 Jian Lu
Jian Lu Yi Wang1
Yi Wang1 Jing Wu
Jing Wu Yusi Duan
Yusi Duan Haifang Zhang
Haifang Zhang Hong Du
Hong Du- 1Department of Clinical Laboratory, The Second Affiliated Hospital of Soochow University, Suzhou, Jiangsu, China
- 2Department of Laboratory Medicine, The Affiliated Guangji Hospital of Soochow University, Suzhou Mental Health Center, Suzhou, Jiangsu, China
Rheumatoid arthritis (RA) is a severe, chronic autoimmune disease affecting approximately 1% of the global population. Research has demonstrated that microorganisms play a crucial role in the onset and progression of RA. This indicates that the disruption of immune homeostasis may originate from mucosal sites, such as the gut and oral cavity. In the intestines of patients in the preclinical stage of RA, an increased abundance of Prevotella species with a strong association to the disease was observed. In the oral cavity, infections by Porphyromonas gingivalis and Aggregatibacter actinomycetemcomitans can mediate the production of anti-citrullinated protein antibodies (ACPAs), potentially contributing to RA pathogenesis. Nevertheless, no single bacterial species has been consistently identified as the primary driver of RA. This review will discuss the connection between gut and oral bacteria in the development of arthritis. Additionally, it explores the role of bacterial extracellular vesicles (bEVs) in inducing inflammation and their potential pathogenic roles in RA.
1 Introduction
Rheumatoid Arthritis (RA) is a clinically significant, refractory autoimmune disease, characterized by uncontrolled inflammation and cartilage destruction in the affected joints, eventually leading to joint dysfunction and deformity (1). Current therapeutic regimens include glucocorticoids, non-steroidal anti-inflammatory drugs (NSAIDs), disease-modifying anti-rheumatic drugs (DMARDs), and small molecule inhibitors. Although these medications can significantly ameliorate patients’ clinical symptoms, they are not curative.
Currently, more than 60% of RA patients do not achieve true remission, placing a substantial burden on social health and the economy. The pathogenesis of RA is complex, and its exact etiology remains unclear, with factors such as smoking, hormones, gut microorganisms, and infections all associated with RA development. At its core, RA is driven by autoimmunity, where the immune system mistakenly attacks self-tissues, particularly synovial joints. This autoimmune response is closely linked to microbiome dysregulation, which is pivotal in shaping immune tolerance and systemic inflammation. The interplay between autoimmunity and microbiome dysregulation provides a critical framework for understanding RA pathogenesis (2). Notably, the dysregulation of the gut and oral microbiota has emerged as a key contributor to RA pathogenesis, potentially through molecular mimicry, epitope spreading, and the activation of autoreactive T and B cells. Recent studies have shown that IgA antibodies against cyclic citrullinated peptides can be detected in RA patients several years before the disease onset. IgA antibodies are primarily found in mucous membranes, indicating that RA could potentially originate in areas such as the intestinal tract, oral cavity, and lung mucosa. Individuals with an elevated risk of developing RA are known to exhibit signs of persistent systemic and mucosal inflammation. Current research links the progression of mucosal dysbiosis, inflammatory processes, and the production of autoantibodies with subsequent phases in the emergence of systemic autoimmunity (3–6).
Antimicrobial drug therapy, such as minocycline or salazosulfadiazine, has proven effective in some RA patients, indicating a close association between the intestinal and oral microbiota and the onset and progression of RA (7, 8). Furthermore, certain medications that modulate intestinal microbiota homeostasis, including biotics (prebiotics, probiotics, and postbiotics) and some traditional Chinese medicine, have demonstrated significant efficacy in RA, further supporting the notion that microbial factors may be important pathogenic elements and therapeutic targets in the development and progression of RA (9–12). Therefore, Understanding the regulatory mechanisms of pathogenic microorganisms in the pathogenesis of RA is crucial for developing new therapeutic strategies and preventive measures. Intervening with relevant pathogenic microorganisms can potentially regulate immune system imbalances, reduce systemic inflammation, and block the development of RA. This study aims to summarize the pathogenic roles and mechanisms of these microorganisms in RA. It will cover both conventional and novel mechanisms, particularly focusing on how bacterial extracellular vesicles (bEVs) modulate the inflammatory response and contribute to the progression of RA.
2 Bacterial factors in RA development
2.1 Intestinal flora imbalance
The imbalance in gut microbiota plays an important role in the pathogenesis of autoimmune diseases (13, 14). In RA, studies have demonstrated that the gut microbiome of patients exhibits an ecological imbalance, characterized by specific microbial features associated with increased intestinal permeability, inflammatory cell infiltration, and the production of anti-citrullinated protein antibodies (ACPAs). In recent years, researchers have employed 16S rRNA and Next Generation Sequencing (NGS) technologies to conduct comprehensive studies on the gut microbiota of RA patients and healthy individuals. The study results demonstrated that the microbial composition in the gut of pre-clinical RA patients changed significantly, coupled with a marked reduction in microbial diversity. Notably, pre-clinical RA patients exhibited a pronounced enrichment of prevotellaceae (15, 16). Similarly, an increased abundance of Prevotella copri and a decreased abundance of Bacteroides species have also been observed in American patients with new-onset RA (17). This pattern has also been noted in Japanese and European cohorts (18). Nii and Maeda et al. revealed that Prevotella copri can induce cytokines associated with Th17 cells, such as IL-6 and IL-23, promoting arthritis development in mice. Moreover, this increase in Prevotella correlates with Th17 cell-mediated mucosal inflammation, consistent with its capacity to drive Th17 immune responses in vitro (19). Notably, anti-rheumatic drug-mediated disease remission was observed to increase microbial richness and diversity (20, 21).
Apart from Prevotella, the abundance of other gut bacteria has also been confirmed to change significantly in patients with RA. Jeong et al. underscored significant differences in microbial distributions from phylum to genus levels between healthy individuals and early RA patients. The phylum Bacteroidetes was enriched in early RA patients, whereas Actinobacteria, such as genus Collinsella, were more prevalent among healthy subjects (22). Zhang et al. observed an elevated presence of Lactobacillus salivarius in the gut, teeth, and saliva of RA patients, alongside a decrease in Haemophilus species (21). Fusobacterium nucleatum (F. nucleatum) was also significantly enriched in RA patients and positively correlated with inflammatory cytokines and disease activity, suggesting its potential involvement in RA inflammation (23). Notably, the intestinal microbiota in RA patients exhibits dynamic changes across different stages of the disease. Collinsella aerofaciens, which has been associated with severe arthritis in experiments, was significantly elevated in the early RA stage, potentially compromising gut barrier integrity and triggering clinical arthritis. In the later stages, specific microbes such as Veillonella parvula, Eggerthella lenta, and Bifidobacterium longum were elevated and associated with increased gut permeability and inflammation (24). Gut bacteriome alterations in RA are summarized in Table 1.
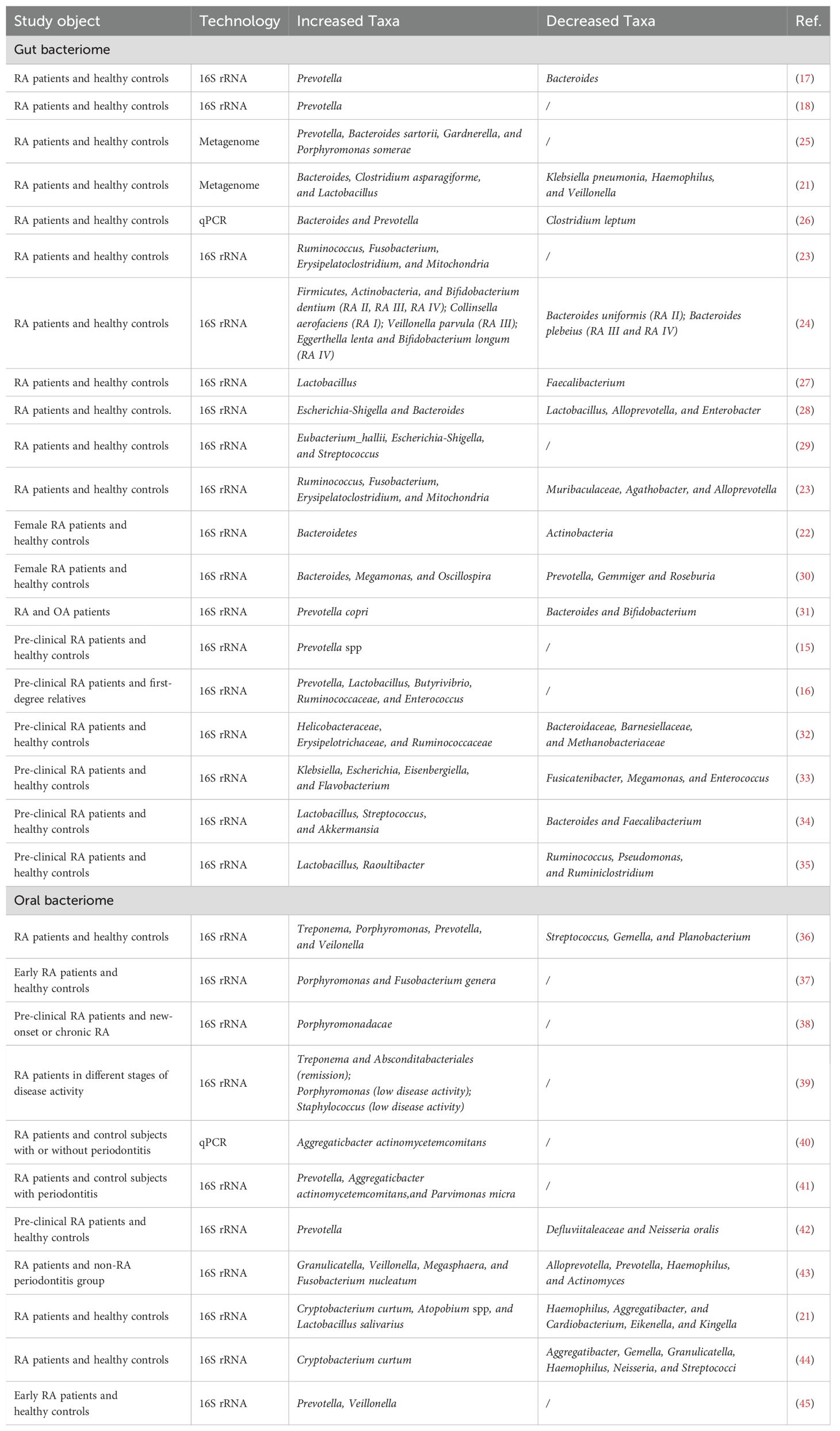
Table 1. Alterations in the bacteriome in RA.
2.2 Oral pathogenic bacterial infection
The initial inflammation in both RA and periodontitis is caused by the activation of intrinsic cells, monocyte macrophages, and dendritic cells. Both diseases exhibit enhanced bone resorption (46). The development of RA may accelerate periodontal tissue destruction, with the extent of periodontal damage being positively correlated with the severity of RA disease activity. Animal experiments have demonstrated that the development of periodontitis exacerbates the progression of RA (47). Notably, the elimination of periodontal lesions significantly attenuated RA disease progression, even in the absence of targeted treatment, suggesting that periodontal disease and RA may share similar pathogenic mechanisms (48, 49).
Porphyromonas gingivalis (P. gingivalis) is a pathogen that colonizes the oral cavity and is commonly associated with periodontal diseases. However, its pathogenic effects extend beyond the oral cavity. P. gingivalis can also contribute to the occurrence of various diseases such as Alzheimer’s disease, colon cancer, and diabetes via the digestive tract and bloodstream (50, 51). In RA, infection with P. gingivalis is closely associated with disease progression (36–38). Studies indicate that RA patients have a significantly higher likelihood of developing periodontal disease compared to non-RA patients, exhibiting more severe periodontal disease progression and a significant positive correlation with arthritis activity (52). Moreover, the majority of RA patients exhibit high levels of oral pathogenic bacterial DNA in their oral plaque and joint synovial fluid. P. gingivalis DNA is among the most easily detectable bacterial nucleic acids, showing a significantly increased detection rate compared to the control group (53, 54). Furthermore, it has been demonstrated that P. gingivalis can exacerbate T cell-driven arthritis through the induction of an antigen-specific Th17 response (55).
Citrullination, an essential post-translational protein modification, is primarily catalyzed by peptidylarginine deiminase (PAD) enzymes. PAD enzymes convert arginine residues on protein peptides to citrulline, which is targeted by RA-specific autoantibodies, thus stimulating self-reactive T cells and exacerbating inflammatory damage in the joints (56). P. gingivalis, a bacterium closely linked to the onset and progression of RA, possesses a unique enzyme known as P. gingivalis peptidylarginine deiminase (PPAD). This enzyme can induce citrullination in both self and host proteins. It is demonstrated that 33% of RA patients exhibit increased reactivity to anti-citrullinated PPAD (anti-cit-PPAD) antibodies prior to clinical disease onset. Furthermore, 77% of RA patients display anti-citrulline-specific immune responses to PPAD-derived peptides. Furthermore, studies have confirmed a correlation between the levels of anti-PPAD antibodies and ACPAs. These findings suggest that P. gingivalis may significantly contribute to RA pathogenesis and progression by promoting ACPAs production (57). In the collagen-induced arthritis (CIA) model, infection with P. gingivalis resulted in earlier onset, a more severe course, and enhanced disease progression. Moreover, compared to the wild-type strain, the ability of PPAD-deficient P. gingivalis to promote disease progression in RA model mice is significantly reduced (58). In addition to inducing protein citrullination, P. gingivalis may contribute to the progression of RA by affecting the intestinal immune system and gut microbiota composition. Fecal microbiota transplantation (FMT) from P. gingivalis-inoculated experimental arthritis mice resulted in more severe joint inflammation compared to FMT from control mice (59). Aggregatibacter actinomycetemcomitans (A.a.) is another pathogenic bacterium that plays a significant role in periodontal disease. This bacterium is notably enriched in the oral cavity of RA patients and has been associated with disease activity (40, 60). While A.a. does not have the ability to directly citrullinate proteins, it can induce high expression of PAD enzymes in neutrophils, thereby promoting self-antigen citrullination, triggering the formation of ACPAs, and exacerbating the autoimmune response in RA (61). Studies have demonstrated that patients infected with A.a. exhibit positive cyclic citrullinated peptide (CCP) antibodies and that A.a. infection is associated with the formation of these antibodies (60). However, the role and mechanisms of A.a. in the onset and development of RA still require further investigation. While early studies propose bidirectional interactions between periodontitis and RA, direct causal evidence linking P. gingivalis and A.a. induced periodontitis to RA progression in murine models is absent. Instead, findings predominantly support RA-driven exacerbation of periodontitis severity (62). This discrepancy may arise from experimental design limitations, such as inducing periodontitis after RA onset in animal studies, which could confound observational outcomes. Additionally, short experimental timelines and inherent differences between animal models and human disease complexity limit translatability. Thus, rigorous longitudinal studies are essential to clarify oral microbiota’s role in RA progression. Oral bacteriome alterations in RA are summarized in Table 1.
3 Pathogenic mechanisms
3.1 Metabolic dysregulation
Gut microbiota dysbiosis has been shown to alter the synthesis of microbial metabolites, leading to immune and metabolic imbalances. The gut microbiota produce Short-Chain Fatty Acids (SCFAs) such as acetate, propionate, and butyrate, and the modulation of these metabolites is closely linked to the onset of autoimmune diseases (63). In animal models of arthritis, SCFAs, particularly butyrate, have been shown to effectively inhibit osteoclast differentiation and prevent bone loss (64, 65). By influencing gene expression, butyrate can promote the differentiation of Treg cells, suppress Th17 cells, down-regulate pro-inflammatory cytokine production, and maintain immune homeostasis (66–68). In addition, butyrate also increases serotonin-derived 5-HIAA, directly activates regulatory B cells (Bregs), and inhibits germinal center B cell and plasmablast differentiation (65). Studies indicate that patients with RA exhibit a significant disruption in butyrate metabolism in the gut, leading to markedly decreased circulating butyrate levels (69). Notably, butyrate metabolism is associated with ACPAs production. Research suggests that the proportion of bacteria involved in butyrate metabolism negatively correlates with ACPAs titers and affected joint counts (66). Due to its ability to modulate immune responses via the Treg/IL-10/Th17 axis, butyrate administration mitigates joint inflammation and bone destruction in mice (70). Thus, gut microbiota-mediated butyrate metabolism might play a pivotal role in RA inflammation, suggesting that targeting butyrate metabolism could be a potential strategy for the clinical treatment of RA.
Additionally, abnormal tryptophan metabolism has been closely associated with the disruption of immune tolerance, potentially triggering the onset of autoimmune diseases such as RA (33, 71). Tryptophan, an essential amino acid, is primarily produced through the breakdown of food by lactobacilli and bifidobacteria and is further metabolized by intestinal bacteria into indole metabolites in the colon (72, 73). Research indicates that indole maintains epithelial cell structure and function, promotes goblet cell differentiation and mucin production, thereby enhancing intestinal barrier function and reducing inflammatory responses (74). Indole acetaldehyde and indole-3-acetic acid can induce CD4+ T cell differentiation into Treg cells through aryl hydrocarbon receptors. Additionally, indole-3-acetic acid can inhibit Th17 cell polarization (75, 76). Metabolomic analysis of fecal samples from patients with RA and healthy individuals revealed significantly decreased levels of downstream tryptophan metabolites in the feces of RA patients, including serotonin, xanthurenic acid, and 3-hydroxyanthranilic acid (3-HAA) (33). Xanthurenic acid exhibits immunosuppressive effects (77), while 3-HAA can inhibit macrophage inflammatory responses (78). Notably, tryptophan metabolite levels in the synovial fluid of RA patients are significantly lower than those in osteoarthritis patients (79), suggesting that tryptophan metabolism may play a crucial role in RA development, with intestinal dysbiosis potentially being a key factor in its onset and progression.
3.2 Molecular mimicry
Research indicates that some pathogenic microorganisms express homologous proteins similar to host proteins, leading to host immune imbalance under specific circumstances, which plays a critical role in the pathogenesis of autoimmune disorders such as RA. One aspect of molecular mimicry in RA involves the citrullination process, catalyzed by PPAD from P. gingivalis, a bacterium associated with periodontal disease and RA. PPAD catalyzes the citrullination of proteins such as vimentin, fibrinogen, and α-enolase, which are autoantigens in RA. This process results in the production of citrullinated proteins recognized by autoreactive T cells, leading to the generation of ACPAs, unique to RA (80). Additionally, microorganisms possess antigenic epitopes similar or identical to those of human cells. This similarity can trigger immune responses against these antigens upon infection. For instance, bacteria such as Escherichia coli and Klebsiella pneumoniae produce peptides like L-ASNase67-81, a segment of bacterial L-asparaginase, which can activate HLA-DRB10401-restricted T cells in early RA patients. This activation leads to the expression of CD154 and the production of cytokines such as IL-2, IL-17A/F, and IFN-γ, crucial in disease progression (81). Moreover, Peptides presented by HLA-DR from N-acetylglucosamine-6-sulfatase and filamin A show significant sequence similarity to epitopes from Prevotella sp. and Butyricimonas sp., which are targeted by T-cells and B-cells as auto-antigens in over half of RA patients. This similarity indicates a crucial role in RA development by triggering autoimmune responses to bacterial antigens (82). Similarly, shared sequences between Collinsella and DRB10401 suggest that Collinsella may also induce RA via molecular mimicry (20). These autoimmune epitopes are prominently found in bacterial species of the Firmicutes and Proteobacteria phyla, potentially having a greater impact on the disease in genetically susceptible individuals (83). Persistent colonization of bacteria with cross-reactive epitopes in hosts with high-risk Human Leukocyte Antigen (HLA) genes could lead to the sustained activation of auto-reactive T cells in the gut, contributing to RA development.
3.3 Altered intestinal permeability
Studies have indicated that disruption of the gut barrier, such as through apoptosis of intestinal epithelial cells caused by microbial infections, can lead to a pro-inflammatory environment and the differentiation of Th17 cells and other T helper cells. In a mouse model of RA, significant impairment of intestinal barrier function was observed before the onset of arthritis. Similarly, elevated serum markers associated with compromised intestinal barrier function in humans before RA onset are linked to an increased risk of developing RA (84). Additionally, there is a noted association between serological markers of intestinal permeability, disease activity, and response to biologic disease-modifying antirheumatic drugs (85). Mechanistically, dysbiosis in the gut microbiota can compromise intestinal mucosal integrity, activate the gut immune system, and trigger the migration of immune cells such as group 3 innate lymphoid cells (ILC3s), follicular helper T cells (Tfh cells), and mucosa-associated invariant T cells (MAITs) to the systemic circulation and joints, ultimately contributing to arthritis (86–90). These findings underscore a significant connection between compromised gut integrity and systemic inflammation in RA.
4 A novel pro-inflammatory factor: bEVs
Bacterial extracellular vesicles (bEVs) are spherical, double-layered vesicles secreted by bacteria, with diameters ranging from approximately 20 to 300 nm. bEVs can carry various effector molecules, including lipopolysaccharides, proteins, and nucleic acids, and participate in interactions between bacteria and host cells (91). Gram-negative and Gram-positive bacteria produce different types of bEVs because of differences in cell wall structure. bEVs secreted by Gram-negative bacteria are termed outer membrane vesicles (OMVs), while those released by Gram-positive bacteria are called cytoplasmic membrane vesicles (CMVs) (92). These bEVs can deliver their contents to target cells through receptor-ligand interactions, membrane fusion, or receptor-mediated endocytosis, thereby modulating the biological behavior of host cells. bEVs have been shown to induce a pro-inflammatory responses, thereby enhancing the host defense mechanism against infections. Additionally, bEVs can influence adaptive immune responses via antigen presentation and T-cell activation (Table 2). Studies indicate that bEVs are involved in the occurrence and progression of various autoimmune diseases (117).
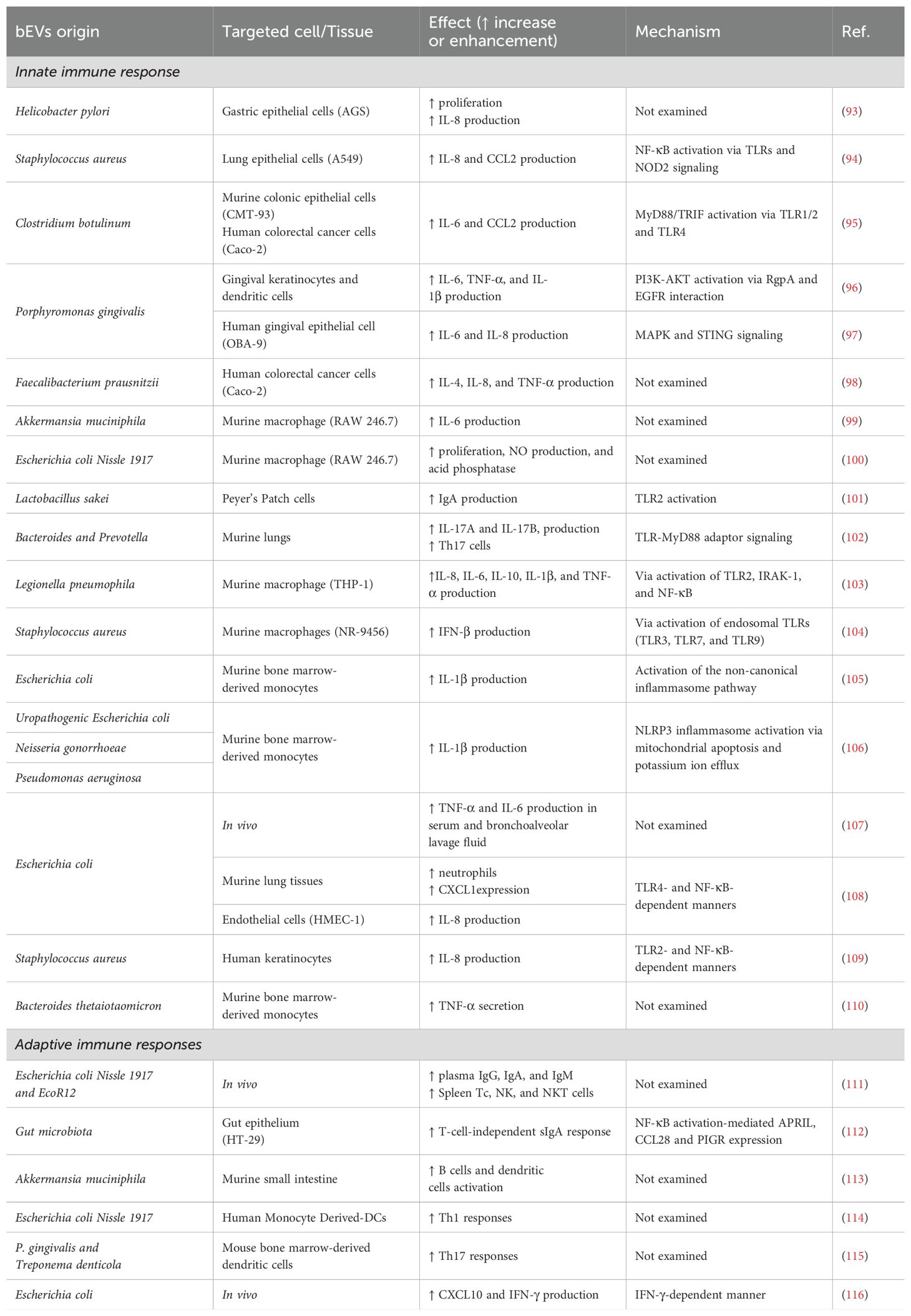
Table 2. Pro-inflammatory effects of bEVs.
4.1 The role of bEVs in promoting innate immune responses
Bacterial extracellular vesicles (bEVs) play a crucial role in host-microbe interactions by disrupting mucosal integrity and internalizing into various cell types, notably epithelial cells, which are primary contact points for bacteria. This interaction induces diverse immune responses, primarily through the activation of Toll-like receptors (TLRs) and other pathogen recognition receptors (PRRs) that recognize pathogen-associated molecular patterns (PAMPs), leading to immune activation. The inflammatory potential of bEVs was first observed when interleukin (IL)-8 was released from gastric epithelial cells stimulated by Helicobacter pylori bEVs (93). Additionally, Staphylococcus aureus can employ bEVs to transport lipoproteins, nucleic acids, and peptidoglycan, which are recognizable by host PRRs. This interaction has been demonstrated in human lung epithelial cells, where bEVs stimulation led to the secretion of cytokines IL-8 and CCL2, along with a strain-specific secretion of the pro-inflammatory cytokine IL-6 (94). Similarly, bEVs from various Clostridium species have been shown to enhance IL-6 and CCL2 expression in mouse colonic epithelial cells and human colorectal cancer cells (95). While TLRs signaling is a well-documented pathway, bEVs also promote inflammatory responses through the dissemination of toxins. For example, gingipains from P. gingivalis bEVs can induce the secretion of IL-6, IL-8, and TNF-α by interacting with the epidermal growth factor receptor within the lipid rafts of keratinocytes (96, 97).
As tissue-resident cells, macrophages are pivotal in intercepting bacterial extracellular vesicles (bEVs) that penetrate the mucosal barrier. Upon contact with the cellular surface or following phagocytosis, these bEVs activate various inflammatory pathways in macrophages, influenced by their specific cargos. Notably, macrophages employ Toll-like receptors (TLRs) to recognize bEVs, thereby initiating the release of inflammatory cytokines. Research by Yang et al. demonstrated that bEVs from Bacteroides and Prevotella species exacerbate bleomycin-induced lung fibrosis. This interaction involves bEVs binding to TLR4 and TLR2 on alveolar macrophages via their lipopolysaccharides (LPS) and lipoproteins, respectively, leading to the secretion of IL-17b, a key cytokine in this fibrosis model (102). Further studies with THP-1 macrophage-like cells have shown that bEVs from L. pneumophila activate TLR2 signaling, leading to the secretion of cytokines, including IL-8, IL-6, IL-10, IL-1β, and TNF-α (103). Beyond proteins, the nucleic acids present on or within bEVs can also engage TLRs. For instance, Staphylococcus aureus bEVs trigger TLR3, TLR7, and TLR9, inducing IFN-β expression in NR-9456 murine macrophages (104). Moreover, bEVs can initiate inflammasome activation, a fundamental inflammatory pathway, as seen with Escherichia coli (E. coli) strains expressing the virulence factor HlyF, which enhances IL-1β release and increases cell death through a non-canonical pathway (105, 118). Such inflammasome activation may also be stimulated by bEVs-induced mitochondrial dysfunction caused by pathogens such as uropathogenic E. coli, P. aeruginosa, and Neisseria gonorrhoeae (106). These findings underscore the diverse and intricate mechanisms by which bEVs, carrying varied cargos, mediate inflammation through both TLR signaling and inflammasome activation, highlighting the complex nature of bEV-mediated immune modulation.
The localized release of chemoattractant molecules is pivotal in recruiting neutrophils to control infections or participate in inflammation. However, the interaction between neutrophils and bacterial extracellular vesicles (bEVs) introduces further complexity to this immune response. Studies have demonstrated that bEVs from E. coli, when administered intraperitoneally, accumulate in various organs and lead to increased leukocyte infiltration, particularly neutrophils (107). Additional research revealed that these bEVs colocalize with endothelial cells in mice lungs, inducing the neutrophil chemoattractant CXCL1 expression. Complementary in vitro experiments with human endothelial cells demonstrated that the CXCL1 homolog, IL-8, is produced through TLR4 in an NF-κB-dependent manner (108). In skin, Staudenmaier et al. observed that bEVs from Staphylococcus aureus induce the release of IL-8 from human keratinocytes through NF-κB signaling, dependent on TLR2 (109). The intricate interaction highlights the multifaceted role of bEVs in modulating host immune responses and underscores the need for additional research to completely understand their mechanisms and implications in host-pathogen dynamics. The pro-inflammatory effects of bEVs on the innate immune response are summarized in Table 2.
Mechanistically, gram-positive and gram-negative bacteria can release bEVs carrying LTA/lipoproteins and LPS, which activate membrane TLR2 and TLR4, resulting in MyD88 and subsequent NF-κB activation, thereby promoting the production of various pro-inflammatory cytokines and chemokines (119, 120). Nucleic acids protected from degradation by bEVs further induce a potent innate immune response by activating endosomal TLR3, TLR7, and TLR9 signaling pathways. Additionally, the stimulator of interferon genes (STING) signaling pathway in the cytoplasm is triggered by DNA carried by bEVs, which further promotes the production of inflammatory factors (97). Once bEVs enter the cells, intracellular receptors such as NOD1 and NOD2 can enhance NF-κB and subsequent IL-6 and IL-8 expression through peptidoglycan activation (121, 122) (Figure 1). Moreover, bEVs within cells can expose bacterial components such as LPS using proteins like GBPs, SNX-10, and Hemolysin to inflammasomes NLRP3/NLRC4, thereby activating caspase-1 and caspase-11 to enhance the release of pro-inflammatory factors (123–126).
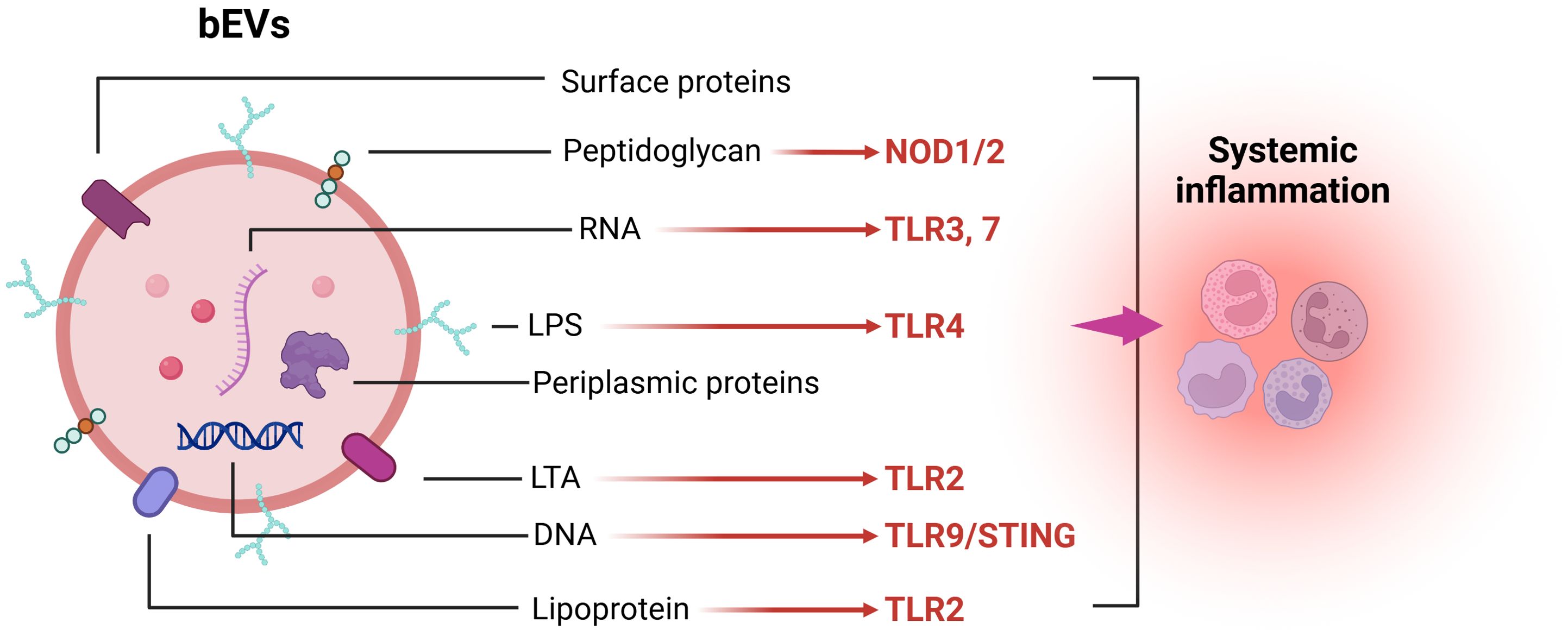
Figure 1. bEVs components and their pattern recognition receptors (PRRs).
4.2 The role of bEVs in promoting adaptive immune responses
Recent studies have elucidated the multifaceted roles of bacterial extracellular vesicles (bEVs) in modulating immune responses, emphasizing their potential in both innate and adaptive immunity. For instance, bEVs can induce an innate immune response and are recognized by antigen-presenting cells, thereby initiating adaptive immune responses. It has been demonstrated that bEVs from E. coli strains can promote the production of plasma IgG, IgA, and IgM and induce a greater proportion of spleen Tc, NK, and NKT cells in healthy suckling rats (111). Additionally, dietary influences on gut microbiota have been shown to affect bEV release, evidenced by increased bEV production in mice fed a high-protein diet, which in turn promotes the expression of immunoglobulin A (IgA)-inducing cytokines and chemokines, thereby enhancing mucosal immunity (112). bEVs can also enter Peyer’s patches and activate immune cells through direct contact. Studies have shown that Akkermansia muciniphila (A. muciniphila) bEVs in the gut lumen can enter Peyer’s patches, activate dendritic cells (DCs), and promote their proliferation. Consequently, with the assistance of DCs, CD69+ B cells and IgA+ secreting plasma cells significantly increase, thereby elevating the concentration of intestinal IgA (113, 127). This process is crucial for reducing pathogenic microorganisms in the gut. The immunomodulatory effects of bEVs from intestinal E. coli strains also underscore their role in defining specific T-helper cell responses. For example, bEVs from the probiotic E. coli Nissle 1917 program dendritic cells (DCs) to drive the pro-inflammatory Th1 response, which is essential for pathogen eradication (114). P. gingivalis and Treponema denticola bEVs stimulate dendritic cells (DCs) to induce TH17 and TH1 cell differentiation, respectively, a process potentially mediated by the increased secretion of IL-6 and IL-12 (115).
In the field of tumor therapy, Escherichia coli-derived bEVs have been shown to target and accumulate in tumor tissues, inducing the production of the anti-tumor cytokine CXCL10 and interferon-gamma (IFN-γ) and exerting their anti-tumor effects in an IFN-γ-dependent manner. Mechanistically, trypsin-treated bEVs failed to induce IFN-γ production, indicating that surface proteins are critical factors in inducing IFN-γ generation (116). Additionally, studies suggest that the expansion of Vγ9Vδ2 T cells induced by bEVs could represent a significant mechanism driving their anti-tumor effects (128). These findings collectively underscore the critical role of bEVs in shaping immune responses, offering promising avenues for therapeutic interventions. The pro-inflammatory effects of bEVs on the adaptive immune response are summarized in Table 2.
5 The pathogenic role of bEVs in RA
Multi-omics analyses of fecal, serum, and synovial fluid samples from RA patients have demonstrated that the gut microbiota can participate in the onset and progression of RA through invasion and metabolite secretion. Under normal circumstances, pathogenic microbes are primarily restricted by the mucosal barrier and rarely translocate into distant organs intercellularly; their primary mode of influence is through the secretion of bEVs, proteins, and metabolites. As critical players in the cross-domain interactions between the host and microbes within pathogenic environments, there is increasing evidence that bEVs participate in the host’s immune regulation by carrying molecules such as peptidoglycans, lipids, proteins, and nucleic acids (129). The presence of bEVs identified in various human biofluids and tissues suggests that these vesicles may contribute to the onset of tissue inflammation under pathological conditions (113, 130, 131).
In patients with RA, studies have identified a significant enrichment of F. nucleatum in fecal samples, with the levels being significantly positively correlated with RA disease activity. The bEVs secreted by F. nucleatum can promote the release of pro-inflammatory cytokines interleukin-8 (IL-8) and tumor necrosis factor (TNF) in colonic epithelial cells through a TLR4-dependent mechanism (132). Additionally, these bEVs can induce epithelial cell death through the FADD-RIPK1-caspase-3 signaling pathway, contributing to the development of intestinal inflammation (133). Furthermore, F. nucleatum bEVs can drive macrophage polarization toward the pro-inflammatory M1 phenotype (134). Interestingly, studies have demonstrated that bEVs secreted by certain gut bacteria can reach synovial tissues, confirming the possible existence of the “gut-joint axis”. Notably, bEVs derived from can migrate to the joints and trigger local inflammatory responses through the virulence factor FadA they carry. Mechanistically, these bEVs can activate the Rab5a-YB-1 signaling axis via FadA, thereby promoting the generation of synovial macrophages and the production of IL-6 and TNF-α (23, 135). These findings suggest that bacterial extracellular vesicles (bEVs) from the gut may cross the mucosal barrier and influence arthritis inflammation through the “gut-joint axis”.
As an oral pathogen closely related to the onset and development of RA, DNA components of P. gingivalis were identified in the synovial fluid of RA patients. However, the presence of P. gingivalis bacteria has not been observed (53). Interestingly, it has been confirmed that bEVs secreted by P. gingivalis can destroy the intestinal barrier (136). These bEVs, which transport bacterial DNA and associated effector molecules, may induce systemic inflammation and facilitate the leakage of intestinal substances into the bloodstream. This mechanism contributes to the pathogenesis of diverse systemic disorders associated with barrier dysfunction (137, 138). It was demonstrated that the ratio of cells to outer membrane vesicles (bEVs) is approximately 1:2,000. Their increased stability is attributed to their resistance to proteases and nucleases. In contrast to the original P. gingivalis source, bEVs demonstrate an enhanced capability to infiltrate deep tissues and elicit an inflammatory response within the host organism (139). Fleetwood and colleagues discovered that P. gingivalis bEVs can penetrate gingival tissue, resulting in tissue damage and inflammation. Stimulation of macrophages by bEVs resulted in the production of a significant amount of inflammatory factors, such as TNFα, IL-12p70, IL-6, IFNβ, and nitric oxide, compared to cells infected with P. gingivalis (140).
It was demonstrated that P. gingivalis’s unique PPAD can citrullinate human proteins, and contributing to the loss of tolerance to citrullinated proteins in RA (57, 141). PPAD not only disrupts amino acid balance but also compromises the body’s immune system, leading to an increased production of citrullinated antibodies against self-antigens (142). Interestingly, studies have indicated that PPAD is primarily released extracellularly through EVs (143). Moreover, PPAD in P. gingivalis bEVs with A-LPS modification can be protected from proteolytic degradation, which is closely associated with the citrullination of proteins (144). Notably, 78 citrullinated proteins have been identified in bEVs derived from the P. gingivalis W83 strain, suggesting a significant association between bEVs and RA (145). Additionally, P. gingivalis bEVs have been demonstrated to stimulate IL-6 and IL-8 secretion in epithelial cells via MAPK and STING pathways (97). P. gingivalis bEVs can also activate neutrophils, inducing degranulation without causing their death, thereby promoting inflammation (146).
Collectively, these findings suggest that bacterial extracellular vesicles (bEVs) from bacteria such as F. nucleatum and P. gingivalis exert a significant pro-inflammatory effect, potentially serving as crucial pathogenic factors in the occurrence and progression of RA (Figure 2).
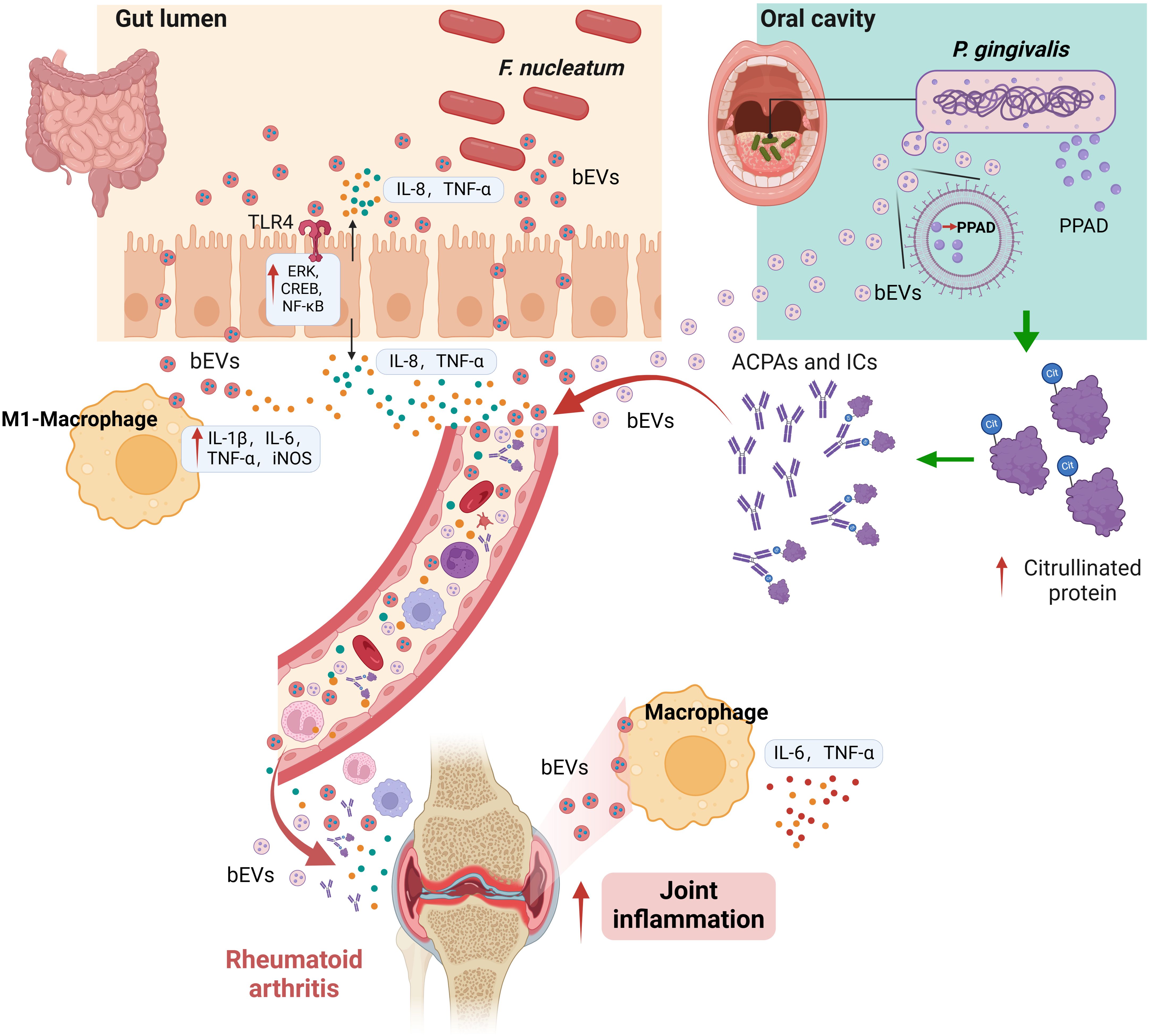
Figure 2. Proposed pathogenic roles of bEVs in RA. bEVs from F. nucleatum in the intestinal tract may enhance pro-inflammatory cytokine release and macrophage polarization, potentially influencing joint inflammation via the gut-joint axis. P. gingivalis bEVs, carrying PPAD, are hypothesized to penetrate mucosal barriers, thereby promoting systemic inflammation and autoimmunity by altering immune responses and protein citrullination. These mechanisms are based on emerging evidence and require further experimental validation.
6 Current challenges and future perspectives
The role of bacterial factors in inflammatory diseases is being increasingly emphasized. Changes in the abundance of bacteria such as Prevotella copri in the gut, as well as infections of pathogenic bacteria like P. gingivalis and Aggregatibacter actinomycetemcomitans in the oral cavity, are considered high-risk and causative factors in the onset of RA. Furthermore, recent studies have also highlighted the crucial role of the gut microbiota in modulating immune responses and contributing to the pathogenesis of other autoimmune diseases (ADs). The gut bacterium Ruminococcus gnavus, enriched in SLE, has been correlated with disease activity and lupus nephritis through its ability to induce the production of anti-double-stranded DNA antibodies. Additionally, specific gut commensals, such as segmented filamentous bacteria (SFB) and members of the Erysipelotrichaceae family, promote autoimmune inflammation by driving the polarization of T helper 17 (TH17) cells via mechanisms involving serum amyloid A (SAA) and interleukin-23 (IL-23) (147). Mechanistically, bacteria that promote the onset and progression of autoimmune diseases can induce inflammation by altering host metabolism, employing molecular mimicry, and disrupting intestinal permeability. These findings underscore the concept that the gut microbiota not only regulates local intestinal immunity but also exerts significant systemic effects by enabling gut-primed immune cells to migrate to extra-intestinal tissues, thereby exacerbating disease activity.
Emerging evidence suggests a potential role of bacterial migration from the oral cavity to the gut, forming an “oral-gut axis” which could play a synergistic role in the pathogenesis of autoimmune diseases (148). For example, oral pathogens such as P. gingivalis can alter the gut microbiome, leading to elevated serum endotoxin levels, increased inflammatory markers, and impaired gut barrier function, ultimately exacerbating arthritis in collagen-induced arthritis (CIA) mice. This interaction underscores the importance of studying the oral-gut axis to elucidate the intricate mechanisms underlying the connection between microbial dysbiosis and rheumatoid arthritis (RA) (149). However, whether these bacteria can reach peripheral organs such as joints through the peripheral circulation, thereby exacerbating arthritis, remains unclear. While some studies have isolated potentially pathogenic bacteria from late-stage joint fluid in RA patients (24), other studies have confirmed the absence of intact bacterial cells in the joints. Therefore, further and more in-depth research is needed to elucidate the exact pathogenic mechanisms of microbial factors in RA.
As research into eukaryotic exosomes advances, additional studies suggest that bacterial extracellular vesicles (bEVs) play a pivotal role in microbe-host cell interactions and disease development. These bEVs have also been shown to regulate both innate and adaptive immune responses, influencing immune reactions associated with RA inflammation. Hence, the discovery of bEVs may offer new insights into how pathogenic microorganisms contribute to and exacerbate inflammatory diseases such as RA. Nevertheless, because the gut and oral cavities harbor a vast array of bacterial species, bEVs derived from diverse microbial communities exhibit tremendous heterogeneity in their molecular cargo and biological activities. This diversity complicates attributing explicit pathogenic or protective functions to bEVs from any single species without detailed molecular profiling. Accordingly, clarifying how bEVs populations vary across different microbial ecosystems—both in healthy individuals and RA patients—would be instrumental in discerning their distinct effects on disease progression. In addition, despite notable progress in bEVs research, the field lacks standardized guidelines akin to the “minimal information for studies of extracellular vesicles” (MISEV) used for eukaryotic vesicles. This gap leads to varied isolation methods, impacting bEVs populations and complicating data interpretation. Moreover, the biogenesis of bEVs from bacteria remains under investigation. Although evidence supports regulated bEVs formation, the processes behind bEVs production and cargo selection remain incompletely understood. Further research is imperative to deepen our understanding of bEVs roles in inter-bacterial and bacteria-host communication.
Although the direct experimental evidence linking bEVs to RA pathogenesis remains limited, the accumulating findings from related fields strongly suggest their potential significance in autoimmune diseases, including RA. These observations support the hypothesis that bEVs may play a crucial role in RA through mechanisms such as immune cell activation, cytokine production, and antigen presentation. As mentioned above, bEVs from P. gingivalis have been shown to carry citrullinated proteins and PPAD, which can disrupt immune tolerance and promote the production of anti-citrullinated protein antibodies (ACPAs). Similarly, bEVs from gut bacteria such as F. nucleatum have been implicated in gut barrier disruption and systemic inflammation, further supporting their potential role in RA pathogenesis. However, further experimental studies are urgently needed to validate these hypotheses and better understand the specific pathways through which bEVs contribute to RA development. Future research should focus on elucidating how bEVs interact with immune cells and tissues within the RA context. For instance, experimental approaches could investigate the cargo composition of RA-specific bEVs, their uptake by synovial macrophages, and their ability to modulate T-cell responses. Additionally, studies exploring the systemic dissemination of bEVs and their impact on joint inflammation could provide critical insights into the “gut-joint” and “oral-joint” axes. By addressing these gaps, future research could establish a more comprehensive understanding of bEVs as potential mediators and therapeutic targets in RA.
Author contributions
JL: Funding acquisition, Investigation, Writing – original draft. YW: Software, Visualization, Writing – original draft. JW: Software, Visualization, Writing – original draft. YD: Investigation, Writing – original draft. HZ: Writing – review & editing. HD: Investigation, Methodology, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was funded by the National Natural Science Foundation of China (NSFC) (No. 82402699), Natural Science Foundation of Jiangsu Province of China (No. BK20240360), Natural Science Foundation of the Jiangsu Higher Education Institutions of China (No. 24KJD320002), Project of Medical Research Fund of Jiangsu Provincial Health Commission (Z2023043), Gusu Talent Program (2023) 024.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Zhang F, Jonsson AH, Nathan A, Millard N, Curtis M, Xiao Q, et al. Deconstruction of rheumatoid arthritis synovium defines inflammatory subtypes. Nature. (2023) 623:616–24. doi: 10.1038/s41586-023-06708-y
2. Lan W, Lu Q, Ma W, Jiang Z, Chen Y, Wang Z, et al. Investigating the causal relationship between the gut microbiome and rheumatoid arthritis: mediating effects of immune cells. J Transl Med. (2025) 23:187. doi: 10.1186/s12967-025-06206-x
3. Finckh A, Gilbert B, Hodkinson B, Bae SC, Thomas R, Deane KD, et al. Global epidemiology of rheumatoid arthritis. Nat Rev Rheumatol. (2022) 18:591–602. doi: 10.1038/s41584-022-00827-y
4. Alivernini S, Firestein GS, McInnes IB. The pathogenesis of rheumatoid arthritis. Immunity. (2022) 55:2255–70. doi: 10.1016/j.immuni.2022.11.009
5. Yang Y, Xia C, Yao C, Ma X, Shen Z, Chen P, et al. Mucosal immunity and rheumatoid arthritis: An update on mechanisms and therapeutic potential. Autoimmun Rev. (2025) 24:103775. doi: 10.1016/j.autrev.2025.103775
6. Holers VM, Demoruelle MK, Kuhn KA, Buckner JH, Robinson WH, Okamoto Y, et al. Rheumatoid arthritis and the mucosal origins hypothesis: protection turns to destruction. Nat Rev Rheumatol. (2018) 14:542–57. doi: 10.1038/s41584-018-0070-0
7. van Vollenhoven RF, Ernestam S, Geborek P, Petersson IF, Cöster L, Waltbrand E, et al. Addition of infliximab compared with addition of sulfasalazine and hydroxychloroquine to methotrexate in patients with early rheumatoid arthritis (Swefot trial): 1-year results of a randomised trial. Lancet. (2009) 374:459–66. doi: 10.1016/s0140-6736(09)60944-2
8. Nayak RR. Fanning the flames of autoimmunity: the microbiome in rheumatic disease. Arthritis Rheumatol. (2022) 74:549–52. doi: 10.1002/art.42057
9. Guan Y, Zhao X, Lu Y, Zhang Y, Lu Y, Wang Y. New bitongling regulates gut microbiota to predict angiogenesis in rheumatoid arthritis via the gut-joint axis: a deep neural network approach. Front Microbiol. (2025) 16:1528865. doi: 10.3389/fmicb.2025.1528865
10. Qin Q, Hu G, Zhou X, Zhu R, Chen J, Zeng K, et al. Therapeutic potential of the probiotic Lactiplantibacillus plantarum BX 62 and its postbiotics in alleviating rheumatoid arthritis in mice. Curr Res Food Sci. (2024) 9:100915. doi: 10.1016/j.crfs.2024.100915
11. Yadav S, Sapra L, Srivastava RK. Polysaccharides to postbiotics: Nurturing bone health via modulating “gut-immune axis. Int J Biol Macromol. (2024) 278:134655. doi: 10.1016/j.ijbiomac.2024.134655
12. Ying ZH, Mao CL, Xie W, Yu CH. Postbiotics in rheumatoid arthritis: emerging mechanisms and intervention perspectives. Front Microbiol. (2023) 14:1290015. doi: 10.3389/fmicb.2023.1290015
13. Mo C, Bi J, Li S, Lin Y, Yuan P, Liu Z, et al. The influence and therapeutic effect of microbiota in systemic lupus erythematosus. Microbiol Res. (2024) 281:127613. doi: 10.1016/j.micres.2024.127613
14. Wu Z, Tian E, Chen Y, Dong Z, Peng Q. Gut microbiota and its roles in the pathogenesis and therapy of endocrine system diseases. Microbiol Res. (2023) 268:127291. doi: 10.1016/j.micres.2022.127291
15. Alpizar-Rodriguez D, Lesker TR, Gronow A, Gilbert B, Raemy E, Lamacchia C, et al. Prevotella copri in individuals at risk for rheumatoid arthritis. Ann Rheum Dis. (2019) 78:590–3. doi: 10.1136/annrheumdis-2018-214514
16. Wells PM, Adebayo AS, Bowyer RCE, Freidin MB, Finckh A, Strowig T, et al. Associations between gut microbiota and genetic risk for rheumatoid arthritis in the absence of disease: a cross-sectional study. Lancet Rheumatol. (2020) 2:e418–27. doi: 10.1016/s2665-9913(20)30064-3
17. Scher JU, Sczesnak A, Longman RS, Segata N, Ubeda C, Bielski C, et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife. (2013) 2:e01202. doi: 10.7554/eLife.01202
18. Maeda Y, Kurakawa T, Umemoto E, Motooka D, Ito Y, Gotoh K, et al. Dysbiosis contributes to arthritis development via activation of autoreactive T cells in the intestine. Arthritis Rheumatol. (2016) 68:2646–61. doi: 10.1002/art.39783
19. Nii T, Maeda Y, Motooka D, Naito M, Matsumoto Y, Ogawa T, et al. Genomic repertoires linked with pathogenic potency of arthritogenic Prevotella copri isolated from the gut of patients with rheumatoid arthritis. Ann Rheum Dis. (2023) 82:621–9. doi: 10.1136/ard-2022-222881
20. Chen J, Wright K, Davis JM, Jeraldo P, Marietta EV, Murray J, et al. An expansion of rare lineage intestinal microbes characterizes rheumatoid arthritis. Genome Med. (2016) 8:43. doi: 10.1186/s13073-016-0299-7
21. Zhang X, Zhang D, Jia H, Feng Q, Wang D, Liang D, et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat Med. (2015) 21:895–905. doi: 10.1038/nm.3914
22. Jeong Y, Kim JW, You HJ, Park SJ, Lee J, Ju JH, et al. Gut microbial composition and function are altered in patients with early rheumatoid arthritis. J Clin Med. (2019) 8(5):693. doi: 10.3390/jcm8050693
23. Hong M, Li Z, Liu H, Zheng S, Zhang F, Zhu J, et al. Fusobacterium nucleatum aggravates rheumatoid arthritis through FadA-containing outer membrane vesicles. Cell Host Microbe. (2023) 31:798–810 e797. doi: 10.1016/j.chom.2023.03.018
24. Cheng M, Zhao Y, Cui Y, Zhong C, Zha Y, Li S, et al. Stage-specific roles of microbial dysbiosis and metabolic disorders in rheumatoid arthritis. Ann Rheum Dis. (2022) 81:1669–77. doi: 10.1136/ard-2022-222871
25. Kishikawa T, Maeda Y, Nii T, Motooka D, Matsumoto Y, Matsushita M, et al. Metagenome-wide association study of gut microbiome revealed novel aetiology of rheumatoid arthritis in the Japanese population. Ann Rheum Dis. (2020) 79:103–11. doi: 10.1136/annrheumdis-2019-215743
26. Rodrigues GSP, Cayres LCF, Gonçalves FP, Takaoka NNC, Lengert AH, Tansini A, et al. Detection of increased relative expression units of bacteroides and prevotella, and decreased clostridium leptum in stool samples from Brazilian rheumatoid arthritis patients: A pilot study. Microorganisms. (2019) 7(10):413. doi: 10.3390/microorganisms7100413
27. Picchianti-Diamanti A, Panebianco C, Salemi S, Sorgi ML, Di Rosa R, Tropea A, et al. Analysis of gut microbiota in rheumatoid arthritis patients: disease-related dysbiosis and modifications induced by etanercept. Int J Mol Sci. (2018) 19(10):2938. doi: 10.3390/ijms19102938
28. Sun Y, Chen Q, Lin P, Xu R, He D, Ji W, et al. Characteristics of gut microbiota in patients with rheumatoid arthritis in shanghai, China. Front Cell Infect Microbiol. (2019) 9:369. doi: 10.3389/fcimb.2019.00369
29. Zhu J, Wang T, Lin Y, Xiong M, Chen J, Jian C, et al. The change of plasma metabolic profile and gut microbiome dysbiosis in patients with rheumatoid arthritis. Front Microbiol. (2022) 13:931431. doi: 10.3389/fmicb.2022.931431
30. Yun H, Wang X, Wei C, Liu Q, Li X, Li N, et al. Alterations of the intestinal microbiome and metabolome in women with rheumatoid arthritis. Clin Exp Med. (2023) 23:4695–706. doi: 10.1007/s10238-023-01161-7
31. Lee JY, Mannaa M, Kim Y, Kim J, Kim GT, Seo YS. Comparative analysis of fecal microbiota composition between rheumatoid arthritis and osteoarthritis patients. Genes (Basel). (2019) 10(10):748. doi: 10.3390/genes10100748
32. Rooney CM, Mankia K, Mitra S, Moura IB, Emery P, Wilcox MH. Perturbations of the gut microbiome in anti-CCP positive individuals at risk of developing rheumatoid arthritis. Rheumatol (Oxford). (2021) 60:3380–7. doi: 10.1093/rheumatology/keaa792
33. Yu D, Du J, Pu X, Zheng L, Chen S, Wang N, et al. The gut microbiome and metabolites are altered and interrelated in patients with rheumatoid arthritis. Front Cell Infect Microbiol. (2021) 11:763507. doi: 10.3389/fcimb.2021.763507
34. Chen Y, Ma C, Liu L, He J, Zhu C, Zheng F, et al. Analysis of gut microbiota and metabolites in patients with rheumatoid arthritis and identification of potential biomarkers. Aging (Albany NY). (2021) 13:23689–701. doi: 10.18632/aging.203641
35. Luo Y, Tong Y, Wu L, Niu H, Li Y, Su LC, et al. Alteration of gut microbiota in individuals at high-risk for rheumatoid arthritis associated with disturbed metabolome and the initiation of arthritis through the triggering of mucosal immunity imbalance. Arthritis Rheumatol. (2023) 75:1736–48. doi: 10.1002/art.42616
36. Liu X, Tian K, Ma X, Wang S, Luo C, Du Q. Analysis of subgingival microbiome of periodontal disease and rheumatoid arthritis in Chinese: A case-control study. Saudi J Biol Sci. (2020) 27:1835–42. doi: 10.1016/j.sjbs.2020.04.040
37. Esberg A, Johansson L, Johansson I, Dahlqvist SR. Oral microbiota identifies patients in early onset rheumatoid arthritis. Microorganisms. (2021) 9(8):1657. doi: 10.3390/microorganisms9081657
38. Kim JW, Jung H, Baek IP, Nam Y, Kang J, Chung MK, et al. Differential effects of periodontal microbiome on the rheumatoid factor induction during rheumatoid arthritis pathogenesis. Sci Rep. (2022) 12:19636. doi: 10.1038/s41598-022-21788-y
39. Kozhakhmetov S, Babenko D, Issilbayeva A, Nurgaziyev M, Kozhakhmetova S, Meiramova A, et al. Oral microbial signature of rheumatoid arthritis in female patients. J Clin Med. (2023) 12(11):3694. doi: 10.3390/jcm12113694
40. Laugisch O, Wong A, Sroka A, Kantyka T, Koziel J, Neuhaus K, et al. Citrullination in the periodontium–a possible link between periodontitis and rheumatoid arthritis. Clin Investig. (2016) 20:675–83. doi: 10.1007/s00784-015-1556-7
41. Corrêa JD, Fernandes GR, Calderaro DC, Mendonça SMS, Silva JM, Albiero ML, et al. Oral microbial dysbiosis linked to worsened periodontal condition in rheumatoid arthritis patients. Sci Rep. (2019) 9:8379. doi: 10.1038/s41598-019-44674-6
42. Tong Y, Zheng L, Qing P, Zhao H, Li Y, Su L, et al. Oral microbiota perturbations are linked to high risk for rheumatoid arthritis. Front Cell Infect Microbiol. (2019) 9:475. doi: 10.3389/fcimb.2019.00475
43. Eriksson K, Lundmark A, Delgado LF, Hu YOO, Fei G, Lee L, et al. Salivary microbiota and host-inflammatory responses in periodontitis affected individuals with and without rheumatoid arthritis. Front Cell Infect Microbiol. (2022) 12:841139. doi: 10.3389/fcimb.2022.841139
44. Lopez-Oliva I, Paropkari AD, Saraswat S, Serban S, Yonel Z, Sharma P, et al. Dysbiotic subgingival microbial communities in periodontally healthy patients with rheumatoid arthritis. Arthritis Rheumatol. (2018) 70:1008–13. doi: 10.1002/art.40485
45. Kroese JM, Brandt BW, Buijs MJ, Crielaard W, Lobbezoo F, Loos BG, et al. Differences in the oral microbiome in patients with early rheumatoid arthritis and individuals at risk of rheumatoid arthritis compared to healthy individuals. Arthritis Rheumatol. (2021) 73:1986–93. doi: 10.1002/art.41780
46. Hascoët E, Blanchard F, Blin-Wakkach C, Guicheux J, Lesclous P, Cloitre A. New insights into inflammatory osteoclast precursors as therapeutic targets for rheumatoid arthritis and periodontitis. Bone Res. (2023) 11:26. doi: 10.1038/s41413-023-00257-w
47. Cantley MD, Haynes DR, Marino V, Bartold PM. Pre-existing periodontitis exacerbates experimental arthritis in a mouse model. J Clin Periodontol. (2011) 38:532–41. doi: 10.1111/j.1600-051X.2011.01714.x
48. Salemi S, Biondo MI, Fiorentino C, Argento G, Paolantonio M, Di Murro C, et al. Could early rheumatoid arthritis resolve after periodontitis treatment only?: case report and review of the literature. Med (Baltimore). (2014) 93:e195. doi: 10.1097/md.0000000000000195
49. Zhao X, Liu Z, Shu D, Xiong Y, He M, Xu S, et al. Association of periodontitis with rheumatoid arthritis and the effect of non-surgical periodontal treatment on disease activity in patients with rheumatoid arthritis. Med Sci Monit. (2018) 24:5802–10. doi: 10.12659/msm.909117
50. Lin J, Huang D, Xu H, Zhan F, Tan X. Macrophages: A communication network linking Porphyromonas gingivalis infection and associated systemic diseases. Front Immunol. (2022) 13:952040. doi: 10.3389/fimmu.2022.952040
51. Seyama M, Yoshida K, Yoshida K, Fujiwara N, Ono K, Eguchi T, et al. Outer membrane vesicles of Porphyromonas gingivalis attenuate insulin sensitivity by delivering gingipains to the liver. Biochim Biophys Acta Mol Basis Dis. (2020) 1866:165731. doi: 10.1016/j.bbadis.2020.165731
52. Joseph R, Rajappan S, Nath SG, Paul BJ. Association between chronic periodontitis and rheumatoid arthritis: a hospital-based case-control study. Rheumatol Int. (2013) 33:103–9. doi: 10.1007/s00296-011-2284-1
53. Martinez-Martinez RE, Abud-Mendoza C, Patiño-Marin N, Rizo-Rodríguez JC, Little JW, Loyola-Rodríguez JP. Detection of periodontal bacterial DNA in serum and synovial fluid in refractory rheumatoid arthritis patients. J Clin Periodontol. (2009) 36:1004–10. doi: 10.1111/j.1600-051X.2009.01496.x
54. Reichert S, Haffner M, Keyßer G, Schäfer C, Stein JM, Schaller HG, et al. Detection of oral bacterial DNA in synovial fluid. J Clin Periodontol. (2013) 40:591–8. doi: 10.1111/jcpe.12102
55. de Aquino SG, Abdollahi-Roodsaz S, Koenders MI, van de Loo FA, Pruijn GJ, Marijnissen RJ, et al. Periodontal pathogens directly promote autoimmune experimental arthritis by inducing a TLR2- and IL-1-driven Th17 response. J Immunol. (2014) 192:4103–11. doi: 10.4049/jimmunol.1301970
56. Rodríguez J, Lafaurie GI, Bautista-Molano W, Chila-Moreno L, Bello-Gualtero JM, Romero-Sánchez C. Adipokines and periodontal markers as risk indicators of early rheumatoid arthritis: a cross-sectional study. Clin Investig. (2021) 25:1685–95. doi: 10.1007/s00784-020-03469-0
57. Jenning M, Marklein B, Ytterberg J, Zubarev RA, Joshua V, van Schaardenburg D, et al. Bacterial citrullinated epitopes generated by Porphyromonas gingivalis infection-a missing link for ACPA production. Ann Rheum Dis. (2020) 79:1194–202. doi: 10.1136/annrheumdis-2019-216919
58. Maresz KJ, Hellvard A, Sroka A, Adamowicz K, Bielecka E, Koziel J, et al. Porphyromonas gingivalis facilitates the development and progression of destructive arthritis through its unique bacterial peptidylarginine deiminase (PAD). PloS Pathog. (2013) 9:e1003627. doi: 10.1371/journal.ppat.1003627
59. Hamamoto Y, Ouhara K, Munenaga S, Shoji M, Ozawa T, Hisatsune J, et al. Effect of Porphyromonas gingivalis infection on gut dysbiosis and resultant arthritis exacerbation in mouse model. Arthritis Res Ther. (2020) 22:249. doi: 10.1186/s13075-020-02348-z
60. Martinsson K, Di Matteo A, Öhman C, Johansson A, Svärd A, Mankia K, et al. Antibodies to leukotoxin A from the periodontal pathogen Aggregatibacter actinomycetemcomitans in patients at an increased risk of rheumatoid arthritis. Front Med (Lausanne). (2023) 10:1176165. doi: 10.3389/fmed.2023.1176165
61. Konig MF, Abusleme L, Reinholdt J, Palmer RJ, Teles RP, Sampson K, et al. Aggregatibacter actinomycetemcomitans-induced hypercitrullination links periodontal infection to autoimmunity in rheumatoid arthritis. Sci Transl Med. (2016) 8:369ra176. doi: 10.1126/scitranslmed.aaj1921
62. Trombone AP, Claudino M, Colavite P, de Assis GF, Avila-Campos MJ, Silva JS, et al. Periodontitis and arthritis interaction in mice involves a shared hyper-inflammatory genotype and functional immunological interferences. Genes Immun. (2010) 11:479–89. doi: 10.1038/gene.2010.13
63. Rasouli-Saravani A, Jahankhani K, Moradi S, Gorgani M, Shafaghat Z, Mirsanei Z, et al. Role of microbiota short-chain fatty acids in the pathogenesis of autoimmune diseases. BioMed Pharmacother. (2023) 162:114620. doi: 10.1016/j.biopha.2023.114620
64. Lucas S, Omata Y, Hofmann J, Böttcher M, Iljazovic A, Sarter K, et al. Short-chain fatty acids regulate systemic bone mass and protect from pathological bone loss. Nat Commun. (2018) 9:55. doi: 10.1038/s41467-017-02490-4
65. Rosser EC, Piper CJM, Matei DE, Blair PA, Rendeiro AF, Orford M, et al. Microbiota-derived metabolites suppress arthritis by amplifying aryl-hydrocarbon receptor activation in regulatory B cells. Cell Metab. (2020) 31:837–851 e810. doi: 10.1016/j.cmet.2020.03.003
66. He J, Chu Y, Li J, Meng Q, Liu Y, Jin J, et al. Intestinal butyrate-metabolizing species contribute to autoantibody production and bone erosion in rheumatoid arthritis. Sci Adv. (2022) 8:eabm1511. doi: 10.1126/sciadv.abm1511
67. Lin L, Zhang K, Xiong Q, Zhang J, Cai B, Huang Z, et al. Gut microbiota in pre-clinical rheumatoid arthritis: From pathogenesis to preventing progression. J Autoimmun. (2023) 141:103001. doi: 10.1016/j.jaut.2023.103001
68. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. (2013) 504:446–50. doi: 10.1038/nature12721
69. Yao Y, Cai X, Zheng Y, Zhang M, Fei W, Sun D, et al. Short-chain fatty acids regulate B cells differentiation via the FFA2 receptor to alleviate rheumatoid arthritis. Br J Pharmacol. (2022) 179:4315–29. doi: 10.1111/bph.15852
70. Hui W, Yu D, Cao Z, Zhao X. Butyrate inhibit collagen-induced arthritis via Treg/IL-10/Th17 axis. Int Immunopharmacol. (2019) 68:226–33. doi: 10.1016/j.intimp.2019.01.018
71. Li Y, Zhang SX, Yin XF, Zhang MX, Qiao J, Xin XH, et al. The gut microbiota and its relevance to peripheral lymphocyte subpopulations and cytokines in patients with rheumatoid arthritis. J Immunol Res. (2021) 2021:6665563. doi: 10.1155/2021/6665563
72. Roager HM, Licht TR. Microbial tryptophan catabolites in health and disease. Nat Commun. (2018) 9:3294. doi: 10.1038/s41467-018-05470-4
73. Shen J, Yang L, You K, Chen T, Su Z, Cui Z, et al. Indole-3-acetic acid alters intestinal microbiota and alleviates ankylosing spondylitis in mice. Front Immunol. (2022) 13:762580. doi: 10.3389/fimmu.2022.762580
74. Cong S, Wang L, Meng Y, Cai X, Zhang C, Gu Y, et al. Saussurea involucrata oral liquid regulates gut microbiota and serum metabolism during alleviation of collagen-induced arthritis in rats. Phytother Res. (2023) 37:1242–59. doi: 10.1002/ptr.7681
75. Wilck N, Matus MG, Kearney SM, Olesen SW, Forslund K, Bartolomaeus H, et al. Salt-responsive gut commensal modulates T(H)17 axis and disease. Nature. (2017) 551:585–9. doi: 10.1038/nature24628
76. Cervantes-Barragan L, Chai JN, Tianero MD, Di Luccia B, Ahern PP, Merriman J, et al. Lactobacillus reuteri induces gut intraepithelial CD4(+)CD8αα(+) T cells. Science. (2017) 357:806–10. doi: 10.1126/science.aah5825
77. Balog A, Varga B, Fülöp F, Lantos I, Toldi G, Vécsei L, et al. Kynurenic acid analog attenuates the production of tumor necrosis factor-α, calgranulins (S100A 8/9 and S100A 12), and the secretion of HNP1-3 and stimulates the production of tumor necrosis factor-stimulated gene-6 in whole blood cultures of patients with rheumatoid arthritis. Front Immunol. (2021) 12:632513. doi: 10.3389/fimmu.2021.632513
78. Lee K, Kwak JH, Pyo S. Inhibition of LPS-induced inflammatory mediators by 3-hydroxyanthranilic acid in macrophages through suppression of PI3K/NF-κB signaling pathways. Food Funct. (2016) 7:3073–82. doi: 10.1039/c6fo00187d
79. Kang KY, Lee SH, Jung SM, Park SH, Jung BH, Ju JH. Downregulation of tryptophan-related metabolomic profile in rheumatoid arthritis synovial fluid. J Rheumatol. (2015) 42:2003–11. doi: 10.3899/jrheum.141505
80. Yang Y, Hong Q, Zhang X, Liu Z. Rheumatoid arthritis and the intestinal microbiome: probiotics as a potential therapy. Front Immunol. (2024) 151:1331486. doi: 10.3389/fimmu.2024.1331486
81. Moten D, Teneva I, Apostolova D, Batsalova T, Dzhambazov B. Molecular mimicry of the rheumatoid arthritis-related immunodominant T-cell epitope within type II collagen (CII260-270) by the bacterial L-asparaginase. Int J Mol Sci. (2022) 23(16):9149. doi: 10.3390/ijms23169149
82. Pianta A, Arvikar SL, Strle K, Drouin EE, Wang Q, Costello CE, et al. Two rheumatoid arthritis-specific autoantigens correlate microbial immunity with autoimmune responses in joints. J Clin Invest. (2017) 127:2946–56. doi: 10.1172/jci93450
83. Negi S, Singh H, Mukhopadhyay A. Gut bacterial peptides with autoimmunity potential as environmental trigger for late onset complex diseases: In-silico study. PloS One. (2017) 12:e0180518. doi: 10.1371/journal.pone.0180518
84. Tajik N, Frech M, Schulz O, Schälter F, Lucas S, Azizov V, et al. Targeting zonulin and intestinal epithelial barrier function to prevent onset of arthritis. Nat Commun. (2020) 11:1995. doi: 10.1038/s41467-020-15831-7
85. Audo R, Sanchez P, Rivière B, Mielle J, Tan J, Lukas C, et al. Rheumatoid arthritis is associated with increased gut permeability and bacterial translocation which are reversed by inflammation control. Rheumatol (Oxford). (2022) 10:keac454. doi: 10.1093/rheumatology/keac454
86. Yang F, Luo X, Zhu W, Li J, Zheng Z, Zhu P. Dysregulation of innate lymphoid cells in patients with active rheumatoid arthritis and mice with collagen-induced arthritis. Mediators Inflammation. (2021) 2021:1915068. doi: 10.1155/2021/1915068
87. Wang T, Rui J, Shan W, Xue F, Feng D, Dong L, et al. Imbalance of Th17, Treg, and helper innate lymphoid cell in the peripheral blood of patients with rheumatoid arthritis. Clin Rheumatol. (2022) 41:3837–49. doi: 10.1007/s10067-022-06315-8
88. Hinrichs AC, Kruize AA, Leavis HL, van Roon JAG. In patients with primary Sjögren’s syndrome innate-like MAIT cells display upregulated IL-7R, IFN-γ, and IL-21 expression and have increased proportions of CCR9 and CXCR5-expressing cells. Front Immunol. (2022) 13:1017157. doi: 10.3389/fimmu.2022.1017157
89. Qi J, Liu C, Bai Z, Li X, Yao G. T follicular helper cells and T follicular regulatory cells in autoimmune diseases. Front Immunol. (2023) 14:1178792. doi: 10.3389/fimmu.2023.1178792
90. Wei X, Niu X. T follicular helper cells in autoimmune diseases. J Autoimmun. (2023) 134:102976. doi: 10.1016/j.jaut.2022.102976
91. Xie J, Haesebrouck F, Van Hoecke L, Vandenbroucke RE. Bacterial extracellular vesicles: an emerging avenue to tackle diseases. Trends Microbiol. (2023) 31:1206–24. doi: 10.1016/j.tim.2023.05.010
92. Furuyama N, Sircili MP. Outer membrane vesicles (OMVs) produced by gram-negative bacteria: structure, functions, biogenesis, and vaccine application. BioMed Res Int. (2021) 2021:1490732. doi: 10.1155/2021/1490732
93. Ismail S, Hampton MB, Keenan JI. Helicobacter pylori outer membrane vesicles modulate proliferation and interleukin-8 production by gastric epithelial cells. Infect Immun. (2003) 71:5670–5. doi: 10.1128/iai.71.10.5670-5675.2003
94. Bitto NJ, Cheng L, Johnston EL, Pathirana R, Phan TK, Poon IKH, et al. Staphylococcus aureus membrane vesicles contain immunostimulatory DNA, RNA and peptidoglycan that activate innate immune receptors and induce autophagy. J Extracell Vesicles. (2021) 10:e12080. doi: 10.1002/jev2.12080
95. Kobayashi N, Abe K, Akagi S, Kitamura M, Shiraishi Y, Yamaguchi A, et al. Membrane Vesicles Derived From Clostridium botulinum and Related Clostridial Species Induce Innate Immune Responses via MyD88/TRIF Signaling in vitro. Front Microbiol. (2022) 13:720308. doi: 10.3389/fmicb.2022.720308
96. Ciaston I, Budziaszek J, Satala D, Potempa B, Fuchs A, Rapala-Kozik M, et al. Proteolytic activity-independent activation of the immune response by gingipains from porphyromonas gingivalis. mBio. (2022) 13:e0378721. doi: 10.1128/mbio.03787-21
97. Uemura Y, Hiroshima Y, Tada A, Murakami K, Yoshida K, Inagaki Y, et al. Porphyromonas gingivalis Outer Membrane Vesicles Stimulate Gingival Epithelial Cells to Induce Pro-Inflammatory Cytokines via the MAPK and STING Pathways. Biomedicines. (2022) 10(10):2643. doi: 10.3390/biomedicines10102643
98. Rabiei N, Ahmadi Badi S, Ettehad Marvasti F, Nejad Sattari T, Vaziri F, Siadat SD. Induction effects of Faecalibacterium prausnitzii and its extracellular vesicles on toll-like receptor signaling pathway gene expression and cytokine level in human intestinal epithelial cells. Cytokine. (2019) 121:154718. doi: 10.1016/j.cyto.2019.05.005
99. Kang CS, Ban M, Choi EJ, Moon HG, Jeon JS, Kim DK, et al. Extracellular vesicles derived from gut microbiota, especially Akkermansia muciniphila, protect the progression of dextran sulfate sodium-induced colitis. PloS One. (2013) 8:e76520. doi: 10.1371/journal.pone.0076520
100. Hu R, Lin H, Li J, Zhao Y, Wang M, Sun X, et al. Probiotic Escherichia coli Nissle 1917-derived outer membrane vesicles enhance immunomodulation and antimicrobial activity in RAW264.7 macrophages. BMC Microbiol. (2020) 20:268. doi: 10.1186/s12866-020-01953-x
101. Yamasaki-Yashiki S, Miyoshi Y, Nakayama T, Kunisawa J, Katakura Y. IgA-enhancing effects of membrane vesicles derived from Lactobacillus sakei subsp. sakei NBRC15893. Biosci Microbiota Food Health. (2019) 38:23–9. doi: 10.12938/bmfh.18-015
102. Yang D, Chen X, Wang J, Lou Q, Lou Y, Li L, et al. Dysregulated lung commensal bacteria drive interleukin-17B production to promote pulmonary fibrosis through their outer membrane vesicles. Immunity. (2019) 50:692–706 e697. doi: 10.1016/j.immuni.2019.02.001
103. Jung AL, Stoiber C, Herkt CE, Schulz C, Bertrams W, Schmeck B. Legionella pneumophila-derived outer membrane vesicles promote bacterial replication in macrophages. PloS Pathog. (2016) 12:e1005592. doi: 10.1371/journal.ppat.1005592
104. Rodriguez BV, Kuehn MJ. Staphylococcus aureus secretes immunomodulatory RNA and DNA via membrane vesicles. Sci Rep. (2020) 10:18293. doi: 10.1038/s41598-020-75108-3
105. David L, Taieb F, Pénary M, Bordignon PJ, Planès R, Bagayoko S, et al. Outer membrane vesicles produced by pathogenic strains of Escherichia coli block autophagic flux and exacerbate inflammasome activation. Autophagy. (2022) 18:2913–25. doi: 10.1080/15548627.2022.2054040
106. Deo P, Chow SH, Han ML, Speir M, Huang C, Schittenhelm RB, et al. Mitochondrial dysfunction caused by outer membrane vesicles from Gram-negative bacteria activates intrinsic apoptosis and inflammation. Nat Microbiol. (2020) 5:1418–27. doi: 10.1038/s41564-020-0773-2
107. Jang SC, Kim SR, Yoon YJ, Park KS, Kim JH, Lee J, et al. In vivo kinetic biodistribution of nano-sized outer membrane vesicles derived from bacteria. Small. (2015) 11:456–61. doi: 10.1002/smll.201401803
108. Lee J, Yoon YJ, Kim JH, Dinh NTH, Go G, Tae S, et al. Outer membrane vesicles derived from escherichia coli regulate neutrophil migration by induction of endothelial IL-8. Front Microbiol. (2018) 9:2268. doi: 10.3389/fmicb.2018.02268
109. Staudenmaier L, Focken J, Schlatterer K, Kretschmer D, Schittek B. Bacterial membrane vesicles shape Staphylococcus aureus skin colonization and induction of innate immune responses. Exp Dermatol. (2022) 31:349–61. doi: 10.1111/exd.14478
110. Hickey CA, Kuhn KA, Donermeyer DL, Porter NT, Jin C, Cameron EA, et al. Colitogenic Bacteroides thetaiotaomicron Antigens Access Host Immune Cells in a Sulfatase-Dependent Manner via Outer Membrane Vesicles. Cell Host Microbe. (2015) 17:672–80. doi: 10.1016/j.chom.2015.04.002
111. Martínez-Ruiz S, Sáez-Fuertes L, Casanova-Crespo S, Rodríguez-Lagunas MJ, Pérez-Cano FJ, Badia J, et al. Microbiota-derived extracellular vesicles promote immunity and intestinal maturation in suckling rats. Nutrients. (2023) 15(21):4701. doi: 10.3390/nu15214701
112. Tan J, Ni D, Taitz J, Pinget GV, Read M, Senior A, et al. Dietary protein increases T-cell-independent sIgA production through changes in gut microbiota-derived extracellular vesicles. Nat Commun. (2022) 13:4336. doi: 10.1038/s41467-022-31761-y
113. Wang X, Lin S, Wang L, Cao Z, Zhang M, Zhang Y, et al. Versatility of bacterial outer membrane vesicles in regulating intestinal homeostasis. Sci Adv. (2023) 9:eade5079. doi: 10.1126/sciadv.ade5079
114. Diaz-Garrido N, Badia J, Baldomà L. Modulation of dendritic cells by microbiota extracellular vesicles influences the cytokine profile and exosome cargo. Nutrients. (2022) 14(2):344. doi: 10.3390/nu14020344
115. Lim Y, Kim HY, An SJ, Choi BK. Activation of bone marrow-derived dendritic cells and CD4(+) T cell differentiation by outer membrane vesicles of periodontal pathogens. J Microbiol. (2022) 14:2123550. doi: 10.1080/20002297.2022.2123550
116. Kim OY, Park HT, Dinh NTH, Choi SJ, Lee J, Kim JH, et al. Bacterial outer membrane vesicles suppress tumor by interferon-γ-mediated antitumor response. Nat Commun. (2017) 8:626. doi: 10.1038/s41467-017-00729-8
117. Caruana JC, Walper SA. Bacterial membrane vesicles as mediators of microbe - microbe and microbe - host community interactions. Front Microbiol. (2020) 11:432. doi: 10.3389/fmicb.2020.00432
118. Murase K, Martin P, Porcheron G, Houle S, Helloin E, Pénary M, et al. HlyF produced by extraintestinal pathogenic escherichia coli is a virulence factor that regulates outer membrane vesicle biogenesis. J Infect Dis. (2016) 213:856–65. doi: 10.1093/infdis/jiv506
119. Fitzgerald KA, Kagan JC. Toll-like receptors and the control of immunity. Cell. (2020) 180:1044–66. doi: 10.1016/j.cell.2020.02.041
120. Lajqi T, Köstlin-Gille N, Hillmer S, Braun M, Kranig SA, Dietz S, et al. Gut microbiota-derived small extracellular vesicles endorse memory-like inflammatory responses in murine neutrophils. Biomedicines. (2022) 10(2):442. doi: 10.3390/biomedicines10020442
121. Cañas MA, Fábrega MJ, Giménez R, Badia J, Baldomà L. Outer membrane vesicles from probiotic and commensal escherichia coli activate NOD1-mediated immune responses in intestinal epithelial cells. Front Microbiol. (2018) 9:498. doi: 10.3389/fmicb.2018.00498
122. Johnston EL, Heras B, Kufer TA, Kaparakis-Liaskos M. Detection of bacterial membrane vesicles by NOD-like receptors. Int J Mol Sci. (2021) 22(3):1005. doi: 10.3390/ijms22031005
123. Santos JC, Dick MS, Lagrange B, Degrandi D, Pfeffer K, Yamamoto M, et al. LPS targets host guanylate-binding proteins to the bacterial outer membrane for non-canonical inflammasome activation. EMBO J. (2018) 37(6):e98089. doi: 10.15252/embj.201798089
124. Finethy R, Luoma S, Orench-Rivera N, Feeley EM, Haldar AK, Yamamoto M, et al. Inflammasome activation by bacterial outer membrane vesicles requires guanylate binding proteins. mBio. (2017) 8(5):e01188-17. doi: 10.1128/mBio.01188-17
125. Chen S, Yang D, Wen Y, Jiang Z, Zhang L, Jiang J, et al. Dysregulated hemolysin liberates bacterial outer membrane vesicles for cytosolic lipopolysaccharide sensing. PloS Pathog. (2018) 14:e1007240. doi: 10.1371/journal.ppat.1007240
126. Wang X, Ni J, You Y, Feng G, Zhang S, Bao W, et al. SNX10-mediated LPS sensing causes intestinal barrier dysfunction via a caspase-5-dependent signaling cascade. EMBO J. (2021) 40:e108080. doi: 10.15252/embj.2021108080
127. Reboldi A, Arnon TI, Rodda LB, Atakilit A, Sheppard D, Cyster JG. IgA production requires B cell interaction with subepithelial dendritic cells in Peyer’s patches. Science. (2016) 352:aaf4822. doi: 10.1126/science.aaf4822
128. Firth J, Sun J, George V, Huang JD, Bajaj-Elliott M, Gustafsson K. Bacterial outer-membrane vesicles promote Vγ9Vδ2 T cell oncolytic activity. Front Immunol. (2023) 14:1198996. doi: 10.3389/fimmu.2023.1198996
129. Arya SB, Collie SP, Parent CA. The ins-and-outs of exosome biogenesis, secretion, and internalization. Trends Cell Biol. (2024) 34:90–108. doi: 10.1016/j.tcb.2023.06.006
130. Xie J, Cools L, Van Imschoot G, Van Wonterghem E, Pauwels MJ, Vlaeminck I, et al. Helicobacter pylori-derived outer membrane vesicles contribute to Alzheimer’s disease pathogenesis via C3-C3aR signalling. J Extracell Vesicles. (2023) 12:e12306. doi: 10.1002/jev2.12306
131. Bittel M, Reichert P, Sarfati I, Dressel A, Leikam S, Uderhardt S, et al. Visualizing transfer of microbial biomolecules by outer membrane vesicles in microbe-host-communication in vivo. J Extracell Vesicles. (2021) 10:e12159. doi: 10.1002/jev2.12159
132. Engevik MA, Danhof HA, Ruan W, Engevik AC, Chang-Graham AL, Engevik KA, et al. Fusobacterium nucleatum secretes outer membrane vesicles and promotes intestinal inflammation. mBio. (2021) 12(2):e02706-20. doi: 10.1128/mBio.02706-20
133. Liu L, Liang L, Yang C, Zhou Y, Chen Y. Extracellular vesicles of Fusobacterium nucleatum compromise intestinal barrier through targeting RIPK1-mediated cell death pathway. Gut Microbes. (2021) 13:1–20. doi: 10.1080/19490976.2021.1902718
134. Chen G, Sun Q, Cai Q, Zhou H. Outer membrane vesicles from fusobacterium nucleatum switch M0-like macrophages toward the M1 phenotype to destroy periodontal tissues in mice. Front Microbiol. (2022) 13:815638. doi: 10.3389/fmicb.2022.815638
135. Ye H, Wang H, Han B, Chen K, Wang X, Ma F, et al. Guizhi Shaoyao Zhimu decoction inhibits neutrophil extracellular traps formation to relieve rheumatoid arthritis via gut microbial outer membrane vesicles. Phytomedicine. (2025) 136:156254. doi: 10.1016/j.phymed.2024.156254
136. Nonaka S, Okamoto R, Katsuta Y, Kanetsuki S, Nakanishi H. Gingipain-carrying outer membrane vesicles from Porphyromonas gingivalis cause barrier dysfunction of Caco-2 cells by releasing gingipain into the cytosol. Biochem Biophys Res Commun. (2024) 707:149783. doi: 10.1016/j.bbrc.2024.149783
137. Farrugia C, Stafford GP, Murdoch C. Porphyromonas gingivalis outer membrane vesicles increase vascular permeability. J Dent Res. (2020) 99:1494–501. doi: 10.1177/0022034520943187
138. Nonaka S, Kadowaki T, Nakanishi H. Secreted gingipains from Porphyromonas gingivalis increase permeability in human cerebral microvascular endothelial cells through intracellular degradation of tight junction proteins. Neurochem Int. (2022) 154:105282. doi: 10.1016/j.neuint.2022.105282
139. O’Brien-Simpson NM, Pathirana RD, Walker GD, Reynolds EC. Porphyromonas gingivalis RgpA-Kgp proteinase-adhesin complexes penetrate gingival tissue and induce proinflammatory cytokines or apoptosis in a concentration-dependent manner. Infect Immun. (2009) 77:1246–61. doi: 10.1128/iai.01038-08
140. Fleetwood AJ, Lee MKS, Singleton W, Achuthan A, Lee MC, O’Brien-Simpson NM, et al. Metabolic remodeling, inflammasome activation, and pyroptosis in macrophages stimulated by porphyromonas gingivalis and its outer membrane vesicles. Front Cell Infect Microbiol. (2017) 7:351. doi: 10.3389/fcimb.2017.00351
141. Gabarrini G, Grasso S, van Winkelhoff AJ, van Dijl JM. Gingimaps: protein localization in the oral pathogen porphyromonas gingivalis. Microbiol Mol Biol Rev. (2020) 84(1):e00032-19. doi: 10.1128/mmbr.00032-19
142. Kriauciunas A, Gleiznys A, Gleiznys D, Janužis ,G. The influence of porphyromonas gingivalis bacterium causing periodontal disease on the pathogenesis of rheumatoid arthritis: systematic review of literature. Cureus. (2019) 11:e4775. doi: 10.7759/cureus.4775
143. Gabarrini G, Chlebowicz MA, Vega Quiroz ME, Veloo ACM, Rossen JWA, Harmsen HJM, et al. Conserved citrullinating exoenzymes in porphyromonas species. J Dent Res. (2018) 97:556–62. doi: 10.1177/0022034517747575
144. Gabarrini G, Heida R, van Ieperen N, Curtis MA, van Winkelhoff AJ, van Dijl JM. Dropping anchor: attachment of peptidylarginine deiminase via A-LPS to secreted outer membrane vesicles of Porphyromonas gingivalis. Sci Rep. (2018) 8:8949. doi: 10.1038/s41598-018-27223-5
145. Larsen DN, Mikkelsen CE, Kierkegaard M, Bereta GP, Nowakowska Z, Kaczmarek JZ, et al. Citrullinome of porphyromonas gingivalis outer membrane vesicles: confident identification of citrullinated peptides. Mol Cell Proteomics. (2020) 19:167–80. doi: 10.1074/mcp.RA119.001700
146. du Teil Espina M, Fu Y, van der Horst D, Hirschfeld C, López-Álvarez M, Mulder LM, et al. Coating and corruption of human neutrophils by bacterial outer membrane vesicles. Microbiol Spectr. (2022) 10:e0075322. doi: 10.1128/spectrum.00753-22
147. Miyauchi E, Shimokawa C, Steimle A, Desai MS, Ohno H. The impact of the gut microbiome on extra-intestinal autoimmune diseases. Nat Rev Immunol. (2023) 23:9–23. doi: 10.1038/s41577-022-00727-y
148. Kunath BJ, De Rudder C, Laczny CC, Letellier E, Wilmes P. The oral-gut microbiome axis in health and disease. Nat Rev Microbiol. (2024) 22:791–805. doi: 10.1038/s41579-024-01075-5
Keywords: gut microbiome, oral microbiome, bacterial extracellular vesicles, rheumatoid arthritis, pathogenesis
Citation: Lu J, Wang Y, Wu J, Duan Y, Zhang H and Du H (2025) Linking microbial communities to rheumatoid arthritis: focus on gut, oral microbiome and their extracellular vesicles. Front. Immunol. 16:1503474. doi: 10.3389/fimmu.2025.1503474
Received: 29 September 2024; Accepted: 31 March 2025;
Published: 16 April 2025.
Edited by:
Constantin Caruntu, Carol Davila University of Medicine and Pharmacy, RomaniaReviewed by:
José Ricardo Jensen, Butantan Institute, BrazilChen-Huan Yu, Chinese Academy of Sciences (CAS), China
Zhengrui Li, Shanghai Jiao Tong University, China
Copyright © 2025 Lu, Wang, Wu, Duan, Zhang and Du. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hong Du, aG9uZ19kdUAxMjYuY29t; Jian Lu, bGpzZGYyeUAxNjMuY29t