 Abhishek Gupta
Abhishek Gupta Avishek Dutta
Avishek Dutta Jayeeta Sarkar1
Jayeeta Sarkar1 Pinaki Sar
Pinaki Sar- 1Environmental Microbiology and Genomics Laboratory, Department of Biotechnology, Indian Institute of Technology Kharagpur, Kharagpur, India
- 2School of Bioscience, Indian Institute of Technology Kharagpur, Kharagpur, India
- 3Department of Geology and Geophysics, Indian Institute of Technology Kharagpur, Kharagpur, India
Sulfate- and iron-reducing heterotrophic bacteria represented minor proportion of the indigenous microbial community of highly acidic, oligotrophic acid mine drainage (AMD), but they can be successfully stimulated for in situ bioremediation of an AMD impacted soil (AIS). These anaerobic microorganisms although played central role in sulfate- and metal-removal, they remained inactive in the AIS due to the paucity of organic carbon and extreme acidity of the local environment. The present study investigated the scope for increasing the abundance and activity of inhabitant sulfate- and iron-reducing bacterial populations of an AIS from Malanjkhand Copper Project. An AIS of pH 3.5, high soluble SO42− (7838 mg/l) and Fe (179 mg/l) content was amended with nutrients (cysteine and lactate). Thorough geochemical analysis, 16S rRNA gene amplicon sequencing and qPCR highlighted the intrinsic metabolic abilities of native bacteria in AMD bioremediation. Following 180 days incubation, the nutrient amended AIS showed marked increase in pH (to 6.6) and reduction in soluble -SO42− (95%), -Fe (50%) and other heavy metals. Concomitant to physicochemical changes a vivid shift in microbial community composition was observed. Members of the Firmicutes present as a minor group (1.5% of total community) in AIS emerged as the single most abundant taxon (∼56%) following nutrient amendments. Organisms affiliated to Clostridiaceae, Peptococcaceae, Veillonellaceae, Christensenellaceae, Lachnospiraceae, Bacillaceae, etc. known for their fermentative, iron and sulfate reducing abilities were prevailed in the amended samples. qPCR data corroborated with this change and further revealed an increase in abundance of dissimilatory sulfite reductase gene (dsrB) and specific bacterial taxa. Involvement of these enhanced populations in reductive processes was validated by further enrichments and growth in sulfate- and iron-reducing media. Amplicon sequencing of these enrichments confirmed growth of Firmicutes members and proved their sulfate- and iron-reduction abilities. This study provided a better insight on ecological perspective of Firmicutes members within the AMD impacted sites, particularly their involvement in sulfate- and iron-reduction processes, in situ pH management and bioremediation.
Introduction
Acid mine drainage (AMD) is considered to be a global environmental problem faced by mining industries due to the biological oxidation of sulfidic minerals (Johnson and Hallberg, 2005; Neculita and Zagury, 2008; Qian et al., 2017). Owing to its highly toxic nature manifested through acidic pH, elevated levels of heavy metals and sulfate, AMD is not only a threat to aquatic and terrestrial ecosystems but considered to be a major contributor in long term degradation of environmental quality (Johnson and Hallberg, 2005; Chandra and Gerson, 2010; Hallberg, 2010). Despite its extreme nature, a diverse range of microorganisms inhabit AMD systems (Méndez-García et al., 2015; Chen et al., 2016; Huang et al., 2016). The most dominant bacterial populations residing in AMD are highly acidophilic, chemolithoautotrophic iron and sulfur oxidizers such as Acidithiobacillus, Leptospirillum, Ferrithrix, and Ferritrophicum etc. (Baker and Banfield, 2003; Chen et al., 2015, 2016; Méndez-García et al., 2015; Huang et al., 2016; Mesa et al., 2017; Teng et al., 2017). These acidophilic, autotrophic and Fe/S oxidizing microorganisms mainly contribute toward AMD generation and were studied extensively for their physiology, molecular mechanisms and ecological relevance (Denef et al., 2010; Kuang et al., 2013; Méndez-García et al., 2014; Chen et al., 2015; Goltsman et al., 2015; Chen et al., 2016), whereas the small heterotrophic populations thriving in the same niches could be of great significance in reducing AMD generation process and attenuating the overall hazard of these systems remain less explored.
Microbial sulfur- and iron-metabolisms through redox transformations coupled with or without energy generation constitute the major biochemical reactions within AMD (Baker and Banfield, 2003; Druschel et al., 2004). These transformation reactions facilitate generation of acidity and contribute toward raising the soluble -sulfate or -iron concentrations, while on the other hand could lead to reversal of such processes and aid to restoration of such environments. Sulfate- and iron-reductions are the two key reactions carried out by heterotrophic sulfate- or iron-reducing bacteria (SRBs or IRBs) that could reverse the AMD generation, metal precipitation and thus decrease the soluble metal concentrations and facilitate in raising the pH of AMD or AMD impacted ecosystems (Kaksonen et al., 2004; Church et al., 2007; Bijmans et al., 2009, 2010; Giloteaux et al., 2013). Bioremediation of AMD or AMD impacted ecosystems have been a subject of intense research in last decades (Kaksonen et al., 2004; Luptakova and Kusnierova, 2005; Church et al., 2007; Hiibel et al., 2008; Becerra et al., 2009; Bijmans et al., 2009; Hiibel et al., 2011; Burns et al., 2012; Moreau et al., 2013; Xingyu et al., 2013; Lefticariu et al., 2015; Sahinkaya et al., 2015; Deng et al., 2016; Zhang et al., 2016; Kefeni et al., 2017). In particular, enhancing the activities of indigenous microorganisms capable of sulfate- and/or iron-reduction and generation of alkalinity have gained interest for developing in situ bioremediation strategies (Neculita et al., 2007; Hiibel et al., 2008, 2011; Becerra et al., 2009; Bijmans et al., 2009; Burns et al., 2012; Xingyu et al., 2013; Lefticariu et al., 2015).
It is interesting to note that AMD or AMD impacted environment harbors SRBs and/or IRBs, but generally with low abundance and they remained metabolically less active at pH < 5.0 (Church et al., 2007; Sánchez-Andrea et al., 2011, 2012a; Giloteaux et al., 2013; Méndez-García et al., 2015). The limited presence and activities of these bacteria in AMD could be due to the presence of low organic carbon/other environmental variables and thermodynamic limitations as dissimilatory sulfate- and/or iron-reduction are energetically expensive (Church et al., 2007; Muyzer and Stams, 2008; Bird et al., 2011; Johnson, 2012; Giloteaux et al., 2013). Nevertheless, metabolic versatility of SRB has been exploited in bioremediation of AMD with different approaches, among which amendment of suitable carbon and electron sources, nitrogen, phosphorus compounds etc. are important (Kaksonen et al., 2004; Church et al., 2007; Neculita et al., 2007; Hiibel et al., 2008, 2011; Becerra et al., 2009; Bijmans et al., 2009; Burns et al., 2012; Xingyu et al., 2013; Zhang and Wang, 2014; Lefticariu et al., 2015; Zhang et al., 2017).
During the past decades, microbiology of AMD has been studied extensively, particularly the cultivation-independent deep sequencing studies have resolved the community composition and biogeochemical functions of previously unknown microorganisms (Bertin et al., 2011; Kuang et al., 2013; Méndez-García et al., 2014; Chen et al., 2015; Goltsman et al., 2015; Hua et al., 2015). In contrast, exploration of AMD communities with special reference to heterotrophic SRBs and IRBs or other metal reducing populations remained less explored (Giloteaux et al., 2013). In situ bioremediation of these hazardous wastes is limited due to paucity of knowledge on the diversity of SRBs/IRBs and factors that promote their activities.
In the present study we aimed to explore the abundance and role of indigenous sulfate- and/or metal-reducing bacterial populations in natural attenuation of an AMD impacted soil designated as AIS. Soil impacted with highly acidic, sulfate- and multiple heavy metal-rich AMD from Asia’s largest open-cast copper mine of Malanjkhand Copper Project (MCP) was used in this study. Microcosm based approach was adopted to promote presence and activities of indigenous sulfate- and/or metal-reducing bacteria using cysteine and lactate as biostimulation agents. A thorough assessment of microbial populations involved in sulfate/metal reduction and their characterization was done through 16S rRNA gene based amplicon sequencing coupled with qPCR and DGGE. The study was structured to answer the following questions: (i) How far it is possible to enhance the presence and activities of indigenous sulfate- and iron-reducing microbial populations present within an AMD impacted soil? (ii) What is the effect of such treatment(s) in the improvement of local physicochemical conditions, particularly the pH, concentrations of soluble -sulfate, -iron and -other heavy metals present therein? and (iii) Is it possible to enrich and cultivate the specific populations responsible for sulfate- and iron-reduction and management of the local physicochemical condition? The study demonstrates a comprehensive composition of microbial community residing in AIS and investigates the scope for in situ bioremediation.
Materials and Methods
Sampling Site
The AMD impacted soil was collected in a sterile container from 5–10 cm below the top layer of a field flooded with AMD from a neighboring sump of Malanjkhand Copper Project (MCP), Balaghat district, Madhya Pradesh, India (N 21° 59.91′, E 080° 41.879′) in the year 2014. The soil is exposed to AMD for over 10 years. The AMD water is released (as overflow) from the adjacent sump which receives AMD continuously from the mine areas. Selected physicochemical parameters such as oxidation reduction potential (ORP), pH and conductivity were measured on-site using multiparameter (Orion Star A329 portable Multiparameter, Thermo Fisher Scientific). All samples were collected following aseptic techniques, stored immediately at 4°C, brought to the laboratory and stored at −80°C till further processing.
Microcosm Preparation
The microcosm setup was prepared with 5 g of AMD contaminated soil (AIS) using 20 ml filter sterilized distilled water in 30 ml glass vial. Three sets of microcosms were prepared. The first microcosm was amended with 0.1% (w/v) cysteine hydrochloride and designated as C. The second microcosm was amended with both 0.1% (w/v) cysteine hydrochloride and 0.1% (w/v) lactate (as sodium lactate), designated as C+L. The third microcosm was not amended with anything extra and designated as H (H stands for H2O, since only filter sterilized distilled water was present with AIS). Killed control was prepared for each setup by adding 2% (w/v) HgCl2 as biocide. The glass vials were sealed with gas-tight rubber stoppers and aluminum crimp seals. To mimic the natural environment nitrogen was not purged into the microcosm vials. The microcosms were incubated in dark for 180 days at 30°C. Each microcosm was set up in duplicate. Since the microcosms were of sacrificial type (i.e., the vial once opened was not reused in the same study) three experimental replicates were prepared: one for 4 months (120 days) incubation and marked as C_4M, C+L_4M, and H_4M; second for 5 months (150 days) incubation and marked as C_5M, C+L_5M, and H_5M and third for 6 months (180 days) incubation and marked as C_6M, C+L_6M, and H_6M. Physicochemical parameters were measured from each microcosm setups (at 120 and 180 days of incubation). Samples were withdrawn from each of the setup in triplicates and used for measuring the physicochemical parameters. The major physicochemical parameters such as pH and ORP of the slurry were measured by Orion Multi parameter (Orion Star A329 portable Multiparameter, Thermo Fisher Scientific). The slurry samples were taken out from the microcosm setup and centrifuged at 4000 rpm to settle down the soil particles. SO42− estimation was performed with the supernatant through BaCl2 turbidometric spectroscopy based method (Chesnin and Yien, 1951) while for Fe2+ estimation, samples were acidified to avoid any oxidation and Fe2+ concentration was measured by Ferrozine method (Viollier et al., 2000). The major elements such as Fe, Cu, As, Cr, Ni, and Zn were estimated from the slurry using atomic absorption spectroscopy (Perkin Elmer). In short, the slurry was centrifuged at 4000 rpm and supernatant was passed through 0.22 μm filter membrane and 2% HNO3 was added to prevent any oxidation.
Metagenome Extraction, Library Preparation, and Sequencing
The microbial diversity analysis based on 16S rRNA gene amplicon targeted sequencing was performed with 6M setups (i.e., with 180 days incubation). Original AIS sample (0_Day) was also used for comparison. From the three microcosms and the 0_Day AIS, samples were withdrawn in triplicates and metagenome was extracted from each of the withdrawn samples using Power Soil DNA Isolation Kit (MoBio laboratories) according to the manufacturer’s protocol. Metagenome from the replicate samples were pooled, mixed thoroughly and used for amplification of V4 region of 16S rRNA gene. V4 region of 16S rRNA gene was amplified with V4 specific primers (Bates et al., 2011). The following amplification conditions: 95°C for 5 min, 35 cycles of 95°C for 40 s, 50°C for 45 s and 72°C for 40 s with final extension at 72°C for 7 min were used for amplification of V4 region. Thereafter amplicons were purified using 2% E-gel (E-Gel SizeSelect II Agarose Gel, Thermo Fisher Scientific) and sequencing was performed with Ion S5TM System (Thermo Fischer Scientific). In order to understand the microbial diversity at 5M setups (i.e., with 150 days incubation), Denaturing gradient gel electrophoresis (DGGE) was performed with H_5M, C+L_5M, and C_5M samples. Metagenome was extracted in triplicates from these setups and were pooled together to amplify the V4 region using GC-clamp forward primer as described above. A DCode Universal Mutation Detection system (Bio-Rad, United States) was used to perform DGGE with similar protocol as described by Paul et al. (2015). The denaturing gradient from 35 to 70% was used for the present study. Twenty-three distinct bands in DGGE profile were excised and eluted by keeping it in 20 μl DNase free PCR water at 4°C for overnight. These gel eluted products were re-amplified by using without GC clamp 515F and 806R primers (V4 region) and were cloned into the pTZ57RT vector for sequencing. EzTaxon1 and SILVA 119 reference database2 were used for the taxonomic assignment of the obtained sequences.
Quantification of Bacterial/Specific Taxa and dsrB Copy Number
Quantification of bacterial abundance and remarkably shifted taxa; Firmicutes, Acidobacteria, Actinobacteria as well as dsrB gene involved in sulfate reduction were performed for all the samples (0_Day, H_6M, C_6M, and C+L_6M). The bacterial abundance was quantified through bacterial specific 16S rRNA gene copy number. Similarly, abundance of Actinobacteria, Acidobacteria, and Firmicutes were quantified through specific 16S rRNA gene specific to these taxa. Copy numbers of functional gene dsrB were also quantified using qPCR based technique to estimate the sulfate-reducing populations. Real-time primers for bacterial 16S rRNA gene was taken from Muyzer et al. (1993), primers specific to Actinobacteria and Firmicutes was taken from Mühling et al. (2008), primer used for Acidobacteria as described by Lee and Cho (2011) and dsrB was taken from Purkamo et al. (2013). The qPCR was performed in Quant Studio 5 Real-Time PCR System (Thermo Fisher Scientific) with Power SYBR green PCR Mastermix (Invitrogen), with a total volume of 10 μl containing primer concentration of 5 picomoles and 2 μl of metagenomic DNA. All the reactions were set in triplicates. The following amplification conditions: 95°C for 10 min, 40 cycles of 95°C for 15 s, 55°C for 30 s and 72°C for 30 s was followed for bacterial and dsrB gene while 63°C, 59°C, and 57°C annealing temperature were used for Actinobacteria, Acidobacteria, and Firmicutes, respectively. Melting curve analysis was run after each assay to check PCR specificity. Bacterial 16S rRNA gene copy numbers were determined in each sample by comparing the amplification result to a standard dilution series ranging from 102 to 108 of plasmid DNA containing the 16S rRNA gene of Achromobacter sp. MTCC 12117. Firmicutes gene copy number was calculated from plasmid DNA containing 16S rRNA gene from Bacillus. Whereas 16S rRNA gene of Actinobacteria and Acidobacteria as well as dsrB gene were cloned from metagenome and different dilution series of plasmid DNA copy number were used to prepare the standard curve for comparing the amplification result. The efficiency of qPCR was calculated using formula E = 10 (−1/ − Slope) – 1. The standard curve was linear for all the taxa specific and dsrB gene. R2 value was greater than 0.993 for all the standard curve while efficiency was ranges from 84 to 112% (Supplementary Table S1).
Enrichment of Firmicutes Specific Members and Their Potential Role in Fe3+ and SO42− Reduction
Firmicutes specific populations were enriched in facultative anaerobic medium (Stieglmeier et al., 2009) and Clostridium specific medium containing following ingredients in g/L NaCl 2.0, K2HPO4 5.0, MgCl2 0.2, ferric citrate 0.2, yeast extract 1.0, lysine 0.5 and cellulose 7.0 at pH 7.0 in 50 ml glass serum vials. Both the media were purged with filtered N2 gas for 15–20 min to remove the oxygen and cysteine HCl (0.025%) was added as a mild reducing agent. Serum bottles were sealed with rubber stoppers. Two ml slurry from both C_6M and C+L_6M was used as inoculum in both the media and incubated at 30°C for 2 weeks. The enrichment was sub-cultured three times in the same media before transferring into sulfate reducing medium (SRM) (modified from Postgate, 1963) and iron reducing medium (IRM) (containing ferric citrate 5 mM, NH4Cl 1.50 g/L, NaH2PO4 0.60 g/L, KCl 0.10 g/L, sodium acetate 2.50 g/L and yeast extract 0.05 g/L). Nitrogen gas was flushed for 15–20 min and cysteine HCl (0.025%) was added as a mild reducing agent in both the media to make the environment anaerobic. The pH of these two media was set to 7.0 using 1N NaOH/1N HCl and incubated at 30°C for 2 weeks. Enriched population was sub-cultured thrice in same media after seeing the visual changes in the media (iron containing medium turned colorless, sulfate reducing medium turned black due to precipitation of iron sulfide). Remaining sulfate and increased iron (Fe2+) concentrations were measured for assessing the reduction of sulfate and iron (Fe3+) using BaCl2 turbidometric method and Ferrozine method, respectively. Briefly, 2 ml samples were taken out and bacterial cells were pelleted down to use supernatant for estimation of SO42− and Fe2+ concentration.
DNA Extraction From Enrichment
Total DNA from enriched populations was extracted from 4 ml of each enrichment. Equal volume of 0.5 M ammonium oxalate was added in iron enrichment to dissolve iron precipitates. The culture was pelleted at high speed for 5 min at room temperature. The cell pellet was dissolved in 500 μL TNE buffer (Tris HCl-10 mM, NaCl-2.0 M, EDTA-1 mM), 1/10 volume silica bead was added and vortexed for 15–20 min. 100 μL lysozyme (100 mg/ml) was added in the cell suspension, vortexed briefly to mix and incubated at 37°C for 2 h. 30 μL proteinase K (20 mg/ml) and 50 μL SDS (10%) were added and incubated at 37°C for 45 min. DNA was then extracted using chloroform:isoamyl alcohol (24:1). DNA pellet was washed twice with ice-cold 70% ethanol and the pellet was air dried. DNA was resuspended in PCR grade water. 16S rRNA gene amplicon from the DNA was prepared as described above for microcosm treatments (see section “Metagenome Extraction, Library Preparation, and Sequencing”). To understand the microbial diversity of these enrichments, amplicon based analysis was performed with Clostridium and facultative enrichments from both C_6M and C+L_6M setups but to identify the main iron and sulfate reducing populations, enrichments from C_6M was considered.
Diversity Analysis and Statistical Tool
Ion Torrent data analysis of V4 region of 16S rRNA gene was performed with QIIME 1.9.1 pipeline (Caporaso et al., 2010). Quality filtering of reads and bioinformatics were performed as described by Gupta et al., 2017. In brief, quality filtering was performed for raw reads to remove primers, sequences with homopolymers run of >6 bp and read length beyond the range of 230–300 bp. Only 3 primer mismatches were allowed due to degeneracy of primer set in this step. Denovo OTU picking was performed with uclust and SILVA 119 reference database3 was used for taxonomy assignments of reads as mentioned in QIIME pipeline. The OTU level analysis was performed by sub-sampling the samples to the lowest number of reads obtained in any of the samples through QIIME 1.9.1 pipeline. Venn diagram was generated in InteractiVenn4 (Heberle et al., 2015) for top 100 OTUs. Microbial metabolic pathways were estimated based on the 16S rRNA gene data from the closed OTU picking method using PICRUSt software package (Langille et al., 2013) on the web-based Galaxy server5. For PICRUSt analysis, Greengenes database6 was used for taxonomy assignment. One-way ANOVA was performed to assess the changes in the microbial diversity between the treatments using PAST software version 3.20 (Hammer et al., 2001). Weighted pair group mean arithmetic (WPGA) based hierarchical clustering was performed with Bray–Curtis distance dissimilarity matrix. Ternary plot was generated using PAST software to assess difference in diversity pattern among the treatments. All the data represented for physicochemical parameters were mean of its triplicates with standard deviation.
Nucleotide Accession Number
Metagenomic sequences are available under the NCBI BioProject ID PRJNA416924. The SRR number for each samples are SRR6320797 (C+L_6M), SRR6320796 (C_6M), SRR6320800 (0_Day), SRR6320884 (H_6M), SRR6320885 (FA_C_6M), SRR6320921 (Clos_C_6M), SRR6320922 (Clos_IRM), SRR6320919 (FA_IRM), SRR6320923 (Clos_SRM), SRR6320920 (FA_SRM), SRR7865998 (FA_C+L) and SRR7865999 (Clos_C+L). Sequence of DGGE bands were submitted in Genbank under accession numbers MH938427-MH938447.
Results
Change in Physicochemical Parameters After the End of Incubation
Nutrient amendments to AIS facilitated a considerable improvement of its physicochemical conditions (Table 1). At the onset of the study (0_Day), major physicochemical parameters of the soil slurry were measured. This sample was found to be of highly acidic (pH 3.51) nature; rich in soluble SO42− (7838 mg/l) and Fe (179 mg/l). Following incubation with nutrients, significant increase in pH (up to pH 6.61) but decrease in ORP (up to 110 mV) were observed coupled with considerable changes in concentrations of SO42−, Fe, Fe2+ and heavy metals. Control set (H_6M) with only water addition showed slight change with respect to the test physicochemical parameters while killed control did not show any shift at all. Incubation with only water (H_6M) could initiate reactions responsible for the observed shift in pH and ORP, presence of nutrients favored such reactions significantly. Following cysteine and cysteine + lactate amendment, soluble sulfate concentration was greatly reduced along with Fe (total Fe as well as Fe2+), Cu, Zn and Ni. Compared to H_6M that showed nearly 50% decrease in SO42− (to that of its initial level), cysteine + lactate addition could led to a 95% reduction. Microcosm amended with only cysteine showed only up to 76% lowering of SO42− (compared to 0_Day). Soluble Fe level presented an interesting trend: concentrations of both total Fe and Fe2+ were enhanced in H_6M (2.3-fold for Fe and 2.5-fold Fe2+) and C_6M (5.6-fold for Fe and 4.8-fold for Fe2+), while the values decreased significantly (0.5-fold for Fe and 0.6-fold for Fe2+) in C+L_6M. Although an overall enrichment experiment showed a strong role of the test nutrients in improving the local physicochemical condition of the AMD impacted soil, lactate + cysteine was identified as a better stimulant than cysteine alone.
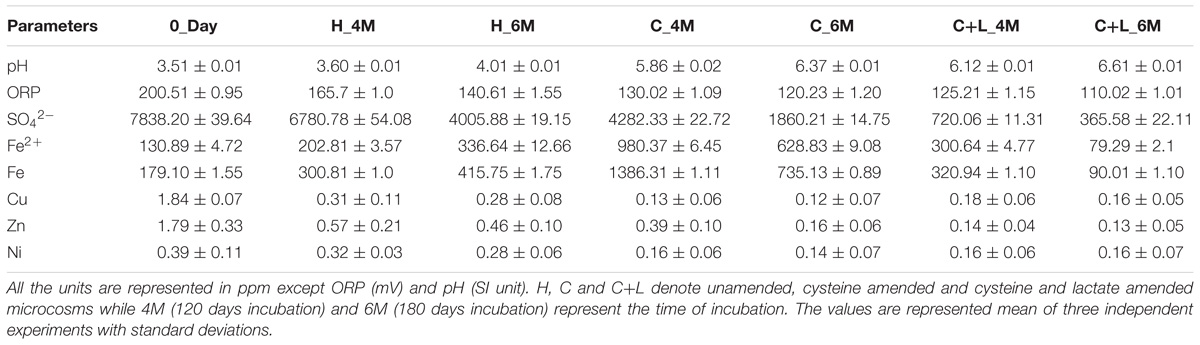
Table 1. Details of physicochemical parameters of the microcosm setup.
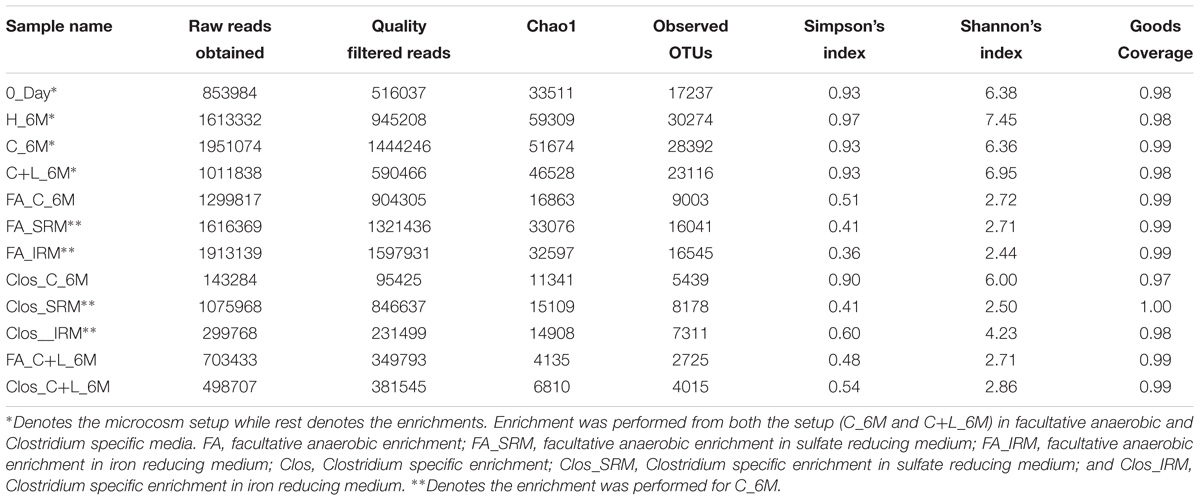
Table 2. Details of 16S rRNA gene reads and non-parametric diversity indices of microbial communities from microcosms and enrichments.
Shift in Microbial Community Composition
16S rRNA gene amplicon sequencing and estimated diversity indices revealed an assessable shift in microbial community composition of AIS following incubation with nutrient amendments (Table 2). Both estimated Chao1 and observed OTUs were increased coupled with distinct shifts in microbial community composition (Table 2). The most abundant bacterial phyla within the AIS at 0_Day were Proteobacteria (42%), WD272 (20%), Actinobacteria (14%), Acidobacteria (11%), Chloroflexi (9%), and Firmicutes (1.5%) (Figure 1). Following incubation, a distinct shift in community composition with great enhancement of Firmicutes coupled with the striking decrease in abundance of Proteobacteria, Acidobacteria, and Actinobacteria were detected (Figure 1). Abundance of the members of Firmicutes affiliated to Clostridia, OPB54, Negativicutes, and Bacilli was increased in both C+L_6M and C_6M. The extent of enhancement of Firmicutes was up to 36.5-fold in C_6M and 35.4-fold in C+L_6M (Figure 1). Proteobacteria [Gammaproteobacteria (35%), Alphaproteobacteria (6%), Betaproteobacteria (1%), and Deltaproteobacteria (0.07%)] that constituted the major phylum at 0_Day was found to be considerably less prevalent within the communities enriched with various amendments (Figure 1). The noteworthy decrease in abundance of Gammaproteobacteria and Alphaproteobacteria was observed in all the setup whereas abundance of Betaproteobacteria and Deltaproteobacteria was increased in C_6M (Figure 1). Members of the phylum Chloroflexi (Ktedonobacteria and KD4-96) also showed a substantiate increase in their abundance in C_6M (19.0%) while it got reduced in C+L_6M (0.03%). The other major classes such as Acidobacteria, Acidimicrobiia, Actinobacteria, and Thermoleophilia showed decrease in their abundance in C_6M and C+L_6M (Figure 1).
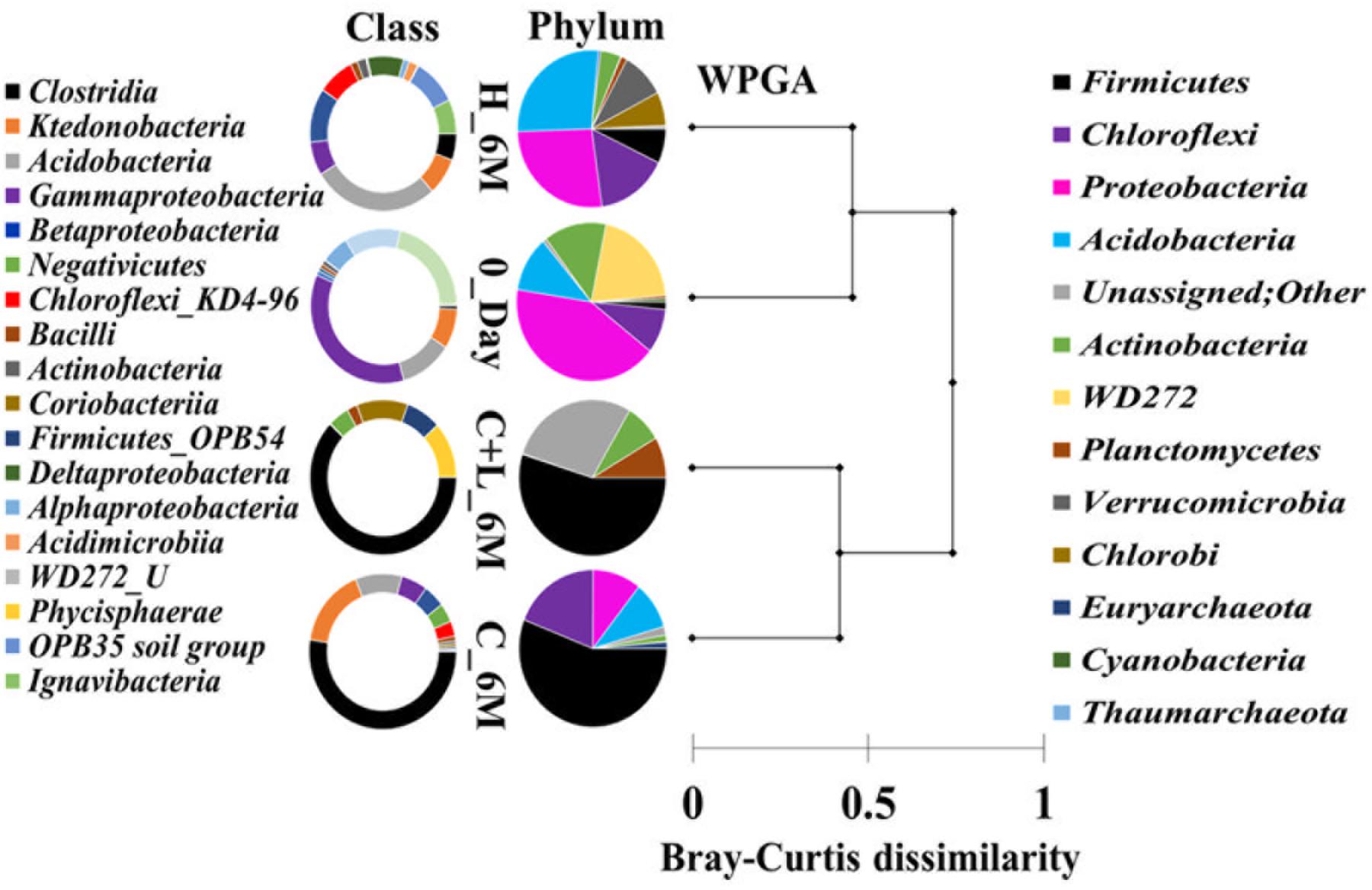
Figure 1. Distribution of relative abundance of 16S rRNA gene sequences detected in microcosm setup (amended and unamended) at phylum-level (cumulative abundance > 0.1%) and class-level (top 18) taxonomic resolution. WPGA based agglomerative hierarchical clustering of treatments on the basis of abundance of phylum using Bray–Curtis dissimilatory matrix is represented. U denotes uncultured member.
Family level analysis within the 0_Day, H_6M, C_6M and C+L_6M microcosms showed increase in abundance of several families. The abundance of facultative and/or strict anaerobic members of Clostridiales (Clostridiaceae 1, Family XIII, Christensenellaceae, Lachnospiraceae, Gracilibacteraceae, Peptococcaceae, Peptostreptococcaceae, Ruminococcaceae, and VadinBB60), Bacillales (Alicyclobacillaceae and Bacillaceae), Veillonellaceae, uncultured OPB54 and Coriobacteriaceae was increased in both C_6M and C+L_6M. Heat map analysis (Figure 2) of the distribution of major genera (also considering taxa classified up to family level) under Clostridiales indicated considerable enhancement in abundance of several taxa commonly attributed to sulfate- and iron-reduction following nutrient amendments. In contrast to this, only water amendment (control; H_6M) allowed enhancement of mostly known taxa involved in iron and sulfur oxidation [Acidobacteriaceae (Subgroup 1), Gallionellaceae, Xanthomonadaceae] and few other taxa such as OPB35 soil group, KD4-96, Ktedonobacteria_JG30-KF-AS9, BSV26 and Cystobacteraceae. Overall the successful enrichment of diverse fermentative and anaerobic populations was achieved, suppressing the growth of acidophilic members following creation of anoxic environment and supply of metabolizable C- and N-sources. A ternary plot was generated to understand the distribution of top 50 genera across 0_Day, C_6M and C+L_6M microcosm samples (Figure 3). The result showed that acidophilic genera such as Ferrithrix, Metallibacterium, Acidobacterium, uncultured –Acidomicrobiales and -Acidobacteriaceae Subgroup 1 etc. were more prevalent at 0_Day. In contrast, taxa affiliated to Firmicutes; Desulfitobacterium, Clostridium Sensu Stricto 1, Desulfosporosinus, Desulfurispora, uncultured -Christensenellaceae, -OPB54, -Clostridiales Family XVII, -Ruminococcaceae, -Lachnospiraceae, -Peptococcaceae etc. capable of sulfate- and iron-reduction dominated in C_6M and C+L_6M. One-way ANOVA analysis confirmed that microbial diversity among the treatments was significantly different (P < 0.05).
Microbial Shift at OTUs Level
In order to understand the dynamics of microbial community composition beyond the taxonomic level, most abundant OTUs (top 100 OTUs) from each of the microcosms were analyzed (Figures 4A–D). Top 100 OTUs from each microcosm contributed 79–85% of the total reads of the respective samples. The interesting finding was that when considering top 100 OTUs of one treatment, the same OTUs in another treatment contributed less percentage of the total reads, clearly indicating the effect of treatments (Figures 4E–H). Venn diagram depicted the pattern of sharing of OTUs among the treatments (Figure 4I) and signified that how the abundance of OTUs was significantly changed during the treatments. Taxonomic identities of these OTUs were determined to find their affiliation to 32 different taxa (Figure 5). Out of 100 OTUs from each of the microcosms, OTUs affiliated to Firmicutes were dominant in C+L_6M (80 OTUs) and C_6M (64 OTUs) while OTUs assigned to Proteobacteria were high in 0_Day (40 OTUs) and H_6M (33 OTUs) (Figure 5). These results were perfectly in line with our taxonomy based observation of increasing abundance of Clostridia in C+L_6M and C_6M. Total 26 OTUs (out of top 100 OTUs) were found to be shared between 0_Day and H_6M (Figure 6A). These common OTUs were affiliated mostly to acidophilic taxa. Among the C_6M and C+L_6M communities 24 shared OTUs were detected and these were affiliated to iron/sulfate reducing, fermentative and anaerobic Firmicutes taxa (Figure 6B).
qPCR Based Quantification of Bacterial/Specific Taxa and dsrB Gene
Quantitative estimation of the major taxa (Firmicutes, Acidobacteria, and Actinobacteria) as well as dsrB gene (involved in dissimilatory sulfate reduction) was performed for 0_Day, C_6M, C+L_6M and H_6M communities using qPCR based approach. Total bacterial 16S rRNA gene copies indicated a marginal reduction in bacterial abundance following microcosm amendments (Figure 7). The estimation of 16S rRNA gene copies for Actinobacteria, Acidobacteria, and Firmicutes corroborated with the amplicon based community data suggesting the decrease in abundance of Actinobacteria and Acidobacteria but increase in Firmicutes following nutrient amendment (Figure 7). The involvement of sulfate reducing bacteria in nutrient amended microcosms was highlighted by a remarkable increase in dsrB gene copy number from 7.8 × 104 to 3.9 × 105–1.0 × 106 (Figure 7).
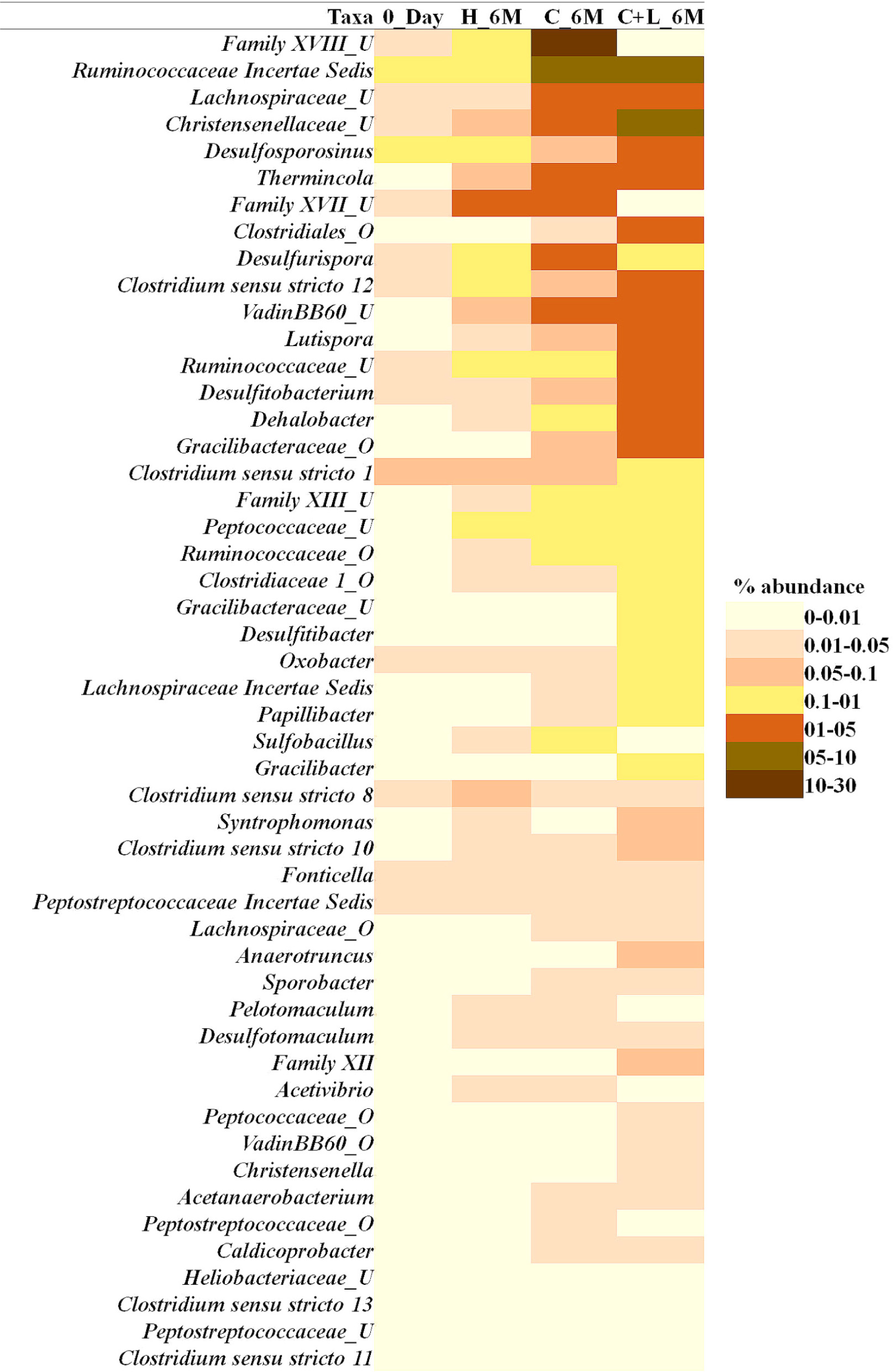
Figure 2. Heat map based relative abundance of distribution of top 50 Clostridiales members in all the microcosm setup. U and O denote uncultured member and others, respectively.
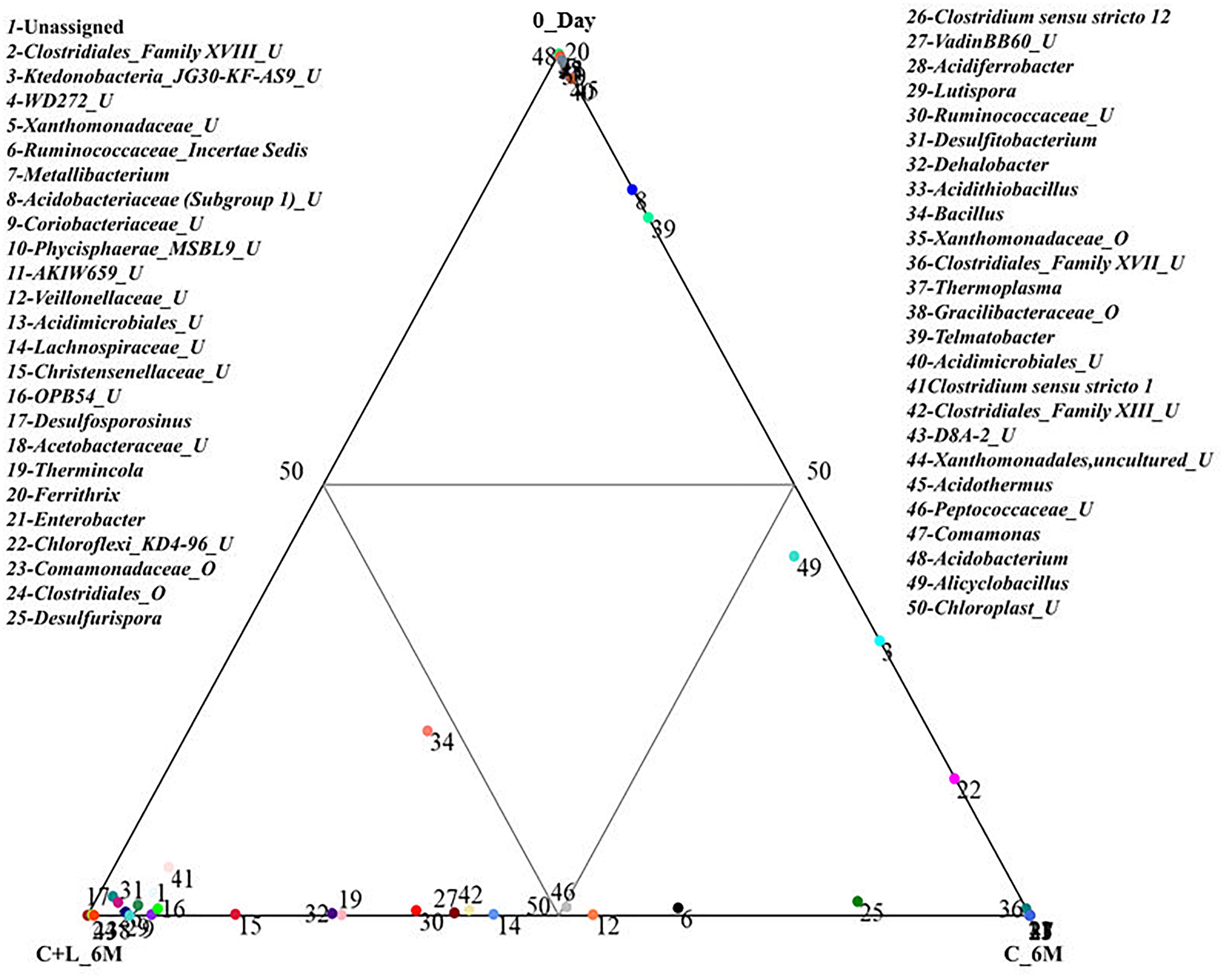
Figure 3. Distribution of taxonomic groups at genera level in 0_Day, C_6M and C+L_6M. The 50 most abundant genera (cumulative abundance > 0.2%) associated with each sample is visualized in ternary plots. The position in the triangle indicates the relative abundance of each taxon among the three samples. U and O denote uncultured member and others, respectively.
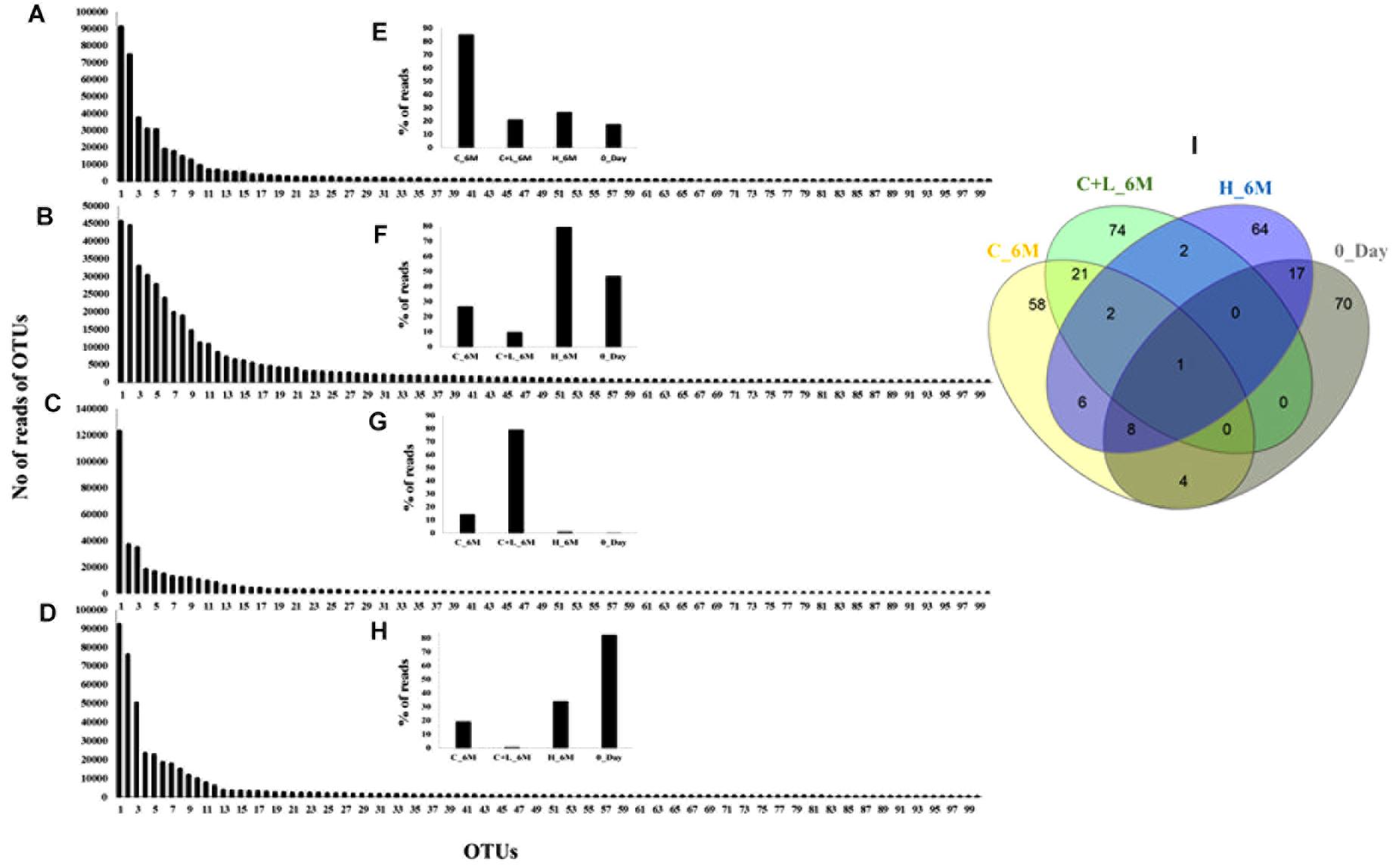
Figure 4. Rank abundance based analysis of top 100 OTUs in the microcosm setup. Rank abundance profile of top 100 OTUs from each sample is depicted (A) C_6M, (B) H_6M, (C) C+L_6M, and (D) 0_Day. (E–H) represent the percent distribution of top 100 OTUs across the samples. (I) Venn diagram shows the shared and unique OTUs across the samples (considering top 100 OTUs from each sample). Detail taxonomic affiliations of 100 OTUs are presented in Supplementary Table S2
PICRUSt Based Functional Prediction of the Community
Metabolic functions of the microbial communities were established through PICRUSt analysis. Using the genome-wide analysis tools integrated in PICRUSt, we could look into the genomic inventories related to sulfate and cysteine metabolism and other major biogeochemical processes of the enriched communities (Supplementary Figure S1). The result showed abundance of genes involved in dissimilatory sulfate metabolism (aprAB and dsrAB), cysteine metabolism (cysteine desulfhydrase, cysteine synthase, cystathionine synthase and cystathionine lyase) along with hydrogenases, metal tolerance/transporter gene for As, Fe, Cu, Zn, Co, etc., nitrogen metabolism and other major categories of metabolic functions. Considerable change in the abundance of dsrAB (involved in dissimilatory sulfate reduction) and cysteine desulfhydrase (involved in cysteine utilization) was observed in nutrient amended microcosms. The analysis clearly indicated that enriched microbial populations were genetically equipped for dissimilatory sulfate reduction following cysteine amendment.
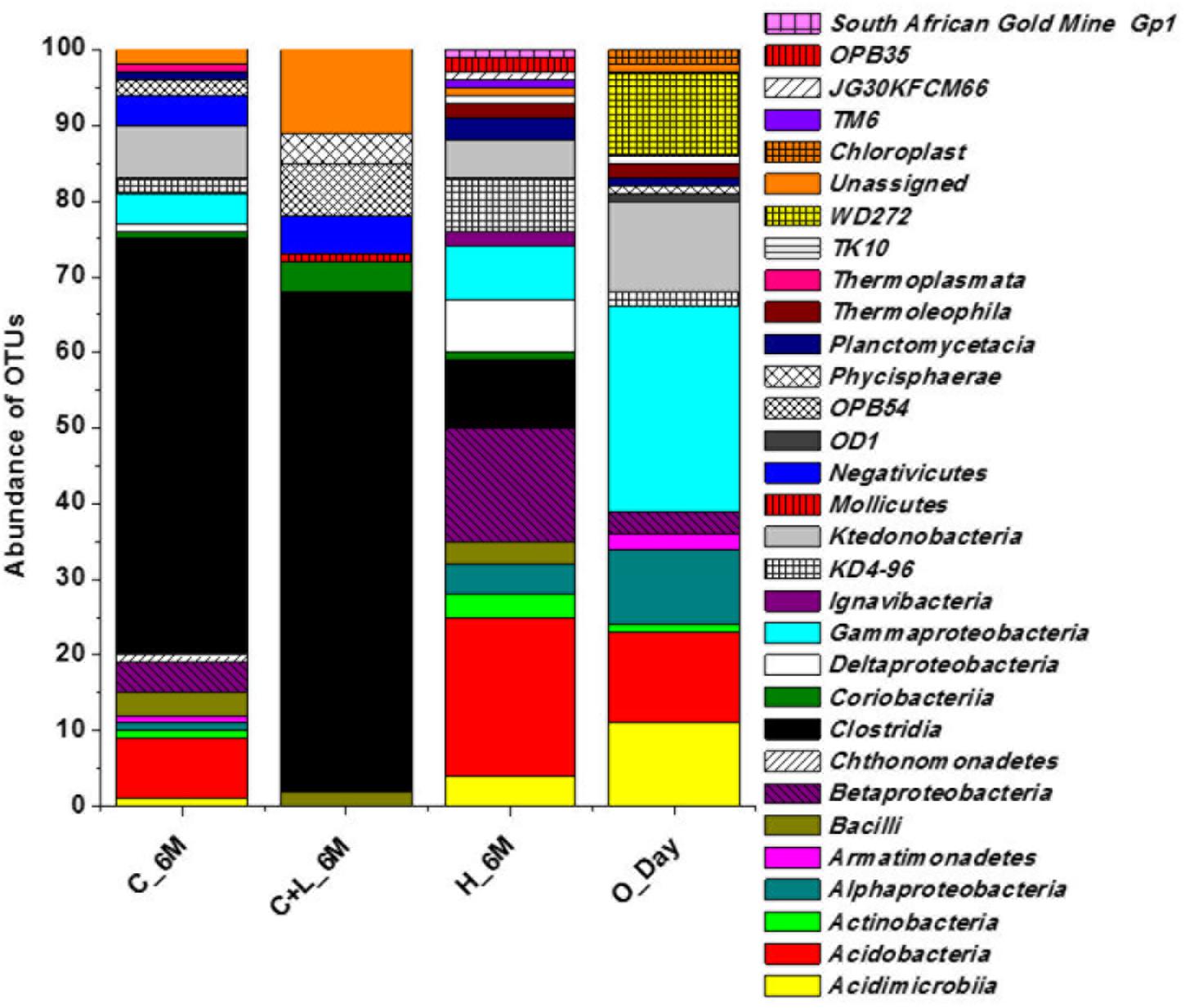
Figure 5. Taxonomic distribution pattern (at class-level) of top 100 OTUs in each setup.
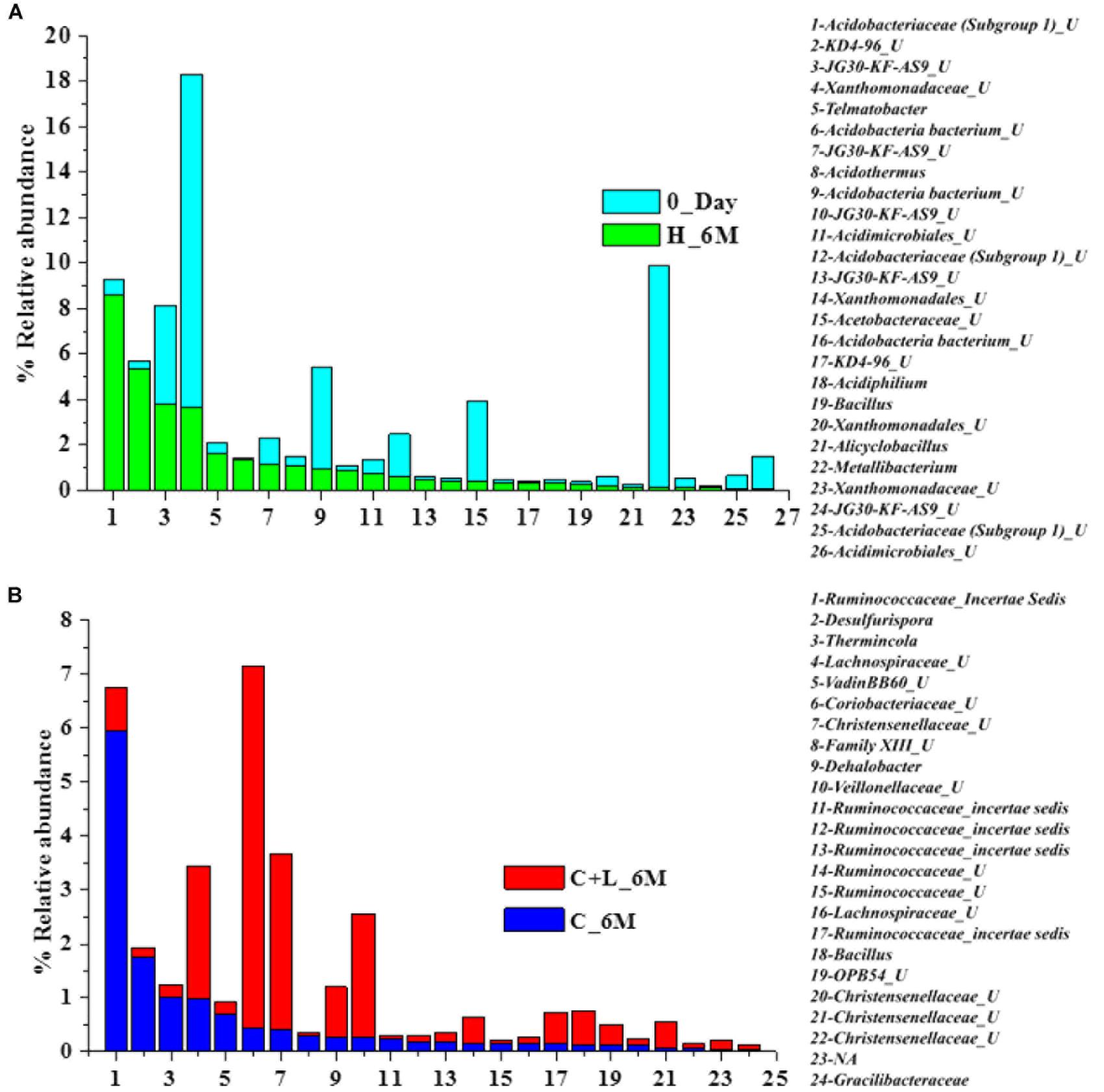
Figure 6. Distribution of shared OTUs (from top 100 OTUs) between the samples (A) 0_Day and H_6M and (B) C+L_6M and C_6M. U denotes uncultured member.
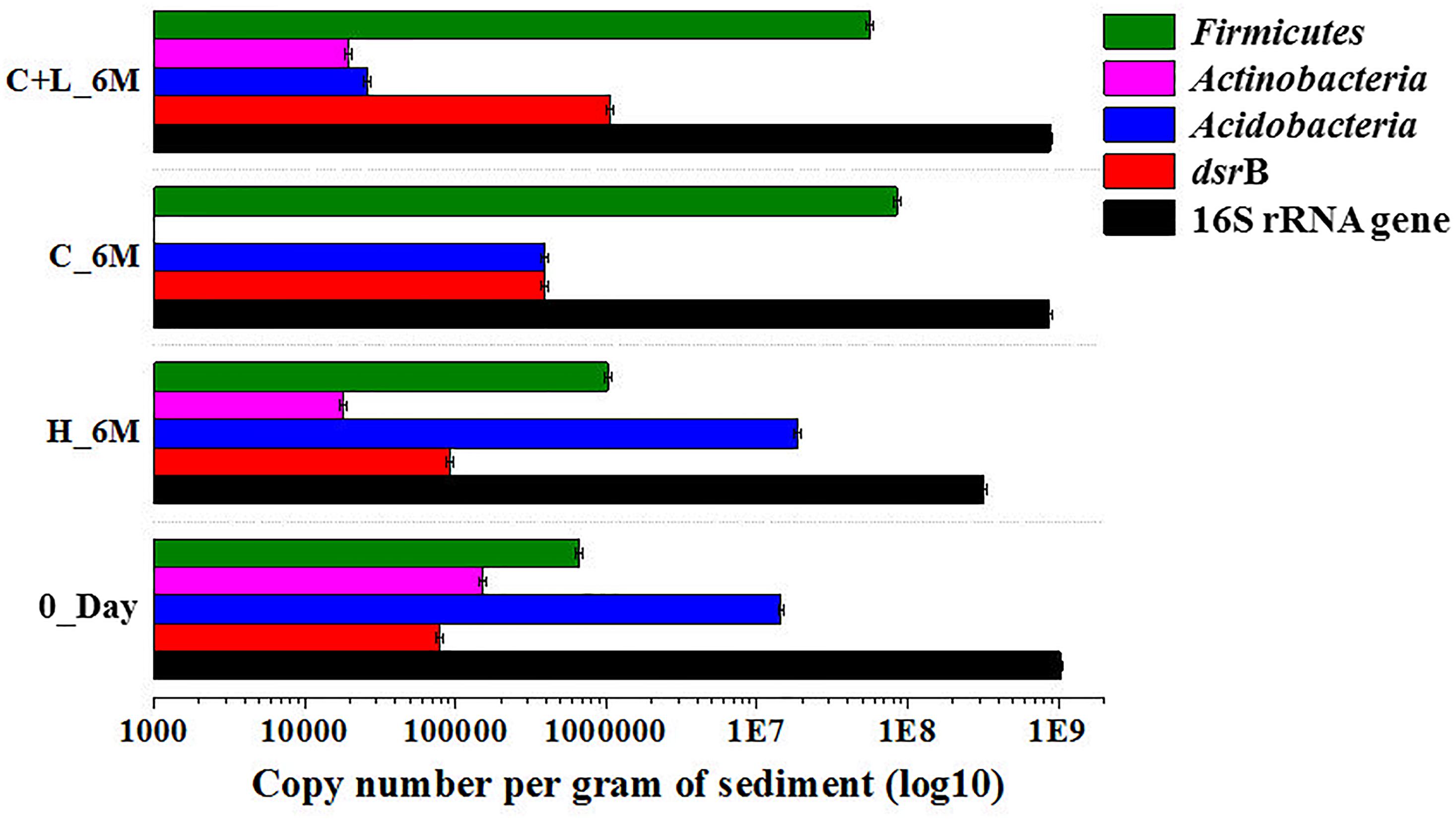
Figure 7. Quantitative PCR based analysis of gene copy number of bacteria/specific taxa and dsrB gene among the microcosm setup.
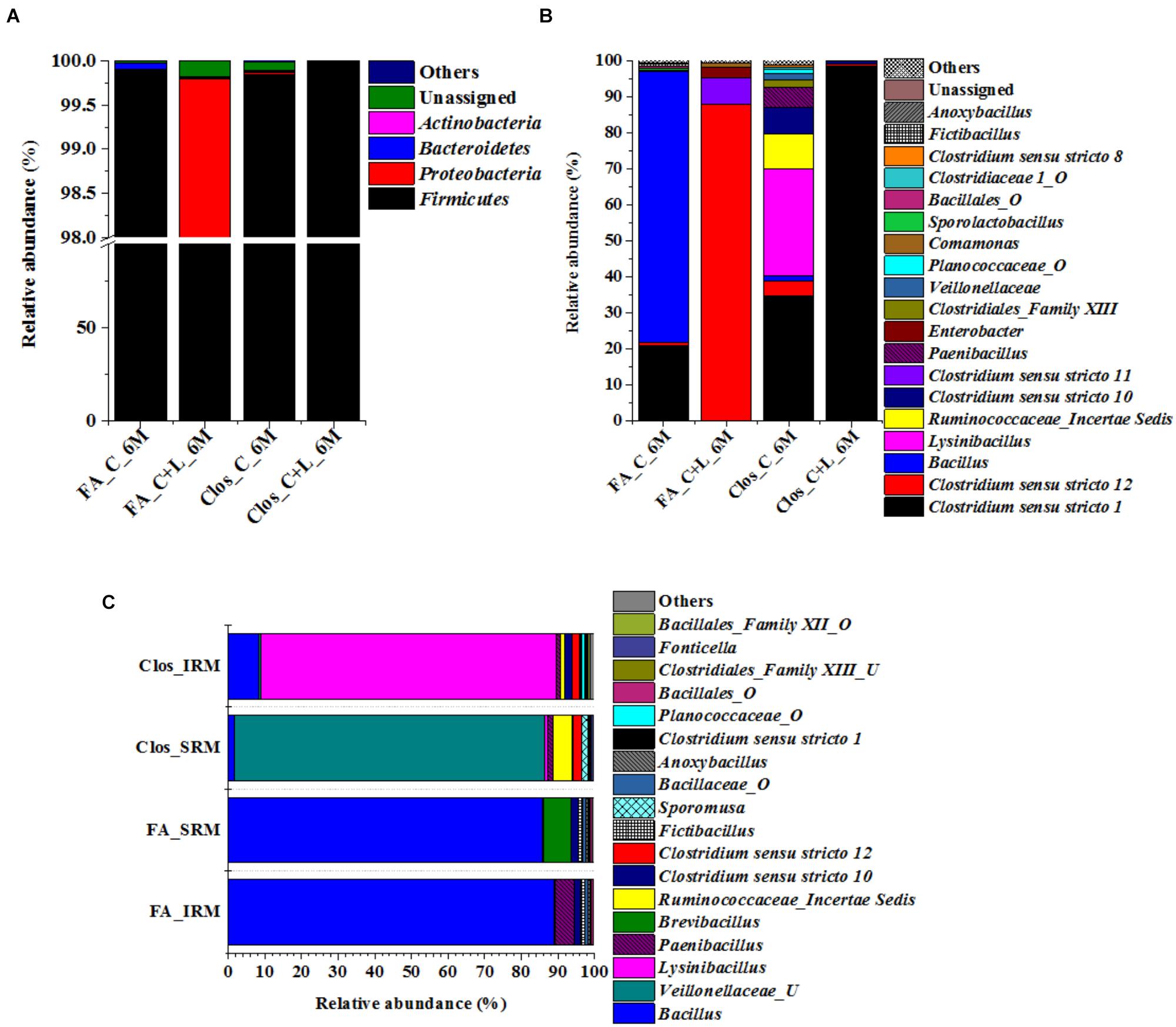
Figure 8. Taxonomic distribution of enrichment setups from C_6M and C+L_6M. (A) Phylum-level distribution of enriched populations in Clostridium specific (Clos) and Facultative anaerobic (FA) enrichment. (B) Distribution of genera in Clostridium specific enrichment and Facultative anaerobic enrichment. (C) Shift in microbial diversity (represented at genera-level) when enriched population of Clos_C_6M and FA_C_6M were subjected to iron and sulfate reducing medium. Clos_IRM: distribution pattern of Clostridium specific enrichment in iron reducing medium; Clos_SRM: distribution pattern of Clostridium specific enrichment in sulfate reducing medium; FA_IRM: distribution pattern of Facultative anaerobic enrichment in iron reducing medium and FA_SRM: distribution pattern of Facultative anaerobic enrichment in iron reducing medium. U and O denote uncultured member and others, respectively.
Enrichment of Firmicutes Members Using Specific Medium From Microcosm Setup
The 16S rRNA gene based investigation indicated that following cysteine amendment abundance of members of Firmicutes were enriched considerably. Most of these taxa were known for their role in anaerobic sulfate- and iron-reduction. Although the qPCR and PICRUSt supported the role of these members in the observed physicochemical changes within our AIS microcosms, final validation of their biogeochemical role was done by enrichment of the Firmicutes members under specific culture conditions. Cultures from both C_6M and C+L_6M microcosms were sub-cultured in two specific media: Clostridium specific and facultative anaerobic. Following three repeated sub-culturing in the respective media, taxonomic identities of the enriched populations were established by 16S rRNA gene based amplicon sequencing (Figure 8A). The results indicated that our culture conditions were highly supportive for the enrichment of Firmicutes members in both the Clostridium specific and facultative anaerobic media. Members of this phylum contributed 95.33–99.85% while Proteobacteria, Actinobacteria, and Bacteroidetes constituted very small populations (Figure 8A). The most dominant genera detected in the Clostridium specific enrichment belonged to Clostridiales and Bacillales members such as Clostridium sensu stricto 1, Lysinibacillus, Ruminococcaceae_incertae sedis, Clostridium sensu stricto 10, Clostridium sensu stricto 12, Paenibacillus, Clostridiales_Family XIII, uncultured Planococcaceae and uncultured Veillonellaceae members (Figure 8B). In the facultative anaerobic medium sub cultured from C_6M microcosm, Bacillus was the most dominant genera (75.70%) followed by Clostridium sensu stricto 1 (20.63%), Clostridium sensu stricto 12 (1.01%) and Sporolactobacillus (0.68%) (Figure 8B). In contrast, Clostridium sensu stricto 12 and Clostridium sensu stricto 11 accounted for more than 95% of the community grown in the same medium but subcultured from C+L_6M (Figure 8B).
Sulfate- and Iron-Reduction Potential of the Microorganisms Enriched in Clostridium Specific and Facultative Anaerobic Media
Bacterial cultures enriched in Clostridium specific and facultative anaerobic media were further inoculated in sulfate reducing and iron reducing media (designated as SRM and IRM) for assessing their potential toward sulfate- and iron-reduction. Following three repeated sub-culturing, 16S rRNA gene amplicon sequencing was done for all the four sets derived originally from C_6M microcosm. The amplicon sequencing result of Clostridium specific enrichment grown in SRM showed that uncultured Veillonellaceae members, Ruminococcaceae incertae sedis and Clostridium sensu stricto 12 accounted for 92.25% (Figure 8C). The same enrichment culture grown in IRM showed the abundance of Lysinibacillus (80.43%) along with Bacillus, uncultured Planococcaceae member, Clostridium sensu stricto 12 and Clostridium sensu stricto 10 (Figure 8C). Similar study performed with facultative anaerobic enrichment indicated proliferation (cumulative abundance of 98%) of Bacillus, Brevibacillus, Clostridium sensu stricto 10, Fictibacillus, Anoxybacillus, and Clostridium sensu stricto 1 in SRM (Figure 8C) while Bacillus, Paenibacillus, Clostridium sensu stricto 10 and Fictibacillus accounted for 97.71% of the IRM culture (Figure 8C). Metabolic abilities of the enriched populations derived from microcosms C_6M and C+L_6M microcosms toward sulfate- and iron-reduction were confirmed by quantitative estimation of SO42− and Fe2+ ions. Nearly complete reduction of SO42− (15 mM) and Fe3+ (5 mM) were noticed following 10–14 days of incubation, confirming their abilities for reduction of these terminal electron acceptors. The formation of black precipitates of iron sulfide in SRM and change in color of the IRM from yellow to light green or colorless with precipitation of Fe (Ferric citrate as redox indicator) (Pan et al., 2017) was also noted (Supplementary Figure S2). In order to confirm the presence of these SRB and IRB after 4 months of incubation where a decline in soluble iron concentration (Fe2+) was observed due to precipitation with sulfide produced by sulfate reducing activity in the treatments, DGGE based microbial community analysis was performed with 5 months incubated microcosms (H_5M, C_5M, and C+L_5M). The banding pattern obtained for C_5M and C+L_5M communities showed enrichment of almost similar types of microbial populations (Figure 9). The enrichment of Clostridium sp., Themincola sp., Bacillus sp., Steroidobacter sp., as well as members of Acidobacteriaceae, Ruminococcaceae, and Coriobacteriaceae in these treatments clearly indicated their potential toward both iron- and sulfate-reduction (Figure 9). These groups were also detected in the same treatments through amplicon based sequencing after 6 months of incubation. These known iron and sulfate reducing populations were also detected in both iron and sulfate reducing media hence confirmed their involvement in reduction of iron and sulfate during 5 months of incubated setups.
Discussion
Geomicrobiology of AMD including the nature of microorganisms and biogeochemical functions of various acidophilic microorganisms is well established. In contrast to that, the broader ecological roles of AMD organisms in terms of the attenuation of the hazardous nature of such acidic environment remain less explored. Our study demonstrated that it is possible to enhance the activities of indigenous sulfate- and iron-reducing bacteria of an AIS to achieve improvement of its major physicochemical parameters desirable for bioremediation. With respect to the major questions we posed during this study, our results proved that (a) it is possible to enhance the abundance and activities of autochthonous sulfate- and iron-reducing bacteria of an AIS and (b) this altered microbial community could lead toward changing the physicochemical conditions favorably, thus decreasing the hazardous nature of the studied sample considerably.
There are reports highlighting the presence of heterotrophic sulfate- and iron-reducing bacteria (Clostridiaceae, Peptococcaceae, and Bacillaceae members) within highly acidic AMD systems (Sánchez-Andrea et al., 2012a,b). Our biostimulation based approach was successful in enhancing the abundance of Firmicutes members capable of anaerobic sulfate-/iron-reduction. In the native AIS, these bacterial taxa constituted only 1.5% which (low abundance of heterotrophic reducing taxa) corroborated the earlier reports on different AMD environments (Chen L.X. et al., 2013; Kuang et al., 2013). Increase in abundance of these anaerobic/facultative anaerobic populations surpassing the acid producing-, sulfur- and metal-oxidizing microorganisms with nutrient amendments was impressive. All these members of the phylum Firmicutes were well known for their facultative to strict anaerobic metabolism, but not so much for sulfate- and iron-reduction except few taxa such as Clostridium, Desulfosporosinus, Desulfotomaculum etc. (Chockalingam and Subramanian, 2006; Church et al., 2007; Sánchez-Andrea et al., 2012b; Pan et al., 2017). The increased abundance of gene encoding dissimilatory sulfite reductase (dsrB) and Firmicutes specific 16S rRNA gene detected in qPCR, reduction of -sulfate/-iron and rise in pH were all in strong agreement. Our results demonstrated that a number of sulfate- and iron-reducing bacterial taxa present in AMD impacted environment can be proliferated and implicated with the desirable reductive processes successfully. The PICRUSt analysis confirmed that the enriched bacterial populations were genetically equipped for dissimilatory sulfate reduction processes.
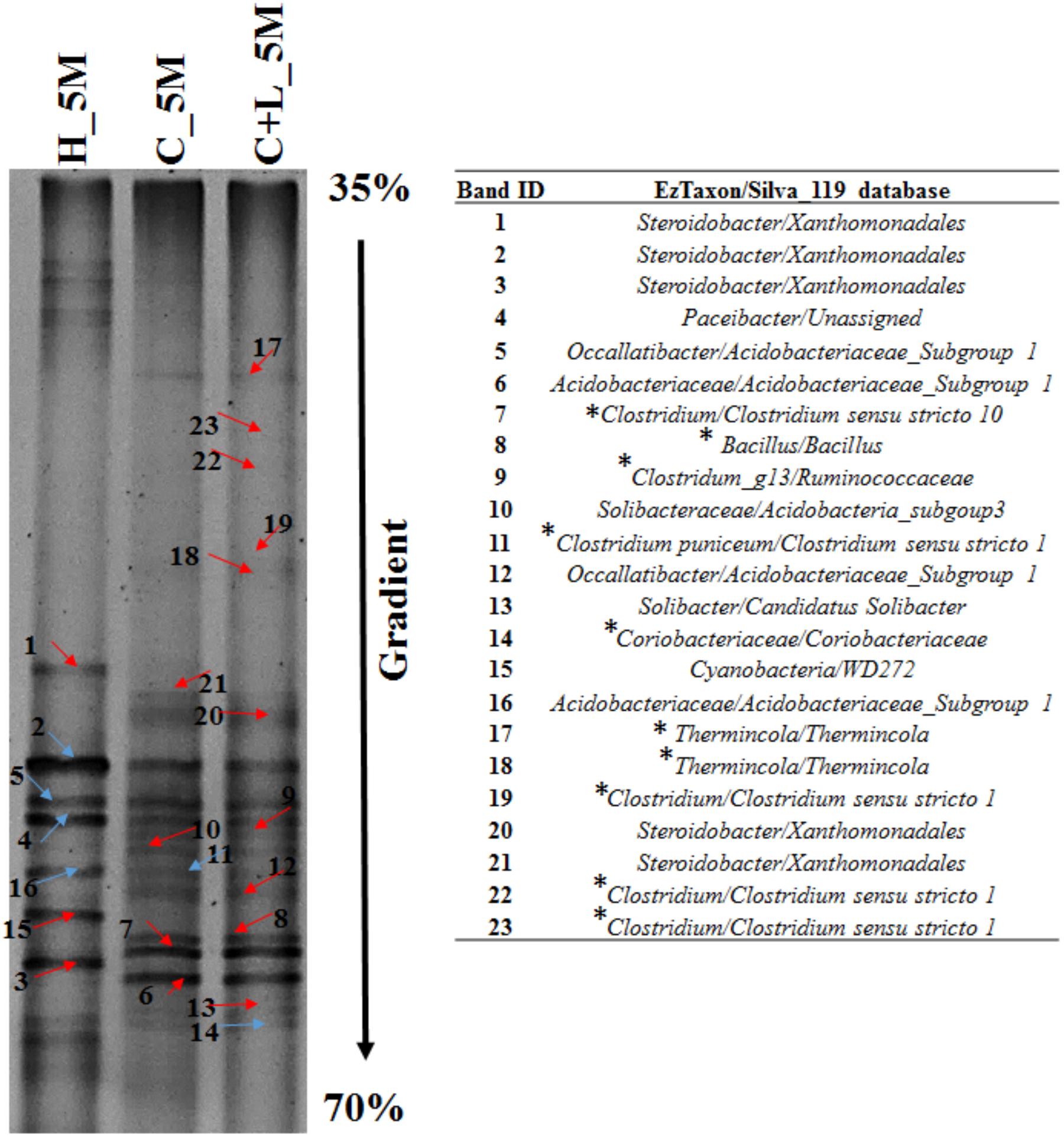
Figure 9. Denaturing gradient gel electrophoresis (DGGE) profile of treatments (H_5M, C_5M, and C+L_5M) at 150 days of incubation. Red arrow indicates distinct band while blue represents common bands. ∗Represents the organisms detected in both treatment and enrichment of C_6M and C+L_6M.
The effect of cysteine (alone or along with lactate) as successful proxy to provide required metabolic resources and thus biostimulate the target groups of microorganisms could be attributed to its dual characteristics. Cysteine could be used by the microbes as a carbon and nitrogen source, and also might act as a reducing agent (that helps in scavenging the dissolved oxygen) to facilitate reduction of iron and sulfate. Microbes catabolize cysteine for their fermentative mode of metabolism through two enzymes (i) cysteine desulfhydrase which produces NH3, pyruvate and H2S and (ii) cystathionine-γ-lyase which utilizes an oxidized form of cysteine (Morra and Dick, 1991). Microbe mediated H2S production was possible from selective enrichment of soil amended with cysteine through cysteine desulfhydrase enzyme. Wang et al. (2000) demonstrated that the genes encoding cysteine desulfhydrase and serine acetyltransferase may be used to develop a metabolically engineered Escherichia coli that can carry out aerobic sulfate reduction. Suitability of carbon sources rich in amino acids, but low in lignin in promoting sulfate reduction was also reported (Coetser et al., 2006). Recently, Zhang et al. (2017) established the role of tryptone and yeast extract in the remediation of mine tailings by promoting the growth of SRB.
Microbial taxa enriched during this study were reported to be of facultative- or strict-anaerobic nature, involved in anaerobic hydrolytic fermentation, cysteine utilization, acetate- and H2S-production and metal reduction (Petrie et al., 2003; Church et al., 2007; Finke and Jørgensen, 2008; Kosaka et al., 2008; Li et al., 2011; Bertel et al., 2012; AlAbbas et al., 2013; Hausmann et al., 2016; Peng et al., 2016; Pan et al., 2017). The major genera identified in this study such as Clostridium, Clostridium sensu stricto members, Lutispora, Sporobacter, Acetanaerobacterium, Caldicoprobacter, Gracilibacter, Oxobacter, Fonticella, Papillibacter, as well as unclassified members of Ruminococcaceae, Lachnospiraceae, and Christensenellaceae were all reported as anaerobic, fermentative, cellulose- and cysteine-metabolizing, acetogenic, and iron reducing members (Grech-Mora et al., 1996; Defnoun et al., 2000; Chen and Dong, 2004; Lee et al., 2006; Shiratori et al., 2008; Bouanane-Darenfed et al., 2011; Chen M. et al., 2013; Fraj et al., 2013; Peng et al., 2016). Presence of these organisms was reported from diverse sulfur-rich environments including hot spring (Zavarzina et al., 2007; Bouanane-Darenfed et al., 2011; Fraj et al., 2013), mine tailings/drainage/soil (Sánchez-Andrea et al., 2011; Gupta et al., 2017), constructed wetland (Lee et al., 2006) and AMD treatments sites (Clarke et al., 2004; Kaksonen et al., 2004; Pruden et al., 2007; Bijmans et al., 2009; Hiibel et al., 2011; Lu et al., 2011; Martins et al., 2011; Sánchez-Andrea et al., 2014; Deng et al., 2016). Considering the known metabolic characteristics of these taxa and their ecological relevance, we could attribute their abundance to the observed sulfate- and iron-reduction. In accordance with previous reports, the other known strict anaerobic sulfate reducing taxa such as Desulfurispora, Desulfotomaculum, Desulfosporosinus, and Desulfitobacterium was also enriched during our study (Kaksonen et al., 2004; Church et al., 2007; Hiibel et al., 2008, 2011; Bijmans et al., 2010). The enhanced abundance of facultative anaerobic fermentative and strictly anaerobic sulfate reducing populations following cysteine amendments highlights the synergistic role of these metabolically dependent organisms confirming the fermentation coupled with sulfate reduction phenomenon (Finke and Jørgensen, 2008). We hypothesize that in the presence of cysteine, fermentative organisms become activated, producing metabolites and deplete the dissolved oxygen rapidly and thereby creating more anoxic niches. Within these anoxic micro-niches strict anaerobic populations proliferate, making use of the sulfate as preferred terminal electron acceptor thus facilitates sulfate reduction and rise in pH (Church et al., 2007). Our attempt to confirm the physiological abilities of the enriched populations toward sulfate- and iron-reduction by using culture media specific for Clostridium and facultative anaerobic bacteria supported the above hypothesis. We were successful in identifying the facultative and strict anaerobic sulfate- and iron-reducing populations with conformity through specific enrichment and deep sequencing.
The potential involvement of individual members of the enriched populations toward reductive processes was validated by a third level of enrichment wherein sulfate- and iron-reducing populations were grown more selectively in two specific media. These sulfate- and iron-reducing bacteria specific enrichments were meant to segregate and identify the organisms responsible for individual terminal electron acceptor utilization (iron as Fe3+ and sulfate as SO42). 16S rRNA gene sequencing of metagenomes retrieved from these enrichments revealed that members of the families Clostridiaceae, as well as Bacillaceae (genera Lysinibacillus, Bacillus and Paenibacillus etc.), Veillonellaceae and Ruminococcaceae etc. specifically contributed toward sulfate- or iron-reduction. Presence of these members in both C_5M and C+L_5M microcosms through DGGE further confirmed their potential of sulfate and iron reduction. Clostridiaceae and Bacillaceae members were previously reported in different AMD bioremediation studies or in sulfate-/iron-reducing enrichments/AMD environment (Clarke et al., 2004; Scala et al., 2006; Hiibel et al., 2008, 2011; Sánchez-Andrea et al., 2011; Yi et al., 2012; Giloteaux et al., 2013; Zhang and Wang, 2016; Zhang et al., 2016). The predominance of metal reducing Pelosinus (member of Veillonellaceae) on lactate amendment was reported by Mosher et al. (2012). Metal reduction and fermentative mode of metabolism of Veillonellaceae members were reported by earlier investigators including the whole genome sequence analysis of uncultured Veillonellaceae strain RU4 that confirmed presence of genes for sulfate reduction as well as polysulfide reduction (Brown et al., 2012; Shah, 2013; Kwon et al., 2016). Zhao et al. (2010) reported the role of Ruminococcus spp. (member of Ruminococcaceae) in sulfate reduction. Thus in our study, these enriched members confirmed their involvement in iron and sulfate reduction.
Conclusion
An acidic, sulfate-, iron- and other heavy metal-rich AMD impacted soil harbored low proportion of heterotrophic, sulfate- and iron-reducing anaerobic bacterial populations. These redox active members can be successfully stimulated by cysteine and lactate amendment. These enriched microbial groups can facilitate dramatic change in physiochemical condition. The microorganisms which got enriched with nutrient amendment belonged to the fermentative and strict anaerobic sulfate- and iron-reducing populations affiliated to Clostridiaceae, Veillonellaceae, Bacillaceae, Ruminococcaceae etc. Increased abundance of these organisms as evident from 16S rRNA amplicon sequencing and taxon-specific qPCR; enhancement of dsrB gene, change in genomic composition suitable for carrying out the required catabolic function corroborated with reduction in soluble sulfate- and iron-reduction and pH management. This study enabled us to gain a better insight on ecological perspective of the members of phylum Firmicutes indigenous to AMD impacted sites and more importantly, their involvement in sulfate- and, iron-reduction processes. The study also demonstrated the suitability of amino acid/protein rich natural substances as potent biostimulation agent for bioremediation of AMD/AMD impacted sites and provided us the specific microbial populations capable of anaerobic sulfate-, and/or iron-reduction which could be used as a potent bioaugmentation agent for future bioremediation applications.
Author Contributions
PS conceived and designed the experiments and arranged funds. AG performed the major experiments. PS and AG were responsible for manuscript preparation. MP, PS, and AG arranged sampling from MCP. AG and JS performed the qPCR. AG and AD performed the bioinformatics analysis for deciphering microbial diversity. AG and AD performed the 16S rRNA gene amplicon sequencing in Ion S5 sequencer.
Funding
The authors are grateful to Department of Biotechnology, Government of India for funding the project (BT/PR 7533/BCE/8/959/2013, Dated 10/12/2013). The authors are thankful to IIT Kharagpur for providing the NGS facility (Ion S5 sequencer) through SGBSI challenge grant (IIT/SRIC/BT/ODM/2015-16/141). AG thanks the Department of Biotechnology, Government of India for providing fellowship under DBT-JRF category (DBT/2014/IITKH/113). Financial support to AD (IIT/ACAD (PGS&R)/F.II/2/14/BS/91R01) and JS (IIT/ACAD(PGS&R)/F.II/2/13/BT/91P01) from IIT Kharagpur and institutional fellowship was acknowledged.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The generous help from Malanjkhand Copper Project, Hindustan Copper Limited authority for sample collections was acknowledged.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.02882/full#supplementary-material
Footnotes
- ^ www.ezbiocloud.net
- ^ www.arb-silva.de/documentation/release-119
- ^ www.arb-silva.de/documentation/release-119
- ^ http://www.interactivenn.net
- ^ https://huttenhower.sph.harvard.edu/galaxy
- ^ http://greengenes.secondgenome.com
References
AlAbbas, F. M., Bhola, S. M., Spear, J. R., Olson, D. L., and Mishra, B. (2013). The shielding effect of wild type iron reducing bacterial flora on the corrosion of linepipe steel. Eng. Fail. Anal. 33, 222–235. doi: 10.1016/j.engfailanal.2013.05.020
Baker, B. J., and Banfield, J. F. (2003). Microbial communities in acid mine drainage. FEMS Microbiol. Ecol. 44, 139–152. doi: 10.1016/S0168-6496(03)00028-X
Bates, S. T., Berg-Lyons, D., Caporaso, J. G., Walters, W. A., Knight, R., and Fierer, N. (2011). Examining the global distribution of dominant archaeal populations in soil. ISME J. 5, 908–917. doi: 10.1038/ismej.2010.171
Becerra, C. A., López-Luna, E. L., Ergas, S. J., and Nüsslein, K. (2009). Microcosm-based study of the attenuation of an acid mine drainage-impacted site through biological sulfate and iron reduction. Geomicrobiol. J. 26, 9–20. doi: 10.1080/014904508025992
Bertel, D., Peck, J., Quick, T. J., and Senko, J. M. (2012). Iron transformations induced by an acid-tolerant Desulfosporosinus species. Appl. Environ. Microbiol. 78, 81–88. doi: 10.1128/AEM.06337-11
Bertin, P. N., Heinrich-Salmeron, A., Pelletier, E., Goulhen-Chollet, F., Arsène-Ploetze, F., Gallien, S., et al. (2011). Metabolic diversity among main microorganisms inside an arsenic-rich ecosystem revealed by meta-and proteo-genomics. ISME J. 5, 1735–1747. doi: 10.1038/ismej.2011.51
Bijmans, M. F., De Vries, E., Yang, C. H., Buisman, N., Cees, J., Lens, P. N., et al. (2010). Sulfate reduction at pH 4.0 for treatment of process and wastewaters. Biotechnol. Prog. 26, 1029–1037. doi: 10.1002/btpr.400
Bijmans, M. F., Dopson, M., Peeters, T. W., Lens, P. N., and Buisman, C. J. (2009). Sulfate reduction at pH 5 in a high-rate membrane bioreactor: reactor performance and microbial community analyses. J. Microbiol. Biotechnol. 19, 698–708. doi: 10.4014/jmb.0809.502
Bird, L. J., Bonnefoy, V., and Newman, D. K. (2011). Bioenergetic challenges of microbial iron metabolisms. Trends Microbiol. 19, 330–340. doi: 10.1016/j.tim.2011.05.001
Bouanane-Darenfed, A., Fardeau, M. L., Grégoire, P., Joseph, M., Kebbouche-Gana, S., Benayad, T., et al. (2011). Caldicoprobacteralgeriensis sp. nov. A new thermophilic anaerobic, xylanolytic bacterium isolated from an Algerian hot spring. Curr. Microbiol. 62, 826–832. doi: 10.1007/s00284-010-9798-9
Brown, S. D., Podar, M., Klingeman, D. M., Johnson, C. M., Yang, Z. K., Utturkar, S. M., et al. (2012). Draft genome sequences for two metal-reducing Pelosinus fermentans strains isolated from a Cr (VI)-contaminated site and for type strain R7. J. Bacteriol. 194, 5147–5148. doi: 10.1128/JB.01174-12
Burns, A. S., Pugh, C. W., Segid, Y. T., Behum, P. T., Lefticariu, L., and Bender, K. S. (2012). Performance and microbial community dynamics of a sulfate-reducing bioreactor treating coal generated acid mine drainage. Biodegradation 23, 415–429. doi: 10.1007/s10532-011-9520-y
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Chandra, A. P., and Gerson, A. R. (2010). The mechanisms of pyrite oxidation and leaching: a fundamental perspective. Surf. Sci. Rep. 65, 293–315. doi: 10.1016/j.surfrep.2010.08.003
Chen, L. X., Hu, M., Huang, L. N., Hua, Z. S., Kuang, J. L., Li, S. J., et al. (2015). Comparative metagenomic and metatranscriptomic analyses of microbial communities in acid mine drainage. ISME J. 9, 1579–1592. doi: 10.1038/ismej.2013.242
Chen, L. X., Huang, L. N., Méndez-García, C., Kuang, J. L., Hua, Z. S., Liu, J., et al. (2016). Microbial communities, processes and functions in acid mine drainage ecosystems. Curr. Opin. Biotechnol. 38, 150–158. doi: 10.1016/j.copbio.2016.01.013
Chen, L. X., Li, J. T., Chen, Y. T., Huang, L. N., Hua, Z. S., Hu, M., et al. (2013). Shifts in microbial community composition and function in the acidification of a lead/zinc mine tailings. Environ. Microbiol. 15, 2431–2444. doi: 10.1111/1462-2920.12114
Chen, M., Cao, F., Li, F., Liu, C., Tong, H., Wu, W., et al. (2013). Anaerobic transformation of DDT related to iron (III) reduction and microbial community structure in paddy soils. J. Agric. Food Chem. 61, 2224–2233. doi: 10.1021/jf305029p
Chen, S., and Dong, X. (2004). Acetanaerobacterium elongatum gen. nov., sp. nov., from paper mill waste water. Int. J. Syst. Evol. Microbiol. 54, 2257–2262. doi: 10.1099/ijs.0.63212-0
Chesnin, L., and Yien, C. H. (1951). Turbidimetric determination of available sulfates. Soil Sci. Soc. Am. J. 15, 149–151. doi: 10.2136/sssaj1951.036159950015000C0032x
Chockalingam, E., and Subramanian, S. (2006). Studies on removal of metal ions and sulphate reduction using rice husk and Desulfotomaculum nigrificans with reference to remediation of acid mine drainage. Chemosphere 62, 699–708. doi: 10.1016/j.chemosphere.2005.05.013
Church, C. D., Wilkin, R. T., Alpers, C. N., Rye, R. O., and McCleskey, R. B. (2007). Microbial sulfate reduction and metal attenuation in pH 4 acid mine water. Geochem. Trans. 8:10. doi: 10.1186/1467-4866-8-10
Clarke, A. M., Kirby, R., and Rose, P. D. (2004). Molecular microbial ecology of lignocellulose mobilisation as a carbon source in mine drainage wastewater treatment. Water 30, 658–661.
Coetser, S. E., Pulles, W., Heath, R. G. M., and Cloete, T. E. (2006). Chemical characterisation of organic electron donors for sulfate reduction for potential use in acid mine drainage treatment. Biodegradation 17, 67–77. doi: 10.1007/s10532-005-7567-3
Defnoun, S., Labat, M., Ambrosio, M., Garcia, J. L., and Patel, B. K. (2000). Papillibacter cinnamivorans gen. nov., sp. nov., a cinnamate-transforming bacterium from a shea cake digester. Int. J. Syst. Evol. Microbiol. 50, 1221–1228. doi: 10.1099/00207713-50-3-1221
Denef, V. J., Mueller, R. S., and Banfield, J. F. (2010). AMD biofilms: using model communities to study microbial evolution and ecological complexity in nature. ISME J. 4, 599–610. doi: 10.1038/ismej.2009.158
Deng, D., Weidhaas, J. L., and Lin, L. S. (2016). Kinetics and microbial ecology of batch sulfidogenic bioreactors for co-treatment of municipal wastewater and acid mine drainage. J. Hazard. Mater. 305, 200–208. doi: 10.1016/j.jhazmat.2015.11.041
Druschel, G. K., Baker, B. J., Gihring, T. M., and Banfield, J. F. (2004). Acid mine drainage biogeochemistry at Iron Mountain, California. Geochem. Trans. 5:13. doi: 10.1185/1467-4866-5-13
Finke, N., and Jørgensen, B. B. (2008). Response of fermentation and sulfate reduction to experimental temperature changes in temperate and Arctic marine sediments. ISME J. 2, 815–829. doi: 10.1038/ISMEJ.2008.2
Fraj, B., Hania, W. B., Postec, A., Hamdi, M., Ollivier, B., and Fardeau, M. L. (2013). Fonticella tunisiensis gen. nov., sp. nov., isolated from a hot spring. Int. J. Syst. Evol. Microbiol. 63, 1947–1950. doi: 10.1099/ijs.0.041947-0
Giloteaux, L., Duran, R., Casiot, C., Bruneel, O., Elbaz-Poulichet, F., and Goñi-Urriza, M. (2013). Three-year survey of sulfate-reducing bacteria community structure in Carnoules acid mine drainage (France), highly contaminated by arsenic. FEMS Microbiol. Ecol. 83, 724–737. doi: 10.1111/1574-6941.12028
Goltsman, D. S. A., Comolli, L. R., Thomas, B. C., and Banfield, J. F. (2015). Community transcriptomics reveals unexpected high microbial diversity in acidophilic biofilm communities. ISME J. 9, 1014–1023. doi: 10.1038/ismej.2014.200
Grech-Mora, I., Fardeau, M. L., Patel, B. K. C., Ollivier, B., Rimbault, A., Prensier, G., et al. (1996). Isolation and characterization of Sporobacter termitidis gen. nov., sp. nov., from the digestive tract of the wood-feeding termite Nasutitermes lujae. Int. J. Syst. Evol. Microbiol. 46, 512–518. doi: 10.1099/00207713-46-2-512
Gupta, A., Dutta, A., Sarkar, J., Paul, D., Panigrahi, M. K., and Sar, P. (2017). Metagenomic exploration of microbial community in mine tailings of Malanjkhand copper project, India. Genom. Data 12, 11–13. doi: 10.1016/j.gdata.2017.02.004
Hallberg, K. B. (2010). New perspectives in acid mine drainage microbiology. Hydrometallurgy 104, 448–453. doi: 10.1016/j.hydromet.2009.12.013
Hammer, Ø., Harper, D. A. T., and Ryan, P. D. (2001). PAST: paleontological statistics software package for education and data analysis. Palaeontol. Electronica 4, 1–9.
Hausmann, B., Knorr, K. H., Schreck, K., Tringe, S. G., del Rio, T. G., Loy, A., et al. (2016). Consortia of low-abundance bacteria drive sulfate reduction-dependent degradation of fermentation products in peat soil microcosms. ISME J. 10, 2365–2375. doi: 10.1038/ismej.2016.42
Heberle, H., Meirelles, G. V., da Silva, F. R., Telles, G. P., and Minghim, R. (2015). InteractiVenn: a web-based tool for the analysis of sets through Venn diagrams. BMC Bioinformatics 16:169. doi: 10.1186/s12859-015-0611-3
Hiibel, S. R., Pereyra, L. P., Breazeal, M. V. R., Reisman, D. J., Reardon, K. F., and Pruden, A. (2011). Effect of organic substrate on the microbial community structure in pilot-scale sulfate-reducing biochemical reactors treating mine drainage. Environ. Eng. Sci. 28, 563–572. doi: 10.1089/ees.2010.0237
Hiibel, S. R., Pereyra, L. P., Inman, L. Y., Tischer, A., Reisman, D. J., Reardon, K. F., et al. (2008). Microbial community analysis of two field-scale sulfate-reducing bioreactors treating mine drainage. Environ. Microbiol. 10, 2087–2097. doi: 10.1111/j.1462-2920.2008.01630.x
Hua, Z. S., Han, Y. J., Chen, L. X., Liu, J., Hu, M., Li, S. J., et al. (2015). Ecological roles of dominant and rare prokaryotes in acid mine drainage revealed by metagenomics and metatranscriptomics. ISME J. 9, 1280–1294. doi: 10.1038/ismej.2014.212
Huang, L. N., Kuang, J. L., and Shu, W. S. (2016). Microbial ecology and evolution in the acid mine drainage model system. Trends Microbiol. 24, 581–593. doi: 10.1016/j.tim.2016.03.004
Johnson, D. B. (2012). Geomicrobiology of extremely acidic subsurface environments. FEMS Microbiol. Ecol. 81, 2–12. doi: 10.1111/j.1574-6941.2011.01293
Johnson, D. B., and Hallberg, K. B. (2005). Acid mine drainage remediation options: a review. Sci. Total Environ. 338, 3–14. doi: 10.1016/j.scitotenv.2004.09.002
Kaksonen, A. H., Plumb, J. J., Robertson, W. J., Franzmann, P. D., Gibson, J. A., and Puhakka, J. A. (2004). Culturable diversity and community fatty acid profiling of sulfate-reducing fluidized-bed reactors treating acidic, metal-containing wastewater. Geomicrobiol. J. 21, 469–480. doi: 10.1080/01490450490505455
Kefeni, K. K., Msagati, T. A., and Mamba, B. B. (2017). Acid mine drainage: prevention, treatment options, and resource recovery: a review. J. Clean. Prod. 151, 475–493. doi: 10.1016/j.jclepro.2017.03.082
Kosaka, T., Kato, S., Shimoyama, T., Ishii, S., Abe, T., and Watanabe, K. (2008). The genome of Pelotomaculum thermopropionicum reveals niche-associated evolution in anaerobic microbiota. Genome Res. 18, 442–448. doi: 10.1101/gr.7136508
Kuang, J. L., Huang, L. N., Chen, L. X., Hua, Z. S., Li, S. J., Hu, M., et al. (2013). Contemporary environmental variation determines microbial diversity patterns in acid mine drainage. ISME J. 7, 1038–1050. doi: 10.1038/ismej.2012.139
Kwon, M. J., O’Loughlin, E. J., Boyanov, M. I., Brulc, J. M., Johnston, E. R., Kemner, K. M., et al. (2016). Impact of organic carbon electron donors on microbial community development under iron-and sulfate-reducing conditions. PLoS One 11:e0146689. doi: 10.1371/journal.pone.0146689
Langille, M. G., Zaneveld, J., Caporaso, J. G., McDonald, D., Knights, D., Reyes, J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821. doi: 10.1038/nbt.2676
Lee, S. H., and Cho, J. C. (2011). Group-specific PCR primers for the phylum Acidobacteria designed based on the comparative analysis of 16S rRNA gene sequences. J. Microbiol. Methods 86, 195–203. doi: 10.1016/j.mimet.2011.05.003
Lee, Y. J., Romanek, C. S., Mills, G. L., Davis, R. C., Whitman, W. B., and Wiegel, J. (2006). Gracilibacter thermotolerans gen. nov., sp. nov., an anaerobic, thermotolerant bacterium from a constructed wetland receiving acid sulfate water. Int. J. Syst. Evol. Microbiol. 56, 2089–2093. doi: 10.1099/ijs.0.64040-0
Lefticariu, L., Walters, E. R., Pugh, C. W., and Bender, K. S. (2015). Sulfate reducing bioreactor dependence on organic substrates for remediation of coal-generated acid mine drainage: field experiments. Appl. Geochem. 63, 70–82. doi: 10.1016/j.apgeochem.2015.08.002
Li, H., Peng, J., Weber, K. A., and Zhu, Y. (2011). Phylogenetic diversity of Fe (III)-reducing microorganisms in rice paddy soil: enrichment cultures with different short-chain fatty acids as electron donors. J. Soils Sediments 11:1234. doi: 10.1007/s11368-011-0371-2
Lu, J., Chen, T., Wu, J., Wilson, P. C., Hao, X., and Qian, J. (2011). Acid tolerance of an acid mine drainage bioremediation system based on biological sulfate reduction. Bioresour. Technol. 102, 10401–10406. doi: 10.1016/j.biortech.2011.09.046
Luptakova, A., and Kusnierova, M. (2005). Bioremediation of acid mine drainage contaminated by SRB. Hydrometallurgy 77, 97–102. doi: 10.1016/j.hydromet.2004.10.019
Martins, M., Santos, E. S., Faleiro, M. L., Chaves, S., Tenreiro, R., Barros, R. J., et al. (2011). Performance and bacterial community shifts during bioremediation of acid mine drainage from two Portuguese mines. Int. Biodeterior. Biodegradation 65, 972–981. doi: 10.1016/j.ibiod.2011.07.006
Méndez-García, C., Mesa, V., Sprenger, R. R., Richter, M., Diez, M. S., Solano, J., et al. (2014). Microbial stratification in low pH oxic and suboxic macroscopic growths along an acid mine drainage. ISME J. 8, 1259–1274. doi: 10.1038/ismej.2013.242
Méndez-García, C., Peláez, A. I., Mesa, V., Sánchez, J., Golyshina, O. V., and Ferrer, M. (2015). Microbial diversity and metabolic networks in acid mine drainage habitats. Front. Microbiol. 6:475. doi: 10.3389/fmicb.2015.00475
Mesa, V., Gallego, J. L., González-Gil, R., Lauga, B., Sánchez, J., Méndez-García, C., et al. (2017). Bacterial, archaeal, and eukaryotic diversity across distinct microhabitats in an acid mine drainage. Front. Microbiol. 8:1756. doi: 10.3389/fmicb.2017.01756
Moreau, J. W., Fournelle, J. H., and Banfield, J. F. (2013). Quantifying heavy metals sequestration by sulfate-reducing bacteria in an Acid mine drainage-contaminated natural wetland. Front. Microbiol. 4:43. doi: 10.3389/fmicb.2013.00043
Morra, M. J., and Dick, W. A. (1991). Mechanisms of H2S production from cysteine and cystine by microorganisms isolated from soil by selective enrichment. Appl. Environ. Microbiol. 57, 1413–1417.
Mosher, J. J., Phelps, T. J., Podar, M., Hurt, R. A., Campbell, J. H., Drake, M. M., et al. (2012). Microbial community succession during lactate amendment and electron acceptor limitation reveals a predominance of metal-reducing Pelosinus spp. Appl. Environ. Microbiol. 78, 2082–2091. doi: 10.1128/AEM.07165-11
Mühling, M., Woolven-Allen, J., Murrell, J. C., and Joint, I. (2008). Improved group-specific PCR primers for denaturing gradient gel electrophoresis analysis of the genetic diversity of complex microbial communities. ISME J. 2, 379–392. doi: 10.1038/ismej.2007.97
Muyzer, G., De Waal, E. C., and Uitterlinden, A. G. (1993). Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl. Environ. Microbiol. 59, 695–700.
Muyzer, G., and Stams, A. J. (2008). The ecology and biotechnology of sulphate-reducing bacteria. Nat. Rev. Microbiol. 6, 441–454. doi: 10.1038/nrmicro1892
Neculita, C. M., and Zagury, G. J. (2008). Biological treatment of highly contaminated acid mine drainage in batch reactors: long-term treatment and reactive mixture characterization. J. Hazard. Mater. 157, 358–366. doi: 10.1016/j.jhazmat.2008.01.002
Neculita, C. M., Zagury, G. J., and Bussière, B. (2007). Passive treatment of acid mine drainage in bioreactors using sulfate-reducing bacteria. J. Environ. Qual. 36, 1–16. doi: 10.2134/jeq2006.0066
Pan, Y., Yang, X., Xu, M., and Sun, G. (2017). The role of enriched microbial consortium on iron-reducing bioaugmentation in sediments. Front. Microbiol. 8:462. doi: 10.3389/fmicb.2017.00462
Paul, D., Kazy, S. K., Gupta, A. K., Pal, T., and Sar, P. (2015). Diversity, metabolic properties and arsenic mobilization potential of indigenous bacteria in arsenic contaminated groundwater of West Bengal, India. PLoS One 10:e0118735. doi: 10.1371/journal.pone.0118735
Peng, Q. A., Shaaban, M., Wu, Y., Hu, R., Wang, B., and Wang, J. (2016). The diversity of iron reducing bacteria communities in subtropical paddy soils of China. Appl. Soil Ecol. 101, 20–27. doi: 10.1016/j.apsoil.2016.01.012
Petrie, L., North, N. N., Dollhopf, S. L., Balkwill, D. L., and Kostka, J. E. (2003). Enumeration and characterization of iron (III)-reducing microbial communities from acidic subsurface sediments contaminated with uranium (VI). Appl. Environ. Microbiol. 69, 7467–7479. doi: 10.1128/AEM.69.12.7467-7479.2003
Postgate, J. R. (1963). Versatile medium for the enumeration of sulfate-reducing bacteria. Appl. Microbiol. 11, 265–267.
Pruden, A., Messner, N., Pereyra, L., Hanson, R. E., Hiibel, S. R., and Reardon, K. F. (2007). The effect of inoculum on the performance of sulfate-reducing columns treating heavy metal contaminated water. Water Res. 41, 904–914. doi: 10.1016/j.watres.2006.11.025
Purkamo, L., Bomberg, M., Nyyssönen, M., Kukkonen, I., Ahonen, L., Kietäväinen, R., et al. (2013). Dissecting the deep biosphere: retrieving authentic microbial communities from packer-isolated deep crystalline bedrock fracture zones. FEMS Microbiol. Ecol. 85, 324–337. doi: 10.1111/1574-6941.12126
Qian, G., Schumann, R. C., Li, J., Short, M. D., Fan, R., Li, Y., et al. (2017). Strategies for reduced acid and metalliferous drainage by pyrite surface passivation. Minerals 7:42. doi: 10.3390/min7030042
Sahinkaya, E., Yurtsever, A., Toker, Y., Elcik, H., Cakmaci, M., and Kaksonen, A. H. (2015). Biotreatment of As-containing simulated acid mine drainage using laboratory scale sulfate reducing upflow anaerobic sludge blanket reactor. Miner. Eng. 75, 133–139. doi: 10.1016/j.mineng.2014.08.012
Sánchez-Andrea, I., Knittel, K., Amann, R., Amils, R., and Sanz, J. L. (2012a). Quantification of Tinto River sediment microbial communities: importance of sulfate-reducing bacteria and their role in attenuating acid mine drainage. Appl. Environ. Microbial. 78, 4638–4645. doi: 10.1128/AEM.00848-12
Sánchez-Andrea, I., Rojas-Ojeda, P., Amils, R., and Sanz, J. L. (2012b). Screening of anaerobic activities in sediments of an acidic environment: Tinto River. Extremophiles 16, 829–839. doi: 10.1007/s00792-012-0478-4
Sánchez-Andrea, I., Rodríguez, N., Amils, R., and Sanz, J. L. (2011). Microbial diversity in anaerobic sediments at Rio Tinto, a naturally acidic environment with a high heavy metal content. Appl. Environ. Microbiol. 77, 6085–6093. doi: 10.1128/AEM.00654-11
Sánchez-Andrea, I., Sanz, J. L., Bijmans, M. F., and Stams, A. J. (2014). Sulfate reduction at low pH to remediate acid mine drainage. J. Hazard. Mater. 269, 98–109. doi: 10.1016/j.jhazmat.2013.12.032
Scala, D. J., Hacherl, E. L., Cowan, R., Young, L. Y., and Kosson, D. S. (2006). Characterization of Fe (III)-reducing enrichment cultures and isolation of Fe (III)-reducing bacteria from the Savannah River site, South Carolina. Res. Microbiol. 157, 772–783. doi: 10.1016/j.resmic.2006.04.001
Shah, M. (2013). Iron Oxide Reduction by a Clostridial Consortium: Insights from Physiological and Genome Analyses. New Brunswick, NJ: Rutgers University–New Brunswick.
Shiratori, H., Ohiwa, H., Ikeno, H., Ayame, S., Kataoka, N., Miya, A., et al. (2008). Lutispora thermophila gen. nov., sp. nov., a thermophilic, spore-forming bacterium isolated from a thermophilic methanogenic bioreactor digesting municipal solid wastes. Int. J. Syst. Evol. Microbiol. 58, 964–969. doi: 10.1099/ijs.0.65490-0
Stieglmeier, M., Wirth, R., Kminek, G., and Moissl-Eichinger, C. (2009). Cultivation of anaerobic and facultatively anaerobic bacteria from spacecraft-associated clean rooms. Appl. Environ. Microbiol. 75, 3484–3491. doi: 10.1128/AEM.02565-08
Teng, W., Kuang, J., Luo, Z., and Shu, W. (2017). Microbial diversity and community assembly across environmental gradients in acid mine drainage. Minerals 7:106. doi: 10.3390/min7060106
Viollier, E., Inglett, P. W., Hunter, K., Roychoudhury, A. N., and Van Cappellen, P. (2000). The ferrozine method revisited: Fe (II)/Fe (III) determination in natural waters. Appl. Geochem. 15, 785–790. doi: 10.1016/S0883-2927(99)00097-9
Wang, C. L., Maratukulam, P. D., Lum, A. M., Clark, D. S., and Keasling, J. D. (2000). Metabolic engineering of an aerobic sulfate reduction pathway and its application to precipitation of cadmium on the cell surface. Appl. Environ. Microbiol. 66, 4497–4502. doi: 10.1128/AEM.66.10.4497-4502.2000
Xingyu, L., Zou, G., Wang, X., Zou, L., Wen, J., Ruan, R., et al. (2013). A novel low pH sulfidogenic bioreactor using activated sludge as carbon source to treat acid mine drainage (AMD) and recovery metal sulfides: pilot scale study. Miner. Eng. 48, 51–55. doi: 10.1016/j.mineng.2012.11.004
Yi, W., Wang, B., and Qu, D. (2012). Diversity of isolates performing Fe (III) reduction from paddy soil fed by different organic carbon sources. Afr. J. Biotechnol. 11, 4407–4417. doi: 10.5897/AJB11.1216
Zavarzina, D. G., Sokolova, T. G., Tourova, T. P., Chernyh, N. A., Kostrikina, N. A., and Bonch-Osmolovskaya, E. A. (2007). Thermincola ferriacetica sp. nov., a new anaerobic, thermophilic, facultatively chemolithoautotrophic bacterium capable of dissimilatory Fe (III) reduction. Extremophiles 11, 1–7. doi: 10.1007/s00792-006-0004-7
Zhang, M., Liu, X., Li, Y., Wang, G., Wang, Z., and Wen, J. (2017). Microbial community and metabolic pathway succession driven by changed nutrient inputs in tailings: effects of different nutrients on tailing remediation. Sci. Rep. 7:474. doi: 10.1038/s41598-017-00580-3
Zhang, M., and Wang, H. (2014). Organic wastes as carbon sources to promote sulfate reducing bacterial activity for biological remediation of acid mine drainage. Miner. Eng. 69, 81–90. doi: 10.1016/j.mineng.2014.07.010
Zhang, M., and Wang, H. (2016). Preparation of immobilized sulfate reducing bacteria (SRB) granules for effective bioremediation of acid mine drainage and bacterial community analysis. Miner. Eng. 92, 63–71. doi: 10.1016/j.mineng.2016.02.008
Zhang, M., Wang, H., and Han, X. (2016). Preparation of metal-resistant immobilized sulfate reducing bacteria beads for acid mine drainage treatment. Chemosphere 154, 215–223. doi: 10.1016/j.chemosphere.2016.03.103
Keywords: acid mine drainage, bioremediation, Firmicutes, biostimulation, quantitative PCR, metagenomics, dissimilatory sulfate reduction
Citation: Gupta A, Dutta A, Sarkar J, Panigrahi MK and Sar P (2018) Low-Abundance Members of the Firmicutes Facilitate Bioremediation of Soil Impacted by Highly Acidic Mine Drainage From the Malanjkhand Copper Project, India. Front. Microbiol. 9:2882. doi: 10.3389/fmicb.2018.02882
Received: 31 December 2017; Accepted: 12 November 2018;
Published: 11 December 2018.
Edited by:
Rajesh K. Sani, South Dakota School of Mines and Technology, United StatesReviewed by:
Virginia Helena Albarracín, Center for Electron Microscopy (CIME), ArgentinaChristopher Anthony Abin, University of Oklahoma, United States
Copyright © 2018 Gupta, Dutta, Sarkar, Panigrahi and Sar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pinaki Sar, c2FycGluYWtpQHlhaG9vLmNvbQ==; cHNhckBidC5paXRrZ3AuYWMuaW4=