 Palash C. Maity*
Palash C. Maity* Antonella Nicolò
Antonella Nicolò Hassan Jumaa
Hassan Jumaa- Institute of Immunology, Ulm University, Ulm, Germany
Expression of the membrane-bound form of the immunoglobulin (Ig) as part of the antigen receptor is indispensable for both the development and the effector function of B cells. Among five known isotypes, IgM and IgD are the common B cell antigen receptors (BCRs) that are co-expressed in naïve B cells. Despite having identical antigen specificity and being associated with the same signaling heterodimer Igα/Igβ (CD79a/CD79b), IgM and IgD-BCR isotypes functionally differ from each other in the manner of antigen binding, the formation of isolated nanoclusters and in their interaction with co-receptors such as CD19 and CXCR4 on the plasma membrane. With recent developments in experimental techniques, it is now possible to investigate the nanoscale organization of the BCR and better understand early events of BCR engagement. Interestingly, the cytoskeleton network beneath the membrane controls the BCR isotype-specific organization and its interaction with co-receptors. BCR triggering results in reorganization of the cytoskeleton network, which is further modulated by isotype-specific signals from co-receptors. For instance, IgD-BCR is closely associated with CXCR4 on mature B cells and this close proximity allows CXCR4 to employ the BCR machinery as signaling hub. In this review, we discuss the functional specificity and nanocluster assembly of BCR isotypes and the consequences of cross-talk between CXCR4 and IgD-BCR. Furthermore, given the role of BCR and CXCR4 signaling in the development and survival of leukemic B cells, we discuss the consequences of the cross-talk between CXCR4 and the BCR for controlling the growth of transformed B cells.
Introduction
B-lymphocytes (B cells) are central to the mammalian humoral immune response, as they produce and secrete immunoglobulins (Igs), also known as antibodies that contribute to neutralization, fixation, and clearance of pathogens. Besides the secreted form of Igs, B cells also express a membrane-bound form of Ig (mIg) as part of the B cell antigen receptor (BCR), which is indispensable for B cell differentiation, survival, and activation (1–3). While it is unclear how in the absence of foreign antigens BCR-derived signals regulate selection and survival of B cells throughout development, it is evident that binding of foreign antigen to mature B cells triggers BCR-dependent proliferation and differentiation of the mature B cells into antibody secreting plasma cells or memory B cells (1, 3–5). Each B cell expresses a unique BCR specificity as a result of the random rearrangement of the IG-gene segments in the course of early developmental stages (6–8). This process generates a highly diverse pool of naïve B cells carrying arrays of specificities, which could theoretically distinguish >1014 different non-self molecular monograms or antigens (9, 10). Upon antigen encounter, the selected BCR specificities are further modified through the process of somatic hypermutation (SHM) within the germinal center (GC) (11–14), thereby resulting in optimized antibodies against invading pathogens.
The unique antigen binding specificity of an antibody is determined by the combination of its heavy chain (HC) and light chain (LC) variable domains (VH and VL, respectively), produced by recombination of the variable (V), diverse (D), and joining (J) segments of the IG gene. A pair of recombination activating genes called RAG1 and RAG2 catalyze the V(D)J recombination during the development of B cells (15). Once generated, the recombined and selected V(D)J rearrangements provide unique antigen binding specificity to the respective B cell (16–19). By alternative splicing of pre-mRNA or class-switch recombination (CSR), a recombined VDJ cassette can be expressed as IgM, IgD, IgG, IgA, or IgE isotypes, by using different constant gene segments. Each secretable isotype possesses different neutralization, fixation, and clearance role (20–23). Although the VH and VL regions determine the antigen binding specificity, the constant region of Ig has an important role in fine-tuning the antigen sensing process (20, 22, 23).
In principle, all the five isotypes can be spliced as the membrane-associated mIg form thereby presenting as BCR on the B cell surface (4). During early development, B cells express only IgM-BCR, while IgD is produced later along with IgM by alternative pre-mRNA splicing at mature B cell stages (6, 24, 25). After encountering an antigen, IgM+IgD+ mature B cells undergo CSR to produce IgG, IgA, or IgE isotypes. Interestingly, B cells do not equally utilize the BCR isotypes. However, the mechanisms regulating this selectivity are not fully understood. For instance, IgA-BCR is relatively common in human but rare in mouse, while IgE-BCR is completely underrepresented in both species (26–28). This might indicate that BCR isotypes possess different affinity for distinct antigens, that they own different signaling capacities or that they are specialized for specific antigen forms (4, 20, 22, 23). In line with these views, the IgG-BCR produces more traction force than IgM-BCR while interacting with membrane-bound antigens, suggesting a specialized role of IgG-BCR to interact with complex or membrane-bound antigens (29, 30). Moreover, the co-existence of IgM and IgD-BCR on naïve recirculating B cells also provokes the hypothesis of a functional difference. However, the specific role of the IgD-BCR remained obscure for a long time. With the advent of cutting edge technology, accumulating evidence points to functional differences between these two BCR isotypes. For instance, it has been found that IgM and IgD-BCRs do differ in antigen sensing, signal commitment, structural flexibility as well as in their nanocluster organization on the plasma membrane (PM) landscape (31–33).
Therefore, it is important to discuss the functional specificities of IgM and IgD-BCRs in light of B cell development (section Altered B cell development), antigen selectivity (section Selective antigen responsiveness), and GC response and affinity maturation (section GC response and affinity maturation). In addition, we explain how nanocluster assembly of different BCR isotypes on mature B cells supports their functional differences (section Characterization of BCR nanoclusters). In light of this isotype-specific segregation, we address the interaction between BCR isotypes and co-receptors as well as the consequences of these processes in B cell activation and B cell-related diseases (section Synchronization effect of chemokine receptor CXCR4).
Functional Specificity of BCR Isotypes
Since mature naïve B cells express both IgM and IgD-BCR on their surface, it has been proposed that these two BCR isotypes are functionally redundant. Several lines of evidence support this view. First, mIgM and mIgD are generated from alternative splicing of the same pre-mRNA thereby having the same variable (VH) region and identical antigen binding specificity. Second, both mIg classes are associated with the Igα/Igß heterodimer (encoded by CD79A and CD79B genes, respectively), for signal initiation and a plethora of common signaling proteins including BLNK (also known as SLP65) Syk, Lyn, Btk, or PLCγ2 to transmit and integrate the intracellular signaling. Lastly, knockout (KO) mouse for either of the isotypes showed relatively weak effect on B cell development indicating that IgM and IgD could compensate for each other's function (34–36). Thus, IgD was thought to be a reserve receptor to ensure functional immune responses as IgD-BCR may activate B cell signaling in case IgM-BCR fails. However, recent studies provide compelling evidences indicating functional segregation of IgM and IgD on the B cell surface. As discussed below, several lines of evidence support this view.
Altered B Cell Development
In the wild type situation, IgM, but not IgD, efficiently associates with the germline encoded surrogate LC composed of VpreB and λ5 to form the pre-BCR, which is expressed at the pre-B cell stage of development (37, 38) (Figure 1). Expression of the pre-BCR triggers LC gene recombination driving B cell development further to the immature stage. Notably, the signaling capacity of the pre-BCR largely relies on μHC rather than on δHC that are usually part of IgM or IgD, respectively (Figure 1). At immature stage, differential poly-adenylation and alternative splicing of the Ig HC pre-mRNA encompassing the recombined VHDJH exon and the downstream IGHM (Cμ) and IGHD (C∂) exons lead to co-expression of IgM and IgD (6, 39). Such developmental regulation of IgM and IgD expression points to a specific non-redundant role of IgD in B cell development or function. In line with this, IgM KO mice (expressing only IgD) show a decrease in the proportion of innate-like B1 B cells that are known to require stronger signaling for their development and to express higher IgM and less IgD than follicular (FO) B cells (40). The marginal zone (MZ) and FO B cells seem to develop normally in the IgM-deficient mice although the number of FO B cells is slightly increased in these mice (33, 35) (Figure 1). In contrast, IgD deficiency results in mild effects in B cell development such as a decrease in the number of FO B cells to variable extent in different animals (34, 36) (Figure 1). Strikingly, IgD deficiency delays affinity maturation of B cells in primary antibody response against protein antigens (36) (discussed in section GC response and affinity maturation).
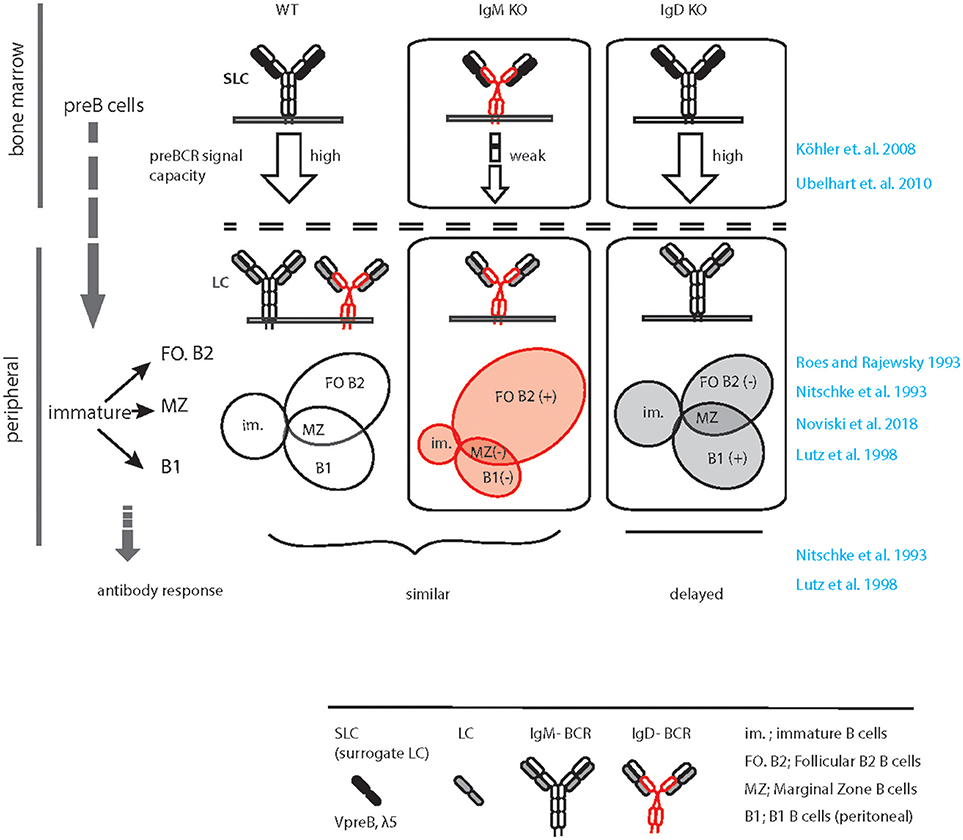
Figure 1. Graphical summary of altered peripheral B cell subsets and immune response in IgM KO and IgD KO animals. Schematic representation of B cell development in WT (left), IgM KO (middle), and IgD KO (right) animals highlighting the defect in preBCR signaling, increased follicular (FO) B cell compartment and decreased B1/ MZ B cell compartment in IgM KO animals. In contrast, follicular (FO) B cell compartment is reduced and B1/ MZ B cell compartment is increased in IgD KO animals. In response to antigen, IgD KO animals show a time delay and decreased serum antibody titer compared to WT animals.
Usually, IgD expression begins at the transitional stage between immature and mature B cell compartments (6). As discussed before, alternative splicing of the HC pre-mRNA containing the Cμ and C∂ exons result in co-expression of IgM and IgD-BCRs. Such co-expression is associated with gradual decrease in IgM expression in mature naïve B cells as compared to immature B cells. Generally, immature B cells express increased amount of IgM while mature naïve B cells express increased amount of IgD together with slightly reduced level of IgM. However, B cells from IgM KO mice express almost 1.5–2.0-fold excess of IgD-BCR on the PM (33). On the contrary, IgD deficiency does not greatly increase IgM expression, although it limits the down-modulation of IgM expression within the mature B cell repertoire. Thus, in IgD KO mice all mature B cells tend to express increased amount of IgM similar to what is observed in wildtype immature B cells (Maity et al., unpublished data). Together, these alterations in IgM and IgD-BCR expression in the mature B cell compartments point to a potential difference in the strength of signals generated through these two BCR isotypes. It is conceivable that the threshold for survival signal is achieved by relatively low amount of IgM-BCR, but high amount of IgD-BCR on the surface is required. Thus, considering the role of BCR-induced signals in selection and survival of B cells, it is tempting to speculate that the signaling through IgM-BCR is stronger than that of the IgD-BCR.
In line with this hypothesis, it has been suggested that increased IgD expression in mature B cell compartment is necessary to down-modulate the increased amount of IgM on immature B cells, thereby setting a variable range of IgM expression across the B cell repertoire (33). Moreover, IgD-BCR induces mild signaling in vivo (33) despite being strongly signaling in vitro (41) and functionally equivalent to IgM ex vivo (unpublished result Maity et al.). Taken together, the elevated amount of IgD-BCR is necessary to enable the survival of IgM-deficient mature B cells, while IgD-deficient mature B cells achieve the signaling threshold required for survival by evading the down-modulation of IgM-BCR. Therefore, IgM expression on mature B cells from IgD-deficient mice is similar to that of the wildtype immature B cells. This suggests that the amount of IgM expressed on immature B cells is sufficient for selection and survival. Furthermore, the expression of IgD-BCR is not necessary for these processes and is most likely required for efficient mature B cell function.
Selective Antigen Responsiveness
Another line of evidence for diverse functional specificity of IgM and IgD comes from their structural differences (32, 36, 42). The N-terminal VH and VL of HC and LC together form the antigen-binding site. The VH/VL and the first constant domain of both HC and LC (CH1, CL) constitute the Fab (fraction antigen binding) fragment, which is joined with the next CH domain through a hinge region. The structure of the hinge region is strikingly different for IgM and IgD (32, 36, 42). While IgM has a smaller hinge region, IgD is characterized by a long hinge region with charged residues and O-linked glycosylation (24, 42). The long hinge region gives IgD more flexibility to orient its antigen binding Fab fragment toward potential antigens. Thus, despite equal binding of monovalent and multivalent antigens, IgD is optimized for responding to multivalent antigen in immune-complexes (36, 42). Using an in vitro reconstitution system in SLP65, Rag2, and λ5 triple knockout (TKO) pro B cells, it was demonstrated that IgD-BCRs, specific for hen egg lysozyme (HEL), or 4-hydroxy 5-Iodo 3-nitrophenylacetyl (NIP), bind and initiate calcium response only to multimeric antigen complexes but not to monovalent antigens (32). In contrast, when expressed as IgM, the same BCRs were found to be responsive to monovalent antigens as well as to multivalent complex antigens. Swapping the hinge region between IgD and IgM also interchanged their specificity toward antigen valency. Remarkably, in the same study it was also shown that anergic B cells characterized by elevated surface IgD:IgM ratio failed to respond to monovalent antigens but remained fully responsive to multivalent complex antigens. Thus, higher IgD expression by anergic B cells is a mechanism to keep them quiescent toward monovalent autoantigens thereby preventing autoreactive responses, while they remain fully active against multivalent foreign immune-complexes thereby mounting proper immune responses. Conversely, higher surface IgM and low surface IgD expression in B1 B cells may allow activation by self-structures, which might be necessary for the house keeping functions of B1 B cells such as removal of cell debris. Simultaneously, B1 B cells retain the capacity to promptly mount innate immune response against common microbial antigens.
Similar evidence is also obtained from a recent study employing a transgenic reporter mouse in which B cell activation is monitored by green fluorescent protein (GFP) expression under the control of Nr4a1 (Nur77), an immediate early response gene of antigen receptor signaling (33, 43). Using this model, the authors showed that IgD is less efficient than IgM in sensing endogenous antigens. While both isotypes can efficiently mediate GC entry, B cells lacking IgM are defective in differentiating into short-lived plasma cells (SLPC) (33). It is therefore conceivable that lowering surface IgM expression on mature FO B cells provides an important mechanism to limit their differentiation to antibody secreting SLPC thereby preventing uncontrolled immune responses to cross-reactive autoantigens that bind at low specificity.
GC Response and Affinity Maturation
The germinal centers (GCs) are densely packed cellular domains within the lymphoid organs that are formed during the immune response (44). Within GC, antigen-specific B cells are selected, enriched and their antigen-binding specificities are improved in a process known as affinity maturation by SHM (13, 45). Since CSR also takes place in GC, the GC reaction is important for the generation of high-affinity antibodies with different effector functions including memory responses (46, 47). During T cell-dependent (TD) immune response, it is believed that a considerable number of FO B cells participates in the GC reaction (44, 48). As such, IgM deficiency neither significantly impacts the development of FO B cells nor it impairs TD-immune responses or affinity maturation through the GC reaction (35). In IgM KO mice, the TD immune responses toward carrier-conjugated monovalent antigens such as 2,4-Dinitrophenol-ovalbumin (DNP-ova) and complex antigen like sheep red blood cell (SRBC) remain unaltered as compared to wildtype counterpart (33, 35). However, a recent study (33) reported that the early class-switch response is defective in IgM-deficient B cells resulting in impaired generation of short-lived IgG1+ plasma cells (SLPC), although the unswitched PCs remained unaltered. Interestingly, this impaired IgG1+ SLPC is intrinsic to IgM-deficient B cells and independent of monoclonal or polyclonal antigens used for immunizing the animals (33).
Unlike IgM deficiency, studies using IgD-deficient mice revealed that absence of IgD leads to the retardation of TD-immune response, antibody production, and affinity maturation as compared to wildtype counterpart (34, 36). In particular, immunization with small-molecule antigens such as DNP-ova or NIP-chicken gammaglobulin (NIP-CG) showed a delayed antibody production and defective affinity maturation in IgD-deficient mice. Although the amounts of different serum Igs, except IgE, were normal in non-immunized IgD-deficient mice, antigen-specific IgG1, and IgG2 serum titers were largely reduced upon immunization (34, 36). As already mentioned, IgD-deficient mice also showed a delay in affinity maturation, i.e., production of high affinity antibodies, against NIP-CG by 3–4 days. On the contrary, using SRBCs as antigen, it was shown that IgD is redundant for GC reaction and immune response (33). Of note, unlike carrier conjugated small-molecules like DNP-ova and NIP-CG, SRBC is a robust polyclonal antigen, which can mount immune response independent of adjuvant. Together, these results suggest that the IgD-BCRs may be required for recruitment of B cells into GC reaction and subsequent affinity maturation during primary immune responses (34, 36).
Notably, studies employing mouse models of autoimmunity revealed that IgM-BCR exaggerates autoantibody production specifically in the absence of IgD (33, 49). For instance, the IgD-deficient lpr mice, a mouse model of systemic autoimmunity, showed elevated production of all different subtypes of IgG (IgG2a, IgG2b, and IgG3) autoantibodies, increased deposition of immune complexes in the kidney and more severe phenotype compared to IgD-sufficient lpr mice (49). Although these mice have elevated abnormal CD4−CD8−–double negative T cells in the spleen and lymph nodes, the severe autoimmunity in IgD-deficient lpr mice suggests a protective role for IgD-BCR in preventing deregulated autoimmune responses induced by IgM-BCR. In line with this, deficiency of IgD in Lyn−/− mice, a commonly used model of systemic Lupus Erythematosus (SLE) also shows enhanced autoantibody production (33). On the contrary, IgM deficiency in Lyn−/− background abrogates this autoantibody generation.
Taken together, the positive effect of IgM on early class-switch and on the generation of SLPC suggests that IgM-BCR may readily induce immune responses to autoantigen and that the presence of IgD-BCR negatively regulates this by attenuating the differentiation of autoreactive B cells into antibody secreting plasma cells. This view is in agreement with the lower threshold for activation of IgM as compared to IgD and with the fact that IgD binds to, but is not activated by, soluble monovalent antigens. Notably, an increased amount of monovalent antigen prevents the activation of IgD-BCR by immune complexes of the same antigen suggesting that IgD-BCR is regulated by the ratio of monovalent to complex antigen (32). Thus, all forms of antigens including autoantigens readily activate the IgD-deficient B cells that express only IgM. Most likely, the selectivity of IgD-BCRs toward antigen complexes and its regulation by soluble monovalent antigens controls the threshold for activation of wildtype B cells (42). Thus, similar to malignant transformation, the manifestation of autoimmunity may be a multistep process, in which the loss of IgD-mediated control together with the loss of a negative regulator, such as Lyn, result in rapid development of autoreactive immune responses. Thus, maintaining IgD at a higher proportion as compared to IgM may well be an important step in the prevention of aberrant outbreak of autoreactivity in wild type animals.
Characterization of BCR Nanoclusters
The above discussion underlines the notion that characterizing the molecular mechanisms of BCR activation is critical for understanding B cell selection, survival as well as abnormal B cell responses toward autoantigens. While it is well-known that, upon binding the cognate antigen, the BCR activates B cell signaling and mediates antigen internalization (50, 51), the mechanism of signal initiation upon antigen binding remained a long-standing debate. Alternate models proposed an antigen-mediated cross-linking of adjacent BCRs or antigen-induced conformational change and rearrangement of BCR clusters (31, 52–54). There are experimental evidences both favoring and opposing these models, which have been reviewed elsewhere (54–56). Intriguingly, for all these models it is necessary to consider the initial state of the BCR prior to antigen binding, which remained a challenge for some time. The ordered assembly of the BCR on the PM in the native state was far below the resolution of confocal microscopes and therefore remained elusive (55). Recent advancement of microscopic techniques, especially the super-resolution techniques enabled the visualization of the nanometer scale organization of receptors on the PM (31, 57–59).
Identifying BCR Nanoclusters by dSTORM
The most commonly used method for visualizing the nanoscale organization is the direct stochastic optical reconstitution microscopy (dSTORM). This method exploits the sparse blinking property of the fluorophores under reducing chemical environment combined with high energy excitation leading to dark state of the fluorophores (60). This induced stochastic optical blinking is recorded with high-speed acquisition system, usually an EM-CCD camera, which ensures splitting, and registering of fluorophore peaks or optical point spread functions (PSFs) into different frames. Finally, the individual frames are computed to obtain the ensemble high-resolution images. Resolutions of a dSTORM image are combined with the efficiency of correctly identifying the PSFs and localizing them with an empirically determined uncertainty (61, 62). Several factors including the samples, their preparation, types of fluorophores, performances of acquisition devices and relative drifts associated to microscope platforms during imaging influence the uncertainty of localizing the PSFs, which in turn determines the resolution. In practice, one could expect a 10-fold improvement of image resolution compared to standard fluorescence microscopy (31, 59, 63).
In recent years, dSTORM was employed by different laboratories to investigate the organization of BCRs on the PM in resting and activated B cells (31, 58, 59). Despite their differences in methods of sample preparation and sample source, the data obtained from dSTORM revealed that the native BCR resides as nanoclusters or protein-islands, and not as individual freely moving entities on the PM.
However, the mechanism of BCR activation by antigen-mimicking anti-BCR antibodies or antigen-independent cytoskeleton remodeling induced by Latrunculin A (LatA) remained controversial in these studies (31, 58, 59). The reasons for these variations are not completely understood and it is conceivable that they are linked to differences in methods and reagents as discussed below.
dSTORM Imaging of Resting and Activated BCRs
In order to image the native organization of BCRs on the PM, every protocol must ensure non-stimulatory conditions and avoid induced clustering or crosslinking. To accomplish the non-stimulatory conditions, the labeling reaction should be performed on ice by using fluorescently labeled probes against the BCRs (31). Additionally, the probes must have equal labeling efficiency and accessibility to potential binding sites on different nanoscale structures starting from monomers to large oligomers (Figure 2). This is strikingly different for antigen based labeling as compared to anti-BCR antibody based labeling. Since antigen-binding is the main role of a BCR, the antigen binding sites are protruded on the top of the BCR molecule. Therefore, fluorescently labeled antigens would equally access and bind to the BCRs regardless of their dense or loose clusters (Figure 2, resting and activated). Indeed, the overall BCR density obtained by dSTORM for both resting and activated B cells remained consistent upon labeling with antigen (31). In contrast, the epitopes for the anti-BCR Fab fragments might be partially buried in BCR oligomers and more accessible for labeling only in dispersed BCR clusters or monomers (Figure 2). Therefore, labeling with Fab fragments might not identify the dissociation of BCR molecules from tight oligomers to dispersed and dissociated smaller units. Instead it detects dispersed smaller units with high density labeling making them indistinguishable from tight oligomers (Figure 2).
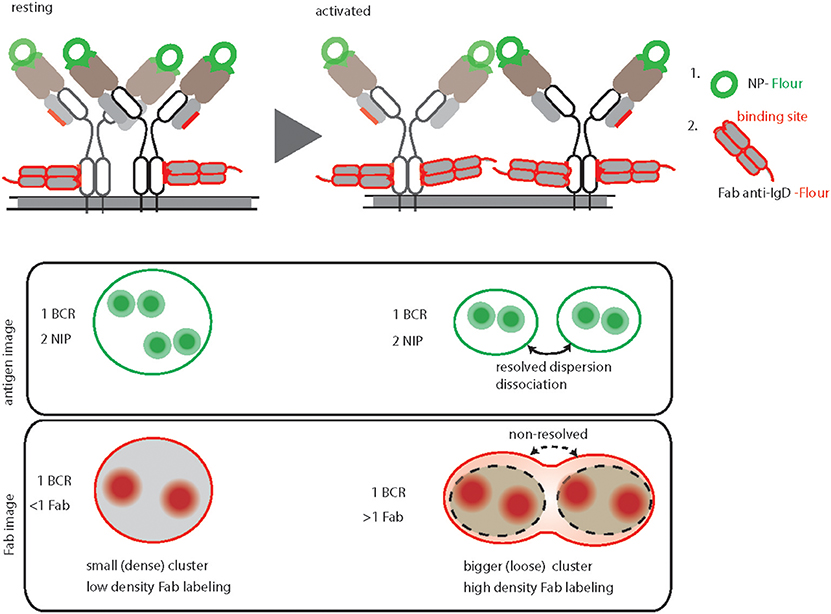
Figure 2. Graphical summary and comparison of antigen-based and anti-BCR Fab fragment-based BCR labeling for dSTORM imaging. Top, schematic representation of IgD-BCR nanoscale organization on resting (left) and activated (right) B cells, demonstrating their equally efficient fluorescent labeled NP-antigen (green) binding in contrast to differential fluorescently labeled anti-IgD Fab fragment (red) binding. The accessible sites for antigen and anti-BCR antibody (anti-IgD Fab fragment) of a NP specific IgD- BCR are highlighted by green and red color, respectively. Bottom, schematic of antigen-based dSTORM imaging compared to Fab fragment-based dSTORM imaging of resting and activated IgD-BCR nanoscale organization, allowing quantification of dissociated BCR units upon activation and non-resolving large clusters of activated BCRs due to increase labeling density, respectively.
The number of fluorophores per cluster is used to deduce the number of BCR molecules within a particular nanoscale domain. This requires a defined staining and fluorophore labeling protocol, which can be linearly correlated to the number of BCR molecules. In the case of antigen staining, the BCR to antigen ratio always remains close to a 1:2 ratio (Figure 2) (31). Unlike small molecules (e.g., NP) or small proteins (e.g., HEL, MW 14.2 kDa), estimating the number of fluorophores attached to a bigger protein molecule such as Fab fragment antibody (MW 50kDa) is somewhat challenging. In addition, staining with Fab fragments generated from a polyclonal antibody is incapable of reporting a linear BCR to fluorophore ratio due to multiple binding sites of the antibody (58, 59).
The protocol of adhering B cells must guarantee the non-stimulatory conditions including native PM organization and preferably untouched receptors. In this regard, settling cells at low temperature (on ice) extensively prevents activation and reorganizations (31). In contrast, adhering cells at 37°C or room temperature (RT) might induce altered membrane organization or BCR internalization resulting in acquisition of intracellular fluorescence during imaging, which may not be excluded by total internal reflection fluorescence (TIRF) mode microscopy (58, 59). Furthermore, activation of surface-adhered B cells by treatment with either anti-BCR antibody or LatA might influence the PM for induced changes and constrain the BCR dynamics (64, 65). Particularly, an induced crowding of the BCR toward the attachment surface seems to be unavoidable when using specific tethering agents to adhere B cells and to simultaneously activate B cells. In contrast, a simplified protocol to stimulate B cells followed by staining, attachment and fixation avoids any further distortions as well as provides opportunities to compare with other microscopy methods (31, 66).
In summary, the dSTORM technology provided evidence for nanoscale protein-islands organization of the BCR in the resting state. However, characterization of the active state of BCRs by dSTORM method still remains challenging. Due to lack of a consensus protocol, it is difficult to compare among different studies. Therefore, the application of super resolution imaging for BCRs or any other immunoreceptors must be updated and rationalized to visualize the native membrane organization.
Isolated Nanoclusters of IgM and IgD-BCRs
Intriguingly, the evidences that BCR molecules are organized in nanoclusters inspired new model of isotype-specific segregation of IgM and IgD on the PM of resting B cells. In turn, two-color dSTORM experiments facilitated the visualization of IgM and IgD-BCRs simultaneously and revealed their independent nanocluster organizations in separate membrane domains (31). Moreover, the size and number of receptors per nanoclusters of IgM and IgD are strikingly different from each other (31, 59). While IgD nanoclusters contain approximately 48 BCRs within a radius of about 240 nm, IgM nanoclusters contain 30 BCRs within a radius of about 150 nm (31, 42). This difference in size is proportional to the relative expression of BCR isotypes on the cell surface, which is usually in the ratio of 65 to 35 for IgD and IgM-BCRs, respectively. In addition, two-color dSTORM also reported an average distance of 300–350 nm that separates individual nanoclusters of IgM and IgD-BCRs (31, 42). This was further supported by two-marker electron microscopy imaging of the B cell PM (31, 42). Notably, the differences in the number of receptors per nanoclusters were also reported in single-color dSTORM using anti-BCR Fab labeling protocol, but the separation between IgM and IgD-BCRs could not be measured by this method (59).
The fact that IgM and IgD BCRs reside in separate membrane domains supports earlier biochemical studies revealing that they can be stimulated independently from each other (67). Indeed, using two-color dSTORM it was shown that the stimulation of one isotype of BCR did not impact the organization of the other isotype, thereby facilitating their independent signal initiation (31, 55). Furthermore, IgD-BCRs reside in lipid raft-like membrane domains and co-localize with glycosyl-phosphatidyl-inositol linked (GPI) protein CD52, while IgM-BCRs reside in non-raft domains prior to activation (68). The raft-like membrane domains are rich in GPI protein, GM1 gangliosides, and other important co-receptors such as CD19 and CXCR4 (42, 69, 70). Indeed, IgD-BCRs co-localize with CD19 and CXCR4 in resting B cells, whereas IgM-BCRs gain proximity with CD19 only upon activation. Interestingly, both CD19 and CXCR4 are considered to be co-activators to BCR signaling, while the receptor phosphatase CD22 acts as an inhibitor to BCR signaling. Notably, CD22 also exists as preformed nanoclusters and its proximity increases with IgM-BCR upon activation (71, 72). These observations are in full agreement with the fact that the translocation of BCRs into lipid rafts is necessary for stronger signaling (73) and that the signaling through IgM-BCR differs from IgD-BCR. In the next section we discuss how the interplay between CXCR4 and BCR isotypes modulates B cell function in healthy and neoplastic condition.
Synchronization Effect of Chemokine Receptor CXCR4
CXCR4 Signaling in Healthy and Malignant B Cells
CXCR4 belongs to the G protein coupled receptor (GPCR) family that selectively binds the CXC family chemokine Stromal Cell-Derived Factor 1 (SDF-1) also known as CXCL12 (74, 75). In response to CXCL12, CXCR4 signaling activates diverse GPCR pathways, resulting in migration, adhesion, and transcriptional activation of downstream target genes. Interestingly, recent findings uncovered functional differences between IgM and IgD BCR isotypes in the context of CXCR4 chemokine receptor signaling (76). IgD-deficient B cells were found to be defective in CXCR4 signaling with no calcium mobilization upon CXCL12-mediated stimulation of CXCR4 and impaired chemotactic migration toward CXCL12 gradient. In contrast, IgM-deficient B cells, expressing only IgD B cells showed no impairments in CXCR4 signaling. Interestingly, stimulation of the co-receptor CD19 with anti-CD19 antibody restores the above-mentioned defects in IgD-deficient B cells. Deeper inspection led to the observation that CXCR4 and CD19 co-localize in the same nanocluster as IgD on the cell membrane but not within IgM nanoclusters (55, 76). Thus, physical separation of these two isotypes on the B cell membrane also implies their functional specificity (42).
In addition to the prominent role of CXCR4 signaling in pro and pre–B cells in the bone marrow, this chemokine receptor also controls the migration of mature B cells into secondary lymphoid tissues (77). Therefore, CXCR4 signaling is of particular importance for lymphomagenesis, infiltration, migration, and retention of leukemic B cells in particular lymphoid tissues (78, 79). For instance, a number of studies pointed out an important role of CXCR4 signaling in Chronic Lymphocytic Leukemia (CLL) (77, 79). CLL is a mature B cell malignancy characterized by clonal accumulation of CD5+ B cells in peripheral blood, bone marrow and secondary lymphoid organs (80, 81). Continuous BCR signaling is considered to be the central pathway mediating the pathogenesis (82–84). It has been shown that ligation of surface IgM-BCR by anti-IgM antibody leads to B cell signaling in CLL, while IgD ligation by similar antibody treatment is unable to activate these cells (85, 86). Interestingly, stimulation using anti-IgM antibodies reduced the chemotaxis of CLL B cells toward CXCL12, while IgD stimulation led to opposite result suggesting IgD dependence of CXCR4 signaling (77, 87).
Often, expression of CXCR4 in neoplastic B cells in CLL is enhanced compared to normal B cells thereby conferring increased functional response to CXCL12. Indeed, CXCR4 overexpression in these neoplastic B cells is regarded as one of the factors responsible for their enhanced migration toward bone marrow niche, enriched with stromal cell derived CXCL12 (77, 83, 88). Furthermore, increased CXCR4 expression on CLL cells also accounts for their resistance to spontaneous or drug-induced apoptosis, providing a protective niche for tumor cells, and making them unresponsive to conventional chemotherapy (79, 88–91).
Apart from deregulated expression, several CXCR4 mutations are common to leukemic B cell and related disorders including CLL and Waldenström macroglobulinemia (WM) (92–94). The most interesting CXCR4 somatic mutations are truncations of the C-terminal tail by 9–12 amino acids. This is also the commonly found germline variation in warts, hypogammaglobulinemia, infections, and myelokathexis (WHIM) syndrome. WHIM-like CXCR4 mutation results in CXCL12 desensitization and sustained CXCR4 signaling in leukemic cells (95), which manifests in the clinical warts-like symptoms in WHIM patients. Furthermore, the WHIM-like CXCR4 mutation accounts for sustained survival signal in leukemic cells and renders them resistant to inhibitors of the Bruton's tyrosine kinase (BTK) (96). Interestingly, BTK is a downstream kinase of BCR, Toll-like receptor (TLR), and CXCR4 (97). BTK inhibition causes impaired CXCR4 signaling and reduces the PM pool of CXCR4, resulting in rapid egress of CLL cells from CXCL12-rich niches and consequently prevents re-entry of CLL cells (78, 98, 99). Thus, simultaneous deactivation of both BCR and CXCR4 signaling reveals the clinical efficacy of BTK inhibitor, demonstrating the complex interplay between BCR and CXCR4 (100, 101).
CXCR4 and Cytoskeleton Remodeling
Similar to other GPCRs, CXCR4 signaling also induces actin cytoskeleton remodeling (76, 102). This links CXCR4 signaling directly to BCR signaling as actin depolymerization by either LatA or CytoD is sufficient to induce robust BCR signaling (31, 65, 103). Furthermore, B cell-specific loss of function mutations in actin binding protein and related regulatory molecules (ABP-1 and WIP) modulates BCR signaling confirming pivotal role of the actin cytoskeleton for signal initiation and processing (50, 104–106). Although the molecular basis for actin cytoskeleton and BCR signaling cross-talk was mechanistically interpreted by picket-fence model, the in vivo trigger of this axis remained elusive. Increasing evidence suggests that the dominant IgD isotype on mature B cells is proximal to CXCR4 and therefore, IgD is required for CXCR4 signaling in mature B cells (Figure 3). This is in line with the observation that, in comparison to the IgM-BCR, the IgD-BCR resides in more actin-dense regions of the PM (Figure 3) (65). Thus, the effect of releasing the BCR from the constraints posed by the actin picket-fence might be greater for IgD than for IgM, although the exposure of B cells to LatA results in the dissociation of BCR oligomers of both classes, IgD and IgM (Figure 3) (31, 68). Together, the differential association of BCR isotypes with chemokine receptors confirms the functional specificity of IgD-type BCR and its role in efficient integration of the migratory cues from lymphoid tissue environment and antigen recognition during an immune response. In parallel, cooperating with CXCL12 induced CXCR4 signaling, IgD ensures low-grade activation of mature B cells in absence of antigen (Figure 3).
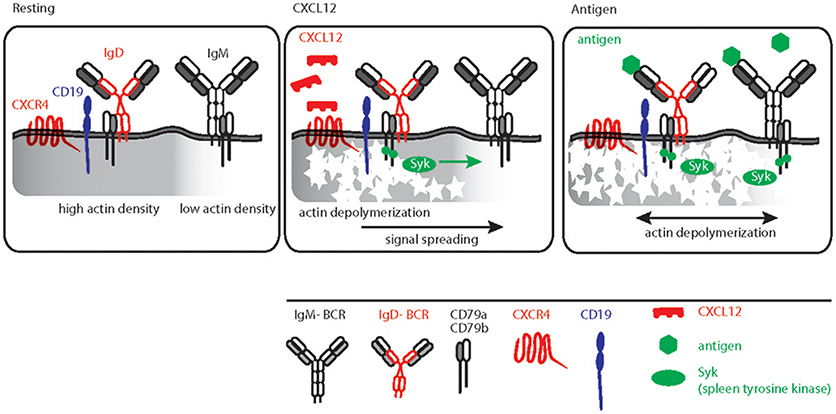
Figure 3. Schematic representation of CXCL12 and antigen triggered B cell activation. Left, resting B cell membrane showing IgD and IgM BCR nanoclusters differentially localized in actin dense (gray shaded) and actin poor regions, respectively. IgD BCRs are residing in close proximity to CD19 and CXCR4. Middle, CXCL12 induced signaling in B cells depicting the sequential triggering of local actin remodeling, recruitment of Syk and IgD BCR activation followed by signal spreading toward actin poor domains. Right, antigen mediated BCR signaling, Syk recruitment and actin remodeling leading to massive actin remodeling and B cell activation.
Conclusion
The existence of different classes of antibodies and their BCR counterparts in mammals is certainly related to their evolutionary conservation and necessity to diversify the repertoire. Nevertheless, the prominent usage of IgM and IgD during B cell development mark them as specialized antigen receptors compared to other isotypes. Overcoming the previous redundancy postulate, we begin to understand functional difference between IgM and IgD. With its structural specificity for multivalent antigens, its isolated nanocluster membrane organization and its coordination with particular co-receptors, the IgD BCR regulates and diversifies B cell responses. However, much more remains to be explored, specifically regarding the role of IgM and IgD in neoplastic B cells and autoimmune diseases. The interplay between CXCR4 and BCR isotypes in leukemic cells and their impact on pathogenesis also remains of particular interest.
Author Contributions
PM, MD, and HJ reviewed the literature and wrote the manuscript. PM and MD prepared the figures. AN contributed to the writing of the CXCR4 segment. All authors read and approved the review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We apologize to the scientists whose work was not cited owing to space limitations. We thank Mr. Marc Young, Dr. Elias Hobeika, and Prof. Michael Reth for critical reading of this manuscript. This study is supported by the IRTG-TRR130 (B cells and beyond), SFB 1279 (Exploration of the human peptidome), SFB 1074 (Experimental Models and Clinical Translation in Leukemia) and the European Research Council advanced grant 694992.
References
1. Lam KP, Kuhn R, Rajewsky K. In vivo ablation of surface immunoglobulin on mature B cells by inducible gene targeting results in rapid cell death. Cell (1997) 90:1073–83. doi: 10.1016/S0092-8674(00)80373-6
2. Pelanda R, Braun U, Hobeika E, Nussenzweig MC, Reth M. B cell progenitors are arrested in maturation but have intact VDJ recombination in the absence of Ig-alpha and Ig-beta. J Immunol. (2002) 169:865–72. doi: 10.4049/jimmunol.169.2.865
3. Srinivasan L, Sasaki Y, Calado DP, Zhang B, Paik JH, DePinho RA, et al. PI3 kinase signals BCR-dependent mature B cell survival. Cell (2009) 139:573–86. doi: 10.1016/j.cell.2009.08.041
4. Tong P, Granato A, Zuo T, Chaudhary N, Zuiani A, Han SS, et al. IgH isotype-specific B cell receptor expression influences B cell fate. Proc Natl Acad Sci USA (2017) 114:E8411–20. doi: 10.1073/pnas.1704962114
5. Rolink AG, Andersson J, Melchers F. Molecular mechanisms guiding late stages of B-cell development. Immunol Rev. (2004) 197:41–50. doi: 10.1111/j.0105-2896.2004.0101.x
6. Gutzeit C, Chen K, Cerutti A. The enigmatic function of IgD: some answers at last. Eur J Immunol. (2018) 48:1101–13. doi: 10.1002/eji.201646547
7. Ralph DK, Matsen FA. Consistency of VDJ rearrangement and substitution parameters enables accurate B cell receptor sequence annotation. PLoS Comput Biol. (2016) 12:e1004409. doi: 10.1371/journal.pcbi.1004409
8. Georgiou G, Ippolito GC, Beausang J, Busse CE, Wardemann H, Quake SR. The promise and challenge of high-throughput sequencing of the antibody repertoire. Nat Biotechnol. (2014) 32:158–68. doi: 10.1038/nbt.2782
9. Schroeder HW. Jr, Cavacini L. Structure and function of immunoglobulins. J Allergy Clin Immunol. (2010) 125:S41–52. doi: 10.1016/j.jaci.2009.09.046
10. Jackson KJ, Kidd MJ, Wang Y, Collins AM. The shape of the lymphocyte receptor repertoire: lessons from the B cell receptor. Front Immunol. (2013) 4:263. doi: 10.3389/fimmu.2013.00263
11. Tarlinton D, Victora G. Editorial overview: germinal centers and memory B-cells: from here to eternity. Curr Opin Immunol. (2017) 45:5–8. doi: 10.1016/j.coi.2017.05.002
12. Gitlin AD, Shulman Z, Nussenzweig MC. Clonal selection in the germinal centre by regulated proliferation and hypermutation. Nature (2014) 509:637–40. doi: 10.1038/nature13300
13. Mesin L, Ersching J, Victora GD. Germinal center B cell dynamics. Immunity (2016) 45:471–82. doi: 10.1016/j.immuni.2016.09.001
14. Dufaud CR, McHeyzer-Williams LJ, McHeyzer-Williams MG. Deconstructing the germinal center, one cell at a time. Curr Opin Immunol. (2017) 45:112–8. doi: 10.1016/j.coi.2017.03.007
15. Ji Y, Resch W, Corbett E, Yamane A, Casellas R, Schatz DG. The in vivo pattern of binding of RAG1 and RAG2 to antigen receptor loci. Cell (2010) 141:419–31. doi: 10.1016/j.cell.2010.03.010
16. Kumar R, Bach MP, Mainoldi F, Maruya M, Kishigami S, Jumaa H, et al. Antibody repertoire diversification through VH gene replacement in mice cloned from an IgA plasma cell. Proc Natl Acad Sci USA. (2015) 112:E450–7. doi: 10.1073/pnas.1417988112
17. Sun A, Novobrantseva TI, Coffre M, Hewitt SL, Jensen K, Skok JA, et al. VH replacement in primary immunoglobulin repertoire diversification. Proc Natl Acad Sci USA. (2015) 112:E458–66. doi: 10.1073/pnas.1418001112
18. Lange MD, Huang L, Yu Y, Li S, Liao H, Zemlin M, et al. Accumulation of VH replacement products in igh genes derived from autoimmune diseases and anti-viral responses in human. Front Immunol. (2014) 5:345. doi: 10.3389/fimmu.2014.00345
19. Liu J, Lange MD, Hong SY, Xie W, Xu K, Huang L, et al. Regulation of VH replacement by B cell receptor-mediated signaling in human immature B cells. J Immunol. (2013) 190:5559–66. doi: 10.4049/jimmunol.1102503
20. Casadevall A, Janda A. Immunoglobulin isotype influences affinity and specificity. Proc Natl Acad Sci USA. (2012) 109:12272–3. doi: 10.1073/pnas.1209750109
21. Chen K, Xu W, Wilson M, He B, Miller NW, Bengten E, et al. Immunoglobulin D enhances immune surveillance by activating antimicrobial, proinflammatory and B cell-stimulating programs in basophils. Nat Immunol. (2009) 10:889–98. doi: 10.1038/ni.1748
22. Torres M, May R, Scharff MD, Casadevall A. Variable-region-identical antibodies differing in isotype demonstrate differences in fine specificity and idiotype. J Immunol. (2005) 174:2132–42. doi: 10.4049/jimmunol.174.4.2132
23. Tudor D, Yu H, Maupetit J, Drillet AS, Bouceba T, Schwartz-Cornil I, et al. Isotype modulates epitope specificity, affinity, and antiviral activities of anti-HIV-1 human broadly neutralizing 2F5 antibody. Proc Natl Acad Sci USA (2012) 109:12680–5. doi: 10.1073/pnas.1200024109
24. Chen K, Cerutti A. The function and regulation of immunoglobulin D. Curr Opin Immunol. (2011) 23:345–52. doi: 10.1016/j.coi.2011.01.006
25. Geisberger R, Lamers M, Achatz G. The riddle of the dual expression of IgM and IgD. Immunology (2006) 118:429–37. doi: 10.1111/j.1365-2567.2006.02386.x
26. Conrad DH, Gibb DR, Sturgill J. Regulation of the IgE response. F1000 Biol Rep. (2010) 2:14. doi: 10.3410/B2-14
27. Sackesen C, van de Veen W, Akdis M, Soyer O, Zumkehr J, Ruckert B, et al. Suppression of B-cell activation and IgE, IgA, IgG1 and IgG4 production by mammalian telomeric oligonucleotides. Allergy (2013) 68:593–603. doi: 10.1111/all.12133
28. Cerutti A, Cols M, Gentile M, Cassis L, Barra CM, He B, et al. Regulation of mucosal IgA responses: lessons from primary immunodeficiencies. Ann N Y Acad Sci. (2011) 1238:132–44. doi: 10.1111/j.1749-6632.2011.06266.x
29. Shaheen S, Wan Z, Li Z, Chau A, Li X, Zhang S, et al. Substrate stiffness governs the initiation of B cell activation by the concerted signaling of PKCbeta and focal adhesion kinase. Elife (2017) 6:e23060. doi: 10.7554/eLife.23060
30. Wang J, Lin F, Wan Z, Sun X, Lu Y, Huang J, et al. Profiling the origin, dynamics, and function of traction force in B cell activation. Sci Signal. (2018) 11:eaai9192. doi: 10.1126/scisignal.aai9192
31. Maity PC, Blount A, Jumaa H, Ronneberger O, Lillemeier BF, Reth M. B cell antigen receptors of the IgM and IgD classes are clustered in different protein islands that are altered during B cell activation. Sci Signal. (2015) 8:ra93. doi: 10.1126/scisignal.2005887
32. Übelhart R, Hug E, Bach MP, Wossning T, Duhren-von Minden M, Horn AH, et al. Responsiveness of B cells is regulated by the hinge region of IgD. Nat Immunol. (2015) 16:534–43. doi: 10.1038/ni.3141
33. Noviski M, Mueller JL, Satterthwaite A, Garrett-Sinha LA, Brombacher F, Zikherman J. IgM and IgD B cell receptors differentially respond to endogenous antigens and control B cell fate. Elife (2018) 7:e35074. doi: 10.7554/eLife.35074
34. Nitschke L, Kosco MH, Kohler G, Lamers MC. Immunoglobulin D-deficient mice can mount normal immune responses to thymus-independent and -dependent antigens. Proc Natl Acad Sci USA (1993) 90:1887–91. doi: 10.1073/pnas.90.5.1887
35. Lutz C, Ledermann B, Kosco-Vilbois MH, Ochsenbein AF, Zinkernagel RM, Kohler G, et al. IgD can largely substitute for loss of IgM function in B cells. Nature (1998) 393:797–801. doi: 10.1038/31716
36. Roes J, Rajewsky K. Immunoglobulin D (IgD)-deficient mice reveal an auxiliary receptor function for IgD in antigen-mediated recruitment of B cells. J Exp Med. (1993) 177:45–55. doi: 10.1084/jem.177.1.45
37. Kohler F, Hug E, Eschbach C, Meixlsperger S, Hobeika E, Kofer J, et al. Autoreactive B cell receptors mimic autonomous pre-B cell receptor signaling and induce proliferation of early B cells. Immunity (2008) 29:912–21. doi: 10.1016/j.immuni.2008.10.013
38. Ohnishi K, Melchers F. The nonimmunoglobulin portion of lambda5 mediates cell-autonomous pre-B cell receptor signaling. Nat Immunol. (2003) 4:849–56. doi: 10.1038/ni959
39. Enders A, Short A, Miosge LA, Bergmann H, Sontani Y, Bertram EM, et al. Zinc-finger protein ZFP318 is essential for expression of IgD, the alternatively spliced Igh product made by mature B lymphocytes. Proc Natl Acad Sci USA. (2014) 111:4513–8. doi: 10.1073/pnas.1402739111
40. Casola S, Otipoby KL, Alimzhanov M, Humme S, Uyttersprot N, Kutok JL, et al. B cell receptor signal strength determines B cell fate. Nat Immunol. (2004) 5:317–27. doi: 10.1038/ni1036
41. Kim KM, Reth M. The B cell antigen receptor of class IgD induces a stronger and more prolonged protein tyrosine phosphorylation than that of class IgM. J Exp Med. (1995) 181:1005–14. doi: 10.1084/jem.181.3.1005
42. Hobeika E, Maity PC, Jumaa H. Control of B cell responsiveness by isotype and structural elements of the antigen receptor. Trends Immunol. (2016) 37:310–20. doi: 10.1016/j.it.2016.03.004
43. Zikherman J, Parameswaran R, Weiss A. Endogenous antigen tunes the responsiveness of naive B cells but not T cells. Nature (2012) 489:160–4. doi: 10.1038/nature11311
44. Kerfoot SM, Yaari G, Patel JR, Johnson KL, Gonzalez DG, Kleinstein SH, et al. Germinal center B cell and T follicular helper cell development initiates in the interfollicular zone. Immunity (2011) 34:947–60. doi: 10.1016/j.immuni.2011.03.024
45. De Silva NS, Klein U. Dynamics of B cells in germinal centres. Nat Rev Immunol. (2015) 15:137–48. doi: 10.1038/nri3804
46. Weisel FJ, Zuccarino-Catania GV, Chikina M, Shlomchik MJ. A temporal switch in the germinal center determines differential output of memory b and plasma cells. Immunity (2016) 44:116–30. doi: 10.1016/j.immuni.2015.12.004
47. Shlomchik MJ, Weisel F. Germinal center selection and the development of memory B and plasma cells. Immunol Rev. (2012) 247:52–63. doi: 10.1111/j.1600-065X.2012.01124.x
48. Wang X, Cho B, Suzuki K, Xu Y, Green JA, An J, et al. Follicular dendritic cells help establish follicle identity and promote B cell retention in germinal centers. J Exp Med. (2011) 208:2497–510. doi: 10.1084/jem.20111449
49. Guo L, Tian J, Guo Z, Zheng B, Han S. The absence of immunoglobulin D B cell receptor-mediated signals promotes the production of autoantibodies and exacerbates glomerulonephritis in murine lupus. Clin Exp Immunol. (2011) 164:227–35. doi: 10.1111/j.1365-2249.2011.04332.x
50. Liu C, Fallen MK, Miller H, Upadhyaya A, Song W. The actin cytoskeleton coordinates the signal transduction and antigen processing functions of the B cell antigen receptor. Front Biol. (2013) 8:475–85. doi: 10.1007/s11515-013-1272-0
51. Spillane KM, Tolar P. Mechanics of antigen extraction in the B cell synapse. Mol Immunol. (2018) 101:319–28. doi: 10.1016/j.molimm.2018.07.018
52. Tolar P, Pierce SK. A conformation-induced oligomerization model for B cell receptor microclustering and signaling. Curr Top Microbiol Immunol. (2010) 340:155–69. doi: 10.1007/978-3-642-03858-7_8
53. Tolar P, Sohn HW, Pierce SK. The initiation of antigen-induced B cell antigen receptor signaling viewed in living cells by fluorescence resonance energy transfer. Nat Immunol. (2005) 6:1168–76. doi: 10.1038/ni1262
54. Yang J, Reth M. The dissociation activation model of B cell antigen receptor triggering. FEBS Lett. (2010) 584:4872–7. doi: 10.1016/j.febslet.2010.09.045
55. Maity PC, Yang J, Klaesener K, Reth M. The nanoscale organization of the B lymphocyte membrane. Biochim Biophys Acta (2015) 1853:830–40. doi: 10.1016/j.bbamcr.2014.11.010
56. Mattila PK, Batista FD, Treanor B. Dynamics of the actin cytoskeleton mediates receptor cross talk: an emerging concept in tuning receptor signaling. J Cell Biol. (2016) 212:267–80. doi: 10.1083/jcb.201504137
57. Huang B, Babcock H, Zhuang X. Breaking the diffraction barrier: super-resolution imaging of cells. Cell (2010) 143:1047–58. doi: 10.1016/j.cell.2010.12.002
58. Lee J, Sengupta P, Brzostowski J, Lippincott-Schwartz J, Pierce SK. The nanoscale spatial organization of B-cell receptors on immunoglobulin M- and G-expressing human B-cells. Mol Biol Cell (2017) 28:511–23. doi: 10.1091/mbc.e16-06-0452
59. Mattila PK, Feest C, Depoil D, Treanor B, Montaner B, Otipoby KL, et al. The actin and tetraspanin networks organize receptor nanoclusters to regulate B cell receptor-mediated signaling. Immunity (2013) 38:461–74. doi: 10.1016/j.immuni.2012.11.019
60. Dempsey GT, Vaughan JC, Chen KH, Bates M, Zhuang X. Evaluation of fluorophores for optimal performance in localization-based super-resolution imaging. Nat Methods (2011) 8:1027–36. doi: 10.1038/nmeth.1768
61. Veatch SL, Machta BB, Shelby SA, Chiang EN, Holowka DA, Baird BA. Correlation functions quantify super-resolution images and estimate apparent clustering due to over-counting. PLoS ONE (2012) 7:e31457. doi: 10.1371/journal.pone.0031457
62. Sengupta P, Jovanovic-Talisman T, Skoko D, Renz M, Veatch SL, Lippincott-Schwartz J. Probing protein heterogeneity in the plasma membrane using PALM and pair correlation analysis. Nat Methods (2011) 8:969–75. doi: 10.1038/nmeth.1704
63. Lillemeier BF, Mörtelmaier MA, Forstner MB, Huppa JB, Groves JT, Davis MM. TCR and Lat are expressed on separate protein islands on T cell membranes and concatenate during activation. Nat Immunol. (2010) 11:90–6. doi: 10.1038/ni.1832
64. Harwood NE, Batista FD. The cytoskeleton coordinates the early events of B-cell activation. Cold Spring Harb Perspect Biol. (2011) 3:a002360. doi: 10.1101/cshperspect.a002360
65. Treanor B, Depoil D, Bruckbauer A, Batista FD. Dynamic cortical actin remodeling by ERM proteins controls BCR microcluster organization and integrity. J Exp Med. (2011) 208:1055–68. doi: 10.1084/jem.20101125
66. Fiala GJ, Kaschek D, Blumenthal B, Reth M, Timmer J, Schamel WW. Pre-clustering of the B cell antigen receptor demonstrated by mathematically extended electron microscopy. Front Immunol. (2013) 4:427. doi: 10.3389/fimmu.2013.00427
67. Gold MR, Matsuuchi L, Kelly RB, DeFranco AL. Tyrosine phosphorylation of components of the B-cell antigen receptors following receptor crosslinking. Proc Natl Acad Sci USA (1991) 88:3436–40. doi: 10.1073/pnas.88.8.3436
68. Klasener K, Maity PC, Hobeika E, Yang J, Reth M. B cell activation involves nanoscale receptor reorganizations and inside-out signaling by Syk. Elife (2014) 3:e02069. doi: 10.7554/eLife.02069
69. Cherukuri A, Cheng PC, Sohn HW, Pierce SK. The CD19/CD21 complex functions to prolong B cell antigen receptor signaling from lipid rafts. Immunity (2001) 14:169–79. doi: 10.1016/S1074-7613(01)00098-X
70. van Zelm MC, Smet J, Adams B, Mascart F, Schandene L, Janssen F, et al. CD81 gene defect in humans disrupts CD19 complex formation and leads to antibody deficiency. J Clin Invest. (2010) 120:1265–74. doi: 10.1172/JCI39748
71. Gasparrini F, Feest C, Bruckbauer A, Mattila PK, Muller J, Nitschke L, et al. Nanoscale organization and dynamics of the siglec CD22 cooperate with the cytoskeleton in restraining BCR signalling. EMBO J. (2016) 35:258–80. doi: 10.15252/embj.201593027
72. Muller J, Obermeier I, Wohner M, Brandl C, Mrotzek S, Angermuller S, et al. CD22 ligand-binding and signaling domains reciprocally regulate B-cell Ca2+ signaling. Proc Natl Acad Sci USA (2013) 110:12402–7. doi: 10.1073/pnas.1304888110
73. Cheng PC, Brown BK, Song W, Pierce SK. Translocation of the B cell antigen receptor into lipid rafts reveals a novel step in signaling. J Immunol. (2001) 166:3693–701. doi: 10.4049/jimmunol.166.6.3693
74. Jacobson O, Weiss ID. CXCR4 chemokine receptor overview: biology, pathology and applications in imaging and therapy. Theranostics (2013) 3:1–2. doi: 10.7150/thno.5760
75. Karpova D, Bonig H. Concise review: CXCR4/CXCL12 signaling in immature hematopoiesis–lessons from pharmacological and genetic models. Stem Cells (2015) 33:2391–9. doi: 10.1002/stem.2054
76. Becker M, Hobeika E, Jumaa H, Reth M, Maity PC. CXCR4 signaling and function require the expression of the IgD-class B-cell antigen receptor. Proc Natl Acad Sci USA (2017) 114:5231–6. doi: 10.1073/pnas.1621512114
77. Davids MS, Burger JA. Cell trafficking in chronic lymphocytic leukemia. Open J Hematol. (2012) 3:3. doi: 10.13055/ojhmt_3_S1_03.120221
78. Chen SS, Chang BY, Chang S, Tong T, Ham S, Sherry B, et al. BTK inhibition results in impaired CXCR4 chemokine receptor surface expression, signaling and function in chronic lymphocytic leukemia. Leukemia (2016) 30:833–43. doi: 10.1038/leu.2015.316
79. O'Hayre M, Salanga CL, Kipps TJ, Messmer D, Dorrestein PC, Handel TM. Elucidating the CXCL12/CXCR4 signaling network in chronic lymphocytic leukemia through phosphoproteomics analysis. PLoS ONE (2010) 5:e11716. doi: 10.1371/journal.pone.0011716
80. Rai KR, Jain P. Chronic lymphocytic leukemia (CLL)-Then and now. Am J Hematol. (2016) 91:330–40. doi: 10.1002/ajh.24282
81. Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Dohner H, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood (2018) 131:2745–60. doi: 10.1182/blood-2017-09-806398
82. Duhren-von Minden M, Ubelhart R, Schneider D, Wossning T, Bach MP, Buchner M, et al. Chronic lymphocytic leukaemia is driven by antigen-independent cell-autonomous signalling. Nature (2012) 489:309–12. doi: 10.1038/nature11309
83. Burger JA, Chiorazzi N. B cell receptor signaling in chronic lymphocytic leukemia. Trends Immunol. (2013) 34:592–601. doi: 10.1016/j.it.2013.07.002
84. Hallek M. Chronic lymphocytic leukemia: 2017 update on diagnosis, risk stratification, and treatment. Am J Hematol. (2017) 92:946–65. doi: 10.1002/ajh.24826
85. Lanham S, Hamblin T, Oscier D, Ibbotson R, Stevenson F, Packham G. Differential signaling via surface IgM is associated with VH gene mutational status and CD38 expression in chronic lymphocytic leukemia. Blood (2003) 101:1087–93. doi: 10.1182/blood-2002-06-1822
86. Mockridge CI, Potter KN, Wheatley I, Neville LA, Packham G, Stevenson FK. Reversible anergy of sIgM-mediated signaling in the two subsets of CLL defined by VH-gene mutational status. Blood (2007) 109:4424–31. doi: 10.1182/blood-2006-11-056648
87. Haerzschel A, Catusse J, Hutterer E, Paunovic M, Zirlik K, Eibel H, et al. BCR and chemokine responses upon anti-IgM and anti-IgD stimulation in chronic lymphocytic leukaemia. Ann Hematol. (2016) 95:1979–88. doi: 10.1007/s00277-016-2788-6
88. Burger JA, Burger M, Kipps TJ. Chronic lymphocytic leukemia B cells express functional CXCR4 chemokine receptors that mediate spontaneous migration beneath bone marrow stromal cells. Blood (1999) 94:3658–67.
89. Busillo JM, Benovic JL. Regulation of CXCR4 signaling. Biochim Biophys Acta (2007) 1768:952–63. doi: 10.1016/j.bbamem.2006.11.002
90. Debnath B, Xu S, Grande F, Garofalo A, Neamati N. Small molecule inhibitors of CXCR4. Theranostics (2013) 3:47–75. doi: 10.7150/thno.5376
91. Teicher BA, Fricker SP. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res. (2010) 16:2927–31. doi: 10.1158/1078-0432.CCR-09-2329
92. Crowther-Swanepoel D, Qureshi M, Dyer MJ, Matutes E, Dearden C, Catovsky D, et al. Genetic variation in CXCR4 and risk of chronic lymphocytic leukemia. Blood (2009) 114:4843–6. doi: 10.1182/blood-2009-07-235184
93. Hernandez PA, Gorlin RJ, Lukens JN, Taniuchi S, Bohinjec J, Francois F, et al. Mutations in the chemokine receptor gene CXCR4 are associated with WHIM syndrome, a combined immunodeficiency disease. Nat Genet. (2003) 34:70–4. doi: 10.1038/ng1149
94. Treon SP, Cao Y, Xu L, Yang G, Liu X, Hunter ZR. Somatic mutations in MYD88 and CXCR4 are determinants of clinical presentation and overall survival in Waldenstrom macroglobulinemia. Blood (2014) 123:2791–6. doi: 10.1182/blood-2014-01-550905
95. Balabanian K, Lagane B, Pablos JL, Laurent L, Planchenault T, Verola O, et al. WHIM syndromes with different genetic anomalies are accounted for by impaired CXCR4 desensitization to CXCL12. Blood (2005) 105:2449–57. doi: 10.1182/blood-2004-06-2289
96. Cao Y, Hunter ZR, Liu X, Xu L, Yang G, Chen J, et al. The WHIM-like CXCR4(S338X) somatic mutation activates AKT and ERK, and promotes resistance to ibrutinib and other agents used in the treatment of Waldenstrom's Macroglobulinemia. Leukemia (2015) 29:169–76. doi: 10.1038/leu.2014.187
97. Hendriks RW, Yuvaraj S, Kil LP. Targeting Bruton's tyrosine kinase in B cell malignancies. Nat Rev Cancer (2014) 14:219–32. doi: 10.1038/nrc3702
98. Byrd JC, Brown JR, O'Brien S, Barrientos JC, Kay NE, Reddy NM, et al. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med. (2014) 371:213–23. doi: 10.1056/NEJMoa1400376
99. de Rooij MF, Kuil A, Geest CR, Eldering E, Chang BY, Buggy JJ, et al. The clinically active BTK inhibitor PCI-32765 targets B-cell receptor- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood (2012) 119:2590–4. doi: 10.1182/blood-2011-11-390989
100. de Gorter DJ, Beuling EA, Kersseboom R, Middendorp S, van Gils JM, Hendriks RW, et al. Bruton's tyrosine kinase and phospholipase Cgamma2 mediate chemokine-controlled B cell migration and homing. Immunity (2007) 26:93–104. doi: 10.1016/j.immuni.2006.11.012
101. Palmesino E, Moepps B, Gierschik P, Thelen M. Differences in CXCR4-mediated signaling in B cells. Immunobiology (2006) 211:377–89. doi: 10.1016/j.imbio.2005.12.003
102. Ganguly S, Saxena R, Chattopadhyay A. Reorganization of the actin cytoskeleton upon G-protein coupled receptor signaling. Biochim Biophys Acta (2011) 1808:1921–9. doi: 10.1016/j.bbamem.2011.04.001
103. Baeker TR, Simons ER, Rothstein TL. Cytochalasin induces an increase in cytosolic free calcium in murine B lymphocytes. J Immunol. (1987) 138:2691–7.
104. Seeley-Fallen MK, Liu LJ, Shapiro MR, Onabajo OO, Palaniyandi S, Zhu X, et al. Actin-binding protein 1 links B-cell antigen receptors to negative signaling pathways. Proc Natl Acad Sci USA (2014) 111:9881–6. doi: 10.1073/pnas.1321971111
105. Song W, Liu C, Upadhyaya A. The pivotal position of the actin cytoskeleton in the initiation and regulation of B cell receptor activation. Biochim Biophys Acta (2014) 1838:569–78. doi: 10.1016/j.bbamem.2013.07.016
Keywords: B cell antigen receptor (BCR), Chemokine receptor 4 (CXCR4), Nanoclusters, Cytoskeleton, B cell malignancies
Citation: Maity PC, Datta M, Nicolò A and Jumaa H (2018) Isotype Specific Assembly of B Cell Antigen Receptors and Synergism With Chemokine Receptor CXCR4. Front. Immunol. 9:2988. doi: 10.3389/fimmu.2018.02988
Received: 12 September 2018; Accepted: 04 December 2018;
Published: 18 December 2018.
Edited by:
Wenxia Song, University of Maryland, College Park, United StatesReviewed by:
Aaron James Marshall, University of Manitoba, CanadaMarcus R. Clark, University of Chicago, United States
Copyright © 2018 Maity, Datta, Nicolò and Jumaa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Palash C. Maity, cGFsYXNoLm1haXR5QHVuaS11bG0uZGU=
Hassan Jumaa, aGFzc2FuLmp1bWFhQHVuaS11bG0uZGU=