 Margarita Bartish
Margarita Bartish Sonia V. del Rincón1,2
Sonia V. del Rincón1,2 Christopher E. Rudd
Christopher E. Rudd H. Uri Saragovi
H. Uri Saragovi- 1Lady Davis Institute, Jewish General Hospital, Translational Center for Research in Cancer, McGill University, Montreal, QC, Canada
- 2Oncology and Experimental Medicine, McGill University, Montreal, QC, Canada
- 3Division of Immuno-Oncology, Research Center Maisonneuve-Rosemont Hospital, Montreal, QC, Canada
- 4Département de Médecine, Université de Montréal, Montreal, QC, Canada
- 5Pharmacology and Therapeutics, and Ophthalmology and Vision Sciences, McGill University, Montreal, QC, Canada
Though a healthy immune system is capable of recognizing and eliminating emergent cancerous cells, an established tumor is adept at escaping immune surveillance. Altered and tumor-specific expression of immunosuppressive cell surface carbohydrates, also termed the “tumor glycocode,” is a prominent mechanism by which tumors can escape anti-tumor immunity. Given their persistent and homogeneous expression, tumor-associated glycans are promising targets to be exploited as biomarkers and therapeutic targets. However, the exploitation of these glycans has been a challenge due to their low immunogenicity, immunosuppressive properties, and the inefficient presentation of glycolipids in a conventional major histocompatibility complex (MHC)-restricted manner. Despite this, a subset of T-cells expressing the gamma and delta chains of the T-cell receptor (γδ T cells) exist with a capacity for MHC-unrestricted antigen recognition and potent inherent anti-tumor properties. In this review, we discuss the role of tumor-associated glycans in anti-tumor immunity, with an emphasis on the potential of γδ T cells to target the tumor glycocode. Understanding the many facets of this interaction holds the potential to unlock new ways to use both tumor-associated glycans and γδ T cells in novel therapeutic interventions.
Introduction
The cornerstone of a healthy immune system is the ability to distinguish “self” from “nonself,” to mount a response to “nonself” while minimizing the reactivity to “self” (1). A tumor originates from cells that remain mostly “self.” Thus, identifying meaningful differences between pathological and healthy cells has been difficult in the dynamic tumor microenvironment (TME). Nonetheless, the erratic pattern of gene expression, altered metabolism, deregulated signaling pathways, and often high mutational burden results in the presentation of neoantigens on the surface of tumor cells. These novel antigens can be recognized by both the innate and adaptive arms of the immune system, although this response can be counteracted by the TME via immunoediting, immunoevasion, and immunosuppression. For example, tumors can exploit inhibitory receptors on T-cells by inducing an unresponsive or exhausted state (2).
Research in the area of novel immunotherapies has been focused on deploying, re-educating or enhancing immune defenses to overcome the suppressive, detrimental TME. The past decade has witnessed a revolution in the application of immunotherapy for the treatment of cancer, resulting in the approval of immune checkpoint blockade (ICB). ICB uses monoclonal antibodies (mAbs) directed against the inhibitory receptors (IRs) present on the surface of T cells, or the natural ligands of IRs, often expressed by cancer cells. The blockade of IR-ligand interactions reduces the inhibitory regulation of T cells. ICB against cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4) and programmed death 1 (PD-1) or the PD-1 ligand (PD-L1) have produced survival benefits for an ever-expanding list of malignancies (3). However, only a limited subset of patients benefit from ICB, and there are, at times, toxic side effects due to inflammation and autoimmunity (4). Thus, there is a need to understand the mechanisms used by cancer cells to suppress and shape immune responses and to involve novel immune cell subsets in the design of anti-tumor targeted therapies. Here, we discuss the role of glycans in the context of immunity.
The changes in gene expression that accompany malignant transformation have a significant impact on the glycome, glycoproteome and glycolipidome—the glycocode of cancer cells—leading to the overexpression and de novo expression of novel glycan epitopes (5). These have been studied extensively in the context of promoting tumor cell-intrinsic aspects of proliferation, signaling and metastasis. Relatively recently, the glycocode of tumor cells has been implicated in suppressing anti-tumor immunity, emerging as a novel immune checkpoint, and, thus, a target for immunotherapy. While now recognized as an axis of immune modulation with druggable and therapeutic potential (6), its potential has remained underdeveloped clinically. Moreover, the subset of immune cells that attack carbohydrate targets remains poorly understood. In this review, we discuss the way in which γδ T cells have the potential to become effectors against carbohydrate moieties on cancer cells.
Glycosylation in the Tumor-Immune Cell Interplay
All cells are covered with a dense coat of glycans, chains of carbohydrates that are covalently attached to proteins or lipids (7). Glycan diversity is immense, stemming from the numerous monosaccharide building blocks that can be assembled into linear or branched chains of various lengths by multiple types of chemical bonds, and diversified further by coupling to proteins, nucleic acids or lipids (8). This diversity creates a unique glycan “landscape” of expression for each cell and constitutes a major aspect of the molecular interface between cells and their environment. Glycans are also important for the transport of nascent proteins to the surface of cells as well as, in a larger context, the maintenance of tissue structure and extracellular matrix organization, cell membrane integrity, cell-cell adhesion, and cellular signaling. To immune cells, surface glycans serve as an identifying feature of a cell, a calling card of sorts (9, 10).
Aberrant glycosylation is a hallmark feature of cancer cells (11–13). Key among the distinguishing features of a tumor's “glycan topography” is the anomalous expression of sialic acid–carrying glycans (sialoglycans) (14). Sialic acids are a family of negatively charged, nine-carbon sugar molecules linked to mucins, extracellular matrix, cell surface glycoproteins (N- and O-linked oligosaccharide chains), or glycolipids by α-2,3; α-2,6 and α-2,8 linkages (15).
Tumor cells are covered with a dense layer of sialoglycans, some of which are uniquely associated with malignancy (16). This coating protects tumor cells from being recognized and eradicated by the immune system, as it can both mask their “non-self” immunogenicity and interfere with immune cell function (17, 18). For instance, elevated sialylation of cancer cells disrupts the interaction of the NK-activating receptor natural killer group 2D (NKG2D) with ligands on the tumor cells, reducing NK-activating signals derived from tumor cells (19). This strategy by tumor cells is reminiscent of sialic acid coatings used by parasites and other pathogens to evade immunity (20). Despite these examples linking protein sialylation to pathology, we note that this post-translational modification is not always deleterious. Sialylation of some proteins is associated with neuroprotective signals (15).
The Sialic Acid-Siglec Axis of Tumor Immunomodulation
As “self-associated molecular patterns” (SAMPs), sialic acids are recognized by sialic acid-binding Ig-type lectins (Siglecs). Twenty years of study document the importance of sialic acids in discriminating “self” and “non-self,” showing the existence of natural antibodies to a variety of sialidase-treated immune cells in human serum [reviewed in (21)]. In humans, the Siglec family comprises 14 members. These are subdivided into the conserved Siglecs:−1 (Sialoadhesin/CD169),−2 (CD22),−4 (Myelin-associated glycoprotein/MAG),−15, and the CD33-related Siglecs−3,−5 to−11,−14 and−16 (22). The Siglecs are composed of modular immunoglobulin-like (Ig-like) domains, usually with the V-like domain at the N-terminus mediating binding to sialic acids. This domain shows a high degree of sequence similarity to other Ig-like domains in the receptor family with the exception of the C-2 set Ig domains near the plasma membrane. The cytoplasmic domains have immunoreceptor tyrosine-based inhibition motifs (ITIMs) that bind to the protein tyrosine phosphatases src homology region 2 domain-containing phosphatases 1 and 2 (SHP-1 and SHP-2). SHP-1 has a clear negative signaling role, while SHP-2 has been shown to play both positive and negative roles in immune cells.
Functionally, Siglec binding to sialic acid facilitates tolerance to cell membrane antigens expressed by the same cell. In B cells, for example, Siglec-sialic acid binding suppresses B cell activation and stimulates B cell apoptosis (23–25). While a key physiological mechanism to prevent autoimmunity, inhibitory Siglec-sialic acid interaction illustrates how an immunological fail-safe can be hijacked by tumors to escape host immunity. The engagement of Siglec-7 and Siglec-9 on NK cells by tumor-associated sialic acids inhibits NK cell activation (26–28). Conversely, the loss of Siglec-7 expression on an NK cell line promotes sustained cytotoxic activity against leukemia cells in vitro (29). Furthermore, the binding of the cancer-associated sialylated glycoform of MUC1 to Siglec-9 on macrophages resulted in their differentiation to immunosuppressive so-called M2 macrophages with upregulated PD-L1 expression (30). Tumor-associated macrophages were also shown to express Siglec-15, and the interaction between Siglec-15 on M2 macrophages and tumor-associated sialyl-Tn (sTn) antigen elevated macrophage-produced TGF-β, a known pleiotropic mediator of pro-tumor responses (31). Recently, tumor-expressed CD24 (a highly sialylated glycoprotein) was shown to hinder the ability of tumor-associated macrophages to phagocytize tumors via binding to Siglec-10 (32). The expression of multiple members of the Siglec family was shown on myeloid-derived suppressor cells (MDSCs) of glioma patients, although the functional consequences of this expression on this already immune suppressive cell type are unclear (33).
The innate and adaptive arms of the immune system can influence each other, thus the effects of sialoglycan-Siglec interactions may indirectly affect the activation status and function of the cells of adaptive immunity. For example, the interaction between sialylated antigens and Siglec-E (murine ortholog of human Siglec-9) on dendritic cells can influence the T cell population, favoring differentiation of antigen-specific regulatory T cells and reducing the numbers of effector T cells (34). Although the expression of Siglecs on normal T cells is low, these can become elevated in tumor-infiltrating lymphocytes (TILs), resulting in suppressed anti-tumor T cell function (35, 36). In melanoma, Haas et al. identified Siglec-9 expression on tumor-infiltrating, but not peripheral, CD8+ cells (36). By disrupting the sialoglycan-Siglec pathway between tumor cells and T cells, Stanczak et al. demonstrated a delay of tumor growth and an increased infiltration of CD8+ T cells in a mouse model of colorectal cancer (35).
Given the role of tumor sialylation in the establishment of an immunosuppressive TME, both sides of the sialoglycan-Siglec axis have been targeted therapeutically. To reduce the sensitivity of immune cells to tumor sialoglycans, Siglec function can be blocked with monoclonal antibodies (mAbs). MAbs have been tested pre-clinically against Siglec-7 (26), Siglec-9 (35), and Siglec-15 (37). A challenge of this approach is to confine the response to the tumor setting, as deregulated immune activation might have detrimental consequences outside of TME. The expression of Siglec-9, for example, is restricted to TILs, thus its systemic inhibition by a mAb is unlikely to impact peripheral cytotoxic CD8+ T cell function. However, Siglec-9 is also abundantly expressed by neutrophils and a blocking antibody could result in their uncontrolled and potentially damaging activation. An alternative direction is to target the causative agents of immunosuppression, namely sialoglycans on the tumor cells, rather than on receptors of immune cells. Stalling de novo sialic acid synthesis, via the use of glycomimetic sialic acid analogs that cannot be attached to the glycan chain, has been shown to reduce the density of sialoglycans on the tumor surface and delay of tumor growth and metastasis (38, 39).
Finally, the sialoglycan coverage on tumor cells might be “shaved” using sialidases, sialic acid trimming enzymes. In the 1960s and 1970s, it was found that injecting a tumor-bearing animal with growth-arrested tumor digests treated with sialidases—a very early form of cancer vaccination—impeded the growth of the pre-existing tumor in mouse (40) and dog (17) cancer models (41). Despite promising results in early trials on advanced patients using sialidase-treated cancer cells to boost the immune response, this form of immunotherapy did not become a standard of care (42, 43). In recent years, in an approach the authors termed precision glycocalyx editing, a pre-clinical study coupled a recombinant sialidase to a therapeutic mAb against the human epidermal growth factor receptor 2 (HER2) (44). The antibody directed the effects of the sialidase to the HER2-expressing tumor cells, simultaneously reducing Siglec-mediated NK cell suppression and exposing the tumor cells to NK cell-mediated antibody-dependent cytotoxicity.
Gangliosides as Part of the Tumor “Glycocode”
Among the key sialic acid-containing glycocompounds found on the surface of tumors are the gangliosides—a family of glycosphingolipids with one or several sialic acid molecules attached to the extracellular carbohydrate chain. Though named after the cell type from which they were first isolated— “Ganglienzellen,” neurons—gangliosides are ubiquitously expressed on the membranes of all eukaryotic cells, typically clustering in cholesterol-rich lipid microdomains or rafts (45). Indeed, evidence suggests that gangliosides co-localize with signaling molecules and adhesion molecules in glyco-signaling domains on the cell surface (46).
It is the unique glycan tree-structure that defines each different ganglioside. In ganglioside nomenclature, the prefix G stands for “ganglio” while the letters M (mono-), D (di-), T (tri-) and Q (quad-) denote the number of sialic acid molecules. Further classification is made on the basis of thin layer chromatography migration and is represented by Arabic numerals and lower case letters reflecting the order of migration of each corresponding type (47). GM1 is expressed on most eukaryotic cells and has a prominent role in the activation of intracellular signals in neuronal and lymphoid cells. In particular, GM1 represents the major ganglioside component of the brain (48), with several key neuronal functions becoming compromised as a consequence of decreasing GM1 levels during, for example, aging (49, 50). In contrast, GD2 and GD3 are almost exclusively expressed in tumor cells (51). As such, GD2 and GD3, are examples of a subset of gangliosides referred to as tumor marker gangliosides (TMGs), a family comprising about 20 different gangliosides present preferentially or almost exclusively and at high density on the cell surface of certain cancers (Table 1).
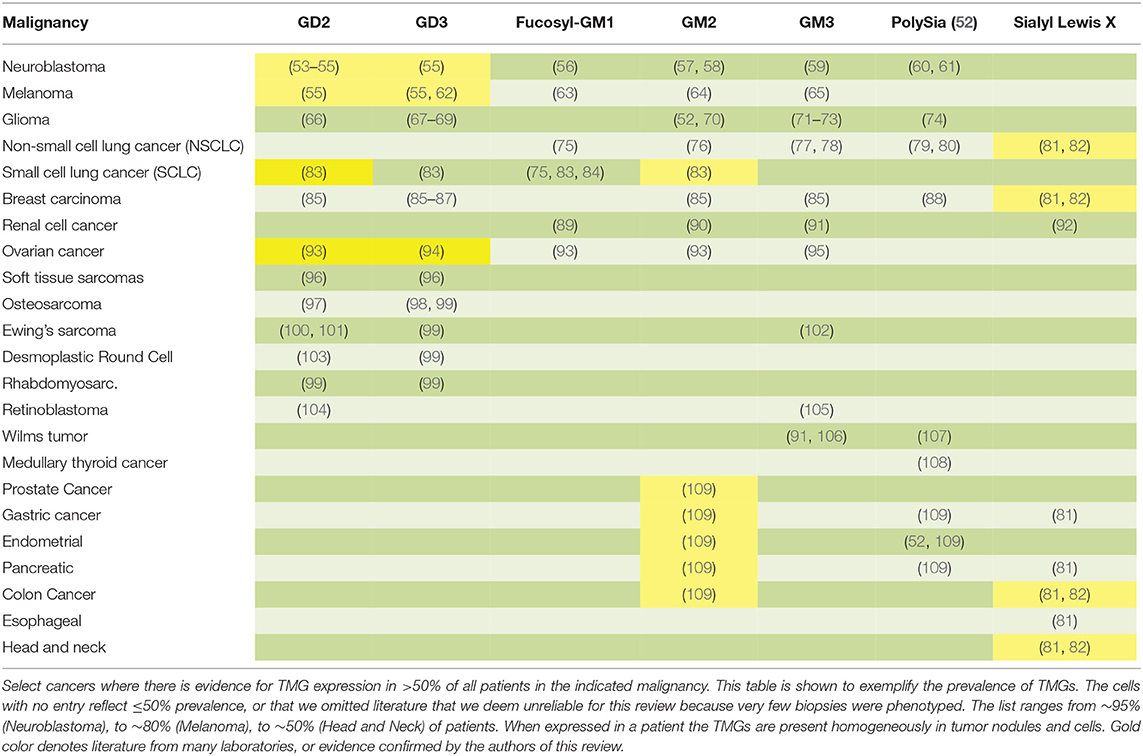
Table 1. Tumor marker ganglioside targets.
Ganglioside biosynthesis begins in the endoplasmic reticulum, with the synthesis of the ceramide precursor, common to all glycosphingolipids, and continues in the Golgi apparatus where the ceramide is converted to glucosylceramide. Sugar residues—galactose, glucose and sialic acids—are added, one by one, catalyzed by specific glycosyltransferases. Some gangliosides can also result from the removal of a sugar or sugar branch by glycosidases. As several enzymes or pathways can generate a ganglioside, their biosynthesis is hard to target. Nonetheless, this strategy has been explored in pre-clinical studies. The inhibition of glucosylceramide synthase, the enzyme which catalyzes the first step in glycosphingolipid synthesis, by N-butyldeoxynojirimycin (NB-DNJ) has been shown to temporarily delay tumor onset in a mouse melanoma model (110). However, prolonged treatment with NB-DNJ is toxic, and in the absence of the inhibitor, ganglioside levels rapidly recovered. Targeting GD3 synthase, the enzyme responsible for the biosynthesis of GD2 and GD3, reduced tumor stem cell functionality, abrogated in vivo tumor formation (111), and interfered with the epithelial-to-mesenchymal transition and metastasis in murine models (112).
Upon export to the plasma membrane, the sphingolipid (ceramide) part of the molecule—two lipid tails consisting of the long-chain amino alcohol sphingosine coupled to a fatty acid—anchors the ganglioside to the cell surface, while the glycan moiety is exposed to the external environment. Apart from the plasma membrane, their main cellular location, gangliosides are also detected in other cellular organelles, including the nuclear envelope (113) and mitochondria (114). Importantly, they can also be actively “shed” and taken up by other cells (115). The excretion of gangliosides from a cell into the extracellular environment is poorly studied, and may be the result of release in the form of exosomes, microparticles, or as micelles given the physical properties of the gangliosides (hydrophobic tail and hydrophilic head). Extracellular gangliosides improve tumorigenicity of poorly tumorigenic cells in mouse models (116) and have been implicated as a mechanism by which tumors suppress immune cell function (117, 118).
Biology and Function of Tumor Marker Gangliosides
As the term “tumor marker” suggests, TMG expression is tightly associated with malignant cells. Table 1 summarizes the cancer types with a high TMG prevalence across patients. However, in addition to their status as biomarkers of malignancy, GD2 and GD3 play active roles during cancer development, with proven links to tumor growth, metastasis, and immune evasion.
Because of their accumulation on the outer leaflet of the plasma membrane (with the sugars being extracellular), gangliosides participate in cellular communication. The carbohydrate “head” of gangliosides can interact with proteins, lipids, and glycans present in the extracellular matrix and on other cells. It can also interact laterally within the membrane to regulate lipid rafts or microdomain formation (119). As components of rafts, gangliosides affect signaling processes during cancer progression. For example, GD2 and GD3 promote ligand-independent activation of wild type receptor tyrosine kinases (RTKs), including EGF-R, TrkA, TrkB, PDGF-R, IGF1-R, MET, as well as cytoplasmic src-related kinases (e.g., Src, Lck) (98, 120, 121). The expression of GD2 and GD3 can therefore be viewed as pro-oncogenic and may be etiological in tumors where oncogenic mutations are not clearly identified.
The pro-tumor roles of tumor gangliosides have been implicated at all stages of tumor development. GD2 expression was linked to breast cancer stem cell phenotypes, while suppression of its biosynthesis in breast cancer cell lines decreased mammosphere formation and tumor initiation (111). In vitro experiments have connected ganglioside production to an increase in cancer cell migration and invasion (98). Conversely, a mAb against GD2 was shown to induce apoptosis in small cell lung cancer cells (83, 122) and an anti-GD3 antibody inhibited the growth of human melanoma cells in vitro (123). Cancer cells devoid of GM2 and GM3 synthases formed avascular tumors, suggesting the involvement of TMGs in angiogenesis during tumor growth (124). Additionally, the exogenous addition of GD3 to glioma cells stimulated VEGF production, suggesting a role for tumor-shed gangliosides in de novo blood vessel formation (125). In patient-derived melanoma cells, ganglioside expression is tightly linked to melanoma aggressiveness and patient survival, with patients expressing GD3 having the shortest survival (126).
Building on the discussion of glycan-containing sialic acids as a segment of the tumor glycocode used by the tumor to evade anti-tumor immunity, GD2 and GD3 can be regarded as immunosuppressive, even in cases where other immune escape proteins such as PD-1 or PD-L1 are absent. Indeed, the observations that TMGs can inhibit antibody production and lymphocyte proliferation were first made decades ago (127, 128). This is increasingly relevant in modern immunotherapy as it is possible that the high failure rate of conventional ICB therapy in melanoma (targeting PD-1 or PD-L1) is associated with high GD2/GD3 expression, a hypothesis that we are evaluating experimentally. As sialic acid-containing compounds, GD2 and GD3 can interact with Siglecs (129–131). Siglec-7, in particular, displays a strong affinity for the α2,8-linked disialic acids found on GD2 and GD3 (132). Additionally, TMGs influence the recruitment and function of immune cells in Siglec-independent ways. TMGs interfere with IL-2/IL-2R binding, key to T cell proliferation (133). They have also been shown to induce apoptosis in T cells (90) and dendritic cells (134), and impair antigen presentation in human monocytes (135). In a tumor model engineered to lack GM3, GM2, GM1, and GD1a, the observed impairment of tumor growth was attributed to a reduction, and decreased activity, of MDSCs (136). Intriguingly, the presence of MDSCs could be restored by exogenous supplementation of gangliosides which suggests a direct connection between tumor-produced gangliosides and the recruitment of immunosuppressive MDSCs to the TME.
TMGs as Therapeutic Targets
In 2009, GD2 was “ranked” by the NCI as 12th in priority of all clinical cancer antigens, with additional three gangliosides (GD3, fucosyl-GM1, and N-acetyl GM3) included in the list of 75 prioritized antigens (137). The high expression of GD2 and GD3 in cancer makes these promising targets for therapeutic intervention. Moreover, when GD2 or GD3 are present in a cancer, tumor cells express them stably and homogeneously, and tumor microheterogeneity with regards to TMG expression has not been reported. GD2 or GD3 persist throughout tumor progression, and expression does not appear to downregulate after chemotherapy, in at least the reported studies of neuroblastoma (138), osteosarcoma (139), and in the ex vivo examination of several cell lines (140).
The etiological role of GD2/GD3 in oncogenesis and immune suppression are additional features that would make these glycolipids ideal therapeutic targets for clinical translation. However, exploiting GD2 or GD3 has been challenging. Monoclonal antibodies against GD2 [Dinutuximab/Ch14.18 mAb (141–143) and 3F8 mAb (144)] and GD3 [BEC2 (145), R24 (146)] achieved partial success in cancer therapy as passive immunity (i.e., the administration of purified antibodies against a target). However, they cause serious adverse effects, such as high-grade visceral pain, that is not blocked by morphine (147). While Ch14.18 mAb, in combination with GM-CSF and IL-2, stimulates antibody-dependent cell-mediated cytotoxicity and improves overall survival in neuroblastoma (141), in clinical trials it had low efficacy or exhibited a low therapeutic index [reviewed in (148)]. More recently, engineered chimeric antigen receptors (CAR) expressing an anti-GD2 mAb sequence in T or NK cells, were used in combination with ICB inhibitors and cytokines (149), but the cells did not persist in circulation and the treatment showed no efficacy (150). The failure is not surprising given that the CAR was engineered from mAbs that also exhibit a low therapeutic index in passive immunity. In addition to the clinical CAR T cell studies performed in neuroblastoma and melanoma, pre-clinically, CAR anti-GD2 T cells have recently been tested against breast cancer (151) and diffuse midline glioma (152).
Historically, the development of anti-GD2 or anti-GD3 vaccines has been tried without success. Glycolipids are poorly immunogenic and are not thought to be processed by antigen presenting cells or presented by MHC antigens. While lipids can be recognized by specialized CD1 (MHC class I-like molecules)–restricted T cells, each ganglioside does not have a unique type of lipid. In fact, the lipid tails can be heterogeneous within a single ganglioside (ranging in carbon chain length, oxidation and saturation state). Hence, only subtle differences exist between normal GM1 and tumor GD2 and GD3 carbohydrate heads, and their lipid tails can be shared or can exhibit heterogeneity across all gangliosides whether they are normal or TMGs. Thus, when using whole TMGs as immunogens, there are concerns with regards to tolerance, cross-reactivity or transient and ineffective immune reactions.
Notwithstanding the aforementioned concerns, initial attempts at developing GD2/GD3 vaccines used native GD2 or GD3 glycolipids chemically conjugated to carriers (153, 154). These were somewhat immunogenic and induced humoral responses that delayed tumor growth in mice via a complement-dependent cytotoxicity (CDC) mechanism. However, anti-ganglioside antibody titers were not long-lasting even after multiple immunizations, and there was no correlation between humoral titer and tumor therapeutic efficacy (147).
The Potential of γδ T Cells in Targeting the “Glycocode”
γδ T cells, expressing the gamma and delta chains of the T-cell receptor (TCR) coupled to the CD3 invariant signaling chains, are a subclass of T lymphocytes whose defining characteristic is their ability to display traits of both the innate and adaptive immune systems (155). They express a TCR whose engagement with its target mediates T cell activation. Human γδ T cell subsets—the subsets in other species will differ—are classified according to the Vδ chain in the TCR. Vδ(1-8) together with one of 6 different Vγ chains (2–5, 8, and 9) forms the mature TCR via V(D)J recombination. In this manner, they generate TCR diversity similarly to conventional αβ T cells.
On the other hand, the TCRs of the γδ type recognize qualitatively distinct antigens, with kinetics, antigen recognition mechanisms and tissue localization fundamentally distinct from αβ T cells. While some γδ T cells can found in circulation, with the Vγ9Vδ2 being the major subset corresponding to about 5% of total CD3+ cells in the periphery (156), the two other main γδ T cell subsets, Vδ1 and Vδ3, are predominantly tissue-resident. Like innate immune cells, γδ T cells recognize targets with broad patterns of pathogen-encoded or dysregulated-self signatures, as opposed to the specificity displayed by the αβ T cells.
Antigen Recognition by γδ T Cells
Unlike αβ T cells, γδ T cells do not rely on peptide presentation by the MHC complex of antigen-presenting cell to become activated. The precise mechanisms behind γδ T cell antigen recognition remains a field of intense research, complicated by the vast array of structurally diverse classes of self and non-self-ligands recognized by the γδ TCR (157). This includes soluble and membrane-bound proteins and peptides of a wide range of sizes, as well as non-protein targets such as phospholipids, non-peptidic antigens and carbohydrates.
Although often referred to as MHC-unrestricted, some of the most well-characterized targets of γδ-TCR include the non-classical class I MHC molecules. The MHC-I related molecules T10 and T22, found specifically in mice, were the first ligands whose binding to the γδ-TCR was confirmed biochemically (158, 159). Recognition of lipids presented by the CD1 family of MHC class I-like proteins was established a few years later (160–162) and is a key aspect of TME-related γδ T cell biology. Vδ1 and Vδ3 T cells can recognize the sphingolipid α-galactosylceramide (α-GalCer) presented by CD1d and, as a consequence, upregulate cytokine production characteristic of Th0 (i.e. IFNγ and IL-4), Th1 (i.e., IFNγ) and Th2 (i.e., IL-4) cells (163, 164). Recently, a third type of non-classical class I MHC molecule, the MHC-related protein 1 (MR1), was shown to be a target of γδ-TCR (165). This protein is involved in the presentation of microbial metabolites related to vitamin B2 biosynthesis and is known to stimulate a special subset of αβ T cells known as mucosal-associated invariant T cells (MAIT cells). Intriguingly, the resolved crystal structure of a γδTCR–MR1–antigen complex revealed a key difference to the previously proposed modes of TCR-ligand recognition. The γδ TCR was found to bind underneath the MR1 antigen-binding cleft, suggesting a new “antigen-agnostic” mode of TCR-target interaction, the biological implications of which are not yet understood.
In addition to targets presented as a part of non-classical MHC molecules, several types of non-peptidic antigens have been described to activate T cells with γδ TCRs. Tumor cell recognition, in particular, is enhanced by the ability of γδ T cells to recognize such antigens, which often are by-products of dysregulated tumor processes (and which do not bind MHC molecules). Phosphoantigens, or phosphorylated non-peptide antigens, are the classical example of this principle. The phosphoantigen isopentenyl pyrophosphate (IPP) accumulates in tumor cells due to the deregulated mevalonate pathway (166), and specifically and potently activates Vγ9Vδ2 T cells (167). Although phosphoantigens were originally thought to activate the γδ TCR directly, new evidence shows that this recognition requires the participation of Ig superfamily family members known as the butyrophilins. In addition to the previously implicated butyrophilin 3A1 (168–170), two recent studies identified butyrophilin 2A1 as key for the recognition of phosphoantigens by γδ T cells (171, 172). Rigau et al. propose a model in which the phosphoantigen production by a target cell modifies a complex composed of butyrophilin 3A1 and butyrophilin 2A1 causing it to co-bind and activate the Vγ9Vδ2 TCR.
In addition to TCR receptor-mediated activation, it should be emphasized that γδ T cells also express other activating receptors, such as the NK cell receptors (173, 174). Building on an early study showing susceptibility to carcinogenesis in the absence of γδ T cells (175), Strid et al. showed that activation of NKG2D receptor by ligand Rae-1 (known as MICA in humans) on tissue-resident Vγ5Vδ1 γδ T cells inhibits skin cancer in a mouse model (176). In addition to skin, the role of NKG2D for γδ T cell activation was further shown in peripheral Vγ9Vδ2 cells (177). In NK cells, the NKG2D receptor has previously been mentioned in this text as being disrupted by elevated levels of sialylation on tumor cells. Given its now known functional roles in γδ T cells, it is thus possible that the immunosuppressive effects of elevated cancer cell sialylation can extend to impaired γδ T cell function.
γδ T Cell Targeting of Carbohydrates in Tumor Gangliosides
The broad repertoire of targets recognizable by the γδ TCR as well as other receptors on the γδ T cells is suggestive of their potential use against specific carbohydrate targets on tumor cells, such as TMGs. However, this is an emergent field and concrete examples of such carbohydrate reactivity remain scarce.
The role of CD1d in γδ T cell activation provides an indirect example in which the spheres of γδ T cells and TMGs intersect. In one study, ovarian tumor-shed GD3 inhibited NKT-cell activation, with GD3 binding with high affinity to both human and murine CD1d. In vivo administration of GD3 suppressed α-GalCer-induced NKT cell activation in a dose-dependent manner, leading to the establishment of an immunosuppressive TME (94). While experimentally proven only in the context of NKT cells, it is possible that anti-GD3 blockade of the GD3-CD1d interaction would free up its recognition by, and subsequent activation of, γδ T cells, providing an additional therapeutic benefit. It should be noted that GD3-CD1d interaction can conversely have an immune activating effect. In melanoma, GD3 has been shown to activate NKT cells in a CD1d-dependent manner (178, 179).
Key to the potential of γδ T cells in targeting tumor-associated glycans is the fact that they do not require MHC presentation of antigens. This is relevant to tumor glycobiology because pure carbohydrates are typically not presented by MHC (180). While αβ T cells can recognize MHC-processed glycopeptides (peptides attached to a glycan), an early study determined the MHC-unrestricted carbohydrate specificity of γδ T cells (181). In addition, some MHC-restricted T-cell epitopes can be unaffected by glycosylation. An H-2Kb-restricted peptide retains an ability to be presented in its glycosylated form (hence, presentation is unaffected by peptide glycosylation) as the tethered carbohydrate fits in the central region of the TCR binding site. Hence, manipulation of γδ or αβ TCRs may yield previously unexploited strategies to target non-protein antigens in an MHC-unrestricted manner.
We recently reported the generation of synthetic GD2 and GD3 carbohydrate head-groups displayed on a multivalent polyamidoamine scaffold (PAMAM-GD2 and PAMAM-GD3). The PAMAM-GDs are lipid-free, water-soluble, inexpensive to produce, well-characterized chemically and structurally (including a crucial β-configuration at the first sugar), and identical to native carbohydrate head-groups on the surface of tumors (182, 183). These products (hereafter, called vaccines) are potent immunogens—when inoculated in mice, they stimulate B- and T cell immunity. The vaccines, as monotherapy, are therapeutic against four aggressive and metastatic syngeneic cancer models, significantly reducing primary tumors, metastatic burden, and importantly extending overall survival. Unexpectedly, this study revealed the expansion of γδ T cells mediated by a pure carbohydrate dendrimer. This occurs rapidly after vaccination in mice (independent of tumor presence) and in tumor-bearing mice (or upon tumor challenge) was followed by the expansion of the CD8+ T cells in vivo. Adoptive transfer of a relatively low number of the T cells isolated after vaccination is also therapeutic in tumor models (182).
The data support the notion that vaccination can expand and activate γδ T cells directly (and perhaps through APCs), which then bypass the immunosuppressive TME and become TILs (Figure 1). Expansion of γδ T cells is detected in tumor-bearing as well as in non-tumor-bearing mice. Hence while the vaccine may also block the immune-suppressive action of TMGs upon T cells this mechanism is unlikely to account for the initial expansion, but may be relevant to anti-tumor efficacy. The initial expansion of γδ T cells is followed by a second wave of expansion and recruitment of CD8+ effector TILs. The ability of γδ T cells to activate other T cell subsets has been shown previously (184, 185). It is possible that the second wave of CD8+ effector TILs recognize neoantigens presented, or shed, by injured or stressed tumor cells. We note that while in vivo T-cell memory generated by glycomimetic vaccines was not evaluated, the anti-TMG humoral immunity matures and class-switches from IgM to IgG, and is a surrogate marker of memory.
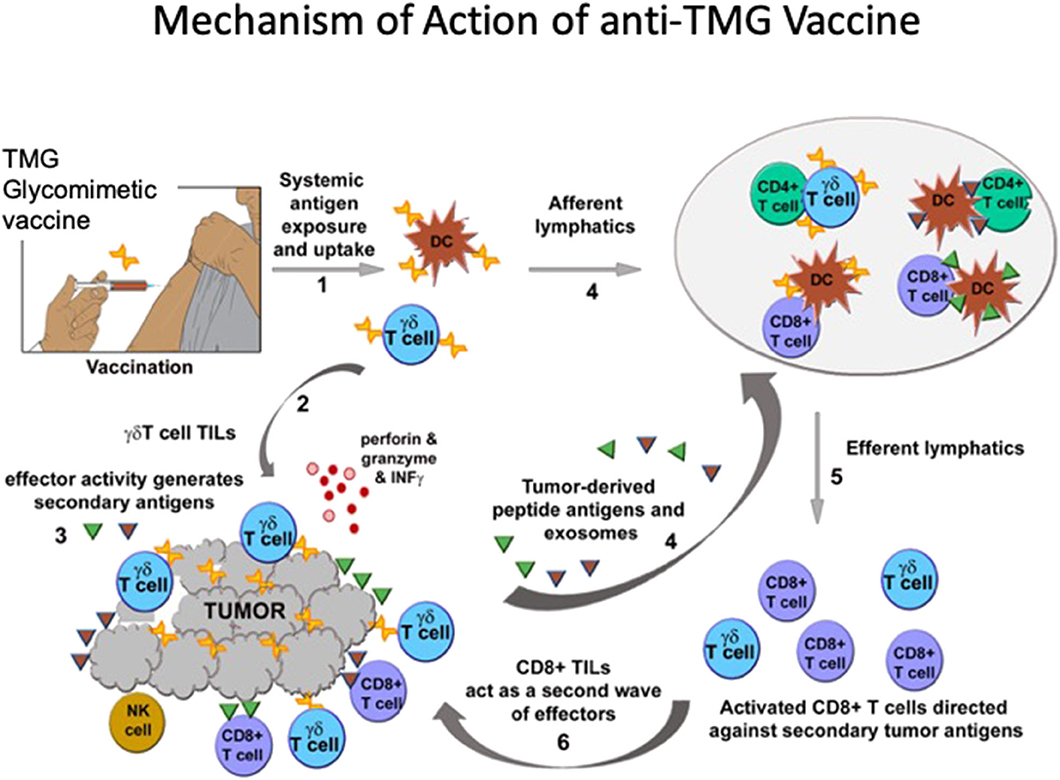
Figure 1. TMG glycomimetic vaccine mechanisms of immune activation. After systemic delivery of the vaccine antigens, (1) there is a rapid expansion of γδ T cells. It is unknown whether γδ TCRs expand by binding directly to the vaccine glycomimetic product, or whether the antigen is presented by DCs. Expansion of γδ T cells is independent of whether mice bear tumors expressing TMGs, so while the vaccine may also block the immune-suppressive action of TMGs upon T cells this mechanism is unlikely to account for the initial expansion. (2) In mice bearing TMG-expressing tumors, vaccination affords a significant increase in γδ T cells TILs. (3) The effector activity generates secondary antigens or neoantigens. (4) Putative neoantigens (not yet identified) circulate and are presented to CD8+ αβ T cells which (5, 6) expand as a second wave mainly comprising CD8+ T cells that also become TILs. The generation of in vivo T-cell memory in glycomimetic vaccines was not evaluated. However, anti-TMG humoral immunity (evaluated as a surrogate marker) matures and class-switches from IgM to IgG.
The mechanism of action of the PAMAM-GD2 and GD3 vaccines and the role of γδ T cells in mediating immunity against TMGs is paradigm-shifting, because virtually all previous experimental and clinical data using vaccines directed against TMGs have focused on humoral immunity rather than on cellular immunity. Such bias was perhaps motivated by the early promising results of using anti-TMG mAbs as therapeutic agents.
The γδ T cells are susceptible to PD-1–mediated inhibition (186, 187), and the tumor models where the vaccines were evaluated express high levels of PD-L1. The high therapeutic efficacy suggests that the vaccines partially overcome the inhibitory effects of PD-L1 upon γδ T cells. Ongoing studies are evaluating whether combination therapy with ICB might augment the anti-cancer effects of vaccines.
Adoptive transfer of T cells isolated from vaccinated mice resulted in the appearance of γδ T cells as TILs, and in a high therapeutic index. However, the study did not evaluate the sequence of γδ TCRs that were expanded, and did not address whether the glycomimetic vaccine products bind directly to the γδ TCRs or are presented via CD1, for example. Moreover, the antigens and the mechanism causing a second wave of CD8+ T cell expansion, whether it is γδ T cell dependent, and whether it is relevant therapeutically, are key for the proper development of a future cancer vaccine. The TMG glycomimetic cancer vaccine is an exciting approach that requires further evaluation of immune-mechanisms and connections between TMG and γδ T cells. Also, it is noteworthy that the concept of harnessing γδ T cells and targeting sugars for cancer therapy has been under examination for non-cancer pathologies ranging from anti-viral, anti-bacteria, and anti-parasitic therapy (188–191) to autoimmune diseases (192).
Therapeutic Targeting of γδ T Cells
The scientific literature regarding the clinical efficacy of γδ T cell therapy is overall positive, supporting the further exploration of their use in a clinical setting (156, 193). Studies performed thus far included patients with hematological malignancies (follicular lymphoma, multiple myeloma and acute myeloid leukemia), and non-hematological tumors, such as renal cell, breast and prostate cancer (194). Vδ1+ cells have shown promising results pre-clinically (195), and the infiltration of these cells correlated with necrotizing tumors and patient survival in melanoma (196). However, the bulk of clinical studies have used Vγ9Vδ2 T-cells due to their relatively high availability in the peripheral blood and their potential to be cultured, expanded and activated ex vivo. To activate the Vγ9Vδ2 T-cells, the butyrophilin-mediated reactivity of the Vγ9Vδ2 TCR to phosphoantigens can be exploited, using chemical compounds to elevate or mimic the expression of phosphoantigens either on tumor cells or on antigen-presenting cells in the TME. Such compounds include aminobisphosphonates (for example pamidronate and zoledronate) or synthetic phosphoantigen analogs (197, 198). The approach offers a useful tool for expansion, but is not necessarily a useful therapeutic approach, because the expanded Vγ9Vδ2 T cells are nearly monoclonal and are not specific for a desired antigen. Indeed, the clinical response in the trials conducted thus far has been minimal [reviewed in (199) and (200)]. Therefore, these experiments are an interrogation of γδ T cell biology, with some examination of safety parameters. Furthermore, the clinical data strongly point to the need for a combinatorial approach with other immune-based therapies for maximum efficacy (193, 201). Even with these limitations, the early results reported are encouraging (193).
CAR γδT cell Therapy
T cells engineered to express chimeric antigen receptors (CARs) comprise a branch of immunotherapy that combines the antigen specificity of monoclonal antibodies with the signaling motifs of receptors to promote the proliferation of cytotoxic effector T cells. CAR therapy has been successfully applied in several types of hematologic malignancies (202–204), and while translation to solid tumors has been somewhat limited there is some success reported with CARs in breast cancer (151) and diffuse midline glioma (152).
It is possible that engineered CAR T cells, just as naturally occurring T cells, are restricted by the immunosuppressive TME. This could impair T cell recruitment, function, and survival. In this context, γδ CAR T cells are an intriguing alternative target. Transduction of γδ T cells with CARs might direct their cytotoxic activity specifically against a tumor antigen, while retaining their other advantageous features such as the ability to cross-present antigen to αβ T cells. Moreover, a key advantage of the non-MHC restricted nature of γδ T cells is that CAR γδ T-cell preparations can be generated and expanded from pooled healthy donors.
To maximize the efficacy of the CAR therapy, an ideal antigen for CAR generation would need to be tumor-specific, highly and stably expressed by all malignant cells, and etiological to tumor development. Highlighting the therapeutic potential of TMGs, one of the first studies to engineer γδ CAR T cells used the GD2-antigen (205). The authors reported that GD2-CARs of both Vδ1 and Vδ2 subsets were expanded in sufficient numbers for clinical studies. The expression of the GD2-CAR by γδ T cells enhanced their innate cytotoxicity by directing its effects against GD2-expressing tumor cells. Further amplifying the anti-tumor immune response, expanded CAR-transduced Vδ2 cells retained the ability to internalize and cross-present tumor antigens to αβ T lymphocytes.
γδ T cells were originally thought to lack memory, and this was a concern in the design of CAR γδ T cell therapies. A lack of memory potentially translates into short-lived anti-tumor responses, but this may be overcome by using the CAR γδ T cell therapy in multiple treatment cycles. Encouragingly, recent data from mouse and human studies suggest that γδ T cells indeed have characteristics reminiscent of memory αβ T lymphocytes, promoting antigen-specific adaptive immunity (206–209). For example, in mice infected with B. pertussis, lung resident memory γδ T cells were shown to expand upon secondary infection with increased production of the cytokine, IL-17 (207). In humans, Vδ1+ and Vδ2+ γδ T cells are evidenced to promote microbial-specific adaptive immunity (208, 209).
Concluding Remarks
The concept of the “glycocode” (6, 210) poses that protein glycosylation—with sialic acids appearing to be key—regulates biological events that are crucial to immunity and cancer progression, comparable to, but beyond, the PD-1–type of checkpoint inhibition. This concept extends to the glycosylation of membrane and matrix proteins, mucins, and gangliosides, where TMGs represent a glyco-immune-checkpoint. Vaccines, antibodies, small molecules, soluble glycoproteins, enzymatic cleavage of sialic acids, or soluble competitors targeting the glyco-immune-checkpoint would be a promising approach to therapy (211).
It will be important to consider γδ T cell biology in the context of strategies targeting the glycocode and sialic acid-containing protein and ganglioside targets. Table 2 shows a comparison of the features of different cancer immunotherapies, including many that have a γδ T cell mechanism of action. Table 2 lists our view of their relative advantages and disadvantages. We present an overview of their current stage of development, which we view as a benchmark of the time that each approach has been in development (factoring time and investment of resources) and the degree of expectation of success. However, based on history, most approaches are expected to perhaps find a narrow niche or indication where they may be of utility. Unfortunately, most will either fail clinically, or will face difficult regulatory hurdles, or become untenable in the marketplace.
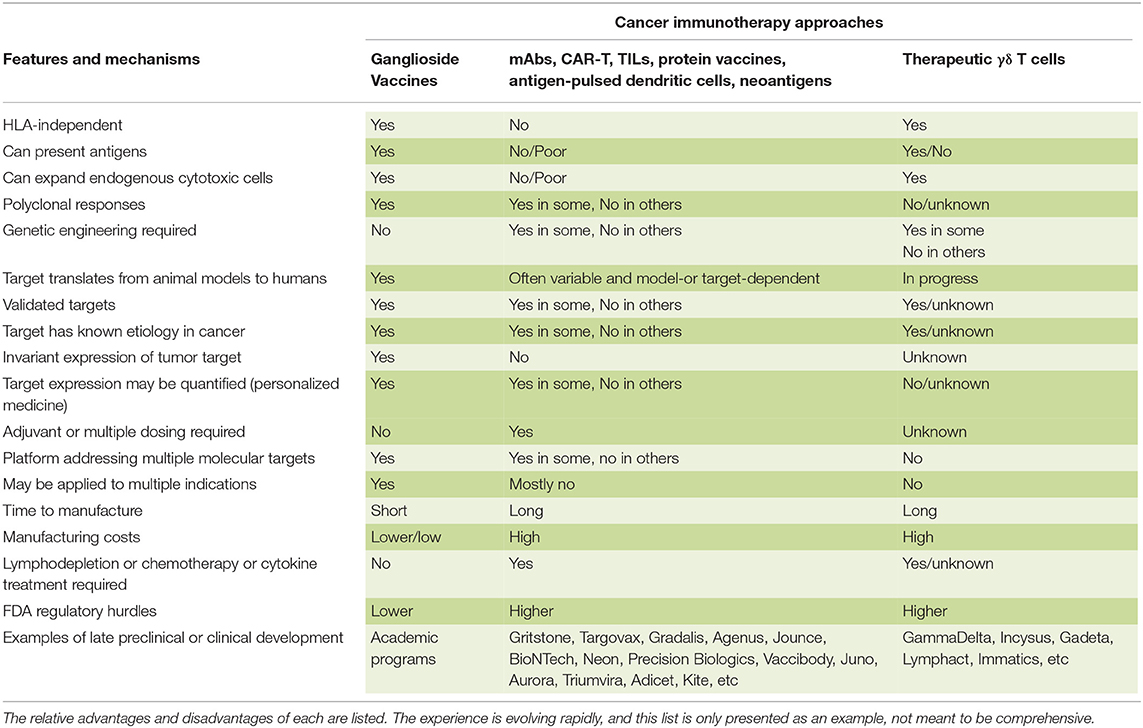
Table 2. Immunotherapeutic approaches involving γδ T cells or γδ TCRs.
Our work in developing glycomimetic vaccines surprisingly resulted in early activation of γδ T cells in vivo, and in high therapeutic efficacy in cancer. In addition to cancer, conceptually, this advance is applicable to therapies for other pathologies (e.g., antivirals) that could benefit from the activation of the innate and the adaptive immune systems by targeting sialic acids and other glycans.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
For this work HS received funding from the Canadian Institutes for Health Research (CIHR) (PJT-162291), the CIHR Proof-of-Principle Tier 1 (POP-1), and the GlycoNet Network of Centers of Excellence. SR was funded by the CIHR (PJT-162260), the McGill Interdisciplinary Initiative in Infection and Immunity (Mi4), and the Cole Foundation. CR was supported by a Canadian Institutes for Health Research Foundation Award (159912).
Conflict of Interest
HS is inventor in patents for anti-TMG vaccines.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Medzhitov R, Janeway CA Jr. Decoding the patterns of self and nonself by the innate immune system. Science. (2002) 296:298–300. doi: 10.1126/science.1068883
2. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. (2015) 15:486–99. doi: 10.1038/nri3862
3. Hargadon KM, Johnson CE, Williams CJ. Immune checkpoint blockade therapy for cancer: an overview of FDA-approved immune checkpoint inhibitors. Int Immunopharmacol. (2018) 62:29–39. doi: 10.1016/j.intimp.2018.06.001
4. Haslam A, Prasad V. Estimation of the percentage of US patients with cancer who are eligible for and respond to checkpoint inhibitor immunotherapy drugs. JAMA Netw Open. (2019) 2:e192535. doi: 10.1001/jamanetworkopen.2019.2535
5. Peixoto A, Relvas-Santos M, Azevedo R, Santos LL, Ferreira JA. Protein glycosylation and tumor microenvironment alterations driving cancer hallmarks. Front Oncol. (2019) 9:380. doi: 10.3389/fonc.2019.00380
6. RodrIguez E, Schetters STT, van Kooyk Y. The tumour glyco-code as a novel immune checkpoint for immunotherapy. Nat Rev Immunol. (2018) 18:204–11. doi: 10.1038/nri.2018.3
8. Ghazarian H, Idoni B, Oppenheimer SB. A glycobiology review: carbohydrates, lectins and implications in cancer therapeutics. Acta Histochem. (2011) 113:236–47. doi: 10.1016/j.acthis.2010.02.004
9. Rabinovich GA, van Kooyk Y, Cobb BA. Glycobiology of immune responses. Ann N Y Acad Sci. (2012) 1253:1–15. doi: 10.1111/j.1749-6632.2012.06492.x
10. Kiessling LL. Chemistry-driven glycoscience. Bioorg Med Chem. (2018) 26:5229–38. doi: 10.1016/j.bmc.2018.09.024
11. Pinho SS, Reis CA. Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer. (2015) 15:540–55. doi: 10.1038/nrc3982
12. Fuster MM, Esko JD. The sweet and sour of cancer: glycans as novel therapeutic targets. Nat Rev Cancer. (2005) 5:526–42. doi: 10.1038/nrc1649
13. Hakomori S. Tumor malignancy defined by aberrant glycosylation and sphingo(glyco)lipid metabolism. Cancer Res. (1996) 56:5309–18.
14. Bull C, Stoel MA, den Brok MH, Adema GJ. Sialic acids sweeten a tumor's life. Cancer Res. (2014) 74:3199–204. doi: 10.1158/0008-5472.CAN-14-0728
15. Gagnon M, Saragovi HU. Gangliosides: therapeutic agents or therapeutic targets? Expert Opin Ther Patents. (2002) 12:1215–23. doi: 10.1517/13543776.12.8.1215
16. Heimburg-Molinaro J, Lum M, Vijay G, Jain M, Almogren A, Rittenhouse-Olson K. Cancer vaccines and carbohydrate epitopes. Vaccine. (2011) 29:8802–26. doi: 10.1016/j.vaccine.2011.09.009
17. Sedlacek HH, Meesmann H, Seiler FR. Regression of spontaneous mammary tumors in dogs after injection of neuraminidase-treated tumor cells. Int J Cancer. (1975) 15:409–16. doi: 10.1002/ijc.2910150307
18. Currie GA, Bagshawe KD. The masking of antigens on trophoblast and cancer cells. Lancet. (1967) 1:708–10. doi: 10.1016/S0140-6736(67)92183-6
19. Cohen M, Elkabets M, Perlmutter M, Porgador A, Voronov E, Apte RN, et al. Sialylation of 3-methylcholanthrene-induced fibrosarcoma determines antitumor immune responses during immunoediting. J Immunol. (2010) 185:5869–78. doi: 10.4049/jimmunol.1001635
20. Wesener DA, Dugan A, Kiessling LL. Recognition of microbial glycans by soluble human lectins. Curr Opin Struct Biol. (2017) 44:168–78. doi: 10.1016/j.sbi.2017.04.002
21. Chen GY, Brown NK, Zheng P, Liu Y. Siglec-G/10 in self-nonself discrimination of innate and adaptive immunity. Glycobiology. (2014) 24:800–6. doi: 10.1093/glycob/cwu068
22. Pearce OM, Laubli H. Sialic acids in cancer biology and immunity. Glycobiology. (2016) 26:111–28. doi: 10.1093/glycob/cwv097
23. Macauley MS, Pfrengle F, Rademacher C, Nycholat CM, Gale AJ, von Drygalski A, et al. Antigenic liposomes displaying CD22 ligands induce antigen-specific B cell apoptosis. J Clin Invest. (2013) 123:3074–83. doi: 10.1172/JCI69187
24. Lanoue A, Batista FD, Stewart M, Neuberger MS. Interaction of CD22 with alpha2,6-linked sialoglycoconjugates: innate recognition of self to dampen B cell autoreactivity? Eur J Immunol. (2002) 32:348–55. doi: 10.1002/1521-4141(200202)32:2<348::AID-IMMU348>3.0.CO;2-5
25. Macauley MS, Paulson JC. Siglecs induce tolerance to cell surface antigens by BIM-dependent deletion of the antigen-reactive B cells. J Immunol. (2014) 193:4312–21. doi: 10.4049/jimmunol.1401723
26. Hudak JE, Canham SM, Bertozzi CR. Glycocalyx engineering reveals a Siglec-based mechanism for NK cell immunoevasion. Nat Chem Biol. (2014) 10:69–75. doi: 10.1038/nchembio.1388
27. Jandus C, Boligan KF, Chijioke O, Liu H, Dahlhaus M, Demoulins T, et al. Interactions between Siglec-7/9 receptors and ligands influence NK cell-dependent tumor immunosurveillance. J Clin Invest. (2014) 124:1810–20. doi: 10.1172/JCI65899
28. Falco M, Biassoni R, Bottino C, Vitale M, Sivori S, Augugliaro R, et al. Identification and molecular cloning of p75/AIRM1, a novel member of the sialoadhesin family that functions as an inhibitory receptor in human natural killer cells. J Exp Med. (1999) 190:793–802. doi: 10.1084/jem.190.6.793
29. Huang CH, Liao YJ, Fan TH, Chiou TJ, Lin YH, Twu YC. A developed NK-92MI cell line with Siglec-7(neg) phenotype exhibits high and sustainable cytotoxicity against leukemia cells. Int J Mol Sci. (2018) 19:1073. doi: 10.3390/ijms19041073
30. Beatson R, Tajadura-Ortega V, Achkova D, Picco G, Tsourouktsoglou TD, Klausing S, et al. The mucin MUC1 modulates the tumor immunological microenvironment through engagement of the lectin Siglec-9. Nat Immunol. (2016) 17:1273–81. doi: 10.1038/ni.3552
31. Takamiya R, Ohtsubo K, Takamatsu S, Taniguchi N, Angata T. The interaction between Siglec-15 and tumor-associated sialyl-Tn antigen enhances TGF-beta secretion from monocytes/macrophages through the DAP12-Syk pathway. Glycobiology. (2013) 23:178–87. doi: 10.1093/glycob/cws139
32. Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW, et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature. (2019) 572:392–6. doi: 10.1038/s41586-019-1456-0
33. Santegoets KCM, Gielen PR, Bull C, Schulte BM, Kers-Rebel ED, Kusters B, et al. Expression profiling of immune inhibitory Siglecs and their ligands in patients with glioma. Cancer Immunol Immunother. (2019) 68:937–49. doi: 10.1007/s00262-019-02332-w
34. Perdicchio M, Ilarregui JM, Verstege MI, Cornelissen LA, Schetters ST, Engels S, et al. Sialic acid-modified antigens impose tolerance via inhibition of T-cell proliferation and de novo induction of regulatory T cells. Proc Natl Acad Sci USA. (2016) 113:3329–34. doi: 10.1073/pnas.1507706113
35. Stanczak MA, Siddiqui SS, Trefny MP, Thommen DS, Boligan KF, von Gunten S, et al. Self-associated molecular patterns mediate cancer immune evasion by engaging Siglecs on T cells. J Clin Invest. (2018) 128:4912–23. doi: 10.1172/JCI120612
36. Haas Q, Boligan KF, Jandus C, Schneider C, Simillion C, Stanczak MA, et al. Siglec-9 regulates an effector memory CD8(+) T-cell subset that congregates in the melanoma tumor microenvironment. Cancer Immunol Res. (2019) 7:707–18. doi: 10.1158/2326-6066.CIR-18-0505
37. Wang J, Sun J, Liu LN, Flies DB, Nie X, Toki M, et al. Siglec-15 as an immune suppressor and potential target for normalization cancer immunotherapy. Nat Med. (2019) 25:656–66. doi: 10.1038/s41591-019-0374-x
38. Bull C, Boltje TJM, Wassink de Graaf AM, van Delft FL, den Brok MH, Adema GJ. Targeting aberrant sialylation in cancer cells using a fluorinated sialic acid analog impairs adhesion, migration, and in vivo tumor growth. Mol Cancer Ther. (2013) 12:1935–46. doi: 10.1158/1535-7163.MCT-13-0279
39. Bull C, Boltje TJ, van Dinther EA, Peters T, de Graaf AM, Leusen JH, et al. Targeted delivery of a sialic acid-blocking glycomimetic to cancer cells inhibits metastatic spread. ACS Nano. (2015) 9:733–45. doi: 10.1021/nn5061964
40. Rios A, Simmons RL. Immunospecific regression of various syngeneic mouse tumors in response to neuraminidase-treated tumor cells. J Natl Cancer Inst. (1973) 51:637–44.
41. Sedlacek HH, Seiler FR. Immunotherapy of neoplastic diseases with neuraminidase: contradictions, new aspects, revised concepts. Cancer Immunol Immunother. (1978) 5:153–63. doi: 10.1007/BF00199623
42. Sedlacek HH, Seiler FR, Schwick HG. Neuraminidase and tumor immunotherapy. Klinische Wochenschrift. (1977) 55:199–214. doi: 10.1007/BF01487712
43. Benjamini E, Rennick DM. Cancer immunotherapy: facts and fancy. CA Cancer J Clin. (1979) 29:362–70. doi: 10.3322/canjclin.29.6.362
44. Xiao H, Woods EC, Vukojicic P, Bertozzi CR. Precision glycocalyx editing as a strategy for cancer immunotherapy. Proc Natl Acad Sci USA. (2016) 113:10304–9. doi: 10.1073/pnas.1608069113
45. Pike LJ. Lipid rafts: bringing order to chaos. J Lipid Res. (2003) 44:655–67. doi: 10.1194/jlr.R200021-JLR200
46. Iwabuchi K, Zhang Y, Handa K, Withers DA, Sinay P, Hakomori S. Reconstitution of membranes simulating “glycosignaling domain” and their susceptibility to lyso-GM3. J Biol Chem. (2000) 275:15174–81. doi: 10.1074/jbc.275.20.15174
47. Svennerholm L. Chromatographlc separation of human brain gangliosides*. J Neurochem. (1963) 10:613–23. doi: 10.1111/j.1471-4159.1963.tb08933.x
48. Wiegandt H. The structure and the function of gangliosides. Angew Chem Int Ed Engl. (1968) 7:87–96. doi: 10.1002/anie.196800871
49. Xie X, Wu G, Lu ZH, Ledeen RW. Potentiation of a sodium-calcium exchanger in the nuclear envelope by nuclear GM1 ganglioside. J Neurochem. (2002) 81:1185–95. doi: 10.1046/j.1471-4159.2002.00917.x
50. Mutoh T, Tokuda A, Miyadai T, Hamaguchi M, Fujiki N. Ganglioside GM1 binds to the Trk protein and regulates receptor function. Proc Natl Acad Sci USA. (1995) 92:5087–91. doi: 10.1073/pnas.92.11.5087
51. Cavdarli S, Delannoy P, Groux-Degroote S. O-acetylated gangliosides as targets for cancer immunotherapy. Cells. (2020) 9:741. doi: 10.3390/cells9030741
52. Elkashef SM, Allison SJ, Sadiq M, Basheer HA, Ribeiro Morais G, Loadman PM, et al. Polysialic acid sustains cancer cell survival and migratory capacity in a hypoxic environment. Sci Rep. (2016) 6:33026. doi: 10.1038/srep33026
53. Hettmer S, Ladisch S, Kaucic K. Low complex ganglioside expression characterizes human neuroblastoma cell lines. Cancer Lett. (2005) 225:141–9. doi: 10.1016/j.canlet.2004.11.036
54. Mujoo K, Cheresh DA, Yang HM, Reisfeld RA. Disialoganglioside GD2 on human neuroblastoma cells: target antigen for monoclonal antibody-mediated cytolysis and suppression of tumor growth. Cancer Res. (1987) 47:1098–104.
55. Cheresh DA, Pierschbacher MD, Herzig MA, Mujoo K. Disialogangliosides GD2 and GD3 are involved in the attachment of human melanoma and neuroblastoma cells to extracellular matrix proteins. J Cell Biol. (1986) 102:688–96. doi: 10.1083/jcb.102.3.688
56. Kozireski-Chuback D, Wu G, Ledeen RW. Developmental appearance of nuclear GM1 in neurons of the central and peripheral nervous systems. Brain Res Dev Brain Res. (1999) 115:201–8. doi: 10.1016/S0165-3806(99)00062-0
57. Cavanna B, Carpo M, Pedotti R, Scarpini E, Meucci N, Allaria S, et al. Anti-GM2 IgM antibodies: clinical correlates and reactivity with a human neuroblastoma cell line. J Neuroimmunol. (1999) 94:157–64. doi: 10.1016/S0165-5728(98)00245-8
58. Vrionis FD, Wikstrand CJ, Fredman P, Mansson JE, Svennerholm L, Bigner DD. Five new epitope-defined monoclonal antibodies reactive with GM2 and human glioma and medulloblastoma cell lines. Cancer Res. (1989) 49:6645–51.
59. Mirkin BL, Clark SH, Zhang C. Inhibition of human neuroblastoma cell proliferation and EGF receptor phosphorylation by gangliosides GM1, GM3, GD1A and GT1B. Cell Prolif. (2002) 35:105–15. doi: 10.1046/j.1365-2184.2002.00228.x
60. Hildebrandt H, Becker C, Gluer S, Rosner H, Gerardy-Schahn R, Rahmann H. Polysialic acid on the neural cell adhesion molecule correlates with expression of polysialyltransferases and promotes neuroblastoma cell growth. Cancer Res. (1998) 58:779–84.
61. Valentiner U, Muhlenhoff M, Lehmann U, Hildebrandt H, Schumacher U. Expression of the neural cell adhesion molecule and polysialic acid in human neuroblastoma cell lines. Int J Oncol. (2011) 39:417–24. doi: 10.3892/ijo.2011.1038
62. Pukel CS, Lloyd KO, Travassos LR, Dippold WG, Oettgen HF, Old LJ. GD3, a prominent ganglioside of human melanoma. Detection and characterisation by mouse monoclonal antibody. J Exp Med. (1982) 155:1133–47. doi: 10.1084/jem.155.4.1133
63. Nicolae CD, Nicolae I. Antibodies against GM1 gangliosides associated with metastatic melanoma. Acta Dermatovenerol Croat. (2013) 21:86–92. Available online at: https://www.google.com/url?sa=t&rct=j&q=&esrc=s&source=web&cd=&cad=rja&uact=8&ved=2ahUKEwi89qDt_u7rAhWCVt8KHfdDCy0QFjAEegQIAxAB&url=https%3A%2F%2Fhrcak.srce.hr%2Ffile%2F157175&usg=AOvVaw2Dh0g9sCEJOlzGbsnZPvU7
64. Hoon DS, Ando I, Sviland G, Tsuchida T, Okun E, Morton DL, et al. Ganglioside GM2 expression on human melanoma cells correlates with sensitivity to lymphokine-activated killer cells. Int J Cancer. (1989) 43:857–62. doi: 10.1002/ijc.2910430520
65. Tsuchida T, Saxton RE, Morton DL, Irie RF. Gangliosides of human melanoma. Cancer. (1989) 63:1166–74. doi: 10.1002/1097-0142(19890315)63:6<1166::AID-CNCR2820630621>3.0.CO;2-5
66. Mennel HD, Bosslet K, Wiegandt H, Sedlacek HH, Bauer BL, Rodden AF. Expression of GD2-epitopes in human intracranial tumors and normal brain. Exp Toxicol Pathol. (1992) 44:317–24. doi: 10.1016/S0940-2993(11)80218-6
67. Lama G, Mangiola A, Proietti G, Colabianchi A, Angelucci C, A DA, et al. Progenitor/stem cell markers in brain adjacent to glioblastoma: GD3 ganglioside and NG2 proteoglycan expression. J Neuropathol Exp Neurol. (2016) 75:134–47. doi: 10.1093/jnen/nlv012
68. Ohkawa Y, Momota H, Kato A, Hashimoto N, Tsuda Y, Kotani N, et al. Ganglioside GD3 enhances invasiveness of gliomas by forming a complex with platelet-derived growth factor receptor alpha and yes kinase. J Biol Chem. (2015) 290:16043–58. doi: 10.1074/jbc.M114.635755
69. Birks SM, Danquah JO, King L, Vlasak R, Gorecki DC, Pilkington GJ. Targeting the GD3 acetylation pathway selectively induces apoptosis in glioblastoma. Neuro Oncol. (2011) 13:950–60. doi: 10.1093/neuonc/nor108
70. Hedberg KM, Mahesparan R, Read TA, Tysnes BB, Thorsen F, Visted T, et al. The glioma-associated gangliosides 3'-isoLM1, GD3 and GM2 show selective area expression in human glioblastoma xenografts in nude rat brains. Neuropathol Appl Neurobiol. (2001) 27:451–64. doi: 10.1046/j.1365-2990.2001.00353.x
71. Fujimoto Y, Izumoto S, Suzuki T, Kinoshita M, Kagawa N, Wada K, et al. Ganglioside GM3 inhibits proliferation and invasion of glioma. J Neurooncol. (2005) 71:99–106. doi: 10.1007/s11060-004-9602-3
72. Noll EN, Lin J, Nakatsuji Y, Miller RH, Black PM. GM3 as a novel growth regulator for human gliomas. Exp Neurol. (2001) 168:300–9. doi: 10.1006/exnr.2000.7603
73. Bassi R, Viani P, Giussani P, Riboni L, Tettamanti G. GM3 ganglioside inhibits endothelin-1-mediated signal transduction in C6 glioma cells. FEBS Lett. (2001) 507:101–4. doi: 10.1016/S0014-5793(01)02966-0
74. Trouillas J, Daniel L, Guigard MP, Tong S, Gouvernet J, Jouanneau E, et al. Polysialylated neural cell adhesion molecules expressed in human pituitary tumors and related to extrasellar invasion. J Neurosurg. (2003) 98:1084–93. doi: 10.3171/jns.2003.98.5.1084
75. Brezicka FT, Olling S, Nilsson O, Bergh J, Holmgren J, Sorenson S, et al. Immunohistological detection of fucosyl-GM1 ganglioside in human lung cancer and normal tissues with monoclonal antibodies. Cancer Res. (1989) 49:1300–5.
76. Yamada T, Bando H, Takeuchi S, Kita K, Li Q, Wang W, et al. Genetically engineered humanized anti-ganglioside GM2 antibody against multiple organ metastasis produced by GM2-expressing small-cell lung cancer cells. Cancer Sci. (2011) 102:2157–63. doi: 10.1111/j.1349-7006.2011.02093.x
77. Blanco R, Dominguez E, Morales O, Blanco D, Martinez D, Rengifo CE, et al. Prognostic significance of N-Glycolyl GM3 ganglioside expression in non-small cell lung carcinoma patients: new evidences. Patholog Res Int. (2015) 2015:132326. doi: 10.1155/2015/132326
78. Hayashi N, Chiba H, Kuronuma K, Go S, Hasegawa Y, Takahashi M, et al. Detection of N-glycolyated gangliosides in non-small-cell lung cancer using GMR8 monoclonal antibody. Cancer Sci. (2013) 104:43–7. doi: 10.1111/cas.12027
79. Tanaka F, Otake Y, Nakagawa T, Kawano Y, Miyahara R, Li M, et al. Prognostic significance of polysialic acid expression in resected non-small cell lung cancer. Cancer Res. (2001) 61:1666–70. Available online at: https://cancerres.aacrjournals.org/content/canres/61/4/1666.full.pdf
80. Tanaka F, Otake Y, Nakagawa T, Kawano Y, Miyahara R, Li M, et al. Expression of polysialic acid and STX, a human polysialyltransferase, is correlated with tumor progression in non-small cell lung cancer. Cancer Res. (2000) 60:3072–80. Available online at: https://cancerres.aacrjournals.org/content/60/11/3072.full-text.pdf
81. Liang JX, Liang Y, Gao W. Clinicopathological and prognostic significance of sialyl Lewis X overexpression in patients with cancer: a meta-analysis. Onco Targets Ther. (2016) 9:3113–25. doi: 10.2147/OTT.S102389
82. Natoni A, Macauley MS, O'Dwyer ME. Targeting selectins and their ligands in cancer. Front Oncol. (2016) 6:93. doi: 10.3389/fonc.2016.00093
83. Yoshida S, Fukumoto S, Kawaguchi H, Sato S, Ueda R, Furukawa K. Ganglioside GD2 in small cell lung cancer cell lines: enhancement of cell proliferation and mediation of apoptosis. Cancer Res. (2001) 61:4244–52. Available online at: https://cancerres.aacrjournals.org/content/61/10/4244.full-text.pdf
84. Watarai S, Kiura K, Shigeto R, Shibayama T, Kimura I, Yasuda T. Establishment of monoclonal antibodies specific for ganglioside GM1: detection of ganglioside GM1 in small cell lung carcinoma cell lines and tissues. J Biochem. (1994) 116:948–54. doi: 10.1093/oxfordjournals.jbchem.a124651
85. Liang YJ, Ding Y, Levery SB, Lobaton M, Handa K, Hakomori SI. Differential expression profiles of glycosphingolipids in human breast cancer stem cells vs. cancer non-stem cells. Proc Natl Acad Sci USA. (2013) 110:4968–73. doi: 10.1073/pnas.1302825110
86. Steenackers A, Vanbeselaere J, Cazet A, Bobowski M, Rombouts Y, Colomb F, et al. Accumulation of unusual gangliosides G(Q3) and G(P3) in breast cancer cells expressing the G(D3) synthase. Molecules. (2012) 17:9559–72. doi: 10.3390/molecules17089559
87. Bobowski M, Vincent A, Steenackers A, Colomb F, Van Seuningen I, Julien S, et al. Estradiol represses the G(D3) synthase gene ST8SIA1 expression in human breast cancer cells by preventing NFkappaB binding to ST8SIA1 promoter. PLoS ONE. (2013) 8:e62559. doi: 10.1371/journal.pone.0062559
88. Wang X, Li X, Zeng YN, He F, Yang XM, Guan F. Enhanced expression of polysialic acid correlates with malignant phenotype in breast cancer cell lines and clinical tissue samples. Int J Mol Med. (2016) 37:197–206. doi: 10.3892/ijmm.2015.2395
89. Das T, Sa G, Hilston C, Kudo D, Rayman P, Biswas K, et al. GM1 and tumor necrosis factor-alpha, overexpressed in renal cell carcinoma, synergize to induce T-cell apoptosis. Cancer Res. (2008) 68:2014–23. doi: 10.1158/0008-5472.CAN-07-6037
90. Biswas K, Richmond A, Rayman P, Biswas S, Thornton M, Sa G, et al. GM2 expression in renal cell carcinoma: potential role in tumor-induced T-cell dysfunction. Cancer Res. (2006) 66:6816–25. doi: 10.1158/0008-5472.CAN-06-0250
91. Sakakibara N, Gasa S, Kamio K, Makita A, Nonomura K, Togashi M, et al. Distinctive glycolipid patterns in Wilms' tumor and renal cell carcinoma. Cancer Lett. (1991) 57:187–92. doi: 10.1016/0304-3835(91)90155-B
92. Borzym-Kluczyk M, Radziejewska I, Cechowska-Pasko M. Increased expression of MUC1 and sialyl Lewis antigens in different areas of clear renal cell carcinoma. Clin Exp Nephrol. (2015) 19:732–7. doi: 10.1007/s10157-014-1013-y
93. Ravindranath MH, Muthugounder S, Presser N, Selvan SR, Santin AD, Bellone S, et al. Immunogenic gangliosides in human ovarian carcinoma. Biochem Biophys Res Commun. (2007) 353:251–8. doi: 10.1016/j.bbrc.2006.12.001
94. Webb TJ, Li X, Giuntoli RL 2nd, Lopez PH, Heuser C, Schnaar RL, et al. Molecular identification of GD3 as a suppressor of the innate immune response in ovarian cancer. Cancer Res. (2012) 72:3744–52. doi: 10.1158/0008-5472.CAN-11-2695
95. Prinetti A, Aureli M, Illuzzi G, Prioni S, Nocco V, Scandroglio F, et al. GM3 synthase overexpression results in reduced cell motility and in caveolin-1 upregulation in human ovarian carcinoma cells. Glycobiology. (2010) 20:62–77. doi: 10.1093/glycob/cwp143
96. Chang HR, Cordon-Cardo C, Houghton AN, Cheung NK, Brennan MF. Expression of disialogangliosides GD2 and GD3 on human soft tissue sarcomas. Cancer. (1992) 70:633–8.
97. Heiner JP, Miraldi F, Kallick S, Makley J, Neely J, Smith-Mensah WH, et al. Localization of GD2-specific monoclonal antibody 3F8 in human osteosarcoma. Cancer Res. (1987) 47:5377–81.
98. Shibuya H, Hamamura K, Hotta H, Matsumoto Y, Nishida Y, Hattori H, et al. Enhancement of malignant properties of human osteosarcoma cells with disialyl gangliosides GD2/GD3. Cancer Sci. (2012) 103:1656–64. doi: 10.1111/j.1349-7006.2012.02344.x
99. Dobrenkov K, Ostrovnaya I, Gu J, Cheung IY, Cheung NK. Oncotargets GD2 and GD3 are highly expressed in sarcomas of children, adolescents, young adults. Pediatr Blood Cancer. (2016) 63:1780–5. doi: 10.1002/pbc.26097
100. Lipinski M, Braham K, Philip I, Wiels J, Philip T, Goridis C, et al. Neuroectoderm-associated antigens on Ewing's sarcoma cell lines. Cancer Res. (1987) 47:183–7.
101. Kailayangiri S, Altvater B, Meltzer J, Pscherer S, Luecke A, Dierkes C, et al. The ganglioside antigen G(D2) is surface-expressed in Ewing sarcoma and allows for MHC-independent immune targeting. Br J Cancer. (2012) 106:1123–33. doi: 10.1038/bjc.2012.57
102. Scursoni AM, Galluzzo L, Camarero S, Lopez J, Lubieniecki F, Sampor C, et al. Detection of N-glycolyl GM3 ganglioside in neuroectodermal tumors by immunohistochemistry: an attractive vaccine target for aggressive pediatric cancer. Clin Dev Immunol. (2011) 2011:245181. doi: 10.1155/2011/245181
103. Modak S, Gerald W, Cheung NK. Disialoganglioside GD2 and a novel tumor antigen: potential targets for immunotherapy of desmoplastic small round cell tumor. Med Pediatr Oncol. (2002) 39:547–51. doi: 10.1002/mpo.10151
104. Portoukalian J, David MJ, Gain P, Richard M. Shedding of GD2 ganglioside in patients with retinoblastoma. Int J Cancer. (1993) 53:948–51. doi: 10.1002/ijc.2910530614
105. Torbidoni AV, Scursoni A, Camarero S, Segatori V, Gabri M, Alonso D, et al. Immunoreactivity of the 14F7 Mab raised against N-Glycolyl GM3 Ganglioside in retinoblastoma tumours. Acta Ophthalmol. (2015) 93:e294–300. doi: 10.1111/aos.12578
106. Scursoni AM, Galluzzo L, Camarero S, Pozzo N, Gabri MR, de Acosta CM, et al. Detection and characterization of N-glycolyated gangliosides in Wilms tumor by immunohistochemistry. Pediatr Dev Pathol. (2010) 13:18–23. doi: 10.2350/08-10-0544.1
107. Roth J, Zuber C, Wagner P, Blaha I, Bitter-Suermann D, Heitz PU. Presence of the long chain form of polysialic acid of the neural cell adhesion molecule in Wilms' tumor. Identification of a cell adhesion molecule as an oncodevelopmental antigen and implications for tumor histogenesis. Am J Pathol. (1988) 133:227–40.
108. Komminoth P, Roth J, Saremaslani P, Matias-Guiu X, Wolfe HJ, Heitz PU. Polysialic acid of the neural cell adhesion molecule in the human thyroid: a marker for medullary thyroid carcinoma and primary C-cell hyperplasia. An immunohistochemical study on 79 thyroid lesions. Am J Surg Pathol. (1994) 18:399–411. doi: 10.1097/00000478-199404000-00008
109. Zhang S, Cordon-Cardo C, Zhang HS, Reuter VE, Adluri S, Hamilton WB, et al. Selection of tumor antigens as targets for immune attack using immunohistochemistry: I. Focus on gangliosides. Int J Cancer. (1997) 73:42–9.
110. Guerrera M, Ladisch S. N-butyldeoxynojirimycin inhibits murine melanoma cell ganglioside metabolism and delays tumor onset. Cancer Lett. (2003) 201:31–40. doi: 10.1016/S0304-3835(03)00459-2
111. Battula VL, Shi Y, Evans KW, Wang RY, Spaeth EL, Jacamo RO, et al. Ganglioside GD2 identifies breast cancer stem cells and promotes tumorigenesis. J Clin Invest. (2012) 122:2066–78. doi: 10.1172/JCI59735
112. Sarkar TR, Battula VL, Werden SJ, Vijay GV, Ramirez-Pena EQ, Taube JH, et al. GD3 synthase regulates epithelial-mesenchymal transition and metastasis in breast cancer. Oncogene. (2015) 34:2958–67. doi: 10.1038/onc.2014.245
113. Ledeen R, Wu G. GM1 in the nuclear envelope regulates nuclear calcium through association with a nuclear sodium-calcium exchanger. J Neurochem. (2007) 103(Suppl. 1):126–34. doi: 10.1111/j.1471-4159.2007.04722.x
114. Sano R, Annunziata I, Patterson A, Moshiach S, Gomero E, Opferman J, et al. GM1-ganglioside accumulation at the mitochondria-associated ER membranes links ER stress to Ca(2+)-dependent mitochondrial apoptosis. Mol Cell. (2009) 36:500–11. doi: 10.1016/j.molcel.2009.10.021
115. Lauc G, Heffer-Lauc M. Shedding and uptake of gangliosides and glycosylphosphatidylinositol-anchored proteins. Biochim Biophys Acta. (2006) 1760:584–602. doi: 10.1016/j.bbagen.2005.11.014
116. Ladisch S, Kitada S, Hays EF. Gangliosides shed by tumor cells enhance tumor formation in mice. J Clin Invest. (1987) 79:1879–82. doi: 10.1172/JCI113031
117. McKallip R, Li R, Ladisch S. Tumor gangliosides inhibit the tumor-specific immune response. J Immunol. (1999) 163:3718–26.
118. Lu P, Sharom FJ. Immunosuppression by YAC-1 lymphoma: role of shed gangliosides. Cell Immunol. (1996) 173:22–32. doi: 10.1006/cimm.1996.0248
119. Krengel U, Bousquet PA. Molecular recognition of gangliosides and their potential for cancer immunotherapies. Front Immunol. (2014) 5:325. doi: 10.3389/fimmu.2014.00325
120. Tong W, Maira M, Gagnon M, Saragovi HU. Ligands binding to cell surface ganglioside GD2 cause Src-dependent activation of N-Methyl-D-aspartate receptor signaling and changes in cellular morphology. PLoS ONE. (2015) 10:e0134255. doi: 10.1371/journal.pone.0134255
121. Cazet A, Bobowski M, Rombouts Y, Lefebvre J, Steenackers A, Popa I, et al. The ganglioside G(D2) induces the constitutive activation of c-Met in MDA-MB-231 breast cancer cells expressing the G(D3) synthase. Glycobiology. (2012) 22:806–16. doi: 10.1093/glycob/cws049
122. Aixinjueluo W, Furukawa K, Zhang Q, Hamamura K, Tokuda N, Yoshida S, et al. Mechanisms for the apoptosis of small cell lung cancer cells induced by anti-GD2 monoclonal antibodies: roles of anoikis. J Biol Chem. (2005) 280:29828–36. doi: 10.1074/jbc.M414041200
123. Dippold WG, Knuth A, Meyer zum Buschenfelde KH. Inhibition of human melanoma cell growth in vitro by monoclonal anti-GD3-ganglioside antibody. Cancer Res. (1984) 44:806–10.
124. Liu Y, Wondimu A, Yan S, Bobb D, Ladisch S. Tumor gangliosides accelerate murine tumor angiogenesis. Angiogenesis. (2014) 17:563–71. doi: 10.1007/s10456-013-9403-4
125. Koochekpour S, Merzak A, Pilkington GJ. Vascular endothelial growth factor production is stimulated by gangliosides and TGF-β isoforms in human glioma cells in vitro. Cancer Lett. (1996) 102:209–15. doi: 10.1016/0304-3835(96)04161-4
126. Tringali C, Silvestri I, Testa F, Baldassari P, Anastasia L, Mortarini R, et al. Molecular subtyping of metastatic melanoma based on cell ganglioside metabolism profiles. BMC Cancer. (2014) 14:560. doi: 10.1186/1471-2407-14-560
127. Agarwal MK, Neter E. Effect of selected lipids and surfactants on immunogenicity of several bacterial antigens. J Immunol. (1971) 107:1448–56.
128. Ladisch S, Gillard B, Wong C, Ulsh L. Shedding and immunoregulatory activity of YAC-1 lymphoma cell gangliosides. Cancer Res. (1983) 43:3808–13.
129. Rapoport E, Mikhalyov I, Zhang J, Crocker P, Bovin N. Ganglioside binding pattern of CD33-related siglecs. Bioorg Med Chem Lett. (2003) 13:675–8. doi: 10.1016/S0960-894X(02)00998-8
130. Nicoll G, Avril T, Lock K, Furukawa K, Bovin N, Crocker PR. Ganglioside GD3 expression on target cells can modulate NK cell cytotoxicity via siglec-7-dependent and -independent mechanisms. Eur J Immunol. (2003) 33:1642–8. doi: 10.1002/eji.200323693
131. Ito A, Handa K, Withers DA, Satoh M, Hakomori S. Binding specificity of siglec7 to disialogangliosides of renal cell carcinoma: possible role of disialogangliosides in tumor progression. FEBS Lett. (2001) 504:82–6. doi: 10.1016/S0014-5793(01)02734-X
132. Yamaji T, Teranishi T, Alphey MS, Crocker PR, Hashimoto Y. A small region of the natural killer cell receptor, Siglec-7, is responsible for its preferred binding to alpha 2,8-disialyl and branched alpha 2,6-sialyl residues. A comparison with Siglec-9. J Biol Chem. (2002) 277:6324–32. doi: 10.1074/jbc.M110146200
133. Lu P, Sharom FJ. Gangliosides are potent immunosuppressors of IL-2-mediated T-cell proliferation in a low protein environment. Immunology. (1995) 86:356–63.
134. Peguet-Navarro J, Sportouch M, Popa I, Berthier O, Schmitt D, Portoukalian J. Gangliosides from human melanoma tumors impair dendritic cell differentiation from monocytes and induce their apoptosis. J Immunol. (2003) 170:3488–94. doi: 10.4049/jimmunol.170.7.3488
135. Heitger A, Ladisch S. Gangliosides block antigen presentation by human monocytes. Biochim Biophys Acta. (1996) 1303:161–8. doi: 10.1016/0005-2760(96)00091-4
136. Wondimu A, Liu Y, Su Y, Bobb D, Ma JS, Chakrabarti L, et al. Gangliosides drive the tumor infiltration and function of myeloid-derived suppressor cells. Cancer Res. (2014) 74:5449–57. doi: 10.1158/0008-5472.CAN-14-0927
137. Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, et al. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res. (2009) 15:5323–37. doi: 10.1158/1078-0432.CCR-09-0737
138. Kramer K, Gerald WL, Kushner BH, Larson SM, Hameed M, Cheung NK. Disaloganglioside GD2 loss following monoclonal antibody therapy is rare in neuroblastoma. Med Pediatr Oncol. (2001) 36:194–6. doi: 10.1002/1096-911X(20010101)36:1<194::AID-MPO1046>3.0.CO;2-B
139. Poon VI, Roth M, Piperdi S, Geller D, Gill J, Rudzinski ER, et al. Ganglioside GD2 expression is maintained upon recurrence in patients with osteosarcoma. Clin Sarcoma Res. (2015) 5:4. doi: 10.1186/s13569-014-0020-9
140. Doronin II, Vishnyakova PA, Kholodenko IV, Ponomarev ED, Ryazantsev DY, et al. Ganglioside GD2 in reception and transduction of cell death signal in tumor cells. BMC Cancer. (2014) 14:295. doi: 10.1186/1471-2407-14-295
141. Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, et al. Children's Oncology, Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med. (2010) 363:1324–34. doi: 10.1056/NEJMoa0911123
142. Saleh MN, Khazaeli MB, Wheeler RH, Allen L, Tilden AB, Grizzle W, et al. Phase I trial of the chimeric anti-GD2 monoclonal antibody ch14.18 in patients with malignant melanoma. Hum Antibodies Hybridomas. (1992) 3:19–24. doi: 10.3233/HAB-1992-3104
143. Handgretinger R, Anderson K, Lang P, Dopfer R, Klingebiel T, Schrappe M, et al. A phase I study of human/mouse chimeric antiganglioside GD2 antibody ch14.18 in patients with neuroblastoma. Eur J Cancer. (1995) 31A:261–7. doi: 10.1016/0959-8049(94)00413-Y
144. Cheung NK, Neely JE, Landmeier B, Nelson D, Miraldi F. Targeting of ganglioside GD2 monoclonal antibody to neuroblastoma. J Nucl Med. (1987) 28:1577–83.
145. McCaffery M, Yao TJ, Williams L, Livingston PO, Houghton AN, Chapman PB. Immunization of melanoma patients with BEC2 anti-idiotypic monoclonal antibody that mimics GD3 ganglioside: enhanced immunogenicity when combined with adjuvant. Clin Cancer Res. (1996) 2:679–86.
146. Bajorin DF, Chapman PB, Wong G, Coit DG, Kunicka J, Dimaggio J, et al. Phase I evaluation of a combination of monoclonal antibody R24 and interleukin 2 in patients with metastatic melanoma. Cancer Res. (1990) 50:7490–5.
147. Navid F, Santana VM, Barfield RC. Anti-GD2 antibody therapy for GD2-expressing tumors. Curr Cancer Drug Targets. (2010) 10:200–9. doi: 10.2174/156800910791054167
148. Keyel ME, Reynolds CP. Spotlight on dinutuximab in the treatment of high-risk neuroblastoma: development and place in therapy. Biologics. (2019) 13:1–12. doi: 10.2147/BTT.S114530
149. Esser R, Muller T, Stefes D, Kloess S, Seidel D, Gillies SD, et al. NK cells engineered to express a GD2 -specific antigen receptor display built-in ADCC-like activity against tumour cells of neuroectodermal origin. J Cell Mol Med. (2012) 16:569–81. doi: 10.1111/j.1582-4934.2011.01343.x
150. Gargett T, Yu W, Dotti G, Yvon ES, Christo SN, Hayball JD, et al. GD2-specific CAR T cells undergo potent activation and deletion following antigen encounter but can be protected from activation-induced cell death by PD-1 blockade. Mol Ther. (2016) 24:1135–49. doi: 10.1038/mt.2016.63
151. Seitz CM, Schroeder S, Knopf P, Krahl AC, Hau J, Schleicher S, et al. GD2-targeted chimeric antigen receptor T cells prevent metastasis formation by elimination of breast cancer stem-like cells. Oncoimmunology. (2020) 9:1683345. doi: 10.1080/2162402X.2019.1683345
152. Mount CW, Majzner RG, Sundaresh S, Arnold EP, Kadapakkam M, Haile S, et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M(+) diffuse midline gliomas. Nat Med. (2018) 24:572–9. doi: 10.1038/s41591-018-0006-x
153. Chapman PB, Morrisey D, Panageas KS, Williams L, Lewis JJ, Israel RJ, et al. Vaccination with a bivalent GM2 and GD2 ganglioside conjugate vaccine: a trial comparing doses of GD2-keyhole limpet hemocyanin. Clin Cancer Res. (2000) 6:4658–62. Available online at: https://clincancerres.aacrjournals.org/content/6/12/4658.full-text.pdf
154. Ragupathi G, Livingston PO, Hood C, Gathuru J, Krown SE, Chapman PB, et al. Consistent antibody response against ganglioside GD2 induced in patients with melanoma by a GD2 lactone-keyhole limpet hemocyanin conjugate vaccine plus immunological adjuvant QS-21. Clin Cancer Res. (2003) 9:5214–20. Available online at: https://clincancerres.aacrjournals.org/content/9/14/5214.full-text.pdf
155. Vantourout P, Hayday A. Six-of-the-best: unique contributions of γδ T cells to immunology. Nat Rev Immunol. (2013) 13:88–100. doi: 10.1038/nri3384
156. Hoeres T, Smetak M, Pretscher D, Wilhelm M. Improving the efficiency of Vγ9Vδ2 T-cell immunotherapy in cancer. Front Immunol. (2018) 9:800. doi: 10.3389/fimmu.2018.00800
157. Born WK, Kemal Aydintug M, O'Brien RL. Diversity of γδ T-cell antigens. Cell Mol Immunol. (2013) 10:13–20. doi: 10.1038/cmi.2012.45
158. Crowley MP, Fahrer AM, Baumgarth N, Hampl J, Gutgemann I, Teyton L, et al. A population of murine γδ T cells that recognize an inducible MHC class Ib molecule. Science. (2000) 287:314–6. doi: 10.1126/science.287.5451.314
159. Wingren C, Crowley MP, Degano M, Chien Y, Wilson IA. Crystal structure of a γδ T cell receptor ligand T22: a truncated MHC-like fold. Science. (2000) 287:310–4. doi: 10.1126/science.287.5451.310
160. Bai L, Picard D, Anderson B, Chaudhary V, Luoma A, Jabri B, et al. The majority of CD1d-sulfatide-specific T cells in human blood use a semiinvariant Vδ1 TCR. Eur J Immunol. (2012) 42:2505–10. doi: 10.1002/eji.201242531
161. Luoma AM, Castro CD, Mayassi T, Bembinster LA, Bai L, Picard D, et al. Crystal structure of Vδ1 T cell receptor in complex with CD1d-sulfatide shows MHC-like recognition of a self-lipid by human γδ T cells. Immunity. (2013) 39:1032–42. doi: 10.1016/j.immuni.2013.11.001
162. Agea E, Russano A, Bistoni O, Mannucci R, Nicoletti I, Corazzi L, et al. Human CD1-restricted T cell recognition of lipids from pollens. J Exp Med. (2005) 202:295–308. doi: 10.1084/jem.20050773
163. Uldrich AP, Le Nours J, Pellicci DG, Gherardin NA, McPherson KG, Lim RT, et al. CD1d-lipid antigen recognition by the γδ TCR. Nat Immunol. (2013) 14:1137–45. doi: 10.1038/ni.2713
164. Mangan BA, Dunne MR, O'Reilly VP, Dunne PJ, Exley MA, O'Shea D, et al. Cutting edge: CD1d restriction and Th1/Th2/Th17 cytokine secretion by human Vδ3 T cells. J Immunol. (2013) 191:30–4. doi: 10.4049/jimmunol.1300121
165. Le Nours J, Gherardin NA, Ramarathinam SH, Awad W, Wiede F, Gully BS, et al. A class of γδ T cell receptors recognize the underside of the antigen-presenting molecule MR1. Science. (2019) 366:1522–7. doi: 10.1126/science.aav3900
166. Tanaka Y, Morita CT, Tanaka Y, Nieves E, Brenner MB, Bloom BR. Natural and synthetic non-peptide antigens recognized by human γδ T cells. Nature. (1995) 375:155–8. doi: 10.1038/375155a0
167. Gober HJ, Kistowska M, Angman L, Jeno P, Mori L, de Libero G. Human T cell receptor γδ cells recognize endogenous mevalonate metabolites in tumor cells. J Exp Med. (2003) 197:163–8. doi: 10.1084/jem.20021500
168. Harly C, Guillaume Y, Nedellec S, Peigne CM, Monkkonen H, Monkkonen J, et al. Key implication of CD277/butyrophilin-3 (BTN3A) in cellular stress sensing by a major human γδ T-cell subset. Blood. (2012) 120:2269–79. doi: 10.1182/blood-2012-05-430470
169. Sandstrom A, Peigne CM, Leger A, Crooks JE, Konczak F, Gesnel MC, et al. The intracellular B30.2 domain of butyrophilin 3A1 binds phosphoantigens to mediate activation of human Vγ9Vδ2 T cells. Immunity. (2014) 40:490–500. doi: 10.1016/j.immuni.2014.03.003
170. Willcox CR, Vantourout P, Salim M, Zlatareva I, Melandri D, Zanardo L, et al. Butyrophilin-like 3 directly binds a human Vγ4+ T cell receptor using a modality distinct from clonally-restricted antigen. Immunity. (2019) 51:813–25.e4. doi: 10.1016/j.immuni.2019.09.006
171. Karunakaran MM, Willcox CR, Salim M, Paletta D, Fichtner AS, Noll A, et al. Butyrophilin-2A1 directly binds germline-encoded regions of the Vγ9Vδ2 TCR and is essential for phosphoantigen sensing. Immunity. (2020) 52:487–98.e6. doi: 10.1016/j.immuni.2020.02.014
172. Rigau M, Ostrouska S, Fulford TS, Johnson DN, Woods K, Ruan Z, et al. Butyrophilin 2A1 is essential for phosphoantigen reactivity by γδ T cells. Science. (2020) 367:aay516. doi: 10.1126/science.aay5516
173. Correia DV, Lopes A, Silva-Santos B. Tumor cell recognition by γδ T lymphocytes: T-cell receptor vs. NK-cell receptors. Oncoimmunology. (2013) 2:e22892. doi: 10.4161/onci.22892
174. Silva-Santos B, Strid J. Working in “NK Mode”: natural killer group 2 member d and natural cytotoxicity receptors in stress-surveillance by γδ T cells. Front Immunol. (2018) 9:851. doi: 10.3389/fimmu.2018.00851
175. Girardi M, Oppenheim DE, Steele CR, Lewis JM, Glusac E, Filler R, et al. Regulation of cutaneous malignancy by γδ T cells. Science. (2001) 294:605–9. doi: 10.1126/science.1063916
176. Strid J, Roberts SJ, Filler RB, Lewis JM, Kwong BY, Schpero W, et al. Acute upregulation of an NKG2D ligand promotes rapid reorganization of a local immune compartment with pleiotropic effects on carcinogenesis. Nat Immunol. (2008) 9:146–54. doi: 10.1038/ni1556
177. Lanca T, Correia DV, Moita CF, Raquel H, Neves-Costa A, Ferreira C, et al. The MHC class Ib protein ULBP1 is a nonredundant determinant of leukemia/lymphoma susceptibility to γδ T-cell cytotoxicity. Blood. (2010) 115:2407–11. doi: 10.1182/blood-2009-08-237123
178. Park JE, Wu DY, Prendes M, Lu SX, Ragupathi G, Schrantz N, et al. Fine specificity of natural killer T cells against GD3 ganglioside and identification of GM3 as an inhibitory natural killer T-cell ligand. Immunology. (2008) 123:145–55. doi: 10.1111/j.1365-2567.2007.02760.x
179. Wu DY, Segal NH, Sidobre S, Kronenberg M, Chapman PB. Cross-presentation of disialoganglioside GD3 to natural killer T cells. J Exp Med. (2003) 198:173–81. doi: 10.1084/jem.20030446
180. Weintraub A. Immunology of bacterial polysaccharide antigens. Carbohydr Res. (2003) 338:2539–47. doi: 10.1016/j.carres.2003.07.008
181. Speir JA, Abdel-Motal UM, Jondal M, Wilson IA. Crystal structure of an MHC class I presented glycopeptide that generates carbohydrate-specific CTL. Immunity. (1999) 10:51–61. doi: 10.1016/S1074-7613(00)80006-0
182. Tong W, Maira M, Roychoudhury R, Galan A, Brahimi F, Gilbert M, et al. Vaccination with tumor-ganglioside glycomimetics activates a selective immunity that affords cancer therapy. Cell Chem Biol. (2019) 26:1013–26.e4. doi: 10.1016/j.chembiol.2019.03.018
183. Tong W, Sprules T, Gehring K, Saragovi HU. Rational design of peptide ligands against a glycolipid by NMR studies. Methods Mol Biol. (2012) 928:39–52. doi: 10.1007/978-1-62703-008-3_4
184. Brandes M, Willimann K, Moser B. Professional antigen-presentation function by human γδ T Cells. Science. (2005) 309:264–8. doi: 10.1126/science.1110267
185. Brandes M, Willimann K, Bioley G, Levy N, Eberl M, Luo M, et al. Cross-presenting human γδ T cells induce robust CD8+ alphabeta T cell responses. Proc Natl Acad Sci USA. (2009) 106:2307–12. doi: 10.1073/pnas.0810059106
186. Castella B, Foglietta M, Sciancalepore P, Rigoni M, Coscia M, Griggio V, et al. Anergic bone marrow Vγ9Vδ2 T cells as early and long-lasting markers of PD-1-targetable microenvironment-induced immune suppression in human myeloma. Oncoimmunology. (2015) 4:e1047580. doi: 10.1080/2162402X.2015.1047580
187. Hoeres T, Holzmann E, Smetak M, Birkmann J, Wilhelm M. PD-1 signaling modulates interferon-gamma production by Gamma Delta (γδ) T-Cells in response to leukemia. Oncoimmunology. (2019) 8:1550618. doi: 10.1080/2162402X.2018.1550618
188. Neves PC, Rudersdorf RA, Galler R, Bonaldo MC, de Santana MG, Mudd PA, et al. CD8+ gamma-delta TCR+ and CD4+ T cells produce IFN-gamma at 5-7 days after yellow fever vaccination in Indian rhesus macaques, before the induction of classical antigen-specific T cell responses. Vaccine. (2010) 28:8183–8. doi: 10.1016/j.vaccine.2010.09.090
189. Neves PC, Santos JR, Tubarao LN, Bonaldo MC, Galler R. Early IFN-gamma production after YF 17D vaccine virus immunization in mice and its association with adaptive immune responses. PLoS ONE. (2013) 8:e81953. doi: 10.1371/journal.pone.0081953
190. Muia RP, Yu H, Prescher JA, Hellman U, Chen X, Bertozzi CR, et al. Identification of glycoproteins targeted by Trypanosoma cruzi trans-sialidase, a virulence factor that disturbs lymphocyte glycosylation. Glycobiology. (2010) 20:833–42. doi: 10.1093/glycob/cwq037
191. Poole J, Day CJ, von Itzstein M, Paton JC, Jennings MP. Glycointeractions in bacterial pathogenesis. Nat Rev Microbiol. (2018) 16:440–52. doi: 10.1038/s41579-018-0007-2
192. Yuki N. Carbohydrate mimicry: a new paradigm of autoimmune diseases. Curr Opin Immunol. (2005) 17:577–82. doi: 10.1016/j.coi.2005.09.004
193. Fisher JP, Heuijerjans J, Yan M, Gustafsson K, Anderson J. γδ T cells for cancer immunotherapy: a systematic review of clinical trials. Oncoimmunology. (2014) 3:e27572. doi: 10.4161/onci.27572
194. Fournie JJ, Sicard H, Poupot M, Bezombes C, Blanc A, Romagne F, et al. What lessons can be learned from γδ T cell-based cancer immunotherapy trials? Cell Mol Immunol. (2013) 10:35–41. doi: 10.1038/cmi.2012.39
195. Maeurer MJ, Martin D, Walter W, Liu K, Zitvogel L, Halusczcak K, et al. Human intestinal Vδ1+ lymphocytes recognize tumor cells of epithelial origin. J Exp Med. (1996) 183:1681–96. doi: 10.1084/jem.183.4.1681
196. Bialasiewicz AA, Ma JX, Richard G. Alpha/beta- and gamma/delta TCR(+) lymphocyte infiltration in necrotising choroidal melanomas. Br J Ophthalmol. (1999) 83:1069–73. doi: 10.1136/bjo.83.9.1069
197. Moulin M, Alguacil J, Gu S, Mehtougui A, Adams EJ, Peyrottes S, et al. Vγ9Vδ2 T cell activation by strongly agonistic nucleotidic phosphoantigens. Cell Mol Life Sci. (2017) 74:4353–67. doi: 10.1007/s00018-017-2583-0
198. Kunzmann V, Bauer E, Feurle J, Weissinger F, Tony HP, Wilhelm M. Stimulation of γδ T cells by aminobisphosphonates and induction of antiplasma cell activity in multiple myeloma. Blood. (2000) 96:384–92. doi: 10.1182/blood.V96.2.384
199. Sebestyen Z, Prinz I, Dechanet-Merville J, Silva-Santos B, Kuball J. Translating γδ (γδ) T cells and their receptors into cancer cell therapies. Nat Rev Drug Discov. (2020) 19:169–84. doi: 10.1038/s41573-019-0038-z
200. Deniger DC, Moyes JS, Cooper LJ. Clinical applications of gamma delta T cells with multivalent immunity. Front Immunol. (2014) 5:636. doi: 10.3389/fimmu.2014.00636
201. Nicol AJ, Tokuyama H, Mattarollo SR, Hagi T, Suzuki K, Yokokawa K, et al. Clinical evaluation of autologous gamma delta T cell-based immunotherapy for metastatic solid tumours. Br J Cancer. (2011) 105:778–86. doi: 10.1038/bjc.2011.293
202. Garfall AL, Maus MV, Hwang WT, Lacey SF, Mahnke YD, Melenhorst JJ, et al. Chimeric antigen receptor T cells against CD19 for multiple myeloma. N Engl J Med. (2015) 373:1040–7. doi: 10.1056/NEJMoa1504542
203. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
204. Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. (2015) 7:303ra139. doi: 10.1126/scitranslmed.aac5415
205. Capsomidis A, Benthall G, Van Acker HH, Fisher J, Kramer AM, Abeln Z, et al. Chimeric antigen receptor-engineered human gamma delta T cells: enhanced cytotoxicity with retention of cross presentation. Mol Ther. (2018) 26:354–65. doi: 10.1016/j.ymthe.2017.12.001
206. Ribot JC, deBarros A, Pang DJ, Neves JF, Peperzak V, Roberts SJ, et al. CD27 is a thymic determinant of the balance between interferon-gamma- and interleukin 17-producing γδ T cell subsets. Nat Immunol. (2009) 10:427–36. doi: 10.1038/ni.1717
207. Misiak A, Wilk MM, Raverdeau M, Mills KH. IL-17-Producing innate and pathogen-specific tissue resident memory γδ T cells expand in the lungs of bordetella pertussis-infected mice. J Immunol. (2017) 198:363–74. doi: 10.4049/jimmunol.1601024
208. Davey MS, Willcox CR, Joyce SP, Ladell K, Kasatskaya SA, McLaren JE, et al. Clonal selection in the human Vδ1 T cell repertoire indicates γδ TCR-dependent adaptive immune surveillance. Nat Commun. (2017) 8:14760. doi: 10.1038/ncomms14760
209. Dimova T, Brouwer M, Gosselin F, Tassignon J, Leo O, Donner C, et al. Effector Vgamma9Vδ2 T cells dominate the human fetal γδ T-cell repertoire. Proc Natl Acad Sci USA. (2015) 112:E556–65. doi: 10.1073/pnas.1412058112
210. Dube DH, Bertozzi CR. Glycans in cancer and inflammation–potential for therapeutics and diagnostics. Nat Rev. (2005) 4:477–88. doi: 10.1038/nrd1751
Keywords: gangliosides, sialic acid, tumor marker ganglioside, γδ T cells, immunotherapy, cancer
Citation: Bartish M, del Rincón SV, Rudd CE and Saragovi HU (2020) Aiming for the Sweet Spot: Glyco-Immune Checkpoints and γδ T Cells in Targeted Immunotherapy. Front. Immunol. 11:564499. doi: 10.3389/fimmu.2020.564499
Received: 21 May 2020; Accepted: 31 August 2020;
Published: 29 September 2020.
Edited by:
Jurgen Kuball, Utrecht University, NetherlandsReviewed by:
Dennis Beringer, University Medical Center Utrecht, NetherlandsChristelle Harly, INSERM U1232 Centre de Recherche en Cancérologie et Immunologie Nantes Angers (CRCINA), France
Copyright © 2020 Bartish, del Rincón, Rudd and Saragovi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: H. Uri Saragovi, dXJpLnNhcmFnb3ZpQG1jZ2lsbC5jYQ==; Margarita Bartish, bWFyZ2FyaXRhLmJhcnRpc2hAbWFpbC5tY2dpbGwuY2E=