 Jelena Milovanovic1,2
Jelena Milovanovic1,2 Aleksandar Arsenijevic1
Aleksandar Arsenijevic1 Bojana Stojanovic1,3
Bojana Stojanovic1,3 Tatjana Kanjevac4
Tatjana Kanjevac4 Dragana Arsenijevic1,5
Dragana Arsenijevic1,5 Gordana Radosavljevic1
Gordana Radosavljevic1 Marija Milovanovic1*Nebojsa Arsenijevic1
Marija Milovanovic1*Nebojsa Arsenijevic1- 1Faculty of Medical Sciences, Center for Molecular Medicine and Stem Cell Research, University of Kragujevac, Kragujevac, Serbia
- 2Department of Histology and Embriology, Faculty of Medical Sciences, University of Kragujevac, Kragujevac, Serbia
- 3Department of Pathophysiology, Faculty of Medical Sciences, University of Kragujevac, Kragujevac, Serbia
- 4Department of Dentistry, Faculty of Medical Sciences, University of Kragujevac, Kragujevac, Serbia
- 5Department of Pharmacy, Faculty of Medical Sciences, University of Kragujevac, Kragujevac, Serbia
A critical role for IL-17, a cytokine produced by T helper 17 (Th17) cells, has been indicated in the pathogenesis of chronic inflammatory and autoimmune diseases. A positive effect of blockade of IL-17 secreted by autoreactive T cells has been shown in various inflammatory diseases. Several cytokines, whose production is affected by environmental factors, control Th17 differentiation and its maintenance in tissues during chronic inflammation. The roles of IL-17 in the pathogenesis of chronic neuroinflammatory conditions, multiple sclerosis (MS), experimental autoimmune encephalomyelitis (EAE), Alzheimer's disease, and ischemic brain injury are reviewed here. The role of environmental stimuli in Th17 differentiation is also summarized, highlighting the role of viral infection in the regulation of pathogenic T helper cells in EAE.
Introduction
Interleukin-17 (IL-17) is the first-described and founder member of the IL-17 family of inflammatory cytokines, which contains six members: IL-17A, IL-17B, IL-17C, IL-17D, IL-17E, and IL-17F. The gene that encodes IL-17A was discovered in 1993 as an RNA transcript homologous to a Herpesvirus Saimiri gene, and the protein, initially called CTLA-8, was cloned (1). However, IL-17 attracted widespread attention in 2005, when two independent groups simultaneously characterized a new population of T helper (Th) CD4+ cells that produced IL-17A, named Th17 (2, 3). T helper CD4+ cells were first marked as the principal source of IL-17, but it was later shown that CD8+ cells also produce this cytokine, and these cells are termed Tc17. Also, several types of innate immune cells such as γδ T, natural killer T (NKT), TCRβ+ natural Th17, and Type 3 innate lymphoid cells (ILC3) produce IL-17 (4). All of these IL-17-producing cells are termed “Type 17” cells.
The proinflammatory activities of IL-17 are key in anti-microbial protection of the host, but uncontrolled IL-17 activity is associated with different immunopathological conditions, autoimmune diseases, and cancer progression (5). A critical role for IL-17R signaling in protection against bacterial and fungal infections, particularly by Candida albicans and Klebsiella pneumoniae, has been described in various studies in mice (6). In humans, mutations in IL-17 signaling genes (ACT1, IL17RA, IL17RC) are associated with chronic mucocutaneous candidiasis (5, 7, 8). The same condition also develops in individuals with AIRE deficiency, a condition accompanied by the production of anti-IL-17 antibodies (9).
Anti-IL-17A antibodies have shown therapeutic effect in various inflammatory diseases. Several anti-IL-17 antibodies have been approved for the treatment of plaque psoriasis (10, 11). Positive effects of IL-17 blockade have been shown in clinical trials of ankylosing spondylitis and psoriatic arthritis (12). Anti-IL17R antibody treatment of Crohn's disease has been shown to worsen the disease (13, 14), whereas targeting cytokines that control the differentiation of Th17 cells and therefore IL-17 secretion with anti-p40 subunit antibodies (Ustekinumab, Briakinumab) and anti-IL-6 receptor antibody (Tocilizumab) showed efficacy (15–17). These findings indicate that IL-17, by maintaining the integrity of the intestinal barrier, plays a dominantly protective role that overcomes its potential for tissue destruction in inflammatory bowel disease (18). Clinical use of antibodies that target IL-17 signaling gave insights into functions of IL-17 in humans.
IL-17R Signaling
The family of IL-17 receptors contains five different receptors (IL-17RA, IL-17RB, IL-17RC, IL-17RD, and IL-17RE) with common a cytoplasmic motif known as the SEFIR domain (19). IL-17 exists either as a homodimer or as a heterodimer, and both forms of the cytokine induce signals through dimeric IL-17RA and IL-17RC receptor complex (5). Binding of IL-17 to its receptor induces activation of several independent signaling pathways mediated by a cytosolic adaptor protein, Act1, and different TRAF proteins (5, 19, 20). IL-17 signaling mediated through TRAF6 and TRAF4 results in the transcription of inflammatory genes. Activation of TRAF6 by binding of IL-17 to its receptor leads to triggering of NF-κB, C/EBPβ, C/EBPδ, and MAPK pathways, while TRAF4 activation in complex with MEKK3 and MEK5 activates ERK5 (21). On the other hand, the mRNA stability of genes controlled by IL-17 is controlled IL-17-activated TRAF2 and TRAF5 (22).
Expression of IL-17R is ubiquitous, but the main targets of IL-17 are non-hematopoietic cells (23). IL-17 signaling induces the production of proinflammatory cytokines (IL-1, IL-6, G-CSF, GM-CSF, and TNF) and chemokines (CXCL1, CXCL2, CXCL5, CCL2, CCL7, CCL20, and IL-8), matrix metalloproteinases (MMP1, MMP3, MMP9, and MMP13), and anti-microbial peptides (β-defensins, S-100 proteins) (24, 25). The biological activities of IL-17 are often the result of synergistic or cooperative effects of IL-17 and other inflammatory cytokines (26). There are several mechanisms of negative regulation of IL-17 signal transduction. The negative regulators of IL-17 signaling are different ubiquitinases, deubiquitinases, kinases, endoribonuclease, and micro RNAs (21).
However, there is tissue-specific IL-17-dependent gene induction (27). In gut epithelium, IL-17 regulates the expression of several molecules that contribute to the preservation of continuous intestinal epithelium. In renal epithelial cells, IL-17 induces the expression of kallikrein 1 (28), while in salivary epithelium, it induces the expression of histatins (29), molecules that are involved in protection against C. albicans. IL-17-mediated osteolysis, which is detected in periodontitis and mouse models of arthritis or periodontal disease, is probably mediated by a receptor activator of NF-κB ligand (RANKL, TNFSF11, or osteoprotegerin ligand, OPGL) whose espression is induced by IL-17 (30, 31).
Differentiation of TH17 Cells
Th17 cells are classified as an inflammatory subset of T helper cells that perform key roles at mucosal surfaces where they mediate protection from bacteria and fungi and also contribute to the regulation of the mutualistic microorganisms that constitute the microbiota (32, 33). However, Th17 cells are also one of the major factors in the pathogenesis of several autoimmune diseases, including autoimmune disease of the central nervous system, Multiple sclerosis (MS) (2, 3, 34–39). The process of differentiation of naive CD4+ cells into Th17 cells is very similar to that of Th1 differentiation, but transcriptional factors that mediate this are distinct and it require stimulation with the cytokines IL-1β, IL-6, IL-21, and TGFβ, which are produced by professional antigen-presenting cells (APCs) (32, 40–47). Cytokines produced by APCs stimulate the JAK-STAT3 axis and upregulate the expression of transcription factors RORγt and RORα, identified as markers of the Th17 lineage (48–52). The differentiation of Th17 cells is reduced in the states of IL-6, IL-21, TGFβ, or RORγt deficiency, which leads to reduced production of Th17 cytokines and impaired defense against extracellular bacteria and fungi but also attenuation of autoimmunity (41, 48, 53). However, an alternative mode for the differentiation of pathogenic Th17 cells in the absence of TGFβ signaling has been described in vivo in Experimental Autoimmune Encephalomyelitis (EAE) (54). Cytokines that induce Th1 and Th2 differentiation are described as the main inhibitors of Th17 differentiation. IL-2 is a key repressor of Th17 differentiation, as it activates transcription factor STAT5 and thus inhibits IL-17 production (55), while inhibition of IL-2 expression in T lymphocytes stimulates Th17 cell development (56, 57).
In animal models of autoimmune diseases, proinflammatory cytokines IL-1β and IL-23 have been shown to be enhancers and stabilizers of partially or completely differentiated effector Th17 cells, which dominantly express corresponding receptors for these cytokines, IL-1R1 and IL-23R (44, 58–61). In line with this observation, transfer of Th17 cells in vitro obtained by exposure to IL-6 and TGFβ does not induce EAE in mouse, while Th17 cells obtained by stimulation of naive cells with IL-1β, IL-6, and IL-23 achieve the pathogenic potential and are able to elicit EAE (55). In fact, it was later shown that IL-6 and TGFβ in Th17 cells induce production of anti-inflammatory cytokine IL-10, while IL-23 has a critical role in the induction of the endogenous cytokine TGFβ3. Suppression of IL-10 production in Th17 cells during their differentiation results in high expressions of T-bet, IL-23R, and GM-CSF, markers of Th17 cells with pathogenic potential (62, 63). Furthermore, IL-1 and IL-23 stimulation through JunB and SOCS family members (64, 65) affects the effector profile of Th17 cells and induces the development of highly pathogenic double-positive IL-17+ IFNγ+ and IL-17+ GM-CSF+ T cells (66–68). These pathogenic double-positive cells originate from Th17 cells. However, IL-23 is not required for the differentiation and maintenance of nonpathogenic Th17 cells in the gut and functional plasticity toward T follicular helper cells (66, 68). The novel genes Gpr65, Toso, and Plzp, identified by the single-cell RNA-sequencing analysis of ex vivo Th17 cells, are found to promote Th17 pathogenicity and to cause EAE and chronic inflammation in the CNS of mice, while CD5 antigen-like (CD5L) attenuates Th17 cell-mediated disease (69, 70).
Before activation, T cells do not express receptors for cytokines IL-1 and IL-23 (58, 71). During the initial phase of Th17 differentiation, IL-6 induces binding of RORγt to the Il1r1 locus and binding of STAT3 to the Il23r locus, leading to the expression of these genes (54). Phosphorylation of STAT3 increases the expression of the conserved miR-183/96/182 cluster, which in turn reduces the expression of Foxo1, a transcription factor that negatively regulates the expressions of IL-1R1 and IL-23R (72). The Major Transcriptional Effector of Notch Signaling, RBPJ, promotes IL-23R expression and induces pathogenicity of Th17 cells (73). The differentiation of Th17 is stabilized by positive feed-forward loop stimulation with IL-1β and IL-23, accompanied by upregulation of IL-1R and IL-23R (50, 74). The hallmark of effector Th17 cells is IL-23R expression, and its signaling promotes the expression of transcriptional factor Blimp-1, which induces the expression of several genes in vivo, leading to the enhanced pathogenicity of Th17 cells (75).
The differentiation of human Th17 cells in vitro, similar to mouse Th17 cells, requires IL-1, IL-6, IL-23, and TGFβ. Initially, a few studies demonstrated that TGFβ was not required, while stimulation with IL-1β, IL-6, and IL-23 was sufficient for induction of human Th17 cell differentiation (76, 77). Later studies showed that TGFβ, IL-23, and IL-1β (or IL-6) were the key factors needed for differentiation of human Th17 cells under serum-free conditions, since serum could be the source of TGFβ or aryl hydrocarbon receptor (AhR) ligands (78, 79). In line with findings regarding Th17 differentiation in mouse, IL-23 is also the key player in the differentiation of human Th17 cells; moreover, human CD4+ T cells express IL-23R before activation and immediately respond to IL-23, while IL-23 signals in accordance with stimulation with IL-1β further upregulate IL-23R expression (78, 80). Described dominant-negative mutations of STAT3 gene, which can be manifested by hyper-immunoglobulin E syndrome, inadequate Th17 cell differentiation, and reduced production of IL-17, supports the roles of STAT3 in the IL-6- and IL-23-mediated process of Th17 differentiation in humans (81, 82).
Environmental Factors That Affect the Pathogenic Potential of Th17 Cells
Different environmental factors modulate the reactions of the immune system and strongly accelerate the pathogenic potential of Th17 cells. T helper cells under Th17 culture condition increase expression of the aryl hydrocarbon receptor (AhR), a ligand-dependent transcription factor that senses environmental toxins and endogenous molecules such as metabolites of tryptophan, and the stimulation of this molecule induces the release of IL-17 and IL-22 by effector Th17 cells (83, 84). The transcription factor, hypoxia-inducible factor 1 (HIF-1), a key metabolic sensor, directly regulates expression of RORγt and IL-17 at the transcriptional level and promotes Th17 differentiation (85, 86). Signaling via kinase complex mTORC1 coordinates metabolic and transcriptional programs that regulate the development of pathogenic Th17 cells (87). Disrupted mTORC1 signaling in Th17 cells leads to upregulated expression of TCF-1 (transcription factor T-cell factor 1) and development of stemness-like features, while transdifferentiation in the Th1 is arrested. Mice with blocked mTORC1 activity are protected from EAE, while their Th17 cells do not express T-bet and IFN-γ (87).
A high-salt diet can enhance the differentiation of Th17 cells and thus contribute to the development of EAE (88, 89). Exposure to a high-salt diet induces the expression of serum glucocorticoid kinase 1 (SGK1), which promotes the expression of IL-23R and thus stabilizes pathogenic Th17 cells and enhances the production of GM-CSF (88, 89). Since mice with T-cell-specific deletion of Sgk1 develop attenuated EAE, without exacerbation after exposure to a high-salt diet, it appears that a high-salt diet modulates EAE severity by its direct effect on T-cell differentiation (88).
Th17 and IL-17 in Multiple Sclerosis and EAE
Multiple sclerosis is a chronic inflammatory disease of the central nervous system (CNS) that is characterized by damage to myelinated axons in the CNS, leading to the loss of myelin sheath. Inflammatory processes that cause myelin damage lead to the destruction of oligodendrocytes and axons, with subsequent axonal loss, and transient or permanent loss of neurologic functions, resulting in various types of disabilities of different severity (90). An overall reduction in CNS volume is very often seen in MS. Localized inflammatory foci can be found in the white matter in almost all areas of CNS, with a considerable number of plaques in the gray matter and anywhere in the CNS parenchyma, including the optic nerves, brainstem, periventricular white matter, and cervical spinal cord (91–93). The course of MS and clinical symptoms are highly variable and unpredictable, varying from a relatively benign illness with minimal impairment to a rapidly evolving and life-threatening disease that requires serious medical treatment (94).
The precise etiology of MS is unknown, but it is considered that both genetic and environmental factors play significant roles in its development (95). The pathogenesis of MS also remains elusive, but it is believed that MS is an autoimmune disease mediated by auto-reactive CD4+ T cells specific for myelin antigens. Autoreactive T cells initiate and perpetuate an inflammatory cascade, resulting in demyelination and axonal loss (96). The huge heterogeneity of disease course in patients with MS and in the histopathological features seen in the CNS indicates that multiple immunopathological pathways contribute to the disease development. Evidence from clinical studies suggests that inflammatory mediators, such as cytokines, play an essential role in the pathogenesis of MS (91, 97).
The pathogenesis of MS has been mostly described by analogy to EAE, an animal model of MS (98–100). In typical EAE induced by immunization with autoantigen, myelin-specific CD4+ T cells are activated in the lymph organs in the periphery, develop encephalitogenic potential, and infiltrate the CNS, where they recognize specific autoantigens presented by local antigen-presenting cells (APCs) and reactivate. The inflammatory process in MS is initiated by binding of pathogen-associated molecular patterns (PAMPs) from pathogens or commensal bacteria and damage-associated molecular patterns (DAMPs) from dead or dying cells to pathogen recognition receptors (PRRs), leading to activation of innate immune cells and production of IL-1, IL-6, IL-12, IL-18, and IL-23, cytokines that promote the differentiation and expansion of encephalitogenic Th1 and Th17 cells (101, 102). Myelin-specific CD4+ T cells that enter the CNS are reactivated and expanded by the IL-1β and IL-23 produced by resident microglia and infiltrating inflammatory monocytes. Encephalityogenic Th1 and Th17 cells in the CNS produce inflammatory cytokines that activate glial cells to produce inflammatory mediators, matrix metalloproteinases, chemokines, and free radicals, which induce myelin damage, leading to manifestations of neurologic deficits (65, 66, 101, 103). One of the main differences between MS and animal models (EAE) is the localization of demyelination. In EAE demyelination, is mainly located in the spinal cord, whereas in MS, this process mainly affects the cerebral and cerebellar cortex (104). The dominant population of T cells in active MS lesions are CD8+ T cells, but in EAE, the primary encephalitogenic T cells and dominant population in CNS infiltrates are CD4+ T cells, with less evidence for the role of CD8+ T cells (105). Neurodegeneration is more typical for MS, while in EAE models, the dominant finding is neuroinflammation (106). In the later stages of MS, neurodegeneration appears to be independent of the inflammatory process, which cannot be found in the acute inflammatory EAE model (107). However, axonal and neuronal loss and demyelination with remyelination can be observed in EAE in Biozzi antibody high (ABH) mice (108). Despite the limitations of the EAE models, the main findings regarding MS pathogenesis have come from EAE studies, as has the design, development, and validation of many therapeutics used for the treatment of MS (109).
Cytokines play roles in the pathogenesis of MS and EAE and in the processes of inducing oligodendrocyte cell death, neuronal dysfunction, and axonal degeneration (110). Th17 cells are considered to be one of the key effectors of autoimmune inflammatory diseases, including MS and experimental disease EAE (2, 111–113). Increased expression of IL-17- and Th17-associated transcripts (Il6, Il17a) has been demonstrated in MS plaques collected at autopsy (114). Further, IL-17 was marked as the highest-ranking gene expressed in the CNS of MS patients at autopsy (114); this was before the discovery of Th17 cells. Also, another report indicated that MS is a primarily IL-17-mediated autoimmune disease (78). Later, the results of various studies showed that a single nucleotide polymorphism (SNP) in IL-23R gene is linked to several human autoimmune diseases, indicating that IL-23 signaling is an essential event in the development of pathogenic Th17 cells. It is known that IL-17 can stimulate the production of other proinflammatory cytokines and chemokines and thus evince a powerful proinflammatory effect (115). The concentration of IL-17 is significantly higher in the serum of MS patients with relapses and remissions than in normal, healthy subjects (116) and is in correlation with disease activity, as demonstrated by magnetic resonance imaging (117). Consistently with the increased concentration of IL-17 in liquor and peripheral blood of MS patients, the proportion of Th17 cells is also increased, especially during relapses, while there is no change in Th1 cells (118, 119). Th17 cells are able to cross the blood–brain barrier, and their presence in MS lesions is associated with enhanced neuroinflammation (120). It has been shown that IFN-γ-producing Th17 cells cross the blood–brain barrier and accumulate in the CNS during the active phase of MS (121). Besides CD4+ T cells, there is evidence that IL-17-producing CD8 T cells contribute to CNS tissue damage in EAE and are also present in the liquor of patients with MS (122, 123). Importantly, it has been documented that the cells that enter the CNS in the first wave of CNS infiltration are Th17 cells (124), followed by infiltration with other immune cells that further promote and sustain tissue inflammation. Also, the presence of IL-17- and IL-22-producing Th cells has been reported in the early stages of MS (125).
Beneficial effects of treatment with rituximab, blocking anti-CD20 antibody, in EAE are associated with decreased production of several cytokines, including IL-17 (126). Neutralization of IL-17 can significantly attenuate the progress of EAE by attenuating the induction of pathogenic cytokines (58). Also, EAE severity was ameliorated in IL-17-deficient animals (123, 127, 128), while the disease was mild, with delayed onset, in RORγt-deficient mice (48).
One study indicates that the beneficial effect of vitamin D supplementation in MS patients is mediated by alleviating the percentage of pathogenic T-cell subsets that produce IL-17 (129). It has also recently been shown that amelioration of MS by dimethyl fumarate is associated with suppression of IL-17+ CD8+ Tc17 cells (130). The beneficial effect of statins in some forms of MS could be due to their effect on Th17 cells (131). Phase IIa study has been conducted in order to investigate possible beneficial effects of Secukinumab, an IL-17A-neutralizing monoclonal antibody. No adverse effects of Secukinumab were detected, while the results of this study indicate that blocking IL-17A with an antibody may reduce MRI lesion activity in MS (132).
There are studies that demonstrate the importance of Th17 cells in EAE and MS, but there is also evidence that indicates that Th1 cells are the main mediators of neuropathology in the EAE model (113, 133). Several reports indicated that IFN-γ-deficient and IFN-γR-deficient mice, as well as anti-IFN-γ-treated mice, develop EAE (134, 135). Also, there are reports showing a protective role for IFN-γ in EAE, mediated by the supression of pathogenic Th17 cells (3). The presence of T cells that coexpress IL-17 and IFN-γ under inflammatory situations has been reported (136, 137). These Th1/Th17 cells were noticed in the CNS of mice with EAE (138). Data obtained from the mouse studies indicate that Th17 cells lacking Il17a generated in vitro are able to induce EAE upon adoptive transfer, similar to wild-type Th17 cells (127, 139). Finally, it seems that there is significant plasticity of Th17 cells, with evidence that lymphocytes obtained from the blood of MS patients have an increased potential to switch from IL-17-secreting Th17 cells to IFN-γ-secreting Th1, also called ex-Th17 cells (121).
Several studies have demonstrated that IL-23, a cytokine essential for the differentiation and expansion of Th17 cells, promotes EAE more robustly than IL-12, a cytokine that stimulates the development of INF-γ-producing Th1 cells (140). IL-23 is a covalent heterodimer of p40 (IL-12) and p19 (IL-23) subunits (70). IL-12 and IL-23 share the p40 subunit. The same cell types, mainly dendritic cells, produce both of these two cytokines, but their relative ratio depends on the nature of stimuli that activate dendritic cells (141). IL-12Rβ2-deficient mice with excluded IL-23 signaling are more susceptible to EAE, develop disease earlier, and have more severe disease, with greater demyelination and CNS inflammation, compared to WT mice (142). This result was contrary to findings in IL-12Rβ1-deficient mice (excluded IL-12 signaling) (143). Also, it has been shown that IL-23, not IL-12, plays the key role in the development of CNS autoimmune inflammation, affecting the subset of memory Th1 cells (144). It has also been shown that IL-23 induces differentiation of highly encephalitogenic Th cells that produce IL-17A (145).
In attempts to clearly define the roles of Th1 and Th17 subpopulations in MS pathogenesis, it was shown that the transfer of Th17 cells induces more severe EAE compared to Th1 cells (58). Another study showed that autoantigen-specific Th1 and Th17 cells were able to induce disease with similar severity but with different pathological findings (133). Th1-mediated neuroinflammation was characterized by macrophage infiltration, while, in Th17-mediated disease, neutrophil predominated in CNS infiltrates (133). Also, it was found that Th17 cells induce mainly brain damage, in contrast to Th1 cells, which dominantly induce spinal cord inflammation (146).
IL-17 mediates EAE development by the stimulation of IL-17R expressed on endothelial cells, astrocytes, microglia, and resident neuroectodermal cells (147). Mouse astrocytes express receptor for IL-17 (148) when stimulated with recombinant IL-17A in vitro, but also, in vivo in the EAE model, they produce various cytokines and chemokines, IL-6, TNFα, CCL2, CCL3, CCL20, CXCL1, CXCL2, CXCL9, CXCL10, and CXCL11 (IP-9) (149–151), that promote the influx of immune cells into the CNS and mediate neuroinflammation. Similarly, human astrocytes cultured with IL-17 in vitro produce IL-6, a cytokine that perpetuates the differentiation of CD4 naive cells into Th17 cells (152). The role of IL-17-mediated activation of astrocytes in EAE pathogenesis was confirmed by attenuation of EAE in animals with blocked IL-17 signaling in astrocytes (152). IL-17 also contributes to EAE development by affecting the activity of NG2+ oligodendrocyte precursor cells (OPCs) (153). Further, in vitro treatment of these cells with IL-17 strongly inhibits the maturation of oligodendrocytes and reduces their survival (154). Another study also indicates that IL-17 mediates apoptosis and inhibits differentiation of oligodendrocytes in vitro (155). IL-17 stimulates the maturation of primary OPCs and their participation in inflammatory processes (156). Microglial cells stimulated in vitro with IL-17 produce inflammatory mediators IL-6 and CXCL2, while only LPS pre-stimulated microglia exert enhanced cytotoxic effects (157). Further, microglial cells co-cultured with Th1/Th17 cells, but not Th1-only cells, produce high amounts of IL-1β, IL-6, and TNF-α, which promote further Th17 differentiation, neuroinflammation, and damage (158). IL-17 disrupts blood–brain barrier (BBB) tight junctions in vitro and in vivo in MS and promotes CNS inflammation (120). In an EAE model, it has been shown that IL-17 disrupts BBB by the induction of oxidative stress in endothelial cells accompanied by down-modulation of the tight junction molecule occludin (159). IL-17A levels are elevated in the CSF of relapsing-remitting MS patients, and this level correlates with the level of BBB dysfunction. Also, the treatment of BBB cell line hCMEC/D3 with a combination of IL-17A and IL-6 reduces the expression of tight junction-associated genes and disrupts monolayer integrity (160). Indirect evidence supports the role of IL-17 in direct neuronal damage. Different neuronal populations express IL-17 receptor (161). Direct contact, resembling immune synapses, of MOG-specific Th17 cells and neurons in demyelinating lesions associated with axonal damage has been shown by confocal, electron, and intravital microscopy, indicating the central role of Th17 cells in neuronal dysfunction (162).
IL-22, a cytokine whose production specifically induces IL-23, contributes to the pathogenicity of Th17 cells (163). It has been reported that IL-22 contributes to MS severity (120) as well as dysregulated expression of IL-22 and its antagonist, IL-22BP (164). Single nucleotide polymorphism in the IL-22R A2 gene is associated with MS risk (91, 165). IL-22 can contribute to MS pathogenesis by enhancing the expression of Fas in oligodendrocytes, resulting in oligodendrocytic apoptosis, and decreasing the expression of FOXP3 in T cells (166). Production of IL-22 is increased during the peak phase of EAE and is decreased during remission (167). However, beside involvement in many neurological inflammations, IL-22 may also be protective (168).
Although Th17 cells and their hallmark cytokines IL-17, IL-22, and IL-23 have been marked as the crucial players in the pathogenesis of MS and EAE, however, mice lacking IL-17 and IL-22 develop EAE (62, 169).
Findings indicating that GM-CSF has the key role in the encephalitogenic potential of Th17 cells in mice (74, 170, 171), specifically, increased levels of GM-CSF in the cerebrospinal fluid and serum of active MS patients with the relapsing-remitting type of the disease and increased secretion of GM-CSF from T cells isolated from the peripheral blood and brain lesion of MS, suggest that GM-CSF also plays an important role in MS development (172, 173). Unlike other cytokines, GM-CSF plays a non-redundant role in EAE development, and its secretion alone is able to provide development of autoaggressive and pathogenic MOG-specific T cells (170). GM-CSF-deficient Th cells are not able to induce EAE, indicating that the encephalitogenic potential of both Th1 and Th17 cells depends on their GM-CSF production (74).
In our previous studies, we have shown that overcoming resistance to induction of EAE with MOG35−55 peptide of BALB/c mice by infection with murine cytomegalovirus (MCMV) (174) or by deletion of ST2 gene (169) is associated with increased production of IL-17 in T cells.
Disease developed by MCMV-infected BALB/c mice is accompanied with an increase in IL-17-positive CD4+ and CD8+ cells in the central nervous system. Brain infiltrates in MCMV-infected BALB/c mice were more significant that in C57BL/6 mice, with a similar number of CD4+ and CD8+ cells, contrary to the dominantly CD4+ cells in C57BL/6 mice, which develop “typical” EAE (105). The encephalitogenic potential of CD4+ T cells in the CNS infiltrates of BALB/c mice is further documented by the detection of CCR6, the key molecule that mediates the initial infiltration of the CNS by Th17 cells (174, 175). Almost equal participation of IFN-γ- and T-bet (Th1)- and IL-17- and RORγt (Th17)-expressing cells was found in the CNS of MCMV-infected MOG35−55 immunized BALB/c mice, in contrast to almost exclusive CNS infiltration with Th1 cells in C57BL/6 mice infected with γHV-68 before EAE induction (174, 176). Further, CNS infiltrates of BALB/c mice infected with MCMV before MOG35−55 immunization contained CD8+ cells that express T1 and T17 transcriptional factors and corresponding cytokines, TNF-α and IFN-γ (Tc1) and IL-17 (Tc17 cells) (174).
Since cerebrospinal fluid of early-stage MS patients contains a greater number of Tc17 cells in comparison with peripheral blood, these cells are considered to be required for the accumulation of Th17 cells in the CNS in MS (177). No inflammatory T1 and T17 cells were found in the CNS of BALB/c mice immunized with MOG35−55 (174), while in the CNS of unimmunized BALB/c mice infected with MCMV neonatally, Tc1 cells (IFN-γ and T-bet+) dominated (178). CD8+ T cells isolated from CNS of MCMV-infected and MOG35−55-immunized mice produced inflammatory cytokines in response to in vitro MOG35−55 peptide stimulation but were not specific for viral epitopes pp89 and m164 (174). These findings indicate that the newly developing autoimmune process in MOG35−55-immunized BALB/c mice previously infected with MCMV attracts a new population of IL-17-producing CD8+ cells that participate in the development of autoimmunity (177). These findings are in line with previous reports that the expansion of myelin-specific CD8+ T cells follows CD4+ T cell-mediated initiation of the autoimmune process in CNS, thus contributing to tissue damage (179). The significant presence of IL-17-, CCR6-, and RORγt-positive CD4+ and CD8+ cells in the CNS of MOG35−55−immunized BALB/c mice with non-productive MCMV infection in contrast to uninfected BALB/c mice immunized with MOG35−55, with negligible number of these cells in the CNS, indicates that MCMV infection probably modulates the activation and differentiation of antigen-presenting cells in the periphery, changing their signature cytokines, and thus, after additional stimulus, enables the development of Th17/Tc17 cells with encephalitogenic potential (174).
Our results indicate that MCMV infection of BALB/c mice significantly affects dendritic cells in peripheral lymph nodes, thus enabling differentiation of encephalitogenic cells (174). In line with the well-known capacity of MCMV to encode an analog of chemokine CCL2 (180) that induces monocyte recruitment and viral dissemination (181), we found a higher percentage of CCR2+ dendritic cells in the peripheral lymph nodes of MCMV-infected mice (174). In contrast with a previous report that MCMV attracts monocytes that acquire immunosuppressive characteristics (182), we found higher percentages of dendritic cell-expressing markers of activation, CD86 and CD40, and Th1-promoting cytokine IL-12, indicating that MCMV infection of BALB/c mice increases the proportion of inflammatory dendritic cells in peripheral lymph nodes and thus enables the development of encephalitogenic T cells (174).
IL-17 in Alzheimer's Disease
Alzheimer's Disease (AD) is the most common neurodegenerative disorder causing cognitive impairment in the elderly (183, 184). The histopathological hallmarks of AD are amyloid plaques in the brain, mainly consisting of fibrillary forms of amyloid β peptide-40 (Aβ-40) and amyloid β peptide-42 (Aβ-42) (185). The fibrillary forms of amyloid β found in the amyloid plaques are obtained by a sequential cleavage from amyloid precursor proteins (186, 187). Highly insoluble Aβ peptides generated in the CNS play a crucial role in the pathogenesis of AD; they activate the complement pathway (168) and stimulate microglia to produce the proinflammatory cytokines and chemokines and thus induce accumulation of inflammatory cells into the CNS (188, 189). This proinflammatory process mediated by microglia leads to neurodegeneration (188, 190), although microglia play a protective role also, due to the clearing of Aβ aggregates by phagocytosis (191). Aβ peptides also increase the production of reactive nitrogen and oxygen species by microglial cells, leading to oxidative stress development, stimulation of Th17 cells, and IL-17 production (192, 193). It appears that the main roles of IL-17 in AD pathogenesis are the attraction of neutrophils and the stimulation of their function. It has been shown that Aβ aggregates mediate the chemotaxis and the recruitment of neutrophils in the CNS of mice overexpressing human mutant amyloid precursor protein (APP), which produce IL-17 and thus amplify neutrophil entry in the CNS (192), although mesenteric lymph nodes of these mice have lower production of IL-17 as a consequence of reduced differentiation of Th17 cells (194). Since neutrophils are the main targets of IL-17 in the CNS but are also very important sources of this cytokine, these cells, by promoting inflammation and CNS tissue damage, could have an important role in the development of AD pathology. Results from in vitro experiments indicate that IL-17 might promote autophagy in neurons and thus induce neurodegeneration (195).
There have been more reports about the role of innate immunity in AD than about adaptive immunity, but increased activation of T and B lymphocytes was recently demonstrated in a triple transgenic mouse model that replicated Aβ and tau neuropathology (196). Moreover, it has been shown that these cells produced high levels of IL-2, TNF-α, IL-17, and GM-CSF, indicating that neurodegeneration in these mice is associated with Th17 polarization (196). Increased expression of IL-17, IL-22, and RORγt has been found in the hippocampus, CSF, and serum of rats after intrathecal injection of Aβ-42 peptide (197). In the same study, Zhang et al. indicated that after disruption of the blood–brain barrier with Aβ-42 injection, Th17 cells enter into the brain (197). Tian et al. reported that postoperative cognitive dysfunction is associated with an enhanced level of IL17A in the hippocampus and suggested that IL-17-mediated damage of the hippocampus leads to Aβ1-42 accumulation and thus probably to cognitive decline (198). Increased expression of RORγt, IL-23, and IL-17 was found in the brains of Aβ-42-injected rats, while Treg-related cytokines TGF-β and IL-35 were decreased (199). Activated Th1 or Th17 cells in the brain produce inflammatory cytokines IFN-γ or IL-17 and thus heighten the inflammatory cascade, recruit and activate immune cells, and promote AD neuropathology (192, 200).
In MS, cytokines released by Th17 cells bind to their receptors on neurons and activate the apoptotic pathway, leading to neurodegeneration (201). Expression of Fas and FasL is also increased in the brain of AD rats (197, 202), and it could be assumed that Th17 cells activate the apoptotic pathway in neurons by Fas/FasL interaction and thus contribute to the development of neurodegeneration in AD (197, 203).
Elevated levels of IL-1β in the brains of AD mice homozygous for a destructive mutation of TLR4 cause up-regulation of IL-17 (204). In a very recent study, it has been shown that the administration of blocking anti-IL-17 antibody decreases the cognitive impairment and neuroinflammation induced by Aβ1−42 injection into cerebral ventricles of adult CD1 mice, as suggested by reduced Aβ1−42, glial fibrillary acidic protein (GFAP), S100 proteins, and inflammatory mediators and cytokines (205). This result supports the previously indicated role of IL-17 and related cytokines in promoting AD neuroinflammation and neurodegeneration (206). On the other hand, there is a study that indicates a protective role for IL-17 in an animal model of AD (207). Intracranially overexpressed IL-17 reduced cerebral amyloid angiopathy and improved anxiety and learning deficits (207). Further, it has recently been shown that ICR mice injected with IL-17 have an improvement in spatial learning as measured by the Morris water maze test, which is associated with the promotion of maturation of already-formed neuroblasts and the inhibition of neuroprogenitor proliferation (208).
The number of both CD4+ and CD8+ T cells in the brain parenchyma and vascular endothelium in humans with AD is higher than in healthy controls (209). Further, naive lymphocytes obtained from AD patients had increased production of Th17-related cytokine IL-21 and had higher expression of Th17 transcription factor RORγt, while monocytes obtained from the same patients produced higher amounts of IL-6 and IL-23 (210). A higher proportion of Th17 cells has been noticed in peripheral blood of patients with mild cognitive impairment due to AD pathology than in subjects with mild cognitive impairment due to pathologies other than AD and healthy controls (211). Also, higher concentrations of IL-17 and IL-23 were detected in the serum of AD patients than in healthy controls (212). IL-17 is reported to be a good plasma biomarker for distinguishing individuals with AD from cognitively healthy control subjects (213). Also, it was reported that the IL-17 concentration in cerebrospinal fluid could be used antemortem for identification of frontotemporal lobar degeneration with tau pathology (214).
It has been proposed that a desirable AD vaccine should induce Th2 and inhibit Th1/Th17 immune responses to Aβ in order to limit or prevent neuroinflammation and subsequent neurodegeneration (215).
A number of reports indicate the important role of IL-17 in AD pathogenesis; however, the precise mechanism of IL-17 upregulation in the CNS of AD patients is not known. It is possible that microbial infection, as was reported for respiratory infection (216) or inadequate immune surveillance in the gut (194), induces higher IL-17 production in the CNS, which later leads to deposition of amyloid-β. However, the opposite sequence of events is possible; that is, deposition of amyloid-β and inadequate clearance stimulate receptors of innate immune cells and induce production of IL-17, which perpetuates AD pathogenesis.
IL-17 in Ischemic Brain Injury
Brain ischemia causes necrosis of the affected CNS tissue due to the loss of nutritional supply (217). Damaged CNS tissue releases damage-associated molecular patterns (DAMPs) that stimulate resident innate immune cells in the CNS, in the first line microglia (218). Activated microglia cells have a dual role: these cells play a beneficial role by phagocytosis of damaged tissue but also release inflammatory mediators TNF-α, IL-1β, IL-6, and IL-17, which enhance inflammation and tissue damage (219). DAMP molecules, released after ischemic brain damage, such as high mobility group 1 box 1 (HMGB1) (220, 221) and peroxiredoxin, induce IL-23 production in microglia/macrophages by activating TLR2 and TLR4, which subsequently induce the expression of IL-17 in other immune cells but also in microglia (222).
Activated immune cells after reperfusion additionally damage CNS tissue and significantly contribute to overall tissue damage after stroke. Adaptive immunity most probably contributes to inflammation development in CNS tissue after ischemia-reperfusion, especially T cells (223, 224). Similar was found in animal models: RAG1(–/–) mice, after stroke induced by transient middle cerebral artery occlusion, developed reduced damage of brain tissue, but detrimental effects of T cells in cerebral ischemia did not depend on antigen recognition or TCR costimulation (225). The presence of Th1 as well as Th17 cells was noticed in the brain lesions in ischemic stroke (226). These cells release proinflammatory cytokines and thus contribute to tissue damage (226).
Waisman et al. indicate IL-17 as one of the molecules that play a particular role in the delayed phase of the postinfarct inflammatory cascade (217). Increased expression of IL-17 mRNA in peripheral blood mononuclear cells was detected in patients after ischemic stroke, and its expression was in correlation with Scandinavian Stroke Scale scores (227). High expression of IL-17 was observed in ischemic injured brain tissue in experimental animals and also in postmortem analyzed human tissues (228–230). Also, higher expression of IL-17 at the mRNA and protein levels has been detected in the penumbral brain tissue 1, 3, and 6 days after reperfusion in mice (231). In an animal model of ischemic stroke, an increased number of IL-17-producing blood mononuclear cells were observed (232). In another study, IL-17 levels were elevated 3 days after reperfusion. This induction of IL-17 production was IL-23-dependent, and γδ T cells were indicated as the main source of IL-17 (233). In this study, it has been shown that IL-17 plays the main role in the stage of tissue damage after infarction, since IL-17-deficient mice had attenuated damage only on day 4 after ischemic insult (234). On the other hand, IL-23p19-deficient mice developed attenuated CNS tissue damage on day one after stroke induction (234), but γδ T-cell-deficient mice still develop brain injury after ischemia reperfusion induction (228). Although there is no clear evidence that Th17 cells play a role in tissue damage after stroke induction, activation of T cells and autoantigen-specific T cells, which exacerbates ischemic brain injury, was noticed in experimental animals (234). Also, an increase in the proportion of Th17 cells and a decrease in Treg cells in the periphery might contribute to CNS tissue damage after ischemia-reperfusion (235). Astrocytes are also marked as a source of IL-17 in inflammatory foci after brain ischemia-reperfusion (228, 236).
IL-17 may contribute to CNS tissue damage by several mechanisms, as described previously in other inflammatory diseases of CNS, affecting the cells that express IL-17 receptor, microglia, endothelial cells, astrocytes, and neurons, as summarized in Table 1. Although ischemia-reperfusion induces necrosis of blood–brain barrier, IL-17 could enhance BBB damage by the disruption of tight junctions (120) and by the promotion of monocyte migration across the BBB through an intracellular adhesion molecule (ICAM) 1-dependent mechanism (159). Levels of inflammatory cytokines IL-1β, TNFα, and matrix metalloproteinases, indicators of BBB damage, are decreased after stroke induction in IL-17-deficient mice (233). Increased expression of IL-17 receptor on neurons has been shown simultaneously with increased expression of IL-17 in CNS tissue after stroke induction, indicating the role of IL-17 in direct neuronal damage (230). This observation is supported by in vitro study (230). IL-17 also enhances autophagy in neurons and thus aggravates neuronal ischemic injuries (237). In synergy with TNF-α released by macrophages, IL-17 stimulates astrocytes to produce CXCL1, which recruits neutrophils into the CNS and thus enhances inflammation and damage (228). Astrocytes stimulated in vitro with TNFα and IL-17A show enhanced expression of several chemokines that have a role in the attraction of other immune cells, CCL20, CXCL2, CXCL9, CXCL10, and CXCL11, (153). IL-17, synergistically with IL-6, induces expression of CCL20 in astrocytes, which is a chemokine that attracts Th17 cells (149).
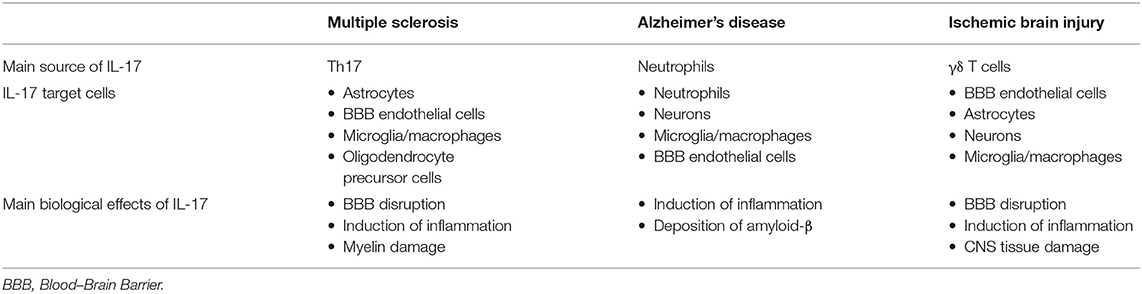
Table 1. The main cellular source of IL-17 and its target cells in chronic inflammatory neurological diseases.
On the other hand, IL-17A induces the expression of molecules that have neuroprotective effects, brain-derived neurotrophic factors (BDNF), glia-derived neurotrophic factors (GDNF), and nerve growth factors (NGF), indicating that IL-17 might have a role in the reduction of damage (157). Recently, it has been shown that recombinant mouse IL-17A significantly attenuates damage of cortical astrocytes after stroke induction in a dose-dependent manner by inhibition of apoptosis (238).
Concluding Comments
Despite a large number of reports that indicate an important or, in some diseases, even indispensable role of IL-17 and Th17-related cytokines in inflammatory and degenerative neurological diseases, the precise mechanism of the pathogenic effect of IL-17 in the CNS is still elusive. Numerous in vivo and in vitro studies identify several types of CNS tissue cells as IL-17 targets and illustrate the effects of stimulation of these cells with IL-17 (summarized in Figure 1). However, the relative contributions of these processes to tissue damage and the development of inflammatory CNS diseases in humans are still undetermined. In order to gain new insights into the role of IL-17 in the pathogenesis and eventual new treatment of neuroinflammatory and neurodegenerative diseases, additional research in this field is required.
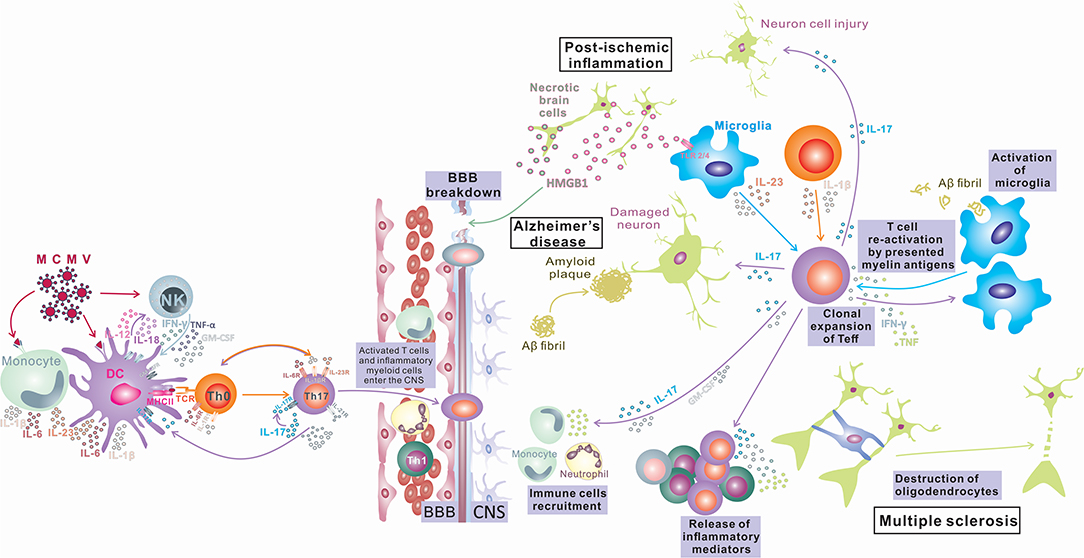
Figure 1. IL-17 in Inflammatory Diseases of the Central Nervous System. Infection in the periphery (viral infections) activates innate immunity (monocytes/macrophages and NK cells) and induces a proinflammatory environment that changes the phenotype of antigen-presenting cells, which differentiate into inflammatory APCs that produce inflammatory cytokines IL-1, IL-6, IL-12, and IL-23. These APCs induce bystander activation of autoreactive T cells and their differentiation toward encephalitogenic T cells (IFN-γ, IL-17, TNF-α, Tbet, RORγt, CXCR3, and CCR6 positive) capable of entering the CNS, where after reactivation in contact with antigens presented by local tissue APCs, they proliferate and produce cytokines (IL-17, GM-CSF) that contribute to BBB disruption and recruitment of other immune cells into the CNS, finally inducing myelin damage (Multiple sclerosis). Peripheral infections can compromise the BBB and lead to an influx of IL-17-producing cells into the CNS. IL-17 can induce damage to neurons by direct cytotoxic effects or by recruitment of neutrophils and induction of inflammation, leading to deposition of amyloid fibrils and plaque formation (Alzheimer's disease). Also, the opposite order of events is possible, where microglia phagocytize amyloid fibrils and induce differentiation of T cells toward IL-17-producing cells, and the released IL-17 damages the BBB, recruits neutrophils, and induces inflammation and neuron damage, which exacerbates amyloid deposition (Alzheimer's disease). CNS tissue damaged by ischemia releases damage-associated molecular patterns (HMGB1) that stimulate microglia to release inflammatory mediators TNF-α, IL-1β, IL-6, and IL-17, which enhance inflammation and tissue damage. Activated microglia also can induce Th17 development. IL-17 released by innate immune cells or Th17 cells can enhance BBB damage, recruit immune cells, and enhance inflammation, inducing direct neuronal damage (postischemic inflammation).
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was funded by grants from the Serbian Ministry of Science and Technological Development, Grants No. ON175069, the Serbian bilateral project with PR China (06/2018), and the Faculty of Medical Sciences, University of Kragujevac (JP18/19 and JP19/19).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Rouvier E, Luciani MF, Mattéi MG, Denizot F, Golstein P. CTLA-8, cloned from an activated T cell, bearing AU-rich messenger RNA instability sequences, and homologous to a Herpesvirus Saimiri gene. J Immunol. (1993) 150:5445–56.
2. Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. (2005) 6:1123–32. doi: 10.1038/ni1254
3. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. (2005) 6:1133–41. doi: 10.1038/ni1261
4. Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol. (2010) 10:479–89. doi: 10.1038/nri2800
5. Amatya N, Garg AV, Gaffen SL. IL-17 signaling: the yin and the yang. Trends Immunol. (2017) 38:310–22. doi: 10.1016/j.it.2017.01.006
6. Gu C, Wu L, Li X. IL-17 family: cytokines, receptors and signaling. Cytokine. (2013) 64:477–85. doi: 10.1016/j.cyto.2013.07.022
7. Puel A, Cypowyj S, Bustamante J, Wright JF, Liu L, Kim HK, et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science. (2011) 332:65–68. doi: 10.1126/science.1200439
8. Boisson B, Wang C, Pedergnana V, Wu L, Cypowyj S, Rybojad M, et al. A biallelic ACT1 mutation selectively abolishes interleukin-17 responses in humans with chronic mucocutaneous candidiasis. Immunity. (2013) 39:676–86. doi: 10.1016/j.immuni.2013.09.002
9. Puel A, Döffinger R, Natividad A, Chrabieh M, Barcenas-Morales G, Picard C, et al. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J Exp Med. (2010) 207:291–7. doi: 10.1084/jem.20091983
10. Sanford M, McKeage K. Secukinumab: first global approval. Drugs. (2015) 75:329–38. doi: 10.1007/s40265-015-0359-0
11. Blauvelt A, Papp KA, Griffiths CEM, Puig L, Weisman J, Dutronc Y, et al. Phase 3 Trials of Ixekizumab in Moderate-to-Severe Plaque Psoriasis. N Engl J Med. (2016) 375:345–56. doi: 10.1056/NEJMoa1512711
12. Miossec P, Kolls JK. Targeting IL-17 and TH17 cells in chronic inflammation. Nat Rev Drug Discov. (2012) 11:763–76. doi: 10.1038/nrd3794
13. Targan SR, Feagan B, Vermeire S, Panaccione R, Melmed GY, Landers C, et al. A Randomized, Double-Blind, Placebo-Controlled Phase 2 Study of Brodalumab in Patients With Moderate-to-Severe Crohn's Disease. Am J Gastroenterol. (2016) 111:1599–607. doi: 10.1038/ajg.2016.298
14. Hueber W, Sands BE, Lewitzky S, Vandemeulebroecke M, Reinisch W, Higgins PD, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn's disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut. (2012) 61:1693–700. doi: 10.1136/gutjnl-2011-301668
15. Panaccione R, Sandborn WJ, Gordon GL, Lee SD, Safdi A, Sedghi S, et al. Briakinumab for treatment of Crohn's disease: results of a randomized trial. Inflamm Bowel Dis. (2015) 21:1329–40. doi: 10.1097/MIB.0000000000000366
16. Ito H, Takazoe M, Fukuda Y, Hibi T, Kusugami K, Andoh A, et al. A pilot randomized trial of a human anti-interleukin-6 receptor monoclonal antibody in active Crohn's disease. Gastroenterology. (2004) 126:989–96. doi: 10.1053/j.gastro.2004.01.012
17. Sandborn WJ, Gasink C, Gao LL, Blank MA, Johanns J, Guzzo C, et al. Ustekinumab induction and maintenance therapy in refractory Crohn's disease. N Engl J Med. (2012) 367:1519–28. doi: 10.1056/NEJMoa1203572
18. Lee JS, Tato CM, Joyce-Shaikh B, Gulen MF, Cayatte C, Chen Y, et al. Interleukin-23-dependent IL-17 production regulates intestinal epithelial permeability. Immunity. (2015) 43:727–38. doi: 10.1016/j.immuni.2015.09.003
19. Novatchkova M, Leibbrandt A, Werzowa J, Neubüser A, Eisenhaber F. The STIR-domain superfamily in signal transduction, development and immunity. Trends Biochem Sci. (2003) 28:226–9. doi: 10.1016/S0968-0004(03)00067-7
20. Leonardi A, Chariot A, Claudio E, Cunningham K, Siebenlist U. CIKS, a connection to IκB kinase and stress-activated protein kinase. Proc Natl Acad Sci USA. (2000) 97:10494–9. doi: 10.1073/pnas.190245697
21. Song X, Qian Y. The activation and regulation of IL-17 receptor mediated signaling. Cytokine. (2013) 62:175–82. doi: 10.1016/j.cyto.2013.03.014
22. Hartupee J, Liu C, Novotny M, Li X, Hamilton T. IL-17 enhances chemokine gene expression through mRNA stabilization. J Immunol. (2007) 179:4135–41. doi: 10.4049/jimmunol.179.6.4135
23. Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol. (2014) 14:585–600. doi: 10.1038/nri3707
24. Ruiz de Morales JMG, Puig L, Daudén E, Cañete JD, Pablos JL, Martin AO, et al. Critical role of interleukin (IL)-17 in inflammatory and immune disorders: an updated review of the evidence focusing in controversies. Autoimmun Rev. (2020) 19:102429. doi: 10.1016/j.autrev.2019.102429
25. Onishi RM, Gaffen SL. Interleukin-17 and its target genes: mechanisms of interleukin-17 function in disease. Immunology. (2010) 129:311–21. doi: 10.1111/j.1365-2567.2009.03240.x
26. Shen F, Gaffen SL. Structure-function relationships in the IL-17 receptor: implications for signal transduction and therapy. Cytokine. (2008) 41:92–104. doi: 10.1016/j.cyto.2007.11.013
27. Chang SH, Dong C. Signaling of interleukin-17 family cytokines in immunity and inflammation. Cell Signal. (2011) 23:1069–75. doi: 10.1016/j.cellsig.2010.11.022
28. Ramani K, Garg AV, Jawale CV, Conti HR, Whibley N, Jackson EK, et al. The kallikrein-kinin system: a novel mediator of IL-17-driven anti-candida immunity in the kidney. PLoS Pathog. (2016) 12:e1005952. doi: 10.1371/journal.ppat.1005952
29. Conti HR, Baker O, Freeman AF, Jang WS, Holland SM, Li RA, et al. New mechanism of oral immunity to mucosal candidiasis in hyper-IgE syndrome. Mucosal Immunol. (2011) 4:448–455. doi: 10.1038/mi.2011.5
30. Kotake S, Udagawa N, Takahashi N, Matsuzaki K, Itoh K, Ishiyama S, et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest. (1999) 103:1345–52. doi: 10.1172/JCI5703
31. Gaffen SL, Hajishengallis G. A new inflammatory cytokine on the block: re-thinking periodontal disease and the Th1/Th2 paradigm in the context of Th17 cells and IL-17. J Dent Res. (2008) 87:817–28. doi: 10.1177/154405910808700908
32. Lee JY, Hall JA, Kroehling L, Wu L, Najar T. Serum amyloid a proteins induce pathogenic Th17 cells and promote inflammatory disease. Cell. (2020) 180:79–91.e16. doi: 10.1016/j.cell.2019.11.026
33. Honda K, Littman DR. The microbiota in adaptive immune homeostasis and disease. Nature. (2016) 535:75–84. doi: 10.1038/nature18848
34. Takatori H, Kanno Y, Chen Z, O'Shea JJ. New complexities in helper T cell fate determination and the implications for autoimmune diseases. Mod Rheumatol. (2008) 18:533–41. doi: 10.3109/s10165-008-0099-z
35. Vaknin-Dembinsky A, Balashov K, Weiner HL. IL-23 is increased in dendritic cells in multiple sclerosis and down-regulation of IL-23 by antisense oligos increases dendritic cell IL-10 production. J Immunol. (2006) 176:7768–74. doi: 10.4049/jimmunol.176.12.7768
36. Wen SR, Liu GJ, Feng RN, Gong FC, Zhong H, Duan SR, et al. Increased levels of IL-23 and osteopontin in serum and cerebrospinal fluid of multiple sclerosis patients. J Neuroimmunol. (2012) 244:94–6. doi: 10.1016/j.jneuroim.2011.12.004
37. McGeachy MJ, Cua DJ, Gaffen SL. The IL-17 family of cytokines in health and disease. Immunity. (2019) 50:892–906. doi: 10.1016/j.immuni.2019.03.021
38. Patel DD, Kuchroo VK. Th17 cell pathway in human immunity: lessons from genetics and therapeutic interventions. Immunity. (2015) 43:1040–51. doi: 10.1016/j.immuni.2015.12.003
39. Stockinger B, Omenetti S. The dichotomous nature of T helper 17 cells. Nat Rev Immunol. (2017) 17:535–44. doi: 10.1038/nri.2017.50
40. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. (2006) 441:235–8. doi: 10.1038/nature04753
41. Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-β induces development of the T(H)17 lineage. Nature. (2006) 441:231–4. doi: 10.1038/nature04754
42. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFβ in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. (2006) 24:179–89. doi: 10.1016/j.immuni.2006.01.001
43. Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. (2007) 8:967–74. doi: 10.1038/ni1488
44. Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, et al. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity. (2009) 17:576–87. doi: 10.1016/j.immuni.2009.02.007
45. Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, et al. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat Immunol. (2011) 12:255–63. doi: 10.1038/ni.1993
46. Komuczki J, Tuzlak S, Friebel E, Hartwig T, Spath S, Rosenstiel P, et al. Fate-Mapping of GM-CSF expression identifies a discrete subset of inflammation-driving T helper cells regulated by cytokines IL-23 and IL-1β. Immunity. (2019) 21:1289–304.e6. doi: 10.1016/j.immuni.2019.04.006
47. McGeachy MJ, Chen Y, Tato CM, Laurence A, Joyce-Shaikh B, Blumenschein WM, et al. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat Immunol. (2009) 10:314–24. doi: 10.1038/ni.1698
48. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. (2006) 126:1121–33. doi: 10.1016/j.cell.2006.07.035
49. Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors RORα and RORγ. Immunity. (2008) 28:29–39. doi: 10.1016/j.immuni.2007.11.016
50. Zhong Z, Wen Z, Darnell JE Jr. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science. (1994) 264:95–8. doi: 10.1126/science.8140422
51. Takeda K, Kaisho T, Yoshida N, Takeda J, Kishimoto T, Akira S. Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: generation and characterization of T cell-specific Stat3-deficient mice. J Immunol. (1998) 161:4652–60.
52. Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, et al. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. (2007) 282:9358–63. doi: 10.1074/jbc.C600321200
53. Eugster HP, Frei K, Kopf M, Lassmann H, Fontana A. IL-6-deficient mice resist myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. Eur J Immunol. (1998) 28:2178–87. doi: 10.1002/(SICI)1521-4141(199807)28:07<2178::AID-IMMU2178>3.0.CO;2-D
54. Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, et al. Generation of pathogenic T(H)17 cells in the absence of TGF-β signalling. Nature. (2010) 467:967–71. doi: 10.1038/nature09447
55. Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. (2007) 26:371–81. doi: 10.1016/j.immuni.2007.02.009
56. Kim HS, Jang SW, Lee W, Kim K, Sohn H, Hwang SS, et al. PTEN drives Th17 cell differentiation by preventing IL-2 production. J Exp Med. (2017) 214:3381–98. doi: 10.1084/jem.20170523
57. Quintana FJ, Jin H, Burns EJ, Nadeau M, Yeste A, Kumar D, et al. Aiolos promotes TH17 differentiation by directly silencing Il2 expression. Nat Immunol. (2012) 13:770–7. doi: 10.1038/ni.2363
58. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. (2005) 201:233–40. doi: 10.1084/jem.20041257
59. Schiffenbauer J, Streit WJ, Butfiloski E, LaBow M, Edwards C III, Moldawer LL. The induction of EAE is only partially dependent on TNF receptor signaling but requires the IL-1 type I receptor. Clin Immunol. (2000) 95:117–23. doi: 10.1006/clim.2000.4851
60. Sutton C, Brereton C, Keogh B, Mills KH, Lavelle EC. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J Exp Med. (2006) 203:1685–91. doi: 10.1084/jem.20060285
61. Matsuki T, Nakae S, Sudo K, Horai R, Iwakura Y. Abnormal T cell activation caused by the imbalance of the IL-1/IL-1R antagonist system is responsible for the development of experimental autoimmune encephalomyelitis. Int Immunol. (2006) 18:399–407. doi: 10.1093/intimm/dxh379
62. McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, et al. TGF-β and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain TH-17 cell-mediated pathology. Nat. Immunol. (2007) 8:1390–7. doi: 10.1038/ni1539
63. Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, et al. Induction and molecular signature of pathogenic TH17 cells. Nat Immunol. (2012) 13:991–9. doi: 10.1038/ni.2416
64. Carr TM, Wheaton JD, Houtz GM, Ciofani M. JunB promotes Th17 cell identity and restrains alternative CD4+ T-cell programs during inflammation. Nat Commun. (2017) 8:301. doi: 10.1038/s41467-017-00380-3
65. Hasan Z, Koizumi SI, Sasaki D, Yamada H, Arakaki N, Fujihara Y, et al. JunB is essential for IL-23-dependent pathogenicity of Th17 cells. Nat Commun. (2017) 8:15628. doi: 10.1038/ncomms15628
66. Yasuda K, Takeuchi Y, Hirota K. Correction to: the pathogenicity of Th17 cells in autoimmune diseases. Semin Immunopathol. (2019) 41:299. doi: 10.1007/s00281-019-00746-3
67. Basu R, Whitley SK, Bhaumik S, Zindl CL, Schoeb TR, Benveniste EN, et al. IL-1 signaling modulates activation of STAT transcription factors to antagonize retinoic acid signaling and control the TH17 cell-iTreg cell balance. Nat Immunol. (2015) 16:286–95. doi: 10.1038/ni.3099
68. Hirota K, Turner JE, Villa M, Duarte JH, Demengeot J, Steinmetz OM, et al. Plasticity of Th17 cells in Peyer's patches is responsible for the induction of T cell-dependent IgA responses. Nat Immunol. (2013) 14:372–9. doi: 10.1038/ni.2552
69. Gaublomme JT, Yosef N, Lee Y, Gertner RS, Yang LV, Wu C, et al. Single-cell genomics unveils critical regulators of Th17 cell pathogenicity. Cell. (2015) 163:1400–12. doi: 10.1016/j.cell.2015.11.009
70. Wang C, Yosef N, Gaublomme J, Wu C, Lee Y, Clish CB, et al. CD5L/AIM regulates lipid biosynthesis and restrains Th17 cell pathogenicity. Cell. (2015) 163:1413–27. doi: 10.1016/j.cell.2015.10.068
71. Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. (2000) 13:715–25. doi: 10.1016/S1074-7613(00)00070-4
72. Ichiyama K, Gonzalez-Martin A, Kim BS, Jin HY, Jin W, Xu W, et al. The MicroRNA-183-96-182 cluster promotes t helper 17 cell pathogenicity by negatively regulating transcription factor foxo1 expression. Immunity. (2016) 44:1284–98. doi: 10.1016/j.immuni.2016.05.015
73. Meyer Zu Horste G, Wu C, Wang C, Cong L, Pawlak M, Lee Y, et al. RBPJ controls development of pathogenic Th17 cells by regulating IL-23 receptor expression. Cell Rep. (2016) 16:392–404. doi: 10.1016/j.celrep.2016.05.088
74. El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. (2011) 12:568–75. doi: 10.1038/ni.2031
75. Jain R, Chen Y, Kanno Y, Joyce-Shaikh B, Vahedi G, Hirahara K, et al. Interleukin-23-induced transcription factor blimp-1 promotes pathogenicity of T helper 17 cells. Immunity. (2016) 44:131–42. doi: 10.1016/j.immuni.2015.11.009
76. Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1β and 6 but not transforming growth factor-β are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. (2007) 8:942–9. doi: 10.1038/ni1496
77. Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. (2007) 8:950–7. doi: 10.1038/ni1497
78. Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)-17 cells requires transforming growth factor-β and induction of the nuclear receptor RORγt. Nat Immunol. (2008) 9:641–9. doi: 10.1038/ni.1610
79. Volpe E, Servant N, Zollinger R, Bogiatzi SI, Hupé P, Barillot E, et al. A critical function for transforming growth factor-β, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat Immunol. (2008) 9:650–7. doi: 10.1038/ni.1613
80. Chen Z, Tato CM, Muul L, Laurence A, O'Shea JJ. Distinct regulation of interleukin-17 in human T helper lymphocytes. Arthritis Rheum. (2007) 56:2936–46. doi: 10.1002/art.22866
81. Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. (2007) 448:1058–62. doi: 10.1038/nature06096
82. Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature. (2008) 452:773–6. doi: 10.1038/nature06764
83. Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature. (2008) 453:65–71. doi: 10.1038/nature06880
84. Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, et al. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature. (2008) 453:106–9. doi: 10.1038/nature06881
85. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell. (2011) 146:772–84. doi: 10.1016/j.cell.2011.07.033
86. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. (2011) 208:1367–76. doi: 10.1084/jem.20110278
87. Karmaus PWF, Chen X, Lim SA, Herrada AA, Nguyen TM, Xu B, et al. Metabolic heterogeneity underlies reciprocal fates of TH17 cell stemness and plasticity. Nature. (2019) 565:101–5. doi: 10.1038/s41586-018-0806-7
88. Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, Kishi Y, et al. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature. (2013) 496:513–7. doi: 10.1038/nature11984
89. Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, et al. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature. (2013) 496:518–22. doi: 10.1038/nature11868
90. Weinshenker BG. Epidemiology of multiple sclerosis. Neurol Clin. (1996) 14:291–308. doi: 10.1016/S0733-8619(05)70257-7
91. Wang K, Song F, Fernandez-Escobar A, Luo G, Wang JH, Sun Y. The properties of cytokines in multiple sclerosis: pros and cons. Am J Med Sci. (2018) 356:552–60. doi: 10.1016/j.amjms.2018.08.018
92. Annunziato F, Cosmi L, Santarlasci V, Maggi L, Liotta F, Mazzinghi B, et al. Phenotypic and functional features of human Th17 cells. J Exp Med. (2007) 204:1849–61. doi: 10.1084/jem.20070663
94. Hauser SL, Oksenberg JR. The neurobiology of multiple sclerosis: genes, inflammation, and neurodegeneration. Neuron. (2006) 52:61–76. doi: 10.1016/j.neuron.2006.09.011
95. Nylander A, Hafler DA. Multiple sclerosis. J Clin Invest. (2012) 122:1180–8. doi: 10.1172/JCI58649
96. Wu GF, Alvarez E. The immunopathophysiology of multiple sclerosis. Neurol Clin. (2011) 29:257–78. doi: 10.1016/j.ncl.2010.12.009
97. Lucchinetti C, Brück W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. (2000) 47:707–17. doi: 10.1002/1531-8249(200006)47:6<707::AID-ANA3>3.0.CO;2-Q
98. Steinman L, Zamvil SS. How to successfully apply animal studies in experimental allergic encephalomyelitis to research on multiple sclerosis. Ann Neurol. (2006) 60:12–21. doi: 10.1002/ana.20913
99. Kuchroo VK, Anderson AC, Waldner H, Munder M, Bettelli E, Nicholson LB. T cell response in experimental autoimmune encephalomyelitis (EAE): role of self and cross-reactive antigens in shaping, tuning, and regulating the autopathogenic T cell repertoire. Ann Rev Immunol. (2002) 20:101–23. doi: 10.1146/annurev.immunol.20.081701.141316
100. Furlan R, Cuomo C, Martino G. Animal models of multiple sclerosis. Methods Mol Biol. (2009) 549:157–73. doi: 10.1007/978-1-60327-931-4_11
101. McGinley AM, Edwards SC, Raverdeau M, Mills KHG. Th17 cells, γδ T cells and their interplay in EAE and multiple sclerosis. J Autoimmun. (2018) 87, 97–108. doi: 10.1016/j.jaut.2018.01.001
102. Mills KH. TLR-dependent T cell activation in autoimmunity. Nat Rev Immunol. (2011) 11:807–22. doi: 10.1038/nri3095
103. Williams KC, Ulvestad E, Hickey WF. Immunology of multiple sclerosis. Clin Neurosci. (1994) 2:229–45.
104. Kutzelnigg A1, Faber-Rod JC, Bauer J, Lucchinetti CF, Sorensen PS, Laursen H, et al. Widespread demyelination in the cerebellar cortex in multiple sclerosis. Brain Pathol. (2007) 17:38–44. doi: 10.1111/j.1750-3639.2006.00041.x
105. Babbe H, Roers A, Waisman A, Lassmann H, Goebels N, Hohlfeld R, et al. Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med. (2000) 192:393–404. doi: 10.1084/jem.192.3.393
106. BD Trapp BD, Nave KA. Multiple sclerosis: an immune or neurodegenerative disorder? Annu Rev Neurosci. (2008) 31:247–69. doi: 10.1146/annurev.neuro.30.051606.094313
107. Rudick RA, Fisher E, Lee JC, Simon J, Jacobs L. Use of the brain parenchymal fraction to measure whole brain atrophy in relapsing-remitting MS. Multiple Sclerosis Collaborative Research Group. Neurology. (1999) 53:1698–704. doi: 10.1212/WNL.53.8.1698
108. Hampton DW, Anderson J, Pryce G, Irvine KA, Giovannoni G, Fawcett JW, et al. An experimental model of secondary progressive multiple sclerosis that shows regional variation in gliosis, remyelination, axonal and neuronal loss. J Neuroimmunol. (2008) 201–202:200–11. doi: 10.1016/j.jneuroim.2008.05.034
109. Farooqi N, Gran B, Constantinescu CS. Are current disease-modifying therapeutics in multiple sclerosis justified on the basis of studies in experimental autoimmune encephalomyelitis? J Neurochem. (2010) 115:829–44. doi: 10.1111/j.1471-4159.2010.06982.x
110. Wujek JR, Bjartmar C, Richer E, Ransohoff RM, Yu M, Tuohy VK, et al. Axon loss in the spinal cord determines permanent neurological disability in an animal model of multiple sclerosis. J Neuropathol Exp Neurol. (2002) 61:23–32. doi: 10.1093/jnen/61.1.23
111. Hofstetter HH, Ibrahim SM, Koczan D, Kruse N, Weishaupt A, Toyka KV, et al. Therapeutic efficacy of IL-17 neutralization in murine experimental autoimmune encephalomyelitis. Cell Immunol. (2005) 237:123–30. doi: 10.1016/j.cellimm.2005.11.002
112. Traugott U, Raine CS, McFarlin DE. Acute experimental allergic encephalomyelitis in the mouse: immunopathology of the developing lesion. Cell. Immunol. (1985) 91:240–54. doi: 10.1016/0008-8749(85)90047-4
113. Dungan LS, McGuinness NC, Boon L, Lynch MA, Mills KH. Innate IFN-γ promotes development of experimental autoimmune encephalomyelitis: a role for NK cells and M1 macrophages. Eur J Immunol. (2014) 44:2903–17. doi: 10.1002/eji.201444612
114. Lock C, Hermans G, Pedotti R, Brendolan A, Schadt E, Garren H, et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med. (2002) 8:500–8. doi: 10.1038/nm0502-500
115. Kolls JK, Linden A. Interleukin-17 family members and inflammation. Immunity. (2004) 21:467–76. doi: 10.1016/j.immuni.2004.08.018
116. Schofield C, Fischer SK, Townsend MJ, Mosesova S, Peng K, Setiadi AF, et al. Characterization of IL-17AA and IL-17FF in rheumatoid arthritis and multiple sclerosis. Bioanalysis. (2016) 8:2317–27. doi: 10.4155/bio-2016-0207
117. Hedegaard CJ, Krakauer M, Bendtzen K, Lund H, Sellebjerg F, Nielsen CH. T helper cell type 1 (Th1), Th2 and Th17 responses to myelin basic protein and disease activity in multiple sclerosis. Immunology. (2008) 125:161–9. doi: 10.1111/j.1365-2567.2008.02837.x
118. Brucklacher-Waldert V, Stuerner K, Kolster M, Wolthausen J, Tolosa E. Phenotypical and functional characterization of T helper 17 cells in multiple sclerosis. Brain. (2009) 132:3329–41. doi: 10.1093/brain/awp289
119. Durelli L, Conti L, Clerico M, Boselli D, Contessa G, Ripellino P, et al. T-helper 17 cells expand in multiple sclerosis and are inhibited by interferon-β. Ann. Neurol. (2009) 65:499–509. doi: 10.1002/ana.21652
120. Kebir H, Kreymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, Bernard M, et al. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med. (2007) 13:1173–5. doi: 10.1038/nm1651
121. Kebir H, Ifergan I, Alvarez JI, Bernard M, Poirier J, Arbour N, et al. Preferential recruitment of interferon-γ-expressing TH17 cells in multiple sclerosis. Ann Neurol. (2009) 66:390–402. doi: 10.1002/ana.21748
122. Schirmer L, Rothhammer V, Hemmer B, Korn T. Enriched CD161high CCR6+ γδ T cells in the cerebrospinal fluid of patients with multiple sclerosis. JAMA Neurol. (2013) 70:345–51. doi: 10.1001/2013.jamaneurol.409
123. Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, et al. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol. (2008) 172:146–55. doi: 10.2353/ajpath.2008.070690
124. Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, et al. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. (2007) 448:484–7. doi: 10.1038/nature05970
125. Wing AC, Hygino J, Ferreira TB, Kasahara TM, Barros PO, Sacramento PM, et al. Interleukin-17-and interleukin-22-secreting myelin-specific CD4+ T cells resistant to corticoids are related with active brain lesions in multiple sclerosis patients. Immunology. (2016) 147:212–20. doi: 10.1111/imm.12552
126. Brod SA. Ingested (Oral) Rituximab Inhibits Eae. Cytokine. (2016) 85:177–83. doi: 10.1016/j.cyto.2016.06.026
127. Segal BM. Th17 cells in autoimmune demyelinating disease. Semin Immunopathol. (2010) 32:71–7. doi: 10.1007/s00281-009-0186-z
128. Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, et al. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. (2006) 177:566–73. doi: 10.4049/jimmunol.177.1.566
129. da Costa DS, Hygino J, Ferreira TB, Kasahara TM, Barros PO, Monteiro C, et al. Vitamin D modulates different IL-17-secreting T cell subsets in multiple sclerosis patients. J Neuroimmunol. (2016) 299:8–18. doi: 10.1016/j.jneuroim.2016.08.005
130. Lückel C, Picard F, Raifer H, Campos Carrascosa L, Guralnik A, Zhang Y, et al. IL-17+ CD8+ T cell suppression by dimethyl fumarate associates with clinical response in multiple sclerosis. Nat Commun. (2019) 10:5722. doi: 10.1038/s41467-019-13731-z
131. Ntolkeras G, Barba C, Mavropoulos A, Vasileiadis GK, Dardiotis E, Sakkas LI, et al. On the immunoregulatory role of statins in multiple sclerosis: the effects on Th17 cells. Immunol Res. (2019) 67:310–324. doi: 10.1007/s12026-019-09089-5
132. Havrdová E, Belova A, Goloborodko A, Tisserant A, Wright A, Wallstroem E, et al. Activity of secukinumab, an anti-IL-17A antibody, on brain lesions in RRMS: results from a randomized, proof-of-concept study. J Neurol. (2016) 263:1287–95. doi: 10.1007/s00415-016-8128-x
133. Kroenke MA, Carlson TJ, Andjelkovic AV, Segal BM. IL-12–and IL-23–modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. J Exp Med. (2008) 205:1535–41. doi: 10.1084/jem.20080159
134. Ferber IA, Brocke S, Taylor-Edwards C, Ridgway W, Dinisco C, Steinman L, et al. Mice with a disrupted IFN-γ gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE). J Immunol. (1996) 156:5–7.
135. Heremans H, Dillen C, Groenen M, Martens E, Billiau A. Chronic relapsing experimental autoimmune encephalomyelitis (CREAE) in mice: enhancement by monoclonal antibodies against interferon-γ. Europ J Immunol. (1996) 26:2393–8. doi: 10.1002/eji.1830261019
136. Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, et al. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol. (2007) 8:639–46. doi: 10.1038/ni1467
137. Lee YK, Mukasa R, Hatton RD, Weaver CT. Developmental plasticity of Th17 and Treg cells. Curr Opin Immunol. (2009) 21:274–80. doi: 10.1016/j.coi.2009.05.021
138. Abromson-Leeman S, Bronson RT, Dorf ME. Encephalitogenic T cells that stably express both T-bet and RORγt consistently produce IFNγ but have a spectrum of IL-17 profiles. J Neuroimmunol. (2009) 215:10–24. doi: 10.1016/j.jneuroim.2009.07.007
139. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol. (2009) 27:485–517. doi: 10.1146/annurev.immunol.021908.132710
140. Touil T, Fitzgerald D, Zhang GX, Rostami AM, Gran B. Pathophysiology of interleukin-23 in experimental autoimmune encephalomyelitis. Drug News Perspect. (2006) 19:77–83. doi: 10.1358/dnp.2006.19.2.977443
141. Gerosa F, Baldani-Guerra B, Lyakh LA, Batoni G, Esin S, Winkler-Pickett RT, et al. Differential regulation of interleukin 12 and interleukin 23 production in human dendritic cells. J Exp Med. (2008) 205:1447–61. doi: 10.1084/jem.20071450
142. Zhang GX, Gran B, Yu S, Li J, Siglienti I, Chen X, et al. Induction of experimental autoimmune encephalomyelitis in IL-12 receptor-β 2-deficient mice: IL-12 responsiveness is not required in the pathogenesis of inflammatory demyelination in the central nervous system. J Immunol. (2003) 170:2153–60. doi: 10.4049/jimmunol.170.4.2153
143. Lyakh L, Trinchieri G, Provezza L, Carra G, Gerosa F. Regulation of interleukin-12/interleukin-23 production and the T-helper 17 response in humans. Immunol Rev. (2008) 226:112–31. doi: 10.1111/j.1600-065X.2008.00700.x
144. Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. (2003) 421:744–8. doi: 10.1038/nature01355
145. Fitzgerald DC, Ciric B, Touil T, Harle H, Grammatikopolou J, Das Sarma J, et al. Suppressive effect of IL-27 on encephalitogenic Th17 cells and the effector phase of experimental autoimmune encephalomyelitis. J Immunol. (2007) 179:3268–75. doi: 10.4049/jimmunol.179.5.3268
146. Stromnes IM, Cerretti LM, Liggitt D, Harris RA, Goverman JM. Differential regulation of central nervous system autoimmunity by T(H)1 and T(H)17 cells. Nat Med. (2008) 14:337–42. doi: 10.1038/nm1715
147. Lin W, Wang N, Zhou K, Su F, Jiang Y, et al. RKIP mediates autoimmune inflammation by positively regulating IL-17R signaling. EMBO Rep. (2018) 19:e44951. doi: 10.15252/embr.201744951
148. Das Sarma J, Ciric B, Marek R, Sadhukhan S, Caruso ML, Shafagh J, et al. Functional interleukin-17 receptor A is expressed in central nervous system glia and upregulated in experimental autoimmune encephalomyelitis. J Neuroinflamm. (2009) 6:14. doi: 10.1186/1742-2094-6-14
149. Meares GP, Ma X, Qin H, Benveniste EN. Regulation of CCL20 expression in astrocytes by IL-6 and IL-17. Glia. (2012) 60:771–81. doi: 10.1002/glia.22307
150. Yi H, Bai Y, Zhu X, Lin L, Zhao L, Wu X, et al. IL-17A induces MIP-1alpha expression in primary astrocytes via Src/MAPK/PI3K/NF-kB pathways:implications for multiple sclerosis. J Neuroimmune Pharmacol. (2014) 9:629–41. doi: 10.1007/s11481-014-9553-1
151. Xiao Y, Jin J, Chang M, Nakaya M, Hu H, Zou Q, et al. TPL2 mediates autoimmune inflammation through activation of the TAK1 axis of IL-17 signaling. J Exp Med. (2014) 211:1689–1702. doi: 10.1084/jem.20132640
152. Elain G, Jeanneau K, Rutkowska A, Mir AK, Dev KK. The selective anti-IL17A monoclonal antibody secukinumab (AIN457) attenuates IL17A-induced levels of IL6 in human astrocytes. Glia. (2014) 62:725–35. doi: 10.1002/glia.22637
153. Kang Z, Altuntas CZ, Gulen MF, Liu C, Giltiay N, Qin H, et al. Astrocyte-restricted ablation of interleukin-17-induced Act1-mediated signaling ameliorates autoimmune encephalomyelitis. Immunity. (2010) 32:414–25. doi: 10.1016/j.immuni.2010.03.004
154. Kang Z, Wang C, Zepp J, Wu L, Sun K, Zhao J, et al. Act1 mediates IL-17-induced EAE pathogenesis selectively in NG2+ glial cells. Nat Neurosci. (2013) 16:1401–8. doi: 10.1038/nn.3505
155. Paintlia MK, Paintlia AS, Singh AK, Singh I. Synergistic activity of interleukin-17 and tumor necrosis factor-alpha enhances oxidative stress-mediated oligodendrocyte apoptosis. J Neurochem. (2011) 116:508–21. doi: 10.1111/j.1471-4159.2010.07136.x
156. Rodgers JM, Robinson AP, Rosler ES, Lariosa-Willingham K, Persons RE, Dugas JC, et al. IL-17A activates ERK1/2 and enhances differentiation of oligodendrocyte progenitor cells. Glia. (2015) 63:768–79. doi: 10.1002/glia.22783
157. Kawanokuchi J, Shimizu K, Nitta A, Yamada K, Mizuno, Takeuchia H, et al. Production and functions of IL-17 in microglia. J Neuroimmunol. (2008) 194:54–61. doi: 10.1016/j.jneuroim.2007.11.006
158. Murphy AC, Lalor SJ, Lynch MA, Mills KH. Infiltration of Th1 and Th17 cells and activation of microglia in the CNS during the course of experimental autoimmune encephalomyelitis. Brain Behav Immun. (2010) 24:641–51. doi: 10.1016/j.bbi.2010.01.014
159. Huppert J, Closhen D, Croxford A, White R, Kulig P, Pietrowski E, et al. Cellular mechanisms of IL-17-induced blood-brain barrier disruption. FASEB J. (2010) 24:1023–34. doi: 10.1096/fj.09-141978
160. Setiadi AF, Abbas AR, Jeet S, Wong K, Bischof A, Peng I, et al. IL-17A is associated with the breakdown of the blood-brain barrier in relapsing-remitting multiple sclerosis. J Neuroimmunol. (2019) 332:147–54. doi: 10.1016/j.jneuroim.2019.04.011
161. Reed MD, Yim YS, Wimmer RD, Kim H, Ryu C, Welch GM, et al. IL-17a promotes sociability in mouse models of neurodevelopmental disorders. Nature. (2020) 577:249–53. doi: 10.1038/s41586-019-1843-6
162. Siffrin V, Radbruch H, Glumm R, Niesner R, Paterka M, Herz J, et al. In vivo imaging of partially reversible Th17 cell-induced neuronal dysfunction in the course of encephalomyelitis. Immunity. (2010) 33:424–36. doi: 10.1016/j.immuni.2010.08.018
163. Kreymborg K, Etzensperger R, Dumoutier L, Haak S, Rebollo A, Buch T, et al. IL-22 is expressed by Th17 cells in an IL-23-dependent fashion, but not required for the development of autoimmune encephalomyelitis. J Immunol. (2007) 179:8098–104. doi: 10.4049/jimmunol.179.12.8098
164. Perriard G, Mathias A, Enz L, Canales M, Schluep M, Gentner M, et al. Interleukin-22 is increased in multiple sclerosis patients and targets astrocytes. J Neuroinflammation. (2015) 12:119. doi: 10.1186/s12974-015-0335-3
165. Beyeen AD, Adzemovic MZ, Ockinger J, Stridh P, Becanovic K, Laaksonen H, et al. IL-22RA2 associates with multiple sclerosis and macrophage effector mechanisms in experimental neuroinflammation. J Immunol. (2010) 185:6883–90. doi: 10.4049/jimmunol.1001392
166. Zhen J, Yuan J, Fu Y, Zhu R, Wang M, Chang H, et al. IL-22 promotes Fas expression in oligodendrocytes and inhibits FOXP3 expression in T cells by activating the NF-κB pathway in multiple sclerosis. Mol Immunol. (2017) 82:84–93. doi: 10.1016/j.molimm.2016.12.020
167. Almolda B, Costa M, Montoya M, González B, Castellano B. Increase in Th17 and Treg lymphocytes and decrease of IL22 correlate with the recovery phase of acute EAE in rat. PLoS ONE. (2011) 6:e27473. doi: 10.1371/journal.pone.0027473
168. Xin N, Namaka MP, Dou C, Zhang Y. Exploring the role of interleukin-22 in neurological and autoimmune disorders. Int Immunopharmacol. (2015) 28:1076–83. doi: 10.1016/j.intimp.2015.08.016
169. Milovanovic M, Volarevic V, Ljujic B, Radosavljevic G, Jovanovic I, Arsenijevic N, et al. Deletion of IL-33R. (ST2) abrogates resistance to EAE in BALB/C mice by enhancing polarization of APC to inflammatory phenotype. PLoS ONE. (2012) 7:e45225. doi: 10.1371/journal.pone.0045225
170. Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, et al. RORγt drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phasof autoimmune neuroinflammation. Nat Immunol. (2011) 12:560–7. doi: 10.1038/ni.2027
171. Haak S, Croxford AL, Kreymborg K, Heppner FL, Pouly S, Becher B, et al. IL-17A and IL-17F do not contribute vitally to autoimmune neuro-inflammation in mice. J Clin Invest. (2009) 119:61–9. doi: 10.1172/JCI35997
172. Carrieri PB, Provitera V, De Rosa T, Tartaglia G, Gorga F, Perrella O. Profile of cerebrospinal fluid and serum cytokines in patients with relapsing-remitting multiple sclerosis: a correlation with clinical activity. Immunopharmacol Immunotoxicol. (1998) 20:373–82. doi: 10.3109/08923979809034820
173. Rasouli J, Ciric B, Imitola J, Gonnella P, Hwang D, Mahajan K, et al. Expression of GM-CSF in T cells is increased in multiple sclerosis and suppressed by IFN-β therapy. J Immunol. (2015) 194:5085–93. doi: 10.4049/jimmunol.1403243
174. Milovanovic J, Popovic B, Milovanovic M, Kvestak D, Arsenijevic A, Stojanovic B, et al. Murine cytomegalovirus infection induces susceptibility to EAE in resistant BALB/c mice. Front Immunol. (2017) 8:192. doi: 10.3389/fimmu.2017.00192
175. Reboldi A, Coisne C, Baumjohann D, Benvenuto F, Bottinelli D, Lira S, et al. C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol. (2009) 10:514–23. doi: 10.1038/ni.1716
176. Casiraghi C, Shanina I, Cho S, Freeman ML, Blackman MA, Horwitz MS. γherpesvirus latency accentuates EAE pathogenesis: relevance to Epstein–Barr virus and multiple sclerosis. PLoS Pathog. (2012) 8:e1002715. doi: 10.1371/journal.ppat.1002715
177. Huber M, Heink S, Pagenstecher A, Reinhard K, Ritter J, Visekruna A, et al. IL-17A secretion by CD8+ T cells supports Th17-mediated autoimmune encephalomyelitis. J Clin Invest. (2013) 123:247–60. doi: 10.1172/JCI63681
178. Bantug GR, Cekinovic D, Bradford R, Koontz T, Jonjic S, Britt WJ. CD8+ T lymphocytes control murine cytomegalovirus replication in the central nervous system of newborn animals. J Immunol. (2008) 181:2111–23. doi: 10.4049/jimmunol.181.3.2111
179. Ji Q, Castelli L, Goverman JM. MHC class I-restricted myelin epitopes are cross-presented by Tip-DCs that promote determinant spreading to CD8? T cells. Nat Immunol. (2013) 14:254–61. doi: 10.1038/ni.2513
180. Saederup N, Aguirre SA, Sparer TE, Bouley DM, Mocarski ES. Murine cytomegalovirus CC chemokine homolog MCK-2 (m131-129) is a determinant of dissemination that increases inflammation at initial sites of infection. J Virol. (2001) 75:9966–76. doi: 10.1128/JVI.75.20.9966-9976.2001
181. Noda S, Aguirre SA, Bitmansour A, Brown JM, Sparer TE, Huang J, et al. Cytomegalovirus MCK-2 controls mobilization and recruitment of myeloid progenitor cells to facilitate dissemination. Blood. (2006) 107:30–8. doi: 10.1182/blood-2005-05-1833
182. Daley-Bauer LP, Wynn GM, Mocarski ES. Cytomegalovirus impairs antiviral CD8+ T cell immunity by recruiting inflammatory monocytes. Immunity. (2012) 37:122–33. doi: 10.1016/j.immuni.2012.04.014
183. Karantzoulis S, Galvin JE. Distinguishing Alzheimer's disease from other major forms of dementia. Expert Rev Neurother. (2011) 11:1579–91. doi: 10.1586/ern.11.155
184. Wray S, Fox NC. Stem cell therapy for Alzheimer's disease: hope or hype? Lancet Neurol. (2016) 15:133–5. doi: 10.1016/S1474-4422(15)00382-8
185. Gouras GK, Olsson TT, Hansson O. β-Amyloid peptides and amyloid plaques in Alzheimer's disease. Neurotherapeutics. (2015) 12:3–11. doi: 10.1007/s13311-014-0313-y
186. Gu L, Tran J, Jiang L, Guo Z. A new structural model of Alzheimer's Aβ42 fibrils based on electron paramagnetic resonance data and Rosetta modeling. J Struct Biol. (2016) 194:61–7. doi: 10.1016/j.jsb.2016.01.013
187. Lyons B, Friedrich M, Raftery M, Truscott R. Amyloid plaque in the human brain can decompose from Aβ(1-40/1-42) by spontaneous nonenzymatic processes. Anal Chem. (2016) 88:2675–84. doi: 10.1021/acs.analchem.5b03891
188. Wang WY, Tan MS, Yu JT, Tan L. Role of pro-inflammatory cytokines released from microglia in Alzheimer's disease. Ann Transl Med. (2015) 3:136. doi: 10.3978/j.issn.2305-5839.2015.03.49
189. Rudinskiy N, Fuerer C, Demurtas D, Zamorano S, De Piano C, Herrmann AG, et al. Amyloid-beta oligomerization is associated with the generation of a typical peptide fragment fingerprint. Alzheimers Dement. (2016) 12:996–1013. doi: 10.1016/j.jalz.2016.03.011
190. Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol. (2010) 6:193–201. doi: 10.1038/nrneurol.2010.17
191. Myhre O, Utkilen H, Duale N, Brunborg G, Hofer T. Metal dyshomeostasis and inflammation in Alzheimer's and Parkinson's diseases: possible impact of environmental exposures. Oxid Med Cell Long. (2013) 2013:726954. doi: 10.1155/2013/726954
192. Zenaro E, Pietronigro E, Della Bianca V, Piacentino G, Constantin G, Budui S, et al. Neutrophils promote Alzheimer's disease-like pathology and cognitive decline via LFA-1 integrin. Nat. Med. (2015) 21:880–6. doi: 10.1038/nm.3913
193. Yin Y, Wen S, Li G, Wang D. Hypoxia enhances stimulating effect of amyloid beta peptide (25–35) for interleukin 17 and T helper lymphocyte subtype 17 upregulation in cultured peripheral blood mononuclear cells. Microbiol Immunol. (2009) 53:281–6. doi: 10.1111/j.1348-0421.2009.00120.x
194. Saksida T, Koprivica I, Vujičić M, Stošić-Grujičić S, Perović M. Impaired IL-17 production in gut-residing immune cells of 5xFAD mice with Alzheimer's disease pathology. J Alzheimers Dis. (2018) 61:619–30. doi: 10.3233/JAD-170538
195. Wang X, Zhang M, Liu H. LncRNA17A regulates autophagy and apoptosis of SH-SY5Y cell line as an in vitro model for Alzheimer's disease. Biosci Biotechnol Biochem. (2019) 83:609–21. doi: 10.1080/09168451.2018.1562874
196. St-Amo I, Bosoi CR, Paré I, Ignatius Arokia Doss PM, Rangachari M, Hébert SS, et al. Peripheral adaptive immunity of the triple transgenic mouse model of Alzheimer's disease. J Neuroinflammation. (2019) 16:3. doi: 10.1186/s12974-018-1380-5
197. Zhang J, Ke KF, Liu Z, Qiu YH, Peng YP. Th17 cell-mediated neuroinflammation is involved in neurodegeneration of aβ1-42-induced Alzheimer's disease model rats. PLoS ONE. (2013) 8:e75786. doi: 10.1371/journal.pone.0075786
198. Tian A, Ma H, Zhang R, Tan W, Wang X, Wang Y, et al. Interleukin17A promotes postoperative cognitive dysfunction by triggering β-amyloid accumulation via the transforming growth factor-β (TGFβ)/smad signaling pathway. PLoS ONE. (2015) 10:e0141596. doi: 10.1371/journal.pone.0141596
199. Zhang Y, Liu M, Sun H, Yin K. Matrine improves cognitive impairment and modulates the balance of Th17/Treg cytokines in a rat model of Aβ1-42-induced Alzheimer's disease. Central-Eur J Immunol. (2016) 40:411. doi: 10.5114/ceji.2015.56961
200. Browne TC, McQuillan K, McManus RM, O'Reilly JA, Mills KH, Lynch MA. IFN-γ production by amyloid β-specific Th1 cells promotes microglial activation and increases plaque burden in a mouse model of Alzheimer's disease. J Immunol. (2013) 190:2241–51. doi: 10.4049/jimmunol.1200947
201. Tzartos JS, Craner MJ, Friese MA, Jakobsen KB, Newcombe J, Esiri MM, et al. IL-21 and IL-21 receptor expression in lymphocytes and neurons in multiple sclerosis brain. Am J Pathol. (2011) 178:794–802. doi: 10.1016/j.ajpath.2010.10.043
202. Tahmasebinia F, Pourgholaminejad A. The role of Th17 cells in auto-inflammatory neurological disorders. Prog Neuropsychopharmacol Biol Psychiatry. (2017) 79:408–16. doi: 10.1016/j.pnpbp.2017.07.023
203. Giuliani F, Goodyer CG, Antel JP, Yong VW. Vulnerability of human neurons to T cell-mediated cytotoxicity. J Immunol. (2003) 171:368–79. doi: 10.4049/jimmunol.171.1.368
204. Jin JJ, Kim HD, Maxwell JA, Li L, Fukuchi K. Toll-like receptor 4-dependent upregulation of cytokines in a transgenic mouse model of Alzheimer's disease. J Neuroinflammation. (2008) 5:23. doi: 10.1186/1742-2094-5-23
205. Cristiano C, Volpicelli F, Lippiello P, Buono B, Raucci F, Piccolo M, et al. Neutralization of interleukin-17 rescues amyloid-β-induced neuroinflammation and memory impairment. Br J Pharmacol. (2019) 176:3544–57. doi: 10.1111/bph.14586
206. Solleiro-Villavicencio H, Rivas-Arancibia S. Effect of chronic oxidative stress on neuroinflammatory response mediated by CD4+T cells in neurodegenerative diseases. Front Cell Neurosci. (2018) 12:114. doi: 10.3389/fncel.2018.00114
207. Yang J, Kou J, Lalonde R, Fukuchi KI. Intracranial IL-17A overexpression decreases cerebral amyloid angiopathy by upregulation of ABCA1 in an animal model of Alzheimer's disease. Brain Behav Immun. (2017) 65:262–73. doi: 10.1016/j.bbi.2017.05.012
208. Tfilin M, Turgeman G. Interleukine-17 administration modulates adult hippocampal neurogenesis and improves spatial learning in mice. J Mol Neurosci. (2019) 69:254–63. doi: 10.1007/s12031-019-01354-4
209. Lemprière S. T cells on patrol in Alzheimer disease. Nat Rev Neurol. (2020) 16:128–9. doi: 10.1038/s41582-020-0317-7
210. Saresella M, Calabrese E, Marventano I, Piancone F, Gatti A, Alberoni M, et al. Increased activity of Th-17 and Th-9 lymphocytes and a skewing of the post-thymic differentiation pathway are seen in Alzheimer's disease. Brain Behav Immunity. (2011) 25:539–47. doi: 10.1016/j.bbi.2010.12.004
211. Oberstein TJ, Taha L, Spitzer P, Hellstern J, Herrmann M, Kornhuber J, et al. Imbalance of circulating Th17 and regulatory T cells in Alzheimer's disease: a case control study. Front Immunol. (2018) 9:1213. doi: 10.3389/fimmu.2018.01213
212. Chen J-M, Jiang G-X, Li Q-W, Zhou Z-M, Cheng Q. Increased serum levels of interleukin-18, -23 and -17 in chinese patients with Alzheimer's disease. Dem Geriat Cogn Disord. (2014) 38:321–9. doi: 10.1159/000360606
213. Doecke JD, Laws SM, Faux NG, Wilson W, Burnham SC. Blood-based protein biomarkers for diagnosis of Alzheimer disease. Arch Neurol. (2012) 69:1318–25. doi: 10.1001/archneurol.2012.1282
214. Hu WT, Chen-Plotkin A, Grossman M, Arnold SE, Clark CM. Novel CSF biomarkers for frontotemporal lobar degenerations. Neurology. (2010) 75:2079–86. doi: 10.1212/WNL.0b013e318200d78d
215. Marciani DJ. Alzheimer's disease vaccine development: a new strategy focusing on immune modulation. J Neuroimmunol. (2015) 287:54–63. doi: 10.1016/j.jneuroim.2015.08.008
216. McManus RM, Higgins SC, Mills KH, Lynch MA. Respiratory infection promotes T cell infiltration and amyloid-β deposition in APP/PS1 mice. Neurobiol Aging. (2014) 35:109–21. doi: 10.1016/j.neurobiolaging.2013.07.025
217. Wissman A, Hauptman J, Regen T. The role of IL-17 in CNS diseases. Acta Neuropathol. (2015) 129:625–37. doi: 10.1007/s00401-015-1402-7
218. Shekhar S, Cunningham MW, Pabbidi MR, Wang S, Booz GW, Fan F, et al. Targeting vascular inflammation in ischemic stroke: recent developments on novel immunomodulatory approaches. Eur J Pharmacol. (2018) 833:531–44. doi: 10.1016/j.ejphar.2018.06.028
219. Kigerl KA, de Rivero Vaccari JP, Dietrich WD, Popovich PG, Keane RW. Pattern recognition receptors and central nervous system repair. Exp Neurol. (2014) 258:5–16. doi: 10.1016/j.expneurol.2014.01.001
220. Hayakawa K, Qiu J, Lo EH. Biphasic actions of HMGB1 signaling in inflammation and recovery after stroke. Ann N Y Acad Sci. (2010) 1207:50–7. doi: 10.1111/j.1749-6632.2010.05728.x
221. Zhang J, Takahashi HK, Liu K, Wake H, Liu R, Maruo T, et al. Anti-high mobility group box-1 monoclonal antibody protects the blood-brain barrier from ischemia-induced disruption in rats. Stroke. (2011) 42:1420–8. doi: 10.1161/STROKEAHA.110.598334
222. Shichita T, Hasegawa E, Kimura A, Morita R, Sakaguchi R, Takada I, et al. Peroxiredoxin family proteins are key initiators of postischemic inflammation in the brain. Nat Med. (2012) 18:911–7. doi: 10.1038/nm.2749
223. Planas AM, Gómez-Choco M, Urra X, Gorina R, Caballero M. Brain-derived antigens in lymphoid tissue of patients with acute stroke. J Immunol. (2012) 188:2156–63. doi: 10.4049/jimmunol.1102289
224. Brait VH, Arumugam TV, Drummond GR, Sobey CG. Importance of T lymphocytes in brain injury, immunodeficiency, and recovery after cerebral ischemia. J Cereb Blood Flow Metab. (2012) 32:598–611. doi: 10.1038/jcbfm.2012.6
225. Kleinschnitz C, Schwab N, Kraft P, Hagedorn I, Dreykluft A, Scrhwarz T, et al. Early detrimental T-cell effects in experimental cerebral ischemia are neither related to adaptive immunity nor thrombus formation. Blood. (2010) 115:3835–42. doi: 10.1182/blood-2009-10-249078
226. Arumugam TV, Granger DN, Mattson MP. Stroke and T-cells. Neuromolecular Med. (2005) 7:229–42. doi: 10.1385/NMM:7:3:229
227. Kostulas N, Pelidou SH, Kivisäkk P, Kostulas V, Link H. Increased IL-1β, IL-8, and IL-17 mRNA expression in blood mononuclear cells observed in a prospective ischemic stroke study. Stroke. (1999) 30:2174–9. doi: 10.1161/01.STR.30.10.2174
228. Gelderblom M, Weymar A, Bernreuther C, Velden J, Arunachalam P, Steinbach K, et al. Neutralization of the IL-17 axis diminishes neutrophil invasion and protects from ischemic stroke. Blood. (2012) 120:3793–802. doi: 10.1182/blood-2012-02-412726
229. Li GZ, Zhong D, Yang LM, Sun B, Zhong ZH, Yin YH, et al. Expression of interleukin-17 in ischemic brain tissue. Scand J Immunol. (2005) 62:481–6. doi: 10.1111/j.1365-3083.2005.01683.x
230. Wang DD, Zhao YF, Wang GY, Sun B, Kong QF, Zhao K, et al. IL-17 potentiates neuronal injury induced by oxygen-glucose deprivation and affects neuronal IL-17 receptor expression. J Neuroimmunol. (2009) 212:17–25. doi: 10.1016/j.jneuroim.2009.04.007
231. Zhang J, Mao X, Zhou T, Cheng X, Lin Y. IL-17A contributes to brain ischemia reperfusion injury through calpain-TRPC6 pathway in mice. Neuroscience. (2014) 274:419–28. doi: 10.1016/j.neuroscience.2014.06.001
232. Li HL, Kostulas N, Huang YM, Xiao BG, van der Meide P, Kostuals V, et al. IL-17 and IFN-γ mRNA expression is increased in the brain and systemically after permanent middle cerebral artery occlusion in the rat. J Neuroimmunol. (2001) 116:5–14. doi: 10.1016/S0165-5728(01)00264-8
233. Shichita T, Sugiyama Y, Ooboshi H, Sugimori H, Nakagawa R, Takada I, et al. Pivotal role of cerebral interleukin-17-producing γδ T cells in the delayed phase of ischemic brain injury. Nat Med. (2009) 15:946–50. doi: 10.1038/nm.1999
234. Jin WN, Gonzales R, Feng Y, Wood K, Chai Z, Dong J-F, et al. Brain Ischemia Induces Diversified Neuroantigen-Specific T-Cell Responses That Exacerbate Brain Injury. Stroke. (2018) 49:1471–1478. doi: 10.1161/STROKEAHA.118.020203
235. Dolati S, Ahmadi M, Khalili M, Taheraghdam AA, Siahmansouri H, Babaloo Z, et al. Peripheral Th17/Treg imbalance in elderly patients with ischemic stroke. Neurol Sci. (2018) 39:647–54. doi: 10.1007/s10072-018-3250-4
236. Zhang Y, Xu D, Qi H, Yuan Y, Liu H, Yao S, et al. Enriched environment promotes post-stroke neurogenesis through NF-κB-mediated secretion of IL-17A from astrocytes. Brain Res. (2018) 1687:20–31. doi: 10.1016/j.brainres.2018.02.030
237. Liu T, Han S, Dai Q, Zheng J, Liu C, Li S, et al. IL-17A-mediated excessive autophagy aggravated neuronal ischemic injuries via Src-PP2B-mTOR pathway. Front Immunol. (2019) 10:2952. doi: 10.3389/fimmu.2019.02952
Keywords: IL-17, Th17, EAE, Alzheimer's disease, ischemic brain injury
Citation: Milovanovic J, Arsenijevic A, Stojanovic B, Kanjevac T, Arsenijevic D, Radosavljevic G, Milovanovic M and Arsenijevic N (2020) Interleukin-17 in Chronic Inflammatory Neurological Diseases. Front. Immunol. 11:947. doi: 10.3389/fimmu.2020.00947
Received: 14 February 2020; Accepted: 22 April 2020;
Published: 03 June 2020.
Edited by:
Nicola Ivan Lorè, IRCCS San Raffaele Scientific Institute, ItalyCopyright © 2020 Milovanovic, Arsenijevic, Stojanovic, Kanjevac, Arsenijevic, Radosavljevic, Milovanovic and Arsenijevic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marija Milovanovic, bWFyaWphcG9zdGFAZ21haWwuY29t