 Yatao Guo1
Yatao Guo1 Yao Zhai2Zhao Zhang1Daixi Li1Zhanwei Wang3Jingquan Li4
Yao Zhai2Zhao Zhang1Daixi Li1Zhanwei Wang3Jingquan Li4 Zilong He5
Zilong He5 Songnian Hu5Yu Kang5*Zhancheng Gao1*
Songnian Hu5Yu Kang5*Zhancheng Gao1*- 1Department of Respiratory and Critical Care Medicine, Peking University People’s Hospital, Beijing, China
- 2University of Technology Sydney, Ultimo, NSW, Australia
- 3Laboratory Medicine, Peking University People’s Hospital, Beijing, China
- 4Institute of Botany, Chinese Academy of Sciences, Beijing, China
- 5Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing, China
Bacterial isolate X39 was isolated from a community-acquired pneumonia patient in Beijing, China. A phylogenetic tree based on rpoB genes and average nucleotide identity data confirmed that isolate X39 belonged to Klebsiella variicola. The genome of K. variicola X39 contained one circular chromosome and nine plasmids. Comparative genomic analyses with other K. variicola isolates revealed that K. variicola X39 contained the most unique genes. Of these unique genes, many were prophages and transposases. Many virulence factors were shared between K. variicola X39 and Klebsiella pneumoniae F1. The pathogenicity of K. variicola X39 was compared with that of K. pneumoniae F1 in an abdominal infection model. The results indicated that K. variicola X39 was less virulent than typical clinical K. pneumoniae F1. The genome of K. variicola X39 also contained some genes involved in plant colonization, nitrogen fixation, and defense against oxidative stress. GFP-labeled K. variicola X39 could colonize maize as an endophytic bacterium. We concluded that K. variicola X39 was a kingdom-crossing strain.
Introduction
The Klebsiella genus consists of pathogens capable of colonizing and infecting humans and animals, as well as bacteria capable of colonizing plants as endophytes (Podschun and Ullmann, 1998). As the leading Klebsiella pathogen, Klebsiella pneumoniae is one of the most common pathogens of hospital-acquired pneumonia (Chung et al., 2011). Nevertheless, with the development of sequencing technology and molecular epidemiology, methods of phylogenetic reconstruction can divide K. pneumoniae into three distinct Klebsiella species: K. pneumoniae, K. quasipneumoniae, and K. variicola (Rosenblueth et al., 2004; Brisse et al., 2014; Maatallah et al., 2014). K. variicola was first proposed as a new Klebsiella species in 2004 based on clinical and plant-associated isolates (Rosenblueth et al., 2004). The species K. singaporensis was first isolated from a single soil isolate in 2004 (Li et al., 2004) and identified as a junior heterotypic synonym of K. variicola (Brisse et al., 2014). K. variicola is able to fix N2 and is abundant in the environment. As the current VITEK automated system is unable to efficiently distinguish the Klebsiella species, some of the clinical Klebsiella isolates previously considered Klebsiella pneumoniae may in fact be Klebsiella variicola or K. quasipneumoniae. For example, former K. pneumoniae 342 was identified as an isolate of K. variicola (Brisse et al., 2014). K. pneumoniae KPC-142 originally reported as K. pneumoniae by VITEK 2 system was actually K. quasipneumoniae subsp. similipneumoniae KPC-142 (Nicolas et al., 2018). Most of the K. variicola strains currently reported were found in the environment, and case reports of K. variicola infection were rare. The potential for clinical K. variicola isolates to colonize plants is poorly understood, and the mechanism of plant-associated Klebsiella isolates as drug-resistance genes reservoirs remains unclear.
In the present study, K. variicola X39 was isolated from the sputum of a community-acquired pneumonia patient and revealed resistance to a variety of antibiotics. We compared the pathogenicity of K. variicola X39 with that of typical clinical K. pneumoniae F1. On the other hand, we confirmed that K. variicola X39 was able to colonize plant roots, stems, and leaves by labeling K. variicola X39 with green fluorescent protein (GFP). Here, we analyzed the genomic characteristics of K. variicola X39 and laid the foundation for understanding the resistance, virulence, endophytic lifestyle of K. variicola.
Materials and Methods
Bacterial Isolation, Clinical Information, Biochemical Identification, and Antimicrobial Susceptibility Testing
An acute renal failure patient with pulmonary infection was admitted to the Department of Nephrology, Peking University People’s Hospital in March 2011. K. pneumoniae (eventually identified as K. variicola) was cultured three times consecutively in the sputum of the patient. Written informed consent was obtained from the patient. The isolate was designated X39 for further study. The patient eventually died of lung infection (Supplementary Figure S1) and heart failure. Biochemical identification of isolate X39 was determined using the VITEK 2 system and API 20E biochemical assays (bioMérieux, Marcy-l’Étoile, France). The antimicrobial susceptibilities of isolate X39 were determined by identifying the minimum inhibitory concentration (MIC) values by the VITEK 2 system, in accordance with the guidelines of the Clinical and Laboratory Standards Institute.
Genome Sequencing and Assembly
Klebsiella variicola X39 was cultured overnight in Luria–Bertani broth at 37°C. We used a QIAamp DNA Mini Kit (QIAGEN, Hilden, Germany) to extract the genomic DNA and determined the quality and concentration of DNA using a NanoDrop spectrometer (Thermo Scientific, Wilmington, MA, United States). A 4- to 10-kb insert library was obtained from the genomic DNA of K. variicola X39 and sequenced by Pacific Bioscience’s (PacBio, Menlo Park, CA, United States) Single Molecule, Real-Time (SMRT) sequencing technology at the Beijing Institute of Genomics, Chinese Academy of Sciences. Sequencing was performed on two SMRT cells, and average coverage was 84×. The pacific sequencing yielded 201,794 high-quality filtered reads with an average length of 3,170 bp, and the N50 read length was 4,556 bp. Hierarchical genome assembly process (HGAP) with SMRT analysis 3.0 was used to assemble the high-quality reads. The preliminary assembly results obtained were compared and analyzed. The chromosomal and plasmid sequences were screened and, respectively, assembled into circular DNA, i.e., the final 0 gap complete sequence.
Genome Annotation
GeneMarkS software was used to predict the protein-encoding genes (Besemer et al., 2001). The GO, KEGG, COG, and NR annotation of each predicted gene was assigned based on the results of BLASTP (E-value ≤10-5; identity ≥40%; coverage ≥40%). TRNAscan-SE version 1.3.1 (Lowe and Eddy, 1997), rRNAmmer version 1.2 (Lagesen et al., 2007), and Rfam (database version 11) (Gardner et al., 2009; Nawrocki et al., 2009) were used to predict the tRNAs, rRNAs, and sRNAs, respectively. Genomic islands (GIs), prophages, and CRISPRS were predicted by IslandPath DIMOB (Hsiao et al., 2005), PHAST (database download date: November 10, 2014) (Zhou et al., 2011), and CRISPRFinder1 (Grissa et al., 2007).
Phylogenetic Analyses and Average Nucleotide Identity Analysis
The complete nucleotide sequence of rpoB genes from other K. pneumoniae, K. quasipneumoniae, and K. variicola isolates (Supplementary Table S1) was downloaded from the GenBank database. The rpoB genes from Klebsiella oxytoca CAV1374 (CP011636.1), Klebsiella oxytoca KONIH1 (CP008788.1), and Escherichia coli K-12-MG1655 (U00096.3) were also included in the phylogenetic analysis. The phylogenetic tree was constructed using PHYML (maximum likelihood, ML) with a Tamura-Nei parameter model and 1,000 bootstrap replications (Mega X) (Kumar et al., 2018). Average nucleotide identity analysis was performed between K. variicola X39 and other Klebsiella isolates (Supplementary Table S1) included in the phylogenetic tree using an online ANI Calculator2 (Yoon et al., 2017).
Comparative Genomic Analyses
Klebsiella variicola X39 was clinically isolated. We further selected two non-clinical K. variicola isolates - K. variicola 342 (Brisse et al., 2014) and K. variicola DX120E (Lin et al., 2015) - and two clinical K. variicola isolates - K. variicola MGH20 and K. variicola MGH40 - whose genome sequences were available in GenBank. The homologous genes were calculated using Cd hit (v4.6.1) with a threshold of default 0.5 (Li and Godzik, 2006). Homologous genes present in all five isolates served as core genes. After removing the core genes, we obtained the unique genes of each isolate.
Pathogenicity Testing
In order to evaluate the pathogenicity of K. variicola X39, we generated an abdominal infection animal model. This study was performed in accordance with the principles of the Basel Declaration and recommendations of the National Laboratory Animal Standardization Technical Committee. The protocol was approved by the Peking University People’s Hospital Institutional Animal Care and Use Committee (2015–27). K. pneumoniae isolate F1 was a clinical strain isolated from a patient with urinary tract infection in our laboratory, where we sequenced its genome (CP026130.1). K. pneumoniae isolate F1 was also included in the study. Eight-week-old female BALB/c mice were obtained from Sibeifu (Beijing, China) Laboratory Animal Science and Technology Co., Ltd. The overnight culture of K. variicola X39 and K. pneumoniae F1 were washed by saline and resuspended in saline. 5 × 109, 5 × 108, 5 × 107, and 5 × 106 CFU/ml were obtained by serial dilution to determine the median lethal dosage (LD50). Six mice were injected at each concentration. Each BALB/c mouse was injected 200 μL bacterial suspension intraperitoneally. Six mice were injected with 200 μL saline each mouse as a negative control group. All mice were monitored daily for survival for 14 days. The LD50 was calculated using the probit regression model of SPSS 24.
K. variicola X39 Competent Cells
The competent cells of K. variicola X39 were prepared as follows: frozen K. variicola X39 was inoculated on a blood agar plate and cultured overnight at 37°C. The isolates were then inoculated in 15 mL precooled ultrapure water and immediately centrifuged at 5,000 rpm for 5 min at 4°C. The supernatant was removed and discarded, and the cell pellet was resuspended in 15 mL precooled ultrapure water. These steps were repeated twice. The density of bacteria was measured by the WGZ-2XJ Bacteria Turbidity Meter (Shanghai Xin Rui, Shanghai, China). The bacterial turbidity was finally concentrated to 3, and the suspension was divided into 100-μL tubes.
Construction of GFP-Labeled K. variicola X39
Vector plasmid puA66 was provided by Professor Uri Alon, which contained GFP and kanamycin-resistance gene kan (Figure 4A). A CTX-M-14 gene promoter was inserted upstream of the GFP open reading frame in the pUA66 plasmid so that the fluorescence intensity of GFP could be directly observed by fluorescence microscopy (Wang et al., 2014). Purified plasmid DNA (1 μg) was added to 100-μL ice-chilled cell suspension and mixed gently. The mixture was bathed in ice for 30 min and then transferred to an ice-chilled electroporation cuvette (1 mm electrode gap, Bio-Rad, Richmond, CA, United States). Electroporation was performed using the Bio-Rad Micropublisher apparatus. Pulse voltage and pulse time were 2.1 kV and 4 ms, respectively. Luria–Bertani medium (900 μL) was added to the electroporation cuvette and mixed gently. The bacterial solution was transferred to a 15-mL sterile centrifuge tube and shaken for 1 h at 190 rpm. The bacterial solution (100 μL) was plated on Mueller–Hinton Agar medium containing 100 mg/mL kanamycin. Colonies were inoculated in 1 mL Luria–Bertani medium, shaken for 3 h, and then observed under an Olympus FV1000MPE multiphoton laser scanning microscope (Tokyo, Japan).
Plant Growth and Inoculation
Maize seeds were soaked in 1% sodium hypochlorite solution for 10 min and then in 75% alcohol for 5 min to kill bacteria on the surface of seeds. Next, the seeds were rinsed repeatedly with sterile water. Then, the seeds were placed on wet sterile papers in a sterile bottle at room temperature, and after about 3 days, the maize roots were approximately 2 cm in length. The germinated seedlings were transferred to a sterile tissue culture bottle (10.8 cm in height and 7.5 cm in diameter) filled with 20 mL 1/2 × Murashige & Skoog medium (Murashige and Skoog, 1962). Next, 2 mL GFP-labeled K. variicola X39 cells (1 × 109 cells/mL) were added to the tissue culture bottle. Phosphate-buffered saline solution (2 mL) was added to the control group.
Colonization of GFP-Labeled K. variicola X39 in Maize
The maize seedlings were retrieved from the 1/2 Murashige & Skoog medium after 3 and 10 days, respectively. The maize root surfaces were rinsed with sterile water and then directly observed under the Olympus FV1000MPE multiphoton laser scanning microscope. The stems were sectioned in the transverse direction, and the leaves and epidermis were separated to observe the colonization of GFP-labeled K. variicola X39.
Results
Genomic Features of K. variicola X39
We assembled the genome sequences of isolate X39 into 10 circular replicons: a 5,641,443-bp chromosome and nine plasmids (Figure 1). The genomic traits of isolate X39 were provided in Table 1. A total of 18 GIs were detected ranging in size from 4528 to 36,309 bp in K. variicola X39 (Supplementary Table S2). GIs005 was related to type IV and type VI secretion system.
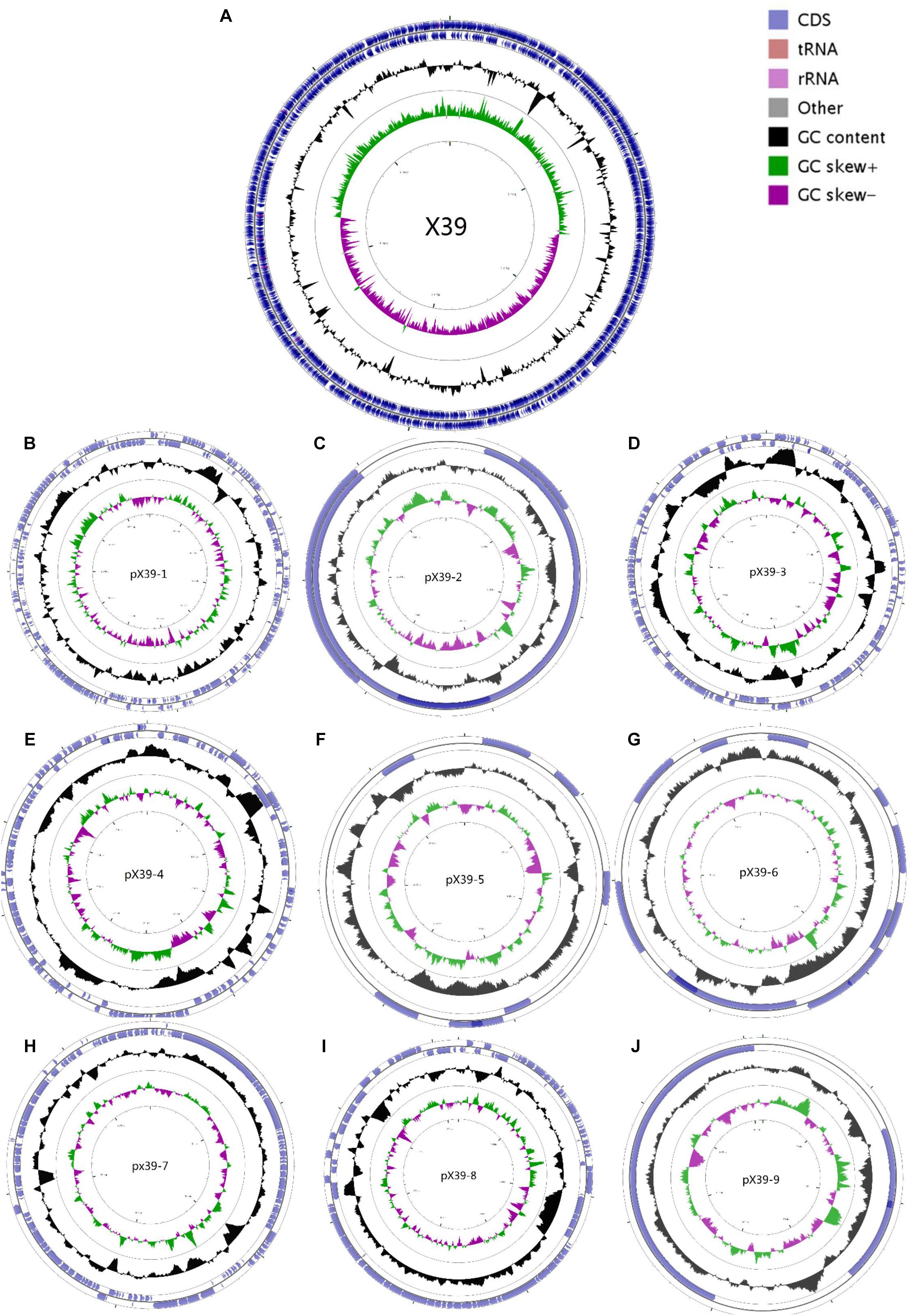
FIGURE 1. Circular maps of the chromosome (A) and nine plasmids (B–J) of K. variicola isolate X39. From the outside to the center: Genes on the forward strand, genes on the reverse strand, GC content, GC skew. The circular maps were generated by CGView v1.0.
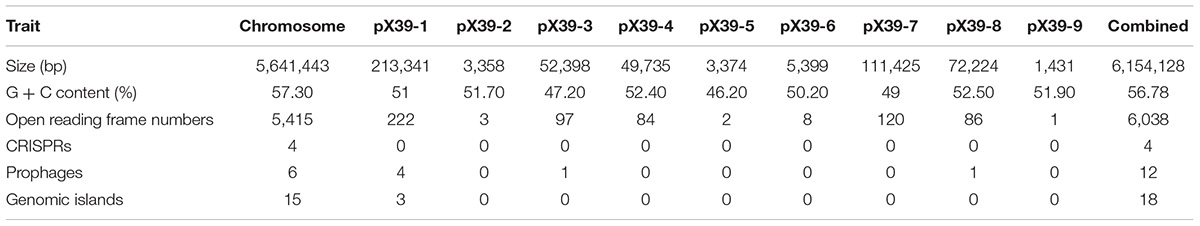
TABLE 1. The genomic traits of K. variicola X39.
We first used the Basic Local Alignment Search Tool (BLAST)3 to evaluate the genome of isolate X39. The results indicated that isolate X39 chromosome was most similar to K. variicola At-22 chromosome with a 99% identity of 90% coverage. The phylogenetic tree based on the rpoB genes showed that K. pneumoniae and K. variicola were clearly separated, and isolate X39 was closely related to K. variicola (Figure 2). The genome of K. variicola X39 has high average nucleotide identities (Goris et al., 2007) above 98% to the available genomes of K. variicola 342, At-22 (Pinto-Tomas et al., 2009), DSM15968, DE120E, GJ1,GJ2,GJ3 (Di et al., 2017), MGH20, MGH40, BIDMC61, E57-7, ID_49, NL49 (Supplementary Table S3), and thus belongs to K. variicola.
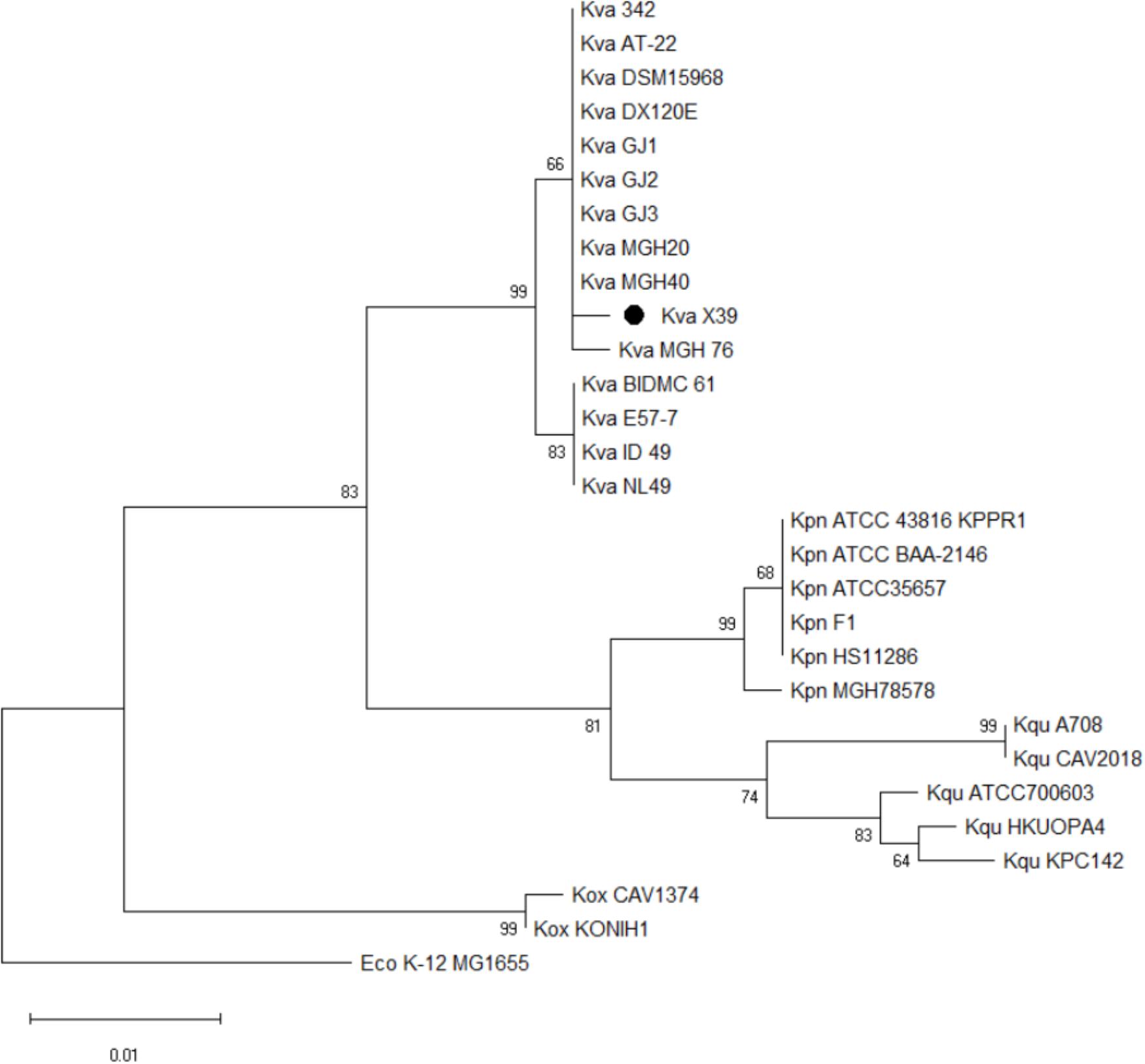
FIGURE 2. Phylogenetic tree based on rpoB genes shows the phylogenetic relationship of isolate X39 (∙) and indicates that it belongs to Klebsiella variicola. Escherichia coli K-12 MG1655 is used as an outgroup.
The largest of the nine plasmids, pX39-1, was most similar to plasmid pNH25.1 of K. pneumoniae strain NH25 with a 99% identity of 50% coverage. The similar regions mainly encoded transposases/recombinases and ABC transporter permease. The plasmid pX39-4 was most similar to plasmid pEcNDM1 of E. coli strain EcNDM1 with a 99% identity of 96% coverage. This similarity was restricted to regions of the plasmids conferring partitioning, conjugative transfer, and replication. The plasmid pX39-8 was most similar to plasmid pE20-qnrS of Klebsiella aerogenes strain E20 with a 99% identity of 87% coverage. The similar regions mainly encoded proteins for plasmid conjugative transfer.
We carried out the functional analysis by Cluster of Orthologous Groups (COGS) (Supplementary Table S4). Of the 5,812 protein-coding genes, 4,082 genes (70.2%) pertained to a COG category. The results revealed that the three major metabolism classes - amino acid transport and metabolism (E), carbohydrate transport and metabolism (G), and inorganic ion transport and metabolism (P) - represented 25.24% of all protein-coding genes. The high proportion of these genes in the genome indicated K. variicola X39 had the inherent potential for efficient uptake of nutrients and competition with surrounding microorganisms (Niazi et al., 2014).
Genomic Features of K. variicola X39 Adaptation to Plants
Klebsiella variicola is often found in plants (Rosenblueth et al., 2004; Lin et al., 2015; Martinez-Romero et al., 2015). A high number of genes that were involved in plant colonization, nitrogen fixation, and defense against oxidative stress were found in the genome of K. variicola X39. Many plant-associated bacteria could produce cellulose to promote adhesion and colonization of plant roots (Romling and Galperin, 2015). The genome of K. variicola X39 contains all genes that can synthesize cellulose (bcsABCD) (X39GM004945–X39GM004948). Nitrogen is one of the indispensable micronutrients for plant growth. K. variicola has been reported to fix nitrogen since it was discovered (Rosenblueth et al., 2004). K. variicola X39 contains all genes encoding the nitrogenase enzyme (nifDHK) (X39GM001181–X39GM001183). After contact with bacteria, plants can produce a range of defense substances including phytoalexins, nitric oxide, and reactive oxygen (Zeidler et al., 2004). Therefore, endophytes must be able to tolerate the reactive oxygen environment during colonization. Accordingly, a series of enzymes and regulatory factors that can help bacteria deal with oxidative stress were found in the genome of K. variicola X39, including superoxide dismutase (sod, X39GM001732, X39GM001742, and X39GM004640), catalase (katG, X39GM001916; katE, X39GM002512; katN, X39GM002637), 10 putative peroxidases, three hydroperoxide reductases (X39GM00328, X39GM003285, and X39GM003608), and 15 putative glutathione S-transferases or glutathione S-transferase domain/family proteins. An AcrAB (X39GM003513-3514) locus, belonging to the RND transporter family and necessary for the export of apple tree phytoalexins and the successful colonization of host plants (Burse et al., 2004), was also identified in the genome of K. variicola X39.
Comparative Genomic Analysis
The putative orthology between K. variicola X39 and the other four K. variicola isolates were determined by comparative genomic analysis. These results revealed that 4,242 gene clusters were shared between them. K. variicola X39 contained the most unique gene clusters (Figure 3A). A total of 1,074 gene clusters were identified as exclusive to K. variicola X39 (Supplementary Table S5). Of these unique gene clusters, 550 genes were from the nine plasmids, and 588 genes were from the chromosome. The COG category of the unique genes showed that K. variicola X39 possessed more genes related to energy production and conversion, amino acid transport and metabolism, coenzyme transport and metabolism, signal transduction mechanisms, defense mechanisms, and mobilome: prophages, transposons compared with other four K. variicola isolates (Figure 3B and Supplementary Table S6). K. variicola X39 possessed at least 37 transposases or putative transposases not found in the other four K. variicola isolates. K. variicola X39 and the other four K. variicola isolates differed in the distribution of genes encoding type IV and type VI secretion systems. Eight type IV secretion proteins and six type VI secretion proteins identified in K. variicola X39 were absent in the other four K. variicola isolates (Supplementary Table S5).
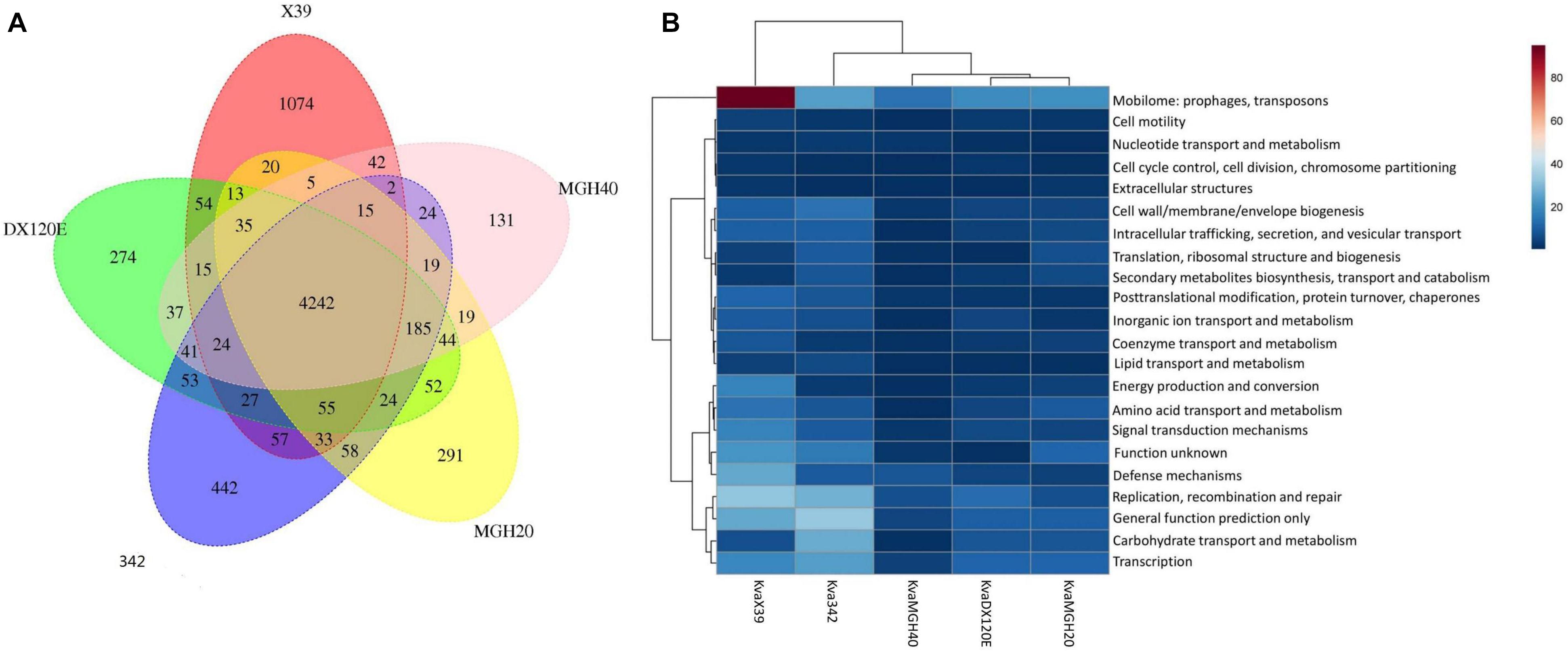
FIGURE 3. (A) Whole-genome comparison of K. variicola X39, K. variicola MGH20, K. variicola MGH40, K. variicola DX120E, and K. variicola 342. (B) The heatmap of the COG category of the unique genes in K. variicola X39, K. variicola MGH20, K. variicola MGH40, K. variicola DX120E, and K. variicola 342. The heatmap was made using MetaboAnalyst 4.0.
Virulence of K. variicola X39
We compared the genome of K. variicola X39 with the Virulence Factors of Pathogenic Bacteria Database (VFDB 2012 update) and identified 155 virulence factors. These virulence factors mainly included some genes related to adhesion, capsule production, and iron acquisition. Siderophores, such as IroN (X39GM002504), aerobactin (X39GM002739), and enterobactin (entABCDEF and fepABCDG), were extensively distributed among K. variicola X39. Simultaneously, we compared the virulence factors of K. variicola X39 with those of typical clinical K. pneumoniae F1 (Supplementary Figure S2 and Supplementary Table S7). The results indicated that K. variicola X39 and K. pneumoniae F1 shared many virulence factors.
Antibiotic Resistance
Klebsiella variicola X39 showed resistance to many antibiotics (Table 2). A series of efflux pumps and β-lactamase genes conferring resistance to a variety of drugs were found in the genome of K. variicola X39. The genome encoded three β-lactamase genes (X39GM000612, X39GM001420, and X39GM002154). Of these, both X39GM000612 and X39GM001420 encode class A extended-spectrum β-lactamase CTX-M-24. X39GM000612 was located in pX39-8, and X39GM001420 was located in the chromosome. Len (X39GM002154) is an intrinsic resistance gene of K. variicola. Qnrs1 (X39GM000620), which was first identified in a clinical isolate of Shigella flexneri 2b in Japan (Hata et al., 2005), was located in pX39-8 and conferred a plasmid-mediated quinolone resistance (Martínez-Martínez et al., 1998). K. variicola X39 also included two efflux pump superfamilies: the major facilitator superfamily, including the RosA/B efflux pump, and the resistance nodulation division superfamily, including the AcrAB–TolC multidrug efflux pump. The RosA/B efflux pump was first reported in Yersinia conferring resistance to cationic antimicrobial peptides (CAMPs) (Bengoechea and Skurnik, 2000). The AcrAB–TolC efflux pump conferred resistance to aminoglycosides, glycylcyclines, macrolides, β-lactams, and acriflavine.
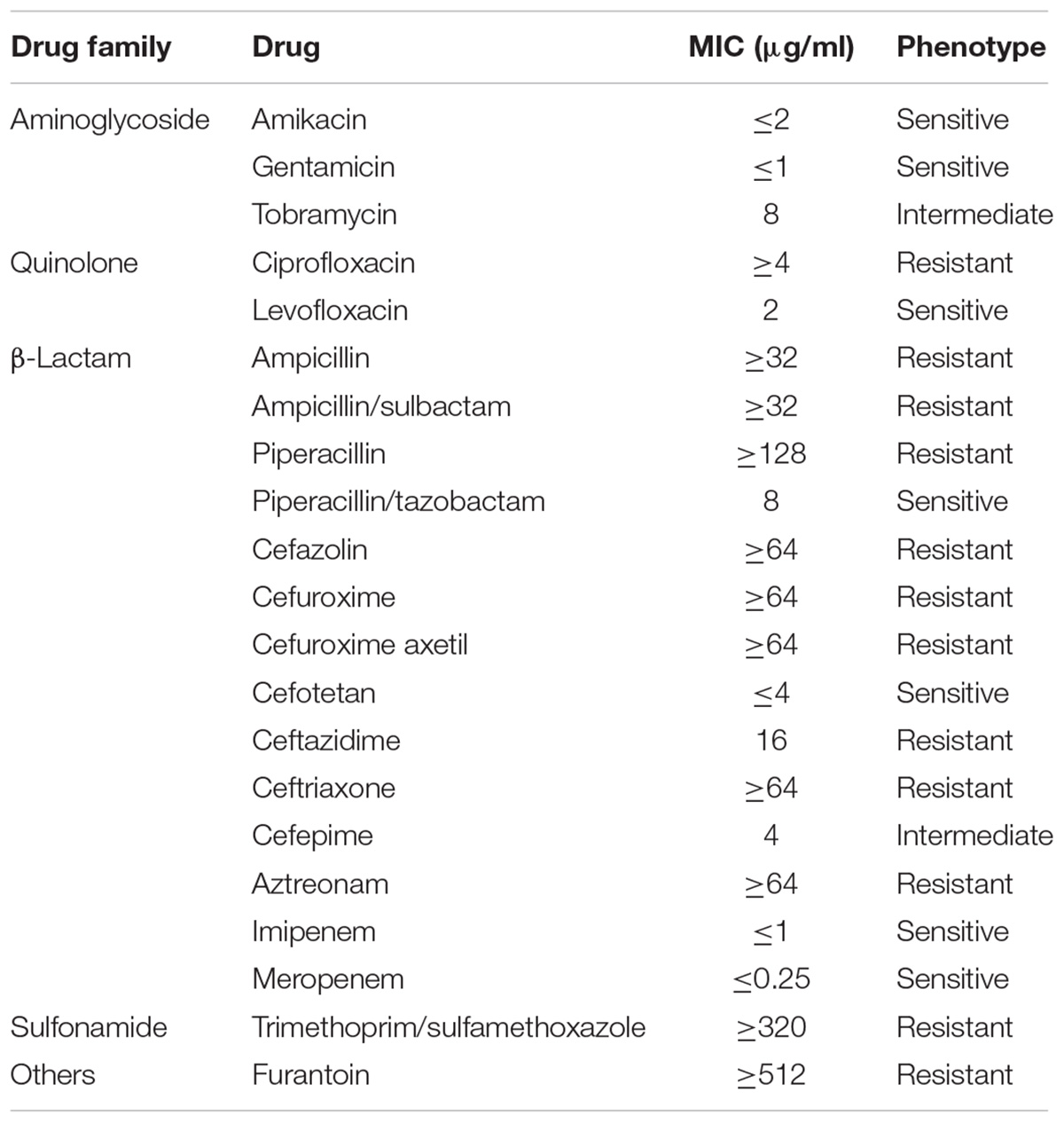
TABLE 2. K. variicola X39 antibiotic-resistance profile.
Pathogenicity of K. variicola X39
We used the BALB/c mice to study the pathogenicity of K. variicola X39. Clinical K. pneumoniae isolate F1 was used as a control group. K. variicola X39 showed an LD50 of 1.97 × 107 CFU in the BALB/c mice, and the LD50 of K. pneumoniae F1 was 7.36 × 106 CFU. The negative control group survived until sacrificed.
Construction of GFP-Labeled K. variicola X39 and Colonization in Maize
Klebsiella variicola X39 itself does not produce green fluorescence (Figure 4B). In order to visually observe whether K. variicola X39 can colonize plants, we transfected K. variicola X39 with a reconstructive plasmid pUA66 that produces GFP (Figure 4C). The GFP-labeled K. variicola X39 was used to inoculate maize seedlings grown in the gnotobiotic system. Three days after inoculation, GFP-labeled K. variicola X39 cells were found to be attached to the root surface and distributed within the cortex (Figure 4D). Ten days after inoculation, transverse sections showed that bacterial cells were distributed inside the stems (Figure 4E). A large number of bacterial cells were also found in leaves (Figure 4F). No bacteria that produced GFP were found in the control group (Figures 4G–I).

FIGURE 4. (A) Construction of plasmid puA66. (B) Confocal image of K. variicola X39. (C) Confocal image of GFP-labeled K. variicola X39. (D) Colonization patterns in root after 3 days of inoculation. (E) Colonization patterns in stem after 10 days of inoculation. (F) Colonization patterns in leaf after 10 days of inoculation. (G–I) Confocal images of the respective control groups.
Discussion
Klebsiella variicola was firstly identified in 2004 in Mexico and consisted of plant isolates and clinical isolates (Rosenblueth et al., 2004). For many years, K. variicola was viewed most commonly as a benign endosymbiont of plants or occasionally an opportunistic pathogen. In our study, we identified isolate X39, which was initially identified as K. pneumoniae by the VITEK 2 system, from the sputum of a community-acquired pneumonia patient. We used PacBio sequencing technology to obtain the whole-genome sequence of isolate X39. Martinez-Romero et al. (2018) used both phylogenetic tree based on rpoB genes and average nucleotide identity to identify a lot of misclassified Klebsiella spp. genomes. A phylogenetic tree based on rpoB genes and average nucleotide identity data confirmed that isolate X39 belonged to K. variicola.
We compared the virulence factors of K. variicola X39 with those of typical clinical K. pneumoniae F1 and found that most virulence factors were common between them. Genomic analysis indicated that K. pneumoniae and K. variicola shared some virulence determinants that were able to cause infections in humans (Andrade et al., 2014; Garza-Ramos et al., 2015a). The pathogenicity of K. variicola X39 was compared with that of K. pneumoniae F1 in an abdominal infection model. The result displayed that the LD50 of K. variicola X39 was higher than clinical K. pneumoniae F1, which indicated that typical clinical K. pneumoniae F1 was more virulent than K. variicola X39. The result was also consistent with the patterns of K. variicola 342 (Fouts et al., 2008). However, another report suggested that K. variicola could cause serious infections including bacteremia and could induce higher mortality compared with K. pneumoniae (Maatallah et al., 2014).
Klebsiella variicola X39 was mainly resistant to quinolone and β-lactam antibiotics. K. variicola X39 contained chromosome-encoded CTX-M-24 and plasmid-encoded CTX-M-24, which were very rare in Enterobacteriaceae. Qnrs1, located in pX39-8, confers a plasmid-mediated quinolone resistance. As far as we know, pX39-8 is the first detected plasmid carrying a qnrs1 gene conferring quinolone resistance in K. variicola isolates of human origin.
Klebsiella variicola is mainly found in the environment, especially in plants. We identified many genes that were involved in plant colonization, nitrogen fixation, and defense against oxidative stress in the genome of K. variicola X39. Endophytes can promote plant growth through nitrogen fixation; the nitrogen fixation capacity of K. variicola X39 was confirmed by acetylene reduction activity (data not shown). We further used the GFP-labeled K. variicola X39 to inoculate seedlings of maize, which is one of the most common crops in northern China. We visually observed obvious colonization in maize roots, stems, and leaves three days and 10 days after inoculation, which illustrated that clinical K. variicola X39 retained the capacity to colonize plants. Comparative genomic analysis between K. variicola X39 and the other K. variicola isolates revealed that K. variicola X39 contained the most unique genes, many of which were prophages and transposases. Prophages can assist the transfer of antimicrobial resistance genes and virulent factors to bacteria genome (Kutateladze and Adamia, 2010). Transposases are mobile genetic elements that are important for bacteria to adapt to the surrounding environment (Vigil-Stenman et al., 2017). These results may explain why K. variicola X39 can adapt to different hosts.
The species name K. variicola means “from different sources,” and recent reports confirmed the rationality of the name (Rosenblueth et al., 2004; Andrade et al., 2014; Davidson et al., 2015; Lin et al., 2015). Through our study, we concluded that K. variicola X39 was a kingdom-crossing bacterium (van Baarlen et al., 2007) that could infect humans as a pathogen and colonize plants as an endophyte. Although K. variicola can fix nitrogen, it should not be used as a biological fertilizer because of its potential pathogenicity. IMI-2 and OXA-181 carbapenemase and New Delhi metallo-β-lactamase have appeared in K. variicola isolates in Europe and in an environmental K. variicola isolate from an urban river in South Korea, respectively (Zurfluh et al., 2015; Di et al., 2017; Hopkins et al., 2017). Plants can serve as a reservoir for K. variicola isolates that were able to opportunistically infect humans or animals, allowing K. variicola to exchange genetic information with other environmental pathogens and gain the advantage of selection, which may also be the reason for the increasing resistance of K. variicola. Recently, a colistin-resistant hypervirulent K. variicola isolate has been isolated from the blood of a patient in Sichuan, China (Lu et al., 2018). The plasmid BLAST results implied that isolate X39 may exchange genes with Enterobacteriaceae. It is difficult to distinguish K. variicola from other Klebsiella species using the current VITEK automated system. Some simple molecular biology methods have been developed to identify K. variicola (Garza-Ramos et al., 2015b; Fonseca et al., 2017). As more K. variicola isolates are identified, the epidemiological characteristics of K. variicola will require further studies.
Data Availability
The sequences of the chromosome and nine plasmids of K. variicola X39 have been deposited in GenBank under accession numbers CP018307 and CP023978–CP023986, respectively.
Author Contributions
ZG, YK, and SH designed the experiments. YG, YZ, ZZ, and DL performed the experiments. ZW collected and identified the isolate. YG and ZH performed isolate sequencing and bioinformatics analysis. JL operated the Olympus FV1000MPE Multiphoton Laser Scanning Microscope. YG wrote the manuscript. All authors reviewed the manuscript and agreed to the publication of this manuscript.
Funding
The study was funded by National Natural Science Foundation of China (81470207) and National Key Research & Development Program of China from Ministry of Science and Technology (2016YFC0903800).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.02428/full#supplementary-material
Footnotes
- ^http://crispr.i2bc.paris-saclay.fr
- ^https://www.ezbiocloud.net/tools/ani
- ^https://blast.ncbi.nlm.nih.gov/Blast.cgi
References
Andrade, B. G., de Veiga Ramos, N., Marin, M. F., Fonseca, E. L., and Vicente, A. C. (2014). The genome of a clinical Klebsiella variicola strain reveals virulence-associated traits and a pl9-like plasmid. FEMS Microbiol. Lett. 360, 13–16. doi: 10.1111/1574-6968.12583
Bengoechea, J. A., and Skurnik, M. (2000). Temperature-regulated efflux pump/potassium antiporter system mediates resistance to cationic antimicrobial peptides in Yersinia. Mol. Microbiol. 37, 67–80.
Besemer, J., Lomsadze, A., and Borodovsky, M. (2001). GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 29,2607–2618.
Brisse, S., Passet, V., and Grimont, P. A. (2014). Description of Klebsiella quasipneumoniae sp. nov., isolated from human infections, with two subspecies, Klebsiella quasipneumoniae subsp. quasipneumoniae subsp. nov. and Klebsiella quasipneumoniae subsp. similipneumoniae subsp. nov., and demonstration that Klebsiella singaporensis is a junior heterotypic synonym of Klebsiella variicola. Int. J. Syst. Evol. Microbiol. 64(Pt 9), 3146–3152. doi: 10.1099/ijs.0.062737-0
Burse, A., Weingart, H., and Ullrich, M. S. (2004). The phytoalexin-inducible multidrug efflux pump AcrAB contributes to virulence in the fire blight pathogen, Erwinia amylovora. Mol. Plant Microbe Interact. 17, 43–54. doi: 10.1094/mpmi.2004.17.1.43
Chung, D. R., Song, J. H., Kim, S. H., Thamlikitkul, V., Huang, S. G., Wang, H., et al. (2011). High prevalence of multidrug-resistant nonfermenters in hospital-acquired pneumonia in Asia. Am. J. Respir. Crit. Care Med. 184, 1409–1417. doi: 10.1164/rccm.201102-0349OC
Davidson, F. W., Whitney, H. G., and Tahlan, K. (2015). Genome sequences of Klebsiella variicola isolates from dairy animals with bovine mastitis from newfoundland, Canada. Genome Announc. 3:e00938-15. doi: 10.1128/genomeA.00938-15
Di, D. Y., Jang, J., Unno, T., and Hur, H. G. (2017). Emergence of Klebsiella variicola positive for NDM-9, a variant of New Delhi metallo-beta-lactamase, in an urban river in South Korea. J. Antimicrob. Chemother. 72, 1063–1067. doi: 10.1093/jac/dkw547
Fonseca, E. L., Ramos, N. D., Andrade, B. G., Morais, L. L., Marin, M. F., and Vicente, A. C. (2017). A one-step multiplex PCR to identify Klebsiella pneumoniae, Klebsiella variicola, and Klebsiella quasipneumoniae in the clinical routine. Diagn. Microbiol. Infect. Dis. 87, 315–317. doi: 10.1016/j.diagmicrobio.2017.01.005
Fouts, D. E., Tyler, H. L., DeBoy, R. T., Daugherty, S., Ren, Q., Badger, J. H., et al. (2008). Complete genome sequence of the N2-fixing broad host range endophyte Klebsiella pneumoniae 342 and virulence predictions verified in mice. PLoS Genet. 4:e1000141. doi: 10.1371/journal.pgen.1000141
Gardner, P. P., Daub, J., Tate, J. G., Nawrocki, E. P., Kolbe, D. L., Lindgreen, S., et al. (2009). Rfam: updates to the RNA families database. Nucleic Acids Res. 37, D136–D140. doi: 10.1093/nar/gkn766
Garza-Ramos, U., Silva-Sanchez, J., Barrios, H., Rodriguez-Medina, N., Martinez-Barnetche, J., and Andrade, V. (2015a). Draft genome sequence of the first hypermucoviscous Klebsiella variicola clinical isolate. Genome Announc. 3:e01352-14. doi: 10.1128/genomeA.01352-14
Garza-Ramos, U., Silva-Sanchez, J., Martinez-Romero, E., Tinoco, P., Pina-Gonzales, M., Barrios, H., et al. (2015b). Development of a multiplex-PCR probe system for the proper identification of Klebsiella variicola. BMC Microbiol. 15:64. doi: 10.1186/s12866-015-0396-6
Goris, J., Konstantinidis, K. T., Klappenbach, J. A., Coenye, T., Vandamme, P., and Tiedje, J. M. (2007). DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57(Pt 1), 81–91. doi: 10.1099/ijs.0.64483-0
Grissa, I., Vergnaud, G., and Pourcel, C. (2007). CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35, W52–W57. doi: 10.1093/nar/gkm360
Hata, M., Suzuki, M., Matsumoto, M., Takahashi, M., Sato, K., Ibe, S., et al. (2005). Cloning of a novel gene for quinolone resistance from a transferable plasmid in Shigella flexneri 2b. Antimicrob. Agents Chemother. 49, 801–803. doi: 10.1128/AAC.49.2.801-803.2005
Hopkins, K. L., Findlay, J., Doumith, M., Mather, B., Meunier, D., D’Arcy, S., et al. (2017). IMI-2 carbapenemase in a clinical Klebsiella variicola isolated in the UK. J. Antimicrob. Chemother. 72, 2129–2131. doi: 10.1093/jac/dkx103
Hsiao, W. W., Ung, K., Aeschliman, D., Bryan, J., Finlay, B. B., and Brinkman, F. S. (2005). Evidence of a large novel gene pool associated with prokaryotic genomic islands. PLoS Genet. 1:e62. doi: 10.1371/journal.pgen.0010062
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Kutateladze, M., and Adamia, R. (2010). Bacteriophages as potential new therapeutics to replace or supplement antibiotics. Trends Biotechnol. 28, 591–595. doi: 10.1016/j.tibtech.2010.08.001
Lagesen, K., Hallin, P., Rodland, E. A., Staerfeldt, H. H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Li, W., and Godzik, A. (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659. doi: 10.1093/bioinformatics/btl158
Li, X., Zhang, D., Chen, F., Ma, J., Dong, Y., and Zhang, L. (2004). Klebsiella singaporensis sp. nov., a novel isomaltulose-producing bacterium. Int. J. Syst. Evol. Microbiol. 54(Pt 6), 2131–2136. doi: 10.1099/ijs.0.02690-0
Lin, L., Wei, C., Chen, M., Wang, H., Li, Y., Li, Y., et al. (2015). Complete genome sequence of endophytic nitrogen-fixing Klebsiella variicola strain DX120E. Stand. Genomic Sci. 10, 22. doi: 10.1186/s40793-015-0004-2
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964.
Lu, Y., Feng, Y., McNally, A., and Zong, Z. (2018). Occurrence of colistin-resistant hypervirulent Klebsiella variicola. J. Antimicrob. Chemother. doi: 10.1093/jac/dky301 [Epub ahead of print].
Maatallah, M., Vading, M., Kabir, M. H., Bakhrouf, A., Kalin, M., Naucler, P., et al. (2014). Klebsiella variicola is a frequent cause of bloodstream infection in the stockholm area, and associated with higher mortality compared to K. pneumoniae. PLoS One 9:e113539. doi: 10.1371/journal.pone.0113539
Martínez-Martínez, L., Pascual, A., and Jacoby, G. A. (1998). Quinolone resistance from a transferable plasmid. Lancet 351, 797–799. doi: 10.1016/s0140-6736(97)07322-4
Martinez-Romero, E., Rodriguez-Medina, N., Beltran-Rojel, M., Silva-Sanchez, J., Barrios-Camacho, H., Perez-Rueda, E., et al. (2018). Genome misclassification of Klebsiella variicola and Klebsiella quasipneumoniae isolated from plants, animals and humans. Salud Publica Mex. 60, 56–62. doi: 10.21149/8149
Martinez-Romero, E., Silva-Sanchez, J., Barrios, H., Rodriguez-Medina, N., Martinez-Barnetche, J., Tellez-Sosa, J., et al. (2015). Draft genome sequences of Klebsiella variicola plant isolates. Genome Announc. 3:e01015-15. doi: 10.1128/genomeA.01015-15
Murashige, T., and Skoog, F. (1962). A revised medium for rapid growth and bio assays with tobacco tissue cultere. Physiol. Plant. 15, 473–497. doi: 10.1111/j.1399-3054.1962.tb08052
Nawrocki, E. P., Kolbe, D. L., and Eddy, S. R. (2009). Infernal 1.0: inference of RNA alignments. Bioinformatics 25, 1335–1337. doi: 10.1093/bioinformatics/btp157
Niazi, A., Manzoor, S., Asari, S., Bejai, S., Meijer, J., and Bongcam-Rudloff, E. (2014). Genome analysis of Bacillus amyloliquefaciens Subsp. plantarum UCMB5113: a rhizobacterium that improves plant growth and stress management. PLoS One 9:e104651. doi: 10.1371/journal.pone.0104651
Nicolas, M. F., Ramos, P. I. P., Marques de Carvalho, F., Camargo, D. R. A., de Fatima Morais Alves, C., Loss de Morais, G., et al. (2018). Comparative genomic analysis of a clinical isolate of Klebsiella quasipneumoniae subsp. similipneumoniae, a KPC-2 and OKP-B-6 beta-lactamases producer harboring two drug-resistance plasmids from Southeast Brazil. Front. Microbiol. 9:220. doi: 10.3389/fmicb.2018.00220
Pinto-Tomas, A. A., Anderson, M. A., Suen, G., Stevenson, D. M., Chu, F. S., Cleland, W. W., et al. (2009). Symbiotic nitrogen fixation in the fungus gardens of leaf-cutter ants. Science 326, 1120–1123. doi: 10.1126/science.1173036
Podschun, R., and Ullmann, U. (1998). Klebsiella spp. as nosocomial pathogens: epidemiology, taxonomy, typing methods, and pathogenicity factors. Clin. Microbiol. Rev. 11, 589–603.
Romling, U., and Galperin, M. Y. (2015). Bacterial cellulose biosynthesis: diversity of operons, subunits, products, and functions. Trends Microbiol. 23, 545–557. doi: 10.1016/j.tim.2015.05.005
Rosenblueth, M., Martinez, L., Silva, J., and Martinez-Romero, E. (2004). Klebsiella variicola, a novel species with clinical and plant-associated isolates. Syst. Appl. Microbiol. 27, 27–35. doi: 10.1078/0723-2020-00261
van Baarlen, P., van Belkum, A., Summerbell, R. C., Crous, P. W., and Thomma, B. P. (2007). Molecular mechanisms of pathogenicity: how do pathogenic microorganisms develop cross-kingdom host jumps? FEMS Microbiol. Rev. 31, 239–277. doi: 10.1111/j.1574-6976.2007.00065.x
Vigil-Stenman, T., Ininbergs, K., Bergman, B., and Ekman, M. (2017). High abundance and expression of transposases in bacteria from the Baltic Sea. ISME J. 11, 2611–2623. doi: 10.1038/ismej.2017.114
Wang, X., Kang, Y., Luo, C., Zhao, T., Liu, L., Jiang, X., et al. (2014). Heteroresistance at the single-cell level: adapting to antibiotic stress through a population-based strategy and growth-controlled interphenotypic coordination. mBio 5:e00942-13. doi: 10.1128/mBio.00942-13
Yoon, S. H., Ha, S. M., Lim, J., Kwon, S., and Chun, J. (2017). A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek 110, 1281–1286. doi: 10.1007/s10482-017-0844-4
Zeidler, D., Zahringer, U., Gerber, I., Dubery, I., Hartung, T., Bors, W., et al. (2004). Innate immunity in Arabidopsis thaliana: lipopolysaccharides activate nitric oxide synthase (NOS) and induce defense genes. Proc. Natl. Acad. Sci. U.S.A. 101, 15811–15816. doi: 10.1073/pnas.0404536101
Zhou, Y., Liang, Y., Lynch, K. H., Dennis, J. J., and Wishart, D. S. (2011). PHAST: a fast phage search tool. Nucleic Acids Res. 39, W347–W352. doi: 10.1093/nar/gkr485
Keywords: Klebsiella variicola, genome, virulence, resistance, endophyte
Citation: Guo Y, Zhai Y, Zhang Z, Li D, Wang Z, Li J, He Z, Hu S, Kang Y and Gao Z (2018) Complete Genomic Analysis of a Kingdom-Crossing Klebsiella variicola Isolate. Front. Microbiol. 9:2428. doi: 10.3389/fmicb.2018.02428
Received: 30 May 2018; Accepted: 21 September 2018;
Published: 09 October 2018.
Edited by:
Vasco Ariston De Carvalho Azevedo, Universidade Federal de Minas Gerais, BrazilReviewed by:
Pablo Ivan Pereira Ramos, Instituto Gonçalo Moniz (IGM), Fiocruz Bahia, BrazilJason Sahl, Northern Arizona University, United States
Copyright © 2018 Guo, Zhai, Zhang, Li, Wang, Li, He, Hu, Kang and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yu Kang, a2FuZ3lAYmlnLmFjLmNu Zhancheng Gao, emNnYW9AYmptdS5lZHUuY24=