 Yinggai Song1,2,3
Yinggai Song1,2,3 Minghao Du4
Minghao Du4 Nickolas Menezes da Silva5,6
Nickolas Menezes da Silva5,6 Ence Yang4
Ence Yang4 Vania A. Vicente5
Vania A. Vicente5 G. Sybren de Hoog2,5,6*
G. Sybren de Hoog2,5,6* Ruoyu Li1,2,3*
Ruoyu Li1,2,3*- 1Department of Dermatology and Venerology, Peking University First Hospital, Beijing, China
- 2Research Center for Medical Mycology, Peking University, Beijing, China
- 3National Clinical Research Center for Skin and Immune Diseases, Beijing, China
- 4Peking University Health Science Center, Beijing, China
- 5Microbiology, Parasitology and Pathology Post-Graduation Program, Department of Pathology, Federal University of Paraná, Curitiba, Brazil
- 6Center of Expertise in Mycology of Radboud University Medical Center, Canisius Wilhelmina Hospital, Nijmegen, Netherlands
Exophiala spinifera, a capsule-producing black yeast, is overrepresented as agent of disseminated infection in humans with inherited dysfunction of the CARD9 gene. In a review of published caspase recruitment domain-containing protein 9 (CARD9) deficiency cases, black fungi were linked to mutations other than those prevalent in yeast and dermatophyte cases, and were found to respond to a larger panel of cytokines. Here, we sequenced and annotated the genomes of BMU 08022 from a patient with CARD9 deficiency and two environmental strains, BMU 00051 and BMU 00047. We performed genomic and transcriptomic analysis for these isolates including published black yeasts genomes, using a combination of long-read (PACBIO) and short-read (Illumina) sequencing technologies with a hybrid assembly strategy. We identified the virulence factors, fitness, and the major genetic and gene expression differences between the strains with RNAseq technology. Genome assembly reached sub-chromosome level with between 12,043 and 12,130 predicted genes. The number of indels identified in the clinical strain was higher than observed in environmental strains. We identify a relatively large core genome of 9,887 genes. Moreover, substantial syntenic rearrangements of scaffolds I and III in the CARD9-related isolate were detected. Seventeen gene clusters were involved in the production of secondary metabolites. PKS-cluster 17 was consistently found to be absent in the clinical strain. Comparative transcriptome analysis demonstrated that 16 single-copy genes were significantly differentially expressed upon incubation in brain-heart infusion broth vs. Sabouraud glucose broth. Most of the single-copy genes upregulated with Brain Heart Infusion (BHI) were transporters. There were 48 unique genes differentially expressed exclusively to the clinical strain in two different media, including genes from various metabolic processes and transcriptional regulation. Up-regulated genes in the clinical strain with Gene Ontology (GO) enrichment are mainly involved in transmembrane transport, biosynthetic process and metabolic process. This study has provided novel insights into understanding of strain-differences in intrinsic virulence of the species and indicated that intraspecific variability may be related to habitat choice. This indicates that strains of E. spinifera are differentially prone to cause infection in susceptible patient populations, and provides clues for future studies exploring the mechanisms of pathogenic and adaptive strategies of black yeasts in immunodeficient patients.
Introduction
Melanized fungi of the order Chaetothyriales are renowned as agents of human infection. The number of cases is low when compared to dermatophytes, Aspergillus, and Candida, but with 86 species in 7 genera that have been proven as potential agents of vertebrate disease (de Hoog et al., 2020) the order ranges fourth in clinical biodiversity, after Onygenales, Eurotiales, and Saccharomycetales. In 19 chaetothyrialean species, systemic or disseminated infection has been observed, which frequently led to death of the patient. The neurotropic species Cladophialophora bantiana is one of the most feared fungi, because it causes brain abscesses in healthy-appearing patients, with a case fatality rate of 65% despite antifungal therapy (Kantarcioglu et al., 2017). Numerous investigations nevertheless suggest that most members of the order – with a possible exception of agents of chromoblastomycosis – are opportunists rather than pathogens. Opportunists are defined as fungi that complete their natural life cycle without involvement of an animal host, being able of unintentional infection only through coincidental similarities between conditions of host tissue and natural habitat. Gostincar et al. (2018) ascribed their infectious ability to polyextremotolerance, which is tolerance to environmental stress supplemented with efficient nutrient scavenging and toxin management. Properties that in pathogenic fungi have a role in virulence may have very different functionalities in the fungus’ natural habitat. Song et al. (2017) noted that hypothesized virulence factors, which enhance infection in truly host-associated fungi like Candida or Histoplasma, in black yeasts may respond adversely to proxies of host resistance, and thus comparable genes can be functionally different between fungal groups.
An important consequence of opportunism is that all strains have an equal chance to be inoculated into the host, and thus clinical strains are not necessarily different from environmental ones. Pathogenic adaptation is then less likely to occur because of absence of host-related transmission. However, after coincidental inoculation, some strains may be more prone to cause symptoms than others, and thus there may be a difference between clinical and environmental strains. Black yeast-like fungi mostly cause local, (sub) cutaneous infections. The host conditions of patients with brain abscesses acquired via the inhalative route are not well understood. For disseminated cases, recent data have shown (Lanternier et al., 2013) that these are often associated with host-inherited mutations in caspase recruitment domain-containing protein 9 (CARD9). This protein plays a role in the dectin pathway regulating innate immunity activating pro-inflammatory cytokines. Since most cases of disseminated disease in black yeast-like fungi were described prior to 2013, it is possible that the majority of these cases occurred in patients with CARD9 dysfunctional innate immunity. Several of the infection types of chaetothyrialean fungi show a certain degree of unexplained endemism, such as the prevalence of Cladophialophora brain abscess in the Indian subcontinent (Chakrabarti et al., 2016). Similarly, the majority of CARD9-associated black fungal cases is found in East Asia: China, Korea, Japan, although the etiologic agents have a global distribution. For an explanation of virulence of members of Chaetothyriales, we thus might expect answers from the interaction of the fungus with windows of opportunity associated with CARD9 regulation and human race.
To study the fungal side of this tripartite relationship, we sequenced the genomes and transcriptomes with different culture conditions of an Exophiala spinifera strain isolated from a patient with a CARD9-related disorder, and compared this with two environmental strains of the same species, and with other published genomes (Supplementary Table S1). We applied state-of-the-art technology, to quantify eventual differences between clinical and environmental strains with maximum precision and to describe the species’ intraspecific variability. A number of genomes of Chaetothyriales were already available, but these were sequenced with somewhat older techniques. One of the aims is therefore to describe the technical and biological variation in these datasets, and determine whether the new techniques provide a better answer to questions as formulated above.
Materials and Methods
Literature Search
Keywords “CARD9” and “fungal infection” were used in PubMed to search the English literature including research articles, reviews and case reports published until November 2019.
Strains and Culture Conditions
Three strains of E. spinifera were analyzed (Table 1): BMU 08022 (ESC1, from a CARD9-deficient patient, Jiangsu, China), BMU00051 (ESE1, from bark, Shenzen, China), and BMU00047 (ESE2, from soil, Colombia). To prepare DNA for genome sequencing, mycelia were harvested from fresh cultures on Sabouraud Dextrose Agar (SDA), frozen in liquid nitrogen, and stored at −80°C until further processing. For RNA extraction, strains were inoculated in Sabouraud Dextrose Broth (SDB) and in Brain Heart Infusion Broth (BHI) (Das et al., 2010) and harvested after 20 h, centrifuged and frozen in liquid nitrogen. Liquid nitrogen was used to grind the samples for DNA/RNA extraction.

Table 1. Isolation data of strains sequenced in this study, and isolates included for comparison.
DNA and RNA Extraction
Genomic DNA was extracted using the MOBIO Power Microbial Maxi DNA Isolation kit (MoBio Laboratories, Carlsbad, CA, United States). DNA was concentrated by QUBIT (Invitrogen, Carlsbad, CA, United States), and an Agilent Bio Analyzer 2100 using a 1000 DNA Chip (Agilent Technologies, Palo Alto, CA, United States), and its quality was determined by electrophoresis on agarose gels. Total RNA was extracted from cultures ground in liquid nitrogen using TRIZOL reagent (Thermo Fisher Scientific, Waltham, MA, United States) according to the manufacturer’s instructions, followed by two phenol (pH 4.6)-chloroform-isoamyl alcohol (25:24:1) extraction steps and two chloroform extraction steps after the initial TRIZOL-chloroform phase separation. RNA pellets were dissolved in 88 μL of nuclease-free water and subjected to genomic DNA digestion with DNase (Qiagen, Hilden, Germany). RNA samples were then concentrated using RNA Clean & Concentrator (Zymo Research, Irvine, CA, United States). RNA quality was confirmed using an Agilent 2100 Bioanalyzer (Agilent). Two micrograms of total RNA per sample was used for cDNA library construction.
cDNA Synthesis
The cDNA synthesis was performed using IMPRON II reverse transcriptase kit (Promega, Madison, WI, United States) with 1 μg of total RNA added according to manufacturer’s instructions. The efficiency of the cDNA synthesis process was assessed by PCR using the following reference genes: β-tubulin and ITS. The integrity of PCR amplification products was verified by electrophoresis on 1.2% agarose gels.
PacBio Sequencing and Raw Assembly
The extracted DNA of three E. spinifera strains were sequenced on the Pacific Biosciences (Pacific Biosciences, Menlo Park, CA, United States) Single Molecule, Real-Time (SMRT) DNA sequencing technology (platform: RS II; chemistry: P6-C4). The raw reads were processed using the standard SMRT analysis pipeline v5.0.1 (Chin et al., 2013). The de novo assembly was carried out following CANU v1.4 assembly protocol (Koren et al., 2017) with QUIVER polishing (Chin et al., 2013). Assemblies were refined by manual curation. Pairwise alignments were carried out using BLASTN v2.6.0+ (Altschul et al., 1990). The draft assemblies were further improved with FINISHERSC (Lam et al., 2015) and polished with ARROW (Chin et al., 2013). For each genome assembly, contigs were examined and removed if redundant (i.e., aligning to any other contig in the same assembly with >90% identity). All contigs containing rDNA repeats were excluded from the above step.
Illumina Sequencing, Read Mapping and Error Correction
Our genome assembly strategy is summarized in Figure 1. In addition to the PACBIO sequencing, we also performed Illumina 150-bp paired-end sequencing for each strain. We examined the raw Illumina reads via FASTQC v0.11.51 and performed adaptor-removal and quality-based trimming by TRIMMOMATIC v0.36 (Bolger et al., 2014). For each strain, the trimmed reads were mapped to the corresponding PACBIO assemblies by BWA 0.7.12 and using BWAMEM v0.7.16 (Li and Durbin, 2009). The resulting read alignments were subsequently processed by SAMTOOLS v1.6 (Li et al., 2009). On the basis of Illumina read alignments, we further performed error correction with PILON v1.22 (Walker et al., 2014) to generate final assemblies for downstream analysis. BUSCO v3 (Simão et al., 2015) was used to assess the completeness of the final assembled genomes.
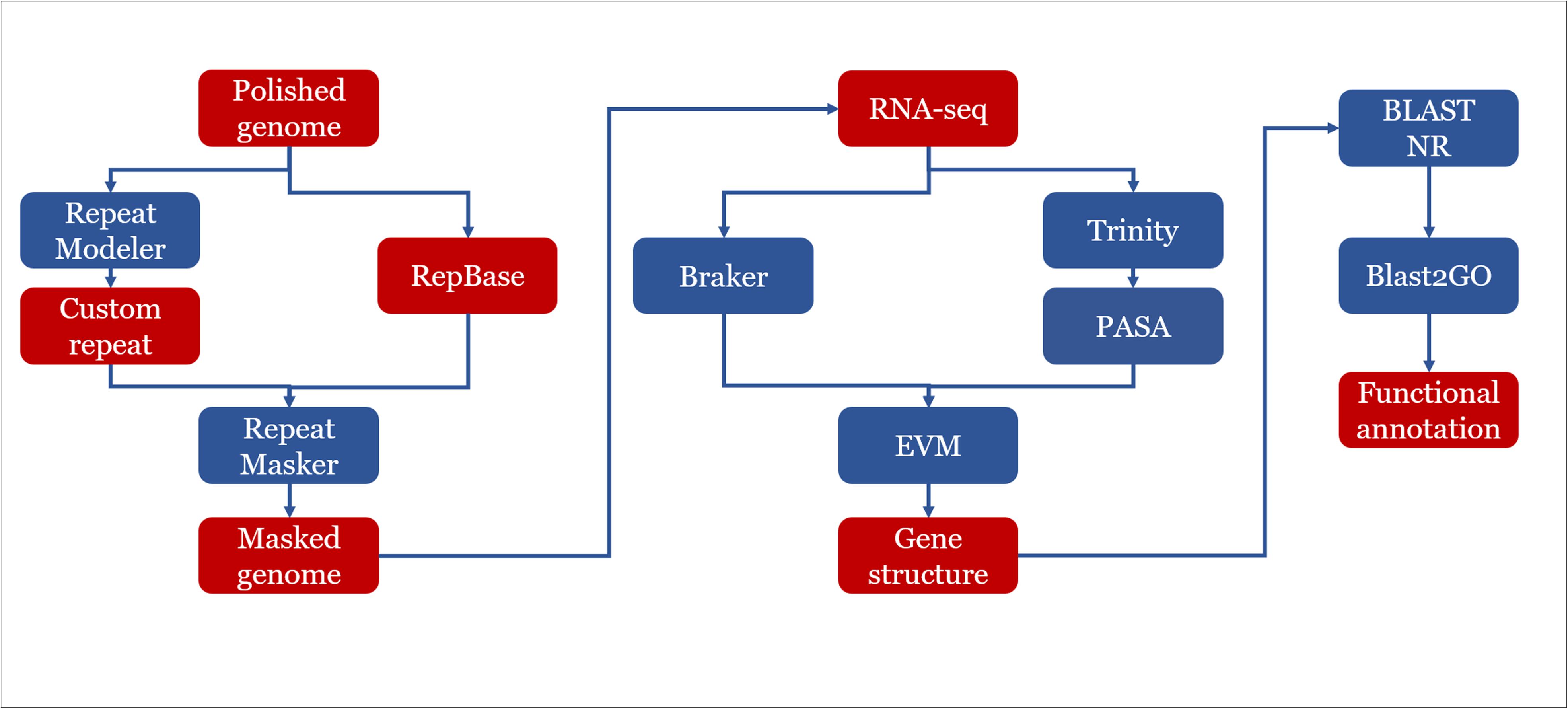
Figure 1. Genome assembly and annotation strategy used in this study. Assembly: the raw reads were processed using the standard SMRT analysis pipeline v5.0. The de novo assembly was carried out following CANU v1.4 assembly protocol with QUIVER polishing. Assemblies were refined by manual curation. Pairwise alignments were carried out using BLASTN v2.6.0+. The draft assemblies were further improved with FINISHERSC and polished with ARROW. Annotation: a hybrid strategy combining ab initio predictions and transcriptomic support (RNA-seq) was applied in gene prediction. Three ab initio gene finders, GENEMARK.HMM, FGENESH, and AUGUSTUS were used.
Repetitive Sequence Prediction and Masking
Repetitive elements were predicted using a de novo approach by applying REPEATMODELER v1.0.112, which includes RECON v1.08 (Bao and Eddy, 2002) and REPEATSCOUT v1.0.5 (Price et al., 2005) to construct a strain-specific repeat library. To further classify repetitive sequences, BLAST v2.2.28 (Camacho et al., 2009) was used to search the repeat library against SWISS-PROT protein database. Sequences with similarities to known proteins were discarded from the repeat library. Repetitive sequences were masked with REPEATMASKER v4.0.72 using the de novo constructed library.
Annotation of rRNA, tRNA and Repeat Elements
The rRNA loci were predicted by using RNAMMER v1.2 (Lagesen et al., 2007) and tRNA by TRNASCAN-SE v2.0 (Lowe and Eddy, 1997). Repeat elements were identified using REPEATMASKER v4.0.62 based on REPBASE LIBRARY 20160829. REPEATMASKER was run with options that skipped the low-complexity DNAs masking for the purpose of gene prediction.
Gene Prediction and Functional Annotation
A hybrid strategy combining ab initio predictions and transcript alignments was applied in annotation of protein coding genes. Firstly, protein coding genes were predicted using BRAKER (Hoff et al., 2016), which was an ab initio approach combined with RNA-seq data. Then, RNA-seq was de novo assembled by using TRINITY (Grabherr et al., 2011). The assembled transcripts were aligned to the reference genome of corresponding strains by PASA pipeline (Haas et al., 2003). Finally, EVIDENCEMODELER (Haas et al., 2008) was used to combine ab initio predictions and transcript alignments into consensus gene structure.
Functional annotation of predicted proteins was done by BLAST (Camacho et al., 2009) similarity search of the predicted proteins against NR database with an e-value of 1e–05. Gene Ontology (GO) terms were retrieved by using Blast2go (Gotz et al., 2008) with default annotation parameters. To further improve the annotations, INTERPROSCAN v5.33-72.0 (Jones et al., 2014) was used to identify known protein domains. BLAST (Camacho et al., 2009) results of NR database and results of the INTERPROSCAN analysis were combined to gain a complete functional annotation of all predicted genes. Genes predicted to encode carbohydrate-active enzymes (CAZYMES) were identified with DBCAN2 (Zhang et al., 2018). Also the software SMURF (Khaldi et al., 2010) was used to predict secondary metabolite gene clusters in three genomes of E. spinifera.
Comparison of Genome Assemblies
The synteny among three strains of E. spinifera was determined using MUMMER v3.23 (Delcher et al., 2003). We used NUCMER implemented in MUMMER v3.23 to align the genomes of the three strains of E. spinifera. In addition, the DNADIFF package in MUMMER v3.23 was used to generate coordinates for the differences among genomes of the three strains analyzed. The alignment statistics, SNPs, indels, inversions, etc., were also calculated from the results of MUMMER. We plotted these features circularly with CIRCOS v0.69.6 (Krzywinski et al., 2009).
Identification of Homology Groups
For nuclear protein-coding genes, we used INPARANOID v4.13 to identify gene homology across the three strains of E. spinifera. We calculated protein length distributions for each homologous group (1:1:1 association among strains) of the strains. The correlation of exon length and intron length of homologous groups (1:1 association between strains) were calculated by pairwise comparisons among three strains.
Identification of Orthologous Genes
Orthologous groups were delimited using ORTHOFINDER v2.2.6 (Emms and Kelly, 2015), in which all predicted protein sequences were compared using a BLAST (Camacho et al., 2009) all-against-all search. The single-copy genes, duplicated genes, and strain-specific genes were extracted from the ORTHOFINDER output. To detect the core genome, specific and shared genes, the data was filtered based on the ORTHOFINDER output. The R package UPSETR (Conway et al., 2017) was used to construct statistics of orthologous groups among the three strains under study.
Identification of Genes Under Positive Selection
Single-copy orthologous proteins were aligned by using CLUSTALO v1.2.44. To obtain the alignments of codons, the corresponding nucleotide-sequence alignments were derived by substituting the respective coding sequences from the protein sequences. For each single-copy orthologous group, genes from the two environmental strains and their orthologous genes in the clinical strain formed sequence triplets. The test for the asymmetric evolution was constituted as a relative rate test between clinical strain and environmental strains on an unrooted tree. The statistical tests were conducted with a codon-based branch-site model using the CODEML program of PAMLv4.9 (Yang, 2007). We compared ω0 and ω1 between single-copy orthologous genes to detect differences in proportion of selected sites in the two clades. Likelihood ratio (LR) was used to test for significance. To do this, two models were applied to the data: model 1 (ω0 = ω1) constrains the two ω values to be equal for the three sequences, and model 2 (ω0 ≠ ω1) estimates the two ω values as free parameters. Collected maximum likelihood values ML1 and ML2 from the two models were used to calculate the LR, LR = 2 (lnML2−lnML1). LR is then compared against the χ2 distribution with one degree of freedom.
Transcriptome Analysis
The raw reads were assessed for quality by FASTQC v0.11.5 and filtered to remove low-quality reads with TRIMMOMATIC v0.36 (Bolger et al., 2014). Filtered reads were mapped to the corresponding reference genome of three E. spinifera strains using STAR v2.5.3a (Dobin et al., 2013). The gene expression levels were calculated using FEATURECOUNTS v1.5.2 (Liao et al., 2014) and normalized based on the FPKM method. Differentially expressed genes were detected by R package DESEQ2 (Love et al., 2014). Differentially expressed genes were filtered by adjusted p-value < 0.05 and | log2 fold change| > 1. GO enrichment analysis was performed by R package TOPGO5. Only 1-1-1 orthologous genes were considered when detecting differentially expressed genes among different strains. For analysis of RNA-seq data, differentially expressed genes were detected by R Bioconductor package DESeq2 with the Benjamini-Hochberg adjusted p-value < 0.05 and fold change >2. Student’s t-tests were performed for the comparisons, and a p-value < 0.05 was considered to represent significance.
Data Availability
All genomes and RNA-seq have been deposited in NCBI. The RNA-seq data can be accessed by PRJNA600825, the PACBIO assemblies (PRJNA600825), Illumina assemblies (PRJNA600036). The RNA-seq is available already (https://www.ncbi.nlm.nih.gov/sra/PRJNA600825).
Results
Literature Data on Black Fungi in CARD9 Patients
An overview was made of published cases of fungal infections in patients with CARD9 deficiencies (Table 2; Glocker et al., 2009; Drewniak et al., 2013; Lanternier et al., 2013, 2015a,b; Gavino et al., 2014, 2016; Drummond et al., 2015; Grumach et al., 2015; Herbst et al., 2015; Jachiet et al., 2015; Alves de Medeiros et al., 2016; Celmeli et al., 2016; Yan et al., 2016; Boudghene et al., 2017; Zhang et al., 2017; Arango-Franco et al., 2018; Cetinkaya et al., 2018; De Bruyne et al., 2018; Zhong et al., 2018; Huang et al., 2019). Sixteen different mutations have been observed. When cases are arranged according to phylogeny of the fungus, three main groups can be recognized: ascomycetous yeasts (Candida: order Saccharomycetales), dermatophytes (order Onygenales), and black fungi (mainly order Chaetothyriales, single representatives of orders Pleosporales and Venturiales). A single proven case with a member of Mucorales has been published. Mutations are not randomly distributed in the three fungal groups (Figure 2; p = 0.001). For example, p.Q295X is found in 13 cases of Candida infection, but was not found among cases by other fungi. The main cytokine impaired was IL-6 (43/50 cases). In Onygenales, this was the only deficient cytokine reported, while, in contrast, in seven cases of Candida infection GM-CSF or TNFα were absent instead. Black fungi were associated with a relatively large panel of missing cytokines, particularly TNFα and IL-1β. The relative absence of cytokines in the three fungal main groups is illustrated in Figure 3.
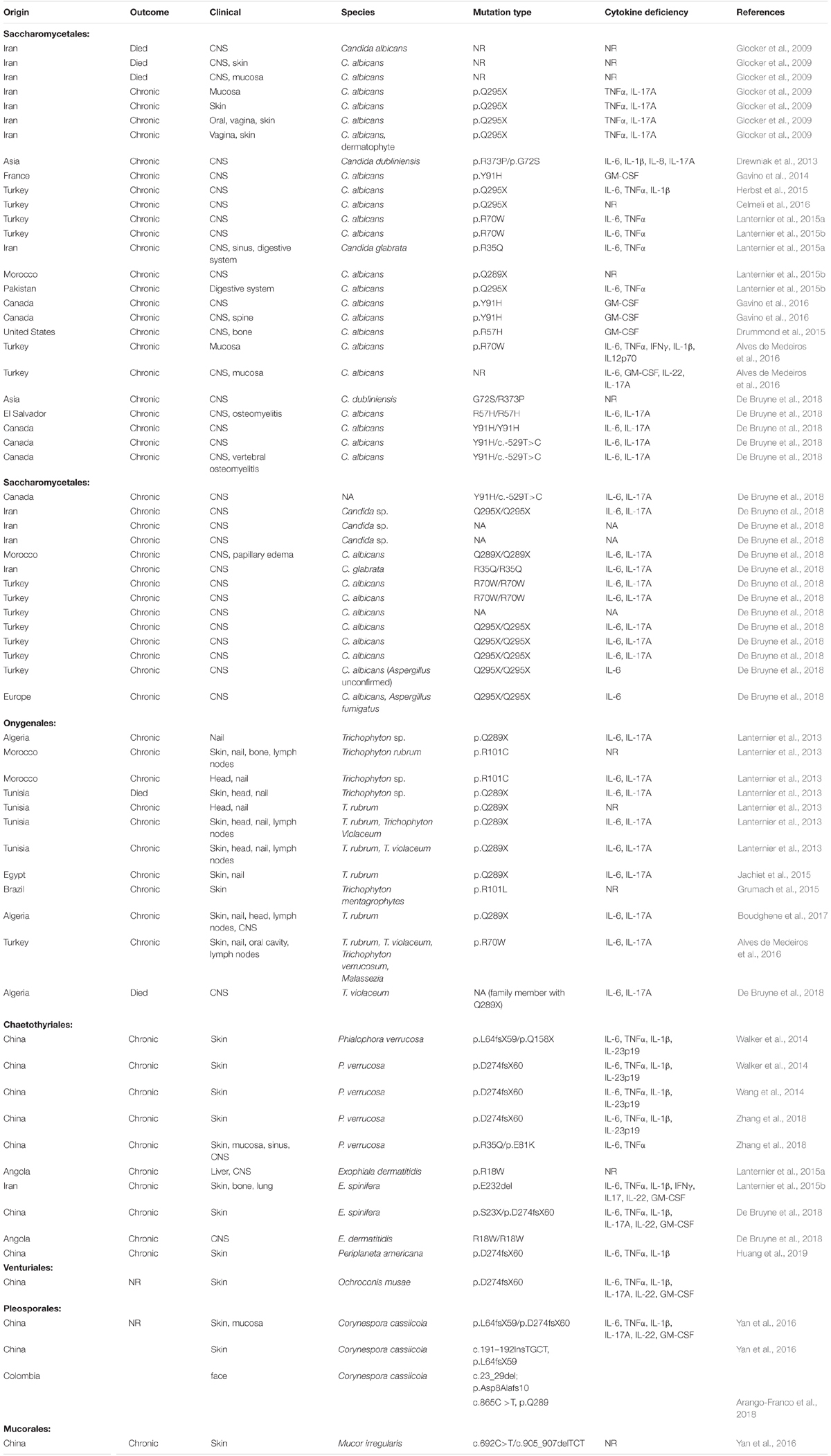
Table 2. Overview of published cases of fungal infection in patients with CARD9 deficiency.
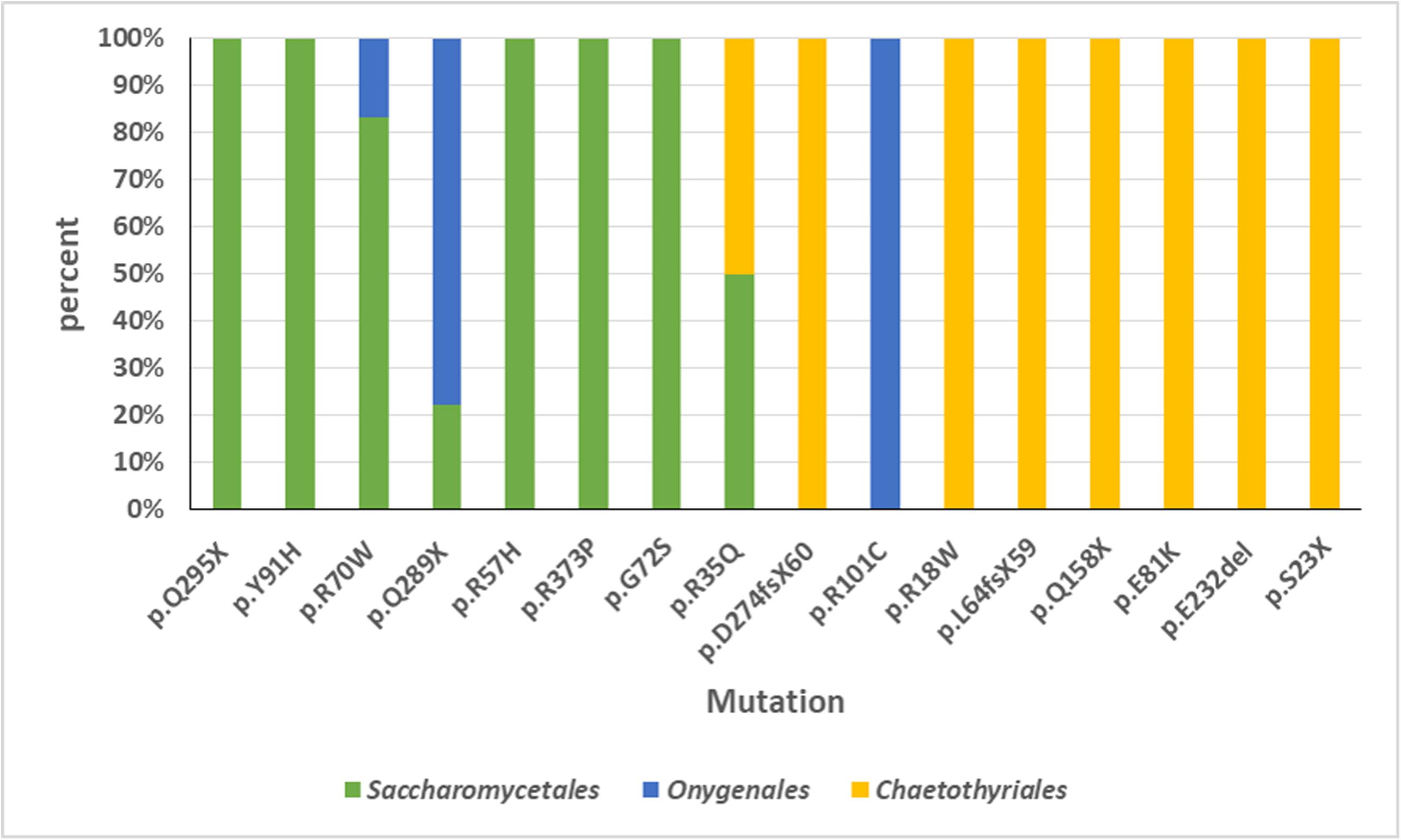
Figure 2. Overview of CARD9 mutations in published cases of fungal infections in patients with CARD9 deficiencies. Sixteen different mutations have been observed. Three main groups can be recognized: ascomycetous yeasts (Candida: order Saccharomycetales), dermatophytes (order Onygenales), and black fungi (mainly order Chaetothyriales, single representatives of orders Pleosporales and Venturiales). A single proven case with a member of Mucorales has been published. Mutations are not randomly distributed in the three fungal groups.
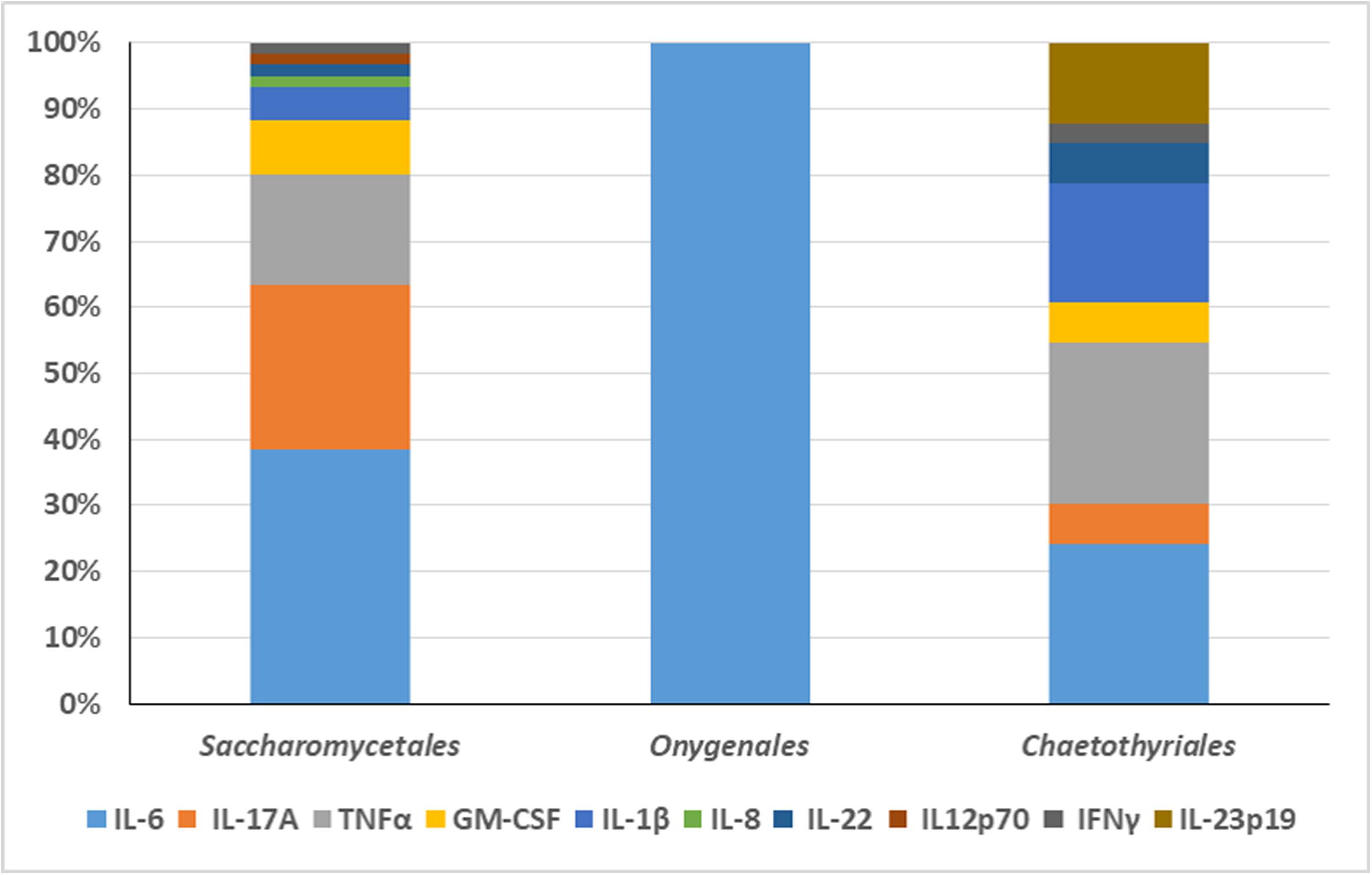
Figure 3. The relative absence of cytokines in the three fungal main groups is illustrated. The main cytokine impaired in Candida infection was IL-6. GM-CSF or TNFα were absent in seven cases of Candida infection. In Onygenales, IL-6 was the only deficient cytokine reported. Black fungi were associated with a relatively large panel of missing cytokines, particularly TNFα and IL-1β.
Genome Sequencing and Assembly
We sequenced the genomes of three strains: BMU 08022 (clinical strain from human skin, ESC1), BMU 00051 (environmental strain from bark, ESE1), and BMU 00047 (environmental strain from soil, ESE2) (Table 1) using a combination of long-read (PACBIO) and short-read (Illumina) sequencing technologies. The third generation sequencing is supplemented by the second generation sequencing, and combined with a variety of assembly software and parameter optimizations (Figure 1). For each genome, PACBIO sequencing provided more than 100 × coverage and Illumina reached 170× coverage. For strain ESC1, we obtained a draft genome assembly of 33,559,924 bp organized into nine contigs with an L90 of 7, and an N50 of 4.0 Mb. The genome of ESE1 was 32,380,025 bp in size and comprised seven contigs with an L90 of 7, and N50 of 4.9 Mb. In ESE2, the genome assembly of 32,696,644 bp contained nine contigs with an L90 of 7, and an N50 4.6 Mb. For all three strains, more than 94% of the Illumina reads were aligned to the draft genome assemblies in post hoc validation (Table 3). The results of genome assembly of the three strains reached sub-chromosome level.
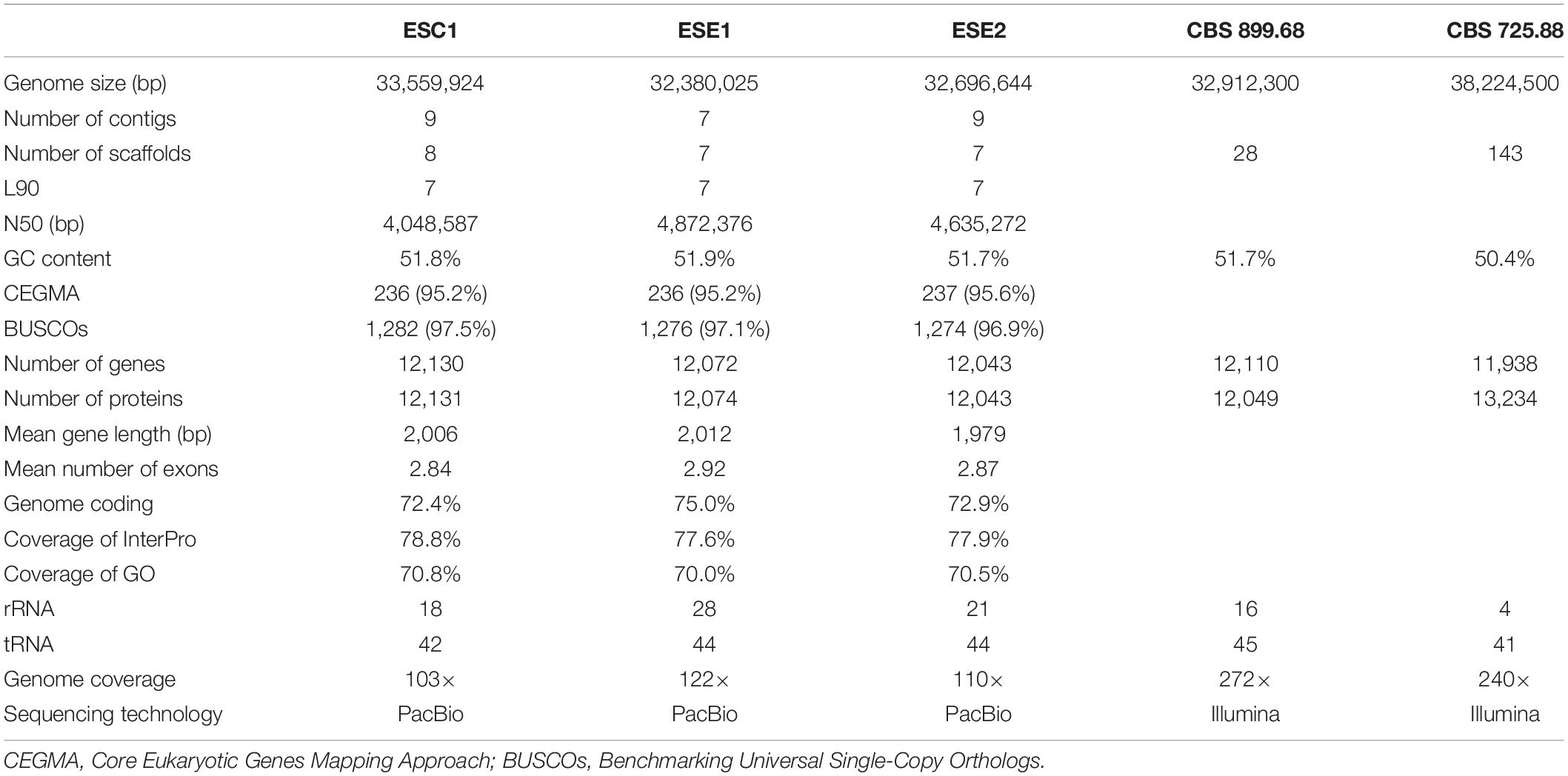
Table 3. Genome assembly of analyzed strains of Exophiala spinifera, compared with deposited genomes of E. spinifera (CBS 899.68) and E. oligosperma (CBS 725.88).
Colinear Analysis
We used whole-genome alignment to identify homologous and strain-specific segments among three E. spinifera strains. We identified a total of 172,269 and 170,807 SNPs for the clinical strain compared to the environmental strains BMU 00051 and BMU 00047, respectively (Table 4). However, the number of SNPs between the environmental strains was 362,264, which indicated that the genome of the clinical strain was intermediate between the two environmental strains from different continents. Likewise, the number of indels identified in the clinical strain was higher than compared with environmental strains (Table 4). There were 105,095 (ESE1) / 104,446 (ESE2) synonymous vs. 67,174 (ESE1) / 66,361 (ESE2) non-synonymous SNPs compared to clinical strain ESC1 (Table 4). We found substantial syntenic rearrangements in scaffolds I and III between ESC1 vs. ESE1 and ESC1 vs. ESE2, while in scaffolds VII and VIII of ESC1 syntenic rearrangements existed with scaffold III of ESE1 (Figure 4). Comparing the genomes of ESC1, ESE1, and ESE2, recombination was observed in scaffolds I and III between the clinical and environmental strains (Figure 4B). Scaffold I of ESC1 is equal to part of scaffolds I and III of the environmental strains, and scaffold III of ESC1 is equal to the remainders of scaffolds I and III of the environmental strains. This is shown in more detail in Figure 4, where scaffold VIII of ESC1 is shown to be comparable to part of scaffold I of the environmental strains, and two areas proved to be inverted between clinical and environmental strains.
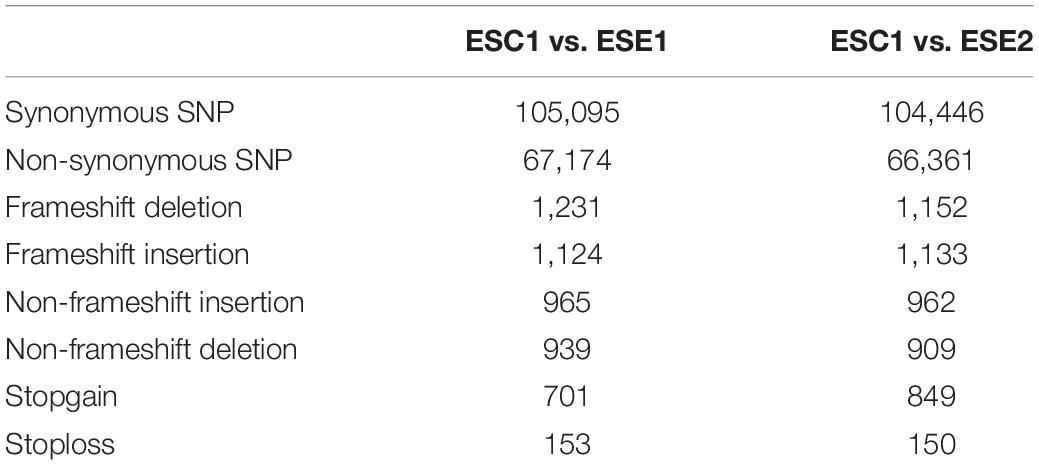
Table 4. Comparison of single nucleotide polymorphisms of clinical vs. environmental strains.
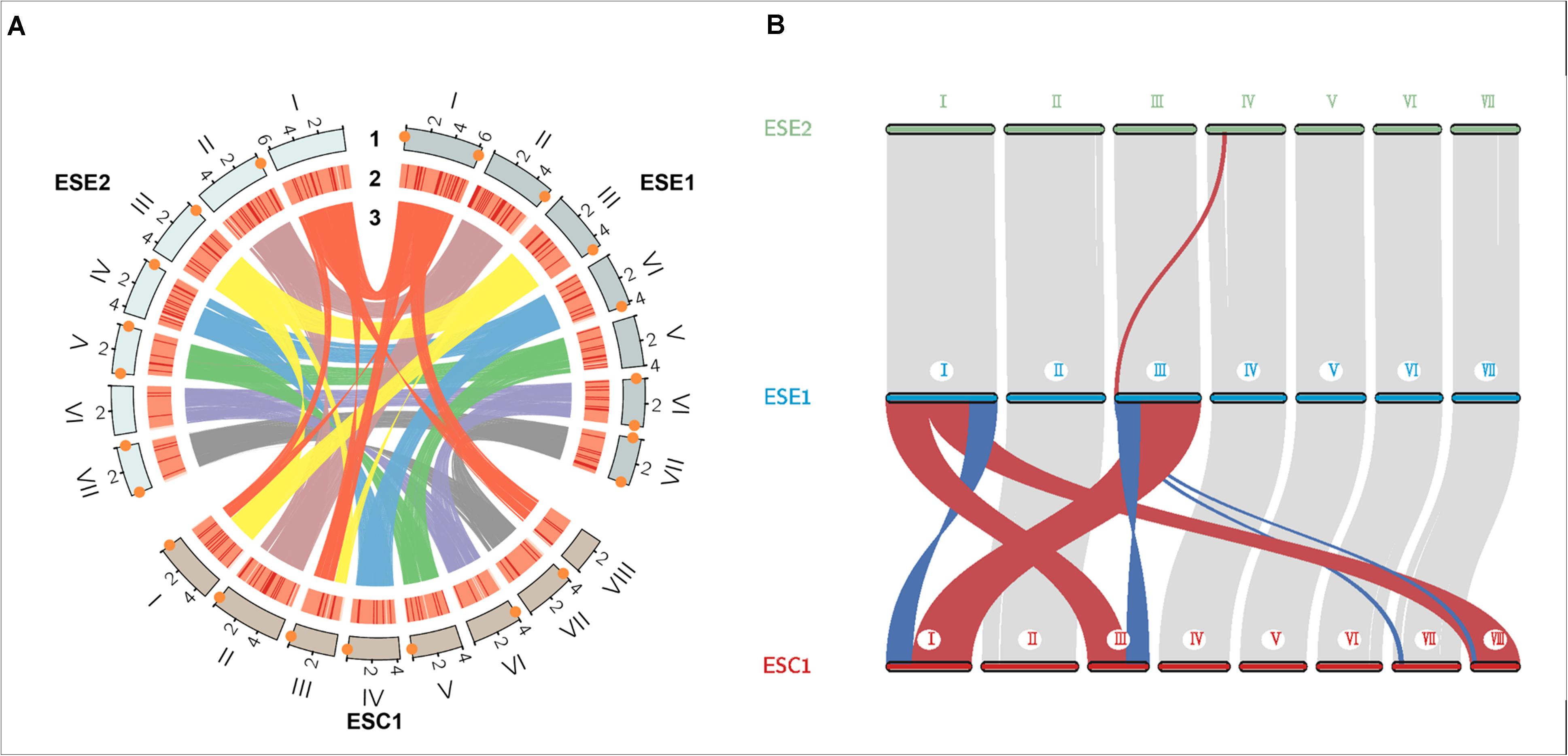
Figure 4. Genome colinear analysis for the three E. spinifera genomes. (A) Recombination is observed between clinical strain ESC1 and environmental strains ESE1 and ESE2 in scaffolds I and III. Track 1, genome position; track 2, gene density; track 3, sequence synteny. Orange diamonds: telomere sequences. (B) Location of sequence rearrangements in scaffolds I and III of ESC1. Red, stands for positive collinearity; Blue, reverse complementary.
Gene Content and Functional Annotation
Genome functional annotation was performed with INTERPROSCAN, resulting in over 59% of genes being protein-coding genes annotated with GO term. A hybrid strategy combining ab initio predictions and transcriptomic support (RNA-seq) was applied in gene prediction. Annotation of the strains yielded 12,131 protein-coding genes for E. spinifera ESC1, 12,074 protein-coding genes for ESE1 and 12,043 for ESE2 (Table 3). A total of 9,887 genes represented the core genome (Supplementary Figure S1). The environmental strains shared more genes (336) than compared with clinical strain ESC1. The clinical strain had 647 specific gene clusters, included transporter, and binding protein. The list of the respective functional annotations is available in Supplementary Table S2. Plotting the protein lengths of the three strains, considerable differences were noted (Figure 5): the distribution pattern of strain-specific genes length varied from clinical strain ESC1 to environmental strain, and ESC1 deviated by having more genes. Single-copy orthologous genes were compared to detect differences in proportion of selected sites, with LR as test of significance. In contrast, homologous introns were weakly correlated between ESC1 and environmental strain (ESE1 and ESE2), with significant length variations, while correlation were present between environmental strains (r2 = 0.57, higher than the other two groups) (Figure 6).
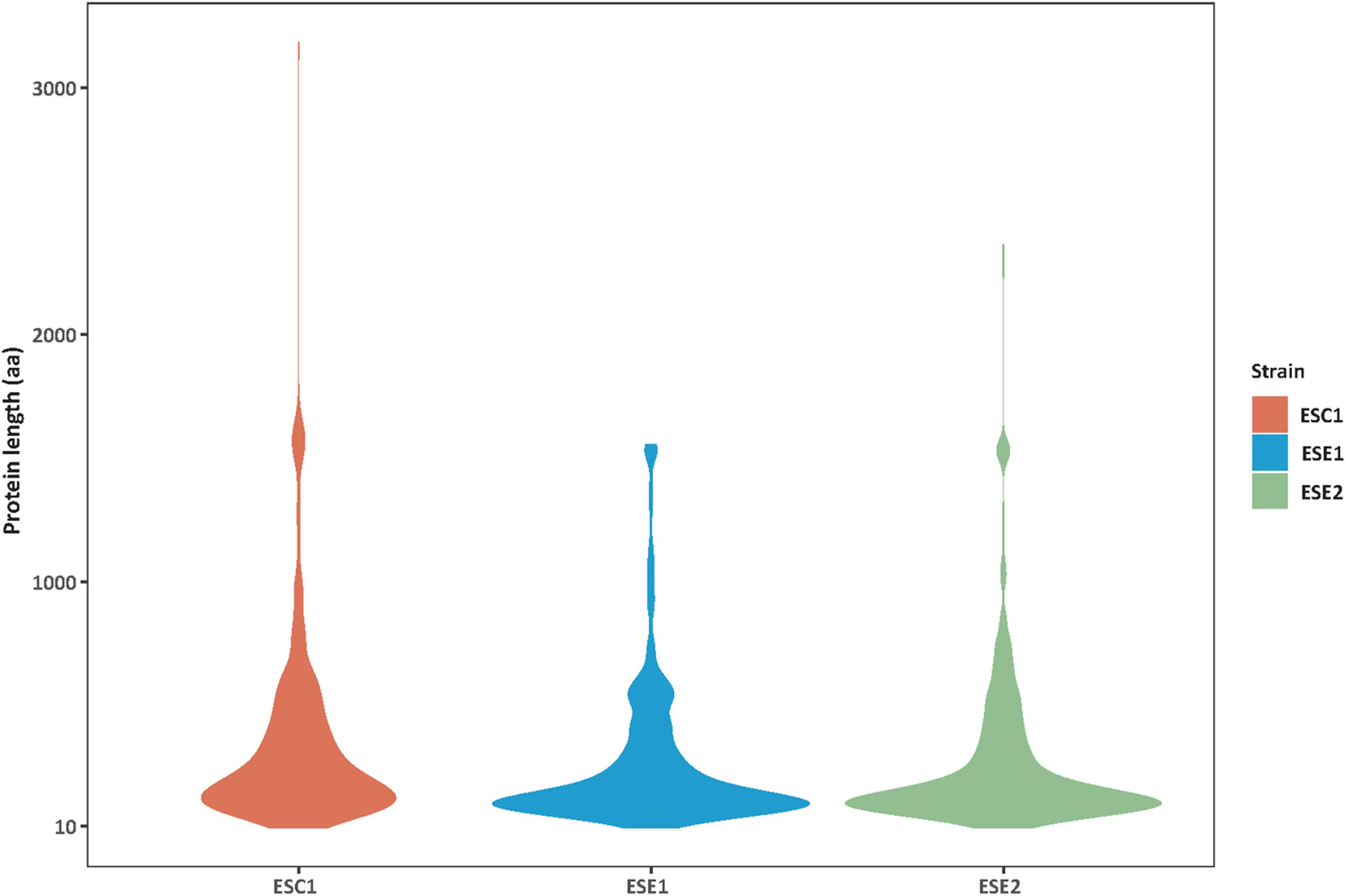
Figure 5. Distribution variability of specific protein length among three strains. The protein lengths (Y-axis) in strains analyzed (n = X-axis) showing an intraspecific variability. Greater differences were found between clinical strain ESC1 and environmental strains ESE1 and ESE2, while the protein length distribution of the two environmental strains was more consistent.
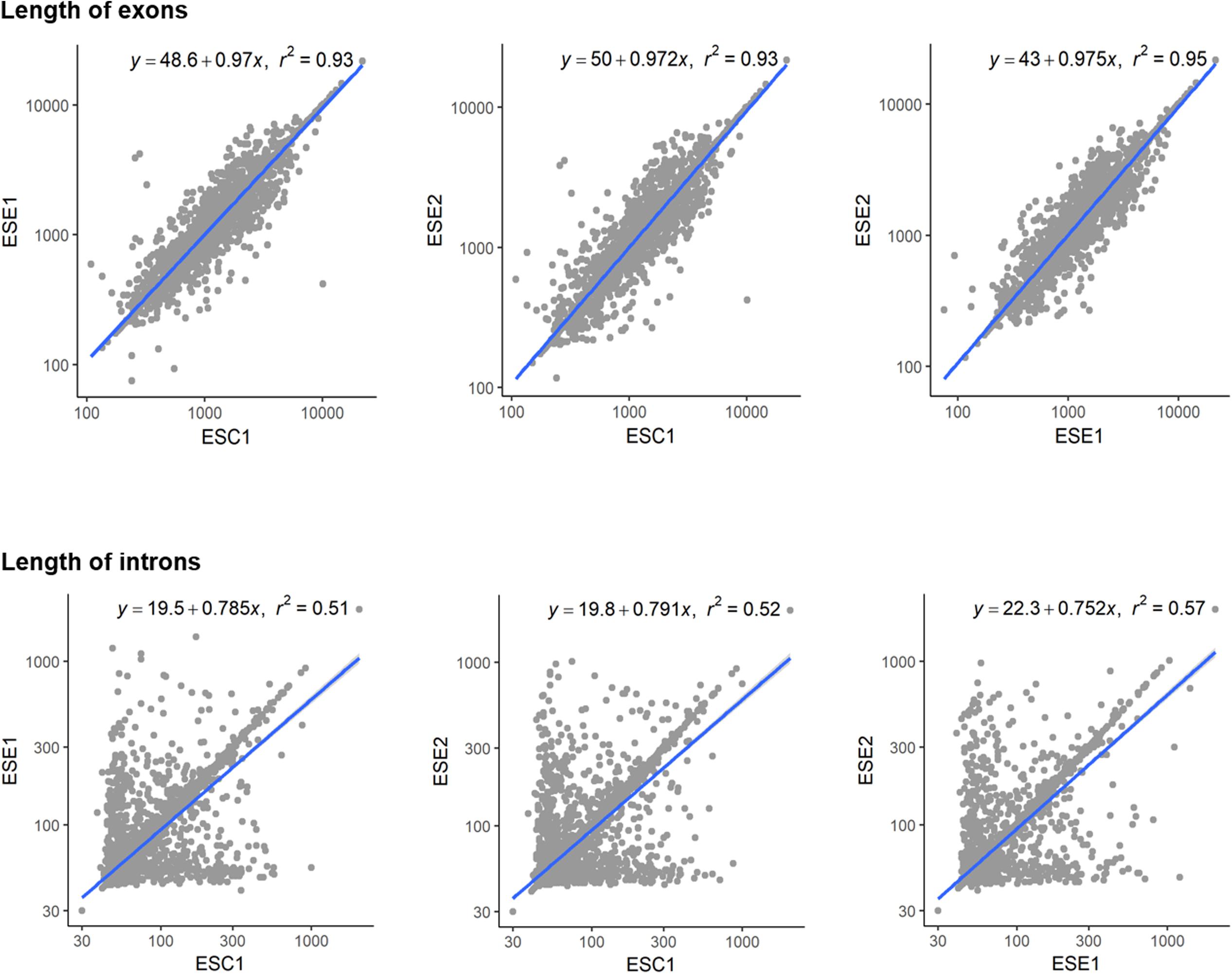
Figure 6. Length variations of introns and exons. The blue lines show trends in exon length distribution, the slopes being different between exons and intron. Positive and negative correlations are shown. Homologous introns were weakly correlated, with significant length variations.
Secondary Metabolite Clusters
We identified a total of 9,329 homologous groups at the gene level (1:1:1 association among three strains). In the assumption that the core genomes with basic metabolic functions have similar biological functions, we focused on secondary metabolites. Seventeen gene clusters were involved in the production of secondary metabolites (Figure 7). Clusters 1, 2, 4, 14, and 16 are PKS-related clusters. Cluster 16 is present in environmental strain ESE2 only. The cluster contains the single-copy gene benzoate 4-monooxygenase cytochrome P450, which catalyzes the benzoate degradation step in the toluene catabolic pathway (Table 5).
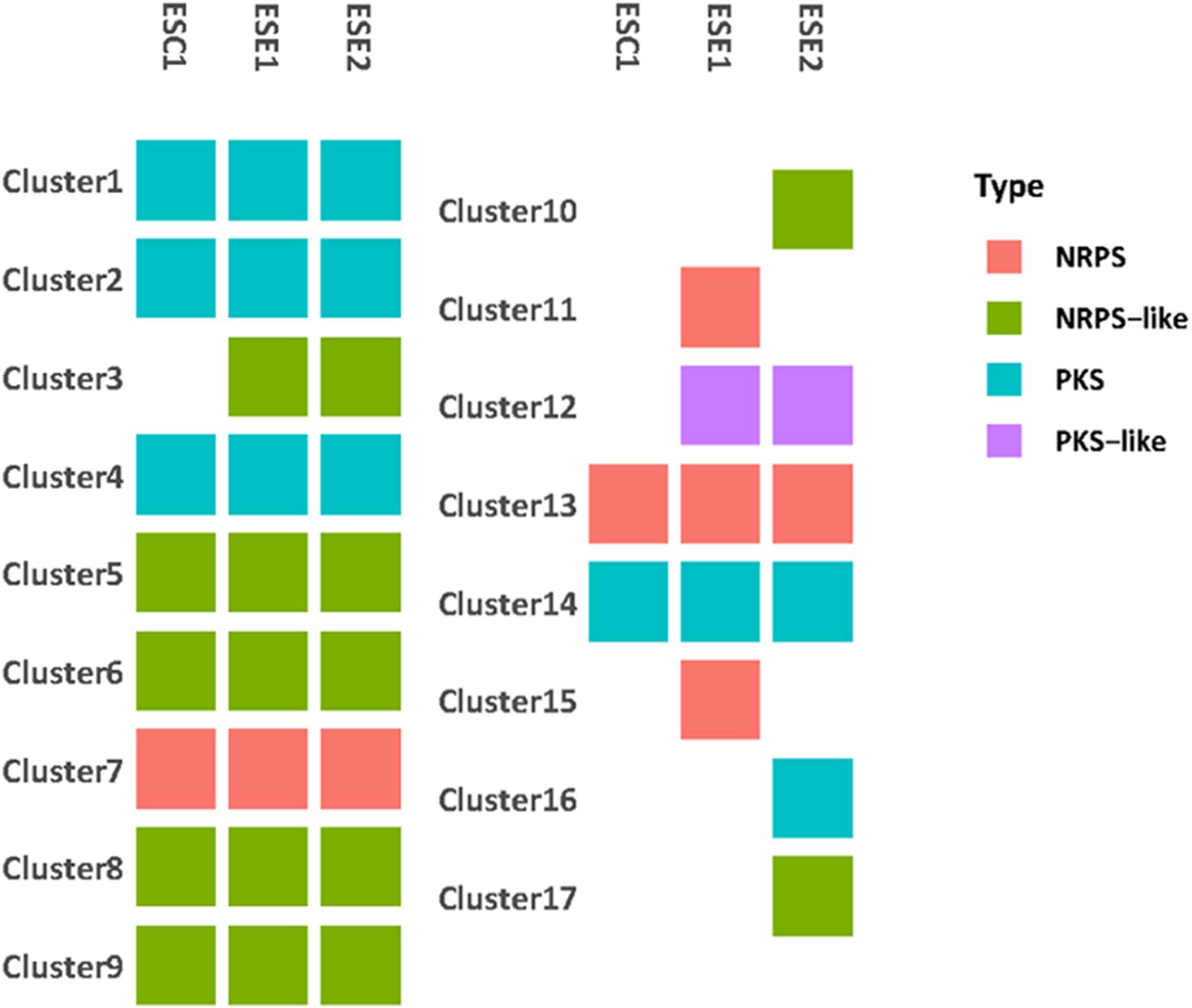
Figure 7. Genome comparison of secondary metabolite clusters of three strains. Seventeen gene clusters were involved in the production of secondary metabolites. Environmental strains ESE1 and ESE2 are predicted to have more secondary metabolite clusters than the clinical strain ESC1.Clusters 1, 2, 4, 14, and 16 are PKS-related clusters. Cluster 16 is present in environmental strain ESE2 only.
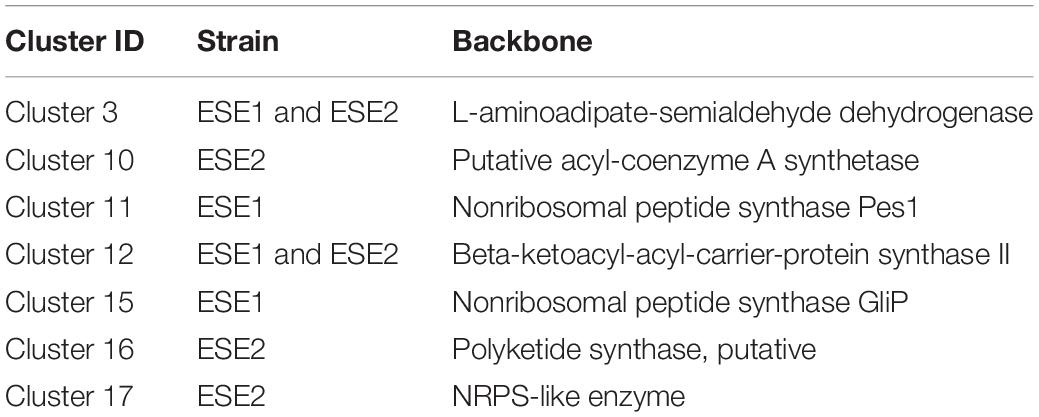
Table 5. Function of specific secondary metabolic gene clusters.
RNA-seq
Sequence clustering at the mRNA level was used to compare the three strains to identify homologous groups among the three strains. For all three strains, poly (A)-enriched, strand-specific RNA-seq from two cultivation conditions (SDB and BHI broth) with different culture degrees was performed. Comparison of differentially expressed genes under different culture conditions demonstrated that ESC1 revealed significant differences in response to the two media in 16 single-copy genes, while the environmental strains ESE1 and ESE2 remained almost indifferent (Figure 8 and Supplementary Table S3).
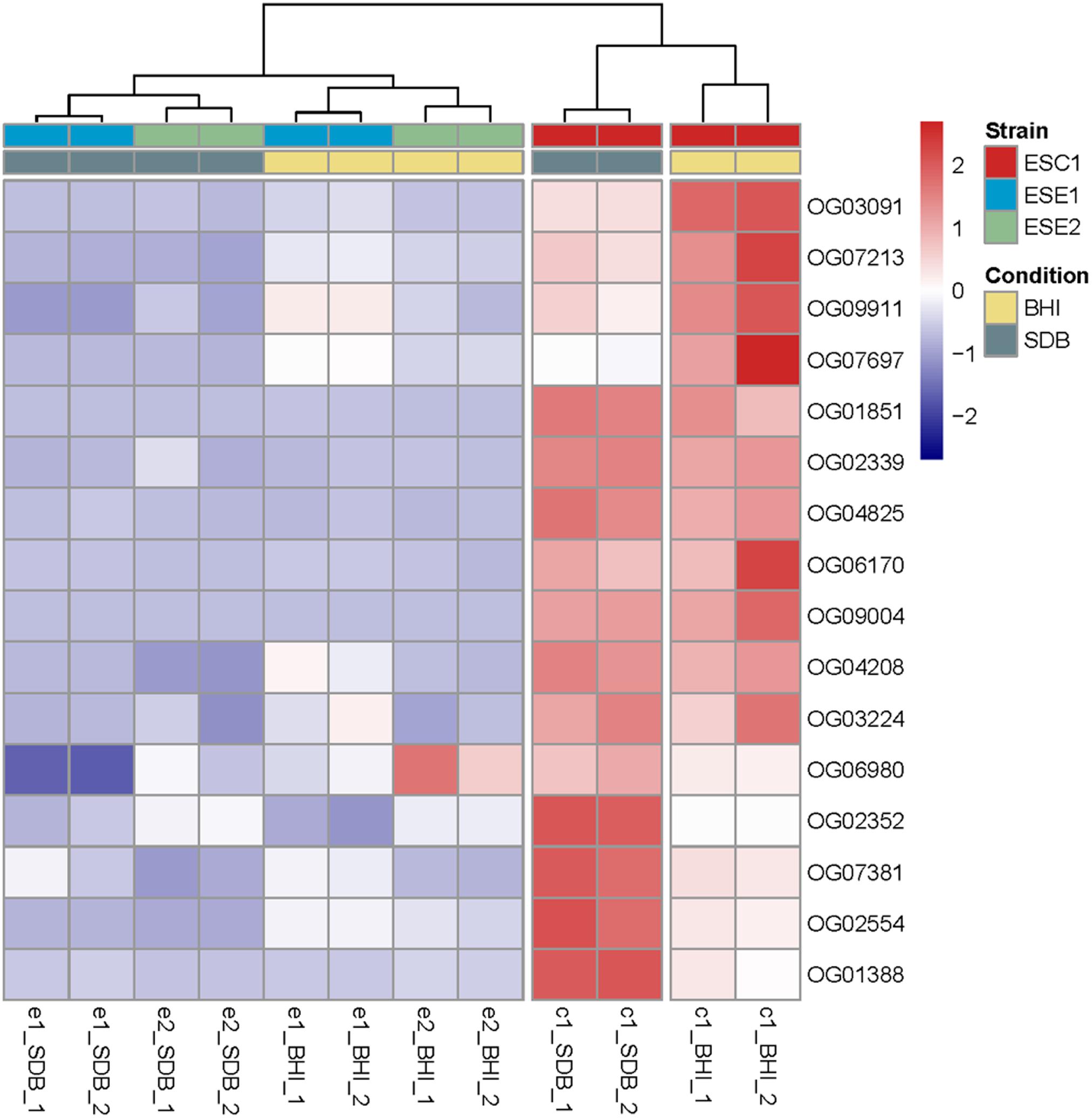
Figure 8. Comparison of differentially expressed genes upon incubation in SDB and BHI broth. Clustering of deviations from zero and shown in a dendrogram by complete linkage hierarchical clustering using Euclidean distance. All tests are shown in duplicate. The results demonstrated that ESC1 revealed significant differences in response to the two media in 16 genes, while the environmental strains ESE1 and ESE2 remained almost indifferent.
There were 48 unique genes (22 in BHI media and 26 genes in SDB media; Table 6) involved in metabolism and transcriptional regulation differentially expressed exclusively in the clinical strain compared to the two environmental strains, including 12 genes without functional annotation. Supplementary Table S4 displays the functional gene content (name, IPR families, KOG, and GO) and respective expression levels in each scenery/media, to enable the visualization/filters of this data. The table includes the gene ID, description, IPR numbers and the GO information. Differentially expressed genes between clinical isolate and environmental isolates under BHI culture are listed in Supplementary Table S5. There were 637 up-regulated genes between ESC1 and ESE1, and 659 up-regulated genes between ESC1 and ESE2. These specific upregulated genes in the clinical strain mainly act in transmembrane transport, translational elongation, ribosome biogenesis, and some other biosynthetic processes (GO enrichment results are summarized in Supplementary Table S6). GO analysis showed that the gene annotations were mainly in three categories: biological processes (BP), molecular function (MF), and cell component (CC). GO categories among three strains showed a similar enrichment distribution (Supplementary Figure S2). However, GO annotations of genes unique to each strain were not identical in BP and MF ontologies. In the biological process, the differentially expressed genes were mainly enriched in response to cellular process and metabolic process. In the MF, differentially expressed genes were mainly related to binding and catalytic activity (Supplementary Figure S3).

Table 6. Unique genes differentially expressed exclusively in the clinical strain.
The correlation between differential expression genes and positive selection genes are listed in Supplementary Table S7. There were 53 positively selected genes that were also up-regulated between ESC1 and ESE1 in BHI culture. Comparing ESC1 and ESE2 under BHI conditions, 53 positively selected genes were also up-regulated in ESC1. A total of 29 positively selected genes were up-regulated in ESC1 when compared with both ESE1 and ESE2 in BHI broth. These genes were involved in positive regulation of translation, regulation of translational termination, and ATP synthesis coupled proton transport.
Genes Under Positive Selection
Models 1 (ω0 = ω1) and 2 (ω0 ≠ ω1) were used to estimate the ω values as free parameters, with LRs as parameters of significance. A total of 29 genes yielded significant values indicating positive selection (p ≤ 0.0001) (Supplementary Table S8). These genes were subjected to GO analysis and were mainly enriched for BP, including cellular response, positive regulation of phosphorylation and transferase activity. Some replication- and transcription-associated genes and several structural protein genes were also discovered under positive selection (Supplementary Table S9).
Discussion
Black yeasts are generally considered to be opportunists; consequently, they should lack any specialized adaptation to the vertebrate host (Seyedmousavi et al., 2018). Infection is coincidentally promoted by factors that enhance survival in the environmental niche of the fungus. Such factors must be present in chaetothyrialean black fungi, given the relatively large number of species reported from infections in humans and in cold-blooded animals. The Chaetothyriales are particularly over-represented in patients with inherited deficiencies in caspase recruitment domain-containing signaling protein 9 (CARD9), a disorder impairing the dectin pathway of innate immunity. This protein enhances pattern recognition receptors to induce NF-κB and MAPK activation, leading to a cytokine cascade. Malfunctioning of the protein decreases protection against infection by larger microbes such as parasites and especially fungi. It has been puzzling why the patients, theoretically being susceptible to any fungal infection, usually carry only a single species. Reviewing the 77 fungal cases published to date (Table 1), a possible explanation might be found in the fact that this protection deficiency appears to be highly specific. Only three groups of fungi are prevalent: Candida spp. (order Saccharomycetales), dermatophytes (order Onygenales) and black fungi of the order Chaetothyriales. A single case of Mucor irregularis has been published. This is the only species of Mucor that is known to cause chronic infections in apparently healthy individuals (Lu et al., 2013), other Mucorales being acute in compromised patients. As noted by Vaezi et al. (2018), there is a regional bias in CARD9 deficiency cases in that the great majority of cases by Candida and dermatophytes are from northern Africa, Turkey, and Iran, while chaetothyrialean cases are particularly encountered in East Asia. Notably, severe and chronic black fungal infections are well-known in China (de Hoog et al., 2020). However, most cases of disseminated fungal infection in “healthy” patients were published before CARD9 deficiency was discovered, thus the possibility is not excluded that more patients had a hidden inherited immune disorder. Remarkably, common opportunists such as Aspergillus or Fusarium are nearly or completely lacking. The over-representation of chaetothyrialean black fungi in this susceptible patient population is also striking because such infections are rare in other patient cohorts, in healthy as well as in otherwise immunocompromised hosts. In black fungi, i.e., members of Chaetothyriales, Pleosporales, and Venturiales, 40% of the mutations were heterozygous. Mutation types tend to be different between the groups of yeasts, dermatophytes, and black fungi (Figure 2): black fungi responded to other mutations than yeasts and dermatophytes. Also the affected cytokines differed between groups.
From these data it is obvious that black fungi, and particularly those belonging to the order Chaetothyriales, respond in a rather specific manner to impairment of the human immune system. Comparing the three main groups (yeasts, dermatophytes, black fungi) with respect to their response to cytokines, quite remarkably, the patients with chronic dermatophyte infections all were reported to have IL-6 deficiency alone, while in black fungi TNFα and often also IL-17A were impaired (Figure 3). In Candida, a more variable picture was observed, but IL-6 deficiency was observed in 23 out of 31 cases. IL-6 is a proinflammatory cytokine controlling T-cell differentiation, particularly Th17 and regulatory T cells (Zhao et al., 2012). These conclusions should however be taken with some care, as cytokine measurements were not consistent between publications.
The data are nevertheless sufficient to surmise that black fungal infections, particularly of members of Chaetothyriales, are not randomly occurring in susceptible patients with impaired immunity in general, but respond specifically to the conditions provided by CARD9 impairment. Because of their pronounced ability to decompose monoaromatic toxins, black yeasts of this order are enriched in the domestic environment (Moreno et al., 2018), and therefore close vicinity to humans is probable. Many species are oligotrophic and have efficient nutrient scavenging systems. Toxin management has been hypothesized to enhance survival strategies in the environment (Gostincar et al., 2018), and industrial pollution by monoaromates might promote growth of these fungi near humans. In this scenario, take-up by susceptible individuals followed by successful infection, may explain their high frequency in CARD9 patients. The cytochrome P-450 gene family has been suggested as a possible factor explaining fungal neurotropism, but our clinical strain lacked PKS cluster 16 which is present in one of the environmental strains (Figure 5 and Table 4) and thus lacks benzoate 4-monooxygenase, an important gene in hydrocarbon catabolism.
Over the past decade, advances in Next Generation Sequencing (NGS) technologies and decreasing sequencing costs allowed an increase in the number of sequenced genomes, with better quality. Long-read sequencing technologies have contributed greatly to comparative genomics among species and can also be applied to study genomics within a species (Kim et al., 2019). The long-read sequencing method, PACBIO, has allowed the ability to obtain complete and accurate sequences. Genome data of our strains are listed in Table 2, showing a range of intraspecific variability of 863,280 bp and 87 genes. These genomes were sequenced by a combined Illumina and PACBIO strategy. We identify a relatively large core genome of 9,887 genes from our three genomes. The two environmental strains shared more genes (336) than a comparison with clinical strain ESC1. The clinical strain had 647 specific gene clusters. Comparing the genomes of E. spinifera sequenced to date, the older Illumina genomes were within the range of the above established species variance. Similarly, comparable numbers of genes were found, suggesting that published genomes are reliable, despite the fact that the number of scaffolds in the Illumina-only genomes was considerably higher.
In our study, the utility of long read sequencing is the possibility to investigate complex fungal genomes and characterize genomic variation. Despite surmised opportunism in black yeasts, our clinical strain of E. spinifera, derived from a CARD9-deficient patient, differed significantly from two environmental strains which originated from different continents. In the environmental/clinical comparison, rearrangements were observed in scaffolds I and III (Figure 4), including two inversions, and ESC1 having an extra scaffold VIII (Figure 4). The rearrangements in this genome area are significant (Figure 4B). In contrast, clinical strain ESC1 was intermediate between the two environmental strains in numbers of SNPs and Indels. This suggests that the genomic rearrangements were a saltational event in an otherwise homogeneous population, as was noted in Candida after passage through a murine host (Forche et al., 2009). If, in contrast to Candida, E. spinifera is an opportunist residing in a non-human habitat, no transmission takes place after infection, and thus mutations are unlikely to be maintained in the population (De Hoog et al., 2018). The possibility cannot be excluded that the rearrangements were acquired during presence in the CARD9-deficient patient. Whether or not these may contribute to increased virulence of the species in an evolutionary step, as suggested by Casadevall (2007), or if they are lost as supposed by De Hoog et al. (2018), requires study of a larger set of genomes and understanding of natural variability of E. spinifera in a wider array of habitats.
E. spinifera shows variable responses with hemolysis (Song et al., 2017). Although these authors noted that this ability did not match with the division environmental/clinical, the possibility is not excluded that hemolytic strains have an enhanced invasive potency for vertebrate hosts. We registered up- or down-regulation of single-copy proteins in response to SDB vs. BHI, and noted that ESC1 showed significant differences with the two media in 16 single-copy genes, while the environmental strains remained almost indifferent; in vitro, ESC1 and ESE1 showed hemolysis, while ESE2 was negative. Most of the single-copy genes upregulated with BHI were transporters in KOG classification. Moreover, there were 48 unique genes differentially expressed exclusively to the clinical strain in two different media, including genes from various metabolic processes and transcriptional regulation. Up-regulated genes in clinical strain in GO enrichment were mainly involved in transmembrane transport, biosynthetic process, and metabolic process.
In this study, significant genomic rearrangements between strains of the same species were demonstrated. Two environmental strains from different continents were largely identical, whereas a clinical strain from a CARD9-deficient patient was different and deviated in the number of genes, even though all strains were phenotypically similar (Song et al., 2017). In general, clinical and environmental strains of a single opportunistic fungus are taken to have comparable infectious abilities, because neither of them is equipped with specialized virulence factors. However, the observed quantitative genetic variability of strains of the single species E. spinifera possibly is associated with a differential chance to cause infection in susceptible patient populations; differences may be small but clinically relevant. We compared our NGS approach with older Illumina techniques, which showed some minor flaws e.g., in the number of scaffolds, but for quantitative description of the genome these data proved to be sufficient. The power of long reads obtained by PACBIO sequencing is to enable assembly of a complete genome sequence, which disclosed significant genomic rearrangements. Main improvement of PACBIO data are expected in qualitative studies of specific gene functions.
Data Availability Statement
The datasets generated for this study can be found in the NCBI. The RNA-seq data can be accessed by PRJNA600825, the PacBio assemblies (PRJNA600825), Illumina assemblies (PRJNA600036). The RNA-seq is available already (https://www.ncbi.nlm.nih.gov/sra/PRJNA600825).
Author Contributions
RL and GS designed the experiments and supervised the data analysis. YS performed the experiments and wrote the relevant portions of the manuscript. MD and EY analyzed the data. NM and VV provided the technical support. All authors discussed the results and commented on the manuscript.
Funding
The authors are grateful for the financial support of International Cooperation and Exchanges Project (NSFC No. 81520108026) from National Natural Science Foundation of China, and National Natural Science Foundation of China (NSFC No. 31670145).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge Prof. Mihai Netea (Radboud University Medical Center) for advice concerning CARD9 prevalence.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.01880/full#supplementary-material
Footnotes
- ^ http://www.bioinformatics.babraham.ac.uk/
- ^ http://www.repeatmasker.org
- ^ http://inparanoid.sbc.su.se/
- ^ http://www.clustal.org/
- ^ https://bioconductor.org/
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Alves de Medeiros, A. K., Lodewick, E., Bogaert, D. J. A., Haerynck, F., Van Daele, S., Lambrecht, B., et al. (2016). Chronic and invasive fungal infections in a family with CARD9 deficiency. J. Clin. Immunol. 36, 204–209. doi: 10.1007/s10875-016-0255-8
Arango-Franco, C. A., Moncada-Vélez, M., Beltrán, C. P., Berrío, I., Mogollón, C., Restrepo, A., et al. (2018). Early-onset invasive infection due to Corynespora cassiicola associated with compound heterozygous CARD9 mutations in a Colombian patient. J. Clin. Immunol. 38, 794–803. doi: 10.1007/s10875-018-0549-0
Bao, Z., and Eddy, S. R. (2002). Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 12, 1269–1276. doi: 10.1101/gr.88502
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Boudghene, O., Amrani, N., Boudghéne Stambouli, K., and Bouali, F. (2017). Dermatophytic disease with deficit in CARD9: a new case with a brain impairment. J. Mycol. Med. 27, 250–253. doi: 10.1016/j.mycmed.2017.01.001
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinform. 10:421. doi: 10.1186/1471-2105-10-421
Casadevall, A. (2007). Determinants of virulence in the pathogenic fungi. Fungal. Biol. Rev. 21, 130–132. doi: 10.1016/j.fbr.2007.02.007
Celmeli, F., Oztoprak, N., Turkkahraman, D., Seyman, D., Mutlu, E., Frede, N., et al. (2016). Successful granulocyte colony-stimulating factor treatment of relapsing Candida albicans meningoencephalitis caused by CARD9 deficiency. Pediatr. Infect. Dis. 35, 428–431. doi: 10.1097/INF.0000000000001028
Cetinkaya, P. G., Ayvaz, D. C., Karaatmaca, B., Gocmen, R., Söylemezoðlu, F., Bainter, W., et al. (2018). A young girl with severe cerebral fungal infection due to card 9 deficiency. Clin. Immunol. 191, 21–26. doi: 10.1016/j.clim.2018.01.002
Chakrabarti, A., Kaur, H., Rudramurthy, S. M., Appannanavar, S. B., Patel, A., Mukherjee, K. K., et al. (2016). Brain abscess due to Cladophialophora bantiana: a review of 124 cases. Med. Mycol. 54, 111–119. doi: 10.1093/mmy/myv091
Chin, C. S., Alexander, D. H., Marks, P., Klammer, A. A., Drake, J., Heiner, C., et al. (2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods. 10, 563–569. doi: 10.1038/nmeth.2474
Conway, J. R., Lex, A., and Gehlenborg, N. (2017). UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics 33, 2938–2940. doi: 10.1093/bioinformatics/btx364
Das, S., Sharma, S., Kar, S., Sahu, S. K., Samal, B., and Mallick, A. (2010). Is inclusion of Sabouraud dextrose agar essential for the laboratory diagnosis of fungal keratitis? Indian J. Ophthalmol. 58, 281–286. doi: 10.4103/0301-4738.64122
De Bruyne, M., Hoste, L., Bogaert, D. J., Van den Bossche, L., Tavernier, S. J., Parthoens, E., et al. (2018). A CARD9 founder mutation disrupts NF-κB signaling by inhibiting BCL10 and MALT1 recruitment and signalosome formation. Front. Immunol. 9:2366. doi: 10.3389/fimmu.2018.02366
De Hoog, G. S., Ahmed, S. A., Danesi, P., Guillot, J., and Gräser, Y. (2018). “Distribution of pathogens and outbreak fungi in the fungal Kingdom,” in Emerging and Epizootic Fungal Infections In Animals, ed. S. Seyedmousavi (Cham: Springer), 3–16.
de Hoog, G. S., Guarro, J., Gené, J., Ahmed, S., Al-Hatmi, A. M. S., Figueras, M. J., et al. (2020). Atlas Of Clinical Fungi, 4th Edn, Utrecht: Westerdijk Fungal Biodiversity Institute.
Delcher, A. L., Salzberg, S. L., and Phillippy, A. M. (2003). Using MUMmer to identify similar regions in large sequence sets. Curr. Protoc. Bioinform. Chapt. 10:Unit1013. doi: 10.1002/0471250953.bi1003s00
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. doi: 10.1093/bioinformatics/bts635
Drewniak, A., Gazendam, R. P., Tool, A. T. J., van Houdt, M., Jansen, M. H., van Hamme, J. L., et al. (2013). Invasive fungal infection and impaired neutrophil killing in human CARD9 deficiency. Blood 121, 2385–2392. doi: 10.1182/blood-2012-08-450551
Drummond, R. A., Collar, A. L., Swamydas, M., Rodriguez, C. A., Lim, J. K., Mendez, L. M., et al. (2015). CARD9-dependent neutrophil recruitment protects against fungal invasion of the central nervous system. PLoS. Pathog. 11:e1005293. doi: 10.1371/journal.ppat.1005293
Emms, D. M., and Kelly, S. (2015). OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16, 157–157. doi: 10.1186/s13059-015-0721-2
Forche, A., Magee, P. T., Selmecki, A., Berman, J., and May, G. (2009). Evolution in Candida albicans populations during a single passage through a mouse host. Genetics 182, 799–811. doi: 10.1534/genetics.109.103325
Gavino, C., Cotter, A., Lichtenstein, D., Lejtenyi, D., Fortin, C., Legault, C., et al. (2014). CARD9 deficiency and spontaneous central nervous system candidiasis: complete clinical remission with GM-CSF therapy. Clin. Infect. Dis. 59, 81–84. doi: 10.1093/cid/ciu215
Gavino, C., Hamel, N., Zeng, J. B., Legault, C., Guiot, M.-C., Chankowsky, J., et al. (2016). Impaired RASGRF1/ERK-mediated GM-CSF response characterizes CARD9 deficiency in French-Canadians. J. Allergy Clin. Immunol. 137, 1178–1188. doi: 10.1016/j.jaci.2015.09.016
Glocker, E.-O., Hennigs, A., Nabavi, M., Schäffer, A. A., Woellner, C., Salzer, U., et al. (2009). A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N. Engl. J. Med. 361, 1727–1735. doi: 10.1056/NEJMoa0810719
Gostincar, C., Stajich, J. E., Zupancic, J., Zalar, P., and Gunde-Cimerman, N. (2018). Genomic evidence for intraspecific hybridization in a clonal and extremely halotolerant yeast. BMC Genom. 19:364. doi: 10.1186/s12864-018-4751-5
Gotz, S., García-Gómez, J. M., Terol, J., Williams, T. D., Nagaraj, S. H., Nueda, M. J., et al. (2008). High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 36, 3420–3435. doi: 10.1093/nar/gkn176
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Grumach, A. S., de Queiroz-Telles, F., Migaud, M., Lanternier, F., Filho, N. R., Palma, S. M. U., et al. (2015). A homozygous CARD9 mutation in a Brazilian patient with deep dermatophytosis. J. Clin. Immunol. 35, 486–490. doi: 10.1007/s10875-015-0170-4
Haas, B. J., Delcher, A. L., Mount, S. M., Wortman, J. R., Smith, R. K. Jr., Hannick, L. I., et al. (2003). Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids. Res. 31, 5654–5666. doi: 10.1093/nar/gkg770
Haas, B. J., Salzberg, S. L., Zhu, W., Pertea, M., Allen, J. E., Orvis, J., et al. (2008). Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9:R7. doi: 10.1186/gb-2008-9-1-r7
Herbst, M., Gazendam, R., Reimnitz, D., Sawalle-Belohradsky, J., Groll, A., Schlegel, P.-G., et al. (2015). Chronic Candida albicans meningitis in a 4-year-old girl with a homozygous mutation in the CARD9 gene (Q295X). Pediatr. Infect. Dis. J. 34, 999–1002. doi: 10.1097/INF.0000000000000736
Hoff, K. J., Lange, S., Lomsadze, A., Borodovsky, M., and Stanke, M. (2016). BRAKER1: unsupervised RNA-seq-based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics 32, 767–769. doi: 10.1093/bioinformatics/btv661
Huang, C., Zhang, Y., Song, Y., Wan, Z., Wang, X., and Li, R. (2019). Phaeohyphomycosis caused by Phialophora americana with CARD9 mutation and 20-year literature review in China. Mycoses 62, 908–919. doi: 10.1111/myc.12962
Jachiet, M., Lanternier, F., Rybojad, M., Bagot, M., Ibrahim, L., Casanova, J.-L., et al. (2015). Posaconazole treatment of extensive skin and nail dermatophytosis due to autosomal recessive deficiency of CARD9. JAMA Dermatol. 151, 192–194. doi: 10.1001/jamadermatol.2014.2154
Jones, P., Binns, D., Chang, H.-Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Kantarcioglu, A. S., Guarro, J., de Hoog, S., Apaydin, H., and Kiraz, N. (2017). An updated comprehensive systematic review of Cladophialophora bantiana and analysis of epidemiology, clinical characteristics, and outcome of cerebral cases. Med. Mycol. 55, 579–604. doi: 10.1093/mmy/myw124
Khaldi, N., Seifuddin, F. T., Turner, G., Haft, D., Nierman, W. C., Wolfe, K. H., et al. (2010). SMURF: genomic mapping of fungal secondary metabolite clusters. Fungal. Genet. Biol. 47, 736–741. doi: 10.1016/j.fgb.2010.06.003
Kim, C., Kim, J., Kim, S., Cook, D. E., Evans, K. S., Andersen, E. C., et al. (2019). Long-read sequencing reveals intra-species tolerance of substantial structural variations and new subtelomere formation in C. elegans. Genome Res. 29, 1023–1035. doi: 10.1101/gr.246082.118
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome. Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Lagesen, K., Hallin, P., Rodland, E. A., Staerfeldt, H. H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Lam, K.-K., LaButti, K., Khalak, A., and Tse, D. (2015). FinisherSC: a repeat-aware tool for upgrading de novo assembly using long reads. Bioinformatics 31, 3207–3209. doi: 10.1093/bioinformatics/btv280
Lanternier, F., Barbati, E., Meinzer, U., Liu, L., Pedergnana, V., Migaud, M., et al. (2015a). Inherited CARD9 deficiency in 2 unrelated patients with invasive Exophiala infection. J. Infect. Dis. 211, 1241–1250. doi: 10.1093/infdis/jiu412
Lanternier, F., Mahdaviani, S. A., Barbati, E., Chaussade, H., Koumar, Y., Levy, R., et al. (2015b). Inherited CARD9 deficiency in otherwise healthy children and adults with Candida species-induced meningoencephalitis, colitis, or both. J. Allergy. Clin. Immunol. 135, 1558–1568. doi: 10.1016/j.jaci.2014.12.1930
Lanternier, F., Pathan, S., Vincent, Q. B., Liu, L., Cypowyj, S., Prando, C., et al. (2013). Deep dermatophytosis and inherited CARD9 deficiency. N. Engl. J. Med. 369, 1704–1714. doi: 10.1056/NEJMoa1208487
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Liao, Y., Smyth, G. K., and Shi, W. (2014). FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. doi: 10.1093/bioinformatics/btt656
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550–550. doi: 10.1186/s13059-014-0550-8
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.955
Lu, X. L., Najafzadeh, M. J., Dolatabadi, S., Ran, Y. P., Gerrits van den Ende, A. H. G., Shen, Y. N., et al. (2013). Taxonomy and epidemiology of Mucor irregularis, agent of chronic cutaneous mucormycosis. Persoonia. 30, 48–56. doi: 10.3767/003158513X665539
Moreno, L. F., Ahmed, A. A. O., Brankovics, B., Cuomo, C. A., Menken, S. B. J., Taj-Aldeen, S. J., et al. (2018). Genomic understanding of an infectious brain disease from the desert. G3 8, 909–922. doi: 10.1534/g3.117.300421
Price, A. L., Jones, N. C., and Pevzner, P. A. (2005). De novo identification of repeat families in large genomes. Bioinformatics 21(Suppl. 1), i351–i358. doi: 10.1093/bioinformatics/bti1018
Seyedmousavi, S., Bosco, S. D. M. G., de Hoog, S., Ebel, F., Elad, D., Gomes, R. R., et al. (2018). Fungal infections in animals: a patchwork of different situations. Med. Mycol. 56, 165–187. doi: 10.1093/mmy/myx104
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Song, Y., Laureijssen-van de Sande, W. W. J., Moreno, L. F., Gerrits van den Ende, B., Li, R., and de Hoog, S. (2017). Comparative ecology of capsular Exophiala species causing disseminated infection in humans. Front. Microbiol. 8:2514. doi: 10.3389/fmicb.2017.02514
Vaezi, A., Fakhim, H., Abtahian, Z., Khodavaisy, S., Geramishoar, M., Alizadeh, A., et al. (2018). Frequency and geographic distribution of CARD9 mutations in patients with severe fungal infections. Front. Microbiol. 9:2434. doi: 10.3389/fmicb.2018.02434
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963
Wang, X., Wang, W., Lin, Z., Wang, X., Li, T., Yu, J., et al. (2014). CARD9 mutations linked to subcutaneous phaeohyphomycosis and TH17 cell deficiencies. J. Allergy. Clin. Immunol. 133, 905–908.e3. doi: 10.1016/j.jaci.2013.09.033
Yan, X. X., Yu, C. P., Fu, X. A., Bao, F. F., Du, D. H., Wang, C., et al. (2016). CARD9 mutation linked to Corynespora cassiicola infection in a Chinese patient. Br. J. Dermatol. 174, 176–179. doi: 10.1111/bjd.14082
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Zhang, H., Yohe, T., Huang, L., Entwistle, S., Wu, P., Yang, Z., et al. (2018). dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 46, W95–W101. doi: 10.1093/nar/gky418
Zhang, R., Wang, X., Wan, Z., and Li, R. (2017). CARD9 mutations and related immunological research of one case with disseminated phaeohyphomycosis. J. Microbiol. Infect. 12, 14–23.
Zhao, X., Boenisch, O., Yeung, M., Mfarrej, B., Yang, S., Turka, L. A., et al. (2012). Critical role of proinflammatory cytokine IL-6 in allograft rejection and tolerance. Am. J. Transplant. 12, 90–101. doi: 10.1111/j.1600-6143.2011.03770.x
Keywords: black yeasts, comparative genomics, transcriptome analysis, intraspecific variability, gene rearrangement, CARD9 deficiency, pathogenicity, virulence
Citation: Song Y, Du M, Menezes da Silva N, Yang E, Vicente VA, Sybren de Hoog G and Li R (2020) Comparative Analysis of Clinical and Environmental Strains of Exophiala spinifera by Long-Reads Sequencing and RNAseq Reveal Adaptive Strategies. Front. Microbiol. 11:1880. doi: 10.3389/fmicb.2020.01880
Received: 22 February 2020; Accepted: 16 July 2020;
Published: 31 July 2020.
Edited by:
Esther Segal, Tel Aviv University, IsraelReviewed by:
Macit Ilkit, Çukurova University, TurkeyKathryn Bushley, University of Minnesota Twin Cities, United States
Copyright © 2020 Song, Du, Menezes da Silva, Yang, Vicente, Sybren de Hoog and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: G. Sybren de Hoog, cy5ob29nQHdpLmtuYXcubmw=; Ruoyu Li, bXljb2xhYkAxMjYuY29t