 Limin Liang1
Limin Liang1 Xuehai Zheng1
Xuehai Zheng1 Wenfang Fan1
Wenfang Fan1 Duo Chen1Zhen Huang1Jiangtao Peng2Jinmao Zhu1Weiqi Tang2
Duo Chen1Zhen Huang1Jiangtao Peng2Jinmao Zhu1Weiqi Tang2 Youqiang Chen1
Youqiang Chen1 Ting Xue1*
Ting Xue1*- 1The Public Service Platform for Industrialization Development Technology of Marine Biological Medicine and Products of the State Oceanic Administration, Center of Engineering Technology Research for Microalga Germplasm Improvement of Fujian, Fujian Key Laboratory of Special Marine Bioresource Sustainable Utilization, Key Laboratory of Developmental and Neural Biology, Southern Institute of Oceanography, College of Life Sciences, Fujian Normal University, Fuzhou, China
- 2Institute of Oceanography, Marine Biotechnology Center, Minjiang University, Fuzhou, China
Schizochytrium sp. is the best natural resource for omega-3 long-chain polyunsaturated fatty acids. We report a high-quality genome sequence of Schizochytrium limacinum SR21, which has a 63 Mb genome size, with a contig N50 of 2.67 Mb and 6,838 protein-coding genes. Phylogenomic and comparative genomic analyses revealed that DHA-producing Schizochytrium and Aurantiochytrium strains were highly similar and possessed similar genes. Analysis of the fatty acid synthase (FAS) for LC-PUFAs production results in the annotation of all genes in map00062 and map01212. A gene cluster and 10 ORFs related to PKS pathway were found in the genome. 1,402 differentially expressed genes (DEGs) of the treated groups (0.5 g/L yeast extract) were identified by comparing with the control groups (1.0 g/L yeast extract) at 36 h. A weighted gene coexpression network analysis revealed that 2 of 7 modules correlated highly with the fatty acid and DHA contents. The DEGs and transcription factors were significantly correlated with fatty acid biosynthesis, including MYB, Zinc Finger and ACOX. The results showed that these hub genes are regulated by genes involved in fatty acid biosynthesis pathways. The results providing an important reference for further research on promoting fatty acid and DHA accumulation in S. limacinum SR21.
Introduction
Schizochytrium sp. is a unicellular fungal-like marine protist found ubiquitously in marine environments and considered as the best natural resource for omega-3 long-chain polyunsaturated fatty acids (LC-PUFAs) (Newell et al., 2019). Docosahexaenoic acid (DHA, 22:6) is a high-value LC-PUFA with strong biological activity for the food, feed, medicine, health care, and pharmaceutical industries (Ambati et al., 2014; Browning et al., 2014; Newell et al., 2019). The traditional commercial source of DHA is fish oil, which is undesirable because of its offensive odor, potential contamination and complex purification process (Mühlroth et al., 2013; Ye et al., 2015). Schizochytrium sp. is considered a noteworthy and satisfactory alternative to fish oil due to the advantages of its fast growth rate, high purity, slight fishy smell, and more than 50% lipid rich in DHA. Many studies have been conducted to optimize the media composition and culture conditions, including nutrient deprivation, strain adaption, temperature shift and genetic engineering to enhance the lipid accumulation efficiency in Schizochytrium sp. (Sakaguchi et al., 2012; Ren et al., 2014, 2015; Sun et al., 2014, 2016). However, Schizochytrium sp. can accumulate lipids up to 50% of its dry weight, with DHA generally constituting 40% or more of the total oils by the above mentioned methods.
Two LC-PUFAs biosynthetic pathways, fatty acid synthase (FAS) and polyketide synthase (PKS) system, have long been speculated to exist in the genomes of Aurantiochytrium sp., Schizochytrium sp., and Thraustochytriidae sp. (Warude et al., 2006; Orikasa and Nishida, 2007). The two major steps in fatty acid biosynthesis are elongation and desaturation carried out by two carbon units in the FAS pathway (Morais et al., 2015). For FAS pathway, Lippmeier et al., 2009 found Δ-5, Δ-6, and Δ-9 elongase activities in Schizochytrium sp. ATCC20888 without the present of Δ-12 desaturation. Ren et al. (2017), detected one elongase and three kinds (Δ-6, Δ-8, and Δ-12) of desaturase activities in Schizochytrium sp. PKS pathway of LC-PUFAs synthesis involve the processing of the saturated 16:0 or 18:0 products by repetitive decarboxylative Claisen ester condensations. This process usually involves 3-ketoacyl synthase (KS), malonyl-CoA acyltransferase (MAT), acyl carrier proteins (ACP), 3-ketoacyl-ACP reductase (KR), enoyl reductase (ER), and dehydrase (DH). Metz (2001), identified 11 regions within the five open reading frames (ORFs) form Shewanella sp., eight of these were related to PKS proteins and three of these to FAS. Meanwhile, three ORFs (orfA, orfB, orfC) involved in the synthesis of LC-PUFAs genes from Schizochytrium sp. were identified and had similar structural and functional regions of genes compared with Shewanella sp. by sequencing analysis and comparison (Metz, 2001). FAS and PKS biosynthetic pathways have strong homologies in the chemical mechanisms involved in chain elongation and precursors (acetyl-CoA, malonyl-CoA) (Kaulmann and Hertweck, 2002). Although clustered genes involved FAS and PKS pathway were analyzed based on the genomic fragment, transcriptomics and incomplete genomic information, the complete FAS and PKS genes of DHA-producing Schizochytrium sp. have not been reported yet by high-quality genomic resources.
In order to uncover the regulatory mechanism of lipid migration and LC-PUFA synthesis, genomics and transcriptomics studies have revealed genes or proteins involved in LC-PUFA biosynthesis (Ren et al., 2017). Scaffolding genome assemblies remain challenging even with the rapidly increasing sequence coverage generated by current next-generation sequence technologies. With scaffolding information, draft genome sequences of four DHA-producing thraustochytrid strains, namely, Schizochytrium sp. CCTCC M209059 (39.09 Mb, scaffold N50 of 595 kb), Aurantiochytrium sp. T66 (43 Mb, scaffold N50 of 1.3 Mb), Schizochytrium sp. Mn4 (65.69 Mb, scaffold N50 of 153 kb) and Thraustochytriidae sp. SW8 (61.67 Mb, scaffold N50 of 127 kb), have been produced and provided some key information to improve our understanding of the molecular mechanisms for LC-PUFA synthesis (Ji et al., 2015; Liu et al., 2016; Song et al., 2018), but incomplete genome assemblies bring some problems for the subsequent study of Schizochytrium sp. LC-PUFA biosynthesis at the DNA level. Meanwhile, high-quality genomic resources will help to breed novel strains of Schizochytrium sp. that could have higher LC-PUFAs and DHA yield in industries. In addition, a systematic analysis of the molecular synthesis and regulatory networks for PUFA biosynthesis in Schizochytrium sp. has not been performed by genomics and transcriptomics.
In this study, to systematically understand the molecular pathway and regulatory network for LC-PUFAs and DHA production in Schizochytrium sp., we used third-generation sequencing (TGS) of the PacBio SEQUEL platform to generate a high-quality genome assembly and annotation of the DHA-producing strain Schizochytrium limacinum SR21 (S. limacinum SR21). In addition, LC-PUFA production could be enhanced under yeast extract starvation, which is the most common stress that occurs during Schizochytrium sp. fermentation. We therefore performed gene family, transcriptome sequencing and weighted correlation network analysis (WGCNA) on stressed cells at six stages of fermentation (12, 24, 36, 48, 60, and 72 h) to reveal additional genes that are potentially involved in the accumulation and regulation of LC-PUFAs and DHA production.
Materials and Methods
Sample Materials, Genomic DNA Extraction, and Genome Assembly
Schizochytrium limacinum SR21 (S. limacinum SR21) was purchased from the American Type Culture Collection (ATCC). The basal fermentation medium contained 5.0 g of glucose, 1.0 g of peptone, 1.0 g of yeast extract, and 1 L of seawater. Cells were grown in 250-mL Erlenmeyer flasks containing 100 mL of medium and incubated at 20°C in an orbital shaker set at 220 rpm. Genomic DNA was isolated from 100 ml of fresh culture. The cell suspension was centrifuged at 8000 rpm for 5 min. The cell pellet was then suspended in approximately 1 mL of medium and pipetted into a 2-mL tube and centrifuged again at 8000 rpm for 5 min. Briefly, 800 μL of 2% mercaptoethanol solution was pipetted into each sample, followed by the addition of 800 μL of 10% w/v CTAB (cetyl/hexadecyl trimethyl ammonium bromide, in 0.7M NaCl solution) and incubation at 56°C for 10 min. After extraction with an equal volume of phenol: chloroform: isoamyl alcohol (25:24:1), the mixture was centrifuged at 12,000 rpm for 10 min twice. The supernatant was dissolved in 0.4 mL of 100 μg/mL RNase and incubated at 37°C for 30 min. An equal volume of chloroform: isoamyl alcohol (24:1) was then centrifuged at 12,000 rpm for 10 min. Genomic DNA was precipitated by adding 2.5 volumes of 100% ethanol and collected by spinning at 12,000 rpm and 4°C for 10 min. After the supernatant was discarded, the resulting genomic DNA pellet was stored in 5.0 mL of 70% cold ethanol at 4°C overnight to allow the impurity to dissolve. Finally, DNA was eluted in 100 μL of 10 mM Tris-HCl by centrifugation at 12,000 rpm for 1 min. The purity and concentration of DNA were analyzed using a NanoDrop 2000 Spectrophotometer (Thermo Scientific, United States).
More than 5 μg of sheared and concentrated DNA was applied to size-selection by the BluePippin system. Approximately 20-kb SMRTbellTM libraries were prepared according to the released protocol from PacBio company. A total of 6.7 Gb subreads were sequenced on the PacBio sequel system, i.e., 106 × coverage of the estimated genome size. After removing the low-quality (containing 10 or more Ns and low-quality bases with quality scores ≤ 7) and redundant reads, the full PacBio subreads were corrected, trimmed and assembled using CANU version 1.7 with parameter corOutCoverage = 80 (Koren et al., 2017). To improve the accuracy, primary contigs were further polished by the Pilon program using 9.4 Gb (150×) Illumina paired-end reads (Walker et al., 2014; Supplementary Table S1). Assessment of genome completeness was performed with BUSCO using Eukaryotic models (Simao et al., 2015).
Genome Annotation
Before annotating the gene structures of the S. limacinum SR21 genome, we identified repeat sequences using multiple programs, including Tandem Repeats Finder, LTR_FINDER, Repeat ProteinMask and RepeatMasker (Huda and Jordan, 2009). Tandem Repeats Finder was employed to search for tandem repeats in our genome assembly using the following parameters: Match = 2, Mismatch = 5, Delta = 7, PM = 60, PI = 10, Minscore = 80, and MaxPerid = 2,000. A de novo repeat library was built by the LTR_FINDER (version 1.0.6). Subsequently, the RepeatMasker was utilized to align our genome sequences onto the Repbase TE (version 3.2.9) to search the known repeat sequences as well as map onto the de novo repeat libraries to identify novel types of repeat sequences (Jurka et al., 2005).
We then performed annotation of the S. limacinum SR21 genome assembly with three approaches, including homology-based, transcriptome-based, and ab initio annotation. We selected several representative species, including Paramecium tetraurelia, Saccharomyces cerevisiae, Symbiodinium kawagutii and Symbiodinium minutum, Chlamydomonas eustigma, Chromochloris zofingiensis, and Micromonas pusilla, to perform the homology annotation (Brussaard et al., 1999; Kellis et al., 2004; Aury et al., 2006; Lin et al., 2015; Kanzaki et al., 2017; Roth et al., 2017). The protein sequences from the abovementioned species were aligned onto our genome sequences utilizing TblastN with an E-value ≤ 1e-5. Genewise 2.2.0 was subsequently employed to predict possible gene structures based on all TblastN results (Smith et al., 2011; Kagale et al., 2014). Total RNA was extracted from control cells for subsequent transcriptome sequencing using an Illumina HiSeq 4000 platform. We utilized Cufflinks (version 2.2.1) to identify the preliminary genes (Trapnell et al., 2012). Moreover, Augustus and Genscan were selected for ab initio annotation using the repeat-masked genome sequences (Thayer et al., 2000; Sommerfeld et al., 2009). Finally, we employed GLEAN software to integrate all genes predicted from the three annotation procedures (Elsik et al., 2007). Functional annotation of the protein-coding genes was carried out by BLASTP with an E-value ≤ 1e-5 to four integrated protein sequence databases: eggNOG, GO, COG, and KEGG (Ashburner et al., 2000; Tatusov et al., 2000; Kanehisa et al., 2014).
Phylogenetic Analysis
To investigate the relationship of S. limacinum SR21 with the other 13 species, we performed phylogenetic analysis using the protein-coding gene from the S. limacinum SR21 genome and other species. Protein sequences of single-copy genes were extracted from 14 species and downloaded from the NCBI and JGI databases, including Arabidopsis thaliana (A. thaliana), Aurantiochytrium limacinum ATCC-MYA-1381 (A. limacinum ATCC-MYA-1381), Chlamydomonas reinhardtii (C. reinhardtii), Fragilariopsis cylindrus (F. cylindrus), Hondaea fermentalgiana (H. fermentalgiana), Phaeodactylum tricornutum (P. tricornutum), Phytophthora infestans (P. infestans), Phytophthora parasitica (P. parasitica), Phytophthora sojae (P. sojae), Saprolegnia parasitica (S. parasitica), Schizochytrium aggregatum-ATCC28209 (S. aggregatum-ATCC28209), Thalassiosira pseudonana (T. pseudonana) and Thraustotheca clavata (T. clavata). The similarities among proteins from all species were searched using the all-to-all manner by BLASTP software with an E-value ≤ 1e-5. Orthofinder software (version 2.27) was used to generate multiple sequence alignment for protein sequences in each single-copy family with default parameters as well as phylogenetic tree construction (Emms and Kelly, 2015). A. thaliana and C. reinhardtii were designated as the outgroup of the phylogenetic tree. The phylogenetic relationships were constructed through superalignment of the coding DNA sequences (CDSs) using the maximum likelihood (ML) method. The CDSs were aligned with the guidance of the protein alignments and then concatenated into the superalignment matrix of each family. We compared the cluster size differences between the ancestor and each species, analyzed the expansion and contraction of the gene families by using CAFE software (version 2.1) (Cristianini and Demuth, 2006).
Culture Conditions and Induction of DHA Biosynthesis
Recent studies showed that yeast extract starvation could enhance lipid production, which is essential for DHA accumulation (Konishi et al., 2011). In our previous study, the total fatty content and DHA production of S. limacinum SR21 drastically increased from 12 to 36 h in basal fermentation medium containing 1.0 or 0.5 g/L yeast extract, respectively. However, the stimulation of total fatty content and DHA production was gradually attenuated above 36 h (Figure 1). Compared with 1.0 g/L yeast extract, 0.5 g/L yeast extract could be conducive to the accumulation of DHA. Therefore, we considered that the attenuation of yeast extract-induced stimulation may be caused by the shortage of carbon sources during cultivation with 0.5 g/L yeast extract. To investigate the influence of yeast extract on lipid production, S. limacinum SR21 was cultured in basal fermentation medium with 0.5 g/L yeast extract. S. limacinum SR21 was cultured in 500-mL beakers in basal fermentation medium under a light intensity of 25 mmol photons m–1 s–2 with a 12 h:12 h light:dark cycle at 20°C. When the cells reached the logarithmic phase (106 cells/mL), the culture was evenly divided into six aliquots of 1000 mL each at different concentrations of yeast extract. They were used as three experimental controls (control, 1.0 g/L yeast extract) and three treatments (treated, 0.5 g/L yeast extract) and were sampled at 12, 24, 36, 48, 60, and 72 h. All the samples were centrifuged at 8,000 rpm, kept at 4°C for 10 min, and then stored at −80°C until subsequent RNA-seq analyses.
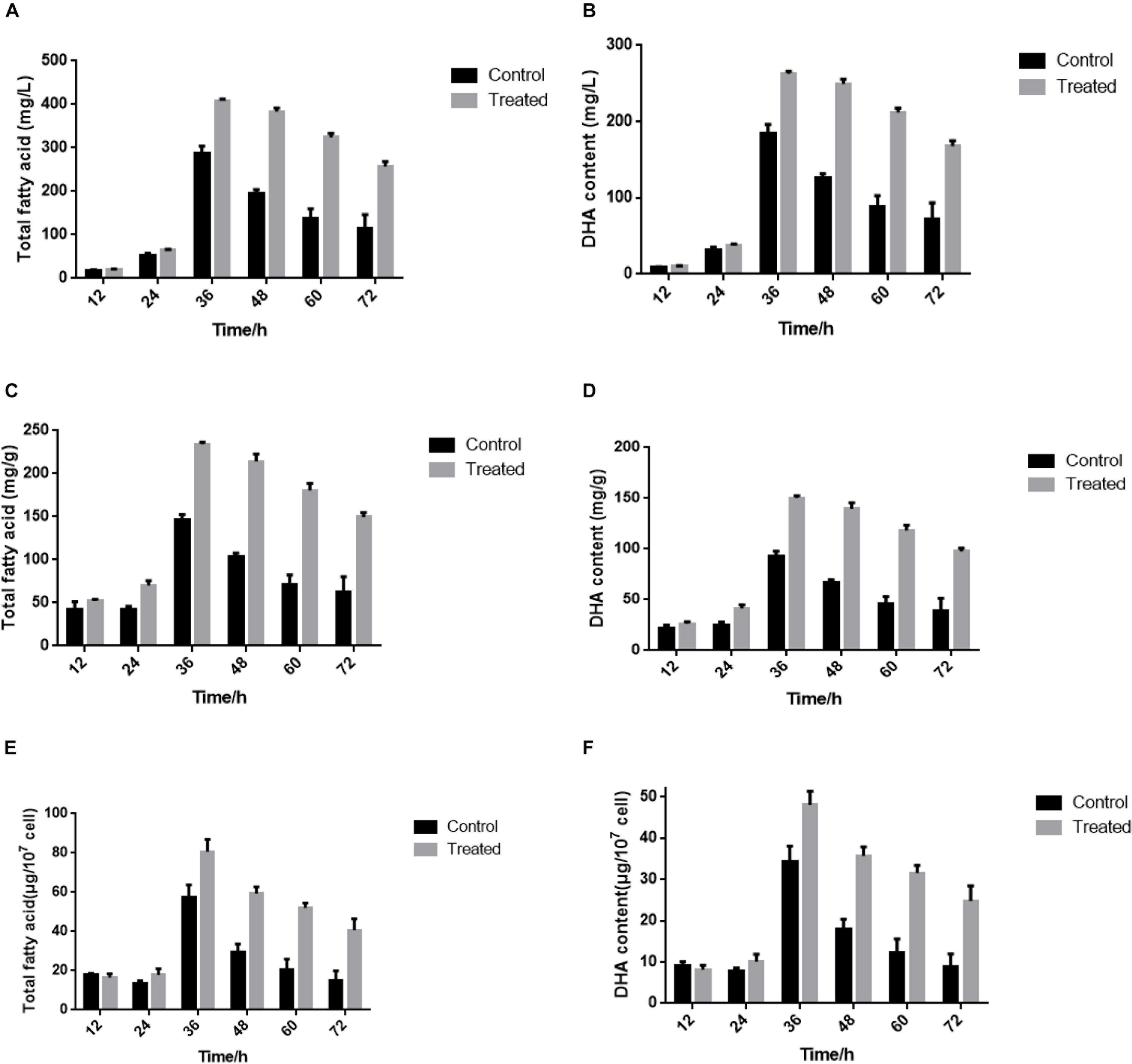
Figure 1. (A,B) Content of total fatty acid and DHA in fresh weight at different cultivation times. (C,D) Content of total fatty acid and DHA in dry weight at different cultivation times. (E,F) Content of total fatty acid and DHA in 107 cell density at different cultivation times. The control groups represent that S. limacinum SR21 was cultured in basal fermentation medium with 1.0 g/L yeast extract. The treated groups represent that S. limacinum SR21 was cultured in basal fermentation medium with 0.5 g/L yeast extract. All experiments were performed in triplicate. Each value presents the mean ± SD. “I” represents error bars for the various determinations (n = 3).
Total RNA Isolation, Library Construction, and Sequencing
Analysis of the influence of yeast extract on lipid production of S. limacinum SR21 was performed in basal fermentation medium at different concentrations of yeast extract. Samples from the six stages were collected at 12, 24, 36, 48, 60, and 72 h. The collected samples were immediately frozen in liquid nitrogen and stored at −80°C until RNA extraction. The samples of stages 12, 24, 36, 48, 60, and 72 h of S. limacinum SR21 were used to construct six libraries. Total RNA from S. limacinum SR21 was extracted using a TransZol Up Plus RNA Kit (Transgen Biotech, Beijing). A NanoDrop 2000 Spectrophotometer (Thermo Scientific, United States) and a 2100 Bioanalyzer (Agilent Technologies, United States) were applied to check the RNA molecule quality, and the absorbance at 260 nm/280 nm was 1.8, and the RIN value was 9.1. cDNA was prepared using the SMARTer PCR cDNA Synthesis Kit (Clontech) from 2 μg of purified RNA. The RNA libraries were sequenced on the Illumina HiSeqTM 2000 sequencing platform.
Expression Quantification and Differential Expression Analysis
Reads originating from RNA-seq were aligned to the reference genome using HISAT2. Fragments per kilobase of transcript per million fragments mapped (FPKM) was adopted to quantify the abundance of assembled transcripts using Stringtie and Ballgown (Pertea et al., 2015, 2016). EdgeR was applied to analyze differentially expressed genes (DEGs) in which the criteria were a twofold change (log2FC > 1 or < -1) in expression level and false discovery rate (FDR) < 0.05 (Nikolayeva and Robinson, 2014). The enrichment analysis of GO terms and KEGG pathways was performed using the online OmicShare tools3. All expressed genes were used as the background. We finally generated a total of 309,962,820 high-quality clean reads. The total mapping ratio of each sample to the genome assembly ranged from 92.78 to 93.86%, and the number of transcribed genes in each sample was predicted to range from 110,625,09 to 112,127,27 (Supplementary Table S2). FPKM values of all transcripts are provided in the Supplementary Table S3.
Weighted Gene Coexpression Network Analysis (WGCNA)
Coexpression analysis was conducted using weighted correlation network analysis (WGCNA) (Langfelder and Horvath, 2008). After selecting 2257 DEGs between control and treatment, we log-transformed these FPKM values using log2 (FPKM value + 1) as recommended on the WGCNA FAQ’s page. We chose a soft power value β (β = 20) to approximate a scale-free network topology to generate a network, guided by a convenient 1-step network construction and module detection function in the R Tutorial1. Then, the Module Eigengene (ME) was calculated, which represents the expression profile of each module. Next, based on the correlation between the ME and trait, we estimated the module-trait relationships to identify highly correlated modules. The module is considered to be associated with traits, where the module-trait relationship value is ≥ 0.4 and P ≤ 0.05. As a basis for identifying hub genes, intramodular connectivity that was founded on the correlation between the ME and the expression profile was detected.
qRT-PCR Validation
Total RNA from the three replicates was extracted using a TransZol Up Plus RNA Kit (Transgen Biotech, Beijing). Primers were designed using Primer Premier 5.0, and the sequences are listed in the Supplementary Table S4. qRT-PCR was performed using the Applied Biosystems 7300 real-time PCR System (Framingham, MA, United States) with SYBR Green PCR Master Mix (TaKaRa) following the procedures described previously (Borecka-Melkusova et al., 2009; Kong et al., 2014). All PCRs were performed in triplicate. 18S rRNA was used as the reference gene (Noda et al., 2004). The relative expression level was quantified by the 2–Δ Δ Ct method.
Results
Genome Assembly and Annotation
A total of 6.7 Gb Pacbio subreads (∼106 × coverage of the estimated genome size) and 9.4 Gb Illumina paired-end reads (∼150 × coverage of the estimated genome size), resulting in approximately 125-fold coverage of the S. limacinum SR21 genome. All reads from the S. limacinum SR21 genome were assembled into 63 Mb, consisting of 52 contigs. The contig N50 is 2.67 Mb, and the longest contig 4,02 Mb (Figure 2 and Table 1) based on the TGS of the PacBio SEQUEL platform. The GC ratio of sequence reads was 49.9%. We further utilized the Benchmarking Universal Single-Copy Orthologs (BUSCO) software to examine the completeness of our present assembly. The results indicated that the assembled genome is of good quality (89.1% completeness) (Supplementary Table S5).
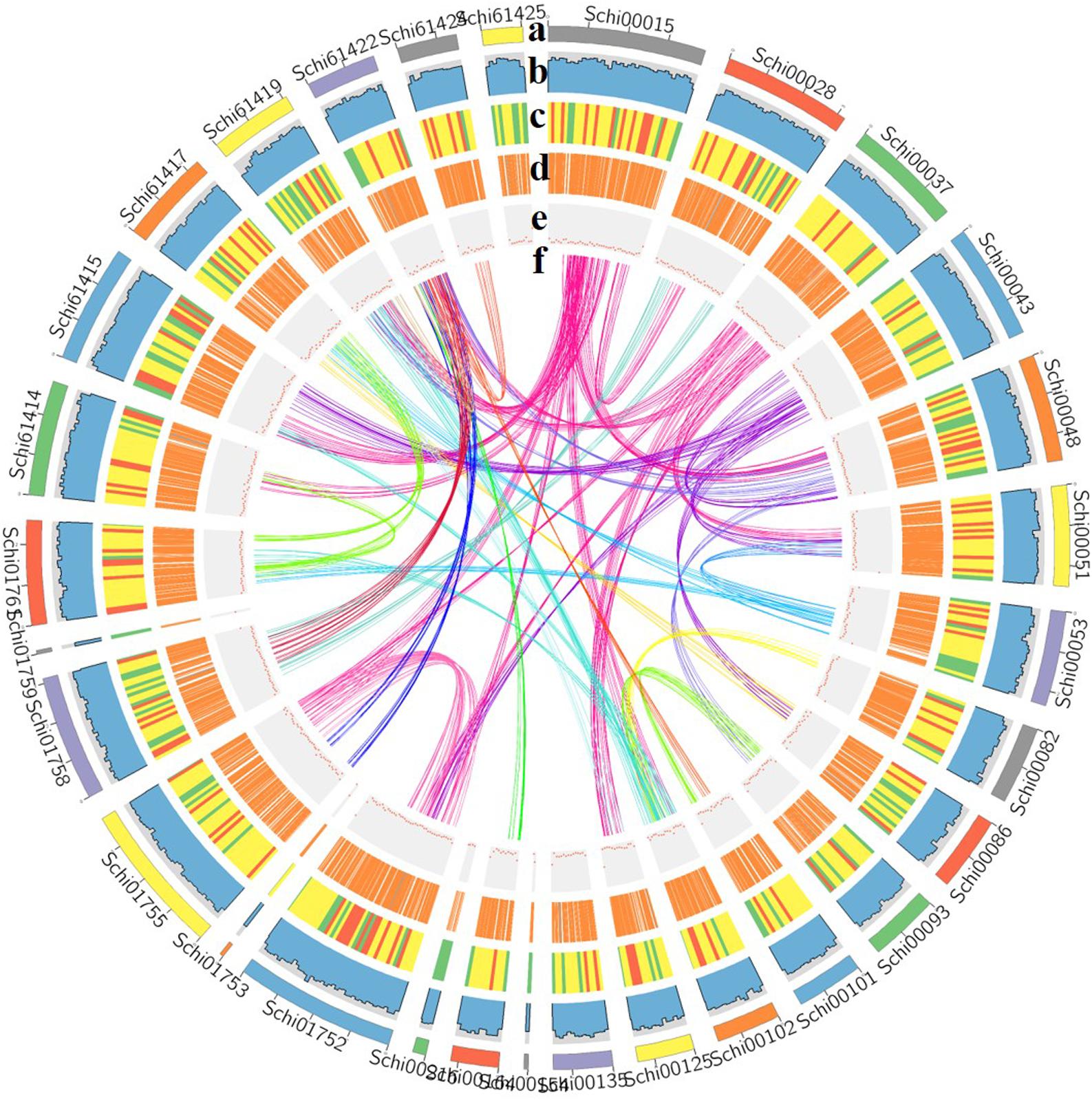
Figure 2. Characterization of the genome [(a) assembled contigs; (b) GC content; (c) gene density; (d) expression values of root- and shoot-expressed genes; (e) percent coverage of TEs in non-overlapping windows; (f) relationship between syntenic blocks].
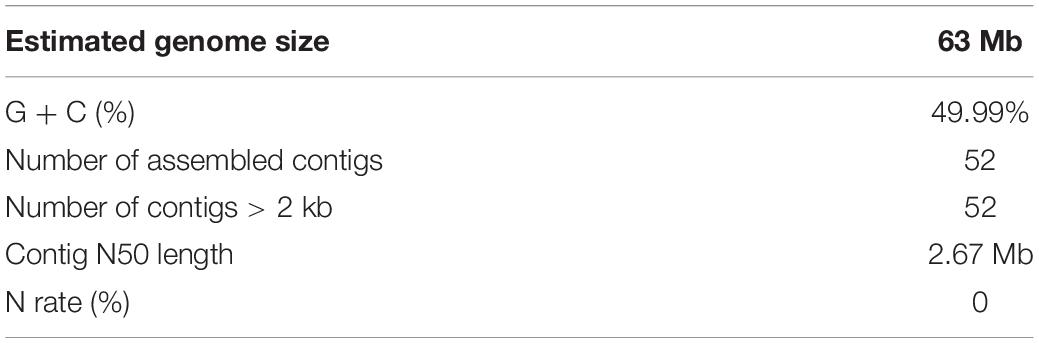
Table 1. Schizochytrium limacinum SR21 genome statistics.
A combination of reference plant protein homology support, transcriptome data, and ab initio gene prediction were used to generate all gene models. All gene models were merged, and redundancy was removed by MAKER, leading to a total of 6,838 protein-coding genes, with an average length of 2.4 kb. The NCBI non-redundant protein (nr) database with an e-value threshold of 1e-5 was used for functional annotation for protein-coding genes using BLASTX and BLASTN. The Blast2GO package was applied to assign ontology and pathway information to protein-coding genes using Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG). Finally, we functionally annotated 6502, 5822, 3692, 5822, and 4341 genes to nr, EggNOG, GO, COG, and KEGG, respectively, leading to 6508 (95.17% of the total) genes with at least one hit in a public databases (Table 2). In total, 482 transcription factors were identified in the S. limacinum SR21 genome, and these genes were classified into 19 families, including 316 protein kinase family proteins, 50 MYB and 42 WD-40 repeat family proteins (Supplementary Table S6).
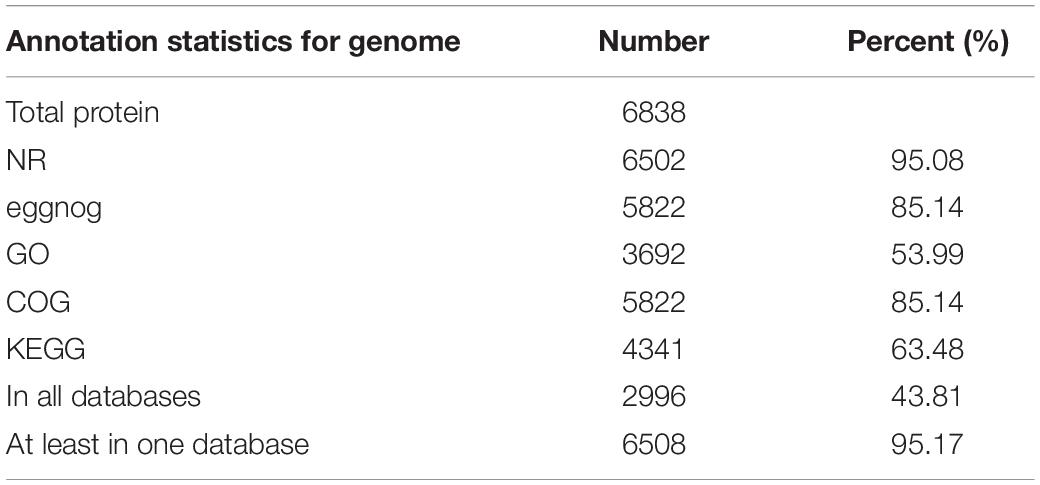
Table 2. Overview of genome annotation.
The complete FAS genes in map00062 and map01212 were annotated, and the function of these genes enables the synthesis of fatty acids and the accumulation of DHA in S. limacinum SR21, including 3-ketoacyl-CoA synthase (KCS), 3-oxoacyl-CoA reductase (KAR), 3-hydroxyacyl-CoA dehydratase (HS1), enoyl-CoA reductase (ER), acyl-coenzyme A thioesterase (ACOT), 3-hydroxyacyl-CoA dehydrogenase (HADH), acetyl-CoA acyltransferase (ACAA), enoyl-CoA hydratase (ECHS), acyl-CoA dehydrogenase (ACDH),Δ-4 desaturase, Δ-5 desaturase, Δ-1 elongase, Δ-3 elongase, Δ-4 elongase, and Δ-6 elongase. Furthermore, we identified a gene cluster and 10 ORFs related to PKS pathway containing domains with homology to those in Shewanella pneumatophori (GenBank accession number U73935.1.), Schizochytrium sp. ATCC_20888 (GenBank accession number AF378327, AF378328, AF378329) and Moritella (GenBank accession number AB025342.1). The gene cluster (4,475 amino acids) of S. limacinum SR21 included typical PKS related domain, which contains the following domains:3-ketoacyl synthase (KS), malonyl-CoA acyltransferase (MAT), acyl carrier proteins (ACP), 3-ketoacyl-ACP reductase (KR), and dehydrase (DH) (Table 3).
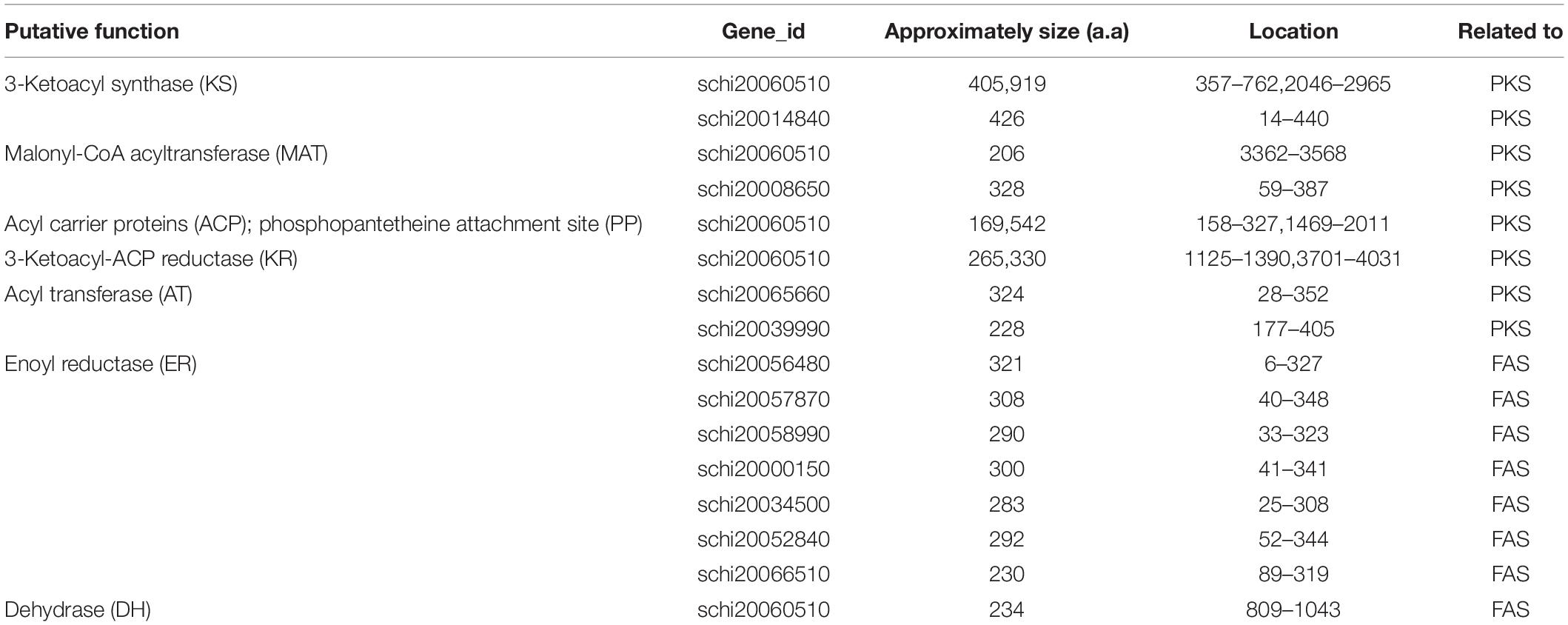
Table 3. Summary of sequence analysis data of Schizochytrium limacinum SR21 genes encoding enzymes of FAS and PKS. a.a., amino acid.
Annotation of Non-coding RNA (ncRNA)
We identified rRNA, tRNA, and snRNA genes in the S. limacinum SR21 genome by searching the Rfam database using BlastN with an E-value ≤ 1e-5 and predicted tRNAs and rRNAs by tRNAscan-SE and RNAmmer, resulting in an S. limacinum SR21 genome with 536 tRNAs, 339 rRNAs, 1 sRNA, 1 lncRNA, 1 ribozyme, and 9 snRNAs (Supplementary Table S7).
Repeat Element (TE) Annotation
Repetitive sequences of the S. limacinum SR21 genome accounted for 12.47% of the assembled genome. Long terminal repeat (LTR) retrotransposons accounted for 1.83% of the genome, including 0.30% Ty1/copia, 0.10% Ty3/gypsy, and 1.44% other. The tandem repeats finder identified over 44,073 tandem repeats, accounting for 4.86% of the S. limacinum SR21 genome (Supplementary Table S8).
Evolution of the S. limacinum SR21 Genome and Gene Family Analysis
We performed comparative genomic analyses among 14 species and detected 22,858 families of homologous genes, and among them, 1,107 gene families were common. Furthermore, 186 of the 1,107 common gene families contained one copy in each plant species. These 186 single-copy orthologous genes were used to construct the phylogenetic tree (Figure 3). The results confirmed that the strains S. limacinum SR21, S. aggregatum-ATCC28209, A. limacinum ATCC-MYA-1381 and H. fermentalgiana clustered together on the phylogenetic tree. A total of 3,931 gene families were identified in the S. limacinum SR21, among which 41 and 3,890 gene families showed expansion and contraction, respectively. A total of 3,890 genes in the contracted families were annotated to KEGG pathways (Supplementary Table S9) and GO terms (Supplementary Table S10), respectively. KEGG analysis found that most of the contracted gene families were clustered in signal transduction, lipid metabolism, environmental adaptation, metabolism of terpenoids and polyketides. GO analysis showed that the expanded orthogroups were related to biological regulation, catalytic activity, developmental process, metabolic process, stimulus response, signaling, and reproductive process.
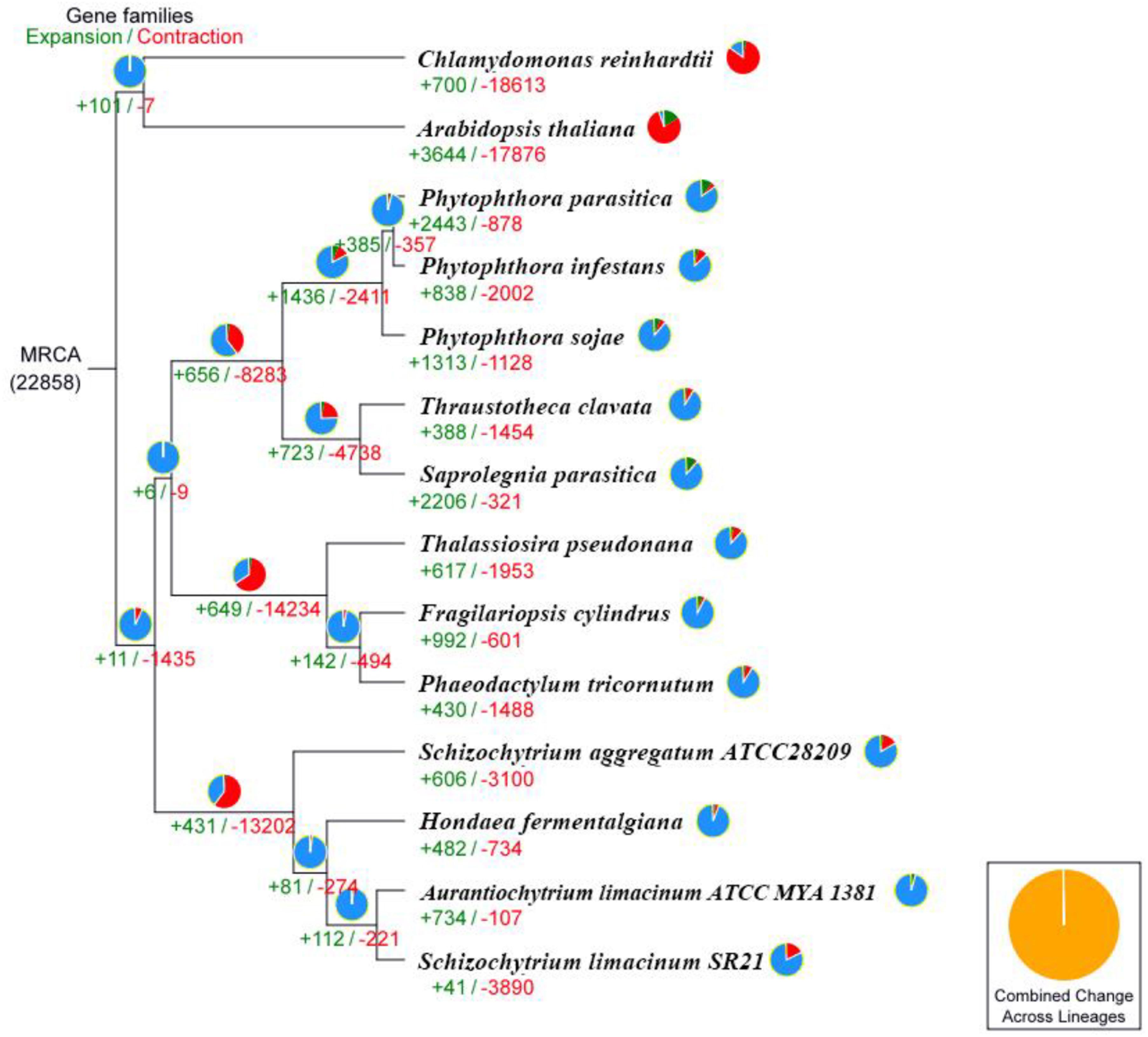
Figure 3. Phylogenetic tree and number of gene families displaying expansion (green) and contraction (red) among 14 plant species. The pie charts show the expanded (green), contracted (red), and conserved (blue) gene family proportions among all gene families. MRCA, most recent common ancestor.
Compared with S. limacinum SR21, A. limacinum, C. reinhardtii, P. tricornutum, and S. aggregatum -ATCC28209, 1936 (33.36%) of 5,803 S. limacinum SR21 gene families were shared by the five species, whereas 224 gene families were unique to S. limacinum SR21 (Figure 4). The 244 unique families are novel gene families in S. limacinum SR21 during long history of evolution. Some of them may be lost in other species, while we believe that there are gene families de novo originated in Schizochytrium sp. GO enrichment analysis of these 224 unique families showed enrichment of immune system processes, biological regulation, metabolic processes, developmental processes and reproductive processes (Supplementary Table S11). KEGG analysis showed enrichment of fatty acid biosynthesis, fatty acid degradation, nitrogen metabolism and metabolic pathways (Supplementary Table S12).
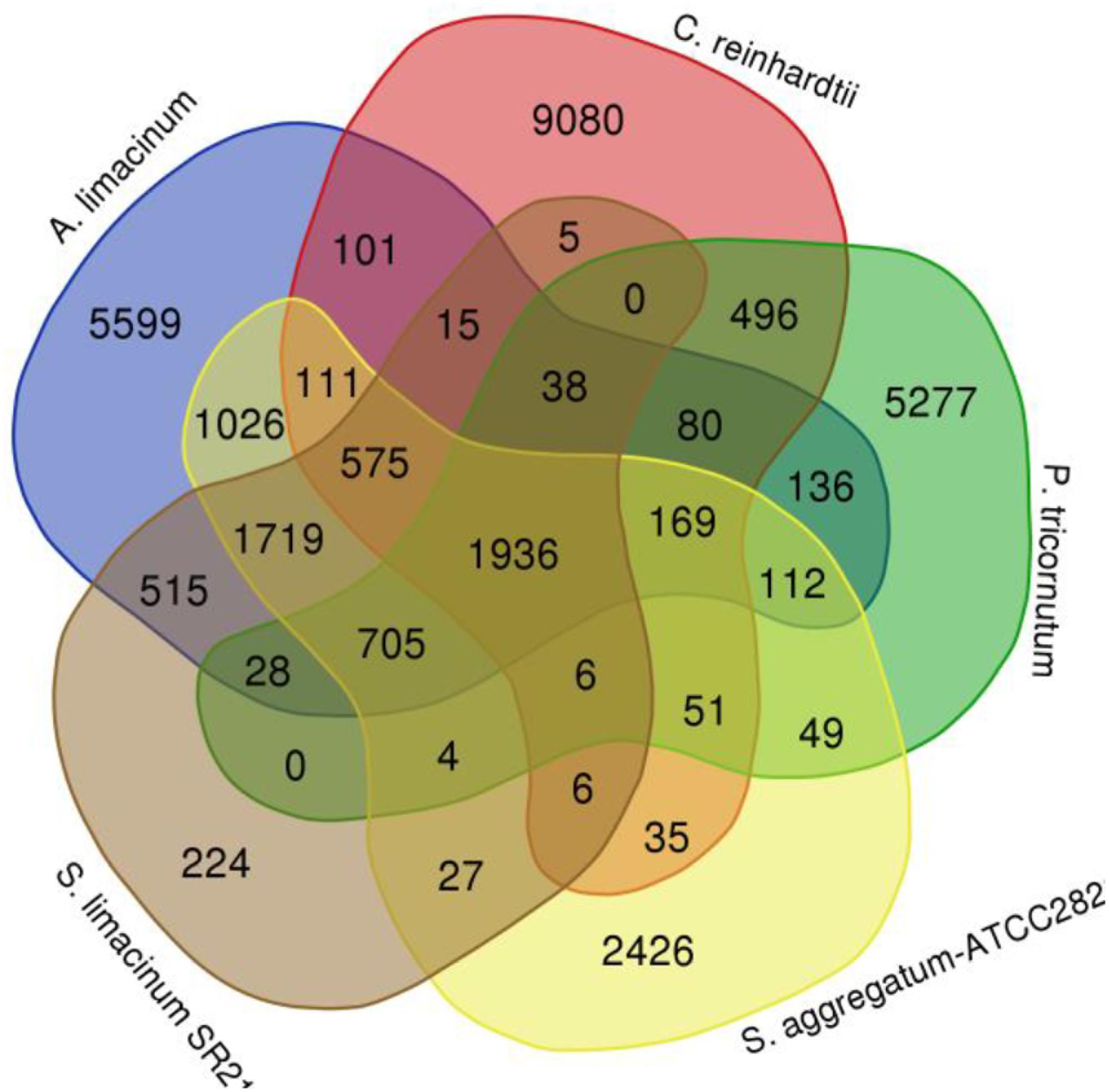
Figure 4. Venn diagram of gene families in Schizochytrium limacinum SR21 and four other species.
Regarding LC-PUFA synthesis, it is mostly thought that polyunsaturated fatty acids in Schizochytrium sp. are synthesized by the FAS and PKS pathway. The desaturation-elongation proteins constitute one of the largest families of transcription factors and are involved in the regulation of the desaturation and elongation of polyunsaturated fatty acids in the FAS pathway. We identified 6 elongase and 5 desaturase subfamilies in these five species, including Δ-4 desaturase, Δ-5 desaturase, Δ-1 elongase, Δ-3 elongase, Δ-4 elongase, Δ-6 elongase, acyl-ACP, acyl-CoA, acyl-MGDG, and acyl-phospholipid. The numbers of the desaturation-elongation gene family of S. limacinum SR21 were much less than those of other species (Figures 5A,B).
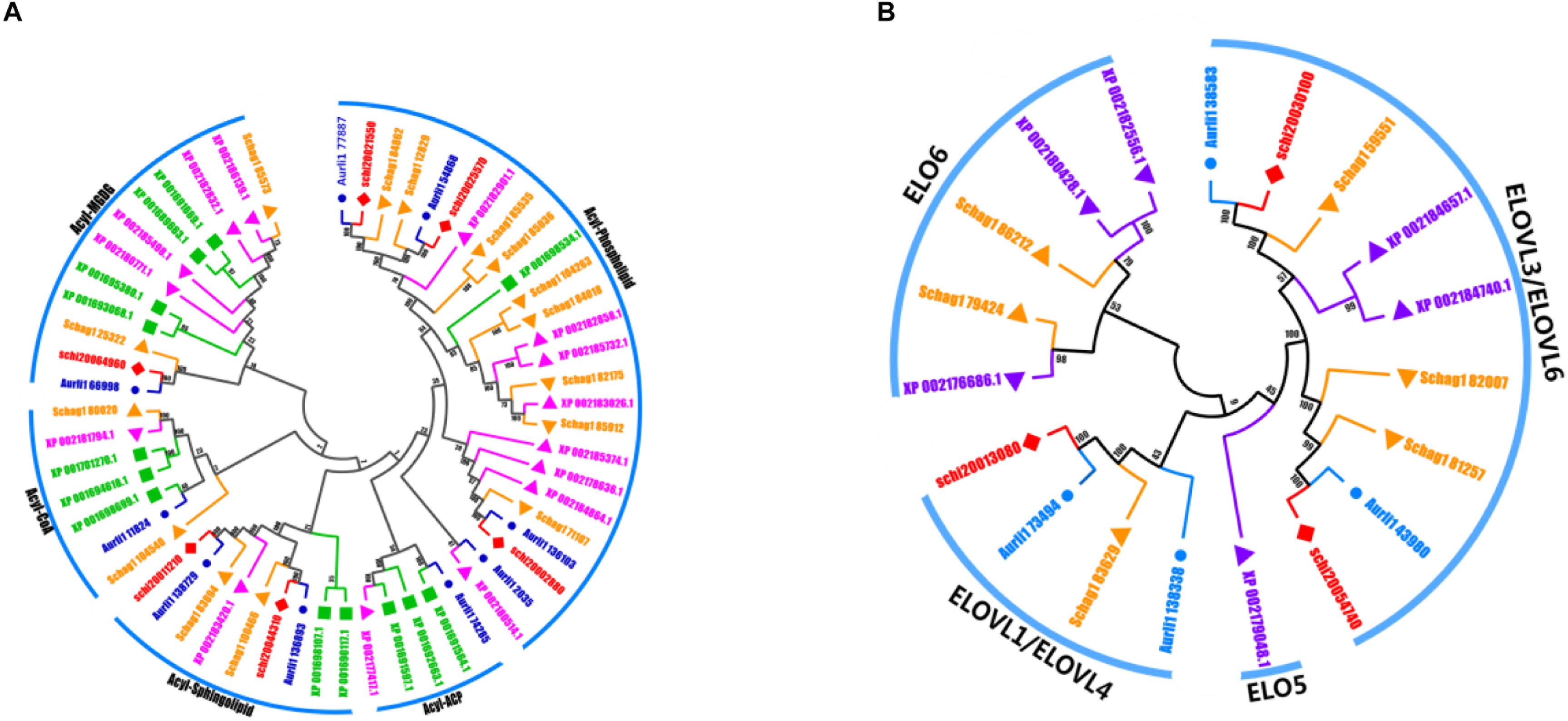
Figure 5. (A) The evolutionary tree and expression values of the desaturation gene family. (B) The evolutionary tree and expression values of the elongation gene family. A. limacinum (Auril), S. limacinum SR21 (schi), P. tricornutum (XP), and S. aggregatum (Schag).
Transcriptome Profiling and Differentially Expressed Genes
Of a total of 6,834 annotated genes, 6,632 (97.04%) genes were expressed in at least in 3 samples with a read count per million (CPM) value larger than 1. The extremely low rate (2.96%) of filtered out genes indicated that gene expression was efficiently detected across all time points. To obtain an overview of the transcriptome profile, principal component analysis (PCA) was performed by normalized log10(FPKM + 1) values. The first principal component (PC1), which explains 49.09% of the total variance, shows clearly different gene expression profiles between the first two early stages (12 and 24 h) and other stages (Figure 6A). The samples from the first two time points were assembled into a cluster that was distinguished from other samples, and some genes were expressed at the highest level at 24 h via the heatmap of gene expression (Figure 6B). Furthermore, to obtain insight into the DEGs involved in DHA biosynthesis and specific responsiveness to the treatment, DEGs of six pairwise comparisons between control and treatment at the same time point (control 12 h vs. treated 12 h, control 24 h vs. treated 24 h, etc.) were focused on (Figure 6C). Among these comparisons, the difference between the two transcriptomes of the control and treatment was greatest at 36 h, especially with respect to downregulated expression, indicating that 36 h might be a key time point in response to DHA biosynthesis. This result was consistent with the phenotype performance of DHA and fatty acid.
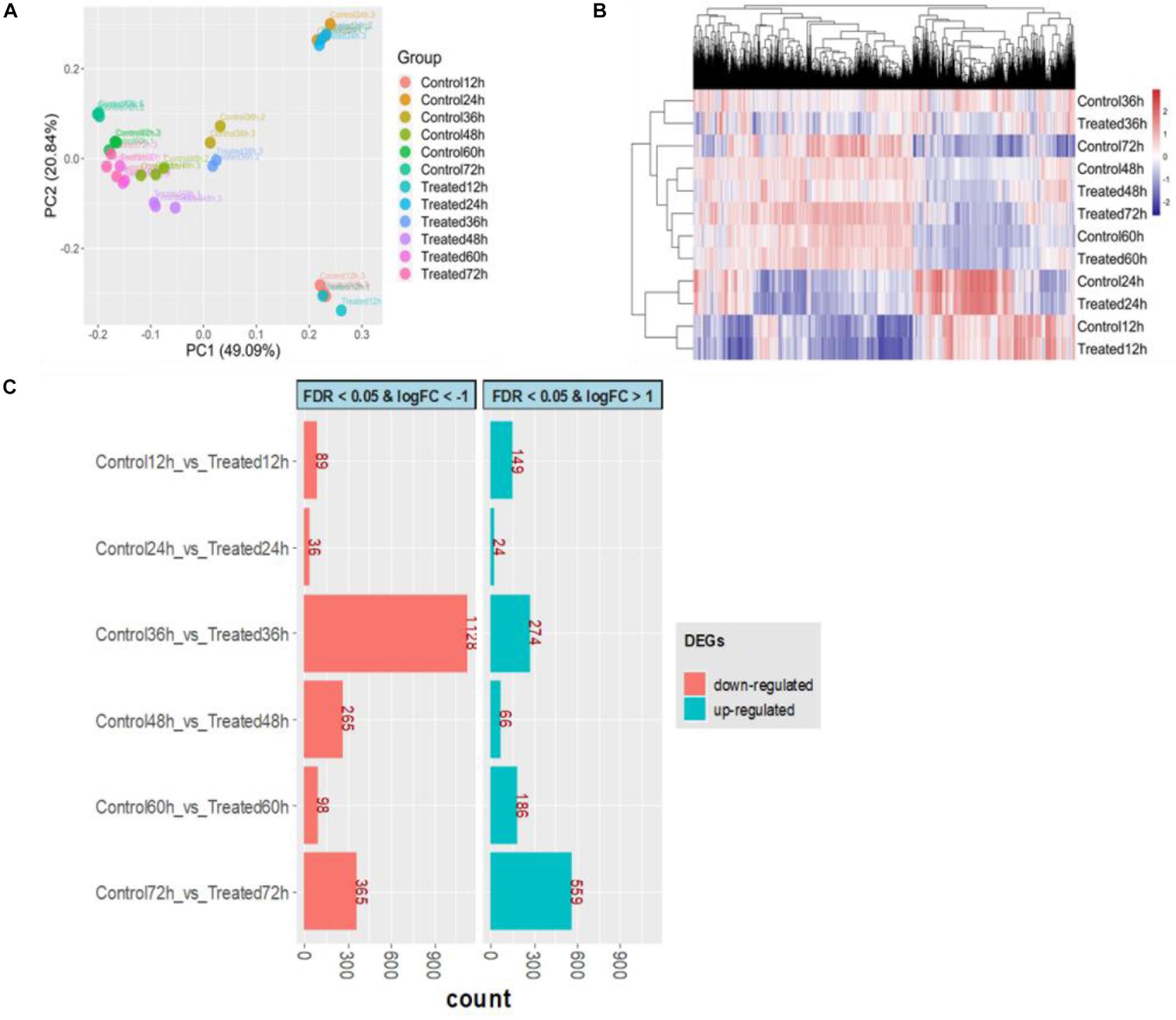
Figure 6. Overview of transcriptional analysis between control and treatment. (A) Principal component analysis for all samples. (B) Hierarchical clustering for transcriptome profile. (C) The number of differentially expressed genes (DEGs); red represents downregulated, and green represents upregulated. The control groups (Control12h, Control24h, Control36h, Control48h, Control60h, Control72h) represent that S. limacinum SR21 was cultured in basal fermentation medium with 1.0 g/L yeast extract at 12, 24, 36, 48, 60, and 72 h, respectively. The treated groups (Treated12h, Treated24h, Treated36h, Treated48h, Treated60h, Treated72h) represent that S. limacinum SR21 was cultured in basal fermentation medium with 0.5 g/L yeast extract at 12, 24, 36, 48, 60, and 72 h, respectively.
By comparing with the control 36-h group, we identified 1,402 DEGs (log2 FC ≥ | 1| and FDR ≤ 0.05) in the treated 36-h group, with 274 upregulated and 1,128 downregulated (Supplementary Table S13). Forty-two transcription factors were identified in these DEGs and were classified into 17 families, including 14 MYB, 5 WRKY, 2 C3H, and 2 C2H2 (Supplementary Table S14). GO term enrichment analysis results varied from GO classification and expression changes in DEGs. In biological processes, the upregulated DEGs were significantly enriched in biosynthetic processes, metabolic processes, stimulus responses and phosphorylation, and the downregulated DEGs were significantly enriched in protein modification processes, signal transduction and protein metabolic processes (Supplementary Table S15). KEGG pathway analysis found that 76 upregulated DEGs and 16 downregulated DEGs were enriched in the biosynthesis of secondary metabolites (Supplementary Table S16).
Gene Coexpression Network Involved in Fatty Acid and DHA Accumulation
The gene coexpression network constructed by WGCNA provides a systems biology approach to understand the gene networks instead of individual genes. Thus, WGCNA was adopted in this study to identify modules representing functional categories. According to the scale-free topology model fit and mean connectivity, a soft threshold power β was set to 20 in our study, which produced an approximate scale-free network with appropriate mean connectivity. After screening, a total of 36 samples and 2257 DEGs were used for coexpression network construction, and 7 modules with module sizes named black (75 genes), blue (535 genes), brown (194 genes), green (104 genes), red (80 genes), turquoise (1,020 genes), and yellow (155 genes) were generated (Figure 7A). Ninety-four genes were outside of those nine modules and are labeled as the gray module. Notably, analysis of the module-trait relationships revealed that the yellow and brown modules were identified as significantly highly expressed and were relevant and consistent with the results of phenotype performance of DHA and fatty acids, especially the yellow module (Figure 7B). The yellow module, DHA and fatty acid were clustered together and were highly positively related (Figure 7C). As shown in the Supplementary Table S17, a total of 9 TFs were annotated in the yellow module, and several TFs highly positively correlated with fatty and DHA accumulation were found among these 6 very-long-chain fatty acid biosynthesis regulatory genes, including three protein kinase family proteins (schi20066080, schi20050050, and schi20028430), 2 MYB (schi20061500 and schi20062190) and 1 Zinc Finger (schi20050340). According to GO enrichment analysis, unigenes in the content-related yellow module were enriched in different metabolic pathways, including lipid oxidation, lipid catabolic processes, fatty acid metabolic processes and lipid biosynthetic processes (Supplementary Table S18). KEGG enrichment analysis in the yellow module was performed to identify the key genes and pathways closely related to fatty acid and DHA accumulation (Supplementary Table S19). As we can see in the Supplementary Table S20, DEGs in the yellow module were significantly enriched in the biosynthesis of secondary metabolites, pyruvate metabolism, fatty acid degradation, and alpha-linolenic acid metabolism.
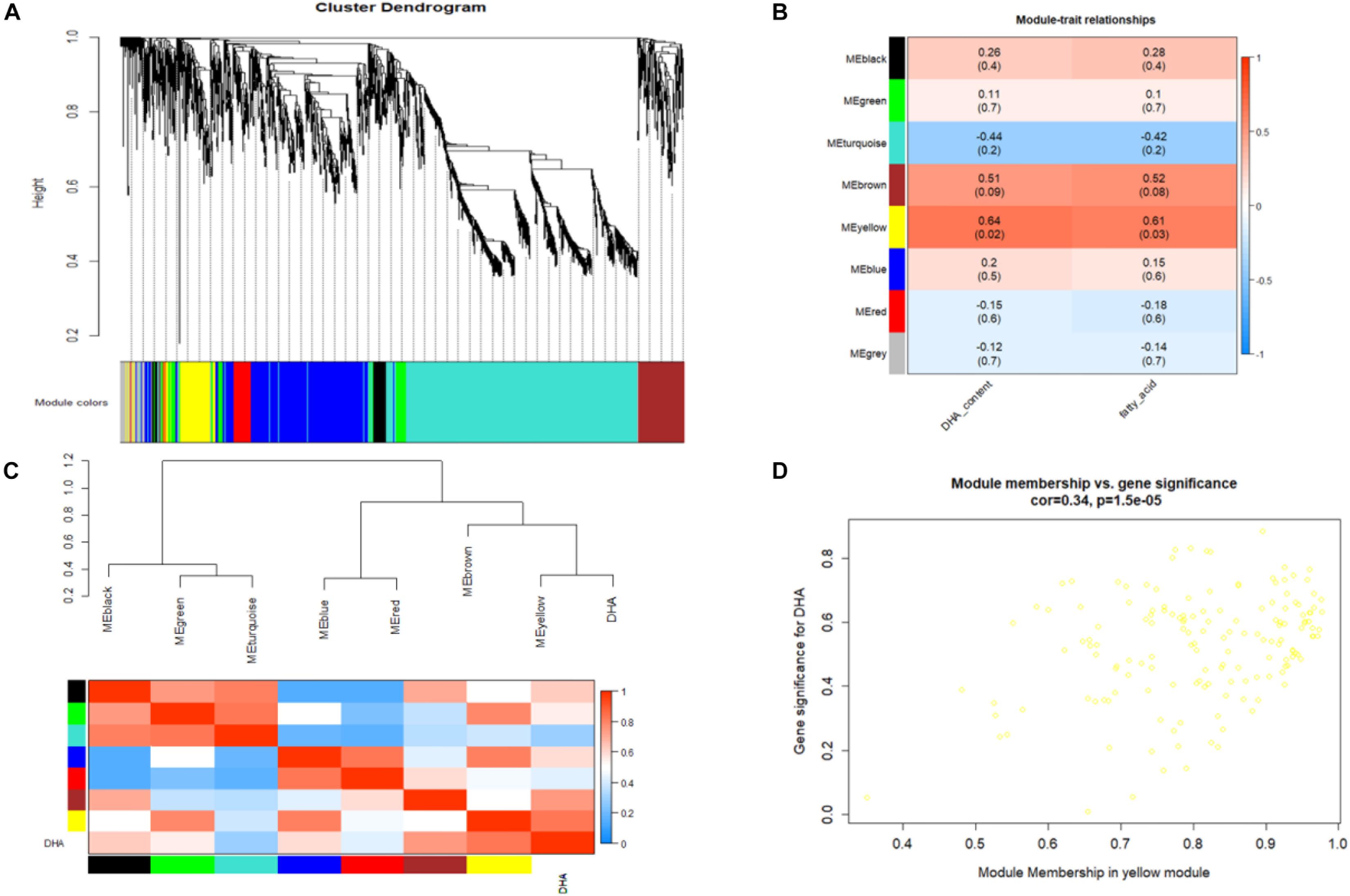
Figure 7. (A) Cluster dendrogram and module assignment from WGCNA. Each color represents a certain gene module. (B) Heatmap of the correlation of WGCNA modules with traits. Red is the correlation between module and trait, and blue is a negative correlation. The module highlighted with dark color represents a significant module associated with traits. (C) The eigengene dendrogram and heatmap for modules with DHA. (D) Correlation between significant genes and module membership in the yellow module.
Analysis of Hub Genes and qRT-PCR Validation
Figure 7D shows that the intramodular connectivity and module membership have a high positive linear correlation in the yellow module. In general, a high module membership corresponds to a high intramodular connectivity. Genes with both of these properties may be important candidate hub genes. In order to identify the hub genes that well-represent the yellow module, we analyzed the module in further detail by GO and KEGG pathway (Supplementary Figure S1). The yellow module has 155 total genes, and the top 30 genes are listed in the Supplementary Table S20 and Supplementary Figure S2. Hub gene analysis identified acyl-CoA oxidase (schi20049930) and N-ethylmaleimide reductase (schi20023280) in the yellow module, which are involved in fatty acid beta-oxidation, very-long-chain fatty acid metabolic processes and oxidation-reduction processes. In order to confirm the accuracy of unigene expression levels, three unigenes from the yellow module and RNA-Seq data were selected for qPCR analysis, and their relative expression levels were compared with FPKM values from RNA-Seq data. The results showed that the expression of all three unigenes measured by qPCR was consistent with the RNA-Seq data (Supplementary Figure S3).
Discussion
Four DHA-producing thraustochytrid strains (Schizochytrium sp. CCTCC M209059, Aurantiochytrium sp. T66, Schizochytrium sp. Mn4, and Thraustochytriidae sp. SW8) have been produced based on next-generation sequencing platforms or a small number of PacBio RS (Ji et al., 2015; Liu et al., 2016; Song et al., 2018). However, short sequencing reads typically cannot span highly repetitive segments of genomes and be assembled into sets of small contigs (scaffold N50 127 kb to 1.3 Mb). In order to produce a high-quality genome, we generated a draft genome assembly with 63 Mb in total length and 52 contigs (>2,000 bp) with a high contig N50 of 2.67 Mb. A large number of protein-coding genes (6,838) was predicted by the gene models built with de novo, homology-based, and experimental data obtained from transcription results. These findings indicated that a high-quality S. limacinum SR21 genome was generated, which provided a valuable reference for understanding the molecular synthesis and regulatory networks for PUFA biosynthesis in Schizochytrium sp.
Based on the concatenated sequence alignment of S. limacinum SR21 and 13 other species, the strains S. limacinum SR21, S. aggregatum-ATCC28209, A. limacinum ATCC-MYA-1381 and H. fermentalgiana clustered together on the phylogenetic tree. A total of 3,931 gene families were identified in the S. limacinum SR21, and the number of contracted gene families (3,890) is significantly greater than that of expanded gene families (41). KEGG and GO analyses showed that the gene families involved glycerophospholipid metabolism, glycerolipid metabolism, cutin, suberine, and wax biosynthesis have significantly contracted in the S. limacinum SR21 genome, which is not directly related to fatty acid biosynthesis.
LC-PUFAs can be synthesized by fatty acid synthases pathway (FAS) and polyketide synthases pathway (PKS). Currently, the two major steps in fatty acid biosynthesis were clarified as elongation and desaturation carried out by an elongase and desaturase in the FAS pathway (Morais et al., 2015). Lippmeier et al. (2009) found Δ-5, Δ-6 and Δ-9 elongase activities in Schizochytrium sp. ATCC20888 without the present of Δ-12 desaturation. Ren et al. (2017), detected one elongase and three kinds (Δ-6, Δ-8, and Δ-12) of desaturase activities in Schizochytrium sp. The missing of some specific enzymes might be the reason of the incomplete genomic information. In our research, we identified two unigenes encoding desaturase (Δ-4 desaturase, Δ-5 desaturase) and four unigenes encoding elongase protein (Δ-1 elongase, Δ-3 elongase, Δ-4 elongase, Δ-6 elongase), and the all genes needed for FAS pathway in map00062 and map01212 were annotated (Supplementary Figure S4). Similar to Lippmeier’s results, we also find Δ-6 elongase which catalyzes C18:4 to C20:4, indicating Schizochytrium was able to convert SDA to ARA. Different from Lippmeier’s and Ren’s results, we annotated Δ-4 desaturase which coverts C22:5 to C22:6, indicating Schizochytrium was able to catalyze DPA to DHA and played important role in the fatty acid synthesis. Maybe it is the reason why Schizochytrium sp. can accumulate rich LC-PUFAs and DHA. However, Schizochytrium sp. is considered as the best natural resource for LC-PUFAs and more than 50% lipid rich in DHA. The questions remain as to how to accumulate rich LC-PUFAs and promote DHA production with the relatively low numbers of desaturase and elongase gene families in S. limacinum SR21 compared with other four species. We assumed that the gene members of these families have stronger catalytic capabilities or that there are some strong transcriptional regulators that regulate the ability of these functional genes to enhance fatty acid synthesis.
Metz (2001), identified 11 domains from Schizochytrium sp. by comparing with Shewanella sp. domains, predicted eight gene products, closely related to the PKS protein domain (Metz, 2001). The prokaryotic Shewanella sp. and eukaryotic Schizochytrium sp. genes have high homology, and the gene structure and functional regions are similar. In PKS pathway for LC-PUFAs synthesis, the pathway catalyzed by polyketide does not require desaturation and elongation of saturated fatty acids, the synthases for PKS are totally different from PKS in both structure and mechanism. They concluded that PUFA synthesis in Schizochytrium sp. is accomplished in part by these PKS enzymes. Until now, although the related domains of the PKS pathway have been cloned and annotated in the comparison with the related domains of the PKS pathway of bacteria, the fatty acid biosynthesis has not been still clarified whether through the FAS or PKS pathway mainly in Schizochytrium sp. We identified one gene cluster and 10 ORFs related to PKS pathway containing domains with homology to those in Shewanella pneumatophori, Schizochytrium sp. ATCC_20888, and Moritella. The gene cluster (4,475 amino acids) of S. limacinum SR21 included typical PKS related domain, including 3 KS, 1 MAT, 6 ACP, 2 KR, and 1 DH (Figure 8). In addition, we also annotated some domains related to PKS and FAS in the genome of S. limacinum SR21, including 1 KS, 7 ER, 2 AT, and 1 MAT (Table 3). These genes are individually distributed on the genome, not in cluster. And the function of these genes needs to be figured out.
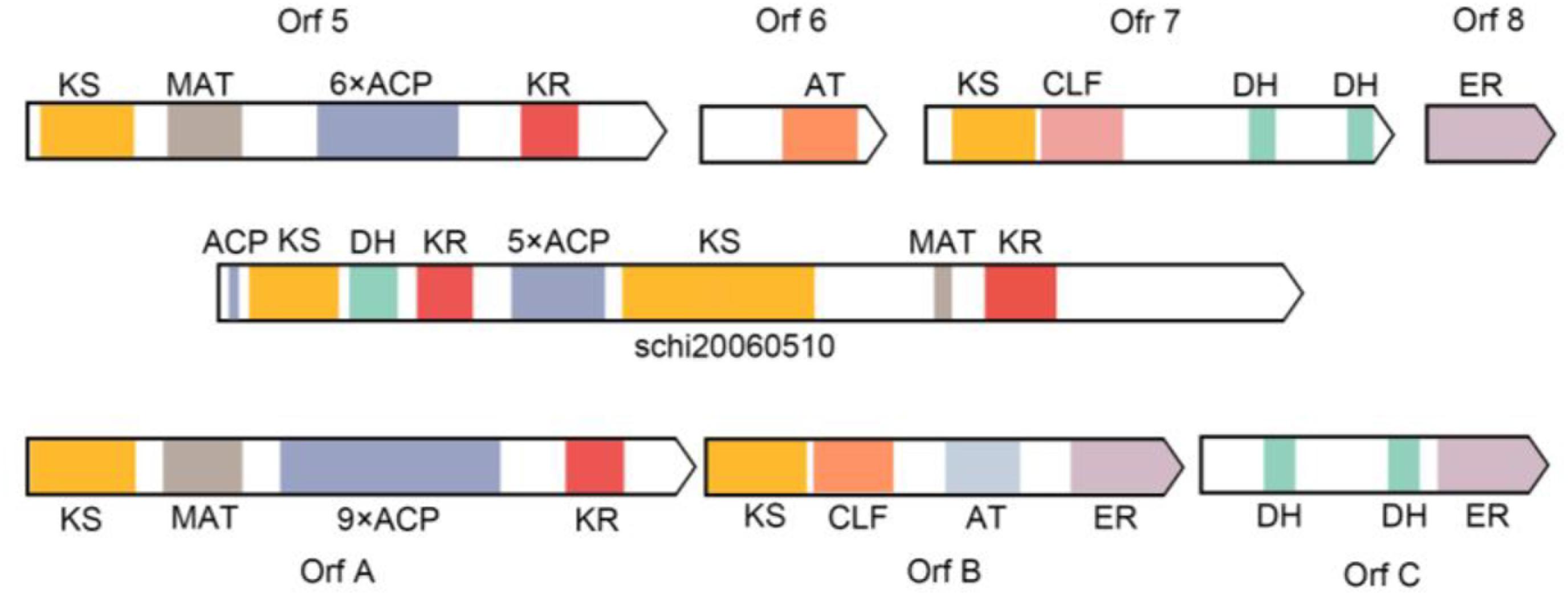
Figure 8. Comparison of putative PKS enzyme domains in Shewanella pneumatophori (GenBank accession number U73935.1.), Schizochytrium sp. ATCC_20888 (GenBank accession number AF378327, AF378328, AF378329) and Schizochytrium limacinum SR21 (schi20060510). 3-ketoacyl synthase (KS), malonyl-CoA acyltransferase (MAT), acyl carrier proteins (ACP), 3-Ketoacyl-ACP reductase (KR), enoyl reductase (ER), and dehydrase (DH).
Yeast extract starvation are the premise of lipid production and DHA accumulation. In our previous study, the total fatty content and DHA production of S. limacinum SR21 drastically increased from 12 to 36 h in basal fermentation medium containing 1.0 or 0.5 g/L yeast extract, respectively. The R2R3-type MYB96 transcription factor is a pivotal regulator of fatty acid elongation, which regulates cuticular wax accumulation under drought conditions in Arabidopsis leaves (Seo et al., 2011). Lee H. G. et al. (2015), reported that MYB96 could directly regulate fatty acid elongation to stimulate accumulation of eicosenoic acid in Arabidopsis seed maturation and development. In our study, most of DEGs were downregulated after nitrogen limitation at 36 h and 42 transcription factors were identified and classified into 17 families in these DEGs, including 14 MYB, 5 WRKY, 2 C3H, and 2 C2H2. A weighted gene coexpression network analysis revealed that 2 of 7 modules correlated highly with the fatty acid and DHA contents, and DEGs and transcription factors were significantly correlated with fatty acid biosynthesis, including MYB, Zinc Finger and ACOX. AtMYB12, AtMYB111, AtMYB11, and MdMYB22 have been shown to be involved in flavonoid biosynthesis and anthocyanin biosynthesis in Arabidopsis, tobacco and apple (Park et al., 2008; Lee K. et al., 2015; Tian et al., 2017). These studies suggest that the identified TFs may be involved in nitrogen limitation-induced accumulation and regulation of LC-PUFAs and DHA production in S. limacinum SR21, likely working together with a transcription factor complex. Interestingly, several studies have suggested a close relationship between fatty acid and TAG synthesis. ACOX (acyl-CoA oxidase) catalyzes the first step in the pathway of peroxisomal fatty acid beta-oxidation, which is part of lipid metabolism, catalyzing the desaturation of acyl-CoAs to 2-trans-enoyl-CoAs (Kim and Kim, 2018). Here, the WGCNA distributed the ACOX1 gene from the fatty acid biosynthetic pathway into the yellow module. Furthermore, qRT-PCR confirmed that ACOX1 was strongly induced by nitrogen limitation treatment. We also found that these DEGs and hub genes are involved in the encoding enzymes of PKS pathway based on the results of transcriptome and weighted gene coexpression network analysis. Therefore, we hypothesize that nitrogen limitation can accumulate DHA and fatty acid contents by activating the fatty acid beta-oxidation process in FAS pathway. The results can provide a basis for distinguishing which pathways of fatty acid biosynthesis is mainly achieved through, and we can regulate FAS pathways to increase the production of fatty acids and DHA in Schizochytrium sp. The results should help improve the accumulation of fatty acid and DHA in Schizochytrium sp. and other microalgae, providing a valuable reference for industrial applications.
Data Availability Statement
The whole genome sequence data reported in this article have been deposited in the Genome Warehose in National Genomics Data Center, Beijing Institute of Genomics (BIG), Chinese Academy of Sciences, under accession number GWHABLD00000000 that is publicly accessible at https://bigd.big.ac.cn/gwh. Raw sequencing data for RNA-seq was used for annotation and biological analyses, and have also been deposited in BIG Sub system under BioProject accession number PRJCA002397 (http://bigd.big.ac.cn).
Author Contributions
YC and TX designed and coordinated the entire project. TX, YC, WT, and JZ together led and performed the entire project. LL, XZ, and ZH performed the collection and processing of samples. TX, XZ, WF, DC, JP, and WT performed the analyses of genome evolution and gene family analyses. TX, LL, XZ, DC, and YC participated in manuscript writing and revision. All authors read and approved the final manuscript.
Funding
This work was supported by the Natural Science Foundation of Fujian Province, China (Grant Number 2017J01622) and the Sugar Crop Research System (Grant Number CARS-170501).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer FQ declared a shared affiliation, with no collaboration, with the authors to the handling editor at the time of review.
Acknowledgments
This manuscript was edited for proper English language by the highly qualified native English-speaking editors at American Journal Experts.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.00687/full#supplementary-material
Footnotes
- ^ https://horvath.genetics.ucla.edu/html/CoexpressionNetwork/Rpackages/WGCNA/Tutorials/
- ^ http://www.omicshare.com/tools
- ^ http://www.omicshare.com/tools
References
Ambati, R. R., Phang, S. M., Ravi, S., and Aswathanarayana, R. G. (2014). Astaxanthin: sources, extraction, stability, biological activities and its commercial applications–a review. Mar. Drugs. 12, 128–152. doi: 10.3390/md12010128
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene ontology: tool for the unification of biology. Nat. Genet. 25, 25–29. doi: 10.1038/75556
Aury, J. M., Jaillon, O., Duret, L., Noel, B., Jubin, C., Porcel, B. M., et al. (2006). Global trends of whole-genome duplications revealed by the ciliate Paramecium tetraurelia. Nature 444, 171–178. doi: 10.1038/nature05230
Borecka-Melkusova, S., Moran, G. P., Sullivan, D. J., Kucharikova, S., Chorvat, D. Jr., and Bujdakova, H. (2009). The expression of genes involved in the ergosterol biosynthesis pathway in Candida albicans and Candida dubliniensis biofilms exposed to fluconazole. Mycoses 52, 118–128. doi: 10.1111/j.1439-0507.2008.01550.x
Browning, L. M., Walker, C. G., Mander, A. P., West, A. L., Gambell, J., Madden, J., et al. (2014). Compared with daily, weekly n-3 PUFA intake affects the incorporation of eicosapentaenoic acid and docosahexaenoic acid into platelets and mononuclear cells in humans. J. Nutr. 144, 667–672. doi: 10.3945/jn.113.186346
Brussaard, C. P., Thyrhaug, R., Marie, D., and Bratbak, G. (1999). Flow cytometric analyses of viral infection in two marine phytoplankton species, Micromonas pusilla (Prasinophyceae) and Phaeocystis pouchetii (Prymnesiophyceae). J. Phycol. 35, 941–948. doi: 10.1046/j.1529-8817.1999.3550941.x
Cristianini, N., and Demuth, J. P. (2006). CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22, 1269–1271. doi: 10.1093/bioinformatics/btl097
Elsik, C. G., Mackey, A. J., Reese, J. T., Milshina, N. V., Roos, D. S., and Weinstock, G. M. (2007). Creating a honey bee consensus gene set. Genome. Biol. 8, R13. doi: 10.1186/gb-2007-8-1-r13
Emms, D. M., and Kelly, S. (2015). OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome. Biol. 16, 157. doi: 10.1186/s13059-015-0721-2
Huda, A., and Jordan, I. K. (2009). Analysis of transposable element sequences using CENSOR and RepeatMasker. Methods. Mol. Biol. 537, 323–336. doi: 10.1007/978-1-59745-251-9_16
Ji, X. J., Mo, K. Q., Ren, L. J., Li, G. L., Huang, J. Z., and Huang, H. (2015). Genome sequence of Schizochytrium sp. CCTCC M209059, an effective producer of docosahexaenoic acid-rich lipids. Genome. Announc. 3, e819–e815. doi: 10.1128/genomeA.00819-15
Jurka, J., Kapitonov, V. V., Pavlicek, A., Klonowski, P., Kohany, O., and Walichiewicz, J. (2005). Repbase update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467. doi: 10.1159/000084979
Kagale, S., Robinson, S. J., Nixon, J., Xiao, R., Huebert, T., Condie, J., et al. (2014). Polyploid evolution of the Brassicaceae during the Cenozoic era. Plant Cell 26, 2777–2791. doi: 10.1105/tpc.114.126391
Kanehisa, M., Goto, S., Sato, Y., Kawashima, M., Furumichi, M., and Tanabe, M. (2014). Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic. Acids. Res. 42, 199–205. doi: 10.1093/nar/gkt1076
Kanzaki, N., Kiontke, K., Tanaka, R., Hirooka, Y., Schwarz, A., Muller-Reichert, T., et al. (2017). Description of two three-gendered nematode species in the new genus Auanema (Rhabditina) that are models for reproductive mode evolution. Sci. Rep. 7, 11135. doi: 10.1038/s41598-017-09871-1
Kaulmann, U., and Hertweck, C. (2002). Biosynthesis of Polyunsaturated Fatty Acids by Polyketide Synthases. Angew. Chem. Int. Edit 41, 1866–1869. doi: 10.1002/1521-3773(20020603)41:11<1866::aid-anie1866>3.0.co;2-3
Kellis, M., Patterson, N., Birren, B., Berger, B., and Lander, E. S. (2004). Methods in comparative genomics: genome correspondence, gene identification and regulatory motif discovery. J. Comput. Biol. 11, 319–355. doi: 10.1089/1066527041410319
Kim, S., and Kim, K. J. (2018). Structural insight into the substrate specificity of acyl-CoA oxidase1 from Yarrowia lipolytica for short-chain dicarboxylyl-CoAs. Biochem. Biophys. Res. Commun. 495, 1628–1634. doi: 10.1016/j.bbrc.2017.11.191
Kong, Q., Yuan, J., Gao, L., Zhao, S., Jiang, W., Huang, Y., et al. (2014). Identification of suitable reference genes for gene expression normalization in qRT-PCR analysis in watermelon. PLoS One 9:e90612. doi: 10.1371/journal.pone.0090612
Konishi, M., Nagahama, T., Fukuoka, T., Morita, T., Imura, T., Kitamoto, D., et al. (2011). Yeast extract stimulates production of glycolipid biosurfactants, mannosylerythritol lipids, by Pseudozyma hubeiensis SY62. J. Biosci. Bioeng. 111, 702–705. doi: 10.1016/j.jbiosc.2011.02.004
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Langfelder, P., and Horvath, S. (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 9:559. doi: 10.1186/1471-2105-9-559
Lee, H. G., Park, B. Y., and Kim, H. U. (2015). MYB96 stimulates C18 fatty acid elongation in Arabidopsis seeds.Plant Biotechnol. Rep. 9, 161. doi: 10.1007/s11816-015-0352-9
Lee, K., Lee, H. G., Yoon, S., Kim, H. U., and Seo, P. J. (2015). The arabidopsis MYB96 transcription factor is a positive regulator of ABSCISIC ACID-INSENSITIVE4 in the control of seed germination. Plant Physiol. 168, 677–689. doi: 10.1104/pp.15.00162
Lin, S., Cheng, S., Song, B., Zhong, X., Lin, X., Li, W., et al. (2015). The Symbiodinium kawagutii genome illuminates dinoflagellate gene expression and coral symbiosis. Science 350, 691–694. doi: 10.1126/science.aad0408
Lippmeier, J. C., Crawford, K. S., Owen, C. B., Rivas, A. A., Metz, J. G., and Apt, K. E. (2009). Characterization of both polyunsaturated fatty acid biosynthetic pathways in Schizochytrium sp. Lipids 7, 621–630. doi: 10.1007/s11745-009-3311-9
Liu, B., Ertesvag, H., Aasen, I. M., Vadstein, O., Brautaset, T., and Heggeset, T. M. (2016). Draft genome sequence of the docosahexaenoic acid producing thraustochytrid Aurantiochytrium sp. T66. Genom. Data. 8, 115–116. doi: 10.1016/j.gdata.2016.04.013
Metz, J. G. (2001). Production of Polyunsaturated Fatty Acids by Polyketide Synthases in Both Prokaryotes and Eukaryotes. Science 293, 290–293. doi: 10.1126/science.1059593
Morais, S., Mourente, G., Martinez, A., Gras, N., and Tocher, D. R. (2015). Docosahexaenoic acid biosynthesis via fatty acyl elongase and Delta4-desaturase and its modulation by dietary lipid level and fatty acid composition in a marine vertebrate. Biochim. Biophys. Acta. 1851, 588–597. doi: 10.1016/j.bbalip.2015.01.014
Mühlroth, A., Li, K., Røkke, G., Winge, P., Olsen, Y., et al. (2013). Pathways of lipid metabolism in marine algae, co-expression network, bottlenecks and candidate genes for enhanced production of EPA and DHA in species of Chromista. Mar. Drugs. 11, 4662–4697. doi: 10.3390/md11114662
Newell, M., Goruk, S., Mazurak, V., Postovit, L., and Field, C. J. (2019). Role of docosahexaenoic acid in enhancement of docetaxel action in patient-derived breast cancer xenografts. Breast Cancer Res. Treat. 177, 1–11. doi: 10.1007/s10549-019-05331-8
Nikolayeva, O., and Robinson, M. D. (2014). edgeR for differential RNA-seq and ChIP-seq analysis: an application to stem cell biology. Methods Mol. Biol. 1150, 45–79. doi: 10.1007/978-1-4939-0512-6_3
Noda, N., Kanno, Y., Kato, N., Kazuma, K., and Suzuki, M. (2004). Regulation of gene expression involved in flavonol and anthocyanin biosynthesis during petal development in lisianthus (Eustoma grandiflorum). Physiol. Plant. 122, 305–313. doi: 10.1111/j.1399-3054.2004.00407.x
Orikasa, Y., and Nishida, T. (2007). Bacterial genes responsible for the biosynthesis of eicosapentaenoic and docosahexaenoic acids and their heterologous expression. Appl. Environ. Microb. 73, 665–670. doi: 10.1128/AEM.02270-06
Park, J. S., Kim, J. B., Cho, K. J., Cheon, C. I., Sung, M. K., Choung, M. G., et al. (2008). Arabidopsis R2R3-MYB transcription factor AtMYB60 functions as a transcriptional repressor of anthocyanin biosynthesis in lettuce (Lactuca sativa). Plant Cell. Rep. 27, 985–994. doi: 10.1007/s00299-008-0521-1
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T., and Salzberg, S. L. (2016). Transcript-level expression analysis of RNA-seq experiments with HISAT. StringTie and Ballgown. Nat. Protoc. 11, 1650–1667. doi: 10.1038/nprot.2016.095
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T. C., Mendell, J. T., and Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. doi: 10.1038/nbt.3122
Ren, L., Hu, X., Zhao, X., Chen, S., Wu, Y., Li, D., et al. (2017). Transcriptomic analysis of the regulation of lipid fraction migration and fatty acid biosynthesis in Schizochytrium sp. Sci. Rep. 7, 3562. doi: 10.1038/s41598-017-03382-9
Ren, L. J., Sun, L. N., Zhuang, X. Y., Qu, L., Ji, X. J., and Huang, H. (2014). Regulation of docosahexaenoic acid production by Schizochytrium sp.: effect of nitrogen addition. Bioprocess. Biosyst. Eng. 37, 865–872. doi: 10.1007/s00449-013-1057-5
Ren, L. J., Zhuang, X. Y., Chen, S. L., Ji, X. J., and Huang, H. (2015). Introduction of omega-3 desaturase obviously changed the fatty acid profile and sterol content of Schizochytrium sp. J. Agric. Food Chem. 63, 9770–9776. doi: 10.1021/acs.jafc.5b04238
Roth, M. S., Cokus, S. J., Gallaher, S. D., Walter, A., Lopez, D., Erickson, E., et al. (2017). Chromosome-level genome assembly and transcriptome of the green alga Chromochloris zofingiensis illuminates astaxanthin production. Proc. Natl. Acad. Sci. U. S. A. 114, E4296–E4305. doi: 10.1111/j.1365-2966.2006.11067.x
Sakaguchi, K., Matsuda, T., Kobayashi, T., Ohara, J., Hamaguchi, R., et al. (2012). Versatile transformation system that is applicable to both multiple transgene expression and gene targeting for Thraustochytrids. Appl. Environ. Microbiol. 78, 3193–3202. doi: 10.1128/AEM.07129-11
Seo, P. J., Lee, S. B., Suh, M. C., Park, M. J., Go, Y. S., and Park, C. M. (2011). The MYB96 transcription factor regulates cuticular wax biosynthesis under drought conditions in Arabidopsis. Plant Cell 23, 1138–1152. doi: 10.2307/41433854
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Smith, J. A., O’Donnell, K., Mount, L. L., Shin, K., Peacock, K., et al. (2011). A novel fusarium species causes a canker disease of the critically endangered conifer. Torreya taxifolia. Plant Dis. 95, 633–639. doi: 10.1094/PDIS-10-10-0703
Sommerfeld, D., Lingner, T., Stanke, M., Morgenstern, B., and Richter, H. (2009). AUGUSTUS at MediGRID: adaption of a bioinformatics application to grid computing for efficient genome analysis. Future Gener. Comput. Syst. 25, 337–345. doi: 10.1016/j.future.2008.05.010
Song, Z., Stajich, J. E., Xie, Y., Liu, X., He, Y., Chen, J., et al. (2018). Comparative analysis reveals unexpected genome features of newly isolated Thraustochytrids strains: on ecological function and PUFAs biosynthesis. BMC Genom. 19:541. doi: 10.1186/s12864-018-4904-6
Sun, L., Ren, L., Zhuang, X., Ji, X., Yan, J., and Huang, H. (2014). Differential effects of nutrient limitations on biochemical constituents and docosahexaenoic acid production of Schizochytrium sp. Bioresour. Technol. 159, 199–206. doi: 10.1016/j.biortech.2014.02.106
Sun, X. M., Ren, L. J., Ji, X. J., Chen, S. L., Guo, D. S., and Huang, H. (2016). Adaptive evolution of Schizochytrium sp. by continuous high oxygen stimulations to enhance docosahexaenoic acid synthesis. Bioresour. Technol. 211, 374–381. doi: 10.1016/j.biortech.2016.03.093
Tatusov, R. L., Galperin, M. Y., Natale, D. A., and Koonin, E. V. (2000). The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 28, 33–36. doi: 10.1093/nar/28.1.33
Thayer, E. C., Bystroff, C., and Baker, D. (2000). Detection of protein coding sequences using a mixture model for local protein amino acid sequence. J. Comput. Biol. 7, 317–327. doi: 10.1089/10665270050081559
Tian, J., Zhang, J., Han, Z.-Y., Song, T.-T., Li, J.-Y., Wang, Y.-R., et al. (2017). McMYB12 transcription factors co-regulate Proanthocyanidin and anthocyanin biosynthesis in Malus crabapple. Sci. Rep. 7, 43715. doi: 10.1038/srep43715
Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., et al. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc 7, 562–578. doi: 10.1038/nprot.2012.016
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963
Warude, D., Joshi, K., and Harsulkar, A. (2006). Polyunsaturated Fatty Acids: Biotechnology. Crit. Rev. Biotechnol. 26, 83–93. doi: 10.1080/07388550600697479
Keywords: Schizochytrium limacinum SR21, genome, transcriptome, fatty acid, DHA
Citation: Liang L, Zheng X, Fan W, Chen D, Huang Z, Peng J, Zhu J, Tang W, Chen Y and Xue T (2020) Genome and Transcriptome Analyses Provide Insight Into the Omega-3 Long-Chain Polyunsaturated Fatty Acids Biosynthesis of Schizochytrium limacinum SR21. Front. Microbiol. 11:687. doi: 10.3389/fmicb.2020.00687
Received: 13 December 2019; Accepted: 25 March 2020;
Published: 16 April 2020.
Edited by:
John R. Battista, Louisiana State University, United StatesReviewed by:
Xiaojin Song, Qingdao Institute of Bioenergy and Bioprocess Technology (CAS), ChinaFeng Qi, Fujian Normal University, China
Copyright © 2020 Liang, Zheng, Fan, Chen, Huang, Peng, Zhu, Tang, Chen and Xue. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ting Xue, eHVldGluZ0Bmam51LmVkdS5jbg==