 Govindarajan Thangavelu
Govindarajan Thangavelu Bruce R. Blazar
Bruce R. Blazar- Division of Blood and Marrow Transplantation, Department of Pediatrics, Masonic Cancer Center, University of Minnesota, Minneapolis, MN, United States
Acute graft-vs.-host disease (GVHD) limits the efficacy of allogeneic hematopoietic stem cell transplantation (allo-HSCT), a main therapy to treat various hematological disorders. Despite rapid progress in understanding GVHD pathogenesis, broad immunosuppressive agents are most often used to prevent and remain the first line of therapy to treat GVHD. Strategies enhancing immune tolerance in allo-HSCT would permit reductions in immunosuppressant use and their associated undesirable side effects. In this review, we discuss the mechanisms responsible for GVHD and advancement in strategies to achieve immune balance and tolerance thereby avoiding GVHD and its complications.
Introduction
Immunological tolerance is a self-regulatory mechanism of the immune system to protect the host from a wide variety of foreign antigens without causing immunopathology such as autoimmunity (1, 2). The mechanisms of immunological tolerance can be divided into central and peripheral tolerance. Central tolerance involves the clonal deletion of self-reactive lymphocytes in the primary lymphoid organs, namely the thymus and bone marrow. Despite its high efficiency, central tolerance often is incomplete due to the escape of self-reactive lymphocytes into the periphery. Hence, there is need of an additional layer of tolerance in the periphery to suppress self-reactive lymphocytes. Peripheral tolerance mechanisms consist of deletion, anergy, ignorance and immune regulation (2, 3).
Although significant progress has been made toward immunological tolerance induction in experimental animal models, translation to the clinic for allogeneic hematopoietic stem cell transplantation (allo-HSCT) remains challenging. One manifestation of tolerance induction failure in allo-HSCT is graft-vs.-host disease (GVHD), a life-threatening complication due to donor T cell recognition of host alloantigens. During GVHD, conditioning regimen induced tissue injury drives proinflammatory processes that support the priming of donor anti-host alloreactive T cells via T cell receptor (TCR) engagement, co-stimulation and cytokine signaling. These inflammatory events are counteracted by anti-inflammatory processes often augmented by proinflammatory cytokines; however, for those that develop GVHD, it is clear that anti-inflammatory compensatory mechanisms are overwhelmed and hence unable to control T-cell activation, differentiation and expansion (4, 5). This review will focus on acute GVHD pathogenic and tolerance mechanisms including as available clinical trial results and conclude with the concept of tissue tolerance. Since GVHD acquisition is a sign of failed tolerance induction, we will not discuss GVHD therapy.
Overview of Alloreactive T-cell Activation, Amplification and Migration
In allo-HSCT, donor CD4+ and CD8+ T cells can receive TCR signals engagement of peptide- major histocompatibility complex (MHC) (termed signal 1) that occurs as a result of major or minor histocompatibility antigen disparities between donor and host. Studies from mouse models revealed that donor CD4+ T cells play a central role in GVHD induction by exhibiting cytolytic activity, producing effector cytokines and helping donor CD8+ T cells to proliferate via IL-2 production (6).
Upon alloantigen activation, CD4+ T cells differentiate into T helper (Th) cell subsets including, most relevant to this review, Th1 (secreting IL-2, IFN-γ), Th2 (secreting IL-4, IL-5, IL-10, IL-13) and Th17 (secreting IL-17A, IL-17F, IL-21, IL-22, TNF) (6). Our group and others have previously provided evidence against the assumption that GVHD is strictly a Th1 driven process (7–9). In our previous study, deletion of IFN-γ in donor inoculum accelerated GVHD lethality, while deletion of IL-4 resulted in reduced GVHD lethality (7). In other studies, Th2 and Th17 subsets were shown to contribute to GVHD severity with different GVHD target organs (8–10). Recently a subset of CD4+ T cells was found to produce GM-CSF that was linked to the support of GVHD pathology by licensing myeloid cells to produce IL-1 and reactive oxygen species (11).
Similar to CD4+ T cells, CD8+ T cells have been implicated as contributing to GVHD in both major and minor histocompatibility models, the former typically in conjunction with CD4+ T cells and contributing to tissue injury, whereas in the latter, CD8+ T cells alone can be sufficient to cause GVHD (12–15). Similar to CD4+ T cells, CD8+ T cells can also differentiate to cytokine producing subsets including Tc1, Tc2, and Tc17 subsets. These CD8+ subsets possess variable capacities to induce acute or chronic GVHD (cGVHD) (16, 17).
A second or co-stimulatory signal (termed signal 2) then is required for full CD4+ and CD8+ T cell activation, expansion, differentiation, survival, and metabolic fitness. Previous studies (18–22) delineated the role of co-stimulatory molecules including CD28 (18), ICOS (CD278) (19), CD40L (CD154), OX40 (CD134) (20), and 4-1BB (CD137) (21). Co-inhibitory molecules can counterbalance co-stimulatory molecules. Cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4; CD152) (23), programmed death−1 (PD-1; CD279) and its ligand (PD-L1; CD274) (24, 25), B and T lymphocyte attenuator (CD272) (26), and B7-H3 (CD276) (27) have been shown to attenuate GVHD lethality. A third signal provided by inflammatory cytokines such as IL-12 or type 1 interferon is required for optimal CD8+ T cell function (28, 29).
An amplifying component of the immune response is ascribed to conditioning-related tissue damage releases damage-associated molecular pattern (DAMPs) and pathogen-associated molecular pattern (PAMPs) molecules (5). These molecules initiate immune responses during the early phases of GVHD and also provide a source of inflammatory cytokines that drive T cell responses. The role of DAMPs in accelerating GVHD lethality was illustrated by the binding of extracellular ATP and subsequent signaling of the purinergic P2X7 and P2Y2 receptors in host antigen presenting cells (APCs) bolstering donor T cell priming and alloreactive responses (30, 31). Conversely, ecto-nucleotidases such as CD39 and CD73, which regulate extra-cellular ATP levels, play suppressive roles in controlling GVHD (32–34). Other DAMPs such as uric acid, IL-33, heparan sulfate, high-mobility group box 1 protein, sialic acid–binding immunoglobulin-type lectins, mitochondrial components, and biglycans fuel GVHD responses (5).
The role of bacterial components in activating APCs and promoting GVHD via PAMPs is well established (35). For example, lipopolysaccharides are toll-like receptor 4 ligand and are implicated in marshaling innate immunity reactions, NF-κB activation, and transcription of pro-inflammatory cytokines genes (35). DAMPs and PAMPs not only contribute to GVHD initiation but also may augment later allogeneic T cell activation, differentiation, and expansion. Priming of allo-reactive donor T cells most often occurs in secondary lymphoid organs through interaction of the TCR with allo-peptide and MHC antigens expressed on host (termed direct allorecognition) or less often, on donor (termed indirect allorecognition) APCs. Both hematopoietic cells and non-hematopoietic cells are involved in alloantigen presentation that promotes and amplifies GVHD responses (36, 37). Recently, neutrophils have also been shown to exacerbate GVHD lethality by releasing reactive oxygen species in the gastrointestinal (GI) tract and surprisingly up-regulating MHC class II antigens (38, 39).
Chemokines guiding the migration of T cells toward GVHD target organs (40) wherein activated T cells mediate targeted tissue cell death via FAS ligand, perforin/granzymes, and releasing pro-inflammatory mediators mainly tumor necrosis factor (TNF-α), interferon (IFN-γ) (5, 41, 42). Other cytokines such as IL-7, IL-15, and IL-6 directly or indirectly support the expansion or activation of the innate and adaptive immune system and have been implicated in exacerbating GVHD lethality (43, 44). To achieve long term tolerance in allo-HSCT settings, strategies to control T cell activation, differentiation, expansion, and homing are critical to allow anti-inflammatory and central and peripheral regulatory events to be dominant over pro-inflammatory mechanisms. The following sections discuss approaches to blunt the distinct stages of GVHD induction (Table 1).
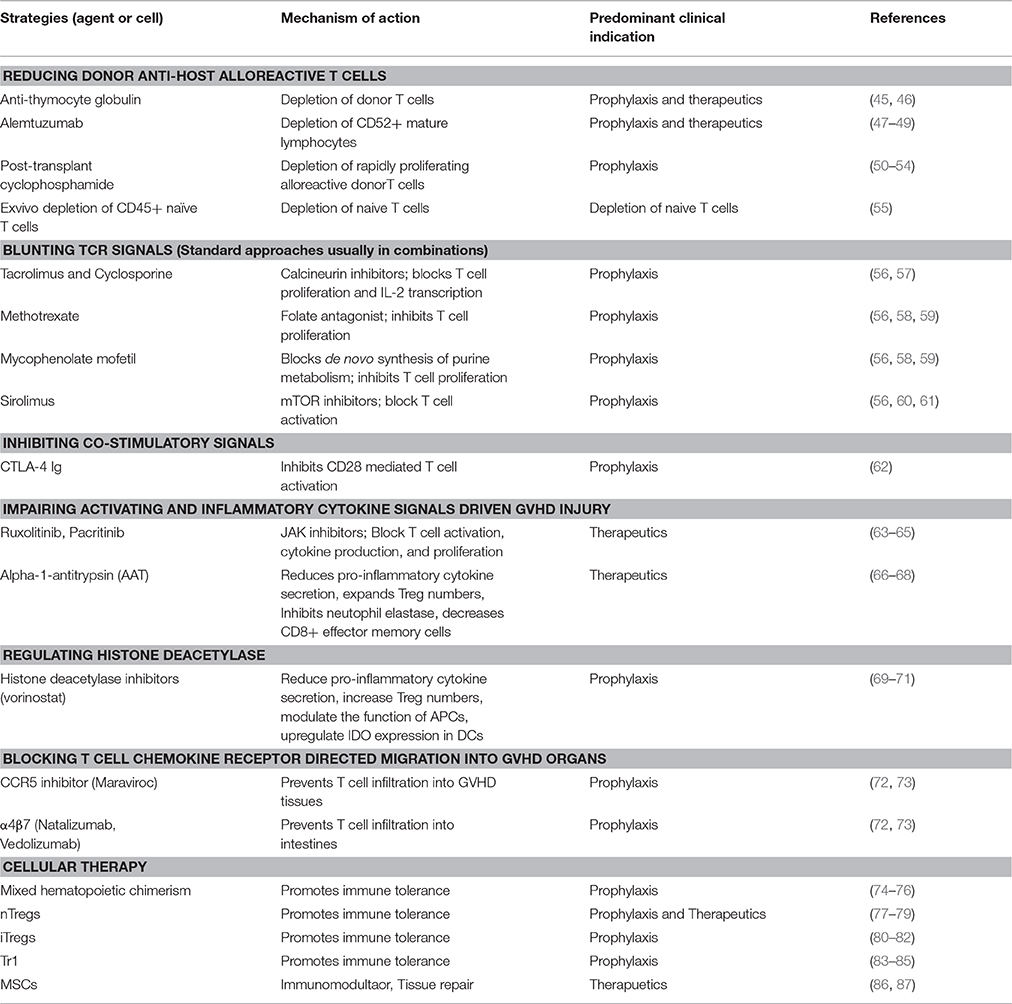
Table 1. Approaches to blunt the distinct stages of GVHD induction.
Reducing Donor Anti-host Alloreactive T Cell Burden
In vitro or in vivo T Cell Depletion
In allo-HSCT, the cellular composition of the graft includes hematopoietic stem cells (HSCs) and a wide variety of cells, which influence engraftment. HSCs restore hematopoietic function, whereas other cell types such as mature T cells promote engraftment by inhibiting graft rejection mediated by recipient immune responses. Although T cells play a central role in the pathogenesis of GVHD, depletion of T cells increases the risk of infection and also of leukemia relapse (88, 89). Donor T cell depletion may be accomplished by in vitro or in vivo strategies. Pan-T cell depletion of the donor grafts can be highly effective but is associated with increased susceptibility to infections and malignancy recurrence due to the relatively long period of time required to reconstitute the immune system (90). In vivo administration of anti-T cell globulin (45, 46) or anti-CD52 mAb, CAMPATH-1 (47–49), reduce the donor T cell burden, while resulting in a state of T cell deficiency.
T cells are broadly classified as naïve vs. antigen experienced memory T cells (TM) (91). Stage of T cell differentiation is a critical factor in determining the capacity of T cells to induce GVHD. For instance, unlike naïve T cells, alloreactive effector and central TM cells failed to induce GVHD in pre-clinical models (92–94). The reduced ability of TM cells to induce GVHD is attributed to their reduced survival, expansion and alloreactivity (95). In a first-in-human trial, depletion of CD45RA+ naïve T cells from peripheral blood stem cells did not reduce the incidence of GVHD (55). Nonetheless, all patients with GVHD uniformly responded to corticosteroids (55). A recent clinical trial (NCT01523223) used a final infusate of highly purified (>94%) CD8+ TM cells to treat relapse after allo-HSCT patients (96). Consistent with the results of pre-clinical models, CD8+ TM infusions are associated with low incidence of GVHD (1 of 15 patients, grade II liver GVHD). Altogether, strategies employing T cell grafts depleted of Tnaive cells may facilitate immune tolerance in allo-HSCT settings by hampering pro-inflammatory responses.
In vivo Post-transplant Cyclophosphamide Induced Alloreactive T Cell Depletion
In a recent approach, cyclophosphamide (Cy) that has both anti-neoplastic and immune modulatory effects, has been used to deplete alloreactive donor T cells and thereby prevent GVHD (50–52). Post-transplant cyclophosphamide (PTCy), typically given for 2 consecutive daily doses between days 3–5 post-transplant in combination with calcineurin inhibitors (CNI) and mycophenolate mofetil (53, 97, 98) or as a single agent (99, 100). Cy, a cytotoxic alkylating agent, specifically targets rapidly proliferating alloreactive T cells because of their impaired ability to replicate their damaged DNA (100–102). On the other hand, Tregs are relatively resistant to PTCy through increased expression of aldehyde dehydrogenase enzyme (103), which converts active to inactive Cy metabolites. The expansion and induction of Tregs promotes peripheral tolerance by suppressing remaining allo-reactive T cells and also hastens immune reconstitution. The final step for achieving long-term tolerance induced by PTCy is mediated by the later stage intrathymic deletion of immature alloreactive donor T cells. In clinical trials, PTCy reduced GVHD in both HLA-matched and partially HLA-mismatched allo-HSCT patients (53, 54). There are multiple ongoing clinical trials (NCT01028716, NCT01349101, NCT01860170, NCT02053545, NCT02065154, NCT02167958, NCT02169791) to investigate the effects of PTCy in conjunction with other agents to prevent GVHD. Overall results of clinical trials have shown a reduction in acute GVHD with a pronounced reduction in cGVHD albeit with organ toxicity, carcinogenicity and increased rates of infections.
Blunting TCR Signals
Standard pharmacological regimens to prevent acute GVHD involve calcineurin inhibitors (CNI), mammalian target of rapamycin (mTOR) inhibitors, and anti-metabolites (5, 56). Calcineurin inhibitors such as tacrolimus or cyclosporine inhibit IL-2 production and subsequently clonal expansion of activated T cells (57). Sirolimus, a lipophilic macrocytic lactone, which binds to FKBP12, and inhibits the mTOR kinase activity, reducing cytokine responses and regulating cell proliferation, survival and metabolism by integrating information from environmental cues including stress signals such as nutrient deprivation (60). TCR, IL-2, CD28, sphingosine-1-phosphate receptor and leptin signals up-regulate the mTORC1 complex. Unlike CNI, sirolimus preferentially supports Tregs generation as Teffectors (Teffs) are mTOR-dependent whereas in vitro or in vivo induced peripheral Tregs and FoxP3 expression are favored by mTORC1 complex inhibition by sirolimus (61). Antimetabolites predominantly methotrexate, a folic acid antagonist and mycophenolate mofetil, an inhibitor of the de novo purine metabolism are being used with other immunosuppressants in allo-HSCT patients (56, 58, 59).
Inhibiting Co-stimulatory Signals
It is well established that the fine-tuned balance between co-stimulation and inhibitory signals dictates immune responses (104, 105). Numerous co-stimulatory and co-inhibitory molecules have been identified and targeted to prevent and reduce various inflammatory diseases including GVHD. Preclinical studies of co-stimulatory and co-inhibitor pathway blockade for GVHD prevention have been comprehensively reviewed recently; the reader is referred to (22). Here we will focus on clinical trial results to prevent GVHD using CTLA4-Ig to block B7/CD28 co-stimulation.
The two-signal model of T cell activation required that both antigen and secondary stimuli are essential for optimal T cell activation (106, 107). The co-stimulatory CD28, identified as a receptor for B7-1 (CD80) ligand and B7-2 (CD86), is constitutively expressed on T cells (108–112). CD28 signals support T cell growth and survival. The co-inhibitory receptor CTLA-4, which also binds to B7-1 and B7-2, serves to temper T cell responses in part by down-regulating CD28 expression.
Linsley and coworkers developed CTLA-4 Ig, consisting of the extracellular CTLA-4 domain, and an immunoglobulin Fc fragment fusion partner to prolong its half-life, as a therapeutic agent that binds and sequesters B7 ligands from CD28 engagement (22). Earlier studies from the 1990s, including from our group, reported the efficacy of CTLA-4 Ig in the prevention of autoimmunity, solid organ allograft rejections and GVHD lethality (113–115) in murine models. The results from these studies laid the foundation for the first clinical trial (NCT01012492) in GVHD using abatacept (humanized CTLA4-Ig fusion protein) that showed a reduced GVHD incidence (62). Phase II studies (NCT01743131) testing the efficacy of abatacept against standard GVHD prophylaxis has been completed for in 7–8/8 HLA matched related or unrelated donor transplants.
The immunomodulatory effect of abatacept was associated with increased expression of PD-1 on T cells of the clinical responders. The role of the PD-1 pathway in inducing immune tolerance and controlling acute GVHD has been well established (24, 116). Although the beneficial effect of abatacept depends on blocking CD28 co-stimulation, it can also interfere with the endogenous CTLA-4 co-inhibition pathway and can lead to unwanted immune responses (117). The advent of fusion proteins or antibodies that block only the CD28 pathway without interfering with CTLA-4 may have an edge over abatacept due to their specificity. Belatacept, a 2 amino acid derivative of abatacept, was developed as a selective co-stimulatory pathway blocker, that has favorable results in renal transplant rejection compared to cyclosporine prophylaxis (118). A CD28 antagonistic antibody, FR104, has been tested in various pre-clinical models (119–121). More recently, in a non-human primate (NHP) GVHD model, compared to CTLA4-Ig or CTLA4-Ig/sirolimus prophylaxis, FR104 or combined FR104/sirolimus prophylaxis delayed the onset of GVHD by controlling T cell activation and proliferation (122). However, there were non-GVHD-related deaths in the FR104/sirolimus-treated NHP due to sepsis. Detailed immunological analysis revealed that T cells from those primates failed to produce IFN-γ. The results from this study still highlight FR104/sirolimus combination as a promising therapy to treat GVHD in human patients due to better infection control compared to NHP.
Impairing Activating and Inflammatory Cytokine Signals Driven GVHD Injury
Immune Activating Cytokines Contributing to GVHD
JAKs are intracellular tyrosine kinases and act as downstream of cytokines, growth factors and hormone signaling. The JAK family members comprises JAK1, JAK2, JAK3, and TYK2 (123). JAK signaling supports the development, proliferation, and activation of T- and B- cells, DCs, macrophages, and neutrophils, all implicated in GVHD pathogenesis.
Ruxolitinib, a selective inhibitor of JAK1 and JAK2 reduced GVHD, associated with decreased proinflammatory cytokine production, Th1 differentiation and increased Tregs proportions (124, 125). Although ruxolitinib has been primarily reported as a treatment for steroid refractory or resistant GVHD (63), a recent study in myelofibrosis patient reported that ruxolitinib, given during peritransplant period, can reduce GVHD (64). Overall, 1 out of 12 patients developed severe (grade III) GVHD without major events during conditioning. However, CMV reactivation was seen in 4 of 6 CMV positive patients and 2 had cytopenias requiring ruxolitinib discontinuation (64). In other studies (126), baricitinib, a best-in-class Jak1/2 inhibitor, blunted IFNγR and IL6R signaling, resulting in complete protection from GVHD lethality as well as the reversal of active GVHD prevents GVHD with 100% survival, and reverses ongoing GVHD with dramatically increased Tregs along with decreased Th1 and Th2 differentiation, MHC class II and B7 ligand expression on APCs (126). Pacritinib is a potent JAK2 inhibitor that can reduce GVHD by sparing iTregs and polarizing T cells toward Th2 differentiation (65). A phase I/II trial (NCT02891603) combining pacritinib with standard immune suppression to prevent GVHD is currently being investigated.
Tofacitinib, a first generation JAK1/JAK3 inhibitor, reduced murine GVHD lethality (127). Antibodies directed to the IL2R common gamma chain that signals via JAK3 and STAT5 reduce proinflammatory cytokine production, CD8+ T cell granzyme B expression and severe GVHD lethality (128). Indeed JAK3 knockout T cells were unable to cause GVHD mortality in sublethally irradiated MHC class II disparate recipients. Pharmacological JAK3 inhibition with WHI-P131 given as prophylaxis ameliorated GVHD severity with a prolonged survival when compared to control mice (129). As many of these reagents are in the clinic including for GVHD prevention they may become part of an in vivo approach to achieve tolerance.
Alpha-1-antitrypsin to Reduce Pro-inflammatory Responses Post-transplant
Alpha-1-antitrypsin (AAT) is an acute phase secretory protein and a serine proteinase inhibitor, elevated during inflammation due to its predominant synthesis in hepatocytes (130, 131). Numerous lines of evidence demonstrated the anti-inflammatory properties of AAT. Studies have shown that the deficiency of AAT aggravated the severity of inflammatory disease, whereas addition of AAT to LPS-stimulated monocytes or mononuclear cells inhibited the release of pro-inflammatory cytokines (132–134). In GVHD patients, there was a negative correlation between AAT levels in donor plasma and occurrence of GVHD (135). Indeed, AAT treatment attenuated the lethality of GVHD in pre-clinical murine models by both increasing IL-10 levels and numbers of Tregs, and reducing the levels of pro-inflammatory cytokines such as IL1-β, TNF-α, and IL-6 (66, 135, 136). This tolerogenic effect of AAT, which induced Tregs expansion, was mediated by an increase in the numbers of CD8+ CD205+ DCs (135). AAT strongly inhibits neutrophil elastase and that may also contribute to reduced GVHD lethality due to the pathogenic role of neutrophils in GVHD (38). In clinical trials (NCT01523821 and NCT01700036), AAT treatment increased the proportion of Tregs and reduced GVHD manifestations (67), while decreasing numbers of CD8+ TM cells (68) in steroid refractory (SR) GVHD patients without clinical toxicity.
Regulating Histone Deacetylase
Histone acetylation epigenetically regulates cell function by modulating gene expression. Acetylation is often associated with transcription activation, while deacetylation is associated with repression. The interplay between histone acetyltransferases (HATs) and histone deacetylases (HDAC) influences histone acetylation to impact numerous cellular functions, including cell differentiation, and apoptosis (56). HDAC inhibitors (HDACi) function an anti-inflammatory agents in autoimmune and inflammatory disorders (137). HDACi, namely vorinostat (SAHA), romidepsin (Istodax) and panobinostat (LBH589), are FDA-approved agents to treat cancers. HDACi treatment ameliorated murine GVHD through upregulation of indoleamine 2,3-dioxygenase (IDO) in DCs, in a STAT-3-dependent pathway (138, 139). Trytophan depletion and/or the generation of tryptophan catabolites has proven to be immune suppressive for murine GVHD (140, 141) as discussed in detail below. A completed phase I/II clinical trial (NCT00810602) of vorinostat with standard GVHD prophylaxis in patients who received matched related donor allo-HSCT reported reduced GVHD with lower levels of plasma IL-1β, TNF-α, IL-6, and IL-8 (69–71). Furthermore, HDACi treatment increased Treg cell numbers and enhanced their function in those patients (71). Extending this treatment to unrelated donor HCT (NCT01790568) also showed vorinostat to be result in a low rate of GVHD (70).
Blocking T Cell Chemokine Receptor Directed Migration Into GVHD Organs
Chemokine receptors control the trafficking of T cells into tissues, where they may be primed, re-stimulated in the case of memory T cells, or cause cytolysis and tissue destruction. Chemokines produced by tissues injured by the conditioning regimen or GVHD itself may result in the elaboration of chemokines that direct the recruitment of specific innate and adaptive immune cells. Chemokine and chemokine receptor interactions that can influence GVHD pathogenesis have been reviewed (142). For example, during tissue damage, the up-regulation of CCR5 directs lymphocyte homing to the inflamed intestine and liver tissues (143–146). In mouse GVHD models, the efficacy of CCR5 blockade was dependent upon the degree of conditioning regimen injury. Whereas, anti-CCR5 mAb prevented T cell homing to Peyer's patches in the absence of conditioning (146), GVHD was accelerated with lethal radiation conditioning due to increased T cell expansion, IFN-γ and TNF-α production, and infiltration into the liver and lung (144, 146). In patients, reduced CCR5 expression correlated with lower GVHD (147, 148). Short-term addition of CCR5 antagonist, maraviroc added to standard GVHD prophylaxis resulted in reduced GI and liver GVHD in allo-HSCT patients given reduced intensity conditioning (72). Compared to this short-term treatment of 1 month (72), the extended course of maraviroc (3 months) was also safe and resulted in a significantly improved survival and higher GVHD-free (73). The relationship between conditioning regimen intensity and efficacy of CCR5 antagonism in allo-HSCT patients is unknown and warrants investigation.
Studies have demonstrated that the expression of gut-homing molecules, including α4β7-integrin and chemokine receptor CCR9, by T cells is required for homing to the intestines. GI injury due to conditioning is a key trigger for GVHD pathogenesis and results in the homing of donor T cells to the injured GI tract. Natalizumab is a potential drug of interest to mitigate GI GVHD due to its selective inhibition against α4 integrins of α4β7. Natalizumab and vedolizumab, a specific anti-α4β7 integrin monoclonal antibody, have been used in for GVHD treatment but not prevention, which have distinct cellular infiltrates and pathophysiologies (149, 150). Homing receptor blockade may potentiate tolerance induction in allo-HSCT as GVHD by precluding immune cell recruitment into GVHD organs and amplification of tissue injury.
Regulating GVHD by Exploiting Cellular Metabolism Mechanisms
Intrinsic T Cell Metabolic Energy Sources Required for GVHD
One way to tailor immune tolerance is to change the metabolic fitness. Immune cells require considerable bioenergy to generate and sustain immune responses against pathogens, allografts, and tumor cells. To accomplish these effector responses, immune cells utilize multiple metabolic pathways. The major metabolic pathways involved in cellular growth and proliferation are tricarboxylic acid (TCA) cycle, glycolysis, amino acids, pentose phosphate, fatty acid synthesis and oxidation (151, 152). Despite their diverse end products, these pathways are interdependent as biosynthesis of one pathway depends on the intermediate products of other pathways.
The TCA cycle takes place in the mitochondria to generate energy through oxidation of acetyl CoA, which is derived from sources such as glucose, fatty acids (FA) and glutamine (151, 152). The end products of the TCA cycle, namely NADH and FADH2 contribute electrons into the electron transport chain (ETC). The ETC is involved in highly efficient ATP generation by supporting oxidative phosphorylation (OXPHOS). Metabolically quiescent cells, like naive T cells, generate energy via OXPHOS by fueling TCA cycle with the available nutrients. However, upon cognate antigen encounter, T cells undergo a metabolic switch from OXPHOS to glycolysis to meet their energy needs (151–153). In glycolysis, extracellular glucose enters the cell through glucose transporters followed by the sequential conversion of glucose to pyruvate and other products by different enzymes. The availability of oxygen in the cell influences the fate of pyruvate. In the case of hypoxia, pyruvate is converted to lactate and NAD+. However, in normoxia, pyruvate is oxidized through the TCA cycle. Glycolysis plays a crucial role in cellular metabolism by providing precursors to other metabolic pathways. For example, cytoplasmic acetyl-CoA, a metabolite of glycolysis, promotes lipid synthesis by generating cholesterol and fatty acids. In a preclinical model, donor T cells shown to increase oxidative phosphorylation in both syngeneic and allogeneic recipients (153, 154). Glycolytic activity was only higher in donor T cells of allogeneic recipients than those of acute GVHD controls or syngeneic BMT recipients, indicating that Teffector cells causing GVHD are more dependent upon glycolysis (154–156). Pharmacological inhibition of mTORC1 or a phosphofructosekinase-2 isoform PFKB3 reduced GVHD lethality (154). Moreover, mice given T cell deficient in the glucose transport glut-1 were unable to induce GVHD (157).
Apart from glycolysis, glucose can also be metabolized via the pentose phosphate pathway (PPP) and glycogen synthesis (151). PPP is comprised of oxidative and non-oxidative branches. The oxidative branch of PPP maintains the cellular redox environment by generating reducing equivalents of NADPH. Whereas, the non-oxidative branch supports cell proliferation by generating required nucleotide and amino acid precursors (151). During GVHD, PPP activity of alloreactive T cells was increased (154).
Fatty acid oxidation (FAO) generates energy by converting FA to acetyl CoA, which enters into the TCA cycle (151, 153). In addition, FAO supports the ETC production of ATP by generating NADH and FADH2. Short and medium chain FA passively diffuse into the mitochondria, whereas the carnitine palmitoyl transferase (CPT) system regulates long chain FA (C14 to C18) metabolism (158). Discordant results have been reported about the activity of FAO in alloreactive T cells with some studies reporting increased FAO (155, 159), while a recent one demonstrated diminished FAO (154). FA synthesis plays a crucial role in sustaining T cell proliferation by generating lipids through utilization of products derived from other metabolic pathways (151, 153). Lipid synthesis is regulated by enzymes such as acetyl-CoA carboxylases (ACC 1 and 2) and fatty acid synthase (151, 153, 154). Deficiency of ACC1 in donor T cells ameliorated GVHD due to impaired de novo FA synthesis (160). Sphingolipids are major components of eukaryotic cell membranes and play a crucial role in cellular survival, proliferation, differentiation and growth arrest. A recent study reported that ceramide, a metabolite of sphingolipids, modulate GVHD lethality (161). Targeting ceramide synthase 6, a ceramide biosynthetic enzyme, by either genetic deletion in donor T cells or pharmacological inhibition ameliorated GVHD due to reduced donor T cell proliferation and Th1 differentiation (161).
Glutamine, a key amino acid and readily available resource in serum, is required for T cell activation (5). Glutamine is involved in nucleotide synthesis and its metabolite glutamate also fosters the TCA cycle, glutathione and amino synthesis (151, 153). In allo-HSCT, donor T cells upregulated glutamine transport channels namely glutaminase 2, phosphoribosyl pyrophosphate amidotransferase, and glutamine-fructose-6-phosphate transaminase to increase the uptake of glutamine (25, 154). Furthermore, only donor T cells from allo-HSCT had increased levels of glutamate products aspartate and ornithine, which indicate that donor T cells can restore the exhausted intermediates of TCA cycle by increasing glutaminolysis (153, 154). Based on these studies, strategies inhibiting glycolysis, fatty acid oxidation, oxidative phosphorylation, or glutaminolysis may be an area of great potential to control GVHD (5).
Extrinsic Regulation of Cellular Metabolism in GVHD
A defense mechanism against GVHD lethality can be conferred by essential amino acid depletion results in a state of metabolic starvation. For example, high host tissue expression of IDO that catabolizes and hence depletes L-tryptophan was critical to reduce colonic GVHD (140). Donor T cell-derived IFN-γ upregulated the expression of IDO in colonic epithelial cells which in turn, diminished T cell proliferation and inflammation (141). Similarly, IDO expression was upregulated in the duodenal epithelial cells of GVHD patients and may be involved in the control of GI GVHD (162). The metabolic products of tryptophan catabolism has been shown to be immune suppressive. Whereas, combined administration of three tryptophan metabolites suppressed GVHD, kynurenines given in this way did not appear to be tolerogenic since GVHD was controlled only during the continuous administration period (141). In other studies, arginine depletion by myeloid-derived suppressor cell production of arginase I or infusion of pegylated L-arginase I was shown to reduce the vigor of the GVHD lethality response (163).
Mammalian hosts harbor a large number and a wide variety of commensal bacteria on surfaces of the body, especially in the GI tract. Commensal bacterial density in the GI ranges from 1011 to 1014 per gram of luminal content (164). The interaction between GI commensals and host immune cells plays a critical role in the development of the immune system and the maintenance of intestinal immune homeostasis. For example, germ-free mice have impaired immune systems with smaller Peyer's patches, lower numbers of IgA-producing plasma cells and lower numbers of CD8+ intraepithelial cells (165, 166). Dysregulation of GI microbiota has been associated with various inflammatory diseases (167–169).
In addition to metabolizing host dietary components, microbes produce their own metabolites that can have substantial immune system effects (170–173). In GVHD mice (174, 175) and patients (176–178), the diversity of the intestinal microbiota is significantly altered, which can be associated with the lethality of the disease. For example, butyrate, a short chain fatty acid microbial metabolite is an HDACi serves as the main energy source of intestinal epithelial cells (IECs) (179, 180). In a mouse model of GVHD, the reduction in intestinal butyrate resulted in decreased histone acetylation within CD326+ IECs (181). Administration of exogenous butyrate mitigated GVHD by increasing both anti-apoptotic and junctional proteins of IECs. This beneficial effect was not found to be mediated by donor Tregs, however the role of host Tregs in this model remains to be explored. Similarly, intragastric gavage of 17 rationally-selected strains of high butyrate–producing Clostridia also reduced GVHD and improved survival (181). A clinical trial (NCT02763033), which aims to increase butyrate levels in the intestines using dietary supplements containing potato-based resistant starch, is ongoing. Overall, these results demonstrate that HDACi can mitigate GVHD lethality.
In addition to butyrate, a recent study (182) reported that allo-HSCT conditioning regimens reduced indole or indole derivatives due to altered intestinal microbiome. Importantly, either supplementation with exogenous indole derivative or colonization of bacteria that can deliver indole metabolites into intestines of allogeneic murine recipients ameliorated GVHD lethality with reduced mucosal damage and pro-inflammatory cytokines (182). Beyond GVHD amelioration, recipient-specific tolerance was developed in donor T cells of recipients that were administered with the tryptophan metabolite and indole derivative, indole-3-carboxaldehyde, found in foods such as collard greens and broccoli.
Tissue Tolerance Mechanisms
It is a known fact that the survival of host against infections depends on the capacity of host's immune system. Recently, the role of parenchymal tissues on reducing disease severity and protecting from immunopathology has been gaining attention as tissues can modulate immune responses (183, 184). In non-infectious disease settings like GVHD, tissue tolerance is defined as an intrinsic and protective mechanism of parenchymal tissue to ameliorate GVHD against ongoing alloimmune responses. Studies from our laboratory demonstrated that the expression of the co-inhibitory molecule by parenchymal tissues promoted tolerance and reduced the lethality of GVHD. Experimental evidence has demonstrated the increased expression of co-inhibitory molecules such as programmed death-1 ligands and B7-H3 on T cells in GVHD targeted tissues (27, 156). The absence of these molecules accelerated GVHD lethality due to augmented T cell effector responses. Thus, co-inhibitory pathways induced during alloresponses serve to dampen alloreactive donor T cell responses and hence GVHD.
Reparative Processes
Emerging data suggest that tissue tolerance can also be mediated through the regeneration of damaged tissues. In a recent study, administration of the Wnt-agonist R-spondin1 mitigated GVHD by protecting intestinal stem cells (ISC) and facilitating repair of the intestinal epithelium (185). In line with this finding, IL-22, which has been shown to activate ISC, enhanced intestinal epithelial regeneration and ameliorated GVHD (186). A phase I/II clinical trial (NCT02406651) is currently investigating the safety and efficacy of use of IL-22 in combination with corticosteroids for the treatment of patients with newly diagnosed GI GVHD.
Tregs and innate lymphoid cells 2 (ILC2) aid tissue repair by secreting amphiregulin, an epidermal growth factor that promotes tissue repair under inflammatory conditions (187). Non-lymphoid cells, in particular, mesenchymal stem cells (MSCs), have also been shown to facilitate tissue repair by polarizing tissue macrophages to the anti-inflammatory phenotype (188). These macrophages help repair tissues through enhanced fibroblastic proliferation and also reduce donor T cell proliferation and so limit GVHD. Furthermore, MSCs promote tissue repair by increasing the proliferation of ILC3 and their subsequent IL-22 production (189). Overall, strategies harnessing tissue tolerance represent a novel and expanding area of research in GVHD.
Cellular Therapies
Infusions of tolerogenic cells are one of the most attractive strategies to achieve long-term immune tolerance in clinical studies due to the long-term persistence of those cells. There are numerous immunoregulatory cells that have been used to induce transplantation tolerance in clinical models, but herein we will focus on Tregs, invariant natural killer T (iNKT) cells (see also Dominik Schneidawind's chapter) and MSCs.
Mixed Hematopoietic Chimerism and Tolerance Induction
Mixed hematopoietic chimerism also has been shown to be to facilitate kidney and liver solid organ graft acceptance in mice and humans (190–193), with high levels causing central deletional tolerance albeit at the risk of GVHD and transient chimerism allowing for peripheral tolerance that begins with Treg mediated mechanisms and transitions into peripheral tolerance likely including deletion of donor alloreactive T cells (74, 194–196). While transient T cell chimerism in hematological malignancy patients can decrease GVHD (75), mixed donor T cell chimerism present on day 90 in allo-HSCT patients receiving a reduced intensity conditioning regimen did not preclude GVHD generation; however the incidence was significantly lower than those with full donor T cell chimerism (35 vs. 61%), providing a platform upon which to tolerance induction may be more likely to be achieved (76).
T Regulatory Cell Infusion for Tolerance Induction
Tregs play a crucial role in maintaining immune homeostasis and tolerance by preventing autoimmunity and immunopathology. Tregs may be derived from the thymus (thymic-derived or natural Tregs (tTregs or nTregs), peripherally derived Tregs (pTregs), and in vitro induced Tregs (iTregs) (197). In this review, we will focus on both basic and clinical studies using different subsets of Tregs for the prevention of GVHD and discuss their limitations.
Thymic-Derived Tregs
Phenotypic features of tTregs include the constitutive expression of CD25, the high- affinity IL-2 receptor, CTLA-4, and Forkhead box P3 (Foxp3), a lineage transcription factor. Adoptive transfer of tTregs has been demonstrated to control allograft rejection and GVHD by limiting alloimmune responses (198–200). Preclinical studies have shown a high efficacy of Treg infusion and GVHD prevention (201–203). In allo-HSCT patients, there was an inverse correlation between Treg frequency and risk of acute GVHD (204).
Translation to the clinic proved challenging due to the low frequency of tTregs (typically 2–3% of CD4+ T cells) in the peripheral blood (205). The phenotypic profile of human tTregs was not as readily demarcated in peripheral blood as in the spleen and lymph nodes of mice. Moreover, compared to non-Treg T cells, Tregs were found to be hyporesponsive resulting in poor expansile properties and a preponderance of contaminating non-Tregs even when the latter represented a minor proportion of input cells. The first acute GVHD prevention clinical studies were reported by two groups (77, 78). In our study (77), umbilical cord blood cells were used as a source of tTregs (NCT00602693). Advantages included ease of tTreg isolation as a result of higher frequency of CD4+CD25bright cells and reduced likelihood of CD25+ Teffs contamination due to fetal microenvironment that minimizes external antigen exposure. Ex vivo expansion permitted tTreg activation, maximizing expansion and suppressor function. GVHD was reduced but not eliminated at Treg:Teff ratios of ≤1:6 in patients receiving cyclosporine A or sirolimus and mycophenolate. In the second study by our group, tTreg expansion was dramatically increased by restimulating tTregs with cell-based artificial antigen presenting cells and when given to patients receiving sirolimus and mycophenolate mofetil, GVHD was virtually eliminated (79). In the study by Martelli and coworkers (78), tTregs were freshly isolated from peripheral blood and allowed to become activated and expanded in vivo prior to the infusion of haploidentical T cells and in the absence of post-transplant immune suppression. GVHD was very low considering the high T cell dose given. Since tTregs could not be detected in peripheral blood beyond ~2 weeks post-transplant, these studies suggest that tTregs have tolerized the donor T cell graft. In other studies, antigen-specific tTregs have been generated and expanded in vitro in rodents (206) and are being tested in the clinic for GVHD prevention.
Tregs rely on IL-2 for their generation, proliferation, lineage stability and survival; however, they are poor producers of IL-2 (207). In patients, ultra-low dose IL-2 given as GVHD prophylaxis days 7–30 resulted in Treg expansion in vivo and no instances of GVHD in 16 pediatric allo-HSCT recipients (208). In patients with cGVHD, low dose IL-2 administration ameliorated cGVHD lethality by preferentially allowing in vivo Treg expansion, increasing the Treg:Teff ratio and thus favoring tolerance (209, 210). Since both Tregs and activated Teffs respond to IL-2, it is currently unknown whether these studies can be extrapolated to the higher risk adult population, which may be benefitted by more selective Treg expansion approaches. For example, two recent studies employed novel approach to selectively target Treg expansion. In one of the studies (211), investigators engineered IL-2 cytokine-receptor orthogonal pairs that interact with one another but not with their natural cytokine and receptor counterparts. Introduction of a mutated IL-2Rβ into T cells that preferentially binds orthogonal but not natural IL-2 enabled the selective cellular targeting of engineered T cells in vitro and in vivo, with limited off-target effects and negligible toxicity, suggesting a clinical strategy to selectively target Tregs in vivo in patients. In a different study, the same group employed complexes of human IL-2 with a unique conformational structure that stabilized IL-2 and promoted preferential STAT5 phosphorylation and Treg expansion (212).
In a non-IL-2 based approach, investigators have used reagents that stimulate death receptor 3 (DR3, TNFRSF25), a member of the tumor necrosis factor (TNF) receptor superfamily primarily expressed on Tregs, lymphoid tissue inducer cells, and NKT cells (213). The natural ligand of DR3, TL1a, is expressed on endothelial cells and APCs (213). An agonistic αDR3 mAb significantly expanded Tregs in vivo and prevented the development of allergic lung inflammation (214) and cardiac allograft survival by increasing the proportion of Tregs (215). Treating donors with αDR3 preferentially allowed Tregs expansion with reduced Tcon activation and those donor T cells reduced GVHD (216). A key role of TNF binding to TNFR2 was discovered to be critical to Treg control of GVHD (217, 218). Collectively, strategies to increase the tTreg/Teffs in vivo represent a promising therapeutic option to reduce GVHD and remain an active area of research.
Inducible Tregs (iTregs)
Although tTreg cellular therapy has great potential in controlling GVHD, higher doses of Tregs are required and it has been challenging to achieve uniform and robust tTreg expansion in clinics. Generation of iTregs is an alternative strategy to overcome the obstacle of limited nTreg cell numbers. Previous studies have established the potency of iTregs in controlling various autoimmune disorders (219, 220). In an experimental GVHD study, antigen-specific iTregs were generated and they were able to reduce GVHD by inhibiting the activation, proliferation and migration of donor T cells (221). The methylation status of the Treg-specific demethylated region (TSDR) of the Foxp3 promoter determines the stability of Tregs by maintaining the stable expression of Foxp3 (222, 223). Unlike tTregs, iTregs are completely methylated at the TSDR and tend to be unstable in GVHD mice (80, 224). Hence, studies have attempted to use various agents such as rapamycin, retinoic acid, and IL-6 blockade to induce and maintain iTregs (224). However, only the usage of sirolimus both in vitro and in vivo was shown to improve CD4+ iTreg stability in a mouse model of GVHD (80). Given the role of iTregs in controlling GVHD, there is an ongoing phase-I trial (NCT01634217) initiated by our institute to test the safety of CD4+ iTregs, generated using sirolimus, TGF-β, and IL-2, when given as GVHD prophylaxis to matched sibling donors along with CNI and mycophenolate thereby reducing the inflammatory environment. Intriguingly, CD4+ tTregs and iTregs were shown to be synergistic in controlling colitis in mice (81). A previous study reported that CD8+ iTregs can be induced by activating CD8+ CD25- T cells with allogeneic CD11c+ DCs, IL-2, TGF-β and retinoic acid. Although CD8+ iTregs expressed higher levels of suppressive molecules like CD39+CD73+, CTLA-4, and granzyme than CD4+ iTregs, there was no difference observed between their in vitro suppressive functions (82). In contrast to their in vitro suppressive functions, CD8+ iTregs are less potent than CD4+ iTregs in controlling GVHD due to their pro-apoptotic phenotype and thus reduced survival but are more effective in eliminating leukemia cells (82, 225). Intriguingly, CD8+ iTreg expression of FoxP3 can be stabilized by JAK2 targeting (226).
Type 1 T Regulatory (Tr1) Cells
Type 1 T regulatory (Tr1) cells are a distinct pTreg subset discovered by Bacchetta and Roncarolo and colleagues in severe combined immune deficiency patients who did not develop GVHD but had anti-host reactive T cell clones that produced high IL-10 and low IL-2 protein (227, 228). Tr1 cells lack constitutive expression of Foxp3, and have been shown to exert immune tolerance mainly via production of cytokines such as IL-10 and TGF-β (229, 230) that can inhibit murine GVHD lethality (228).
Using novel transgenic mice, Hill's group recently reported that Tr1 cells are the dominant immunoregulatory cells after allo-HSCT due to defective tTreg homeostasis (231). Infusion of Tr1 cells reduced GVHD, while Tr1 deficiency aggravated GVHD lethality. Murine and human Tr1 cells are typically generated by alloantigenic stimulator cell exposure in the presence of high IL-10 (83, 228, 229, 232). As with antigen-specific tTregs, Tr1 cells may have a reduced capacity for global immunosuppression due to their allospecificity. A recently completed phase-I trial demonstrated the feasibility of host-specific donor Tr1 therapy in GVHD patients. Infusions of Tr1 cells reduced GVHD, enhanced immune reconstitution and promoted tolerance induction (84). There are ongoing clinical trials testing the efficacy of Tr1 cell therapy in controlling autoimmunity and other inflammatory disorders (230).
Invariant Natural Killer T (iNKT) Cells
iNKT cells are a rare lineage of immunomodulatory cells and they produce large quantities of anti-inflammatory cytokines such as IL-4 and IL-10 (85). Numerous lines of evidence have highlighted the potency of iNKT cells in promoting immune tolerance in GVHD (233). Studies from the early 2000s demonstrated that a combined regimen of fractionated total hematopoietic irradiation and depletion with anti-T cell antibodies reduced GVHD in rodent models (234, 235). The protective effect against GVHD was meditated by the expansion of host immunoregulatory iNKT cells, which secreted IL-4 and supported donor Treg proliferation (234–236). Pharmacological approaches to expand iNKT cells, using a synthetic iNKT TCR ligand, α-galactosylceramide (alphaGalCer), also attenuated GVHD (237). An important consideration in these studies was the usage of reduced conditioning regimens that may help in the survival of host iNKT cells and their expansion. However, using lethally irradiated GVHD mouse models, Negrin's group demonstrated that the lethality of GVHD could be mitigated by adoptive transfer of low numbers of recipient-type, donor-type, or third party iNKT cells (238–240). These studies shed light on the role of iNKT cells in expanding both donor Tregs and myeloid derived suppressor cells (MDSCs). Interestingly, the protective effects of iNKT cell and donor Treg expansions were dependent on MDSCs and thus, crosstalk between these distinct cell populations promoted immune tolerance in GVHD settings. Results from these experimental models led to the initiation of a phase-II trial (NCT01379209) in GVHD patients. This clinical trial used a single dose of RGI-2001, the liposomal formulation of α-GalCer to expand iNKT cells. While there was reduced GVHD and increased expansion of Tregs observed in some patients, iNKT cells were very low in number and difficult to detect in the peripheral blood (241). Clinical studies testing infusions of iNKT cells hold promise to control GVHD.
Mesenchymal Stem Cells (MSCs)
Therapeutic infusions of MSCs are one of the leading options to treat GVHD. Although MSCs are rare non-hematopoietic cells in bone marrow, these cells are easy to isolate and can be expanded rapidly in vitro due to their multipotent and self-renewable properties (205). Immunomodulatory effects of MSCs in attenuating GVHD are mediated by secretion of cytokines (IL-6, TGF-β), soluble receptors (PDL-1, PDL-2) and effector molecules (nitric oxide, PGE2). MSCs also downregulate a wide range of chemokine (CCL1, CCL3, CCL8, CCL17, CCL22) expressions on donor T cells to limit T cell effector migration into target tissues (86, 205). The suppressive capacity of MSCs is enhanced by IFN-γ produced during GVHD, which up-regulates PDL-1 and IDO expression on MSCs to control T cell activation (205, 242). In other studies, high host anti-donor cytotoxic T lymphocyte (CTL) activity serves to eliminate donor MSCs and at the same time induce IDO and immune suppression by perforin-dependent host CTL mediated donor MSC apoptosis (243). Additionally, MSCs participate in the reparative process of tissue by promoting angiogenesis, regeneration, and remodeling (205). These properties have led to multiple clinical trials (NCT03158896, NCT00284986, NCT00361049, NCT00366145, NCT02336230) exploring the use of MSC infusion as an adjunctive strategy for GVHD prevention (87).
Concluding Remarks
Recently, there have been significant advances in the field of allo-HSCT to treat GVHD. Early phase studies involving AAT, HDACi and co-stimulation blockade have shown promising results, although randomized clinical trials and longer follow-up will be required to validate these existing results. Adoptive cellular therapies are powerful strategies to achieve peripheral tolerance swiftly in allo-HSCT recipients by blunting the inflammatory component of GVHD. Clinical trials using tTregs have reported promising results, but the long-term effects of Tregs on immune responses against infections and tumors have yet to be determined. To reduce non-specific immunosuppression and increase potency of antigen-specific suppression, generation of antigen-specific Tregs by a variety of approaches including engineering Tregs using chimeric antigen receptors (CAR) or designated T-cell receptors reactive against antigens present in GVHD organs may be an attractive approach. The first clinical trial evaluating CAR Treg therapy in the prevention of organ transplant rejection is expected to start by next year. Gene augmentation and gene editing techniques may be employed to direct Tregs to particular GVHD organs such as the gut or to increase Treg stability under inflammatory conditions. Renewed efforts are required to gain insight into tolerance induction in allo-HSCT and to develop safe and effective strategies to combat GVHD.
Author Contributions
GT wrote the manuscript. BB assisted in the writing and editing of the manuscript.
Funding
This work was supported by grants from the National Institutes of Health, National Institute of Allergy and Infectious Diseases, National Heart, Lung, and Blood Institute, and National Cancer Institute R01 HL56067, HL1181879, and AI34495 (BB). GT was supported by the Canadian Institutes of Health Research (CIHR) fellowship.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Luo X, Miller SD, Shea LD. Immune tolerance for autoimmune disease and cell transplantation. Annu Rev Biomed Eng. (2016) 18:181–205. doi: 10.1146/annurev-bioeng-110315-020137
2. Bluestone JA. Mechanisms of tolerance. Immunol Rev. (2011) 241:5–19. doi: 10.1111/j.1600-065X.2011.01019.x
3. Ezekian B, Schroder PM, Freischlag K, Yoon J, Kwun J, Knechtle SJ. Contemporary Strategies and Barriers to Transplantation Tolerance. Transplantation. (2018) 102:1213–22. doi: 10.1097/TP.0000000000002242
4. Zeiser R, Blazar BR. Preclinical models of acute and chronic graft-versus-host disease: how predictive are they for a successful clinical translation? Blood. (2016) 127:3117–26. doi: 10.1182/blood-2016-02-699082
5. Zeiser R, Blazar BR. Acute graft-versus-host disease - biologic process, prevention, and therapy. N Engl J Med. (2017) 377:2167–79. doi: 10.1056/NEJMra1609337
6. Coghill JM, Sarantopoulos S, Moran TP, Murphy WJ, Blazar BR, Serody JS. Effector CD4+ T cells, the cytokines they generate, and GVHD: something old and something new. Blood. (2011) 117:3268–76. doi: 10.1182/blood-2010-12-290403
7. Murphy WJ, Welniak LA, Taub DD, Wiltrout RH, Taylor PA, Vallera DA, et al. Differential effects of the absence of interferon-gamma and IL-4 in acute graft-versus-host disease after allogeneic bone marrow transplantation in mice. J Clin Invest. (1998) 102:1742–8. doi: 10.1172/JCI3906
8. Yi T, Chen Y, Wang L, Du G, Huang D, Zhao D, et al. Reciprocal differentiation and tissue-specific pathogenesis of Th1, Th2, and Th17 cells in graft-versus-host disease. Blood. (2009) 114:3101–12. doi: 10.1182/blood-2009-05-219402
9. Nikolic B, Lee S, Bronson RT, Grusby MJ, Sykes M. Th1 and Th2 mediate acute graft-versus-host disease, each with distinct end-organ targets. J Clin Invest. (2000) 105:1289–98. doi: 10.1172/JCI7894
10. Carlson MJ, West ML, Coghill JM, Panoskaltsis-Mortari A, Blazar BR, Serody JS. In vitro-differentiated TH17 cells mediate lethal acute graft-versus-host disease with severe cutaneous and pulmonary pathologic manifestations. Blood. (2009) 113:1365–74. doi: 10.1182/blood-2008-06-162420
11. Tugues S, Amorim A, Spath S, Martin-Blondel G, Schreiner B, De Feo D, et al. Graft-versus-host disease, but not graft-versus-leukemia immunity, is mediated by GM-CSF-licensed myeloid cells. Sci Transl Med. (2018) 10:eaat8410. doi: 10.1126/scitranslmed.aat8410
12. Korngold R, Sprent J. Lethal graft-versus-host disease after bone marrow transplantation across minor histocompatibility barriers in mice. Prevention by removing mature T cells from marrow. J Exp Med. (1978) 148:1687–98. doi: 10.1084/jem.148.6.1687
13. Sprent J, Schaefer M, Lo D, Korngold R. Properties of purified T cell subsets. II. In vivo responses to class I vs. class II H-2 differences. J Exp Med. (1986) 163:998–1011. doi: 10.1084/jem.163.4.998
14. Sprent J, Schaefer M, Lo D, Korngold R. Functions of purified L3T4+ and Lyt-2+ cells in vitro and in vivo. Immunol Rev. (1986) 91:195–218. doi: 10.1111/j.1600-065X.1986.tb01489.x
15. Korngold R, Sprent J. Variable capacity of L3T4+ T cells to cause lethal graft-versus-host disease across minor histocompatibility barriers in mice. J Exp Med. (1987) 165:1552–64. doi: 10.1084/jem.165.6.1552
16. Fowler DH, Breglio J, Nagel G, Eckhaus MA, Gress RE. Allospecific CD8+ Tc1 and Tc2 populations in graft-versus-leukemia effect and graft-versus-host disease. J Immunol. (1996) 157:4811–21.
17. Hill GR, Olver SD, Kuns RD, Varelias A, Raffelt NC, Don AL, et al. Stem cell mobilization with G-CSF induces type 17 differentiation and promotes scleroderma. Blood. (2010) 116:819–28. doi: 10.1182/blood-2009-11-256495
18. Blazar BR, Taylor PA, Boyer MW, Panoskaltsis-Mortari A, Allison JP, Vallera DA. CD28/B7 interactions are required for sustaining the graft-versus-leukemia effect of delayed post-bone marrow transplantation splenocyte infusion in murine recipients of myeloid or lymphoid leukemia cells. J Immunol. (1997) 159:3460–73.
19. Taylor PA, Panoskaltsis-Mortari A, Freeman GJ, Sharpe AH, Noelle RJ, Rudensky AY, et al. Targeting of inducible costimulator (ICOS) expressed on alloreactive T cells down-regulates graft-versus-host disease (GVHD) and facilitates engraftment of allogeneic bone marrow (BM). Blood. (2005) 105:3372–80. doi: 10.1182/blood-2004-10-3869
20. Blazar BR, Sharpe AH, Chen AI, Panoskaltsis-Mortari A, Lees C, Akiba H, et al. Ligation of OX40 (CD134) regulates graft-versus-host disease (GVHD) and graft rejection in allogeneic bone marrow transplant recipients. Blood. (2003) 101:3741–8. doi: 10.1182/blood-2002-10-3048
21. Blazar BR, Kwon BS, Panoskaltsis-Mortari A, Kwak KB, Peschon JJ, Taylor PA. Ligation of 4-1BB (CDw137) regulates graft-versus-host disease, graft-versus-leukemia, and graft rejection in allogeneic bone marrow transplant recipients. J Immunol. (2001) 166:3174–83. doi: 10.4049/jimmunol.166.5.3174
22. Kean LS, Turka LA, Blazar BR. Advances in targeting co-inhibitory and co-stimulatory pathways in transplantation settings: the Yin to the Yang of cancer immunotherapy. Immunol Rev. (2017) 276:192–212. doi: 10.1111/imr.12523
23. Blazar BR, Taylor PA, Panoskaltsis-Mortari A, Sharpe AH, Vallera DA. Opposing roles of CD28:B7 and CTLA-4:B7 pathways in regulating in vivo alloresponses in murine recipients of MHC disparate T cells. J Immunol. (1999) 162:6368–77.
24. Blazar BR, Carreno BM, Panoskaltsis-Mortari A, Carter L, Iwai Y, Yagita H, et al. Blockade of programmed death-1 engagement accelerates graft-versus-host disease lethality by an IFN-gamma-dependent mechanism. J Immunol. (2003) 171:1272–7. doi: 10.4049/jimmunol.171.3.1272
25. Saha A, O'Connor RS, Thangavelu G, Lovitch SB, Dandamudi DB, Wilson CB, et al. Programmed death ligand-1 expression on donor T cells drives graft-versus-host disease lethality. J Clin Invest. (2016) 126:2642–60. doi: 10.1172/JCI85796
26. Albring JC, Sandau MM, Rapaport AS, Edelson BT, Satpathy A, Mashayekhi M, et al. Targeting of B and T lymphocyte associated (BTLA) prevents graft-versus-host disease without global immunosuppression. J Exp Med. (2010) 207:2551–9. doi: 10.1084/jem.20102017
27. Veenstra RG, Flynn R, Kreymborg K, McDonald-Hyman C, Saha A, Taylor PA, et al. B7-H3 expression in donor T cells and host cells negatively regulates acute graft-versus-host disease lethality. Blood. (2015) 125:3335–46. doi: 10.1182/blood-2014-09-603357
28. Curtsinger JM, Lins DC, Mescher MF. Signal 3 determines tolerance versus full activation of naive CD8 T cells: dissociating proliferation and development of effector function. J Exp Med. (2003) 197:1141–51. doi: 10.1084/jem.20021910
29. Curtsinger JM, Mescher MF. Inflammatory cytokines as a third signal for T cell activation. Curr Opin Immunol. (2010) 22:333–40. doi: 10.1016/j.coi.2010.02.013
30. Wilhelm K, Ganesan J, Muller T, Durr C, Grimm M, Beilhack A, et al. Graft-versus-host disease is enhanced by extracellular ATP activating P2X7R. Nat Med. (2010) 16:1434–8. doi: 10.1038/nm.2242
31. Klambt V, Wohlfeil SA, Schwab L, Hulsdunker J, Ayata K, Apostolova P, et al. A Novel Function for P2Y2 in myeloid recipient-derived cells during graft-versus-host disease. J Immunol. (2015) 195:5795–804. doi: 10.4049/jimmunol.1501357
32. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. (2007) 204:1257–65. doi: 10.1084/jem.20062512
33. Del Papa B, Pierini A, Sportoletti P, Baldoni S, Cecchini D, Rosati E, et al. The NOTCH1/CD39 axis: a Treg trip-switch for GvHD. Leukemia. (2016) 30:1931–4. doi: 10.1038/leu.2016.87
34. Tsukamoto H, Chernogorova P, Ayata K, Gerlach UV, Rughani A, Ritchey JW, et al. Deficiency of CD73/ecto-5'-nucleotidase in mice enhances acute graft-versus-host disease. Blood. (2012) 119:4554–64. doi: 10.1182/blood-2011-09-375899
35. Toubai T, Mathewson ND, Magenau J, Reddy P. Danger signals and graft-versus-host disease: current understanding and future perspectives. Front Immunol. (2016) 7:539. doi: 10.3389/fimmu.2016.00539
36. Markey KA, Banovic T, Kuns RD, Olver SD, Don AL, Raffelt NC, et al. Conventional dendritic cells are the critical donor APC presenting alloantigen after experimental bone marrow transplantation. Blood. (2009) 113:5644–9. doi: 10.1182/blood-2008-12-191833
37. Koyama M, Kuns RD, Olver SD, Raffelt NC, Wilson YA, Don AL, et al. Recipient nonhematopoietic antigen-presenting cells are sufficient to induce lethal acute graft-versus-host disease. Nat Med. (2011) 18:135–42. doi: 10.1038/nm.2597
38. Schwab L, Goroncy L, Palaniyandi S, Gautam S, Triantafyllopoulou A, Mocsai A, et al. Neutrophil granulocytes recruited upon translocation of intestinal bacteria enhance graft-versus-host disease via tissue damage. Nat Med. (2014) 20:648–54. doi: 10.1038/nm.3517
39. Hulsdunker J, Ottmuller KJ, Neeff HP, Koyama M, Gao Z, Thomas OS, et al. Neutrophils provide cellular communication between ileum and mesenteric lymph nodes at graft-versus-host disease onset. Blood. (2018) 131:1858–69. doi: 10.1182/blood-2017-10-812891
40. Zeiser R, Socie G, Blazar BR. Pathogenesis of acute graft-versus-host disease: from intestinal microbiota alterations to donor T cell activation. Br J Haematol. (2016) 175:191–207. doi: 10.1111/bjh.14295
41. Baker MB, Altman NH, Podack ER, Levy RB. The role of cell-mediated cytotoxicity in acute GVHD after MHC-matched allogeneic bone marrow transplantation in mice. J Exp Med. (1996) 183:2645–56. doi: 10.1084/jem.183.6.2645
42. Graubert TA, DiPersio JF, Russell JH, Ley TJ. Perforin/granzyme-dependent and independent mechanisms are both important for the development of graft-versus-host disease after murine bone marrow transplantation. J Clin Invest. (1997) 100:904–11. doi: 10.1172/JCI119606
43. Thiant S, Yakoub-Agha I, Magro L, Trauet J, Coiteux V, Jouet JP, et al. Plasma levels of IL-7 and IL-15 in the first month after myeloablative BMT are predictive biomarkers of both acute GVHD and relapse. Bone Marrow Transplant. (2010) 45:1546–52. doi: 10.1038/bmt.2010.13
44. Chen X, Das R, Komorowski R, Beres A, Hessner MJ, Mihara M, et al. Blockade of interleukin-6 signaling augments regulatory T-cell reconstitution and attenuates the severity of graft-versus-host disease. Blood. (2009) 114:891–900. doi: 10.1182/blood-2009-01-197178
45. Mohty M. Mechanisms of action of antithymocyte globulin: T-cell depletion and beyond. Leukemia. (2007) 21:1387–94. doi: 10.1038/sj.leu.2404683
46. Finke J, Bethge WA, Schmoor C, Ottinger HD, Stelljes M, Zander AR, et al. Standard graft-versus-host disease prophylaxis with or without anti-T-cell globulin in haematopoietic cell transplantation from matched unrelated donors: a randomised, open-label, multicentre phase 3 trial. Lancet Oncol. (2009) 10:855–64. doi: 10.1016/S1470-2045(09)70225-6
47. Hale G, Waldmann H. Risks of developing Epstein-Barr virus-related lymphoproliferative disorders after T-cell-depleted marrow transplants. CAMPATH Users. Blood. (1998) 91:3079–83.
48. Kottaridis PD, Milligan DW, Chopra R, Chakraverty RK, Chakrabarti S, Robinson S, et al. In vivo CAMPATH-1H prevents graft-versus-host disease following nonmyeloablative stem cell transplantation. Blood. (2000) 96:2419–25.
49. Schnitzler M, Hasskarl J, Egger M, Bertz H, Finke J. Successful treatment of severe acute intestinal graft-versus-host resistant to systemic and topical steroids with alemtuzumab. Biol Blood Marrow Transplant. (2009) 15:910–8. doi: 10.1016/j.bbmt.2009.04.002
50. Mayumi H, Umesue M, Nomoto K. Cyclophosphamide-induced immunological tolerance: an overview. Immunobiology. (1996) 195:129–39. doi: 10.1016/S0171-2985(96)80033-7
51. Luznik L, Jalla S, Engstrom LW, Iannone R, Fuchs EJ. Durable engraftment of major histocompatibility complex-incompatible cells after nonmyeloablative conditioning with fludarabine, low-dose total body irradiation, and posttransplantation cyclophosphamide. Blood. (2001) 98:3456–64. doi: 10.1182/blood.V98.12.3456
52. Luznik L, Engstrom LW, Iannone R, Fuchs EJ. Posttransplantation cyclophosphamide facilitates engraftment of major histocompatibility complex-identical allogeneic marrow in mice conditioned with low-dose total body irradiation. Biol Blood Marrow Transplant. (2002) 8:131–8. doi: 10.1053/bbmt.2002.v8.pm11939602
53. Luznik L, O'Donnell PV, Symons HJ, Chen AR, Leffell MS, Zahurak M, et al. HLA-haploidentical bone marrow transplantation for hematologic malignancies using nonmyeloablative conditioning and high-dose, posttransplantation cyclophosphamide. Biol Blood Marrow Transplant. (2008) 14:641–50. doi: 10.1016/j.bbmt.2008.03.005
54. Luznik L, Bolanos-Meade J, Zahurak M, Chen AR, Smith BD, Brodsky R, et al. High-dose cyclophosphamide as single-agent, short-course prophylaxis of graft-versus-host disease. Blood. (2010) 115:3224–30. doi: 10.1182/blood-2009-11-251595
55. Bleakley M, Heimfeld S, Loeb KR, Jones LA, Chaney C, Seropian S, et al. Outcomes of acute leukemia patients transplanted with naive T cell-depleted stem cell grafts. J Clin Invest. (2015) 125:2677–89. doi: 10.1172/JCI81229
56. Choi SW, Reddy P. Current and emerging strategies for the prevention of graft-versus-host disease. Nat Rev Clin Oncol. (2014) 11:536–47. doi: 10.1038/nrclinonc.2014.102
57. Hiraoka A, Ohashi Y, Okamoto S, Moriyama Y, Nagao T, Kodera Y, et al. Phase III study comparing tacrolimus (FK506) with cyclosporine for graft-versus-host disease prophylaxis after allogeneic bone marrow transplantation. Bone Marrow Transplant. (2001) 28:181–5. doi: 10.1038/sj.bmt.1703097
58. Ram R, Storb R. Pharmacologic prophylaxis regimens for acute graft-versus-host disease: past, present and future. Leuk Lymphoma. (2013) 54:1591–601. doi: 10.3109/10428194.2012.762978
59. Inagaki J, Kodama Y, Fukano R, Noguchi M, Okamura J. Mycophenolate mofetil for treatment of steroid-refractory acute graft-versus-host disease after pediatric hematopoietic stem cell transplantation. Pediatr Transplant. (2015) 19:652–8. doi: 10.1111/petr.12545
60. Chapman NM, Chi H. mTOR links environmental signals to T cell fate decisions. Front Immunol. (2014) 5:686. doi: 10.3389/fimmu.2014.00686
61. Battaglia M, Stabilini A, Roncarolo MG. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood. (2005) 105:4743–8. doi: 10.1182/blood-2004-10-3932
62. Koura DT, Horan JT, Langston AA, Qayed M, Mehta A, Khoury HJ, et al. In vivo T cell costimulation blockade with abatacept for acute graft-versus-host disease prevention: a first-in-disease trial. Biol Blood Marrow Transplant. (2013) 19:1638–49. doi: 10.1016/j.bbmt.2013.09.003
63. Zeiser R, Burchert A, Lengerke C, Verbeek M, Maas-Bauer K, Metzelder SK, et al. Ruxolitinib in corticosteroid-refractory graft-versus-host disease after allogeneic stem cell transplantation: a multicenter survey. Leukemia. (2015) 29:2062–8. doi: 10.1038/leu.2015.212
64. Kröger N, Abd Kadir SSS, Zabelina T, Wolschke C, Ayuk FA, Badbaran A, et at. ruxolitinib during peritransplant period for myelofibrosis patients undergoing allogeneic stem cell transplantation reduces acute graft-versus-host disease. Blood. (2016) 128:2242.
65. Betts BC, Bastian D, Iamsawat S, Nguyen H, Heinrichs JL, Wu Y, et al. Targeting JAK2 reduces GVHD and xenograft rejection through regulation of T cell differentiation. Proc Natl Acad Sci USA. (2018) 115:1582–7. doi: 10.1073/pnas.1712452115
66. Tawara I, Sun Y, Lewis EC, Toubai T, Evers R, Nieves E, et al. Alpha-1-antitrypsin monotherapy reduces graft-versus-host disease after experimental allogeneic bone marrow transplantation. Proc Natl Acad Sci USA. (2012) 109:564–9. doi: 10.1073/pnas.1117665109
67. Marcondes AM, Hockenbery D, Lesnikova M, Dinarello CA, Woolfrey A, Gernsheimer T, et al. Response of steroid-refractory acute GVHD to alpha1-antitrypsin. Biol Blood Marrow Transplant. (2016) 22:1596–601. doi: 10.1016/j.bbmt.2016.05.011
68. Magenau JM, Goldstein SC, Peltier D, Soiffer RJ, Braun T, Pawarode A, et al. alpha1-Antitrypsin infusion for treatment of steroid-resistant acute graft-versus-host disease. Blood. (2018) 131:1372–9. doi: 10.1182/blood-2017-11-815746
69. Choi SW, Braun T, Chang L, Ferrara JL, Pawarode A, Magenau JM, et al. Vorinostat plus tacrolimus and mycophenolate to prevent graft-versus-host disease after related-donor reduced-intensity conditioning allogeneic haemopoietic stem-cell transplantation: a phase 1/2 trial. Lancet Oncol. (2014) 15:87–95. doi: 10.1016/S1470-2045(13)70512-6
70. Choi SW, Braun T, Henig I, Gatza E, Magenau J, Parkin B, et al. Vorinostat plus tacrolimus/methotrexate to prevent GVHD after myeloablative conditioning, unrelated donor HCT. Blood. (2017) 130:1760–7. doi: 10.1182/blood-2017-06-790469
71. Choi SW, Gatza E, Hou G, Sun Y, Whitfield J, Song Y, et al. Histone deacetylase inhibition regulates inflammation and enhances Tregs after allogeneic hematopoietic cell transplantation in humans. Blood. (2015) 125:815–9. doi: 10.1182/blood-2014-10-605238
72. Reshef R, Luger SM, Hexner EO, Loren AW, Frey NV, Nasta SD, et al. Blockade of lymphocyte chemotaxis in visceral graft-versus-host disease. N Engl J Med. (2012) 367:135–45. doi: 10.1056/NEJMoa1201248
73. Reshef R, Ganetsky A, Acosta EP, Blauser R, Crisalli L, McGraw J, et al. Extended CCR5 blockade for graft-versus-host disease prophylaxis improves outcomes of reduced intensity unrelated donor hematopoietic cell transplantation: a phase II clinical trial. Biol Blood Marrow Transplant. (2018) S1083-8791(18)30601-3. doi: 10.1016/j.bbmt.2018.09.034. [Epub ahead of print].
74. Leventhal J, Abecassis M, Miller J, Gallon L, Ravindra K, Tollerud DJ, et al. Chimerism and tolerance without GVHD or engraftment syndrome in HLA-mismatched combined kidney and hematopoietic stem cell transplantation. Sci Transl Med. (2012) 4:124ra28. doi: 10.1126/scitranslmed.3003509
75. Lowsky R, Takahashi T, Liu YP, Dejbakhsh-Jones S, Grumet FC, Shizuru JA, et al. Protective conditioning for acute graft-versus-host disease. N Engl J Med. (2005) 353:1321–31. doi: 10.1056/NEJMoa050642
76. Mohty M, Avinens O, Faucher C, Viens P, Blaise D, Eliaou JF. Predictive factors and impact of full donor T-cell chimerism after reduced intensity conditioning allogeneic stem cell transplantation. Haematologica. (2007) 92:1004–6. doi: 10.3324/haematol.10971
77. Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood. (2011) 117:1061–70. doi: 10.1182/blood-2010-07-293795
78. Di Ianni M, Falzetti F, Carotti A, Terenzi A, Castellino F, Bonifacio E, et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood. (2011) 117:3921–8. doi: 10.1182/blood-2010-10-311894
79. Brunstein CG, Miller JS, McKenna DH, Hippen KL, DeFor TE, Sumstad D, et al. Umbilical cord blood-derived T regulatory cells to prevent GVHD: kinetics, toxicity profile, and clinical effect. Blood. (2016) 127:1044–51. doi: 10.1182/blood-2015-06-653667
80. Zhang P, Tey SK, Koyama M, Kuns RD, Olver SD, Lineburg KE, et al. Induced regulatory T cells promote tolerance when stabilized by rapamycin and IL-2 in vivo. J Immunol. (2013) 191:5291–303. doi: 10.4049/jimmunol.1301181
81. Haribhai D, Williams JB, Jia S, Nickerson D, Schmitt EG, Edwards B, et al. A requisite role for induced regulatory T cells in tolerance based on expanding antigen receptor diversity. Immunity. (2011) 35:109–22. doi: 10.1016/j.immuni.2011.03.029
82. Heinrichs J, Li J, Nguyen H, Wu Y, Bastian D, Daethanasanmak A, et al. CD8(+) Tregs promote GVHD prevention and overcome the impaired GVL effect mediated by CD4(+) Tregs in mice. Oncoimmunology. (2016) 5:e1146842. doi: 10.1080/2162402X.2016.1146842
83. Bacchetta R, Sartirana C, Levings MK, Bordignon C, Narula S, Roncarolo MG. Growth and expansion of human T regulatory type 1 cells are independent from TCR activation but require exogenous cytokines. Eur J Immunol. (2002) 32:2237–45. doi: 10.1002/1521-4141(200208)32:8<2237::AID-IMMU2237>3.0.CO;2-2
84. Bacchetta R, Lucarelli B, Sartirana C, Gregori S, Lupo Stanghellini MT, Miqueu P, et al. Immunological outcome in haploidentical-HSC transplanted patients treated with il-10-anergized donor T cells. Front Immunol. (2014) 5:16. doi: 10.3389/fimmu.2014.00016
85. Mavers M, Maas-Bauer K, Negrin RS. Invariant natural killer T cells as suppressors of graft-versus-host disease in allogeneic hematopoietic stem cell transplantation. Front Immunol. (2017) 8:900. doi: 10.3389/fimmu.2017.00900
86. Ren G, Zhang L, Zhao X, Xu G, Zhang Y, Roberts AI, et al. Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell. (2008) 2:141–50. doi: 10.1016/j.stem.2007.11.014
87. Amorin B, Alegretti AP, Valim V, Pezzi A, Laureano AM, da Silva MA, et al. Mesenchymal stem cell therapy and acute graft-versus-host disease: a review. Hum Cell. (2014) 27:137–50. doi: 10.1007/s13577-014-0095-x
88. Ho VT, Soiffer RJ. The history and future of T-cell depletion as graft-versus-host disease prophylaxis for allogeneic hematopoietic stem cell transplantation. Blood. (2001) 98:3192–204. doi: 10.1182/blood.V98.12.3192
89. Marmont AM, Horowitz MM, Gale RP, Sobocinski K, Ash RC, van Bekkum DW, et al. T-cell depletion of HLA-identical transplants in leukemia. Blood. (1991) 78:2120–30.
90. Bacigalupo A, Lamparelli T, Bruzzi P, Guidi S, Alessandrino PE, di Bartolomeo P, et al. Antithymocyte globulin for graft-versus-host disease prophylaxis in transplants from unrelated donors: 2 randomized studies from Gruppo Italiano Trapianti Midollo Osseo (GITMO). Blood. (2001) 98:2942–7. doi: 10.1182/blood.V98.10.2942
91. Dutton RW, Bradley LM, Swain SL. T cell memory. Annu Rev Immunol. (1998) 16:201–23. doi: 10.1146/annurev.immunol.16.1.201
92. Chen BJ, Cui X, Sempowski GD, Liu C, Chao NJ. Transfer of allogeneic CD62L- memory T cells without graft-versus-host disease. Blood. (2004) 103:1534–41. doi: 10.1182/blood-2003-08-2987
93. Chen BJ, Deoliveira D, Cui X, Le NT, Son J, Whitesides JF, et al. Inability of memory T cells to induce graft-versus-host disease is a result of an abortive alloresponse. Blood. (2007) 109:3115–23. doi: 10.1182/blood-2006-04-016410
94. Dutt S, Baker J, Kohrt HE, Kambham N, Sanyal M, Negrin RS, et al. CD8+CD44(hi) but not CD4+CD44(hi) memory T cells mediate potent graft antilymphoma activity without GVHD. Blood. (2011) 117:3230–9. doi: 10.1182/blood-2010-10-312751
95. Huang W, Chao NJ. Memory T cells: a helpful guard for allogeneic hematopoietic stem cell transplantation without causing graft-versus-host disease. Hematol Oncol Stem Cell Ther. (2017) 10:211–9. doi: 10.1016/j.hemonc.2017.05.006
96. Muffly L, Sheehan K, Armstrong R, Jensen K, Tate K, Rezvani AR, et al. Infusion of donor-derived CD8(+) memory T cells for relapse following allogeneic hematopoietic cell transplantation. Blood Adv. (2018) 2:681–90. doi: 10.1182/bloodadvances.2017012104
97. McCurdy SR, Kasamon YL, Kanakry CG, Bolanos-Meade J, Tsai HL, Showel MM, et al. Comparable composite endpoints after HLA-matched and HLA-haploidentical transplantation with post-transplantation cyclophosphamide. Haematologica. (2017) 102:391–400. doi: 10.3324/haematol.2016.144139
98. Robinson TM, O'Donnell PV, Fuchs EJ, Luznik L. Haploidentical bone marrow and stem cell transplantation: experience with post-transplantation cyclophosphamide. Semin Hematol. (2016) 53:90–7. doi: 10.1053/j.seminhematol.2016.01.005
99. Kanakry CG, Bolanos-Meade J, Kasamon YL, Zahurak M, Durakovic N, Furlong T, et al. Low immunosuppressive burden after HLA-matched related or unrelated BMT using posttransplantation cyclophosphamide. Blood. (2017) 129:1389–93. doi: 10.1182/blood-2016-09-737825
100. Kanakry CG, Tsai HL, Bolanos-Meade J, Smith BD, Gojo I, Kanakry JA, et al. Single-agent GVHD prophylaxis with posttransplantation cyclophosphamide after myeloablative, HLA-matched BMT for AML, ALL, and MDS. Blood. (2014) 124:3817–27. doi: 10.1182/blood-2014-07-587477
101. Al-Homsi AS, Roy TS, Cole K, Feng Y, Duffner U. Post-transplant high-dose cyclophosphamide for the prevention of graft-versus-host disease. Biol Blood Marrow Transplant. (2015) 21:604–11. doi: 10.1016/j.bbmt.2014.08.014
102. Ganguly S, Ross DB, Panoskaltsis-Mortari A, Kanakry CG, Blazar BR, Levy RB, et al. Donor CD4+ Foxp3+ regulatory T cells are necessary for posttransplantation cyclophosphamide-mediated protection against GVHD in mice. Blood. (2014) 124:2131–41. doi: 10.1182/blood-2013-10-525873
103. Kanakry CG, Ganguly S, Zahurak M, Bolanos-Meade J, Thoburn C, Perkins B, et al. Aldehyde dehydrogenase expression drives human regulatory T cell resistance to posttransplantation cyclophosphamide. Sci Transl Med. (2013) 5:211ra157. doi: 10.1126/scitranslmed.3006960
104. Sharpe AH. Mechanisms of costimulation. Immunol Rev. (2009) 229:5–11. doi: 10.1111/j.1600-065X.2009.00784.x
105. Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. (2013) 13:227–42. doi: 10.1038/nri3405
106. Lafferty KJ, Cunningham AJ. A new analysis of allogeneic interactions. Aust J Exp Biol Med Sci. (1975) 53:27–42. doi: 10.1038/icb.1975.3
107. Jenkins MK, Schwartz RH. Antigen presentation by chemically modified splenocytes induces antigen-specific T cell unresponsiveness in vitro and in vivo. J Exp Med. (1987) 165:302–19. doi: 10.1084/jem.165.2.302
108. Linsley PS, Brady W, Grosmaire L, Aruffo A, Damle NK, Ledbetter JA. Binding of the B cell activation antigen B7 to CD28 costimulates T cell proliferation and interleukin 2 mRNA accumulation. J Exp Med. (1991) 173:721–30. doi: 10.1084/jem.173.3.721
109. Linsley PS, Clark EA, Ledbetter JA. T-cell antigen CD28 mediates adhesion with B cells by interacting with activation antigen B7/BB-1. Proc Natl Acad Sci USA. (1990) 87:5031–5. doi: 10.1073/pnas.87.13.5031
110. Hathcock KS, Laszlo G, Dickler HB, Bradshaw J, Linsley P, Hodes RJ. Identification of an alternative CTLA-4 ligand costimulatory for T cell activation. Science. (1993) 262:905–7. doi: 10.1126/science.7694361
111. Azuma M, Ito D, Yagita H, Okumura K, Phillips JH, Lanier LL, et al. B70 antigen is a second ligand for CTLA-4 and CD28. Nature. (1993) 366:76–9. doi: 10.1038/366076a0
112. Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol Rev. (2009) 229:12–26. doi: 10.1111/j.1600-065X.2009.00770.x
113. Finck BK, Linsley PS, Wofsy D. Treatment of murine lupus with CTLA4Ig. Science. (1994) 265:1225–7. doi: 10.1126/science.7520604
114. Turka LA, Linsley PS, Lin H, Brady W, Leiden JM, Wei RQ, et al. T-cell activation by the CD28 ligand B7 is required for cardiac allograft rejection in vivo. Proc Natl Acad Sci USA. (1992) 89:11102–5. doi: 10.1073/pnas.89.22.11102
115. Blazar BR, Taylor PA, Linsley PS, Vallera DA. In vivo blockade of CD28/CTLA4: B7/BB1 interaction with CTLA4-Ig reduces lethal murine graft-versus-host disease across the major histocompatibility complex barrier in mice. Blood. (1994) 83:3815–25.
116. Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. (2010) 236:219–42. doi: 10.1111/j.1600-065X.2010.00923.x
117. Poirier N, Mary C, Dilek N, Hervouet J, Minault D, Blancho G, et al. Preclinical efficacy and immunological safety of FR104, an antagonist anti-CD28 monovalent Fab' antibody. Am J Transplant. (2012) 12:2630–40. doi: 10.1111/j.1600-6143.2012.04164.x
118. Vincenti F, Rostaing L, Grinyo J, Rice K, Steinberg S, Gaite L, et al. Belatacept and long-term outcomes in kidney transplantation. N Engl J Med. (2016) 374:333–43. doi: 10.1056/NEJMoa1506027
119. Haanstra KG, Dijkman K, Bashir N, Bauer J, Mary C, Poirier N, et al. Selective blockade of CD28-mediated T cell costimulation protects rhesus monkeys against acute fatal experimental autoimmune encephalomyelitis. J Immunol. (2015) 194:1454–66. doi: 10.4049/jimmunol.1402563
120. Hippen KL, Watkins B, Tkachev V, Lemire AM, Lehnen C, Riddle MJ, et al. Preclinical testing of antihuman CD28 Fab' antibody in a novel nonhuman primate small animal rodent model of xenogenic graft-versus-host disease. Transplantation. (2016) 100:2630–9. doi: 10.1097/TP.0000000000001465
121. Poirier N, Dilek N, Mary C, Ville S, Coulon F, Branchereau J, et al. FR104, an antagonist anti-CD28 monovalent fab' antibody, prevents alloimmunization and allows calcineurin inhibitor minimization in nonhuman primate renal allograft. Am J Transplant. (2015) 15:88–100. doi: 10.1111/ajt.12964
122. Watkins BK, Tkachev V, Furlan SN, Hunt DJ, Betz K, Yu A, et al. CD28 blockade controls T cell activation to prevent graft-versus-host disease in primates. J Clin Invest. (2018) 128:3991–4007. doi: 10.1172/JCI98793
123. Yamaoka K, Saharinen P, Pesu M, Holt VE 3rd, Silvennoinen O, O'Shea JJ. The Janus kinases (Jaks). Genome Biol. (2004) 5:253. doi: 10.1186/gb-2004-5-12-253
124. Schroeder MA, Choi J, Staser K, DiPersio JF. The role of janus kinase signaling in graft-versus-host disease and graft versus leukemia. Biol Blood Marrow Transplant. (2018) 24:1125–34. doi: 10.1016/j.bbmt.2017.12.797
125. Spoerl S, Mathew NR, Bscheider M, Schmitt-Graeff A, Chen S, Mueller T, et al. Activity of therapeutic JAK 1/2 blockade in graft-versus-host disease. Blood. (2014) 123:3832–42. doi: 10.1182/blood-2013-12-543736
126. Choi J, Cooper ML, Staser K, Ashami K, Vij KR, Wang B, et al. Baricitinib-induced blockade of interferon gamma receptor and interleukin-6 receptor for the prevention and treatment of graft-versus-host disease. Leukemia. (2018) 32:2483–94. doi: 10.1038/s41375-018-0123-z
127. Okiyama N, Furumoto Y, Villarroel VA, Linton JT, Tsai WL, Gutermuth J, et al. Reversal of CD8 T-cell-mediated mucocutaneous graft-versus-host-like disease by the JAK inhibitor tofacitinib. J Invest Dermatol. (2014) 134:992–1000. doi: 10.1038/jid.2013.476
128. Hechinger AK, Smith BA, Flynn R, Hanke K, McDonald-Hyman C, Taylor PA, et al. Therapeutic activity of multiple common gamma-chain cytokine inhibition in acute and chronic GVHD. Blood. (2015) 125:570–80. doi: 10.1182/blood-2014-06-581793
129. Cetkovic-Cvrlje M, Roers BA, Waurzyniak B, Liu XP, Uckun FM. Targeting Janus kinase 3 to attenuate the severity of acute graft-versus-host disease across the major histocompatibility barrier in mice. Blood. (2001) 98:1607–13. doi: 10.1182/blood.V98.5.1607
130. Silverman EK, Sandhaus RA. Clinical practice. Alpha1-antitrypsin deficiency. N Engl J Med. (2009) 360:2749–57. doi: 10.1056/NEJMcp0900449
131. Carrell RW. alpha 1-Antitrypsin: molecular pathology, leukocytes, and tissue damage. J Clin Invest. (1986) 78:1427–31. doi: 10.1172/JCI112731
132. Ekeowa UI, Marciniak SJ, Lomas DA. alpha(1)-antitrypsin deficiency and inflammation. Expert Rev Clin Immunol. (2011) 7:243–52. doi: 10.1586/eci.10.95
133. Janciauskiene S, Larsson S, Larsson P, Virtala R, Jansson L, Stevens T. Inhibition of lipopolysaccharide-mediated human monocyte activation, in vitro, by alpha1-antitrypsin. Biochem Biophys Res Commun. (2004) 321:592–600. doi: 10.1016/j.bbrc.2004.06.123
134. Ehlers MR. Immune-modulating effects of alpha-1 antitrypsin. Biol Chem. (2014) 395:1187–93. doi: 10.1515/hsz-2014-0161
135. Marcondes AM, Karoopongse E, Lesnikova M, Margineantu D, Welte T, Dinarello CA, et al. alpha-1-Antitrypsin (AAT)-modified donor cells suppress GVHD but enhance the GVL effect: a role for mitochondrial bioenergetics. Blood. (2014) 124:2881–91. doi: 10.1182/blood-2014-04-570440
136. Brennan TV, Lin L, Huang X, Cardona DM, Li Z, Dredge K, et al. Heparan sulfate, an endogenous TLR4 agonist, promotes acute GVHD after allogeneic stem cell transplantation. Blood. (2012) 120:2899–908. doi: 10.1182/blood-2011-07-368720
137. Hancock WW, Akimova T, Beier UH, Liu Y, Wang L. HDAC inhibitor therapy in autoimmunity and transplantation. Ann Rheum Dis. (2012) 71 (Suppl. 2):i46–54. doi: 10.1136/annrheumdis-2011-200593
138. Sun Y, Chin YE, Weisiger E, Malter C, Tawara I, Toubai T, et al. Cutting edge: Negative regulation of dendritic cells through acetylation of the nonhistone protein STAT-3. J Immunol. (2009) 182:5899–903. doi: 10.4049/jimmunol.0804388
139. Reddy P, Sun Y, Toubai T, Duran-Struuck R, Clouthier SG, Weisiger E, et al. Histone deacetylase inhibition modulates indoleamine 2,3-dioxygenase-dependent DC functions and regulates experimental graft-versus-host disease in mice. J Clin Invest. (2008) 118:2562–73. doi: 10.1172/JCI34712
140. Jasperson LK, Bucher C, Panoskaltsis-Mortari A, Taylor PA, Mellor AL, Munn DH, et al. Indoleamine 2,3-dioxygenase is a critical regulator of acute graft-versus-host disease lethality. Blood. (2008) 111:3257–65. doi: 10.1182/blood-2007-06-096081
141. Jasperson LK, Bucher C, Panoskaltsis-Mortari A, Mellor AL, Munn DH, Blazar BR. Inducing the tryptophan catabolic pathway, indoleamine 2,3-dioxygenase (IDO), for suppression of graft-versus-host disease (GVHD) lethality. Blood. (2009) 114:5062–70. doi: 10.1182/blood-2009-06-227587
142. Wysocki CA, Panoskaltsis-Mortari A, Blazar BR, Serody JS. Leukocyte migration and graft-versus-host disease. Blood. (2005) 105:4191–9. doi: 10.1182/blood-2004-12-4726
143. Serody JS, Burkett SE, Panoskaltsis-Mortari A, Ng-Cashin J, McMahon E, Matsushima GK, et al. T-lymphocyte production of macrophage inflammatory protein-1alpha is critical to the recruitment of CD8(+) T cells to the liver, lung, and spleen during graft-versus-host disease. Blood. (2000) 96:2973–80.
144. Wysocki CA, Burkett SB, Panoskaltsis-Mortari A, Kirby SL, Luster AD, McKinnon K, et al. Differential roles for CCR5 expression on donor T cells during graft-versus-host disease based on pretransplant conditioning. J Immunol. (2004) 173:845–54. doi: 10.4049/jimmunol.173.2.845
145. Murai M, Yoneyama H, Harada A, Yi Z, Vestergaard C, Guo B, et al. Active participation of CCR5(+)CD8(+) T lymphocytes in the pathogenesis of liver injury in graft-versus-host disease. J Clin Invest. (1999) 104:49–57. doi: 10.1172/JCI6642
146. Murai M, Yoneyama H, Ezaki T, Suematsu M, Terashima Y, Harada A, et al. Peyer's patch is the essential site in initiating murine acute and lethal graft-versus-host reaction. Nat Immunol. (2003) 4:154–60. doi: 10.1038/ni879
147. Bogunia-Kubik K, Duda D, Suchnicki K, Lange A. CCR5 deletion mutation and its association with the risk of developing acute graft-versus-host disease after allogeneic hematopoietic stem cell transplantation. Haematologica. (2006) 91:1628–34.
148. Ma Q, Gooley TA, Storb RF. CCR5 expression on cells from HLA-matched unrelated marrow donors and graft-versus-host disease. Biol Blood Marrow Transplant. (2010) 16:132–3. doi: 10.1016/j.bbmt.2009.05.017
149. Floisand Y, Lundin KEA, Lazarevic V, Kristiansen JD, Osnes LTN, Tjonnfjord GE, et al. Targeting integrin alpha4beta7 in steroid-refractory intestinal graft-versus-host disease. Biol Blood Marrow Transplant. (2017) 23:172–5. doi: 10.1016/j.bbmt.2016.10.009
150. Bukauskas A, Griskevicius L, Peceliunas V. Lessons learned from early experiences with vedolizumab for steroid-refractory acute graft- versus-host disease with gastrointestinal involvement. Biol Blood Marrow Transplant. (2017) 23:1597. doi: 10.1016/j.bbmt.2017.05.028
151. O'Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. (2016) 16:553–65. doi: 10.1038/nri.2016.70
152. Geltink RIK, Kyle RL, Pearce EL. Unraveling the complex interplay between T cell metabolism and function. Annu Rev Immunol. (2018) 36:461–88. doi: 10.1146/annurev-immunol-042617-053019
153. Nguyen HD, Kuril S, Bastian D, Yu XZ. T-cell metabolism in hematopoietic cell transplantation. Front Immunol. (2018) 9:176. doi: 10.3389/fimmu.2018.00176
154. Nguyen HD, Chatterjee S, Haarberg KM, Wu Y, Bastian D, Heinrichs J, et al. Metabolic reprogramming of alloantigen-activated T cells after hematopoietic cell transplantation. J Clin Invest. (2016) 126:1337–52. doi: 10.1172/JCI82587
155. Gatza E, Wahl DR, Opipari AW, Sundberg TB, Reddy P, Liu C, et al. Manipulating the bioenergetics of alloreactive T cells causes their selective apoptosis and arrests graft-versus-host disease. Sci Transl Med. (2011) 3:67ra8. doi: 10.1126/scitranslmed.3001975
156. Saha A, Aoyama K, Taylor PA, Koehn BH, Veenstra RG, Panoskaltsis-Mortari A, et al. Host programmed death ligand 1 is dominant over programmed death ligand 2 expression in regulating graft-versus-host disease lethality. Blood. (2013) 122:3062–73. doi: 10.1182/blood-2013-05-500801
157. Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. (2014) 20:61–72. doi: 10.1016/j.cmet.2014.05.004
158. Raud B, Roy DG, Divakaruni AS, Tarasenko TN, Franke R, Ma EH, et al. Etomoxir actions on regulatory and memory T cells are independent of Cpt1a-mediated fatty acid oxidation. Cell Metab. (2018) 28:504–15 e7. doi: 10.1016/j.cmet.2018.06.002
159. Byersdorfer CA, Tkachev V, Opipari AW, Goodell S, Swanson J, Sandquist S, et al. Effector T cells require fatty acid metabolism during murine graft-versus-host disease. Blood. (2013) 122:3230–7. doi: 10.1182/blood-2013-04-495515
160. Raha S, Raud B, Oberdorfer L, Castro CN, Schreder A, Freitag J, et al. Disruption of de novo fatty acid synthesis via acetyl-CoA carboxylase 1 inhibition prevents acute graft-versus-host disease. Eur J Immunol. (2016) 46:2233–8. doi: 10.1002/eji.201546152
161. Sofi MH, Heinrichs J, Dany M, Nguyen H, Dai M, Bastian D, et al. Ceramide synthesis regulates T cell activity and GVHD development. JCI Insight. (2017) 2:91701. doi: 10.1172/jci.insight.91701
162. Ratajczak P, Janin A, Peffault de Larour R, Koch L, Roche B, Munn D, et al. IDO in human gut graft-versus-host disease. Biol Blood Marrow Transplant. (2012) 18:150–5. doi: 10.1016/j.bbmt.2011.08.002
163. Highfill SL, Rodriguez PC, Zhou Q, Goetz CA, Koehn BH, Veenstra R, et al. Bone marrow myeloid-derived suppressor cells (MDSCs) inhibit graft-versus-host disease (GVHD) via an arginase-1-dependent mechanism that is up-regulated by interleukin-13. Blood. (2010) 116:5738–47. doi: 10.1182/blood-2010-06-287839
164. Hill DA, Artis D. Intestinal bacteria and the regulation of immune cell homeostasis. Annu Rev Immunol. (2010) 28:623–67. doi: 10.1146/annurev-immunol-030409-101330
165. Hamada H, Hiroi T, Nishiyama Y, Takahashi H, Masunaga Y, Hachimura S, et al. Identification of multiple isolated lymphoid follicles on the antimesenteric wall of the mouse small intestine. J Immunol. (2002) 168:57–64. doi: 10.4049/jimmunol.168.1.57
166. Kamada N, Seo SU, Chen GY, Nunez G. Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol. (2013) 13:321–35. doi: 10.1038/nri3430
167. Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. (2009) 461:1282–6. doi: 10.1038/nature08530
168. Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. (2012) 336:1268–73. doi: 10.1126/science.1223490
169. Varelias A, Ormerod KL, Bunting MD, Koyama M, Gartlan KH, Kuns RD, et al. Acute graft-versus-host disease is regulated by an IL-17-sensitive microbiome. Blood. (2017) 129:2172–85. doi: 10.1182/blood-2016-08-732628
170. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. (2013) 504:451–5. doi: 10.1038/nature12726
171. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. (2013) 341:569–73. doi: 10.1126/science.1241165
172. Kim M, Qie Y, Park J, Kim CH. Gut microbial metabolites fuel host antibody responses. Cell Host Microbe. (2016) 20:202–14. doi: 10.1016/j.chom.2016.07.001
173. Fujiwara H, Docampo MD, Riwes M, Peltier D, Toubai T, Henig I, et al. Microbial metabolite sensor GPR43 controls severity of experimental GVHD. Nat Commun. (2018) 9:3674. doi: 10.1038/s41467-018-06048-w
174. Jenq RR, Ubeda C, Taur Y, Menezes CC, Khanin R, Dudakov JA, et al. Regulation of intestinal inflammation by microbiota following allogeneic bone marrow transplantation. J Exp Med. (2012) 209:903–11. doi: 10.1084/jem.20112408
175. Jenq RR, Taur Y, Devlin SM, Ponce DM, Goldberg JD, Ahr KF, et al. Intestinal blautia is associated with reduced death from graft-versus-host disease. Biol Blood Marrow Transplant. (2015) 21:1373–83. doi: 10.1016/j.bbmt.2015.04.016
176. Shono Y, Docampo MD, Peled JU, Perobelli SM, Velardi E, Tsai JJ, et al. Increased GVHD-related mortality with broad-spectrum antibiotic use after allogeneic hematopoietic stem cell transplantation in human patients and mice. Sci Transl Med. (2016) 8:339ra71. doi: 10.1126/scitranslmed.aaf2311
177. Shono Y, van den Brink MRM. Gut microbiota injury in allogeneic haematopoietic stem cell transplantation. Nat Rev Cancer. (2018) 18:283–95. doi: 10.1038/nrc.2018.10
178. Taur Y, Coyte K, Schluter J, Robilotti E, Figueroa C, Gjonbalaj M, et al. Reconstitution of the gut microbiota of antibiotic-treated patients by autologous fecal microbiota transplant. Sci Transl Med. (2018) 10:eaap9489. doi: 10.1126/scitranslmed
179. Donohoe DR, Garge N, Zhang X, Sun W, O'Connell TM, Bunger MK, et al. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. (2011) 13:517–26. doi: 10.1016/j.cmet.2011.02.018
180. Ganapathy V, Thangaraju M, Prasad PD, Martin PM, Singh N. Transporters and receptors for short-chain fatty acids as the molecular link between colonic bacteria and the host. Curr Opin Pharmacol. (2013) 13:869–74. doi: 10.1016/j.coph.2013.08.006
181. Mathewson ND, Jenq R, Mathew AV, Koenigsknecht M, Hanash A, Toubai T, et al. Gut microbiome-derived metabolites modulate intestinal epithelial cell damage and mitigate graft-versus-host disease. Nat Immunol. (2016) 17:505–13. doi: 10.1038/ni.3400
182. Swimm A, Giver CR, DeFilipp Z, Rangaraju S, Sharma A, Ulezko Antonova A, et al. Indoles derived from intestinal microbiota act via type I interferon signaling to limit Graft-versus-Host-Disease. Blood. (2018) 132:2506–19. doi: 10.1182/blood-2018-03-838193
183. Wu SR, Reddy P. Tissue tolerance: a distinct concept to control acute GVHD severity. Blood. (2017) 129:1747–52. doi: 10.1182/blood-2016-09-740431
184. Matzinger P, Kamala T. Tissue-based class control: the other side of tolerance. Nat Rev Immunol. (2011) 11:221–30. doi: 10.1038/nri2940
185. Takashima S, Kadowaki M, Aoyama K, Koyama M, Oshima T, Tomizuka K, et al. The Wnt agonist R-spondin1 regulates systemic graft-versus-host disease by protecting intestinal stem cells. J Exp Med. (2011) 208:285–94. doi: 10.1084/jem.20101559
186. Lindemans CA, Calafiore M, Mertelsmann AM, O'Connor MH, Dudakov JA, Jenq RR, et al. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature. (2015) 528:560–4. doi: 10.1038/nature16460
187. Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S, et al. A distinct function of regulatory T cells in tissue protection. Cell. (2015) 162:1078–89. doi: 10.1016/j.cell.2015.08.021
188. Bouchlaka MN, Moffitt AB, Kim J, Kink JA, Bloom DD, Love C, et al. Human mesenchymal stem cell-educated macrophages are a distinct high IL-6-producing subset that confer protection in graft-versus-host-disease and radiation injury models. Biol Blood Marrow Transplant. (2017) 23:897–905. doi: 10.1016/j.bbmt.2017.02.018
189. van Hoeven V, Munneke JM, Cornelissen AS, Omar SZ, Spruit MJ, Kleijer M, et al. Mesenchymal stromal cells stimulate the proliferation and IL-22 production of group 3 innate lymphoid cells. J Immunol. (2018) 201:1165–73. doi: 10.4049/jimmunol.1700901
190. Ildstad ST, Sachs DH. Reconstitution with syngeneic plus allogeneic or xenogeneic bone marrow leads to specific acceptance of allografts or xenografts. Nature. (1984) 307:168–70. doi: 10.1038/307168a0
191. Sykes M, Sheard MA, Sachs DH. Graft-versus-host-related immunosuppression is induced in mixed chimeras by alloresponses against either host or donor lymphohematopoietic cells. J Exp Med. (1988) 168:2391–6. doi: 10.1084/jem.168.6.2391
192. Sharabi Y, Sachs DH. Mixed chimerism and permanent specific transplantation tolerance induced by a nonlethal preparative regimen. J Exp Med. (1989) 169:493–502. doi: 10.1084/jem.169.2.493
193. Kawai T, Cosimi AB, Spitzer TR, Tolkoff-Rubin N, Suthanthiran M, Saidman SL, et al. HLA-mismatched renal transplantation without maintenance immunosuppression. N Engl J Med. (2008) 358:353–61. doi: 10.1056/NEJMoa071074
194. Scandling JD, Busque S, Shizuru JA, Engleman EG, Strober S. Induced immune tolerance for kidney transplantation. N Engl J Med. (2011) 365:1359–60. doi: 10.1056/NEJMc1107841
195. Morris H, DeWolf S, Robins H, Sprangers B, LoCascio SA, Shonts BA, et al. Tracking donor-reactive T cells: Evidence for clonal deletion in tolerant kidney transplant patients. Sci Transl Med. (2015) 7:272ra10. doi: 10.1126/scitranslmed.3010760
196. Zuber J, Sykes M. Mechanisms of mixed chimerism-based transplant tolerance. Trends Immunol. (2017) 38:829–43. doi: 10.1016/j.it.2017.07.008
197. Abbas AK, Benoist C, Bluestone JA, Campbell DJ, Ghosh S, Hori S, et al. Regulatory T cells: recommendations to simplify the nomenclature. Nat Immunol. (2013) 14:307–8. doi: 10.1038/ni.2554
198. Walsh PT, Taylor DK, Turka LA. Tregs and transplantation tolerance. J Clin Invest. (2004) 114:1398–403. doi: 10.1172/JCI200423238
199. Bluestone JA, Tang Q. Treg cells-the next frontier of cell therapy. Science. (2018) 362:154–5. doi: 10.1126/science.aau2688
200. Dominguez-Villar M, Hafler DA. Regulatory T cells in autoimmune disease. Nat Immunol. (2018). doi: 10.1038/s41590-018-0120-4
201. Taylor PA, Noelle RJ, Blazar BR. CD4(+)CD25(+) immune regulatory cells are required for induction of tolerance to alloantigen via costimulatory blockade. J Exp Med. (2001) 193:1311–8. doi: 10.1084/jem.193.11.1311
202. Hoffmann P, Ermann J, Edinger M, Fathman CG, Strober S. Donor-type CD4(+)CD25(+) regulatory T cells suppress lethal acute graft-versus-host disease after allogeneic bone marrow transplantation. J Exp Med. (2002) 196:389–99. doi: 10.1084/jem.20020399
203. Cohen JL, Trenado A, Vasey D, Klatzmann D, Salomon BL. CD4(+)CD25(+) immunoregulatory T Cells: new therapeutics for graft-versus-host disease. J Exp Med. (2002) 196:401–6. doi: 10.1084/jem.20020090
204. Rezvani K, Mielke S, Ahmadzadeh M, Kilical Y, Savani BN, Zeilah J, et al. High donor FOXP3-positive regulatory T-cell (Treg) content is associated with a low risk of GVHD following HLA-matched allogeneic SCT. Blood. (2006) 108:1291–7. doi: 10.1182/blood-2006-02-003996
205. Blazar BR, MacDonald KPA, Hill GR. Immune regulatory cell infusion for graft-versus-host disease prevention and therapy. Blood. (2018) 131:2651–60. doi: 10.1182/blood-2017-11-785865
206. Semple K, Yu Y, Wang D, Anasetti C, Yu XZ. Efficient and selective prevention of GVHD by antigen-specific induced Tregs via linked-suppression in mice. Biol Blood Marrow Transplant. (2011) 17:309–18. doi: 10.1016/j.bbmt.2010.12.710
207. Chinen T, Kannan AK, Levine AG, Fan X, Klein U, Zheng Y, et al. An essential role for the IL-2 receptor in Treg cell function. Nat Immunol. (2016) 17:1322–33. doi: 10.1038/ni.3540
208. Kennedy-Nasser AA, Ku S, Castillo-Caro P, Hazrat Y, Wu MF, Liu H, et al. Ultra low-dose IL-2 for GVHD prophylaxis after allogeneic hematopoietic stem cell transplantation mediates expansion of regulatory T cells without diminishing antiviral and antileukemic activity. Clin Cancer Res. (2014) 20:2215–25. doi: 10.1158/1078-0432.CCR-13-3205
209. Matsuoka K, Koreth J, Kim HT, Bascug G, McDonough S, Kawano Y, et al. Low-dose interleukin-2 therapy restores regulatory T cell homeostasis in patients with chronic graft-versus-host disease. Sci Transl Med. (2013) 5:179ra43. doi: 10.1126/scitranslmed.3005265
210. Koreth J, Kim HT, Jones KT, Lange PB, Reynolds CG, Chammas MJ, et al. Efficacy, durability, and response predictors of low-dose interleukin-2 therapy for chronic graft-versus-host disease. Blood. (2016) 128:130–7. doi: 10.1182/blood-2016-02-702852
211. Sockolosky JT, Trotta E, Parisi G, Picton L, Su LL, Le AC, et al. Selective targeting of engineered T cells using orthogonal IL-2 cytokine-receptor complexes. Science. (2018) 359:1037–42. doi: 10.1126/science.aar3246
212. Trotta E, Bessette PH, Silveria SL, Ely LK, Jude KM, Le DT, et al. A human anti-IL-2 antibody that potentiates regulatory T cells by a structure-based mechanism. Nat Med. (2018) 24:1005–14. doi: 10.1038/s41591-018-0070-2
213. Croft M. The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol. (2009) 9:271–85. doi: 10.1038/nri2526
214. Schreiber TH, Wolf D, Tsai MS, Chirinos J, Deyev VV, Gonzalez L, et al. Therapeutic Treg expansion in mice by TNFRSF25 prevents allergic lung inflammation. J Clin Invest. (2010) 120:3629–40. doi: 10.1172/JCI42933
215. Wolf D, Schreiber TH, Tryphonopoulos P, Li S, Tzakis AG, Ruiz P, et al. Tregs expanded in vivo by TNFRSF25 agonists promote cardiac allograft survival. Transplantation. (2012) 94:569–74. doi: 10.1097/TP.0b013e318264d3ef
216. Kim BS, Nishikii H, Baker J, Pierini A, Schneidawind D, Pan Y, et al. Treatment with agonistic DR3 antibody results in expansion of donor Tregs and reduced graft-versus-host disease. Blood. (2015) 126:546–57. doi: 10.1182/blood-2015-04-637587
217. Leclerc M, Naserian S, Pilon C, Thiolat A, Martin GH, Pouchy C, et al. Control of GVHD by regulatory T cells depends on TNF produced by T cells and TNFR2 expressed by regulatory T cells. Blood. (2016) 128:1651–9. doi: 10.1182/blood-2016-02-700849
218. Chopra M, Biehl M, Steinfatt T, Brandl A, Kums J, Amich J, et al. Exogenous TNFR2 activation protects from acute GvHD via host T reg cell expansion. J Exp Med. (2016) 213:1881–900. doi: 10.1084/jem.20151563
219. Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, et al. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med. (2004) 199:1455–65. doi: 10.1084/jem.20040139
220. Zhang H, Podojil JR, Chang J, Luo X, Miller SD. TGF-beta-induced myelin peptide-specific regulatory T cells mediate antigen-specific suppression of induction of experimental autoimmune encephalomyelitis. J Immunol. (2010) 184:6629–36. doi: 10.4049/jimmunol.0904044
221. Li J, Heinrichs J, Haarberg K, Semple K, Veerapathran A, Liu C, et al. HY-specific induced regulatory t cells display high specificity and efficacy in the prevention of acute graft-versus-host disease. J Immunol. (2015) 195:717–25. doi: 10.4049/jimmunol.1401250
222. Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature. (2010) 463:808–12. doi: 10.1038/nature08750
223. Huehn J, Polansky JK, Hamann A. Epigenetic control of FOXP3 expression: the key to a stable regulatory T-cell lineage? Nat Rev Immunol. (2009) 9:83–9. doi: 10.1038/nri2474
224. Beres A, Komorowski R, Mihara M, Drobyski WR. Instability of Foxp3 expression limits the ability of induced regulatory T cells to mitigate graft versus host disease. Clin Cancer Res. (2011) 17:3969–83. doi: 10.1158/1078-0432.CCR-10-3347
225. Agle K, Vincent BG, Piper C, Belle L, Zhou V, Shlomchik W, et al. Bim regulates the survival and suppressive capability of CD8(+) FOXP3(+) regulatory T cells during murine GVHD. Blood. (2018) 132:435–47. doi: 10.1182/blood-2017-09-807156
226. Iamsawat S, Daenthanasanmak A, Voss JH, Nguyen H, Bastian D, Liu C, et al. Stabilization of Foxp3 by targeting JAK2 enhances efficacy of CD8 induced regulatory T cells in the prevention of graft-versus-host disease. J Immunol. (2018) 201:2812–23. doi: 10.4049/jimmunol.1800793
227. Bacchetta R, de Waal Malefijt R, Yssel H, Abrams J, de Vries JE, Spits H, et al. Host-reactive CD4+ and CD8+ T cell clones isolated from a human chimera produce IL-5, IL-2, IFN-gamma and granulocyte/macrophage-colony-stimulating factor but not IL-4. J Immunol. (1990) 144:902–8.
228. Bacchetta R, Bigler M, Touraine JL, Parkman R, Tovo PA, Abrams J, et al. High levels of interleukin 10 production in vivo are associated with tolerance in SCID patients transplanted with HLA mismatched hematopoietic stem cells. J Exp Med. (1994) 179:493–502. doi: 10.1084/jem.179.2.493
229. Gregori S, Passerini L, Roncarolo MG. Clinical Outlook for Type-1 and FOXP3(+) T regulatory cell-based therapy. Front Immunol. (2015) 6:593. doi: 10.3389/fimmu.2015.00593
230. Gregori S, Roncarolo MG. Engineered T regulatory type 1 cells for clinical application. Front Immunol. (2018) 9:233. doi: 10.3389/fimmu.2018.00233
231. Zhang P, Lee JS, Gartlan KH, Schuster IS, Comerford I, Varelias A, et al. Eomesodermin promotes the development of type 1 regulatory T (TR1) cells. Sci Immunol. (2017) 2:eaah7152. doi: 10.1126/sciimmunol.aah7152
232. Groux H, O'Garra A, Bigler M, Rouleau M, Antonenko S, de Vries JE, et al. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. (1997) 389:737–42. doi: 10.1038/39614
233. Van Kaer L, Wu L. Therapeutic potential of invariant natural killer T cells in autoimmunity. Front Immunol. (2018) 9:519. doi: 10.3389/fimmu.2018.00519
234. Lan F, Zeng D, Higuchi M, Higgins JP, Strober S. Host conditioning with total lymphoid irradiation and antithymocyte globulin prevents graft-versus-host disease: the role of CD1-reactive natural killer T cells. Biol Blood Marrow Transplant. (2003) 9:355–63. doi: 10.1016/S1083-8791(03)00108-3
235. Lan F, Zeng D, Higuchi M, Huie P, Higgins JP, Strober S. Predominance of NK1.1+TCR alpha beta+ or DX5+TCR alpha beta+ T cells in mice conditioned with fractionated lymphoid irradiation protects against graft-versus-host disease: “natural suppressor” cells. J Immunol. (2001) 167:2087–96. doi: 10.4049/jimmunol.167.4.2087
236. Pillai AB, George TI, Dutt S, Strober S. Host natural killer T cells induce an interleukin-4-dependent expansion of donor CD4+CD25+Foxp3+ T regulatory cells that protects against graft-versus-host disease. Blood. (2009) 113:4458–67. doi: 10.1182/blood-2008-06-165506
237. Morecki S, Panigrahi S, Pizov G, Yacovlev E, Gelfand Y, Eizik O, et al. Effect of KRN7000 on induced graft-vs-host disease. Exp Hematol. (2004) 32:630–7. doi: 10.1016/j.exphem.2004.04.005
238. Leveson-Gower DB, Olson JA, Sega EI, Luong RH, Baker J, Zeiser R, et al. Low doses of natural killer T cells provide protection from acute graft-versus-host disease via an IL-4-dependent mechanism. Blood. (2011) 117:3220–9. doi: 10.1182/blood-2010-08-303008
239. Schneidawind D, Pierini A, Alvarez M, Pan Y, Baker J, Buechele C, et al. CD4+ invariant natural killer T cells protect from murine GVHD lethality through expansion of donor CD4+CD25+FoxP3+ regulatory T cells. Blood. (2014) 124:3320–8. doi: 10.1182/blood-2014-05-576017
240. Schneidawind D, Baker J, Pierini A, Buechele C, Luong RH, Meyer EH, et al. Third-party CD4+ invariant natural killer T cells protect from murine GVHD lethality. Blood. (2015) 125:3491–500. doi: 10.1182/blood-2014-11-612762
241. Chen YB, Efebera YA, Johnston L, Ball ED, Avigan D, Lekakis LJ, et al. Increased Foxp3(+)Helios(+) regulatory T cells and decreased acute graft-versus-host disease after allogeneic bone marrow transplantation in patients receiving sirolimus and RGI-2001, an activator of invariant natural killer T cells. Biol Blood Marrow Transplant. (2017) 23:625–34. doi: 10.1016/j.bbmt.2017.01.069
242. Davies LC, Heldring N, Kadri N, Le Blanc K. Mesenchymal stromal cell secretion of programmed death-1 ligands regulates T cell mediated immunosuppression. Stem Cells. (2017) 35:766–76. doi: 10.1002/stem.2509
Keywords: graft-vs.-host disease, immune tolerance, alpha-1 antitrypsin, allogeneic hematopoietic stem cell transplantation, T regulatory cells
Citation: Thangavelu G and Blazar BR (2019) Achievement of Tolerance Induction to Prevent Acute Graft-vs.-Host Disease. Front. Immunol. 10:309. doi: 10.3389/fimmu.2019.00309
Received: 30 October 2018; Accepted: 06 February 2019;
Published: 06 March 2019.
Edited by:
Everett Meyer, Stanford University, United StatesReviewed by:
Birgit Sawitzki, Charité Medical University of Berlin, GermanyAlain Le Moine, Free University of Brussels, Belgium
Copyright © 2019 Thangavelu and Blazar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bruce R. Blazar, YmxhemEwMDFAdW1uLmVkdQ==