 Zeynep Öztürk
Zeynep Öztürk Cahir J. O’Kane
Cahir J. O’Kane Juan José Pérez-Moreno
Juan José Pérez-Moreno- Department of Genetics, University of Cambridge, Cambridge, United Kingdom
The physical continuity of axons over long cellular distances poses challenges for their maintenance. One organelle that faces this challenge is endoplasmic reticulum (ER); unlike other intracellular organelles, this forms a physically continuous network throughout the cell, with a single membrane and a single lumen. In axons, ER is mainly smooth, forming a tubular network with occasional sheets or cisternae and low amounts of rough ER. It has many potential roles: lipid biosynthesis, glucose homeostasis, a Ca2+ store, protein export, and contacting and regulating other organelles. This tubular network structure is determined by ER-shaping proteins, mutations in some of which are causative for neurodegenerative disorders such as hereditary spastic paraplegia (HSP). While axonal ER shares many features with the tubular ER network in other contexts, these features must be adapted to the long and narrow dimensions of axons. ER appears to be physically continuous throughout axons, over distances that are enormous on a subcellular scale. It is therefore a potential channel for long-distance or regional communication within neurons, independent of action potentials or physical transport of cargos, but involving its physiological roles such as Ca2+ or organelle homeostasis. Despite its apparent stability, axonal ER is highly dynamic, showing features like anterograde and retrograde transport, potentially reflecting continuous fusion and breakage of the network. Here we discuss the transport processes that must contribute to this dynamic behavior of ER. We also discuss the model that these processes underpin a homeostatic process that ensures both enough ER to maintain continuity of the network and repair breaks in it, but not too much ER that might disrupt local cellular physiology. Finally, we discuss how failure of ER organization in axons could lead to axon degenerative diseases, and how a requirement for ER continuity could make distal axons most susceptible to degeneration in conditions that disrupt ER continuity.
Introduction
Endoplasmic reticulum (ER) is a membrane-bound organelle found throughout the cytoplasm of all eukaryotic cells. It typically comprises more than half of the total animal cell membranes (Alberts et al., 2002), or 35% of cytoplasmic volume at any one time (Valm et al., 2017). Light and electron microscopy (EM), and membrane dye labeling, suggest that uniquely among organelles, the ER appears pervasive and continuous throughout cells, including neurons (Tsukita and Ishikawa, 1976; Terasaki et al., 1994; Wu et al., 2017; Yalçın et al., 2017) (Figure 1). Physically connected to the nuclear envelope, and sharing with it a common lumen, ER includes rough and smooth ER domains. The first is called “rough” due to the density of ribosomes on its surface, which are involved in synthesis of proteins for export from the cytosol. Rough ER is found mainly around the nucleus, organized in a network of membrane sheets that envelop a lumen with a constant distance between sheets, and interconnected by spiral ramps (Terasaki et al., 2013). Smooth ER, with few ribosomes, is found in peripheral parts of the cell, as a network of interconnected cylindrical tubules, with occasional and irregularly spaced sheets (with a constant lumen size like rough ER) or cisternae with a larger and less regularly shaped lumen. This variability in ER morphology influences ER function. Tubules are regions for lipid synthesis and contacts with other organelles; cisternae, with a larger lumen, in addition have more capacity for calcium storage (Schwarz and Blower, 2016). Therefore, the extent and spatial distribution of different ER morphologies depends on cell type and cell demands.
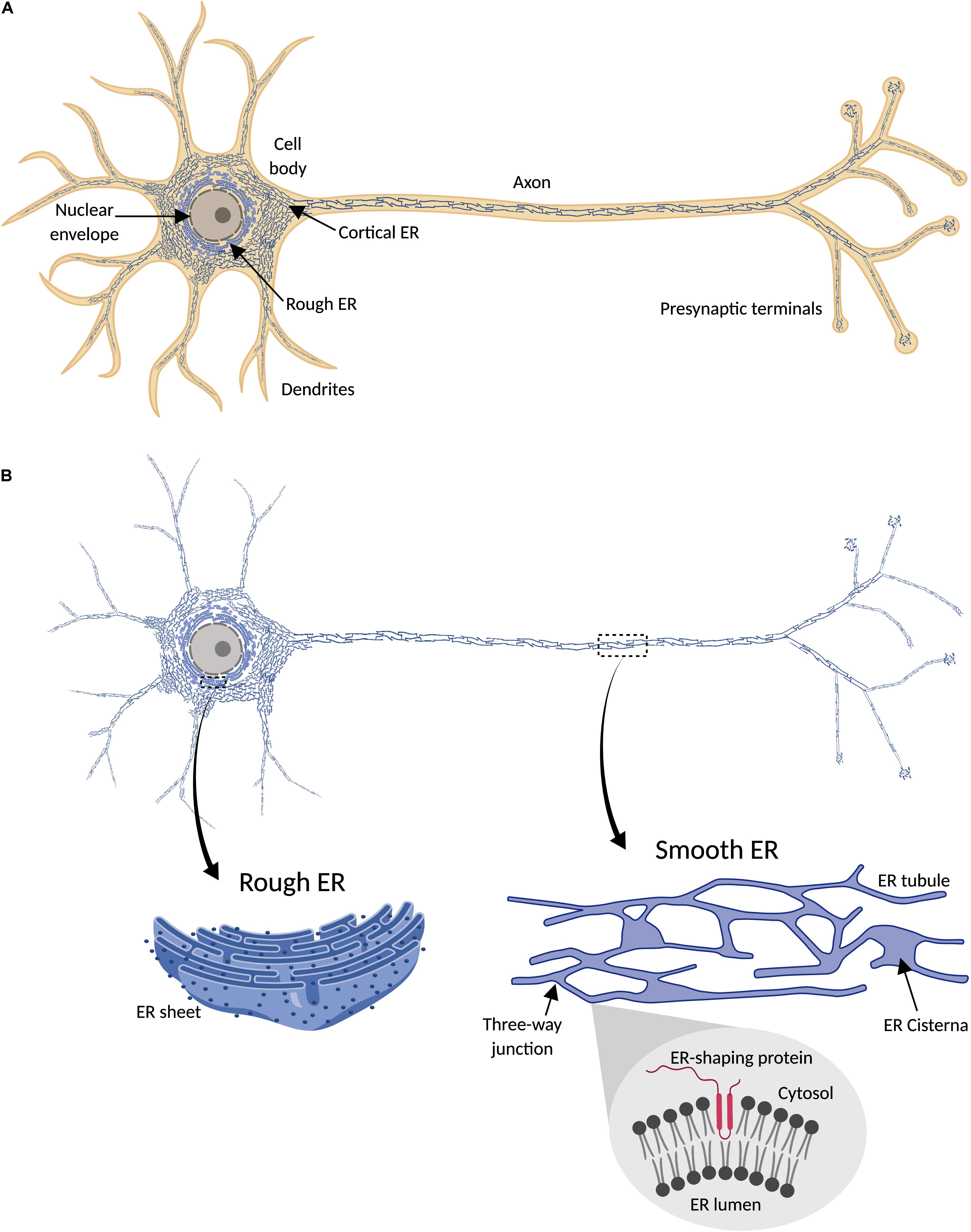
Figure 1. Endoplasmic reticulum (ER) distribution in neuronal cells. (A) ER network (blue) is continuously distributed throughout the cytosol (orange) in neurons, including cell body, axon, presynaptic terminals, and most dendrites. (B) Since ER forms a continuous structure that follows the shape of the cell, it has been termed “a neuron within a neuron” (Berridge, 1998, 2002). In the cell body, continuous with the nuclear envelope, is found rough ER, characterized by its sheet-like morphology and the presence of ribosomes (dark blue points) attached to the ER membrane (magnified detail on the left). Peripheral ER (smooth) includes cortical ER in the cell body and dendritic and axonal ER, and is mainly formed by a network of interconnected tubules, with occasional sheets or cisternae (magnified detail on the right). Tubulation of ER membrane is physically promoted by ER-shaping proteins, which share a characteristic intramembrane domain inserted in the cytosolic face of the ER membrane (magnified detail on the bottom right).
Rough ER is specialized for protein export from the cytosol. Most exported proteins have an N-terminal signal sequence that directs them to rough ER, where they are co-translationally transported across the ER membrane, folded in the ER lumen, and sorted for transport to another intracellular membrane compartment or the extracellular secretory pathway. Type I transmembrane proteins, with an extracellular or luminal N-terminus, follow similar pathways, but remain embedded in the ER membrane, due to an internal transmembrane domain that prevents the rest of the protein from being exported (Schwarz and Blower, 2016). Smooth ER, on the other hand, plays important roles in both lipid (Jacquemyn et al., 2017) and glucose (Marini et al., 2016; Müller et al., 2018) metabolism, as well as in Ca2+ dynamics (Schwarz and Blower, 2016).
Until recently, axonal ER received scant attention since its characterization by electron microscopy some four decades ago e.g., Tsukita and Ishikawa (1976), partly due to the lack of molecular markers and the difficulty of resolving it using conventional osmium staining in thin EM sections. However, both these hurdles have recently been overcome e.g., Villegas et al. (2014) and Yalçın et al. (2017). Axonal ER has the characteristics of a largely smooth tubular ER network, which must share many of its functions with the better characterized equivalent network in non-neuronal cells, but with specialized roles and constraints that reflect the long and narrow dimensions of axons. In this review, we wish to focus on the behavior and function of axonal ER, drawing both on work in axons, but also examining how our broader knowledge of the role of smooth tubular ER might apply to neurons, particularly axons. As background to our review, we have summarized proteins with roles related to tubular ER function, and which of them have been observed in axons (Table 1).
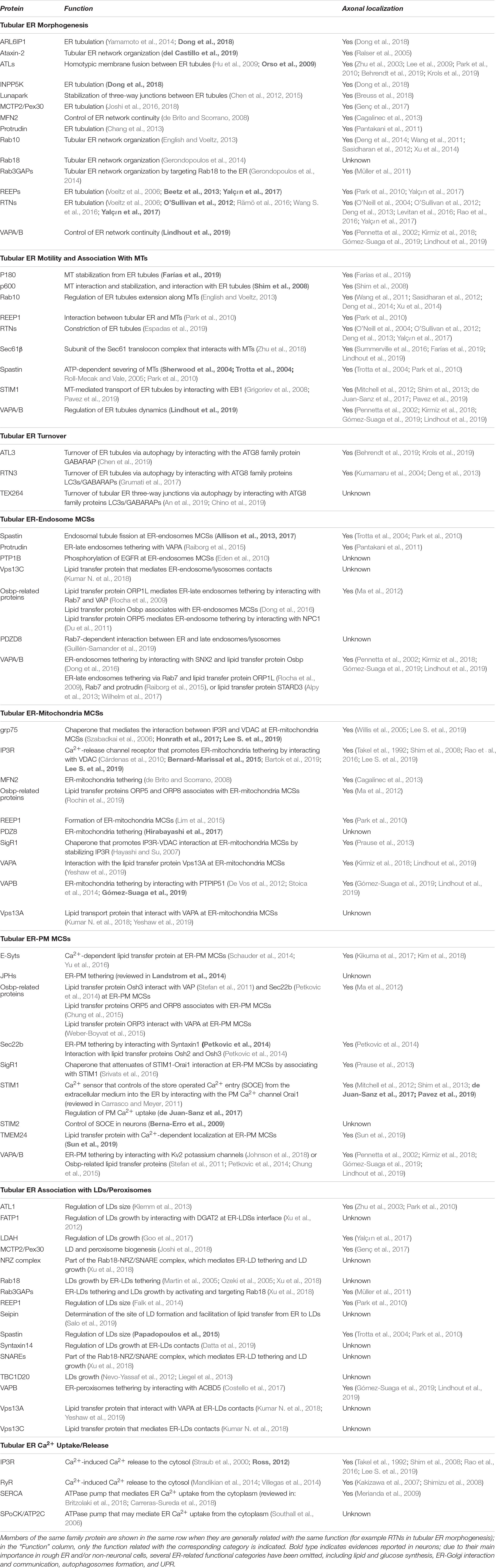
Table 1. Functional classification of tubular ER-related proteins and localization in axons.
Roles of Endoplasmic Reticulum
ER and Lipid Metabolism
The majority of phospholipids, sterols, sphingolipids, and neutral lipids are synthesized in smooth ER and distributed from there to other cellular compartments. Controlled trafficking of these lipids from/to ER is required to maintain the lipid composition of membranes such as mitochondrial or plasma membrane (PM) (Fagone and Jackowski, 2009; Muallem et al., 2017). Two different transport mechanisms mediate the transfer of lipids between ER and other cellular organelles: vesicular transport is dependent on membrane budding and fusion via the vesicular transport machinery, whereas non-vesicular lipid transport is mediated by ER membrane contact sites (MCSs) with other organelles (Figure 2). In addition, biogenesis of lipid droplets (LDs) (for storage of sterols and triglycerides) and peroxisomes (involved in lipid decomposition through β-oxidation) initiates in the ER. LDs form initially in the ER lipid bilayer, and bud from the ER membrane as they grow. Although it is not still clear how sites of LD formation are determined, some of the ER proteins associated with them have been identified (e.g., seipin) (Szymanski et al., 2007; Salo et al., 2016, 2019) or Syntaxin14 (Datta et al., 2019). These and other proteins, which tether MCS between ER and LDs (discussed below), and control fatty-acid-induced droplet growth, may have roles in determining the sites of nascent LDs. Similarly, peroxisome biogenesis also seems to originate at specific ER domains. In yeast, both peroxisomes and LDs form and remain associated with the same ER subdomains, which contain Pex30 protein (Joshi et al., 2016, 2018), suggesting a link between LD and peroxisome biogenesis at the ER membrane. Although the existence of LDs in neurons has been debated, there is some evidence for their presence, including in axons (reviewed in Pennetta and Welte, 2018).
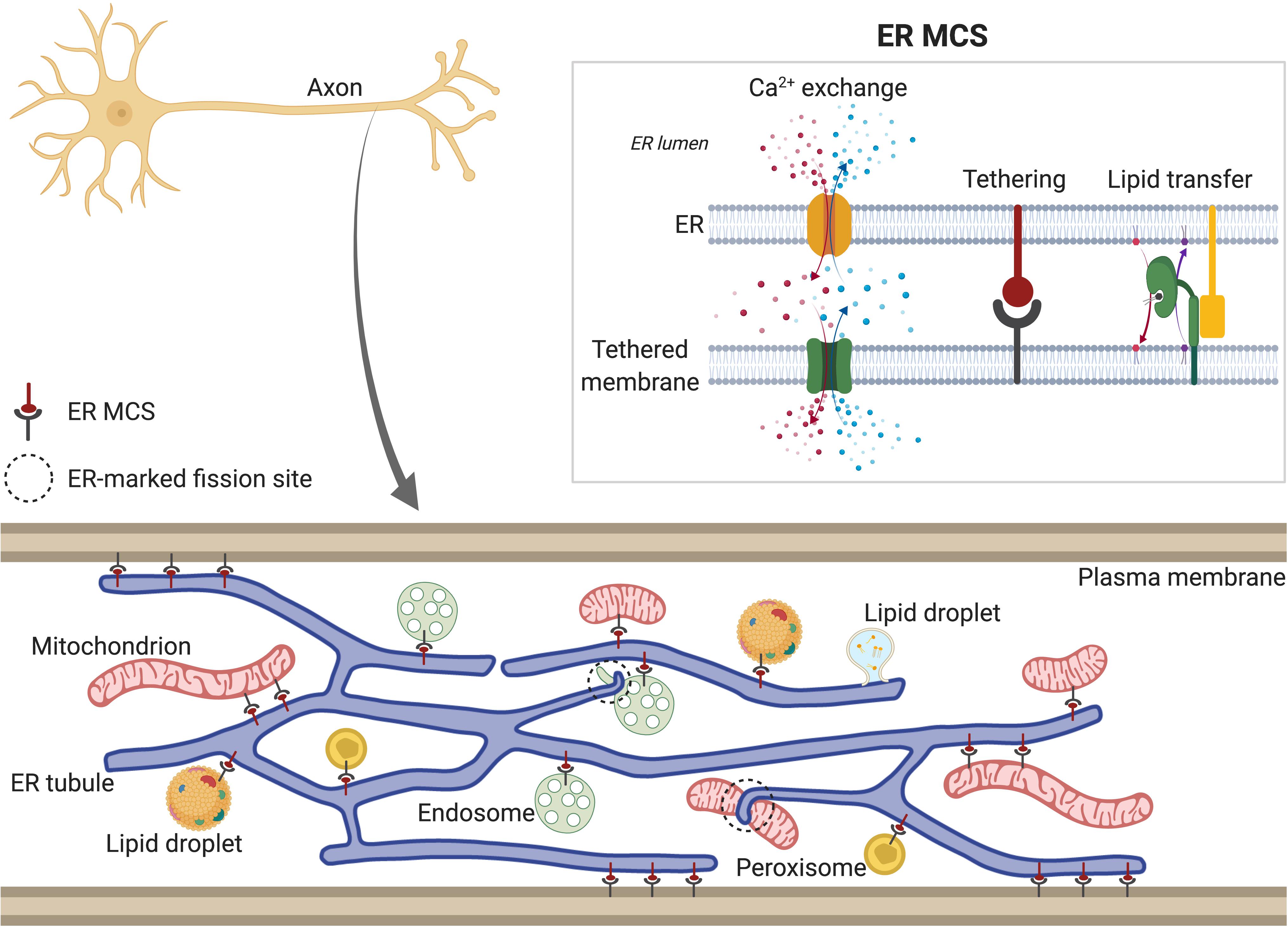
Figure 2. Tubular ER membrane contact sites (MCSs). Tubular ER membrane forms contacts with other membranes in the cell, including plasma membrane (PM), Golgi, mitochondria, endosomes, lipid droplets (LDs) and peroxisomes. Axonal ER (bottom) also contains MCSs, except with Golgi, due to its absence in axons. ER MCSs can regulate fission of some of the tethered organelles, such as endosomes and mitochondria (dotted circles). ER MCSs (top right) are formed by interactions between ER membrane proteins and proteins found in the other cellular membranes (tethering). This keeps both membranes close enough to allow transfer of lipids and Ca2+. Depending on the specific channels localized at each MCS (see main text for details), Ca2+ can be released from ER lumen to the other compartment (red) or from other compartment to ER lumen (represented in blue). In both scenarios, Ca2+ is released locally to the intramembrane space and then taken up by the acceptor compartment. ER MCSs also mediate non-vesicular and bidirectional transport of lipids, which are shuttled between membranes by lipid transfer proteins (green).
ER and Glucose Metabolism
Intracellular glucose is phosphorylated to glucose-6-phosphate (G6P) to be used in metabolism and produce ATP. In tissues like liver, G6P is taken up from cytosol into the ER and dephosphorylated there by G6Pase-α to be returned to cytosol when necessary. Deficiency of G6Pase-α causes glycogen storage disease (Chou et al., 2015). Another G6Pase isozyme, G6Pase-β, is required in astrocytes for glucose storage in their ER lumen. Knockdown of G6Pase-β causes reduction not only of glucose accumulation, but also of Ca2+ regulation, and ATP production in the ER of astrocytes. It has been proposed that ER may serve as a glucose intracellular highway. G6Pase-β would facilitate accumulation of glucose to the ER lumen, where it would be free to diffuse and be protected from further metabolism, until returning to the cytosol through ER glucose transporters, and being phosphorylated to G6P to enter glycolysis (Müller et al., 2018). However, it has recently been argued that glucose transport through the ER is too slow (around 550–3,700 times lower than glucose consumption) to be quantitatively relevant for nutrient delivery (Dienel, 2019), and so the physiological role of glucose in the ER lumen is not fully resolved.
ER and Calcium Dynamics
Release and re-uptake of Ca2+ to/from the cytosol mediate initiation and termination of many cellular responses to signals. ER is a major calcium store and manages Ca2+ homeostasis by either behaving as a local Ca2+ sink, or a store that releases Ca2+ to other cellular compartments (Raffaello et al., 2016), controlling processes such as mitochondrial homeostasis and function (Britti et al., 2018). ER can buffer excess presynaptic cytosolic Ca2+ during repetitive firing, by uptake via the SERCA calcium ATPase (Sanyal et al., 2005) or potentially also the SPoCk ATPase (Southall et al., 2006). Conversely it can also supply Ca2+ to the cytosol via channels including inositol tris-phosphate receptors (IP3R) or ryanodine receptors (RyR) (reviewed by Ross, 2012). A specific role of ER Ca2+ has been recently shown in mammalian neurons, where at presynaptic terminals, ER luminal Ca2+ can promote PM Ca2+ uptake via the ER Ca2+ sensor STIM1, a process required for presynaptic Ca2+ flux and exocytosis (de Juan-Sanz et al., 2017). Depletion of either the vesicle-associated membrane protein-associated proteins A (VAPA) and B (VAPB), which are ER membrane receptors, or the cytoplasmic VAP-associated protein Secernin-1 (SCRN1), reduces presynaptic Ca2+ influx and synaptic vesicle cycling (Lindhout et al., 2019). Also, in Drosophila neurons, the ER-resident Ca2+ sensor MCTP (multiple C2 domain and transmembrane region protein) promotes release of synaptic vesicles (Genç et al., 2017). Therefore, maintenance of ER Ca2+ appears to be crucial for proper synaptic function.
A continuous ER network can also support regional or long-distance Ca2+ signaling or homeostasis. Ca2+ signals can propagate through the cytosol by Ca2+-induced Ca2+ release from ER, and thus mediate regional and/or global communication within the cell, analogous to but slower than action potential propagation in the PM. Ca2+-induced Ca2+ release can be mediated by IP3R or RyR receptors, and be potentiated by elevated cytosolic Ca2+ (Straub et al., 2000; Ross, 2012). We know little of the occurrence or roles of propagating Ca2+ waves in axons, but a few cases are known. For example, a propagating elevation of cytosolic Ca2+ is seen after axonal injury in the early stages of Wallerian degeneration (Vargas et al., 2015). A back-propagating Ca2+ wave, which depends on ER Ca2+ stores, is also required for the regenerative response to axon injury in dorsal root ganglion (DRG) neurons (Cho et al., 2013). Long-range Ca2+ waves also play a role in inhibitory signaling among outgrowing neurites to ensure that only a single neurite will form an axon, although a role for ER in this has not been shown (Takano et al., 2017). All these are situations in which a local event must be communicated to induce responses in other parts of the cell or axon, and where ER continuity can potentially underpin this communication.
The ER lumen can also act as an intracellular highway for Ca2+, allowing “Ca2+ tunneling”. When luminal Ca2+ is released to the cytosol, it must be replenished. The fastest route for replenishment across significant intracellular distances is diffusion through the ER lumen, where there is relatively little Ca2+ buffering, leaving Ca2+ free to diffuse throughout the lumen of the ER network. This has been shown in non-neuronal cells, including pancreatic acinar cells, Xenopus oocytes (reviewed in Petersen et al., 2017) and HeLa cells (Courjaret et al., 2018), but has not been investigated in neurons.
Axonal ER
Presynaptic terminals can lie up to 1 m from the cell body in human neurons. How can axons mediate communication, and be physically maintained, across this distance? Action potentials at the PM carry long-range signals, and the microtubule (MT) network transports physical cargoes (Hirokawa and Takemura, 2005). A third potential channel for communication along axons is ER, which appears physically continuous throughout neurons (Tsukita and Ishikawa, 1976; Terasaki et al., 1994; Wu et al., 2017; Yalçın et al., 2017) (Figure 1), and has therefore been termed a “neuron within a neuron” (Berridge, 1998, 2002).
An important role for tubular ER is also implied by the genetics of some neurological disorders (Table 2). For instance, mutations in proteins that regulate tubular ER organization are causative for hereditary spastic paraplegia (HSP) and other axonopathies (Hübner and Kurth, 2014; Liberski and Blackstone, 2017). Gradual accumulation of abnormally clustered tubular ER is also found in areas surrounding amyloid plaques in Alzheimer’s disease (AD) brains (Sharoar et al., 2016). Mutation of proteins associated with membrane contacts between ER and mitochondria can also cause diverse neurological defects, including AD, amyotrophic lateral sclerosis (ALS), Parkinson’s disease (PD) or Charcot-Marie-Tooth disease (CMT) (Bernard-Marissal et al., 2018). To understand the impact of axonal ER in neurodegeneration, it is first essential to understand how its organization and dynamics are regulated, and the consequences of disrupting these processes.

Table 2. Tubular ER-related proteins associated with neurological disorders.
Axonal ER Organization
Local ER organization is mediated by a group of evolutionary conserved protein families found in the ER membrane, each one specialized in regulating particular aspects of ER morphology. Axonal ER is mostly composed of tubular ER (Figure 1). Therefore, to understand axonal ER biology, it is critical to know how the tubular ER network is regulated. It is well established that reticulons (RTNs) and REEP proteins drive formation of ER tubules (Voeltz et al., 2006; Beetz et al., 2013). In yeast, these two protein families are together essential for most peripheral ER tubules (Hu et al., 2008), although in other organisms like Drosophila, the absence of RTNs and REEPs is not enough to abolish ER tubule formation in axons (Yalçın et al., 2017), indicating that additional proteins are involved in formation of ER tubules in neurons. RTNs and REEPs are proposed to control ER tubulation through their hydrophobic intramembrane domains, which insert in the cytosolic face of the ER lipid bilayer, distorting it and inducing local curvature of the bilayer (Shibata et al., 2009) (Figure 1). Also, Pex30, a yeast protein enriched at sites of nascent LDs and peroxisomes on ER, is found in tubules and the edges of sheets, as are RTNs. Pex30 has a reticulon homology domain (RHD), as does its closest human homolog, MCTP2. Expression of Pex30 (Joshi et al., 2016) or MCTP2 RHD restores ER tubules in yeast lacking RTNs (Joshi et al., 2018). Therefore, in addition to their role in lipid metabolism, Pex30/MCTP2 may have a redundant role with RTNs and REEPs in tubular ER formation.
Other proteins that can shape tubular ER include ADP-ribosylation factor-like 6 interacting protein 1 (ARL6IP1) (Yamamoto et al., 2014) and protrudin (Chang et al., 2013). Both are found in tubular ER and possess hairpin-loop domains like RTNs and REEPs. However, the presence of a short polar stretch in the middle of their potential hairpins suggest that they span the entire ER membrane. Nevertheless, there is evidence for roles for both ARL6IP1 and protrudin in shaping tubular ER. In Drosophila larval motor neurons, ARL6IP1 knockdown disrupts ER distribution at axon terminals (Fowler and O’Sullivan, 2016). In addition, ARL6IP1 recruits INPP5K, an inositol 5-phosphatase enriched in newly formed ER tubules. Loss of ARL6IP1 or INPP5K leads to an increase in ER sheets (Dong et al., 2018), consistent with the idea that ARL6IP1 promotes formation of ER tubules. Also, protrudin depletion in mammalian cells alters ER network morphology, promoting the extension of ER sheets (Chang et al., 2013), again consistent with a role in promoting formation of ER tubules.
Axonal ER forms a network of interconnected tubular structures. Fusion between ER tubules is mediated by the atlastins (ATLs), a family of GTPases that mediate fusion between ER membranes in a GTP-dependent manner (Orso et al., 2009). As in yeast and mammalian cell culture models (Hu et al., 2009; Kornak et al., 2014; Wang S. et al., 2016), depletion of ATL in Drosophila neurons leads to tubular ER fragmentation and unbranched ER tubules (Orso et al., 2009). Similar branching defects have also been reported in C. elegans neurons mutated in atln-1, the ATL1 ortholog (Liu et al., 2019). In addition to their GTPase catalytic domain, ATLs also possess intramembrane domains, similar to those found in RTNs and REEPs, that are not only essential for ATL membrane fusion activity, but also could explain why ATL1 drives the generation of membrane tubules from proteoliposomes in vitro (Betancourt-Solis et al., 2018).
Another protein with an ER intramembrane domain is the M1 form of the MT-severing protein spastin (Park et al., 2010), which interacts with RTNs (Mannan et al., 2006), REEPs and ATLs (Park et al., 2010). However, there is not yet any evidence for a direct role for spastin in promoting formation of ER tubules. Similarly, a potential role of FAM134B protein in ER tubulation is not clear. FAM134B contains a predicted RHD, and its loss produces expanded ER sheets. Also, via its RHD, FAM134B binds to liposomes and produces highly curved proteoliposomes. However, FAM134B is mainly found in perinuclear ER, where it has been proposed to promote curvature at the edges of sheets (Khaminets et al., 2015; Bhaskara et al., 2019).
ER-shaping proteins that mediate tubulation, such as RTNs and REEPs, localize not only in tubular ER, but also at the edges of ER sheets (Shibata et al., 2010), both regions with high curvature. Another ER-resident protein whose localization is related to its function is Lunapark. This protein is directly involved in shaping the ER tubular network by stabilizing three-way junctions between ER tubules, where it also localizes (Chen et al., 2012, 2015). Absence of Lunapark in mammalian cells increases the amount of ER sheets, but does not abolish the formation of an ER tubular network (Wang S. et al., 2016; Zhou et al., 2018), suggesting that although required to stabilize the three-way junctions, Lunapark is not essential to develop ER tubules.
Roles in shaping tubular ER are not confined to proteins with intramembrane hairpin domains. For example, the small GTPases Rab10 and Rab18 regulate tubular ER morphology: depletion of Rab10 produces expansion of cisternal ER and fewer ER tubules (English and Voeltz, 2013), and loss of Rab18 from ER tubules, by depletion of the Rab3GAP complex (which is also a Rab18 GEF), causes fragmentation of the ER tubular network and spread of ER sheets (Gerondopoulos et al., 2014). Another example is the RNA-binding protein Ataxin-2, also associated with ER, whose depletion causes formation of ER aggregates in the peripheral ER network of Drosophila neurons (del Castillo et al., 2019). Mitofusin-2 (MFN2) is found at both mitochondria and ER membranes, being particularly enriched at the latter. A role for ER MFN2 in ER tubulation has been proposed, since it is required to keep luminal continuity at the peripheral ER in mammalian cells (de Brito and Scorrano, 2008). VAPA/B are also found at ER membrane and required for axonal ER continuity (Lindhout et al., 2019).
Finally, one characteristic feature of axonal ER is its very small diameter – ER tubules in most cells frequently has a diameter of ∼60 nm (varying between 25 and 90 nm), but in axons, ER tubules have a diameter on average around 40 nm, and often becoming small enough for the lumen not to be even visible (Wu et al., 2017; Yalçın et al., 2017; Terasaki, 2018). The reasons for this specialization are unclear, but the predicted consequences include limited capacity to either release or sequester Ca2+, as well as limitations on both Ca2+ tunneling and glucose tunneling. Therefore, while ER continuity appears to be important for its function, there also appear to be reasons to constrain lumen continuity.
Axonal ER Membrane Contact Sites (MCSs)
The continuity of the axonal ER network suggests a role for it in communicating throughout neurons, but also provides capacity to regulate cellular processes at local or regional levels. Tubular ER has MCSs with nearly every membranous organelle, including PM (Elbaz and Schuldiner, 2011; Phillips and Voeltz, 2016; Wu et al., 2018) (Figure 2). This is also true for axonal ER, where focused ion-beam scanning electron microscopy (FIB-SEM) reveals membrane domains in close proximity to other organelles (Wu et al., 2017). Although most of our understanding of ER MCSs comes from yeast and mammalian non-neuronal cells (Eisenberg-Bord et al., 2016), the molecular machinery associated with these MCSs is present in axons (Bernard-Marissal et al., 2018). ER MCSs are not mere physical attachments between ER and other organelles; depending on both the organelle and their function, they can be enriched for different proteins. For example, some MCSs are related to Ca2+ exchange, others for lipid exchange (Eisenberg-Bord et al., 2016). Impairment of these MCSs disrupts organellar communication and can cause diverse cellular defects, including in organelle dynamics (Friedman et al., 2011; Mandikian et al., 2014; Rowland et al., 2014), lipid metabolism (Bian et al., 2017; Hua et al., 2017) or Ca2+ levels (Bernard-Marissal et al., 2015; de Juan-Sanz et al., 2017). Here, we summarize the different MCS known between ER and other organelles, and those MCSs reported in neurons.
ER-Mitochondria MCS
In mouse neurons, axonal ER tubules typically contact around 5% of mitochondrial surface (Wu et al., 2017). Although it is still unknown which signals control the formation and maintenance of these MCSs, we know about some molecules located there. ER-shaping protein REEP1 is reported not only at ER membrane, but also at mitochondrial membrane, and to be required for ER-mitochondria interactions in non-neuronal cells (Lim et al., 2015). A role for MFNs in ER-mitochondria tethering has been also reported in non-neuronal cells (de Brito and Scorrano, 2008), and they are proposed to have a similar role in neurons, where they are required for proper mitochondrial dynamics (Züchner et al., 2004). Another tethering complex in neuronal cells comprises VAPB (at ER membrane) bound to the mitochondrial protein tyrosine phosphatase-interacting protein 51 (PTPIP51) (De Vos et al., 2012; Stoica et al., 2014). The VAPB-PTPIP51 tether has been specifically reported in presynaptic regions; neuronal activity increases presynaptic VAPB-PTPIP51 MCSs; and loss of either VAPB or PTPIP51 reduces synaptic activity, suggesting an important role for VAPB-PTPIP51 MCSs in synaptic function (Gómez-Suaga et al., 2019). Also in neurons, the ER membrane protein PDZD8 promotes ER-mitochondria MCSs (Hirabayashi et al., 2017).
ER-mitochondria MCSs are required for diverse processes, including controlling mitochondrial Ca2+ levels. ER is a major Ca2+ source, and mitochondria can take up Ca2+ from ER via ER-mitochondria MCSs, where Ca2+ exits the ER lumen through the IP3R, which is required for maintaining ER-mitochondria MCSs (Bartok et al., 2019). IP3R is stabilized by the ER chaperone Sigma receptor-1 (SigR1) (Hayashi and Su, 2007), and binds to the voltage-dependent anion channel (VDAC) on mitochondrial outer membrane (Cárdenas et al., 2010). IP3R-VDAC interaction is stabilized by the molecular chaperone glucose-regulated protein 75 (grp75) (Szabadkai et al., 2006). After exiting the ER lumen, Ca2+ is translocated into mitochondria by calcium uniporters. Mitochondrial Ca2+ levels control ATP production by activation of key metabolic enzymes in the Krebs cycle (Gellerich et al., 2010), and at high levels, promote apoptosis by opening the mitochondrial permeability transition pore (mPTP) and allowing release of mitochondrial contents to the cytosol (Baumgartner et al., 2009). Therefore, ER-mitochondria MCSs have a critical role in mitochondrial function by controlling mitochondrial Ca2+ levels. Enrichment and function of VDAC, IP3R, SigR1 or grp75 proteins at ER-mitochondria MCSs has been specifically reported in neurons (Shoshan-Barmatz et al., 2004; Higo et al., 2010; Mavlyutov et al., 2010), including in axons (Bernard-Marissal et al., 2015).
Some of the enzymes required for the biosynthesis of mitochondrial lipids, such as phospholipids or cholesterol, are found in the ER membrane, and interestingly, enriched at ER-mitochondria MCSs, where lipids are exchanged between both organelles (Vance, 2014). Mitochondrial function (Tasseva et al., 2013) and balanced fusion and fission (Steenbergen et al., 2005) are dependent on specific lipid composition, and therefore, lipid exchange at ER-mitochondria MCSs may be critical in regulating these processes. The Vps13 family of lipid-transfer proteins are localized to a variety of intracellular membrane contacts; of these, Vps13A (vacuolar protein sorting-associated protein 13A), which directly interacts with VAPA (Yeshaw et al., 2019), is found at mammalian ER-mitochondrial MCSs (Kumar N. et al., 2018), although it is not known how much it contributes to lipid transport there (Petrungaro and Kornmann, 2019). Oxysterol binding protein (Osbp)-related proteins ORP5 and ORP8 are also found at ER-mitochondria MCS, where they mediate non-vesicular transport of phosphatidylserine from the ER to mitochondria in mammalian cells (Rochin et al., 2019).
Mitochondrial fusion and fission events are driven by membrane-bound GTPases like MFNs and dynamin related protein (DRP-1) (Hoppins et al., 2007). In addition to the above-mentioned role for MFNs in tethering ER-mitochondria MCSs, DRP-1 recruitment to mitochondria occurs after ER tubules mark the mitochondrial constriction site (Friedman et al., 2011), suggesting a critical role for ER-mitochondria MCSs in mitochondrial dynamics. Moreover, a role for tubular ER in this process is supported by the fact that knockdown of key ER-shaping proteins such as ATL3, RTN1, RTN4, or Lunapark produces structural alteration of mitochondria in human cells (Milani et al., 2018; Krols et al., 2019). These evidence comes from non-neuronal cells, but since ER-shaping proteins control tubular ER in axons (see above), it is reasonable to expect that axonal ER regulates mitochondrial dynamics similarly. Another role attributed to ER-mitochondria MCSs and pending exploration in neurons, is the formation of autophagosomes (Hamasaki et al., 2013), which mediate the fundamental cellular process of autophagy (Mizushima et al., 2011).
ER-PM MCS
Contacts between large flat ER cisternae and PM are mostly seen in neuronal cell bodies, but also in axons, where between 0.5 and 1% of the PM surface contacts ER (Wu et al., 2017). Depending on function, these MCSs can be mediated by various ER membrane proteins, allowing exchange of particular lipids or Ca2+ between ER and PM membranes (Saheki and Camilli, 2017).
Ca2+ exchange at ER-PM MCSs can be mediated by different molecular complexes. In non-neuronal cells, the ER Ca2+ sensor STIM1 binds a store-operated channel (SOC), the PM Ca2+ channel Orai1, in a process stimulated by Ca2+ depletion in ER to induce Ca2+ influx from the extracellular medium (Carrasco and Meyer, 2011). In contrast to its stabilizing role on the ER-Ca2+ release channel IP3R (see above), SigR1 associates with STIM1 and attenuates its interaction with Orai1 (Srivats et al., 2016). In neurons, the role described for STIM1 seems to be mostly performed by its paralog STIM2 (Berna-Erro et al., 2009), whereas STIM1 activation in presynaptic terminals locally modulates presynaptic function, impacting activity-driven Ca2+ entry and release probability (de Juan-Sanz et al., 2017).
PM voltage-gated K+ channels (Kv) and ER RyR Ca2+ channels also accumulate at ER-PM MCSs in neurons (Mandikian et al., 2014), controlling local Ca2+ homeostasis and the formation of these MCSs (Fox et al., 2015; Kirmiz et al., 2018). Other molecules related to ER-PM MCSs are junctophilins (JPHs), ER membrane proteins that bind the PM through their N-terminal domain. In addition to tethering both membranes, JPHs have a role in PM-ER Ca2+ signaling, potentially by their interaction with Ca2+ channels and/or facilitating the interaction between Ca2+ channels from both membranes (Landstrom et al., 2014). Although this role has been mainly studied in muscle junctional membrane complexes, which are formed between the sarcoplasmic reticulum and T-tubule membranes, there is evidence supporting a similar role for JPHs in neurons (Moriguchi et al., 2006; Sahu et al., 2019).
ER-PM MCSs also allow transport of lipids. Similar to mitochondria, enzymes for the biosynthesis of some PM lipids are found in ER membranes. This compartmentalization of lipid metabolic enzymes facilitates regulation of the lipid composition of both membranes (Lauwers et al., 2016), which affects key cellular processes such as the actin cytoskeleton, membrane protein recruitment, and vesicle trafficking (Toker, 2002). Extended synaptotagmins (E-Syts), integral ER membrane proteins, bind to PM phosphatidyl inositol 4,5 bisphosphate (PI(4,5)P2) and transfer glycerophospholipids between ER and PM in a Ca2+-dependent manner (Yu et al., 2016; Bian et al., 2017) harboring lipids within their SMP (synaptotagmins-like-mitochondrial-lipid binding protein) domain (Schauder et al., 2014; Jeong et al., 2017). In Drosophila neurons, E-Syt localizes at axonal ER, and is required for proper neurotransmission and synaptic growth. However, at least in the presynaptic region, E-Syt may not be required for lipid homeostasis, since synaptic balance of phospholipids PI(4,5)P2 and PI(3)P appears normal in E-Syt mutants (Kikuma et al., 2017). Transmembrane protein 24 (TMEM24), also regulated by Ca2+ and with SMP domains, is enriched at ER-PM MCSs, where it acts as a tether and transfers phospholipids in mammalian neurons, including in axons (Sun et al., 2019).
As at MCSs between ER and other organelles, VAP proteins mediate tethering between ER and PM membranes, indicating a general role for these proteins in the formation of ER MCS. VAPs can recruit diverse proteins with lipid-binding domains to the ER by binding to FFAT [diphenylalanine (FF) in an acidic tract] motifs in these proteins, allowing exchange of phospholipids, ceramides or sterols between both membranes (Murphy and Levine, 2016). Moreover, the SNARE proteins Sec22b (ER-resident) and Syntaxin1 (PM protein) mediate non-fusogenic MCSs between ER and PM in neurons. This tether contributes to PM expansion in growth cones (Petkovic et al., 2014), a process thought to be controlled by non-vesicular lipid transfer (Gupton and Gertler, 2010). Since Sec22 interacts with lipid transfer proteins Osh2 and Osh3 (oxysterol-binding homology proteins 2 and 3; phosphoinositide-binding proteins) in yeast, it is proposed that Sec22b-Syntaxin1 interaction may regulate neuronal PM growth by controlling lipid synthesis and transfer (Petkovic et al., 2014).
ER MCS and Intracellular Membrane Trafficking
Endocytosed molecules from the PM are delivered to endosomes, where they are sorted for traffic to other compartments, for example to lysosomes for degradation (Elkin et al., 2016). ER and endosomes form dynamic MCSs, where both organelles are linked to MTs (Friedman et al., 2013). ER-endosome MCSs are reported in axons, where ER contacts tubulovesicular structures (early endosomes), multivesicular bodies (late endosomes) and lysosomes (Wu et al., 2017). MCSs between ER and endosomes can be mediated by VAP and lipid-binding proteins, allowing exchange of lipids like cholesterol or phospholipids (Eden, 2016). One lipid-transfer protein associated with these MCSs is Vps13C (Kumar N. et al., 2018). In addition, endosomal cargoes can interact with ER membrane proteins, as revealed by the interaction between the ER-localized protein tyrosine phosphatase PTP1B and the endosome-associated epidermal growth factor receptor (EGFR). In this interaction, PTP1B dephosphorylates EGFR and leads its degradation (Eden et al., 2010). Similar than its role on mitochondrial dynamics, ER contacts define the position of endosome fission (Rowland et al., 2014; Hoyer et al., 2018). No evidence of lipid transfer between ER and endosomes have been reported in axons yet. However, a role for tubular ER (including in neurons) in regulating endosomal dynamics has been proposed. The ER-shaping protein and MT-severing ATPase spastin, is required to initiate endosomal tubule fission at ER-endosomal MCSs. Failure in this process causes increased endosomal tubulation (Allison et al., 2013) and abnormal lysosomal morphology (Allison et al., 2017).
Both LDs and peroxisomes, with roles in lipid storage and lipid degradation respectively, can be generated from ER (Joshi et al., 2018). Moreover, the distribution and function of these two organelles depend on their contacts with ER (Ohsaki et al., 2017; Farré et al., 2018). ER-peroxisome contacts can be mediated by VAP proteins and the endosomal-associated protein acyl-coenzyme A–binding domain protein 5 (ACBD5) (Costello et al., 2017; Hua et al., 2017). In neurons, overexpression of ACBD5 produces diminished peroxisomal long-range movements, along with accumulation of peroxisomes in dendrites and axons (Wang Y. et al., 2018).
During LD biogenesis, membrane continuity is observed between LD and ER, forming membrane bridges. In addition, LDs also associate with the ER, at sites where protein complexes tether the membranes of both organelles. Molecules identified at these tethers include a complex formed by diacylglycerol O-acyltransferase 2 (DGAT2) (in LD membrane) and the fatty acid transport protein FATP1 (in ER membrane) (Xu et al., 2012); one formed by LD-associated Rab18 with the ER-associated proteins NAG-RINT1-ZW10 (NRZ complex) and Syntaxin18, Use1 and BNIP1 (SNARE proteins) (Xu et al., 2018); and tethering mediated by Vps13A and Vps13C (Kumar N. et al., 2018). At least one of them, Vps13A, interacts with the ER-membrane protein VAP-A (Yeshaw et al., 2019). These tethers, which are involved in LD biogenesis, have not been reported in neurons yet. Nevertheless, ER membrane proteins REEP1 (Falk et al., 2014) and spastin (Papadopoulos et al., 2015) are associated with LDs in neurons, controlling LD distribution and size. A similar role has been reported in non-neuronal cells for additional ER membrane proteins, such as ATL (Klemm et al., 2013), seipin (Wang H. et al., 2016; Salo et al., 2019), LD-associated hydrolase (LDAH) (Goo et al., 2017) or Syntaxin14 (Datta et al., 2019). Interestingly, ATL (Zhu et al., 2003), seipin (Ito et al., 2008) and LD-associated hydrolase (LDAH) Drosophila ortholog (Thiel et al., 2013; Yalçın et al., 2017), localize at neuronal ER tubules. Therefore, they may perform a similar role in LD biogenesis in neurons.
In cell bodies and in non-neuronal cells, ER exchanges material with the Golgi apparatus via both vesicle trafficking, and non-vesicular transport mediated by MCSs (Sassano and Agostinis, 2019). These contacts between ER and Golgi also depend on VAP and lipid-binding proteins, allowing exchange of lipids such as ceramides or phospholipids. Although canonical Golgi structures have not been observed outside the cell body, Golgi-derived structures, called Golgi outposts, are found in dendrites (Gardiol et al., 1999; Pierce et al., 2001; Horton and Ehlers, 2003). Golgi outposts have been also reported in axons during synapse development (Sytnyk, 2003) and in mature axons (González et al., 2016), mediating vesicular trafficking with ER, and revealing a machinery for the local processing of membrane proteins. Whether axonal ER and Golgi outposts also possess MCS remains unknown.
Axonal ER Dynamics
During cell development, ER material is organized and distributed through the cell (Bobinnec et al., 2003). For example, in axon growth cones, dynamic ER tubules drive ER network extension into the cell periphery (Dailey and Louis, 1989; Farías et al., 2019). During these stages, ER tubule distribution is not “sculpted” to remain unalterable, but remains highly dynamic even in differentiated cells and neurons. New tubules grow from old ones, old tubules retract, new junctions appear, and cisternae relocate. Remarkably, this dynamic nature is evolutionary conserved, being observed in plants (Stefano and Brandizzi, 2017), yeast (Du et al., 2004) and animal cells (Lee and Chen, 1988; Nixon-Abell et al., 2016; Schroeder et al., 2018), indicating that the capacity of tubular ER to change its distribution plays a critical role in cell responses to physiological changes [i.e., ER stress (Schuck et al., 2009) or Ca2+ levels (Ribeiro et al., 2000; Brough et al., 2005), or to control processes such as cell motility (Hernandez et al., 2006) or cell division (Bobinnec et al., 2003; Lu et al., 2009)]. The continuity of the ER network within the confined dimensions of axons obscures some features of ER dynamics, but anterograde and retrograde transport of ER tubules can be seen superimposed on the apparent stability of the continuous network (Yalçın et al., 2017). Since the axonal ER network is dynamic, since the ER assembly mechanisms appear to be quite efficient at maintaining a continuous network and avoiding gaps even when ER tubule numbers are significantly reduced, and since there appear to be no physical constraints on an even denser network of ER tubules (Yalçın et al., 2017), we suggest the existence of homeostatic mechanisms to achieve the physiological density and continuity of ER tubules. These mechanisms would ensure the presence of enough ER tubules to maintain continuity by fusing and joining up gaps in the network, while avoiding the presence of excess ER tubules; such mechanisms would depend on a pool of free mobile tubules through which excess tubules can be transported away, or required tubules delivered.
MT-Mediated Tubular ER Dynamics
Although some components of the ER-shaping machinery are sufficient to generate tubules in vitro (i.e., RTNs), MTs are additionally required in vivo (Terasaki et al., 1986; Dailey and Louis, 1989; Lu et al., 2009; Farías et al., 2019). In animal cells, peripheral ER tends to co-align with MTs (Terasaki et al., 1986; Kimura et al., 2017; Farías et al., 2019), which drive the movement of ER tubules. Two different mechanisms have been described for how ER tubules move alongside MTs (Figure 3). One of these involves movement of individual ER tubules on extending MTs, mediated by the tip attachment complex (TAC). In TACs, the tip of an ER tubule is attached to the tip of a MT plus end, through a complex containing the ER protein STIM1 and the MT-associated protein EB1. This binding leads to the ER tubule movement when the MT grows or retracts (Grigoriev et al., 2008; Pavez et al., 2019). The other mechanism mediating ER tubule movement along MTs, faster and more frequent than TAC, is ER sliding, driven by MT motor proteins such as kinesin-1 and dynein (Woźniak et al., 2009). In ER sliding, the tip of an ER tubule binds to stable (acetylated) MTs, and ER movements result from ER transport along the MT. Since this movement is MT-motor dependent, it does not necessarily correlate with MT growth or shrinkage (Lee and Chen, 1988; Waterman-Storer and Salmon, 1998; Woźniak et al., 2009). TAC-mediated movement and ER sliding are independent of each other, for example, ER sliding is still observed when STIM1 or EB1 are mutated (Grigoriev et al., 2008), or in the presence of nocodazole, which depolymerizes non-acetylated MTs (Terasaki et al., 1986; Waterman-Storer and Salmon, 1998).
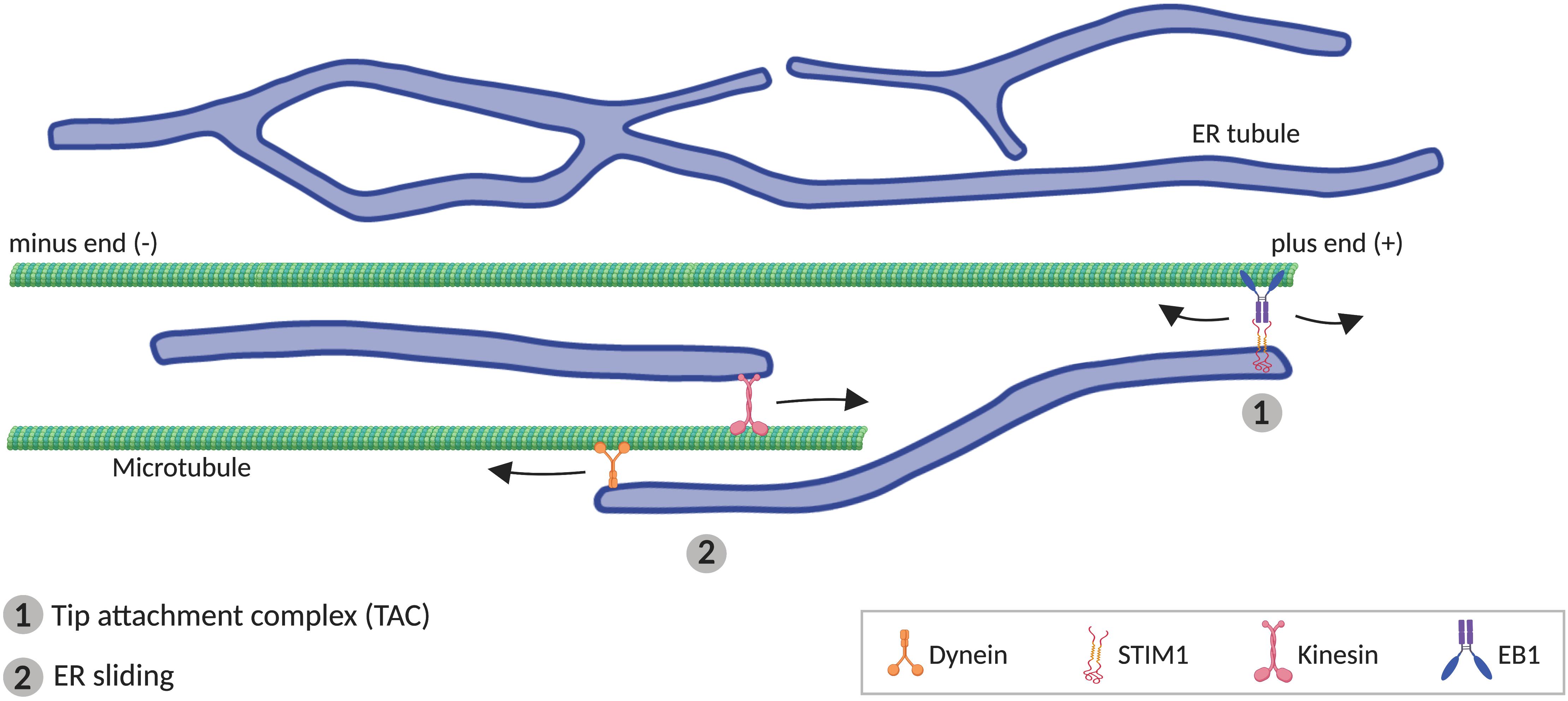
Figure 3. Microtubule (MT)-mediated transport of ER tubules. (1) Tip attachment complex (TAC) is formed between a MT plus end and the tip of an ER tubule through STIM1-EB1 interaction, resulting in movement of the ER tubule when the MT grows or retracts. (2) In ER sliding mechanism, the tip of an ER tubule associates to MTs, where motor proteins kinesin and dynein mediates, respectively, anterograde and retrograde movement of the ER tubule.
STIM2 and EB3, paralogs of STIM1 and EB1 respectively, are preferentially expressed in the central nervous system (Nakagawa et al., 2000; Skibinska-Kijek et al., 2009), and they also physically bind each other (Grigoriev et al., 2008), what suggests that STIM2-EB3 complex may mediates TAC in neurons. However, although knockdown of both EB1 and EB3 reduces dendritic ER expansion, it does not affect axonal ER distribution (Farías et al., 2019), suggesting that TAC mechanism may not be essential for axonal ER transport. Conversely, knockdown of kinesin-1 or dynein disrupts anterograde or retrograde transport, respectively, of ER tubule along the axon (Farías et al., 2019), supporting a critical role for ER sliding in axonal ER transport. In spite of this, it is still not clear which ER molecules serve as adaptors. One component of the complex that binds ER and MTs may be Protein 600 (p600), which appears associated with MTs and ER tubules in neurons (Shim et al., 2008). In addition, several integral ER membrane proteins are able to bind MTs. One of them is kinectin, proposed as an ER adaptor for kinesin-1 (Kumar et al., 2006), mediating ER extension (Zhang et al., 2010a). However, kinectin associates with ER sheets, and is excluded from axons (Farías et al., 2019). P180 protein, which contains a kinectin-homologous region and two MT-binding domains, also can bind kinesin-1 (Diefenbach et al., 2004), and interestingly, its distribution is not only restricted to the ER cisternae in the soma. P180 is also found at axonal ER tubules, from where it stabilizes MTs (Farías et al., 2019). Similarly, Sec61β, a subunit of the Sec61 translocon complex, is present in both soma and axon (Farías et al., 2019), and interacts directly with MTs (Zhu et al., 2018). Also, REEP1 and spastin can bind MTs, promoting respectively ER alignment along MTs (Park et al., 2010) and MT severing (Roll-Mecak and Vale, 2005). Nevertheless, the potential role for these ER proteins in ER transport remains to be explored.
Axonal ER is mostly composed of interconnected ER tubules, with occasional small sheets or cisternae (Wu et al., 2017; Yalçın et al., 2017). Antagonistically to tubular ER-shaping proteins, ER membrane protein Climp63 promotes formation of ER sheets, stabilizing them through homodimeric luminal bridges (Shibata et al., 2010). Interestingly, Climp63 mediates static interactions with MTs, stabilizing the ER network (Verkhovsky et al., 2005).
ER-Shaping Proteins and Tubular ER Motility
Proteins that shape or regulate tubular ER also influence ER tubule motility, although the mechanisms for this are not always clear. For example, Rab10 depletion decreases both extension frequency and fusion efficiency of ER tubules (English and Voeltz, 2013). Rab10 is enriched at the leading edge of nearly half of all dynamic ER tubules, and these dynamic domains track along MTs (English and Voeltz, 2013), suggesting a direct role for Rab10 in regulating tubular ER movements along MTs. VAPA/B knockdown impairs tubular ER movements in axons (Lindhout et al., 2019), potentially by affecting ER MCSs, which may destabilize the ER network. Also, RTN overexpression reduces retraction speed and increases fission frequency of ER tubules (Espadas et al., 2019); since RTN is distributed along the tubular ER network, its role in ER dynamics might simply result from its effect on the physical properties of the ER tubules.
ER MCS Dynamics
ER movements also result from transport of ER-associated organelles along MTs. As they traffic, endosomes and mitochondria can keep their MCS with ER, which results in changes in the position and morphology of the tethered ER tubules (Wu et al., 2018). This may be a way to ensure that during traffic events, ER tubules still can regulate these organelles, including their fission, lipid composition, or Ca2+ levels. The number and the size of ER MCS are highly dynamic, and dependent on physiological conditions. For example, Ca2+ levels can regulate ER-PM lipid exchange through the control of E-Syt tethering and function (Yu et al., 2016; Bian et al., 2017), and also STIM1 activation, promoting STIM1-Orai1 tethering to mediate Ca2+ exchange between ER and PM (Carrasco and Meyer, 2011). Key cellular functions have also been associated with ER-mitochondria MCS dynamics. For example, mitochondrial ATP production depends on mitochondrial Ca2+ uptake at ER-mitochondria MCSs. The importance of balanced ER-mitochondrial MCSs is illustrated by the fact that excessive mitochondrial Ca2+ uptake from ER can turn detrimental for the cell, triggering excessive opening of the mPTP and causing apoptosis (Paillard et al., 2013).
ER Turnover
To maintain ER homeostasis, the ER network requires a regulated system to remove excessive ER expansion. In addition, removing ER could facilitate network remodeling. Most of our knowledge about ER autophagy (ER-phagy) is related with the unfolded protein response (UPR), a degradative system that allows the removal of excessive accumulated misfolded and unfolded proteins in the ER lumen (Lindholm et al., 2017), and therefore, mostly associated with rough ER. An additional role for ER-phagy is turnover and clearance of ER (Grumati et al., 2018). This process is regulated by integral ER proteins, which act as ER-phagy receptors, targeting ER fragments to autophagosomes for lysosomal degradation. To date, six different ER-phagy receptors (see below) have been identified. Each of them specifically binds to ATG8 family members, such as LC3 and GABARAP, which are found in the membrane of incipient autophagosomes (Birgisdottir et al., 2013). Consistent with the hypothesis that ER-phagy can be activated locally to regulate ER excess, different ER-phagy receptors are found at different ER network domains. For rough ER, an ER-phagy receptor role has been reported for Sec62, a member of the translocon complex (Fumagalli et al., 2016), and for cell-cycle progression gene 1 (CCPG1), which is activated by the UPR (Smith et al., 2018). Similarly, ER-phagy receptor FAM134B has been linked to turnover of perinuclear ER cisterna (Khaminets et al., 2015; Bhaskara et al., 2019). Remarkably, the molecules identified as tubular ER-phagy receptors are tubular ER-shaping proteins. A long RTN3 isoform, which contains several LC3-interacting regions in its large amino-terminal domain, can promote degradation of ER tubules (Grumati et al., 2017). A similar role for ATL3 has also been observed (Chen et al., 2019). Recently, the predicted single-pass transmembrane ER protein, TEX264, was identified as an ER-phagy receptor (An et al., 2019; Chino et al., 2019), specifically mediating the degradation of ER tubule three-way junctions (An et al., 2019). All these ER-phagy receptors have been studied in non-neuronal mammalian cells, and their roles in neurons remain to be explored. More comprehensive details about ER-phagy process can be found in recent specific reviews (Grumati et al., 2018; Wilkinson, 2019).
ER and Axonal Neurodegeneration
The physiological roles of ER discussed above make it an obvious potential site of degenerative pathology, for example from impaired lipid, Ca2+, or organelle homeostasis. However, its physical properties, in particular its continuity, offer additional vulnerabilities for pathology. A summary of the evidence relating tubular ER and neurological disorders is presented in Table 2. In this section, we will cover the relationship between axonal ER and some neurodegenerative diseases, by looking at causative mutant proteins and their potential roles in disease mechanisms (summarized in Figure 4).
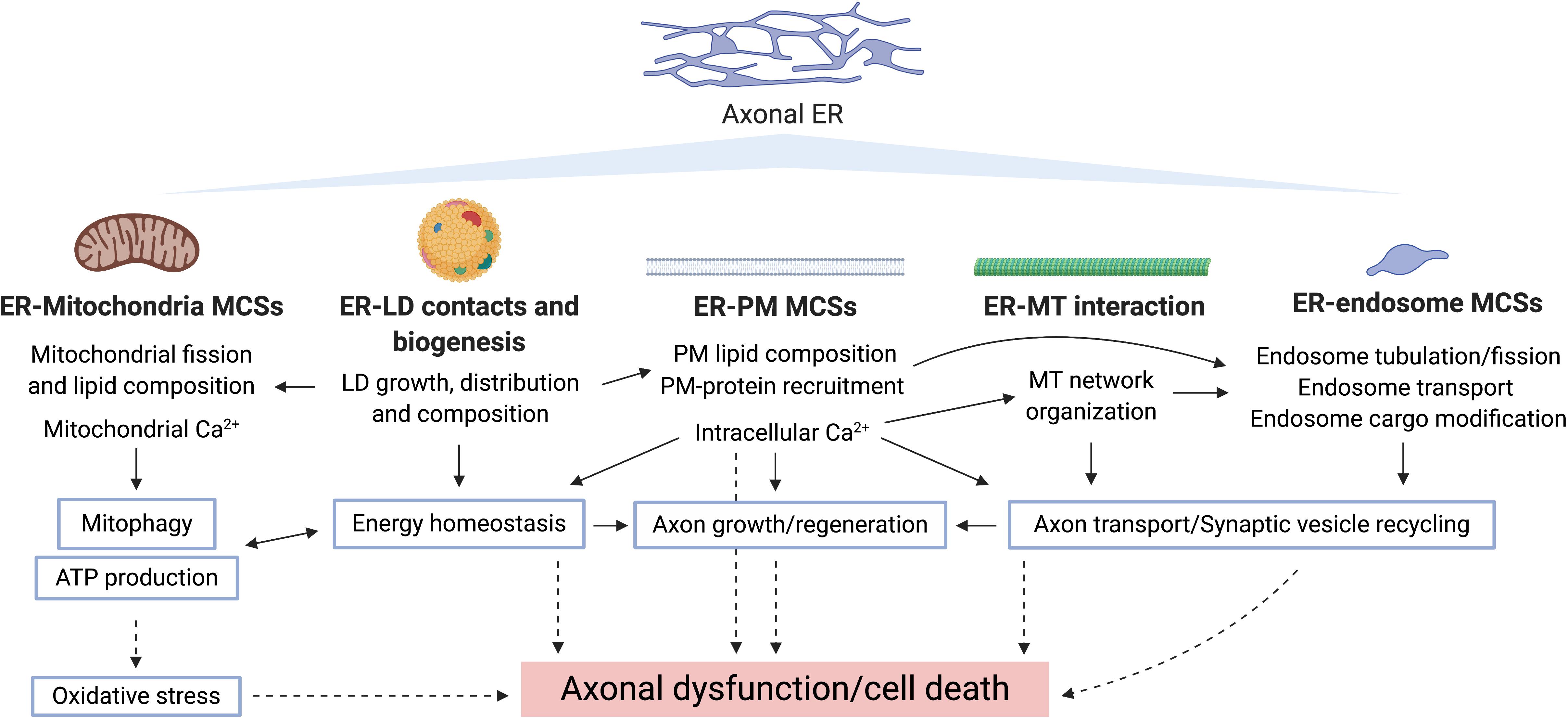
Figure 4. Potential routes underlying axonal ER involvement in neurodegeneration. Bold type shows pathways by which axonal ER may influence other cellular compartments/organelles, where the specific processes regulated in each case are indicated. Blue-border text boxes show processes indirectly regulated by ER. Arrows show regulation of the indicated process. Broken lines show consequences of disrupting the corresponding process.
Hereditary Spastic Paraplegias (HSPs)
The HSPs are a group of inherited, progressive, heterogeneous diseases, predominantly characterized by degeneration of longer upper motor neurons (those with somata in the brain, and axons projecting down the spinal cord). In “pure” HSPs, the main symptoms are lower limb weakness and spasticity. Additional symptoms are observed in “complicated” HSPs, including cerebellar ataxia, peripheral neuropathy, or optic atrophy, among other symptoms. Over 80 distinct spastic gait genetic loci (SPG1-80 and others) are so far known to be causative for HSP. These genes implicate cellular pathways such as membrane shaping and trafficking, mitochondrial function, lipid and protein metabolism, and axon development (Blackstone, 2018; Boutry et al., 2019). Remarkably, around half of HSP patients carry mutations affecting the ER-shaping proteins spastin, ATL1, RTN2 or REEP1 (Boutry et al., 2019), suggesting a relationship between ER modeling and axon maintenance. Other SPG proteins, e.g., REEP2 (Esteves et al., 2014), ARL6IP1, Rab3GAP2 (Novarino et al., 2014), or seipin (Windpassinger et al., 2004) affect ER morphogenesis, and others are also localized on ER (Liberski and Blackstone, 2017), supporting the idea that ER maintenance is essential for axonal maintenance. Mutations in Vps13D, required for mitochondrial size and clearance (Anding et al., 2018), are also causative for recessive spastic paraplegia or ataxia (Gauthier et al., 2018; Seong et al., 2018), thus suggesting that lipid transfer to or from the ER might be vulnerable in HSP, although the exact subcellular compartments where Vps13D acts are still unknown. There is experimental evidence for roles of several ER-localized SPG proteins in neurite or axon outgrowth (Chen et al., 2000; GrandPré et al., 2000; Gil et al., 2012). However, at least in the pure HSPs, axon growth and development must be largely unaffected, and the mechanisms that lead to degeneration are not necessarily the same ones that impair neurite outgrowth. In addition to their roles in tubular ER morphogenesis, some of the ER-shaping proteins related to HSP also have roles in other cellular processes, such as lipid metabolism, Ca2+ signaling or regulation of MTs.
Both ATL1 and seipin regulate LD growth (Szymanski et al., 2007; Klemm et al., 2013; Wang H. et al., 2016; Salo et al., 2019). In spite of the little information about LD in axons, they are implicated in membrane biogenesis and cellular signaling. Therefore, their role in axons may be relevant in pathogenesis (Pennetta and Welte, 2018).
REEP1 coprecipitates and co-aligns with polymerized MTs, indicating that it might help to modulate coupling of the tubular ER network with MT dynamics (Park et al., 2010). In cortical motor neuron cell bodies, REEP1 knockout shows reduced complexity of the peripheral ER network, producing a reduced number of ER structures with increased length (Beetz et al., 2013). REEP1 is also found in axonal growth cones (Park et al., 2010), and its knockdown results in neurite outgrowth defects and degeneration (Lim et al., 2015). Moreover, REEP1 is detected at ER-mitochondria MCSs, suggesting that mitochondrial disruption might be due to disrupted tubular ER organization, which affects lipid and Ca2+ exchange between ER and mitochondria through contacts (Lim et al., 2015).
Spastin is a MT-severing protein that also has roles in ER-endosome interactions and LD maturation (Papadopoulos et al., 2015; Allison et al., 2017). In zebrafish, knockdown of spastin causes reduced outgrowth in motor neuron axons and connectivity problems, as well as apoptosis in other neuron types (Wood et al., 2006). In an iPSC (induced pluripotent stem cell) model of spastin-linked HSP, axons displayed swellings with accumulation of mitochondria and tau, suggesting axonal transport problems (Denton et al., 2014). Spastin and ATL1 might function in axonal regeneration by mediating amounts of ER and MTs in growing axon tips (Levitan et al., 2016). All these data suggest that ER dynamics is critical in neurodegeneration and affects not only ER functions but also mitochondria, endosomes, and LDs, perhaps through ER MCSs with them.
ARL6IP1 knockdown results in abnormal branching of spinal motor neuron axons and a curly tail phenotype in zebrafish, suggesting a role for ARL6IP1 in neuronal development (Novarino et al., 2014). In Drosophila, knockdown of ARL6IP1, which causes tubular ER loss and elongated mitochondria in presynaptic terminals, also leads to a progressive locomotor deficit (Fowler and O’Sullivan, 2016).
Warburg Micro Syndrome (WRBM)
WRBM is an autosomal recessive, developmental brain disorder with symptoms including intellectual disability, microcephaly, cataracts in early childhood, and lower limb spasticity (Warburg et al., 1993). Mutations in Rab18, Rab3GAP1, Rab3GAP2, and TBC1D20 cause WRBM (Bem et al., 2011; Borck et al., 2011; Handley et al., 2013; Liegel et al., 2013). RAB3GAP1 and RAB3GAP2 form a heterodimer that (despite the name) is a guanine nucleotide exchange factor that activates Rab18 (Gerondopoulos et al., 2014), and TBC1D20 has been reported as a GTP-activating protein that inactivates Rab18, suggesting a common disease mechanism involving Rab18 signaling (Handley et al., 2015). These proteins have roles in ER organization, ER-LD tethering, secretion, and autophagy (including in neurons) (Nevo-Yassaf et al., 2012; Liegel et al., 2013; Gerondopoulos et al., 2014; Sidjanin et al., 2016; Xu et al., 2018; Dejgaard and Presley, 2019; Nian et al., 2019). Recruitment of Rab18 to ER by Rab3GAP is essential for a normal ER tubular network, and this is disrupted by disease-causing mutations (Gerondopoulos et al., 2014), making ER a potential site for WRBM pathology. WRBM presents as a complicated spastic paraplegia that progressively spreads to the upper body. Therefore, the paraplegia symptoms could potentially be caused by ER defects as in HSP, with additional symptoms due to other roles of Rab18, Rab3GAP and TBC1D20.
Alzheimer’s Disease (AD)
Alzheimer’s disease is the most common neurodegenerative disease, causing a decline in memory, difficulties in speaking, writing, understanding, identifying objects, and disorientation. Its main molecular hallmarks are extracellular beta-amyloid plaques, intracellular tau tangles, dystrophic neurites (DNs) and dysfunctional mitochondria (Scheltens et al., 2016; Kumar K. et al., 2018). Mutations in amyloid precursor protein (APP), and in the proteases that generate beta-amyloid from APP, Presenilin-1 (PS1, PSEN1) and Presenilin-2 (PS2, PSEN2) can cause early-onset AD, and apolipoprotein E/E4 (APOE4), a protein implicated in lipid metabolism and inflammation, is a strong genetic risk factor for late-onset AD (Van Cauwenberghe et al., 2016; Kumar K. et al., 2018).
Dysfunctional tubular ER in DNs is a feature of AD pathology (Sharoar et al., 2016). Mutations in Rab10 are associated with AD (Ridge et al., 2018). Rab10 binds to both MTs and tubular ER (Shim et al., 2008), potentially mediating interaction between them; therefore, proper axonal ER dynamics and transport may be processes that are vulnerable in AD. The tubular ER-shaping proteins RTN3 and RTN4B interact with β-secretase BACE1 (β-amyloid converting enzyme 1) (He et al., 2004), and negatively modulate BACE1 cleavage of APP during release of amyloid β peptides (Shi et al., 2009; Araki et al., 2013). RTN3 but not RTN1 is preferentially localized in DNs (Shi et al., 2017), and transgenic mice that overexpress RTN3 accumulate DNs and show impaired learning and memory and synaptic plasticity (Hu et al., 2007), suggesting that DNs with RTN3 aggregates (RTN3 immunoreactive DNs, or RIDNs) can cause cognitive dysfunction in AD. Additional ER shaping proteins like REEP2 and REEP5 also localize to RIDNs, but not other structural or ER stress-related proteins, suggesting that RIDN formation is due to accumulation of tubular ER in axons (Sharoar et al., 2016). In addition, aging may facilitate RIDN formation; in AD mouse brains, RIDN formation with abnormal ER tubules in axons increases over time (Sharoar et al., 2016).
Alterations in tubular ER network may also affect ER-mitochondria contacts in AD pathology; PS1 and PS2 are highly enriched in ER-mitochondria MCSs (Area-Gomez et al., 2009). In fact, amyloid β production is particularly high at ER-mitochondria MCSs, causing increased ER-mitochondria tethering and related functions such as lipid and Ca2+ exchange (Hedskog et al., 2013; Schreiner et al., 2014). Altered ER Ca2+ homeostasis is becoming a central player in AD pathology. Beta-amyloid plaques impair neuronal Ca2+ homeostasis (Kuchibhotla et al., 2008). Aβ oligomers are capable of forming Ca2+ permeable channels in the PM, and this alters Ca2+ homeostasis by elevating Ca2+ in the cytosol, resulting in cytotoxicity (Demuro and Parker, 2013). Also, wild type PS1/2, independent of secretase activity, can form Ca2+-permeable ion channels in ER membrane to leak Ca2+ to the cytosol, whereas in PS1/2 mutants the ER is overloaded with Ca2+, similar to aging neurons (Tu et al., 2006). In this context, several ER proteins involved in Ca2+ signaling (IP3R, RyR, SERCA, SigR1, STIM1, and STIM2) appear related with AD pathogenesis (Table 2), supporting a critical role for ER Ca2+ in AD. Also, both AD patients and mouse AD models show decreased MFN2 protein levels, correlating with disruption of mitochondria dynamics (Wang et al., 2009; Manczak et al., 2011), and increasing ER-mitochondria MCSs ameliorates AD defects in a Drosophila model (Garrido-Maraver et al., 2019).
Amyotrophic Lateral Sclerosis (ALS)
Amyotrophic lateral sclerosis is a fatal neurodegenerative disease with upper and lower motor neuron death, leading to progressive weakness and atrophy of muscles and to paralysis (Hardiman et al., 2017). Most ALS cases are sporadic, but some are familial (Beleza-Meireles and Al-Chalabi, 2009). Almost half of the familial cases are due to mutations affecting superoxide dismutase (SOD1), TAR DNA-binding protein 43 (TDP-43), fused in sarcoma (FUS) and dipeptide repeat proteins derived from the C9ORF72 gene. Another ALS gene encodes VAPB (Nishimura et al., 2004; Ling et al., 2013), which like VAPA, localizes to ER and mediates tethering between ER and other organelles (Murphy and Levine, 2016), and regulates tubular ER morphogenesis and dynamics (Lindhout et al., 2019). Both VAPA and VAPB expression is reduced in ALS patients, SOD1-ALS mice, and in HeLa cells. ALS-causative mutation of VAPB produces accumulation of VAPB in the cytosol and VAPB aggregates that contain ER tubules (still continuous with the ER and linked to mitochondria outer-membrane). Neurons expressing this VAPB mutant allele show increased cell death (Teuling et al., 2007). In addition to its association with ALS, the same VAPB mutation has been found in spinal muscular atrophy (SMA) patients (Nishimura et al., 2004). SMA is caused by degeneration of alpha motor neurons in the spinal cord, producing progressive muscle paralysis and atrophy (Tisdale and Pellizzoni, 2015).
ER MCSs appear to be important in ALS pathology. In motor neurons isolated from ALS patients and in the spinal cord of SOD1-ALS mice, a proapoptotic and mitophagy-associated protein BNIP1 (BCL2 interacting protein 1) was found as an ALS risk gene, and the expression level of BNIP1 was lowered compared to controls (Karch et al., 2018). BNIP1 plays a role in ER network organization by mediating ER membrane fusion (Nakajima et al., 2004). Also, mutations in SigR1, a chaperone that regulates ER Ca2+ channels (Hayashi and Su, 2007; Srivats et al., 2016), are associated with ALS (Al-Saif et al., 2011; Prause et al., 2013). Ataxin-2, an RNA-binding protein required for proper tubular ER morphogenesis and dynamics (del Castillo et al., 2019), is associated with ALS (Elden et al., 2010). Depletion of ataxin-2 causes disruption of peripheral ER morphology, more tubular instead of cisternal, and abnormal LD structure together with defective mitochondria morphology in C. elegans embryos (del Castillo et al., 2019).
Parkinson’s Disease (PD)
Parkinson’s disease is a common neurodegenerative disorder linked to aging, with symptoms like tremor, rigid muscles, bradykinesia and postural abnormalities. It can be caused by both genetic and environmental factors. A common feature in any form of PD is loss of dopaminergic neurons, which have long unmyelinated axons, with accumulation of intraneuronal Lewy body inclusions (Poewe et al., 2017). Several crucial cellular pathways, such as ER function, protein degradation, Ca2+ signaling and intracellular trafficking, have been linked to PD (Cherubini and Wade-Martins, 2017).
Managing Ca2+ homeostasis is one of the main roles of tubular ER. Mutation of PINK1, a PD causative gene, increases mitochondrial defects such as loss of membrane potential, increased size, and reduced ATP levels, all of which are rescued in PD cell models by inhibition of mitochondria calcium uniporters, which take up Ca2+ released from ER (Marongiu et al., 2009). Also, mutations in α-synuclein cause familial dominant forms of PD (Meade et al., 2019); mutant α-synuclein can disrupt ER-mitochondria tethering by binding to VAPB (De Vos et al., 2012; Paillusson et al., 2017). Therefore, ER-mitochondria contacts may potentially affect the severity of PD symptoms, and offer potential strategies for therapy. LRRK2 (leucine rich repeat kinase 2) is another PD causative gene. In astrocytes, mutant LRRK2 localizes to ER membrane where it suppresses activity of the Ca2+ ATPase SERCA, causing Ca2+ depletion in ER, and overload of Ca2+ in mitochondria via induced ER-mitochondria MCSs, eventually resulting in dysfunctional mitochondria (Lee J.H. et al., 2019). This mechanism could potentially also operate in neurons and axons. Mutations in Vps13C, encoding a lipid-transfer protein localized at both ER-endosome and ER-LD MCSs (Kumar N. et al., 2018), are associated with PD, and cause mitochondrial dysfunction and PINK1/Parkin-dependent mitophagy (Lesage et al., 2016; Schormair et al., 2018).
Loss of MFN2 results in progressive and retrograde degeneration of dopaminergic neurons in the nigrostriatal circuit in mice, increasing mitochondrial fragmentation and decreasing mitochondrial transport (Pham et al., 2012). MFN2 plays a role in mitophagy mediated by PINK1-Parkin, and knockout of MFN2 in dopaminergic neurons leads to impaired localization of Parkin and axonal loss (Tanaka et al., 2010; Lee et al., 2012; Chen and Dorn, 2013). These data also suggest potential roles of ER and ER tethering in PD mechanisms, as mitophagy occurs via ER-mitochondria physical contacts (Böckler and Westermann, 2014; Puri et al., 2019). Moreover, in Drosophila and iPSC PD models, increases in ER-Mitochondria MCSs cause abnormal lipid trafficking, depleting phosphatidylserine from ER. This ER lipid defect impairs sleep patterns (Valadas et al., 2018), one of the non-motor symptoms associated with PD (Munhoz et al., 2015).
MCTP2 gene, a human homolog of Pex30, with potential roles in tubular ER formation (Joshi et al., 2018), was found as a risk factor for early onset PD development (Latourelle et al., 2009), suggesting that disrupted ER organization in dopaminergic axons might facilitate PD development.
Huntington’s Disease (HD)
Huntington’s disease is an autosomal dominant, progressive, and neurodegenerative disease, with cognitive decline and defects in motor coordination. The cause of the disease is a CAG repeat expansion in the HTT1 gene, encoding huntingtin, a ubiquitously expressed cytoplasmic protein. Mostly spiny projection neurons (SPNs) degenerate in HD, but it is not clearly known why mutant Huntington (mHTT) protein selectively targets these neurons (Bates et al., 2015). The function of wild type Huntingtin (wHTT) protein is not fully understood, but its multiple protein-protein interaction sites indicate that it might have a scaffolding role (Ochaba et al., 2014).
Disrupted Ca2+ signaling is also observed in HD disease models (Mackay et al., 2018), suggesting that disordered Ca2+ handling makes SPNs more vulnerable to Ca2+-mediated cell death. Ca2+ disruption also causes aberrant synaptic plasticity (Hidalgo and Arias-Cavieres, 2016). Binding of mHTT to IP3R increases receptor responsiveness to IP3, resulting in enhanced Ca2+ release (Tang et al., 2003), which may ultimately lead to apoptosis. The enhanced Ca2+ leak also depletes ER Ca2+ stores, triggering SOC replenishment of ER Ca2+ (Mackay et al., 2018). This interaction between mHTT and IP3R might be a cause for neurodegeneration. Consistent with this, IP3R blockers prevent enhanced glutamate-mediated cell death in mouse striatal neurons (Tang et al., 2005), and in mouse neuronal cell culture. Moreover, IP3R knockdown prevents synapse loss, and inhibition of SOC channels rescues spine loss (Wu J. et al., 2016). Also, a repeat expansion in the gene encoding JPH3 associates to HD-like 2 (Holmes et al., 2001), which phenocopies HD (Schneider and Bird, 2016). JPH3 promotes ER-PM MCSs, controlling Ca2+ communication in hippocampal neurons (Moriguchi et al., 2006). Therefore, ER roles in Ca2+ handling appear to be important for HD pathology and could present potential therapeutic targets.
Mutations in Vps13A are causative for chorea-acanthocytosis (Ueno et al., 2001; Shen et al., 2017), a neurological syndrome with a HD-like phenotype (Jung et al., 2011). Since Vps13A is a lipid-transfer protein found at ER MCSs with mitochondria and LDs (Kumar N. et al., 2018), this suggests lipid transfer between ER and other organelles as a primary vulnerability in the disease. However, Vps13A also mediates ER MCS tethering (Kumar N. et al., 2018; Yeshaw et al., 2019), and therefore Vps13A dysfunction could indirectly affect Ca2+ communication.
Peripheral Neuropathies and ER
Peripheral neuropathies are a group of disorders characterized by injury or pathology in the peripheral nervous system (Callaghan et al., 2015). They are heterogenous, with a mix of genetic and acquired causes, clinical presentations, and pathologies. Patients can have motor insufficiency, sensory abnormalities, or both, depending on the disease. Mechanisms implicated in their pathologies include dysregulation of glucose pathway by hyperglycemia, dysfunction of mitochondria, reactive oxygen toxicity, impairments in inflammatory signaling pathways, axonal transport defects, and disrupted K+ and Na+ channels (Cashman and Höke, 2015). In addition, dysfunctional ER can also contribute. For example, ER stress is one cause of some peripheral neuropathies (Shin et al., 2010; Lupachyk et al., 2013; Inceoglu et al., 2015). Dysfunctional SOC or ER-mitochondria MCSs, which in turn may produce ER stress, can also cause peripheral neuropathies. We will discuss the importance of smooth/tubular ER-associated processes, highlighting the critical role of disrupted Ca2+ homeostasis.
Charcot-Marie-Tooth disorder (CMT) [also known as hereditary motor and sensory neuropathy (HMSN)] is one of the most common inherited neurological disorders, with distal weakness and muscle atrophy, presenting with demyelination (CMT type 1) and axonal loss (CMT type 2) (Shy and Patzkó, 2011). CMT type 1A (CMT1A) is a peripheral neuropathy caused by duplications or mutations in peripheral myelin protein 22 (PMP22), a tetra-span membrane protein (Juneja et al., 2019). These lead to demyelination, increased Schwann cell number and axonal loss, although the mechanisms that cause disease are not understood. Recently, it has been found that PMP22 controls Ca2+ homeostasis by interacting with the ER membrane protein STIM1, which results in increased Ca2+ influx through SOC channels (Vanoye et al., 2019). This supports an important role for ER Ca2+ in the pathogenesis of CMT1A, although any role disrupted by PMP mutations would likely be associated with Schwann cells and not with neuronal ER. The peripheral neuropathy CMT2A affects the peripheral and central nervous system, resulting in axon degeneration and progressive sensory loss in patients (Lee et al., 2017). CMT2A occurs as mostly autosomal dominant (Züchner et al., 2004), but sometimes recessive (Iapadre et al., 2018) or semi-dominant (Piscosquito et al., 2015) due to mutations in MFN2. As MFN2 localizes at both mitochondrial outer membrane and ER membrane, mediating tethering of both organelles (de Brito and Scorrano, 2008), ER-mitochondria MCSs might be important in the molecular mechanism of CMT2A. In fact, the connectivity between ER and mitochondria correlates with the severity of the disease, and the reinforcement of these contacts prevents some axonal defects caused by MFN2 mutation (Bernard-marissal et al., 2019; Larrea et al., 2019). Neurons and fibroblasts cultured from patients carrying MFN2 mutations show increased numbers of mitochondria in axons, which appear swollen and degenerating (Verhoeven et al., 2006; Amiott et al., 2008). Moreover, MFN2 mutant iPSC-derived motor neurons, obtained from CMT2A patients, show disrupted mitochondrial trafficking and cytoskeletal arrangement like patients have, although the mitochondrial morphology is unaffected (Saporta et al., 2015). Therefore, MFN2 mutations might cause CMT2A pathogenesis through disrupting ER-mitochondria tethering and its functions. This is supported by other recent findings that altered ER-mitochondria MCSs leads to CMT axonopathy (Bernard-marissal et al., 2019), and interestingly, increase of these contact sites promote axon regeneration (Lee S. et al., 2019). Together, this evidence suggests a critical role for ER Ca2+ in CMT, particularly in regulating mitochondrial function.
Other tubular ER-related proteins have also been linked to CMT pathologies. The junctophilin JPH1 mediates ER-PM tethering (Golini et al., 2011) and activates RyR channels to release Ca2+ from sarcoplasmic reticulum (Hirata et al., 2006). Remarkably, its paralogues JPH3 and JPH4 enable functional coupling of PM Ca2+ channels with RyRs in the ER of hippocampal neurons (Moriguchi et al., 2006; Sahu et al., 2019). Deficiency of GADP1 (ganglioside-induced differentiation-associated protein 1), an outer mitochondrial membrane protein associated with CMT, affects Ca2+ homeostasis by reducing SOC (Pla-Martín et al., 2013). Interestingly, it has been reported that overexpression of JPH1 rescues the SOC defects in GADP1-silenced cells, and that mutation of both GADP1 and JPH1 inhibits SOC activity. In fact, patients where these two genes are mutated show a more severe CMT than those where only GADP1 is mutated (Pla-Martín et al., 2015). Another example is the ER chaperone SigR1, which modulates the Ca2+ release channels IP3R and STIM1 (Hayashi and Su, 2007; Srivats et al., 2016). Mutation in SigR1 can result in CMT (Li et al., 2015). In addition, mutation of REEP1 can cause not only HSP, but also spinal CMT, affecting lower motor neurons (Beetz et al., 2012). Mutation of the ER protein seipin is also associated with CMT as well as HSP (Auer-Grumbach et al., 2005). This protein mediates ER-LD tethering (Salo et al., 2016, 2019), and gain-of-toxic-function mutation impairs synaptic neurotransmission (Wei et al., 2014).
Hereditary sensory neuropathies (HSNs) are neurological disorders characterized by degeneration of sensory neurons, with distal sensory loss in the lower limbs. HSNs are classified in eight phenotypically diverse types, which relate to specific genomic mutations and inheritance pattern (Schwartzlow and Kazamel, 2019). Mutations associated to HSNs affect functionally heterogeneous genes, making difficult to find a common pathway affected in patients (Rotthier et al., 2012). Several of the genes associated to the disease directly relate to the ER. Mutations in FAM134B can cause HSN type II, which shows autosomal recessive inheritance and manifests at early childhood. FAM134B is found at the cis-Golgi network, regulating the morphology of this structure (Kurth et al., 2009), but also at the membrane of perinuclear ER, where acts as a ER-phagy receptor, mediating ER sequestration into autophagosomes (Khaminets et al., 2015). The most common HSN is HSN type I, presenting a progressive autosomal dominant pattern of inheritance and causing a slowly progressive neuronal degeneration (Schwartzlow and Kazamel, 2019). HSN type I can be caused by mutations in SPTL1 or SPTL2 genes, which respectively encodes for subunits 1 and 2 of the enzyme serine palmitoyltransferase (SPT). This enzyme is found at the ER membrane, and is required for the synthesis of sphingolipids (Lowther et al., 2012), which present a key role in cell signal transduction (Hannun and Obeid, 2018). However, as for FAM134B, the current evidences do not support SPT distribution at axonal ER, and therefore, it remains unknown whether these proteins may have a role in axonal ER regulation. Other HSN type I-associated mutations affect the tubular ER-shaping proteins ATL1 (Guelly et al., 2011) and ATL3 (Fischer et al., 2014; Kornak et al., 2014). The ATL3 sensory neuropathy-causing mutation, ATL3Y192C, produces a less profuse ER network and prevents ATL localization at the axonal ER (Behrendt et al., 2019). Moreover, this mutation causes an increased number of ER-mitochondria MCSs, altering mitochondrial dynamics and distribution (Krols et al., 2019). This evidence suggests potential mechanisms by which defects in tubular ER may produce HSN.
Diabetic neuropathies are a type of neurodegenerative disease that mainly target sensory axons, autonomous axons and to some extent, motor axons (Feldman et al., 2019). Common symptoms are numbness and paresthesia, and feet are affected earlier than hands more severely together with autonomic or motor neuropathies (Charnogursky et al., 2014). ER-mitochondria MCSs play an essential role in diabetic neuropathies. They are regulated by glucose levels, acting as a nutrient sensor to switch between fat and glucose oxidation (Theurey and Rieusset, 2017). High glucose levels disrupt both integrity and function of ER-mitochondria MCSs in hepatic cells, inducing mitochondrial fission and impaired respiration (Theurey et al., 2016). This glucose-dependent regulation of ER-mitochondria MCSs is defective in the liver of insulin-resistant obese mice (Theurey et al., 2016), which show increased ER-mitochondria MCSs, causing mitochondrial dysfunction through mitochondrial Ca2+ overload (Arruda et al., 2014). Pancreatic beta-cells from patients with type 2 diabetes show reduced ER-mitochondria MCSs, and a similar effect is observed after fatty acid overload (Thivolet et al., 2017). Interestingly, ER-mitochondria MCSs are reduced in pro-opiomelanocortin neurons of diabetic mice, and reduction of ER-mitochondria MCSs in the same neurons, via ablation of MFN2, induces leptin resistance and obesity (Schneeberger et al., 2013). In addition, type 1 diabetic rats show reduced ER Ca2+ content in DRG neurons, possibly due to decreased levels of the ER Ca2+ ATPase SERCA (Verkhratsky and Fernyhough, 2008). Therefore, regulation of Ca2+ homeostasis through ER-Mitochondria MCSs is critical in diabetes pathogenesis, including the associated peripheral neuropathy. In rat cortical neurons rats, diabetes-induced neuritic dystrophy and cognitive dysfunction correlate with formation of aggregates of the tubular ER-shaping protein RTN3, similar to that observed in AD (see above). Aggregation of RTN3 prevents its inhibitory effect on BACE1, leading in turn to aggregation of amyloid β and diabetic dementia (Zhao et al., 2013). However, it remains to be explored whether similar defects are associated with diabetes in peripheral nerves.
Human immunodeficiency virus (HIV)-associated sensory neuropathy commonly develops during HIV infection, characterized by numbness and hypersensitivity of the feet and lower legs, and above the knees in more severe cases (Mangus et al., 2014). In primary DRG neurons, HIV coat protein gp120 causes axonal degeneration by inducing rapid large IP3-mediated Ca2+ release from ER. Decreasing ER Ca2+ storage promotes neuronal resistance to gp120-induced cell death (Höke et al., 2009), suggesting a crucial role for axonal ER.
Chemotherapy-induced peripheral neuropathy is a frequent side effect of chemotherapy (Zajaczkowska̧ et al., 2019). Dysregulation of Ca2+ homeostasis, where ER has a key role, contributes to the pain caused by chemotherapy-induced peripheral neuropathy (Siau and Bennett, 2006). Ca2+ signaling from ER to mitochondria may have an important role, since a selective SigR1 antagonist reduces hypersensitivity and the proportion of patients developing severe chemotherapy-induced peripheral neuropathy (Bruna and Velasco, 2018).
Discussion
Neurodegeneration is a complex and multifactorial process causing progressive neurological diseases. The study of genes that influence these conditions provides information about the biology of neurons and their pathologies, and is a catalyst for development of therapeutic strategies. An increasing body of evidence implicates dysfunctional ER in many neurological disorders that affect axons. In rough ER, ER stress is caused by accumulation of misfolded/unfolded proteins, which activates UPR to restore homeostasis, but if not resolved, causes neuronal degeneration and death (Hetz and Saxena, 2017). However, axonal ER, which is mostly tubular and smooth and undertakes very little protein export, is also linked to neurodegeneration. Tubular ER is directly involved in the regulation of many cellular processes, such as lipid biosynthesis and degradation, Ca2+ signaling, or organelles and MT dynamics. In axons, ER forms a mostly tubular network that is pervasive and continuous, and in human axons up to 1 m in length, physically linking the cell body with presynaptic terminals. Therefore, neuronal cells are particularly vulnerable to suffer defects when tubular ER morphology or tubular ER MCSs goes wrong. Mutations in genes encoding both tubular ER-shaping proteins and proteins related with ER MCSs, are observed in patients with different disorders (i.e., HSP, CMT, WRBM, PD, AD) (Table 2), where different types of neurons are preferentially affected. Interestingly, mutations affecting ER morphology may indirectly cause altered ER MCSs, potentially explaining why mutations in genes related to either ER morphology or ER MCSs produce similar neuronal defects. The pervasive and continuous nature of the ER suggests an important role for tubular ER continuity and/or local presence in axons. Evidence also suggests a critical role for ER Ca2+, particularly in regulating mitochondrial function. Defects in ER Ca2+ handling are associated with the pathogenic mechanisms of different neurological diseases (Table 2), and some of these ER Ca2+ defects relate to ER-mitochondria MCSs.
Tubular ER shows a highly dynamic nature, and the transport of ER tubules depends on the MT network. Therefore, it is not surprising that long cells, as neurons are, appear preferentially affected in patients with mutations in genes related with tubular ER dynamics. Therefore, proper axonal ER dynamics (including ER MCSs) and transport may be crucial to managing local changes/responses within the neuron.
Axonal ER has been largely neglected as an organelle since its early characterization around four decades ago. We are only starting to learn about its biological importance, and there is growing evidence for it as a potential site of pathology, and hence a potential therapeutic target, particularly for neurological disorders that affect distal longer axons. The preferential susceptibility of distal axons to disease-causing mutations in ER-shaping proteins points to functional importance of its specialized architecture. Exciting areas for study in the coming years include both the mechanisms that adapt the dynamic tubular ER network to the dimensions of axons – its physical continuity, narrow lumen, density of tubules in the cytosol – and the physiological roles of these architectural features, and how impairment of these functions can lead to degeneration. The genetics of the HSP diseases has opened windows both on possible disease mechanisms, and a fascinating area of basic biology. Understanding the basic biology of axonal ER will lead to new insights on the mechanisms of axon degeneration, and conversely understanding the disease mechanisms will lead to new insights in basic biology of axonal ER. Studies of the basic cell biology and the disease mechanisms will feed each other over the coming years.
Author Contributions
ZÖ wrote the first draft of the manuscript and designed the figures. CO’K helped draft and edit the manuscript. JP-M wrote the first draft, edited the manuscript, and designed and created the figures.
Funding
ZÖ is funded by the Republic of Turkey Ministry of National Education Scholarship (MEB1416) from Turkish Embassy. JP-M is supported by Marie Skłodowska-Curie fellowship 745007 from the European Union Horizon 2020 Research and Innovation Program. The work of CO’K is funded by grant BB/S001212/1 from the UK Biotechnology and Biological Sciences Research Council, and grant MR/S011226/1 from the UK Medical Research Council.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
All figures were created with BioRender.
References
Akizu, N., Cantagrel, V., Zaki, M. S., Al-Gazali, L., Wang, X., Rosti, R. O., et al. (2015). Biallelic mutations in SNX14 cause a syndromic form of cerebellar atrophy and lysosome-autophagosome dysfunction. Nat. Genet. 47, 528–534. doi: 10.1038/ng.3256
Alberts, B., Johnson, A., Lewis, J., Raff, M., Roberts, K., and Walter, P. (2002). “The endoplasmic reticulum,” in Molecular Biology of the Cell, ed. B. Alberts, (New York, NY: Garland Science).
Aligianis, I. A., Johnson, C. A., Gissen, P., Chen, D., Hampshire, D., Hoffmann, K., et al. (2005). Mutations of the catalytic subunit of RAB3GAP cause Warburg Micro syndrome. Nat. Genet. 37, 221–223. doi: 10.1038/ng1517
Aligianis, I. A., Morgan, N. V., Mione, M., Johnson, C. A., Rosser, E., Hennekam, R. C., et al. (2006). Mutation in Rab3 GTPase-activating protein (RAB3GAP) noncatalytic subunit in a kindred with martsolf syndrome. Am. J. Hum. Genet. 78, 702–707. doi: 10.1086/502681
Allison, R., Edgar, J. R., Pearson, G., Rizo, T., Newton, T., Günther, S., et al. (2017). Defects in ER-endosome contacts impact lysosome function in hereditary spastic paraplegia. J. Cell Biol. 216, 1337–1355. doi: 10.1083/jcb.201609033
Allison, R., Lumb, J. H., Fassier, C., Connell, J. W., Ten Martin, D., Seaman, M. N. J., et al. (2013). An ESCRT–spastin interaction promotes fission of recycling tubules from the endosome. J. Cell Biol. 202, 527–543. doi: 10.1083/jcb.201211045
Alpy, F., Rousseau, A., Schwab, Y., Legueux, F., Stoll, I., Wendling, C., et al. (2013). STARD3 or STARD3NL and VAP form a novel molecular tether between late endosomes and the ER. J. Cell Sci. 126, 5500–5512. doi: 10.1242/jcs.139295
Al-Saif, A., Al-Mohanna, F., and Bohlega, S. (2011). A mutation in sigma-1 receptor causes juvenile amyotrophic lateral sclerosis. Ann. Neurol. 70, 913–919. doi: 10.1002/ana.22534
Amiott, E. A., Lott, P., Soto, J., Kang, P. B., McCaffery, J. M., DiMauro, S., et al. (2008). Mitochondrial fusion and function in Charcot–Marie–Tooth type 2A patient fibroblasts with mitofusin 2 mutations. Exp. Neurol. 211, 115–127. doi: 10.1016/j.expneurol.2008.01.010
An, H., Ordureau, A., Paulo, J. A., Shoemaker, C. J., Denic, V., and Harper, J. W. (2019). TEX264 is an endoplasmic reticulum-resident ATG8-interacting protein critical for er remodeling during nutrient stress. Mol. Cell 74:891-908.e10. doi: 10.1016/j.molcel.2019.03.034
Anding, A. L., Wang, C., Chang, T. K., Sliter, D. A., Powers, C. M., Hofmann, K., et al. (2018). Vps13D encodes a ubiquitin-binding protein that is required for the regulation of mitochondrial size and clearance. Curr. Biol. 28, 287–295. doi: 10.1016/j.cub.2017.11.064
Araki, W., Oda, A., Motoki, K., Hattori, K., Itoh, M., Yuasa, S., et al. (2013). Reduction of β-amyloid accumulation by reticulon 3 in transgenic mice. Curr. Alzheimer Res. 10, 135–142. doi: 10.2174/1567205011310020003
Area-Gomez, E., de Groof, A. J. C., Boldogh, I., Bird, T. D., Gibson, G. E., Koehler, C. M., et al. (2009). Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am. J. Pathol. 175, 1810–1816. doi: 10.2353/ajpath.2009.090219
Arruda, A. P., Pers, B. M., Parlakgül, G., Güney, E., Inouye, K., and Hotamisligil, G. S. (2014). Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat. Med. 20, 1427–1435. doi: 10.1038/nm.3735
Auer-Grumbach, M., Schlotter-Weigel, B., Lochmüller, H., Strobl-Wildemann, G., Auer-Grumbach, P., Fischer, R., et al. (2005). Phenotypes of the N88S Berardinelli-Seip congenital lipodystrophy 2 mutation. Ann. Neurol. 57, 415–424. doi: 10.1002/ana.20410
Bartok, A., Weaver, D., Golenár, T., Nichtova, Z., Katona, M., Bánsághi, S., et al. (2019). IP3 receptor isoforms differently regulate ER-mitochondrial contacts and local calcium transfer. Nat. Commun. 10:3726. doi: 10.1038/s41467-019-11646-11643
Bates, G. P., Dorsey, R., Gusella, J. F., Hayden, M. R., Kay, C., Leavitt, B. R., et al. (2015). Huntington disease. Nat. Rev. Dis. Prim. 1, 1–21. doi: 10.1038/nrdp.2015.5
Baumgartner, H. K., Gerasimenko, J. V., Thorne, C., Ferdek, P., Pozzan, T., Tepikin, A. V., et al. (2009). Calcium elevation in mitochondria is the main Ca2+ requirement for mitochondrial permeability transition pore (mPTP) opening. J. Biol. Chem. 284, 20796–20803. doi: 10.1074/jbc.M109.025353
Beetz, C., Koch, N., Khundadze, M., Zimmer, G., Nietzsche, S., Hertel, N., et al. (2013). A spastic paraplegia mouse model reveals REEP1-dependent ER shaping. J. Clin. Invest. 123, 4273–4282. doi: 10.1172/JCI65665
Beetz, C., Pieber, T. R., Hertel, N., Schabhüttl, M., Fischer, C., Trajanoski, S., et al. (2012). Exome sequencing identifies a REEP1 mutation involved in distal hereditary motor neuropathy type v. Am. J. Hum. Genet. 91, 139–145. doi: 10.1016/j.ajhg.2012.05.007
Behrendt, L., Kurth, I., and Kaether, C. (2019). A disease causing ATLASTIN 3 mutation affects multiple endoplasmic reticulum-related pathways. Cell. Mol. Life Sci. 76, 1433–1445. doi: 10.1007/s00018-019-03010-x
Beleza-Meireles, A., and Al-Chalabi, A. (2009). Genetic studies of amyotrophic lateral sclerosis: controversies and perspectives. Amyotroph. Lateral Scler. 10, 1–14. doi: 10.1080/17482960802585469
Bem, D., Yoshimura, S.-I., Nunes-Bastos, R., Bond, F. F., Kurian, M. A., Rahman, F., et al. (2011). Loss-of-function mutations in RAB18 cause warburg micro syndrome. Am. J. Hum. Genet. 88, 499–507. doi: 10.1016/j.ajhg.2011.03.012
Berna-Erro, A., Braun, A., Kraft, R., Kleinschnitz, C., Schuhmann, M. K., Stegner, D., et al. (2009). STIM2 regulates capacitive Ca2+ entry in neurons and plays a key role in hypoxic neuronal cell death. Sci. Signal. 2, 1–11. doi: 10.1126/scisignal.2000522
Bernard-Marissal, N., Chrast, R., and Schneider, B. L. (2018). Endoplasmic reticulum and mitochondria in diseases of motor and sensory neurons: a broken relationship? Cell Death Dis. 9, 1–16. doi: 10.1038/s41419-017-0125-121
Bernard-marissal, N., Hameren, G., Van Juneja, M., Pellegrino, C., and Louhivuori, L. (2019). Altered interplay between endoplasmic reticulum and mitochondria in Charcot – Marie – Tooth type 2A neuropathy. Proc. Natl. Acad. Sci. U.S.A. 116, 2328–2337. doi: 10.1073/pnas.1810932116
Bernard-Marissal, N., Médard, J. J., Azzedine, H., and Chrast, R. (2015). Dysfunction in endoplasmic reticulum-mitochondria crosstalk underlies SIGMAR1 loss of function mediated motor neuron degeneration. Brain 138, 875–890. doi: 10.1093/brain/awv008
Berridge, M. J. (2002). The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium 32, 235–249. doi: 10.1016/S0143416002001823
Betancourt-Solis, M. A., Desai, T., and McNew, J. A. (2018). The atlastin membrane anchor forms an intramembrane hairpin that does not span the phospholipid bilayer. J. Biol. Chem. 293, 18514–18524. doi: 10.1074/jbc.RA118.003812
Bhaskara, R. M., Grumati, P., Garcia-Pardo, J., Kalayil, S., Covarrubias-Pinto, A., Chen, W., et al. (2019). Curvature induction and membrane remodeling by FAM134B reticulon homology domain assist selective ER-phagy. Nat. Commun. 10:2370. doi: 10.1038/s41467-019-10345-10343
Bian, X., Saheki, Y., and De Camilli, P. (2017). Ca2+ releases E-Syt1 autoinhibition to couple ER-plasma membrane tethering with lipid transport. EMBO J. 37, 219–234. doi: 10.15252/embj.201797359
Birgisdottir, A. B., Lamark, T., and Johansen, T. (2013). The LIR motif – crucial for selective autophagy. J. Cell Sci. 126, 3237–3247. doi: 10.1242/jcs.126128
Blackstone, C. (2018). Converging cellular themes for the hereditary spastic paraplegias. Curr. Opin. Neurobiol. 51, 139–146. doi: 10.1016/j.conb.2018.04.025
Bobinnec, Y., Marcaillou, C., Morin, X., and Debec, A. (2003). Dynamics of the endoplasmic reticulum during early development of Drosophila melanogaster. Cell Motil. Cytoskeleton 54, 217–225. doi: 10.1002/cm.10094
Böckler, S., and Westermann, B. (2014). Mitochondrial ER contacts are crucial for mitophagy in yeast. Dev. Cell 28, 450–458. doi: 10.1016/j.devcel.2014.01.012
Bojarski, L., Pomorski, P., Szybinska, A., Drab, M., Skibinska-Kijek, A., Gruszczynska-Biegala, J., et al. (2009). Presenilin-dependent expression of STIM proteins and dysregulation of capacitative Ca2+ entry in familial Alzheimer’s disease. Biochim. Biophys. Acta - Mol. Cell Res. 1793, 1050–1057. doi: 10.1016/j.bbamcr.2008.11.008
Borck, G., Wunram, H., Steiert, A., Volk, A. E., Körber, F., Roters, S., et al. (2011). A homozygous RAB3GAP2 mutation causes Warburg Micro syndrome. Hum. Genet. 129, 45–50. doi: 10.1007/s00439-010-0896-892
Boutry, M., Morais, S., and Stevanin, G. (2019). Update on the genetics of spastic paraplegias. Curr. Neurol. Neurosci. Rep. 19, 1–8.
Breuss, M. W., Nguyen, A., Song, Q., Nguyen, T., Stanley, V., James, K. N., et al. (2018). Mutations in LNPK, encoding the endoplasmic reticulum junction stabilizer lunapark, cause a recessive neurodevelopmental syndrome. Am. J. Hum. Genet. 103, 296–304. doi: 10.1016/j.ajhg.2018.06.011
Britti, E., Delaspre, F., Tamarit, J., and Ros, J. (2018). Mitochondrial calcium signalling and neurodegenerative diseases. Neuronal Signal. 2:NS20180061. doi: 10.1042/NS20180061
Britzolaki, A., Saurine, J., Flaherty, E., Thelen, C., and Pitychoutis, P. M. (2018). The SERCA2: a gatekeeper of neuronal calcium homeostasis in the brain. Cell. Mol. Neurobiol. 38, 981–994. doi: 10.1007/s10571-018-0583-588
Brough, D., Schell, M. J., and Irvine, R. F. (2005). Agonist-induced regulation of mitochondrial and endoplasmic reticulum motility. Biochem. J. 392, 291–297. doi: 10.1042/bj20050738
Bruna, J., and Velasco, R. (2018). Sigma-1 receptor: a new player in neuroprotection against chemotherapy-induced peripheral neuropathy. Neural Regen. Res. 13, 775–778. doi: 10.4103/1673-5374.232459
Cagalinec, M., Safiulina, D., Liiv, M., Liiv, J., Choubey, V., Wareski, P., et al. (2013). Principles of the mitochondrial fusion and fission cycle in neurons. J. Cell Sci. 126, 2187–2197. doi: 10.1242/jcs.118844
Callaghan, B. C., Price, R. S., Chen, K. S., and Feldman, E. L. (2015). The importance of rare subtypes in diagnosis and treatment of peripheral neuropathy. JAMA Neurol. 72, 1510. doi: 10.1001/jamaneurol.2015.2347
Cárdenas, C., Miller, R. A., Smith, I., Bui, T., Molgó, J., Müller, M., et al. (2010). Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 142, 270–283. doi: 10.1016/J.CELL.2010.06.007
Carpanini, S. M., McKie, L., Thomson, D., Wright, A. K., Gordon, S. L., Roche, S. L., et al. (2014). A novel mouse model of Warburg Micro syndrome reveals roles for RAB18 in eye development and organisation of the neuronal cytoskeleton. Dis. Model. Mech. 7, 711–722. doi: 10.1242/dmm.015222
Carrasco, S., and Meyer, T. (2011). STIM proteins and the endoplasmic reticulum-plasma membrane junctions. Annu. Rev. Biochem. 80, 973–1000. doi: 10.1146/annurev-biochem-061609-165311
Carreras-Sureda, A., Pihán, P., and Hetz, C. (2018). Calcium signaling at the endoplasmic reticulum: fine-tuning stress responses. Cell Calcium 70, 24–31. doi: 10.1016/j.ceca.2017.08.004
Cashman, C. R., and Höke, A. (2015). Mechanisms of distal axonal degeneration in peripheral neuropathies. Neurosci. Lett. 596, 33–50. doi: 10.1016/j.neulet.2015.01.048
Chang, J., Lee, S., and Blackstone, C. (2013). Protrudin binds atlastins and endoplasmic reticulum-shaping proteins and regulates network formation. Proc. Natl. Acad. Sci. U.S.A. 110, 14954–14959. doi: 10.1073/PNAS.1307391110
Charnogursky, G., Lee, H., and Lopez, N. (2014). “Diabetic neuropathy,” in Handbook of Clinical Neurology, eds G. Nappi and M. Moskowitz, (Amsterdam: Elsevier), 773–785. doi: 10.1016/B978-0-7020-4087-0.00051-56
Chen, M. S., Huber, A. B., Van Der Haar, M. E. D., Frank, M., Schnell, L., Spillmann, A. A., et al. (2000). Nogo-A is a myelin-associated neurite outgrowth inhibitor and an antigen for monoclonal antibody IN-1. Nature 403, 434–439. doi: 10.1038/35000219
Chen, Q., Xiao, Y., Chai, P., Zheng, P., Teng, J., and Chen, J. (2019). ATL3 is a tubular er-phagy receptor for GABARAP- mediated selective autophagy report ATL3 is a tubular ER-phagy receptor for GABARAP-mediated selective autophagy. Curr. Biol. 29:846-855.e6. doi: 10.1016/j.cub.2019.01.041
Chen, S., Desai, T., McNew, J. A., Gerard, P., Novick, P. J., and Ferro-Novick, S. (2015). Lunapark stabilizes nascent three-way junctions in the endoplasmic reticulum. Proc. Natl. Acad. Sci. 112, 418–423. doi: 10.1073/pnas.1423026112
Chen, S., Novick, P., and Ferro-Novick, S. (2012). ER network formation requires a balance of the dynamin-like GTPase Sey1p and the Lunapark family member Lnp1p. Nat. Cell Biol. 14, 707–716. doi: 10.1038/ncb2523
Chen, Y., and Dorn, G. W. (2013). PINK1-Phosphorylated mitofusin 2 is a parkin receptor for culling damaged mitochondria. Science 340, 471–475. doi: 10.1126/science.1231031
Cheng, C.-Y., Wu, J.-C., Tsai, J.-W., Nian, F.-S., Wu, P.-C., Kao, L.-S., et al. (2015). ENU mutagenesis identifies mice modeling Warburg Micro Syndrome with sensory axon degeneration caused by a deletion in Rab18. Exp. Neurol. 267, 143–151. doi: 10.1016/j.expneurol.2015.03.003
Cherubini, M., and Wade-Martins, R. (2017). Convergent pathways in Parkinson’s disease. Cell Tissue Res. 373, 79–90. doi: 10.1007/s00441-017-2700-2702
Cheung, K. H., Shineman, D., Müller, M., Cárdenas, C., Mei, L., Yang, J., et al. (2008). Mechanism of Ca2+ Disruption in Alzheimer’s disease by presenilin regulation of InsP3 receptor channel gating. Neuron 58, 871–883. doi: 10.1016/j.neuron.2008.04.015
Chino, H., Hatta, T., Natsume, T., and Mizushima, N. (2019). Intrinsically disordered protein TEX264 mediates ER-phagy. Mol. Cell 74:909-921.e6. doi: 10.1016/j.molcel.2019.03.033
Cho, Y., Sloutsky, R., Naegle, K. M., and Cavalli, V. (2013). Injury-Induced HDAC5 nuclear export is essential for axon regeneration. Cell 155:894. doi: 10.1016/j.cell.2013.10.004
Chou, J. Y., Jun, H. S., and Mansfield, B. C. (2015). Type I glycogen storage diseases: disorders of the glucose-6-phosphatase/glucose-6-phosphate transporter complexes. J. Inherit. Metab. Dis. 38, 511–519. doi: 10.1007/s10545-014-9772-x
Chung, J., Torta, F., Masai, K., Lucast, L., Czapla, H., Tanner, L. B., et al. (2015). PI4P/phosphatidylserine countertransport at ORP5- and ORP8-mediated ER - Plasma membrane contacts. Science 349, 428–432. doi: 10.1126/science.aab1370
Conroy, J., McGettigan, P., Murphy, R., Webb, D., Murphy, S. M., McCoy, B., et al. (2014). A novel locus for episodic ataxia: UBR4 the likely candidate. Eur. J. Hum. Genet. 22, 505–510. doi: 10.1038/ejhg.2013.173
Costello, J. L., Castro, I. G., Hacker, C., Schrader, T. A., Metz, J., Zeuschner, D., et al. (2017). ACBD5 and VAPB mediate membrane associations between peroxisomes and the ER. J. Cell Biol. 216, 331–342. doi: 10.1083/jcb.201607055
Courjaret, R., Dib, M., and Machaca, K. (2018). Spatially restricted subcellular Ca2+ signaling downstream of store-operated calcium entry encoded by a cortical tunneling mechanism. Sci. Rep. 8, 1–13. doi: 10.1038/s41598-018-29562-29569
Dailey, E., and Louis, S. (1989). Dynamics of the endoplasmic reticulum organelles in growth cones of cultured and other membranous neurons. J. Neurosci. 9, 1897–1909. doi: 10.1523/JNEUROSCI.09-06-01897.1989
Datta, S., Liu, Y., Hariri, H., Bowerman, J., and Henne, W. M. (2019). Cerebellar ataxia disease–associated Snx14 promotes lipid droplet growth at ER–droplet contacts. J. Cell Biol. 218, 1335–1351. doi: 10.1083/JCB.201808133
de Brito, O. M., and Scorrano, L. (2008). Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610. doi: 10.1038/nature07534
De Gregorio, C., Delgado, R., Ibacache, A., Sierralta, J., and Couve, A. (2017). Drosophila Atlastin in motor neurons is required for locomotion and presynaptic function. J. Cell Sci. 130, 3507–3516. doi: 10.1242/jcs.201657
de Juan-Sanz, J., Holt, G. T., Schreiter, E. R., de Juan, F., Kim, D. S., and Ryan, T. A. (2017). Axonal endoplasmic reticulum Ca2+ content controls release probability in CNS nerve terminals. Neuron 93, 867.e–881.e. doi: 10.1016/j.neuron.2017.01.010
De Vos, K. J., Mórotz, G. M., Stoica, R., Tudor, E. L., Lau, K.-F., Ackerley, S., et al. (2012). VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum. Mol. Genet. 21, 1299–1311. doi: 10.1093/hmg/ddr559
Dejgaard, S. Y., and Presley, J. F. (2019). Rab18: new insights into the function of an essential protein. Cell. Mol. Life Sci. 76, 1935–1945. doi: 10.1007/s00018-019-03050-3053
del Castillo, U., Gnazzo, M. M., Turpin, C. G. S., Nguyen, K. C. Q., Semaya, E., Lam, Y., et al. (2019). Conserved role for Ataxin-2 in mediating ER dynamics. Traffic 20, 1–12. doi: 10.1111/tra.12647
Del Prete, D., Checler, F., and Chami, M. (2014). Ryanodine receptors: physiological function and deregulation in Alzheimer disease. Mol. Neurodegener. 9, 1–15. doi: 10.1186/1750-1326-9-21
Demuro, A., and Parker, I. (2013). Cytotoxicity of intracellular A 42 Amyloid oligomers involves Ca2+ release from the endoplasmic reticulum by stimulated production of inositol trisphosphate. J. Neurosci. 33, 3824–3833. doi: 10.1523/JNEUROSCI.4367-12.2013
Deng, C. Y., Lei, W. L., Xu, X. H., Ju, X. C., Liu, Y., and Luo, Z. G. (2014). JIP1 mediates anterograde transport of Rab10 cargos during neuronal polarization. J. Neurosci. 34, 1710–1723. doi: 10.1523/JNEUROSCI.4496-13.2014
Deng, M., He, W., Tan, Y., Han, H., Hu, X., Xia, K., et al. (2013). Increased expression of reticulon 3 in neurons leads to reduced axonal transport of β site amyloid precursor protein-cleaving enzyme. J. Biol. Chem. 288, 30236–30245. doi: 10.1074/jbc.M113.480079
Denton, K. R., Lei, L., Grenier, J., Rodionov, V., Blackstone, C., and Li, X.-J. (2014). Loss of Spastin function results in disease-specific axonal defects in human pluripotent stem cell-based models of hereditary spastic paraplegia. Stem Cells 32, 414–423. doi: 10.1002/stem.1569
Desai-Shah, M., and Cooper, R. L. (2009). Different mechanisms of Ca2+ regulation that influence synaptic transmission: comparison between crayfish and drosophila neuromuscular junctions. Synapse 63, 1100–1121. doi: 10.1002/syn.20695
Diefenbach, R. J., Diefenbach, E., Douglas, M. W., and Cunningham, A. L. (2004). The ribosome receptor, p180, interacts with kinesin heavy chain, KIF5B. Biochem. Biophys. Res. Commun. 319, 987–992. doi: 10.1016/j.bbrc.2004.05.069
Dienel, G. A. (2019). The “protected” glucose transport through the astrocytic endoplasmic reticulum is too slow to serve as a quantitatively-important highway for nutrient delivery. J. Neurosci. Res. 97, 854–862. doi: 10.1002/jnr.24432
Djurovic, S., Le Hellard, S., Kähler, A. K., Jönsson, E. G., Agartz, I., Steen, V. M., et al. (2009). Association of MCTP2 gene variants with schizophrenia in three independent samples of Scandinavian origin (SCOPE). Psychiatry Res. 168, 256–258. doi: 10.1016/j.psychres.2008.08.007
Dong, R., Saheki, Y., Swarup, S., Lucast, L., Harper, J. W., and De Camilli, P. (2016). Endosome-ER contacts control actin nucleation and retromer function through VAP-dependent regulation of PI4P. Cell 166, 408–423. doi: 10.1016/j.cell.2016.06.037
Dong, R., Zhu, T., Benedetti, L., Gowrishankar, S., Deng, H., Cai, Y., et al. (2018). The inositol 5-phosphatase INPP5K participates in the fine control of ER organization. J. Cell Biol. 217, 3577–3592. doi: 10.1083/jcb.201802125
Du, X., Kumar, J., Ferguson, C., Schulz, T. A., Ong, Y. S., Hong, W., et al. (2011). A role for oxysterol-binding protein-related protein 5 in endosomal cholesterol trafficking. J. Cell Biol. 192, 121–135. doi: 10.1083/jcb.201004142
Du, Y., Ferro-Novick, S., and Novick, P. (2004). Dynamics and inheritance of the endoplasmic reticulum. J. Cell Sci. 117, 2871–2878. doi: 10.1242/jcs.01286
Dupuis, L., Gonzalez de Aguilar, J. L., Di Scala, F., Rene, F., De Tapia, M., Pradat, P. F., et al. (2002). Nogo provides a molecular marker for diagnosis of amyotrophic lateral sclerosis. Neurobiol. Dis. 10, 358–365. doi: 10.1006/nbdi.2002.0522
Eden, E. R. (2016). The formation and function of ER-endosome membrane contact sites. Biochim. Biophys. Acta - Mol. Cell Biol. Lipids 1861, 874–879. doi: 10.1016/j.bbalip.2016.01.020
Eden, E. R., White, I. J., Tsapara, A., and Futter, C. E. (2010). Membrane contacts between endosomes and ER provide sites for PTP1B-epidermal growth factor receptor interaction. Nat. Cell Biol. 12, 267–272. doi: 10.1038/ncb2026
Eisenberg-Bord, M., Shai, N., Schuldiner, M., and Bohnert, M. (2016). A tether is a tether is a tether: tethering at membrane contact sites. Dev. Cell 39, 395–409. doi: 10.1016/j.devcel.2016.10.022
Elbaz, Y., and Schuldiner, M. (2011). Staying in touch: the molecular era of organelle contact sites. Trends Biochem. Sci. 36, 616–623. doi: 10.1016/j.tibs.2011.08.004
Elden, A. C., Kim, H. J., Hart, M. P., Chen-Plotkin, A. S., Johnson, B. S., Fang, X., et al. (2010). Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466, 1069–1075. doi: 10.1038/nature09320
Elkin, S. R., Lakoduk, A. M., and Schmid, S. L. (2016). Endocytic pathways and endosomal trafficking: a primer. Wiener Medizinische Wochenschrift 166, 196–204. doi: 10.1007/s10354-016-0432-437
English, A. R., and Voeltz, G. K. (2013). Rab10 GTPase regulates ER dynamics and morphology. Nat. Cell Biol. 15, 169–178. doi: 10.1038/ncb2647
Espadas, J., Pendin, D., Bocanegra, R., Escalada, A., Misticoni, G., Trevisan, T., et al. (2019). Dynamic constriction and fission of endoplasmic reticulum membranes by reticulon. Nat. Commun. 10:5327. doi: 10.1038/s41467-019-13327-13327
Esteves, T., Durr, A., Mundwiller, E., Loureiro, J. L., Boutry, M., Gonzalez, M. A., et al. (2014). Loss of association of REEP2 with membranes leads to hereditary spastic paraplegia. Am. J. Hum. Genet. 94, 268–277. doi: 10.1016/j.ajhg.2013.12.005
Fagone, P., and Jackowski, S. (2009). Membrane phospholipid synthesis and endoplasmic reticulum function. J. Lipid Res. 50, S311–S316. doi: 10.1194/jlr.R800049-JLR200
Falk, J., Rohde, M., Bekhite, M. M., Neugebauer, S., Hemmerich, P., Kiehntopf, M., et al. (2014). Functional mutation analysis provides evidence for a role of REEP1 in lipid droplet biology. Hum. Mutat. 35, 497–504. doi: 10.1002/humu.22521
Farías, G. G., Fréal, A., Tortosa, E., Stucchi, R., Pan, X., Portegies, S., et al. (2019). Feedback-Driven mechanisms between microtubules and the endoplasmic reticulum instruct neuronal polarity. Neuron 102:184-201.e8. doi: 10.1016/j.neuron.2019.01.030
Farré, J., Mahalingam, S. S., Proietto, M., and Subramani, S. (2018). Peroxisome biogenesis, membrane contact sites, and quality control. EMBO Rep. 20:e46864. doi: 10.15252/embr.201846864
Feldman, E. L., Callaghan, B. C., Pop-Busui, R., Zochodne, D. W., Wright, D. E., Bennett, D. L., et al. (2019). Diabetic neuropathy. Nat. Rev. Dis. Prim. 5:41. doi: 10.1038/s41572-019-0092-91
Fink, K. L., López-Giráldez, F., Kim, I. J., Strittmatter, S. M., and Cafferty, W. B. J. (2017). Identification of intrinsic Axon growth modulators for intact CNS neurons after injury. Cell Rep. 18, 2687–2701. doi: 10.1016/j.celrep.2017.02.058
Fischer, D., Schabhüttl, M., Wieland, T., Windhager, R., Strom, T. M., and Auer-Grumbach, M. (2014). A novel missense mutation confirms ATL3 as a gene for hereditary sensory neuropathy type 1. Brain 137, 91–92. doi: 10.1093/brain/awu091
Fowler, P. C., and O’Sullivan, N. C. (2016). ER-shaping proteins are required for ER and mitochondrial network organisation in motor neurons. Hum. Mol. Genet. 25, 2827–2837. doi: 10.1093/hmg/ddw139
Fox, P. D., Haberkorn, C. J., Akin, E. J., Seel, P. J., Krapf, D., and Tamkun, M. M. (2015). Induction of stable ER-plasma-membrane junctions by Kv2.1 potassium channels. J. Cell Sci. 128, 2096–2105. doi: 10.1242/jcs.166009
Friedman, J. R., DiBenedetto, J. R., West, M., Rowland, A. A., and Voeltz, G. K. (2013). Endoplasmic reticulum–endosome contact increases as endosomes traffic and mature. Mol. Biol. Cell 24, 1030–1040. doi: 10.1091/mbc.e12-10-0733
Friedman, J. R., Lackner, L. L., West, M., DiBenedetto, J. R., Nunnari, J., and Voeltz, G. K. (2011). ER tubules mark sites of mitochondrial division. Science 334, 358–362. doi: 10.1126/science.1207385
Fumagalli, F., Noack, J., Bergmann, T. J., Presmanes, E. C., Pisoni, G. B., Fasana, E., et al. (2016). Translocon component Sec62 acts in endoplasmic reticulum turnover during stress recovery. Nat. Cell Biol. 18, 1173–1184. doi: 10.1038/ncb3423
Gardiol, A., Racca, C., and Triller, A. (1999). Dendritic and postsynaptic protein synthetic machinery. J. Neurosci. 19, 168–179. doi: 10.1523/jneurosci.19-01-00168.1999
Garrido-Maraver, J., Samantha, H. Y. L., and Martins, M. L. (2019). Forcing contacts between mitochondria and the endoplasmic reticulum extends lifespan in a Drosophila model of Alzheimer’s disease. bioRxiv [Preprint]
Gauthier, J., Meijer, I. A., Lessel, D., Mencacci, N. E., Krainc, D., Hempel, M., et al. (2018). Recessive mutations in VPS13D cause childhood onset movement disorders. Ann. Neurol. 83, 1089–1095. doi: 10.1002/ana.25204
Gellerich, F. N., Gizatullina, Z., Trumbeckaite, S., Nguyen, H. P., Pallas, T., Arandarcikaite, O., et al. (2010). The regulation of OXPHOS by extramitochondrial calcium. Biochim. Biophys. Acta - Bioenerg. 1797, 1018–1027. doi: 10.1016/j.bbabio.2010.02.005
Genç, Ö, Dickman, D. K., Ma, W., Tong, A., Fetter, R. D., and Davis, G. W. (2017). MCTP is an ER-resident calcium sensor that stabilizes synaptic transmission and homeostatic plasticity. eLife 6:e22904. doi: 10.7554/eLife.22904
Gerber, S., Alzayady, K. J., Burglen, L., Brémond-Gignac, D., Marchesin, V., Roche, O., et al. (2016). Recessive and Dominant de Novo ITPR1 mutations cause Gillespie syndrome. Am. J. Hum. Genet. 98, 971–980. doi: 10.1016/j.ajhg.2016.03.004
Gerondopoulos, A., Bastos, R. N., Yoshimura, S., Anderson, R., Carpanini, S., Aligianis, I., et al. (2014). Rab18 and a Rab18 GEF complex are required for normal ER structure. J. Cell Biol. 205, 707–720. doi: 10.1083/jcb.201403026
Gil, J. E., Kim, E., Kim, I. S., Ku, B., Park, W. S., Oh, B. H., et al. (2012). Phosphoinositides differentially regulate protrudin localization through the FYVE domain. J. Biol. Chem. 287, 41268–41276. doi: 10.1074/jbc.M112.419127
Golini, L., Chouabe, C., Berthier, C., Cusimano, V., Fornaro, M., Bonvallet, R., et al. (2011). Junctophilin 1 and 2 proteins interact with the L-type Ca 2+ channel dihydropyridine receptors (DHPRs) in skeletal muscle. J. Biol. Chem. 286, 43717–43725. doi: 10.1074/jbc.M111.292755
Gómez-Suaga, P., Pérez-Nievas, B. G., Glennon, E. B., Lau, D. H. W., Paillusson, S., Mórotz, G. M., et al. (2019). The VAPB-PTPIP51 endoplasmic reticulum-mitochondria tethering proteins are present in neuronal synapses and regulate synaptic activity. Acta Neuropathol. Commun. 7:35. doi: 10.1186/s40478-019-0688-684
González, C., Cánovas, J., Fresno, J., Couve, E., Court, F. A., and Couve, A. (2016). Axons provide the secretory machinery for trafficking of voltage-gated sodium channels in peripheral nerve. Proc. Natl. Acad. Sci. U.S.A. 113, 1823–1828. doi: 10.1073/pnas.1514943113
Goo, Y. H., Son, S. H., and Paul, A. (2017). Lipid droplet-associated hydrolase promotes lipid droplet fusion and enhances ATGL degradation and triglyceride accumulation. Sci. Rep. 7, 1–13. doi: 10.1038/s41598-017-02963-y
GrandPré, T., Nakamura, F., Vartanlan, T., and Strittmatter, S. M. (2000). Identification of the Nogo inhibitor of axon regeneration as a Reticulon protein. Nature 403, 439–444. doi: 10.1038/35000226
Grigoriev, I., Gouveia, S. M., van der Vaart, B., Demmers, J., Smyth, J. T., Honnappa, S., et al. (2008). STIM1 Is a MT-Plus-End-Tracking Protein Involved in Remodeling of the ER. Curr. Biol. 18, 177–182. doi: 10.1016/j.cub.2007.12.050
Grumati, P., Dikic, I., and Stolz, A. (2018). ER-phagy at a glance. J. Cell Sci. 131:jcs217364. doi: 10.1242/jcs.217364
Grumati, P., Morozzi, G., Ho, S., Mari, M., Harwardt, M.-L. I., Yan, R., et al. (2017). Full length RTN3 regulates turnover of tubular endoplasmic reticulum via selective autophagy. eLife 6:e25555. doi: 10.7554/eLife.25555
Guelly, C., Zhu, P. P., Leonardis, L., Papić, L., Zidar, J., Schabhüttl, M., et al. (2011). Targeted high-throughput sequencing identifies mutations in atlastin-1 as a cause of hereditary sensory neuropathy type i. Am. J. Hum. Genet. 88, 99–105. doi: 10.1016/j.ajhg.2010.12.003
Guillén-Navarro, E., Sánchez-Iglesias, S., Domingo-Jiménez, R., Victoria, B., Ruiz-Riquelme, A., Rábano, A., et al. (2013). A new seipin-associated neurodegenerative syndrome. J. Med. Genet. 50, 401–409. doi: 10.1136/jmedgenet-2013-101525
Guillén-Samander, A., Bian, X., and de Camilli, P. (2019). PDZD8 mediates a Rab7-dependent interaction of the ER with late endosomes and lysosomes. Proc. Natl. Acad. Sci. U.S.A. 116, 22619–22623. doi: 10.1073/pnas.1913509116
Gupton, S. L., and Gertler, F. B. (2010). Integrin signaling switches the cytoskeletal and exocytic machinery that drives neuritogenesis. Dev. Cell 18, 725–736. doi: 10.1016/j.devcel.2010.02.017
Gutierrez-Merino, C., Lopez-Guerrero, A. M., Alvarez-Barrientos, A., Martin-Romero, F. J., Mata, A. M., Berrocal, M., et al. (2018). STIM1 deficiency is linked to Alzheimer’s disease and triggers cell death in SH-SY5Y cells by upregulation of L-type voltage-operated Ca2+ entry. J. Mol. Med. 96, 1061–1079. doi: 10.1007/s00109-018-1677-y
Hamasaki, M., Furuta, N., Matsuda, A., Nezu, A., Yamamoto, A., Fujita, N., et al. (2013). Autophagosomes form at ER-mitochondria contact sites. Nature 495, 389–393. doi: 10.1038/nature11910
Handley, M. T., Carpanini, S. M., Mali, G. R., Sidjanin, D. J., Aligianis, I. A., Jackson, I. J., et al. (2015). Warburg Micro syndrome is caused by RAB18 deficiency or dysregulation. Open Biol. 5:150047. doi: 10.1098/rsob.150047
Handley, M. T., Morris-Rosendahl, D. J., Brown, S., Macdonald, F., Hardy, C., Bem, D., et al. (2013). Mutation Spectrum in RAB 3 GAP 1, RAB 3 GAP 2, and RAB 18 and Genotype-Phenotype correlations in warburg micro syndrome and martsolf syndrome. Hum. Mutat. 34, 686–696. doi: 10.1002/humu.22296
Hannun, Y. A., and Obeid, L. M. (2018). Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 19, 175–191. doi: 10.1038/nrm.2017.107
Hardiman, O., Al-Chalabi, A., Chio, A., Corr, E. M., Logroscino, G., Robberecht, W., et al. (2017). Amyotrophic lateral sclerosis. Nat. Rev. Dis. Prim. 3:17071. doi: 10.1038/nrdp.2017.71
Hayashi, T., and Su, T.-P. (2007). Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 131, 596–610. doi: 10.1016/j.cell.2007.08.036
Hazan, J., Fonknechten, N., Mavel, D., Paternotte, C., Samson, D., Artiguenave, F., et al. (1999). Spastin, a new AAA protein, is altered in the most frequent form of autosomal dominant spastic paraplegia. Nat. Genet. 23, 296–303. doi: 10.1038/15472
He, W., Lu, Y., Qahwash, I., Hu, X.-Y., Chang, A., and Yan, R. (2004). Reticulon family members modulate BACE1 activity and amyloid-β peptide generation. Nat. Med. 10, 959–965. doi: 10.1038/nm1088
Hedskog, L., Pinho, C. M., Filadi, R., Ronnback, A., Hertwig, L., Wiehager, B., et al. (2013). Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proc. Natl. Acad. Sci. U.S.A. 110, 7916–7921. doi: 10.1073/pnas.1300677110
Hernandez, M. V., Davies Sala, M. G., Balsamo, J., Lilien, J., and Arregui, C. O. (2006). ER-bound PTP1B is targeted to newly forming cell-matrix adhesions. J. Cell Sci. 119, 1233–1243. doi: 10.1242/jcs.02846
Hetz, C., and Saxena, S. (2017). ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 13, 477–491. doi: 10.1038/nrneurol.2017.99
Hidalgo, C., and Arias-Cavieres, A. (2016). Calcium, reactive oxygen species, and synaptic plasticity. Physiology 31, 201–215. doi: 10.1152/physiol.00038.2015
Higo, T., Hamada, K., Hisatsune, C., Nukina, N., Hashikawa, T., Hattori, M., et al. (2010). Mechanism of ER stress-induced brain damage by IP3 receptor. Neuron 68, 865–878. doi: 10.1016/j.neuron.2010.11.010
Hirabayashi, Y., Kwon, S.-K., Paek, H., Pernice, W. M., Paul, M. A., Lee, J., et al. (2017). ER-mitochondria tethering by PDZD8 regulates Ca2+ dynamics in mammalian neurons. Science 358, 623–630. doi: 10.1126/science.aan6009
Hirata, Y., Brotto, M., Weisleder, N., Chu, Y., Lin, P., Zhao, X., et al. (2006). Uncoupling store-operated Ca2+ entry and altered Ca2+ release from sarcoplasmic reticulum through silencing of junctophilin genes. Biophys. J. 90, 4418–4427. doi: 10.1529/biophysj.105.076570
Hirokawa, N., and Takemura, R. (2005). Molecular motors and mechanisms of directional transport in neurons. Nat. Rev. Neurosci. 6, 201–214. doi: 10.1038/nrn1624
Höke, A., Morris, M., and Haughey, N. J. (2009). GPI-1046 protects dorsal root ganglia from gp120-induced axonal injury by modulating store-operated calcium entry. J. Peripher. Nerv. Syst. 14, 27–35. doi: 10.1111/j.1529-8027.2009.00203.x
Holmes, S. E., O’hearn, E., Rosenblatt, A., Callahan, C., Hwang, H. S., Ingersoll-Ashworth, R. G., et al. (2001). A repeat expansion in the gene encoding junctophilin-3 is associated with Huntington disease-like 2. Nat. Genet. 29, 377–378. doi: 10.1038/ng760
Honrath, B., Metz, I., Bendridi, N., Rieusset, J., Culmsee, C., and Dolga, A. M. (2017). Glucose-regulated protein 75 determines ER–mitochondrial coupling and sensitivity to oxidative stress in neuronal cells. Cell Death Discov. 3, 1–13. doi: 10.1038/cddiscovery.2017.76
Hoppins, S., Lackner, L., and Nunnari, J. (2007). The machines that divide and fuse mitochondria. Annu. Rev. Biochem. 76, 751–780. doi: 10.1146/annurev.biochem.76.071905.090048
Horton, A. C., and Ehlers, M. D. (2003). Dual modes of endoplasmic reticulum-to-Golgi transport in dendrites revealed by live-cell imaging. J. Neurosci. 23, 6188–6199. doi: 10.1523/jneurosci.23-15-06188.2003
Hoyer, M. J., Chitwood, P. J., Ebmeier, C. C., Striepen, J. F., Qi, R. Z., Old, W. M., et al. (2018). A novel class of ER membrane proteins regulates ER-associated endosome fission. Cell 175:254-265.e14. doi: 10.1016/j.cell.2018.08.030
Hu, J., Shibata, Y., Voss, C., Shemesh, T., Li, Z., Coughlin, M., et al. (2008). Membrane proteins of the Endoplasmic reticulum induce high-curvature tubules. Science 319, 1247–1250. doi: 10.1126/science.1153634
Hu, J., Shibata, Y., Zhu, P.-P., Voss, C., Rismanchi, N., Prinz, W. A., et al. (2009). A class of dynamin-like GTPases involved in the generation of the tubular ER network. Cell 138, 549–561. doi: 10.1016/j.cell.2009.05.025
Hu, X., Shi, Q., Zhou, X., He, W., Yi, H., Yin, X., et al. (2007). Transgenic mice overexpressing reticulon 3 develop neuritic abnormalities. EMBO J. 26, 2755–2767. doi: 10.1038/sj.emboj.7601707
Hua, R., Cheng, D., Coyaud, É, Freeman, S., Di Pietro, E., Wang, Y., et al. (2017). VAPs and ACBD5 tether peroxisomes to the ER for peroxisome maintenance and lipid homeostasis. J. Cell Biol. 216, 367–377. doi: 10.1083/jcb.201608128
Hübner, C. A., and Kurth, I. (2014). Membrane-shaping disorders: a common pathway in axon degeneration. Brain 137, 3109–3121. doi: 10.1093/brain/awu287
Hyrskyluoto, A., Pulli, I., Törnqvist, K., Huu Ho, T., Korhonen, L., and Lindholm, D. (2013). Sigma-1 receptor agonist PRE084 is protective against mutant huntingtin-induced cell degeneration: involvement of calpastatin and the NF-κB pathway. Cell Death Dis. 4, 1–9. doi: 10.1038/cddis.2013.170
Iapadre, G., Morana, G., Vari, M. S., Pinto, F., Lanteri, P., Tessa, A., et al. (2018). A novel homozygous MFN2 mutation associated with severe and atypical CMT2 phenotype. Eur. J. Paediatr. Neurol. 22, 563–567. doi: 10.1016/j.ejpn.2017.12.020
Inceoglu, B., Bettaieb, A., Trindade Da Silva, C. A., Lee, K. S. S., Haj, F. G., and Hammock, B. D. (2015). Endoplasmic reticulum stress in the peripheral nervous system is a significant driver of neuropathic pain. Proc. Natl. Acad. Sci. U.S.A. 112, 9082–9087. doi: 10.1073/pnas.1510137112
Ito, D., Fujisawa, T., Iida, H., and Suzuki, N. (2008). Characterization of seipin/BSCL2, a protein associated with spastic paraplegia 17. Neurobiol. Dis. 31, 266–277. doi: 10.1016/j.nbd.2008.05.004
Jacquemyn, J., Cascalho, A., and Goodchild, R. E. (2017). The ins and outs of endoplasmic reticulum-controlled lipid biosynthesis. EMBO Rep. 18, 1905–1921. doi: 10.15252/embr.201643426
Jeong, H., Park, J., Jun, Y., and Lee, C. (2017). Crystal structures of Mmm1 and Mdm12-Mmm1 reveal mechanistic insight into phospholipid trafficking at ER-mitochondria contact sites. Proc. Natl. Acad. Sci. U.S.A. 114, E9502–E9511. doi: 10.1073/pnas.1715592114
Johnson, B., Leek, A. N., Solé, L., Maverick, E. E., Levine, T. P., and Tamkun, M. M. (2018). Kv2 potassium channels form endoplasmic reticulum/plasma membrane junctions via interaction with VAPA and VAPB. Proc. Natl. Acad. Sci. U.S.A. 115, E7331–E7340. doi: 10.1073/pnas.1805757115
Joshi, A. S., Huang, X., Choudhary, V., Levine, T. P., Hu, J., and Prinz, W. A. (2016). A family of membrane-shaping proteins at ER subdomains regulates pre-peroxisomal vesicle biogenesis. J. Cell Biol. 215, 515–529. doi: 10.1083/jcb.201602064
Joshi, A. S., Nebenfuehr, B., Choudhary, V., Satpute-Krishnan, P., Levine, T. P., Golden, A., et al. (2018). Lipid droplet and peroxisome biogenesis occur at the same ER subdomains. Nat. Commun. 9:2940. doi: 10.1038/s41467-018-05277-5273
Juneja, M., Burns, J., Saporta, M. A., and Timmerman, V. (2019). Challenges in modelling the Charcot-Marie-Tooth neuropathies for therapy development. J. Neurol. Neurosurg. Psychiatry 90, 58–67. doi: 10.1136/jnnp-2018-318834
Jung, H. H., Danek, A., and Walker, R. H. (2011). Neuroacanthocytosis syndromes. Orphanet J. Rare Dis. 6:68. doi: 10.1186/1750-1172-6-68
Kakizawa, S., Kishimoto, Y., Hashimoto, K., Miyazaki, T., Furutani, K., Shimizu, H., et al. (2007). Junctophilin-mediated channel crosstalk essential for cerebellar synaptic plasticity. EMBO J. 26, 1924–1933. doi: 10.1038/sj.emboj.7601639
Karch, C. M., Wen, N., Fan, C. C., Yokoyama, J. S., Kouri, N., Ross, O. A., et al. (2018). Selective genetic overlap between amyotrophic lateral sclerosis and diseases of the frontotemporal dementia spectrum. JAMA Neurol. 75, 860–875. doi: 10.1001/jamaneurol.2018.0372
Karnezis, T., Mandemakers, W., McQualter, J. L., Zheng, B., Ho, P. P., Jordan, K. A., et al. (2004). The neurite outgrowth inhibitor Nogo A is involved in autoimmune-mediated demyelination. Nat. Neurosci. 7, 736–744. doi: 10.1038/nn1261
Khaminets, A., Heinrich, T., Mari, M., Grumati, P., Huebner, A. K., Akutsu, M., et al. (2015). Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 522, 354–358. doi: 10.1038/nature14498
Kikuma, K., Li, X., Kim, D., Sutter, D., and Dickman, D. K. (2017). Extended synaptotagmin localizes to presynaptic ER and promotes neurotransmission and synaptic growth in drosophila. Genetics 207, 993–1006. doi: 10.1534/genetics.117.300261
Kim, K. W., Tang, N. H., Piggott, C. A., Andrusiak, M. G., Park, S., Zhu, M., et al. (2018). Expanded genetic screening in caenorhabditis elegans identifies new regulators and an inhibitory role for NAD + in axon regeneration. eLife 7:e39756. doi: 10.7554/eLife.39756
Kimura, K., Mamane, A., Sasaki, T., Sato, K., Takagi, J., Niwayama, R., et al. (2017). Endoplasmic-reticulum-mediated microtubule alignment governs cytoplasmic streaming. Nat. Cell Biol. 19, 399–406. doi: 10.1038/ncb3490
Kirmiz, M., Palacio, S., Thapa, P., King, A. N., Sack, J. T., and Trimmer, J. S. (2018). Remodeling neuronal ER–PM junctions is a conserved nonconducting function of Kv2 plasma membrane ion channels. Mol. Biol. Cell 29, 2410–2432. doi: 10.1091/mbc.E18-05-0337
Klemm, R. W., Norton, J. P., Cole, R. A., Li, C. S., Park, S. H., Crane, M. M., et al. (2013). A Conserved role for atlastin GTPases in regulating lipid droplet size. Cell Rep. 3, 1465–1475. doi: 10.1016/j.celrep.2013.04.015
Kornak, U., Mademan, I., Schinke, M., Voigt, M., Krawitz, P., Hecht, J., et al. (2014). Sensory neuropathy with bone destruction due to a mutation in the membrane-shaping atlastin GTPase 3. Brain 137, 683–692. doi: 10.1093/brain/awt357
Krajnak, K., and Dahl, R. (2018). A new target for Alzheimer’s disease: a small molecule SERCA activator is neuroprotective in vitro and improves memory and cognition in APP/PS1 mice. Bioorganic Med. Chem. Lett. 28, 1591–1594. doi: 10.1016/j.bmcl.2018.03.052
Krols, M., Asselbergh, B., Rycke, R. De, Winter, V. De, Seyer, A., Müller, F., et al. (2019). Sensory neuropathy-causing mutations in ATL3 affect ER – mitochondria contact sites and impair axonal mitochondrial distribution. Hum. Mol. Genet. 28, 615–627. doi: 10.1093/hmg/ddy352
Kuchibhotla, K. V., Goldman, S. T., Lattarulo, C. R., Wu, H.-Y., Hyman, B. T., and Bacskai, B. J. (2008). Aβ plaques lead to aberrant regulation of Calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron 59, 214–225. doi: 10.1016/J.NEURON.2008.06.008
Kumamaru, E., Kuo, C. H., Fujimoto, T., Kohama, K., Zeng, L. H., Taira, E., et al. (2004). Reticulon3 expression in rat optic and olfactory systems. Neurosci. Lett. 356, 17–20. doi: 10.1016/j.neulet.2003.11.009
Kumar, J., Yu, H., and Sheetz, M. (2006). Kinectin, an essential anchor for kinesin-driven vesicle motility. Science 267, 1834–1837. doi: 10.1126/science.7892610
Kumar, K., Kumar, A., Keegan, R. M., and Deshmukh, R. (2018). Recent advances in the neurobiology and neuropharmacology of Alzheimer’s disease. Biomed. Pharmacother. 98, 297–307. doi: 10.1016/j.biopha.2017.12.053
Kumar, N., Leonzino, M., Hancock-Cerutti, W., Horenkamp, F. A., Li, P., Lees, J. A., et al. (2018). VPS13A and VPS13C are lipid transport proteins differentially localized at ER contact sites. J. Cell Biol. 217, 3625–3639. doi: 10.1083/jcb.201807019
Kurth, I., Pamminger, T., Hennings, J. C., Soehendra, D., Huebner, A. K., Rotthier, A., et al. (2009). Mutations in FAM134B, encoding a newly identified Golgi protein, cause severe sensory and autonomic neuropathy. Nat. Genet. 41, 1179–1181. doi: 10.1038/ng.464
Landstrom, A. P., Beavers, D. L., and Wehrens, X. H. T. (2014). The junctophilin family of proteins: from bench to bedside. Trends Mol. Med. 20, 353–362. doi: 10.1016/j.molmed.2014.02.004
Larrea, D., Pera, M., Gonnelli, A., Quintana-Cabrera, R., Akman, H. O., Guardia-Laguarta, C., et al. (2019). MFN2 mutations in Charcot–Marie–Tooth disease alter mitochondria-associated ER membrane function but do not impair bioenergetics. Hum. Mol. Genet. 28:1782. doi: 10.1093/HMG/DDZ008
Latourelle, J. C., Pankratz, N., Dumitriu, A., Wilk, J. B., Goldwurm, S., Pezzoli, G., et al. (2009). Genomewide association study for onset age in Parkinson disease. BMC Med. Genet. 10:98. doi: 10.1186/1471-2350-10-98
Lauwers, E., Goodchild, R., and Verstreken, P. (2016). Membrane lipids in presynaptic function and disease. Neuron 90, 11–25. doi: 10.1016/j.neuron.2016.02.033
Lee, C., and Chen, L. B. (1988). Dynamic behavior of endoplasmic reticulum in living cells. Cell 54, 37–46. doi: 10.1016/0092-8674(88)90177-8
Lee, J. H., Han, J. H., Kim, H., Park, S. M., Joe, E. H., and Jou, I. (2019). Parkinson’s disease-associated LRRK2-G2019S mutant acts through regulation of SERCA activity to control ER stress in astrocytes. Acta Neuropathol. Commun. 7:68. doi: 10.1186/s40478-019-0716-714
Lee, S., Wang, W., Hwang, J., Namgung, U., and Min, K.-T. (2019). Increased ER–mitochondria tethering promotes axon regeneration. Proc. Natl. Acad. Sci. U.S.A. 116, 16074–16079. doi: 10.1073/pnas.1818830116
Lee, M., Paik, S. K., Lee, M.-J., Kim, Y.-J., Kim, S., Nahm, M., et al. (2009). Drosophila Atlastin regulates the stability of muscle microtubules and is required for synapse development. Dev. Biol. 330, 250–262. doi: 10.1016/j.ydbio.2009.03.019
Lee, M., Park, C., Chung, H.-K., Kim, H. J., Choi, Y., Yoo, J. H., et al. (2017). Cerebral white matter abnormalities in patients with charcot-marie-tooth disease. Ann. Neurol. 81, 147–151. doi: 10.1002/ana.24824
Lee, S., Sterky, F. H., Mourier, A., Terzioglu, M., Cullheim, S., Olson, L., et al. (2012). Mitofusin 2 is necessary for striatal axonal projections of midbrain dopamine neurons. Hum. Mol. Genet. 21, 4827–4835. doi: 10.1093/hmg/dds352
Lesage, S., Drouet, V., Majounie, E., Deramecourt, V., Jacoupy, M., Nicolas, A., et al. (2016). Loss of VPS13C function in Autosomal-recessive parkinsonism causes mitochondrial dysfunction and increases PINK1/Parkin-dependent mitophagy. Am. J. Hum. Genet. 98, 500–513. doi: 10.1016/j.ajhg.2016.01.014
Levitan, E. S., Weiner, A. T., Deitcher, D. L., Gheres, K. W., Zhou, C., Rolls, M. M., et al. (2016). Spastin, atlastin, and ER relocalization are involved in axon but not dendrite regeneration. Mol. Biol. Cell 27, 3245–3256. doi: 10.1091/mbc.e16-05-0287
Li, X., Hu, Z., Liu, L., Xie, Y., Zhan, Y., Zi, X., et al. (2015). A SIGMAR1 splice-site mutation causes distal hereditary motor neuropathy. Neurology 84, 2430–2437. doi: 10.1212/WNL.0000000000001680
Liberski, P. P., and Blackstone, C. (2017). “Hereditary spastic paraplegia,” in Neurodegeneration, eds A. Schapira, Z. Wszolek, T. M. Dawson, and N. Wood, (Hoboken, NJ: John Wiley & Sons), 161–178. doi: 10.1002/9781118661895.ch15
Licker, V., Turck, N., Kövari, E., Burkhardt, K., Côte, M., Surini-Demiri, M., et al. (2014). Proteomic analysis of human substantia nigra identifies novel candidates involved in parkinson’s disease pathogenesis. Proteomics 14, 784–794. doi: 10.1002/pmic.201300342
Liegel, R. P., Handley, M. T., Ronchetti, A., Brown, S., Langemeyer, L., Linford, A., et al. (2013). Loss-of-function mutations in TBC1d20 cause cataracts and male infertility in blind sterile mice and Warburg micro syndrome in humans. Am. J. Hum. Genet. 93, 1001–1014. doi: 10.1016/j.ajhg.2013.10.011
Lim, Y., Cho, I.-T. I. T., Schoel, L. J., Cho, G., and Golden, J. A. (2015). Hereditary spastic paraplegia-linked REEP1 modulates endoplasmic reticulum/mitochondria contacts. Ann. Neurol. 78, 679–696. doi: 10.1002/ana.24488
Lindholm, D., Korhonen, L., Eriksson, O., and Kõks, S. (2017). Recent insights into the role of unfolded protein response in ER stress in health and disease. Front. Cell Dev. Biol. 5:48. doi: 10.3389/fcell.2017.00048
Lindhout, F. W., Cao, Y., Kevenaar, J. T., Bodzȩta, A., Stucchi, R., Boumpoutsari, M. M., et al. (2019). VAP-SCRN1 interaction regulates dynamic endoplasmic reticulum remodeling and presynaptic function. EMBO J. 38:e101345. doi: 10.15252/embj.2018101345
Ling, S.-C., Polymenidou, M., and Cleveland, D. W. W. (2013). Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438. doi: 10.1016/j.neuron.2013.07.033
Liu, X., Guo, X., Niu, L., Li, X., Sun, F., Hu, J., et al. (2019). Atlastin-1 regulates morphology and function of endoplasmic reticulum in dendrites. Nat. Commun. 10:568. doi: 10.1038/s41467-019-08478-8476
Lowther, J., Naismith, J. H., Dunn, T. M., and Campopiano, D. J. (2012). Structural, mechanistic and regulatory studies of serine palmitoyltransferase. Biochem. Soc. Trans. 40, 547–554. doi: 10.1042/BST20110769
Lu, L., Ladinsky, M. S., and Kirchhausen, T. (2009). Cisternal organization of the endoplasmic reticulum during mitosis. Mol. Biol. Cell 20, 3471–3480. doi: 10.1091/mbc.E09
Lupachyk, S., Watcho, P., Stavniichuk, R., Shevalye, H., and Obrosova, I. G. (2013). Endoplasmic reticulum stress plays a key role in the pathogenesis of diabetic peripheral neuropathy. Diabetes 62, 944–952. doi: 10.2337/db12-0716
Ma, Z., Liu, Z., and Huang, X. (2012). Membrane phospholipid asymmetry counters the adverse effects of sterol overloading in the Golgi membrane of Drosophila. Genetics 190, 1299–1308. doi: 10.1534/genetics.111.137687
Mackay, J. P., Nassrallah, W. B., and Raymond, L. A. (2018). Cause or compensation?-Altered neuronal Ca 2+ handling in Huntington’s disease. CNS Neurosci. Ther. 24, 301–310. doi: 10.1111/cns.12817
Maksimova, N., Hara, K., Nikolaeva, I., Chun-Feng, T., Usui, T., Takagi, M., et al. (2010). Neuroblastoma amplified sequence gene is associated with a novel short stature syndrome characterised by optic nerve atrophy and Pelger-Huët anomaly. J. Med. Genet. 47, 538–548. doi: 10.1136/jmg.2009.074815
Manczak, M., Calkins, M. J., and Reddy, P. H. (2011). Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: implications for neuronal damage. Hum. Mol. Genet. 20, 2495–2509. doi: 10.1093/hmg/ddr139
Mandikian, D., Bocksteins, E., Parajuli, L. K., Bishop, H. I., Cerda, O., Shigemoto, R., et al. (2014). Cell type-specific spatial and functional coupling between mammalian brain Kv2.1 K+ channels and ryanodine receptors. J. Comp. Neurol. 522, 3555–3574. doi: 10.1002/cne.23641
Mangus, L. M., Dorsey, J. L., Laast, V. A., Ringkamp, M., Ebenezer, G. J., Hauer, P., et al. (2014). Unraveling the pathogenesis of HIV peripheral neuropathy: insights from a simian immunodeficiency virus macaque model. ILAR J. 54, 296–303. doi: 10.1093/ilar/ilt047
Mannan, A. U., Boehm, J., Sauter, S. M., Rauber, A., Byrne, P. C., Neesen, J., et al. (2006). Spastin, the most commonly mutated protein in hereditary spastic paraplegia interacts with Reticulon 1 an endoplasmic reticulum protein. Neurogenetics 7, 93–103. doi: 10.1007/s10048-006-0034-34
Marini, C., Ravera, S., Buschiazzo, A., Bianchi, G., Orengo, A. M., Bruno, S., et al. (2016). Discovery of a novel glucose metabolism in cancer: the role of endoplasmic reticulum beyond glycolysis and pentose phosphate shunt. Sci. Rep. 6, 1–13. doi: 10.1038/srep25092
Marongiu, R., Spencer, B., Crews, L., Adame, A., Patrick, C., Trejo, M., et al. (2009). Mutant Pink1 induces mitochondrial dysfunction in a neuronal cell model of Parkinson’s disease by disturbing calcium flux. J. Neurochem. 108, 1561–1574. doi: 10.1111/j.1471-4159.2009.05932.x
Martin, S., Driessen, K., Nixon, S. J., Zerial, M., and Parton, R. G. (2005). Regulated localization of Rab18 to lipid droplets: effects of lipolytic stimulation and inhibition of lipid droplet catabolism. J. Biol. Chem. 280, 42325–42335. doi: 10.1074/jbc.M506651200
Mavlyutov, T. A., Epstein, M. L., Andersen, K. A., Ziskind-Conhaim, L., and Ruoho, A. E. (2010). The sigma-1 receptor is enriched in postsynaptic sites of C-terminals in mouse motoneurons. An anatomical and behavioral study. Neuroscience 167, 247–255. doi: 10.1016/j.neuroscience.2010.02.022
Meade, R. M., Fairlie, D. P., and Mason, J. M. (2019). Alpha-synuclein structure and Parkinson’s disease – lessons and emerging principles. Mol. Neurodegener. 14, 1–14. doi: 10.1186/s13024-019-0329-321
Merianda, T. T., Lin, A. C., Lam, J. S. Y., Vuppalanchi, D., Willis, D. E., Karin, N., et al. (2009). A functional equivalent of endoplasmic reticulum and Golgi in axons for secretion of locally synthesized proteins. Mol. Cell. Neurosci. 40, 128–142. doi: 10.1016/j.mcn.2008.09.008
Miki, Y., Tanji, K., Mori, F., and Wakabayashi, K. (2015). Sigma-1 receptor is involved in degradation of intranuclear inclusions in a cellular model of Huntington’s disease. Neurobiol. Dis. 74, 25–31. doi: 10.1016/j.nbd.2014.11.005
Milani, M., Cohen, G. M., and Varadarajan, S. (2018). ER shaping proteins regulate mitochondrial fission, outer membrane permeabilization and apoptosis. bioRxiv [Preprint] doi: 10.1101/340448
Mishina, M., Ishiwata, K., Ishii, K., Kitamura, S., Kimura, Y., Kawamura, K., et al. (2005). Function of sigma 1 receptors in Parkinson’s disease. Acta Neurol. Scand. 112, 103–107. doi: 10.1111/j.1600-0404.2005.00432.x
Mishina, M., Ohyama, M., Ishii, K., Kitamura, S., Kimura, Y., Oda, K. I., et al. (2008). Low density of sigma 1 receptors in early Alzheimer’s disease. Ann. Nucl. Med. 22, 151–156. doi: 10.1007/s12149-007-0094-z
Misko, A., Jiang, S., Wegorzewska, I., Milbrandt, J., and Baloh, R. H. (2010). Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J. Neurosci. 30, 4232–4240. doi: 10.1523/JNEUROSCI.6248-09.2010
Mitchell, C. B., Gasperini, R. J., Small, D. H., and Foa, L. (2012). STIM1 is necessary for store-operated calcium entry in turning growth cones. J. Neurochem. 122, 1155–1166. doi: 10.1111/j.1471-4159.2012.07840.x
Mizushima, N., Yoshimori, T., and Ohsumi, Y. (2011). The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 27, 107–132. doi: 10.1146/annurev-cellbio-092910-154005
Montenegro, G., Rebelo, A. P., Connell, J., Allison, R., Babalini, C., D’Aloia, M., et al. (2012). Mutations in the ER-shaping protein reticulon 2 cause the axon-degenerative disorder hereditary spastic paraplegia type 12. J. Clin. Invest. 122, 538–544. doi: 10.1172/JCI60560
Mori, T., Hayashi, T., and Su, T.-P. (2012). Compromising -1 receptors at the Endoplasmic reticulum render cytotoxicity to physiologically relevant concentrations of dopamine in a nuclear factor- B/Bcl-2-dependent mechanism: potential relevance to Parkinson’s disease. J. Pharmacol. Exp. Ther. 341, 663–671. doi: 10.1124/jpet.111.190868
Moriguchi, S., Nishi, M., Komazaki, S., Sakagami, H., Miyazaki, T., Masumiya, H., et al. (2006). Functional uncoupling between Ca2+ release and afterhyperpolarization in mutant hippocampal neurons lacking junctophilins. Proc. Natl. Acad. Sci. U.S.A. 103, 10811–10816. doi: 10.1073/pnas.0509863103
Muallem, S., Chung, W. Y., Jha, A., and Ahuja, M. (2017). Lipids at membrane contact sites: cell signaling and ion transport. EMBO Rep. 18, 1893–1904. doi: 10.15252/embr.201744331
Müller, M., Pym, E. C. G., Tong, A., and Davis, G. W. (2011). Rab3-GAP controls the progression of Synaptic Homeostasis at a late stage of vesicle release. Neuron 69, 749–762. doi: 10.1016/j.neuron.2011.01.025
Müller, M. S., Fouyssac, M., and Taylor, C. W. (2018). Effective Glucose uptake by human astrocytes requires its sequestration in the Endoplasmic reticulum by Glucose-6-Phosphatase-β. Curr. Biol. 28:3481-3486.e4. doi: 10.1016/j.cub.2018.08.060
Munhoz, R. P., Moro, A., Silveira-Moriyama, L., and Teive, H. A. (2015). Non-motor signs in Parkinson’s disease: a review. Arq. Neuropsiquiatr. 73, 454–462. doi: 10.1590/0004-282X20150029
Murphy, S. E., and Levine, T. P. (2016). VAP, a Versatile access point for the Endoplasmic Reticulum: review and analysis of FFAT-like motifs in the VAPome. Biochim. Biophys. Acta - Mol. Cell Biol. Lipids 1861, 952–961. doi: 10.1016/j.bbalip.2016.02.009
Nakagawa, H., Koyama, K., Murata, Y., Morito, M., Akiyama, T., and Nakamura, Y. (2000). EB3, a novel member of the EB1 family preferentially expressed in the central nervous system, binds to a CNS-specific APC homologue. Oncogene 19, 210–216. doi: 10.1038/sj.onc.1203308
Nakajima, K., Hirose, H., Taniguchi, M., Kurashina, H., Arasaki, K., Nagahama, M., et al. (2004). Involvement of BNIP1 in apoptosis and endoplasmic reticulum membrane fusion. EMBO J. 23, 3216–3226. doi: 10.1038/sj.emboj.7600333
Nevo-Yassaf, I., Yaffe, Y., Asher, M., Ravid, O., Eizenberg, S., Henis, Y. I., et al. (2012). Role for TBC1D20 and Rab1 in Hepatitis C virus replication via interaction with lipid Droplet-bound nonstructural protein 5A. J. Virol. 86, 6491–6502. doi: 10.1128/jvi.00496-412
Nian, F.-S., Li, L.-L., Cheng, C.-Y., Wu, P.-C., Lin, Y.-T., Tang, C.-Y., et al. (2019). Rab18 Collaborates with Rab7 to Modulate Lysosomal and Autophagy Activities in the Nervous System: an Overlapping Mechanism for Warburg Micro Syndrome and Charcot-Marie-Tooth Neuropathy Type 2B. Mol. Neurobiol. 56, 6095–6105. doi: 10.1007/s12035-019-1471-z
Nishimura, A. L., Mitne-Neto, M., Silva, H. C. A., Richieri-Costa, A., Middleton, S., Cascio, D., et al. (2004). A mutation in the Vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet. 75, 822–831. doi: 10.1086/425287
Nixon-Abell, J., Obara, C. J., Weigel, A. V., Li, D., Legant, W. R., Xu, C. S., et al. (2016). Increased spatiotemporal resolution reveals highly dynamic dense tubular matrices in the peripheral ER. Science 354:aaf3928. doi: 10.1126/science.aaf3928
Novarino, G., Fenstermaker, A. G., Zaki, M. S., Hofree, M., Silhavy, J. L., Heiberg, A. D., et al. (2014). Exome sequencing links corticospinal motor neuron disease to common Neurodegenerative disorders. Science 343, 506–511. doi: 10.1126/science.1247363
Ochaba, J., Lukacsovich, T., Csikos, G., Zheng, S., Margulis, J., Salazar, L., et al. (2014). Potential function for the Huntingtin protein as a scaffold for selective autophagy. Proc. Natl. Acad. Sci. U.S.A. 111, 16889–16894. doi: 10.1073/pnas.1420103111
Ohsaki, Y., Sołtysik, K., and Fujimoto, T. (2017). “The lipid Droplet and the Endoplasmic reticulum,” in Organelle Contact Sites: From Molecular Mechanism to Disease, eds M. Tagaya and T. Simmen, (Singapore: Springer Singapore), 111–120. doi: 10.1007/978-981-10-4567-7_8
O’Neill, P., Whalley, K., and Ferretti, P. (2004). Nogo and Nogo-66 receptor in human and chick: implications for development and regeneration. Dev. Dyn. 231, 109–121. doi: 10.1002/dvdy.20116
Orem, B. C., Pelisch, N., Williams, J., Nally, J. M., and Stirling, D. P. (2017). Intracellular calcium release through IP3R or RyR contributes to secondary axonal degeneration. Neurobiol. Dis. 106, 235–243. doi: 10.1016/j.nbd.2017.07.011
Orso, G., Pendin, D., Liu, S., Tosetto, J., Moss, T. J., Faust, J. E., et al. (2009). Homotypic fusion of ER membranes requires the dynamin-like GTPase atlastin. Nature 460, 978–983. doi: 10.1038/nature08280
O’Sullivan, N. C., Jahn, T. R., Reid, E., and O’Kane, C. J. (2012). Reticulon-like-1, the Drosophila orthologue of the hereditary spastic paraplegia gene reticulon 2, is required for organization of endoplasmic reticulum and of distal motor axons. Hum. Mol. Genet. 21, 3356–3365. doi: 10.1093/hmg/dds167
Ozeki, S., Cheng, J., Tauchi-Sato, K., Hatano, N., Taniguchi, H., and Fujimoto, T. (2005). Rab18 localizes to lipid droplets and induces their close apposition to the endoplasmic reticulum-derived membrane. J. Cell Sci. 118, 2601–2611. doi: 10.1242/jcs.02401
Paillard, M., Tubbs, E., Thiebaut, P. A., Gomez, L., Fauconnier, J., Da Silva, C. C., et al. (2013). Depressing mitochondria-reticulum interactions protects cardiomyocytes from lethal hypoxia-reoxygenation injury. Circulation 128, 1555–1565. doi: 10.1161/CIRCULATIONAHA.113.001225
Paillusson, S., Gomez-Suaga, P., Stoica, R., Little, D., Gissen, P., Devine, M. J., et al. (2017). α-Synuclein binds to the ER–mitochondria tethering protein VAPB to disrupt Ca2+ homeostasis and mitochondrial ATP production. Acta Neuropathol. 134, 129–149. doi: 10.1007/s00401-017-1704-z
Pantakani, D. V. K., Czyzewska, M. M., Sikorska, A., Bodda, C., and Mannan, A. U. (2011). Oligomerization of ZFYVE27 (Protrudin) is necessary to promote neurite extension. PLoS One 6:e0029584. doi: 10.1371/journal.pone.0029584
Papadopoulos, C., Orso, G., Mancuso, G., Herholz, M., Gumeni, S., Tadepalle, N., et al. (2015). Spastin binds to lipid droplets and affects lipid metabolism. PLoS Genet. 11:e1005149. doi: 10.1371/journal.pgen.1005149
Park, J. S., Halegoua, S., Kishida, S., and Neiman, A. M. (2015). A conserved function in phosphatidylinositol metabolism for mammalian Vps13 family proteins. PLoS One 10:e0124836. doi: 10.1371/journal.pone.0124836
Park, S. H., Zhu, P.-P., Parker, R. L., and Blackstone, C. (2010). Hereditary spastic paraplegia proteins REEP1, spastin, and atlastin-1 coordinate microtubule interactions with the tubular ER network. J. Clin. Invest. 120, 1097–1110. doi: 10.1172/JCI40979
Pavez, M., Thompson, A. C., Arnott, H. J., Mitchell, C. B., D’Atri, I., Don, E. K., et al. (2019). STIM1 is required for remodelling of the endoplasmic reticulum and microtubule cytoskeleton in steering growth cones. J. Neurosci. 39, 5095–5114. doi: 10.1523/JNEUROSCI.2496-18.2019
Pei, J. J., Sersen, E., Iqbal, K., and Grundke-Iqbal, I. (1994). Expression of protein phosphatases (PP-1, PP-2A, PP-2B and PTP-1B) and protein kinases (MAP kinase and P34cdc2) in the hippocampus of patients with Alzheimer disease and normal aged individuals. Brain Res. 655, 70–76. doi: 10.1016/0006-8993(94)91598-91599
Pennetta, G., Hiesinger, P. R., Fabian-Fine, R., Meinertzhagen, I. A., and Bellen, H. J. (2002). Drosophila VAP-33A directs bouton formation at neuromuscular junctions in a dosage-dependent manner. Neuron 35, 291–306. doi: 10.1016/s0896-6273(02)00769-9
Pennetta, G., and Welte, M. A. (2018). Emerging links between lipid droplets and motor neuron diseases. Dev. Cell 45, 427–432. doi: 10.1016/j.devcel.2018.05.002
Petersen, O. H., Courjaret, R., and Machaca, K. (2017). Ca2+ tunnelling through the ER lumen as a mechanism for delivering Ca2+ entering via store-operated Ca2+ channels to specific target sites. J. Physiol. 595, 2999–3014. doi: 10.1113/JP272772
Petkovic, M., Jemaiel, A., Daste, F., Specht, C. G., Izeddin, I., Vorkel, D., et al. (2014). The SNARE Sec22b has a non-fusogenic function in plasma membrane expansion. Nat. Cell Biol. 16, 434–444. doi: 10.1038/ncb2937
Petrungaro, C., and Kornmann, B. (2019). Lipid exchange at ER-mitochondria contact sites: a puzzle falling into place with quite a few pieces missing. Curr. Opin. Cell Biol. 57, 71–76. doi: 10.1016/j.ceb.2018.11.005
Pham, A. H., Meng, S., Chu, Q. N., and Chan, D. C. (2012). Loss of Mfn2 results in progressive, retrograde degeneration of dopaminergic neurons in the nigrostriatal circuit. Hum. Mol. Genet. 21, 4817–4826. doi: 10.1093/hmg/dds311
Phillips, M. J., and Voeltz, G. K. (2016). Structure and function of ER membrane contact sites with other organelles. Nat. Rev. Mol. Cell Biol. 17, 69–82. doi: 10.1038/nrm.2015.8
Pierce, J. P., Mayer, T., and McCarthy, J. B. (2001). Evidence for a satellite secretory pathway in neuronal dendritic spines. Curr. Biol. 11, 351–355. doi: 10.1016/S0960-9822(01)00077-X
Piscosquito, G., Saveri, P., Magri, S., Ciano, C., Di Bella, D., Milani, M., et al. (2015). Mutational mechanisms in MFN2 -related neuropathy: compound heterozygosity for recessive and semidominant mutations. J. Peripher. Nerv. Syst. 20, 380–386. doi: 10.1111/jns.12145
Pla-Martín, D., Calpena, E., Lupo, V., Márquez, C., Rivas, E., Sivera, R., et al. (2015). Junctophilin-1 is a modifier gene of GDAP1-related Charcot-Marie-Tooth disease. Hum. Mol. Genet. 24, 213–229. doi: 10.1093/hmg/ddu440
Pla-Martín, D., Rueda, C. B., Estela, A., Sánchez-Piris, M., González-Sánchez, P., Traba, J., et al. (2013). Silencing of the Charcot-Marie-Tooth disease-associated gene GDAP1 induces abnormal mitochondrial distribution and affects Ca2+ homeostasis by reducing store-operated Ca2+ entry. Neurobiol. Dis. 55, 140–151. doi: 10.1016/j.nbd.2013.03.010
Poewe, W., Seppi, K., Tanner, C. M., Halliday, G. M., Brundin, P., Volkmann, J., et al. (2017). Parkinson disease. Nat. Rev. Dis. Prim. 3:17013. doi: 10.1038/nrdp.2017.13
Prause, J., Goswami, A., Katona, I., Roos, A., Schnizler, M., Bushuven, E., et al. (2013). Altered localization, abnormal modification and loss of function of sigma receptor-1 in amyotrophic lateral sclerosis. Hum. Mol. Genet. 22, 1581–1600. doi: 10.1093/hmg/ddt008
Pulst, S., Nechiporuk, A., Nechiporuk, T., Gispert, S., Chen, X., Lopes-cendes, I., et al. (1996). Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat. Genet. 14, 269–276. doi: 10.1038/ng1196-269
Puri, R., Cheng, X.-T., Lin, M.-Y., Huang, N., and Sheng, Z.-H. (2019). Mul1 restrains Parkin-mediated mitophagy in mature neurons by maintaining ER-mitochondrial contacts. Nat. Commun. 10:3645. doi: 10.1038/s41467-019-11636-11635
Raffaello, A., Mammucari, C., Gherardi, G., and Rizzuto, R. (2016). Calcium at the center of cell signaling: interplay between Endoplasmic reticulum. Mitochondria, and Lysosomes. Trends Biochem. Sci. 41, 1035–1049. doi: 10.1016/j.tibs.2016.09.001
Raiborg, C., Wenzel, E. M., Pedersen, N. M., Olsvik, H., Schink, K. O., Schultz, S. W., et al. (2015). Repeated ER-endosome contacts promote endosome translocation and neurite outgrowth. Nature 520, 234–238. doi: 10.1038/nature14359
Ralser, M., Nonhoff, U., Albrecht, M., Lengauer, T., Wanker, E. E., Lehrach, H., et al. (2005). Ataxin-2 and huntingtin interact with endophilin-A complexes to function in plastin-associated pathways. Hum. Mol. Genet. 14, 2893–2909. doi: 10.1093/hmg/ddi321
Rämö, O., Kumar, D., Gucciardo, E., Joensuu, M., Saarekas, M., Vihinen, H., et al. (2016). NOGO-A/RTN4A and NOGO-B/RTN4B are simultaneously expressed in epithelial, fibroblast and neuronal cells and maintain ER morphology. Sci. Rep. 6, 1–14. doi: 10.1038/srep35969
Rao, K., Stone, M. C., Weiner, A. T., Gheres, K. W., Zhou, C., Deitcher, D. L., et al. (2016). Spastin, atlastin, and ER relocalization are involved in axon but not dendrite regeneration. Mol. Biol. Cell 27, 3245–3256. doi: 10.1091/mbc.E16-05-0287
Ratnaparkhi, A., Lawless, G. M., Schweizer, F. E., Golshani, P., and Jackson, G. R. (2008). A Drosophila model of ALS: human ALS-associated mutation in VAP33A suggests a dominant negative mechanism. PLoS One 3:e002334. doi: 10.1371/journal.pone.0002334
Ribeiro, C. M. P., McKay, R. R., Hosoki, E., Bird, G. S. J., and Putney, J. W. (2000). Effects of elevated cytoplasmic calcium and protein kinase C on endoplasmic reticulum structure and function in HEK293 cells. Cell Calcium 27, 175–185. doi: 10.1054/ceca.2000.0108
Ridge, P. G., Karch, C. M., Hsu, S., Arano, I., Teerlink, C. C., Ebbert, M. T. W., et al. (2018). Linkage, whole genome sequence, and biological data implicate variants in RAB10 in Alzheimer’s disease resilience. Genome Med. 10, 1–14. doi: 10.1186/s13073-018-0516-7
Rocha, N., Kuijl, C., Van Der Kant, R., Janssen, L., Houben, D., Janssen, H., et al. (2009). Cholesterol sensor ORP1L contacts the ER protein VAP to control Rab7-RILP-p150Glued and late endosome positioning. J. Cell Biol. 185, 1209–1225. doi: 10.1083/jcb.200811005
Rochin, L., Sauvanet, C., Jääskeläinen, E., Houcine, A., Kivelä, A., Ma, X., et al. (2019). ORP5 regulates transport of lipids and calcium to Mitochondria at Endoplasmic reticulum-mitochondria membrane contact sites. bioRxiv [Preprint] doi: 10.1101/695577
Roll-Mecak, A., and Vale, R. D. (2005). The Drosophila homologue of the hereditary spastic paraplegia protein, spastin, severs and disassembles microtubules. Curr. Biol. 15, 650–655. doi: 10.1016/j.cub.2005.02.029
Ross, W. N. (2012). Understanding calcium waves and sparks in central neurons. Nat. Rev. Neurosci. 13, 157–168. doi: 10.1038/nrn3168
Rotthier, A., Baets, J., Timmerman, V., and Janssens, K. (2012). Mechanisms of disease in hereditary sensory and autonomic neuropathies. Nat. Rev. Neurol. 8, 73–85. doi: 10.1038/nrneurol.2011.227
Rowland, A. A., Chitwood, P. J., Phillips, M. J., and Voeltz, G. K. (2014). ER contact sites define the position and timing of endosome fission. Cell 159, 1027–1041. doi: 10.1016/j.cell.2014.10.023
Ryskamp, D., Wu, J., Geva, M., Kusko, R., Grossman, I., Hayden, M., et al. (2017). The sigma-1 receptor mediates the beneficial effects of pridopidine in a mouse model of Huntington disease. Neurobiol. Dis. 97, 46–59. doi: 10.1016/j.nbd.2016.10.006
Saheki, Y., and Camilli, P. D. (2017). Endoplasmic reticulum – Plasma membrane contact sites. Annu. Rev. Biochem. 86, 659–684. doi: 10.1146/annurev-biochem-
Sahu, G., Wazen, R.-M., Colarusso, P., Chen, S. R. W., Zamponi, G. W., and Turner, R. W. (2019). Junctophilin proteins tether a Cav1-RyR2-KCa3.1 tripartite complex to regulate neuronal excitability. Cell Rep. 28:2427-2442.e6. doi: 10.1016/j.celrep.2019.07.075
Sakane, A., Manabe, S., Ishizaki, H., Tanaka-Okamoto, M., Kiyokage, E., Toida, K., et al. (2006). Rab3 GTPase-activating protein regulates synaptic transmission and plasticity through the inactivation of Rab3. Proc. Natl. Acad. Sci. U.S.A. 103, 10029–10034. doi: 10.1073/pnas.0600304103
Salo, V. T., Belevich, I., Li, S., Karhinen, L., Vihinen, H., Vigouroux, C., et al. (2016). Seipin regulates ER–lipid droplet contacts and cargo delivery. EMBO J. 35, 2699–2716. doi: 10.15252/embj.201695170
Salo, V. T., Li, S., Vihinen, H., Hölttä-Vuori, M., Szkalisity, A., Horvath, P., et al. (2019). Seipin facilitates Triglyceride flow to lipid droplet and counteracts droplet Ripening via Endoplasmic reticulum contact. Dev. Cell 50:478-493.e9. doi: 10.1016/j.devcel.2019.05.016
Sanyal, S., Consoulas, C., Kuromi, H., Basole, A., Mukai, L., Kidokoro, Y., et al. (2005). Analysis of conditional paralytic mutants in Drosophila sarco-endoplasmic reticulum calcium ATPase reveals novel mechanisms for regulating membrane excitability. Genetics 169, 737–750. doi: 10.1534/genetics.104.031930
Saporta, M. A., Dang, V., Volfson, D., Zou, B., Xie, X., Simon, A., et al. (2015). Axonal Charcot–Marie–Tooth disease patient-derived motor neurons demonstrate disease-specific phenotypes including abnormal electrophysiological properties. Exp. Neurol. 263, 190–199. doi: 10.1016/j.expneurol.2014.10.005
Sasidharan, N., Sumakovic, M., Hannemann, M., Hegermann, J., Liewal, J. F., Olendrowitz, C., et al. (2012). RAB-5 and RAB-10 cooperate to regulate neuropeptide release in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 109, 18944–18949. doi: 10.1073/pnas.1203306109
Sassano, M. L., and Agostinis, P. (2019). Staying in touch: taking a closer look at ER-Golgi contact sites. J. Cell Biol. 218, 729–731. doi: 10.1083/jcb.201901039
Schauder, C. M., Wu, X., Saheki, Y., Narayanaswamy, P., Torta, F., Wenk, M. R., et al. (2014). Structure of a lipid-bound extended synaptotagmin indicates a role in lipid transfer. Nature 510, 552–555. doi: 10.1038/nature13269
Scheltens, P., Blennow, K., Breteler, M. M. B., de Strooper, B., Frisoni, G. B., Salloway, S., et al. (2016). Alzheimer’s disease. Lancet 388, 505–517. doi: 10.1016/S0140-6736(15)01124-1121
Schneeberger, M., Dietrich, M. O., Sebastián, D., Imbernón, M., Castaño, C., Garcia, A., et al. (2013). Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell 155, 172–187. doi: 10.1016/j.cell.2013.09.003
Schneider, S. A., and Bird, T. (2016). Huntington’s disease, Huntington’s disease look-alikesı, and benign hereditary chorea: what’s new? Mov. Disord. Clin. Pract. 3, 342–354. doi: 10.1002/mdc3.12312
Schormair, B., Kemlink, D., Mollenhauer, B., Fiala, O., Machetanz, G., Roth, J., et al. (2018). Diagnostic exome sequencing in early-onset Parkinson’s disease confirms VPS13C as a rare cause of autosomal-recessive Parkinson’s disease. Clin. Genet. 93, 603–612. doi: 10.1111/cge.13124
Schreiner, B., Hedskog, L., Wiehager, B., and Ankarcrona, M. (2014). Amyloid-β peptides are generated in Mitochondria-associated Endoplasmic reticulum membranes. J. Alzheimer’s Dis. 43, 369–374. doi: 10.3233/JAD-132543
Schroeder, L. K., Barentine, A. E. S., Merta, H., Schweighofer, S., Zhang, Y., Baddeley, D., et al. (2018). Dynamic nanoscale morphology of the ER surveyed by STED microscopy. J. Cell Biol. 218, 83–96. doi: 10.1083/jcb.201809107
Schuck, S., Prinz, W. A., Thorn, K. S., Voss, C., and Walter, P. (2009). Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J. Cell Biol. 187, 525–536. doi: 10.1083/jcb.200907074
Schwartzlow, C., and Kazamel, M. (2019). Hereditary sensory and autonomic neuropathies: adding more to the classification. Curr. Neurol. Neurosci. Rep. 19:52. doi: 10.1007/s11910-019-0974-973
Schwarz, D. S., and Blower, M. D. (2016). The endoplasmic reticulum: structure, function and response to cellular signaling. Cell. Mol. Life Sci. 73, 79–94. doi: 10.1007/s00018-015-2052-2056
Scott, L. J., Muglia, P., Kong, X. Q., Guan, W., Flickinger, M., Upmanyu, R., et al. (2009). Genome-wide association and meta-analysis of bipolar disorder in individuals of European ancestry. Proc. Natl. Acad. Sci. U.S.A. 106, 7501–7506. doi: 10.1073/pnas.0813386106
Seong, E., Insolera, R., Dulovic, M., Kamsteeg, E. J., Trinh, J., Brüggemann, N., et al. (2018). Mutations in VPS13D lead to a new recessive ataxia with spasticity and mitochondrial defects. Ann. Neurol. 83, 1075–1088. doi: 10.1002/ana.25220
Sharoar, M. G., Shi, Q., Ge, Y., He, W., Hu, X., Perry, G., et al. (2016). Dysfunctional tubular endoplasmic reticulum constitutes a pathological feature of Alzheimer’s disease. Mol. Psychiatry 21, 1263–1271. doi: 10.1038/mp.2015.181
Shen, Y., Liu, X., Long, X., Han, C., Wan, F., Fan, W., et al. (2017). Novel VPS13a gene mutations identified in patients diagnosed with chorea-acanthocytosis (chAc): case presentation and literature review. Front. Aging Neurosci. 9:95. doi: 10.3389/fnagi.2017.00095
Sherwood, N. T., Sun, Q., Xue, M., Zhang, B., and Zinn, K. (2004). Drosophila spastin regulates synaptic microtubule networks and is required for normal motor function. PLoS Biol. 2:e429. doi: 10.1371/journal.pbio.0020429
Shi, Q., Ge, Y., He, W., Hu, X., and Yan, R. (2017). RTN1 and RTN3 protein are differentially associated with senile plaques in Alzheimer’s brains. Sci. Rep. 7, 1–9. doi: 10.1038/s41598-017-05504-5509
Shi, Q., Prior, M., He, W., Tang, X., Hu, X., and Yan, R. (2009). Reduced Amyloid deposition in mice overexpressing RTN3 is adversely affected by preformed dystrophic neurites. J. Neurosci. 29, 9163–9173. doi: 10.1523/JNEUROSCI.5741-08.2009
Shibata, Y., Hu, J., Kozlov, M. M., and Rapoport, T. A. (2009). Mechanisms shaping the membranes of cellular organelles. Annu. Rev. Cell Dev. Biol. 25, 329–354. doi: 10.1146/annurev.cellbio.042308.113324
Shibata, Y., Shemesh, T., Prinz, W. A., Palazzo, A. F., Kozlov, M. M., and Rapoport, T. A. (2010). Mechanisms determining the morphology of the peripheral ER. Cell 143, 774–788. doi: 10.1016/j.cell.2010.11.007
Shim, S., Zheng, J. Q., and Ming, G. L. (2013). A critical role for STIM1 in filopodial calcium entry and axon guidance. Mol. Brain 6, 1–14. doi: 10.1186/1756-6606-6-51
Shim, S. Y., Wang, J., Asada, N., Neumayer, G., Tran, H. C., Ishiguro, K.-I., et al. (2008). Protein 600 is a Microtubule/Endoplasmic reticulum-associated protein in CNS neurons. J. Neurosci. 28, 3604–3614. doi: 10.1523/jneurosci.5278-07.2008
Shimizu, H., Fukaya, M., Yamasaki, M., Watanabe, M., Manabe, T., and Kamiya, H. (2008). Use-dependent amplification of presynaptic Ca2+ signaling by axonal ryanodine receptors at the hippocampal mossy fiber synapse. Proc. Natl. Acad. Sci. U.S.A. 105, 11998–12003. doi: 10.1073/pnas.0802175105
Shin, Y. K., Jang, S. Y., Lee, H. K., Jung, J., Suh, D. J., Seo, S. Y., et al. (2010). Pathological adaptive responses of schwann cells to endoplasmic reticulum stress in bortezomib-induced peripheral neuropathy. Glia 58, 1961–1976. doi: 10.1002/glia.21065
Shoshan-Barmatz, V., Zalk, R., Gincel, D., and Vardi, N. (2004). Subcellular localization of VDAC in mitochondria and ER in the cerebellum. Biochim. Biophys. Acta - Bioenerg. 1657, 105–114. doi: 10.1016/j.bbabio.2004.02.009
Shukla, A., Upadhyai, P., Shah, J., Neethukrishna, K., Bielas, S., and Girisha, K. M. (2017). Autosomal recessive spinocerebellar ataxia 20: report of a new patient and review of literature. Eur. J. Med. Genet. 60, 118–123. doi: 10.1016/j.ejmg.2016.11.006
Shy, M. E., and Patzkó, Á (2011). Axonal Charcot–Marie–Tooth disease. Curr. Opin. Neurol. 24, 475–483. doi: 10.1097/WCO.0b013e32834aa331
Siau, C., and Bennett, G. J. (2006). Dysregulation of cellular calcium homeostasis in chemotherapy-evoked painful peripheral neuropathy. Anesth. Analg. 102, 1485–1490. doi: 10.1213/01.ane.0000204318.35194.ed
Sidjanin, D. J., Park, A. K., Ronchetti, A., Martins, J., and Jackson, W. T. (2016). TBC1D20 mediates autophagy as a key regulator of autophagosome maturation. Autophagy 12, 1759–1775. doi: 10.1080/15548627.2016.1199300
Skibinska-Kijek, A., Wisniewska, M. B., Gruszczynska-Biegala, J., Methner, A., and Kuznicki, J. (2009). Immunolocalization of STIM1 in the mouse brain. Acta Neurobiol. Exp. 69, 413–428.
Smith, M. D., Harley, M. E., Kemp, A. J., Von Kriegsheim, A., Behrends, C., Wilkinson, S., et al. (2018). CCPG1 is a non-canonical autophagy Cargo receptor essential for ER-Phagy and Pancreatic ER ARTICLE CCPG1 is a non-canonical autophagy Cargo receptor essential for ER-Phagy and pancreatic ER proteostasis. Dev. Cell 44:217-232.e11. doi: 10.1016/j.devcel.2017.11.024
Solowska, J. M., Morfini, G., Falnikar, A., Himes, B. T., Brady, S. T., Huang, D., et al. (2008). Quantitative and functional analyses of spastin in the nervous system: implications for hereditary spastic paraplegia. J. Neurosci. 28, 2147–2157. doi: 10.1523/JNEUROSCI.3159-07.2008
Southall, T. D., Terhzaz, S., Cabrero, P., Chintapalli, V. R., Evans, J. M., Dow, J. A. T., et al. (2006). Novel subcellular locations and functions for secretory pathway Ca 2+ /Mn 2+ -ATPases. Physiol. Genomics 26, 35–45. doi: 10.1152/physiolgenomics.00038.2006
Srivats, S., Balasuriya, D., Pasche, M., Vistal, G., Michael Edwardson, J., Taylor, C. W., et al. (2016). Sigma1 receptors inhibit store-operated Ca2+ entry by attenuating coupling of STIM1 to Orai1. J. Cell Biol. 213, 65–79. doi: 10.1083/jcb.201506022
Steenbergen, R., Nanowski, T. S., Beigneux, A., Kulinski, A., Young, S. G., and Vance, J. E. (2005). Disruption of the phosphatidylserine decarboxylase gene in mice causes embryonic lethality and mitochondrial defects. J. Biol. Chem. 280, 40032–40040. doi: 10.1074/jbc.M506510200
Stefan, C. J., Manford, A. G., Baird, D., Yamada-Hanff, J., Mao, Y., and Emr, S. D. (2011). Osh proteins regulate phosphoinositide metabolism at ER-plasma membrane contact sites. Cell 144, 389–401. doi: 10.1016/j.cell.2010.12.034
Stefano, G., and Brandizzi, F. (2017). Advances in plant ER architecture and dynamics. Plant Physiol. 176, 178–186. doi: 10.1104/pp.17.01261
Stoica, R., De Vos, K. J., Paillusson, S., Mueller, S., Sancho, R. M., Lau, K. F., et al. (2014). ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 5:3996. doi: 10.1038/ncomms4996
Stone, M. C., Rao, K., Gheres, K. W., Kim, S., Tao, J., La Rochelle, C., et al. (2012). Normal Spastin gene dosage is specifically required for Axon regeneration. Cell Rep. 2, 1340–1350. doi: 10.1016/j.celrep.2012.09.032
Straub, S. V., Giovannucci, D. R., and Yule, D. I. (2000). Calcium wave propagation in pancreatic acinar cells: functional interaction of inositol 1,4,5-trisphosphate receptors, ryanodine receptors, and mitochondria. J. Gen. Physiol. 116, 547–560.
Summerville, J., Faust, J., Fan, E., Pendin, D., Daga, A., Formella, J., et al. (2016). The effects of ER morphology on synaptic structure and function in Drosophila melanogaster. J. Cell Sci. 129, 1635–1648. doi: 10.1242/jcs.184929
Sun, E. W., Guillén-Samander, A., Bian, X., Wu, Y., Cai, Y., Messa, M., et al. (2019). Lipid transporter TMEM24/C2CD2L is a Ca 2+ -regulated component of ER–plasma membrane contacts in mammalian neurons. Proc. Natl. Acad. Sci. U.S.A. 116:201820156. doi: 10.1073/pnas.1820156116
Sun, S., Zhang, H., Liu, J., Popugaeva, E., Xu, N. J., Feske, S., et al. (2014). Reduced synaptic STIM2 expression and impaired store-operated calcium entry cause destabilization of mature spines in mutant presenilin mice. Neuron 82, 79–93. doi: 10.1016/j.neuron.2014.02.019
Sytnyk, V. (2003). Trans-Golgi network delivery of synaptic proteins in synaptogenesis. J. Cell Sci. 117, 381–388. doi: 10.1242/jcs.00956
Szabadkai, G., Bianchi, K., Várnai, P., De Stefani, D., Wieckowski, M. R., Cavagna, D., et al. (2006). Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 175, 901–911. doi: 10.1083/jcb.200608073
Szymanski, K. M., Binns, D., Bartz, R., Grishin, N. V., Li, W.-P., Agarwal, A. K., et al. (2007). The lipodystrophy protein seipin is found at endoplasmic reticulum lipid droplet junctions and is important for droplet morphology. Proc. Natl. Acad. Sci. U.S.A. 104, 20890–20895. doi: 10.1073/pnas.0704154104
Takano, T., Wu, M., Nakamuta, S., Naoki, H., Ishizawa, N., Namba, T., et al. (2017). Discovery of long-range inhibitory signaling to ensure single axon formation. Nat. Commun. 8, 1–17. doi: 10.1038/s41467-017-00044-42
Takel, K., Stukenbrok, H., Metcalf, A., Mignery, G. A., Sudhof, T. C., Volpe, P., et al. (1992). Ca2+ stores in Purkinje neurons: Endoplasmic reticulum subcompartments demonstrated by the heterogeneous distribution of the InsP3 receptor, Ca2+-ATPase, and calsequestrin. J. Neurosci. 12, 489–505. doi: 10.1523/jneurosci.12-02-00489.1992
Tanaka, A., Cleland, M. M., Xu, S., Narendra, D. P., Suen, D.-F., Karbowski, M., et al. (2010). Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 191, 1367–1380. doi: 10.1083/jcb.201007013
Tang, T.-S., Slow, E., Lupu, V., Stavrovskaya, I. G., Sugimori, M., Llinas, R., et al. (2005). Disturbed Ca2+ signaling and apoptosis of medium spiny neurons in Huntington’s disease. Proc. Natl. Acad. Sci. U.S.A. 102, 2602–2607. doi: 10.1073/pnas.0409402102
Tang, T.-S., Tu, H., Chan, E. Y. W., Maximov, A., Wang, Z., Wellington, C. L., et al. (2003). Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1. Neuron 39, 227–239. doi: 10.1016/s0896-6273(03)00366-0
Tasseva, G., Bai, H. D., Davidescu, M., Haromy, A., Michelakis, E., and Vance, J. E. (2013). Phosphatidylethanolamine deficiency in mammalian mitochondria impairs oxidative phosphorylation and alters mitochondrial morphology. J. Biol. Chem. 288, 4158–4173. doi: 10.1074/jbc.M112.434183
Terasaki, M. (2018). Axonal endoplasmic reticulum is very narrow. J. Cell Sci. 131:jcs210450. doi: 10.1242/jcs.210450
Terasaki, M., Chen, L. B., and Fujiwara, K. (1986). Microtubules and the endoplasmic reticulum are highly interdependent structures. J. Cell Biol. 103, 1557–1568. doi: 10.1083/JCB.103.4.1557
Terasaki, M., Shemesh, T., Kasthuri, N., Klemm, R. W., Schalek, R., Hayworth, K. J., et al. (2013). Stacked endoplasmic reticulum sheets are connected by helicoidal membrane motifs. Cell 154, 285–296. doi: 10.1016/j.cell.2013.06.031
Terasaki, M., Slater, N. T., Fein, A., Schmidek, A., and Reese, T. S. (1994). Continuous network of endoplasmic reticulum in cerebellar Purkinje neurons. Proc. Natl. Acad. Sci. U.S.A. 91, 7510–7514. doi: 10.1073/pnas.91.16.7510
Teuling, E., Ahmed, S., Haasdijk, E., Demmers, J., Steinmetz, M. O., Akhmanova, A., et al. (2007). Motor neuron disease-associated mutant vesicle-associated membrane protein-associated protein (VAP) B recruits wild-type VAPs into endoplasmic reticulum-derived tubular aggregates. J. Neurosci. 27, 9801–9815. doi: 10.1523/JNEUROSCI.2661-07.2007
Theurey, P., and Rieusset, J. (2017). Mitochondria-associated membranes response to nutrient availability and role in Metabolic diseases. Trends Endocrinol. Metab. 28, 32–45. doi: 10.1016/j.tem.2016.09.002
Theurey, P., Tubbs, E., Vial, G., Jacquemetton, J., Bendridi, N., Chauvin, M. A., et al. (2016). Mitochondria-associated endoplasmic reticulum membranes allow adaptation of mitochondrial metabolism to glucose availability in the liver. J. Mol. Cell Biol. 8, 129–143. doi: 10.1093/jmcb/mjw004
Thiel, K., Heier, C., Haberl, V., Thul, P. J., Oberer, M., Lass, A., et al. (2013). The evolutionarily conserved protein CG9186 is associated with lipid droplets, required for their positioning and for fat storage. J. Cell Sci. 126, 2198–2212. doi: 10.1242/jcs.120493
Thivolet, C., Vial, G., Cassel, R., Rieusset, J., and Madec, A. M. (2017). Reduction of endoplasmic reticulum-mitochondria interactions in beta cells from patients with type 2 diabetes. PLoS One 12:e0182027. doi: 10.1371/journal.pone.0182027
Thomas, A. C., Williams, H., Setõ-Salvia, N., Bacchelli, C., Jenkins, D., O’Sullivan, M., et al. (2014). Mutations in SNX14 cause a distinctive autosomal-recessive cerebellar ataxia and intellectual disability syndrome. Am. J. Hum. Genet. 95, 611–621. doi: 10.1016/j.ajhg.2014.10.007
Tisdale, S., and Pellizzoni, L. (2015). Disease mechanisms and therapeutic approaches in spinal muscular atrophy. J. Neurosci. 35, 8691–8700. doi: 10.1523/JNEUROSCI.0417-15.2015
Toker, A. (2002). Phosphoinositides and signal transduction. Cell. Mol. Life Sci. Mol. Life Sci. 59, 761–779. doi: 10.1007/s00018-002-8465-z
Trotta, N., Orso, G., Rossetto, M. G., Daga, A., and Broadie, K. (2004). The hereditary spastic paraplegia gene, spastin, regulates microtubule stability to modulate synaptic structure and function. Curr. Biol. 14, 1135–1147. doi: 10.1016/j.cub.2004.06.058
Tsai, S. Y. A., Pokrass, M. J., Klauer, N. R., Nohara, H., and Su, T. P. (2015). Sigma-1 receptor regulates Tau phosphorylation and axon extension by shaping p35 turnover via myristic acid. Proc. Natl. Acad. Sci. U.S.A. 112, 6742–6747. doi: 10.1073/pnas.1422001112
Tsukita, S., and Ishikawa, H. (1976). Three-dimensional distribution of smooth endoplasmic reticulum in myelinated axons. J. Electron Microsc. 25, 141–149. doi: 10.1093/oxfordjournals.jmicro.a050013
Tu, H., Nelson, O., Bezprozvanny, A., Wang, Z., Lee, S.-F., Hao, Y.-H., et al. (2006). Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell 126, 981–993. doi: 10.1016/J.CELL.2006.06.059
Ueno, S., Maruki, Y., Nakamura, M., Tomemori, Y., Kamae, K., Tanabe, H., et al. (2001). The gene encoding a newly discovered protein, chorein, is mutated in chorea-acanthocytosis. Nat. Genet. 28, 121–122. doi: 10.1038/88825
Vajda, F., Jordi, N., Dalkara, D., Joly, S., Christ, F., Tews, B., et al. (2015). Cell type-specific Nogo-A gene ablation promotes axonal regeneration in the injured adult optic nerve. Cell Death Differ. 22, 323–335. doi: 10.1038/cdd.2014.147
Valadas, J. S., Esposito, G., Vandekerkhove, D., Miskiewicz, K., Deaulmerie, L., Raitano, S., et al. (2018). ER lipid defects in neuropeptidergic neurons impair sleep patterns in Parkinson’s disease. Neuron 98:1155-1169.e6. doi: 10.1016/j.neuron.2018.05.022
Valm, A. M., Cohen, S., Legant, W. R., Melunis, J., Hershberg, U., Wait, E., et al. (2017). Applying systems-level spectral imaging and analysis to reveal the organelle interactome. Nature 546, 162–167. doi: 10.1038/nature22369
Van Cauwenberghe, C., Van Broeckhoven, C., and Sleegers, K. (2016). The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet. Med. 18, 421–430. doi: 10.1038/gim.2015.117
Van De Leemput, J., Chandran, J., Knight, M. A., Holtzclaw, L. A., Scholz, S., Cookson, M. R., et al. (2007). Deletion at ITPR1 underlies ataxia in mice and spinocerebellar ataxia 15 in humans. PLoS Genet. 3:e0030108. doi: 10.1371/journal.pgen.0030108
Vance, J. E. (2014). MAM (mitochondria-associated membranes) in mammalian cells: lipids and beyond. Biochim. Biophys. Acta - Mol. Cell Biol. Lipids 1841, 595–609. doi: 10.1016/j.bbalip.2013.11.014
Vanoye, C. G., Sakakura, M., Follis, R. M., Trevisan, A. J., Narayan, M., Li, J., et al. (2019). Peripheral myelin protein 22 modulates store-operated calcium channel activity, providing insights into Charcot-Marie-Tooth disease etiology. J. Biol. Chem. 294, 12054–12065. doi: 10.1074/jbc.RA118.006248
Vargas, M. E., Yamagishi, Y., Tessier-Lavigne, M., and Sagasti, A. (2015). Live imaging of Calcium dynamics during Axon degeneration reveals two functionally distinct phases of calcium influx. J. Neurosci. 35, 15026–15038. doi: 10.1523/jneurosci.2484-15.2015
Verhoeven, K., Claeys, K. G., Züchner, S., Schröder, J. M., Weis, J., Ceuterick, C., et al. (2006). MFN2 mutation distribution and genotype/phenotype correlation in Charcot-Marie-Tooth type 2. Brain 129, 2093–2102. doi: 10.1093/brain/awl126
Verkhovsky, A. B., Klopfenstein, D. R., and Hauri, H.-P. (2005). Phosphorylation Controls CLIMP-63–mediated anchoring of the Endoplasmic reticulum to microtubules. Mol. Biol. Cell 16, 1928–1937. doi: 10.1091/mbc.E04
Verkhratsky, A., and Fernyhough, P. (2008). Mitochondrial malfunction and Ca2+ dyshomeostasis drive neuronal pathology in diabetes. Cell Calcium 44, 112–122. doi: 10.1016/j.ceca.2007.11.010
Vieira, M. N. N., Lyra e Silva, N. M., Ferreira, S. T., and De Felice, F. G. (2017). Protein tyrosine phosphatase 1B (PTP1B): a potential target for Alzheimer’s therapy? Front. Aging Neurosci. 9:7. doi: 10.3389/fnagi.2017.00007
Villegas, R., Martinez, N. W., Lillo, J., Pihan, P., Hernandez, D., Twiss, J. L., et al. (2014). Calcium release from intra-axonal endoplasmic reticulum leads to axon degeneration through mitochondrial dysfunction. J. Neurosci. 34, 7179–7189. doi: 10.1523/JNEUROSCI.4784-13.2014
Voeltz, G. K., Prinz, W. A., Shibata, Y., Rist, J. M., and Rapoport, T. A. (2006). A class of membrane proteins shaping the tubular Endoplasmic reticulum. Cell 124, 573–586. doi: 10.1016/j.cell.2005.11.047
Wakil, S. M., Alhissi, S., Dossari, H., Al Alqahtani, A., Shibin, S., Melaiki, B. T., et al. (2019). Truncating ARL6IP1 variant as the genetic cause of fatal complicated hereditary spastic paraplegia. BMC Med. Genet. 20:119. doi: 10.1186/s12881-019-0851-6
Wallen, Z. D., Chen, H., Hill-Burns, E. M., Factor, S. A., Zabetian, C. P., and Payami, H. (2018). Plasticity-related gene 3 (LPPR1) and age at diagnosis of Parkinson disease. Neurol. Genet. 4:e271. doi: 10.1212/nxg.0000000000000271
Wang, H., Becuwe, M., Housden, B. E., Chitraju, C., Porras, A. J., Graham, M. M., et al. (2016). Seipin is required for converting nascent to mature lipid droplets. eLife 5:e16582. doi: 10.7554/elife.16582
Wang, S., Tukachinsky, H., Romano, F. B., and Rapoport, T. A. (2016). Cooperation of the ER-shaping proteins atlastin, lunapark, and reticulons to generate a tubular membrane network. eLife 5:e18605. doi: 10.7554/eLife.18605
Wang, L., Gao, J., Liu, J., Siedlak, S. L., Torres, S., Fujioka, H., et al. (2018). Mitofusin 2 regulates Axonal transport of Calpastatin to prevent neuromuscular synaptic elimination in Skeletal muscles. Cell Metab. 28:400-414.e8. doi: 10.1016/j.cmet.2018.06.011
Wang, Y., Metz, J., Costello, J. L., Passmore, J., Schrader, M., Schultz, C., et al. (2018). Intracellular redistribution of neuronal peroxisomes in response to ACBD5 expression. PLoS One 13:e0209507. doi: 10.1371/journal.pone.0209507
Wang, T., Liu, Y., Xu, X.-H., Deng, C.-Y., Wu, K.-Y., Zhu, J., et al. (2011). Lgl1 activation of Rab10 promotes Axonal membrane trafficking underlying neuronal polarization. Dev. Cell 21, 431–444. doi: 10.1016/j.devcel.2011.07.007
Wang, X., Su, B., Lee, H.-G., Li, X., Perry, G., Smith, M. A., et al. (2009). Impaired balance of Mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 29, 9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009
Warburg, M., Sjö, O., Fledelius, H. C., and Pedersen, S. A. (1993). Autosomal recessive microcephaly, microcornea, congenital cataract, mental retardation, optic atrophy, and hypogenitalism. Micro syndrome. Am. J. Dis. Child. 147, 1309–1312. doi: 10.1001/archpedi.1993.02160360051017
Waterman-Storer, C. M., and Salmon, E. D. (1998). Endoplasmic reticulum membrane tubules are distributed by microtubules in living cells using three distinct mechanisms. Curr. Biol. 8, 798–806.
Weber-Boyvat, M., Kentala, H., Lilja, J., Vihervaara, T., Hanninen, R., Zhou, Y., et al. (2015). OSBP-related protein 3 (ORP3) coupling with VAMP-associated protein A regulates R-Ras activity. Exp. Cell Res. 331, 278–291. doi: 10.1016/j.yexcr.2014.10.019
Wei, S., Soh, S. L.-Y., Xia, J., Ong, W.-Y., Pang, Z. P., and Han, W. (2014). Motor neuropathy-associated mutation impairs Seipin functions in neurotransmission. J. Neurochem. 129, 328–338. doi: 10.1111/jnc.12638
Wiessner, M., Roos, A., Munn, C. J., Viswanathan, R., Whyte, T., Cox, D., et al. (2017). Mutations in INPP5K, encoding a Phosphoinositide 5-Phosphatase, cause congenital muscular dystrophy with cataracts and mild cognitive impairment. Am. J. Hum. Genet. 100, 523–536. doi: 10.1016/j.ajhg.2017.01.024
Wilhelm, L. P., Wendling, C., Védie, B., Kobayashi, T., Chenard, M., Tomasetto, C., et al. (2017). STARD 3 mediates endoplasmic reticulum-to-endosome cholesterol transport at membrane contact sites. EMBO J. 36, 1412–1433. doi: 10.15252/embj.201695917
Wilkinson, S. (2019). ER-phagy: shaping up and destressing the endoplasmic reticulum. FEBS J. 286, 2645–2663. doi: 10.1111/febs.14932
Willis, D., Ka, W. L., Zheng, J. Q., Chang, J. H., Smit, A., Kelly, T., et al. (2005). Differential transport and local translation of cytoskeletal, injury-response, and neurodegeneration protein mRNAs in axons. J. Neurosci. 25, 778–791. doi: 10.1523/JNEUROSCI.4235-04.2005
Windpassinger, C., Auer-Grumbach, M., Irobi, J., Patel, H., Petek, E., Hörl, G., et al. (2004). Heterozygous missense mutations in BSCL2 are associated with distal hereditary motor neuropathy and Silver syndrome. Nat. Genet. 36, 271–276. doi: 10.1038/ng1313
Wood, J. D., Landers, J. A., Bingley, M., McDermott, C. J., Thomas-McArthur, V., Gleadall, L. J., et al. (2006). The microtubule-severing protein Spastin is essential for axon outgrowth in the zebrafish embryo. Hum. Mol. Genet. 15, 2763–2771. doi: 10.1093/hmg/ddl212
Woźniak, M. J., Bola, B., Brownhill, K., Yang, Y.-C., Levakova, V., and Allan, V. J. (2009). Role of kinesin-1 and cytoplasmic dynein in Endoplasmic reticulum movement in VERO cells. J. Cell Sci. 122, 1979–1989. doi: 10.1242/jcs.041962
Wu, B., Yamaguchi, H., Lai, F. A., and Shen, J. (2013). Presenilins regulate calcium homeostasis and presynaptic function via ryanodine receptors in hippocampal neurons. Proc. Natl. Acad. Sci. U.S.A. 110, 15091–15096. doi: 10.1073/pnas.1304171110
Wu, H., Carvalho, P., and Voeltz, G. K. (2018). Here, there, and everywhere: the importance of ER membrane contact sites. Science. 361:eaan5835. doi: 10.1126/science.aan5835
Wu, J., Ryskamp, D. A., Liang, X., Egorova, P., Zakharova, O., Hung, G., et al. (2016). Enhanced store-operated calcium entry leads to striatal synaptic loss in a Huntington’s disease mouse model. J. Neurosci. 36, 125–141. doi: 10.1523/JNEUROSCI.1038-15.2016
Wu, Q., Sun, X., Yue, W., Lu, T., Ruan, Y., Chen, T., et al. (2016). RAB18, a protein associated with Warburg Micro syndrome, controls neuronal migration in the developing cerebral cortex. Mol. Brain 9, 1–12. doi: 10.1186/s13041-016-0198-192
Wu, Y., Whiteus, C., Xu, C. S., Hayworth, K. J., Weinberg, R. J., Hess, H. F., et al. (2017). Contacts between the endoplasmic reticulum and other membranes in neurons. Proc. Natl. Acad. Sci. U.S.A. 114, E4859–E4867. doi: 10.1073/pnas.1701078114
Xu, D., Li, Y., Wu, L., Li, Y., Zhao, D., Yu, J., et al. (2018). Rab18 promotes lipid droplet (LD) growth by tethering the ER to LDs through SNARE and NRZ interactions. J. Cell Biol. 217, 975–995. doi: 10.1083/jcb.201704184
Xu, N., Zhang, S. O., Cole, R. A., McKinney, S. A., Guo, F., Haas, J. T., et al. (2012). The FATP1-DGAT2 complex facilitates lipid droplet expansion at the ER-lipid droplet interface. J. Cell Biol. 198, 895–911. doi: 10.1083/jcb.201201139
Xu, X. H., Deng, C. Y., Liu, Y., He, M., Peng, J., Wang, T., et al. (2014). MARCKS regulates membrane targeting of Rab10 vesicles to promote axon development. Cell Res. 24, 576–594. doi: 10.1038/cr.2014.33
Yalçın, B., Zhao, L., Stofanko, M., O’Sullivan, N. C., Kang, Z. H., Roost, A., et al. (2017). Modeling of axonal endoplasmic reticulum network by spastic paraplegia proteins. eLife 6:e23882. doi: 10.7554/eLife.23882
Yamamoto, Y., Yoshida, A., Miyazaki, N., Iwasaki, K., and Sakisaka, T. (2014). Arl6IP1 has the ability to shape the mammalian ER membrane in a reticulon-like fashion. Biochem. J. 458, 69–79. doi: 10.1042/BJ20131186
Yang, Z., Huh, S. U., Michelle Drennan, J., Kathuria, H., Martinez, J. S., Tsuda, H., et al. (2012). Drosophila Vap-33 is required for axonal localization of dscam isoforms. J. Neurosci. 32, 17241–17250. doi: 10.1523/JNEUROSCI.2834-12.2012
Yeshaw, W. M., van der Zwaag, M., Pinto, F., Lahaye, L. L., Faber, A. I. E., Gómez-Sánchez, R., et al. (2019). Human VPS13A is associated with multiple organelles and influences mitochondrial morphology and lipid droplet motility. eLife 8:e43561. doi: 10.7554/eLife.43561
Yu, H., Liu, Y., Gulbranson, D. R., Paine, A., Rathore, S. S., and Shen, J. (2016). Extended synaptotagmins are Ca 2+ -dependent lipid transfer proteins at membrane contact sites. Proc. Natl. Acad. Sci. U.S.A. 113, 4362–4367. doi: 10.1073/pnas.1517259113
Zajaczkowska̧, R., Kocot-Kȩpska, M., Leppert, W., Wrzosek, A., Mika, J., and Wordliczek, J. (2019). Mechanisms of chemotherapy-induced peripheral neuropathy. Int. J. Mol. Sci. 20:1451. doi: 10.3390/ijms20061451
Zhang, C., Wu, B., Beglopoulos, V., Wines-Samuelson, M., Zhang, D., Dragatsis, I., et al. (2009). Presenilins are essential for regulating neurotransmitter release. Nature 460, 632–636. doi: 10.1038/nature08177
Zhang, X., Tee, Y. H., Heng, J. K., Zhu, Y., Hu, X., Margadant, F., et al. (2010a). Kinectin-mediated endoplasmic reticulum dynamics supports focal adhesion growth in the cellular lamella. J. Cell Sci. 123, 3901–3912. doi: 10.1242/jcs.069153
Zhang, X., Zhou, J. Y., Chin, M. H., Schepmoes, A. A., Petyuk, V. A., Weitz, K. K., et al. (2010b). Region-specific protein abundance changes in the brain of MPTP-Lnduced parkinson’s disease mouse model. J. Proteome Res. 9, 1496–1509. doi: 10.1021/pr901024z
Zhao, B., Pan, B., Shen, S., Sun, X., Hou, Z., Yan, R., et al. (2013). Diabetes-induced central Neuritic dystrophy and cognitive deficits are associated with the formation of Oligomeric reticulon-3 via oxidative stress. J. Biol. Chem. 288, 15590–15599. doi: 10.1074/jbc.M112.440784
Zhao, X., Alvarado, D., Rainier, S., Lemons, R., Hedera, P., Weber, C. H., et al. (2001). Mutations in a newly identified GTPase gene cause autosomal dominant hereditary spastic paraplegia. Nat. Genet. 29, 326–331. doi: 10.1038/ng758
Zhao, Y., Tan, W., Sheng, W., and Li, X. (2016). Identification of biomarkers associated with Alzheimer’s disease by bioinformatics analysis. Am. J. Alzheimers. Dis. Other Demen. 31, 163–168. doi: 10.1177/1533317515588181
Zhou, X., He, Y., Huang, X., Guo, Y., Li, D., and Hu, J. (2018). Reciprocal regulation between lunapark and atlastin facilitates ER three-way junction formation. Protein Cell. 10, 510–525. doi: 10.1007/s13238-018-0595-597
Zhou, Y., Carmona, S., Muhammad, A. K. M. G., Bell, S., Landeros, J., Vazquez, M., et al. (2019). Restoring mitofusin balance prevents axonal degeneration in a Charcot-Marie-Tooth type 2A model. J. Clin. Invest. 129, 1756–1771. doi: 10.1172/JCI124194
Zhu, P. P., Patterson, A., Lavoie, B., Stadler, J., Shoeb, M., Patel, R., et al. (2003). Cellular localization, oligomerization, and membrane association of the hereditary spastic paraplegia 3A (SPG3A) protein atlastin. J. Biol. Chem. 278, 49063–49071. doi: 10.1074/jbc.M306702200
Zhu, P. P., Soderblom, C., Tao-Cheng, J. H., Stadler, J., and Blackstone, C. (2006). SPG3A protein atlastin-1 is enriched in growth cones and promotes axon elongation during neuronal development. Hum. Mol. Genet. 15, 1343–1353. doi: 10.1093/hmg/ddl054
Zhu, Y., Zhang, G., Lin, S., Shi, J., Zhang, H., and Hu, J. (2018). Sec61β facilitates the maintenance of endoplasmic reticulum homeostasis by associating microtubules. Protein Cell 9, 616–628. doi: 10.1007/s13238-017-0492-495
Züchner, S., Mersiyanova, I. V., Muglia, M., Bissar-Tadmouri, N., Rochelle, J., Dadali, E. L., et al. (2004). Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 36, 449–451. doi: 10.1038/ng1341
Keywords: endoplasmic reticulum, hereditary spastic paraplegia, axonal transport, neurodegeneration, smooth ER, organelle contact sites, calcium stores
Citation: Öztürk Z, O’Kane CJ and Pérez-Moreno JJ (2020) Axonal Endoplasmic Reticulum Dynamics and Its Roles in Neurodegeneration. Front. Neurosci. 14:48. doi: 10.3389/fnins.2020.00048
Received: 23 October 2019; Accepted: 13 January 2020;
Published: 29 January 2020.
Edited by:
Tomas Luis Falzone, National Scientific and Technical Research Council (CONICET), ArgentinaReviewed by:
Christian Andreas Huebner, Friedrich Schiller University Jena, GermanyYang Hu, Stanford University, United States
Copyright © 2020 Öztürk, O’Kane and Pérez-Moreno. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cahir J. O’Kane, Yy5va2FuZUBnZW4uY2FtLmFjLnVr; Juan José Pérez-Moreno, anVhbmpvLnBlcmV6LW1vcmVub0BnZW4uY2FtLmFjLnVr