 Marta Vuerich
Marta Vuerich Samiran Mukherjee
Samiran Mukherjee Simon C. Robson
Simon C. Robson Maria Serena Longhi
Maria Serena Longhi- 1Department of Anesthesia, Critical Care & Pain Medicine, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, United States
- 2Division of Gastroenterology, Department of Medicine, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, United States
Inflammatory bowel disease (IBD) is a serious inflammatory condition of the gastrointestinal tract. Crohn's disease (CD) and ulcerative colitis (UC) are two of the most common IBD manifestations and are both associated with unfettered inflammation, often refractory to conventional immunosuppressive treatment. In both conditions, imbalance between effector and regulatory cell immune responses has been documented and is thought to contribute to disease pathogenesis. Purinergic signaling is a known modulator of systemic and local inflammation and growing evidences point to extracellular ATP/adenosine imbalance as a key determinant factor in IBD-associated immune dysregulation. In vitro and pre-clinical studies suggest a role for both ATP (P2) and adenosine (P1) receptors in dictating onset and severity of the disease. Moreover, our experimental data indicate ENTPD1/CD39 and CD73 ectoenzymes as pivotal modulators of intestinal inflammation, with clear translational importance. Here we will provide an updated overview of the current knowledge on the role of the purinergic signaling in modulating immune responses in IBD. We will also review and discuss the most promising findings supporting the use of purinergic-based therapies to correct immune dysregulation in CD and UC.
Introduction
Healthy tissues contain negligible levels of extracellular nucleotides and nucleosides; whereas inflammatory sites are characterized by accumulation of extracellular ATP and adenosine.
Release of nucleotides in the extracellular environment triggers P2 receptors activation on target cells. The P2 receptor family includes seven P2X and eight P2Y members, classified based on their desensitization time and affinity for the ligand. P2 receptors are virtually present on all immune cells, are mainly activated by extracellular ATP and have been generally described as mediators of inflammatory processes (1).
Once released in the extracellular environment, nucleotides can be rapidly hydrolyzed into nucleosides by specific ectonucleotidases. Ectonucleotidases are expressed on the surface of different immune cell types and belong to several enzymatic families, which have been functionally and structurally characterized (2, 3). The prototype member of the NTPDase family is ENTPD1/CD39, a rate-limiting ectoenzyme that hydrolyzes ATP into AMP, which is then further degraded into adenosine by the ecto-5′-nucleotidase/CD73 (2).
Once generated, extracellular adenosine can activate P1 receptors (adenosine receptors) on target cells. Adenosine receptors are classified into four subtypes (A1, A2A, A2B, and A3) and consist of G-coupled, 7-transmembrane spanning receptors expressed by a wide range of immune cells. Adenosine receptors have been mainly associated with immunoregulatory functions (4). Figure 1 shows how the purinergic signaling modulates immune responses during inflammation.
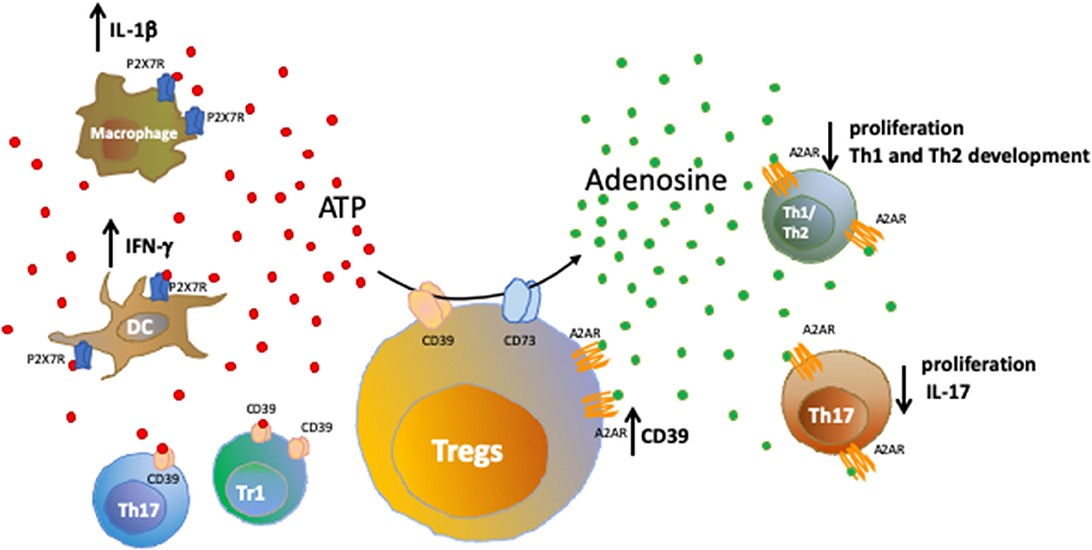
Figure 1. Purinergic signaling in immune cells. Healthy tissues contain negligible levels of extracellular nucleotides; whereas inflammatory sites are characterized by accumulation of extracellular ATP that binds purinergic receptors on target cells, further amplifying inflammatory responses. We show here the effects resulting from the activation of ATP-P2X7R axis in macrophages and dendritic cells. Extracellular nucleotides can be converted into nucleosides by ectonucleotidases present on Tregs, T regulatory type-1 (Tr1) cells and a subset of Th17-cells. The activity of CD39 and CD73 ectoenzymes expressed by Tregs and converting pro-inflammatory ATP into anti-inflammatory adenosine is shown. Upon binding to the A2A receptor (A2AR), extracellular adenosine suppresses effector T cell responses, leading to reduced cell proliferation, limited Th1 and Th2 development and control of IL-17 production by Th17-cells. Notably, A2AR is expressed also by Tregs and its activation promotes CD39 expression.
In this review, we will discuss the role of the purinergic signaling, with a focus on P1 and P2 receptors and on ENTPD1/CD39 and CD73 ectoenzymes, in the context of inflammatory bowel disease (IBD). We will report the most important findings obtained from human studies and experimental models of colitis, and we will also highlight potential novel therapeutic approaches, such as administration of exogenous recombinant ectonucleotidases and targeting of specific intracellular pathways that interfere with purinergic signaling.
IBD is a chronic inflammatory condition of the gastrointestinal tract associated with altered gut microbial composition, disrupted mucosa structure and systemic biochemical alterations (5, 6). IBD most frequent manifestations include Crohn's disease (CD) and ulcerative colitis (UC), which are diagnosed based on the localization of intestinal inflammation and clinical symptoms (5, 7).
Mounting evidence has indicated Th17-cells as the main effector players involved in IBD tissue damage and several additional studies have reported the role of purinergic signaling alterations in the immunopathogenesis of the disease.
Recommended standard therapies for IBD include corticosteroids (7), immunosuppressive drugs (8, 9), amino-salicylates (10, 11), and biological agents like infliximab (12). These currently available treatments, however, are often associated with adverse effects and limited therapeutic efficacy, this emphasizing the need for novel and more effective therapies. Given the role played in the modulation of immune responses, the purinergic signaling might represent a potential therapeutic target.
P2X Receptor Family Members - P2X7
Alterations of purinergic signaling play an important role in promoting tissue inflammation in IBD and several evidences support the involvement of P2X receptors and specifically the P2X7 receptor (P2X7R).
Systemic P2X7R inhibition by administration of the selective inhibitor A740003 or brilliant blue G, prevents the development of TNBS-induced colitis in rat models (13). Similarly, P2X7R deletion is protective in murine models of colitis (14).
P2X7R activation has been also associated with the death of enteric neurons that lead to impaired colon motility (Figure 2A). In rat models of ulcerative colitis, P2X7R co-localizes with immunoreactive cells in the myenteric plexus (15). The decrease in neuronal density in the myenteric plexus has a positive correlation with P2X7R expression in different neuronal populations present in the area (15). P2X7R stimulation triggers pannexin-1 (Panx1) channels opening and inflammasome activation, including the Asc adaptor protein and caspase cleavage, which results in neuronal death. Accordingly, in murine models of colitis, selective inhibition of each component of the P2X7R-inflammasome axis significantly dampens the inflammation while Panx1 inhibition reduces the colonic dysfunction in vivo (21).
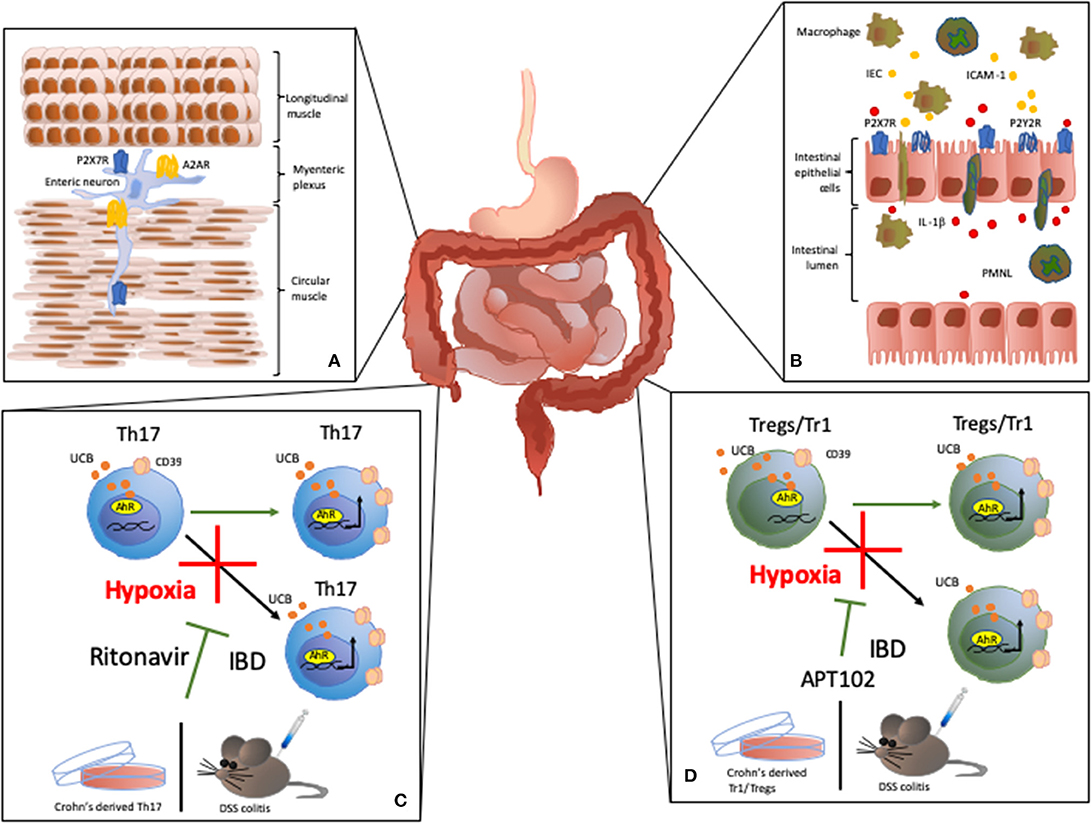
Figure 2. Effects of purinergic signaling modulation in IBD. (A) Effects on colon motility: P2X7R activity has been associated with death of enteric neurons that leads to impaired colon motility in IBD (15). A2AR activity modulates the inhibitory effect of adenosine on colonic motility during experimental colitis. (B) P2Y receptor (P2Y2R) expression and activation on intestinal epithelial cells (IEC) increases the expression of chemoattractive proteins, like ICAM-1, in IEC, promoting macrophage transepithelial migration and adhesion (16). In the inflamed lamina propria and gut epithelium, P2X7R activation triggers IL-1β secretion by IEC in response to polymorphonuclear leukocyte (PMNL) transmigration (17). (C,D) In Tr1, Treg, and Th17-cells, CD39 expression is induced upon activation of aryl hydrocarbon receptor (AhR), an intracellular receptor for pollutants, toxins and endogenous compounds, like unconjugated bilirubin (UCB). UCB boosts cell immunoregulatory properties in vitro and ameliorates the course of experimental colitis in vivo (18). However, in Crohn's disease or experimental colitis, Treg, Tr1, and Th17-cells are refractory to the regulatory effects of UCB due to a deleterious effect of hypoxia (19). Notably, we found that HIF-1α inhibition or ABC transporters blockade obtained upon administration of the antiretroviral ritonavir, limits the detrimental effects of hypoxia in Th17-cells in vitro in in vivo (19). Similarly, administration of the ADPase APT102, restores the response of Treg and Tr1-cells to the regulatory effects of UCB and ameliorates experimental colitis in vivo (20).
Additional evidence has indicated that in a model of 2,4-dinitrobenzenesulfonic-acid (DNBS)-induced colitis P2X7R is upregulated in the neuromuscular layer; notably, increase in electrically induced contractions was recorded in colonic preparations, obtained from colitic mice, and exposed to A804598, a selective P2X7R antagonist (22).
P2X7R expression promotes intestinal inflammation also by inducing different subsets of effector cells, including mast cells. Increased numbers of mast cells were found in the colon of CD patients and experimental murine models of colitis (23). Mast cell-deficient or P2X7R−/− mast cell-reconstituted mice present lower susceptibility to inflammation as compared to the wild type counterpart. Interestingly, P2X7R-mediated activation of mast cells triggers release of pro-inflammatory cytokines, chemokines, and leukotrienes that recruit neutrophils in the inflamed area (23). Further, P2X7R activation has been linked to death and retention of regulatory T-cells (Tregs) in the mesenteric lymph nodes impairing gut immune tolerance (14).
The inflammatory process in the gut is linked to polymorphonuclear leukocyte (PMNL) transmigration into the mucosa leading to increased IL-1β production by intestinal epithelial cells (IEC). Interestingly, in CD patients, P2X7R is overexpressed in the inflamed lamina propria and gut epithelium, where P2X7R activation triggers IL-1β secretion by IEC in response to transmigration of PMNL (17) (Figure 2B).
In the intestinal mucosa, P2X7R is also present on macrophages and dendritic cells where the expression of this receptor positively correlates with IFN-γ, TNF-α, and IL-1-β levels, this leading to epithelial cells apoptosis. Concomitantly, lower concentrations of IL-10 have been also detected (24). In vitro experiments, conducted on human colonic mucosa strips, have revealed that blockade of Panx1 and P2X7R significantly reduce crypt damage, pro-inflammatory cytokine release, loss of tight junctions, and cell permeability (25).
It has been hypothesized that the presence of a gain of function single nucleotide polymorphism or the loss of function SNPs (His155Tyr, Arg307Gln, and Glu496Ala) affect P2X7R activity and could be associated with susceptibility to CD. However, no significant differences were noted among the subjects carrying the polymorphisms in terms of disease incidence (26). A phase II clinical trial on patients with moderate to severe CD evaluated the use of a selective P2X7R antagonist, AZD9056, as a potential therapeutic approach. Despite not having effects on the levels of inflammatory biomarkers, oral administration of AZD9056 induced an overall improvement in the disease symptoms (27). Further investigations are therefore needed to develop P2X7R antagonists that effectively interfere with the inflammatory response; in this regard, several compounds have been proposed and these include Pyroglutamide-Based P2X7R Antagonists (28).
P2X7R, however, plays also an important role in the regulation of follicular T helper cell density in Peyer's patches (29) while favoring the generation of metabolic homeostasis by sensing microbiota-derived ATP (30).
P2Y Receptors
The G-coupled associated P2Y receptors (P2YR) are also involved in IBD immunopathogenesis. In vitro experiments using human nerve-gut preparations and mouse colonic sensory neurons, showed that P2YR activation triggers visceral nociceptors stimulation that can lead to the visceral pain associated with inflammation (31).
Colonic tissue isolated either from IBD patients or mice with experimental colitis displays higher expression of P2Y2 receptor (P2Y2R) when compared to healthy controls. Further investigations have revealed that increase in P2Y2R expression depends on the activity of the C/EBPβ transcription factor, which is also upregulated in IEC in murine models of colitis (32). Investigations conducted on human colon cell lines and colonic tissue from CD and UC patients, revealed that during intestinal inflammation P2YR expression is also regulated through a NF-kB p65-dependent mechanism (33). Activation of P2YR, and particularly P2Y2R, increases the expression of chemoattractive proteins, like ICAM-1, in IEC; this promoting macrophage transepithelial migration and adhesion (16) (Figure 2B).
On the other hand, experiments conducted in murine models of dextran sulfate sodium (DSS) colitis showed that administration of the P2Y2R agonist 2-thioUTP reduces the disease activity index and histological scores. These evidences suggest a role for P2Y2R in the remission phase of IBD (34).
Another study identified a specific activity of P2Y6R that was found to regulate CXCL8 expression in IEC, promoting neutrophil recruitment and inflammatory responses (35). In contrast, in murine models of DSS colitis, P2Y6R deletion has been associated with extensive intestinal inflammation, resulting from increased recruitment of Th17/Th1 lymphocytes in the gut mucosa (36).
In humans, P2Y6R is expressed in a wide range of inflammatory cells and expression levels increase in activated CD4+ and CD8+ T-cells. Because of the pro-inflammatory activity, P2Y6R has been implicated in the pathogenesis of IBD-mediated intestinal damage (37).
ENTPD1/CD39 and CD73 Ectoenzymes
The involvement of ENTPD1/CD39 and CD73 ectoenzymes in the modulation of intestinal inflammation has been extensively studied.
In experimental mouse models of colitis, the impact of CD39 expression strictly depends on the model considered as well as on the cell populations involved in the tissue damage. In the setting of trinitro-benzene-sulfonic-acid (TNBS)-induced colitis in humanized mice, Goettel et al., demonstrated that activation of the transcription factor aryl hydrocarbon receptor (AhR)—an intracellular receptor for pollutants, toxins, and endogenous compounds—by indole-3′-carbonyl-thiazole-4-carboxylic-acid-methyl-ester, induced Tregs and this was linked to CD39 upregulation (38). In contrast, a more favorable course of TNBS-induced colitis has been observed in CD39-null mice when compared to wild type controls (39). In the context of DSS colitis, ENTPD1/CD39 and CD73 expression on activated macrophages was found to limit inflammation, either by directly hydrolyzing pro-inflammatory extracellular ATP into adenosine or by indirectly promoting Treg development (40). Further, in the same experimental model, CD39 deletion exacerbated colitis as reflected by heightened disease activity index, higher levels of pro-inflammatory markers and histological evidence of tissue injury.
Notably, in humans, the presence of SNPs associated with lower levels of CD39 expression correlate with increased susceptibility to CD (41); whereas increased CD39 levels in peripheral blood Tregs are associated with clinical and endoscopic remission (42). Recently Huang and colleagues reported multiple cellular defects in pediatric colitis and IBD, including impaired cyclic AMP-response signaling, infiltration of phosphodiesterase 4B and TNF-expressing macrophages, platelet aggregation and decrease in CD39-expressing intraepithelial T-cells (43); notably, administration of the phosphodiesterase inhibitor dipyridamole ameliorated colitis symptoms in a pilot study (43).
CD39 is known to play an important role in the immunosuppressive activity of suppressor Th17-cells (supTh17). This cell population derives from iTregs upon exposure to Th17 polarizing conditions. SupTh17-cells display high expression of CD39 and actively contribute to the production of adenosine. Compared to bona fide pathogenic Th17, supTh17-cells display higher expression of the enzyme adenosine deaminase and lower expression of A2A receptor (A2AR), these features making these cells refractory to the inhibitory effects of adenosine (44).
We recently found that, in human Th17-cells, CD39 expression is induced upon activation of AhR via unconjugated bilirubin (UCB). Exposure to UCB boosts Th17 immunoregulatory properties in vitro and ameliorates the course of experimental DSS colitis in vivo (18). Furthermore, Th17-cells from CD patients are refractory to the immunosuppressive effects of UCB. This lack of response is directly dependent on high levels of hypoxia-inducible-factor-1alpha (HIF-1α) that promotes ABC transporters to favor UCB exit from the cells and therefore limiting AhR activation. Interestingly, blockade of HIF-1α or ABC transporters limits the detrimental effects of hypoxia both in vitro and in vivo (19) (Figure 2C). Further, human CD39 overexpression or administration of APT102—the extracellular domain with improved ADPase activity of human nucleoside triphosphate diphosphohydrolase-3 (CD39L3), a member of the CD39 family—restores AhR-mediated regulatory effects on CD-derived Tregs in vitro and prevents hypoxia-related damage in experimental colitis in vivo (20) (Figure 2D).
There is however evidence that in IBD patients with active disease, CD73 expression in CD4+ T-cells is associated with a pro-inflammatory Th17-cell phenotype; based on this evidence, CD73 could be therefore used as a marker to monitor disease activity during treatment (45).
A2AR
In IBD, the beneficial effects of adenosine generation by CD39 and CD73 ectoenzymes, have been supported by a wealth of studies. In rat models of chronic experimental colitis, administration of two different selective adenosine deaminase inhibitors significantly improved the course of disease (46). Data revealed that both compounds significantly decreased the inflammatory parameters and the beneficial effect was abrogated in the presence of pharmacological blockade of A2AR or A3 receptor (A3R), suggesting a protective role for both these receptors (46).
A study on the effects of electroacupuncture on visceral pain in a murine model of TNBS-colitis, showed that the beneficial effect of the treatment was linked to increased expression of A1R, A2AR, and A3R and to decreased expression of A2B receptor (A2BR) in colonic tissue. The antalgic effect was mediated by inhibition of release of the pro-inflammatory factors substance P (SP) and IL-1β. This salutary effect was partially abrogated in the presence of adenosine receptors antagonists (47).
A2AR activity has been associated with amelioration of spontaneous ileitis and administration of the A2AR agonist ATL-146e significantly reduced the intestinal mucosa inflammation, leukocyte infiltration in the gut and release of proinflammatory cytokines (48). A2AR plays also an important role in modulating colonic motility. In this regard, exposure of rat colonic longitudinal muscle preparations to the receptor antagonist ZM 241385 increases transmural electrical stimulation-induced contractions, whereas exposure to the receptor agonist CGS 21680, triggered a concentration-dependent reduction of contractile responses. Interestingly, these modulatory functions are further enhanced in preparations derived from animals exposed to DNBS-induced colitis (49). Similarly, in rat ileum/jejunum preparations, CGS 21680 administration prevented the TNBS-related inhibition of acetylcholine-induced contractions and A2BR antagonist PSB-1115 inhibited the contraction-decreasing effect of TNBS. The effect was even enhanced in response to combinatorial treatment with CGS 21680 and PSB-1115, both used at subthreshold concentrations (50). The beneficial role of A2AR in the context of experimental colitis has been also supported by the observation that administration of the A2AR agonist polydeoxyribonucleotide (PDRN), ameliorated the clinical symptoms and promoted tissue repair in two models developed in Sprague-Dawley rats. PDRN administration significantly reduced the circulating levels of pro-inflammatory cytokines, along with decrease in malondialdehyde and myeloperoxidase activity (51). A newly synthetized polar A2AR agonist, in which polar groups were introduced to prevent peroral absorption and subsequent systemic side effects, has been proposed as a potential treatment for IBD. Preliminary experiments conducted in rat ileum/jejunum preparations showed a significant improvement of the impaired acetylcholine-induced contractions and this beneficial effect was boosted by A2BR selective antagonists (52). Importantly, administration of this compound in a rat model of oxazolone-induced colitis limited weight loss and decreased levels of TNF-α in colonic tissue (53).
In humans, overexpression of the miRNA-16 has been reported in UC patients. A recent study identified a correlation between miRNA-16 overexpression and A2AR downregulation in the colonic mucosa of active UC patients. In the same study it was also reported that miRNA-16 inhibits the expression of the A2AR gene by acting at the post-transcriptional level; this effect being mediated upon engagement of the NF-κB pathway (54).
A2BR
Despite the widely described immunoregulatory effects of adenosine, there is evidence supporting a pro-inflammatory role of A2BR in the context of intestinal inflammation.
Experiments conducted in murine models of colitis induced by DSS, TNBS, and Salmonella typhimurium showed a protective effect of A2BR knockout that was associated with lower neutrophil responses, although cell recruitment to the inflammatory site was not impacted (55). Accordingly, administration of the A2BR selective antagonist ATL-801 in DSS-treated wild type or piroxicam-treated IL-10−/− mice, significantly lowered severity of colitis along with levels of pro-inflammatory cytokines (56). Engagement of A2BR has been also linked to the damage associated with intestinal ischemia reperfusion (I/R) injury and hypoxia. In this regard, administration of the A2BR antagonist PSB-1115 results in protection of the intestinal epithelial structure in a murine model of intestinal I/R and in an in vitro model of acute hypoxia (57). Combinatorial administration of PSB-601—another A2BR antagonist—and the A2AR agonist PSB-0777 was found to limit the TNBS-induced contractile disruption in rat ileum/jejunum preparations (52). On the other hand, deletion of A2BR in IEC has been reported having a protective role (55). As an example, an epithelial-specific A2BR deletion resulted in a milder form of experimental colitis, when compared to wild type controls. Further, in vitro studies have shown that the receptor activation on epithelial cells enhances a specific barrier repair response by inducing phosphorylation of vasodilator-stimulated phosphoprotein (VASP) (58).
A3R
The A3R has been also implicated in the modulation of intestinal inflammation. In this context, there have been antithetical reports, providing evidences for either a protective or pathogenic role for this receptor during intestinal inflammation.
In a murine model of experimental colitis induced by intrarectal administration of DNBS, the beneficial effect of adenosine deaminase inhibitors was abrogated in the presence of A2AR and A3R antagonists, suggesting a protective role for both receptors (46). Further, inhibition of visceral pain by electroacupuncture in mice with TNBS-induced colitis, are accompanied by upregulation of A3R along with A2AR and A1R (47). Conversely, a study conducted in a murine model of DSS-induced colitis, revealed a pathogenic effect for the A3R. The impact of A3R deletion was evaluated on the clinical course of experimental colitis and on colon motility that was assessed upon measurement of artificial bead-expulsion, stool-frequency and FITC-dextran transit (59). Interestingly, A3R deficiency protected from DSS-induced tissue damage, limiting the CD4+-cell infiltration in the colon and preserving colon motility (59). The pathogenic role of A3R was also suggested by a clinical study that reported higher levels of A3R in PBMCs from patients with different autoimmune disorders, including CD, when compared to healthy subjects (60).
However, the effects of A3R expression strictly depend on the cells, in which the receptor is expressed. A study conducted on human colonic epithelial cells reported that A3R activation inhibits NF-kB signaling pathway leading to inhibition of IL-8 and IL-1β pro-inflammatory cytokines (61). In line with this observation, the use of the A3R agonist N(6)-(3-iodobenzyl)-adenosine-5-N-methyluronamide in a rat chronic model of TNBS-induced colitis showed beneficial effects on the course of the disease. Interestingly, the receptor inhibitor limited colitis-induced upregulation of other pro-inflammatory purinergic receptors like P2X1, P2X4, P2X7, P2Y2, P2Y6, as well as A2AR and A2BR (62).
Importance of the adenosinergic signaling has been further supported by the findings of Aherne et al. who showed that increased intestinal adenosine levels resulting from epithelial specific deletion of equilibrative nucleoside transporter 2 protected from inflammation in mice with experimental colitis (63).
Supplementary Table 1 summarizes the effects of purinergic receptors in IBD pathophysiology.
Concluding Remarks
Mounting clinical evidence and research data support the involvement of purinergic signaling alterations in IBD pathogenesis, with imbalances in the ATP/adenosine ratio being regarded as underlying immunological dysregulation in this condition. As already observed in other autoimmune conditions, promising therapeutic candidates based on adenosine or ENTPD1/CD39 and CD73 ectonucleotidases have been identified.
Boosting CD39 expression either by inducing AhR-signaling or by administering exogenous ADPase, which displays ectoenzymatic activity comparable to human CD39, showed important immunoregulatory effects in vitro and in vivo, in experimental colitis models. Encouraging pre-clinical data support also the use of selective A2AR agonists, in association with specific inhibition of A2BR. Inhibition of P2X7R-mediated responses has been also associated with beneficial effects.
Taken together, the currently available evidences, implicate the use of purinergic-mediated strategies as adjunctive treatments to correct immune dysregulation in IBD patients.
Author Contributions
MV wrote the manuscript. SM helped drafting some sections of the manuscript. SR and ML reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work has been supported by the National Institute of Health (R01 DK108894 to ML and R21 CA164970 to SR); the Department of Defense Award W81XWH-16-0464 (to SR); and by the AASLD Pilot Research Award and the Department of Anesthesia Seed Grant Award (to ML).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.01882/full#supplementary-material
References
1. Brake AJ, Wagenbach MJ, Julius D. New structural motif for ligand-gated ion channels defined by an ionotropic ATP receptor. Nature. (1994) 371:519–23. doi: 10.1038/371519a0
2. Yegutkin GG. Enzymes involved in metabolism of extracellular nucleotides and nucleosides: functional implications and measurement of activities. Crit Rev Biochem Mol Biol. (2014) 49:473–97. doi: 10.3109/10409238.2014.953627
3. Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: novel checkpoint inhibitor targets. Immunol Rev. (2017) 276:121–44. doi: 10.1111/imr.12528
4. Burnstock G. Purinergic signalling: from discovery to current developments. Exp Physiol. (2014) 99:16–34. doi: 10.1113/expphysiol.2013.071951
5. Mulder DJ, Noble AJ, Justinich CJ, Duffin JM. A tale of two diseases: the history of inflammatory bowel disease. J Crohns Colitis. (2014) 8:341–8. doi: 10.1016/j.crohns.2013.09.009
6. Khan I, Ullah N, Zha L, Bai Y, Khan A, Zhao T, et al. Alteration of gut microbiota in inflammatory bowel disease (IBD): cause or consequence? IBD treatment targeting the gut microbiome. Pathogens. (2019) 8:126. doi: 10.3390/pathogens8030126
7. Wijmenga C. Expressing the differences between crohn disease and ulcerative colitis. PLoS Med. (2005) 2:e230. doi: 10.1371/journal.pmed.0020230
8. van Dieren JM, Kuipers EJ, Samsom JN, Nieuwenhuis EE, van der Woude CJ. Revisiting the immunomodulators tacrolimus, methotrexate, and mycophenolate mofetil: their mechanisms of action and role in the treatment of IBD. Inflamm Bowel Dis. (2006) 12:311–27. doi: 10.1097/01.MIB.0000209787.19952.53
9. Willot S, Noble A, Deslandres C. Methotrexate in the treatment of inflammatory bowel disease: an 8-year retrospective study in a canadian pediatric IBD center. Inflamm Bowel Dis. (2011) 17:2521–6. doi: 10.1002/ibd.21653
10. Habens F, Srinivasan N, Oakley F, Mann DA, Ganesan A, Packham G. Novel sulfasalazine analogues with enhanced NF-kB inhibitory and apoptosis promoting activity. Apoptosis. (2005) 10:481–91. doi: 10.1007/s10495-005-1877-0
11. Nugent SG, Kumar D, Rampton DS, Evans DF. Intestinal luminal pH in inflammatory bowel disease: possible determinants and implications for therapy with aminosalicylates and other drugs. Gut. (2001) 48:571–7. doi: 10.1136/gut.48.4.571
12. Levin AD, Wildenberg ME, van den Brink GR. Mechanism of action of anti-TNF therapy in inflammatory bowel disease. J Crohns Colitis. (2016) 10:989–97. doi: 10.1093/ecco-jcc/jjw053
13. Marques CC, Castelo-Branco MT, Pacheco RG, Buongusto F, do Rosario A Jr, Schanaider A, et al. Prophylactic systemic P2X7 receptor blockade prevents experimental colitis. Biochim Biophys Acta. (2014) 1842:65–78. doi: 10.1016/j.bbadis.2013.10.012
14. Figliuolo VR, Savio LEB, Safya H, Nanini H, Bernardazzi C, Abalo A, et al. P2X7 receptor promotes intestinal inflammation in chemically induced colitis and triggers death of mucosal regulatory T cells. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:1183–94. doi: 10.1016/j.bbadis.2017.03.004
15. da Silva MV, Marosti AR, Mendes CE, Palombit K, Castelucci P. Differential effects of experimental ulcerative colitis on P2X7 receptor expression in enteric neurons. Histochem Cell Biol. (2015) 143:171–84. doi: 10.1007/s00418-014-1270-6
16. Langlois C, Gendron FP. Promoting MPhi transepithelial migration by stimulating the epithelial cell P2Y(2) receptor. Eur J Immunol. (2009) 39:2895–905. doi: 10.1002/eji.200939369
17. Cesaro A, Brest P, Hofman V, Hebuterne X, Wildman S, Ferrua B, et al. Amplification loop of the inflammatory process is induced by P2X7R activation in intestinal epithelial cells in response to neutrophil transepithelial migration. Am J Physiol Gastrointest Liver Physiol. (2010) 299:G32–42. doi: 10.1152/ajpgi.00282.2009
18. Longhi MS, Vuerich M, Kalbasi A, Kenison JE, Yeste A, Csizmadia E, et al. Bilirubin suppresses Th17 immunity in colitis by upregulating CD39. JCI Insight. (2017) 2:e92791. doi: 10.1172/jci.insight.92791
19. Xie A, Robles RJ, Mukherjee S, Zhang H, Feldbrugge L, Csizmadia E, et al. HIF-1alpha-induced xenobiotic transporters promote Th17 responses in crohn's disease. J Autoimmun. (2018) 94:122–33. doi: 10.1016/j.jaut.2018.07.022
20. Robles RJ, Mukherjee S, Vuerich M, Xie A, Harshe R, Cowan PJ, et al. Modulation of CD39 and exogenous APT102 correct immune dysfunction in experimental colitis and crohn's disease. J Crohns Colitis. (2019) 14:818–30. doi: 10.1093/ecco-jcc/jjz182
21. Gulbransen BD, Bashashati M, Hirota SA, Gui X, Roberts JA, MacDonald JA, et al. Activation of neuronal P2X7 receptor-pannexin-1 mediates death of enteric neurons during colitis. Nat Med. (2012) 18:600–4. doi: 10.1038/nm.2679
22. Antonioli L, Giron MC, Colucci R, Pellegrini C, Sacco D, Caputi V, et al. Involvement of the P2X7 purinergic receptor in colonic motor dysfunction associated with bowel inflammation in rats. PLoS ONE. (2014) 9:e116253. doi: 10.1371/journal.pone.0116253
23. Kurashima Y, Amiya T, Nochi T, Fujisawa K, Haraguchi T, Iba H, et al. Extracellular ATP mediates mast cell-dependent intestinal inflammation through P2X7 purinoceptors. Nat Commun. (2012) 3:1034. doi: 10.1038/ncomms2023
24. Neves AR, Castelo-Branco MT, Figliuolo VR, Bernardazzi C, Buongusto F, Yoshimoto A, et al. Overexpression of ATP-activated P2X7 receptors in the intestinal mucosa is implicated in the pathogenesis of crohn's disease. Inflamm Bowel Dis. (2014) 20:444–57. doi: 10.1097/01.MIB.0000441201.10454.06
25. Diezmos EF, Markus I, Perera DS, Gan S, Zhang L, Sandow SL, et al. Blockade of pannexin-1 channels and purinergic P2X7 receptors shows protective effects against cytokines-induced colitis of human colonic mucosa. Front Pharmacol. (2018) 9:865. doi: 10.3389/fphar.2018.00865
26. Haas SL, Ruether A, Singer MV, Schreiber S, Bocker U. Functional P2X7 receptor polymorphisms (His155Tyr, Arg307Gln, Glu496Ala) in patients with crohn's disease. Scand J Immunol. (2007) 65:166–70. doi: 10.1111/j.1365-3083.2006.01876.x
27. Eser A, Colombel JF, Rutgeerts P, Vermeire S, Vogelsang H, Braddock M, et al. Safety and efficacy of an oral inhibitor of the purinergic receptor P2X7 in adult patients with moderately to severely active crohn's disease: a randomized placebo-controlled, double-blind, phase iia study. Inflamm Bowel Dis. (2015) 21:2247–53. doi: 10.1097/MIB.0000000000000514
28. Homerin G, Jawhara S, Dezitter X, Baudelet D, Dufrenoy P, Rigo B, et al. Pyroglutamide-based P2X7 receptor antagonists targeting inflammatory bowel disease. J Med Chem. (2019). 63:2074–94. doi: 10.1021/acs.jmedchem.9b00584
29. Proietti M, Cornacchione V, Rezzonico Jost T, Romagnani A, Faliti CE, Perruzza L, et al. ATP-gated ionotropic P2X7 receptor controls follicular T helper cell numbers in peyer's patches to promote host-microbiota mutualism. Immunity. (2014) 41:789–801. doi: 10.1016/j.immuni.2014.10.010
30. Perruzza L, Gargari G, Proietti M, Fosso B, D'Erchia AM, Faliti CE, et al. T follicular helper cells promote a beneficial gut ecosystem for host metabolic homeostasis by sensing microbiota-derived extracellular ATP. Cell Rep. (2017) 18:2566–75. doi: 10.1016/j.celrep.2017.02.061
31. Hockley JR, Tranter MM, McGuire C, Boundouki G, Cibert-Goton V, Thaha MA, et al. P2Y receptors sensitize mouse and human colonic nociceptors. J Neurosci. (2016) 36:2364–76. doi: 10.1523/JNEUROSCI.3369-15.2016
32. Degagne E, Turgeon N, Moore-Gagne J, Asselin C, Gendron FP. P2Y(2) receptor expression is regulated by C/EBPbeta during inflammation in intestinal epithelial cells. FEBS J. (2012) 279:2957–65. doi: 10.1111/j.1742-4658.2012.08676.x
33. Degagne E, Grbic DM, Dupuis AA, Lavoie EG, Langlois C, Jain N, et al. P2Y2 receptor transcription is increased by NF-kappa B and stimulates cyclooxygenase-2 expression and PGE2 released by intestinal epithelial cells. J Immunol. (2009) 183:4521–9. doi: 10.4049/jimmunol.0803977
34. Degagne E, Degrandmaison J, Grbic DM, Vinette V, Arguin G, Gendron FP. P2Y2 receptor promotes intestinal microtubule stabilization and mucosal re-epithelization in experimental colitis. J Cell Physiol. (2013) 228:99–109. doi: 10.1002/jcp.24109
35. Grbic DM, Degagne E, Larrivee JF, Bilodeau MS, Vinette V, Arguin G, et al. P2Y6 receptor contributes to neutrophil recruitment to inflamed intestinal mucosa by increasing CXC chemokine ligand 8 expression in an AP-1-dependent manner in epithelial cells. Inflamm Bowel Dis. (2012) 18:1456–69. doi: 10.1002/ibd.21931
36. Salem M, El Azreq MA, Pelletier J, Robaye B, Aoudjit F, Sevigny J. Exacerbated intestinal inflammation in P2Y6 deficient mice is associated with Th17 activation. Biochim Biophys Acta Mol Basis Dis. (2019) 1865:2595–605. doi: 10.1016/j.bbadis.2019.06.019
37. Somers GR, Hammet FM, Trute L, Southey MC, Venter DJ. Expression of the P2Y6 purinergic receptor in human T cells infiltrating inflammatory bowel disease. Lab Invest. (1998) 78:1375–83.
38. Goettel JA, Gandhi R, Kenison JE, Yeste A, Murugaiyan G, Sambanthamoorthy S, et al. AHR activation is protective against colitis driven by T cells in humanized mice. Cell Rep. (2016) 17:1318–29. doi: 10.1016/j.celrep.2016.09.082
39. Kunzli BM, Berberat PO, Dwyer K, Deaglio S, Csizmadia E, Cowan P, et al. Variable impact of CD39 in experimental murine colitis. Dig Dis Sci. (2011) 56:1393–403. doi: 10.1007/s10620-010-1425-9
40. Weinhage T, Dabritz J, Brockhausen A, Wirth T, Bruckner M, Belz M, et al. Granulocyte macrophage colony-stimulating factor-activated CD39+/CD73+ murine monocytes modulate intestinal inflammation via induction of regulatory T cells. Cell Mol Gastroenterol Hepatol. (2015) 1:433–49.e1. doi: 10.1016/j.jcmgh.2015.04.005
41. Friedman DJ, Kunzli BM, A-Rahim YI, Sevigny J, Berberat PO, Enjyoji K, et al. From the cover: CD39 deletion exacerbates experimental murine colitis and human polymorphisms increase susceptibility to inflammatory bowel disease. Proc Natl Acad Sci U S A. (2009) 106:16788–93. doi: 10.1073/pnas.0902869106
42. Gibson DJ, Elliott L, McDermott E, Tosetto M, Keegan D, Byrne K, et al. Heightened expression of CD39 by regulatory T lymphocytes is associated with therapeutic remission in inflammatory bowel disease. Inflamm Bowel Dis. (2015) 21:2806–14. doi: 10.1097/MIB.0000000000000566
43. Huang B, Chen Z, Geng L, Wang J, Liang H, Cao Y, et al. Mucosal profiling of pediatric-onset colitis and IBD reveals common pathogenics and therapeutic pathways. Cell. (2019) 179:1160–76.e24. doi: 10.1016/j.cell.2019.10.027
44. Longhi MS, Moss A, Bai A, Wu Y, Huang H, Cheifetz A, et al. Characterization of human CD39+ Th17 cells with suppressor activity and modulation in inflammatory bowel disease. PLoS ONE. (2014) 9:e87956. doi: 10.1371/journal.pone.0087956
45. Doherty GA, Bai A, Hanidziar D, Longhi MS, Lawlor GO, Putheti P, et al. CD73 is a phenotypic marker of effector memory Th17 cells in inflammatory bowel disease. Eur J Immunol. (2012) 42:3062–72. doi: 10.1002/eji.201242623
46. Antonioli L, Fornai M, Colucci R, Awwad O, Ghisu N, Tuccori M, et al. The blockade of adenosine deaminase ameliorates chronic experimental colitis through the recruitment of adenosine A2A and A3 receptors. J Pharmacol Exp Ther. (2010) 335:434–42. doi: 10.1124/jpet.110.171223
47. Hou T, Xiang H, Yu L, Su W, Shu Y, Li H, et al. Electroacupuncture inhibits visceral pain via adenosine receptors in mice with inflammatory bowel disease. Purinergic Signal. (2019) 15:193–204. doi: 10.1007/s11302-019-09655-4
48. Odashima M, Bamias G, Rivera-Nieves J, Linden J, Nast CC, Moskaluk CA, et al. Activation of A2A adenosine receptor attenuates intestinal inflammation in animal models of inflammatory bowel disease. Gastroenterology. (2005) 129:26–33. doi: 10.1053/j.gastro.2005.05.032
49. Antonioli L, Fornai M, Colucci R, Ghisu N, Blandizzi C, Del Tacca M. A2A receptors mediate inhibitory effects of adenosine on colonic motility in the presence of experimental colitis. Inflamm Bowel Dis. (2006) 12:117–22. doi: 10.1097/01.MIB.0000198535.13822.a9
50. Michael S, Warstat C, Michel F, Yan L, Muller CE, Nieber K. Adenosine A(2A) agonist and A(2B) antagonist mediate an inhibition of inflammation-induced contractile disturbance of a rat gastrointestinal preparation. Purinergic Signal. (2010) 6:117–24. doi: 10.1007/s11302-009-9174-y
51. Pallio G, Bitto A, Pizzino G, Galfo F, Irrera N, Squadrito F, et al. Adenosine receptor stimulation by polydeoxyribonucleotide improves tissue repair and symptomology in experimental colitis. Front Pharmacol. (2016) 7:273. doi: 10.3389/fphar.2016.00273
52. El-Tayeb A, Michael S, Abdelrahman A, Behrenswerth A, Gollos S, Nieber K, et al. Development of polar adenosine A2A receptor agonists for inflammatory bowel disease: synergism with A2B antagonists. ACS Med Chem Lett. (2011) 2:890–5. doi: 10.1021/ml200189u
53. Antonioli L, El-Tayeb A, Pellegrini C, Fornai M, Awwad O, Giustarini G, et al. Anti-inflammatory effect of a novel locally acting A2A receptor agonist in a rat model of oxazolone-induced colitis. Purinergic Signal. (2018) 14:27–36. doi: 10.1007/s11302-017-9591-2
54. Tian T, Zhou Y, Feng X, Ye S, Wang H, Wu W, et al. MicroRNA-16 is putatively involved in the NF-kappaB pathway regulation in ulcerative colitis through adenosine A2a receptor (A2aAR) mRNA targeting. Sci Rep. (2016) 6:30824. doi: 10.1038/srep30824
55. Kolachala VL, Vijay-Kumar M, Dalmasso G, Yang D, Linden J, Wang L, et al. A2B adenosine receptor gene deletion attenuates murine colitis. Gastroenterology. (2008) 135:861–70. doi: 10.1053/j.gastro.2008.05.049
56. Kolachala V, Ruble B, Vijay-Kumar M, Wang L, Mwangi S, Figler H, et al. Blockade of adenosine A2B receptors ameliorates murine colitis. Br J Pharmacol. (2008) 155:127–37. doi: 10.1038/bjp.2008.227
57. Yang Y, Qiu Y, Wang W, Xiao W, Liang H, Zhang C, et al. Adenosine A2B receptor modulates intestinal barrier function under hypoxic and ischemia/reperfusion conditions. Int J Clin Exp Pathol. (2014) 7:2006–18.
58. Aherne CM, Saeedi B, Collins CB, Masterson JC, McNamee EN, Perrenoud L, et al. Erratum: epithelial-specific A2B adenosine receptor signaling protects the colonic epithelial barrier during acute colitis. Mucosal Immunol. (2015) 8:699. doi: 10.1038/mi.2015.41
59. Ren T, Grants I, Alhaj M, McKiernan M, Jacobson M, Hassanain HH, et al. Impact of disrupting adenosine A3 receptors ( AR) on colonic motility or progression of colitis in the mouse. Inflamm Bowel Dis. (2011) 17:1698–713. doi: 10.1002/ibd.21553
60. Ochaion A, Bar-Yehuda S, Cohen S, Barer F, Patoka R, Amital H, et al. The anti-inflammatory target A3 adenosine receptor is over-expressed in rheumatoid arthritis, psoriasis and crohn's disease. Cell Immunol. (2009) 258:115–22. doi: 10.1016/j.cellimm.2009.03.020
61. Ren T, Qiu Y, Wu W, Feng X, Ye S, Wang Z, et al. Activation of adenosine A3 receptor alleviates TNF-alpha-induced inflammation through inhibition of the NF-kappaB signaling pathway in human colonic epithelial cells. Mediators Inflamm. (2014) 2014:818251. doi: 10.1155/2014/818251
62. Guzman J, Yu JG, Suntres Z, Bozarov A, Cooke H, Javed N, et al. ADOA3R as a therapeutic target in experimental colitis: proof by validated high-density oligonucleotide microarray analysis. Inflamm Bowel Dis. (2006) 12:766–89. doi: 10.1097/00054725-200608000-00014
Keywords: adenosine receptor, P2 receptor, ectonucleotidase, crohn's disease, ulcerative colitis
Citation: Vuerich M, Mukherjee S, Robson SC and Longhi MS (2020) Control of Gut Inflammation by Modulation of Purinergic Signaling. Front. Immunol. 11:1882. doi: 10.3389/fimmu.2020.01882
Received: 10 March 2020; Accepted: 13 July 2020;
Published: 25 September 2020.
Edited by:
Davide Ferrari, University of Ferrara, ItalyReviewed by:
Luca Antonioli, University of Pisa, ItalyFrancesco Di Virgilio, University of Ferrara, Italy
Copyright © 2020 Vuerich, Mukherjee, Robson and Longhi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Simon C. Robson, c3JvYnNvbkBiaWRtYy5oYXJ2YXJkLmVkdQ==; Maria Serena Longhi, bWxvbmdoaUBiaWRtYy5oYXJ2YXJkLmVkdQ==