 Wurihan
Wurihan Gezi
Gezi Elisa Brambilla2
Elisa Brambilla2 Yixin Shi
Yixin Shi Bianca Sclavi
Bianca Sclavi Morigen
Morigen- 1State Key Laboratory of Reproductive Regulation and Breeding of Grassland Livestock, School of Life Sciences, Inner Mongolia University, Hohhot, China
- 2LBPA, UMR 8113, CNRS, ENS Paris-Saclay, Cachan, France
- 3School of Life Sciences, Arizona State University, Tempe, AZ, United States
The uvrB gene belongs to the SOS network, encoding a key component of the nucleotide excision repair. The uvrB promoter region contains three identified promoters with four LexA binding sites, one consensus and six potential DnaA binding sites. A more than threefold increase in transcription of the chromosomal uvrB gene is observed in both the ΔlexA ΔsulA cells and dnaAA345S cells, and a fivefold increase in the ΔlexA ΔsulA dnaAA345S cells relative to the wild-type cells. The full activity of the uvrB promoter region requires both the uvrBp1-2 and uvrBp3 promoters and is repressed by both the DnaA and LexA proteins. LexA binds tightly to LexA-box1 at the uvrBp1-2 promoter irrespective of the presence of DnaA and this binding is important for the control of the uvrBp1-2 promoter. DnaA and LexA, however, compete for binding to and regulation of the uvrBp3 promoter in which the DnaA-box6 overlaps with LexA-box4. The transcription control of uvrBp3 largely depends on DnaA-box6. Transcription of other SOS regulon genes, such as recN and dinJ, is also repressed by both DnaA and LexA. Interestingly, the absence of LexA in the presence of the DnaAA345S mutant leads to production of elongated cells with incomplete replication, aberrant nucleoids and slow growth. We propose that DnaA is a modulator for maintenance of genome integrity during the SOS response by limiting the expression of the SOS regulon.
Introduction
The uvrB gene encodes the UvrB protein, one of the key components of the NER system (Truglio et al., 2006). NER repair is a versatile pathway that recognizes a wide range of DNA lesions by the concerted function of the UvrABC proteins (Pruteanu and Baker, 2009). The uvrB gene belongs to the SOS regulon (Howard-Flanders et al., 1966). SOS is a global response to DNA damage in which RecA filaments bound on ssDNA promote self-cleavage of the LexA protein. Cleavage of LexA induces expression of the SOS genes, resulting in an arrest of cell division for the time required to repair the damages (Walter, 1996). LexA regulates the SOS regulon by binding to the LexA-box and thus preventing gene expression during normal growth. The LexA-box has the following consensus sequence TACTG(TA)5CAGTA (Walker, 1984), having a conserved trimer of CTG on the left and another trimer of CAG on the right with a variable sequence of spacers; the spacing between “CTG” and “CAG” is invariable at 10 nucleotides (Fernandez De Henestrosa et al., 2000; Wade et al., 2005). LexA contains two domains: an N-terminal winged helix-turn-helix (wHTH) DNA-binding domain and a C-terminal dimer with a latent protease domain (Zhang et al., 2010). In response to DNA damage RecA-ssDNA-ATP filaments are formed and the auto-proteolytic activity of LexA at the C-terminal domain is activated by interacting with the filaments. The degradation of LexA opens the promoter region for RNA polymerase (RNAP) recruitment and the start of transcription.
The DnaA protein initiates chromosomal replication in bacteria by interacting with 9-mer consensus sequences of TTA/TTNCACA, the DnaA-boxes, at the origin for replication (Kornberg and Baker, 1992; Schaper and Messer, 1995). DnaA has a high affinity for ATP and ADP (Sekimizu et al., 1987), and ATP-DnaA is active for the initiation of replication whereas ADP-DnaA is inactive (Sekimizu et al., 1987). DnaA is also a transcription factor, repressing transcription by binding to DnaA-boxes in the promoter regions, such as those found at the promoters for the dnaA gene itself (Atlung et al., 1985; Braun et al., 1985), the mioC gene (Lother et al., 1985) and the nrd operon (Tuggle and Fuchs, 1986; Speck et al., 1999; Olliver et al., 2010) while transcription of the polA gene is stimulated by DnaA in stationary phase (Quinones et al., 1997).
The E. coli uvrB promoter region contains three promoters, namely, uvrBp1, uvrBp2, and uvrBp3 (Sancar et al., 1982). A LexA-box with the AACTGTTTTTTTATCCAGTA sequence has been identified between the -35 and -10 regions of uvrBp2 (Fernandez De Henestrosa et al., 2000). Interestingly, the uvrBp3 promoter contains DnaA boxes (Arikan et al., 1986) which constitute the DARS1 (DnaA reactivation site) site consisting of three DnaA-boxes, where inactive ADP-DnaA is reactivated to form active ATP-DnaA (Fujimitsu et al., 2009). Here, we show that the uvrB promoters are repressed by both DnaA and LexA by specifically binding to its promoter region in either a competitive or an independent manner. Interestingly, two other genes of the SOS regulon, recN and dinJ, are also found to be repressed by both DnaA and LexA. The simultaneous absence of LexA- and DnaA-dependent repression leads to production of elongated cells with incomplete DNA replication with abnormal nucleoids and slow growth. It is likely that regulation of gene expression by DnaA maintains genome integrity during the SOS response in Escherichia coli.
Materials and Methods
Bacterial Strains
All bacterial strains used in this study are derived from the E. coli K12 listed in Table 1. The ΔsulA::neo allele was transferred into the MC4100 by P1 transduction (Miller et al., 1992) and resulting in MC4100ΔsulA::neo. The cat gene was PCR amplified using the plasmid pKD3 as template and primer 582 and 583 as listed in Supplementary Table S2 and inserted into MC4100ΔsulA::neo mutant to replace the chromosomal lexA gene through homologous recombination by One-step Chromosomal Gene Inactivation method (Datsenko and Wanner, 2000), resulting in MC4100ΔsulA::neoΔlexA::cat double mutant. The neo or/and cat genes were removed by the FRT site-specific recombination as described previously (Datsenko and Wanner, 2000), resulting in MC4100ΔsulA or/and MC4100ΔsulAΔlexA double mutant. The cat gene was also PCR amplified using pKD3 as template and primers 48 and 49 listed in Supplementary Table S2, then inserted behind the chromosomal uvrB gene in MC4100 and MC4100ΔsulAΔlexA cells by the method mentioned above. The cat gene was replaced by pCE36 using the FRT site-specific recombination in the cells mentioned, resulting in insertion of the lacZ…neo fusion behind the chromosomal uvrB gene as described previously (Datsenko and Wanner, 2000; Ellermeier et al., 2002). As a result, MC4100uvrB-lacZ…neo, MC4100ΔsulA uvrB-lacZ…neo and MC4100ΔsulAΔlexA uvrB-lacZ…neo cells were constructed. The dnaAA345S…cat allele was transferred into the MC4100uvrB-lacZ…neo, MC4100ΔsulA uvrB-lacZ…neo and MC4100ΔsulAΔlexA uvrB-lacZ…neo cells by P1 transduction (Miller et al., 1992). The lexA3…tet allele was P1 transduced into MC4100 and dnaAA345S…cat cells carrying uvrB-lacZ…neo fusion, respectively (Miller et al., 1992). For construction of recN-lacZ…neo and dinJ-lacZ…neo, the cat gene was PCR amplified using the plasmid pKD3 as template and primers 1229 and 1230 for recN-lacZ…neo, primers 1235 and 1236 for dinJ-lacZ…neo as listed in Supplementary Table S2. After insertion of the cat gene down-stream of the chromosomal recN or dinJ gene in MC4100 cells, the cat gene was replaced by pCE36 using the FRT site-specific recombination, resulting in insertion of the lacZ…neo fusion behind the chromosomal recN or dinJ gene as described previously (Datsenko and Wanner, 2000; Ellermeier et al., 2002). The recN-lacZ…neo or dinJ-lacZ…neo allele was P1 transduced into dnaAA345S, ΔsulAΔlexA, ΔsulAΔlexA dnaAA345S and lexA3 dnaAA345S cells, resulting in dnaAA345S recN-lacZ…neo, ΔsulAΔlexA recN-lacZ…neo, ΔsulAΔlexA dnaAA345S recN-lacZ…neo, lexA3 dnaAA345S recN-lacZ…neo, or dnaAA345S dinJ-lacZ…neo, ΔsulAΔlexA dinJ-lacZ…neo, ΔsulAΔlexA dnaAA345S dinJ-lacZ…neo and lexA3 dnaAA345S dinJ-lacZ…neo. DH5α was used as a host for the preparation of plasmid DNA. The WM2287 strain containing the pdnaA116 plasmid was used for DnaA purification (Schaper and Messer, 1995) and BL21-Gold (DE3) for His6-LexA protein expression and purification.
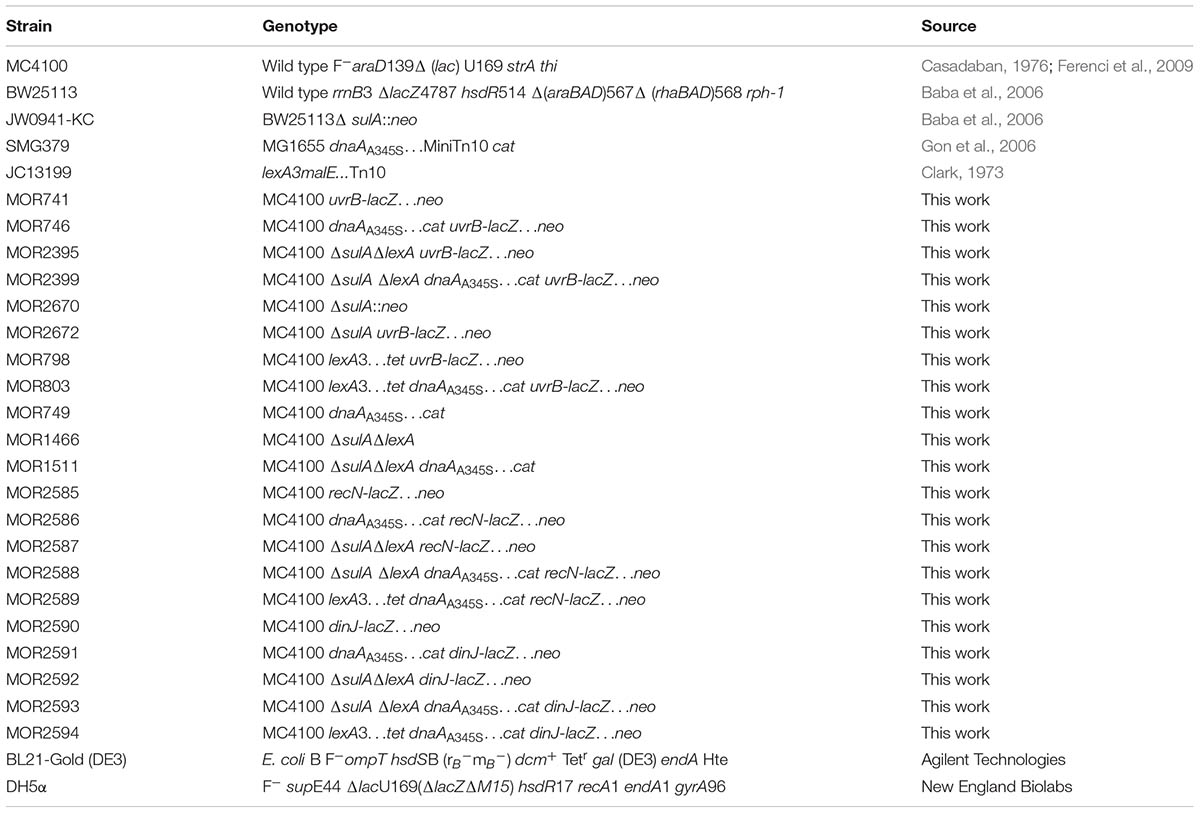
TABLE 1. Bacterial strains.
Growth Media and Conditions
Cells were grown at 37°C in LB (Bertani, 1951) or ABTGcasa medium (Morigen et al., 2005). When necessary, 100 μg/ml of ampicillin, 30 μg/ml of chloramphenicol, 15 μg/ml of tetracycline and 50 μg/ml of kanamycin were added.
Plasmid Constructions
All plasmids used in this study are listed in Table 2 and the primers including their descriptions are listed in Supplementary Table S2. The uvrBp1-3 promoter was PCR amplified using chromosomal DNA from the wild-type BW25113 cells as template and primers 54 and 57. The uvrBp1-3 PCR fragment was inserted in front of the promoterless lacZ gene on pTAC3953 (Brondsted and Atlung, 1994) at BamHI and HindIII sites, resulting in plasmid puvrBp1-3-lacZ. Using the same template, the uvrBp1-2 promoter region was amplified by primers 79 and 57, and the uvrBp3 promoter region by primers 54 and 71. The PCR fragment for each promoter was then inserted into pTAC3953 at the BamHI and HindIII sites (Brondsted and Atlung, 1994), leading to the construction of plasmids puvrBp1-2-lacZ and puvrBp3-lacZ. The uvrBp1-3-lacZ fusion was PCR amplified by primers 54 and 1131 using puvrBp1-3-lacZ as template. The PCR fragment was then inserted into a low copy plasmid, MiniR1 which is about 1–2 copies per the chromosomal ter site (Morigen et al., 2001) at the BamHI and HindIII sites, resulting in MiniR1-uvrBp1-3-lacZ (shown as R1-uvrBp1-3 for short). The uvrBp1-2-lacZ fusion was PCR amplified by primers 79 and 1350 using puvrBp1-2-lacZ as template. The PCR fragment was then inserted into MiniR1 at the BamHI and BglII sites, resulting in MiniR1-uvrBp1-2-lacZ (R1-uvrBp1-2 for short). The uvrBp3-lacZ fusion was PCR amplified by primers 54 and 1350 using puvrBp3-lacZ as a template. The PCR fragment was inserted into MiniR1 at BamHI and BglII sites, resulting in MiniR1-uvrBp3-lacZ (R1-uvrBp3 for short). The DH5α cells were transformed with the resulting ligation. The lexA gene was PCR amplified using the chromosomal DNA from the wild-type BW25113 cells as template and primers 578 and 579. The PCR fragment for lexA was inserted into pET28a (EMD Biosciences) at the NcoI and XhoI sites, resulting in pET28a-his6-lexA which produces His6-LexA protein fusion under IPTG induction. All constructions were sequenced to make sure the plasmid constructions were correct.

TABLE 2. Plasmids.
Site-Directed Mutagenesis
Point mutation was generated using a site-directed mutagenesis kit (TransGen Biotech, Beijing, China) as described previously (Rousseau et al., 2013). The mutations (from TG to GC) in LexA-box1 in the uvrBp1-2 promoter were generated by site-directed mutagenesis using the puvrBp1-2-lacZ plasmid as template and the pair of primers 1214 and 1215. Similarly, using the puvrBp3 plasmid as template, the mutations (from TG to CA) in DnaA-box6 in uvrBp3 promoter, were generated by the pair of primers 1210 and 1211.
Purification of Proteins
The DnaA protein was over-expressed in WM2287/pdnaA116 cells (Krause et al., 1997) and purified as described previously (Olliver et al., 2010). The BL21-Gold (DE3)/pET28a-his6-lexA cells were grown at 37°C in 200 ml of LB medium. At OD600 = 0.6, IPTG with 0.3 mM of final concentration was added and incubated for 2 h. The cells were harvested and washed with PBS once, resuspended in 10 ml of the lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole), and sonicated. The whole cell lysate was mixed with His-Select Ni-NTA slurry (Qiagen) and His6-LexA was purified according to the manufacturer instructions. Purity of the His6-LexA protein sample was detected by staining with INSTANT BLUE (Expedeon) after SDS-PAGE (Supplementary Figure S1) gel-electrophoresis following the manufacturer instructions. The His6-LexA protein concentration was determined by BCA assay (Thermo Scientific) and stored at -80°C after imidazole was removed by dialysis in 1xPBS buffer (0.137 M NaCl, 0.027 M KCl, 0.01 M Na2HPO4, 0.0018 M KH2PO4).
β-Galactosidase Activity Assay
Exponentially growing cells (1 ml) at 37°C in ABTGcasa medium were collected at OD450 = 0.1, 0.2, 0.3, 0.4, and 0.5, then mixed with cold toluene (0.1 ml) and kept on ice immediately. For measurement of β-galactosidase activity, 0.2 ml toluene-treated sample was added to 1 ml Z buffer (40 mM NaH2PO4, 60 mM Na2HPO4, 10 mM KCl, 1 mM MgSO4 and 50 mM β-mercaptoethanol, pH 7.0) containing 0.66 mg/ml o-nitrophenyl-β-D-galactopyranoside. The reaction was performed at 30°C until the color changed to yellow and stopped by addition of 0.5 ml 1 M Na2CO3, and the absorbance at OD420 was measured. The β-galactosidase activity was calculated by 1000∗OD420/reaction time (min) ∗OD450∗0.2 ml (Miller, 1972).
DNase I Footprinting Assays
The uvrB promoter region (523bp) was PCR amplified using chromosomal DNA from the wild-type BW25113 cells as template and primer 828 and 829 for the DNase I footprinting assays. In the PCR reaction, the primer 828 was 5′ labeled with [γ-32P] ATP (GE Healthcare) by T4 polynucleotide kinase (New England Biolabs). The PCR product was purified with the Bio-Spin 6 Columns (Bio-Rad) according to the manufacturer instructions. Approximately 1 nmol of labeled DNA and increasing amounts (final concentration was 50, 100, 200, 300, and 600 nM) of ATP-DnaA or His6-LexA protein were mixed in a 10 μl reaction buffer containing 1 mM DTT, 0.5 mg/ml Ac-BSA, 20 mM HEPES pH 7.6, 50 mM K-glu, 5 mM MgCl2 and 3 mM of ATP (Sigma-Aldrich). The reaction mixture was incubated at 37°C for 15 min. Then, 4 mg/ml of DNase I diluted in digestion buffer (25 mM Tris pH 7.5, mM MgCl2, 1 mM CaCl2, 2 mM DTT and 100 mM KCl) was added and the mixture was incubated at 37°C for 20 s. To determine the protection patterns of the uvrB promoter DNA by two proteins (DnaA and LexA), the second protein was added with final concentrations of 50, 100, 200, 300, and 600 nM after 10 min incubation with the first protein with final concentration of 200 nM, and incubated for 10 min, then digested by DNase I for 30 s. The DNase I digestion was stopped by addition of an equal volume of formamide loading buffer (90% formamide, 1 × TBE, bromophenol blue, xylene cyanol, calf thymus non-specific DNA). Samples were incubated for 5 min at 95°C and analyzed by 6% acrylamide in denaturing conditions (8 M urea and 1 × TBE buffer) by comparison with a DNA sequence ladder generated with the same primers using a A+G reaction as described previously (Maxam and Gilbert, 1980). After electrophoresis, gels were dried and autoradiographed.
UV Irradiation
Cells were exponentially grown at 37°C in 50 ml of ABTGcasa medium, 20 ml of cell culture at OD450 = 0.08 was irradiated in an open petri-dish with 50 J/M2 of UV, then cells were grown in flask at 37°C. Sampling and measurement of β-galactosidase activity was carried out as mentioned above.
Flow Cytometry
Exponentially growing cells in ABTGcasa medium at 37°C were treated with 300 μg/ml rifampicin and 10 μg/ml cephalexin for 4–5 generations. Initiation of DNA replication is inhibited by rifampicin which allows ongoing replication finish while cell division is blocked by cephalexin at the time of addition of the drugs (Skarstad et al., 1986; Boye and Lobner-Olesen, 1991). Cells were fixed in 70% ethanol and stained in Hoechst 33258 (Invitrogen) for 30 min, then analyzed by flow cytometry (LSR Fortessa, BD).
Confocal Fluorescence Microscopy
Exponentially growing cells in ABTGcasa medium at 37°C were harvested, fixed in 70% ethanol, visualized under a Zeiss LSM710 Confocal microscope with 100×/1.4 Plam-Apo at 405 nm laser excitation after staining in Hoechst 33258 for 30 min. Images were scanned by a PMP detector and analyzed with the ZEN 2011 (black version) software to measure cell size and nucleoid distribution.
Results
Both DnaA and LexA Repress Expression of the uvrB Gene
A global transcriptional analysis by using Affymetrix GeneChip E. coli Genome 2.0 arrays showed that expression of the uvrB gene increased 2.7 (±0.9)-fold in a dnaAA345S mutant relative to the wild-type cells (Morigen and Skarstad, unpublished data). The result suggests that DnaA could directly be involved in control of the uvrB gene expression since DnaAA345S binds to DnaA-boxes with a lower affinity compared to wild-type DnaA. The dnaAA345S mutant is a suppressor for a mutant lacking four of the redoxins involved in Nrd activity (Ortenberg et al., 2004) and the purified DnaAA345S protein is defective for ATP binding in vitro (Gon et al., 2006). The dnaAA345S mutant is also found to result in under-replication and larger cell mass with slower growth (Ortenberg et al., 2004; Gon et al., 2006). In order to determine the regulatory effect of DnaA on transcription of the uvrB gene, the lacZ reporter gene was inserted downstream of the chromosomal uvrB gene, resulting in an uvrB-lacZ derivative of the MC4100 strain lacking the chromosomal lacZ gene (Casadaban, 1976; Ferenci et al., 2009). Subsequently, a dnaAA345S allele was transferred to the uvrB-lacZ strain by P1 transduction. The uvrB gene belongs to the SOS regulon which is regulated by LexA (Howard-Flanders et al., 1966). It should be noted that the LexA protein is essential for cell growth but the growth defect in the absence of LexA can be suppressed by deletion of the sulA gene (George et al., 1975). Thus, we removed the lexA gene by constructing the ΔlexA ΔsulA double mutant. To understand how DnaA interacts with LexA in the control of uvrB expression, the uvrB-lacZ allele was P1 transduced into the ΔlexA ΔsulA, ΔlexA ΔsulA dnaAA345S, lexA3 and lexA3 dnaAA345S cells including the ΔsulA mutant as a control. The level of transcription from the uvrB promoter region was measured by the β-galactosidase activity assay in exponentially growing cells. Transcription from the uvrB promoter was 3.5-fold higher in the dnaAA345S cells compared with that of the wild-type cells (Figure 1A), suggesting that DnaA represses uvrB expression. Not surprisingly, transcription from the uvrB promoter region in the ΔsulA mutant was about the same as that in the wild-type cells, while it was 3.3-fold higher in the ΔlexAΔsulA mutant compared to the control, in agreement with previous work (Sancar et al., 1982), indicating that LexA represses uvrB expression. Interestingly, uvrB transcription was further increased, to 5.2-fold, in the ΔlexAΔsulA dnaAA345S triple mutant (Figure 1A), implying that the repression by DnaA and LexA of uvrB expression is additive and thus might be independent of an interaction between the two proteins. The conclusion is supported by 1.6-fold increase of uvrB expression in the ΔsulAΔlexA dnaAA345S cells compared to that in the ΔsulAΔlexA cells (Figure 1A inset). The LexA3 mutant protein is not self-cleavable or largely resistant to cleavage and thus binds to the uvrB promoter region regardless of whether the SOS response is on or off (Little et al., 1980; Markham et al., 1981). Transcription from the uvrB promoter region in the lexA3 mutant was about the same as that in the wild-type cells, while it was 3.6-fold higher in the lexA3 dnaAA345S strain (Figure 1B). These results support the idea that DnaA-dependent repression of uvrB expression is independent of LexA activity. We conclude that in the absence of the SOS response both DnaA and LexA repress transcription of the uvrB gene and function independently of each other.
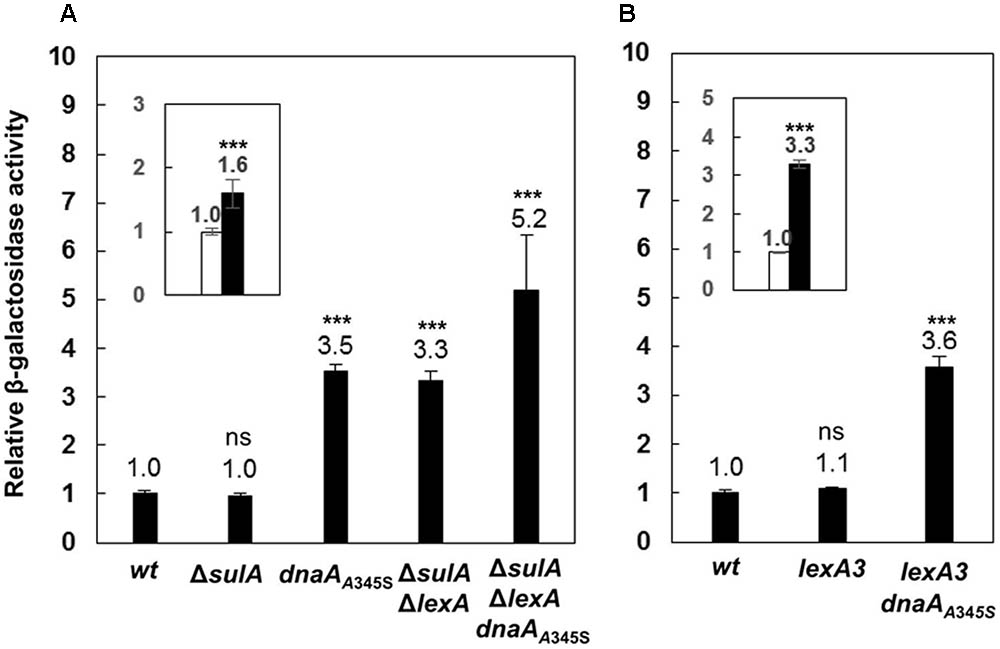
FIGURE 1. The DnaA and LexA proteins repress expression of the uvrB gene independently. (A) The wild-type, ΔsulA, dnaAA345S, ΔsulAΔlexA and ΔsulAΔlexA dnaAA345S cells carrying uvrB-lacZ fusion on their chromosomes were exponentially grown in ABTGcasa medium at 37°C. The β-galactosidase activity from uvrB-lacZ fusion in the cells was determined by Miller method (Miller, 1972), and relative expressions of uvrB-lacZ in all mutants to that in the wild-type (14 U) were calculated. The inset indicates the relative expression of uvrB-lacZ in the ΔsulAΔlexA dnaAA345S cells against that in the ΔsulAΔlexA cells. (B) The β-galactosidase activity in exponentially growing lexA3 and lexA3dnaAA345S cells carrying uvrB-lacZ fusion was determined as described in the legend to (A). The inset indicates the relative expression of uvrB-lacZ in the lexA3dnaAA345S cells against that in the lexA3 cells. The values shown at top of the bars are the average of three individual experiments, and the standard errors are shown. ∗∗∗Stands for P-value ≤ 0.01, ∗∗for 0.01 < P-value ≤ 0.05, and ns represents P-value > 0.05.
The uvrB Promoter Region Contains Seven Potential DnaA-Boxes and Four LexA-Boxes
In order to understand how LexA and DnaA function in the control of uvrB transcription, we searched for the sequences corresponding to DnaA-boxes and LexA-boxes in the uvrB promoter region. The uvrB promoter region was previously shown to contain three DnaA-boxes (Fujimitsu et al., 2009) and one LexA-box (Van Den Berg et al., 1985). By our analysis, four additional DnaA-boxes and three potential LexA-boxes were identified in the region (Figure 2). All these DnaA-boxes were renamed as DnaA-box1, 2, 3, 4, 5, 6, and 7 from the proximal to the distal site relative to the transcription start site of the uvrBp1 promoter (Figure 4A). The characterized LexA-boxes were called LexA-box1, 2, 3, and 4, also in the same orientation (Figure 2). LexA-box1 is closest to the consensus, having the conserved CTG trimer on the left and the CAG trimer on the right end, with nine ATs out of ten bases between these two trimers. LexA-box2, 3, and 4, however, have a CTG on the left but do not have CAG on the right end, having an AT-rich sequence in the middle. LexA-box1 is located between the -35 and -10 sites of the uvrBp2 promoter (Figure 4A) (Sancar et al., 1982), overlapping with DnaA-box1. LexA-box2 and 3 are found between the uvrBp2 and uvrBp3 promoters (Figure 4A), and LexA-box4 is found within the DARS1 (Fujimitsu et al., 2009), overlapping with DnaA-box5 and 6 on the uvrBp3 promoter (Figures 2, 4A). DnaA-box2, 3 and 4 are located between uvrBp2 and uvrBp3, and three other DnaA-boxes (DnaA-box5, 6, and 7) are found in the uvrBp3 promoter (Figure 2), composing the DARS1 region (Fujimitsu et al., 2009). The presence of DnaA-boxes in the uvrB promoter region support the idea that DnaA might be directly involved in transcription control of the uvrB gene and also suggest a possible cooperative or competitive interaction with LexA via their overlapping binding sites (Van Den Berg et al., 1985).
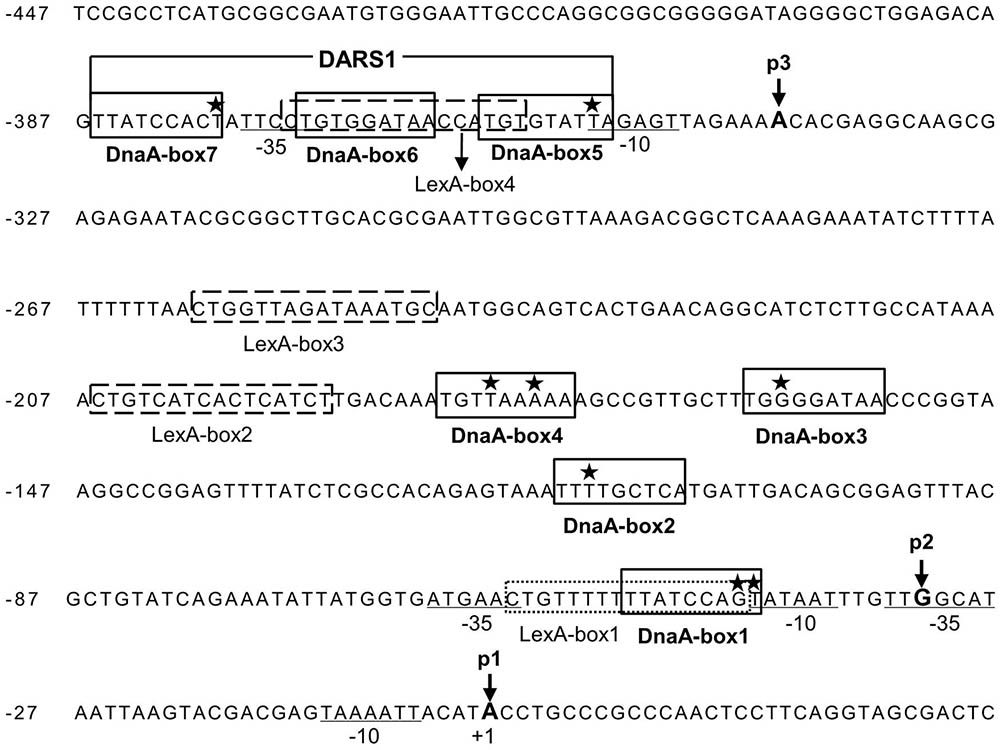
FIGURE 2. The uvrB promoter cluster contains three promoters, seven potential DnaA-boxes and four LexA-boxes. Seven potential DnaA binding sites (DnaA-box1, 2, 3, 4, 5, 6, and 7) in the uvrB promoter region are boxed and labeled. DARS1 site consists of DnaA-box 5, 6, and 7. The stars represent the mismatched nucleotides to the consensus DnaA-box (TTA/TTNCACA). The potential LexA-boxes are boxed with a dashed line and labeled. The –35 and –10 sites of the promoters are underlined and labeled. The positions of nucleotides at the transcriptional starting site from promoter uvrBp1, p2 and p3 are indicated by p1, p2 and p3 with arrow, respectively, while p1 is numbered as +1 (Sancar et al., 1982; Arikan et al., 1986). The letters in bold indicate the transcriptional starting sites (+1) from different promoters.
DnaA Interferes With Binding of LexA to LexA-Box2 and 3 but Not to LexA-Box1
As described above, we have shown that both DnaA and LexA repress transcription from the uvrB promoter cluster, which contains seven potential DnaA-boxes and four LexA-boxes. Now the questions are: (i) do DnaA and/or LexA bind to these potential DnaA-boxes and/or LexA-boxes? (ii) do DnaA and LexA compete for their binding sites in the uvrB promoter region? To address these questions, we performed in vitro DNase I footprinting experiments and determined the binding patterns of DnaA and LexA to the uvrB promoter cluster. A PCR amplified fragment (523 bp) of the uvrB promoter cluster was used in these experiments. The DnaA protein was purified as described previously (Olliver et al., 2010) and His6-LexA was purified as described in Materials and Methods (Supplementary Figure S1). A protection pattern of the uvrB promoter cluster by increasing concentrations of LexA was detected in the presence or absence of DnaA. As shown in Figure 3, LexA protections of LexA-box2, 3, and 4 increased as a function of its concentration whereas LexA-box1 became protected at the lowest concentration of LexA. These results are in agreement with the differences in the LexA-box2, 3, and 4 sequences relative to the consensus sequence of LexA-box1. In the presence of DnaA, the LexA protections to LexA-box2, 3, and 4 were weakened or abolished in a DnaA concentration dependent manner while binding to LexA-box1 remained strong (Figure 3). For the DARS site, which has the LexA-box4 overlapping with DnaA-box5 and 6, the LexA protection was clear. While the overlap of the protection of the two proteins at the DARS site makes it difficult to determine whether LexA is still bound in the presence of DnaA, the appearance of the DnaA protections at the same concentration as in the absence of LexA suggests that the former can bind even in the presence of the latter and could thus displace it (Figure 3). These results suggest that LexA binds to LexA-box1 with high affinity even in the presence of high concentrations of DnaA but not to LexA-box2, 3, and 4. High concentrations of DnaA weaken the binding of LexA to its low affinity boxes. The competition of DnaA for its binding sites with LexA is not necessarily dependent on the fact that DnaA-boxes overlap with the LexA-boxes, possibly due to the ability of ATP-DnaA to form oligomeric structures.
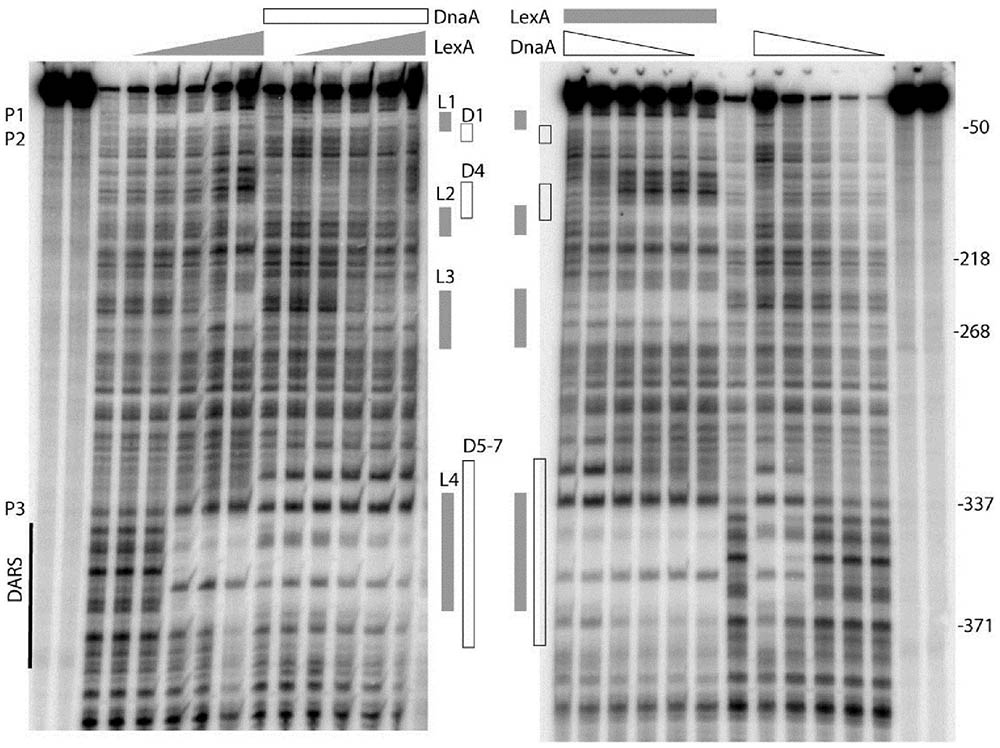
FIGURE 3. DnaA and LexA protect the uvrB promoter region in either competitive or independent manner. The uvrB promoter fragment (–513 to +12 relative to the transcription starting site of uvrBp1) was generated by PCR using the upstream oligonucleotide labeled with gamma 32p-ATP by T4 kinase at the 5′ end. The labeled DNA was either incubated at 37°C with increasing concentrations (50, 100, 200, 300, and 600 nM) of LexA (left) or ATP-DnaA (right) protein. After 10 min incubation of either the ATP-DnaA or the LexA protein with the labeled DNA, the sample had the other protein. The DNA was cleaved by DNase I for 20–30 s after 10 min of incubation with the second protein. The reaction was stopped by addition of an equal volume of formamide gel loading buffer and placed on ice. The gray boxes show the sites protected by the LexA protein. The white boxes correspond to the protection by ATP-DnaA. The protected DnaA-boxes are indicated as D, the LexA-boxes are as L. The DARS site is also shown. P1, P2, and P3 represent promoter uvrBp1, uvrBp2, uvrBp3, respectively. The right and left most two lanes are for ladder.
Strong protections at DARS1 were found for ATP-DnaA (Figure 3). The DnaA protections of these sites were concentration dependent and such protections were not found to be changed in the presence of LexA. To further understand how DnaA and LexA function in the control of uvrB gene expression, we investigated the interaction between DnaA and LexA. The bacterial two-hybrid analysis showed that DnaA did not interact directly with LexA (data not shown).
The Full Activity of the uvrB Promoter Region Requires Both the uvrBp1-2 and uvrBp3 Promoters
The uvrB promoter region has three characterized promoters, namely uvrBp1, uvrBp2, uvrBp3 (Sancar et al., 1982), forming a cluster of uvrB promoters (Figure 4A). To determine the roles of these different promoters in the transcription control of the uvrB gene, each promoter was inserted in front of the lacZ gene into the MiniR1 plasmid. MiniR1 is a low copy plasmid, having 1–2 copies per the chromosomal ter site (Morigen et al., 2001). The resultant plasmids carry a uvrBp1-3-lacZ (for short as R1-uvrBp1-3), uvrBp1-2-lacZ (R1-uvrBp1-2) or uvrBp3-lacZ (R1-uvrBp3) fusion as illustrated in Figure 4A. The R1-uvrBp1-3 construct includes all three promoters. Each plasmid was introduced into the wild-type MC4100 cells or the dnaAA345S, ΔlexAΔsulA or ΔlexAΔsulA dnaAA345S derivatives (Figure 4). Promoter activity was then measured by the β-galactosidase activity assay in exponentially growing cells. Transcription from uvrBp1-2 accounted for 30% of the activity of the full-length promoter region while uvrBp3 accounted for 20% (Figure 4B). The results suggest that full activity of the uvrB promoter requires both the uvrBp1-2 and uvrBp3 promoters.

FIGURE 4. The full activity of the uvrB promoter requires the uvrBp1-2 and uvrBp3 promoters. (A) Illustration for insertion of individual uvrB promoter in front of the lacZ gene into MiniR1 plasmid. Construction of the MiniR1-uvrBp1-3-lacZ (R1-uvrBp1-3), MiniR1-uvrBp1-2-lacZ (R1-uvrBp1-2) or MiniR1-uvrBp3-lacZ (R1-uvrBp3) plasmid was as mentioned in section “Materials and Methods.” Promoter uvrBp1-2 contains promoter 1 and 2, ranging from –77 to +33; promoter uvrBp3 starts from –567 and ends at –280; Promoter uvrBp1-3 consists of promoter 1, 2, and 3, including the region from –567 to +33, the whole cluster of the uvrB promoters. The nucleotide positions are as indicated in Figure 2. The gray arrows represent the lacZ gene. The open rectangles represent LexA-boxes, the open triangles represent DnaA-boxes with orientation, the hatched rectangles represent LexA-boxes overlapping with DnaA-box. The filled arrows indicate positions of the promoters and orientation of transcriptions. The p1, p2, and p3 represent the uvrB promoter 1, 2, and 3. The wild-type, dnaAA345S, ΔsulAΔlexA and ΔsulAΔlexA dnaAA345S cells were transformed by plasmid R1-uvrBp1-3, R1-uvrBp1-2, and R1-uvrBp3, respectively. The resultant transformants were exponentially grown in ABTGcasa medium at 37°C. Activity of the individual plasmid-borne uvrB promoter was measured as the β-galactosidase activity in the cells by Miller method (Miller, 1972). Activity of individual promoter relative to that of R1-uvrBp1-3 (229 U) in the wild-type cells is illustrated (B). Relative activity of promoter R1-uvrBp1-2 (C), or R1-uvrBp3 (D) in the dnaAA345S, ΔsulAΔlexA and ΔsulAΔlexA dnaAA345S cells compared with that in the wild-type cells (70 U for uvrBp1-2 and 44 U for R1-uvrBp3) is illustrated. The insets indicate the relative activity of promoters in the ΔsulAΔlexA dnaAA345S cells against that in the ΔsulAΔlexA cells. The values shown at top of the bars are the average of three individual experiments, and the standard errors are shown. ∗∗∗Stands for P-value ≤ 0.01, ∗∗for 0.01 < P-value ≤ 0.05, and ns represents P-value > 0.05.
Transcription From the Plasmid-Borne uvrBp1-2 or uvrBp3 Is Repressed by DnaA and LexA Independently
To further clarify the function of DnaA and LexA in the control of the uvrBp1-2 or uvrBp3 promoter activity, transcription from the plasmid-borne uvrBp1-2-lacZ construct was measured in the dnaAA345S, ΔsulAΔlexA and ΔsulAΔlexA dnaAA345S mutant strains as described above. As shown in Figure 4C, transcription in the dnaAA345S or ΔsulAΔlexA strains was about twofold higher relative to that in the wild-type cells. The transcription level further increased to 3.6-fold of wild type in the ΔlexAΔsulA dnaAA345S cells. The increase in transcription from uvrBp1-2 in the ΔsulAΔlexA dnaAA345S cells compared to that in the ΔsulAΔlexA cells was clear (Figure 4C inset). These results suggest that uvrBp1-2 promoter activity is tightly regulated by both LexA and DnaA, consistent with the presence of LexA- and DnaA-boxes in the promoters. Similarly, transcription from the plasmid-borne uvrBp3 was twofold higher in the dnaAA345S or ΔsulAΔlexA cells compared with that in the wild-type cells, and a slight further increase was also found in the ΔlexAΔsulA dnaAA345S mutant (Figure 4D) although the increase was not significant (Figure 4D inset). The results indicate that both DnaA and LexA repress the uvrB expression and function independently.
Repression of the uvrBp3 Promoter Is Largely Dependent on the DnaA-Box6
The footprinting analysis showed that both DnaA and LexA bound to DnaA-box5, 6, 7 and LexA-box4 of the uvrBp3 promoter. To determine the role of such binding sites in the control of the uvrBp3 promoter, we mutated the TG in DnaA-box6 to CA on the uvrBp3 plasmid (a derivative of pTAC3953 as described in section “Materials and Methods”) by site-directed mutagenesis, leading to the uvrBp3CA plasmid (Figure 5A). The mutations also changed the LexA-box4 since DnaA-box6 overlaps with LexA-box4 (Figure 5A) and destroyed the DARS1 site where ATP-DnaA is formed (Fujimitsu et al., 2009). It was found that transcription from uvrBp3CA was 5.9-fold higher relative to that from uvrBp3 in the wild-type cells (Figure 5B). Compared with uvrBp3, transcription from uvrBp3CA in the dnaAA345S cells was 6.3-fold greater (Figure 5B), indicating that mutation of the site can still influence transcription in the absence of full DnaA activity, probably by influencing LexA binding. In the ΔlexAΔsulA cells, transcription from uvrBp3CA was 13.4-fold higher relative to the activity of uvrBp3 (Figure 5B). Clearly, in the absence of LexA the mutation of the DnaA-box6 results in a dramatic change in transcription, suggesting that strong repression by the wild type ATP-DnaA is decreased due to the mutations. These same mutations can also impair ATP-DnaA regeneration activity at the DARS1, leading to a decrease in accumulation of ATP-DnaA compared with the wild type plasmid (Fujimitsu et al., 2009).

FIGURE 5. DnaA-box6 in promoter uvrBp3 and LexA-box1 in uvrBp1-2 are important for control of the promoters. Mutations (TG to CA) in DnaA-box6 on plasmid uvrBp3 (A) and mutations (TG to GC) in LexA-box1 on plasmid uvrBp1-2 (C) were introduced, resulting in plasmid puvrBp3CA-lacZ (indicated as uvrBp3CA for short) (A) and puvrBp1-2GC-lacZ (as uvrBp1-2GC) (C). The wild-type, dnaAA345S, ΔsulAΔlexA and ΔsulAΔlexA dnaAA345S cells were transformed with plasmid uvrBp3CA and uvrBp1-2GC, respectively. Relative activity of the mutated promoter uvrBp3CA (B) or uvrBp1-2GC (D) in the wild-type and the mutant cells was compared with that of uvrBp3 or uvrBp1-2 after measurement of the β-galactosidase activity as mentioned in the legend to Figure 2. The open bars stand for uvrBp3 or uvrBp1-2, the filled bars for uvrBp3CA or uvrBp1-2GC. The values shown at top of the bars are the average of three individual experiments, and standard errors are shown. ∗∗∗Stands for P-value ≤ 0.01, ∗∗for 0.01 < P-value ≤ 0.05, and ns represents P-value > 0.05.
LexA-Box1 Is Important for Control of uvrBp1-2 Transcription
The uvrBp1-2 promoter contains LexA-box1 and DnaA-box1. LexA-box1 remains strongly protected by LexA in the presence of high concentrations of DnaA. To understand the role of LexA-box1 in the regulation of uvrBp1-2 transcription, we mutated TG to GC in LexA-box1, resulting in plasmid uvrBp1-2GC (Figure 5C). These mutations scrambled LexA-box1 since the conserved trimer CTG on the left of LexA-box1 is destroyed. Transcription from uvrBp1-2GC was about threefold higher of that from uvrBp1-2 in the wild-type cells (Figure 5D), indicating that LexA-box1 is important for the control of promoter activity. This result also suggests that binding of LexA to the mutated LexA-box1GC is weakened. However, transcription from uvrBp1-2GC did not change relative to that from uvrBp1-2 in the dnaAA345S, ΔlexAΔsulA and ΔlexAΔsulA dnaAA345S cells (Figure 5D). The results indicate that the loss of repression in the mutant strains is thus the same in the presence or absence of the LexA-box1 mutation. This is expected in the case of the ΔlexAΔsulA strain, however, it is surprising in the dnaAA345S and ΔlexAΔsulA dnaAA345S cells since the LexA-box1 mutation should not influence binding by DnaA and a further increase in expression would be expected when DnaA is mutated. It is thus possible that this change in DNA sequence might also affect DnaA oligomerization or RNAP binding.
UV Irradiation Increases Transcription of the uvrB Gene
We found that transcription of the chromosomal uvrB gene was increased 3.2-fold in the uvrB-lacZ strain after UV irradiation (Figure 6A) in the wild-type cells. The level of expression did not significantly change in the ΔlexAΔsulA cells after UV irradiation (Figure 6A). These results confirm that the uvrB gene is one of the SOS genes which are regulated by the LexA protein (Howard-Flanders et al., 1966), responding to UV-induced DNA damage. Interestingly, uvrB gene transcription increased less, 1.8-fold, in the dnaAA345S cells indicating that DnaA’s decreased repressor activity results in a decreased change in gene expression in the presence of LexA. This is not the case in the ΔlexAΔsulA dnaAA345S cells after UV treatment (Figure 6A), indicating that LexA is required for UV-dependent SOS induction but not DnaA. These results are consistent with the need of both DnaA and LexA to maintain a high level of repression in the absence of UV treatment.
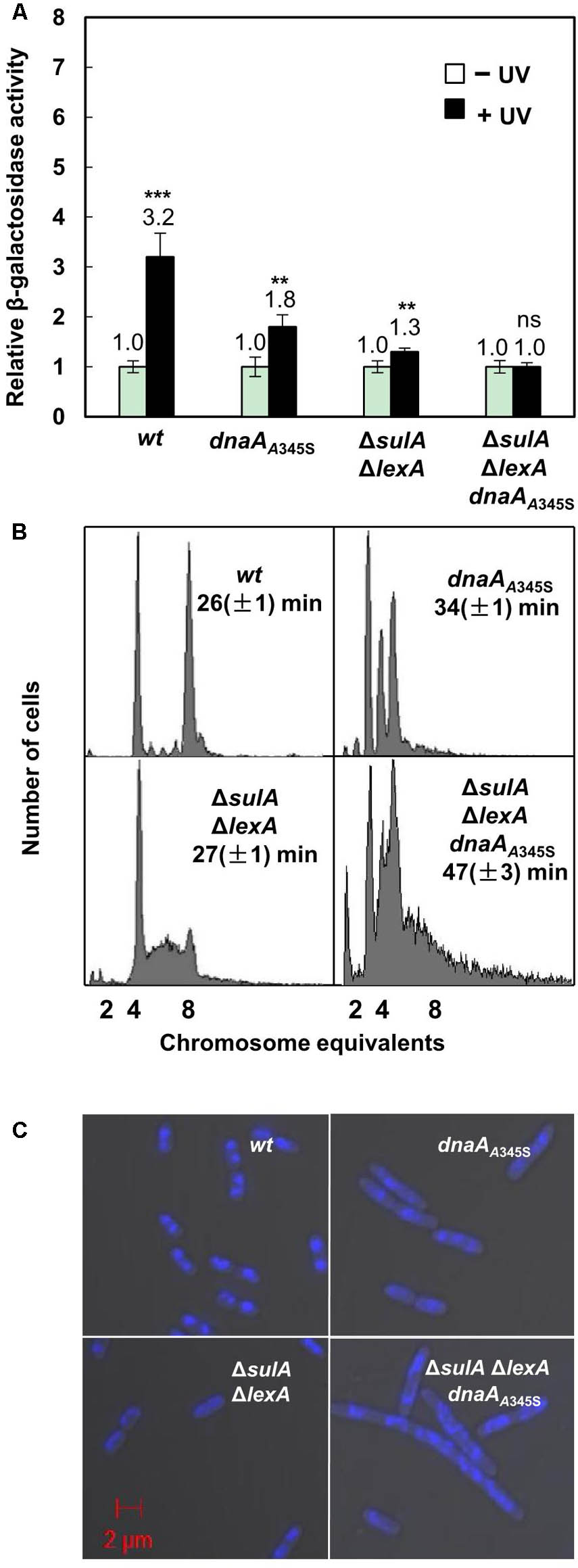
FIGURE 6. The simultaneous absence of LexA- and DnaA-repression leads to formation of elongated cells with incomplete replication and aberrant nucleoids. (A) Exponentially growing wild-type and mutant cells mentioned in the legend to Figure 1A except ΔsulA in ABTGcasa medium at 37°C were treated with UV (50 J/M2) at OD450 = 0.08. The expression of the uvrB gene was assayed as the β-galactosidase activity described in the legend to Figure 1. The filled bars represent expression after UV treatment relative to that from non-treated cells as indicated by the open bars. The values shown at top of the bars are the average of three individual experiments, and the standard errors are shown. ∗∗∗Stands for P-value ≤ 0.01, ∗∗for 0.01 < P-value ≤ 0.05, and ns represents P-value > 0.05. (B) Exponentially growing cells were treated for 4–5 generations with rifampicin and cephalexin to inhibit both initiation of replication and cell division but allowing ongoing replication finish. Then cells were analyzed by flow cytometer after staining with Hoechst 33258 for 30 min. The X-axis indicates chromosome equivalents per cell, the Y-axis represents the number of cells measured. Each measurement includes 10000 cells. The doubling time and genotype of the cells are shown. (C) Exponentially growing cells were harvested and fixed in 70% ethanol. Cells after staining in Hoechst 33258 for 30 min were visualized by Zeiss LSM710 confocal microscope as described in section “Materials and Methods.” The blue structures indicate nucleoids and the red scale bar represents 2 μm.
The Simultaneous Absence of Both LexA- and DnaA-Dependent Repression on Transcription Results in Elongated Cells With Incomplete DNA Replication
To clarify the role of DnaA-dependent repression on SOS-response genes and its link with DNA replication, we compared nucleoids and cell size in the dnaAA345S, ΔlexAΔsulA and ΔlexAΔsulA dnaAA345S cells with that of the wild-type cells. Flow cytometry analysis showed that the wild-type cells had four or eight fully replicated chromosomes, after rifampicin and cephalexin treatment (Figure 6B). The DNA histogram of dnaAA345S showed well-separated two-, three- and four-chromosome peaks (Figure 6B), indicating that the mutant cells contain fully replicated chromosomes although initiation of replication is asynchronous (Gon et al., 2006). However, the ΔlexAΔsulA cell culture showed that a portion of the cells contained a DNA amount between four- and eight-chromosome after rifampicin and cephalexin treatment, indicating that a portion of ΔlexAΔsulA cells has incomplete replication, probably due to overexpression of DNA repair proteins slowing down the replication forks (Figure 6B). The phenotype of incomplete replication was worsened in the ΔlexAΔsulA dnaAA345S cells. Some cells had only one-chromosome while other cells had more DNA than eight-chromosome equivalents (Figure 6B). These results underline the need for the wild-type DnaA to control both initiation of DNA replication and gene expression when the SOS response is activated. These results suggest that the ΔlexAΔsulA dnaAA345S cells have more serious DNA damage, even in the absence of UV irradiation, since severe incomplete replication can be caused from lack of replication elongation or/and partial degradation of the DNA (Skarstad and Boye, 1993; Morigen et al., 2003).
Exponentially growing cells were fixed in 70% ethanol and visualized under Zeiss LSM710 Confocal microscope. As shown in Figures 6B,C, both the wild-type and ΔlexAΔsulA cells were 2.4 ∼ 3.0 μm in length with a similar doubling time of 26 ∼ 27 min, containing mostly well-compacted one or two nucleoids. The dnaAA345S cells were about 4.5 μm in length with a doubling time of 34 min and more nucleoids per cell. The ΔlexAΔsulA dnaAA345S cells were further elongated (about 5.5 μm) with a slower growth, and their multi-nucleoids were not well-compacted (Figures 6B,C and Supplementary Figure S2). These results together suggest that the simultaneous absence of both LexA- and DnaA-dependent repression of gene transcription results in production of elongated cells with incomplete replication of DNA, aberrant nucleoids and slower growth. It is likely that DnaA-dependent repression of gene transcription during the SOS response is required to prevent over-expression of the SOS regulon genes to maintain genome integrity.
Transcriptions of the recN and dinJ Genes Are Also Repressed by Both DnaA and LexA
To test whether DnaA is also involved in regulation of other SOS regulon genes (Finch et al., 1985; Ruangprasert et al., 2014), we searched for DnaA-box and LexA-box in the dinJ and recN genes and found that both genes had LexA-boxes and a DnaA-box around the transcription start sites as shown in Supplementary Figure S3. We then inserted the lacZ reporter gene downstream of the chromosomal recN or dinJ genes, resulting in recN-lacZ or dinJ-lacZ derivatives of the MC4100 strain. The recN-lacZ or dinJ-lacZ allele was transferred to the dnaAA345S, ΔlexAΔsulA, ΔlexAΔsulA dnaAA345S and lexA3 cells. As shown in Figures 7A,B, transcriptions of both the recN and dinJ genes were about 2 ∼ 3-fold higher in the dnaAA345S, ΔlexAΔsulA and lexA3 dnaAA345S cells, respectively, indicating that transcription of recN or dinJ is repressed by both DnaA and LexA, and that the DnaA- or LexA-dependent repression of gene expression is independent. As expected, the increase in transcription of recN or dinJ was strengthened in the ΔlexAΔsulA dnaAA345S cells and it was significant relative to that in the ΔsulAΔlexA cells (Figure 7 insets), suggesting that repression from DnaA and LexA is additive. The observation is supported by the presence of an overlapping LexA-box with a DnaA-box in the recN or dinJ promoter (Fernandez De Henestrosa et al., 2000). We conclude that DnaA is, indeed, involved in control of other genes of the SOS regulon.
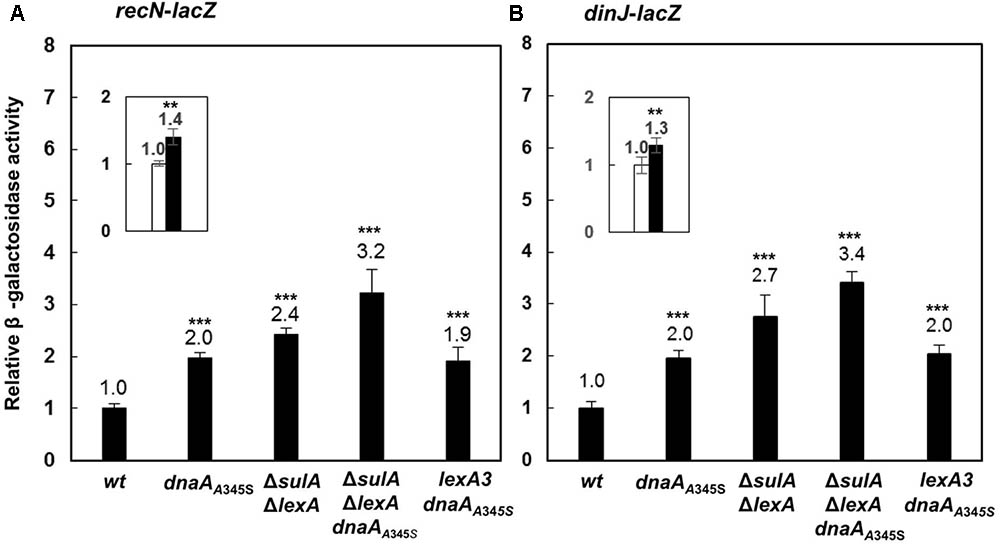
FIGURE 7. The DnaA and LexA proteins reppress expression of the recN or dinJ gene independently. The wild-type, dnaAA345S, ΔsulAΔlexA, ΔsulAΔlexA dnaAA345S and lexA3 dnaAA345S cells carrying recN-lacZ (A) or dinJ-lacZ (B) fusion on their chromosomes were exponentially grown in ABTGcasa medium at 37°C. The β-galactosidase activity from recN-lacZ (13 U for wild-type) (A) or dinJ-lacZ (13 U for wild-type) (B) fusion in the cells was determined as described in the legend to Figure 1. The insets indicate the relative expression of renN-lacZ or dinJ-lacZ in the ΔsulA ΔlexA dnaAA345S cells against that in the ΔsulAΔlexA cells. The values shown at top of the bars are the average of three individual experiments, and the standard errors are shown. ∗∗∗Stands P-value ≤ 0.01, ∗∗ for 0.01 < P-value ≤ 0.05, ns represents P-value > 0.05.
Discussion
The UvrB protein is a very important protein in the response to DNA damage in prokaryotic cells, performing NER with UvrA and UvrC (Truglio et al., 2006). UvrB paralogs have been found in all organisms (Sancar and Reardon, 2004) and the NER repair system plays a central role in maintaining genome integrity. Defects in NER cause several lines of diseases in humans including skin cancers (Lehmann, 2003). However, the control mechanism of uvrB gene expression remains elusive. The results presented here show that the DNA replication initiator, the DnaA protein, and the SOS regulator LexA, regulate the expression of the uvrB gene by interacting with the uvrB promoter region. The regulation mode by both DnaA and LexA applies to the expression control of several SOS genes, and may be conserved in Gram-negative bacteria. However, this hypothesis requires further experiments to be confirmed.
DnaA and LexA Regulate Transcription of the uvrB Gene By Binding to Their Specific Sites
We have shown that transcription from the uvrB promoters is repressed by the presence of both the wild-type LexA and DnaA proteins (Figure 1A). The full activity of the uvrB promoter cluster requires both uvrBp1-2 and uvrBp3 promoters and is repressed by both DnaA and LexA in an additive manner (Figures 4B–D). The DNase I footprinting experiments show that both proteins bind to the promoter region and that LexA-box1 in uvrBp1-2 is the strongest LexA-box from which LexA is not easily displaced by increasing amounts of ATP-DnaA, unlike what is observed at the other LexA-box in this region (Figure 3). LexA-box1 has a typical consensus sequence containing both CTG and CAG trimers at the two ends (Walker, 1984; Fernandez De Henestrosa et al., 2000; Wade et al., 2005) while LexA-box2, 3, and 4 do not. Mutations of LexA-box1 and DnaA-box6 in fact show a strong effect on promoter activity in vivo (Figure 5). The results indicate that binding of LexA or DnaA to LexA-boxes or DnaA-boxes in the uvrB promoter region results in a direct control of promoter activity.
The uvrBp3 promoter is interesting because it completely overlaps with the DARS1 sequence. While the DARS1 sequence is not essential, in its absence initiation of DNA replication occurs at an increased cell mass (Fujimitsu et al., 2009). Binding of LexA to DARS1 and RNA polymerase to the uvrBp3 promoter could both compete with DnaA binding and thus interfere with the regeneration of ATP-DnaA both in the absence (intact LexA) and the presence (RNAP binding) of the SOS response. This can explain the increased loss of repression by the DnaA-box6 mutation in the ΔlexAΔsulA strain (Figure 5B).
DnaA and LexA May Coordinate DNA Replication With DNA Repair
LexA-dependent regulation of DARS1 activity is only one of several processes resulting in an increased ATP-DnaA to ADP-DnaA ratio following DNA damage. Upon prolonged stress, fork stalling and blockage of DNA replication, ATP-DnaA accumulates in the cell (Kurokawa et al., 1999). Hydrolysis of the ATP bound to DnaA is mediated by the RIDA process, which requires ongoing DNA replication (Katayama et al., 1998). When the DNA replication forks stall in the presence of DNA damage ATP-DnaA can thus accumulate in the cell and bind to the other sites on the genome. The longer DNA replication has been stalled, the more ATP-DnaA has accumulated in the cell. This would ensure that the expression of DNA repair proteins by the SOS response is limited when the DNA replication forks have stalled and there is less DNA per cell. Furthermore, expression of the dnaA gene is induced by DNA damage in a RecA and LexA-dependent manner despite the absence of a LexA-box at the dnaA promoter region (Quinones et al., 1991). In these conditions SeqA has been shown to play a key role in cell survival, possibly by limiting over-initiation of DNA replication by increased amounts of ATP-DnaA in the presence of stalled replication forks (Sutera and Lovett, 2006). The increase in ATP-DnaA by decreased RIDA, increased gene expression and increased DARS1 activity occur at the same time as loss of repression by LexA. This may appear to be inconsistent, since they both repress gene expression at several shared targets. However, it is possible that upon DNA damage LexA cleavage results in a rapid loss of repression while the increase in ATP-DnaA occurs with a time delay, due in part to protein synthesis, resulting in a pulse of transcription activity, which, however, is proportional to the number of replication forks, due the amount of ATP-DnaA-dependent repression observed in the absence of DNA damage. It appears to be a situation similar to the hyperinitiation stress observed during oxidative damage (Charbon et al., 2014), but as a sensible response to the problem.
DnaA-Dependent Transcription Repression During the SOS Response Is Required for Maintaining Genome Integrity: A Model for Control of the SOS Regulon by LexA and DnaA
As a summary, we propose a model for the control of the SOS regulon by DnaA and LexA (Figure 8). Both DnaA and LexA repress expression of the SOS genes when the SOS response is off. When the SOS response is triggered due to DNA damage, LexA is self-cleaved (Little et al., 1980), consequently LexA-repression is removed to derepress expression of the SOS genes to repair the DNA damage (Sancar et al., 1982). During the SOS response, DnaA-dependent repression still works and largely limits the expression of the SOS genes, resulting in cells with normal nucleoids and growth rate but minor incomplete replication. The latter can be the indication of the DNA repair process since it includes controlled nicking and digestions of the DNA. The simultaneous absence of DnaA- and LexA-repression leads to cell elongation with serious incomplete replication, uncompacted nucleoids and slow growth (Figures 6B,C), possibly as a result of a further increase in expression of SOS dependent genes (Figures 1A, 7A,B). Obviously, the high level expression of the SOS genes is harmful, causing physiological problems in different cell processes. Among these problems, incomplete DNA replication can be caused by either lack of replication elongation or partial degradation of the DNA (Skarstad and Boye, 1993; Morigen et al., 2003) as a result of DNA damage. It is reasonable to consider that an excess of DNA repair proteins might “repair” DNA regions where the repairs are unwanted, resulting in DNA damage. Indeed, for example overproduction of DinB has been shown to be lethal (Margara et al., 2016). It is likely that DnaA-dependent repression of the transcription of SOS genes during the SOS response is required to prevent over-expression of the SOS genes to maintain genome integrity.
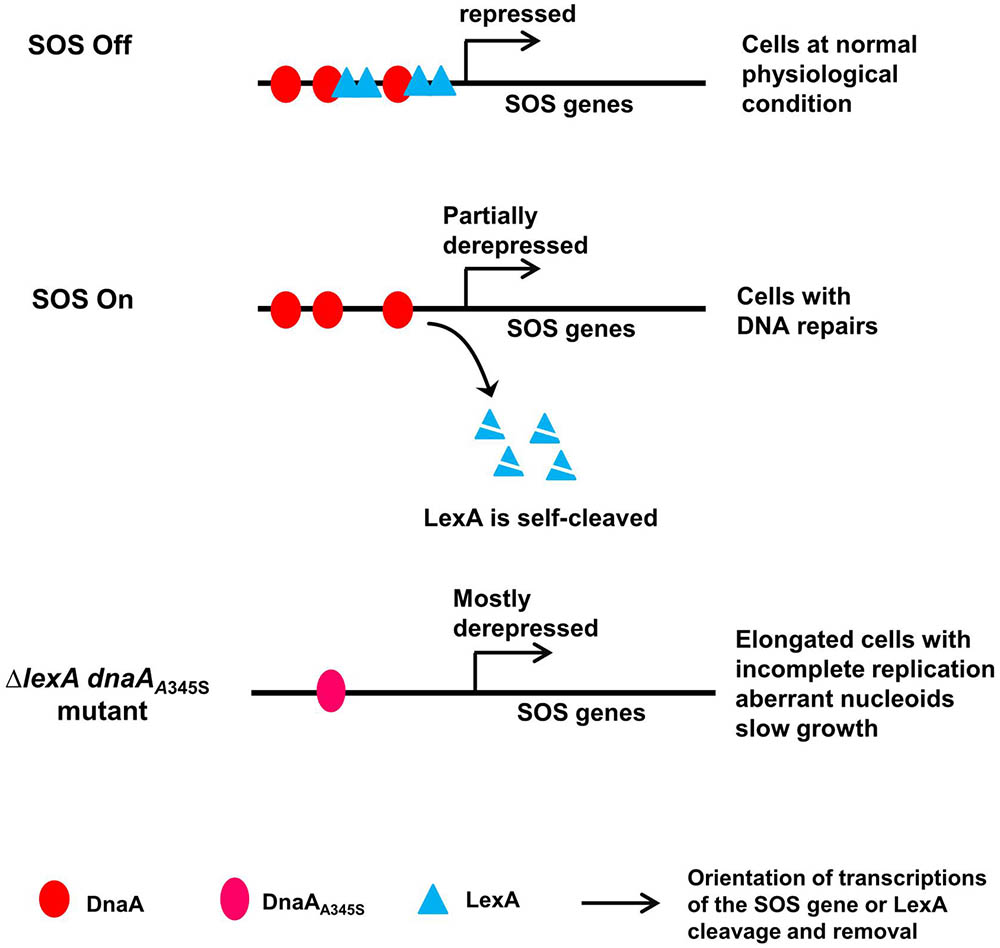
FIGURE 8. A model for control of the SOS regulon by DnaA and LexA. In the proposed model, both DnaA and LexA repress expression of the SOS genes when SOS is off. When SOS is on, LexA is self-cleaved, but DnaA-repression still works, leading to partial expression of the SOS genes and consequent cells with normal nucleoids and growth. In the simultaneous absence of LexA- and DnaA-repression, the SOS genes are mostly derepressed, forming elongated cells with incomplete replication, aberrant nucleoids and slow growth. The red ovals represent the DnaA protein, the cyan triangles represent the LexA protein, the pink oval represents the DnaAA345S mutant protein, arrows indicate orientation of transcriptions of the SOS gene or LexA cleavage and removal as indicated.
Interestingly, 13 DnaA-boxes are found in potential LexA-boxes on the E. coli chromosomes (Fernandez De Henestrosa et al., 2000). In further analysis, the overlapping LexA-box with a DnaA-box in the uvrB or recN promoter was found in several Gram-negative bacteria including Salmonella typhimurium, Serratia marcescens, Citrobacter rodentium, Klebsiella pneumoniae and Yersinia enterocolitica (Supplementary Table S1). These results suggest that DnaA is likely involved in regulation of the SOS regulon, reducing expression of the SOS genes during the SOS response in a manner that is coupled with DNA replication. The control mode may be conserved within Gram-negative bacteria.
Author Contributions
M, W, and BS conceived and designed the experiments. W, G, EB, SW, and HS performed the experiments. M, W, BS, LF, and YS analyzed the data. M and BS provided reagents. M, W, and BS wrote the manuscript. All authors read the manuscript.
Funding
This work was supported by grants from the National Natural Science Foundation of China “NSFC” (Grant No. 31560245 to M); the Natural Science Foundation of Inner Mongolia (Grant No. 20102009 to M), and the Program of Higher-level Talents of Inner Mongolia University “SPH-IMU” (Grant No. Z20090107 to M).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Erik Boye for his critical reading and comments. We also thank “National BioResource Project (NIG, Japan): E. coli” for providing strains.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.01212/full#supplementary-material
Abbreviations
ADP/ATP-DnaA, ADP or ATP binds DnaA; bla, ampicillin resistance gene; cat, chloramphenicol resistance gene; DARS, DnaA reactivation site; DnaA-box, DnaA binding site; FRT, FLP recognition target; LexA-box, LexA binding site; neo, kanamycin resistance gene; NER, nucleotide excision repair; RIDA, regulatory inactivation of DnaA; ssDNA, single strand DNA; tet, tetracycline resistant gene.
References
Arikan, E., Kulkarni, M. S., Thomas, D. C., and Sancar, A. (1986). Sequences of the E. coli uvrB gene and protein. Nucleic Acids Res. 14, 2637–2650. doi: 10.1093/nar/14.6.2637
Atlung, T., Clausen, E. S., and Hansen, F. G. (1985). Autoregulation of the dnaA gene of Escherichia coli K12. Mol. Gen. Genet. 200, 442–450. doi: 10.1007/BF00425729
Baba, T., Ara, T., Hasegawa, M., Takai, Y., Okumura, Y., Baba, M., et al. (2006). Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2:2006.0008. doi: 10.1038/msb4100050
Bertani, G. (1951). Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J. Bacteriol. 62, 293–300.
Boye, E., and Lobner-Olesen, A. (1991). Bacterial growth control studied by flow cytometry. Res. Microbiol. 142, 131–135. doi: 10.1016/0923-2508(91)90020-B
Braun, R. E., O’day, K., and Wright, A. (1985). Autoregulation of the DNA replication gene dnaA in E. coli K-12. Cell 40, 159–169.
Brondsted, L., and Atlung, T. (1994). Anaerobic regulation of the hydrogenase 1 (hya) operon of Escherichia coli. J. Bacteriol. 176, 5423–5428. doi: 10.1128/jb.176.17.5423-5428.1994
Casadaban, M. J. (1976). Transposition and fusion of the lac genes to selected promoters in Escherichia coli using bacteriophage lambda and Mu. J. Mol. Biol. 104, 541–555. doi: 10.1016/0022-2836(76)90119-4
Charbon, G., Bjørn, L., Mendozachamizo, B., Frimodtmøller, J., and Løbnerolesen, A. (2014). Oxidative DNA damage is instrumental in hyperreplication stress-induced inviability of Escherichia coli. Nucleic Acids Res. 42, 13228–13241. doi: 10.1093/nar/gku1149
Clark, A. J. (1973). Recombination deficient mutants of E. coli and other bacteria. Annu. Rev. Genet. 7, 67–86. doi: 10.1146/annurev.ge.07.120173.000435
Datsenko, K. A., and Wanner, B. L. (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645. doi: 10.1073/pnas.120163297
Ellermeier, C. D., Janakiraman, A., and Slauch, J. M. (2002). Construction of targeted single copy lac fusions using lambda Red and FLP-mediated site-specific recombination in bacteria. Gene 290, 153–161. doi: 10.1016/S0378-1119(02)00551-6
Ferenci, T., Zhou, Z., Betteridge, T., Ren, Y., Liu, Y., Feng, L., et al. (2009). Genomic sequencing reveals regulatory mutations and recombinational events in the widely used MC4100 lineage of Escherichia coli K-12. J. Bacteriol. 191, 4025–4029. doi: 10.1128/JB.00118-09
Fernandez De Henestrosa, A. R., Ogi, T., Aoyagi, S., Chafin, D., Hayes, J. J., Ohmori, H., et al. (2000). Identification of additional genes belonging to the LexA regulon in Escherichia coli. Mol. Microbiol. 35, 1560–1572. doi: 10.1046/j.1365-2958.2000.01826.x
Finch, P. W., Chambers, P., and Emmerson, P. T. (1985). Identification of the Escherichia coli recN gene product as a major SOS protein. J. Bacteriol. 164, 653–658.
Fujimitsu, K., Senriuchi, T., and Katayama, T. (2009). Specific genomic sequences of E. coli promote replicational initiation by directly reactivating ADP-DnaA. Genes Dev. 23, 1221–1233. doi: 10.1101/gad.1775809
George, J., Castellazzi, M., and Buttin, G. (1975). Prophage induction and cell division in E. coli. III. Mutations sfiA and sfiB restore division in tif and lon strains and permit the expression of mutator properties of tif. Mol. Gen. Genet. 140, 309–332.
Gon, S., Camara, J. E., Klungsoyr, H. K., Crooke, E., Skarstad, K., and Beckwith, J. (2006). A novel regulatory mechanism couples deoxyribonucleotide synthesis and DNA replication in Escherichia coli. EMBO J. 25, 1137–1147. doi: 10.1038/sj.emboj.7600990
Howard-Flanders, P., Boyce, R. P., and Theriot, L. (1966). Three loci in Escherichia coli K-12 that control the excision of pyrimidine dimers and certain other mutagen products from DNA. Genetics 53, 1119–1136.
Katayama, T., Kubota, T., Kurokawa, K., Crooke, E., and Sekimizu, K. (1998). The initiator function of DnaA protein is negatively regulated by the sliding clamp of the E. coli chromosomal replicase. Cell 94, 61–71.
Kornberg, A., and Baker, T. A. (1992). “Initiation from the E. coli chromosomal origin (oriC),” in DNA Replication, 2nd Edn, eds A. Kornberg and T. A. Baker (New York, NY: W. H. Freeman), 524–536.
Krause, M., Ruckert, B., Lurz, R., and Messer, W. (1997). Complexes at the replication origin of Bacillus subtilis with homologous and heterologous DnaA protein. J. Mol. Biol. 274, 365–380. doi: 10.1006/jmbi.1997.1404
Kurokawa, K., Nishida, S., Emoto, A., Sekimizu, K., and Katayama, T. (1999). Replication cycle-coordinated change of the adenine nucleotide-bound forms of DnaA protein in Escherichia coli. EMBO J. 18, 6642–6652. doi: 10.1093/emboj/18.23.6642
Lehmann, A. R. (2003). DNA repair-deficient diseases, xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. Biochimie 85, 1101–1111. doi: 10.1016/j.biochi.2003.09.010
Little, J. W., Edmiston, S. H., Pacelli, L. Z., and Mount, D. W. (1980). Cleavage of the Escherichia coli lexA protein by the recA protease. Proc. Natl. Acad. Sci. U.S.A. 77, 3225–3229. doi: 10.1073/pnas.77.6.3225
Lother, H., Kölling, R., Kücherer, C., and Schauzu, M. (1985). dnaA protein-regulated transcription: effects on the in vitro replication of Escherichia coli minichromosomes. EMBO J. 4, 555–560.
Margara, L. M., Fernandez, M. M., Malchiodi, E. L., Argarana, C. E., and Monti, M. R. (2016). MutS regulates access of the error-prone DNA polymerase Pol IV to replication sites: a novel mechanism for maintaining replication fidelity. Nucleic Acids Res. 44, 7700–7713. doi: 10.1093/nar/gkw494
Markham, B. E., Little, J. W., and Mount, D. W. (1981). Nucleotide sequence of the lexA gene of Escherichia coli K-12. Nucleic Acids Res. 9, 4149–4161. doi: 10.1093/nar/9.16.4149
Maxam, A. M., and Gilbert, W. (1980). Sequencing end-labeled DNA with base-specific chemical cleavages. Methods Enzymol. 65, 499–560. doi: 10.1016/S0076-6879(80)65059-9
Miller, J. H. (1972). “Analysis of β-galactosidase,” in Experiments in Molecular Genetics, 1st Edn, ed. J. H. Miller (New York, NY: Cold Spring Harbor Laboratory Press), 352–355.
Miller, R., Ripp, S., Replicon, J., Ogunseitan, O., and Kokjohn, T. A. (1992). “Virus-mediated gene transfer in freshwater environments,” in Gene Transfers and Environment, 1st Edn, ed. M. J. Gauthier (Berlin: Springer), 51–62.
Morigen, Boye, E., Skarstad, K., and Lobner-Olesen, A. (2001). Regulation of chromosomal replication by DnaA protein availability in Escherichia coli: effects of the datA region. Biochim. Biophys. Acta 1521, 73–80. doi: 10.1016/S0167-4781(01)00292-5
Morigen, Lobner-Olesen, A., and Skarstad, K. (2003). Titration of the Escherichia coli DnaA protein to excess datA sites causes destabilization of replication forks, delayed replication initiation and delayed cell division. Mol. Microbiol. 50, 349–362. doi: 10.1046/j.1365-2958.2003.03695.x
Morigen, Molina, F., and Skarstad, K. (2005). Deletion of the datA site does not affect once-per-cell-cycle timing but induces rifampin-resistant replication. J. Bacteriol. 187, 3913–3920. doi: 10.1128/JB.187.12.3913-3920.2005
Olliver, A., Saggioro, C., Herrick, J., and Sclavi, B. (2010). DnaA-ATP acts as a molecular switch to control levels of ribonucleotide reductase expression in Escherichia coli. Mol. Microbiol. 76, 1555–1571. doi: 10.1111/j.1365-2958.2010.07185.x
Ortenberg, R., Gon, S., Porat, A., and Beckwith, J. (2004). Interactions of glutaredoxins, ribonucleotide reductase, and components of the DNA replication system of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 101, 7439–7444. doi: 10.1073/pnas.0401965101
Pruteanu, M., and Baker, T. A. (2009). Proteolysis in the SOS response and metal homeostasis in Escherichia coli. Res. Microbiol. 160, 677–683. doi: 10.1016/j.resmic.2009.08.012
Quinones, A., Jueterbock, W. R., and Messer, W. (1991). DNA lesions that block DNA replication are responsible for the DnaA induction caused by DNA damage. Mol. Gen. Genet. 231, 81–87. doi: 10.1007/BF00293825
Quinones, A., Wandt, G., Kleinstauber, S., and Messer, W. (1997). DnaA protein stimulates polA gene expression in Escherichia coli. Mol. Microbiol. 23, 1193–1202. doi: 10.1046/j.1365-2958.1997.2961658.x
Rousseau, A., Mcewen, A. G., Poussin-Courmontagne, P., Rognan, D., Nomine, Y., Rio, M. C., et al. (2013). TRAF4 is a novel phosphoinositide-binding protein modulating tight junctions and favoring cell migration. PLoS Biol. 11:e1001726. doi: 10.1371/journal.pbio.1001726
Ruangprasert, A., Maehigashi, T., Miles, S. J., Giridharan, N., Liu, J. X., and Dunham, C. M. (2014). Mechanisms of toxin inhibition and transcriptional repression by Escherichia coli DinJ-YafQ. J. Biol. Chem. 289, 20559–20569. doi: 10.1074/jbc.M114.573006
Sancar, A., and Reardon, J. T. (2004). Nucleotide excision repair in E. coli and man. Adv. Protein Chem. 69, 43–71. doi: 10.1016/S0065-3233(04)69002-4
Sancar, G. B., Sancar, A., Little, J. W., and Rupp, W. D. (1982). The uvrB gene of Escherichia coli has both lexA-repressed and lexA-independent promoters. Cell 28, 523–530. doi: 10.1016/0092-8674(82)90207-0
Schaper, S., and Messer, W. (1995). Interaction of the initiator protein DnaA of Escherichia coli with its DNA target. J. Biol. Chem. 270, 17622–17626. doi: 10.1074/jbc.270.29.17622
Sekimizu, K., Bramhill, D., and Kornberg, A. (1987). ATP activates DnaA protein in initiating replication of plasmids bearing the origin of the E. coli chromosome. Cell 50, 259–265. doi: 10.1016/0092-8674(87)90221-2
Skarstad, K., and Boye, E. (1993). Degradation of individual chromosomes in recA mutants of Escherichia coli. J. Bacteriol. 175, 5505–5509. doi: 10.1128/jb.175.17.5505-5509.1993
Skarstad, K., Boye, E., and Steen, H. B. (1986). Timing of initiation of chromosome replication in individual Escherichia coli cells. EMBO J. 5, 1711–1717.
Speck, C., Weigel, C., and Messer, W. (1999). ATP- and ADP-DnaA protein, a molecular switch in gene regulation. EMBO J. 18, 6169–6176. doi: 10.1093/emboj/18.21.6169
Sutera, V. A. Jr., and Lovett, S. T. (2006). The role of replication initiation control in promoting survival of replication fork damage. Mol. Microbiol. 60, 229–239. doi: 10.1111/j.1365-2958.2006.05093.x
Truglio, J. J., Croteau, D. L., Van Houten, B., and Kisker, C. (2006). Prokaryotic nucleotide excision repair: the UvrABC system. Chem. Rev. 106, 233–252. doi: 10.1021/cr040471u
Tuggle, C. K., and Fuchs, J. A. (1986). Regulation of the operon encoding ribonucleotide reductase in Escherichia coli: evidence for both positive and negative control. EMBO J. 5, 1077–1085.
Van Den Berg, E. A., Geerse, R. H., Memelink, J., Bovenberg, R. A., Magnee, F. A., and Van De Putte, P. (1985). Analysis of regulatory sequences upstream of the E. coli uvrB gene; involvement of the DnaA protein. Nucleic Acids Res. 13, 1829–1840. doi: 10.1093/nar/13.6.1829
Wade, J. T., Reppas, N. B., Church, G. M., and Struhl, K. (2005). Genomic analysis of LexA binding reveals the permissive nature of the Escherichia coli genome and identifies unconventional target sites. Genes Dev. 19, 2619–2630. doi: 10.1101/gad.1355605
Walker, G. C. (1984). Mutagenesis and inducible responses to deoxyribonucleic acid damage in Escherichia coli. Microbiol. Rev. 48, 60–93.
Walter, G. C. (1996). “The SOS response of Escherichia coli,” in Escherichia coli and Salmonella typhimurium: Cellular and Molecular Biology, eds F. C. NeNeidhardt, R. Curtiss III, J. L. Ingraham, E. C. C. Lin, K. B. Low, and B. Magasanik (Washington, DC: American Society for Microbiology Press), 1400–1416.
Keywords: DnaA, LexA, uvrB gene expression, SOS regulon, regulation
Citation: Wurihan, Gezi, Brambilla E, Wang S, Sun H, Fan L, Shi Y, Sclavi B and Morigen (2018) DnaA and LexA Proteins Regulate Transcription of the uvrB Gene in Escherichia coli: The Role of DnaA in the Control of the SOS Regulon. Front. Microbiol. 9:1212. doi: 10.3389/fmicb.2018.01212
Received: 09 March 2018; Accepted: 17 May 2018;
Published: 18 June 2018.
Edited by:
Daniela De Biase, Sapienza Università di Roma, ItalyReviewed by:
Eduard Torrents, Institute for Bioengineering of Catalonia, SpainAlan Leonard, Florida Institute of Technology, United States
Tove Atlung, Roskilde University, Denmark
Copyright © 2018 Wurihan, Gezi, Brambilla, Wang, Sun, Fan, Shi, Sclavi and Morigen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bianca Sclavi, c2NsYXZpQGxicGEuZW5zLWNhY2hhbi5mcg== Morigen, bW9yaWdlbm1AaG90bWFpbC5jb20=