 Ming-Hsun Tsai
Ming-Hsun Tsai Li-Mei Pai
Li-Mei Pai Chien-Kuo Lee
Chien-Kuo Lee- 1Graduate Institute of Immunology, National Taiwan University College of Medicine, Taipei, Taiwan
- 2Department of Biochemistry and Molecular Biology, Chang Gung University, Taoyuan, Taiwan
- 3Molecular Medicine Research Center, Chang Gung University, Taoyuan, Taiwan
- 4Liver Research Center, Chang Gung Memorial Hospital, Taoyuan, Taiwan
Type I interferon (IFN-I) is induced during innate immune response and is required for initiating antiviral activity, growth inhibition, and immunomodulation. STAT1, STAT2, and STAT3 are activated in response to IFN-I stimulation. STAT1, STAT2, and IRF9 form ISGF3 complex which transactivates downstream IFN-stimulated genes and mediates antiviral response. However, the role of STAT3 remains to be characterized. Here, we review the multiple actions of STAT3 on suppressing IFN-I responses, including blocking IFN-I signaling, downregulating the expression of ISGF3 components, and antagonizing the transcriptional activity of ISGF3. Finally, we discuss the evolution of the suppressive activity of STAT3 and the therapeutic potential of STAT3 inhibitors in host defense against viral infections and IFN-I-associated diseases.
STAT3, an Overlooked Signal Mediator of IFN-I
STAT3 was originally identified as acute-phase response factor (APRF) that is activated by IL-6 and binds to the promoters of acute-phase protein genes in hepatocytes to regulate inflammatory responses (1–4). It is now known that STAT3 is widely expressed in different cells and is activated by an array of cytokines and growth factors to mediate various activities (5–8). Total body ablation of STAT3 results in embryonic lethality (9, 10), whereas tissue-specific knockout of STAT3 reveals multiple functions in immune system, including regulation of homeostasis of immune cells, such as B-cells (11) and granulocytes (12), survival and/or proliferation of thymocytes (13), and differentiation of plasma cells (14), Th2 (15), Treg (16), and follicular helper T (Tfh) cells (17). Although STAT3 controls RORγt expression and Th17 development (18, 19), it, however, is not required for generation of type 3 innate lymphoid cells (ILC3) whose master regulator is also RORγt (20).
Like other STAT proteins, the activity of STAT3 is also regulated by acetylation, methylation and other post-translational modification such as SUMOylation in addition to phosphorylation (21, 22). STAT3 is activated primarily by tyrosine phosphorylation at Y705. In addition, serine phosphorylation at S727 is required for full transactivation ability of STAT3. However, unphosphorylated STAT3 can still form dimers through its N-terminal domain (NTD) and induce transcription (23). Moreover, acetylation of STAT3 plays a positive role in STAT3 transcription ability. Mutation of STAT3 at lysine residue K685, a p300-acetylation site, inhibits dimerization of STAT3 (24, 25). In addition to K685, K49, and K87 at NTD of STAT3 can be acetylated by p300 in response to IL-6, which affects STAT3 downstream gene expression (26). Other than acetylation, K140 and K49 of STAT3 can be methylated and negatively and positively modulate STAT3-mediated transcription, respectively (27, 28). Furthermore, removal of sumoylation of STAT3 at K451 enhances its transcriptional activity by enhancing phosphorylation states (29). Together, these results suggest that multiple post-translational modifications of STAT3 modulate its activation and gene transcription.
Engagement of IFN-I to IFN receptor complex leads to activation of STAT1, STAT2, and STAT3 by tyrosine phosphorylation. STAT1 and STAT2 are considered to be the primordial signal mediators of IFN-I, as genetic ablation (30–33), hypomorphic mutation (33–35) or functional inactivation (35–38) of either molecule severely impairs the induction of IFN-stimulated genes (ISG) and IFN-I-mediated antiviral response in mice and humans. Nevertheless, STAT2 has also been reported to negatively regulate IFN-I response, either by constitutive phosphorylation at T387 to block ISGF3 formation and its DNA binding (39) or by IFN-I-induced phosphorylation at S287 (40) or S734 (41) with mechanisms yet to be defined. Moreover, STAT2 can serve as an adaptor to bridge the interaction between USP18 and IFNAR2, which inhibits ligand binding to the receptor, resulting in decreased receptor dimerization and signaling (42). These results suggest an emerging role of IFN-I signaling mediators in negative feedback regulation of IFN-I response.
While STAT3 is activated by IFN-I stimulation in various cell types in addition to STAT1 and STAT2 (43–47), the actual functions and biological significance of STAT3 in IFN-I response are less appreciated, probably due to relatively transient activation compared to STAT1 (3, 48, 49), impaired IFN-I-mediated, STAT3-dependent transcriptional activity (50, 51) or a dispensable role in some IFN-I-mediated activities (52–54). For example, IFN-I-induced growth stimulatory activity in the absence of either STAT1 or STAT2 is independent of STAT3 (52). STAT3 is non-essential and cannot compensate the loss of STAT1 for IFN-I- or IFN-II-induced antiviral response in U3A, a human epithelial cell line (53). Moreover, during Tfh differentiation, STAT3 is not involved in IFN-I-induced, STAT1-mediated upregulation of the key transcription factor Bcl6 in CD4 T cells (54). In fact, it has also been shown that low or no IFN-I-activated STAT3 is found in CD4 T cells of patients with relapsing remitting multiple sclerosis (55) and in normal melanocytes or melanoma cells from patients (56). However, it is still unclear why STAT3 is selectively activated by IFN-I in different cell types.
However, it becomes clear that STAT3 is not just activated by IFN-I, it also regulates IFN activity. In fact, emerging evidence suggests that STAT3 may function to fine-tune IFN-I response (57–60). Gain- and loss-of-function analyses suggest that STAT3 negatively regulates IFN-α-induced ISG expression and antiviral activity (60). STAT3KO Tfh cells displays a markedly elevated levels of a number of ISGs in addition to T-bet expression, resembling a Th1-like effector phenotype, which leads to impaired germinal center (GC) formation and antibody production during LCMV infection (61). Blockade of IFN-I signaling rescues the defect in LCMV-infected mice, suggesting a requirement of STAT3 for Tfh cell and a fine balance in signaling pathways following acute viral infection. Similarly, knockdown of STAT3 in highly STAT3 activated diffuse large B cell lymphoma (DLBCL) cells results in increased expression of several ISGs (58). Inhibition of STAT3 synergizes with lenalidomide, an IFN-I inducing agent, in suppressing the growth of DLBCL by augmenting IFN-I-induced cytotoxicity. In addition to ISGs, STAT3 deficiency also upregulates IFN-I production, which may contribute to enhanced antiviral response (60) and improved chemotherapeutic activity in tumors (62). Interestingly, STAT1 deficiency in murine macrophage results in a sustained activation of STAT3 in response to combined stimulation of TLR and IFN, leading to repressed production of cytokines like TNF and IL-12 (63). STAT3 depletion restores TLR-dependent inflammatory response in the absence of STAT1, suggesting a functional cross-regulation of STAT1 and STAT3 and an anti-inflammatory role of STAT3 in IFN and TLR response. Together, these studies suggest that STAT3 may function as one of the key regulatory molecules for IFN response.
Several potential mechanisms have been described to illustrate the direct and indirect actions of STAT3 to suppress IFN-I response. For example, STAT3 may sequester STAT1 and prevent it from forming functional homodimers, STAT3 can cooperate with repressors to inhibit ISGF3 binding to DNA, and STAT3 can directly reduce the expression of ISGF3 components. STAT3 can also induce a suppressor to attenuate IFN-I signaling or a miRNA to reduce the expression of ISGF3 components to indirectly block IFN-I response. These mechanisms will be elaborated further in the following section.
Mechanisms of Negative Regulation of IFN-I by STAT3
Sequestration of STAT1
Engagement of IFN-I to IFN receptor triggers the activation of STAT1, STAT2, and STAT3 and the formation of ISGF3 heterotrimer, consisting of STAT1, STAT2, and IRF9, and homodimers of STAT1:STAT1 or STAT3:STAT3 and heterodimer of STAT1:STAT3. It is conceivable that increased amounts of activated STAT3 can compete for binding with STAT1 to prevent the formation of STAT1:STAT1 homodimer. Indeed, overexpression of IFN-α-activated STAT3 inhibits STAT1-dependent gene expression, thereby downregulating the induction of proinflammatory cytokines, including CXCL9 and CXCL10 and a transcription factor IRF1, probably by sequestering activated STAT1 into STAT1:STAT3 heterodimers and reducing DNA binding of STAT1:STAT1 homodimers (57). Interestingly, increased amounts of activated STAT3 does not reduce ISGF3-driven expression of OAS and Mx2 genes. Instead, STAT3 seems to positively regulate their expression. Conversely, in mouse embryonic fibroblast cells lacking STAT3, IL-6 mediates IFNγ-like response with increased expression of MHC class II and antiviral state by inducing prolonged STAT1 activation and shifting homodimers of STAT1 or STAST3 and heterodimer of STAT1:STAT3 toward STAT1 homodimer only (64). Under physiological conditions, prolonged IFN-α treatment also affects STAT3-dependent IL-6 signaling by promoting formation of STAT1:STAT3 and STAT1:STAT1 complex, resulting in downregulation of STAT3 targets such as Bcl-XL, Mcl-1, and survivin and increased apoptosis (65). Likewise, IFN-α priming results in acquisition of pro-inflammatory function of IL-10, leading to expression of IFN-γ-inducible, STAT1-depedent genes (66). Therefore, IFN-I signaling can be antagonized by STAT3, at least, in two ways: to compete for STAT1 association and for DNA binding (Figure 1A).
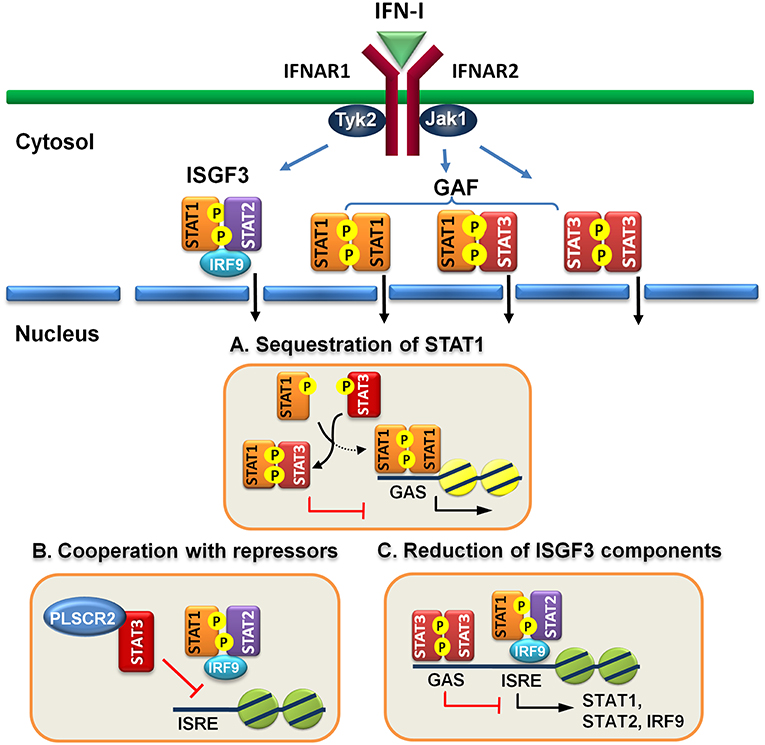
Figure 1. Direct regulatory mechanisms of STAT3 for IFN-I response. (A) Activated STAT3 sequesters activated STAT1 to form heterodimers and prevents STAT1 from forming functional homodimers to transactivate downstream genes. (B) STAT3 cooperates with other repressors, such as PLSCR2 to prevent ISGF3 from binding to DNA. (C) STAT3 binds directly to the promoters of ISGF3 components, including STAT1, STAT2, and IRF9 and suppress their expression.
Cooperation With Repressors
STAT3 is reported to interact with many nuclear proteins to either activate or suppress the functions of STAT3 in transactivation ability and signaling pathways (67). For example, PDZ and LIM domain 2 (PDLIM2), a nuclear E3 ligase, which can interact with STAT3 and terminate its transcription by promoting degradation of STAT3 (68). HDAC1/2 forms a complex with SIN3 transcription regulator homolog A (Sin3a) to suppress the transcriptional activity of STAT3:STAT3 dimers through deacetylation (69, 70). In addition, DAXX directly interacts with STAT3 in the nucleus, leading to suppressing STAT3-mediated transactivation, probably through recruitment of a DNA methyltransferase (71, 72). STAT3 activation is therefore tightly regulated at multiple levels to prevent hyperactivation of STAT3-mediated pathologies, including cancers, autoimmune and inflammatory disorders (73).
However, these proteins are mainly limited to the control of the functions of STAT3 per se. Recently, we found that STAT3 interacts and cooperates with phospholipid scramblase 2 (PLSCR2) to negatively regulate IFN-I response (59). PLSCR2 is also an ISG and is predominantly located to the nucleus. PLSCR2 does not affect IFN-I-dependent phosphorylation of STAT1 and STAT2, nuclear translocation of activated STATs, or assembly of ISGF3 complex. Instead, PLSCR2 suppresses the recruitment of ISGF3 to ISRE of ISGs, in a STAT3-dependent manner, to fine-tune IFN-I response (Figure 1B). PLSCR2 deficiency results in increased ISG expression and antiviral activity. Mutations in palmitoylation motif of PLSCR2 impairs the interactions between PLSCR2 and STAT3, leading to blockade of the suppressive activity. Interestingly, expression profile analysis reveals that in addition to ISGs, genes involved in inflammatory response are also highly enriched in PLSCR2KO cells in response to IFN-I stimulation (59). This is consistent with enhanced inflammatory response upon TLR stimulation (74, 75) and in bowl diseases in the absence of STAT3 due to incapability of inducing SOCS3, a negative regulator of cytokine signaling (76–78) and the lack of signaling through the receptor of an anti-inflammatory cytokine such as IL-10 (79).
Reduction of ISGF3 Components
Constitutive STAT3 activation by autocrine production of IL-6 and IL-10 is found in activated B cell-like (ABC) diffused large B cell lymphoma cell lines (DLBCL) (80, 81). Genome-wide analysis has identified ~2,200 STAT3 direct target genes which control different aspects of B cells, including activation, survival, proliferation, differentiation, and migration (58). STAT3 also regulates multiple oncogenic signaling pathways, including NF-κB, a cell-cycle checkpoint, PI3K/AKT/mTORC1, and STAT3 itself. Interestingly, constitutive STAT3 activation also suppresses the expression of ISGF3 components, such as IRF9, STAT1 and STAT2, and IRF7, a transcription factor for IFN-I production, through direct binding to the promoters of the genes (58). Therefore, STAT3 is able to directly reduce the levels of critical components in signaling pathway to weaken IFN-I response (Figure 1C).
MicroRNAs (miRs) are known to regulate IFN signaling pathways (82, 83). IFN-γ-activated STAT1 binds directly to the promoter of miR155 and induces its expression (84). Moreover, virus infection-induced miR155 also targets SOCS1 to feedback promote IFN-I-mediated antiviral activity (85). Therefore, STAT1 activation positively regulates its function by up-regulating miR-155 and down-regulating a STAT inhibitory factor SOCS1. STAT3, on other hand, stimulates miR-221/222 expression, which in turn targets PDLIM2 to stabilize and increase STAT3 levels (86). Knockdown of miR-221/222 results in upregulation of members of the IFN-α signaling pathway, including STAT1, STAT2, IRF9, and several ISGs in a human glioma cell line (87). Virus infection-induced upregulation of miR221 in peritoneal macrophages negatively regulates innate antiviral response against VSV (88). Therefore, STAT3-dependent transcription of miR-221/222 may function to downregulate STAT1 and STAT2 expression indirectly to antagonize IFN-I response (Figure 2A).
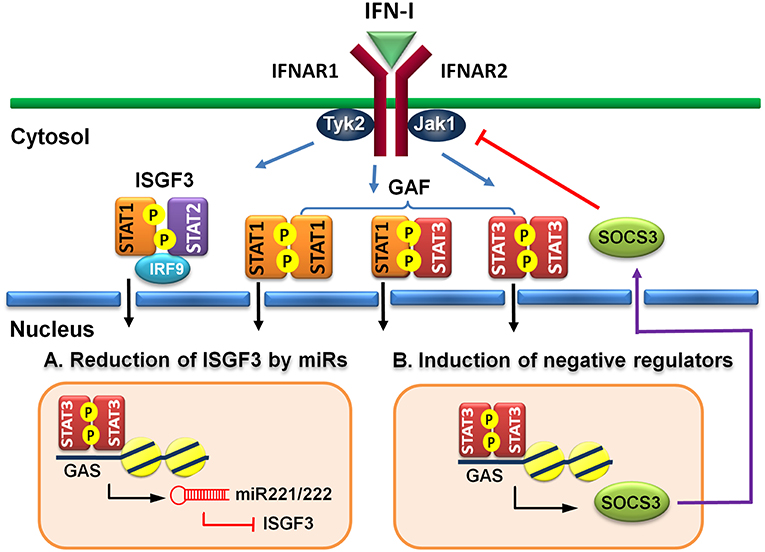
Figure 2. Indirect regulatory mechanisms of STAT3 for IFN-I responses. (A) STAT3 induces microRNA miR-221/222 to target ISGF3 components, including STAT1, STAT2, and IRF9 to reduce their de novo protein synthesis. (B) STAT3 induces a negative regulator, such as SOCS3 to block IFN-I signaling.
Induction of Negative Regulators
Suppressors of cytokine signaling (SOCS) family proteins are ISGs that feedback regulate cytokine signaling through various ways, including blocking receptor docking by activated STATs, inhibiting JAK kinase activity, and promoting degradation of activated JAKs and receptors (89, 90). IFN-I induces SOCS1 and SOCS3 expression in a STAT1- and STAT3-dependent manner, respectively (91). In addition to feedback regulation, the signaling pathways of STAT1 and STAT3 can cross-regulate by the induced SOCS1 and SOCS3 reciprocally (92, 93). Constitutive expression of SOCS3 inhibits IFN-α-induced STAT1 phosphorylation, ISG expression and anti-proliferative activity (94). HSV-1 (95) or IAV (96) infection-induced SOCS3 is responsible for the suppression of signaling and production of IFN-I and impaired antiviral response. Moreover, hepatic SOCS3 expression is also strongly associated with non-responsiveness to IFN-I therapy in HCV patients (97). Therefore, STAT3 can indirectly cross-regulate STAT1-mediated signaling and ISG expression at multiple layers of feedback control through induced SOCS3 (Figure 2B).
Viral Strategies to Exploit STAT3
Given that STAT3 can exert negative effects on IFN-I response, it is conceivable that viruses may exploit STAT3 to evade IFN-I-mediated antiviral immunity to facilitate their replication. Indeed, porcine epidemic diarrhea virus is shown to trigger STAT3 activation via stimulated EGFR signaling to enhance virus replication in an intestinal epithelial cell line (98). Inhibitors or siRNA to EGFR result in augmented expression of IFN-I and ISG genes and decreased viral yield. Similar results are also observed using the same approaches to block STAT3 activation, suggesting that attenuation of antiviral activity by EGFR activation requires STAT3 signaling pathway. EGFR- and IFN-signaling crosstalk is also known to play a role in regulating HCV replication (99). Erlotinib, an EGFR inhibitor, and IFN-α synergize to inhibit HCV infection in a hepatoma cell line (100). While STAT3 silencing or inhibition suppresses HCV infection, SOCS silencing impairs the synergistic antiviral activity of IFN-α and erlotinib. Therefore, EGFR may impair IFN antiviral response by suppressing SOCS3 expression, which relieves SOCS3-mediated antagonism of STAT3, thereby promoting virus replication (100).
Although virus targeting and inhibiting STAT3 seems to be counterintuitive because of its negative role in IFN-I response (57–60), several viruses are reported to degrade STAT3 protein or suppress its functions. For example, the V protein of Mumps virus (MuV) catalyzes proteasomal degradation of STAT1 and STAT3, resulting in blockade of IFN-I, IFN-II, and prevention of the responses to interleukin-6 and v-Src signals and induction of apoptosis in STAT3-dependent multiple myeloma cells and transformed murine fibroblasts (101). Hepatitis E virus (HEV) viral ORF3 protein (pORF3) blocks the nuclear translocation of p-STAT3, probably by impeding endocytosis of EGFR, resulting in downregulation of STAT3-stimulated acute-phase gene-driven reporter activity (102). While, the absence of STAT3 during MuV infection may reduce pro-inflammatory activity of IL-6 and IFN-γ, functional blockade of STAT3 by HEV may result in downregulation of the acute-phase response, a major determinant of inflammation in the host.
In fact, several viruses also boost or attenuate STAT3 functions to perturb immune response, alter cell architecture and tissue organization, prevent apoptosis or trigger cellular transformation to facilitate their replication, which have been thoroughly reviewed elsewhere (103, 104) and will not be discussed further.
Evolutionarily Conserved Feedback Regulation by STAT
The cytokine receptor (CytoR)-JAK-STAT is a highly conserved signaling pathway, which expands extensively in bilateria during early vertebrate evolution and is concurrent with the development of adaptive immune system (105, 106). In early jawed vertebrates, the regulation of IFN has been established through two rounds of whole-genome duplication that occurs between invertebrates and vertebrates to provide expanded signaling molecules, such as positive regulators, JAKs, STATs and IRFs, and negative regulators, protein inhibitor of activated STAT (PIAS) and SOCS (107).
There is only one STAT protein in Drosophila that is stat92E which is required for normal development of several tissues, including embryonic segmentation, imaginal discs, blood cells, and germ cells (108). A crustacean Pm-STAT is also identified from giant tiger prawn (Penaeus monodon) and the phosphorylated form of Pm-STAT is increased in lymphoid organ of the shrimp following white spot syndrome virus (WSSV) infection (109). RNA silencing of Lv-STAT in whiteleg shrimp (Litopenaeus vannamei) significantly reduces the copy number of WSSV and the mortality caused by WSSV infection (110). Moreover, treatment of a specific inhibitor of STAT3 (S3I-201) in hematopoietic tissue of crayfish (Cherax quadricarinatus) also decreases WSSV titers, suggesting a proviral role of the invertebrate STAT protein (110).
In addition to shrimp, an Ec-STAT3 identified from orange-spotted grouper (Epinephelus coioides) is activated and induced to translocate into nucleus following Singapore grouper iridovirus (SGIV) infection. Inhibition of Ec-STAT3 by RNA silencing or a small molecule inhibitor decreases SGIV replication and induces cell cycle arrest and downregulates the expression of prosurvival genes, such as Bcl-2, Bcl-xL, and Bax. Ec-STAT3 is also activated in fish by red-spotted grouper nervous necrosis virus (RGNNV). While inhibition of Ec-STAT3 does not affect RGNNV replication, virus infection-induced vacuolation and autophagy are significantly increased (111). Moreover, the expression of several proinflammatory factors, including TNFα, IL-1β, and IL-8, is also mediated by Ec-STAT3 during infection.
Therefore, these results suggest that microbial infection-triggered STAT signaling pathway is well conserved from invertebrates to vertebrates and that proviral and prosurvial role of STAT3 are probably also preserved from shrimp, fish to mammals, although the feedback regulation of IFN-I response by STAT3 in shrimp and fish remains unclear.
Is STAT3 Druggable or Undruggable?
As an oncogene and is activated in a wide range of malignant cells, STAT3 is considered to be one of the most important therapeutic targets, particularly, for cancers (112). However, there are still lack of clinically available STAT3 inhibitors due to insufficient potency and/or selectivity (113, 114). Currently, the STAT3 inhibitors under development include three categories: small molecule compounds, peptide-based molecules, and double-stranded oligodeoxynucleotide decoy (ODN-decoy) (115). Usually, small molecule compounds and peptide-based inhibitors are designed in a way to effectively reduce tyrosine phosphorylation and/or dimerization of STAT3 and block STAT3-mediated transcriptional activity. For example, SH2 domain of STAT3 is the main target of STAT3 inhibitors which are known to occupy or dock to pY-Tyr705-binding pocket. However, it is also highly homologous to SH2 domain of STAT1. Likewise, the DBD of STAT3 is also highly similar to that of STAT1. Therefore, the chances of cross-reacting and simultaneous suppression of the activity of STAT1 and STAT3 by an inhibitor are high. Indeed, inhibitors like Stattic and S3I-201 that target STAT3 SH2 domain are also reported to inhibit STAT1 phosphorylation and nuclear translocation (116, 117).
Since both STAT3 and STAT1 can recognize the same GAS element of some genes, it comes as no surprise that STAT3 DBD-targeting decoy ODN also binds STAT1 and reduces STAT1-dependent IFNγ-induced cell death (118). Therefore, the inhibitors targeting to either SH2 or DBD domain of STAT3 are likely to suppress STAT1-mediated signaling. In addition, targeting upstream of STAT3, such as JAKs, is also likely to exert a board spectrum of inhibitory effect on different STAT proteins. For example, AG490, AZD1480, and Sorafenib are also able to potently suppress STAT1 phosphorylation in addition to STAT3 phosphorylation (119–121).
When STAT3 inhibitors are used to boost antiviral responses, it is anticipated that STAT1-mediated antiviral responses will also be attenuated due to cross-reactivity. Nevertheless, several STAT3 inhibitors have been shown to enhance antiviral response in vitro or in vivo and display some therapeutic potential. For example, pretreatment of S3I-201 can reduce viral replication in human cytomegalovirus (HCMV), varicella-zoster virus, SGIV, or vesicular stomatitis virus (122–126). Moreover, Stattic can also reduce HCMV replication (126). Although these studies usually attribute the reduced viral replication to the proviral role of STAT3, it is also likely that reduced SOCS3 expression derepresses STAT1 to contribute to antiviral responses.
Despite the concerns raised in many literatures about the potential problems in developing STAT3 inhibitors, some of them are already in clinical trials, including 3 inhibitors targeting to SH2 of STAT3 and 1 inhibitor as an antisense ODN (127). Therefore, there is no definitely answer yet as STAT3 is druggable or not. Nevertheless, different approaches may need to be implanted to provide viable solutions for developing drugs that can selectively block the activity of STAT3.
Conclusions and Perspectives
While accumulating evidence suggests that STAT3 is a negative regulator of the IFN-I response, it only represents one of many other regulatory mechanisms for IFN-I signaling (128), suggesting the importance of balancing the pluripotent activities of IFN-I. In fact, this notion becomes evident with the emerging type I interferonopathies of autoinflammatory diseases, which are Mendelian disorders associated with an up-regulation of IFN-I signaling as a novel type of human inborn errors of immunity, including Aicardi-Goutières syndromes (AGS), familial chilblain lupus (FCL), bilateral striatal necrosis (BSN), STING-associated vasculopathy with onset in infancy (SAVI)…etc. (129). Mutations in STAT3 is known to cause many diseases in humans, including loss-of-function-induced autosomal dominant hyper IgE syndrome (AD-HIES), gain-of-function-induced malignancies and autoimmunity (73). However, it remains to be determined if functional deficiency of STAT3 will lead to type I interferonopathies.
Targeting STAT3 as a therapeutic approach for cancers and other diseases must take the multifaceted functions of STAT3 into consideration. As shown in this review that STAT3 not just possesses proviral and prosurvival activities, it also exhibits seemingly paradoxical roles in inflammation (130) and immune modulation (5, 131, 132). Moreover, due to sequence conservation between STAT1 and STAT3, developing highly selective drugs for STAT3 without affecting STAT1 is desirable, which is also the challenge for precision medicine to deal with these diseases.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by research grants from the Ministry of Science and Technology Taiwan (MOST 102-2320-B-002-030-MY3) and (MOST 105-2911-I-002-509), National Health Research Institutes, Taiwan (NHRI-EX107-10632SI) and the Excellent Translational Medicine Research Projects of National Taiwan University College of Medicine and National Taiwan University Hospital to C-KL and Chang Gung Memorial Hospital (CMRPD1H0191) to L-MP.
References
1. Akira S, Nishio Y, Inoue M, Wang XJ, Wei S, Matsusaka T, et al. Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell. (1994) 77:63–71. doi: 10.1016/0092-8674(94)90235-6
2. Lutticken C, Wegenka UM, Yuan J, Buschmann J, Schindler C, Ziemiecki A, et al. Association of transcription factor APRF and protein kinase Jak1 with the interleukin-6 signal transducer gp130. Science. (1994) 263:89–92. doi: 10.1126/science.8272872
3. Raz R, Durbin JE, Levy DE. Acute phase response factor and additional members of the interferon-stimulated gene factor 3 family integrate diverse signals from cytokines, interferons, and growth factors. J Biol Chem. (1994) 269:24391–5.
4. Zhong Z, Wen Z, Darnell JE Jr. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science. (1994) 264:95–8. doi: 10.1126/science.8140422
5. Hillmer EJ, Zhang H, Li HS, Watowich SS. STAT3 signaling in immunity. Cytokine Growth Factor Rev. (2016) 31:1–15. doi: 10.1016/j.cytogfr.2016.05.001
6. Huynh J, Chand A, Gough D, Ernst M. Therapeutically exploiting STAT3 activity in cancer - using tissue repair as a road map. Nat Rev Cancer. (2019) 19:82–96. doi: 10.1038/s41568-018-0090-8
8. Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer. (2014) 14:736–46. doi: 10.1038/nrc3818
9. Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N, et al. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc Natl Acad Sci USA. (1997) 94:3801–4. doi: 10.1073/pnas.94.8.3801
10. Raz R, Lee CK, Cannizzaro LA, d'Eustachio P, Levy DE. Essential role of STAT3 for embryonic stem cell pluripotency. Proc Natl Acad Sci USA. (1999) 96:2846–51. doi: 10.1073/pnas.96.6.2846
11. Chou WC, Levy DE, Lee CK. STAT3 positively regulates an early step in B-cell development. Blood. (2006) 108:3005–11. doi: 10.1182/blood-2006-05-024430
12. Lee C, Raz R, Gimeno R, Gertner R, Wistinghausen B, Takeshita K, et al. STAT3 is a negative regulator of granulopoiesis but is not required for G-CSF-dependent differentiation. Immunity. (2002) 17:63. doi: 10.1016/S1074-7613(02)00336-9
13. Sano S, Takahama Y, Sugawara T, Kosaka H, Itami S, Yoshikawa K, et al. Stat3 in thymic epithelial cells is essential for postnatal maintenance of thymic architecture and thymocyte survival. Immunity. (2001) 15:261–73. doi: 10.1016/S1074-7613(01)00180-7
14. Diehl SA, Schmidlin H, Nagasawa M, van Haren SD, Kwakkenbos MJ, Yasuda E, et al. STAT3-mediated up-regulation of BLIMP1 Is coordinated with BCL6 down-regulation to control human plasma cell differentiation. J Immunol. (2008) 180:4805–15. doi: 10.4049/jimmunol.180.7.4805
15. Stritesky GL, Muthukrishnan R, Sehra S, Goswami R, Pham D, Travers J, et al. The transcription factor STAT3 is required for T helper 2 cell development. Immunity. (2011) 34:39–49. doi: 10.1016/j.immuni.2010.12.013
16. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. (2006) 441:235–8. doi: 10.1038/nature04753
17. Eddahri F, Denanglaire S, Bureau F, Spolski R, Leonard WJ, Leo O, et al. Interleukin-6/STAT3 signaling regulates the ability of naive T cells to acquire B-cell help capacities. Blood. (2009) 113:2426–33. doi: 10.1182/blood-2008-04-154682
18. Harris TJ, Grosso JF, Yen HR, Xin H, Kortylewski M, Albesiano E, et al. Cutting edge: an in vivo requirement for STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J Immunol. (2007) 179:4313–7. doi: 10.4049/jimmunol.179.7.4313
19. Mathur AN, Chang HC, Zisoulis DG, Stritesky GL, Yu Q, O'Malley JT, et al. Stat3 and Stat4 direct development of IL-17-secreting Th cells. J Immunol. (2007) 178:4901–7. doi: 10.4049/jimmunol.178.8.4901
20. Guo X, Qiu J, Tu T, Yang X, Deng L, Anders RA, et al. Induction of innate lymphoid cell-derived interleukin-22 by the transcription factor STAT3 mediates protection against intestinal infection. Immunity. (2014) 40:25–39. doi: 10.1016/j.immuni.2013.10.021
21. Zhuang S. Regulation of STAT signaling by acetylation. Cell Signal. (2013) 25:1924–31. doi: 10.1016/j.cellsig.2013.05.007
22. Qi QR, Yang ZM. Regulation and function of signal transducer and activator of transcription 3. World J Biol Chem. (2014) 5:231–9. doi: 10.4331/wjbc.v5.i2.231
23. Yang J, Stark GR. Roles of unphosphorylated STATs in signaling. Cell Res. (2008) 18:443–51. doi: 10.1038/cr.2008.41
24. Wang R, Cherukuri P, Luo J. Activation of Stat3 sequence-specific DNA binding and transcription by p300/CREB-binding protein-mediated acetylation. J Biol Chem. (2005) 280:11528–34. doi: 10.1074/jbc.M413930200
25. Yuan ZL, Guan YJ, Chatterjee D, Chin YE. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science. (2005) 307:269–73. doi: 10.1126/science.1105166
26. Ray S, Boldogh I, Brasier AR. STAT3 NH2-terminal acetylation is activated by the hepatic acute-phase response and required for IL-6 induction of angiotensinogen. Gastroenterology. (2005) 129:1616–32. doi: 10.1053/j.gastro.2005.07.055
27. Yang J, Huang J, Dasgupta M, Sears N, Miyagi M, Wang B, et al. Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proc Natl Acad Sci USA. (2010) 107:21499–504. doi: 10.1073/pnas.1016147107
28. Dasgupta M, Dermawan JK, Willard B, Stark GR. STAT3-driven transcription depends upon the dimethylation of K49 by EZH2. Proc Natl Acad Sci USA. (2015) 112:3985–90. doi: 10.1073/pnas.1503152112
29. Zhou Z, Wang M, Li J, Xiao M, Chin YE, Cheng J, et al. SUMOylation and SENP3 regulate STAT3 activation in head and neck cancer. Oncogene. (2016) 35:5826–38. doi: 10.1038/onc.2016.124
30. Durbin JE, Hackenmiller R, Simon MC, Levy DE. Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell. (1996) 84:443–50. doi: 10.1016/S0092-8674(00)81289-1
31. Meraz MA, White JM, Sheehan KC, Bach EA, Rodig SJ, Dighe AS, et al. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell. (1996) 84:431–42. doi: 10.1016/S0092-8674(00)81288-X
32. Park C, Li S, Cha E, Schindler C. Immune response in Stat2 knockout mice. Immunity. (2000) 13:795–804. doi: 10.1016/S1074-7613(00)00077-7
33. Hambleton S, Goodbourn S, Young DF, Dickinson P, Mohamad SM, Valappil M, et al. STAT2 deficiency and susceptibility to viral illness in humans. Proc Natl Acad Sci USA. (2013) 110:3053–8. doi: 10.1073/pnas.1220098110
34. Chen LS, Wei PC, Liu T, Kao CH, Pai LM, Lee CK. STAT2 hypomorphic mutant mice display impaired dendritic cell development and antiviral response. J Biomed Sci. (2009) 16:22. doi: 10.1186/1423-0127-16-22
35. Dupuis S, Jouanguy E, Al-Hajjar S, Fieschi C, Al-Mohsen IZ, Al-Jumaah S, et al. Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat Genet. (2003) 33:388–91. doi: 10.1038/ng1097
36. Dupuis S, Dargemont C, Fieschi C, Thomassin N, Rosenzweig S, Harris J, et al. Impairment of mycobacterial but not viral immunity by a germline human STAT1 mutation. Science. (2001) 293:300–3. doi: 10.1126/science.1061154
37. Sampaio EP, Bax HI, Hsu AP, Kristosturyan E, Pechacek J, Chandrasekaran P, et al. A novel STAT1 mutation associated with disseminated mycobacterial disease. J Clin Immunol. (2012) 32:681–9. doi: 10.1007/s10875-012-9659-2
38. Tsumura M, Okada S, Sakai H, Yasunaga S, Ohtsubo M, Murata T, et al. Dominant-negative STAT1 SH2 domain mutations in unrelated patients with Mendelian susceptibility to mycobacterial disease. Hum Mutat. (2012) 33:1377–87. doi: 10.1002/humu.22113
39. Wang Y, Nan J, Willard B, Wang X, Yang J, Stark GR. Negative regulation of type I IFN signaling by phosphorylation of STAT2 on T387. EMBO J. (2017) 36:202–12. doi: 10.15252/embj.201694834
40. Steen HC, Nogusa S, Thapa RJ, Basagoudanavar SH, Gill AL, Merali S, et al. Identification of STAT2 serine 287 as a novel regulatory phosphorylation site in type I interferon-induced cellular responses. J Biol Chem. (2013) 288:747–58. doi: 10.1074/jbc.M112.402529
41. Steen HC, Kotredes KP, Nogusa S, Harris MY, Balachandran S, Gamero AM. Phosphorylation of STAT2 on serine-734 negatively regulates the IFN-alpha-induced antiviral response. J Cell Sci. (2016) 129:4190–9. doi: 10.1242/jcs.185421
42. Arimoto KI, Lochte S, Stoner SA, Burkart C, Zhang Y, Miyauchi S, et al. STAT2 is an essential adaptor in USP18-mediated suppression of type I interferon signaling. Nat Struct Mol Biol. (2017) 24:279–89. doi: 10.1038/nsmb.3378
43. Gonzalez-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nat Rev Immunol. (2012) 12:125–35. doi: 10.1038/nri3133
44. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. (2014) 14:36–49. doi: 10.1038/nri3581
45. Katze MG, He Y, Gale M Jr. Viruses and interferon: a fight for supremacy. Nat Rev Immunol. (2002) 2:675–87. doi: 10.1038/nri888
46. Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. (2005) 5:375–86. doi: 10.1038/nri1604
47. van Boxel-Dezaire AH, Rani MR, Stark GR. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity. (2006) 25:361–72. doi: 10.1016/j.immuni.2006.08.014
48. Tomic J, Lichty B, Spaner DE. Aberrant interferon-signaling is associated with aggressive chronic lymphocytic leukemia. Blood. (2011) 117:2668–80. doi: 10.1182/blood-2010-05-285999
49. van Boxel-Dezaire AH, Zula JA, Xu Y, Ransohoff RM, Jacobberger JW, Stark GR. Major differences in the responses of primary human leukocyte subsets to IFN-beta. J Immunol. (2010) 185:5888–99. doi: 10.4049/jimmunol.0902314
50. Icardi L, Lievens S, Mori R, Piessevaux J, De Cauwer L, De Bosscher K, et al. Opposed regulation of type I IFN-induced STAT3 and ISGF3 transcriptional activities by histone deacetylases (HDACS) 1 and 2. FASEB J. (2012) 26:240–9. doi: 10.1096/fj.11-191122
51. Icardi L, Mori R, Gesellchen V, Eyckerman S, De Cauwer L, Verhelst J, et al. The Sin3a repressor complex is a master regulator of STAT transcriptional activity. Proc Natl Acad Sci USA. (2012) 109:12058–63. doi: 10.1073/pnas.1206458109
52. Gimeno R, Lee CK, Schindler C, Levy DE. Stat1 and Stat2 but not Stat3 arbitrate contradictory growth signals elicited by alpha/beta interferon in T lymphocytes. Mol Cell Biol. (2005) 25:5456–65. doi: 10.1128/MCB.25.13.5456-5465.2005
53. Horvath CM, Darnell JE, Jr. The antiviral state induced by alpha interferon and gamma interferon requires transcriptionally active Stat1 protein. J Virol. (1996) 70:647–50.
54. Nakayamada S, Poholek AC, Lu KT, Takahashi H, Kato M, Iwata S, et al. Type I IFN induces binding of STAT1 to Bcl6: divergent roles of STAT family transcription factors in the T follicular helper cell genetic program. J Immunol. (2014) 192:2156–66. doi: 10.4049/jimmunol.1300675
55. Zula JA, Green HC, Ransohoff RM, Rudick RA, Stark GR, van Boxel-Dezaire AH. The role of cell type-specific responses in IFN-beta therapy of multiple sclerosis. Proc Natl Acad Sci USA. (2011) 108:19689–94. doi: 10.1073/pnas.1117347108
56. Humpolikova-Adamkova L, Kovarik J, Dusek L, Lauerova L, Boudny V, Fait V, et al. Interferon-alpha treatment may negatively influence disease progression in melanoma patients by hyperactivation of STAT3 protein. Eur J Cancer. (2009) 45:1315–23. doi: 10.1016/j.ejca.2009.01.009
57. Ho HH, Ivashkiv LB. Role of STAT3 in type I interferon responses. Negative regulation of STAT1-dependent inflammatory gene activation. J Biol Chem. (2006) 281:14111–8. doi: 10.1074/jbc.M511797200
58. Lu L, Zhu F, Zhang M, Li Y, Drennan AC, Kimpara S, et al. Gene regulation and suppression of type I interferon signaling by STAT3 in diffuse large B cell lymphoma. Proc Natl Acad Sci USA. (2018) 115:E498-e505. doi: 10.1073/pnas.1715118115
59. Tsai MH, Lee CK. STAT3 cooperates with phospholipid scramblase 2 to suppress type I interferon response. Front Immunol. (2018) 9:1886. doi: 10.3389/fimmu.2018.01886
60. Wang WB, Levy DE, Lee CK. STAT3 negatively regulates type I IFN-mediated antiviral response. J Immunol. (2011) 187:2578–85. doi: 10.4049/jimmunol.1004128
61. Ray JP, Marshall HD, Laidlaw BJ, Staron MM, Kaech SM, Craft J. Transcription factor STAT3 and type I interferons are corepressive insulators for differentiation of follicular helper and T helper 1 cells. Immunity. (2014) 40:367–77. doi: 10.1016/j.immuni.2014.02.005
62. Yang H, Yamazaki T, Pietrocola F, Zhou H, Zitvogel L, Ma Y, et al. STAT3 Inhibition enhances the therapeutic efficacy of immunogenic chemotherapy by stimulating type 1 interferon production by cancer cells. Cancer Res. (2015) 75:3812–22. doi: 10.1158/0008-5472.CAN-15-1122
63. Kim HS, Kim DC, Kim HM, Kwon HJ, Kwon SJ, Kang SJ, et al. STAT1 deficiency redirects IFN signalling toward suppression of TLR response through a feedback activation of STAT3. Sci Rep. (2015) 5:13414. doi: 10.1038/srep13414
64. Costa-Pereira AP, Tininini S, Strobl B, Alonzi T, Schlaak JF, Is'harc H, et al. Mutational switch of an IL-6 response to an interferon-gamma-like response. Proc Natl Acad Sci USA. (2002) 99:8043–7. doi: 10.1073/pnas.122236099
65. Thyrell L, Arulampalam V, Hjortsberg L, Farnebo M, Grander D, Pokrovskaja Tamm K. Interferon alpha induces cell death through interference with interleukin 6 signaling and inhibition of STAT3 activity. Exp Cell Res. (2007) 313:4015–24. doi: 10.1016/j.yexcr.2007.08.007
66. Sharif MN, Tassiulas I, Hu Y, Mecklenbrauker I, Tarakhovsky A, Ivashkiv LB. IFN-alpha priming results in a gain of proinflammatory function by IL-10: implications for systemic lupus erythematosus pathogenesis. J Immunol. (2004) 172:6476–81. doi: 10.4049/jimmunol.172.10.6476
67. Matsuda T, Muromoto R, Sekine Y, Togi S, Kitai Y, Kon S, et al. Signal transducer and activator of transcription 3 regulation by novel binding partners. World J Biol Chem. (2015) 6:324–32. doi: 10.4331/wjbc.v6.i4.324
68. Tanaka T, Yamamoto Y, Muromoto R, Ikeda O, Sekine Y, Grusby MJ, et al. PDLIM2 inhibits T helper 17 cell development and granulomatous inflammation through degradation of STAT3. Sci Signal. (2011) 4:ra85. doi: 10.1126/scisignal.2001637
69. Yang XJ, Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol. (2008) 9:206–18. doi: 10.1038/nrm2346
70. Marie IJ, Chang HM, Levy DE. HDAC stimulates gene expression through BRD4 availability in response to IFN and in interferonopathies. J Exp Med. (2018) 2018:520. doi: 10.1084/jem.20180520
71. Muromoto R, Nakao K, Watanabe T, Sato N, Sekine Y, Sugiyama K, et al. Physical and functional interactions between Daxx and STAT3. Oncogene. (2006) 25:2131–6. doi: 10.1038/sj.onc.1209235
72. Puto LA, Reed JC. Daxx represses RelB target promoters via DNA methyltransferase recruitment and DNA hypermethylation. Genes Dev. (2008) 22:998–1010. doi: 10.1101/gad.1632208
73. Vogel TP, Milner JD, Cooper MA. The ying and yang of STAT3 in human disease. J Clin Immunol. (2015) 35:615–23. doi: 10.1007/s10875-015-0187-8
74. Murray PJ. Understanding and exploiting the endogenous interleukin-10/STAT3-mediated anti-inflammatory response. Curr Opin Pharmacol. (2006) 6:379–86. doi: 10.1016/j.coph.2006.01.010
75. Hu X, Chakravarty SD, Ivashkiv LB. Regulation of interferon and Toll-like receptor signaling during macrophage activation by opposing feedforward and feedback inhibition mechanisms. Immunol Rev. (2008) 226:41–56. doi: 10.1111/j.1600-065X.2008.00707.x
76. Suzuki A, Hanada T, Mitsuyama K, Yoshida T, Kamizono S, Hoshino T, et al. CIS3/SOCS3/SSI3 plays a negative regulatory role in STAT3 activation and intestinal inflammation. J Exp Med. (2001) 193:471–81. doi: 10.1084/jem.193.4.471
77. Takeda K, Clausen BE, Kaisho T, Tsujimura T, Terada N, Forster I, et al. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. (1999) 10:39–49. doi: 10.1016/S1074-7613(00)80005-9
78. Melillo JA, Song L, Bhagat G, Blazquez AB, Plumlee CR, Lee C, et al. Dendritic cell (DC)-specific targeting reveals Stat3 as a negative regulator of DC function. J Immunol. (2010) 184:2638–45. doi: 10.4049/jimmunol.0902960
79. Murray PJ. The primary mechanism of the IL-10-regulated antiinflammatory response is to selectively inhibit transcription. Proc Natl Acad Sci USA. (2005) 102:8686–91. doi: 10.1073/pnas.0500419102
80. Ding BB, Yu JJ, Yu RY, Mendez LM, Shaknovich R, Zhang Y, et al. Constitutively activated STAT3 promotes cell proliferation and survival in the activated B-cell subtype of diffuse large B-cell lymphomas. Blood. (2008) 111:1515–23. doi: 10.1182/blood-2007-04-087734
81. Lam LT, Wright G, Davis RE, Lenz G, Farinha P, Dang L, et al. Cooperative signaling through the signal transducer and activator of transcription 3 and nuclear factor-{kappa}B pathways in subtypes of diffuse large B-cell lymphoma. Blood. (2008) 111:3701–13. doi: 10.1182/blood-2007-09-111948
82. Forster SC, Tate MD, Hertzog PJ. MicroRNA as type I interferon-regulated transcripts and modulators of the innate immune response. Front Immunol. (2015) 6:334. doi: 10.3389/fimmu.2015.00334
83. Kohanbash G, Okada H. MicroRNAs and STAT interplay. Semin Cancer Biol. (2012) 22:70–5. doi: 10.1016/j.semcancer.2011.12.010
84. Kutty RK, Nagineni CN, Samuel W, Vijayasarathy C, Hooks JJ, Redmond TM. Inflammatory cytokines regulate microRNA-155 expression in human retinal pigment epithelial cells by activating JAK/STAT pathway. Biochem Biophys Res Commun. (2010) 402:390–5. doi: 10.1016/j.bbrc.2010.10.042
85. Wang P, Hou J, Lin L, Wang C, Liu X, Li D, et al. Inducible microRNA-155 feedback promotes type I IFN signaling in antiviral innate immunity by targeting suppressor of cytokine signaling 1. J Immunol. (2010) 185:6226–33. doi: 10.4049/jimmunol.1000491
86. Liu S, Sun X, Wang M, Hou Y, Zhan Y, Jiang Y, et al. A microRNA 221- and 222-mediated feedback loop maintains constitutive activation of NFkappaB and STAT3 in colorectal cancer cells. Gastroenterology. (2014) 147:847–59 e11. doi: 10.1053/j.gastro.2014.06.006
87. Zhang C, Han L, Zhang A, Yang W, Zhou X, Pu P, et al. Global changes of mRNA expression reveals an increased activity of the interferon-induced signal transducer and activator of transcription (STAT) pathway by repression of miR-221/222 in glioblastoma U251 cells. Int J Oncol. (2010) 36:1503–12. doi: 10.3892/ijo_00000637
88. Du H, Cui S, Li Y, Yang G, Wang P, Fikrig E, et al. MiR-221 negatively regulates innate anti-viral response. PLoS ONE. (2018) 13:e0200385. doi: 10.1371/journal.pone.0200385
89. Alexander WS, Hilton DJ. The role of suppressors of cytokine signaling (SOCS) proteins in regulation of the immune response. Annu Rev Immunol. (2004) 22:503–29. doi: 10.1146/annurev.immunol.22.091003.090312
90. Yasukawa H, Sasaki A, Yoshimura A. Negative regulation of cytokine signaling pathways. Ann Rev Immunol. (2000) 18:143–64. doi: 10.1146/annurev.immunol.18.1.143
91. Qin H, Niyongere SA, Lee SJ, Baker BJ, Benveniste EN. Expression and functional significance of SOCS-1 and SOCS-3 in astrocytes. J Immunol. (2008) 181:3167–76. doi: 10.4049/jimmunol.181.5.3167
92. Croker BA, Krebs DL, Zhang JG, Wormald S, Willson TA, Stanley EG, et al. SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol. (2003) 4:540–5. doi: 10.1038/ni931
93. Hong F, Jaruga B, Kim WH, Radaeva S, El-Assal ON, Tian Z, et al. Opposing roles of STAT1 and STAT3 in T cell-mediated hepatitis: regulation by SOCS. J Clin Invest. (2002) 110:1503–13. doi: 10.1172/JCI0215841
94. Sakai I, Takeuchi K, Yamauchi H, Narumi H, Fujita S. Constitutive expression of SOCS3 confers resistance to IFN-alpha in chronic myelogenous leukemia cells. Blood. (2002) 100:2926–31. doi: 10.1182/blood-2002-01-0073
95. Yokota S, Yokosawa N, Okabayashi T, Suzutani T, Miura S, Jimbow K, et al. Induction of suppressor of cytokine signaling-3 by herpes simplex virus type 1 contributes to inhibition of the interferon signaling pathway. J Virol. (2004) 78:6282–6. doi: 10.1128/JVI.78.12.6282-6286.2004
96. Pauli EK, Schmolke M, Wolff T, Viemann D, Roth J, Bode JG, et al. Influenza A virus inhibits type I IFN signaling via NF-kappaB-dependent induction of SOCS-3 expression. PLoS Pathog. (2008) 4:e1000196. doi: 10.1371/journal.ppat.1000196
97. Kim KA, Lin W, Tai AW, Shao RX, Weinberg E, De Sa Borges CB, et al. Hepatic SOCS3 expression is strongly associated with non-response to therapy and race in HCV and HCV/HIV infection. J Hepatol. (2009) 50:705–11. doi: 10.1016/j.jhep.2008.12.021
98. Yang L, Xu J, Guo L, Guo T, Zhang L, Feng L, et al. Porcine epidemic diarrhea virus-induced epidermal growth factor receptor activation impairs the antiviral activity of type I interferon. J Virol. (2018) 92:17. doi: 10.1128/JVI.02095-17
99. Schoggins JW. Regulating interferon antiviral activity: a role for epidermal growth factor receptor. Hepatology. (2013) 58:1200–2. doi: 10.1002/hep.26486
100. Lupberger J, Duong FH, Fofana I, Zona L, Xiao F, Thumann C, et al. Epidermal growth factor receptor signaling impairs the antiviral activity of interferon-alpha. Hepatology. (2013) 58:1225–35. doi: 10.1002/hep.26404
101. Ulane CM, Rodriguez JJ, Parisien JP, Horvath CM. STAT3 ubiquitylation and degradation by mumps virus suppress cytokine and oncogene signaling. J Virol. (2003) 77:6385–93. doi: 10.1128/JVI.77.11.6385-6393.2003
102. Chandra V, Kar-Roy A, Kumari S, Mayor S, Jameel S. The hepatitis E virus ORF3 protein modulates epidermal growth factor receptor trafficking, STAT3 translocation, and the acute-phase response. J Virol. (2008) 82:7100–10. doi: 10.1128/JVI.00403-08
103. Roca Suarez AA, Van Renne N, Baumert TF, Lupberger J. Viral manipulation of STAT3: Evade, exploit, and injure. PLoS Pathog. (2018) 14:e1006839. doi: 10.1371/journal.ppat.1006839
104. Kuchipudi SV. The complex role of STAT3 in viral infections. J Immunol Res. (2015) 2015:272359. doi: 10.1155/2015/272359
105. Liongue C, Taznin T, Ward AC. Signaling via the CytoR/JAK/STAT/SOCS pathway: emergence during evolution. Mol Immunol. (2016) 71:166–75. doi: 10.1016/j.molimm.2016.02.002
106. Liongue C, Sertori R, Ward AC. Evolution of cytokine receptor signaling. J Immunol. (2016) 197:11–8. doi: 10.4049/jimmunol.1600372
107. Secombes CJ, Zou J. Evolution of interferons and interferon receptors. Front Immunol. (2017) 8:209. doi: 10.3389/fimmu.2017.00209
108. Arbouzova NI, Zeidler MP. JAK/STAT signalling in Drosophila: insights into conserved regulatory and cellular functions. Development. (2006) 133:2605–16. doi: 10.1242/dev.02411
109. Chen WY, Ho KC, Leu JH, Liu KF, Wang HC, Kou GH, et al. WSSV infection activates STAT in shrimp. Dev Compar Immunol. (2008) 32:1142–50. doi: 10.1016/j.dci.2008.03.003
110. Wen R, Li F, Li S, Xiang J. Function of shrimp STAT during WSSV infection. Fish Shellfish Immunol. (2014) 38:354–60. doi: 10.1016/j.fsi.2014.04.002
111. Huang Y, Huang X, Yang Y, Wang W, Yu Y, Qin Q. Involvement of fish signal transducer and activator of transcription 3 (STAT3) in nodavirus infection induced cell death. Fish Shellfish Immunol. (2015) 43:241–8. doi: 10.1016/j.fsi.2014.12.031
112. Huynh J, Etemadi N, Hollande F, Ernst M, Buchert M. The JAK/STAT3 axis: a comprehensive drug target for solid malignancies. Semin Cancer Biol. (2017) 45:13–22. doi: 10.1016/j.semcancer.2017.06.001
113. Xiong A, Yang Z, Shen Y, Zhou J, Shen Q. Transcription factor STAT3 as a novel molecular target for cancer prevention. Cancers. (2014) 6:926–57. doi: 10.3390/cancers6020926
114. Wong ALA, Hirpara JL, Pervaiz S, Eu JQ, Sethi G, Goh BC. Do STAT3 inhibitors have potential in the future for cancer therapy? Expert Opin Investig Drugs. (2017) 26:883–7. doi: 10.1080/13543784.2017.1351941
115. Wake MS, Watson CJ. STAT3 the oncogene - still eluding therapy? FEBS J. (2015) 282:2600–11. doi: 10.1111/febs.13285
116. Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol. (2006) 13:1235–42. doi: 10.1016/j.chembiol.2006.09.018
117. Lin L, Benson DM Jr., DeAngelis S, Bakan CE, Li PK, Li C, et al. A small molecule, LLL12 inhibits constitutive STAT3 and IL-6-induced STAT3 signaling and exhibits potent growth suppressive activity in human multiple myeloma cells. Int J Cancer. (2012) 130:1459–69. doi: 10.1002/ijc.26152
118. Tadlaoui Hbibi A, Laguillier C, Souissi I, Lesage D, Le Coquil S, Cao A, et al. Efficient killing of SW480 colon carcinoma cells by a signal transducer and activator of transcription (STAT) 3 hairpin decoy oligodeoxynucleotide–interference with interferon-gamma-STAT1-mediated killing. FEBS J. (2009) 276:2505–15. doi: 10.1111/j.1742-4658.2009.06975.x
119. Derenzini E, Lemoine M, Buglio D, Katayama H, Ji Y, Davis RE, et al. The JAK inhibitor AZD1480 regulates proliferation and immunity in Hodgkin lymphoma. Blood Cancer J. (2011) 1:e46. doi: 10.1038/bcj.2011.46
120. Kobayashi A, Tanizaki Y, Kimura A, Ishida Y, Nosaka M, Toujima S, et al. AG490, a Jak2 inhibitor, suppressed the progression of murine ovarian cancer. Eur J Pharmacol. (2015) 766:63–75. doi: 10.1016/j.ejphar.2015.09.039
121. Martin del Campo SE, Levine KM, Mundy-Bosse BL, Grignol VP, Fairchild ET, Campbell AR, et al. The Raf kinase inhibitor sorafenib inhibits JAK-STAT signal transduction in human immune cells. J Immunol. (2015) 195:1995–2005. doi: 10.4049/jimmunol.1400084
122. Marozin S, Altomonte J, Munoz-Alvarez KA, Rizzani A, De Toni EN, Thasler WE, et al. STAT3 inhibition reduces toxicity of oncolytic VSV and provides a potentially synergistic combination therapy for hepatocellular carcinoma. Cancer Gene Ther. (2015) 22:317–25. doi: 10.1038/cgt.2015.23
123. Sen N, Che X, Rajamani J, Zerboni L, Sung P, Ptacek J, et al. Signal transducer and activator of transcription 3 (STAT3) and survivin induction by varicella-zoster virus promote replication and skin pathogenesis. Proc Natl Acad Sci USA. (2012) 109:600–5. doi: 10.1073/pnas.1114232109
124. Huang X, Huang Y, Yang Y, Wei S, Qin Q. Involvement of fish signal transducer and activator of transcription 3 (STAT3) in SGIV replication and virus induced paraptosis. Fish Shellfish Immunol. (2014) 41:308–16. doi: 10.1016/j.fsi.2014.09.011
125. Reitsma JM, Terhune SS. Inhibition of cellular STAT3 synergizes with the cytomegalovirus kinase inhibitor maribavir to disrupt infection. Antiviral Res. (2013) 100:321–7. doi: 10.1016/j.antiviral.2013.09.011
126. Reitsma JM, Sato H, Nevels M, Terhune SS, Paulus C. Human cytomegalovirus IE1 protein disrupts interleukin-6 signaling by sequestering STAT3 in the nucleus. J Virol. (2013) 87:10763–76. doi: 10.1128/JVI.01197-13
127. Johnson DE, O'Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol. (2018) 15:234–48. doi: 10.1038/nrclinonc.2018.8
128. Arimoto KI, Miyauchi S, Stoner SA, Fan JB, Zhang DE. Negative regulation of type I IFN signaling. J Leukocyte Biol. (2018) 103:1099–116. doi: 10.1002/JLB.2MIR0817-342R
129. Rodero MP, Crow YJ. Type I interferon-mediated monogenic autoinflammation: the type I interferonopathies, a conceptual overview. J Exp Med. (2016) 213:2527–38. doi: 10.1084/jem.20161596
130. Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta. (2011) 1813:878–88. doi: 10.1016/j.bbamcr.2011.01.034
131. Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. (2007) 7:41–51. doi: 10.1038/nri1995
Keywords: type I interferon (IFN-I), STAT3, phospholipid scramblase 2, antiviral immunity, SOCS3
Citation: Tsai M-H, Pai L-M and Lee C-K (2019) Fine-Tuning of Type I Interferon Response by STAT3. Front. Immunol. 10:1448. doi: 10.3389/fimmu.2019.01448
Received: 08 January 2019; Accepted: 10 June 2019;
Published: 26 June 2019.
Edited by:
Fabrizio Mattei, Istituto Superiore di Sanità (ISS), ItalyReviewed by:
Ana Maria Gamero, Temple University, United StatesMarkus Johannes Hofer, University of Sydney, Australia
Copyright © 2019 Tsai, Pai and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chien-Kuo Lee, bGVlY2tAbnR1LmVkdS50dw==