement may be present subclinically more often than recognized, as even in JDM patients without clinical cardiac dysfunction abnormal ECG and echocardiographic findings are relatively common (31–33). Conduction abnormalities and myocarditis have been reported, and systolic and diastolic dysfunction was found after long-term follow-up (34–37). Cardiac complications are thought to result from myocarditis and coronary artery disease as well as involvement of the small vessels of the myocardium (38). Involvement of the gut or neural system are rare complications of JDM and are also thought to result from an underlying small vessel angiopathy or vasculitis (39–41). Intestinal consequences of the small vessel angiopathy include ulceration, perforation, hemorrhage, pneumatosis intestinalis and malabsorption (42–44).
Vasculopathy
The pathologic changes underlying symptoms and tissue damage in the skin, muscles, and vital organs have a common factor: in all the affected tissues typical vasculopathic changes are observed, which include loss of capillaries (capillary dropout), perivascular inflammation, and (occlusive) small vessel angiopathy (21, 45). In a recently reported French JDM cohort of 116 patients, vasculopathy-related complications were the main cause of admission to the intensive care unit, illustrating the severity and relevance of vascular involvement in JDM (46). These complications include life-threatening disorders like systemic capillary leak syndrome, recently also described in 3 patients with JDM (47).
Deposition of complement, immune complexes and anti-endothelial antibodies is thought to play an important role in endothelial damage and subsequent capillary dropout (48–54). Clinically, the severity of vasculopathy and the disease phenotype have also been linked. The presence of prominent vascular injury in muscle biopsies identified a subgroup of patients with more severe clinical presentation and outcomes, including profound muscle weakness, limb edema and gastrointestinal involvement (55). This suggests that local vasculopathic changes can reflect systemic vasculopathy and the resulting clinical symptoms. Nailfold capillaroscopy, a commonly and easily used indicator of disease activity in clinical practice, is also based on this principle. The pathologic changes observed in nailfold capillaries, such as capillary dropout, branching and dilatation, likely reflect the systemic blood vessel abnormalities. Loss of end row nailfold capillaries is significantly associated with clinical disease activity scores for muscle and skin and can thus be used as a marker of skin and muscle activity. Nailfold capillaroscopy is especially suited as a non-invasive tool to follow up changes in disease activity over time in patients (56–59).
Taken together, JDM is a truly systemic disease in which not only the muscles and skin are affected, but also vital organs can be involved. The presence of typical vasculopathic changes in the various affected tissues points toward a central role for systemic endothelial dysfunction in the pathogenesis of JDM.
Monitoring of Disease Activity and Tissue Involvement
During clinical follow-up, monitoring of disease activity is crucial to determine the rate of medication tapering or to assess the requirement for intensification of immunosuppressive therapy. Next to clinical evaluation, various tools have been investigated for monitoring of disease activity, among which autoantibodies and other circulating biomarkers, and histopathologic evaluation of muscle biopsies, as well as several imaging techniques.
Clinical Assessment
The primary and most important evaluation of disease activity involves clinical assessment by experienced clinicians and health care professionals. Over the past years, several scoring tools have been devised for internationally standardized evaluation of disease activity (60). The most commonly used tools are now the childhood myositis assessment scale (CMAS), manual muscle testing of 8 muscle groups (MMT-8), physician's and patient's global assessment on a visual analog scale (PGA), cutaneous assessment tool (CAT), cutaneous dermatomyositis disease area and severity index (CDASI), disease activity score (DAS), myositis disease activity assessment tool (MDAAT) and childhood health assessment questionnaire (CHAQ) (61–69). Combined scoring systems are currently being developed (70). The Pediatric Rheumatology International Trials Organization (PRINTO) has composed criteria for defining clinically inactive disease (71). A recent re-evaluation of these PRINTO criteria showed that skin disease may be underestimated as a factor in the assessment of disease activity (72).
Clinical measures of disease activity, however, have limited capacity to detect low-grade inflammation in the tissues which does not cause overt symptoms, but may still contribute to tissue damage in the long term. Moreover, it is challenging to differentiate between various underlying causes of symptoms by clinical assessment. For example, muscle weakness may result from an ongoing inflammatory process, from medication side effects (e.g., steroid myopathy), muscle damage or effects of immobility. Biological assessment of the affected tissues and organs can therefore be helpful or even necessary to aid clinical decision-making concerning medication dose and additional interventions.
Biomarkers for Disease Course, Activity, and Tissue Involvement
Laboratory investigation of blood is a minimally invasive and time-efficient procedure, especially compared to muscle biopsy and some of the imaging methods. It is therefore particularly suited as a method for serial sampling during clinical follow-up. Laboratory investigation can be used for measurement of autoantibodies and for biomarkers related to disease activity and specific (extra)muscular symptoms.
Autoantibodies
Antibodies found in myositis include myositis-specific autoantibodies (MSA), relatively specific to myositis, and myositis-associated antibodies (MAA), which are observed both in myositis and other connective tissue diseases (6). In the past years, different disease phenotypes have been linked to the presence of autoantibodies and particularly myositis-specific autoantibodies (16). The frequencies of autoantibodies in juvenile patients differ substantially from adult DM patients (73). Anti-TIF1 (p155/140) and anti-NXP2 (p140 or MJ) are the most commonly identified autoantibodies in Caucasian JDM patients (20–35 and 16–23%, respectively) (28, 73–76). Anti-TIF1 is associated with skin ulceration, photosensitive skin rashes, lipodystrophy, and edema (24, 75–78), whereas anti-NXP2 is associated with a severe disease course with more profound muscle involvement, calcinosis, gastrointestinal ulceration, joint contractures, and dysphonia (75, 77, 79, 80). A recently identified myositis specific autoantibody which is especially frequent in the Asian JDM population, is anti-MDA5 (CADM-140) (81). It is found in 33% of Asian JDM patients, compared to 7% of Caucasian patients (8, 82). Patients with anti-MDA5 have a higher risk of developing ILD than patients without these antibodies. This anti-MDA5 conferred risk is seen in both Asian and Caucasian JDM cohorts, although the risk difference appears to be more pronounced in Asian cohorts (8, 83). Common symptoms in Caucasian patients with anti-MDA5 antibodies include oral and cutaneous ulceration, arthritis, and milder muscle disease with fewer histologic abnormalities and a higher remission rate off medication after 2 years of follow-up (76, 82, 84, 85). Less frequently identified autoantibodies in the juvenile population include anti-Mi2 (4–10%) and anti-amino-acyl-tRNA synthetase antibodies such as anti-Jo-1 (1–3%) and anti-SAE (<1%). Anti-SRP and anti-HMG-CoA-reductase (Anti-HMGCR) autoantibodies, both accounting for <3% of juvenile myositis patients, are associated with a necrotizing type of myopathy with severe muscle weakness (73, 76, 86, 87).
It remains unclear whether each MSA reflects a distinct pathologic process, influencing the type and severity of disease phenotype and tissue involvement. Notably, autoantibodies against Jo-1, TIF1, SRP, and Mi-2 are not only informative at disease onset, but their levels have been found to correlate with disease activity during follow-up in the context of rituximab treatment (88). This highlights that perhaps autoantibodies should be measured during or soon after the first clinic visit as their levels may decline and become undetectable in remission.
A last and different (not myositis-specific) category of autoantibodies identified in JDM comprises autoantibodies against components of endothelial cells, which are thought to contribute to capillary loss. These anti-endothelial cell autoantibodies (AECA) were detected in 76% of JDM patients, as opposed to 30% of control patients (49). Twenty-two candidate target autoantigens for AECA were identified in JDM plasma, 17 of which were proteins associated with antigen processing and protein trafficking (50). Identification of autoantibody targets may provide novel insights into the auto-immune process and self-antigens involved in JDM.
Biomarkers for Systemic Inflammation and Muscle Disease Activity
Reliable assessment of disease activity during follow-up can be aided by laboratory markers that represent systemic and/or local inflammation. Especially for detection of low-grade inflammation and for differentiation between various causes of muscle weakness, laboratory investigation can be a helpful or even necessary tool.
So far, reliable and validated laboratory markers for disease activity and tissue involvement in JDM are still lacking. A large number of proteins in plasma, serum, and urine as well as circulating immune cell subsets have been investigated as potential biomarkers for (tissue-specific) disease activity in patients with JDM (Tables 1, 2). In theory, every biological parameter that can be measured, could serve as a biomarker. To be suited for use in clinical practice however, a biomarker has to meet additional criteria, such as being reliable, robust, relatively stable and easy to measure. In the following paragraphs we highlight all biological markers that have been associated with disease activity in JDM, regardless of their suitability for use in clinical practice, as some of these identified markers may still contribute to the understanding of the immunopathogenesis of JDM. However, it is important to note that due to the rarity of the disease, many of these studies were carried out in small cohorts of <30 patients (as outlined in Tables 1, 2). Insights based on such small numbers have limitations in a heterogeneous disease like JDM. Therefore, validation of identified markers in larger cohorts is crucial before implementation into clinical practice.
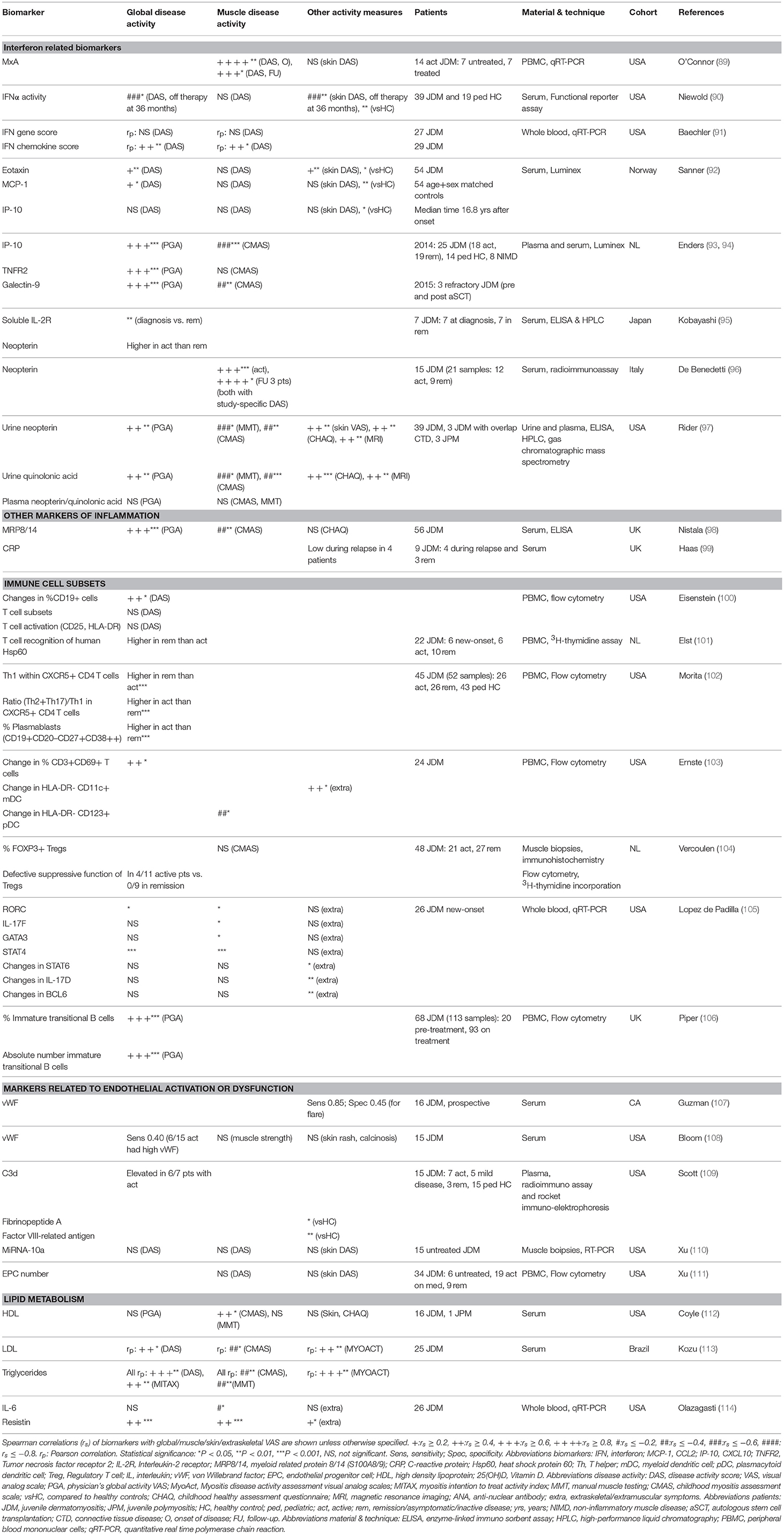
Table 1. Biomarkers for disease activity in JDM cohorts.
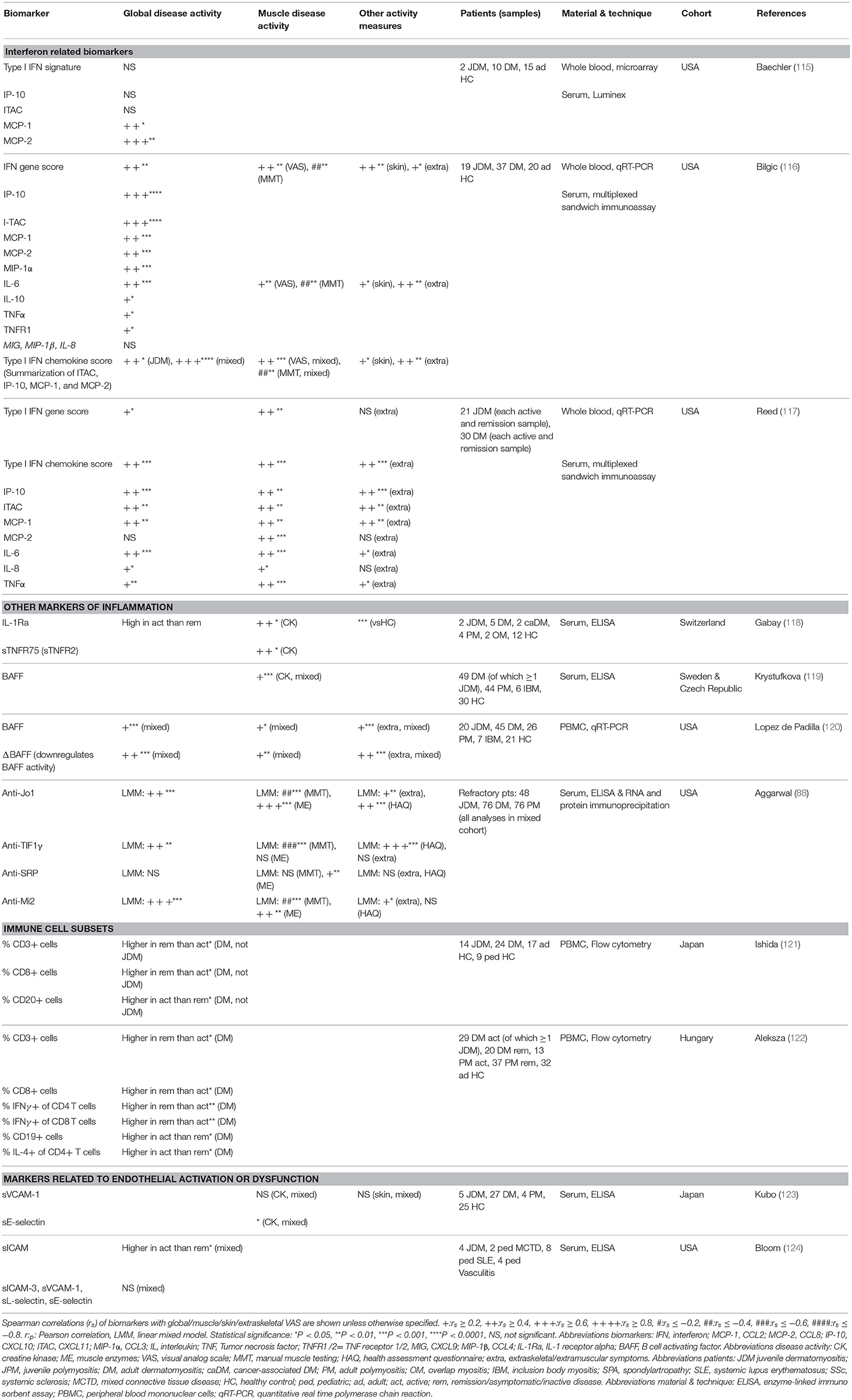
Table 2. Biomarkers for disease activity in mixed cohorts containing patients with JDM.
Currently used laboratory markers
The markers that are currently used in clinical practice, AST, ALT, LDH, aldolase and in particular creatine kinase activity (CK), do not correlate as well with disease activity in JDM as in DM (125–127). At diagnosis, any one muscle enzyme was only elevated in 80–86% of patients with JDM and CK was found to be elevated in only 61–64% of patients (125, 128). In almost 20% of patients the most abnormal measurement of CK was not elevated above normal values (28). Low muscle enzymes at first presentation may be associated with delayed diagnosis (129). During follow-up, CK may underestimate disease activity due to suppressed release by corticosteroids, circulating inhibitors of CK activity, or loss of muscle mass (127, 130–132). On the other hand, CK and aldolase can be elevated in steroid myopathy and are therefore not reliable as markers for disease activity requiring more potent immunosuppression (133). However, according to recent consensus guidelines, these muscle enzymes are still regarded as an important monitoring tool (134, 135).
Markers related to the interferon signature
An important group of investigated biomarkers is related to the type 1 interferon (IFN) signature, which has been demonstrated in the peripheral blood and muscle biopsies of JDM patients (136, 137). Activated plasmacytoid dendritic cells (pDC) are generally thought to be the main producers of the type 1 IFNs (IFNα and IFNβ) in JDM. This notion may be challenged by a recent study measuring circulating IFNα with a highly sensitive assay and investigating the cellular source of IFNα in several systemic inflammatory diseases. JDM patients had higher levels of circulating IFNα than patients with systemic lupus erythematosus (SLE), but lower levels than patients with monogenic interferonopathies. However, neither isolated circulating pDC nor other circulating immune cell subsets from JDM patients expressed more IFNα than cells from healthy controls, suggesting that a non-circulating cellular source may be responsible for IFNα production in JDM (138).
Due to the lack of available methods to measure circulating IFNα and IFNβ until recently, the type 1 IFN signature, consisting of genes upregulated in response to IFNα or IFNβ stimulation, was used as a surrogate marker of type 1 IFN levels. The type 1 IFN signature in whole blood of three mixed DM and JDM cohorts correlated weakly to moderately with global disease activity [spearman r (rs) = 0.33–0.44] and muscle activity (rs = 0.44–0.47), while single IFN signature related serum chemokines MCP-1, IP-10 (CXCL10) and ITAC (CXCL11) had moderate to strong correlations with global (rs = 0.42–0.66), muscle (rs = 0.44–0.50), and extraskeletal disease activity (rs = 0.42–0.55) (115– 117). MxA expression in PBMC, also used as a surrogate for the IFN signature, had a very strong correlation with muscle disease activity of JDM patients (rs = 0.80) at disease onset, but not with skin disease activity (89). IFNα activity measured by a functional reporter assay was also higher in JDM patients than controls (90).
Recently, IP-10, TNF receptor 2 (TNFR2) and galectin-9 were found to strongly correlate with global disease activity (r = 0.60–0.75) in two studies by Enders et al. (93, 94) IP-10, together with MCP-1 and eotaxin, was also higher in 54 JDM patients a median of 17 years after disease onset than matched healthy controls (92). TNFR2 correlated with CK in a mixed IIM cohort (rs = 0.55) (118). Galectin-9 was recently identified as a biomarker for the IFN signature in SLE and anti-phospholipid syndrome (139). IP-10, TNFR2 and galectin-9 are promising biomarkers for disease activity, as they can potently discriminate between active disease and remission even during treatment (93, 94). After stem cell transplantation and concomitant eradication of circulating immune cells, their levels stayed high over several months, which suggests that these proteins are not primarily produced by circulating immune cells, but rather by non-circulating immune or tissue cells, just as IFNα (94, 138). IP-10 and galectin-9 are currently being validated as biomarkers for disease activity in two large international JDM cohorts.
One of the best investigated biomarkers so far in JDM is neopterin, a catabolic product of guanosine triphosphate, which was previously shown to be a marker of immune activation that can be induced by stimulation with IFNγ (140). In the first study identifying serum neopterin as a biomarker for JDM, neopterin levels correlated strongly with muscle strength impairment in 15 JDM patients (rs = 0.68) (96). Elevated serum neopterin levels at diagnosis compared to remission were confirmed in an independent cohort (95). In a juvenile myositis validation cohort, plasma neopterin (n = 13), and quinolonic acid (n = 24), however, did not correlate with myositis disease activity measures (97). Urine neopterin (n = 45) moderately correlated with global (rs = 0.42), muscle (rs = 0.50–0.62) and skin activity (rs = 0.49), and edema on MRI (rs = 0.55). Urine quinolonic acid also correlated with global and muscle activity and edema on MRI (rs = 0.45–0.61) (97). Despite these efforts of validation, neopterin has not been widely implemented into clinical practice as a biomarker for disease activity in JDM.
Other inflammatory mediators
Next to type 1 IFN-related markers, other inflammatory mediators have been studied as biomarkers for JDM. The innate TLR4 ligand myeloid related protein 8/14 (MRP8/14 or S100A8/9), originally found to be elevated in patients with systemic-onset juvenile idiopathic arthritis (JIA), correlated moderately to strongly with global and muscle disease activity in a large cohort of 56 JDM patients (rs = 0.55–0.65) (98, 141). Another marker adopted from studies in JIA, the soluble IL-2 receptor, was elevated at disease onset compared to remission (95, 142). Serum/plasma levels of the more conventional pro-inflammatory cytokines IL-6, IL-8, and TNFα also moderately correlated with global (rs = 0.19–0.46) and muscle disease activity (rs = 0.35–0.52) in three mixed JDM and DM cohorts (116, 117). Remarkably, CRP levels did not increase during disease flares (99). BAFF and especially its antagonistic non-cleavable form ΔBAFF, both important for survival and maturation of B cells, moderately correlated with global, muscle and extraskeletal VAS (rs = 0.27–0.54), and CK (rs = 0.37) in two mixed IIM cohorts (119, 120).
Markers related to vasculopathy and cardiovascular risk
Due to the vasculopathic component of JDM, markers related to endothelial activation and dysfunction were explored for their association with disease activity. Von Willebrand factor (vWF) was increased during most periods of active disease in a prospective cohort study, but did not reliably predict disease flares in another study (107, 108). sICAM-1, a marker of endothelial activation, was higher during active disease than remission in a combined cohort of juvenile patients with various systemic autoimmune diseases. VCAM-1, sICAM-3, and L-selectin did not correlate with disease activity, although expression of MiRNA-10a in JDM muscle, which is negatively associated with VCAM-1 expression, showed a correlative trend with muscle and global DAS [Pearson r (rp) = −0.45] (110, 123, 124). C3d and fibrinopeptide A, which are related to vasculopathic changes, were higher in JDM patients with active disease than in remission (109). Endothelial progenitor cell numbers did not differ between JDM patients and controls and did not correlate with disease activity (111).
In view of the increased cardiovascular risk in JDM patients, the lipid profile has been investigated in relation to disease activity (41). Serum HDL negatively correlated with muscle activity (rs = −0.54), but not global or skin activity (112). Triglyceride levels correlated strongly with global disease activity assessed by DAS (rs = 0.61) and LDL was higher in patients with a higher disease activity (113). Gene expression of the adipokine resistin in PBMC was also upregulated in JDM patients compared to controls and moderately correlated with global and muscle disease activity (rs = 0.51 and rs = 0.50, respectively) (114). These results indicate that the cardiovascular risk profile is more pronounced in JDM patients with active disease.
Circulating immune cell subsets as biomarkers for disease activity
Among the circulating immune cell subsets, T cells and B cells have been studied most extensively in relation to disease activity in JDM. In two mixed cohorts of JDM and DM patients, the frequency of T cells, and especially CD8+ and IFNγ-producing T cells, was decreased during active disease, while the frequency of B cells and IL-4 producing CD4+ T cells was increased compared to remission (121, 122). This may suggest a shifted balance toward a T helper 2 (Th2) type immune response. In cohorts with only JDM patients, total B cell numbers were also increased compared to controls and changes in B cell frequencies accompanied changes in disease activity (rs = 0.47) (100, 106). Within the B cell compartment, numbers and frequencies of circulating immature transitional B cells correlated strongly with global disease activity (rs = 0.69–0.71). Compared to healthy pediatric controls, these specialized B cells were highly proliferative, had a prominent IFN signature and produced less of their regulatory signature cytokine IL-10 (106). Plasmablast frequencies were also increased during active disease compared to remission (102).
Several T cell subsets have been studied in JDM. In 26 new-onset JDM patients the blood gene expression of Th17-related genes, such as RORC and IL-17F, Th1-related genes, including STAT4, and Th2-related genes, including GATA3 and STAT6, was studied in relation to disease activity. RORC, IL-17F, STAT4, and GATA3 positively correlated with muscle activity and RORC and STAT4 correlated with global activity. This would suggest that the immune response is not specifically skewed toward a certain T helper response. However, at baseline, JDM patients had higher gene expression of Th17 related cytokines IL-23, IL-17F, IL-6, and IL-21 than DM patients, indicating that the Th17 pathway may play a more prominent role in the pathogenesis of JDM than DM. Changes in BCL6, a transcription factor for follicular helper T cells, correlated negatively with a change in extramuscular activity (105). Within CXCR5+ follicular helper T cells, the Th1 subset was decreased in active JDM compared to remission and controls, and Th2 and Th17 subsets were increased in JDM compared to controls (102). Regulatory T cell frequencies in muscle biopsies did not correlate with muscle activity, but suppressive activity of circulating Tregs may be impaired during active disease (104). Finally, global disease activity correlated moderately with the activation status of circulating T cells assessed by CD69 expression (rs = 0.43), but not with CD25 and HLA-DR expression (100, 103). The expansion and functional alteration of particular B cell and CD4+ T cell subsets, coinciding with changes in disease activity, hints toward the involvement of these cell subsets in the pathogenesis of JDM.
In conclusion, many circulating, either soluble or cellular, markers have been studied for their relation with muscle and global disease activity. Correlations with disease activity were only moderate for most markers, and some of these molecules are relatively unstable in blood samples or complicated to measure, rendering them unsuited for use in clinical practice. The highest correlations with disease activity were found for markers related to the IFN signature, the lipid profile, for MRP8/14, and immature transitional B cells. However, most of these biomarkers were identified in small patient cohorts and except for neopterin, so far none have been reproduced or thoroughly validated in independent and large JDM cohorts. Neopterin was investigated in a validation cohort, but its correlation with disease activity could only be confirmed in urine, not in plasma. Galectin-9 and IP-10 are currently being validated in two international cohorts and are promising biomarkers for implementation in clinical practice due to their high sensitivity and stability in serum.
Biomarkers for Extramuscular Disease Activity
Next to markers for global and muscle disease activity, biomarkers for involvement of specific tissues and organs have been investigated. Four studies by Kobayashi et al. have focused on biomarkers for ILD, and specifically the rapid progressive (RP-ILD) and chronic ILD type, in a Japanese JDM cohort. Not only the presence, but also the level of anti-MDA5 was a sensitive and specific marker for ILD, with the highest levels found in patients with RP-ILD (8, 143, 144). In addition, BAFF, APRIL, KL-6, and IL-18 levels were higher in patients with RP-ILD compared to chronic ILD and JDM patients without ILD (145). KL-6 was prognostic for ILD, as it stayed high in patients with persistent damage on HRCT (144). Biomarkers for cardiac involvement were tested in a Norwegian JDM cohort, a median of 17 years after diagnosis. Eotaxin and MCP-1 were elevated in patients with cardiac dysfunction and correlated moderately to strongly with systolic and diastolic dysfunction especially in patients with persistently active disease (rs = 0.45–0.65) (146). In the same cohort, a reduced heart rate variability, which is an indicator of cardiac disease, correlated moderately with ESR, hsCRP, and also MCP-1 and eotaxin levels (rs = 0.29–0.47) (147). Next to the autoantibody NXP2, which is prognostic for the development of calcinosis, phosphorylated matrix Gla protein was shown to be higher in patients with calcinosis than without calcinosis (79, 148). Reduced osteocalcin levels were found to be predictive of reduced bone mass, even before start of steroids (149). The presence of the TNFα-308A allele is associated with a more severe disease in JDM. However, apparent associations with this allele are likely to reflect the association with ancestral haplotype 8.1 due to linkage disequilibrium and should be interpreted with this in mind (150). Patients with this genotype are reported to show prolonged symptoms requiring ≥36 months of immunosuppressive therapy, a higher incidence of pathologic calcifications, increased production of TNFα by peripheral blood mononuclear cells in vitro and JDM muscle fibers in vivo, a higher IFNα activity and a higher rate of complications arising from occlusion of capillaries. Vascular occlusion has been linked to higher levels of the anti-angiogenic thrombospondin-1 (90, 151–154). In summary, a number of potential biomarkers for extramuscular disease activity has been identified, and especially for ILD and cardiac dysfunction the biomarkers seem promising. Validation in independent cohorts will have to confirm their potential as biomarkers for these extramuscular symptoms.
Histopathology of Muscle and Skin Biopsies
The diagnostic criteria for JDM by Peter and Bohan encompass histopathological findings consistent with DM: “necrosis of myofibers, phagocytosis, regeneration with basophils, large vesicular sarcolemmal nuclei, and prominent nucleoli, atrophy in a perifascicular distribution, variation in fiber size and an inflammatory exudate, often perivascular” (155, 156). For a long time, muscle biopsies were therefore taken as part of routine diagnostic workup. However, with evolving diagnostic options and more specialized trained pediatric rheumatologists muscle biopsies are currently not always considered a necessity for diagnosis (135).
One of the main problems hindering standardized evaluation of muscle biopsies was the lack of an internationally agreed upon scoring tool. An international consensus group of pediatric rheumatologists and pathologists developed such a tool, which encompasses 4 histopathological scoring domains: inflammatory, vascular, muscle fiber and connective tissue changes (157). The scoring tool has now been validated in an independent cohort consisting of 55 patients and was found to correlate with clinical measures of disease activity, including CMAS, PGA, and MMT-8 (rs = 0.40–0.62) (45). Muscle biopsy scores may also have prognostic potential: in combination with MSA group, these scores were found to predict the risk of remaining on treatment over time, based on analysis of muscle biopsies from 101 JDM patients (158).
The most common findings in muscle biopsy specimens in JDM compared to healthy individuals or patients with non-inflammatory muscle diseases, are profound upregulation of MHC I expression on muscle fibers, increased expression of integrins and complement and membrane attack complex deposition on capillaries and perimysial large vessels, a type 1 IFN signature and immune cell infiltrates consisting mostly of mature pDC, memory CD4+ T cells, and B cells (48, 52, 159–169). (Figure 1) The IFN signature, measured by expression of MxA, correlated with muscle disease activity (166). In skin biopsies similar features are found, with the additional presence of diffuse mast cell infiltration (164).
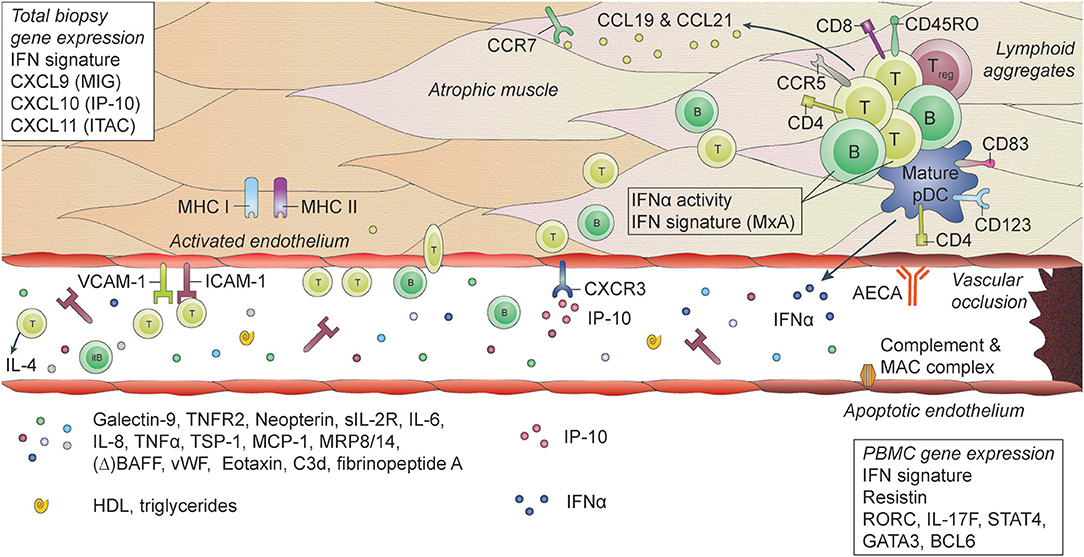
Figure 1. Histopathological features and biomarkers in JDM. JDM is characterized by vasculopathic changes in the tissues, with loss and dysfunction of endothelial cells, leading to capillary dropout and subsequent atrophy of muscle fibers. The exact chain of events leading to loss of blood vessels and muscle fibers is not known, but it is thought that both overexpression of MHC-I (and MHC-II) on myocytes and endothelial damage are early events in the cascade (159, 170). They result in the first attraction of immune cells to the tissue, probably by a stress response of the myocytes and endothelial cells, leading to a first production of chemoattractants. The immune cell infiltrates, which can be organized in lymphoid structures, consist mostly of CD4+ and CD8+ memory T cells, B cells, mature plasmacytoid dendritic cells (pDC) and monocytes. CD4+ and CD8+ T cells are considered responsible for direct killing of muscle cells. pDC are considered the main producers of type I interferons (IFNs), which explains the IFN-I signature that is found in the muscles of JDM patients. Some typical IFN-inducible chemokines, CXCL9 (MIG), CXCL10 (IP-10), and CXCL11 (ITAC), are known for their angiostatic properties. The receptor for these cytokines, CXCR3, is upregulated on endothelial cells in JDM muscle, which may be one of the factors contributing to endothelial dysfunction (137). Other factors include anti-endothelial circulating antibodies (AECA), complement and membrane attack complex (MAC) deposition on endothelial cells. Endothelial cells in muscle also express high levels of ICAM-1 and VCAM-1, which further enables extravasation of immune cells into the tissues and promotes a positive feedback loop resulting in further tissue damage. Not only immune cells in the tissues, but also circulating immune cells show a type I IFN signature and increased IFNα activity. Various circulating markers reflecting immune activation and endothelial activation or distress are increased during active disease in JDM and can potentially be used as biomarkers for disease activity.
Several studies have suggested associations between histopathological findings in muscle biopsies and disease duration before the biopsy or disease severity at a later time point. Biopsy specimens taken after a short duration of untreated disease (<2 months), showed higher expression of VCAM-1 (which correlated with higher serum soluble VCAM-1) and expression of genes involved in stress response and protein turnover, whereas biopsies taken after more than 2 months of untreated disease had more pDC infiltration, higher expression of genes involved in the immune response and vascular remodeling and more apoptosis-related markers (171–173). Thus, it should be taken into account that histological findings can depend on the disease duration before the biopsy. In addition, these findings may indicate that endothelial activation is an early feature of JDM, which precedes immune cell infiltration and vasculopathy.
The degree of vasculopathy and vascular injury (as defined by marked capillary dropout, increased direct immunofluorescent arterial staining and lymphocytic vasculitis, amongst others) was associated with a more severe and chronic disease, with severe or persistent weakness, low remission rates at 12 months requiring additional treatment, subcutaneous edema, and chronic ulcerative disease of the skin and gastrointestinal tract (21, 55, 165). The degree of vasculopathy was also correlated with the expression of angiostatic chemokines MIG, IP-10 and ITAC (137). This indicates that the degree of vascular injury may be one of the most important factors determining long-term disease outcomes and that it is related to the IFN signature.
Not only the type of immune cell infiltration, but also the organization of immune cells in the muscle is of significance in JDM. Organization of immune infiltrates in lymphocytic aggregates or lymphoid follicle–like structures with dendritic cells and T cells, as compared to diffuse infiltrates, was associated with a more severe disease course and less response to treatment (174). MHC I expression, one of the most prominent and early histological features in JDM, did not correlate with clinical features of the disease (159, 160, 175).
The importance of thorough and standardized assessment of tissue involvement is underlined by the fact that even in cases with amyopathic DM, with normal EMG and MRI findings, the muscle biopsy can show focal endomysial lymphocyte and macrophage aggregates and 90% positivity for HLA class I in the sarcolemma (176). Unrecognized, low-grade muscle inflammation may be undertreated, resulting in a larger risk of long term damage. However, muscle biopsy is not routinely performed for children with JDM in all centers and therefore in future, biomarkers which are measureable in blood and correlate with biopsy features would represent a major advance.
Immunopathogenic Implications: Interferons and Vasculopathy
From the biological research conducted in JDM so far, it has become clear that IFNs and their signature play an important role in the immunopathogenesis of JDM (Figure 1). The IFN signature is detectable in muscle fibers, myogenic precursor cells, endothelial cells, skin and several circulating cell subsets of patients with JDM and could point toward a viral etiology (89, 106, 167). Although it has never been demonstrated definitively, several studies suggest that infections may be more common before onset of JDM (177–180). Not only are IFNs potent drivers of (auto)inflammation, they may also be anti-angiogenic factors that could directly or indirectly contribute to endothelial damage and loss in JDM: directly by inhibiting angiogenesis and disrupting the vascular network organization and indirectly by inducing several other angiostatic factors such as galectin-9, IP-10, and ITAC (137, 181–186). In addition, type 1 IFNs inhibit the generation of myotubes and induce atrophy-associated genes in differentiated myotubes. Human skeletal muscle cells can also produce large quantities of IP-10 upon stimulation with IFNγ and TNFα (186, 187).
Rather than being produced by circulating immune cells, IFNs are probably mainly produced within inflamed tissues. Satellite cells, active myogenic cells and endothelial cells in JDM muscle strongly express IFNβ (167). The notion that non-circulating cells within tissues are responsible for IFN production also fits observations by Rodero et al. (138). In particular within muscle of JDM patients the dysbalance between angiogenic and angiostatic factors can contribute to endothelial loss (137, 188). Endothelial cells in JDM muscle downregulate genes related to vessel development, cell adhesion and migration, which are essential for angiogenesis (167). Downregulation of these genes is likely a key event in the development of vasculopathy. Next to being a target of the inflammation, the endothelium may also play an active role in the inflammatory process. In biopsies from JDM patients endothelial cells express inflammatory features, such as high levels of adhesion molecules ICAM-1 and VCAM-1, and produce cytokines and chemokines (161). These can facilitate the attraction and invasion of immune cells into tissues, thereby supporting the inflammatory process and subsequent damage. IP-10 and ITAC were the most highly upregulated genes in endothelial cells from JDM muscle and correlated with the degree of vasculopathy (137, 167). Endothelium-derived IP-10 can even stabilize the interaction between T cells and endothelial cells, thereby possibly contributing to the chronicity of T cell infiltration (189). Recently, a new function has been ascribed to endothelial cells as “semi-professional” antigen presenting cells, which act as sentinels for antigens, and possibly self-antigens, in tissues and facilitate T cell trafficking into these tissues (190, 191). The high expression of MHC molecules on endothelial cells in JDM muscle may support the notion that this process is involved in JDM (160, 175). Although the exact mechanisms of interaction between immune cells and endothelial cells in JDM are still elusive, they may be more elaborate than so far recognized.
Conclusions and Future Perspectives for Biomarker Research
JDM is a multisystem disease. Not only the skin and skeletal muscles are affected, but also other organ systems and tissues such as the lungs, heart and intestines are frequently (subclinically) involved and may be under-evaluated. Vasculopathy due to loss and dysfunction of endothelial cells as a result of the inflammatory process is thought to underlie the symptoms in most of these organs and tissues. Monitoring of disease activity and damage in all of these affected tissues is important during clinical follow-up, as these are key determinants for the long-term outcomes of patients. Tools for monitoring of tissue activity and damage include histopathological investigation of biopsies, and laboratory testing of blood for specific biomarkers as well as several imaging methods. Each of these methods has their strengths and weaknesses and can be of value for specific diagnostic questions at disease onset or during follow-up, as outlined in the consensus-based recommendations for the management of JDM (135, 192). There is still a need for minimally invasive, but at the same time sensitive and specific diagnostic methods that correlate well with clinical symptoms or reflect low-grade, local inflammation. Tissue-specific biomarkers can therefore be of great value as a monitoring tool.
To be able to identify sensitive, robust and reliable biomarkers or develop monitoring tools, it is of key importance to set up well-defined and large prospective patient cohorts, with a thorough longitudinal collection of a standardized clinical dataset assessing disease activity and organ involvement, paired with collection of patient material (193). Such a dataset is required to ensure a strict definition of active and inactive disease [e.g., as proposed by Almeida et al. (72)]. An important consideration for a successful biomarker study is the timing of data and sample collection: depending on the purpose of the biomarker, time points before start of immunosuppressive treatment, before each adjustment of medication, during flares, at paired time points during active and inactive disease or even at regular intervals of max 3–4 months may be crucial to reliably investigate the potency of a biomarker.
Next to the “classical” statistical approach, comparing patients with active disease and patients in remission (cross-sectionally or in paired samples), new computational approaches providing analysis methods that can integrate longitudinal data from multiple patients and multiple (bio)markers or scoring tools should be considered. These methods take into account the fluctuating nature of a relapsing-remitting disease such as JDM and are therefore better suited to test the reliability of a tool that will be used for longitudinal follow-up in clinical practice (194, 195).
To achieve implementation of a marker or tool into clinical practice, both clinical and technical validation in independent cohorts is of utmost importance. Only few markers prove to be stable, reliable and easy to measure, which are key features for a marker or tool to be suited for implementation into clinical practice. Also the invasiveness of the method should be taken into account. Ideally, a period of experimental implementation can demonstrate the added value and feasibility of a marker or tool in clinical practice. To achieve all this in a large group of JDM patients to ensure sufficient statistical power, international networks with well-established collaborations are fundamental.
Eventually, monitoring of disease activity with a reliable tool can be used to guide treatment and thereby facilitate precision medicine, with high dose therapy when indicated but also preventing overtreatment. This may reduce both the duration of active disease and thereby the disease-inflicted damage, and medication side effects, which will benefit the long-term outcomes on various domains, such as muscle weakness, organ damage, cardiopulmonary fitness, and quality of life. Next to facilitating personalized treatment strategies, newly identified biomarkers may also provide insights into the immunopathogenesis of JDM and provide new treatment targets. For instance, new treatment strategies targeting the IFN signature, such as anti-IFN antibodies (sifalimumab) or JAK-inhibition (ruxolitinib) have been shown to reduce the IFN signature in blood and muscle of adult dermatomyositis patients, and may therefore be promising new strategies for patients with JDM (186, 196, 197). Several studies discussed in this review suggest a strong link between the IFN signature and vasculopathy; and vasculopathy has been related to disease severity. Targeting the IFN signature may thus benefit vascularization in JDM and thereby improve outcomes.
Author Contributions
JW collected literature and wrote the manuscript draft. FvW and AvR-K supervised JW, outlined the manuscript focus and revised the manuscript. CD and LW critically revised the manuscript.
Funding
JW is funded by the Bas Stichting, the Princess Beatrix Muscle Foundation and Innovatiefonds Zorgverzekeraars. LW and CD are supported by the NIHR Biomedical Research Centre at Great Ormond Street Hospital and by grants from Arthritis Research UK (21953), Cure-JM, and The Myositis Association.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Meyer A, Meyer N, Schaeffer M, Gottenberg JE, Geny B, Sibilia J. Incidence and prevalence of inflammatory myopathies: a systematic review. Rheumatology (2015) 54:50–63. doi: 10.1093/rheumatology/keu289
2. Miller FW, Cooper RG, Vencovsky J, Rider LG, Danko K, Wedderburn LR, et al. Genome-wide association study of dermatomyositis reveals genetic overlap with other autoimmune disorders. Arthritis Rheum. (2013) 65:3239–47. doi: 10.1093/med/9780199732920.003.0012
3. Miller FW, Chen W, O'Hanlon TP, Cooper RG, Vencovsky J, Rider LG, et al. Genome-wide association study identifies HLA 8.1 ancestral haplotype alleles as major genetic risk factors for myositis phenotypes. Genes Immun. (2015) 16:470–80. doi: 10.1038/gene.2015.28
4. Rothwell S, Cooper RG, Lundberg IE, Miller FW, Gregersen PK, Bowes J, et al. Dense genotyping of immune-related loci in idiopathic inflammatory myopathies confirms HLA alleles as the strongest genetic risk factor and suggests different genetic background for major clinical subgroups. Ann Rheum Dis. (2016) 75:1558–66. doi: 10.1136/annrheumdis-2015-208119
5. Wedderburn LR, Rider LG. Juvenile dermatomyositis: new developments in pathogenesis, assessment and treatment. Best Pract Res Clin Rheumatol. (2009) 23:665–78. doi: 10.1016/j.berh.2009.07.007
6. Lundberg IE, Tjarnlund A, Bottai M, Werth VP, Pilkington C, de Visser M, et al. 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Ann Rheum Dis. (2017) 76:1955–64. doi: 10.1136/annrheumdis-2017-211468
7. Lundberg IE, Tjarnlund A, Bottai M, Werth VP, Pilkington C, de Visser M, et al. 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Arthritis Rheumatol. (2017) 69:2271–82. doi: 10.1002/art.40320
8. Kobayashi N, Takezaki S, Kobayashi I, Iwata N, Mori M, Nagai K, et al. Clinical and laboratory features of fatal rapidly progressive interstitial lung disease associated with juvenile dermatomyositis. Rheumatology (2015) 54:784–91. doi: 10.1093/rheumatology/keu385
9. Huber AM, Mamyrova G, Lachenbruch PA, Lee JA, Katz JD, Targoff IN, et al. Early illness features associated with mortality in the juvenile idiopathic inflammatory myopathies. Arthritis Care Res. (2014) 66:732–40. doi: 10.1002/acr.22212
10. Feldman BM, Rider LG, Reed AM, Pachman LM. Juvenile dermatomyositis and other idiopathic inflammatory myopathies of childhood. Lancet (2008) 371:2201–12. doi: 10.1016/S0140-6736(08)60955-1
11. Huber A, Feldman BM. Long-term outcomes in juvenile dermatomyositis: how did we get here and where are we going? Curr Rheumatol Rep. (2005) 7:441–6. doi: 10.1007/s11926-005-0048-1
12. Gerami P, Walling HW, Lewis J, Doughty L, Sontheimer RD. A systematic review of juvenile-onset clinically amyopathic dermatomyositis. Br J Dermatol. (2007) 157:637–44. doi: 10.1111/j.1365-2133.2007.08055.x
13. Plamondon S, Dent PB. Juvenile amyopathic dermatomyositis: results of a case finding descriptive survey. J Rheumatol. (2000) 27:2031–4.
14. Oberle EJ, Bayer ML, Chiu YE, Co DO. How often are pediatric patients with clinically amyopathic dermatomyositis truly amyopathic? Pediatr Dermatol. (2017) 34:50–7. doi: 10.1111/pde.13013
15. Mamyrova G, Kishi T, Targoff IN, Ehrlich A, Curiel RV, Rider LG. Features distinguishing clinically amyopathic juvenile dermatomyositis from juvenile dermatomyositis. Rheumatology (2018) 57:1956–1963. doi: 10.1093/rheumatology/key190
16. Rider LG, Nistala K. The juvenile idiopathic inflammatory myopathies: pathogenesis, clinical and autoantibody phenotypes, and outcomes. J Intern Med. (2016) 280:24–38. doi: 10.1111/joim.12444
17. Rider LG. The heterogeneity of juvenile myositis. Autoimmun Rev. (2007) 6:241–7. doi: 10.1016/j.autrev.2006.08.009
18. Huber AM, Lang B, LeBlanc CM, Birdi N, Bolaria RK, Malleson P, et al. Medium- and long-term functional outcomes in a multicenter cohort of children with juvenile dermatomyositis. Arthritis Rheum. (2000) 43:541–9. doi: 10.1002/1529-0131(200003)43:3<541::AID-ANR9>3.0.CO;2-T
19. Fisler RE, Liang MG, Fuhlbrigge RC, Yalcindag A, Sundel RP. Aggressive management of juvenile dermatomyositis results in improved outcome and decreased incidence of calcinosis. J Am Acad Dermatol. (2002) 47:505–11. doi: 10.1067/mjd.2002.122196
20. Holzer U, van Royen-Kerkhof A, van der Torre P, Kuemmerle-Deschner J, Well C, Handgretinger R, et al. Successful autologous stem cell transplantation in two patients with juvenile dermatomyositis. Scand J Rheumatol. (2010) 39:88–92. doi: 10.3109/03009740903096622
21. Crowe WE, Bove KE, Levinson JE, Hilton PK. Clinical and pathogenetic implications of histopathology in childhood polydermatomyositis. Arthritis Rheum. (1982) 25:126–39. doi: 10.1002/art.1780250203
22. Huemer C, Kitson H, Malleson PN, Sanderson S, Huemer M, Cabral DA, et al. Lipodystrophy in patients with juvenile dermatomyositis–evaluation of clinical and metabolic abnormalities. J Rheumatol. (2001) 28:610–5.
23. Verma S, Singh S, Bhalla AK, Khullar M. Study of subcutaneous fat in children with juvenile dermatomyositis. Arthritis Rheum. (2006) 55:564–8. doi: 10.1002/art.22108
24. Bingham A, Mamyrova G, Rother KI, Oral E, Cochran E, Premkumar A, et al. Predictors of acquired lipodystrophy in juvenile-onset dermatomyositis and a gradient of severity. Medicine (2008) 87:70–86. doi: 10.1097/MD.0b013e31816bc604
25. Pouessel G, Deschildre A, Le Bourgeois M, Cuisset J-M, Catteau B, Karila C, et al. The lung is involved in juvenile dermatomyositis. Pediatr Pulmonol. (2013) 48:1016–25. doi: 10.1002/ppul.22742
26. Richardson AE, Warrier K, Vyas H. Respiratory complications of the rheumatological diseases in childhood. Arch Dis Child (2016) 101:752–8. doi: 10.1136/archdischild-2014-306049
27. Kobayashi S, Higuchi K, Tamaki H, Wada Y, Wada N, Kubo M, et al. Characteristics of juvenile dermatomyositis in Japan. Acta Paediatr Jpn Overseas Ed. (1997) 39:257–62.
28. Shah M, Mamyrova G, Targoff IN, Huber AM, Malley JD, Rice MM, et al. The clinical phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine (2013) 92:25–41. doi: 10.1097/MD.0b013e31827f264d
29. Sanner H, Aalokken TM, Gran JT, Sjaastad I, Johansen B, Flato B. Pulmonary outcome in juvenile dermatomyositis: a case-control study. Ann Rheum Dis. (2011) 70:86–91. doi: 10.1136/ard.2010.131433
30. Morinishi Y, Oh-Ishi T, Kabuki T, Joh K. Juvenile dermatomyositis: clinical characteristics and the relatively high risk of interstitial lung disease. Mod Rheumatol. (2007) 17:413–7. doi: 10.3109/s10165-007-0610-y
31. Cantez S, Gross GJ, MacLusky I, Feldman BM. Cardiac findings in children with juvenile Dermatomyositis at disease presentation. Pediatr Rheumatol Online J. (2017) 15:54. doi: 10.1186/s12969-017-0182-0
32. Pachman LM, Cooke N. Juvenile dermatomyositis: a clinical and immunologic study. J Pediatr (1980) 96:226–34. doi: 10.1016/S0022-3476(80)80807-9
33. Na S-J, Kim SM, Sunwoo IN, Choi Y-C. Clinical characteristics and outcomes of juvenile and adult dermatomyositis. J Korean Med Sci. (2009) 24:715–21. doi: 10.3346/jkms.2009.24.4.715
34. Constantin T, Ponyi A, Orban I, Molnar K, Derfalvi B, Dicso F, et al. National registry of patients with juvenile idiopathic inflammatory myopathies in Hungary–clinical characteristics and disease course of 44 patients with juvenile dermatomyositis. Autoimmunity (2006) 39:223–32. doi: 10.1080/08916930600622819
35. Shehata R, al-Mayouf S, al-Dalaan A, al-Mazaid A, al-Balaa S, Bahabri S. Juvenile dermatomyositis: clinical profile and disease course in 25 patients. Clin Exp Rheumatol. (1999) 17:115–118.
36. Schwartz T, Sanner H, Gjesdal O, Flato B, Sjaastad I. In juvenile dermatomyositis, cardiac systolic dysfunction is present after long-term follow-up and is predicted by sustained early skin activity. Ann Rheum Dis. (2014) 73:1805–10. doi: 10.1136/annrheumdis-2013-203279
37. Schwartz T, Sanner H, Husebye T, Flato B, Sjaastad I. Cardiac dysfunction in juvenile dermatomyositis: a case-control study. Ann Rheum Dis. (2011) 70:766–71. doi: 10.1136/ard.2010.137968
38. Schwartz T, Diederichsen LP, Lundberg IE, Sjaastad I, Sanner H. Cardiac involvement in adult and juvenile idiopathic inflammatory myopathies. RMD open (2006) 2:e000291. doi: 10.1136/rmdopen-2016-000291
39. Jimenez C, Rowe PC, Keene D. Cardiac and central nervous system vasculitis in a child with dermatomyositis. J Child Neurol (1994) 9:297–300. doi: 10.1177/088307389400900315
40. Ramanan A V, Sawhney S, Murray KJ. Central nervous system complications in two cases of juvenile onset dermatomyositis. Rheumatology (2001) 40:1293–8. doi: 10.1093/rheumatology/40.11.1293
41. Silverberg JI, Kwa L, Kwa MC, Laumann AE, Ardalan K. Cardiovascular and cerebrovascular comorbidities of juvenile dermatomyositis in US children: an analysis of the National Inpatient Sample. Rheumatology (2018) 57:694–702. doi: 10.1093/rheumatology/kex465
42. Robinson AB, Hoeltzel MF, Wahezi DM, Becker ML, Kessler EA, Schmeling H, et al. Clinical characteristics of children with juvenile dermatomyositis: the Childhood Arthritis and Rheumatology Research Alliance Registry. Arthritis Care Res. (2014) 66:404–10. doi: 10.1002/acr.22142
43. Laskin BL, Choyke P, Keenan GF, Miller FW, Rider LG. Novel gastrointestinal tract manifestations in juvenile dermatomyositis. J Pediatr. (1999) 135:371–4. doi: 10.1016/S0022-3476(99)70137-X
44. Mamyrova G, Kleiner DE, James-Newton L, Shaham B, Miller FW, Rider LG. Late-onset gastrointestinal pain in juvenile dermatomyositis as a manifestation of ischemic ulceration from chronic endarteropathy. Arthritis Rheum. (2007) 57:881–4. doi: 10.1002/art.22782
45. Varsani H, Charman SC, Li CK, Marie SKN, Amato AA, Banwell B, et al. Validation of a score tool for measurement of histological severity in juvenile dermatomyositis and association with clinical severity of disease. Ann Rheum Dis. (2015) 74:204–10. doi: 10.1136/annrheumdis-2013-203396
46. Besancon A, Brochard K, Dupic L, Gitiaux C, Delville M, Krid S, et al. Presentations and outcomes of juvenile dermatomyositis patients admitted to intensive care units. Rheumatology (2017) 56:1814–6. doi: 10.1093/rheumatology/kex267
47. Meneghel A, Martini G, Birolo C, Tosoni A, Pettenazzo A, Zulian F. Life-threatening systemic capillary leak syndrome in juvenile dermatomyositis. Rheumatology (2017) 56:1822–3. doi: 10.1093/rheumatology/kex262
48. Goncalves FGP, Chimelli L, Sallum AME, Marie SKN, Kiss MHB, Ferriani VPL. Immunohistological analysis of CD59 and membrane attack complex of complement in muscle in juvenile dermatomyositis. J Rheumatol. (2002) 29:1301–7.
49. Yu HH, Chang HM, Chiu CJ, Yang YH, Lee JH, Wang LC, et al. Detection of anti-p155/140, anti-p140, and antiendothelial cells autoantibodies in patients with juvenile dermatomyositis. J Microbiol Immunol Infect. (2016) 49:264–70. doi: 10.1016/j.jmii.2014.05.012
50. Karasawa R, Tamaki M, Sato T, Tanaka M, Nawa M, Yudoh K, et al. Multiple target autoantigens on endothelial cells identified in juvenile dermatomyositis using proteomics. Rheumatology (2018) 57:671–6. doi: 10.1093/rheumatology/kex468
51. Whitaker JN, Engel WK. Vascular deposits of immunoglobulin and complement in idiopathic inflammatory myopathy. N Engl J Med. (1972) 286:333–8. doi: 10.1056/NEJM197202172860701
52. Kissel JT, Mendell JR, Rammohan KW. Microvascular deposition of complement membrane attack complex in dermatomyositis. N Engl J Med. (1986) 314:329–34. doi: 10.1056/NEJM198602063140601
53. Kissel JT, Halterman RK, Rammohan KW, Mendell JR. The relationship of complement-mediated microvasculopathy to the histologic features and clinical duration of disease in dermatomyositis. Arch Neurol. (1991) 48:26–30. doi: 10.1001/archneur.1991.00530130034016
54. Emslie-Smith AM, Engel AG. Microvascular changes in early and advanced dermatomyositis: a quantitative study. Ann Neurol. (1990) 27:343–56. doi: 10.1002/ana.410270402
55. Gitiaux C, De Antonio M, Aouizerate J, Gherardi RK, Guilbert T, Barnerias C, et al. Vasculopathy-related clinical and pathological features are associated with severe juvenile dermatomyositis. Rheumatology (2016) 55:470–9. doi: 10.1093/rheumatology/kev359
56. Schmeling H, Stephens S, Goia C, Manlhiot C, Schneider R, Luthra S, et al. Nailfold capillary density is importantly associated over time with muscle and skin disease activity in juvenile dermatomyositis. Rheumatology (2011) 50:885–93. doi: 10.1093/rheumatology/keq407
57. Feldman BM, Rider LG, Dugan L, Miller FW, Schneider R. Nailfold capillaries as indicators of disease activity in juvenile idiopathic inflammatory myopathies (JIIM). Arthritis Rheum. (1999) 42:S181.
58. Smith RL, Sundberg J, Shamiyah E, Dyer A, Pachman LM. Skin involvement in juvenile dermatomyositis is associated with loss of end row nailfold capillary loops. J Rheumatol. (2004) 31:1644–9.
59. Bertolazzi C, Cutolo M, Smith V, Gutierrez M. State of the art on nailfold capillaroscopy in dermatomyositis and polymyositis. Semin Arthritis Rheum. (2017) 47:432–44. doi: 10.1016/j.semarthrit.2017.06.001
60. Rider LG, Werth VP, Huber AM, Alexanderson H, Rao AP, Ruperto N, et al. Measures of adult and juvenile dermatomyositis, polymyositis, and inclusion body myositis: physician and Patient/Parent Global Activity, Manual Muscle Testing (MMT), Health Assessment Questionnaire (HAQ)/Childhood Health Assessment Questionnaire (C-HAQ). Arthritis Care Res. (2011) 63(Suppl. 1):S118–57.
61. Lovell DJ, Lindsley CB, Rennebohm RM, Ballinger SH, Bowyer SL, Giannini EH, et al. Development of validated disease activity and damage indices for the juvenile idiopathic inflammatory myopathies. II The Childhood Myositis Assessment Scale (CMAS): a quantitative tool for the evaluation of muscle function The Juvenile Dermatomyositis, D. Arthritis Rheum. (1999) 42:2213–9.
62. Rider LG, Koziol D, Giannini EH, Jain MS, Smith MR, Whitney-Mahoney K, et al. Validation of manual muscle testing and a subset of eight muscles for adult and juvenile idiopathic inflammatory myopathies. Arthritis Care Res. (2010) 62:465–72. doi: 10.1002/acr.20035
63. Rider LG, Feldman BM, Perez MD, Rennebohm RM, Lindsley CB, Zemel LS, et al. Development of validated disease activity and damage indices for the juvenile idiopathic inflammatory myopathies: I. Physician, parent, and patient global assessments Juvenile Dermatomyositis Disease Activity Collaborative Study Group. Arthritis Rheum. (1997) 40:1976–83. doi: 10.1002/art.1780401109
64. Huber AM, Dugan EM, Lachenbruch PA, Feldman BM, Perez MD, Zemel LS, et al. Preliminary validation and clinical meaning of the Cutaneous Assessment Tool in juvenile dermatomyositis. Arthritis Rheum. (2008) 59:214–21. doi: 10.1002/art.23340
65. Huber AM, Lachenbruch PA, Dugan EM, Miller FW, Rider LG. Alternative scoring of the Cutaneous Assessment Tool in juvenile dermatomyositis: results using abbreviated formats. Arthritis Rheum. (2008) 59:352–6. doi: 10.1002/art.23313
66. Bode RK, Klein-Gitelman MS, Miller ML, Lechman TS, Pachman LM. Disease activity score for children with juvenile dermatomyositis: reliability and validity evidence. Arthritis Rheum. (2003) 49:7–15. doi: 10.1002/art.10924
67. Huber AM, Hicks JE, Lachenbruch PA, Perez MD, Zemel LS, Rennebohm RM, et al. Validation of the Childhood Health Assessment Questionnaire in the juvenile idiopathic myopathies. Juvenile Dermatomyositis Disease Activity Collaborative Study Group. J Rheumatol. (2001) 28:1106–11.
68. Tiao J, Feng R, Berger EM, Brandsema JF, Coughlin CC, Khan N, et al. Evaluation of the reliability of the Cutaneous Dermatomyositis Disease Area and Severity Index and the Cutaneous Assessment Tool-Binary Method in juvenile dermatomyositis among paediatric dermatologists, rheumatologists and neurologists. Br J Dermatol. (2017) 177:1086–92. doi: 10.1111/bjd.15596
69. Sultan SM, Allen E, Cooper RG, Agarwal S, Kiely P, Oddis C V, et al. Interrater reliability and aspects of validity of the myositis damage index. Ann Rheum Dis. (2011) 70:1272–6. doi: 10.1136/ard.2010.142117
70. McCann LJ, Kirkham JJ, Wedderburn LR, Pilkington C, Huber AM, Ravelli A, et al. Development of an internationally agreed minimal dataset for juvenile dermatomyositis (JDM) for clinical and research use. Trials 16:268. doi: 10.1186/s13063-015-0784-0
71. Lazarevic D, Pistorio A, Palmisani E, Miettunen P, Ravelli A, Pilkington C, et al. The PRINTO criteria for clinically inactive disease in juvenile dermatomyositis. Ann Rheum Dis. (2013) 72:686–93. doi: 10.1136/annrheumdis-2012-201483
72. Almeida B, Campanilho-Marques R, Arnold K, Pilkington CA, Wedderburn LR, Nistala K. Analysis of published criteria for clinically inactive disease in a large juvenile dermatomyositis cohort shows that skin disease is underestimated. Arthritis Rheumatol. (2015) 67:2495–502. doi: 10.1002/art.39200
73. Betteridge ZE, Gunawardena H, McHugh NJ. Novel autoantibodies and clinical phenotypes in adult and juvenile myositis. Arthritis Res Ther. (2011) 13:209. doi: 10.1186/ar3275
74. Rider LG, Miller FW. Deciphering the clinical presentations, pathogenesis, and treatment of the idiopathic inflammatory myopathies. JAMA (2011) 305:183–90. doi: 10.1001/jama.2010.1977
75. Rider LG, Shah M, Mamyrova G, Huber AM, Rice MM, Targoff IN, et al. The myositis autoantibody phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine (2013) 92:223–43. doi: 10.1097/MD.0b013e31829d08f9
76. Tansley SL, Simou S, Shaddick G, Betteridge ZE, Almeida B, Gunawardena H, et al. Autoantibodies in juvenile-onset myositis: their diagnostic value and associated clinical phenotype in a large UK cohort. J Autoimmun. (2017) 84:55–64. doi: 10.1016/j.jaut.2017.06.007
77. Gunawardena H, Wedderburn LR, North J, Betteridge Z, Dunphy J, Chinoy H, et al. Clinical associations of autoantibodies to a p155/140 kDa doublet protein in juvenile dermatomyositis. Rheumatology (2008) 47:324–8. doi: 10.1093/rheumatology/kem359
78. Habers GEA, Huber AM, Mamyrova G, Targoff IN, O'Hanlon TP, Adams S, et al. Brief Report: association of myositis autoantibodies, clinical features, and environmental exposures at illness onset with disease course in juvenile myositis. Arthritis Rheumatol. (2016) 68:761–8. doi: 10.1002/art.39466
79. Tansley SL, Betteridge ZE, Shaddick G, Gunawardena H, Arnold K, Wedderburn LR, et al. Calcinosis in juvenile dermatomyositis is influenced by both anti-NXP2 autoantibody status and age at disease onset. Rheumatology (2014) 53:2204–8. doi: 10.1093/rheumatology/keu259
80. Gunawardena H, Wedderburn LR, Chinoy H, Betteridge ZE, North J, Ollier WER, et al. Autoantibodies to a 140-kd protein in juvenile dermatomyositis are associated with calcinosis. Arthritis Rheum. (2009) 60:1807–14. doi: 10.1002/art.24547
81. Sato S, Hirakata M, Kuwana M, Suwa A, Inada S, Mimori T, et al. Autoantibodies to a 140-kd polypeptide, CADM-140, in Japanese patients with clinically amyopathic dermatomyositis. Arthritis Rheum. (2005) 52:1571–6. doi: 10.1002/art.21023
82. Tansley SL, Betteridge ZE, Gunawardena H, Jacques TS, Owens CM, Pilkington C, et al. Anti-MDA5 autoantibodies in juvenile dermatomyositis identify a distinct clinical phenotype: a prospective cohort study. Arthritis Res Ther. (2016) 16:R138. doi: 10.1186/ar4600
83. Hoshino K, Muro Y, Sugiura K, Tomita Y, Nakashima R, Mimori T. Anti-MDA5 and anti-TIF1-γ antibodies have clinical significance for patients with dermatomyositis. Rheumatology (2010) 49:1726–33. doi: 10.1093/rheumatology/keq153
84. Hall JC, Casciola-Rosen L, Samedy L-A, Werner J, Owoyemi K, Danoff SK, et al. Anti-melanoma differentiation-associated protein 5-associated dermatomyositis: expanding the clinical spectrum. Arthritis Care Res. (2013) 65:1307–15. doi: 10.1002/acr.21992
85. Fiorentino D, Chung L, Zwerner J, Rosen A, Casciola-Rosen L. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol. (2011) 65:25–34. doi: 10.1016/j.jaad.2010.09.016
86. Tansley SL, Betteridge ZE, Simou S, Jacques TS, Pilkington C, Wood M, et al. Anti-HMGCR autoantibodies in juvenile idiopathic inflammatory myopathies identify a rare but clinically important subset of patients. J Rheumatol. (2017) 44:488–492. doi: 10.3899/jrheum.160871
87. Kishi T, Rider LG, Pak K, Barillas-Arias L, Henrickson M, McCarthy PL, et al. Association of anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase autoantibodies with DRB1*07:01 and severe myositis in juvenile myositis patients. Arthritis Care Res. (2017) 69:1088–94. doi: 10.1002/acr.23113
88. Aggarwal R, Oddis CV, Goudeau D, Koontz D, Qi Z, Reed AM, et al. Autoantibody levels in myositis patients correlate with clinical response during B cell depletion with rituximab. Rheumatology (2016) 55:991–9. doi: 10.1093/rheumatology/kev444
89. O'Connor KA, Abbott KA, Sabin B, Kuroda M, Pachman LM. MxA gene expression in juvenile dermatomyositis peripheral blood mononuclear cells: association with muscle involvement. Clin Immunol. (2006) 120:319–25.
90. Niewold TB, Kariuki SN, Morgan GA, Shrestha S, Pachman LM. Elevated serum interferon-alpha activity in juvenile dermatomyositis: associations with disease activity at diagnosis and after thirty-six months of therapy. Arthritis Rheum. (2009) 60:1815–24. doi: 10.1002/art.24555
91. Baechler EC, Bilgic H, Reed AM. Type I interferon pathway in adult and juvenile dermatomyositis. Arthritis Res Ther. (2011) 13:249. doi: 10.1186/ar3531
92. Sanner H, Schwartz T, Flato B, Vistnes M, Christensen G, Sjaastad I. Increased levels of eotaxin and MCP-1 in juvenile dermatomyositis median 16.8 years after disease onset; associations with disease activity, duration and organ damage. PLoS ONE (2014) 9:e92171. doi: 10.1371/journal.pone.0092171
93. Bellutti Enders F, van Wijk F, Scholman R, Hofer M, Prakken BJ, van Royen-Kerkhof A, et al. Correlation of CXCL10, tumor necrosis factor receptor type II, and galectin 9 with disease activity in juvenile dermatomyositis. Arthritis Rheumatol. (2014) 66:2281–9. doi: 10.1002/art.38676
94. Enders FB, Delemarre EM, Kuemmerle-Deschner J, van der Torre P, Wulffraat NM, Prakken BP, et al. Autologous stem cell transplantation leads to a change in proinflammatory plasma cytokine profile of patients with juvenile dermatomyositis correlating with disease activity. Ann Rheum Dis. (2015) 74:315–7. doi: 10.1136/annrheumdis-2014-206287
95. Kobayashi I, Ono S, Kawamura N, Okano M, Kobayashi K. Elevated serum levels of soluble interleukin-2 receptor in juvenile dermatomyositis. Pediatr Int. (2001) 43:109–11. doi: 10.1046/j.1442-200x.2001.01367.x
96. De Benedetti F, De Amici M, Aramini L, Ruperto N, Martini A. Correlation of serum neopterin concentrations with disease activity in juvenile dermatomyositis. Arch Dis Child. (1993) 69:232–5. doi: 10.1136/adc.69.2.232
97. Rider LG, Schiffenbauer AS, Zito M, Lim KL, Ahmed A, Zemel LS, et al. Neopterin and quinolinic acid are surrogate measures of disease activity in the juvenile idiopathic inflammatory myopathies. Clin Chem. (2002) 48:1681–8.
98. Nistala K, Varsani H, Wittkowski H, Vogl T, Krol P, Shah V, et al. Myeloid related protein induces muscle derived inflammatory mediators in juvenile dermatomyositis. Arthritis Res Ther. (2013) 15:R131. doi: 10.1186/ar4311
99. Haas RH, Dyck RF, Dubowitz V, Pepys MB. C-reactive protein in childhood dermatomyositis. Ann Rheum Dis. (1982) 41:483–5. doi: 10.1136/ard.41.5.483
100. Eisenstein DM, O'Gorman MR, Pachman LM. Correlations between change in disease activity and changes in peripheral blood lymphocyte subsets in patients with juvenile dermatomyositis. J Rheumatol. (1997) 24:1830–2.
101. Elst EF, Klein M, de Jager W, Kamphuis S, Wedderburn LR, van der Zee R, et al. Hsp60 in inflamed muscle tissue is the target of regulatory autoreactive T cells in patients with juvenile dermatomyositis. Arthritis Rheum. (2008) 58:547–55. doi: 10.1002/art.23202
102. Morita R, Schmitt N, Bentebibel S-E, Ranganathan R, Bourdery L, Zurawski G, et al. Human blood CXCR5(+)CD4(+) T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity (2011) 34:108–21. doi: 10.1016/j.immuni.2010.12.012
103. Ernste FC, Crowson CS, de Padilla CL, Hein MS, Reed AM. Longitudinal peripheral blood lymphocyte subsets correlate with decreased disease activity in juvenile dermatomyositis. J Rheumatol. (2013) 40:1200–11. doi: 10.3899/jrheum.121031
104. Vercoulen Y, Bellutti Enders F, Meerding J, Plantinga M, Elst EF, Varsani H, et al. Increased presence of FOXP3+ regulatory T cells in inflamed muscle of patients with active juvenile dermatomyositis compared to peripheral blood. PLoS ONE (2014) 9:e105353. doi: 10.1371/journal.pone.0105353
105. Lopez De Padilla CM, Crowson CS, Hein MS, Pendegraft RS, Strausbauch MA, Niewold TB, et al. Gene expression profiling in blood and affected muscle tissues reveals differential activation pathways in patients with new-onset juvenile and adult dermatomyositis. J Rheumatol. (2017) 44:117–24. doi: 10.3899/jrheum.160293
106. Piper CJM, Wilkinson MGL, Deakin CT, Otto GW, Dowle S, Duurland CL, et al. CD19(+)CD24(hi)CD38(hi) B cells are expanded in juvenile dermatomyositis and exhibit a pro-inflammatory phenotype after activation through toll-like receptor 7 and interferon-alpha. Front Immunol. (2018) 9:1372. doi: 10.3389/fimmu.2018.01372
107. Guzman J, Petty RE, Malleson PN. Monitoring disease activity in juvenile dermatomyositis: the role of von Willebrand factor and muscle enzymes. J Rheumatol. (1994) 21:739–43.
108. Bloom BJ, Tucker LB, Miller LC, Schaller JG. von Willebrand factor in juvenile dermatomyositis. J Rheumatol. (1995) 22:320–5.
109. Scott JP, Arroyave C. Activation of complement and coagulation in juvenile dermatomyositis. Arthritis Rheum. (1987) 30:572–6. doi: 10.1002/art.1780300513
110. Xu D, Huang C-C, Kachaochana A, Morgan GA, Bonaldo MF, Soares MB, et al. MicroRNA-10a regulation of proinflammatory mediators: an important component of untreated juvenile dermatomyositis. J Rheumatol. (2016) 43:161–8. doi: 10.3899/jrheum.141474
111. Xu D, Kacha-Ochana A, Morgan GA, Huang C-C, Pachman LM. Endothelial progenitor cell number is not decreased in 34 children with Juvenile Dermatomyositis: a pilot study. Pediatr Rheumatol Online J. (2017) 15:42. doi: 10.1186/s12969-017-0171-3
112. Coyle K, Rother KI, Weise M, Ahmed A, Miller FW, Rider LG. Metabolic abnormalities and cardiovascular risk factors in children with myositis. J Pediatr. (2009) 155:882–7. doi: 10.1016/j.jpeds.2009.06.009
113. Kozu KT, Silva CA, Bonfa E, Sallum AM, Pereira RMR, Viana VS, et al. Dyslipidaemia in juvenile dermatomyositis: the role of disease activity. Clin Exp Rheumatol. (2013) 31:638–44.
114. Olazagasti JM, Hein M, Crowson CS, de Padilla CL, Peterson E, Baechler EC, et al. Adipokine gene expression in peripheral blood of adult and juvenile dermatomyositis patients and their relation to clinical parameters and disease activity measures. J Inflamm. (2015) 12:29. doi: 10.1186/s12950-015-0075-2
115. Baechler EC, Bauer JW, Slattery CA, Ortmann WA, Espe KJ, Novitzke J, et al. An interferon signature in the peripheral blood of dermatomyositis patients is associated with disease activity. Mol Med. (2007) 13:59–68. doi: 10.2119/2006-00085.Baechler
116. Bilgic H, Ytterberg SR, Amin S, McNallan KT, Wilson JC, Koeuth T, et al. Interleukin-6 and type I interferon-regulated genes and chemokines mark disease activity in dermatomyositis. Arthritis Rheum. (2009) 60:3436–46. doi: 10.1002/art.24936
117. Reed AM, Peterson E, Bilgic H, Ytterberg SR, Amin S, Hein MS, et al. Changes in novel biomarkers of disease activity in juvenile and adult dermatomyositis are sensitive biomarkers of disease course. Arthritis Rheum. (2012) 64:4078–86. doi: 10.1002/art.34659
118. Gabay C, Gay-Croisier F, Roux-Lombard P, Meyer O, Maineti C, Guerne PA, et al. Elevated serum levels of interleukin-1 receptor antagonist in polymyositis/dermatomyositis. A biologic marker of disease activity with a possible role in the lack of acute-phase protein response. Arthritis Rheum. (1994) 37:1744–51. doi: 10.1002/art.1780371206
119. Krystufkova O, Vallerskog T, Helmers SB, Mann H, Putova I, Belacek J, et al. Increased serum levels of B cell activating factor (BAFF) in subsets of patients with idiopathic inflammatory myopathies. Ann Rheum Dis. (2009) 68:836–43. doi: 10.1136/ard.2008.091405
120. Lopez De Padilla CM, McNallan KT, Crowson CS, Bilgic H, Bram RJ, Hein MS, et al. BAFF expression correlates with idiopathic inflammatory myopathy disease activity measures and autoantibodies. J Rheumatol. (2013) 40:294–302. doi: 10.3899/jrheum.120555
121. Ishida T, Matsumoto Y, Ohashi M, Sasaki R. Analysis of lymphocyte subpopulations in peripheral blood in adult and juvenile cases of dermatomyositis. J Dermatol. (1993) 20:30–4. doi: 10.1111/j.1346-8138.1993.tb03825.x
122. Aleksza M, Szegedi A, Antal-Szalmas P, Irinyi B, Gergely L, Ponyi A, et al. Altered cytokine expression of peripheral blood lymphocytes in polymyositis and dermatomyositis. Ann Rheum Dis. (2005) 64:1485–9. doi: 10.1136/ard.2003.017715
123. Kubo M, Ihn H, Yamane K, Yazawa N, Kikuchi K, Soma Y, et al. Increased serum levels of soluble vascular cell adhesion molecule-1 and soluble E-selectin in patients with polymyositis/dermatomyositis. Br J Dermatol. (2000) 143:392–8. doi: 10.1046/j.1365-2133.2000.03668.x
124. Bloom BJ, Miller LC, Blier PR. Soluble adhesion molecules in pediatric rheumatic diseases. J Rheumatol. (2002) 29:832–6.
125. McCann LJ, Juggins AD, Maillard SM, Wedderburn LR, Davidson JE, Murray KJ, et al. The Juvenile Dermatomyositis National Registry and Repository (UK and Ireland)–clinical characteristics of children recruited within the first 5 yr. Rheumatology (2006) 45:1255–60. doi: 10.1093/rheumatology/kel099
126. Pachman LM, Hayford JR, Chung A, Daugherty CA, Pallansch MA, Fink CW, et al. Juvenile dermatomyositis at diagnosis: clinical characteristics of 79 children. J Rheumatol. (1998) 25:1198–204.
127. Targoff IN. Laboratory manifestations of polymyositis/dermatomyositis. Clin Dermatol. (1988) 6:76–92. doi: 10.1016/0738-081X(88)90051-X
128. Mathiesen PR, Zak M, Herlin T, Nielsen SM. Clinical features and outcome in a Danish cohort of juvenile dermatomyositis patients. Clin Exp Rheumatol. (2010) 28:782–9.
129. Ntusi NBA, Heckmann JM. Myopathy with a normal creatine kinase level in juvenile myopathic dermatomyositis. S Afr Med J. (2010) 100:24–5.
130. Hinderks GJ, Frolich J. Low serum creatine kinase values associated with administration of steroids. Clin Chem. (1979) 25:2050–1.
131. Kagen LJ, Aram S. Creatine kinase activity inhibitor in sera from patients with muscle disease. Arthritis Rheum. (1987) 30:213–7. doi: 10.1002/art.1780300212
132. Rider LG, Miller FW. Laboratory evaluation of the inflammatory myopathies. Clin Diagn Lab Immunol. (1995) 2:1–9.
133. Naim MY, Reed AM. Enzyme elevation in patients with juvenile dermatomyositis and steroid myopathy. J Rheumatol. (2006) 33:1392–4.
134. Hinze CH, Oommen PT, Dressler F, Urban A, Weller-Heinemann F, Speth F, et al. Development of practice and consensus-based strategies including a treat-to-target approach for the management of moderate and severe juvenile dermatomyositis in Germany and Austria. Pediatr Rheumatol Online J (2018) 16:40. doi: 10.1186/s12969-018-0257-6
135. Enders FB, Bader-Meunier B, Baildam E, Constantin T, Dolezalova P, Feldman BM, et al. Consensus-based recommendations for the management of juvenile dermatomyositis. Ann Rheum Dis. (2017) 76:329–40. doi: 10.1136/annrheumdis-2016-209247
136. Rice GI, Melki I, Fremond M-L, Briggs TA, Rodero MP, Kitabayashi N, et al. Assessment of type I interferon signaling in pediatric inflammatory disease. J Clin Immunol. (2017) 37:123–32. doi: 10.1007/s10875-016-0359-1
137. Fall N, Bove KE, Stringer K, Lovell DJ, Brunner HI, Weiss J, et al. Association between lack of angiogenic response in muscle tissue and high expression of angiostatic ELR-negative CXC chemokines in patients with juvenile dermatomyositis: possible link to vasculopathy. Arthritis Rheum. (2005) 52:3175–80. doi: 10.1002/art.21303
138. Rodero MP, Decalf J, Bondet V, Hunt D, Rice GI, Werneke S, et al. Detection of interferon alpha protein reveals differential levels and cellular sources in disease. J Exp Med. (2017) 214:1547–55. doi: 10.1084/jem.20161451
139. van den Hoogen LL, van Roon JAG, Mertens JS, Wienke J, Lopes AP, de Jager W, et al. Galectin-9 is an easy to measure biomarker for the interferon signature in systemic lupus erythematosus and antiphospholipid syndrome. Ann Rheum Dis. (2018) 77:1810–14. doi: 10.1136/annrheumdis-2018-213497
140. Wachter H, Fuchs D, Hausen A, Reibnegger G, Werner ER. Neopterin as marker for activation of cellular immunity: immunologic basis and clinical application. Adv Clin Chem. (1989) 27:81–141. doi: 10.1016/S0065-2423(08)60182-1
141. Holzinger D, Frosch M, Kastrup A, Prince FHM, Otten MH, Van Suijlekom-Smit LWA, et al. The Toll-like receptor 4 agonist MRP8/14 protein complex is a sensitive indicator for disease activity and predicts relapses in systemic-onset juvenile idiopathic arthritis. Ann Rheum Dis. (2012) 71:974–80. doi: 10.1136/annrheumdis-2011-200598
142. Silverman ED, Laxer RM, Nelson DL, Rubin LA. Soluble interleukin-2 receptor in juvenile rheumatoid arthritis. J Rheumatol. (1991) 18:1398–402.
143. Kobayashi I, Okura Y, Yamada M, Kawamura N, Kuwana M, Ariga T. Anti-melanoma differentiation-associated gene 5 antibody is a diagnostic and predictive marker for interstitial lung diseases associated with juvenile dermatomyositis. J Pediatr. (2011) 158:675–7. doi: 10.1016/j.jpeds.2010.11.033
144. Kobayashi I, Ono S, Kawamura N, Okano M, Miyazawa K, Shibuya H, et al. KL-6 is a potential marker for interstitial lung disease associated with juvenile dermatomyositis. J Pediatr. (2001) 138:274–6. doi: 10.1067/mpd.2001.110324
145. Kobayashi N, Kobayashi I, Mori M, Sato S, Iwata N, Shigemura T, et al. Increased serum B cell activating factor and a proliferation-inducing ligand are associated with interstitial lung disease in patients with juvenile dermatomyositis. J Rheumatol. (2015) 42:2412–8. doi: 10.3899/jrheum.140977
146. Schwartz T, Sjaastad I, Flato B, Vistnes M, Christensen G, Sanner H. In active juvenile dermatomyositis, elevated eotaxin and MCP-1 and cholesterol levels in the upper normal range are associated with cardiac dysfunction. Rheumatology (2014) 53:2214–22. doi: 10.1093/rheumatology/keu256
147. Barth Z, Nomeland Witczak B, Schwartz T, Gjesdal K, Flato B, Koller A, et al. In juvenile dermatomyositis, heart rate variability is reduced, and associated with both cardiac dysfunction and markers of inflammation: a cross-sectional study median 13.5 years after symptom onset. Rheumatology (2016) 55:535–43. doi: 10.1093/rheumatology/kev376
148. van Summeren MJH, Spliet WGM, van Royen-Kerkhof A, Vermeer C, Lilien M, Kuis W, et al. Calcinosis in juvenile dermatomyositis: a possible role for the vitamin K-dependent protein matrix Gla protein. Rheumatology (2008) 47:267–71. doi: 10.1093/rheumatology/kem360
149. Reed A, Haugen M, Pachman LM, Langman CB. Abnormalities in serum osteocalcin values in children with chronic rheumatic diseases. J Pediatr. (1990) 116:574–80. doi: 10.1016/S0022-3476(05)81605-1
150. Chinoy H, Salway F, John S, Fertig N, Tait BD, Oddis C V, et al. Tumour necrosis factor-alpha single nucleotide polymorphisms are not independent of HLA class I in UK Caucasians with adult onset idiopathic inflammatory myopathies. Rheumatology (2007) 46:1411–6. doi: 10.1093/rheumatology/kem145y
151. Lutz J, Huwiler KG, Fedczyna T, Lechman TS, Crawford S, Kinsella TR, et al. Increased plasma thrombospondin-1 (TSP-1) levels are associated with the TNF alpha-308A allele in children with juvenile dermatomyositis. Clin Immunol. (2002) 103:260–3. doi: 10.1006/clim.2001.5212
152. Niewold TB, Kariuki SN, Morgan GA, Shrestha S, Pachman LM. Gene-gene-sex interaction in cytokine gene polymorphisms revealed by serum interferon alpha phenotype in juvenile dermatomyositis. J Pediatr. (2010) 157:653–7. doi: 10.1016/j.jpeds.2010.04.034
153. Pachman LM, Liotta-Davis MR, Hong DK, Kinsella TR, Mendez EP, Kinder JM, et al. TNFalpha-308A allele in juvenile dermatomyositis: association with increased production of tumor necrosis factor alpha, disease duration, and pathologic calcifications. Arthritis Rheum. (2000) 43:2368–77. doi: 10.1002/1529-0131(200010)43:10<2368::AID-ANR26>3.0.CO;2-8
154. Pachman LM, Fedczyna TO, Lechman TS, Lutz J. Juvenile dermatomyositis: the association of the TNF alpha-308A allele and disease chronicity. Curr Rheumatol Rep. (2001) 3:379–86. doi: 10.1007/s11926-996-0007-5
155. Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. (1975) 292:344–7. doi: 10.1056/NEJM197502132920706
156. Bohan A, Peter JB. Polymyositis and dermatomyositis (second of two parts). N Engl J Med. (1975) 292:403–7. doi: 10.1056/NEJM197502202920807
157. Wedderburn LR, Varsani H, Li CKC, Newton KR, Amato AA, Banwell B, et al. International consensus on a proposed score system for muscle biopsy evaluation in patients with juvenile dermatomyositis: a tool for potential use in clinical trials. Arthritis Rheum. (2007) 57:1192–201. doi: 10.1002/art.23012
158. Deakin CT, Yasin SA, Simou S, Arnold KA, Tansley SL, Betteridge ZE, et al. Muscle biopsy findings in combination with myositis-specific autoantibodies aid prediction of outcomes in juvenile dermatomyositis. Arthritis Rheumatol. (2016) 68:2806–16. doi: 10.1002/art.39753
159. Li CKC, Varsani H, Holton JL, Gao B, Woo P, Wedderburn LR. MHC Class I overexpression on muscles in early juvenile dermatomyositis. J Rheumatol. (2004) 31:605–9.
160. Sallum AME, Kiss MHB, Silva CAA, Wakamatsu A, Sachetti S, Lotufo S, et al. MHC class I and II expression in juvenile dermatomyositis skeletal muscle. Clin Exp Rheumatol. (2009) 27:519–26.
161. Sallum AME, Marie SKN, Wakamatsu A, Sachetti S, Vianna MAAG, Silva CAA, et al. Immunohistochemical analysis of adhesion molecule expression on muscle biopsy specimens from patients with juvenile dermatomyositis. J Rheumatol. (2004) 31:801–7.
162. Sakuta R, Murakami N, Jin Y, Nagai T, Nonaka I, Nishino I. Diagnostic significance of membrane attack complex and vitronectin in childhood dermatomyositis. J Child Neurol. (2005) 20:597–602. doi: 10.1177/08830738050200071201
163. Tezak Z, Hoffman EP, Lutz JL, Fedczyna TO, Stephan D, Bremer EG, et al. Gene expression profiling in DQA1*0501+ children with untreated dermatomyositis: a novel model of pathogenesis. J Immunol. (2002) 168:4154–63. doi: 10.4049/jimmunol.168.8.4154
164. Shrestha S, Wershil B, Sarwark JF, Niewold TB, Philipp T, Pachman LM. Lesional and nonlesional skin from patients with untreated juvenile dermatomyositis displays increased numbers of mast cells and mature plasmacytoid dendritic cells. Arthritis Rheum. (2010) 62:2813–22. doi: 10.1002/art.27529
165. Wargula JC, Lovell DJ, Passo MH, Bove KE, Santangelo JD, Levinson JE. What more can we learn from muscle histopathology in children with dermatomyositis/polymyositis? Clin Exp Rheumatol. (2006) 24:333–43.
166. Soponkanaporn S, Deakin CT, Schutz PW, Marshall LR, Yasin SA, Johnson CM, et al. Expression of myxovirus-resistance protein A: a possible marker of muscle disease activity and autoantibody specificities in juvenile dermatomyositis. Neuropathol Appl Neurobiol. (2018). [Epub ahead of print].
167. Gitiaux C, Latroche C, Weiss-Gayet M, Rodero MP, Duffy D, Bader-Meunier B, et al. Myogenic progenitor cells exhibit type i interferon-driven proangiogenic properties and molecular signature during juvenile dermatomyositis. Arthritis Rheumatol. (2018) 70:134–45. doi: 10.1002/art.40328
168. Minetti C, Gattorno M, Repetto S, Gregorio A, Pedemonte M, Assereto S, et al. Chemokine receptor CCR7 is expressed in muscle fibers in juvenile dermatomyositis. Biochem Biophys Res Commun. (2005) 333:540–543. doi: 10.1016/j.bbrc.2005.05.146
169. Lopez de Padilla CM, Vallejo AN, McNallan KT, Vehe R, Smith SA, Dietz AB, et al. Plasmacytoid dendritic cells in inflamed muscle of patients with juvenile dermatomyositis. Arthritis Rheum. (2007) 56:1658–68.
170. Englund P, Nennesmo I, Klareskog L, Lundberg IE. Interleukin-1alpha expression in capillaries and major histocompatibility complex class I expression in type II muscle fibers from polymyositis and dermatomyositis patients: important pathogenic features independent of inflammatory cell clusters in muscle. Arthritis Rheum. (2002) 46:1044–55. doi: 10.1002/art.10140
171. Kim E, Cook-Mills J, Morgan G, Sredni ST, Pachman LM. Increased expression of vascular cell adhesion molecule 1 in muscle biopsy samples from juvenile dermatomyositis patients with short duration of untreated disease is regulated by miR-126. Arthritis Rheum. (2012) 64:3809–17. doi: 10.1002/art.34606
172. Chen YW, Shi R, Geraci N, Shrestha S, Gordish-Dressman H, Pachman LM. Duration of chronic inflammation alters gene expression in muscle from untreated girls with juvenile dermatomyositis. BMC Immunol. (2008) 9:43. doi: 10.1186/1471-2172-9-43
173. Zhao Y, Fedczyna TO, McVicker V, Caliendo J, Li H, Pachman LM. Apoptosis in the skeletal muscle of untreated children with juvenile dermatomyositis: impact of duration of untreated disease. Clin Immunol. (2007) 125:165–72. doi: 10.1016/j.clim.2007.06.011
174. Lopez De Padilla CM, Vallejo AN, Lacomis D, McNallan K, Reed AM. Extranodal lymphoid microstructures in inflamed muscle and disease severity of new-onset juvenile dermatomyositis. Arthritis Rheum. (2009) 60:1160–72. doi: 10.1002/art.24411
175. Shinjo SK, Sallum AME, Silva CA, Marie SKN. Skeletal muscle major histocompatibility complex class I and II expression differences in adult and juvenile dermatomyositis. Clinics (2012) 67:885–90. doi: 10.6061/clinics/2012(08)05
176. Iglesias E, Jou C, Bou R, Anton J. [Importance of muscle biopsy in the diagnosis of juvenile dermatomyositis]. An Pediatr. (2014) 80:e25–6. doi: 10.1016/j.anpedi.2013.04.017
177. Mamyrova G, Rider LG, Ehrlich A, Jones O, Pachman LM, Nickeson R, et al. (2017). Environmental factors associated with disease flare in juvenile and adult dermatomyositis. Rheumatology 56:1342–7. doi: 10.1093/rheumatology/kex162
178. Manlhiot C, Liang L, Tran D, Bitnun A, Tyrrell PN, Feldman BM. Assessment of an infectious disease history preceding juvenile dermatomyositis symptom onset. Rheumatology (2008) 47:526–9. doi: 10.1093/rheumatology/ken038
179. Pachman LM, Lipton R, Ramsey-Goldman R, Shamiyeh E, Abbott K, Mendez EP, et al. History of infection before the onset of juvenile dermatomyositis: results from the National Institute of Arthritis and Musculoskeletal and Skin Diseases Research Registry. Arthritis Rheum. (2005) 53:166–72. doi: 10.1002/art.21068
180. Pachman LM, Litt DL, Rowley AH, Hayford JR, Caliendo J, Heller S, et al. Lack of detection of enteroviral RNA or bacterial DNA in magnetic resonance imaging-directed muscle biopsies from twenty children with active untreated juvenile dermatomyositis. Arthritis Rheum. (1995) 38:1513–8. doi: 10.1002/art.1780381019
181. Jia H, Thelwell C, Dilger P, Bird C, Daniels S, Wadhwa M. Endothelial cell functions impaired by interferon in vitro: Insights into the molecular mechanism of thrombotic microangiopathy associated with interferon therapy. Thromb Res. (2018) 163:105–16. doi: 10.1016/j.thromres.2018.01.039
182. Aanhane E, Schulkens IA, Heusschen R, Castricum K, Leffler H, Griffioen AW, et al. Different angioregulatory activity of monovalent galectin-9 isoforms. Angiogenesis (2018) 21:545–55. doi: 10.1007/s10456-018-9607-8
183. O'Brien MJ, Shu Q, Stinson WA, Tsou P-S, Ruth JH, Isozaki T, et al. A unique role for galectin-9 in angiogenesis and inflammatory arthritis. Arthritis Res Ther. (2018) 20:31. doi: 10.1186/s13075-018-1519-x
184. Heusschen R, Schulkens IA, van Beijnum J, Griffioen AW, Thijssen VL. Endothelial LGALS9 splice variant expression in endothelial cell biology and angiogenesis. Biochim Biophys Acta. (2014) 1842:284–92. doi: 10.1016/j.bbadis.2013.12.003
185. Campanella GS V, Colvin RA, Luster AD. CXCL10 can inhibit endothelial cell proliferation independently of CXCR3. PLoS ONE (2017) 5:e12700. doi: 10.1371/journal.pone.0012700
186. Ladislau L, Suarez-Calvet X, Toquet S, Landon-Cardinal O, Amelin D, Depp M, et al. JAK inhibitor improves type I interferon induced damage: proof of concept in dermatomyositis. Brain (2018) 141:1609–21. doi: 10.1093/brain/awy105
187. Crescioli C, Sottili M, Bonini P, Cosmi L, Chiarugi P, Romagnani P, et al. Inflammatory response in human skeletal muscle cells: CXCL10 as a potential therapeutic target. Eur J Cell Biol. (2012) 91:139–49. doi: 10.1016/j.ejcb.2011.09.011
188. Nagaraju K, Rider LG, Fan C, Chen Y-W, Mitsak M, Rawat R, et al. Endothelial cell activation and neovascularization are prominent in dermatomyositis. J Autoimmune Dis. (2006) 3:2. doi: 10.1186/1740-2557-3-2
189. Sorensen EW, Lian J, Ozga AJ, Miyabe Y, Ji SW, Bromley SK, et al. CXCL10 stabilizes T cell-brain endothelial cell adhesion leading to the induction of cerebral malaria. JCI Insight (2018) 3:98911. doi: 10.1172/jci.insight.98911
190. Fu H, Kishore M, Gittens B, Wang G, Coe D, Komarowska I, et al. Self-recognition of the endothelium enables regulatory T-cell trafficking and defines the kinetics of immune regulation. Nat Commun. (2014) 5:3436. doi: 10.1038/ncomms4436
191. Pober JS, Merola J, Liu R, Manes TD. Antigen presentation by vascular cells. Front Immunol. (2017) 8:1907. doi: 10.3389/fimmu.2017.01907
192. Huber AM, Robinson AB, Reed AM, Abramson L, Bout-Tabaku S, Carrasco R, et al. Consensus treatments for moderate juvenile dermatomyositis: beyond the first two months. Results of the second Childhood Arthritis and Rheumatology Research Alliance consensus conference. Arthritis Care Res. (2012) 64:546–53. doi: 10.1002/acr.20695
193. McCann LJ, Pilkington CA, Huber AM, Ravelli A, Appelbe D, Kirkham JJ, et al. Development of a consensus core dataset in juvenile dermatomyositis for clinical use to inform research. Ann Rheum Dis. (2018) 77:241–50. doi: 10.1136/annrheumdis-2017-212141
194. van Dijkhuizen E. P., Deakin C. T., Wedderburn L. R., De Iorio M. (2017). Modelling disease activity in juvenile dermatomyositis: a Bayesian approach. Stat Methods Med Res. (2017). doi: 10.1177/0962280217713233
195. Lim LSH, Pullenayegum E, Moineddin R, Gladman DD, Silverman ED, Feldman BM. Methods for analyzing observational longitudinal prognosis studies for rheumatic diseases: a review and worked example using a clinic-based cohort of juvenile dermatomyositis patients. Pediatr Rheumatol Online J. (2017) 15:18. doi: 10.1186/s12969-017-0148-2
196. Higgs BW, Zhu W, Morehouse C, White WI, Brohawn P, Guo X, et al. A phase 1b clinical trial evaluating sifalimumab, an anti-IFN-alpha monoclonal antibody, shows target neutralisation of a type I IFN signature in blood of dermatomyositis and polymyositis patients. Ann Rheum Dis. (2014) 73:256–62. doi: 10.1136/annrheumdis-2012-202794
Keywords: juvenile dermatomyositis, tissue inflammation, vasculopathy, disease monitoring, biomarkers, interferon signature, autoantibodies, personalized medicine
Citation: Wienke J, Deakin CT, Wedderburn LR, van Wijk F and van Royen-Kerkhof A (2018) Systemic and Tissue Inflammation in Juvenile Dermatomyositis: From Pathogenesis to the Quest for Monitoring Tools. Front. Immunol. 9:2951. doi: 10.3389/fimmu.2018.02951
Received: 28 September 2018; Accepted: 30 November 2018;
Published: 18 December 2018.
Edited by:
Ann Marie Reed, Duke University, United StatesReviewed by:
David A. Hafler, Yale University, United StatesHanna Kim, National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), United States
Copyright © 2018 Wienke, Deakin, Wedderburn, van Wijk and van Royen-Kerkhof. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Annet van Royen-Kerkhof, YS52YW5yb3llbkB1bWN1dHJlY2h0Lm5s