Abstract
Significant advances have been made uncovering the factors that render neurons vulnerable in Parkinson's disease (PD). However, the critical pathogenic events leading to cell loss remain poorly understood, complicating the development of disease-modifying interventions. Given that the cardinal motor symptoms and pathology of PD involve the loss of dopamine (DA) neurons of the substantia nigra pars compacta (SNc), a majority of the work in the PD field has focused on this specific neuronal population. PD however, is not a disease of DA neurons exclusively: pathology, most notably in the form of Lewy bodies and neurites, has been reported in multiple regions of the central and peripheral nervous system, including for example the locus coeruleus, the dorsal raphe nucleus and the dorsal motor nucleus of the vagus. Cell and/or terminal loss of these additional nuclei is likely to contribute to some of the other symptoms of PD and, most notably to the non-motor features. However, exactly which regions show actual, well-documented, cell loss is presently unclear. In this review we will first examine the strength of the evidence describing the regions of cell loss in idiopathic PD, as well as the order in which this loss occurs. Secondly, we will discuss the neurochemical, morphological and physiological characteristics that render SNc DA neurons vulnerable, and will examine the evidence for these characteristics being shared across PD-affected neuronal populations. The insights raised by focusing on the underpinnings of the selective vulnerability of neurons in PD might be helpful to facilitate the development of new disease-modifying strategies and improve animal models of the disease.
Introduction
Parkinson's disease (PD) was first described two centuries ago in An essay on the shaking palsy (1). Since then, great strides have been made in understanding the disease basics. However—as with many other neurodegenerative disorders—there is still no disease modifying treatment for PD. Unfortunately, progress has been slow, and a thorough understanding of the pathological processes has been elusive.
PD as a clinical diagnosis is characterized by the detection of significant motor deficits (including bradykinesia, resting tremor, and rigidity) due, in large part, to a loss of dopamine (DA)-containing neurons of the substantia nigra pars compacta (SNc). The SNc is a neuronal population projecting to the caudate and putamen and is critical for regulation of basal ganglia circuitry. At clinical presentation, it has been estimated that 40–60% of SNc DA neurons have already degenerated (2, 3). The clinical features of the disease are diverse and include substantial non-motor features including, autonomic and olfactory dysfunction, constipation, sleep disturbances, depression, and anxiety (4–6).
The diagnostic criteria for PD have been recently re-defined by the International Parkinson and Movement Disorder Society (MDS), with the MDS Clinical Diagnostic Criteria for Parkinson's disease [MDS-PD Criteria (7)]. A diagnosis is made when there is documented parkinsonism (defined as bradykinesia, with tremor at rest and/or rigidity), followed by the exclusion of other possible causes of parkinsonism, and with additional supporting criteria, including olfactory dysfunction or cardiac sympathetic denervation [see (7)]. The recent nature of this re-evaluation illustrates both the heterogeneity of PD expression, and the difficulties encountered in defining it.
In ≈70% of the ‘clinically typical PD cases’, the hallmark pathological finding is the presence of Lewy pathology (LP) in the SNc (4, 5)—however, LP is also found across the central, peripheral, and enteric nervous system (CNS, PNS, and ENS) (6). This includes both Lewy bodies and Lewy neurites: both similar cellular inclusions, formed predominantly of aggregated α-synuclein, but also including a large number of different molecules, proteins and organelles, such as ubiquitin, tubulin, neurofilaments, lipids, and mitochondria (8).
In considering the broad localization of LP and the origins of the various symptoms of PD, a critical point to consider is the dysfunction and loss of neurons in regions of the CNS and PNS, other than the SNc. There have been, indeed, many studies concluding that cholinergic neurons in the pedunculopontine nucleus (PPN), noradrenergic neurons of the locus coeruleus (LC), cholinergic neurons of the nucleus basalis of Meynert (NBM) and of the dorsal motor nucleus of the vagus (DMV), and serotonergic neurons of the raphe nuclei (RN) are lost in PD. The strength of the evidence for actual neuronal cell body loss in these regions is highly variable and is one of the questions addressed in the present review. The fact that the diagnostic criteria for PD have over time been refined adds another layer of complexity to the task of identifying the origin of the diverse symptoms of PD. Presently, PD is classified into either primary or secondary subtypes. Primary parkinsonism includes genetic and idiopathic forms of the disease and secondary parkinsonism includes forms induced by drugs, infections, toxins, vascular defects, brain trauma or tumors or metabolic dysfunctions. This second subtype of PD is also sometimes called atypical parkinsonism when concomitant to progressive supranuclear palsy, multiple system atrophy or corticobasal degeneration, for example.
Since pathology is likely to emerge through different processes depending of PD subtypes, and since modern classification was non-existent when a substantial part of the research literature was produced, attempting to reach clear general vision of various pathophysiological markers and their link to disease progression for each sub-type of PD presents a significant challenge. This review will primarily focus on idiopathic PD, since this category represents the large majority of cases and is likely to represent most of the subjects examined in studies where PD type was not provided.
Another main hurdle in PD research is that the chain of events that leads to the death of neurons is still not clear. The fact that pathology is thought to begin years/decades before the appearance of symptoms might, in part, explain this lack of progress.
PD has been considered to exist as either a strictly monogenetic or environmentally-triggered disease, as well as a mixture of the two. The pathological mechanisms at the core of each form have been proposed to converge in causing cellular stress secondary to mitochondrial dysfunction, perturbed proteostasis and elevated oxidative stress. A major conundrum is that at first glance, these factors alone fail to explain why PD pathology is restricted to very limited subsets of brain nuclei. Therefore, a key question is what do these PD sensitive neurons have in common and what is it about them that renders them more vulnerable compared to neurons from other brain regions?
A better understanding of the fundamental nature of cell loss and cellular dysfunction in the parkinsonian brain is required to develop critically needed, novel, therapeutic strategies. In this review, we aim to re-evaluate the evidence for cell loss in PD, then to highlight the common characteristics that could explain their selective vulnerability.
Physiopathology of parkinson's disease
The focus on SNc DA neurons has brought significant advances in our understanding of PD pathophysiology, as well as of the signaling pathways that lead to DA neuron death. Studies using DA neuron selective toxins such as 6-OHDA and MPTP, as well as investigations of gene products mutated in familial forms of the disease (including α-synuclein, Parkin, Pink1, LRRK2, DJ-1, and GBA1), have been instrumental to better understand some of the key dysfunctional processes implicated in the disease. These include protein clearance (9–11), mitochondrial turnover (12–14), ROS management (15, 16), and inflammation (17, 18). Perturbations of these processes have been proposed to underlie distinct physiological dysfunctions in PD-vulnerable neurons (19). Nonetheless, since the first introduction of Levodopa in the 1950s and the development of deep-brain stimulation in the 1990s, increased understanding of PD pathophysiology has not yet permitted the discovery of disease-modifying therapies.
As stated previously, PD is more than just a disease of DA and the SNc. Non-motor symptoms—including a reduced sense of smell, constipation, orthostatic hypotension, sleep disturbances, depression, and anxiety—are likely to be due to impaired function and/or loss of non-DA neurons (20). There has thus been a growing interest in better understanding the implications of other regions of the CNS and PNS in the progression of PD pathology. In the early 2000s, pioneering work by Braak and colleagues defined stages in PD based on the appearance of LP in various regions of the nervous system, correlating their findings to the symptomatic progression of the disease (21–23). Most notably, LP was detected in the dorsal IX/X motor nuclei, the intermediate reticular zone, the medulla oblongata, the pontine tegmentum, the caudal RN, the gigantocellular reticular nucleus, the coeruleus–subcoeruleus complex, the pars compacta of the substantia nigra, the basal prosencephalon, the mesocortex, and the neocortex. However, multiple lines of evidence suggest that LP is not systematically seen in the PD brain and LP is also documented in healthy individuals (24). Also, in some cases of PD, and most notably in early-onset genetic forms, loss of SNc DA neurons has been reported to occur in the absence of detectable LP (25–27).
Although the role of LP in the pathogenesis of PD has been the subject of much debate (28), the detection of LP has remained central in investigations of the key brain regions and circuits underlying PD pathophysiology. In this context, it may be useful to focus attention on brain and PNS regions that show documented cell death and/or axonal degeneration, irrespective of the presence or absence of LP. This could perhaps provide new perspectives on the actual, more proximate, causes of the major symptoms of the disease and their progression. Relevant to the present point, in their most recent and insightful work, Braak and Tredici write, “We ascribed the same weight to axonopathy and nerve cell dysfunction (presumably attributable, but not limited, to the presence of Lewy pathology) as to neuronal death because the development of pathology together with neurotransmitter loss, axonal, and somatodendritic dysfunction in multiple neuronal populations could prove to be more stressful for involved neurons over time than premature cell death within a select neuronal population” (6).
Where and when does neuronal loss appear in PD?
Loss of neurons in the brain is thought to occur in the context of normal aging. For example, there have been multiple publications reporting significant age-dependent decline in neuron number in the SNc (29–37), as well as in regions such as the PPN (38), and LC (39, 40). Above and beyond such cell loss associated with normal aging, a key question is where in the brain can one find substantial neuronal loss in PD?
Although numerous publications have referred to cell loss occurring in many CNS and PNS regions in the context of PD, we believed it germane to re-evaluate the published scientific literature addressing this question.
To do so, we took great care to find work concentrating on neuronal loss and not only denervation [as is common for the heart, for example (41–43)]. We found 90 primary research articles reporting PD-specific cell loss in the following regions (Table 1): the SNc, VTA, amygdala, cortex, DMV, hypothalamus, laterodorsal tegmental nucleus, LC, NBM, OB, oral pontine reticular nucleus, PPN, pre-supplementary motor cortex, RN, supraoptic nucleus, sympathetic/parasympathetic ganglia, and thalamus. These original articles span from 1953 to 2015. The techniques used to quantify cell loss varied, and we have classified them accordingly. Across all regions examined, 14 of the examinations were defined as observational, 39 as implicating manual counting, 18 used computer-assisted counting, and 26 used stereological counting methods. While informative, the value of observational studies can be considered limited given their lack of precision and the fact that they are greatly influenced by the observer. Lack of bias is also difficult to assure in studies involving manual counting. This technique is also unable to assure that a cell is not being counted twice if present in two subsequent sections. Other techniques such as computer-assisted counting were developed to improve on these aforementioned methods, however, these are also limited in that they often lack rigorous systematic sampling, are sensitive to tissue shrinkage, and are often unable to account for local tissue thickness, or for cells damaged on slice edges. These issues are systematically addressed using modern stereological counting techniques. Another issue to consider is that many of the studies included in this review, including those employing stereology, either did not use age-matched controls, or did not state whether counting was conducted blind to diagnosis. Yet another apparent feature of this literature is the diversity of method iterations used, the varying number of brain regions assessed in each study and, importantly, the stage or type of PD studied (and how this was defined). Here, we will discuss the evidence of cell loss (if not otherwise stated, relative to healthy control cases), ordering the regions in subsections according to the strength of the evidence (Table 1).
Table 1
| Regions | Publications (reference #) | Technique | N (ctrl) | Loss of neurons (%) | Comparison group info (healthy controls unless stated otherwise) | Blinded/age matched | Stated diagnosis, scale of severity, disease duration (expressed in range or mean, when available) | Other regions counted | Correlations (with disease severity, duration or age) |
|---|---|---|---|---|---|---|---|---|---|
| Substantia nigra pars compacta (SNc) | Greenfield and Bosanquet (44) | o | 19 (22) | Some | – | Not stated | iPA, <1–20 years | LC | – |
| Pakkenberg and Brody (45) | m | 10 (10) | 66 | Healthy controls and two young controls | Not stated/Yes | iPA | – | – | |
| Bernheimer et al. (46) | o | 69 (0) | Some | No healthy controls, compared to type of PD and Huntington's disease | Not stated/Yes | PD, H&Y, 1–47 years | – | – | |
| Rajput and Rozdilsky (47) | o | 6 (1) | Some | – | Not stated | iPA, H&Y, 3–18 years | LC, DMV, Cortex, Hypothalamus, Intermediolateral spinal cord, sympathetic ganglia | – | |
| Gaspar and Gray (48) | o | 32 (6) | Some | – | Yes/Yes | iPD, 2–23 years | LC, NBM | – | |
| Tagliavini et al. (49) | o | 6 (5) | Some | – | Not stated/Yes | iPD, 5–13 years | NBM | – | |
| Chan-Palay (50) | o | 9 (22) | Some | – | Yes/Not stated | PD | NBM | – | |
| Gibb and Lees (51) | m | 34 (–) | – | No healthy controls, compared young and old onset | Not stated | PD, 1–34 years | – | – | |
| Hirsch et al. (52) | c | 4 (3) | 77 | – | Not stated | PD | A10, A8, CGS | – | |
| German et al. (53) | c | 5 (3) | 61 | – | Not stated/Yes | PD, 5–27 years | VTA | – | |
| Rinne et al. (54) | s | 12 (18) | 60 | – | Not stated/Yes | iPD, H&Y II–V | – | Yes | |
| Zweig et al. (55) | o | 6 (8) | Mild to severe | Not compared—estimation | Not stated/Yes | PD, 5–14 years | PPN, DR, NBM | – | |
| Gibb et al. (56) | m | 6 (6) | 75 | – | Not stated | PD | – | – | |
| Halliday et al. (57) | c | 4 (4) | 68 | – | Not stated/Yes | PD | SNc + LC, RN, PPN, DMV | Yes (dementia score) | |
| Fearnley and Lees (31) | m | 20 (36) | 20–90 | – | Not stated/Yes | PD, 1.5–38 years | – | Yes (also in controls) | |
| Pakkenberg et al. (58) | s | 7 (7) | 66 | – | Not stated/Yes | PD, 4–16 years | – | – | |
| Paulus and Jellinger (59) | m | 39 (14) | 59 | – | Not stated/Yes | PD, H&Y III–V, 1–31 years | LC, DRN, NBM | – | |
| Xuereb et al. (60) | o | 5 (5) | Some | – | Not stated/Yes | PD | Thalamus (multiple nuclei) | – | |
| Moller (61) | c | 3 (3) | 80 | – | Not stated/Yes | PD | – | – | |
| Zweig et al. (62) | m | 13 (14) | Some | – | Yes | PD, H&Y 4.5, 11 years | LC, VTA, NBM | – | |
| Mouatt-Prigent et al. (63) | c | 4 (3) | 76 | – | Not stated/Yes | iPD | VTA | – | |
| Ma et al. (64) | s | 4 (7) | 70 | – | Not stated | PD | – | – | |
| Halliday et al. (65) | s | 11 (15) | 37–75 | – | Not stated/Yes | PD, 1–18 years | – | Yes | |
| Ma et al. (66) | c | 20 (8) | 76 | – | Not stated/Yes | PD | – | – | |
| Ma et al. (67) | s | 12 (12) | 55 | – | Not stated/Yes | PD, H&Y III–V, 3–17 years | – | Yes | |
| Damier et al. (68) | c | 5 (5) | 86 | – | Not stated | iPD | VTA | Yes | |
| Henderson et al. (69) | c | 9 (8) | 69 | – | Not stated/Yes | PD, H&Y II–V, 3–17 years | Centromedian–Parafascicular Complex, mediodorsal or anterior principal nucleus | – | |
| Zarrow et al. (70) | m | 19 (13) | 78 | Healthy controls, AD | Not stated/Yes | iPD, 12.4 years | LC, NBM | ||
| Greffard et al. (71) | o | 14 (5) | 50 | – | Not stated/Yes | iPD, UPDRS3 = 53, 8.5 years | – | Yes | |
| Rudow et al. (35) | s | 8 (23) | ~80 vs. young, ~75 vs. old controls | Young, middle aged and old healthy controls | Not stated/Yes | PD, 7–20 years | –. | Yes, in controls | |
| Beach et al. (72) | o | 66 (87) | some | Healthy controls, ILDB, DLB, ADLB, ADNLB | Yes/Not stated | PD + DLB, UPDRS = 41, 10.6 years | – | – | |
| Karachi et al. (73) | s | 12 (8) | 69–88 | – | Yes | PD, UPDRS | PPN | – | |
| Milber et al. (74) | s | 13 (17) | 70 | Healthy controls, iLBD | Yes/Not stated | PD, Braak stage I–VI, 8.3 years. | – | Yes in iLBD | |
| Kordower et al. (75) | s | 28 (9) | 50–90 | – | Yes | PD, 1–27 years | – | Yes | |
| Dijkstra et al. (76) | s | 24 (12) | 56 | Healthy controls, iLBD | Yes | PD and iLBD, Braak stage 0–VI, H&Y, 13.6 years | – | Yes | |
| Kraemmer et al. (77) | m | 4 (0) | – | No healthy controls, compare to AD, CJD, CBS, NPH | Yes/Not stated | PD and DLB, 2–4 years | – | – | |
| Cheshire et al. (78) | s | 44 (17) | 75 | – | Yes | PD, LID severity, 14.8 years | RN | – | |
| Iacono et al. (79) | s | 6 (6) | 82 | – | Yes | iPD and iLDB, Braak stage I–IV, H&Y 2–5, | – | – | |
| Total | 38 | o10, m8, c8, s12 | 612 (452) | ||||||
| Locus coeruleus (LC) | Rajput and Rozdilsky (47) | o | 6 (1) | Some | – | Not stated | iPA H&Y, 3–18 years | SN, DMV, Cortex, Hypothalamus, Intermediolateral spinal cord, sympathetic ganglia | – |
| Gaspar and Gray (48) | o | 32 (6) | Some | – | Yes | iPD, 2–23 years | SNc, NBM | – | |
| Hirsch et al. (52) | c | 4 (3) | 55 | – | Not stated | PD | SNc, A10, A8 | – | |
| Chan-Palay and Asan (80) | c | 6 (3) | 31–94* | – | Not stated/Yes | PD | – | – | |
| Zweig et al. (55) | o | 6 (8) | Mild to severe | Not compared—estimation | Not stated/Yes | PD, 5–14 years | PPN, SNc, DR, NBM | – | |
| Halliday et al. (57) | c | 4 (4) | 68 | – | Not stated/Yes | PD | SNc + LC, RN, PPN, DMV | – | |
| Gai et al. (81) | c | 6 (5) | 74 | – | Not stated/Yes | iPD, 5–30 years | PPN, LTN, OPN, RN | Yes | |
| Paulus and Jellinger (59) | m | 37 (12) | 63 | – | Not stated/Yes | PD, H&Y III–V, 1–31 years | SNc, DRN, NBM | – | |
| German et al. (82) | c | 6 (7) | 21–93 | Healthy controls, AD, down-syndrome | Not stated/Yes | PD, 5–16 years | – | – | |
| Patt and Gerhard (83) | o | 8 (8) | Some | – | Not stated | PD | – | – | |
| Zweig et al. (62) | m | 13 (14) | 46–69 | – | Yes/Yes | PD, H&Y 4.5, 11 years | SNc, VTA, NBM | – | |
| Hoogendijk et al. (84) | c | 5 (5) | 39 NS | Healthy controls, AD, ALS | Not stated/Yes | PD, 7 years | – | – | |
| Bertrand et al. (85) | c | 11 (6) | 58–78 | – | Not stated | PD | – | Yes | |
| Zarrow et al. (70) | m | 19 (13) | 83 | Healthy controls, AD | Not stated/Yes | iPD, 12.4 years | SNc, NBM | – | |
| Brunnstrom et al. (86) | m | 25 (0) | Mild-severe | Healthy controls, AD | Yes/Not stated | DLB and PD dementia | – | – | |
| McMillan et al. (87) | m | 7 (8) | 71–88 | Healthy controls, AD, DLB | Yes | PD, 7–25 years | – | – | |
| Dugger et al. (88) | c | 21 (11) | Some | – | Not stated/Yes | LBD, 8.4 years | PPN | – | |
| Del Tredici and Braak (89) | o | 5 (1) | Some | – | Not stated | PD, H&Y 3–5, 7–15 years | – | – | |
| Total | 18 | o5, m4, c9, s0 | 221 (115) | – | |||||
| *31 w/o dementia, 48 w/dementia, 94 if Non-responsive to L-dopa | |||||||||
| Nucleus basalis of meynert (NBM) | Arendt et al. (90) | m | 5 (14) | 70 | – | Not stated/Yes | Postencephalitic PD | – | – |
| Candy et al. (91) | m | 5 (5) | Some | Healthy controls, AD | Not stated | PD | – | – | |
| Nakano and Hirano (92) | m | 2 (5) | 90 | – | Not stated/Yes | PD-dementia complex of Guam, 4–5 years | – | – | |
| Whitehouse et al. (93) | m | 12 (10) | 45–71 | – | Yes | iPD, 4–26 years | – | – | |
| Gaspar and Gray (48) | m | 32 (6) | 36 | – | Yes | iPD, 2–23 years | SNc, LC | – | |
| Nakano and Hirano (94) | m | 11 (13) | 60 | – | Not stated/Yes | PD, 1–17 years | – | – | |
| Tagliavini et al. (49) | m | 6 (5) | 46–69 | – | Not stated/Yes | iPD, 5–13 years | SNc | – | |
| Perry et al. (95) | m | 4 (8) | 17–72 | Healthy controls, AD | Not stated/Yes | PD | – | – | |
| Rogers et al. (96) | m | 4 (5) | Some | Healthy controls, PSP, Creutzfeldt-Jakob disease, ALS, MS and AD (+ individual cases of other diseases) | Not stated/Yes | PD | – | – | |
| Chan-Palay (50) | m | 9 (22) | ~50 | Healthy controls, AD | Yes/Not stated | PD | SNc | – | |
| Paulus and Jellinger (59) | m | 40 (17) | Some | – | Not stated/Yes | PD, H&Y III–V, 1–31 years | SNc, LC, DRN | – | |
| Zweig et al. (62) | o | 13 (14) | Some | – | Yes | PD, H&Y 4.5, 11 years | LC, SNc, VTA | – | |
| Zarrow et al. (70) | m | 19 (13) | 37 | Healthy controls, AD | Not stated/Yes | iPD, 12.4 years | SNc, LC | – | |
| Total | 13 | o1, m12, c0, s0 | 162 (137) | ||||||
| Pedunculopontine nucleus (PPN) | Hirsch et al. (97) | c | 6 (4) | 57 | Healthy controls, supranuclear palsy | Not stated | PD | – | – |
| Jellinger (98) | m | 14 (15) | 53 | – | Not stated/Yes | PD, 10 years | – | – | |
| Zweig et al. (55) | m | 4 (8) | 46–69 | – | Not stated/Yes | PD, 10–14 years | – | – | |
| Halliday et al. (57) | c | 4 (4) | 57 | – | Not stated/Yes | PD | SNc + LC, RN, DMV | – | |
| Gai et al. (81) | c | 6 (5) | 43 | – | Not stated/Yes | iPD, 5–30 years | LTN, OPN, RN, LC | Yes | |
| Rinne et al. (99) | s | 11 (9) | 40 | – | Not stated/Yes | PD, H&Y 2.5 and 5, 9.3 years | – | Yes | |
| Schmeichel et al. (100) | m | 13 (11) | 65 | Healthy controls, MSA | Yes/Not stated | DLB, 3–16 years | Laterodorsal tegmental nucleus | – | |
| Karachi et al. (73) | s | 12 (8) | 31–38 | – | Yes | PD, UPDRS 0–IV | SN | – | |
| Dugger et al. (88) | c | 21 (11) | Some | – | Not stated/Yes | LBD, 8.4 years | LC | – | |
| Hepp et al. (101) | s | 9 (9) | 41 | Healthy controls, DLB | Yes | PD, Braak stage IV–VI, H&Y IV–V, 8–26 years | – | – | |
| Pienaar et al. (102) | s | 8 (5) | 50 | – | Yes | PD, Braak stage II–IV, 6–13 years | – | – | |
| Total | 11 | o0, m3, c4, s4 | 108 (89) | ||||||
| Hypothalamus | Rajput and Rozdilsky (47) | o | 6 (1) | None | – | Not stated | iPA, H&Y, 3–18 years | SN, LC, DMV, Cortex, intermediolateral spinal cord, sympathetic ganglia | – |
| Kremer (103) | m | 8 (15) | None | – | Not stated | PD | – | – | |
| Kremer and Bots (104) | m | 8 (7) | None | – | Not stated/Yes | iPD, 4–17 years | – | – | |
| Purba et al. (105) | m | 6 (6) | 20 | – | Not stated/Yes | PD | – | – | |
| Nakamura et al. (106) | m | 8 (6) | None | – | Not stated/Yes | iPD | – | – | |
| Ansorge et al. (107) | m | 7 (8) | 12–29 | – | Not stated/Yes | PD, 18 years | – | – | |
| Hoogendijk et al. (108) | m | 12 (6) | None | – | Yes | iPD | – | – | |
| Fronczek et al. (109) | c | 9 (9) | 45 | – | Yes | PD, late-stage | – | – | |
| Thannickal et al. (110) | s | 10 (5) | 50 | – | Not stated/Yes | PD, H&Y I–V, 4–23 years | – | Yes | |
| Total | 9 | o1, m6, c1, s1 | 74 (63) | ||||||
| Dorsal motor nucleus of the vagus nerve (DMV) | Eadie (111) | m | 8 (5) | 30 | – | Not stated/Yes | PD | Hypoglossal nuclei, nucleus ambiguus | – |
| Rajput and Rozdilsky (47) | o | 6 (1) | Some | – | Not stated | iPA, H&Y, 3–18 years | SN, LC, Cortex, Hypothalamus, Intermediolateral spinal cord, sympathetic ganglia | – | |
| Halliday et al. (112) | c | 4 (4) | 77 | – | Not stated | PD | RN | – | |
| Halliday et al. (57) | c | 4 (4) | 77 | – | Not stated/Yes | PD | SNc + LC, RN, PPN | – | |
| Saper et al. (113) | m | 5 (5) | 60 | – | Not stated | PD, 2–16 years | – | – | |
| Gai et al. (114) | s | 8 (6) | 55 | – | Not stated/Yes | PD, 5–24 years | Hypoglossal nucleus | Yes | |
| Benarroch et al. (115) | o | 14 (12) | 50 | – | Yes/Not stated | PD or LBD, 10 years | Nucleus ambiguus | – | |
| Total: | 7 | o2, m2, c2, s1 | 49 (37) | ||||||
| Raphe nuclei (RN) | Yamamoto and Hirano (116) | m | 2 (1) | 50–90 | – | Not stated/Yes | iPD | – | – |
| Halliday et al. (112) | c | 4 (4) | 0 dorsal-56 median | – | Not stated | PD | DMV | – | |
| Halliday et al. (57) | c | 4 (4) | 0 dorsal-44 obscurus-60 median | – | Not stated/Yes | PD | SNc + LC, PPN, DMV | – | |
| Gai et al. (81) | c | 6 (5) | 76 | – | Not stated/Yes | iPD, 5–30 years | PPN, LTN, OPN, LC | – | |
| Paulus and Jellinger (59) | m | 23 (6) | 37 | – | Not stated/Yes | PD, H&Y III–V, 1–31 years | SNc, LC, RN, NBM | – | |
| Benarroch et al. (117) | m | 14 (12) | 60–67 | – | Yes | DLB, 5–20 years | – | – | |
| Cheshire et al. (78) | s | 44 (17) | None | – | Yes | PD, LID severity, 14.8 years | SNc | – | |
| Total: | 7 | o0, m3, c3, s1 | 97 (49) | ||||||
| Ventral Tegmental Area (VTA) | Javoy-Agid et al. (118) | m | 2 (2) | 77 | – | Not stated | PD | – | – |
| Hirsch et al. (52) | c | 4 (3) | 48 | – | Not stated | PD | SNc, A10, A8, CGS | – | |
| German et al. (53) | c | 5 (3) | 42 | – | Not stated/Yes | PD, 5–27 years | SNc | – | |
| Zweig et al. (62) | m | 13 (14) | Some | – | Yes | PD, H&Y 4.5, 11 years | LC, SNc, NBM | – | |
| Mouatt-Prigent et al. (63) | c | 4 (3) | Some | – | Not stated/Yes | iPD | SNc | – | |
| Dymecki et al. (119) | m | 7 (6) | 41–62 | – | Not stated/Yes | PD, long-term | – | – | |
| McRitchie et al. (120) | s | 3 (3) | 31 | – | Not stated/Yes | iPD, 1–27 years | A8, A10 | – | |
| Damier et al. (68) | c | 5 (5) | 46 | – | Not stated | iPD | SNc | Yes | |
| Total | 8 | o0, m3, c4, s1 | 43 (39) | ||||||
| Olfactory bulb (OB) | Pearce et al. (121) | m | 7 (7) | 57 | – | Not stated/Yes | PD, 8–19 years | – | – |
| Huisman et al. (122) | s | 10 (10) | Increase of 100 | – | Not stated/Yes | PD, 4–23 years | – | – | |
| Huisman et al. (123) | s | 20 (19) | Increase of 100 in female | – | Yes | iPD, 3–30 years | – | – | |
| Mundinano et al. (124) | s | 6 (15) | Increase Of 100 | – | Not stated/Yes | PD, Braak stage II–V | – | – | |
| Total | 4 | o0, m1, c0, s3 | 43 (51) | ||||||
| Thalamus | Xuereb et al. (60) | m | 5 (5) | None | – | Not stated/Yes | PD | Thalamus (multiple nuclei) | – |
| Henderson et al. (125) | c | 9 (10) | 40–55 | – | Not stated/Yes | PD, H&Y II–V, 7.2 years | Caudal intralaminar nuclei, limbic thalamic nuclei | – | |
| Henderson et al. (69) | s | 9 (8) | 50–70 | – | Not stated/Yes | PD, H&Y II–V, 3–17 years | SNc, Centromedian–parafascicular complex, mediodorsal or anterior principal nucleus | – | |
| Halliday et al. (126) | s | 9 (9) | None | – | Not stated/Yes | PD, H&Y II–V, 9 years | Motor thalamus, Cortex | – | |
| Total | 4 | o0, m1, c1, s2 | 32 (32) | ||||||
| Sympathic/parasympathic ganglia | Rajput and Rozdilsky (47) | o | 6 (1) | Some | – | Not stated | iPA, H&Y, 3–18 years | SN, LC, DMV, Cortex, Hypothalamus | – |
| Wakabayashi and Takahashi (127) | m | 25 (25) | 31–43 | – | Not stated/Yes | PD | – | – | |
| Benarroch et al. (115) | o | 14 (12) | None | – | Yes/Not stated | PD or LBD, 10 years | DMV, nucleus ambiguus | – | |
| Total: | 3 | o2, m1, c0, s0 | 45 (38) | ||||||
| Cortex | Rajput and Rozdilsky (47) | o | 6 (1) | None | – | Not stated | iPA, H&Y, 3–18 years | SN, LC, DMV, Hypothalamus, Intermediolateral spinal cord, sympathetic ganglia | – |
| Pedersen et al. (128) | s | 10 (12) | None | – | Not stated/Yes | PD, 2–25 years | – | – | |
| Total | 2 | o1, m0, c0, s1 | 16 (13) | ||||||
| Pre-supplementary and premotor cortex | MacDonald and Halliday (129) | m | 5 (5) | 32–45 | – | Yes | PD, 10–17 years | – | – |
| Halliday et al. (126) | s | 9 (9) | None | – | Not stated/Yes | PD, H&Y II–V, 9 years | Motor thalamus | – | |
| Total | 2 | o0, m1, c0, s1 | 14 (14) | ||||||
| Amygdala, corticomedial complex | Harding et al. (130) | s | 18 (16) | 30 | – | Yes | PD, 13 years | – | – |
| Hippocampus | Joelving et al. (131) | s | 8 (8) | None | – | Not stated/Yes | PD, 2–25 years | – | – |
| Laterodorsal tegmental nucleus (LTN) | Gai et al. (81) | c | 6 (5) | 41 | – | Not stated/Yes | iPD, 5–30 years | PPN, OPN, RN, LC | Yes |
| Oral pontine reticular nucleus (OPN) | Gai et al. (81) | c | 6 (5) | 41 | – | Not stated/Yes | iPD, 5–30 years | PPN, LTN, RN, LC | Yes |
List of 90 studies quantifying the loss of neurons in the brain in PD.
Included in the table are the technique used for quantification (o, observation; m, manual c, computer assisted; s, stereological counting), the number of subjects and controls (ctrl) studied, the estimated % loss of neurons, any particularity in the comparison group, mention if studies were performed blind and with age-matched controls, the stated diagnosis, scale of severity and disease duration when mentioned and note on other regions counted. Where an average value of loss was not given by authors, this number was calculated from available data. Bold values indicates total numbers per region. *Indicates details which are given at the end of that section.
Substantia nigra pars compacta
Loss of SNc DA neurons in PD is indisputable. Here we found 38 studies addressing this directly with a total of 612 brains. However, if we consider the methods used, we found that 10 of these studies were observational, 8 involved manual counting methods, 8 used computer-assisted methods, and 12 used stereology. Considering stereological methods as best practice for unbiased evaluation of cell number, 181 brains were quantified as such for SNc: still a large number. The average cell loss reported for studies involving stereological methods is ~68%. The definition and clinical stage of PD in most studies varied greatly, especially in reporting. For example, for the 12 studies using stereological methods, three papers (74, 76, 79) staged each case according to the Braak staging (to be expected given that Braak staging only came about in the early 2000s). In the same 12 studies, the age “since disease onset” varied between 1 and 27 years when stated, the Hoehn and Yahr ratings (H&Y, used to describe the progression severity of PD symptoms) varied between 2 to 5 and the UPDSR score (that includes H&Y rating, symptoms and quality-of-life scores) was also on occasion provided. A correlation with disease duration/severity was found in 10 studies. It is relevant here to mention that some authors, including Gibb et al. (56) have discussed the selective vulnerability of restricted sub-regions within the SNc. These data are important and relevant to the progression of the field; however, we found this distinction absent in the majority of the work we examined.
Methodology and Scales of PD Progression
We searched the scientific literature using the search engines and databases of PubMed, Google Scholar and Science Direct. The following search terms were used: “PD,” and “cell loss,” “cell death,” or “reduced cell/neuron number.” Furthermore, these terms were used in combination with brain structure keywords: “SNc,” “VTA,” “LC,” “Raphe,” “DMV,” “PPN,” “NBM,” and “enteric system” (“ENS”), and “gut.” Review and original article abstracts were screened, then, where appropriate, read. Where any direct or indirect claim for cell loss was found (rather than only the presence of LP), the claim was followed to its original source.
The Hoehn and Yahr scale (H&Y) is a widely used clinical rating scale, which defines broad categories of motor function in PD (where 1 is the least severe, and 5, most severe symptoms) (132).
Braak staging is a method of classifying the progression of PD pathology and symptoms based on the presence of Lewy pathology (where 1 represents initial pathology in the brain stem, and 6, severe pathology including the neocortex) (21).
Pedunculopontine nucleus and locus coeruleus
The evidence for cell loss for both the PPN (11 studies), containing cholinergic neurons and the LC (18 studies), containing noradrenergic neurons, is also relatively strong.
For the PPN, four studies used stereological methods. In these four studies, the average loss of cholinergic PPN neurons was 41% and the range of PD stages amongst the subjects evaluated was broad. For example, in Rinne et al. (99), the PD cases ranged from a H&Y rating of 2.5 to 5; in Karachi et al. (73), UDPRS score was used, and in both Hepp et al. (101), and Pienaar et al. (102), the PD cases were between Braak stages 4 and 6 and between 2 and 4, respectively. Although sample sizes were relatively small in these two last studies, nine and eight, respectively, it is somewhat surprising that in the most advanced PD group, loss of cholinergic PPN neurons was not higher than for less advanced PD subjects, contrarily to the report by Rinne et al. (99).
Surprisingly, we found no study quantifying loss of LC neurons using stereological counting methods. For the LC, 221 brains were studied, with cell loss ranging from “some” to 94%. Five of the studies were based on observational quantifications, 4 on manual counting and 9 used computer-assisted counting. In these 18 papers, when stated, the H&Y score was between 3 and 5, and disease duration was between 1 and 31 years. A correlation of the extent of cell loss with disease duration was found in two of these studies (81, 85).
Dorsal motor nucleus of the vagus, raphe nuclei, nucleus basalis of meynert and ventral tegmental area
Substantial cell loss has been documented in the DMV, containing cholinergic neurons, with 7 studies evaluating this loss in 49 cases. Of these, only one study (114) used stereology, where they reported 55% neuronal loss in eight PD cases, ranging from 5 to 24 years post-diagnosis and reported correlation with disease duration/severity.
The importance of re-evaluating cell loss is PD is apparent when considering the serotonergic RN. For these nuclei, which are considered by many authors to be lost in PD, we found 7 papers describing neuronal loss varying between 0 to 90%. Cheshire et al. however, using stereology in 44 late-stage PD subjects, found no cell loss in the dorsal raphe nucleus (78). In the NBM, containing cholinergic neurons, we found 13 papers, 12 using manual counting methods, and one observational, which estimated an average neuronal loss of between “some” to 72%. No correlation with disease duration was reported. The high prevalence of concomitant PD and Alzheimer's disease (AD) might explain why cell loss varied so much for this region. Surprisingly, only 8 studies directly evaluated neuronal loss in the VTA, a dopaminergic region often considered to be only modestly affected in PD. Of these, one study used stereology (120) to evaluate the loss of neurons in 3 cases of PD (or 6 including PD with a secondary diagnosis) that were between 1 and 27 years post-diagnosis and reported an average neuronal loss of 31%. One paper reported correlation of the extent of cell loss with disease duration (68).
Thalamus, hypothalamus, olfactory bulb
Four studies reported neuronal loss in thalamic nuclei, with 2 using stereology (69, 126). In (69), 9 subjects with H&Y disease ratings between 2 and 5 statistically significant loss of 30–40% was reported in the centromedian-parafasicular complex. However, no loss was found in the motor thalamus in 9 subjects with similar H&Y disease ratings in the work of Halliday et al. (126). Neuronal loss has also sometimes been reported in the hypothalamus (9 studies), with one using stereology; Thannickal et al. (110) reported a 50% cell loss in 10 PD cases, with increased loss with disease severity. Olfactory dysfunction is now well established as an early symptom of PD. Four studies evaluating cell loss in the olfactory bulb were reported. One of these (121) described a 57% decrease in neuronal number (identified as cells with “a prominent nucleolus surrounded by Nissl substance”), while the others (122–124), using stereology, reported a 100% increase in the number of TH-positive neurons.
Peripheral nervous system, spinal cord and other brain regions
Though there is substantial evidence for LP occurring in the ENS (133), we did not find any study reporting direct—quantitative evidence—for neuronal loss in the gut. Though it has been inferred that ENS glial cell loss is occurring (134), there is evidence that neuronal loss in the gut is not associated with PD (135). Of note, a publication often cited in support of neuronal loss in the ENS (115) shows, in fact, neuronal loss in the DMV. With regards to the spinal cord, published evidence is also scarce; of the studies most relevant here, Wakabayashi et al. (127), using manual counting methods, described a loss of 31% and 43% respectively in the 2nd and 9th thoracic segments of the intermediolateral of the spinal cord. For the amygdala, the pre-supplementary motor cortex, several other cortical regions, the laterodorsal tegmental nucleus and the oral pontine reticular nucleus, we found only single studies supporting loss, with stereology used for the amygdala (30% loss) (130), and cortex (10% loss) (130) (see Table 1).
Regional order of cell loss?
In summary, it seems clear that there is some level of cell loss in PD in restricted regions including the SNc, LC, NBM, PPN, DMV, VTA, and probably the RN. However—because of the lack of data for some regions, the variety of techniques used to count neurons, potentially numerous unintentional sources of bias, and because of the inconsistency in criteria used for subject sampling—firm conclusions are somewhat limited. In particular, it is difficult to conclude on the relative extent and temporal order of cell loss in these different brain regions as a function of disease progression, information that would be critical to advance the field. Indeed, a direct comparison of the extent of neuronal loss in different regions examined in different studies is hazardous, even if stereological studies were to be selected. Interestingly, of the 38 studies we identified evaluating cell loss in the SNc, only 5 of these also looked at the VTA, and of these only 1 used stereology. Given the importance of the difference in vulnerability of these two nuclei, a systematic evaluation of the extent of loss of these neurons in PD would be very informative. But even if as a technique, stereology mitigates for most of the classic biases, it is still unable to account for the variation in subject sampling, i.e., variation in disease duration, sex and age, unless these criteria were considered in a similar way for each study. Unfortunately, this has not, thus far, been the case. In conclusion, it seems clear that stereological studies comparing multiple regions in the same subjects and these regions in subjects at different stages of PD are critically needed to advance the field.
What are the common features shared by neurons affected in PD?
Although, as mentioned previously, the evidence for the extent of cell loss in regions other than the SNc in the PD brain is not always sufficiently documented, it is clear that some level of cell loss occurs in a limited subset of regions beyond the SNc (Figure 1A), or, to the least, that neuronal functions including neurotransmission are perturbed in multiple neuronal circuits. It is therefore of great interest to identify some of the biological features that distinguish neuronal subgroups in terms of their basal vulnerability to some of the cellular stresses that are invoked to trigger PD, including altered proteostasis (due to lysosomal and/or proteosomal impairment), mitochondrial dysfunction, and sustained oxidant stress (including from highly reactive DA metabolites).
Figure 1
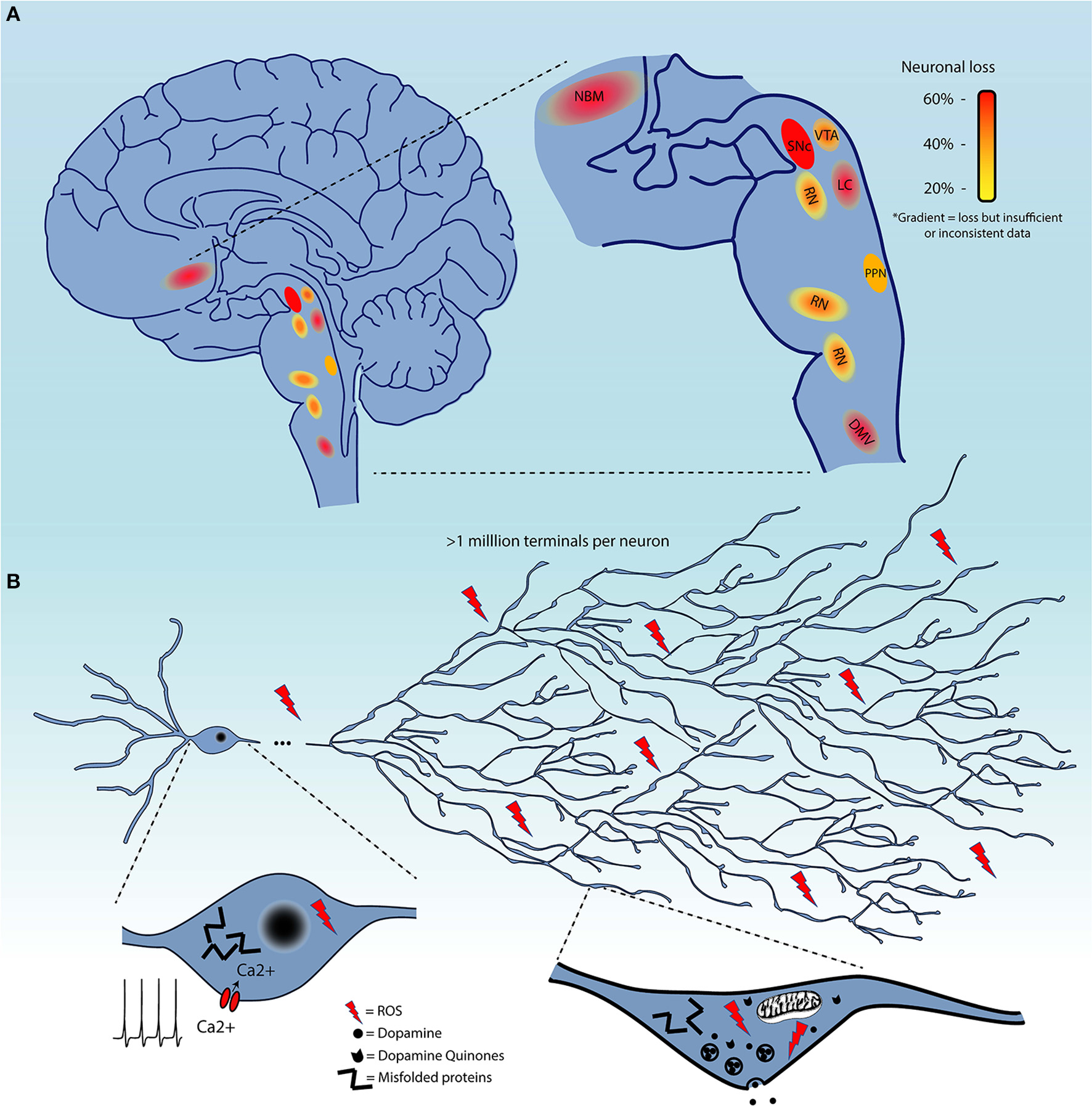
(A) Schematic representation of brain regions demonstrating cell loss in Parkinson's disease. These are color-coded based on the evidence of cell loss. Red = 60%, orange = 40%, and yellow = 20%. Color gradients indicate uncertainty in the extent of this cell loss. (B) Summary of the converging hypotheses that may explain the origins of the selective vulnerability of neurons in Parkinson's disease. This includes the exceptionally large axonal arbor of PD-affected neurons, their electrophysiological properties, including calcium-dependent pacemaking, and high levels of oxidant stress in the somatodendritic and axonal domain, all thought to be contributing to cellular dysfunction and cell loss. Pathological protein aggregation and reactive dopamine quinones are considered as additional precipitating factors.
Several groups have been tackling this question by interrogating the characteristics that render neurons, starting with those of the SNc, particularly vulnerable to degeneration / cell death (136–138). It is likely that some shared functional or structural properties are responsible for selective vulnerability of affected nuclei, as opposed to features truly unique to SNc DA neurons. The causative characteristic(s) should be present in all affected neurons, but also be absent in neurons that do not degenerate or that degenerate much later in the disease. Four main converging hypotheses on selective vulnerability in PD have been gaining attention lately (Figure 1B), related to DA toxicity, iron-content, autonomous pacemaking and axonal arborization size. The next section will explore the likelihood that these hypotheses can explain why select neuronal populations are particularly vulnerable in PD.
Dopamine toxicity
Firstly, it has been suggested that DA neurons in general are most at risk because they produce DA as a neurotransmitter, a molecule that can be toxic in certain conditions through the generation of reactive quinones during its oxidation (139). This oxidation has been proposed to be implicated in the production of neuromelanin in SNc DA neurons. These DA quinones have been shown to interact with and negatively impact the function of mitochondrial protein complexes I, III, and V (140) and of other proteins such as tyrosine hydroxylase, the DA transporter and α-synuclein (141, 142). Such reactive by-products can promote mitochondrial dysfunction, pathological aggregation of proteins such as α-synuclein and oxidative stress (143). Increasing the vesicular packaging of DA accordingly reduces the vulnerability of DA neurons, while down-regulating vesicular packaging has the opposite effect (144–147). Although highly relevant, this phenomenon alone does not readily explain the differential vulnerability of different dopaminergic neuron subgroups (such as SNc vs. VTA) and cannot contribute to the potential vulnerability of non-dopaminergic neurons in PD. Also, in the context of DA-induced toxicity, it is puzzling that levodopa therapy, acting to increase DA synthesis, does not appear to accelerate cell loss (148, 149). For these reasons, even if DA toxicity most certainly contributes to degeneration of SNc DA neurons, it is certainly not the sole factor driving neuronal death in PD.
Iron content
Secondly, iron content is thought to also be an important contributor to the selective vulnerability of SNc DA neurons. Iron is known to be able to generate ROS by the Fenton reaction and has been shown to accumulate with age in SNc (150–152). Since the mitochondrial electron transport chain relies on iron sulfur clusters for its function and since it is believed that SNc neurons have particularly high bioenergetic demands (136, 138, 153), elevated iron content could in part underlie elevated and sustained mitochondrial activity. Another interesting feature of iron in SNc DA neurons is that it can be chelated by neuromelanin, which renders it unavailable for mitochondrial function. Even if the affinity of iron for neuromelanin is much lower than for other iron binding proteins such as ferritin, it is possible that accumulation of neuromelanin and loss of ferritin concentration with age impacts gradually mitochondrial function, which could eventually promote cell death. However, data about potential iron content and iron-binding protein concentration changes in PD is still a matter of debate (154, 155). In addition, data is lacking on iron levels in other brain regions presenting cell death in PD. In fact, the only other region studied in this context has been the LC, which did not show high iron relative to the SNc (156–159).
Autonomous pacemaking
A third highly attractive hypothesis to explain the vulnerability of SNc DA neurons has its origins in the fact that these neurons demonstrate autonomous pacemaking. Many receptors/channels can potentially modulate the excitability and survival of DA neurons (160). The fact that pacemaking activity in SNc DA neurons is accompanied by slow oscillations in intracellular calcium concentrations, caused by the opening of voltage-dependent Cav1 plasma membrane calcium channels (Cav1.1 and 1.3) has recently renewed interest to this topic. In the Cav1 family, Cav1.3 has been suggested to be of particular interest because its voltage-sensitivity and inactivation properties allow a subset of the calcium channels to always stay open during pacemaking, causing extensive calcium entry (137). These oscillations have a positive contribution to cell physiology because they help maintain pacemaking and directly promote mitochondrial oxidative phosphorylation (OXPHOS) (161). However, by doing so, they have been proposed to also promote chronically high levels of ROS production (162, 163). Along with a reduction in mitochondrial function with age, chronically elevated oxidative stress has been proposed to be a causative factor in the decline of neuronal survival (164). Interestingly, CaV currents and autonomous pacemaking are also a feature of LC and DMV neurons (162, 163), and have been hypothesized to be involved in their vulnerability. The fact that other neuronal populations also expressing Cav1.3 such as hippocampal neurons (165) and striatal spiny projection neurons (166) do not degenerate in PD highlights the possibility that the particular vulnerability of SNc DA neurons is due to a combination of physiological phenotypes and not only intracellular calcium oscillations. Intriguingly, recent post-mortem studies showed that there was no decrease in Cav1.3 mRNA level in early or late stage PD in human SNc compared to controls (166, 167), despite significant loss of SNc neurons. Finally, in addition to CaV channels, ATP sensitive potassium channels (K-ATP) have also been reported to regulate the excitability and vulnerability of SNc DA neurons (168).
Axonal arborization size
A fourth hypothesis proposes that neurons such as those of the SNc are particularly vulnerable because of the massive scale of their axonal arborization, leading to very high numbers of axon terminals, elevated energetic requirements, and chronically high oxidant stress. Indeed, it has been shown that SNc DA neurons have an exuberant and highly arborized axonal arborization with estimates upwards of a million neurotransmitter release sites per SNc DA neuron in humans (136, 169): this would make them some of the most highly arborized neurons in the nervous system. This characteristic has the potential to place a very large bioenergetic burden on these cells, leaving little margin for additional bioenergetic stress (136, 138, 153). Related to this, it has been calculated that the ATP requirement for propagation of one action potential grows exponentially with the level of branching (170). In a recent publication (138), we demonstrated in vitro that reducing the axonal arbor size of SNc DA neurons to a size more similar to that of VTA DA neurons using the axonal guidance factor Semaphorin 7A, was sufficient to greatly reduce basal OXPHOS and reduce their vulnerability to toxins including MPP+ and rotenone. Although as previously discussed, the extent of neuronal loss is still unclear for many neuronal populations, it does seem likely that most neuronal nuclei affected in PD include neurons that are relatively few in number, but all possess long and profuse unmyelinated axonal arbors and a large number of axonal terminals (171–176). However, comparative data evaluating axonal arbor size amongst these populations and in populations of neurons that do not degenerate in PD is presently lacking. An interesting possible exception to this hypothesis could be striatal cholinergic interneurons, which were previously estimated in rats to present 500,000 axonal varicosities (177, 178), but have not been reported to degenerate in PD. This estimate was obtained by dividing the estimated number of terminals by the estimated number of cholinergic interneurons in the striatum, which was based on the total number of striatal neurons and the proportion of cholinergic interneurons. Considering recent stereological counting of the number of neurons in the rat striatum, it is possible that the total number of terminals estimated for striatal cholinergic neurons may have been overestimated by a factor of six (179). Based on this report, axonal arborization size of striatal cholinergic interneurons would be less than half of that of SNc neurons. Careful quantitative and comparative studies are clearly needed.
A global bioenergetic failure hypothesis
One commonality between these four hypotheses is that they all suggest that vulnerable neurons are under intense mitochondrial/bioenergetic demand. This could alter the oxidative stress response by depleting antioxidants like glutathione (GSH), as previously suggested to occur in the PD brain (180–182). This stress could also, at a certain point, place the cells in a situation in which the rate of OXPHOS required to sustain neurotransmitter release and cellular excitability leaves too little of the cell's resources to sustain other key cellular functions such as degradation of damaged or misfolded proteins (137). This could lead to preferential dysregulation of axon terminals, triggering a dying back cascade culminating later in cell death (3, 183, 184). Approximately half of the oxygen consumed by mitochondria in SNc DA neurons appears to be used by activity-dependent cellular processes such as firing and neurotransmitter release (138). In this context, axon terminal degeneration seen early in the disease, prior to cell death, could be in part an attempt by stressed neurons to adapt to such excessively high metabolic needs. Such a dying back process could also lead to increased amounts of damaged axonal proteins to manage, potentially promoting their accumulation in intracellular inclusions. Since α-synuclein is highly concentrated in axon terminals, it is possible that retraction of axonal processes in a cell where protein degradation systems are overwhelmed, promotes creation of pathological aggregates of this protein, thus accelerating cell death. Interestingly, lysosomal defects secondary to GBA1 gene mutations are present in up to 10% of PD patients. This gene encodes a glucocerebrosidase responsible for breaking down lysosomal glucolipid. When GBA1 is mutated, the level of glucolipid and of misfolded proteins increases in neurons. This is likely to represent a particular challenge for highly arborized neurons such as those of the SNc, perhaps explaining why such mutations are now considered the greatest genetic risk factor for PD (185–191). Similarly, mutations in gene products implicated in mitophagy and mitochondrial antigen presentation (PARK2, PINK1) (192, 193), oxidative stress response (PARK7) (194, 195), or vesicular trafficking (LRRK2) (196, 197) are present in familial forms of PD and their detrimental impact on cellular functions could also represent larger challenges for highly arborized and energetically ambitious neurons.
Toward better treatments of PD
In the context of the hypotheses discussed here regarding the origin of the selective vulnerability of neurons in PD, novel strategies to promote survival and preservation of cellular functions amongst challenged neuronal populations could possibly come from approaches that aim to reduce mitochondrial burden by either reducing neuronal metabolic needs or optimizing mitochondrial function. As an example, the CaV1.3 channel inhibitor isradipine is presently in phase 3 clinical trial and could possibly reduce the calcium- and activity-related metabolic stress of SNc DA neurons leading to neuroprotection (198). Other promising molecules could come from the repurposing of drugs used to treat diabetes and other metabolic diseases. One example is exenatide, a glucagon-like-peptide-1 agonist that has the property to increase glucose-induced insulin secretion, to prevent the rise of ROS and prevent decreases of mitochondrial function in diet-induced obese mice (199). This agonist was found to reduce the loss of DA neurons in the MPTP mouse model (200) and a recent clinical trial has shown improved motor function after 60 days of administration to PD patients (201). Overexpression of the mitochondrial deacetylase SIRT3 has also recently been shown in two studies to reduce basal OXPHOS by DA neurons and to protect SNc neurons in rodent models of PD (202, 203). With further discoveries of the underlying causes of the intrinsic vulnerability of neurons in the PD brain and PNS, multiple other strategies may soon be devised to address some of the specific challenges faced by energetically challenged neurons.
In conclusion, although the presently available data strongly argue that multiple populations of neurons are affected in PD and degenerate to varying extents, new work is needed to provide a more systematic, comparative, and time-dependent quantification of neuronal loss in this disease. More comprehensive and convincing data on cell death and axon terminal dysfunction in PD will likely provide additional impetus for new work aiming to solve the long-awaited challenge of identifying disease-modifying therapeutic approaches for this incapacitating and ill-treated disorder.
Statements
Author contributions
NG and SB performed the litterature review. NG, SB, and L-ET wrote the manuscript. NG and SB contributed equally to this work.
Acknowledgments
This work was supported by a grant from the Brain Canada and Krembil Foundations, as well as by the Canadian Institutes of Health Research. We would like to express our gratitude to Ms. Sarah Ouellet for her help preparing the illustrations and to Drs. Edward Fon, Michael Schlossmacher and Birgit Liss who graciously provided feedback on an earlier version of this manuscript. We also thank Dr. Heiko Braak for helping us identify some key litterature.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
- AD
Alzheimer Disease
- ADLB
Alzheimer's Disease with Lewy bodies
- ADNLB
Alzheimer's Disease with no Lewy bodies
- ALS
Amyotrophic Lateral Sclerosis
- CBS
corticobasal syndrome
- CGS
central gray substance
- CJD
Creutzfeldt-Jakob disease
- ctrl
control
- DLB
dementia with Lewy bodies
- H&Y
Hoehn and Yahr scale
- iPA
idiopathic paralysis agitans
- LBD or iLBD
Lewy body disease or idiopathic Lewy body disease
- LDB or iLDB
dementia with Lewy bodies or idiopathic dementia with Lewy bodies
- LID
levodopa (L-dopa)–induced dyskinesias
- MS
multiple sclerosis
- MSA
multiple system atrophy
- NPH
normal pressure hydrocephalus
- PD or iPD
Parkinson's disease or idiopathic Parkinson's Disease
- PSP
progressive supranuclear palsy
- UPDRS
unified Parkinson disease rating scale.
Abbreviations
References
1.
ParkinsonJ. An essay on the shaking palsy. (1817). J Neuropsychiatry Clin Neurosci. (2002) 14:223–36; discussion: 2. 10.1176/jnp.14.2.223
2.
ChengHCUlaneCMBurkeRE. Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol. (2010) 67:715–25. 10.1002/ana.21995
3.
BurkeREO'MalleyK. Axon degeneration in Parkinson's disease. Exp Neurol. (2013) 246:72–83. 10.1016/j.expneurol.2012.01.011
4.
LewyFH. Paralysis agitans. In: Handbuch der Neurologie, hsg. von Max Lewandowsky, Dritter Band, Spezielle Neurologie II. Für Später VormerkenVol. 3, ed LewandowskyM. (Berlin: Springer), (1912). p. 920–958.
5.
TretiakoffC. Contribution a l'Etude de l'Anatomiepathologique du Locus Niger de Soemmering Avec Quelques Deductions Relatives a la Pathologenie des Troubles Dutonus Musculaire de Lamaladiede Parkinson, thèse, Paris (1919).
6.
BraakHDelTredici K. Neuropathological staging of brain pathology in sporadic Parkinson's disease: Separating the wheat from the Chaff. J Parkinsons Dis. (2017) 7(s1):S71–S85. 10.3233/JPD-179001
7.
PostumaRBBergDSternMPoeweWOlanowCWOertelWet al. MDS clinical diagnostic criteria for Parkinson's disease. Mov Disord. (2015) 30:1591–601. 10.1002/mds.26424
8.
ShultsCW. Lewy bodies. Proc Natl Acad Sci USA. (2006) 103:1661–8. 10.1073/pnas.0509567103
9.
VilchezDSaezIDillinA. The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat Commun. (2014) 5:5659. 10.1038/ncomms6659
10.
MercadoGCastilloVSotoPSidhuA. ER stress and Parkinson's disease: pathological inputs that converge into the secretory pathway. Brain Res. (2016) 1648(Pt B):626–32. 10.1016/j.brainres.2016.04.042
11.
CaiYArikkathJYangLGuoMLPeriyasamyPBuchS. Interplay of endoplasmic reticulum stress and autophagy in neurodegenerative disorders. Autophagy. (2016) 12:225–44. 10.1080/15548627.2015.1121360
12.
TrubanDHouXCaulfieldTRFieselFCSpringerW. PINK1, Parkin, and Mitochondrial Quality Control: What can we Learn about Parkinson's Disease Pathobiology?J Parkinsons Dis. (2017) 7:13–29. 10.3233/JPD-160989
13.
GiannoccaroMPLaMorgia CRizzoGCarelliV. Mitochondrial DNA and primary mitochondrial dysfunction in Parkinson's disease. Mov Disord. (2017) 32:346–63. 10.1002/mds.26966
14.
BoseABealMF. Mitochondrial dysfunction in Parkinson's disease. J Neurochem. (2016) 139(Suppl. 1):216–31. 10.5607/en.2015.24.2.103
15.
HauserDN1HastingsTG. Mitochondrial dysfunction and oxidative stress in Parkinson's disease and monogenic parkinsonism. Neurobiol Dis. (2013) 51:35–42. 10.1016/j.nbd.2012.10.011
16.
JiangTSunQChenS. Oxidative stress: A major pathogenesis and potential therapeutic target of antioxidative agents in Parkinson's disease and Alzheimer's disease. Prog. Neurobiol. (2016) 147:1–19. 10.1016/j.pneurobio.2016.07.005
17.
TiwariPCPalR. The potential role of neuroinflammation and transcription factors in Parkinson disease. Dialogues Clin Neurosci. (2017) 19:71–80.
18.
JoshiNSinghS. Updates on immunity and inflammation in Parkinson disease pathology. J Neurosci Res. (2017) 96:379–90. 10.1002/jnr.24185
19.
DudaJPotschkeCLissB. Converging roles of ion channels, calcium, metabolic stress, and activity pattern of Substantia nigra dopaminergic neurons in health and Parkinson's disease. J Neurochem. (2016) 139(Suppl. 1):156–78. 10.1111/jnc.13572
20.
PfeifferRF. Non-motor symptoms in Parkinson's disease. Parkinsonism Relat Disord. (2016) 22(Suppl. 1):S119–22. 10.1016/j.parkreldis.2015.09.004
21.
BraakHDelTredici KRubUdeVos RAJansenSteur ENBraakE. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging (2002) 24:197–211. 10.1016/S0197-4580(02)00065-9
22.
BraakHDelTredici KBratzkeHHamm-ClementJSandmann-KeilDRübU. Staging of the intracerebral inclusion body pathology associated with idiopathic Parkinson's disease (preclinical and clinical stages). J Neurol. (2002) 249:iii1–5. 10.1007/s00415-002-1301-4
23.
BraakHGhebremedhinERubUBratzkeHDelTredici K. Stages in the development of Parkinson's disease-related pathology. Cell Tissue Res. (2004) 318:121–34. 10.1007/s00441-004-0956-9
24.
MarkesberyWRJichaGALiuHSchmittFA. Lewy body pathology in normal elderly subjects. J Neuropathol Exp Neurol. (2009) 68:816–22. 10.1097/NEN.0b013e3181ac10a7
25.
SasakiSShirataAYamaneKIwataM. Parkin-positive autosomal recessive juvenile Parkinsonism with α-synuclein-positive inclusions. Neurology. (2004) 63:678–82. 10.1212/01.WNL.0000134657.25904.0B
26.
HayashiSWakabayashiKIshikawaANagaiHSaitoMMaruyamaMet al. An autopsy case of autosomal-recessive juvenile parkinsonism with a homozygous exon 4 deletion in the parkin gene. Mov Disord. (2000) 15:884–8. 10.1002/1531-8257(200009)15:5<884::AID-MDS1019>3.0.CO;2-8
27.
SchneiderSAAlcalayRN. Neuropathology of genetic synucleinopathies with parkinsonism: Review of the literature. Mov Disord. (2017) 32:1504–23. 10.1002/mds.27193
28.
SurmeierDJObesoJAHallidayGM. Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci. (2017) 18:101–13. 10.1038/nrn.2016.178
29.
BuchmanASShulmanJMNagSLeurgansSEArnoldSEMorrisMCet al. Nigral pathology and parkinsonian signs in elders without Parkinson disease. Ann Neurol. (2012) 71:258–66. 10.1002/ana.22588
30.
CabelloCRThuneJJPakkenbergHPakkenbergB. Ageing of substantia nigra in humans: cell loss may be compensated by hypertrophy. Neuropathol Appl Neurobiol. (2002) 28:283–91. 10.1046/j.1365-2990.2002.00393.x
31.
FearnleyJMLeesAJ. Ageing and Parkinson's disease: substantia nigra regional selectivity. Brain (1991) 114 (Pt 5):2283–301.
32.
FedorowHTriblFHallidayGGerlachMRiedererPDoubleKL. Neuromelanin in human dopamine neurons: comparison with peripheral melanins and relevance to Parkinson's disease. Prog Neurobiol. (2005) 75:109–24. 10.1016/j.pneurobio.2005.02.001
33.
MaSYRoyttMCollanYRinneJO. Unbiased morphometrical measurements show loss of pigmented nigral neurones with ageing. Neuropathol Appl Neurobiol. (1999) 25:394–9. 10.1046/j.1365-2990.1999.00202.x
34.
NaoiMMaruyamaW. Cell death of dopamine neurons in aging and Parkinson's disease. Mech Ageing Dev. (1999) 111:175–88.
35.
RudowGO'BrienRSavonenkoAVResnickSMZondermanABPletnikovaOet al. Morphometry of the human substantia nigra in ageing and Parkinson's disease. Acta neuropathologica (2008) 115:461–70. 10.1007/s00401-008-0352-8
36.
StarkAKPakkenbergB. Histological changes of the dopaminergic nigrostriatal system in aging. Cell Tissue Res. (2004) 318:81–92. 10.1007/s00441-004-0972-9
37.
ThiessenBRajputAHLavertyWDesaiH. Age, environments, and the number of substantia nigra neurons. Adv Neurol. (1990) 53:201–6.
38.
RansmayrGFaucheuxBNowakowskiCKubisNFederspielSKaufmannWet al. Age-related changes of neuronal counts in the human pedunculopontine nucleus. Neurosci Lett. (2000) 288:195–8. 10.1016/S0304-3940(00)01244-1
39.
LohrJBJesteDV. Locus ceruleus morphometry in aging and schizophrenia. Acta Psychiatr Scand. (1988) 77:689–97. 10.1111/j.1600-0447.1988.tb05189.x
40.
ShibataESasakiMTohyamaKKanbaraYOtsukaKEharaSet al. Age-related changes in locus ceruleus on neuromelanin magnetic resonance imaging at 3 Tesla. Magn Reson Med Sci. (2006) 5:197–200. 10.2463/mrms.5.197
41.
GoldsteinDS. Cardiac denervation in patients with Parkinson disease. Cleve Clin J Med. (2007) 74(Suppl. 1):S91–4. 10.3949/ccjm.76.s2.10
42.
WongKKRaffelDMKoeppeRAFreyKABohnenNIGilmanS. Pattern of cardiac sympathetic denervation in idiopathic Parkinson disease studied with 11C hydroxyephedrine PET. Radiology (2012) 265:240–7. 10.1148/radiol.12112723
43.
OrimoSOkaTMiuraHTsuchiyaKMoriFWakabayashiKet al. Sympathetic cardiac denervation in Parkinson's disease and pure autonomic failure but not in multiple system atrophy. J Neurol Neurosurg Psychiatry. (2002) 73:776–7. 10.1136/jnnp.73.6.776
44.
GreenfieldJGBosanquetFD. The brain-stem lesions in Parkinsonism. J Neurol Neurosurg Psychiatry (1953) 16:213–26. 10.1136/jnnp.16.4.213
45.
PakkenbergHBrodyH. The number of nerve cells in the substantia nigra in paralysis agitans. Acta Neuropathol. (1965) 5:320–4. 10.1007/BF00686528
46.
BernheimerHBirkmayerWHornykiewiczOJellingerKSeitelbergerF. Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. J Neurol Sci. (1973) 20:415–55. 10.1016/0022-510X(73)90175-5
47.
RajputAHRozdilskyB. Dysautonomia in Parkinsonism: a clinicopathological study. J Neurol Neurosurg Psychiatry (1976) 39:1092–100. 10.1136/jnnp.39.11.1092
48.
GasparPGrayF. Dementia in idiopathic Parkinson's disease. A neuropathological study of 32 cases. Acta Neuropathol. (1984) 64:43–52. 10.1007/BF00695605
49.
TagliaviniFPilleriGBourasCConstantinidisJ. The basal nucleus of Meynert in idiopathic Parkinson's disease. Acta Neurol Scandinav. (1984) 70:20–8. 10.1111/j.1600-0404.1984.tb00798.x
50.
Chan-PalayV. Galanin hyperinnervates surviving neurons of the human basal nucleus of Meynert in dementias of Alzheimer's and Parkinson's disease: a hypothesis for the role of galanin in accentuating cholinergic dysfunction in dementia. J Comp Neurol. (1988) 273:543–57. 10.1002/cne.902730409
51.
GibbWRLeesAJ. A comparison of clinical and pathological features of young- and old-onset Parkinson's disease. Neurology (1988) 38:1402–6. 10.1212/WNL.38.9.1402
52.
HirschEGraybielAMAgidYA. Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson's disease. Nature (1988) 334:345–8. 10.1038/334345a0
53.
GermanDCManayeKSmithWKWoodwardDJSaperCB. Midbrain dopaminergic cell loss in Parkinson's disease: computer visualization. Ann Neurol. (1989) 26:507–14. 10.1002/ana.410260403
54.
RinneJORummukainenJPaljarviLRinneUK. Dementia in Parkinson's disease is related to neuronal loss in the medial substantia nigra. Ann Neurol. (1989) 26:47–50. 10.1002/ana.410260107
55.
ZweigRMJankelWRHedreenJCMayeuxRPriceDL. The pedunculopontine nucleus in Parkinson's disease. Ann Neurol. (1989) 26:41–6. 10.1002/ana.410260106
56.
GibbWRFearnleyJMLeesAJ. The anatomy and pigmentation of the human substantia nigra in relation to selective neuronal vulnerability. Adv Neurol. (1990) 53:31–4.
57.
HallidayGMLiYWBlumbergsPCJohTHCottonRGHowePRet al. Neuropathology of immunohistochemically identified brainstem neurons in Parkinson's disease. Ann Neurol. (1990) 27:373–85. 10.1002/ana.410270405
58.
PakkenbergBMollerAGundersenHJMouritzenDam APakkenbergH. The absolute number of nerve cells in substantia nigra in normal subjects and in patients with Parkinson's disease estimated with an unbiased stereological method. J Neurol Neurosurg Psychiatry. (1991) 54:30–3. 10.1136/jnnp.54.1.30
59.
PaulusWJellingerK. The neuropathologic basis of different clinical subgroups of Parkinson's disease. J Neuropathol Exp Neurol. (1991) 50:743–55. 10.1097/00005072-199111000-00006
60.
XuerebJHPerryRHCandyJMPerryEKMarshallEBonhamJR. Nerve cell loss in the thalamus in Alzheimer's disease and Parkinson's disease. Brain (1991) 114 (Pt 3):1363–79.
61.
MollerA. Mean volume of pigmented neurons in the substantia nigra in Parkinson's disease. Acta Neurol Scand Suppl. (1992) 137:37–9. 10.1111/j.1600-0404.1992.tb05036.x
62.
ZweigRMCardilloJECohenMGiereSHedreenJC. The locus ceruleus and dementia in Parkinson's disease. Neurology (1993) 43:986–91. 10.1212/WNL.43.5.986
63.
Mouatt-PrigentAAgidYHirschEC. Does the calcium binding protein calretinin protect dopaminergic neurons against degeneration in Parkinson's disease?Brain Res. (1994) 668:62–70. 10.1016/0006-8993(94)90511-8
64.
MaSYCollanYRoyttaMRinneJORinneUK. Cell counts in the substantia nigra: a comparison of single section counts and disector counts in patients with Parkinson's disease and in controls. Neuropathol Appl Neurobiol. (1995) 21:10–7. 10.1111/j.1365-2990.1995.tb01023.x
65.
HallidayGMMcRitchieDACartwrightHPamphlettRHelyMAMorrisJG. Midbrain neuropathology in idiopathic Parkinson's disease and diffuse Lewy body disease. J Clin Neurosci. (1996) 3:52–60. 10.1016/S0967-5868(96)90083-1
66.
MaSYRinneJOCollanYRoyttaMRinneUK. A quantitative morphometrical study of neuron degeneration in the substantia nigra in Parkinson's disease. J Neurol Sci. (1996) 140:40–5. 10.1016/0022-510X(96)00069-X
67.
MaSYRoyttaMRinneJOCollanYRinneUK. Correlation between neuromorphometry in the substantia nigra and clinical features in Parkinson's disease using disector counts. J Neurol Sci. (1997) 151:83–7. 10.1016/S0022-510X(97)00100-7
68.
DamierPHirschECAgidYGraybielAM. The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson's disease. Brain (1999) 122 (Pt 8):1437–48. 10.1093/brain/122.8.1437
69.
HendersonJMCarpenterKCartwrightHHallidayGM. Degeneration of the centre median-parafascicular complex in Parkinson's disease. Ann Neurol. (2000) 47:345–52. 10.1002/1531–8249(200003)47:3<345::AID-ANA10>3.0.CO
70.
ZarrowCLynessSAMortimerJAChuiHC. Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in alzheimer and Parkinson diseases. Arch Neurol. (2003) 60:337–41. 10.1001/archneur.60.3.337
71.
GreffardSVernyMBonnetAMBeinisJYGallinariCMeaumeSet al. Motor score of the Unified Parkinson Disease Rating Scale as a good predictor of Lewy body-associated neuronal loss in the substantia nigra. Arch Neurol. (2006) 63:584–8. 10.1001/archneur.63.4.584
72.
BeachTGAdlerCHLueLSueLIBachalakuriJHenry-WatsonJet al. Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol. (2009) 117:613–34. 10.1007/s00401-009-0538-8
73.
KarachiCGrabliDBernardFATandeDWattiezNBelaidHet al. Cholinergic mesencephalic neurons are involved in gait and postural disorders in Parkinson disease. J Clin Invest. (2010) 120:2745–54. 10.1172/JCI42642
74.
MilberJMNoorigianJVMorleyJFPetrovitchHWhiteLRossGWet al. Lewy pathology is not the first sign of degeneration in vulnerable neurons in Parkinson disease. Neurology (2012) 79:2307–14. 10.1212/WNL.0b013e318278fe32
75.
KordowerJHOlanowCWDodiyaHBChuYBeachTGAdlerCHet al. Disease duration and the integrity of the nigrostriatal system in Parkinson's disease. Brain (2013) 136(Pt 8):2419–31. 10.1093/brain/awt192
76.
DijkstraAAVoornPBerendseHWGroenewegenHJNetherlandsBrain BRozemullerAJet al. Stage-dependent nigral neuronal loss in incidental Lewy body and Parkinson's disease. Mov Disord. (2014) 29:1244–51. 10.1002/mds.25952
77.
KraemmerJKovacsGGPerju-DumbravaLPirkerSTraub-WeidingerTPirkerW. Correlation of striatal dopamine transporter imaging with post mortem substantia nigra cell counts. Mov Disord (2014) 29:1767–73. 10.1002/mds.25975
78.
CheshirePAytonSBertramKLLingHLiAMcLeanCet al. Serotonergic markers in Parkinson's disease and levodopa-induced dyskinesias. Mov Disord. (2015) 30:796–804. 10.1002/mds.26144
79.
IaconoDGeraci-ErckMRabinMLAdlerCHSerranoGBeachTGet al. Parkinson disease and incidental Lewy body disease: Just a question of time?Neurology (2015) 85:1670–9. 10.1212/WNL.0000000000002102
80.
Chan-PalayVAsanE. Alterations in catecholamine neurons of the locus coeruleus in senile dementia of the Alzheimer type and in Parkinson's disease with and without dementia and depression. J Comp Neurol. (1989) 287:373–92. 10.1002/cne.902870308
81.
GaiWPHallidayGMBlumbergsPCGeffenLBBlessingWW. Substance P-containing neurons in the mesopontine tegmentum are severely affected in Parkinson's disease. Brain (1991) 114 (Pt 5):2253–67. 10.1093/brain/114.5.2253
82.
GermanDCManayeKFWhiteCL3rdWoodwardDJMcIntireDDSmithWKet al. Disease-specific patterns of locus coeruleus cell loss. Ann Neurol. (1992) 32:667–76. 10.1002/ana.410320510
83.
PattSGerhardL. A Golgi study of human locus coeruleus in normal brains and in Parkinson's disease. Neuropathol Appl Neurobiol. (1993) 19:519–23. 10.1111/j.1365-2990.1993.tb00480.x
84.
HoogendijkWJPoolCWTroostDvanZwieten ESwaabDF. Image analyser-assisted morphometry of the locus coeruleus in Alzheimer's disease, Parkinson's disease and amyotrophic lateral sclerosis. Brain (1995) 118 (Pt 1):131–43. 10.1093/brain/118.1.131
85.
BertrandELechowiczWSzpakGMDymeckiJ. Qualitative and quantitative analysis of locus coeruleus neurons in Parkinson's disease. Folia Neuropathol. (1997) 35:80–6.
86.
BrunnstromHFribergNLindbergEEnglundE. Differential degeneration of the locus coeruleus in dementia subtypes. Clin Neuropathol. (2011) 30:104–10. 10.5414/NPP30104
87.
McMillanPJWhiteSSFranklinAGreenupJLLeverenzJBRaskindMAet al. Differential response of the central noradrenergic nervous system to the loss of locus coeruleus neurons in Parkinson's disease and Alzheimer's disease. Brain Res. (2011) 1373:240–52. 10.1016/j.brainres.2010.12.015
88.
DuggerBNMurrayMEBoeveBFParisiJEBenarrochEEFermanTJet al. Neuropathological analysis of brainstem cholinergic and catecholaminergic nuclei in relation to rapid eye movement (REM) sleep behaviour disorder. Neuropathol Appl Neurobiol. (2012) 38:142–52. 10.1111/j.1365-2990.2011.01203.x
89.
DelTredici KBraakH. Dysfunction of the locus coeruleus-norepinephrine system and related circuitry in Parkinson's disease-related dementia. J Neurol Neurosurg Psychiatry (2013) 84:774–83. 10.1136/jnnp-2011-301817
90.
ArendtTBiglVArendtATennstedtA. Loss of neurons in the nucleus basalis of Meynert in Alzheimer's disease, paralysis agitans and Korsakoff's Disease. Acta Neuropathol. (1983) 61:101–8. 10.1007/BF00697388
91.
CandyJMPerryRHPerryEKIrvingDBlessedGFairbairnAFet al. Pathological changes in the nucleus of Meynert in Alzheimer's and Parkinson's diseases. J Neurol Sci. (1983) 59:277–89. 10.1016/0022-510X(83)90045-X
92.
NakanoIHiranoA. Neuron loss in the nucleus basalis of Meynert in parkinsonism-dementia complex of Guam. Ann Neurol. (1983) 13:87–91. 10.1002/ana.410130118
93.
WhitehousePJHedreenJCWhiteCL3rdPriceDL. Basal forebrain neurons in the dementia of Parkinson disease. Ann Neurol. (1983) 13:243–8. 10.1002/ana.410130304
94.
NakanoIHiranoA. Parkinson's disease: Neuron loss in the nucleus basalis withouth concomitant Alzheimer's Disease. Ann Neurol. (1984) 15:415–8. 10.1002/ana.410150503
95.
PerryEKCurtisMDickDJCandyJMAtackJRBloxhamCAet al. Cholinergic correlates of cognitive impairment in Parkinson's disease: comparisons with Alzheimer's disease. J Neurol Neurosurg Psychiatry (1985) 48:413–21. 10.1136/jnnp.48.5.413
96.
RogersJDBroganDMirraSS. The nucleus basalis of Meynert in neurological disease: a quantitative morphological study. Ann Neurol. (1985) 17:163–70. 10.1002/ana.410170210
97.
HirschECGraybielAMDuyckaertsCJavoy-AgidF. Neuronal loss in the pedunculopontine tegmental nucleus in Parkinson disease and in progressive supranuclear palsy. Proc Natl Acad Sci USA. (1987) 84:5976–80. 10.1073/pnas.84.16.5976
98.
JellingerK. The pedunculopontine nucleus in Parkinson's disease, progressive supranuclear palsy and Alzheimer's disease. J Neurol Neurosurg Psychiatry (1988) 51:540–3. 10.1136/jnnp.51.4.540
99.
RinneJOMaSYLeeMSCollanYRoyttaM. Loss of cholinergic neurons in the pedunculopontine nucleus in Parkinson's disease is related to disability of the patients. Parkinsonism Related Disord. (2008) 14:553–7. 10.1016/j.parkreldis.2008.01.006
100.
SchmeichelAMBuchhalterLCLowPAParisiJEBoeveBWSandroniPet al. Mesopontine cholinergic neuron involvement in Lewy body dementia and multiple system atrophy. Neurology (2008) 70:368–73. 10.1212/01.wnl.0000298691.71637.96
101.
HeppDHRuiterAMGalisYVoornPRozemullerAJMBerendseHWet al. Pedunculopontine cholinergic cell loss in hallucinating Parkinson disease patients but not in dementia with Lewy bodies patients. J Neuropathol Exp Neurol. (2013) 72:1162–70. 10.1097/NEN.0000000000000014
102.
PienaarISElsonJLRaccaCNelsonGTurnbullDMMorrisCM. Mitochondrial abnormality associates with type-specific neuronal loss and cell morphology changes in the pedunculopontine nucleus in Parkinson disease. Am J Pathol. (2013) 183:1826–40. 10.1016/j.ajpath.2013.09.002
103.
KremerHP. The hypothalamic lateral tuberal nucleus: normal anatomy and changes in neurological diseases. Prog Brain Res. (1992) 93:249–61. 10.1016/S0079-6123(08)64576-8
104.
KremerHPBotsGT. Lewy bodies in the lateral hypothalamus: do they imply neuronal loss?Mov Disord. (1993) 8:315–20.
105.
PurbaJSHofmanMASwaabDF. Decreased number of oxytocin-immunoreactive neurons in the paraventricular nucleus of the hypothalamus in Parkinson's disease. Neurology (1994) 44:84–9. 10.1212/WNL.44.1.84
106.
NakamuraSOhnishiKNishimuraMSuenagaTAkiguchiIKimuraJet al. Large neurons in the tuberomammillary nucleus in patients with Parkinson's disease and multiple system atrophy. Neurology (1996) 46:1693–6. 10.1212/WNL.46.6.1693
107.
AnsorgeODanielSEPearceRK. Neuronal loss and plasticity in the supraoptic nucleus in Parkinson's disease. Neurology (1997) 49:610–3. 10.1212/WNL.49.2.610
108.
HoogendijkWJPurbaJSHofmanMAdeVos RAJansenENSwaabDF. Depression in Parkinson's disease is not accompanied by more corticotropin-releasing hormone expressing neurons in the hypothalamic paraventricular nucleus. Biol Psychiatry (1998) 43:913–7. 10.1016/S0006-3223(97)00338-7
109.
FronczekROvereemSLeeSYHegemanIMvanPelt JvanDuinen SGet al. Hypocretin (orexin) loss in Parkinson's disease. Brain (2007) 130(Pt 6):1577–85. 10.1093/brain/awm090
110.
ThannickalTCLaiYYSiegelJM. Hypocretin (orexin) cell loss in Parkinson's disease. Brain (2007) 130(Pt 6):1586–95. 10.1093/brain/awm097
111.
EadieMJ. The pathology of certain medullary nuclei in parkinsonism. Brain (1963) 86:781–92. 10.1093/brain/86.4.781
112.
HallidayGMBlumbergsPCCottonRGBlessingWWGeffenLB. Loss of brainstem serotonin- and substance P-containing neurons in Parkinson's disease. Brain Res. (1990) 510:104–7. 10.1016/0006-8993(90)90733-R
113.
SaperCBSorrentinoDMGermanDCdeLacalle S. Medullary catecholaminergic neurons in the normal human brain and in Parkinson's disease. Ann Neurol. (1991) 29:577–84. 10.1002/ana.410290602
114.
GaiWPBlumbergsPCGeffenLBBlessingWW. Age-related loss of dorsal vagal neurons in Parkinson's disease. Neurology (1992) 42:2106–11. 10.1212/WNL.42.11.2106
115.
BenarrochEESchmeichelAMSandroniPLowPAParisiJE. Involvement of vagal autonomic nuclei in multiple system atrophy and Lewy body disease. Neurology (2006) 66:378–83. 10.1212/01.wnl.0000196638.98781.bb
116.
YamamotoTHiranoA. Nucleus raphe dorsalis in parkinsonism-dementia complex of Guam. Acta Neuropathol. (1985) 67:296–9. 10.1007/BF00687815
117.
BenarrochEESchmeichelAMSandroniPParisiJELowPA. Rostral raphe involvement in Lewy body dementia and multiple system atrophy. Acta Neuropathol. (2007) 114:213–20. 10.1007/s00401-007-0260-3
118.
Javoy-AgidFRubergMTaquetHBokobzaBAgidYGasparPet al. Biochemical neuropathology of Parkinson's disease. Adv Neurol. (1984) 40:189–98.
119.
DymeckiJLechowiczWBertrandESzpakGM. Changes in dopaminergic neurons of the mesocorticolimbic system in Parkinson's disease. Folia Neuropathol. (1996) 34:102–6.
120.
McRitchieDACartwrightHRHallidayGM. Specific A10 dopaminergic nuclei in the midbrain degenerate in Parkinson's disease. Exp Neurol. (1997) 144:202–13. 10.1006/exnr.1997.6418
121.
PearceRKHawkesCHDanielSE. The anterior olfactory nucleus in Parkinson's disease. Mov Disord. (1995) 10:283–7. 10.1002/mds.870100309
122.
HuismanEUylingsHBHooglandPV. A 100% increase of dopaminergic cells in the olfactory bulb may explain hyposmia in Parkinson's disease. Mov Disord. (2004) 19:687–92. 10.1002/mds.10713
123.
HuismanEUylingsHBHooglandPV. Gender-related changes in increase of dopaminergic neurons in the olfactory bulb of Parkinson's disease patients. Mov Disord. (2008) 23:1407–13. 10.1002/mds.22009
124.
MundinanoICCaballeroMCOrdonezCHernandezMDiCaudoCMarcillaIet al. Increased dopaminergic cells and protein aggregates in the olfactory bulb of patients with neurodegenerative disorders. Acta Neuropathol. (2011) 122:61–74. 10.1007/s00401-011-0830-2
125.
HendersonJMCarpenterKCartwrightHHallidayGM. Loss of thalamic intralaminar nuclei in progressive supranuclear palsy and Parkinson's disease: clinical and therapeutic implications. Brain. (2000) 123 (Pt 7):1410–21. 10.1093/brain/123.7.1410
126.
HallidayGMMacdonaldVHendersonJM. A comparison of degeneration in motor thalamus and cortex between progressive supranuclear palsy and Parkinson's disease. Brain (2005) 128(Pt 10):2272–80. 10.1093/brain/awh596
127.
WakabayashiKTakahashiH. Neuropathology of autonomic nervous system in Parkinson's disease. Eur Neurol. (1997) 38(Suppl. 2):2–7. 10.1159/000113469
128.
PedersenKMMarnerLPakkenbergHPakkenbergB. No global loss of neocortical neurons in Parkinson's disease: a quantitative stereological study. Mov Disord. (2005) 20:164–71. 10.1002/mds.20289
129.
MacDonaldVHallidayGM. Selective loss of pyramidal neurons in the pre-supplementary motor cortex in Parkinson's disease. Mov Disord. (2002) 17:1166–73. 10.1002/mds.10258
130.
HardingAJStimsonEHendersonJMHallidayGM. Clinical correlates of selective pathology in the amygdala of patients with Parkinson's disease. Brain (2002) 125(Pt 11):2431–45. 10.1093/brain/awf251
131.
JoelvingFCBilleskovRChristensenJRWestMPakkenbergB. Hippocampal neuron and glial cell numbers in Parkinson's disease–a stereological study. Hippocampus (2006) 16:826–33. 10.1002/hipo.20212
132.
HoehnMMYahrMD. Parkinsonism: onset, progression and mortality. Neurology (1967) 17:427–42. 10.1212/WNL.17.5.427
133.
KlingelhoeferLReichmannH. Pathogenesis of Parkinson disease–the gut-brain axis and environmental factors. Nat Rev Neurol. (2015) 11:625–36. 10.1038/nrneurol.2015.197
134.
SingaramCAshrafWGaumnitzEATorbeyCSenguptaAPfeifferRet al. Dopaminergic defect of enteric nervous system in Parkinson's disease patients with chronic constipation. Lancet (1995) 346:861–4. 10.1016/S0140-6736(95)92707-7
135.
AnnerinoDMArshadSTaylorGMAdlerCHBeachTGGreeneJG. Parkinson's disease is not associated with gastrointestinal myenteric ganglion neuron loss. Acta Neuropathol. (2012) 124:665–80. 10.1007/s00401-012-1040-2
136.
BolamJPPissadakiEK. Living on the edge with too many mouths to feed: Why dopamine neurons die. Mov Disord. (2012) 27:1478–83. 10.1002/mds.25135
137.
SurmeierDJSchumackerPTGuzmanJDIlijicEYangBZampeseE. Calcium and Parkinson's disease. Biochem Biophys Res Commun. (2017) 483:1013–9. 10.1016/j.bbrc.2016.08.168
138.
PacelliCGiguereNBourqueMJLevesqueMSlackRSTrudeauLE. Elevated mitochondrial bioenergetics and axonal arborization size are key contributors to the vulnerability of dopamine neurons. Curr Biol. (2015) 25:2349–60. 10.1016/j.cub.2015.07.050
139.
Segura-AguilarJParisIMunozPFerrariEZeccaLZuccaFA. Protective and toxic roles of dopamine in Parkinson's disease. J Neurochem. (2014) 129:898–915. 10.1111/jnc.12686
140.
VanLaar VSMishizenAJCascioMHastingsTG. Proteomic identification of dopamine-conjugated proteins from isolated rat brain mitochondria and SH-SY5Y cells. Neurobiol Dis. (2009) 34:487–500. 10.1016/j.nbd.2009.03.004
141.
XuYStokesAHRoskoskiRJrVranaKE. Dopamine, in the presence of tyrosinase, covalently modifies and inactivates tyrosine hydroxylase. J Neurosci Res. (1998) 54:691–7. 10.1002/(SICI)1097-4547(19981201)54:5<691::AID-JNR14>3.0.CO
142.
WhiteheadREFerrerJVJavitchJAJusticeJB. Reaction of oxidized dopamine with endogenous cysteine residues in the human dopamine transporter. J Neurochem. (2001) 76:1242–51. 10.1046/j.1471-4159.2001.00125.x
143.
MosharovEVLarsenKEKanterEPhillipsKAWilsonKSchmitzYet al. Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron (2009) 62:218–29. 10.1016/j.neuron.2009.01.033
144.
LohrKMBernsteinAIStoutKADunnARLazoCRAlterSPet al. Increased vesicular monoamine transporter enhances dopamine release and opposes Parkinson disease-related neurodegeneration in vivo. Proc Natl Acad Sci USA. (2014) 111:9977–82. 10.1073/pnas.1402134111
145.
PiflCRajputAReitherHBlesaJCavadaCObesoJAet al. Is Parkinson's disease a vesicular dopamine storage disorder? Evidence from a study in isolated synaptic vesicles of human and nonhuman primate striatum. J Neurosci. (2014) 34:8210–8. 10.1523/JNEUROSCI.5456-13.2014
146.
CaudleWMRichardsonJRWangMZTaylorTNGuillotTSMcCormackALet al. Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. J Neurosci. (2007) 27:8138–48. 10.1523/JNEUROSCI.0319-07.2007
147.
GuillotTSMillerGW. Protective actions of the vesicular monoamine transporter 2 (VMAT2) in monoaminergic neurons. Mol Neurobiol. (2009) 39:149–70. 10.1007/s12035-009-8059-y
148.
FahnS. Does levodopa slow or hasten the rate of progression of Parkinson's disease?J Neurol. (2005) 252(Suppl. 4):iv37–42. 10.1007/s00415-005-4008-5
149.
LipskiJNisticoRBerrettaNGuatteoEBernardiGMercuriNB. L-DOPA: a scapegoat for accelerated neurodegeneration in Parkinson's disease?Prog Neurobiol. (2011) 94:389–407. 10.1016/j.pneurobio.2011.06.005
150.
HaackeEMMiaoYLiuMHabibCAKatkuriYLiuTet al. Correlation of putative iron content as represented by changes in R2* and phase with age in deep gray matter of healthy adults. J Magn Reson Imaging (2010) 32:561–76. 10.1002/jmri.22293
151.
DaughertyARazN. Age-related differences in iron content of subcortical nuclei observed in vivo: a meta-analysis. Neuroimage (2013) 70:113–21. 10.1016/j.neuroimage.2012.12.040
152.
BilgicBPfefferbaumARohlfingTSullivanEVAdalsteinssonE. MRI estimates of brain iron concentration in normal aging using quantitative susceptibility mapping. Neuroimage (2012) 59:2625–35. 10.1016/j.neuroimage.2011.08.077
153.
RenYLiuWJiangHJiangQFengJ. Selective vulnerability of dopaminergic neurons to microtubule depolymerization. J Biol Chem. (2005) 280:34105–12. 10.1074/jbc.M503483200
154.
FriedmanAGalazka-FriedmanJBaumingerER. Iron as a trigger of neurodegeneration in Parkinson's diseaseHandb Clin Neurol. (2007) 83:493–505. 10.1016/S0072-9752(07)83023-5
155.
Sian-HulsmannJMandelSYoudimMBRiedererP. The relevance of iron in the pathogenesis of Parkinson's disease. J Neurochem. (2011) 118:939–57. 10.1111/j.1471-4159.2010.07132.x
156.
KostaPArgyropoulouMIMarkoulaSKonitsiotisS. MRI evaluation of the basal ganglia size and iron content in patients with Parkinson's disease. J Neurol. (2006) 253:26–32. 10.1007/s00415-005-0914-9
157.
ZeccaLShimaTStroppoloAGojCBattistonGAGerbasiRet al. Interaction of neuromelanin and iron in substantia nigra and other areas of human brain. Neuroscience (1996) 73:407–15. 10.1016/0306-4522(96)00047-4
158.
ZuccaFABelleiCGiannelliSTerreniMRGalloriniMRizzioEet al. Neuromelanin and iron in human locus coeruleus and substantia nigra during aging: consequences for neuronal vulnerability. J Neural Transm. (2006) 113:757–67. 10.1007/s00702-006-0453-2
159.
ZeccaLStroppoloAGattiATampelliniDToscaniMGalloriniMet al. The role of iron and copper molecules in the neuronal vulnerability of locus coeruleus and substantia nigra during aging. Proc Natl Acad Sci USA. (2004) 101:9843–8. 10.1073/pnas.0403495101
160.
MichelPPAlvarez-FischerDGuerreiroSHildAHartmannAHirschEC. Role of activity-dependent mechanisms in the control of dopaminergic neuron survival. J Neurochem. (2007) 101:289–97. 10.1111/j.1471-4159.2006.04424.x
161.
GuzmanJNSanchez-PadillaJWokosinDKondapalliJIlijicESchumackerPTet al. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature (2010) 468:696–700. 10.1038/nature09536
162.
GoldbergJAGuzmanJNEstepCMIlijicEKondapalliJSanchez-PadillaJet al. Calcium entry induces mitochondrial oxidant stress in vagal neurons at risk in Parkinson's disease. Nat Neurosci. (2012) 15:1414–21. 10.1038/nn.3209
163.
Sanchez-PadillaJGuzmanJNIlijicEKondapalliJGaltieriDJYangBet al. Mitochondrial oxidant stress in locus coeruleus is regulated by activity and nitric oxide synthase. Nat Neurosci (2014) 17:832–40. 10.1038/nn.3717
164.
ReeveASimcoxETurnbullD. Ageing and Parkinson's disease: Why is advancing age the biggest risk factor?Ageing Res Rev. (2014) 14:19–30. 10.1016/j.arr.2014.01.004
165.
HellJWWestenbroekREWarnerCAhlijanianMKPrystayWGilbertMMet al. Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel alpha 1 subunits. J Cell Biol. (1993) 123:949–62. 10.1083/jcb.123.4.949
166.
HurleyMJGentlemanSMDexterDT. Calcium CaV1 channel subtype mRNA expression in Parkinson's disease examined by in situ hybridization. J Mol Neurosci. (2015) 55:715–24. 10.1007/s12031-014-0410-8
167.
SchiemannJSchlaudraffFKloseVBingmerMSeinoSMagillPJet al. K-ATP channels in dopamine substantia nigra neurons control bursting and novelty-induced exploration. Nat Neurosci. (2012) 15:1272–80. 10.1038/nn.3185
168.
LissBHaeckelOWildmannJMikiTSeinoSRoeperJ. K-ATP channels promote the differential degeneration of dopaminergic midbrain neurons. Nat Neurosci. (2005) 8:1742–51. 10.1038/nn1570
169.
MatsudaWFurutaTNakamuraKCHiokiHFujiyamaFAraiRet al. Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatumJ. Neurosci. (2009) 29:444–53. 10.1523/JNEUROSCI.4029-08.2009
170.
PissadakiEKBolamJP. The energy cost of action potential propagation in dopamine neurons: clues to susceptibility in Parkinson's disease. Front Comput Neurosci. (2013) 7:13. 10.3389/fncom.2013.00013
171.
TakakusakiKShiroyamaTYamamotoTKitaiST. Cholinergic and noncholinergic tegmental pedunculopontine projection neurons in rats revealed by intracellular labeling. J Comp Neurol. (1996) 371:345–61. 10.1002/(SICI)1096-9861(19960729)371:3<345::AID-CNE1>3.0.CO;2-2
172.
GaoKMasonP. Somatodendritic and axonal anatomy of intracellularly labeled serotonergic neurons in the rat medulla. J Comp Neurol. (1997) 389:309–28. 10.1002/(SICI)1096-9861(19971215)389:2<309::AID-CNE9>3.0.CO;2-5
173.
GaoHGlatzerNRWilliamsKWDerbenevAVLiuDSmithBN. Morphological and electrophysiological features of motor neurons and putative interneurons in the dorsal vagal complex of rats and mice. Brain Res. (2009) 1291:40–52. 10.1016/j.brainres.2009.07.024
174.
GagnonDParentM. Distribution of VGLUT3 in highly collateralized axons from the rat dorsal raphe nucleus as revealed by single-neuron reconstructions. PLoS ONE. (2014) 9:e87709. 10.1371/journal.pone.0087709
175.
ChazalGMaW. An ultrastructural analysis of serotoninergic neurons in the nucleus raphe magnus of the rat. Neuroscience. (1989) 33:301–10. 10.1016/0306-4522(89)90210-8
176.
JonesBEYangTZ. The efferent projections from the reticular formation and the locus coeruleus studied by anterograde and retrograde axonal transport in the rat. J Comp Neurol. (1985) 242:56–92. 10.1002/cne.902420105
177.
ContantCUmbriacoDGarciaSWatkinsKCDescarriesL. Ultrastructural characterization of the acetylcholine innervation in adult rat neostriatum. Neuroscience (1996) 71:937–47. 10.1016/0306-4522(95)00507-2
178.
ZhouFMWilsonCJDaniJA. Cholinergic interneuron characteristics and nicotinic properties in the striatum. J Neurobiol. (2002) 53:590–605. 10.1002/neu.10150
179.
MeitzenJPflepsenKRSternCMMeiselRLMermelsteinPG. Measurements of neuron soma size and density in rat dorsal striatum, nucleus accumbens core and nucleus accumbens shell: differences between striatal region and brain hemisphere, but not sex. Neurosci Lett. (2011) 487:177–81. 10.1016/j.neulet.2010.10.017
180.
SianJDexterDTLeesAJDanielSAgidYJavoy-AgidFet al. Alterations in glutathione levels in Parkinson's disease and other neurodegenerative disorders affecting basal ganglia. Ann Neurol. (1994) 36:348–55. 10.1002/ana.410360305
181.
PearceRKOwenADanielSJennerPMarsdenCD. Alterations in the distribution of glutathione in the substantia nigra in Parkinson's disease. J Neural Transm. (1997) 104:661–77. 10.1007/BF01291884
182.
SoficELangeKWJellingerKRiedererP. Reduced and oxidized glutathione in the substantia nigra of patients with Parkinson's disease. Neurosci Lett. (1992) 142:128–30. 10.1016/0304-3940(92)90355-B
183.
TagliaferroPBurkeRE. Retrograde axonal degeneration in Parkinson disease. J Parkinsons Dis. (2016) 6:1–15. 10.3233/JPD-150769
184.
O'MalleyKL. The role of axonopathy in Parkinson's disease. Exp Neurobiol. (2010) 19:115–9. 10.5607/en.2010.19.3.115
185.
KinghornKJAsghariAMCastillo-QuanJI. The emerging role of autophagic-lysosomal dysfunction in Gaucher disease and Parkinson's disease. Neural Regener Res. (2017) 12:380–4. 10.4103/1673-5374.202934
186.
WongYCKraincD. Lysosomal trafficking defects link Parkinson's disease with Gaucher's disease. Mov Disord. (2016) 31:1610–8. 10.1002/mds.26802
187.
Migdalska-RichardsASchapiraAH. The relationship between glucocerebrosidase mutations and Parkinson disease. J Neurochem. (2016) 139(Suppl. 1):77–90. 10.1111/jnc.13385
188.
SidranskyENallsMAAaslyJOAharon-PeretzJAnnesiGBarbosaERet al. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med. (2009) 361:1651–61. 10.1056/NEJMoa0901281
189.
GeggMEBurkeDHealesSJCooperJMHardyJWoodNWet al. Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann Neurol. (2012) 72:455–63. 10.1002/ana.23614
190.
MurphyKEHallidayGM. Glucocerebrosidase deficits in sporadic Parkinson disease. Autophagy (2014) 10:1350–1. 10.4161/auto.29074
191.
DehayBMartinez-VicenteMCaldwellGACaldwellKAYueZCooksonMRet al. Lysosomal impairment in Parkinson's disease. Mov Disord. (2013) 28:725–32. 10.1002/mds.25462
192.
AshrafiGSchwarzTL. PINK1- and PARK2-mediated local mitophagy in distal neuronal axons. Autophagy (2015) 11:187–9. 10.1080/15548627.2014.996021
193.
MatheoudDSugiuraABellemare-PelletierALaplanteARondeauCChemaliMet al. Parkinson's disease-related proteins PINK1 and parkin repress mitochondrial antigen presentation. Cell (2016) 166:314–27. 10.1016/j.cell.2016.05.039
194.
ShengCHengXZhangGXiongRLiHZhangSet al. DJ-1 deficiency perturbs microtubule dynamics and impairs striatal neurite outgrowth. Neurobiol Aging (2013) 34:489–98. 10.1016/j.neurobiolaging.2012.04.008
195.
DiNottia MMasciulloMVerrigniDPetrilloSModoniARizzoVet al. DJ-1 modulates mitochondrial response to oxidative stress: clues from a novel diagnosis of PARK7. Clin Genet. (2017) 92:18–25. 10.1111/cge.12841
196.
MacLeodDDowmanJHammondRLeeteTInoueKAbeliovichA. The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron (2006) 52:587–93. 10.1016/j.neuron.2006.10.008
197.
YueMHinkleKMDaviesPTrushinaEFieselFCChristensonTAet al. Progressive dopaminergic alterations and mitochondrial abnormalities in LRRK2 G2019S knock-in mice. Neurobiol Dis. (2015) 78:172–95. 10.1016/j.nbd.2015.02.031
198.
SurmeierDJHallidayGMSimuniT. Calcium, mitochondrial dysfunction and slowing the progression of Parkinson's disease. Exp Neurol. (2017) 298(Pt B):202–9. 10.1016/j.expneurol.2017.08.001
199.
WangZHouLHuangLGuoJZhouX. Exenatide improves liver mitochondrial dysfunction and insulin resistance by reducing oxidative stress in high fat diet-induced obese mice. Biochem Biophys Res Commun. (2017) 486:116–23. 10.1016/j.bbrc.2017.03.010
200.
LiYPerryTKindyMSHarveyBKTweedieDHollowayHWet al. GLP-1 receptor stimulation preserves primary cortical and dopaminergic neurons in cellular and rodent models of stroke and Parkinsonism. Proc Natl Acad Sci USA. (2009) 106:1285–90. 10.1073/pnas.0806720106
201.
AthaudaDMaclaganKSkeneSSBajwa-JosephMLetchfordDChowdhuryKet al. Exenatide once weekly versus placebo in Parkinson's disease: a randomised, double-blind, placebo-controlled trial. Lancet (2017) 390:1664–75. 10.1016/S0140-6736(17)31585-4
202.
GleaveJAArathoonLRTrinhDLizalKEGiguereNBarberJHMet al. Sirtuin 3 rescues neurons through the stabilisation of mitochondrial biogenetics in the virally-expressing mutant α-synuclein rat model of parkinsonism. Neurobiol Dis. (2017) 106:133–46. 10.1016/j.nbd.2017.06.009
203.
ShiHDengHXGiusDSchumackerPTSurmeierDJMaYC. Sirt3 protects dopaminergic neurons from mitochondrial oxidative stress. Hum Mol Genet. (2017) 26:1915–26. 10.1093/hmg/ddx100
Summary
Keywords
Parkinson, vulnerability, dopamine, cell death, neurodegeneration
Citation
Giguère N, Burke Nanni S and Trudeau L-E (2018) On Cell Loss and Selective Vulnerability of Neuronal Populations in Parkinson's Disease. Front. Neurol. 9:455. doi: 10.3389/fneur.2018.00455
Received
05 April 2018
Accepted
29 May 2018
Published
19 June 2018
Volume
9 - 2018
Edited by
Graziella Madeo, National Institutes of Health (NIH), United States
Reviewed by
Paolo Gubellini, Centre National de la Recherche Scientifique (CNRS), France; Nicolas Xavier Tritsch, Langone Medical Center, New York University, United States
Updates
Copyright
© 2018 Giguère, Burke Nanni and Trudeau.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Louis-Eric Trudeau louis-eric.trudeau@umontreal.ca
This article was submitted to Movement Disorders, a section of the journal Frontiers in Neurology
†Co-first authors.
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.