 Irene Bianconi1†
Irene Bianconi1† Silvia D’Arcangelo1†
Silvia D’Arcangelo1† Alfonso Esposito1Mattia Benedet1Elena Piffer1Grazia Dinnella2Paola Gualdi3Michele Schinella3Ermanno Baldo2
Alfonso Esposito1Mattia Benedet1Elena Piffer1Grazia Dinnella2Paola Gualdi3Michele Schinella3Ermanno Baldo2 Claudio Donati4
Claudio Donati4 Olivier Jousson1*
Olivier Jousson1*- 1Centre for Integrative Biology, University of Trento, Trento, Italy
- 2Trentino Cystic Fibrosis Support Centre, Rovereto Hospital, Rovereto, Italy
- 3Operative Unit of Clinical Pathology, Rovereto Hospital, Rovereto, Italy
- 4Centro Ricerca e Innovazione, Fondazione Edmund Mach, San Michele all’Adige, Italy
Background: During its persistence in cystic fibrosis (CF) airways, P. aeruginosa develops a series of phenotypic changes by the accumulation of pathoadaptive mutations. A better understanding of the role of these mutations in the adaptive process, with particular reference to the development of multidrug resistance (MDR), is essential for future development of novel therapeutic approaches, including the identification of new drug targets and the implementation of more efficient antibiotic therapy. Although several whole-genome sequencing studies on P. aeruginosa CF lineages have been published, the evolutionary trajectories in relation to the development of antimicrobial resistance remain mostly unexplored to date. In this study, we monitored the adaptive changes of P. aeruginosa during its microevolution in the CF airways to provide an innovative, genome-wide picture of mutations and persistent phenotypes and to point out potential novel mechanisms allowing survival in CF patients under antibiotic therapy.
Results: We obtained whole genome sequences of 40 P. aeruginosa clinical CF strains isolated at Trentino Regional Support CF Centre (Rovereto, Italy) from a single CF patient over an 8-year period (2007–2014). Genotypic analysis of the P. aeruginosa isolates revealed a clonal population dominated by the Sequence Type 390 and three closely related variants, indicating that all members of the population likely belong to the same clonal lineage and evolved from a common ancestor. While the majority of early isolates were susceptible to most antibiotics tested, over time resistant phenotypes increased in the persistent population. Genomic analyses of the population indicated a correlation between the evolution of antibiotic resistance profiles and phylogenetic relationships, and a number of putative pathoadaptive variations were identified.
Conclusion: This study provides valuable insights into the within-host adaptation and microevolution of P. aeruginosa in the CF lung and revealed the emergence of an MDR phenotype over time, which could not be comprehensively explained by the variations found in known resistance genes. Further investigations on uncharacterized variations disclosed in this study should help to increase our understanding of the development of MDR phenotype and the poor outcome of antibiotic therapies in many CF patients.
Introduction
Pseudomonas aeruginosa is the most prevalent of all potential opportunistic nosocomial pathogens causing pulmonary and bloodstream infection with high mortality rates of up to 50% (Osmon et al., 2004). Cystic fibrosis (CF) patients infected with P. aeruginosa are exposed to increased morbidity and mortality (Lyczak et al., 2000; Chmiel and Davis, 2003). Progressive airway infection in CF subjects typically begins with recurrent P. aeruginosa infections that are often cleared by antibiotics and subsequently replaced by other strains that can establish permanent chronic infections in the respiratory tract (Folkesson et al., 2012). Long-term colonization of the CF airway is sustained by P. aeruginosa lineages which are clonal to the initially acquired strain (Bragonzi et al., 2009) and can persist in the airway of a patient for years and even decades (Jelsbak et al., 2007; Cramer et al., 2010).
Convergence of phenotypic changes among clinical isolates with a high level of initial phenotypic diversity within a single patient and between multiple strains from the same sample has been previously reported (Workentine et al., 2013; Jeukens et al., 2014; Caballero et al., 2015; Darch et al., 2015; Williams et al., 2015). Most common phenotypic changes during persistent infections include the conversion to a mucoid phenotype, loss of pigmentation, motility, secreted and cell-associated virulence factors, emergence of small colony variants (SCVs), use of alternative metabolic pathways, increased mutation rate, and the acquisition of multidrug resistance (Hogardt and Heesemann, 2010; Folkesson et al., 2012; Sousa and Pereira, 2014; Sommer et al., 2016).
A set of 24 genes in three different P. aeruginosa lineages have been reported to have undergone similar changes in their respective gene expression profiles (Huse et al., 2010), confirming the occurrence of convergent evolution in CF, in which different lineages tend to develop the same adaptive traits independently, due to the stressful CF lung environment. In another study (Marvig et al., 2015), the genomes of 474 longitudinal P. aeruginosa isolates collected from 34 CF patients for an average 5-year period were sequenced; they found 52 common genes mutated in all different lineages confirming that P. aeruginosa strains of various genetic backgrounds adapt to the CF airway following a convergent evolutive process. On the other hand, a high level of phenotypic diversity within a single patient and also between multiple isolates of the same sputum sample has also been reported (Huse et al., 2010; Folkesson et al., 2012). Although most P. aeruginosa infections have a clonal origin, persistent infections are characterized by an adaptive radiation process diversifying the initial clone in various morphotypes (Hogardt and Heesemann, 2010), and leading to the occurrence of subpopulations within the same host (Cullen and McClean, 2015).
As mentioned above, during chronic infection, the bacterium may develop a multidrug resistance (MDR) phenotype by the accumulation of pathoadaptive mutations or the acquisition of mobile elements by horizontal gene transfer (Breidenstein et al., 2011). Estimates indicate that 25–45% of adult CF patients are chronically infected with MDR P. aeruginosa within their airway (Lechtzin et al., 2006). Whole-genome sequencing (WGS) can help to point out potential molecular mechanisms of antibiotic resistance and has already proved to be able to successfully predict antimicrobial susceptibility in several pathogens (Stoesser et al., 2013; Zankari et al., 2013). Nevertheless, despite the availability of >2,000 P. aeruginosa genomes, knowledge on the evolution of genomic features associated with persistence and antimicrobial resistance is still incomplete.
In the present work, we sequenced the whole genome of 40 P. aeruginosa isolates from a single CF patient spanning an 8-year period. We analyzed the population in terms of clonality of the isolates, phylogenetic relationships, polymorphisms in the core genome and variations in the accessory genome. We also characterized the antibiotic resistance profile of the isolates and checked for mutations and variations in antibiotic resistance genes among the population. A better understanding of the evolutionary dynamics occurring during chronic airway infections in CF patients and of the genetic adaptation of strains to the CF lung environment should provide clues for preventive measures or novel therapies to control CF infections in the future.
Materials and Methods
Bacterial Strains
Pseudomonas aeruginosa strains were isolated from the sputum of a single adult male CF patient, aged 24 at the beginning of sampling. The patient carries the heterozygous CFTR mutations ΔF508 and G542X, and he was treated at the Trentino Regional Support CF Centre and the Operative Unit of Clinical Pathology (Hospital of Rovereto, Italy) (Supplementary Figure S1 and Supplementary Table S1).
A total of 40 P. aeruginosa strains were isolated over an 8-year period (2007–2014). To evaluate both the suitability of the material from the lower respiratory tract of the patient (sputum) and the putative pathogens present, a microscopic examination was carried out using Bartlett’s criteria. Sputum samples were fluidized with the addition of dithiothreitol and plated on blood agar and MacConkey agar plates. Pseudomonas aeruginosa was identified using the API ID32GN kit (bioMérieux, Bagno a Ripoli, Italy). Strains from the same sputum samples showing a different colony morphology were collected and characterized separately (Bianconi et al., 2016; D’Arcangelo, 2017).
Antimicrobial Susceptibility Testing
MIC values for all 40 strains were determined by reference broth microdilution using TREK Sensititre custom panel ITGNEGF (Thermo-Fisher, Waltham, MA, United States). The following 12 antibiotics belonging to nine different classes were tested: amikacin, gentamicin, imipenem, meropenem, doripenem, ceftazidime, cefepime, ciprofloxacin, levofloxacin, fosfomycin, colistin, and a combination of piperacillin/tazobactam.
Mucoid Phenotype
Isolates were inoculated on Pseudomonas Isolation Agar (BD, Franklin Lakes, NJ, United States) for 48–72 h at 37°C. Strains producing alginate were considered positive (Bragonzi et al., 2006).
Biofilm Formation Assay
Biofilm formation was assessed by the crystal violet staining assay with minor modifications (O’Toole, 2011). Briefly, pellets of overnight cultures were collected by centrifugation and re-suspended in M63 liquid medium (Amresco, Solon, OH, United States) to a final OD600 of 0.005 and aliquoted in a 96-well U-bottom microtiter plate (Costar, Washington, DC, United States). After 24 h of static incubation at 37°C, supernatants were removed, and OD600 was measured. Cells attached to the microtiter wells were washed and stained with an aqueous solution of 0.1% crystal violet. Cells were washed and resuspended in 30% acetic acid. OD550 of resuspended biofilm was determined and normalized to the OD600 of the corresponding planktonic cells. The assay was repeated at least three times per isolate.
DNA Extraction, Genome Sequencing, and Assembly
The genome sequences of the isolates were previously published under accession numbers MAUO00000000 – MBMR00000000 (Supplementary Table S2) (Bianconi et al., 2016).
Genomic DNA was extracted from overnight cultures in LB Broth (Oxoid, Sigma-Aldrich, Darmstadt, Germany) at 37°C with continuous shaking, using the DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions for Gram-negative bacteria. Sequencing libraries were prepared using the Nextera XT DNA Library Preparation Kit (Illumina, San Diego, CA, United States) with default settings and sequenced on the Illumina MiSeq platform. De novo assembly of draft genomes was carried out using SPAdes version 3.1.0 (Bankevich et al., 2012), with the following options: –careful on and k-mer sizes set as 21,33,55,77,99,127. To further improve the quality of the assemblies, raw reads were mapped on the contigs using Bowtie2 (Langmead and Salzberg, 2012), and contigs with less than three reads mapping and/or with coverage below one were removed. Draft genomes were reordered using Mauve (Darling et al., 2010) with P. aeruginosa DK2 strain sequence used as a reference.
Genotyping, Genome Annotation, Pangenome, and SNPs Analysis
MLST 1.8 (Larsen et al., 2012) was used for genotyping of the population from de novo assembled genomes. An eBURST analysis (eBURST V3, Feil et al., 2004) was carried out to estimate the relatedness of Sequence Types (STs). In silico determination of the O-antigen was performed using the Pseudomonas aeruginosa serotypes (PAst) script1. Genome annotation was performed with Prokka automatic annotation tool v1.11 (Seemann, 2014), and analysis of the core and accessory genomes was carried out using Roary with default settings (Page et al., 2015). An accessory genome presence matrix was created from the standard Roary output file and isolates’ values were plotted ordered by isolation date with the ggpubr package inside R/Bioconductor environment2. To assess the number of gene acquisition/loss events, all acquired genes (n = 542) were pooled to a gene dataset composed by the accessory genes of the first isolated strains TNCF_3 and TNCF_4M (n = 624); each isolate was compared with these two datasets. The barplot was generated using the ggpubr package as above.
SNPs were identified using Snippy3 (Seemann, 2015). This software infers polymorphisms at the nucleotide level by aligning the unassembled reads to a reference genome (in this study we used P. aeruginosa DK2) and it calls the software SnpEff to annotate and predict the effect of each mutation (Cingolani et al., 2012), dividing them into low (e.g., synonym mutations), moderate (e.g., inframe deletion/insertion) and high (e.g., stop gained).
Plasmids and Genomic Islands Prediction
To detect the presence of plasmids from NGS data, the software PlasmidSeeker was used (Roosaare et al., 2018). The algorithm allows the identification of plasmids using the information of plasmid k-mer coverage, which is expected to be higher than that of the genome. For each set of reads we used the respective assembled genome. In addition, we performed two separate analyses using the reference genomes of PAO1 and PA14. The plasmids k-mer database consists of >8,500 known plasmids from several species, 14 of which are specific for P. aeruginosa.
The occurrence and distribution of genomic islands (GIs) were inferred using IslandViewer4 (Bertelli et al., 2017). Prokka output files in GenBank format were uploaded to the IslandViewer web server. To obtain a set of homologous protein clusters (PCs), the proteins found by IslandViewer were clustered using CD-HIT (Fu et al., 2012), with both coverage and identity percentage set at 95%. A genome map of the distribution of the islands in each isolate was drawn using the package genoplotR. Also, PCs were mapped against a custom dataset of 23 previously characterized GIs using tblastn. Ubiquitous PCs that could not be assigned to any of the known GIs were searched on the whole non-redundant NCBI protein database using blastp, restricting the analysis to P. aeruginosa sequences. To determine the occurrence of a phylogenetic signal in the distribution of non-ubiquitous PCs, a phylo.d analysis (R-package caper) (Fritz and Purvis, 2010) was performed.
Type II Toxin–Antitoxin (TA) Systems Detection
Putative type II toxin–antitoxin (TA) loci were identified by using the online server TAfinder4 (Shao et al., 2011; Xie et al., 2018). Newly assembled genomes were previously annotated with the online tool CDSeasy5. Output GenBank files were uploaded in TAfinder and analyzed using default parameters.
Phylogenetic Tree Construction
A rooted phylogenetic tree, based on the distribution of SNPs in the core genome, was then obtained using BEAST V1.8.3 (Drummond et al., 2012). A Markov Chain Monte Carlo chain was run for 9 × 108 steps using the uncorrelated relaxed clock model (Drummond et al., 2006) with lognormal distribution and gtr substitution model with gamma-distributed rates, four categories, shape parameter α. After a burn-in of 4 × 108 steps, trees were recorded every 10.000 steps, and the consensus tree was determined using TreeAnnotator. To evaluate the amount of recombining sites, the phylogenetic tree, together with the alignment of core genes obtained by Roary, were analyzed using clonalframeML (Didelot and Wilson, 2015).
Detection of Phylogenetic Signal and Molecular Evolution Analysis
The phylo.d function within the R package was used to measure the strength of phylogenetic signal in binary traits6. Two simulated null models were used to standardize the phylogenetic signal: phylogenetic randomness and Brownian threshold.
Genes with multiple SNPs were initially considered to be under non-neutral selection. This statement was further verified with the following procedure: (i) the alignment of each homologous gene with reference sequence was exported in FASTA format; (ii) the dN/dS ratio was determined using codeml as implemented in PAML package (Yang, 2007) and the likelihood of four different models (two neutral or nearly neutral, M1a and M7, respectively, and the two corresponding positive selection models M2a and M8) was determined; (iii) the likelihood ratio test, using as null model the neutral one, was performed to determine which model better fitted the observed data (Anisimova and Gascuel, 2006; Yang, 2006). The procedure was implemented in a conservative way to detect non-neutral selection in highly similar sequences, using as null model the neutral one.
Results
Whole-Genome Sequencing and in silico MLST Genotyping
In this study, we sequenced 40 P. aeruginosa isolates from a CF patient over an 8-year period. The average number of contigs per genome was 101 (range 53–356). Mean size of the draft genomes was 6.6 Mb (range 6.545–6.653). Mean G+C content was 66,28% and the genomes encoded for an average of 6,151 putative ORFs. Details of the genomes obtained are provided in Table 1 and preliminarily described in Bianconi et al. (2016).
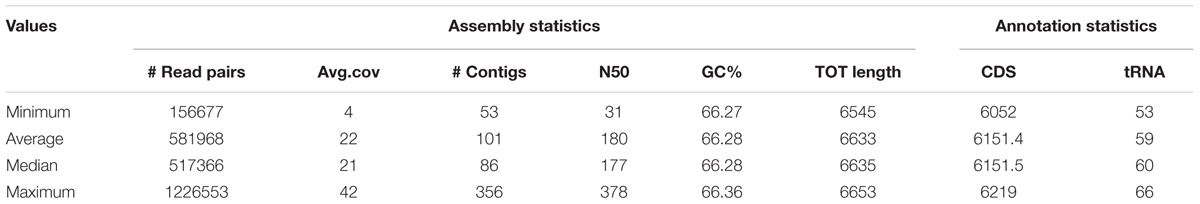
Table 1. Genomic features and statistics of the 40 P. aeruginosa CF isolates.
In silico MLST analysis revealed a population dominated by a previously characterized Sequence Type (ST390, 30 isolates), and three new ST variants (Figure 1): ST1864 (six isolates: TNCF_16, TNCF_85, TNCF_88M, TNCF_101, TNCF_133_1, TNCF_151M), ST1923 (three isolates: TNCF_155_1, TNCF_165, TNCF_176) and ST1863 (one isolate: TNCF_69). ST390 (allelic profile: acs39-aro5-gua1-mut3-nuo4-pps46-trp56) was previously detected in six isolates: in a sputum sample from a CF patient (Canada, in 2004), in an environmental sample (France, in 2009), and in four unspecified isolates7. The novel variants were deposited in the PubMLST database8.
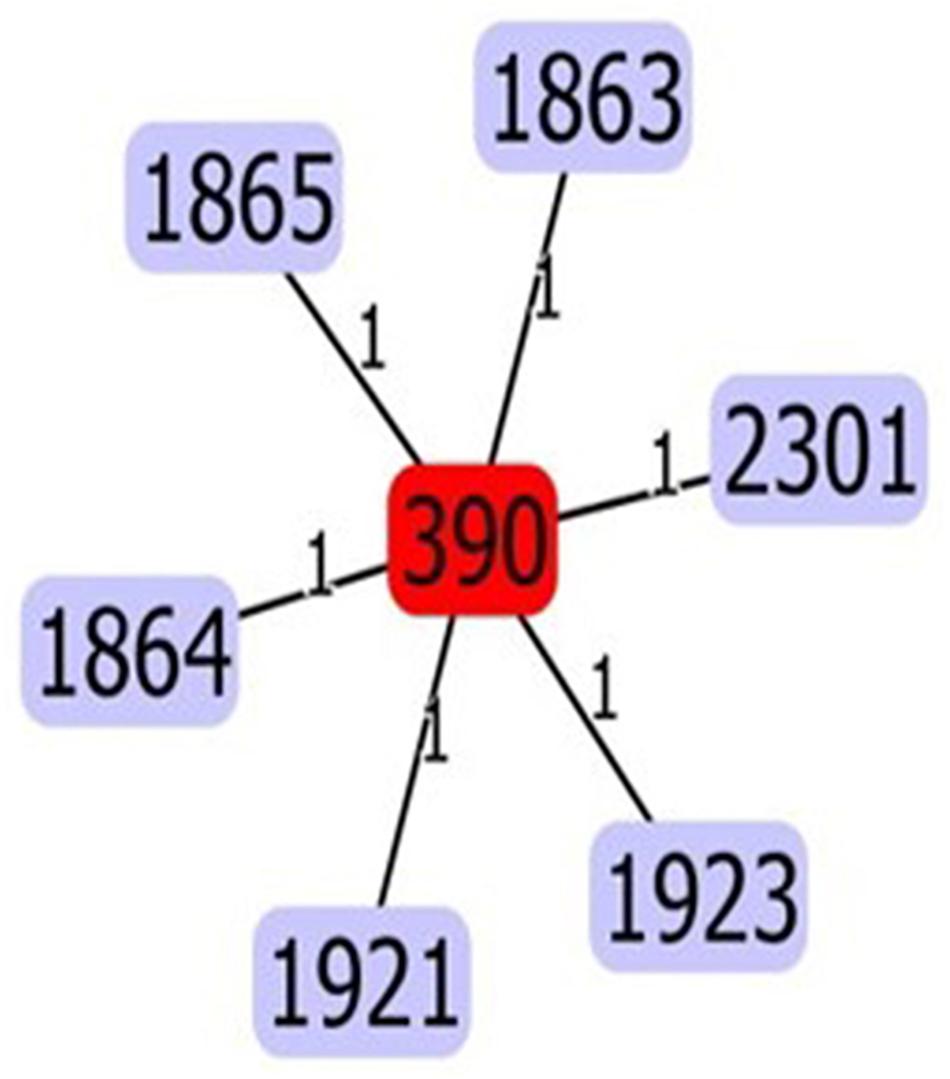
Figure 1. eBURST analysis of the sequence types found in the P. aeruginosa population. The five variants (ST1863, ST1864, ST1865, ST1921, ST2301, and ST1923) differed from ST390 at a single locus. Variants ST1863, ST1864, ST1923 were discovered in the present study.
Phylogenetic Analyses
To reconstruct the evolutionary history of the lineage and to examine the relationships between the isolates we analyzed the distribution of SNPs in the core genome using BEAST. The tree topology (Figure 2) indicates that the population is composed of two main clusters separated by a sizeable basal split: the first one comprised nearly all the early isolates (13 out of a total of 17 isolates sampled between 2007 and 2008), whereas the majority of the late isolates branched within the second cluster (17 out of a total of 23 isolates sampled between 2010 and 2014). The most recent common ancestor of the former clade is dated by BEAST in 1988 ± 19.9 years, and in 1992 ± 14.8 years for the latter, while the date of the divergence of the two clades was estimated to be 1979 ± 21 years. Such high standard deviations thus do not allow to infer divergence times in the population. Regardless of their placement in the phylogenetic tree, all late isolates had longer branches compared to early ones, indicating a more substantial genomic diversity of the formers and/or a different evolutionary rate between the two groups of isolates. The six ST1864 isolates appear to be polyphyletic and fall in the cluster with ST390 early isolates, while the three ST1923 isolates and the ST1863 one group in the cluster with the late ST390 isolates sampled between 2010 and 2014.
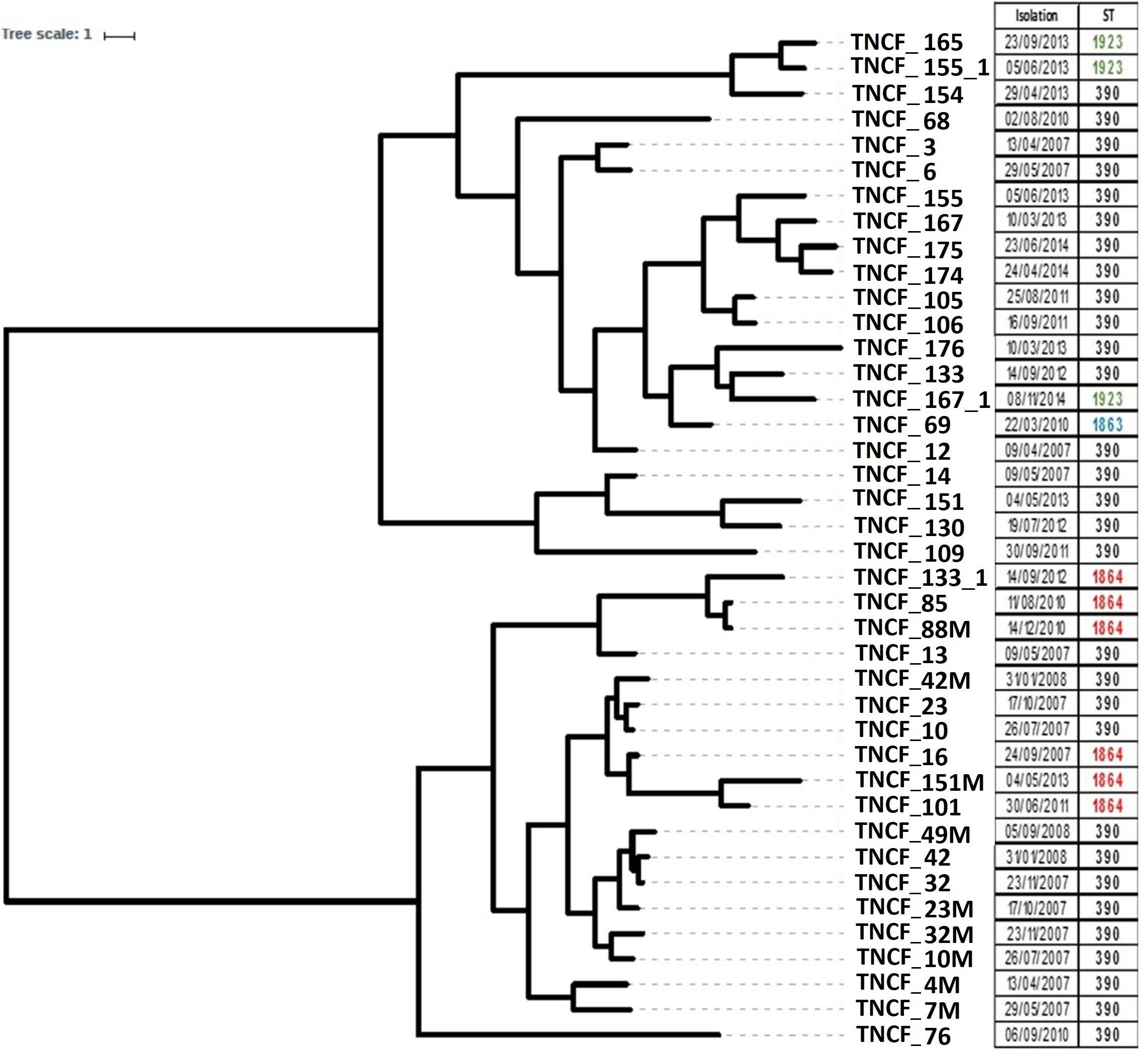
Figure 2. Rooted phylogeny of the P. aeruginosa population. The tree was built on core genome SNPs obtained using BEAST V1.8 with 10 independent runs of 109 steps using the relaxed clock model. Different strains isolated from the same sputum sample share the same isolation date. The right panel reports the date of sampling and the Sequence Type of each isolate.
Excluding homoplasic sites with ClonalFrameML (Supplementary Figure S2), a number of branches collapsed, suggesting that most of the variability within the population is due to horizontal transfer and/or convergent evolution.
Core and Accessory Genome Analyses
Previous studies have shown that the genomes of P. aeruginosa isolates are usually highly conserved (Klockgether et al., 2011, 2013). Analyses of the core and accessory genome of the population confirmed the high homogeneity of these strains revealed by in silico MLST. The pan-genome of the population comprised 7,035 genes, with nearly 85% of the genes shared between all the isolates (5,972 genes). Among these, 5608 were core genes present in 39–40 isolates, and 364 were soft core genes found in at least 38 isolates (Figure 3). A total of 1,166 genes (16%) belonged to the accessory genome (Figure 4A). Among these, 624 were shell genes present in the initial isolates (TNCF_3 or TNCF_4M), while 542 were acquired in the course of time (2,766 days). Among the latter genes, 320 were found to be present only in one isolate (TNCF_6). The average number of accessory genes per genome was 500, with TNCF_12 being the isolate with the smallest accessory genome (394 genes) and TNCF_6 the one with the largest one (523 genes), consistent with the greater length of its genome. In order to get insight into the dynamics of acquisition/loss of accessory genes, we assessed for each isolate the number of genes belonging to each class. As shown in Figure 4B, the general trend in the microevolution of this population leans toward a higher number of gene loss events compared to gene acquisition. The accessory genomes mapped on the phylogenetic tree (Supplementary Figure S3) showed an extensive collapse at the root, and the presence of three major lineages: one includes 31 out of 40 isolates (the bulk of the population); a second lineage comprises isolates TNCF_23, TNCF_10 and TNCF_42M, which formed a monophyletic group also in the SNP-based tree; the third lineage includes TNCF_16, TNCF_7M, TNCF_4M, TNCF_10M, TNCF_32M, TNCF_151M. TNCF_6 also resulted in being the strain with the most diverse accessory genome, in agreement with its high number of unique regions.
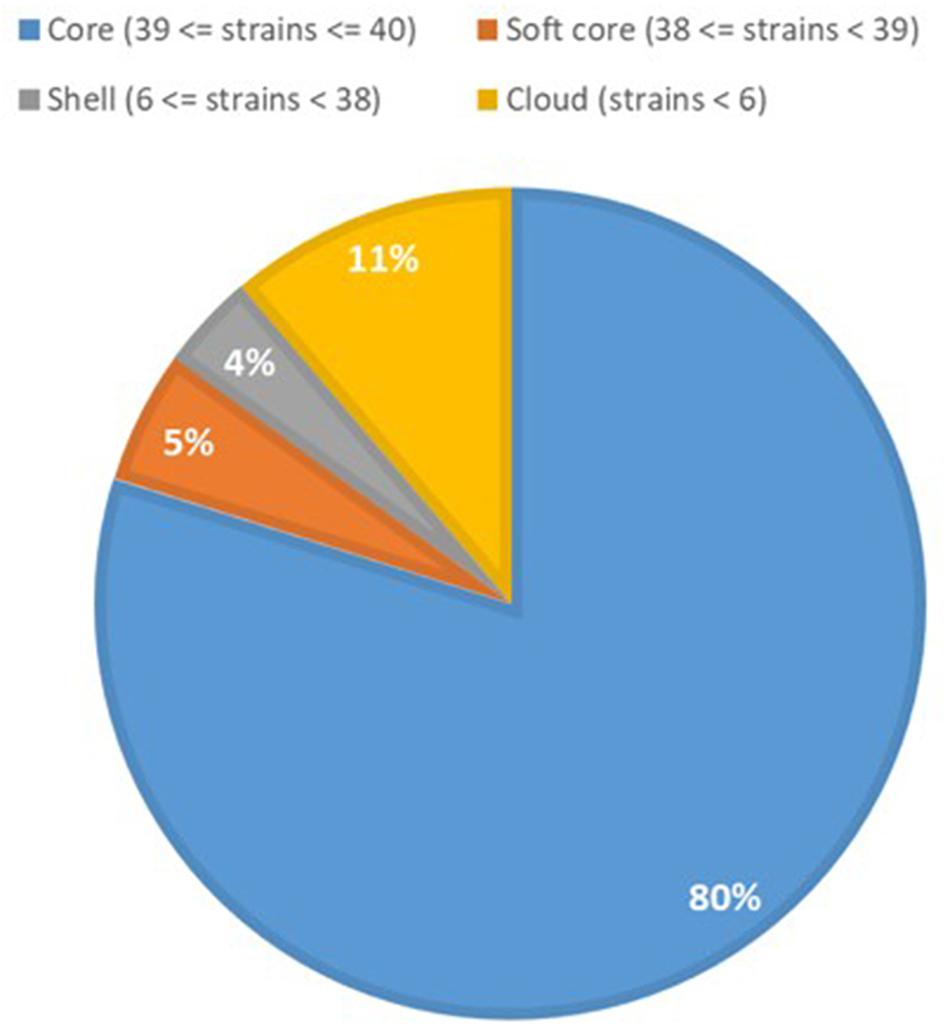
Figure 3. Core and accessory genome proportions in the P. aeruginosa population. Genomic portions present in 38 or 39 isolates out of 40 (softcore) were considered as core genes in the text due to the draft assembly of the genomes.
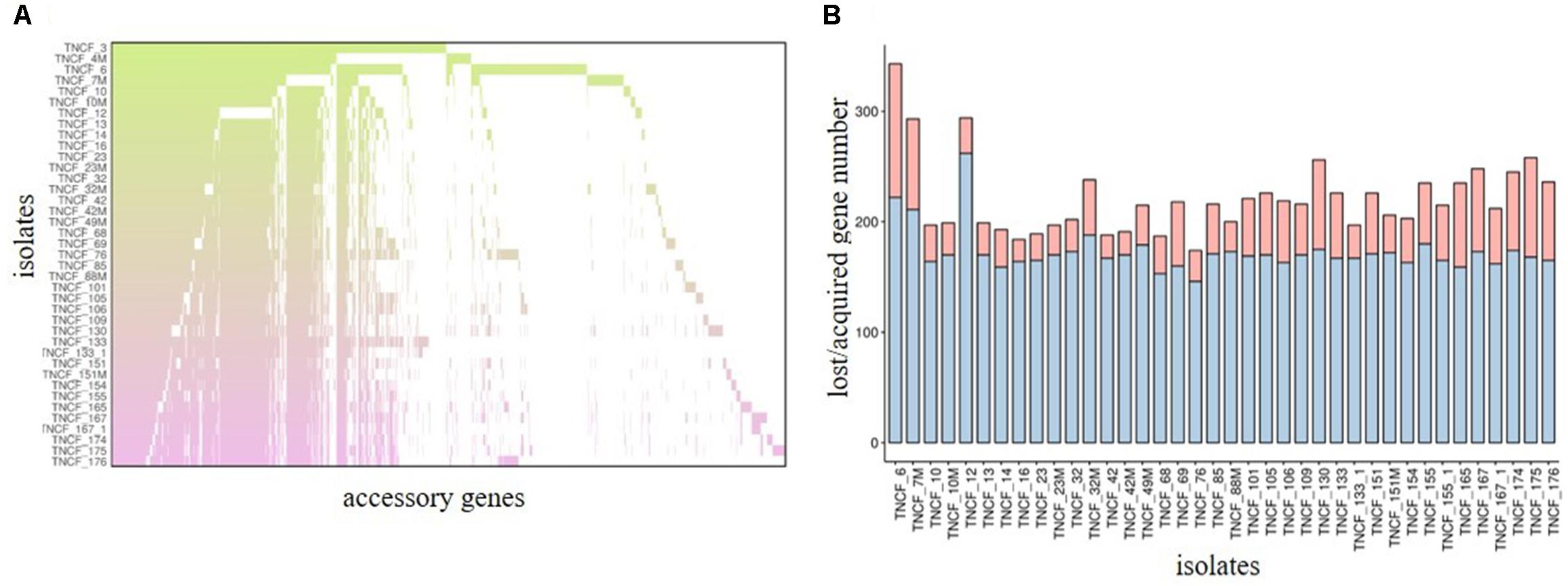
Figure 4. Analyses of the accessory genome. (A) Distribution of accessory genes in the population. Each row represents an isolate and each column represents a gene (gene labels not shown). The gradient represents the time (in days) starting from the isolation date of the first two strains (TNCF_3 and TNCF_4M). (B) Loss or acquisition of accessory genes in the population. Barplots represent the acquired/lost genes (in red and blue, respectively) for each isolate in comparison with the two first isolates TNCF_3 and TNCF_4M.
Clustering of isolates based on the composition of the accessory genome (Supplementary Figure S4) revealed a group of 11 highly similar genomes comprising both early and late isolates, eight belonging to the dominant ST390 type (TNCF_10, TNCF_13, TNCF_23, TNCF_23M, TNCF_32, TNCF_42, TNCF_42M, TNCF_49M) and three belonging to the derivate ST1864 (TNCF_16, TNCF_88M and TNCF_133_1). Accordingly, the number of accessory genes in each genome of this cluster was remarkably homogeneous, ranging from 488 to 511 genes in TNCF_16 and TNCF_10, respectively.
Identification of Plasmids and Genomic Islands
The analyses performed using PAO1, PA14 and each assembled genome as reference detected only one plasmid, belonging to the pKLC102 family, which was ubiquitous in the population with the exception of isolate TNCF_12.
The distribution of PCs was strongly skewed toward the two extremes, i.e., most of the PCs were either ubiquitous or isolate-specific; 87 PCs, belonging to six GIs, were ubiquitous in the population (Figure 5). The tblastn analysis performed on our custom dataset of known GIs detected four PCs (with identity > 80% and e-value < 4.7 e80) putatively belonging to the island LESGI-1 (Winstanley et al., 2009), two PCs (id > 85%, e-value < 1.7 e46) were assigned to LESGI-3 (Winstanley et al., 2009), and three PCs (id > 90%, e-value < 5.3 e80) were assigned to the island PAGI-2 (Klockgether et al., 2007).
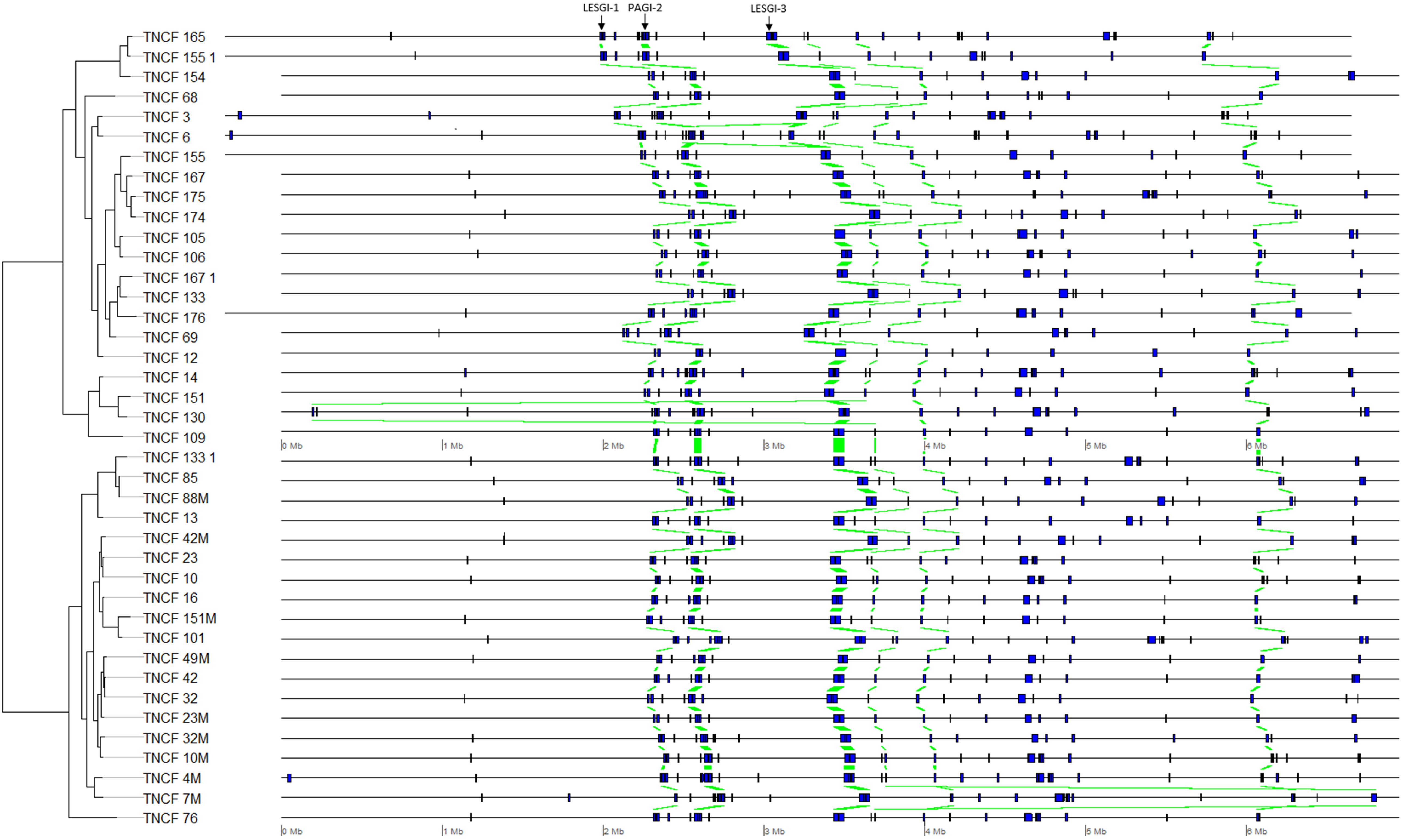
Figure 5. Genomic map showing the position of each genomic island on each genome. Homologous regions are highlighted in green.
Twenty-one ubiquitous PCs, belonging to the three yet unidentified GIs found in the collection, were mapped against the non-redundant NCBI database restricting the search to P. aeruginosa. Most PCs matched with hypothetical proteins, and only six PCs matched with the following annotated proteins: YqaJ-like viral recombinase domain cl09232 (WP_031641785), recombination protein RecT (WP_019682741), resolvase (EJY58266), serine recombinase (WP_070330427), DNA ligase [NAD(+)] LigA (WP_003114675), N-acetyltransferase (WP_003096122).
Type II Toxin–Antitoxin Systems
Most genes belonging to the TA systems were found to be ubiquitous in the population. Non-ubiquitous TA genes belonged to domains COG1733 (found in 37 isolates), pfam13420 (38 isolates), pfam00392 (39) and PRK10151 (which was found only in one isolate, TNCF_7M). TA families Abr-like (COG4456 domain), vapC (cd009981 domain) and RHH-like were present in nearly all isolates (39). Genes belonging to the Xre-like family were the most abundant with at least four genes in each genome (Supplementary Figure S5).
The total number of TA genes found in the isolates ranged from 30 (in TNCF_12) to 36 (in TNCF_101), and the average number of genes was 34. There was no difference among early and late isolates in terms of presence or abundance of TA families or domains, (Supplementary Figure S6).
Variants and SNPs Analysis
We looked at the distribution of variants in the core genome of the population, using DK2 as a reference, as this strain resulted in being the closest relative to this population (data not shown). Data corresponding to insertions/deletions was not considered as the draft nature of the genomes could bias the output of the program. Each isolate contained a total of 27,249 ± 3,300 single nucleotide variations. In particular, variants per genome ranged from 12,983 to 29,744 in the isolates TNCF_7M and TNCF_165, respectively, with a median number of 28,668 nucleotide variations per genome (as in TNCF_23M and TNCF_32) (Supplementary Table S2).
To investigate polymorphisms with a presumable impact on the adaptive process of the isolates, we extracted non-synonymous polymorphisms classified as high impact variants (i.e., variants resulting in protein truncation, loss of function or triggering non-sense-mediated decay). Considering the complete genome sequence of the isolates (i.e., both core and accessory genomes), the number of high impact variations considerably varied in the population, ranging from six in the early isolate TNCF_7M to 167 in the late isolate TNCF_175.
To determine whether the majority of variants fall into few functional categories or are randomly distributed among all of them, we performed a Cluster of Orthologous Groups (COG) analysis, wherein we assigned the genes in which high-impact variants were found to their respective COG categories. The percentage of genes assigned to each COG is shown in Supplementary Figure S7. Mutations occurred primarily in genes involved in transcription (8%), followed by amino acid transport and metabolism, inorganic ion transport and metabolism, signal transduction mechanisms and general functions (7% each). A large proportion of genes (28%) were not assigned to any COG category but were classified as genes with unknown function. The remaining mutated genes were distributed into all functional groups, with proportions reflecting those found in the analysis of whole genomes.
Polymorphisms in the Core Genome of the Population
We then extended the analysis to variants in the core genome with high and moderate impact (non-disruptive variants that might change protein effectiveness). These ranged from 132 (TNCF_7M) to 394 (TNCF_167, TNCF_175) per genome, with a mean of 270 (Supplementary Table S2). Moreover, an analysis of the distribution of variants in the population (Supplementary Figure S8) revealed a cluster of 13 isolates (TNCF_10, TNCF_13, TNCF_16, TNCF_23, TNCF_23M, TNCF_32, TNCF_42, TNCF_42M, TNCF_49M, TNCF_85, TNCF_88M, TNCF_101, TNCF_133_1) with a high similarity in their variants pattern. One isolate, TNCF_7M, stands out in the distance matrix, probably due to the lowest number of SNPs present in the genome.
The analysis also highlighted the diverse subpopulations putatively derived from the differentiation of a first infecting strain, e.g., the isolates belonging to the ST1864 cluster, which showed a marked similarity, with the only exception of the isolated TNCF_151M, which was quite divergent compared to the other strains.
Eight genes belonging to the core genome carried high-impact variants. Interestingly, four of them have unknown function. The gene encoding respiratory nitrate reductase subunit gamma nail carried high impact variants in three closely related isolates, one early, TNCF_14, and two lates, TNCF_130 and TNCF_151. Isolates TNCF_133 and TNCF_167_1 had high impact variants of two genes encoding a polyprotein signal peptidase lspA and a hypothetical protein, and two hypothetical proteins, respectively. Other genes with high-impact variants were those encoding the ABC transporter-binding protein aaltP in TNCF_3, the copper resistance protein A precursor pcoA in TNCF_175 and a gene with unknown function in TNCF_174.
Within the whole population, a total of 675 different genes carried mutations with moderate impact within the core genome and the distribution of these variants was consistent with population structure (Supplementary Figure S8). Focusing on the moderate impact polymorphisms in the core genome, we found 20 genes with three or more of such variations in the population (Supplementary Table S3), which might be indicative of loci under selective pressure (Table 2). Remarkably, 8 of them were genes with a putative or unknown function. The pyoverdine biosynthesis gene pvdL carried a total of five moderate-impact SNPs in all 40 genomes analyzed, as well as the DNA gyrase subunit B gyrB with five variants in 36 genomes. Remarkably two amino acid substitutions, E468-D and S749-P, were found in isolates resistant to fluoroquinolones. The topoisomerase IV subunit B parE gene showed three different variations within the population, including R30-C and H59-R in the late MDR isolates. The two-component regulator system signal sensor kinase pmrB gene showed three different variations among the population; glnE and rsme (yggJ) carried three different variants, while cbrA and gltB, nagE, aruF and pilL had three moderate variants in five, four, and three different genomes, respectively.

Table 2. Core genes with the highest number of SNPs in the population.
We found that at least three genes, gyrB, parE, and pmrB, involved in the resistance to different classes of antibiotics carried mutations potentially affecting the protein function (Supplementary Table S2).
Among the 20 most variable genes (with at least three variations within the population) selected for the analysis of adaptive evolution, nine resulted in being under non-neutral pressure (namely gyrB and parE, pilL, nagE, gltB, gidA, and three genes encoding hypothetical proteins) (Table 2). For 10 of them, the neutral (null) hypothesis could not be rejected. For one gene, pvdL, it was not possible to perform the test, because its sequence was incomplete, probably due to the draft nature of the genomes. All genes for which the likelihood ratio test rejected the null hypothesis had an omega (dN/dS) > 1, suggesting they are under positive selection.
Phenotypic Analyses
The mucoid phenotype, a characteristic marker of adaptation to the CF lung, was present in 12 strains (30%), mainly early and intermediate isolates, while 17 isolates (42.5%) produced a detectable amount of biofilm (Supplementary Table S4).
For the mucoid phenotype, there was a significant inverse correlation (ρ = -0.407, p = 0.009) with the time of isolation, whereas Spearman’s coefficient indicated no significant correlation (ρ = 0.202, p = 0.211) between biofilm formation and time of sampling. A significant correlation between alginate production and biofilm formation was observed (Supplementary Table S5).
Antibiotic Resistance Profiles
Antibiotic susceptibility tests were performed for all isolates of the population (Figure 6). Twelve different antibiotics belonging to nine classes were tested (see the section “Materials and Methods”). All the isolates were susceptible to colistin, whereas nearly all the early isolates were susceptible to almost all the antibiotics tested. On the other hand, resistant and multidrug-resistant (MDR) phenotypes dramatically increased over time in the persistent population (Supplementary Figure S9).
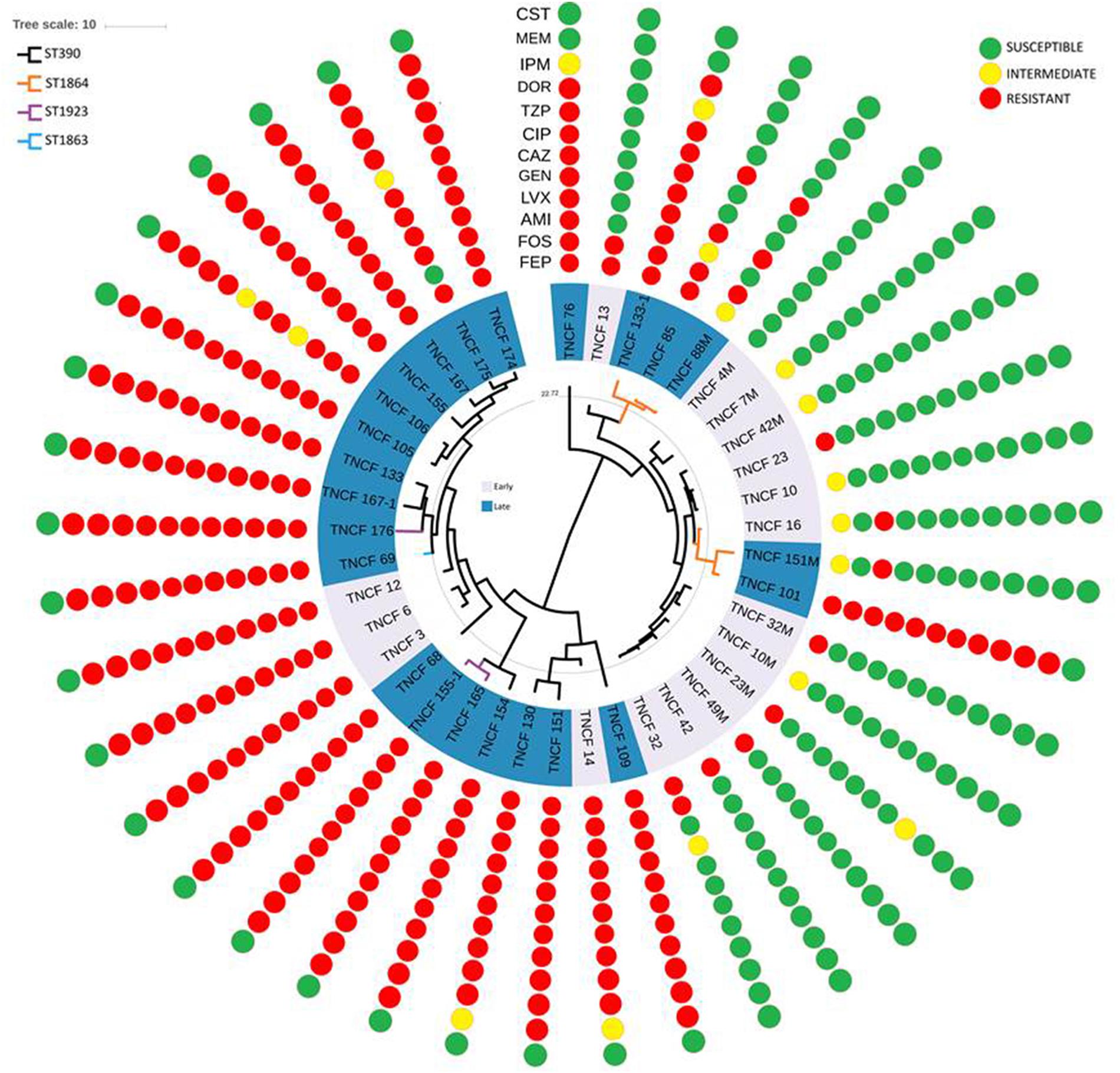
Figure 6. Rooted phylogeny of the P. aeruginosa population with antibiotic susceptibility profiles. Resistance and susceptibility were determined according to the EUCAST clinical breakpoint tables. Resistant, intermediate and susceptible isolates are highlighted in red, yellow, and green, respectively. AMI, amikacin; GEN, gentamicin; IPM, imipenem; MEM, meropenem; DOR, doripenem; CAZ, cefepime; FEP, ceftazidime; CIP, ciprofloxacin; LVX, levofloxacin; FOS, fosfomycin; CST, colistin; TZP, piperacillin/tazobactam.
The vast majority of the 17 strains isolated in 2007 and 2008 were susceptible to nearly all the antibiotics tested, in particular, TNCF_4M was found susceptible to all the antibiotics, while TNCF_7M, TNCF_10, TNCF_10M, and TNCF_42M had an intermediate resistance profile only to cefepime. Four strains were found to have an MDR phenotype: TNCF_3, TNCF_6, TNCF_12, and TNCF_14 were resistant to all antibiotics tested with the exception of colistin.
Among 23 strains isolated between 2010 and 2014, only two of them, TNCF_88M and TNCF_151M, both belonging to ST1864, did not show an MDR phenotype: TNCF_88M, isolated on December 2010, was susceptible to eight antibiotics, with an intermediate profile to cefepime and resistant to ciprofloxacin, levofloxacin, fosfomycin, and piperacillin/tazobactam; whereas TNCF_151M, isolated on May 2013, was found to be susceptible to 10 antibiotics tested, with an intermediate profile to Cefepime and resistant only to amikacin.
The exacerbation of patient’s lung conditions started approximately at the time of predominance of MDR, and biofilm-forming isolates (Supplementary Figures S1, S9; Supplementary Table S4). In the latest stages of infection, the selected antibiotic therapy consisted of a combination of glycopeptides, aminoglycosides, and carbapenems.
As a mucoid phenotype and biofilm formation are often associated with an increase of antibiotic resistance, Spearman’s correlation coefficient was applied to calculate the correlation between the three phenotypes (Supplementary Table S4). MDR was negatively significantly correlated with mucoidy and positively correlated with biofilm formation.
Genomic analyses of the population showed a correlation between the evolution of antibiotic resistance profiles, MLST sequence types, and phylogenetic relationships (Figure 6). We further investigated the MDR isolates in comparison to the susceptible ones, looking specifically at the coding sequences of P. aeruginosa genes involved in antibiotic resistance and susceptibility. As described above, non-synonymous mutations were found in gyrB, parE and pmrB; additional variants present in the MDR isolates compared to the susceptible ones were: ΔC437 in the outer membrane porin gene oprD leading to a frameshift mutation in nearly all MDR isolates, P51-S in MexG and N170-S in the topoisomerase IV subunit A ParC (Table 3 and Supplementary Table S6).
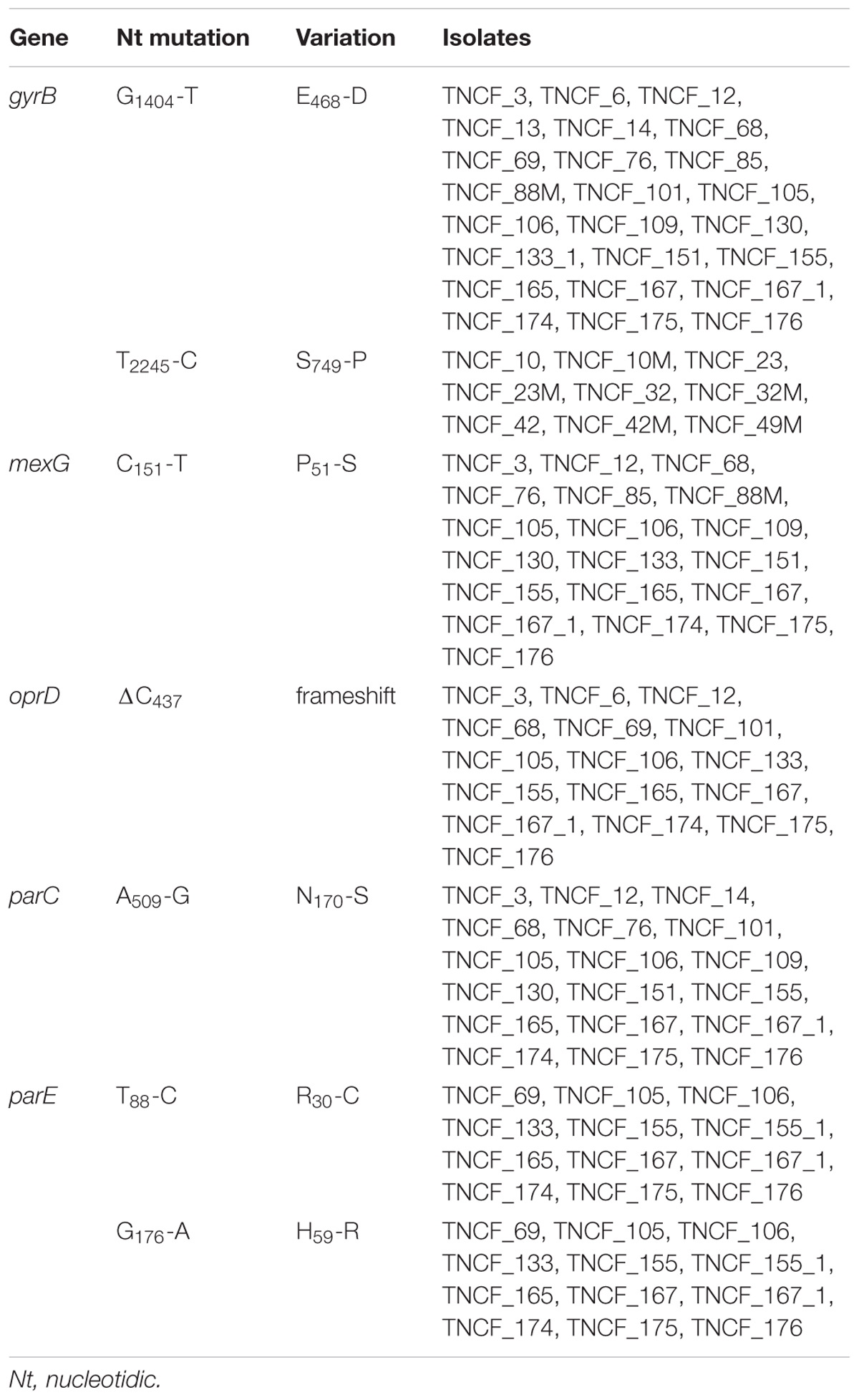
Table 3. Variations in genes involved in antibiotic resistance.
Discussion
Pseudomonas aeruginosa undergoes a characteristic evolutionary adaptation during chronic infection in the CF lung. Previous genomic studies have revealed extensive strain genetic variability within patients, and adaptation to the CF lung environment is recognized to play a crucial role in the diversification of P. aeruginosa (Winstanley et al., 2016).
The occurrence of a dominant Sequence Type in the population together with a small number of closely related genotypes indicates that all isolates likely belong to the same clonal lineage and possibly evolved from a single ancestral colonizing strain. This is in contrast with what has been previously reported in other studies, in which either different lineages coexist within CF patients (Feliziani et al., 2014; Caballero et al., 2015; Williams et al., 2015) or a first infecting lineage is replaced by a new one (Rau et al., 2010; Bianconi et al., 2015). In this patient, a single dominant clonal lineage apparently persisted in the airways for at least 8 years, allowing us to perform a detailed study of the microevolution and adaptive process of the bacterium in the course of chronic infection.
We report the presence of two main lineages in the population, with one of them being the most prevalent in the latest stage of infection (Figure 1). Remarkably, this cluster comprised most multi-drug resistant isolates. Although most of the P. aeruginosa infections have a clonal origin, persistent infections are characterized by an adaptive radiation process leading to a marked diversification of the bacterial clones (Cullen and McClean, 2015). The adaptive process most likely occurs independently in the different individuals of the bacterial population, resulting in the occurrence of clonal subpopulations within the same host (Breidenstein et al., 2011). We have previously characterized genotypically and phenotypically an extensive collection of P. aeruginosa clinical strains isolated from chronic and acute infections (Ballarini et al., 2012). One acute infection isolate (VrPa97), from a wound swab and isolated at the hospital of Verona, Italy, was found to belong to the same genotype (ST390) as that of the isolates analyzed in the present work, thus supporting the idea that very closely related strains can cause acute infections and evolve into highly adapted phenotypes after selection and persistence in the CF lung environment.
A number of comparative genomic studies of P. aeruginosa have shown that most inter-strain diversity is due to the presence of regions of genomic plasticity (RGPs) or GIs often acquired by horizontal gene transfer (Mathee et al., 2008; Kung et al., 2010; Pohl et al., 2014). Plasmid pKLC102 plus six GIs were detected in the population. pKLC102 was previously found in strains of very different origins; this highly motile integrative and conjugative element is almost ubiquitous in P. aeruginosa (Klockgether et al., 2007). It was found to coexist in both episomal and chromosome-integrated forms in P. aeruginosa clone C strains (Klockgether et al., 2004). Three GIs could not be assigned to any previously characterized island, whereas the other ones could be assigned to LESGI-1, PAGI-2, and LESGI-3. LESGI-1 and LESGI-3 were first identified and characterized in the epidemic P. aeruginosa strain LESB58 (Winstanley et al., 2009). LESGI-1 contains phage- and transposon-related genes, in addition to coding sequences showing similarity with proteins of non-pseudomonad species (Winstanley et al., 2009). LESGI-3 island has a mosaic structure and presents a significant homology to PAGI-2, PAGI-3, PAGI-5, and PAPI-1 (Winstanley et al., 2009; Silby et al., 2011). To date, it was described as a unique island of the LESB58 strain, since its particular structure was not found in other strains (Fothergill et al., 2010; Jani et al., 2016). However, even if three PCs showed high identity with both LESGI-3 and PAGI-2, two additional ones with an exclusive assignment to LESGI-3 were found in a different genomic region. It is, therefore, more straightforward to assign the first genomic region to PAGI-2 and the second genomic region, located about 1 Mb downstream, to LESGI-3, rather than speculating on a duplication event of LESGI-3. PAGI-2 was initially described in P. aeruginosa clone C isolates (Larbig et al., 2002). This island is divided into two main phage modules, one coding for chromosome-partitioning proteins (soj) and another containing a bacteriophage P4-related integrase (Klockgether et al., 2007). PAGI-2 is present in approximately 40% of P. aeruginosa strains (Klockgether et al., 2007).
Annotation of SNPs across the sequenced genomes allowed the identification of a number of moderate or high impact mutations in coding sequences (Table 2). Mutations in virulence genes, like pyoverdine biosynthesis gene pvdL and pilin biosynthesis gene chpA (pilL), are known as hallmarks of adaptation of P. aeruginosa strains to the CF airway environment (Winstanley et al., 2016). Siderophore-mediated processes have been linked with virulence regulation of P. aeruginosa and its pathogenic potential (Lamont et al., 2002) and non-synonymous mutations were found in pvdL in the evolutionary pathway leading to a hypermutable CF DK2 clone (Marvig et al., 2014). However, in a study conducted on 36 CF patients, pyoverdine was undetectable in one-third of sputa positive for P. aeruginosa (Martin et al., 2011). The non-synonymous variations of pvdL found in the present study have not been characterized to date, and thus their potential role in the defect in the biosynthesis and functionality of the siderophore system cannot be assessed yet. The presence of pili is necessary for the initial colonization and the translocation of type III exotoxins (Hogardt and Heesemann, 2013), but adapted and late CF isolates accumulate mutations in genes associated with pili, and loss of motility is associated with a higher risk of persistent infections (Bragonzi et al., 2009; Bianconi et al., 2015).
Several mutations in genes associated with antibiotic resistance were detected. The outer membrane protein OprD is involved in the resistance against β-lactams and carbapenems (Li et al., 2012). The protein contains 16 β-strands connected by short loops, eight of which are external loops (Huang et al., 1995). The deletion present in the MDR isolates of the population disrupts the protein just before the fourth loop, and it has been reported that the deletion of loops 3 and 4 results in a non-stable expression of the protein (Huang et al., 1995). In P. aeruginosa repression or inactivation of OprD or a reduced protein expression contribute to moderate resistance to imipenem (Quale et al., 2006; Rodriguez-Martinez et al., 2009; Poole, 2011), and carbapenem-resistant strains are often defective in expression of OprD (Naenna et al., 2010). Moreover, P. aeruginosa PA14 mutants for oprD showed an enhanced fitness in a murine model of mucosal colonization and dissemination (Skurnik et al., 2013).
Several variations were found in the genes encoding DNA gyrase subunit B (GyrB) and topoisomerase IV subunit B (ParE), which are both significant contributors to the acquisition of resistance to fluoroquinolones (Lee et al., 2005). Five different variants were detected for gyrB in 36 genomes. In particular, amino acid substitutions E468-D and S749-P were found in isolates resistant to fluoroquinolones, whereas the gene encoding ParE showed three different variations within the population, including R30-C and H59-R in the late MDR isolates (Table 3). According to the likelihood ratio test, the null hypothesis of neutrality was rejected for both genes, suggesting that they are under non-neutral selection in the population (Table 2). The amino acid substitution E468-D in GyrB was reported to be associated to a high level of resistance to ciprofloxacin in clinical isolates (Lee et al., 2005; Caballero et al., 2015). This variation is within the quinolone resistance-determining regions (QRDR) of the protein, while the mutations in parE are outside the QRDR domain. Nonetheless, non-synonymous variations in gyrB and parE have a low frequency in clinical isolates, and they are described to have only a complementary role in fluoroquinolone resistance (Lee et al., 2005; Lister et al., 2009).
In a recent study (Bruchmann et al., 2013), most of the P. aeruginosa clinical isolates analyzed carried mutations either in gyrA or gyrB or in both gyrA and parC. Mutations in parC gene, encoding topoisomerase IV subunit A, along with those described above in gyrB, are known to confer resistance to fluoroquinolones (Mouneimné et al., 1999; Lee et al., 2005), while clinical isolates with mutations in the QRDR of gyrA and parC show high levels of fluoroquinolone resistance (Lister et al., 2009). A well-characterized double threonine-serine variation in gyrA and parC genes responsible for high-level resistance to fluoroquinolones (Agnello et al., 2016; Fuzi et al., 2017) was not found in the this study. However, while gyrA carried a G75-A variation only in the seven strains belonging to the ST1864, we identified a yet uncharacterized N170-S substitution in parC, present only in the MDR isolates, but this variation is outside the QRDR domain.
The pmrB gene encoding the two-component regulatory system signal sensor kinase showed three different variants within the population. PmrB is part of the two-component regulatory system PmrA/PmrB involved in resistance to polymyxins and other cationic antimicrobial peptides in P. aeruginosa (Moskowitz et al., 2004, 2012; Barrow and Kwon, 2009). Considering that variations found in this population were not directly associated with an MDR phenotype and that this gene was not found to be under non-neutral selective pressure (likelihood ratio test p-value > 0.05), we can assume that the mutations are neutral.
MexG, a component of the efflux pump MexGHI-OpmD, carries an amino acid substitution, P51-S, in nearly all the MDR isolates; mutations in this pump are involved in the resistance to tetracycline and ticarcillin-clavulanate and the export of non-antibiotic compounds, including vanadium (Aendekerk et al., 2002). However, the P51-S variant has not been reported to date. Further investigations are thus needed to assess its putative role in antibiotic resistance.
We observed a significant trend to lose the mucoid phenotype over time in the population, and this phenotype did not correlate with the antibiotic resistance profile of the isolates.
Pseudomonas aeruginosa is known to lose mucoid phenotypes in the latest stages of CF lung disease, because the production of alginate represents a high-energy cost for the bacterium (Hogardt and Heesemann, 2010; Sousa and Pereira, 2014). On another hand, conversion to a mucoid phenotype may promote biofilm formation, which is a crucial factor for the persistence of P. aeruginosa in CF airways (Sousa and Pereira, 2014) and a sessile lifestyle can efficaciously increase tolerance and resistance to antibiotics (Häussler, 2010; Kostakioti et al., 2013). Biofilm formation assay showed that the isolates grew as sessile community mainly in the intermediate/late stage of infection (Supplementary Table S4). These observations were confirmed by Spearman’s correlation between phenotypes and time. We checked if the differences observed in biofilm formation could be due to mutations in the dual-regulator system PA1226–PA1413 (Heacock-Kang et al., 2018), suppressing the ability of P. aeruginosa to form biofilm in vitro. No difference in the two genes between poor and good biofilm producers was found in this study.
Conclusion
The analysis of the genomes of this longitudinal collection of 40 closely related P. aeruginosa strains belonging to the same lineage and isolated from a single CF patient over a long-term period provides new insights that increase our knowledge on the adaptation and microevolution of this pathogen inside its human host. The study revealed the emergence of a MDR phenotype over time that could not be comprehensively explained by mutations found in known resistance genes. Further investigations on uncharacterized variations disclosed in this study should help to increase our understanding of the development of MDR phenotypes and the poor outcome of antibiotic therapies in many CF patients.
Availability of Data and Material
Data analyzed and discussed in this article are included in the article itself or the additional files. Genome sequence data have been deposited at DDBJ/ENA/GenBank under accession numbers MAUO00000000 – MBMR00000000.
Ethics Statement
Strains were isolated during routine monitoring of the CF patient at the Trentino CF Support Centre, Hospital of Rovereto, Italy. The patient provided informed consent in accordance with the Declaration of Helsinki before sampling of strains and storage of clinical data. Written informed consent for publication of clinical details was obtained from the patient. A copy of the consent form is available for review by the Editor of this journal.
Author Contributions
OJ and EB jointly designed the study. EB, PG, GD, and MS isolated and identified the samples and interpreted the data. IB, SD’A, and EP prepared the sequencing libraries. IB, SD’A, AE, MB, CD, and OJ analyzed and interpreted the data. IB, AE, and OJ wrote the manuscript. All authors have read and approved the final version of the manuscript.
Funding
This work was supported by the Associazione Trentina Fibrosi Cistica (Trento, Italy), donations “in memory of Mauro Conotter” and “in memory of Sara Zini.”
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Part of the results presented herein are derived from the Ph.D. thesis of SD’A at the University of Trento, Italy. This represents the only medium these results have appeared in, and is in line with the author’s university policy. The thesis can be accessed online at http://eprints-phd.biblio.unitn.it/. We wish to thank Veronica de Sanctis, Roberto Bertorelli, and Paola Fassan from the LaBSSAH – CIBIO Next Generation Sequencing Facility of the University of Trento for bacterial genome sequencing, and Katy Bailey and Silvano Piazza (CIBIO Bioinformatics Core Facility) for their support on bioinformatics analyses. We also would like to thank Adrian Tett for providing useful edits on this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.03242/full#supplementary-material
Footnotes
- ^ https://github.com/Sandramses/PAst
- ^ https://www.rdocumentation.org/packages/ggpubr
- ^ https://github.com/tseemann/snippy
- ^ http://202.120.12.133/TAfinder/index.php
- ^ http://202.120.12.133/STEP/STEP_CDSeasy.php
- ^ https://www.rdocumentation.org/packages/caper/versions/0.5.2/topics/phylo.d
- ^ https://pubmlst.org/bigsdb?db=pubmlst_paeruginosa_isolates&page=profiles
- ^ https://pubmlst.org/
References
Aendekerk, S., Ghysels, B., Cornelis, P., and Baysse, C. (2002). Characterization of a new efflux pump, MexGHI-OpmD, from Pseudomonas aeruginosa that confers resistance to vanadium. Microbiology 148, 2371–2381. doi: 10.1099/00221287-148-8-2371
Agnello, M., Finkel, S. E., and Wong-Beringer, A. (2016). Fitness cost of fluoroquinolone resistance in clinical isolates of Pseudomonas aeruginosa differs by type III secretion genotype. Front. Microbiol. 7:1591. doi: 10.3389/fmicb.2016.01591
Anisimova, M., and Gascuel, O. (2006). Approximate likelihood-ratio test for branches: a fast, accurate, and powerful alternative. Syst. Biol. 55, 539–552. doi: 10.1080/10635150600755453
Ballarini, A., Scalet, G., Kos, M., Cramer, N., Wiehlmann, L., and Jousson, O. (2012). Molecular typing and epidemiological investigation of clinical populations of Pseudomonas aeruginosa using an oligonucleotide-microarray. BMC Microbiol. 12:152. doi: 10.1186/1471-2180-12-152
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Barrow, K., and Kwon, D. H. (2009). Alterations in two-component regulatory systems of phoPQ and pmrAB are associated with polymyxin B resistance in clinical isolates of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 53, 5150–5154. doi: 10.1128/AAC.00893-09
Bertelli, C., Laird, M. R., Williams, K. P., Lau, B. Y., Hoad, G., Winsor, G. L., et al. (2017). IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45, W30–W35. doi: 10.1093/nar/gkx343
Bianconi, I., Arcangelo, S. D., Benedet, M., Bailey, K. E., Esposito, A., Piffer, E., et al. (2016). Draft genome sequences of 40 Pseudomonas aeruginosa clinical strains isolated from the sputum of a single cystic fibrosis patient over an 8-year period. Genome Announc. 4:e1205–16. doi: 10.1128/genomeA.01205-16.Copyright
Bianconi, I., Jeukens, J., Freschi, L., Alcalá-Franco, B., Facchini, M., Boyle, B., et al. (2015). Comparative genomics and biological characterization of sequential Pseudomonas aeruginosa isolates from persistent airways infection. BMC Genomics 16:1105. doi: 10.1186/s12864-015-2276-8
Bragonzi, A., Paroni, M., Nonis, A., Cramer, N., Montanari, S., Rejman, J., et al. (2009). Pseudomonas aeruginosa microevolution during cystic fibrosis lung infection establishes clones with adapted virulence. Am. J. Respir. Crit. Care Med. 180, 138–145. doi: 10.1164/rccm.200812-1943OC
Bragonzi, A., Wiehlmann, L., Klockgether, J., Cramer, N., Worlitzsch, D., Döring, G., et al. (2006). Sequence diversity of the mucABD locus in Pseudomonas aeruginosa isolates from patients with cystic fibrosis. Microbiology 152, 3261–3269. doi: 10.1099/mic.0.29175-0
Breidenstein, E. B. M., de la Fuente-Núez, C., and Hancock, R. E. W. (2011). Pseudomonas aeruginosa: all roads lead to resistance. Trends Microbiol. 19, 419–426. doi: 10.1016/j.tim.2011.04.005
Bruchmann, S., Dötsch, A., Nouri, B., Chaberny, I. F., and Häussler, S. (2013). Quantitative contributions of target alteration and decreased drug accumulation to Pseudomonas aeruginosa fluoroquinolone resistance. Antimicrob. Agents Chemother. 57, 1361–1368. doi: 10.1128/AAC.01581-12
Caballero, J. D., Clark, S. T., Coburn, B., Zhang, Y., Wang, P. W., Donaldson, S. L., et al. (2015). Selective sweeps and parallel pathoadaptation drive Pseudomonas aeruginosa evolution in the cystic fibrosis lung. mBio 6:e981–15. doi: 10.1128/mBio.00981-15
Chmiel, J. F., and Davis, P. B. (2003). State of the art: why do the lungs of patients with cystic fibrosis become infected and why can’t they clear the infection? Respir. Res. 4:8. doi: 10.1186/1465-9921-4-8
Cingolani, P., Platts, A., Wang, L. L., Coon, M., Nguyen, T., Wang, L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92. doi: 10.4161/fly.19695
Cramer, N., Wiehlmann, L., and Tümmler, B. (2010). Clonal epidemiology of Pseudomonas aeruginosa in cystic fibrosis. Int. J. Med. Microbiol. 300, 526–533. doi: 10.1016/j.ijmm.2010.08.004
Cullen, L., and McClean, S. (2015). Bacterial adaptation during chronic respiratory infections. Pathog 4, 66–89. doi: 10.3390/pathogens4010066
D’Arcangelo, S. (2017). Persistence and Adaptation of Pseudomonas aeruginosa in Cystic Fibrosis Airway. Ph. D thesis, University of Trento, Trento.
Darch, S. E., McNally, A., Harrison, F., Corander, J., Barr, H. L., Paszkiewicz, K., et al. (2015). Recombination is a key driver of genomic and phenotypic diversity in a Pseudomonas aeruginosa population during cystic fibrosis infection. Sci. Rep. 5:7649. doi: 10.1038/srep07649
Darling, A. E., Mau, B., and Perna, N. T. (2010). Progressivemauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. doi: 10.1371/journal.pone.0011147
Didelot, X., and Wilson, D. J. (2015). ClonalFrameML: efficient inference of recombination in whole bacterial genomes. PLoS Comput. Biol. 11:e1004041. doi: 10.1371/journal.pcbi.1004041
Drummond, A. J., Ho, S. Y. W., Phillips, M. J., and Rambaut, A. (2006). Relaxed phylogenetics and dating with confidence. PLoS Biol. 4:e88. doi: 10.1371/journal.pbio.0040088
Drummond, A. J., Suchard, M. A., Xie, D., and Rambaut, A. (2012). Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973. doi: 10.1093/molbev/mss075
Feil, E. J., Li, B. C., Aanensen, D. M., Hanage, W. P., and Spratt, B. G. (2004). eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J. Bacteriol. 186, 1518–1530. doi: 10.1128/JB.186.5.1518-1530.2004
Feliziani, S., Marvig, R. L., Luján, A. M., Moyano, A. J., Di Rienzo, J. A., Krogh Johansen, H., et al. (2014). Coexistence and within-host evolution of diversified lineages of hypermutable Pseudomonas aeruginosa in long-term cystic fibrosis infections. PLoS Genet. 10:e1004651. doi: 10.1371/journal.pgen.1004651
Folkesson, A., Jelsbak, L., Yang, L., Johansen, H. K., Ciofu, O., Hoiby, N., et al. (2012). Adaptation of Pseudomonas aeruginosa to the cystic fibrosis airway: an evolutionary perspective. Nat. Rev. Microbiol. 10, 841–851. doi: 10.1038/nrmicro2907
Fothergill, J. L., White, J., Foweraker, J. E., Walshaw, M. J., Ledson, M. J., Mahenthiralingam, E., et al. (2010). Impact of Pseudomonas aeruginosa genomic instability on the application of typing methods for chronic cystic fibrosis infections. J. Clin. Microbiol. 48, 2053–2059. doi: 10.1128/JCM.00019-10
Fritz, S. A., and Purvis, A. (2010). Selectivity in mammalian extinction risk and threat types: a new measure of phylogenetic signal strength in binary traits. Conserv. Biol. 24, 1042–1051. doi: 10.1111/j.1523-1739.2010.01455.x
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Fuzi, M., Szabo, D., and Csercsik, R. (2017). Double-serine fluoroquinolone resistance mutations advance major international clones and lineages of various multi-drug resistant bacteria. Front. Microbiol. 8:2261. doi: 10.3389/fmicb.2017.02261
Häussler, S. (2010). Multicellular signalling and growth of Pseudomonas aeruginosa. Int. J. Med. Microbiol. 300, 544–548. doi: 10.1016/j.ijmm.2010.08.006
Heacock-Kang, Y., Zarzycki-Siek, J., Sun, Z., Poonsuk, K., Bluhm, A. P., Cabanas, D., et al. (2018). Novel dual regulators of Pseudomonas aeruginosa essential for productive biofilms and virulence. Mol. Microbiol. 109, 401–414. doi: 10.1111/mmi.14063
Hogardt, M., and Heesemann, J. (2010). Adaptation of Pseudomonas aeruginosa during persistence in the cystic fibrosis lung. Int. J. Med. Microbiol. 300, 557–562. doi: 10.1016/j.ijmm.2010.08.008
Hogardt, M., and Heesemann, J. (2013). Microevolution of Pseudomonas aeruginosa to a chronic pathogen of the cystic fibrosis lung. Curr. Top. Microbiol. Immunol. 358, 91–118. doi: 10.1007/82_2011_199
Huang, H., Jeanteur, D., Pattus, F., and Hancock, R. E. W. (1995). Membrane topology and site-specific mutagenesis of Pseudomonas aeruginosa porin OprD. Mol. Microbiol. 16, 931–941. doi: 10.1111/j.1365-2958.1995.tb02319.x
Huse, H. K., Kwon, T., Zlosnik, J. E. A., Speert, D. P., Marcotte, E. M., and Whiteley, M. (2010). Parallel evolution in Pseudomonas aeruginosa over 39,000 generations in vivo. mBio 1:19910. doi: 10.1128/mBio.0019910
Jani, M., Mathee, K., and Azad, R. K. (2016). Identification of novel genomic islands in liverpool epidemic strain of Pseudomonas aeruginosa using segmentation and clustering. Front. Microbiol. 7:1210. doi: 10.3389/fmicb.2016.01210
Jelsbak, L., Johansen, H. K., Frost, A. L., Thøgersen, R., Thomsen, L. E., Ciofu, O., et al. (2007). Molecular epidemiology and dynamics of Pseudomonas aeruginosa populations in lungs of cystic fibrosis patients. Infect. Immun. 75, 2214–2224. doi: 10.1128/IAI.01282-06
Jeukens, J., Boyle, B., Kukavica-Ibrulj, I., Ouellet, M. M., Aaron, S. D., Charette, S. J., et al. (2014). Comparative genomics of isolates of a Pseudomonas aeruginosa epidemic strain associated with chronic lung infections of cystic fibrosis patients. PLoS One 9:e87611. doi: 10.1371/journal.pone.0087611
Klockgether, J., Cramer, N., Wiehlmann, L., Davenport, C. F., and Tümmler, B. (2011). Pseudomonas aeruginosa genomic structure and diversity. Front. Cell. Infect. Microbiol. 2:150. doi: 10.3389/fmicb.2011.00150
Klockgether, J., Miethke, N., Kubesch, P., Bohn, Y. S., Brockhausen, I., Cramer, N., et al. (2013). Intraclonal diversity of the Pseudomonas aeruginosa cystic fibrosis airway isolates TBCF10839 and TBCF121838: distinct signatures of transcriptome, proteome, metabolome, adherence and pathogenicity despite an almost identical genome sequence. Environ. Microbiol. 15, 191–210. doi: 10.1111/j.1462-2920.2012.02842.x
Klockgether, J., Reva, O., Larbig, K., and Tümmler, B. (2004). Sequence analysis of the mobile genome island pKLC102 of Pseudomonas aeruginosa C. J. Bacteriol. 186, 518–534. doi: 10.1128/JB.186.2.518-534.2004
Klockgether, J., Würdemann, D., Reva, O., Wiehlmann, L., and Tümmler, B. (2007). Diversity of the abundant pKLC102/PAGI-2 family of genomic islands in Pseudomonas aeruginosa. J. Bacteriol. 189, 2443–2459. doi: 10.1128/JB.01688-06
Kostakioti, M., Hadjifrangiskou, M., and Hultgren, S. J. (2013). Bacterial biofilms: development, dispersal, and therapeutic strategies in the dawn of the postantibiotic era. Cold Spring Harb. Perspect. Med. 3:a010306. doi: 10.1101/cshperspect.a010306
Kung, V. L., Ozer, E. A., and Hauser, A. R. (2010). The accessory genome of Pseudomonas aeruginosa. Microbiol. Mol. Biol. Rev. 74, 621–641. doi: 10.1128/MMBR.00027-10
Lamont, I. L., Beare, P. A., Ochsner, U., Vasil, A. I., and Vasil, M. L. (2002). Siderophore-mediated signaling regulates virulence factor production in Pseudomon aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 99, 7072–7077. doi: 10.1073/pnas.092016999
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Larbig, K. D., Christmann, A., Johann, A., Klockgether, J., Hartsch, T., Merkl, R., et al. (2002). Gene islands integrated into tRNAGly genes confer genome diversity on a Pseudomonas aeruginosa clone. J. Bacteriol. 184, 6665–6680. doi: 10.1128/JB.184.23.6665-6680.2002
Larsen, M. V., Cosentino, S., Rasmussen, S., Friis, C., Hasman, H., Marvig, R. L., et al. (2012). Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 50, 1355–1361. doi: 10.1128/JCM.06094-11
Lechtzin, N., John, M., Irizarry, R., Merlo, C., Diette, G. B., and Boyle, M. P. (2006). Outcomes of adults with cystic fibrosis infected with antibiotic-resistant Pseudomonas aeruginosa. Respiration 73, 27–33. doi: 10.1159/000087686
Lee, J. K., Lee, Y. S., Park, Y. K., and Kim, B. S. (2005). Alterations in the GyrA and GyrB subunits of topoisomerase II and the ParC and ParE subunits of topoisomerase IV in ciprofloxacin-resistant clinical isolates of Pseudomonas aeruginosa. Int. J. Antimicrob. Agents 25, 290–295. doi: 10.1016/j.ijantimicag.2004.11.012
Li, H., Luo, Y. F., Williams, B. J., Blackwell, T. S., and Xie, C. M. (2012). Structure and function of OprD protein in Pseudomonas aeruginosa: from antibiotic resistance to novel therapies. Int. J. Med. Microbiol. 302, 63–68. doi: 10.1016/j.ijmm.2011.10.001
Lister, P. D., Wolter, D. J., and Hanson, N. D. (2009). Antibacterial-resistant Pseudomonas aeruginosa: clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin. Microbiol. Rev. 22, 582–610. doi: 10.1128/CMR.00040-09
Lyczak, J. B., Cannon, C. L., and Pier, G. B. (2000). Establishment of Pseudomonas aeruginosa infection: lessons from a versatile opportunist. Microb. Infect. 2, 1051–1060. doi: 10.1016/S1286-4579(00)01259-4
Martin, L. W., Reid, D. W., Sharples, K. J., and Lamont, I. L. (2011). Pseudomonas siderophores in the sputum of patients with cystic fibrosis. Biometals 24, 1059–1067. doi: 10.1007/s10534-011-9464-z
Marvig, R. L., Damkiær, S., Hossein Khademi, S. M., Markussen, T. M., Molin, S., and Jelsbak, L. (2014). Within-host evolution of pseudomonas aeruginosa reveals adaptation toward iron acquisition from hemoglobin. mBio 5:e966–14. doi: 10.1128/mBio.00966-14
Marvig, R. L., Sommer, L. M., Molin, S., and Johansen, H. K. (2015). Convergent evolution and adaptation of Pseudomonas aeruginosa within patients with cystic fibrosis. Nat. Genet. 47, 57–65. doi: 10.1038/ng.3148
Mathee, K., Narasimhan, G., Valdes, C., Qiu, X., Matewish, J. M., Koehrsen, M., et al. (2008). Dynamics of Pseudomonas aeruginosa genome evolution. Proc. Natl. Acad. Sci. U.S.A. 105, 3100–3105. doi: 10.1073/pnas.0711982105
Moskowitz, S. M., Brannon, M. K., Dasgupta, N., Pier, M., Sgambati, N., Miller, A. K., et al. (2012). PmrB mutations promote polymyxin resistance of Pseudomonas aeruginosa isolated from colistin-treated cystic fibrosis patients. Antimicrob. Agents Chemother. 56, 1019–1030. doi: 10.1128/AAC.05829-11
Moskowitz, S. M., Ernst, R. K., and Miller, S. I. (2004). PmrAB, a two-component regulatory system of Pseudomonas aeruginosa that modulates resistance to cationic antimicrobial peptides and addition of aminoarabinose to lipid a. J. Bacteriol. 186, 575–579. doi: 10.1128/JB.186.2.575-579.2004
Mouneimné, H., Robert, J., Jarlier, V., and Cambau, E. (1999). Type II topoisomerase mutations in ciprofloxacin-resistant strains of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 43, 62–66. doi: 10.1128/AAC.43.1.62
Naenna, P., Noisumdaeng, P., Pongpech, P., and Tribuddharat, C. (2010). Detection of outer membrane porin protein, an imipenem influx channel, in Pseudomonas aeruginosa clinical isolates. Southeast Asian J. Trop. Med. Public Health 41, 614–624.
Osmon, S., Ward, S., Fraser, V. J., and Kollef, M. H. (2004). Hospital mortality for patients with bacteremia due to staphylococcus aureus or Pseudomonas aeruginosa. Chest 125, 607–616. doi: 10.1378/chest.125.2.607
O’Toole, G. A. (2011). Microtiter dish biofilm formation assay. J. Vis. Exp. 47:2437. doi: 10.3791/2437
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Pohl, S., Klockgether, J., Eckweiler, D., Khaledi, A., Schniederjans, M., Chouvarine, P., et al. (2014). The extensive set of accessory Pseudomonas aeruginosa genomic components. FEMS Microbiol. Lett. 356, 235–241. doi: 10.1111/1574-6968.12445
Poole, K. (2011). Pseudomonas aeruginosa: resistance to the max. Front. Microbiol. 2:65. doi: 10.3389/fmicb.2011.00065
Quale, J., Bratu, S., Gupta, J., and Landman, D. (2006). Interplay of efflux system, ampC, and oprD expression in carbapenem resistance of Pseudomonas aeruginosa clinical isolates. Antimicrob. Agents Chemother. 50, 1633–1641. doi: 10.1128/AAC.50.5.1633-1641.2006
Rau, M. H., Hansen, S. K., Johansen, H. K., Thomsen, L. E., Workman, C. T., Nielsen, K. F., et al. (2010). Early adaptive developments of Pseudomonas aeruginosa after the transition from life in the environment to persistent colonization in the airways of human cystic fibrosis hosts. Environ. Microbiol. 12, 1643–1658. doi: 10.1111/j.1462-2920.2010.02211.x
Rodriguez-Martinez, J. M., Poirel, L., and Nordmann, P. (2009). Molecular epidemiology and mechanisms of carbapenem resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 53, 4783–4788. doi: 10.1128/AAC.00574-09
Roosaare, M., Puustusmaa, M., Möls, M., Vaher, M., and Remm, M. (2018). PlasmidSeeker: identification of known plasmids from bacterial whole genome sequencing reads. PeerJ 6:e4588. doi: 10.7717/peerj.4588
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Seemann, T. (2015). Snippy: Rapid Haploid Variant Calling and Core SNP Phylogeny. Available at: https://github.com/tseemann/snippy
Shao, Y., Harrison, E. M., Bi, D., Tai, C., He, X., Ou, H. Y., et al. (2011). TADB: a web-based resource for Type 2 toxin-antitoxin loci in bacteria and archaea. Nucleic Acids Res. 39, D606–D611. doi: 10.1093/nar/gkq908
Silby, M. W., Winstanley, C., Godfrey, S. A. C., Levy, S. B., and Jackson, R. W. (2011). Pseudomonas genomes: diverse and adaptable. FEMS Microbiol. Rev. 35, 652–680. doi: 10.1111/j.1574-6976.2011.00269.x
Skurnik, D., Roux, D., Cattoir, V., Danilchanka, O., Lu, X., Yoder-Himes, D. R., et al. (2013). Enhanced in vivo fitness of carbapenem-resistant oprD mutants of Pseudomonas aeruginosa revealed through high-throughput sequencing. Proc. Natl. Acad. Sci. U.S.A. 110, 20747–20752. doi: 10.1073/pnas.1221552110
Sommer, L. M., Alanin, M. C., Marvig, R. L., Nielsen, K. G., Høiby, N., von Buchwald, C., et al. (2016). Bacterial evolution in PCD and CF patients follows the same mutational steps. Sci. Rep. 6:28732. doi: 10.1038/srep28732
Sousa, A. M., and Pereira, M. O. (2014). Pseudomonas aeruginosa diversification during infection development in cystic fibrosis lungs-A Review. Pathog 3, 680–703. doi: 10.3390/pathogens3030680
Stoesser, N., Batty, E. M., Eyre, D. W., Morgan, M., Wyllie, D. H., Del Ojo Elias, C., et al. (2013). Predicting antimicrobial susceptibilities for Escherichia coli and Klebsiella pneumoniae isolates using whole genomic sequence data. J. Antimicrob. Chemother. 68, 2234–2244. doi: 10.1093/jac/dkt180
Williams, D., Evans, B., Haldenby, S., Walshaw, M. J., Brockhurst, M. A., Winstanley, C., et al. (2015). Divergent, coexisting Pseudomonas aeruginosa lineages in chronic cystic fibrosis lung infections. Am. J. Respir. Crit. Care Med. 191, 775–785. doi: 10.1164/rccm.201409-1646OC
Winstanley, C., Langille, M. G. I., Fothergill, J. L., Kukavica-Ibrulj, I., Paradis-Bleau, C., Sanschagrin, F., et al. (2009). Newly introduced genomic prophage islands are critical determinants of in vivo competitiveness in the liverpool epidemic strain of pseudomonas aeruginosa. Genome Res. 19, 12–23. doi: 10.1101/gr.086082.108
Winstanley, C., O’Brien, S., and Brockhurst, M. A. (2016). Pseudomonas aeruginosa evolutionary adaptation and diversification in cystic fibrosis chronic lung infections. Trends Microbiol. 24, 327–337. doi: 10.1016/j.tim.2016.01.008
Workentine, M. L., Sibley, C. D., Glezerson, B., Purighalla, S., Norgaard-Gron, J. C., Parkins, M. D., et al. (2013). Phenotypic heterogeneity of Pseudomonas aeruginosa populations in a cystic fibrosis patient. PLoS One 8:e60225. doi: 10.1371/journal.pone.0060225
Xie, Y., Wei, Y., Shen, Y., Li, X., Zhou, H., Tai, C., et al. (2018). TADB 2.0: an updated database of bacterial type II toxin-antitoxin loci. Nucleic Acids Res. 46, D749–D753. doi: 10.1093/nar/gkx1033
Yang, Z. (2006). Computational molecular evolution. Oxford Ser. Ecol. Evol. 14:357. doi: 10.1093/acprof:oso/9780198567028.001.0001
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Keywords: Pseudomonas aeruginosa, persistence, cystic fibrosis, genome evolution, antibiotic resistance
Citation: Bianconi I, D’Arcangelo S, Esposito A, Benedet M, Piffer E, Dinnella G, Gualdi P, Schinella M, Baldo E, Donati C and Jousson O (2019) Persistence and Microevolution of Pseudomonas aeruginosa in the Cystic Fibrosis Lung: A Single-Patient Longitudinal Genomic Study. Front. Microbiol. 9:3242. doi: 10.3389/fmicb.2018.03242
Received: 18 October 2018; Accepted: 13 December 2018;
Published: 11 January 2019.
Edited by:
Feng Gao, Tianjin University, ChinaReviewed by:
Miklos Fuzi, Semmelweis University, HungaryDinesh Sriramulu, Independent Researcher, Chennai, India
Copyright © 2019 Bianconi, D’Arcangelo, Esposito, Benedet, Piffer, Dinnella, Gualdi, Schinella, Baldo, Donati and Jousson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Olivier Jousson, b2xpdmllci5qb3Vzc29uQHVuaXRuLml0; am91c3NvbkBzY2llbmNlLnVuaXRuLml0
†These authors have contributed equally to this work