 Yijie Zhang†
Yijie Zhang† Haiying Li
Haiying Li Jiang Wu
Jiang Wu Haitao Shen
Haitao Shen Xiang Li
Xiang Li Gang Chen
Gang Chen- Brain and Nerve Research Laboratory, Department of Neurosurgery, The First Affiliated Hospital of Soochow University, Suzhou, China
Cerebral ischemia is a severe, acute condition, normally caused by cerebrovascular disease, and results in high rates of disability, and death. Phagoptosis is a newly recognized form of cell death caused by phagocytosis of viable cells, and has been reported to contribute to neuronal loss in brain tissue after ischemic stroke. Previous data indicated that exposure of phosphatidylserine to viable neurons could induce microglial phagocytosis of such neurons. Phosphatidylserine can be reversibly exposed to viable cells as a result of a calcium-activated phospholipid scramblase named TMEM16F. TMEM16F-mediated phospholipid scrambling on platelet membranes is critical for hemostasis and thrombosis, which plays an important role in Scott syndrome and has been confirmed by much research. However, few studies have investigated the association between TMEM16F and phagocytosis in ischemic stroke. In this study, a middle-cerebral-artery occlusion/reperfusion (MCAO/R) model was used in adult male Sprague-Dawley rats in vivo, and cultured neurons were exposed to oxygen-glucose deprivation/reoxygenation (OGD/R) to simulate cerebral ischemia-reperfusion (I/R) injury in vitro. We found that the protein level of TMEM16F was significantly increased at 12 h after I-R injury both in vivo and in vitro, and reversible phosphatidylserine exposure was confirmed in neurons undergoing I/R injury in vitro. Additionally, we constructed a LV-TMEM16F-RNAi transfection system to suppress the expression of TMEM16F during and after cerebral ischemia. As a result, TMEM16F knockdown alleviated motor function injury and decreased the microglial phagocytosis of viable neurons in the penumbra through inhibiting the “eat-me” signal phosphatidylserine. Our data indicate that reducing neuronal phosphatidylserine-exposure via deficiency of TMEM16F blocks phagocytosis of neurons and rescues stressed-but-still-viable neurons in the penumbra, which may contribute to reducing infarct volume and improving functional recovering.
Introduction
Cerebral ischemia is a major cause of human neurological morbidity and mortality and is associated with a poor prognosis worldwide. It is caused by an interrupted blood supply to the brain and can result in severe neurodegeneration and vascular dementia in patients (1). According to the World Health Organization (WHO) data, the incidence of stroke in China is increasing at an annual rate of 8.7%. The annual death toll of stroke is over two million and about 85% of these patients died from ischemic stroke, which seriously threaten peoples' lives and affects their quality of life (2, 3).
Neurons in different ischemic regions experience different death patterns. Inflation, as a form of necrosis that is currently considered to be unprogrammed, often occurs in the ischemic core, where the reduction of blood flow is most pronounced. However, in areas of partial ischemia (the penumbra) and in areas around the infarct (peri-infarct), neurons stressed by ischemia, or its consequences are lost only after some delay, which usually in several programmed death modes such as neuronal apoptosis, PARP-1 dependent necrosis, and iron death, provides an opportunity for therapeutic interference hours or days after the stroke (4, 5).
Phagocytosis is normally secondary to the target cell dying by other means, such as apoptosis (6, 7). However, in viable phosphatidylserine (PS)-exposed cells, phagocytosis can directly cause cell death and such death is referred to as “primary phagocytosis,” with the defining characteristic that inhibition of phagocytosis can prevent cell death. Primary phagocytosis normally does not initiate cell death but rather executes cell death via phagocytosis, which may be induced by exposure of eat-me signals (such as PS) on viable target cells (4).
Recent data indicate that phagocytosis can execute the death of stressed-but-viable neurons during inflammation and neuropathology (8–10). Importantly, neurons in peri-infarct areas have been shown to expose PS in a reversible manner (11); these neurons can survive long-term if the phagocytosis is blocked. During the inflammation process, the PS exposed to neurons is bound by MFG-E8 (Milk fat globule EGF-like factor-8), which can induce phagocytosis via the microglial vitronectin receptor. Thus, blocking exposed PS, MFG-E8 or the vitronectin receptor can prevent neuronal loss and leave viable neurons without inhibiting inflammation. It has been reported that deficiency of MFG-E8 or MerTK (MER receptor tyrosine kinase, a receptor on the microglia) could prevent neuronal loss and improve physical function (10, 12). Therefore, our present study was designed to identify whether blocking PS exposure is enough to inhibit phagocytosis.
Calcium-activated phospholipid scramblase, also known as TMEM16F protein (13, 14), plays an important role in reversible PS-exposure. We therefore hypothesized that TMEM16F may contribute to the PS-exposure of viable neurons in the penumbra after transient cerebral ischemia, and thus promote phagocytosis. Our data showed that phagocytosis induced neuronal death after ischemia; knocking down the expression of TMEM16F could therefore prevent neuronal loss and mitigate long-term functional deficits.
Materials and Methods
Animals and Ethics
Adult male Sprague-Dawley (SD) rats weighing 300–350 g were purchased from the Animal Center of the Chinese Academy of Sciences (Shanghai, China). All experimental rats had adequate food and water, and they were housed in a quiet humidity- and temperature-controlled environment under a regular light/dark schedule (12/12 h light/dark cycle with humidity of 60 ± 5% and temperature of 22 ± 3°C). Primary neuronal cultures in vitro were prepared using 16–18-days-old pregnant SD rats. We strived to reduce the use of animals and relieved their pain as much as possible. The animal experimental protocols were approved by the Animal Care and Use Committee of Soochow University and performed in accordance with the National Institute of Health's guidelines (NIH Publication No. 8023, revised 2011).
MCAO Model Establishment
Rats were subjected to 2 h right middle cerebral artery occlusion (MCAO) using a modified intraluminal filament technique that was first put forward and interpreted by Koizumi et al. (15) in rats. The rats were weighed, and then administered an intra-peritoneal injection of 4% chloral hydrate (0.1 ml/10 g i.p.) as an anesthetic agent (16, 17). After the anesthesia, the right common carotid artery (CCA), external carotid artery (ECA), and internal carotid artery (ICA) of rats were exposed through an incision in the middle on the neck. Subsequently, the proximal CCA and ECA were ligated, and an arterial clip was placed on the distal end of the CCA in order to block the blood flow to prevent bleeding when the filament was inserted. A filament with a diameter of 0.38 mm, whose tip was rounded by heating and coating with 0.01% poly-L-lysine, was inserted into the right CCA. The filament was advanced 18–20 mm further above the bifurcation until there was resistance, reaching, and occluding the ostium of the right middle cerebral artery (MCA). After that, the incision of the ICA was ligated, and the filament was secured in place for 2 h, after which the wound was closed, and the animals were allowed to awaken. Body temperature was maintained at 36.5–37.5°C using a heating pad during the procedure. Two hours post-occlusion, the filament was slowly withdrawn under anesthesia and animals were then returned to their cages for reperfusion (MCAO/R) (18, 19). Animals were assessed for functional impairment using the modified Bederson grading system to verify accurate occlusion of the middle cerebral artery when the rats were awakened (20, 21). Motor deficits were graded from 0 to 4. A score of 0 was given for no visible neurological deficits; a score of 1 was given for forelimb flexion; a score of 2 was given for contralateral weak forelimb grip (the operator places the animal on an absorbent pad and gently pulls the tail); a score of 3 was given for circling to the paretic side only when the tail was stimulated; and a score of 4 was given for spontaneous circling (20, 22, 23). Finally, animals with positive performances were included in our study.
Cell Culture
Primary rat cortical neurons were obtained and cultured as described previously (24). Briefly, cortical neurons were prepared from embryonic-day-18 brains. Then, cortical neurons were digested with 0.25% trypsin-EDTA solution for 5 min at 37°C. The dissociated neurons were seeded on to six-well plates (Corning, USA) precoated with poly-D-lysine (Sigma, USA), and were cultured in Neurobasal medium containing 2% B-27, 0.5 mM of GlutaMAX, 50 U/ml of penicillin, and 50 U/ml of streptomycin (all from Invitrogen, Grand Island, NY, USA) under humidified air containing 5% CO2 at 37°C. The medium was renewed every 2 days until cell confluency was reached.
Experiment Design
To verify the protein level of TMEM16F after ischemic attack (Experiment 1; Supplementary Figure 1A), rats under sham or MCAO/R surgeries were included. Animals experiencing MCAO/R were sacrificed at different intervals (6, 12, 24, 48, 72 h, or 7 d after MCAO/R). Then, brain tissue samples were obtained for analysis. We made a coronal cut at 3 and 9 mm from the front of the frontal lobe, taking a 6 mm thick brain tissue block. Regions from this section that corresponded to the ischemic core and penumbra were dissected. We then made a longitudinal cut (from top to bottom) ~2 mm from the sagittal suture through the right hemisphere. This was done to avoid mesial hemispheric structures, which are supplied primarily by the anterior cerebral artery. We then made a transverse diagonal cut at approximately at the “2 o'clock” position (as shown in Supplementary Figure 2) to separate the core (striatum and overlying cortex) from the penumbra (adjacent cortex) (25). The penumbra tissues surrounding the infarcted core of six rats from each group were extracted and frozen at −80°C until subsequent Western blot analysis. Additionally, we also confirmed the protein level of TMEM16F in vitro. Primary neuron cells were randomly and equally divided into eight groups, namely a control group and seven intervention groups including 2, 4, 6, 12, 24, 48, and 72 h after oxygen and oxygen-glucose deprivation reoxygenation (OGD/R). At the appropriate times after OGD/R for each group, cellular proteins were extracted for Western blot analysis.
To investigate the function of TMEM16F after transient cerebral ischemia in vivo (Experiment 2; Supplementary Figure 1B), specific lentivirus (LV-RNAi) against TMEM16F was applied to knock down TMEM16F. Rats were divided into the following four groups: sham group (sham), MCAO/R group, MCAO/R+ LV-negative control group (NC), and MCAO/R+ LV-TMEM16F-RNAi group (LV-RNAi). The MCAO/R surgery was performed 5 d after the transfection of LV-RNAi in vivo. At 12 h after MCAO/R, rats were sacrificed and the brain samples were collected for analysis. Rats in the neurobehavior groups were tested at 0-, 3-, 5-, 7-, and 14-days intervals after MCAO/R. To confirm the function of TMEM16F and its possible mechanism (Experiment 2; Supplementary Figure 1B), the following in vitro studies were conducted. Based on the interventions 96 h before OGD/R, the primary cultured neurons were divided into the following five groups: control group (Control), OGD/R group, OGD/R+ HitransG A reagent (HA, a reagent that could enhance transfection efficacy), OGD/R+HA+ LV-negative control group (NC), and OGD/R+ HA+LV-TMEM16F-RNAi group (LV-RNAi). At 12 h after OGD/R, cells were used for testing. In addition, at 12 and 24 h after OGD/R, cells that were not transfected with lentivirus were collected for flow cytometry analysis. Details of mortality and exclusion of experimental rats are shown in Supplementary Table 1.
Establishing an Oxygen-Glucose Deprivation/Reoxygenation Model in vitro
Briefly, Neurobasal medium was replaced with DMEM (GIBCO, Carlsbad, CA, USA) and cells were transferred to a 5% CO2 and 95% N2 atmospheric incubator for 2 h at 37°C. After that, neurons were cultured in Neurobasal medium again and maintained in a 5% CO2 atmospheric incubator for the indicated time periods. Control groups were cultured in Neurobasal medium in a 5% CO2 atmospheric incubator for the same period. The pH of culture medium was maintained at 7.2 (26).
Construction of LV-TMEM16F-RNAi System
In vivo
SD male rats (260–280 g body weight) were anesthetized with 4% chloral hydrate (0.1 ml/10 g) via an intra-peritoneal injection. Cortical injection of LV-TMEM16F-RNAi-GFP or LV-NC-GFP was carried out using a stereotaxic instrument. Each rat was subjected to three cortical injections as follows: AP +1.2 (site 1), −0.3 (site 2), −1.8 (site 3); ML +3.0; DV −3.0 from the skull. All of the target points were in the right hemisphere (i.e., ipsilateral to the MCAO). Two microliters of lentivirus suspension containing 9 × 108 TU/ml were injected into each point at a rate of 0.2 μl/min (27, 28). The needle was slowly removed 5 min after completion of each injection. The rats were allowed 5 days of recovery before being subjected to the MCAO/R surgery.
In vitro
Cultured neurons were transfected with the LV-TMEM16F-RNAi-GFP or LV-NC-GFP combined with HitransG A reagent (HA), which could enhance transfection efficacy (GeneChem, China), according to the manufacturer's instructions. At 16 h after transfection, the medium was replaced and the neurons were incubated for another 4 days for further analysis.
TTC Staining
The remaining rats were put to death by cutting off their cervical spines; their brains were quickly removed, and five 2 mm thick coronal slices were cut from each brain using razor blades. The slices were immersed in a physiological solution containing 2% (w/v) 2,3,5-triphenyltetrazolium hydrochloride (TTC) and incubated in a 37°C incubator for 15 min in the dark (29, 30). The infarcted tissue was verified by the complete loss of TTC staining, contrasting with the red-stained viable tissue. The area of infarction was determined by ImageJ software (Rawak Software Inc., Stuttgart, Germany).
Western Blot Analysis
Western blot analysis was performed as in previous studies (31, 32). Briefly, the brain samples of rats or extracted neurons were mechanically lysed in a RIPA lysate buffer (Beyotime, China). The protein concentrations were measured with the bicinchoninic acid (BCA) method using a specific assay kit (Beyotime, China). The protein samples (30 μg/lane) were loaded onto a 10% SDS-polyacrylamide gel, separated, and then electrophoretically transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore Corporation, USA). The membrane was blocked with 5% bovine serum albumin (BSA, BioSharp, China) at 25°C for 1 h. Next, the membrane was incubated with a primary antibody against TMEM16F (Cloud-Clone Corp., PAF813Hu01, 1:1,000 dilution) at 4°C overnight. The primary antibody against β-tubulin (Cell Signaling Technology, Danvers, USA) was diluted 1:5,000 and served as a loading control. Lastly, the membranes were incubated with a horseradish peroxidase-linked secondary antibody (goat anti-rabbit IgG-HRP, Cell Signaling Technology, Danvers, USA) for 2 h at 37°C and were washed three times with PBST (PBS + 0.1% Tween 20). Finally, the membrane was revealed with an enhanced chemiluminescence (ECL) kit (Beyotime Institute of Biotechnology) and the relative quantities of proteins were analyzed with ImageJ software (Rawak Software Inc., Stuttgart, Germany). The details of the resource identifiers for antibodies are shown in Supplementary Table 2.
Immunofluorescent Analysis
The brain tissue samples were fixed in 4% paraformaldehyde, embedded in paraffin, and cut into 4 μm sections. The cultured neurons were fixed with 4% paraformaldehyde. Brain sections were treated with citrate antigen retrieval solution at 100°C followed by blocking with 1% BSA and incubated with primary antibodies at 4°C overnight and corresponding secondary antibodies at 25°C for another 1 h. Light was avoided throughout the whole experimental procedure. The titers of primary antibodies used in immunofluorescence were as follows: rabbit polyclonal anti-TMEM16F antibody (Cloud-clone Corp., PAF813Hu01, 1:100 dilution), mouse monoclonal anti-NeuN antibody (Abcam, ab 104,224, 1:200 dilution), goat monoclonal anti-Iba1 antibody (ab 5076, 1:200 dilution), mouse monoclonal anti-MAP2 antibody (ab11267, 1:200 dilution), and goat monoclonal anti-GFAP antibody (ab535,54,1:200 dilution). Secondary antibodies were purchased from Life Technologies and included the following: Alexa Fluor-488 donkey anti-rabbit IgG antibody (A21206), Alexa Fluor-488 donkey anti-goat IgG antibody (A11055), Alexa Fluor-555 donkey anti-mouse IgG antibody (A31570), and Alexa Fluor-555 donkey anti-goat IgG antibody (A21432). Normal rabbit IgG, normal mouse IgG, and normal goat IgG were used as negative controls for the immunofluorescence assay. Nuclei were stained with DAPI mounting medium. Finally, the sections and cells were observed by a fluorescence microscope (OLYMPUS BX50/BXFLA/DP70; Olympus Co., Tokyo, Japan). The details of the resource identifiers for antibodies are shown in Supplementary Table 2.
pSIVA Binding and Flow Cytometry
Flow Cytometry
After OGD/R, cells (1 × 106 cell per well in a six-well plate) were treated according to the manufacturer's protocol. Briefly, ~6 × 106 cells were harvested from tissue culture plates and centrifuged at 1,500 rpm for 5 min at room temperature. Medium supernatant was removed, and cells were washed once in PBS. Cells were then resuspended in 1X binding buffer at a concentration of 1 × 106–1 × 107/ml. We transferred 100 μl of the resuspended cells (1 × 105 to 1 × 106 cells) to each vial that would be run, and both 5 μl of pSIVA-IANBD (Novus Biologicals, NBP2-29382) and 5 μl of propidium iodide (PI) were added. Samples were incubated at room temperature for 20 min in the dark, then 400 μl of 1X binding buffer was added to each tube, and the samples were analyzed by flow cytometry using a Beckman Coulter flow cytometer (Mississauga, Ontario).
pSIVA Immunofluorescent Analysis
pSIVA-IANBD was dialyzed into medium and applied to the neurons at 10–20 ul/ml concentration, according to the protocols. In this test, fluorescently tagged annexin binds to externalized phosphatidylserine exposed on cell membranes, allowing for monitoring changes that occur at different stages of apoptosis in living cells (33). After 15 min incubation, the cells were fixed and immunostained using MAP2 or TMEM16F antibody. Then, they were incubated with Alexa Fluor 555 conjugated anti-mouse antibody, as described above. In particular, the exposed PS binding with pSIVA did not need extra staining with secondary anti-body and could be observed directly using a green fluorescence filter set. Nuclei were stained with DAPI mounting medium. Cells were observed by a fluorescence microscope (OLYMPUS BX50/BXFLA/DP70; Olympus Co., Tokyo, Japan).
Neurobehavioral Evaluation
At 24 h after reperfusion, rats were tested for behavioral impairment by a scoring system and subjected to the rotarod test (for motor function), the adhesive-removal test (for sensorimotor function), and the Morris Water maze test (for lateralized skilled forelimb reaching) (34).
Rotarod Test
An accelerating rotarod test was employed before and at 3, 5, 7, and 14 days after MCAO to measure the motor function of rats (35). The diameter of the rotarod spindle was 10 cm. The speed of the spindle was increased from 4 to 30 rpm in 60 s and 30 rpm was maintained for a maximum of 300 s. When the rats lost their balance and fell off the rotarod, it triggered the sensor and the time was recorded. Through separation by two panels to prevent the rats from disturbing each other, three rats were able to run at the same time. Before testing, each rat received three training sessions per day for three consecutive days and the last three trials served as the baseline. Then, all rats received a test trial on an accelerating rotarod at all testing days after MCAO/R.
Adhesive-Removal Test
The adhesive removal test was performed as described previously (34). Briefly, the rats were removed from their home cage and trained to be familiar with the testing environment. Then, two small pieces of adhesive-backed paper dots were glued to the wrist of each forelimb. The rats were then gently returned to their testing cages and they typically contacted and then removed each stimulus one at a time using their teeth. The time required to contact and remove both stimuli from each limb was recorded in five trials per day for 3 days separately. If the rats were able to contact the dots and remove those within 10 s at the end of training, then the rats were included in the experimental group. Then, all rats received a test trial at all testing days after MCAO/R.
Morris Water Maze Test
Briefly, we trained the rats in the Morris water maze for 3–6 days (three trials a day) before the formal test. The tests were performed on days 22–26 after MCAO/R in order to evaluate long-term neurobehavioral impairment (36–38). The arena was 50-cm high and 180 cm in diameter and was filled with water to a height of 30 cm at 20–22°C. The platform was placed ~2 cm below the water surface. The starting point for each rat changed every day. Each test continued until the rat found the platform and stood on it for 2 s, or until 60 s had elapsed. After each test, the rats were placed on the platform for 20 s. During testing for each rat, we recorded its swimming speed, latency to find the hidden platform, and the path length of the swimming path to reach the hidden platform (39).
Neurological Scores
At 24 h after MCAO/R, neurological tests were conducted on six rats per group to assess behavioral impairments. Behavioral performance was scored according to a previously published scoring system (20), which was a revised version of the Bederson score.
Statistical Analysis
All data are expressed as the mean ± SEM. GraphPad Prism 7 (GraphPad Software San Diego, CA, USA) was used for statistical analysis. Differences between groups were determined with two-tailed Student's t-tests or Mann-Whitney U-tests for comparisons between two groups or with a one-way analysis of variance (ANOVA) followed by Tukey's post-hoc tests for comparisons of more than two groups. Statistical significance was determined as P < 0.05 unless otherwise stated.
Results
General Observations
No significant differences were found in the body temperature, body weight, heartbeat, or mean arterial blood pressure of rats in any of the MCAO/R experimental groups (Supplementary Figure 3) (40). No animals died in the sham group (0/12), and the mortality rate of the rats after MCAO/R induction was 18.10% (19/105).
TMEM16F Protein Level Increases in Penumbra Neurons After Ischemic Injury
To explore the expression of TMEM16F in brain tissue after MCAO/R, we detected the protein level of TMEM16F by western blotting in vivo (Figure 1A) and in vitro (Figure 1B). After MCAO/R modeling in rats, protein level of TMEM16F were significantly elevated at 6 h and peaked at 12 h (Figures 1A,B). In cultured primary neurons after OGD/R modeling, the protein level of TMEM16F was also significantly increased at 6 h and peaked by 12 h. Immunostaining of brain tissues after MCAO/R demonstrated that the protein level of TMEM16F was significantly elevated at 12 h compared with the sham group (Figure 1C). In addition, double immunofluorescence staining was performed to distinguish changes in cell type-specificity of TMEM16F expression after ischemia. The results showed that TMEM16F was mainly expressed in neurons under ischemic conditions and had a lower expression in microglia, whereas it was hardly detected in astrocytes (Figure 2A).
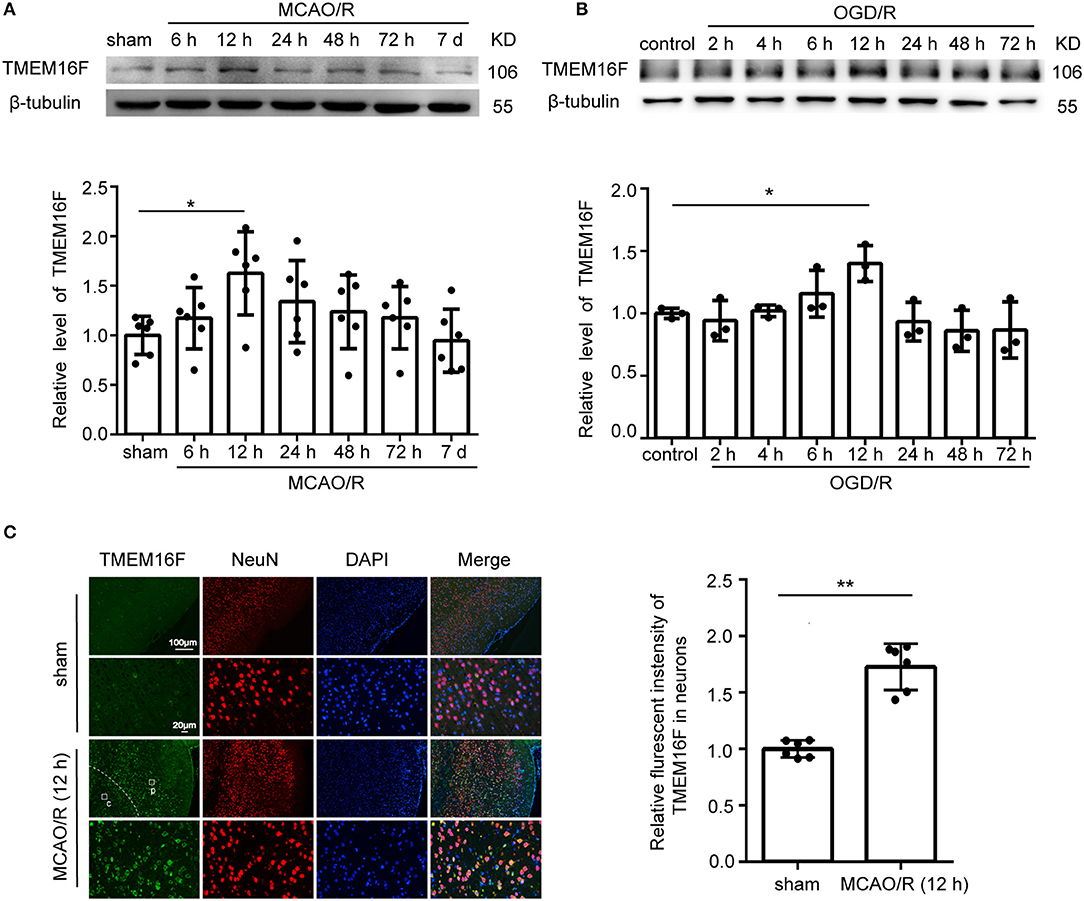
Figure 1. TMEM16F protein level increases after ischemic insult. (A) Western blot analysis and quantification of the protein level of TMEM16F in brain tissue around the penumbra after MCAO/R (n = 6, *P < 0.05). (B) Western blot analysis and quantification of the protein level of TMEM16F in cultured neurons after OGD/R treatment (n = 3, *P < 0.05). (C) Double immunofluorescence analysis was performed with TMEM16F antibodies (green) and a neuronal marker (NeuN, red) in brain sections. (p) represents the penumbra, and (c) represents the ischemic core. Nuclei were fluorescently labeled with DAPI (blue) (n = 6, **P < 0.01). In (A–C), mean values for the sham group were normalized to 1.0. Data are presented as mean ± SEM. Differences were calculated with ordinary one-way ANOVA for (A,B) and Student's t-tests for (C).
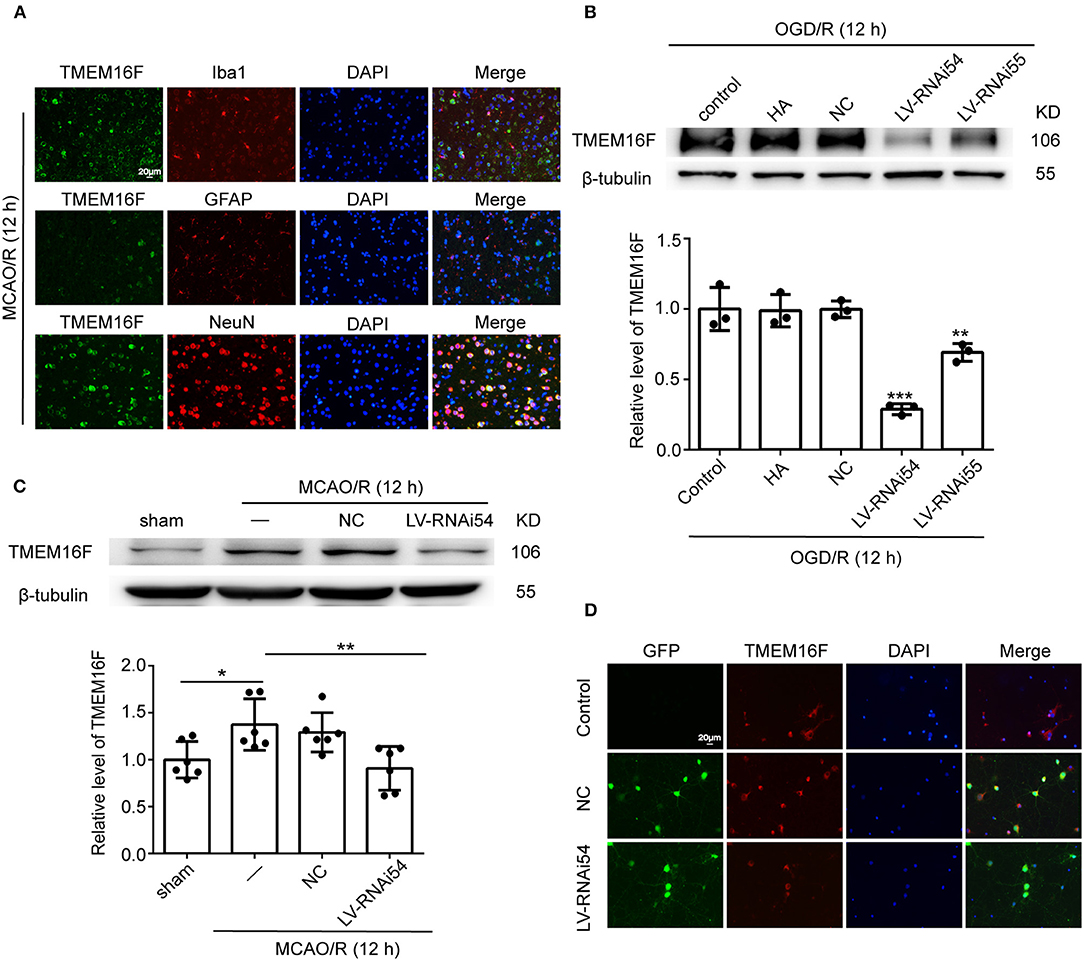
Figure 2. LV-TMEM16F-RNAi transfection downregulates TMEM16F in rat brain. (A) Double immunofluorescence analysis was performed with TMEM16F antibodies (green), a neuronal marker (NeuN, red), a microglia marker (Iba1, red), and an astrocyte marker (GFAP, red) in brain sections. Nuclei were fluorescently labeled with DAPI (blue) (scale bar = 20 μm). (B) Transfection efficiency of TMEM16F lentivirus (LV-RNAi) in cultured neurons. Quantifications of protein levels of TMEM16F in various groups are shown (n = 3, LV-RNAi54 vs. control/HA/NC; ***P < 0.001, LVRNAi55 vs. control/HA/NC, **P < 0.01). (C) Transfection efficiency of TMEM16F lentivirus (LV) in the rat brain. Quantifications of the protein level of TMEM16F in various groups are shown (n = 6, LV-RNAi54 vs. MCAO/R, **P < 0.01; MCAO/R/NC vs. sham, *P < 0.05). (D) Transfection efficiency of TMEM16F lentivirus (LV-RNAi) in cultured neurons was shown with immunofluorescence analysis. HitransG A (HA) is a reagent that could enhance transfection efficacy. In (B,C), the mean values for the sham/control group were normalized to 1.0. Data are presented as mean ± SEM. Differences were calculated with ordinary one-way ANOVA.
TMEM16F Downregulation Reduces Motor Deficits After Ischemia
To further identify the role of TMEM16F in ischemic stroke, we adopted LV-RNAi against TMEM16F to decrease its expression. The interfering effects of LV-TMEM16F-RNAi were confirmed by western blotting both in vivo and in vitro (Figures 2B,C), and the transfection rate was observed using a microscopy in vitro experiment (Figure 2D). Two kinds of lentivirus targeted at TMEM16F were involved in the in vitro experiment, and we found LV-RNAi54 had better knockdown efficacy compared with LV-RNAi55 (Figure 2B). Thus, we chose LV-RNAi54 as the transfection reagent for further experiments. TMEM16F downregulation alleviated the sensory and motor deficits caused by MCAO/R, as suggested by the results of the adhesive-removal test (Figure 3A), rotarod test (Figure 3B), neurobehavioral scores (Figure 3C), and Morris water maze test (Figures 3D,E).
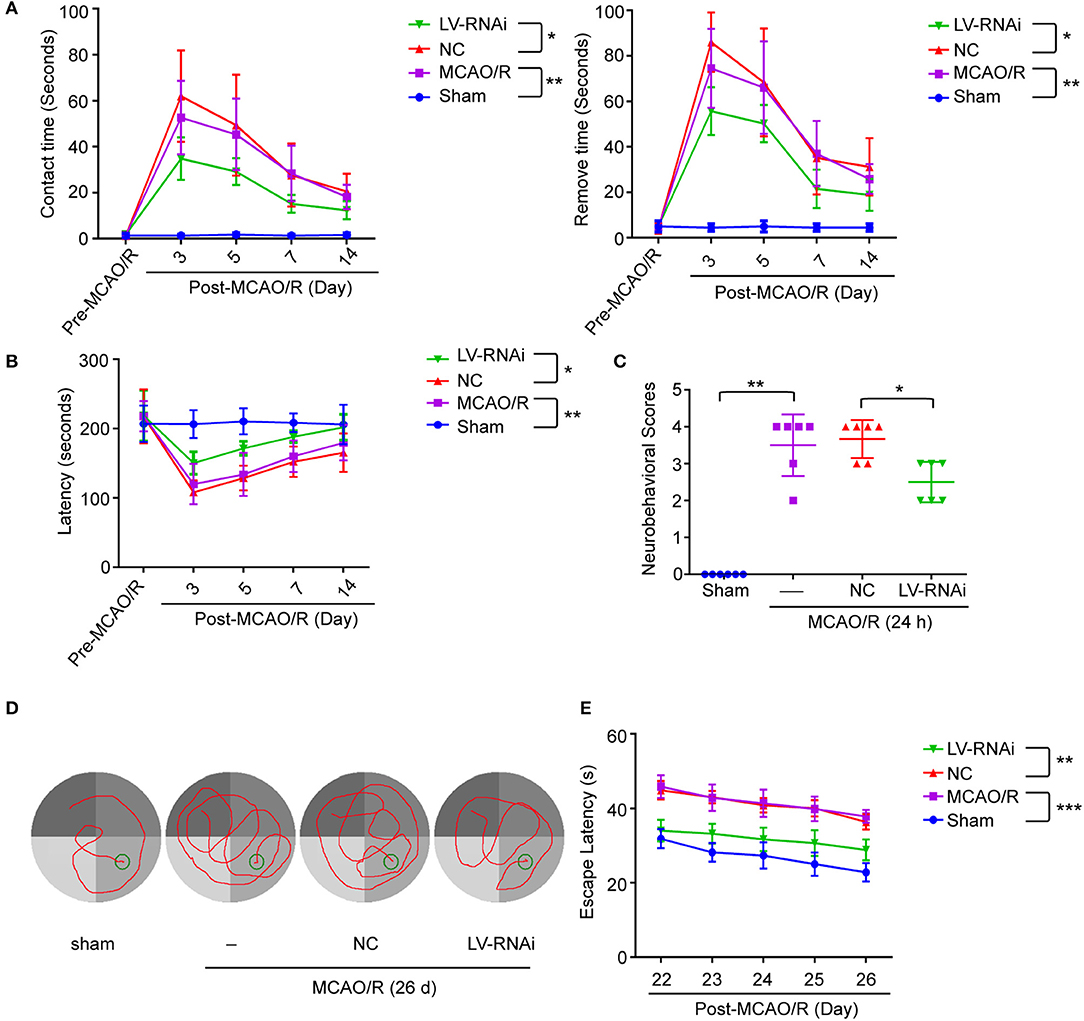
Figure 3. TMEM16F deficiency reduces motor deficits after focal cerebral ischemia. TMEM16F-deficiency rats show (A) shorter contact-time and removal-time in adhesive-removal tests at 3, 5, and 7 days after MCAO/R. (LV-RNAi vs. LV-NC at 3 d, P < 0.001; at 5 d, P < 0.01; at 7 d, P < 0.01) (n = 6, *P < 0.05, **P < 0.01). (B) longer latency time in rotarod test at 3,5,7, and 14 days. (LV-RNAi vs. LV-NC at 3 d, P < 0.001; at 5 d, P < 0.01; at 7 d, P < 0.01; at 14 d, P < 0.01) (n = 6, *P < 0.05, **P < 0.01). (C) lower scores in neurobehavior test (n = 6, *P < 0.05, **P < 0.01). (D) The typical swim path of rats in the Morris water maze test at 26 d after MCAO/R. (E) Time to reach the submerged platform in the water maze 22–26 d after MCAO/R. (LV-RNAi vs. LV-NC at all time points, P < 0.001) (n = 6, **P < 0.01, ***P < 0.001). Data are presented as mean ± SEM. Differences were calculated with two-way ANOVA for (A,B,E) and ordinary one-way ANOVA for (C).
Neurons Can Expose PS in a Reversible Manner After OGD/R
We found that the exposed PS was labeled by pSIVA-IANBD reagent with green fluorescence, distributed on the cell membrane of neurons, including the cell body as well as axons and dendrites, and co-located with TMEM16F at 12 h after OGD/R (Figures 4A,B). OGD/R caused extensive neuronal exposure of the eat-me signal PS, which is known to induce phagocytic uptake by microglia. However, at 24 h after OGD/R, the number of PS-exposing neurons (pSIVA+, PI–) was reduced significantly, and the proportion of healthy neurons (pSIVA–, PI–) increased (Figures 4C,D), compared with OGD/R 12 h. Additionally, there was no difference in the proportion of dead neurons between the two groups, which demonstrated that this PS exposure had been reversed to some extent in the absence of neuronal death or loss compared with OGD/R 12 h, indicating transient PS exposure on viable cells and that these neurons would live long-term without activated microglia joining in. Combined with the previous results, we speculate that knockdown of the expression of TMEM16F after ischemic injury probably protects the stressed-but-still-alive neurons in penumbra from undergoing phagocytosis by active microglia. Areas at the edge of the infarct (the penumbra) may have a chance to revert to healthy tissue when the PS exposure of viable neurons is inhibited.
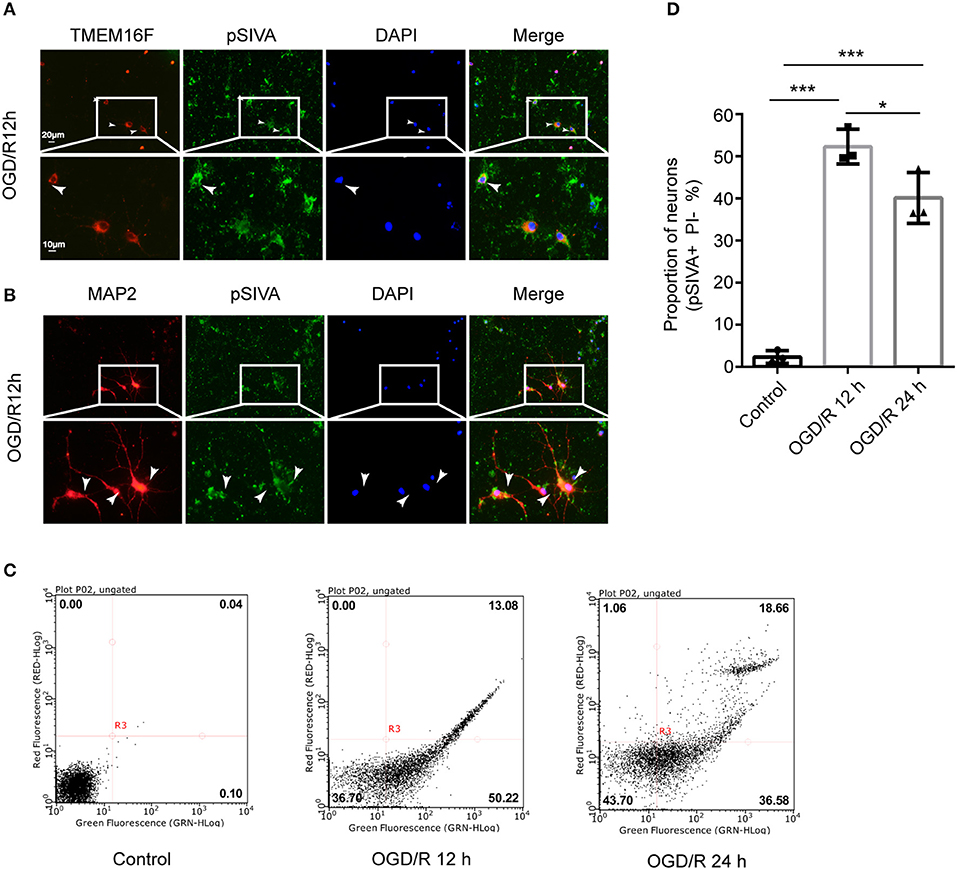
Figure 4. Neurons can expose PS in a reversible manner after OGD/R. (A) Cultured neurons were fluorescently labeled with TMEM16F antibodies (red) and an exposed PS marker (pSIVA, green). Nuclei were fluorescently labeled with DAPI (blue) (n = 3). (B) Cultured neurons were fluorescently labeled with neuronal antibodies (MAP2, red) and a PS marker (pSIVA, green). Nuclei were fluorescently labeled with DAPI (blue) (n = 3). (C) After OGD/R 12 and 24 h, PS-exposure were detected with pSIVA/PI double staining after incubation. (D) Proportion of PS-exposed neurons was compared between different groups (n = 3, OGD/R 12 h and OGD/R 24 h vs. control, ***P < 0.001; OGD/R 12 h vs. OGD/R 24 h, *P < 0.05). Data are presented as mean ± SEM. Differences were calculated with ordinary one-way ANOVA.
Furthermore, in order to investigate whether PS exposure on the surface of neurons was reduced after knocking down TMEM16F, we collected cells after transfection with LV-TMEM16F-RNAi and performed flow cytometry, but the results were not satisfactory. We preferred the lentivirus for intervention because of its excellent and stable intervention effectiveness. However, in in vitro experiments, the transfection period of lentivirus was too long, inevitably affecting cellular status. Additionally, coupled with the effects of lentivirus transfection and OGD/R intervention, the neurons' status at this time was not good enough for flow cytometry analysis and could interfere with the experimental results. Short-term intervention methods (such as transfection with a designated plasmid) should be considered in future research. Other experimental methods should also be taken into account for measuring the PS exposure of attached neurons, as flow cytometry is not ideal.
TMEM16F Deficiency Reduces Phagocytosis of Neurons After Focal Brain Ischemia
The numbers of microglia in the ischemic penumbra were determined by Iba-1-positive cell counts (12). A significant proportion of activated microglia had an amoeboid morphology which was characterized by darker cell bodies and thickened retracted processes, indicating a phagocytic phenotype (41). Additionally, the ramified/resting microglia were characterized by light round or oval cell bodies with fine symmetrical extended processes that were easy to distinguish (42). Few activated microglia were found at 1 d after ischemia, but a strong response was visible in the penumbra at 3 d, occurring in both the TMEM16F knockdown group and untreated animals (Figure 5A). This time-course of microglial recruitment and activation was similar to the time course of neuronal loss, as well as the recruitment of microglia in the core. However, at 3 d after ischemia there was a 11.8 ± 2.0% reduction in the number of active microglia in the TMEM16F knockdown group compared with the untreated group (Figure 5B), indicating that the phagocytic activity of microglia was reduced in TMEM16F-deficient animals. No differences were observed at other time points.
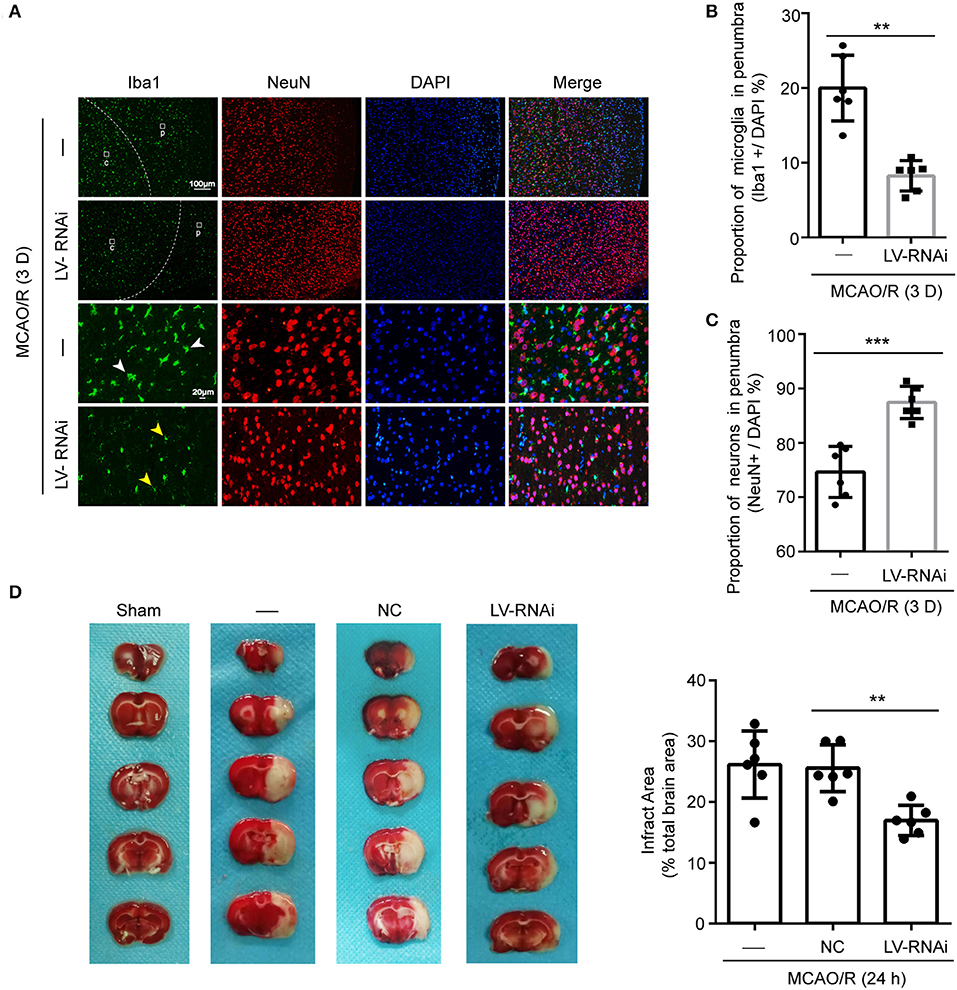
Figure 5. TMEM16F participates in phagocytosis of penumbra neurons after focal ischemia. (A) Macrophage/microglia activation in the penumbra of untreated and LV-RNAi transfected rats at 3 days after MCAO/R. Yellow arrowheads indicate ramified microglia, and white arrowheads indicate amoeboid microglia. (p) represents the penumbra and (c) represents the ischemic core. (B) Proportion of macrophages/microglia in the penumbra, indicating phagocytic activity: TMEM16F knockdown animals displayed decreased levels of phagocytic microglia at day 3 (n = 6, **P < 0.01). (C) Proportion of surviving neurons in the penumbra at 3 days after MCAO/R (n = 6, ***P < 0.001). (D) Triphenyltetrazolium chloride (TTC) staining showing the effects of TMEM16F intervention on neuronal loss after MCAO/R 24 h (n = 6, **P < 0.01). In (B–D), data are presented as mean ± SEM. Differences were calculated with Student's t-tests for (B,C) and ordinary one-way ANOVA for (D).
Furthermore, cell counts based on the neuronal nuclear antigen, NeuN, showed no loss of neurons at 1 d after ischemia, suggesting that neurons had not yet been phagocytosed. At 3 d, a few neurons had been lost in the penumbra (Figure 5C), but the proportion of neurons remaining in TMEM16F knockdown animals was significantly higher (untreated group: 74.7 ± 2.0%; TMEM16F knockdown: 87.5 ± 1.2%). Additionally, at the histological level, shown by TTC staining, deficiency of TMEM16F reduced the ischemic volume after MCAO/R (Figure 5D), indicating that suppressing the expression of TMEM16F could prevent stressed neurons from being phagocytosed by active microglia.
Discussion
Microglia are the professional phagocytes of the brain and are able to rapidly phagocytose dead or dying neurons within hours (8, 43), which has been considered to be a beneficial process. However, microglia can also phagocytose stressed-but-viable neurons that reversibly expose to PS because of the calcium-activated scramblase TMEM16F (13, 44) or temporary lowering of ATP. Our data indicated that the deficiency of TMEM16F, which is required for reversible PS exposure, could prevent stressed-but-still alive neurons in a pre-infract area from being mistaken by microglia, thus blocking phagoptosis and further improving functional outcome after focal cerebral ischemia.
The affected area after cerebral ischemia can be divided into the core and the penumbra. It is generally assumed that in the central core of the ischemic area, which suffers from the highest level of ischemic injury, the cell death is oncotic/necrotic, whereas the penumbra experiences a lower level of ischemia in which neurons are functionally depressed but still viable at early time points (4). Besides, we should notice that the activation of microglia in the penumbra leads to a decrease in neuronal density over time, which means the hypoperfused penumbra may progress to infarct lesions and lead to more severe physical dysfunction (45). Therefore, timely rescue of neurons in the ischemic penumbra is of great significance for post-stroke treatment.
Phagoptosis, also called primary phagocytosis, is a recently recognized form of cell death caused by phagocytosis of viable cells that results in their destruction (46). The process of phagocytosis is normally initiated by the release of attractive signals from the target cell (referred to as “come-get-me”/“find-me” signals) leading to chemotaxis of a nearby macrophage (47). Upon reaching the target cell, the macrophage recognizes cell-surface signals on the target cell (“eat-me” signals), which then induce its uptake. The best characterized “eat-me” signal is the cell-surface exposure of phosphatidylserine PS (8, 10, 48, 49). In healthy cells, PS is found almost exclusively on the inner leaflet of the plasma membrane. However, PS can be exposed on the cell surface under certain conditions. PS exposure can occur as a result of: (i) apoptosis (when PS exposure occurs secondary to calcium elevation, ATP depletion, oxidative stress, or vesicle fusion); (ii) necrosis (due to plasma membrane rupture, calcium elevation or ATP depletion); (iii) calcium elevation (which stimulates the scramblase and inhibits the translocase); (iv) ATP depletion (which inhibits the translocase); (v) oxidative stress (which stimulates the scramblase and inhibits the translocase); or (vi) fusion of intracellular vesicles with plasma membrane (46, 50).
Studies have shown that PS-exposure not only occurs on the surface of cells as an early sign of cell death, but that it can also occur on the surface of viable cells in a reversible manner. It is provoked by exposure of “eat-me” signal PS on viable cells as a result of the activation of sublethal stimuli, potentially causing their phagocytosis when in the presence of phagocytes. For example, stressed but viable neurons can reversibly expose PS, but this only results in phagocytosis if activated microglia are present at the time of PS exposure (12).
In healthy cells that are not activated, PS is found exclusively on the inner leaflet of the plasma membrane, because the aminophospholipid translocase (also called “Flippases,” such as ATP11C and ATP8A) removes PS from the outer leaflet. However, a second enzyme, the phospholipid scramblase, can cause PS exposure by randomizing phospholipid distribution between the inner and outer leaflets (50, 51). In activated neurons, increased intracellular Ca2+ transiently activates TMEM16F to scramble phospholipids, probably inactivating flippase activity of ATP11C/ATP8A (Figure 6). When the stimulus is removed and cells return to a normal state, a decrease of intracellular Ca2+ inactivates TMEM16F, and the flippase restores the asymmetrical PS distribution (52, 53). We have shown here that OGD/R induces an excessive accumulation of intracellular Ca2+, which leads to the activation of TMEM16F, whose protein level reaches a peak at 12 h in vitro. Using pSIVA-IANBD reagent with green fluorescence to label exposed PS, we found that the exposed PS was distributed on the cell membrane of neurons and could co-locate with TMEM16F. Then we collected the cells at 12 and 24 h after OGD/R for flow cytometry analysis. Compared with 12 h reperfusion, the proportion of PS-exposed neurons was significantly decreased and that of healthy cells was increased after 24 h reperfusion without increasing cell death, suggesting a transient PS exposure on stressed-but-viable cells. In this study, microglia were not co-cultured with neurons, so we speculate that when activated microglia do not appear, stressed neurons can return to a normal state and survive longer.
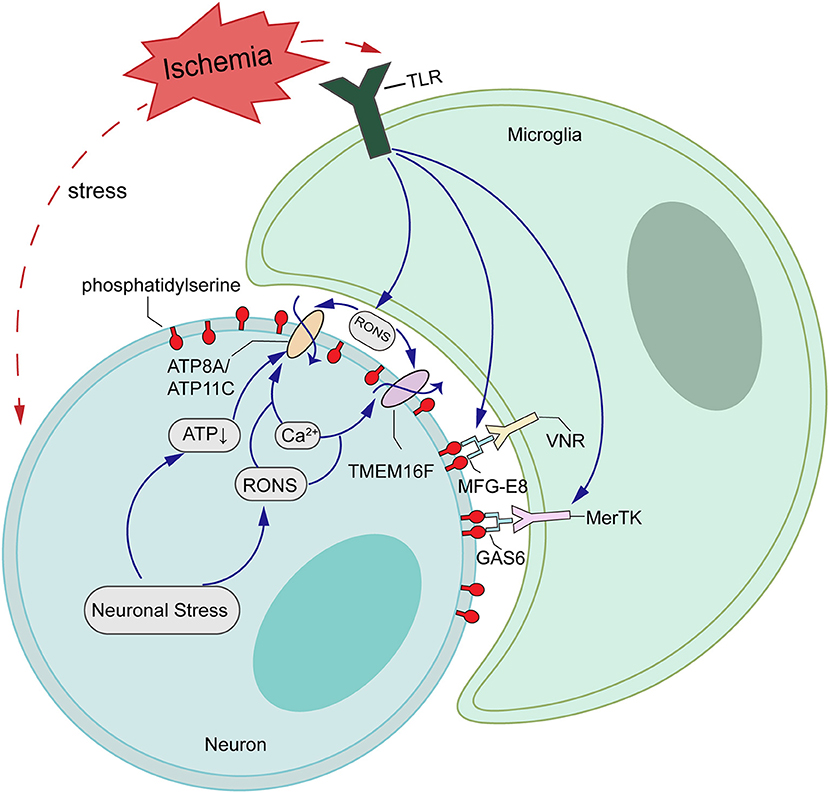
Figure 6. Schematic representations of potential mechanisms of TMEM16F actions in ischemia-reperfusion (I/R) injury. Ischemic stroke upregulates the expression of TMEM16F, which is activated by loaded intracellular Ca2+ and functions as a scramblase, inducing PS-exposure of stressed-but-alive neurons in the penumbra. Additionally, the flippases, such as ATP8A/ATP11C, are likely inactivated by the increasing Ca2+ or ATP depletion. When the stimuli are removed and cells return to a normal state, a decrease in intracellular Ca2+ inactivates TMEM16F, and the flippases restore the asymmetrical PS distribution.
Accordingly, other studies have shown that viable neurons in the postischemic brain can be labeled by annexin V, indicating that PS exposure is reversible at 4 h after transient ischemia. At the same time, the neuronal marker MAP2 is co-localized with annexin V staining, which means the neuronal structure still remains intact. However, by day 1, annexin V-positive cells had a neuronlike morphology, but lost MAP2 staining, suggesting that these cells had already suffered irreversible ischemic injury (11). These neurons can stay alive if activated microglia do not appear (54). We can infer from this that there may be a critical time point between 4 and 24 h at which neurons could not maintain structural integrity anymore and eventually move toward apoptosis. We found that the expression of TMEM16F, a Ca2+-activated phospholipid scramblase, was significantly elevated at 6 h after transient focal ischemia in rats and peaked at 12 h. At this point the scramblase was highly activated and probably increased the exposure of PS, as well as the risk of being phagocytosed.
Microglia also play an important role in this process. PS-exposed neurons are recognized either via the opsonin MFG-E8 and vitronectin receptors on microglia (10) or by the opsonin Gas6 and MerTK (Mer receptor tyrosine kinase) receptors on microglia (8, 9, 55). It appears that inflammatory activation of microglia impairs their ability to discriminate between apoptotic and viable neurons for phagocytosis, resulting in phagoptosis during inflammation (56). Microglia fail to recognize cells that should be removed and phagocytize transiently exposed PS neurons via the microglial vitronectin receptor MFG-E8, leading to an increase in the infarct size. A study had reported that transient brain ischemia caused a delayed neuronal loss, accompanied by microglial phagocytosis of neurons, which was prevented by genetic inactivation of either MFG-E8 or MerTK (12). Only a few activated microglia were found at 1 d after ischemia in our study, but a strong response was visible in the penumbra at 3 d, indicating that the phagocytosis of neurons was delayed by at least 72 h, which is consistent with previous research (41). We also found the proportion of Iba1+ cells were significantly decreased in penumbra after knocking down TMEM16F. In accordance with phagocytosis of viable neurons, deficiency in TMEM16F may protect neurons from phagocytic uptake and lead to a reduction in the infarct size after transient cerebral ischemia in vivo.
It has been reported that expression of TMEM16F is significantly higher in macrophages, where it supports phagocytic activity and cell death (57). Recent studies find that TMEM16F plays an important role in regulating spinal microglia function in neuropathic pain states (58) and spinal cord injury (59). Likely due to a different disease model and the microglia accounting for only 20% of brain tissue, TMEM16F was mainly expressed on the neuronal membrane instead of the microglia in our study after MCAO/R. Further studies are needed to identify whether TMEM16F deficiency also diminishes the branch motility and phagocytic capacity of microglia and contributes to viable neuronal survival in the penumbra after transient cerebral ischemia.
There were some technical limitations to the present study. First, we only used adult male rats to establish the MCAO/R model in vivo, so there may be a problem of gender bias. In vitro, we chose primary neurons to incubate and used a conditional neuronal basal medium, without mixing microglia, so the direct neuron-microglia contact could not be investigated. Additionally, our study explored only the role of TMEM16F after MCAO/R and, in pathological conditions, the downstream factors PS-binding protein MFG-E8 and vitronectin receptors such as MerTK and the relationship between them were not involved. Flippases such as ATP8A/ATP11C probably also contributed to reversible PS exposure cooperated with scramblase TMEM16F. Therefore, the cooperation between scramblase and flippases should be considered. Furthermore, there may be a TMEM16F-MFG-E8 and MerTK pathway in phagocytosis of stressed-but-viable neurons, which needs further research.
In summary, our study revealed that TMEM16F could be upregulated after transient cerebral ischemia and increase phagocytosis by microglia. Stressed neurons could expose PS in a reversible manner; deficiency of this phospholipid scramblase may protect stressed-but-viable neurons from being phagocytosed by activated microglia in the penumbra and impede the expansion of the infarct area. In the ischemic penumbra in patients, neuronal loss was detected several days or months post-stroke (60–62), which was consistent with our data. This important time course between the increase of neuronal PS exposure and the initiation of phagocytosis by activated microglia should be considered and may provide potential therapeutic targets.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Ethics Statement
The animal study was reviewed and approved by the Animal Care and Use Committee of Soochow University.
Author Contributions
YZ, HL, and XL (3rd Author) designed the study, collected and analyzed data, and wrote the manuscript. JW, TX, and JW collected and analyzed data and revised the manuscript. MS and GC supervised the experiments. HS and XL (8th Author) contributed to the study design and revised the manuscript. All authors gave final approval of the manuscript.
Funding
This work was supported by National Key R&D Program of China under Grant (2018YFC1312600 and 2018YFC1312601), National Natural Science Foundation of China under Grant (81830036, 81771254, and 81771255), Jiangsu Provincial Medical Youth Talent under Grant (QNRC2016728), Natural Science Foundation of Jiangsu Province under Grant (BK20170363 and BK20180204), Suzhou Key Medical Centre under Grant (Szzx201501), Scientific Department of Jiangsu Province under Grant (BE2017656), Suzhou Science and Technology under Grant (SS2019056), Jiangsu Commission of Health under Grant (K2019001), Gusu Health Personnel Training Project under Grant (GSWS2019030), and Suzhou Government under Grant (SYS2019045).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.01144/full#supplementary-material
References
1. Trout AL, Kahle MP, Roberts JM, Marcelo A, de Hoog L, Boychuk JA, et al. Perlecan domain-V enhances neurogenic brain repair after stroke in mice. Transl Stroke Res. (2020). doi: 10.1007/s12975-020-00800-5. [Epub ahead of print].
2. Lo EH. A new penumbra: transitioning from injury into repair after stroke. Nat Med. (2008) 14:497–500. doi: 10.1038/nm1735
3. Karl JM, Alaverdashvili M, Cross AR, Whishaw IQ. Thinning, movement, and volume loss of residual cortical tissue occurs after stroke in the adult rat as identified by histological and magnetic resonance imaging analysis. Neuroscience. (2010) 170:123–37. doi: 10.1016/j.neuroscience.2010.06.054
4. Fricker M, Tolkovsky AM, Borutaite V, Coleman M, Brown GC. Neuronal cell death. Physiol Rev. (2018) 98:813–80. doi: 10.1152/physrev.00011.2017
5. Datta A, Sarmah D, Mounica L, Kaur H, Kesharwani R, Verma G, et al. Cell death pathways in ischemic stroke and targeted pharmacotherapy. Transl Stroke Res. (2020). doi: 10.1007/s12975-020-00806-z. [Epub ahead of print].
6. Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol. (2002) 2:965–75. doi: 10.1038/nri957
7. Ravichandran KS. “Recruitment signals” from apoptotic cells: invitation to a quiet meal. Cell. (2003) 113:817–20. doi: 10.1016/S0092-8674(03)00471-9
8. Neher JJ, Neniskyte U, Zhao JW, Bal-Price A, Tolkovsky AM, Brown GC. Inhibition of microglial phagocytosis is sufficient to prevent inflammatory neuronal death. J Immunol. (2011) 186:4973–83. doi: 10.4049/jimmunol.1003600
9. Neniskyte U, Neher JJ, Brown GC. Neuronal death induced by nanomolar amyloid beta is mediated by primary phagocytosis of neurons by microglia. J Biol Chem. (2011) 286:39904–13. doi: 10.1074/jbc.M111.267583
10. Fricker M, Neher JJ, Zhao JW, Thery C, Tolkovsky AM, Brown GC. MFG-E8 mediates primary phagocytosis of viable neurons during neuroinflammation. J Neurosci. (2012) 32:2657–66. doi: 10.1523/JNEUROSCI.4837-11.2012
11. Mari C, Karabiyikoglu M, Goris ML, Tait JF, Yenari MA, Blankenberg FG. Detection of focal hypoxic-ischemic injury and neuronal stress in a rodent model of unilateral MCA occlusion/reperfusion using radiolabeled annexin V. Eur J Nucl Med Mol Imaging. (2004) 31:733–9. doi: 10.1007/s00259-004-1473-5
12. Neher JJ, Emmrich JV, Fricker M, Mander PK, Thery C, Brown GC. Phagocytosis executes delayed neuronal death after focal brain ischemia. Proc Natl Acad Sci USA. (2013) 110:E4098–107. doi: 10.1073/pnas.1308679110
13. Suzuki J, Fujii T, Imao T, Ishihara K, Kuba H, Nagata S. Calcium-dependent phospholipid scramblase activity of TMEM16 protein family members. J Biol Chem. (2013) 288:13305–16. doi: 10.1074/jbc.M113.457937
14. Suzuki J, Denning DP, Imanishi E, Horvitz HR, Nagata S. Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science. (2013) 341:403–6. doi: 10.1126/science.1236758
15. Koizumi J-i, Yoshida Y, Nishigaya K, Kanai H, Ooneda G. Experimental studies of ischemic brain edema effect of recirculation of the blood flow after ischemia on post-ischemic brain edema. Nosotchu. (1989) 11:11–7. doi: 10.3995/jstroke.11.11
16. Gaowa S, Bao N, Da M, Qiburi Q, Ganbold T, Chen L, et al. Traditional Mongolian medicine Eerdun Wurile improves stroke recovery through regulation of gene expression in rat brain. J Ethnopharmacol. (2018) 222:249–60. doi: 10.1016/j.jep.2018.05.011
17. Lv Y, Ge L, Zhao Y. Effect and mechanism of SHED on ulcer wound healing in Sprague-Dawley rat models with diabetic ulcer. Am J Transl Res. (2017) 9:489–98.
18. Wang R, Wang H, Liu Y, Chen D, Wang Y, Rocha M, et al. Optimized mouse model of embolic MCAO: from cerebral blood flow to neurological outcomes. J Cereb Blood Flow Metab. (2020) 2020:271678X20917625. doi: 10.1177/0271678X20917625
19. Cirillo C, Brihmat N, Castel-Lacanal E, Le Friec A, Barbieux-Guillot M, Raposo N, et al. Post-stroke remodeling processes in animal models and humans. J Cereb Blood Flow Metab. (2020) 40:3–22. doi: 10.1177/0271678X19882788
20. Menzies SA, Hoff JT, Betz AL. Middle cerebral artery occlusion in rats: a neurological and pathological evaluation of a reproducible model. Neurosurgery. (1992) 31:100–6. doi: 10.1227/00006123-199207000-00014
21. Balkaya M, Cho S. Optimizing functional outcome endpoints for stroke recovery studies. J Cereb Blood Flow Metab. (2019) 39:2323–42. doi: 10.1177/0271678X19875212
22. Kuts R, Frank D, Gruenbaum BF, Grinshpun J, Melamed I, Knyazer B, et al. A novel method for assessing cerebral edema, infarcted zone and blood-brain barrier breakdown in a single post-stroke rodent brain. Front Neurosci. (2019) 13:1105. doi: 10.3389/fnins.2019.01105
23. Boyko M, Ohayon S, Goldsmith T, Douvdevani A, Gruenbaum BF, Melamed I, et al. Cell-free DNA–a marker to predict ischemic brain damage in a rat stroke experimental model. J Neurosurg Anesthesiol. (2011) 23:222–8. doi: 10.1097/ANA.0b013e31821b536a
24. Shen H, Liu C, Zhang D, Yao X, Zhang K, Li H, et al. Role for RIP1 in mediating necroptosis in experimental intracerebral hemorrhage model both in vivo and in vitro. Cell Death Dis. (2017) 8:e2641. doi: 10.1038/cddis.2017.58
25. Ashwal S, Tone B, Tian HR, Cole DJ, Pearce WJ. Core and penumbral nitric oxide synthase activity during cerebral ischemia and reperfusion. Stroke. (1998) 29:1037–46. doi: 10.1161/01.STR.29.5.1037
26. Wang Z, Higashikawa K, Yasui H, Kuge Y, Ohno Y, Kihara A, et al. FTY720 protects against ischemia-reperfusion injury by preventing the redistribution of tight junction proteins and decreases inflammation in the subacute phase in an experimental stroke model. Transl Stroke Res. (2020). doi: 10.1007/s12975-020-00789-x. [Epub ahead of print].
27. Zhou L, Li F, Xu HB, Luo CX, Wu HY, Zhu MM, et al. Treatment of cerebral ischemia by disrupting ischemia-induced interaction of nNOS with PSD-95. Nat Med. (2010) 16:1439–43. doi: 10.1038/nm.2245
28. Liu Y, Xue X, Zhang H, Che X, Luo J, Wang P, et al. Neuronal-targeted TFEB rescues dysfunction of the autophagy-lysosomal pathway and alleviates ischemic injury in permanent cerebral ischemia. Autophagy. (2019) 15:493–509. doi: 10.1080/15548627.2018.1531196
29. Zhang H, Pan Q, Xie Z, Chen Y, Wang J, Bihl J, et al. Implication of microRNA503 in brain endothelial cell function and ischemic stroke. Transl Stroke Res. (2020). doi: 10.1007/s12975-020-00794-0. [Epub ahead of print].
30. Zhang Q, Yang H, Gao H, Liu X, Li Q, Rong R, et al. PSD-93 interacts with SynGAP and promotes SynGAP ubiquitination and ischemic brain injury in mice. Transl Stroke Res. (2020). doi: 10.1007/s12975-020-00795-z. [Epub ahead of print].
31. Wang Z, Chen Z, Yang J, Yang Z, Yin J, Zuo G, et al. Identification of two phosphorylation sites essential for annexin A1 in blood-brain barrier protection after experimental intracerebral hemorrhage in rats. J Cereb Blood Flow Metab. (2017) 37:2509–25. doi: 10.1177/0271678X16669513
32. Yuan ZF, Tang YM, Xu XJ, Li SS, Zhang JY. 10-Hydroxycamptothecin induces apoptosis in human neuroblastoma SMS-KCNR cells through p53, cytochrome c and caspase 3 pathways. Neoplasma. (2016) 63:72–9. doi: 10.4149/neo_2016_009
33. Kim YE, Chen J, Langen R, Chan JR. Monitoring apoptosis and neuronal degeneration by real-time detection of phosphatidylserine externalization using a polarity-sensitive indicator of viability and apoptosis. Nat Protoc. (2010) 5:1396–405. doi: 10.1038/nprot.2010.101
34. Zhang L, Chen J, Li Y, Zhang ZG, Chopp M. Quantitative measurement of motor and somatosensory impairments after mild (30 min) and severe (2 h) transient middle cerebral artery occlusion in rats. J Neurol Sci. (2000) 174:141–6. doi: 10.1016/S0022-510X(00)00268-9
35. Chen J, Li Y, Wang L, Zhang Z, Lu D, Lu M, et al. Therapeutic benefit of intravenous administration of bone marrow stromal cells after cerebral ischemia in rats. Stroke. (2001) 32:1005–11. doi: 10.1161/01.STR.32.4.1005
36. Ma S, Wang J, Wang Y, Dai X, Xu F, Gao X, et al. Diabetes mellitus impairs white matter repair and long-term functional deficits after cerebral ischemia. Stroke. (2018) 49:2453–63. doi: 10.1161/STROKEAHA.118.021452
37. Li X, Li J, Qian J, Zhang D, Shen H, Li X, et al. Loss of ribosomal RACK1 (Receptor for Activated Protein Kinase C 1) induced by phosphorylation at T50 alleviates cerebral ischemia-reperfusion injury in rats. Stroke. (2018) 2018:STROKEAHA118022404. doi: 10.1161/STROKEAHA.118.022404
38. Wang R, Pu H, Ye Q, Jiang M, Chen J, Zhao J, et al. Transforming growth factor beta-activated kinase 1-dependent microglial and macrophage responses aggravate long-term outcomes after ischemic stroke. Stroke. (2020) 51:975–85. doi: 10.1161/STROKEAHA.119.028398
39. Shen H, Chen Z, Wang Y, Gao A, Li H, Cui Y, et al. Role of neurexin-1beta and neuroligin-1 in cognitive dysfunction after subarachnoid hemorrhage in rats. Stroke. (2015) 46:2607–15. doi: 10.1161/STROKEAHA.115.009729
40. Wang Z, Chen Z, Yang J, Yang Z, Yin J, Duan X, et al. Treatment of secondary brain injury by perturbing postsynaptic density protein-95-NMDA receptor interaction after intracerebral hemorrhage in rats. J Cereb Blood Flow Metab. (2019) 39:1588–601. doi: 10.1177/0271678X18762637
41. Bohatschek M, Kloss CU, Kalla R, Raivich G. In vitro model of microglial deramification: ramified microglia transform into amoeboid phagocytes following addition of brain cell membranes to microglia-astrocyte cocultures. J Neurosci Res. (2001) 64:508–22. doi: 10.1002/jnr.1103
42. Wixey JA, Lee KM, Miller SM, Goasdoue K, Colditz PB, Tracey Bjorkman S, et al. Neuropathology in intrauterine growth restricted newborn piglets is associated with glial activation and proinflammatory status in the brain. J Neuroinflammation. (2019) 16:5. doi: 10.1186/s12974-018-1392-1
43. Fuhrmann M, Bittner T, Jung CK, Burgold S, Page RM, Mitteregger G, et al. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer's disease. Nat Neurosci. (2010) 13:411–3. doi: 10.1038/nn.2511
44. Suzuki J, Umeda M, Sims PJ, Nagata S. Calcium-dependent phospholipid scrambling by TMEM16F. Nature. (2010) 468:834–8. doi: 10.1038/nature09583
45. Back T. Pathophysiology of the ischemic penumbra–revision of a concept. Cell Mol Neurobiol. (1998) 18:621–38.
46. Brown GC, Neher JJ. Eaten alive! Cell death by primary phagocytosis: “phagoptosis.” Trends Biochem Sci. (2012) 37:325–32. doi: 10.1016/j.tibs.2012.05.002
47. Ravichandran KS. Beginnings of a good apoptotic meal: the find-me and eat-me signaling pathways. Immunity. (2011) 35:445–55. doi: 10.1016/j.immuni.2011.09.004
48. Jitkaew S, Witasp E, Zhang S, Kagan VE, Fadeel B. Induction of caspase- and reactive oxygen species-independent phosphatidylserine externalization in primary human neutrophils: role in macrophage recognition and engulfment. J Leukoc Biol. (2009) 85:427–37. doi: 10.1189/jlb.0408232
49. Tyurina YY, Basova LV, Konduru NV, Tyurin VA, Potapovich AI, Cai P, et al. Nitrosative stress inhibits the aminophospholipid translocase resulting in phosphatidylserine externalization and macrophage engulfment: implications for the resolution of inflammation. J Biol Chem. (2007) 282:8498–509. doi: 10.1074/jbc.M606950200
50. Neher JJ, Neniskyte U, Brown GC. Primary phagocytosis of neurons by inflamed microglia: potential roles in neurodegeneration. Front Pharmacol. (2012) 3:27. doi: 10.3389/fphar.2012.00027
51. Fadok VA, de Cathelineau A, Daleke DL, Henson PM, Bratton DL. Loss of phospholipid asymmetry and surface exposure of phosphatidylserine is required for phagocytosis of apoptotic cells by macrophages and fibroblasts. J Biol Chem. (2001) 276:1071–7. doi: 10.1074/jbc.M003649200
52. Leventis PA, Grinstein S. The distribution and function of phosphatidylserine in cellular membranes. Ann Rev Biophys. (2010) 39:407–27. doi: 10.1146/annurev.biophys.093008.131234
53. Segawa K, Nagata S. An apoptotic “Eat Me” signal: phosphatidylserine exposure. Trends Cell Biol. (2015) 25:639–50. doi: 10.1016/j.tcb.2015.08.003
54. Brown GC, Neher JJ. Microglial phagocytosis of live neurons. Nat Rev Neurosci. (2014) 15:209–16. doi: 10.1038/nrn3710
55. Varga DP, Menyhart A, Posfai B, Csaszar E, Lenart N, Cserep C, et al. Microglia alter the threshold of spreading depolarization and related potassium uptake in the mouse brain. J Cereb Blood Flow Metab. (2020) 2020:271678X19900097. doi: 10.1177/0271678X19900097
56. McArthur S, Cristante E, Paterno M, Christian H, Roncaroli F, Gillies GE, et al. Annexin A1: a central player in the anti-inflammatory and neuroprotective role of microglia. J Immunol. (2010) 185:6317–28. doi: 10.4049/jimmunol.1001095
57. Ousingsawat J, Wanitchakool P, Schreiber R, Kunzelmann K. Contribution of TMEM16F to pyroptotic cell death. Cell Death Dis. (2018) 9:300. doi: 10.1038/s41419-018-0373-8
58. Batti L, Sundukova M, Murana E, Pimpinella S, De Castro Reis F, Pagani F, et al. TMEM16F regulates spinal microglial function in neuropathic pain states. Cell Rep. (2016) 15:2608–15. doi: 10.1016/j.celrep.2016.05.039
59. Zhao J, Gao QY. TMEM16F inhibition limits pain-associated behavior and improves motor function by promoting microglia M2 polarization in mice. Biochem Biophys Res Commun. (2019) 517:603–10. doi: 10.1016/j.bbrc.2019.07.070
60. Guadagno JV, Jones PS, Aigbirhio FI, Wang D, Fryer TD, Day DJ, et al. Selective neuronal loss in rescued penumbra relates to initial hypoperfusion. Brain. (2008) 131:2666–78. doi: 10.1093/brain/awn175
61. Nakagawara J, Sperling B, Lassen NA. Incomplete brain infarction of reperfused cortex may be quantitated with iomazenil. Stroke. (1997) 28:124–32. doi: 10.1161/01.STR.28.1.124
Keywords: cerebral ischemia, TMEM16F, phagocytosis, neuronal cell death, microglia
Citation: Zhang Y, Li H, Li X, Wu J, Xue T, Wu J, Shen H, Li X, Shen M and Chen G (2020) TMEM16F Aggravates Neuronal Loss by Mediating Microglial Phagocytosis of Neurons in a Rat Experimental Cerebral Ischemia and Reperfusion Model. Front. Immunol. 11:1144. doi: 10.3389/fimmu.2020.01144
Received: 06 March 2020; Accepted: 11 May 2020;
Published: 07 July 2020.
Edited by:
Xiaoxing Xiong, Renmin Hospital of Wuhan University, ChinaReviewed by:
Guy Charles Brown, University of Cambridge, United KingdomBhakta Prasad Gaire, University of Maryland, Baltimore, United States
Copyright © 2020 Zhang, Li, Li, Wu, Xue, Wu, Shen, Li, Shen and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Meifen Shen, c21mODE2NUAxNjMuY29t; Gang Chen, bmp1X25ldXJvc3VyZ2VyeUAxNjMuY29t
†These authors have contributed equally to this work