 Zhibin Lv
Zhibin Lv Shunshan Jin
Shunshan Jin Hui Ding
Hui Ding Quan Zou
Quan Zou- 1Institute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu, China
- 2Department of Neurology, Heilongjiang Province Land Reclamation Headquarters General Hospital, Harbin, China
- 3Center for Informational Biology, University of Electronic Science and Technology of China, Chengdu, China
To gain insight into the malfunction of the Golgi apparatus and its relationship to various genetic and neurodegenerative diseases, the identification of sub-Golgi proteins, both cis-Golgi and trans-Golgi proteins, is of great significance. In this study, a state-of-art random forests sub-Golgi protein classifier, rfGPT, was developed. The rfGPT used 2-gap dipeptide and split amino acid composition for the feature vectors and was combined with the synthetic minority over-sampling technique (SMOTE) and an analysis of variance (ANOVA) feature selection method. The rfGPT was trained on a sub-Golgi protein sequence data set (137 sequences), with sequence identity less than 25%. For the optimal rfGPT classifier with 93 features, the accuracy (ACC) was 90.5%; the Matthews correlation coefficient (MCC) was 0.811; the sensitivity (Sn) was 92.6%; and the specificity (Sp) was 88.4%. The independent testing scores for the rfGPT were ACC = 90.6%; MCC = 0.696; Sn = 96.1%; and Sp = 69.2%. Although the independent testing accuracy was 4.4% lower than that for the best reported sub-Golgi classifier trained on a data set with 40% sequence identity (304 sequences), the rfGPT is currently the top sub-Golgi protein predictor utilizing feature vectors without any position-specific scoring matrix and its derivative features. Therefore, the rfGPT is a more practical tool, because no sequence alignment is required with tens of millions of protein sequences. To date, the rfGPT is the Golgi classifier with the best independent testing scores, optimized by training on smaller benchmark data sets. Feature importance analysis proves that the non-polar and aliphatic residues composition, the (aromatic residues) + (non-polar, aliphatic residues) dipeptide and aromatic residues composition between NH2-termial and COOH-terminal of protein sequences are the three top biological features for distinguishing the sub-Golgi proteins.
Introduction
The Golgi apparatus (GA) is an important organelle in eukaryotic cells, because lipids and different types of proteins are modified, packaged, and transported in vesicles to different destinations (Rhee et al., 2005). The GA comprises three main parts (Xu and Esko, 2009): cis-Golgi, medial, and trans-Golgi. The cis-Golgi receives proteins and then delivers them to the medial section for protein biosynthesis. The trans-Golgi releases the biosynthesized proteins from the medial section. The proteins in the cis-region of the GA are called cis-Golgi proteins, whereas trans-Golgi proteins are in the trans-Golgi part (Pfeffer, 2001).
Malfunction of the GA can disrupt protein biosynthesis in the medial part, which can lead to neurodegenerative diseases, such Parkinson's (Fujita et al., 2006; Yang J. et al., 2016) and Alzheimer's (Gonatas et al., 1998; Yang et al., 2015). A key step in the understanding of GA function is to determine whether a protein is a sub-Golgi protein (cis-Golgi or trans-Golgi). Such determinations will improve comprehension of the mechanisms for GA dysfunction and provide clues for disease treatment and more effective drug research and development (Gunther et al., 2018).
In the past few years, several protein subcellular locations and protein type prediction tools, including sub-Golgi protein identification tools (Teasdale and Yuan, 2002; Van Dijk et al., 2008; Chou et al., 2010; Ding et al., 2011, 2013; Jiao et al., 2014; Lin et al., 2014; Nikolovski et al., 2014; Jiao and Du, 2016a,b; Yang R. et al., 2016; Ahmad et al., 2017; Wang et al., 2017; Rahman et al., 2018; Ahmad and Hayat, 2019; Wuritu et al., 2019), have been developed using various machine learning algorithms, including increment diversity Mahalanobis discriminant (IDMD) (Ding et al., 2011), support vector machine (SVM) (Ding et al., 2013, 2017; Jiao et al., 2014; Lin et al., 2014; Jiao and Du, 2016a,b), random forest (RF) (Ding et al., 2016a,b; Yang R. et al., 2016; Yu et al., 2017; Liu et al., 2018), and K nearest neighbor algorithm (KNN) (Ahmad et al., 2017; Ahmad and Hayat, 2019), among others. To generate feature vectors for sub-Golgi protein identification, protein amino acid composition (AAC) (Rahman et al., 2018), k-gapped dipeptide composition (k-gapDC) (Ding et al., 2011, 2013), pseudo amino acid composition (PseAAC) (Jiao et al., 2014; Liu et al., 2015), and protein sequences evolutionary information (e.g., position-specific scoring matrix, PSSM) and their derivative features (Yang et al., 2014; Jiao and Du, 2016a,b; Yang R. et al., 2016; Ahmad et al., 2017; Rahman et al., 2018) have been used. Because the extensively used training benchmark data sets (Ding et al., 2013; Yang R. et al., 2016) are unbalanced in sub-Golgi protein classes, a synthetic minority over-sampling technique (SMOTE) has been adopted to obtain class-balanced data sets for training (Yang R. et al., 2016; Ahmad et al., 2017; Wan et al., 2017; Rahman et al., 2018; Ahmad and Hayat, 2019). Diversified feature selection methods, including analysis of variance (ANOVA) (Ding et al., 2013; Jiao and Du, 2016a), minimal redundancy-maximal relevance (mRMR) (Jiao and Du, 2016b; Wang S. P. et al., 2018), maximum relevance-maximum distance (MRMD) (Zou et al., 2016a,b), RF/Wrapper (Pan et al., 2018; Rahman et al., 2018), multi-voting for feature selection (Ahmad and Hayat, 2019), and lasso (Liu et al., 2016), among others, have been used to remove redundant features and improve the prediction accuracy with as few features as possible (Yu et al., 2016; Zhu et al., 2017, 2018; Kuang et al., 2018; Wang H. et al., 2018).
Two widely used benchmark-training data sets have resulted in different optimization models with various independent testing prediction scores. For the benchmark data set of Ding (137 sequences with 25% sequence identity; Ding et al., 2013), Jiao and Du (2016b) applied 49-dimensional features of positional-specific physicochemical properties (PSPCP, a derived feature from PSSM) to train their best SVM model. They achieved jackknife cross-validation results with accuracy (ACC) of 91.2%; Matthew correlation coefficient (MCC) of 0.793; sensitivity (Sn) of 99.0%; and specificity (Sp) of 73.8%, whereas the independent prediction accuracy of their classifier was 87.1%. The best predictor built on the benchmark data set of Yang (304 sequences with 40% sequence identity) (Yang R. et al., 2016) was developed by Ahmad and Hayat (2019). They carefully selected 180-dimensional features from the combined features of split amino acid composition (SAAC), 3-gap dipeptide composition, and PSSM with its derivative features to obtain a designed KNN classifier with good jackknife cross-validation scores (ACC = 94.9%; MCC = 0.90; Sn = 97.2%; Sp = 92.6%) and good independent testing scores (ACC = 94.0%; MCC = 0.84; Sn = 81.5%; Sp = 96.9%).
To our best knowledge, all high-profile sub-Golgi protein predictors trained on either benchmark data sets are constructed on the basis of a PSSM and its derived feature vectors, whose acquisition requires the use of a position-specific iterative basic local alignment search tool to align sub-Golgi protein sequences with a protein database (Jiao and Du, 2016a,b; Rahman et al., 2018; Ahmad and Hayat, 2019). Then, a secondary data transformation is performed (Altschul et al., 1997) in which data are usually converted into a 20 by 20 matrix with average values in each feature dimension (Jiao and Du, 2016a,b; Yang R. et al., 2016; Ahmad et al., 2017; Rahman et al., 2018). The sequence alignment is typically time-consuming, particularly when the protein database for alignment is large and the computing power is limited.
In this paper, instead of using PSSM and its derived features, the focus was on constructing an efficient sub-Golgi protein RF classifier, namely rfGPT, based only on amino acid and dipeptide composition-based feature vectors. Related studies (Li et al., 2016; Luo et al., 2016; Tang et al., 2018; Zhang et al., 2018a,b) have demonstrated the effectiveness of composition and dipeptide and amino acid composition-based features for solving bioinformatics problems. The rfGPT with 55-dimensional features of 2-gap dipeptide composition attained better jackknife cross-validation scores (ACC = 91.1%; MCC = 0.823; Sn = 87.4%; Sp = 94.7%) and better independent testing results (ACC = 89.1%; MCC = 0.631; Sn = 53.8%; Sp = 98.0%) than those classifiers trained on the same data set (Ding et al., 2013; Jiao and Du, 2016a,b). Therefore, to date, the rfGPT is the best sub-Golgi predictor trained from the benchmark data set of Ding via SMOTE (Ding et al., 2013). For further improvement of the rfGPT, 59 2-gap dipeptide composition features selected through ANOVA technology were fused with SAAC features to form 119 new dimensional features, which were then secondarily selected via ANOVA for rfGPT optimization. Ultimately, the rfGPT with 93 dimensional features [59 2-gap dipeptide composition (DC) sub-features plus 34 SAAC sub-features] was the best predictor, with jackknife cross-validation scores of ACC = 90.5%; MCC = 0.811; Sn = 92.6%; and Sp = 88.4%, and independent test scores of ACC = 90.6%; MCC = 0.696; Sn = 96.1%; and Sp = 69.2%.
Materials and Methods
Data Sets
To train models for sub-Golgi protein identification, two benchmark-training data sets are widely used. One data set, D1 in this text, was constructed by Ding et al. (2013), and the other, D2 in this text, was constructed by Yang R. et al. (2016). Before D1 was developed, Ding et al. constructed a smaller data set (D0) which was used once and never used again (Ding et al., 2011). In this work, the data set D1 was downloaded from http://lin-group.cn/server/SubGolgi/data and used to train the sub-Golgi protein classifier. The D1 data set consisted of 137 Golgi-resident protein sequences, with 42 cis-Golgi and 95 trans-Golgi proteins. The D1 data set was selected for model training primarily because the sequence identity was <25%. Thus, the D1 data set contained less sequence noise and redundancy than the D2 data set.
For testing the optimized model, an independent data set D3 provided by Ding et al. (2013) was applied. The D3 data set has been adopted by most of the key researchers in previously reported sub-Golgi predictors (Ding et al., 2013; Jiao and Du, 2016b; Yang R. et al., 2016; Ahmad et al., 2017; Rahman et al., 2018; Ahmad and Hayat, 2019). The D3 data set is generally used only for independent testing and contains 64 test sequences, including 13 cis-Golgi and 51 trans-Golgi protein sequences. The D3 data set is available at http://lin-group.cn/server/SubGolgi/data.
Modeling Overview
The entire rfGPT modeling process is illustrated in Figure 1. Compared with previous predictors, the major difference of the rfGPT used in this study was that only extracted features from amino acid and dipeptide composition were used. In this study, the 2-gapped dipeptide composition profile and SAAC were adopted. Ding et al. (2013) verified the validity of the 2-gapped dipeptide composition profile for sub-Golgi prediction. The SAAC considers that the location of a Golgi protein is related to the composition of amino acid residues at the N-terminal and C-terminal of a protein sequence (Paulson and Colley, 1989). As shown in Figure 1, the 400 dimensions (400D) 2-gapDC features extracted from D1 were used to generate a class-balanced data set via ANOVA and SMOTE, which was then fed into a RF model for optimization and estimation by jackknife cross-validation and independent testing. In this step, an optimized prediction model was sought, whose selected features were then combined with the SAAC features as new features of a new model for further optimization. After the secondary feature selection via ANOVA and SMOTE, the new optimal model was evaluated through jackknife cross-validation and independent testing.
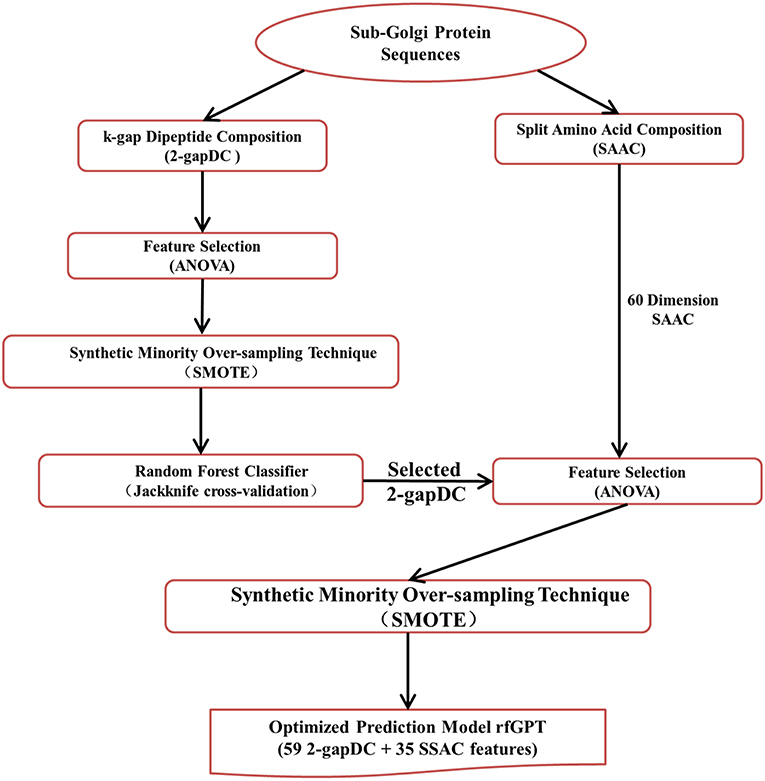
Figure 1. Modeling framework of the state-of-art random forests sub-Golgi protein classifier. ANOVA: analysis of variance.
Feature Extraction
The methods for feature extraction used for sub-Golgi classification are divided into three categories: (1) amino acid and peptide composition and their derived features; (2) PSSM and its derived features; and (3) features combined with amino acid residue physical and chemical properties. In this research, the derived features of category 1 were adopted because they are simple and convenient for feature extraction, namely, to calculate the frequency of peptide and amino acid components. The following two AAC features were adopted.
k-Gapped Dipeptides Composition
In general, the composition of adjacent dipeptides can only reflect the short-range structure of the protein sequence. The dipeptide composition in the larger interval may better reflect the tertiary structure of the protein. In biology, interval residues are more important than adjacent residues. Especially in some common structures, such as helices and plates, two non-adjacent residues are joined by hydrogen bonds (Lin et al., 2015; Wang et al., 2019). The k-gap dipeptides composition (k-gapDC) is an indirect mathematical description of the biological significance, which has been extensively utilized for sub-Golgi protein classification and other bioinformatics fields (Xu et al., 2018; Agrawal et al., 2019; Akbar et al., 2019; Wang et al., 2019). For the k-gapDC, the frequency of a dipeptide separated by k positions is determined, which is then divided by the total number of k-gapped dipeptides; thus, a protein sequence is transformed into a 400D feature vector. The 2-gapDC features were utilized in this work.
Split Amino Acid Composition
It has been proved that the N-terminal and C-terminal of protein sequences can act as signal-anchor domains for subcellular locations, e.g., glycosyltransferases all have a short NH2-terminalcytoplasmic tail, a 16-20-amino acid signal-anchor domain, and an extended stem region which is followed by the large COOH-terminal catalytic domain (Paulson and Colley, 1989). Another example is that lysine at position 329 within a C-terminal dilysine motif is crucial for the endoplasmic reticulum localization of human SLC35B4 (Bazan et al., 2018). All of these inspire us to used split amino acid composition for sub-Golgi protein identification. The split amino acid composition was proposed by Chou (Chou and Shen, 2007), which converts variable-length protein sequences into fixed-length amino acids for feature representation. In SAAC, a protein sequence is initially segmented into different parts, and then the amino acid frequency of each independent part is calculated. In the current work, the protein sequences were split into three segments: 30 N-terminal residues, 30 C-terminal residues, and the intermediate-block residues, which are the sequences between N-terminal and C-terminal parts. A 60D feature vector was obtained from the SAAC instead of the traditional 20D amino acid component. The details of the SAAC feature extraction are described as follows. Considering the length of protein sequence L and the three segments [NSeg (N-terminal), ISeg (intermediate block), and CSeg (C-terminal)] with the lengths Xn, L – Xn – Xc, and Xc (Xn = Xc = 30), respectively, the SAAC feature vector [f1, f2, ··· , f60] is generated by the following formulas:
•
•
•
AA : amino acid residue;
N(AA) : the numbers of AA in different segments.
L: the length of protein sequence;
Xn: the residues numbers of N-terminal segments;
Xc: the residues numbers of C-terminal segments.
fi: the ith SAAC feature vector element, it is one of the 20 amino acid residue frequency in a segment.
Feature Selection
Feature selection is conducted to remove redundant information and to overcome over-fitting in machine learning modeling. A variety of feature selection techniques (Ding et al., 2013; Jiao et al., 2014; Zeng et al., 2015, 2016, 2018; Jiao and Du, 2016a,b; Yang R. et al., 2016; Ahmad et al., 2017; Rahman et al., 2018; Ahmad and Hayat, 2019; Liu Y. et al., 2019; Zhang X. et al., 2019) have been important for sub-Golgi protein identification and for other areas of bioinformatics. ANOVA ranks the importance of features in terms of the ratio of the variance of data within a category to the variance between categories. The larger the value of the ratio is, the more important the feature is. The details for the use of ANOVA as a feature selection technique have been presented previously (Ding et al., 2013; Jiao and Du, 2016a) and are not repeated here. In this study, the ANOVA module from the famous Scikit-learn machine learning tool kit was used for feature selection (https://scikit-learn.org/).
Synthetic Minority Over-sampling Technique
The D1 benchmark data set is imbalanced, with the cis-Golgi protein and trans-Golgi protein sequences ratio of 0.44. Such an imbalance has a significant impact on the acceptability of the application, because the classifiers can be overly suitable for the majority classes. In this case, the prediction accuracy may seem high, but the results may be unacceptable, as minority groups may be completely/partially ignored. To solve this problem, the very effective SMOTE was proposed by Chawla et al. (2002). SMOTE helps to balance unbalanced data sets by creating “synthetic” minority class examples rather than by oversampling with replacement, and is employed by various sub-Golgi classifiers trained on benchmark data set D2 (Yang R. et al., 2016; Ahmad et al., 2017; Rahman et al., 2018; Ahmad and Hayat, 2019). As this manuscript was prepared, the use of SMOTE with benchmark data set D1 had not yet been reported. In this research, the SMOTE module implemented was from http://imbalanced-learn.org.
Evaluation Metrics
Testing Methods
The jackknife cross-validation is a leave-one-out cross-validation method for testing the efficiency of protein classification (Chou and Shen, 2006) and is executed in the following steps. A training data set with T items is separated into two parts. For each run, one part consists of T−1 item for model training, and the remaining part contains one item for testing. This process is repeated T times, and all the items sampled in the training data set act as a testing sample only once. Jackknife cross-validation is a time-consuming method, particularly for large data sets, but the method is robust with small variance. In this article, the benchmark data set D1 collected by Ding et al. (2013) was used for the jackknife cross-validation.
In independent testing, a completely different data set from the training data set is used to evaluate the trained model. Once the model is built with the training data set, tests are performed on the independent data set to evaluate the model. In this article, the independent data set D3 collected by Ding et al. (2013) was used for model performance evaluation.
Performance Metrics
Four standard metrics were used to evaluate the proposed models: ACC, Sn, Sp, and MCC. The metrics are previously described (Wei et al., 2017a,b; Chen et al., 2018; Su et al., 2018; Feng et al., 2019; Zhang S. et al., 2019) and were calculated as follows:
•
•
•
•
where TP is a true positive, TN is a true negative, FP is a false positive, and FN is a false negative.
Classifier
Support vector machine (SVM) (Ding et al., 2011, 2013; Feng et al., 2013; Lin et al., 2014; Jiao and Du, 2016a,b; Zeng et al., 2017; Rahman et al., 2018; Chen et al., 2019; Dao et al., 2019; Liu B. et al., 2019), K-nearest neighbor (KNN) (Ahmad et al., 2017; Ahmad and Hayat, 2019), and random forests (RF) (Yang R. et al., 2016; Pan et al., 2017; Ru et al., 2019; Su et al., 2019; Zheng et al., 2019) classifiers have been used to identify sub-Golgi proteins and for other fields. In this study, RF was selected for modeling because it is a powerful machine-learning tool and facilitates analysis of feature importance. Previously, Yang R. et al. (2016) selected 55 features from composite features (3-gapDC + PSSM derived features) to optimize their random forest classifier. The jackknife cross-validation scores using data set D2 were ACC = 88.5%; MCC = 0.765; Sn = 88.9%; and Sp = 88.0%, and for the independent testing, the scores were ACC = 93.8%; MCC = 0.821; Sn = 92.3%; and Sp = 94.1% (Yang R. et al., 2016). However, those results are somewhat confusing, because other sub-Golgi predictors have lower independent test scores than those for the jackknife cross-validation. To date, no sub-Golgi RF predictor has been trained from benchmark data set D1. In this study, the random forest classification model in the Scikit-learn tool kit (https://scikit-learn.org/) was applied for the implementation, testing, and evaluation of the rfGPT classifier and for the analysis of feature importance.
Results and Discussion
Performance of Random Forests Classifier Without Feature Selection
Table 1 shows the performance of the rfGPT using various extracted features. In the models with the SMOTE technique, the cross-validation scores improved remarkably for ACC, MCC, Sn, and Sp. For example, based on 460D SAAC + 2-gapDC features and SMOTE, the scores of the rfGPT were ACC = 90.5%; MCC = 0.817; Sn = 96.8%; and Sp = 84.2%, which were increases of 20, 132, 44, 2.2, and 171.6%, respectively, compared with the rfGPT without SMOTE. Although the SMOTE technique does improve the recognition rate of minority classes, the accuracy of the independent testing for the rfGPT with diverse features ranged from 78.1 to 82.8%, with little improvement with SMOTE (Table 1). For the other metrics (MCC, Sn, Sp), the case was the same. Thus, other techniques are needed to improve the generalization prediction model. In this paper, to obtain a better rfGPT with fewer features, ANOVA feature selection was used to eliminate redundant features.
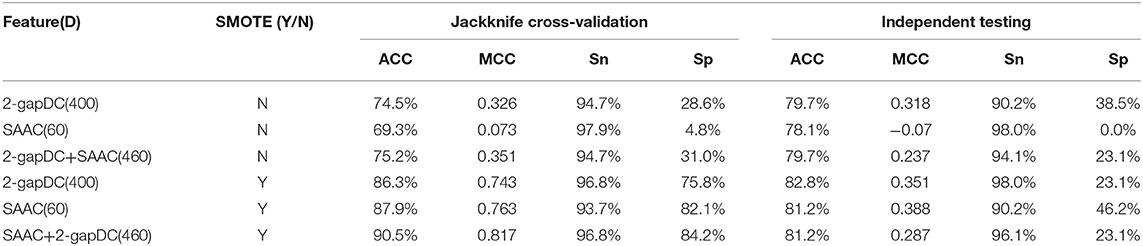
Table 1. Jackknife cross-validation and independent testing results after training on the benchmark data set D1 without feature selection.
Classifier Optimizing via ANOVA Feature Selection
To obtain the optimized classifier, the ANOVA feature selection method was first conducted for 400 2-gapDC features. One hundred sub-data sets containing 1, 2, … and 100 2-gapDC features generated separately after ANOVA feature selection were used for training 100 corresponding RF classifiers. For all 100 classifiers, jackknife cross-validation and independence testing were conducted. Figure 2A shows the accuracy of the cross-validation and independent tests of the 100 classifiers with varying numbers of features. Except for the models with nine and ten selected features, the average accuracy of the jackknife cross-validation of the other models was higher than that of the independent test results. Based on the jackknife cross-validation, the best-trained model with the highest accuracy was the classifier with 59 selected features (rfGPT_1), whereas the classifier with 55 selected features (rfGPT_2) had the highest independent testing accuracy results.
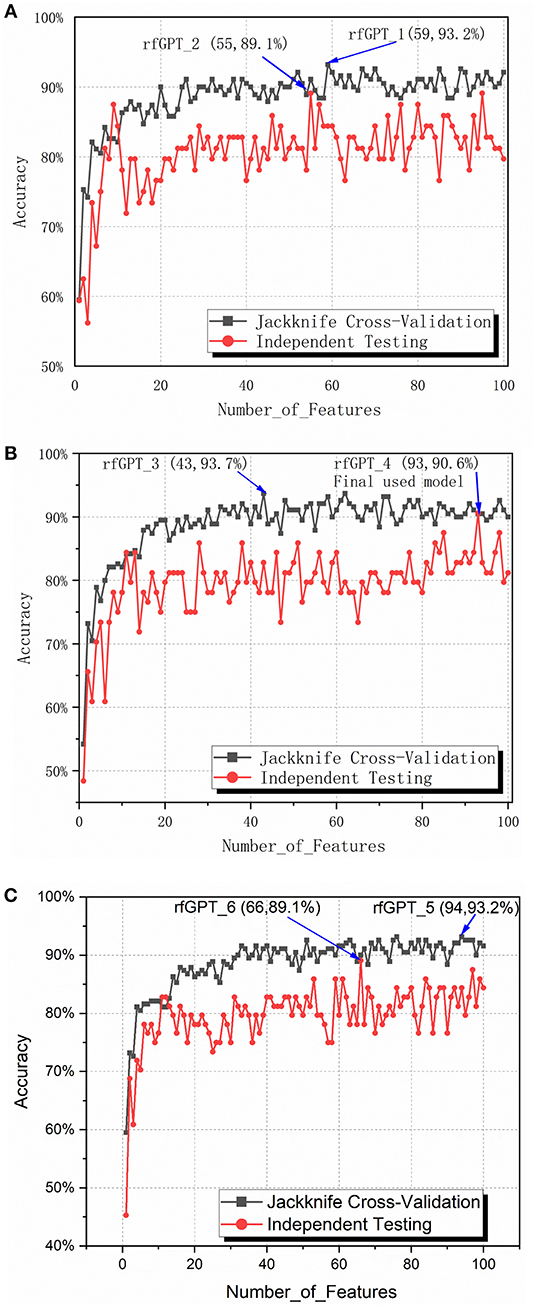
Figure 2. Jackknife cross-validation and independent testing accuracy of the random forest classifier with the number of features varied: (A) 2-gap dipeptide composition (2-gapDC) features (B) 59 selected 2-gapDC features + 60 split amino acid composition (SAAC) features, and (C) 55 selected 2-gapDC features + 60 SAAC features.
The performance scores of both classifiers are listed in Table 2. The jackknife cross-validation scores of rfGPT_2 (ACC = 91.1%; MCC = 0.823; Sn = 94.7%; Sp = 87.4%) were slightly lower than those of rfGPT_1 (ACC = 93.2%; MCC = 86.4%; Sn = 94.7%; Sp = 91.6%). However, rfGPT_2 had the better predictive performance on the independent test sets with scores of ACC = 89.1%; MCC = 0.631; Sn = 98%; and Sp = 53.8%, which were as much as 5.6, 35, 8.3, 10, and 16% larger than the corresponding values of rfGPT_1 (ACC = 84.4%; MCC = 0.466; Sn = 94.1%; Sp = 46.2%). The 89.1% independent testing accuracy of rfGPT_2 was an increase of 2.2% compared with the best SVM sub-Golgi classifier (Jiao and Du, 2016b) trained on the same benchmark data set (D1). The accuracy of 93.2% for rfGPT_1 and 91.1% for rfGPT_2 from the jackknife cross-validations was an increase of 9.0 and 6.5%, respectively, compared with that of the RF classifier obtained by Yang et al. which was trained on benchmark data set D2 (Yang R. et al., 2016).
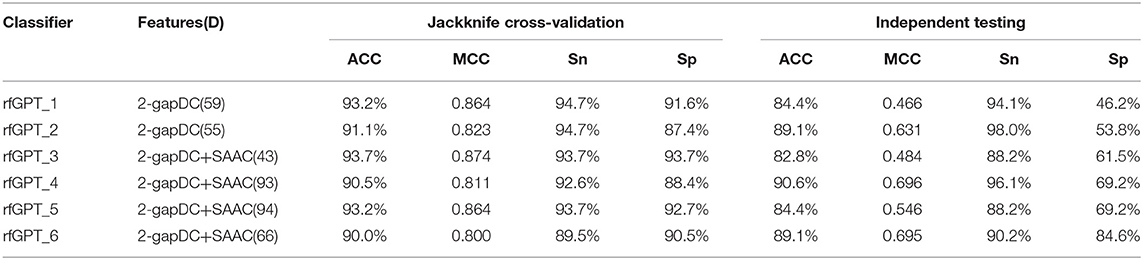
Table 2. The best evaluation scores from jackknife cross-validation and independent testing of different models with various feature types and feature numbers.
For further optimization, the 59 2-gapDC features of rfGPT_1 obtained in the previous step were combined with 60 SAAC features to form 119-dimensional (2-gapDC + SAAC) composite features, and then ANOVA was used to construct 100 data sets with selected 1, 2, ··· and 100 features for building 100 classifiers. The jackknife cross-validation and independent test results for these models are shown in Figure 2B and Table 2. For the cross-validation performance, classifier rfGPT_3 with 43 features was better than classifier rfGPT_4 with 93 features. However, for independent testing, the predictive metric of rfGPT_4 with ACC = 90.6%; MCC = 0.696; Sn = 96.1%; and Sp = 69.2% exceeded that of rfGPT_3 with ACC = 84.4%; MCC = 0.466; Sn = 88.2%; and Sp = 61.5%; the increases were 7.3%, 49, 8.3, 9.0, and 13%, respectively.
Optimization was also performed by combining the 55 2-gapDC features of rfGPT_2 with SAAC features to form 115-dimensional features for 100 new models with various features. The cross-validation and independent testing accuracy scores are revealed in Figure 2C. The scores for rfGPT_5 and rfGPT_6 are shown in Table 2. The independent accuracy of both models was inferior to that of rfGPT_4 (Table 2).
Because most cross-validation and independent testing scores of the classifier rfGPT_4 were superior to those of other models in Table 2, rfGPT_4 was designated as the final sub-Golgi model for prediction.
Feature Importance Analysis
To analyze the importance of the features selected for rfGPT_4, the feature importance function of the Scikit-learn RF model was exploited (Figure 3). As shown in Figure 3A, 59 2-gapDC features and 34 SAAC features were adopted in rfGPT_4, and their importance to the classification of Golgi proteins was 72.4 and 27.6%, respectively. Figure 3B shows the ranking of the 93 features by importance value and the cumulative importance score by importance value order. Among the combined features, the single feature importance was diverse and ranged from 0.16 to 3.64%. Figure 3C shows the importance order of the first 25 specific features, which accounted for 50% of the importance for the rfGPT. Only four of the top 25 features (which included 21 2-gapDC features and 4 SAAC features) had an importance value of more than 3% (Figure 3C).
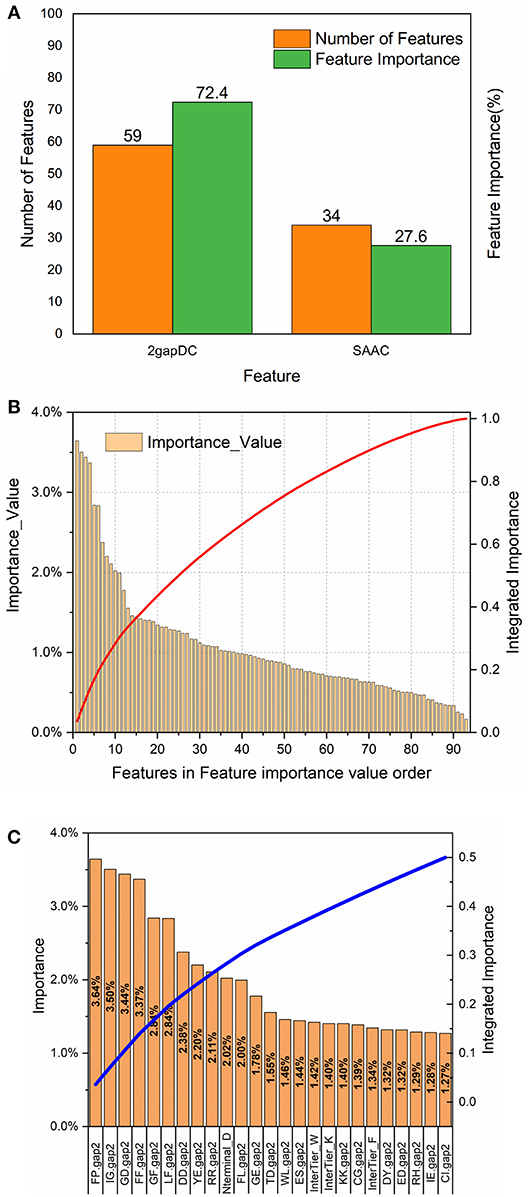
Figure 3. Feature importance analysis of random forests sub-Golgi classifier, rfGPT _4: (A) importance of feature types (B) the ranking orders of 93 features for rfGPT_4 and their integrated importance (red line), and (C) the importance of the top 25 features, which accounted for 50% of the integrated importance (blue line). The A1A2.gap2 means the composition of dipeptide A1A2. A1 or A2 is one of the 20 amino acid residues. Nterminal_D means the composition of amino acid residues D (aspartate) in NH2-terminal of protein sequence. InterTier_K, interTier_W, and interTier_F mean K(lysine), W(tryptophan), and F(phenylalanine) amino acid residues composition of the inter-tier between NH2-terminal and COOH-terminal of protein sequence.
To further analyze the feature bio-meaning, the feature importance values are assigned to different types of amino acid residues, that is aromatic residues, non-polar, and aliphatic residues, polar and non-charged residues, positively charged residues, and negatively charged residues. For instance, FP.gap2 feature as shown in Figure 3C means the composition frequency of dipeptide, which consists of F (phenylalanine) and P (proline) amino acid residence. The importance value 3.64% for FP.gap2 feature is divided by 2 to allocate 1.72% to aromatic residues type and non-polar and aliphatic residues type. Other features importance values are handled in the same way to assign importance value to five type amino acid residues (see Table S1). It finds out that the importance value of non-polar and aliphatic residues, aromatic residues, negatively charged residues, positively charged residues, polar, and non-charged residues are 30%, 24%, 21%, 13% and 12%, respectively. The non-polar and aliphatic property of amino acid residues plays the most critical role in sub-Golgi protein identification, and then the next is aromatic, negatively charged, positively charged, and polar and non-charged in turn. The importance values of the first three properties add up to 75%, so it concludes that to discriminate cis or trans sub-Golgi protein is mainly determined by the non-polar and aliphatic residues, aromatic residues, and negatively charged residues composition frequency.
For 2-gap DC features, the first three most important features are FP.gap2 (3.64%), IG.gap2 (3.50%), and GD.gap2 (3.44%), and five different residue types combined with each other generate 25 type dipeptides, whose feature importance values are listed in Figure 3C and Table S2. The (aromatic residues) + (non-polar, aliphatic residues) dipeptide, (non-polar, aliphatic residues) + (non-polar, aliphatic reduces residues) dipeptide and the (non-polar, aliphatic residues) + (aromatic residues) with the importance values as 8.54%, 8.18%, and 7.36%, respectively, are the top three important features for sub-Golgi classification.
For SAAC features, the protein sequence is segmented into three parts: N-terminal segment, C-terminal segment and the Interblock between N-terminal and C-terminal, whose amino acid composition frequency feature is labeled as Nterminal_A, Cterminal_A and InterTier_A (A represents one of the 20 amino acid residues; see Figure 3C and Table S3). The importance values of N-terminal features, C-terminal features, and Interblock features are 6.43%, 8.81%, and 12.37%, separately. The first three important values of 5 types residues of each block is aromatic residues of Interblock (5.05%), non-polar and aliphatic residues of C-terminal (3.13%), and negatively charged residues of N-terminal (3.00%). The D (aspartate) residues composition of N-terminal, as shown in Figures 3C, is the most important SAAC feature for sub-Golgi classification, but the aromatic residues composition frequency features of the Interblock seem even more important (see Table S3).
To sum up the above, the non-polar and aliphatic residues composition, the (aromatic residues) + (non-polar, aliphatic residues) dipeptide and aromatic residues composition between NH2-termial and COOH-terminal of protein sequences are three top biological features for distinguishing the sub-Golgi proteins.
Metrics Comparison With Existing Predictors
Ten optimized sub-Golgi classifiers that have been developed are presented in Table 3. Three separate data sets (D0, D1, D2), and four machine learning algorithms (IDMD, SVM, KNN, RF) were exploited to train these sub-Golgi classifiers, and one common independent data set was used to evaluate the various sub-Golgi classifiers. A total of six classifiers adopted the PSSM and its derived features for sub-Golgi prediction. Ahmad et al. (2017), training on the D2 data set with 40% sequence identity, achieved the highest independent testing scores (ACC = 94.8%; MCC = 0.86; Sn = 93.9%; Sn = 94.0%) for a classifier; the KNN sub-Golgi classifier with 83 composited features. In contrast to the KNN sub-Golgi classifier of Ahmad et al. the ultimate classifier rfGPT_4 in this paper was trained on the benchmark data set D1 with 25% sequence identity and contained 93 features, without any PSSM and its derivative features. Therefore, the rfGPT_4 is more practical, because the time-consuming sequence alignment step to obtain the PSSM and its derivatives scores using the Position-Specific Iterative Basic Local Alignment Search Tool is avoided. In addition, rfGPT_4 is currently the model with the best independent testing scores for training on data set D1 and is a state-of-art sub-Golgi classifier with only dipeptide and amino acid composition features.
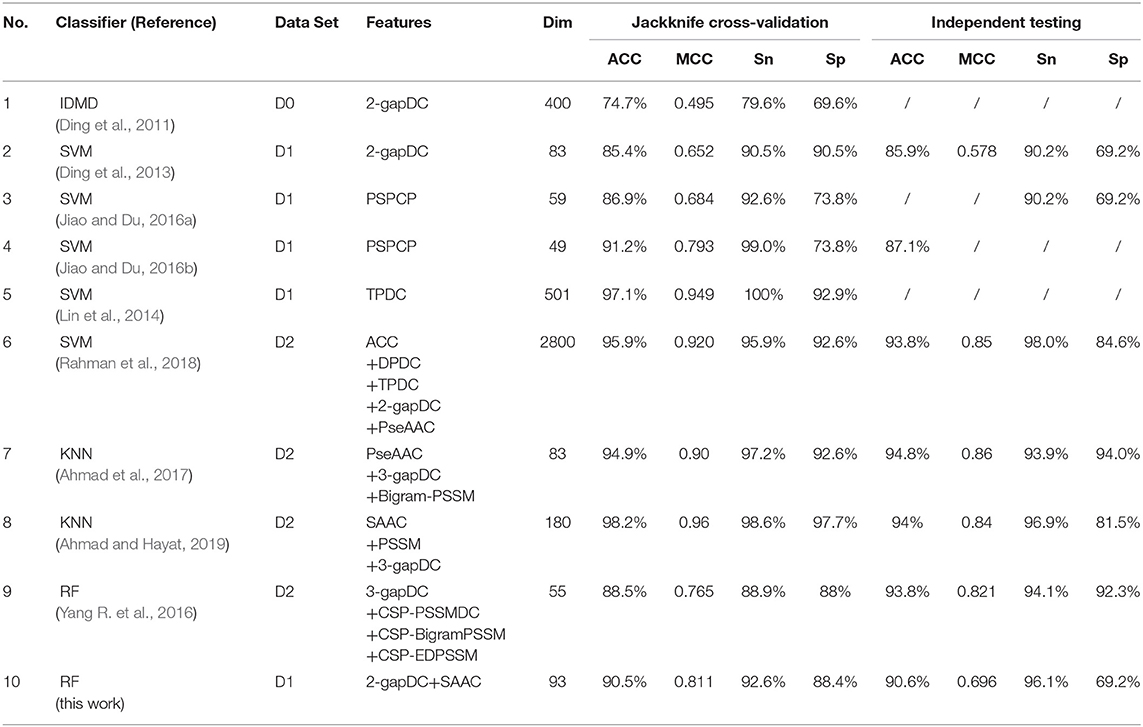
Table 3. Jackknife cross-validation and independent testing scores list for reported sub-Golgi protein classifiers.
Conclusions
In this work, an optimized rfGPT classifier for sub-Golgi protein type (cis and trans) identification was developed. The rfGPT classifier was derived from a random forests machine-learning algorithm, followed by implementation of the SMOTE to overcome a severe imbalance in the training data set and selection of optimal-related features using an ANOVA feature selection technique. The independent testing scores (ACC = 90.6%; MCC = 0.696; Sn = 96.1%; Sp = 69.2%) of the rfGPT ranked it as the one of the top sub-Golgi predictors. The feature importance analysis proves that the non-polar and aliphatic residues composition, the (aromatic residues) + (non-polar, aliphatic residues) dipeptide and aromatic residues composition for block between NH2-termial and COOH-terminal of protein sequence are the top biological features, which play the key role for sub-Golgi proteins identification.
As compared with previous reported sub-Golgi protein classifiers, the rfGPT is with only dipeptide and amino acid residue composition features, which exempted sequence alignment from the procedure. Also, the rfGPT adopted random forests algorithm is easier for feature analysis and for revealing the key bio-factors of sub-Golgi protein classification. However, the rfGPT had an independent prediction accuracy (from a training data set with 25% sequence identity) that was 4.4% lower than that for the best of the reported sub-Golgi protein identifiers (based on the 40% sequence identity data set) and rfGPT uses more features.
The expectation is to build a more general data set of Golgi protein sequences to train the rfGPT model and to realize a more advanced sub-Golgi classifier of the features. In the future, extreme learning (Li et al., 2019) and deep learning (Long et al., 2017; Yu et al., 2018; Lv et al., 2019; Wei et al., 2019; Zhang Z. et al., 2019; Zou et al., 2019) methods will be tested on this problem.
Data Availability
Publicly available datasets were analyzed in this study. This data can be found here: http://lin-group.cn/server/subGolgi2.
Author Contributions
ZL and SJ were responsible for experiments and manuscripts preparation. HD participated in discussions. QZ worked as supervisor for all procedures.
Funding
This work was supported by the National Key R&D Program of China (2018YFC0910405), the Natural Science Foundation of China (No. 61922020, No. 61771331), and the Scientific Research Foundation in Shenzhen (JCYJ20180306172207178).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbioe.2019.00215/full#supplementary-material
Abbreviations
D/Dim, dimension; D0/D1/D2/D3, data sets; IDMD, increment diversity Mahalanobis discriminant; SVM, supporting vector machine; KNN, K-nearest neighbors; RF, random forests; 2-gapDC, 2-gap dipeptide composition; 3-gapDC, 3-gap dipeptide composition; DPDC, Dipeptide compostion; TPDC, Tripeptide composition; AAC, amino acid composition; SAAC, split amino acid composition; PseAAC, pseudo amino acid composition; PSPCP, positional-specific physicochemical properties derived feature from PSSM; PSSM, position-specific scoring matrix; PSSMDC, PSSM-Dipeptide Composition; BigramPSSM, Bi-gram features directly extracted from PSSM; EDPSSM, Evolutionary Difference PSSM; CSP, Common Spatial Patterns; SMOTE, synthetic minority over-sampling technique; ACC, accuracy; MCC, Matthew correlation coefficient; Sn, Sensitivity; Sp, Specificity.
References
Agrawal, P., Kumar, S., Singh, A., Raghava, G. P. S., and Singh, I. K. (2019). NeuroPIpred: a tool to predict, design and scan insect neuropeptides. Sci. Rep. 9:12. doi: 10.1038/s41598-019-41538-x
Ahmad, J., and Hayat, M. (2019). MFSC: multi-voting based feature selection for classification of Golgi proteins by adopting the general form of Chou's PseAAC components. J. Theoret. Biol. 463, 99–109. doi: 10.1016/j.jtbi.2018.12.017
Ahmad, J., Javed, F., and Hayat, M. (2017). Intelligent computational model for classification of sub-Golgi protein using oversampling and fisher feature selection methods. Artif. Intell. Med. 78, 14–22. doi: 10.1016/j.artmed.2017.05.001
Akbar, S., Hayat, M., Kabir, M., and Iqbal, M. (2019). iAFP-gap-SMOTE: an efficient feature extraction scheme gapped dipeptide composition is coupled with an oversampling technique for identification of antifreeze proteins. Lett. Organic Chem. 16, 294–302. doi: 10.2174/1570178615666180816101653
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Bazan, B., Wiktor, M., Maszczak-Seneczko, D., Olczak, T., Kaczmarek, B., and Olczak, M. (2018). Lysine at position 329 within a C-terminal dilysine motif is crucial for the ER localization of human SLC35B4. PLoS ONE 13:e0207521. doi: 10.1371/journal.pone.0207521
Chawla, N. V., Bowyer, K. W., Hall, L. O., and Kegelmeyer, W. P. (2002). SMOTE: synthetic minority over-sampling technique. J. Artif. Intell. Res. 16, 321–357. doi: 10.1613/jair.953
Chen, W., Feng, P., Liu, T., and Jin, D. (2018). Recent advances in machine learning methods for predicting heat shock proteins. Curr. Drug. Metab. 20:224–228. doi: 10.2174/1389200219666181031105916
Chen, W., Lv, H., Nie, F., and Lin, H. (2019). i6mA-Pred: identifying DNA N6-methyladenine sites in the rice genome. Bioinformatics 35, 2796–2800. doi: 10.1093/bioinformatics/btz015
Chou, K.-C., and Shen, H.-B. (2006). Hum-PLoc: a novel ensemble classifier for predicting human protein subcellular localization. Biochem. Biophys. Res. Commun. 347, 150–157. doi: 10.1016/j.bbrc.2006.06.059
Chou, K.-C., and Shen, H.-B. (2007). Recent progress in protein subcellular location prediction. Analyt. Biochem. 370, 1–16. doi: 10.1016/j.ab.2007.07.006
Chou, W.-C., Yin, Y., and Xu, Y. (2010). GolgiP: prediction of Golgi-resident proteins in plants. Bioinformatics 26, 2464–2465. doi: 10.1093/bioinformatics/btq446
Dao, F. Y., Lv, H., Wang, F., Feng, C. Q., Ding, H., Chen, W., et al. (2019). Identify origin of replication in Saccharomyces cerevisiae using two-step feature selection technique. Bioinformatics 35, 2075–2083. doi: 10.1093/bioinformatics/bty943
Ding, H., Guo, S.-H., Deng, E.-Z., Yuan, L.-F., Guo, F.-B., Huang, J., et al. (2013). Prediction of Golgi-resident protein types by using feature selection technique. Chemometr. Intell. Lab. Syst. 124, 9–13. doi: 10.1016/j.chemolab.2013.03.005
Ding, H., Liu, L., Guo, F.-B., Huang, J., and Lin, H. (2011). Identify golgi protein types with modified mahalanobis discriminant algorithm and pseudo amino acid composition. Protein Peptide Lett. 18, 58–63. doi: 10.2174/092986611794328708
Ding, Y., Tang, J., and Guo, F. (2016a). Identification of Protein–Protein Interactions via a novel matrix-based sequence representation model with amino acid contact information. Int. J. Molecul. Sci. 17:1623. doi: 10.3390/ijms17101623
Ding, Y., Tang, J., and Guo, F. (2016b). Predicting protein-protein interactions via multivariate mutual information of protein sequences. BMC Bioinform. 17:398. doi: 10.1186/s12859-016-1253-9
Ding, Y., Tang, J., and Guo, F. (2017). Identification of drug-target interactions via multiple information integration. Inform. Sci. 418–419, 546–560. doi: 10.1016/j.ins.2017.08.045
Feng, C. Q., Zhang, Z. Y., Zhu, X. J., Lin, Y., Chen, W., Tang, H., et al. (2019). iTerm-PseKNC: a sequence-based tool for predicting bacterial transcriptional terminators. Bioinformatics 35, 1469–1477. doi: 10.1093/bioinformatics/bty827
Feng, P. M., Chen, W., Lin, H., and Chou, K. C. (2013). iHSP-PseRAAAC: identifying the heat shock protein families using pseudo reduced amino acid alphabet composition. Anal. Biochem. 442, 118–125. doi: 10.1016/j.ab.2013.05.024
Fujita, Y., Ohama, E., Takatama, M., Al-Sarraj, S., and Okamoto, K. (2006). Fragmentation of Golgi apparatus of nigral neurons with α-synuclein-positive inclusions in patients with Parkinson's disease. Acta Neuropathol. 112, 261–265. doi: 10.1007/s00401-006-0114-4
Gonatas, N. K., Gonatas, J. O., and Stieber, A. (1998). The involvement of the Golgi apparatus in the pathogenesis of amyotrophic lateral sclerosis, Alzheimer's disease, and ricin intoxication. Histochem. Cell Biol. 109, 591–600. doi: 10.1007/s004180050257
Gunther, T., Tulipano, G., Dournaud, P., Bousquet, C., Csaba, Z., Kreienkamp, H. J., et al. (2018). International union of basic and clinical pharmacology. CV. Somatostatin receptors: structure, function, ligands, and new nomenclature. Pharmacol. Rev. 70, 763–835. doi: 10.1124/pr.117.015388
Jiao, Y., Du, P., and Su, X. (2014). “Predicting Golgi-resident proteins in plants by incorporating N-terminal transmembrane domain information in the general form of Chou's pseudoamino acid compositions,” in: 2014 8th International Conference on Systems Biology (ISB) (Qingdao), 226–229.
Jiao, Y.-S., and Du, P.-F. (2016a). Predicting Golgi-resident protein types using pseudo amino acid compositions: approaches with positional specific physicochemical properties. J. Theoret. Biol. 391, 35–42. doi: 10.1016/j.jtbi.2015.11.009
Jiao, Y.-S., and Du, P.-F. (2016b). Prediction of Golgi-resident protein types using general form of Chou's pseudo-amino acid compositions: approaches with minimal redundancy maximal relevance feature selection. J. Theoret. Biol. 402, 38–44. doi: 10.1016/j.jtbi.2016.04.032
Kuang, L., Yu, L., Huang, L., Wang, Y., Ma, P., Li, C., et al. (2018). A personalized QoS prediction approach for CPS service recommendation based on reputation and location-aware collaborative filtering. Sensors 18:1556. doi: 10.3390/s18051556
Li, D., Luo, L., Zhang, W., Liu, F., and Luo, F. (2016). A genetic algorithm-based weighted ensemble method for predicting transposon-derived piRNAs. BMC Bioinform. 17:329. doi: 10.1186/s12859-016-1206-3
Li, Y., Niu, M., and Zou, Q. (2019). ELM-MHC: an improved MHC identification method with extreme learning machine algorithm. J. Proteome Res. 18, 1392–1401. doi: 10.1021/acs.jproteome.9b00012
Lin, H., Ding, H., and Chen, W. (2014). Prediction of golgi-resident protein types using computational method. Bentham Sci. 1, 174–193. doi: 10.2174/9781608058624114010011
Lin, H., Liu, W. X., He, J., Liu, X. H., Ding, H., and Chen, W. (2015). Predicting cancerlectins by the optimal g-gap dipeptides. Sci. Rep. 5:16964. doi: 10.1038/srep16964
Liu, B., Chen, J., Guo, M., and Wang, X. (2019). Protein remote homology detection and fold recognition based on Sequence-Order Frequency Matrix. IEEE/ACM Transact. Comput. Biol. Bioinform. 16, 292–300. doi: 10.1109/TCBB.2017.2765331
Liu, B., Fang, Y., Huang, D.-S., and Chou, K.-C. (2018). iPromoter-2L: a two-layer predictor for identifying promoters and their types by multi-window-based PseKNC. Bioinformaitcs 34, 33–40. doi: 10.1093/bioinformatics/btx579
Liu, B., Liu, F., Wang, X., Chen, J., Fang, L., and Chou, K.-C. (2015). Pse-in-One: a web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res. 43, W65–W71. doi: 10.1093/nar/gkv458
Liu, X., Yang, J., Zhang, Y., Fang, Y., Wang, F., Wang, J., et al. (2016). A systematic study on drug-response associated genes using baseline gene expressions of the Cancer Cell Line Encyclopedia. Sci. Rep. 6:22811. doi: 10.1038/srep22811
Liu, Y., Wang, X., and Liu, B. (2019). A comprehensive review and comparison of existing computational methods for intrinsically disordered protein and region prediction. Briefings Bioinform. 20, 330–346. doi: 10.1093/bib/bbx126
Long, H. X., Wang, M., and Fu, H. Y. (2017). Deep convolutional neural networks for predicting hydroxyproline in proteins. Curr. Bioinform. 12, 233–238. doi: 10.2174/1574893612666170221152848
Luo, L., Li, D., Zhang, W., Tu, S., Zhu, X., and Tian, G. (2016). Accurate prediction of transposon-derived piRNAs by integrating various sequential and physicochemical features. PLoS ONE 11:e0153268. doi: 10.1371/journal.pone.0153268
Lv, Z. B., Ao, C. Y., and Zou, Q. (2019). Protein function prediction: from traditional classifier to deep learning. Proteomics 19:1900119. doi: 10.1002/pmic.201900119
Nikolovski, N., Shliaha, P. V., Gatto, L., Dupree, P., and Lilley, K. S. (2014). Label-free protein quantification for plant golgi protein localization and abundance. Plant Physiol. 166, 1033–1043. doi: 10.1104/pp.114.245589
Pan, Y., Liu, D., and Deng, L. (2017). Accurate prediction of functional effects for variants by combining gradient tree boosting with optimal neighborhood properties. PLoS ONE 12:e0179314. doi: 10.1371/journal.pone.0179314
Pan, Y. W., Zixiang, Z. W, and Deng, L. (2018). Computational identification of binding energy hot spots in protein-RNA complexes using an ensemble approach. Bioinformatics 34, 1473–1480. doi: 10.1093/bioinformatics/btx822
Paulson, J. C., and Colley, K. J. (1989). Glycosyltransferases. Structure, localization, and control of cell type-specific glycosylation. J Biol Chem. 264, 17615–17618.
Pfeffer, S. R. (2001). Constructing a Golgi complex. J. Cell Biol. 155, 873–875. doi: 10.1083/jcb.200109095
Rahman, M. S., Rahman, M. K., Kaykobad, M., and Rahman, M. S. (2018). isGPT: an optimized model to identify sub-Golgi protein types using SVM and Random Forest based feature selection. Artif. Intell. Med. 84, 90–100. doi: 10.1016/j.artmed.2017.11.003
Rhee, S. W., Starr, T., Forsten-Williams, K., and Storrie, B. (2005). The steady-state distribution of glycosyltransferases between the golgi apparatus and the endoplasmic reticulum is approximately 90:10. Traffic 6, 978–990. doi: 10.1111/j.1600-0854.2005.00333.x
Ru, X. Q., Li, L. H., and Zou, Q. (2019). Incorporating distance-based top-n-gram and random forest to identify electron transport proteins. J. Proteome Res. 18, 2931–2939. doi: 10.1021/acs.jproteome.9b00250
Su, R., Liu, X., Wei, L., and Zou, Q. (2019). Deep-resp-forest: a deep forest model to predict anti-cancer drug response. Methods 166, 91–102. doi: 10.1016/j.ymeth.2019.02.009
Su, R., Wu, H., Xu, B., Liu, X., and Wei, L. (2018). Developing a multi-dose computational model for drug-induced hepatotoxicity prediction based on toxicogenomics data. IEEE/ACM Transact. Comput. Biol. Bioinform. 16, 1231–1239. doi: 10.1109/TCBB.2018.2858756
Tang, G., Shi, J., Wu, W., Yue, X., and Zhang, W. (2018). Sequence-based bacterial small RNAs prediction using ensemble learning strategies. BMC Bioinform. 19:503. doi: 10.1186/s12859-018-2535-1
Teasdale, R. D., and Yuan, Z. (2002). Prediction of Golgi Type II membrane proteins based on their transmembrane domains. Bioinformatics 18, 1109–1115. doi: 10.1093/bioinformatics/18.8.1109
Van Dijk, A. D. J., Van Der Krol, A. R., Ter Braak, C. J. F., Bosch, D., and Van Ham, R. C. H. J. (2008). Predicting sub-Golgi localization of type II membrane proteins. Bioinformatics 24, 1779–1786. doi: 10.1093/bioinformatics/btn309
Wan, S., Duan, Y., and Zou, Q. (2017). HPSLPred: an ensemble multi-label classifier for human protein subcellular location prediction with imbalanced source. Proteomics 17:1700262. doi: 10.1002/pmic.201700262
Wang, H., Liu, C., and Deng, L. (2018a). Enhanced prediction of hot spots at protein-protein interfaces using extreme gradient boosting. Sci. Rep. 8:14285. doi: 10.1038/s41598-018-32511-1
Wang, S. P., Zhang, Q., Lu, J., and Cai, Y. D. (2018b). Analysis and prediction of nitrated tyrosine sites with the mRMR method and support vector machine algorithm. Curr. Bioinform. 13, 3–13. doi: 10.2174/1574893611666160608075753
Wang, X., Li, H., Gao, P., Liu, Y., and Zeng, W. (2019). Combining support vector machine with dual g-gap dipeptides to discriminate between acidic and alkaline enzymes. Lett. Organic Chem. 16, 325–331. doi: 10.2174/1570178615666180925125912
Wang, Y., Ding, Y., Guo, F., Wei, L., and Tang, J. (2017). Improved detection of DNA-binding proteins via compression technology on PSSM information. PLoS ONE 12:e0185587. doi: 10.1371/journal.pone.0185587
Wei, L., Su, R., Wang, B., Li, X., Zou, Q., and Gao, X. (2019). Integration of deep feature representations and handcrafted features to improve the prediction of N 6-methyladenosine sites. Neurocomputing 324, 3–9. doi: 10.1016/j.neucom.2018.04.082
Wei, L., Wan, S., Guo, J., and Wong, K. K. (2017a). A novel hierarchical selective ensemble classifier with bioinformatics application. Artif. Intell. Med. 83, 82–90. doi: 10.1016/j.artmed.2017.02.005
Wei, L., Xing, P., Zeng, J., Chen, J., Su, R., and Guo, F. (2017b). Improved prediction of protein–protein interactions using novel negative samples, features, and an ensemble classifier. Artif. Intell. Med. 83, 67–74. doi: 10.1016/j.artmed.2017.03.001
Wuritu, Y., Xiao-Juan, Z., Jian, H., Hui, D., and Hao, L. (2019). A brief survey of machine learning methods in protein sub-golgi localization. Curr. Bioinform. 14, 234–240. doi: 10.2174/1574893613666181113131415
Xu, D., and Esko, J. D. (2009). A Golgi-on-a-chip for glycan synthesis. Nat. Chem. Biol. 5:612. doi: 10.1038/nchembio0909-612
Xu, L., Liang, G., Wang, L., and Liao, C. (2018). A novel hybrid sequence-based model for identifying anticancer peptides. Genes 9:158. doi: 10.3390/genes9030158
Yang, J., Grunewald, S., Xu, Y., and Wan, X. F. (2014). Quartet-based methods to reconstruct phylogenetic networks. BMC Syst. Biol. 8:21. doi: 10.1186/1752-0509-8-21
Yang, J., Huang, T., Petralia, F., Long, Q., Zhang, B., Argmann, C., et al. (2015). Synchronized age-related gene expression changes across multiple tissues in human and the link to complex diseases. Sci. Rep. 5:15145. doi: 10.1038/srep15145
Yang, J., Huang, T., Song, W. M., Petralia, F., Mobbs, C. V., Zhang, B., et al. (2016). Discover the network underlying the connections between aging and age-related diseases. Sci. Rep. 6:32566. doi: 10.1038/srep32566
Yang, R., Zhang, C., Gao, R., and Zhang, L. (2016). A novel feature extraction method with feature selection to identify golgi-resident protein types from imbalanced data. Int. J. Molecul. Sci. 17:218. doi: 10.3390/ijms17020218
Yu, L., Ma, X., Zhang, L., Zhang, J., and Gao, L. (2016). Prediction of new drug indications based on clinical data and network modularity. Sci. Rep. 6:32530. doi: 10.1038/srep32530
Yu, L., Su, R., Wang, B., Zhang, L., Zou, Y., Zhang, J., et al. (2017). Prediction of novel drugs for hepatocellular carcinoma based on multi-source random walk. Ieee-Acm Transact. Comput. Biol. Bioinform. 14, 966–977. doi: 10.1109/TCBB.2016.2550453
Yu, L., Sun, X., Tian, S. W., Shi, X. Y., and Yan, Y. L. (2018). Drug and nondrug classification based on deep learning with various feature selection strategies. Curr. Bioinform. 13, 253–259. doi: 10.2174/1574893612666170125124538
Zeng, X., Liao, Y., Liu, Y., and Zou, Q. (2016). Prediction and validation of disease genes using hetesim scores. IEEE/ACM Transact. Computat. Biol. Bioinform. 14, 687–695. doi: 10.1109/TCBB.2016.2520947
Zeng, X., Liao, Y., Liu, Y., and Zou, Q. (2017). Prediction and validation of disease genes using hetesim scores. IEEE/ACM Transact. Comput. Biol. Bioinform. 14, 687–695.
Zeng, X., Yuan, S., Huang, X., and Zou, Q. (2015). Identification of cytokine via an improved genetic algorithm. Front. Comput. Sci. 9, 643–651. doi: 10.1007/s11704-014-4089-3
Zeng, X. X., Liu, L., Lu, L. Y., and Zou, Q. (2018). Prediction of potential disease-associated microRNAs using structural perturbation method. Bioinformatics 34, 2425–2432. doi: 10.1093/bioinformatics/bty112
Zhang, S., Zhang, T., and Liu, C. (2019). Prediction of apoptosis protein subcellular localization via heterogeneous features and hierarchical extreme learning machine. Sar Qsar Environ. Res. 30, 209–228. doi: 10.1080/1062936X.2019.1576222
Zhang, W., Qu, Q., Zhang, Y., and Wang, W. (2018a). The linear neighborhood propagation method for predicting long non-coding RNA–protein interactions. Neurocomputing 273, 526–534. doi: 10.1016/j.neucom.2017.07.065
Zhang, W., Yue, X., Tang, G., Wu, W., Huang, F., and Zhang, X. (2018b). SFPEL-LPI: sequence-based feature projection ensemble learning for predicting LncRNA-protein interactions. PLoS Comput. Biol. 14:e1006616. doi: 10.1371/journal.pcbi.1006616
Zhang, X., Zou, Q., Rodriguez-Paton, A., and Zeng, C. B. (2019). Meta-path methods for prioritizing candidate disease miRNAs. IEEE/ACM Transact. Comput. Biol. Bioinform. 16, 283–291. doi: 10.1109/TCBB.2017.2776280
Zhang, Z., Zhao, Y., Liao, X., Shi, W., Li, K., Zou, Q., et al. (2019). Deep learning in omics: a survey and guideline. Brief. Funct. Genom. 18, 41–57. doi: 10.1093/bfgp/ely030
Zheng, N., Wang, K., Zhan, W., and Deng, L. (2019). Targeting virus-host protein interactions: feature extraction and machine learning approaches. Curr. Drug Metabol. 20, 177–184. doi: 10.2174/1389200219666180829121038
Zhu, P. F., Xu, Q., Hu, Q. H., Zhang, C. Q., and Zhao, H. (2018). Multi-label feature selection with missing labels. Pattern Recogn. 74, 488–502. doi: 10.1016/j.patcog.2017.09.036
Zhu, P. F., Zhu, W. C., Hu, Q. H., Zhang, C. Q., and Zuo, W. M. (2017). Subspace clustering guided unsupervised feature selection. Pattern Recogn. 66, 364–374. doi: 10.1016/j.patcog.2017.01.016
Zou, Q., Wan, S., Ju, Y., Tang, J., and Zeng, X. (2016a). Pretata: predicting TATA binding proteins with novel features and dimensionality reduction strategy. Bmc Systems Biol. 10:114. doi: 10.1186/s12918-016-0353-5
Zou, Q., Xing, P., Wei, L., and Liu, B. (2019). Gene2vec: gene subsequence embedding for prediction of mammalian N6-Methyladenosine sites from mRNA. RNA 25, 205–218. doi: 10.1261/rna.069112.118
Keywords: random forests, sub-Golgi protein classifier, ANOVA feature selection, split amino acid composition, k-gap dipeptide, synthetic minority over-sampling
Citation: Lv Z, Jin S, Ding H and Zou Q (2019) A Random Forest Sub-Golgi Protein Classifier Optimized via Dipeptide and Amino Acid Composition Features. Front. Bioeng. Biotechnol. 7:215. doi: 10.3389/fbioe.2019.00215
Received: 23 July 2019; Accepted: 22 August 2019;
Published: 04 September 2019.
Edited by:
Yudong Cai, Shanghai University, ChinaReviewed by:
Tao Zeng, Shanghai Institutes for Biological Sciences (CAS), ChinaZhiwen Yu, South China University of Technology, China
Copyright © 2019 Lv, Jin, Ding and Zou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Quan Zou, em91cXVhbkBuY2xhYi5uZXQ=
†These authors have contributed equally to this work