 Shiqi Zhang1,2†
Shiqi Zhang1,2† Tao Zeng3†
Tao Zeng3† Bin Hu4†
Bin Hu4† Yu-Hang Zhang5
Yu-Hang Zhang5 Kaiyan Feng6
Kaiyan Feng6 Lei Chen7
Lei Chen7 Zhibin Niu8
Zhibin Niu8 Jianhao Li4
Jianhao Li4 Tao Huang5*
Tao Huang5* Yu-Dong Cai1*
Yu-Dong Cai1*- 1School of Life Sciences, Shanghai University, Shanghai, China
- 2Department of Biostatistics, University of Copenhagen, Copenhagen, Denmark
- 3Shanghai Research Center for Brain Science and Brain-Inspired Intelligence, Shanghai, China
- 4State Key Laboratory of Livestock and Poultry Breeding, Guangdong Public Laboratory of Animal Breeding and Nutrition, Guangdong Key Laboratory of Animal Breeding and Nutrition, Institute of Animal Science, Guangdong Academy of Agricultural Sciences, Guangzhou, China
- 5Shanghai Institute of Nutrition and Health, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, China
- 6Department of Computer Science, Guangdong AIB Polytechnic, Guangzhou, China
- 7College of Information Engineering, Shanghai Maritime University, Shanghai, China
- 8College of Intelligence and Computing, Tianjin University, Tianjin, China
DNA methylation is an essential epigenetic modification for multiple biological processes. DNA methylation in mammals acts as an epigenetic mark of transcriptional repression. Aberrant levels of DNA methylation can be observed in various types of tumor cells. Thus, DNA methylation has attracted considerable attention among researchers to provide new and feasible tumor therapies. Conventional studies considered single-gene methylation or specific loci as biomarkers for tumorigenesis. However, genome-scale methylated modification has not been completely investigated. Thus, we proposed and compared two novel computational approaches based on multiple machine learning algorithms for the qualitative and quantitative analyses of methylation-associated genes and their dys-methylated patterns. This study contributes to the identification of novel effective genes and the establishment of optimal quantitative rules for aberrant methylation distinguishing tumor cells with different origin tissues.
Introduction
DNA methylation is an essential epigenetic modification for multiple biological processes (Gao et al., 2017). It is characterized by the formation of 5-methylcytosine in the CpG site with the control of DNA methyltransferases (Moore et al., 2013). Recent studies have discovered that non-CpG methylation functions as an expression regulator in mammals (Guo et al., 2014; Zhang et al., 2017). However, the primary role of this process in mammals remains elusive. Since DNA methylation was considered a regulator in gene expression in the 1970's (Holliday and Pugh, 1975), numerous studies have investigated methylation-associated mechanisms, and functions. Ample solid evidence suggests that DNA methylation is involved in essential developmental events, such as X-chromosome inactivation and genomic imprinting. Current knowledge is that DNA methylation in mammals acts as an epigenetic mark of transcriptional repression.
During pathologic progression, tumors are deemed to be a genetic, and epigenetic disease. Classic genetic and epigenetic alterations co-determine tumor initiation and progression (Zhou et al., 2016). Aberrant levels of DNA methylation can be observed in various types of tumor cells. With the increasing recognition of tumorigenesis, altered DNA methylation has been described as a basic “cancer driver” event (Campan et al., 2011) that can be divided into two types, namely, hypomethylation and hypermethylation. In general, the over-activation of proto-oncogenes caused by DNA hypomethylation is a major dysfunctional process during tumorigenesis (Renaud et al., 2015, 2016; Good et al., 2018). Meanwhile, abnormal hypermethylation in CpG islands of tumor suppressor gene promoter (e.g., PTEN and p16) could lead to gene silencing and tumor initiation (Marzese et al., 2014; Cui et al., 2015; De La Rosa et al., 2017). The methylation abnormally and indirectly induces tumorigenesis in other DNA regions, such as repetitive sequences (Hur et al., 2014; Burns, 2017; Chen et al., 2017c). Hence, studies on DNA methylation are warranted to provide new and feasible tumor therapies.
Divergent methylation patterns are intensely associated with cell differentiation (Farlik et al., 2016). Even in a single cell line, methylation patterns may be dynamic among different stages (Kaaij et al., 2013; Petell et al., 2016), and this situation is common for tumor cells. In accordance with the initial original organs and tissues, tumors can be divided into different subtypes with different genome-wide methylation patterns. Therefore, a part of particular methylation patterns should be recognized as epigenetic marks for specific tumor sites (Sahm et al., 2017). For example, mucin is a macromolecular glycoprotein secreted mainly by goblet cells, which act as a protective barrier (Pelaseyed et al., 2014), and hypomethylation of mucin gene MUC5AC is considered a feature in colorectal cancers (Renaud et al., 2015, 2016). Another research also reveals that BRCA1, an essential tumor-suppressor gene, is highly associated with breast and ovarian cancer when the promoter undergoes hypermethylation (Evans et al., 2018). Hence, DNA methylation is supposed to emerge as a tumor-specific marker with large potentiality.
Most conventional studies considered single-gene methylation or specific loci as biomarkers for tumorigenesis. However, the entire genome-scale methylated modification has not been fully revealed. Tumor is a typical type of disease with high heterogeneity and individual difference. Thus, the combination of multiple sites with methylation patterns can highly increase the accuracy and sensitivity of markers. Hence, in this study, we proposed and compared two novel computational approaches involving multiple algorithms, namely, Monte Carlo feature selection (MCFS; Draminski et al., 2008), minimum redundancy maximum relevance (mRMR; Peng et al., 2005), and repeated incremental pruning to produce error reduction (RIPPER; Cohen, 1995), for the qualitative and quantitative analyses of methylation-associated genes and their dys-methylated patterns. This study contributes to the identification of novel effective genes and the establishment of optimal quantitative rules for methylation distinguishing tumor cells with different origin tissues.
Materials and Methods
Dataset
We downloaded the methylation profiles of 1,022 cell lines from Gene Expression Omnibus under accession number GSE68379 (Iorio et al., 2016). In each cell line, the methylation levels of 485,512 probes were measured. We applied the KNN method to impute the missing values. The R function impute.knn from package impute (https://bioconductor.org/packages/impute/) was used, and K was set to 10. Of note, there were actually very few missing values in this dataset, where the highest missing value percentage of the samples was about 0.1%. Therefore, we used the default parameter of K (10) and did not try other values. The 1,022 cell lines were from 13 tissues, and the sample sizes of 13 tissues are listed in Table 1. We determined whether the cell lines from different tissues differ in methylation level.

Table 1. Sample sizes of 13 tissues.
Feature Selection
We proposed two novel feature selection schemes for detecting specific signatures to distinguish methylation-related genes in tumor cells. We use mRMR (Peng et al., 2005) and MCFS (Draminski et al., 2008) to evaluate each feature, select the candidate features, and then use the support vector machine (SVM; Cortes and Vapnik, 1995) and other alternative algorithms to train the subsets of features in the incremental feature selection (IFS; Liu and Setiono, 1998) to identify specific signatures for screening tumor cells.
Selection of Important Features
Each cell line is represented by more than 480,000 methylation features. Clearly, it is impossible that all of them are essential for classifying cell lines into correct tissues. Thus, we first adopt mutual information (MI) to select essential features. The mutual information (MI) between x and y is defined as follows:
where p(x) represents marginal probabilistic density of x and p(x, y) indicates joint probabilistic density of x and y. For each feature, the MI value to class labels is calculated. It is widely accepted that features with high MI values are highly related to class labels, thereby giving key contributions for classification. Thus, we can select important features by setting a threshold for MI value. Features with MI values higher than the threshold are selected for further evaluation. They will be assessed by the following two feature selection methods.
Minimum Redundancy and Maximum Relevance
Remaining features are analyzed by mRMR (Peng et al., 2005). As a feature filtering method, mRMR requires two optimal targets on the highest relevance among selected feature subsets, namely, the maximum relevance between feature sets and labels and the minimum redundancy between features themselves (Peng et al., 2005). Such evaluations are all based on MI values. The output of mRMR contains a feature list, which sorts features according to maximum relevance and minimum redundancy. The list is generated by selecting a feature with maximum relevance to labels and minimum redundancy to already-selected features one by one and adding it to the current feature list.
Monte Carlo Feature Selection
Remaining features are also evaluated by MCFS (Draminski et al., 2008). This method has been applied as a classical feature selection method for dealing with many biological problems. MCFS is a random sampling-based feature selection method. In specific, MCFS trains multiple decision trees in a bootstrap sample set and a subset of randomly selected features (e.g., m features from the original M features, and m < < M). For a specific feature subset, samples with this subset of features can compose p bootstrap training sets. Thus, p decision trees can be obtained through training and evaluation. Assuming that this process is repeated t times, we can finally obtain p × t decision trees.
Relative importance (RI) is a score used to define how features are performed in each constructed classifier from the p × t decision trees. The RI score for a feature g is calculated as follows:
where wAcc is the weighted accuracy calculated by the mean sensitivity of all decision classes, ng(τ) is a node involving feature g in decision tree τ, IG(ng(τ)) is the information gain of ng(τ), no.in τ is the number of samples in decision tree τ, and no.in ng(τ) is the number of training samples in node ng(τ). In addition, u and v are two different weighting factors for adjusting different optimal contributions. After features has been assigned RI scores, a feature list can be generated by the decreasing order of their RI scores.
In this study, we used the MCFS program retrieved from http://www.ipipan.eu/staff/m.draminski/mcfs.html. Default parameters were used to execute such program, where p = 2000, t = 5, and u = v = 1.
Incremental Feature Selection
In the descending ordered feature list generated by MCFS or mRMR, we perform IFS to filter out a set of optimal features for accurately distinguishing different sample groups/classes (Liu and Setiono, 1998). We construct a series of feature subsets with an interval of 10 from the ranked feature list F by MCFS or mRMR. We generate m feature subsets , where the i-th feature subset contains the top 10 × i features in F. All feature subsets are tested by building and evaluating the SVM classifier (or other alternative methods such as rule-based approaches) using 10-fold cross-validation. The feature subset with the best performance is called the optimal feature subset.
Supervised Classifier
The supervised classifiers for IFS include “black-box” classifier SVM, interpretable rule learning classifier RIPPER (Cohen, 1995), and PART algorithm (Frank and Witten, 1998).
Support Vector Machine
SVM is a supervised learning algorithm based on statistical learning theory (Cortes and Vapnik, 1995; Chen et al., 2017b, 2018a; Che et al., 2020; Zhou et al., 2020a,b). It uses kernel techniques (such as Gaussian kernels) to map the original data from a low-dimensional non-linear space to a high-dimensional linear space and then fits the hyperplane in the high-dimensional space with the largest margin between the two classes of samples by using a linear function. We use the sequential minimal optimization (SMO) algorithm in software Weka for SVM classifier training with default parameters. The kernel was a polynomial function, the regularization parameter C was one.
Rule Learning Classifier RIPPER
We also use RIPPER (Cohen, 1995), a learner proposed by William that can generate classification rules to classify samples from different tumor cells. RIPPER can learn interpretable classifications for predicting new data in accordance with IF-ELSE rules. RIPPER learns all rules for each sample class. After learning rules for one class, RIPPER moves to learn the rules for the next class. RIPPER starts from the minority sample class and then to the second minority sample class until the dominant class. The “JRip” tool, implementing RIPPER algorithm, in Weka is used. Default parameters are adopted, where the parameter to determine the amount of data used for pruning is set to three.
Rule Learning Classifier PART
Different from the RIPPER algorithm that builds a full decision tree, the PART algorithm (Frank and Witten, 1998) learns rules by repeatedly generating partial decision trees. It uses a separate-and-conquer strategy to build a rule, removes the instance covered by this rule, and continues to generate rules recursively until all instances are covered. Compared with RIPPER, PART is simpler and does not need any global optimization. To quickly implement PART algorithm, we directly use the tool “PART” in Weka.
SMOTE
As indicated in Table 1, the analyzed dataset consists of different numbers of cell lines from different tissues; thus, it is an imbalanced data. Therefore, we use the synthetic minority over-sampling technique (SMOTE) to obtain approximate balanced data ahead of classifier construction (Chawla et al., 2002). SMOTE produces new samples for the minor class iteratively until the size of the minor class can be equal to that of the major class. The tool “SMOTE” in Weka is used to produce new samples for each minor class (tissue); thus, the numbers of cell lines for all tissues are equal finally, that is, the number of samples in each class (tissue) is 198. The main parameter that determines the number of nearest neighbors in the same class for a selected sample is set to three.
Performance Measurement
As a balanced measurement, the Matthew's correlation coefficient (MCC; Matthews, 1975; Gorodkin, 2004) is used to evaluate and compare the classifier performance. Originally, MCC is designed for binary classification and has wide applications (Chen et al., 2017a,b; Zhao et al., 2018, 2019; Cui and Chen, 2019; Li et al., 2019), as proposed by Matthews in 1975 (Matthews, 1975). We adopte the multi-class version of MCC proposed by Gorodkin (Gorodkin, 2004) because our analyzed dataset contains more than two classes (i.e., tissues), and such MCC is calculated as follows:
where cov(·, ·) stands for the covariance of two matrices, X is a 0-1 matrix indicating the predicted class of each sample, and Y is a 0–1 matrix representing the actual classes of all samples. Such multi-class version of MCC has been widely used in the performance evaluation of multi-class classifiers (Salari et al., 2014; Schmuker et al., 2014; Zhang et al., 2019); thus, the multi-class version of MCC is still called MCC for convenience. In addition, we also report the accuracy of each class and over accuracy (ACC) for reference.
Results
In this study, we analyze the methylation data of cell lines in 13 tissues. The entire procedures are shown in Figure 1. Of the 485,512 methylation features, we first calculate their MI values to class labels. By setting the threshold 0.2 to MI value, 20,451 features remain, which are provided in Table S1. Then, these features are analyzed by mRMR and MCFS methods, respectively, producing two feature lists, which are available in Tables S1, S2, respectively. Then, on the basis of each feature list, we use IFS combined with a particular classifier to determine the optimal feature set and related classification models or rules.
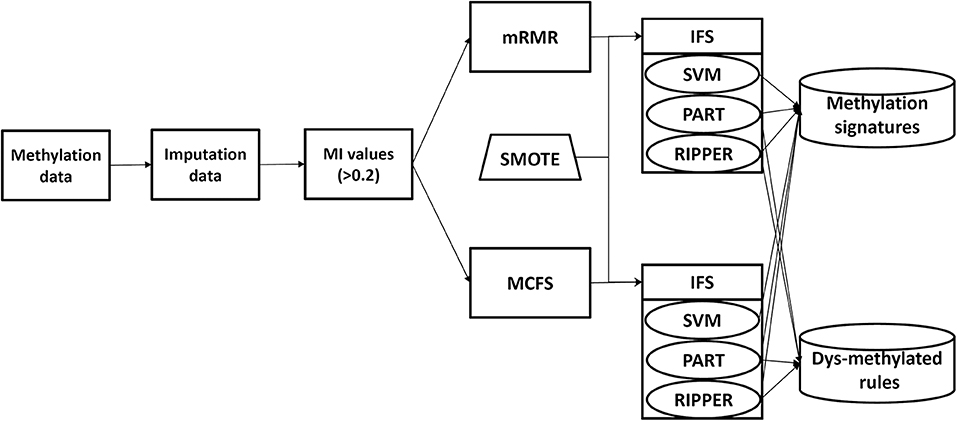
Figure 1. Analysis framework.
Tumor Cell Classification Based on Ranked Features by mRMR
We initially generate a series of feature subsets from the ranked feature list by mRMR and then run the IFS with SVM, RIPPER, and PART to capture optimal features for classifying different tumor cell samples. The performance these classifiers with different numbers of features is listed in Table S3. For an easy observation, an IFS curve is plotted for each classifier with the number of features as X-axis and MCC as the Y-axis, as shown in Figure 2. The highest MCC value generated by the SVM is 0.958 when using top-ranked 1,910 features, the optimal MCC value generated by the PART is 0.741 when using top-ranked 910 features, and the best MCC obtained by RIPPER is 0.703 when using top-ranked 2,810 features. The ACCs corresponding to above MCCs are 0.963, 0.768, and 0.735, respectively. Above results are listed in Table 2. Furthermore, we also count the accuracy of each tissue yielded by above three classifiers, which are illustrated in Figure 3. All accuracies yielded by SVM are over 0.900, whereas only two and one tissues receive the accuracies over 0.900 for PART and RIPPER, respectively. All these results show that the “black-box” classifier SVM performs better than rule-based classifiers. However, rule-based classifiers can learn readable rules for making an interpretable prediction. The PART algorithm generates 72 classification rules, as shown in Table S4, and RIPPER learns 47 classification rules, as shown in Table S5.
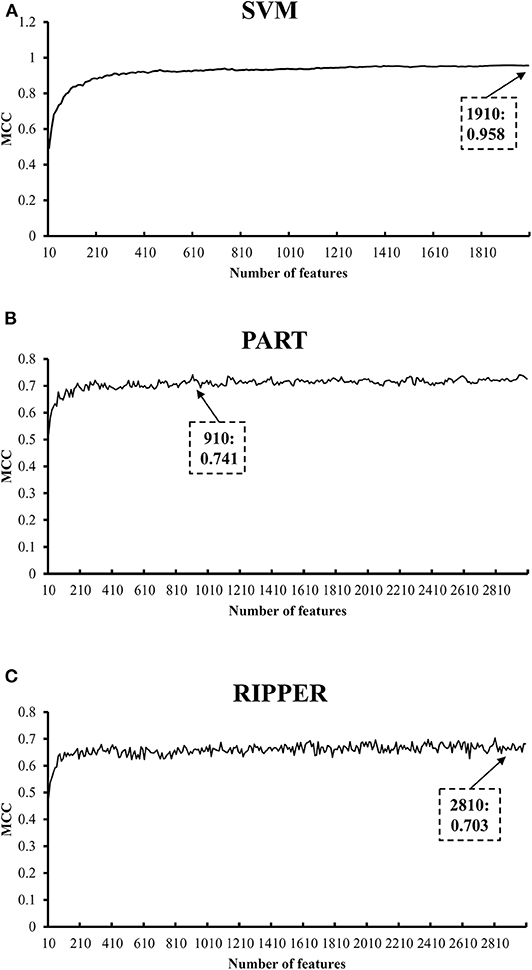
Figure 2. IFS curves with SVM, PART, and RIPPER based on mRMR-ranked features. (A) IFS with SVM. When top 1910 features are used, SVM gives the best MCC of 0.958. (B) IFS with PART. When top 910 features are used, PART gives the best MCC of 0.741. (C) IFS with RIPPER. When top 2810 features are used, RIPPER gives the best MCC of 0.703.

Table 2. Performance of IFS with SVM, PART, and RIPPER based on mRMR-ranked features for classifying tumor cells from different tissues.
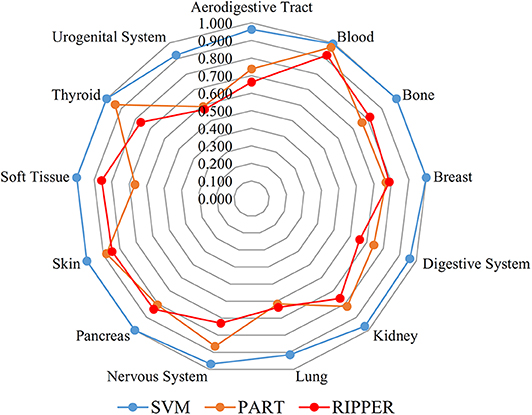
Figure 3. Radar chart to show the performance of the best SVM, PART and RIPPER classifiers on 13 tissues based on the feature list yielded by mRMR.
Tumor Cell Classification Based on Ranked Features by MCFS
We also carry out a similar analysis pipeline on the ranked features from MCFS. The performance of three classifiers on different numbers of features is listed in Table S6. Likewise, an IFS curve is plotted for each classifier, as shown in Figure 4. The best MCCs generated by SVM, PART, and RIPPER are 0.963, 0.770, and 0.716 when using top-ranked 3,600, 1,950, and 2,580 features, respectively, as listed in Table 3. The corresponding ACCs are 0.967, 0.795, and 0.746, respectively (see Table 3). Furthermore, the performance on 13 tissues of these three classifiers is shown in Figure 5. All accuracies generated by SVM are higher than 0.900, whereas for PART and RIPPER, there are only four and three accuracies over 0.900, respectively. SVM also outperforms rule learning classifiers PART and RIPPER. However, one advantage of PART and RIPPER is that they can learn interpretable rules for human understanding. PART learns 80 classification rules (Table S7), and RIPPER learns 48 classification rules (Table S8).
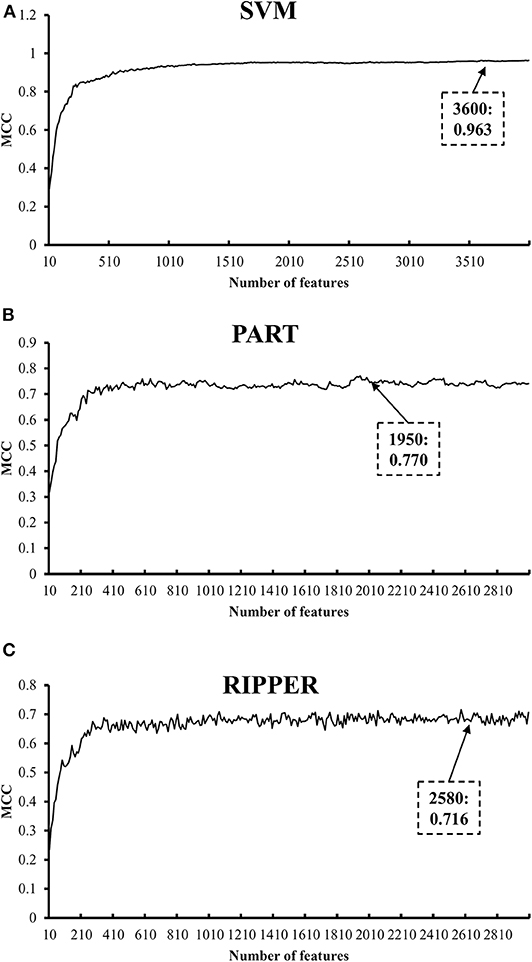
Figure 4. IFS curves with SVM, PART, and RIPPER based on MCFS-ranked features. (A) IFS with SVM. When top 3600 features are used, SVM gives the best MCC of 0.963. (B) IFS with PART. When top 1950 features are used, PART gives the best MCC of 0.770. (C) IFS with RIPPER. When top 2580 features are used, RIPPER gives the best MCC of 0.716.

Table 3. Performance of IFS with SVM, RART, and RIPPER based on MCFS-ranked features for classifying tumor cells from different tissues.
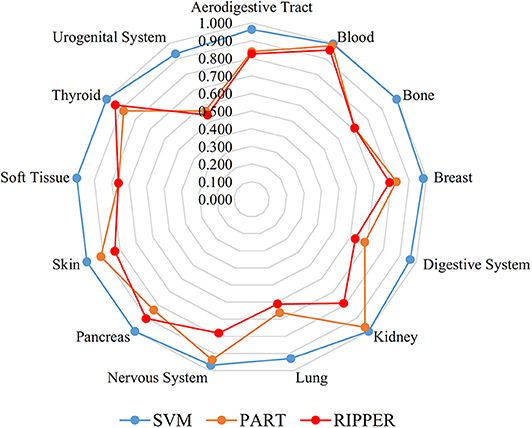
Figure 5. Radar chart to show the performance of the best SVM, PART, and RIPPER classifiers on 13 tissues based on the feature list yielded by MCFS.
Discussion
Established by World Health Organization (WHO), the classification scheme of tumor has been amended several times over the past decades. Scholars attempt to analyze the major characteristic of each type of tumor to provide solid guidance for clinical diagnosis and to avoid misclassification with mimic entities. For instance, as the most common digestive tract malignancies, misdiagnosis of metastatic colorectal cancer is highly responsible for the primary resistance of immune checkpoint inhibitors, displaying microsatellite instability, or defective mismatch repair (Cohen et al., 2019). Furthermore, the diagnosis of tumor with the good deal of insight of DNA methylation should improve the preciseness compared with traditional methods (Sahm et al., 2017). In accordance with our approach and analysis, we detected various methylation patterns of genes and association rules in different cell lines that can be used as the candidate signatures to distinguish 13 tumor subgroups corresponding to particular origin tissues. All predicted candidate signatures have reported that the aberrant methylations occurred and attributed to tumor initiation and progression. A summary and discussion on these signatures are presented in the following section.
Candidate Methylation Signatures Discriminating Origin Tissues of Tumor Cells
The first list of genes has been obtained by the MCFS and SVM algorithms. In accordance with the related results, MIR142 was predicted as one of the most potential genes for tumor classification. In general, the dysfunctions of MIR142 attribute to tumorigenesis and angiogenesis. MIR142 specifically expresses and plays a critical role in various hematopoietic cell lines (Rivkin et al., 2017). Hypermethylation-induced silencing of MIR142 promotes the progression of hepatocellular carcinoma via failing to suppress TGF-β expression (Yu et al., 2017). Similarly, the downregulation of MIR142 induced by promoter hypermethylation participates in thyroid follicular tumor initiation (Colamaio et al., 2015). Recent relevant studies have also confirmed that DNA methylation in MIR142 promoter can be recognized as a novel biomarker for T cell lymphoma (Sandoval et al., 2015). BZRAP1 encodes an associated protein of translocator protein, which regulates the flow of cholesterol into mitochondria. Translocator protein presents different expression patterns in different types of tumor (Bhoola et al., 2018). Although few studies directly concentrated on the function of BZRAP1 methylation, BZRAP1 may be a potential marker for tumor classification considering the relationship between BZRAP1 and translocator proteins. Another gene, IFFO1, is widely methylated in ovarian tumor. Compared with normal blood samples, significant hypo-methylation on IFFO1 promoter is a potentially high-sensitive biomarker for ovarian tumor diagnosis. In addition, hyper-methylation of IFFO1 represses its expression in non-small-cell lung cancer (Feng et al., 2017).
Then, we applied another computational algorithm combining mRMR and SVM to predict differentially methylated gene candidates. The predictable ability of mRMR has been validated with high efficacy and accuracy. Recently, the mRMR algorithm has been applied to identify deriver genes of clear cell renal cell carcinoma (Li et al., 2018). We actually obtained a large group of tumor-associated methylated genes through the mRMR algorithm. Similar to the above MCFS method, BZRAP1 and IFFO1 also appeared in the feature list. Numerous studies have revealed the contribution of methylation on tumorigenesis, implying the accuracy and efficiency of our two analysis pipelines. On the basis of our results, MARVELD2 was predicted to show methylation diversity in tumor cells. MARVELD2 encodes an essential tight junction-associated member protein named “tricellulin.” In general, this protein expresses in tricellular junctions and contributes to the stability of epithelial cell layers. Hence, abnormal MARVELD2 expression always associates with various types of carcinoma pathogenesis. Early in 2011, the expression of MARVELD2 is evidently decreased in every stage of squamous cell carcinoma (Kondoh et al., 2011). Recent studies have further revealed that MARVELD2 is frequently overexpressed in hepatocellular carcinoma cells but downregulated in pancreatic carcinomas cells (Kojima and Sawada, 2012; Korompay et al., 2012; Somoracz et al., 2014). In consideration of the relationship between gene expression and methylation, this evidence could suggest the methylation diversity of MARVELD2 in different tumor types. LDOC1 is an important tumor-suppressor gene that mainly contributes to the regulation of transcriptional response mediated by the nuclear factor kappa B (Griesinger et al., 2017). Hyper-methylation causes LDOC1 silencing in multiple tumor types, such as cervical cancer (Buchholtz et al., 2013), lung cancer (Lee et al., 2019), and oral squamous cell carcinoma (Lee et al., 2013), implying the accuracy and efficacy of our prediction. MGAT1, a member of the glycosyltransferase family, acts as a Medial–Golgi enzyme that mediates the synthesis of complex N-glycans. A previous report confirmed that MGAT1 contributes to tumor migration and invasion (Beheshti Zavareh et al., 2012). As an important obesity-associated gene (Johansson et al., 2010), differential methylation of MGAT1 is associated with obesity risk (Voisin et al., 2015). Considering the strict relationship between obesity and the digestive system, MGAT1 might act as a candidate methylated marker for the digestive system. Moreover, MGAT1 is hyper-methylated in head and neck squamous cell carcinomas (Hwang et al., 2013). Another splicing regulator gene, ESRP2, was also predicted to present methylation diversity in tumor cells. In general, such gene is mainly expressed in various types of epithelial cells. For its particular methylation status, ESRP2 is overexpressed as induced by gene hypo-methylation in ovarian cancer and breast cancer (Heilmann et al., 2017; Jeong et al., 2017). Therefore, ESRP2 methylation might act as a novel diagnosis standard for these cancer sites, thereby validating the efficacy and accuracy of our analysis methods.
Candidate Methylation Patterns Discriminating the Origin Tissues of Tumor Cells
For the predicted features generated by the mRMR and MCFS algorithms, we apply two typical decision tree algorithms, namely, RIPPER and PART, to reveal the potentially associated methylation rules. For each rule group, we choose a few representative rules, as listed in Table 4, for detailed discussion as shown below.
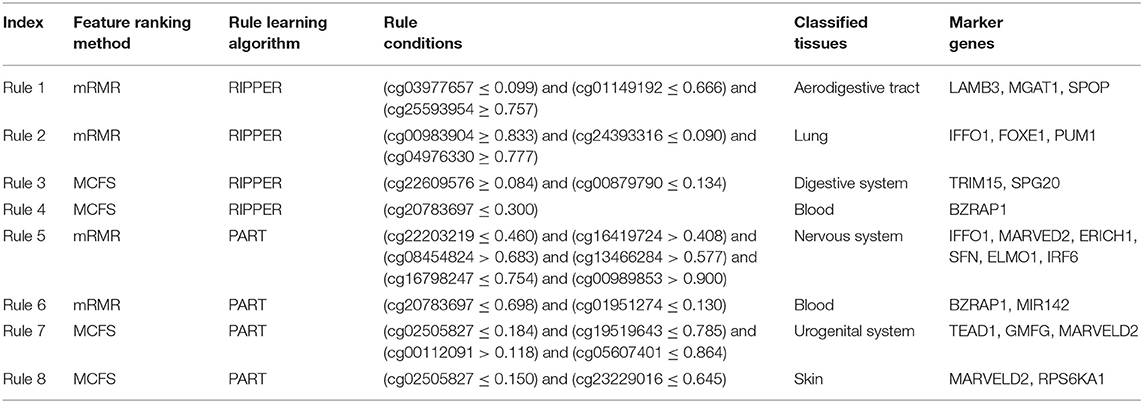
Table 4. Representative rules for classifying tumor cells from different tissues.
Combining the mRMR and RIPPER algorithms, we obtain 47 associated rules, and ample recent reports can validate the accuracy and efficacy of these identified rules. For instance, the combination of three gene methylation status, namely, LAMB3 (cg03977657) and MGAT1 (cg01149192) hypomethylation, and SPOP (cg25593954) hypermethylation, is a specific feature of digestive tract and respiratory tract tumor (Rule 1 in Table 4). LAMB3 is a component of laminin-5, an essential extracellular glycoprotein contributing to the most biological processes of basement membrane, including cell migration (Santamato et al., 2011), signal transduction (Filla et al., 2014), and tumorigenesis (Rani et al., 2013). Early in 2011, hypomethylation induced by abnormal overexpression of LAMB3 contributes to gastric tumor procession (Kwon et al., 2011). SPOP methylation rate is correlated with colorectal tumor survival (Zhi et al., 2016). A study on colorectal tumor has validated that the upregulation of the hedgehog signaling pathway in colorectal tumor mediated by SPOP hypermethylation promotes tumor migration (Zhi et al., 2016). Another rule (Rule 2 in Table 4) for lung tumor classification also verifies the efficacy of our results. Three differentially methylated genes, IFFO1, FOXE1, and PUM1, were predicted as signatures for lung tumor. IFFO1 methylation participates in non-small-cell lung cancer (Feng et al., 2017), and PUM1 is an RNA-binding protein gene that participates in multiple biological processes, such as translational regulation (Lin et al., 2019) and cell development (Lin et al., 2018). Various recent studies have illustrated that PUM1 functions in lung tumor. PUM1 can inhibit the proliferation of non-small-cell lung cancer cells via targeted by MiR-411-5p (Xia et al., 2018) and can mediate the interaction between p27 and MiR-221, which leads to the deterioration of non-small-cell lung cancer (Fernandez et al., 2015). Therefore, the hypermethylation of PUM1 is an important epigenetic characteristic for non-small-cell lung cancer diagnosis.
A total of 48 rules are obtained using the MCFS and RIPPER algorithms. Taking methylation rules for the classification of digestive system tumor as an example (Rule 3 in Table 4), differentially methylated genes TRIM15 (cg00879790), and SPG20 (cg22609576) are identified as signatures. TRIM15 is an essential focal adhesion protein mainly distributed in the duodenum and the small intestine (Fagerberg et al., 2014). In general, such gene function acts as important regulatory component in biological processes, including focal adhesion turnover and cell migration (Uchil et al., 2014). TRIM15 contributes to various types of digestive system tumor, including colon tumor (Lee et al., 2015) and gastric adenocarcinoma (Chen et al., 2018b). Moreover, specifically abnormal hypermethylation on TRIM15 has been detected in the gastric cancer genome (Cheng et al., 2014), confirming the potential of TRIM15 methylation as a candidate signature for gastric cancer diagnosis. Another candidate gene, SPG20, is a potential epigenetic signature for colorectal cancer (Rezvani et al., 2017). Hypermethylation-induced SPG20 silencing directly contributes to the cytokinesis of colorectal cancer cells (Lind et al., 2011). This evidence validates the efficiency of this rule. In addition, according to another rule (Rule 4 in Table 4), gene BZRAP1 was used to contribute to the identification of blood samples. Hypomethylation of such gene is positively correlated with the blood samples. BZRAP1 has been identified in various blood cells especially in monocytes (Yasui et al., 2007; Jyonouchi et al., 2011). Therefore, the hypomethylation of such gene as a biomarker for blood tissues (blood cells) is quite reasonable.
Similarly, 72 rules are generated by the mRMR and PART algorithms. Substantial evidence supports the accuracy of these rules. For instance, we extract a rule (Rule 5 in Table 4) of methylation pattern for nervous system tumor, where IFFO1 (cg22203219), MARVED2 (cg16419724), ERICH1 (cg08454824), SFN (cg13466284), ELMO1 (cg16798247), and IRF6 (cg00989853) were identified as candidate signatures. Among them, SFN and ELMO1 have been widely reported to associate with nervous system tumor process. The hypermethylation of SFN is a reliable biomarker for neuroblastic tumor diagnosis (Banelli et al., 2005, 2010). ELMO1 encodes a cell motility protein that contributes to glioma cell invasion. Recent research has also confirmed that ELMO1 presents abnormal methylated status in glioblastoma (Michaelsen et al., 2018). Furthermore, a specific rule (Rule 6 in Table 4) for blood uses two effective parameters, BZRAP1 (cg20783697) and MIR142 (cg01951274). As for BZRAP1, the hypomethylation of such gene has been discussed above to be correlated with blood samples, validating such rule. As for microRNA142, it and microRNA-29a have been identified as potential biomarkers for myeloid differentiation and acute myeloid leukemia, which would be regarded as a potential biomarker for the identification of blood tissue.
Finally, for the combination use of MCFS and PART algorithms, 80 rules are generated by the MCFS and PART algorithms. These rules can be validated by recent publications. For instance, we use the dys-methylation status of TEAD1 (cg00112091), GMFG (cg05607401), and MARVELD2 (cg02505827) as the diagnostic signatures for urogenital system tumor (Rule 7 in Table 4). Methylation of the MARVELD2 gene could be used to classify multiple different tumor types (Wang et al., 2009). With regard the relationship between MARVELD2 and urogenital system tumor, this gene is highly expressed in the epididymal epithelium and contributes to its integrity (Mandon and Cyr, 2015). Hence, the mutation on MARVELD2 may influence urogenital tumorigenesis. Meanwhile, GMFG is a member of the glia maturation factor family, and it has been validated to mediate angiogenesis by regulating the expression of STAT3 and VEGF (Zuo et al., 2013). Recent literature has confirmed that GMFG might contribute to the migration and invasion of ovarian cancer cells (Zuo et al., 2014). TEAD1, a ubiquitous transcriptional factor, acts as a transcriptional repressor in placental cells (Kessler et al., 2008). Hence, its increased level of methylation may lead to transcriptional alterations, further inducing tumorigenesis. Another specific rule (Rule 8 in Table 4), involving MARVELD2 (cg02505827) and RPS6KA1 (cg23229016), contributes to the identification of skin tissue. As discussed above, MARVELD2 has been reported to contribute to the classification of multiple tumor subtypes at methylation level (Wang et al., 2009), including skin cancer (Jonckheere and Van Seuningen, 2018). As for RPS6KA1, the hypomethylation of such gene has also been reported to be functionally correlated with tumorigenesis in skin by interacting with gene RB1 (Mcevoy et al., 2014).
Limited by the page restrictions of this article, we are unable to discuss all results. Nevertheless, we have shown the efficiency of our computational methods for identifying novel tumor-specific epigenetic signatures. We widely validate the accuracy or relevance of our highly ranked methylation signatures and associated rules via literature studies. Our analysis method provides new insights into the precancerous diagnosis of different tumor types.
Functional Enrichment Analysis for the Common Genes From mRMR and MCFS
Based on the feature list yielded by mRMR, SVM with top 1,910 features provides the best performance, whereas SVM with top 3,600 features produces the best performance based on the list generated by MCFS. A Venn diagram is plotted in Figure 6 to show the difference of these two feature subsets. There are 1,013 common features (methylation sites), corresponding to 470 genes, which are provided in Table S9. For capturing more biological or pathogen understanding on these common marker genes, we carry on the functional enrichment analysis on GO and KEGG. Results are provided in Table S10.
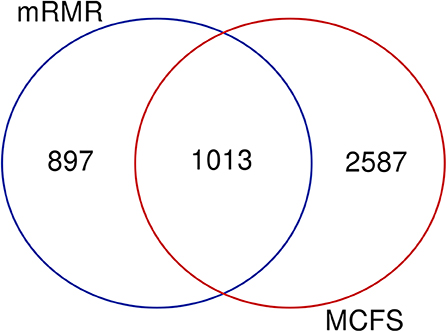
Figure 6. Venn diagram to show two marker gene sets yielded by mRMR and MCFS.
On one hand, for gene ontology enrichment, GO: 0098609 (cell-cell adhesion), GO:0007155 (cell adhesion), and GO: 0022610 (biological adhesion) are the top GO (BP) terms for the enrichment pattern of common marker genes. According to recent publications, early in 1998, the inactivation of E-cadherin-mediated cell adhesion has been reported to participate in the progression of multiple cancer subtypes (Hirohashi, 1998). Further detailed studies confirm that cell-cell adhesion plays irreplaceable roles for the tumorigenesis, although the expression level and detailed contributions are actually not all the same in various cancer subtypes (Birchmeier et al., 1993), e.g., in primary and metastatic lung cancer (Böhm et al., 1994). Next, we identify various GO (CC) terms describing the cell-cell junction, such as GO: 0030054 (cell junction), GO: 0005911 (cell-cell junction), and membrane associated GO terms, including GO: 0044459 (plasma membrane part), GO: 0031226 (intrinsic component of plasma membrane), and GO: 0005887 (integral component of plasma membrane). As analyzed above, cell adhesion is a quite important biological processes for identification and discrimination on different cancer subtypes (Birchmeier et al., 1993). Considering that cell junction is functionally correlated with cell-cell adhesion (Kametani and Takeichi, 2007), it is also reasonable for marker genes to enrich in these related functions. Plasma membrane has also been reported to participate in multiple cancer subtypes (Leth-Larsen et al., 2010), especially in breast cancer (Razandi et al., 2000) and colon cancer (Kakugawa et al., 2002). Furthermore, for GO (MF) terms, GTPase function associated GO terms have been widely screened out, including GTPase regulator activity GO:0005096 (GTPase activator activity), GO:0017048 (Rho GTPase binding), and GO:0051020 (GTPase binding). GTPase function and its related biological processes have been identified in multiple cancer subtypes (Wang et al., 2003; Sethakorn and Dulin, 2013), and have been confirmed to play different regulatory roles for tumorigenesis in different cancer subtypes (Wang et al., 2003).
On the other hand, for KEGG pathways, the top KEGG pathways are just the same as the top biological processes describing the cell junction and adhesion hsa04520 (adhesions junction) and hsa04510 (Focal adhesion). There are other key pathways found, e.g., hsa04015 (Rap1 signaling pathway) and hsa04151 (PI3K-Akt signaling pathway). According to recent publications (Kooistra et al., 2007), Rap1 together with its regulatory pathways have been identified as a key regulator for cell-cell junction formation, so that, it is quite reasonable to regard Rap1 signaling pathway as a discriminative pathway for different cancer subtypes. As for PI3K-Akt signaling pathway, it is actually one of the most famous tumor associated pathways, which has been identified to be pathogenic in multiple tumor subtypes, including breast cancer (Berns et al., 2007), B-cell lymphoma (Lannutti et al., 2011) and endocrine tumor (Robbins and Hague, 2016). Many studies confirm that actually in different tumor subtypes, the activation status and drive contribution of such pathway on tumorigenesis may be not always the same (Boyault et al., 2012).
Conclusions
This study investigates the methylation data of tumor cell lines from 13 tissues. Several machine leaning algorithms are employed to provide deep insights into the data. Some methylation-associated genes and their dys-methylated patterns are extracted. The genes may be novel biomarkers for discriminating different tumor cell lines and the patterns can provide a clear picture on the methylation levels of tumor cell lines in different tissues. The findings reported in this study may be novel materials for the study of tumor cell lines.
Data Availability Statement
The datasets for this study can be found in the Gene Expression Omnibus [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE68379].
Author Contributions
TH and Y-DC designed the study. SZ, TZ, BH, and LC performed the experiments. SZ, Y-HZ, KF, ZN, and JL analyzed the results. SZ, TZ, and BH wrote the manuscript. All authors contributed to the research and reviewed the manuscript.
Funding
This research was supported by the Shanghai Municipal Science and Technology Major Project [2017SHZDZX01], National Key R&D Program of China [2018YFC0910403], National Natural Science Foundation of China [31701151, 61701298], Natural Science Foundation of Shanghai [17ZR1412500], Shanghai Sailing Program [16YF1413800], the Youth Innovation Promotion Association of Chinese Academy of Sciences (CAS) [2016245], the fund of the key Laboratory of Stem Cell Biology of Chinese Academy of Sciences [201703], Key-Area Research and Development Program of Guangdong Province (2018B020203003), Guangzhou science and technology planning project (201707020007).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbioe.2020.00507/full#supplementary-material
Table S1. Ranked features by mRMR.
Table S2. Ranked features by MCFS.
Table S3. Performance of IFS with SVM, PART, and RIPPER when using different numbers of features ranked by mRMR.
Table S4. Learned classification rules by PART based on the ranked features by mRMR.
Table S5. Learned classification rules by RIPPER based on the ranked features by mRMR.
Table S6. Performance of IFS with SVM, PART, and RIPPER when using different numbers of features ranked by MCFS.
Table S7. Learned classification rules by PART based on the ranked features by MCFS.
Table S8. Learned classification rules by RIPPER based on the ranked features by MCFS.
Table S9. Common features (methylation sites) and their corresponding genes detected by mRMR and MCFS.
Table S10. Functional enrichment analysis of common genes detected by mRMR and MCFS.
References
Banelli, B., Bonassi, S., Casciano, I., Mazzocco, K., Di Vinci, A., Scaruffi, P., et al. (2010). Outcome prediction and risk assessment by quantitative pyrosequencing methylation analysis of the SFN gene in advanced stage, high-risk, neuroblastic tumor patients. Int. J. Cancer 126, 656–668. doi: 10.1002/ijc.24768
Banelli, B., Gelvi, I., Di Vinci, A., Scaruffi, P., Casciano, I., Allemanni, G., et al. (2005). Distinct CpG methylation profiles characterize different clinical groups of neuroblastic tumors. Oncogene 24, 5619–5628. doi: 10.1038/sj.onc.1208722
Beheshti Zavareh, R., Sukhai, M. A., Hurren, R., Gronda, M., Wang, X., Simpson, C. D., et al. (2012). Suppression of cancer progression by MGAT1 shRNA knockdown. PLoS ONE 7:e43721. doi: 10.1371/journal.pone.0043721
Berns, K., Horlings, H. M., Hennessy, B. T., Madiredjo, M., Hijmans, E. M., Beelen, K., et al. (2007). A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 12, 395–402. doi: 10.1016/j.ccr.2007.08.030
Bhoola, N. H., Mbita, Z., Hull, R., and Dlamini, Z. (2018). Translocator protein (TSPO) as a potential biomarker in human cancers. Int. J. Mol. Sci. 19:2176. doi: 10.3390/ijms19082176
Birchmeier, W., Weidner, K., Hülsken, J., and Behrens, J. (1993). Molecular mechanisms leading to cell junction (cadherin) deficiency in invasive carcinomas. Semin. Cancer Biol. 4, 231–239.
Böhm, M., Totzeck, B., Birchmeier, W., and Wieland, I. (1994). Differences of E-cadherin expression levels and patterns in primary and metastatic human lung cancer. Clin. Exp. Metasta. 12, 55–62. doi: 10.1007/BF01784334
Boyault, S., Drouet, Y., Navarro, C., Bachelot, T., Lasset, C., Treilleux, I., et al. (2012). Mutational characterization of individual breast tumors: TP53 and PI3K pathway genes are frequently and distinctively mutated in different subtypes. Breast Cancer Res. Treat. 132, 29–39. doi: 10.1007/s10549-011-1518-y
Buchholtz, M. L., Juckstock, J., Weber, E., Mylonas, I., Dian, D., and Bruning, A. (2013). Loss of LDOC1 expression by promoter methylation in cervical cancer cells. Cancer Invest. 31, 571–577. doi: 10.3109/07357907.2013.845671
Burns, K. H. (2017). Transposable elements in cancer. Nat. Rev. Cancer Vol. 17, 415–424. doi: 10.1038/nrc.2017.35
Campan, M., Moffitt, M., Houshdaran, S., Shen, H., Widschwendter, M., Daxenbichler, G., et al. (2011). Genome-scale screen for DNA methylation-based detection markers for ovarian cancer. PLoS ONE 6:e28141. doi: 10.1371/journal.pone.0028141
Chawla, N. V., Bowyer, K. W., Hall, L. O., and Kegelmeyer, W. P. (2002). SMOTE: synthetic minority over-sampling technique. J. Artif. Intell. Res. 16, 321–357. doi: 10.1613/jair.953
Che, J., Chen, L., Guo, Z.-H., Wang, S., and Aorigele (2020). Drug target group prediction with multiple drug networks. Comb. Chem. High Throughput Screen. 23, 274–284. doi: 10.2174/1386207322666190702103927
Chen, L., Chu, C., Zhang, Y.-H., Zheng, M.-Y., Zhu, L., Kong, X., et al. (2017a). Identification of drug-drug interactions using chemical interactions. Curr. Bioinform. 12, 526–534. doi: 10.2174/1574893611666160618094219
Chen, L., Pan, X., Hu, X., Zhang, Y. H., Wang, S., Huang, T., et al. (2018a). Gene expression differences among different MSI statuses in colorectal cancer. Int. J. Cancer 143, 1731–1740. doi: 10.1002/ijc.31554
Chen, L., Wang, S., Zhang, Y. H., Li, J., Xing, Z. H., Yang, J., et al. (2017b). Identify key sequence features to improve CRISPR sgRNA efficacy. IEEE Access 5, 26582–26590. doi: 10.1109/ACCESS.2017.2775703
Chen, W., Lu, C., and Hong, J. (2018b). TRIM15 exerts anti-tumor effects through suppressing cancer cell invasion in gastric adenocarcinoma. Med. Sci. Monit. 24, 8033–8041. doi: 10.12659/MSM.911142
Chen, Y., Widschwendter, M., and Teschendorff, A. E. (2017c). Systems-epigenomics inference of transcription factor activity implicates aryl-hydrocarbon-receptor inactivation as a key event in lung cancer development. Genome Biol. 18:236. doi: 10.1186/s13059-017-1366-0
Cheng, Y., Yan, Z., Liu, Y., Liang, C., Xia, H., Feng, J., et al. (2014). Analysis of DNA methylation patterns associated with the gastric cancer genome. Oncol. Lett. 7, 1021–1026. doi: 10.3892/ol.2014.1838
Cohen, R., Hain, E., Buhard, O., Guilloux, A., Bardier, A., Kaci, R., et al. (2019). Association of primary resistance to immune checkpoint inhibitors in metastatic colorectal cancer with misdiagnosis of microsatellite instability or mismatch repair deficiency status. JAMA Oncol. 5, 551–555. doi: 10.1001/jamaoncol.2018.4942
Cohen, W. W. (1995). “Fast effective rule induction,” in: Twelfth International Conference on Machine Learning (Tahoe City, CA). doi: 10.1016/B978-1-55860-377-6.50023-2
Colamaio, M., Puca, F., Ragozzino, E., Gemei, M., Decaussin-Petrucci, M., Aiello, C., et al. (2015). miR-142-3p down-regulation contributes to thyroid follicular tumorigenesis by targeting ASH1L and MLL1. J. Clin. Endocrinol. Metab. 100, 59–69. doi: 10.1210/jc.2014-2280
Cortes, C., and Vapnik, V. (1995). Support-vector networks. Mach. Learn. 20, 273–297. doi: 10.1007/BF00994018
Cui, C., Gan, Y., Gu, L., Wilson, J., Liu, Z., Zhang, B., et al. (2015). P16-specific DNA methylation by engineered zinc finger methyltransferase inactivates gene transcription and promotes cancer metastasis. Genome Biol. 16:252. doi: 10.1186/s13059-015-0819-6
Cui, H., and Chen, L. (2019). A binary classifier for the prediction of EC numbers of enzymes. Curr. Proteomics 16, 381–389. doi: 10.2174/1570164616666190126103036
De La Rosa, J., Weber, J., Friedrich, M. J., Li, Y., Rad, L., Ponstingl, H., et al. (2017). A single-copy sleeping beauty transposon mutagenesis screen identifies new PTEN-cooperating tumor suppressor genes. Nat. Genet. 49, 730–741. doi: 10.1038/ng.3817
Draminski, M., Rada-Iglesias, A., Enroth, S., Wadelius, C., Koronacki, J., and Komorowski, J. (2008). Monte carlo feature selection for supervised classification. Bioinformatics 24, 110–117. doi: 10.1093/bioinformatics/btm486
Evans, D. G. R., Van Veen, E. M., Byers, H. J., Wallace, A. J., Ellingford, J. M., Beaman, G., et al. (2018). A Dominantly inherited 5′ UTR variant causing methylation-associated silencing of BRCA1 as a cause of breast and ovarian cancer. Am. J. Hum. Genet. 103, 213–220. doi: 10.1016/j.ajhg.2018.07.002
Fagerberg, L., Hallstrom, B. M., Oksvold, P., Kampf, C., Djureinovic, D., Odeberg, J., et al. (2014). Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteomics 13, 397–406. doi: 10.1074/mcp.M113.035600
Farlik, M., Halbritter, F., Müller, F., Choudry, F. A., Ebert, P., Klughammer, J., et al. (2016). DNA methylation dynamics of human hematopoietic stem cell differentiation. Cell. Stem Cell 19, 808–822. doi: 10.1016/j.stem.2016.10.019
Feng, N., Wang, Y., Zheng, M., Yu, X., Lin, H., Ma, R. N., et al. (2017). Genome-wide analysis of DNA methylation and their associations with long noncoding RNA/mRNA expression in non-small-cell lung cancer. Epigenomics 9, 137–153. doi: 10.2217/epi-2016-0120
Fernandez, S., Risolino, M., Mandia, N., Talotta, F., Soini, Y., Incoronato, M., et al. (2015). miR-340 inhibits tumor cell proliferation and induces apoptosis by targeting multiple negative regulators of p27 in non-small cell lung cancer. Oncogene 34, 3240–3250. doi: 10.1038/onc.2014.267
Filla, M. S., Clark, R., and Peters, D. M. (2014). A syndecan-4 binding peptide derived from laminin 5 uses a novel PKCepsilon pathway to induce cross-linked actin network (CLAN) formation in human trabecular meshwork (HTM) cells. Exp. Cell. Res. 327, 171–182. doi: 10.1016/j.yexcr.2014.07.035
Frank, E., and Witten, I. H. (1998). “Generating accurate rule sets without global optimization,” in Fifteenth International Conference on Machine Learning (Hamilton).
Gao, F., Niu, Y., Sun, Y. E., Lu, H., Chen, Y., Li, S., et al. (2017). De novo DNA methylation during monkey pre-implantation embryogenesis. Cell. Res. 27, 526–539. doi: 10.1038/cr.2017.25
Good, C. R., Panjarian, S., Kelly, A. D., Madzo, J., Patel, B., Jelinek, J., et al. (2018). TET1-Mediated hypomethylation activates oncogenic signaling in triple-negative breast cancer. Cancer Res. 78, 4126–4137. doi: 10.1158/0008-5472.CAN-17-2082
Gorodkin, J. (2004). Comparing two K-category assignments by a K-category correlation coefficient. Comput. Biol. Chem. 28, 367–374. doi: 10.1016/j.compbiolchem.2004.09.006
Griesinger, A. M., Witt, D. A., Grob, S. T., Georgio Westover, S. R., Donson, A. M., Sanford, B., et al. (2017). NF-kappaB upregulation through epigenetic silencing of LDOC1 drives tumor biology and specific immunophenotype in Group A ependymoma. Neuro Oncol. 19, 1350–1360. doi: 10.1093/neuonc/nox061
Guo, J., Su, Y., Shin, J. H., Shin, J., Li, H., Xie, B., et al. (2014). Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat. Neurosci. 17, 215–222. doi: 10.1038/nn.3607
Heilmann, K., Toth, R., Bossmann, C., Klimo, K., Plass, C., and Gerhauser, C. (2017). Genome-wide screen for differentially methylated long noncoding RNAs identifies Esrp2 and lncRNA Esrp2-as regulated by enhancer DNA methylation with prognostic relevance for human breast cancer. Oncogene 36, 6446–6461. doi: 10.1038/onc.2017.246
Hirohashi, S. (1998). Inactivation of the E-cadherin-mediated cell adhesion system in human cancers. Am. J. Pathol. 153, 333–339. doi: 10.1016/S0002-9440(10)65575-7
Holliday, R., and Pugh, J. E. (1975). DNA modification mechanisms and gene activity during development. Science 187, 226–232. doi: 10.1126/science.1111098
Hur, K., Cejas, P., Feliu, J., Moreno-Rubio, J., Burgos, E., Boland, C. R., et al. (2014). Hypomethylation of long interspersed nuclear element-1 (LINE-1) leads to activation of proto-oncogenes in human colorectal cancer metastasis. Gut 63, 635–646. doi: 10.1136/gutjnl-2012-304219
Hwang, S., Mahadevan, S., Qadir, F., Hutchison, I. L., Costea, D. E., Neppelberg, E., et al. (2013). Identification of FOXM1-induced epigenetic markers for head and neck squamous cell carcinomas. Cancer 119, 4249–4258. doi: 10.1002/cncr.28354
Iorio, F., Knijnenburg, T. A., Vis, D. J., Bignell, G. R., Menden, M. P., Schubert, M., et al. (2016). A landscape of pharmacogenomic interactions in cancer. Cell 166, 740–754. doi: 10.1016/j.cell.2016.06.017
Jeong, H. M., Han, J., Lee, S. H., Park, H. J., Lee, H. J., Choi, J. S., et al. (2017). ESRP1 is overexpressed in ovarian cancer and promotes switching from mesenchymal to epithelial phenotype in ovarian cancer cells. Oncogenesis 6:e389. doi: 10.1038/oncsis.2017.87
Johansson, A., Marroni, F., Hayward, C., Franklin, C. S., Kirichenko, A. V., Jonasson, I., et al. (2010). Linkage and genome-wide association analysis of obesity-related phenotypes: association of weight with the MGAT1 gene. Obesity 18, 803–808. doi: 10.1038/oby.2009.359
Jonckheere, N., and Van Seuningen, I. (2018). Integrative analysis of the cancer genome atlas and cancer cell lines encyclopedia large-scale genomic databases: MUC4/MUC16/MUC20 signature is associated with poor survival in human carcinomas. J. Trans. Med. 16:259. doi: 10.1186/s12967-018-1632-2
Jyonouchi, H., Geng, L., Streck, D. L., and Toruner, G. A. (2011). Children with autism spectrum disorders (ASD) who exhibit chronic gastrointestinal (GI) symptoms and marked fluctuation of behavioral symptoms exhibit distinct innate immune abnormalities and transcriptional profiles of peripheral blood (PB) monocytes. J. Neuroimmunol. 238, 73–80. doi: 10.1016/j.jneuroim.2011.07.001
Kaaij, L. T., Van De Wetering, M., Fang, F., Decato, B., Molaro, A., Van De Werken, H. J., et al. (2013). DNA methylation dynamics during intestinal stem cell differentiation reveals enhancers driving gene expression in the villus. Genome Biol. Evol. 14:R50. doi: 10.1186/gb-2013-14-5-r50
Kakugawa, Y., Wada, T., Yamaguchi, K., Yamanami, H., Ouchi, K., Sato, I., et al. (2002). Up-regulation of plasma membrane-associated ganglioside sialidase (Neu3) in human colon cancer and its involvement in apoptosis suppression. Proc. Natl. Acad. Sci. U. S. A. 99, 10718–10723. doi: 10.1073/pnas.152597199
Kametani, Y., and Takeichi, M. (2007). Basal-to-apical cadherin flow at cell junctions. Nat. Cell. Biol. 9, 92–98. doi: 10.1038/ncb1520
Kessler, C. A., Bachurski, C. J., Schroeder, J., Stanek, J., and Handwerger, S. (2008). TEAD1 inhibits prolactin gene expression in cultured human uterine decidual cells. Mol. Cell. Endocrinol. 295, 32–38. doi: 10.1016/j.mce.2008.08.007
Kojima, T., and Sawada, N. (2012). Regulation of tight junctions in human normal pancreatic duct epithelial cells and cancer cells. Ann. N. Y. Acad. Sci. 1257, 85–92. doi: 10.1111/j.1749-6632.2012.06579.x
Kondoh, A., Takano, K., Kojima, T., Ohkuni, T., Kamekura, R., Ogasawara, N., et al. (2011). Altered expression of claudin-1, claudin-7, and tricellulin regardless of human papilloma virus infection in human tonsillar squamous cell carcinoma. Acta Otolaryngol. 131, 861–868. doi: 10.3109/00016489.2011.562537
Kooistra, M. R., Dubé, N., and Bos, J. L. (2007). Rap1: a key regulator in cell-cell junction formation. J. Cell. Sci. 120, 17–22. doi: 10.1242/jcs.03306
Korompay, A., Borka, K., Lotz, G., Somoracz, A., Torzsok, P., Erdelyi-Belle, B., et al. (2012). Tricellulin expression in normal and neoplastic human pancreas. Histopathology 60, E76–86. doi: 10.1111/j.1365-2559.2012.04189.x
Kwon, O. H., Park, J. L., Kim, M., Kim, J. H., Lee, H. C., Kim, H. J., et al. (2011). Aberrant up-regulation of LAMB3 and LAMC2 by promoter demethylation in gastric cancer. Biochem. Biophys. Res. Commun. 406, 539–545. doi: 10.1016/j.bbrc.2011.02.082
Lannutti, B. J., Meadows, S. A., Herman, S. E., Kashishian, A., Steiner, B., Johnson, A. J., et al. (2011). CAL-101, a p110δ selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood 117, 591–594. doi: 10.1182/blood-2010-03-275305
Lee, C. H., Wong, T. S., Chan, J. Y., Lu, S. C., Lin, P., Cheng, A. J., et al. (2013). Epigenetic regulation of the X-linked tumour suppressors BEX1 and LDOC1 in oral squamous cell carcinoma. J. Pathol. 230, 298–309. doi: 10.1002/path.4173
Lee, C. H., Yang, J. R., Chen, C. Y., Tsai, M. H., Hung, P. F., Chen, S. J., et al. (2019). Novel STAT3 inhibitor LDOC1 targets phospho-JAK2 for degradation by interacting with LNX1 and regulates the aggressiveness of lung cancer. Cancers 11:63. doi: 10.3390/cancers11010063
Lee, O. H., Lee, J., Lee, K. H., Woo, Y. M., Kang, J. H., Yoon, H. G., et al. (2015). Role of the focal adhesion protein TRIM15 in colon cancer development. Biochim. Biophys. Acta 1853, 409–421. doi: 10.1016/j.bbamcr.2014.11.007
Leth-Larsen, R., Lund, R. R., and Ditzel, H. J. (2010). Plasma membrane proteomics and its application in clinical cancer biomarker discovery. Mol. Cell. Proteomics 9, 1369–1382. doi: 10.1074/mcp.R900006-MCP200
Li, J., Lu, L., Zhang, Y., Liu, M., Chen, L., Huang, T., et al. (2019). Identification of synthetic lethality based on a functional network by using machine learning algorithms. J. Cell. Biochem. 120, 405–416. doi: 10.1002/jcb.27395
Li, Z. C., Zhai, G., Zhang, J., Wang, Z., Liu, G., Wu, G. Y., et al. (2018). Differentiation of clear cell and non-clear cell renal cell carcinomas by all-relevant radiomics features from multiphase CT: a VHL mutation perspective. Eur. Radiol. 29, 3996–4007. doi: 10.1007/s00330-018-5872-6
Lin, K., Qiang, W., Zhu, M., Ding, Y., Shi, Q., Chen, X., et al. (2019). Mammalian pum1 and pum2 control body size via translational regulation of the cell cycle inhibitor Cdkn1b. Cell. Rep. 26, 2434–2450.e2436. doi: 10.1016/j.celrep.2019.01.111
Lin, K., Zhang, S., Shi, Q., Zhu, M., Gao, L., Xia, W., et al. (2018). Essential requirement of mammalian pumilio family in embryonic development. Mol. Biol. Cell. 29, 2922–2932. doi: 10.1091/mbc.E18-06-0369
Lind, G. E., Raiborg, C., Danielsen, S. A., Rognum, T. O., Thiis-Evensen, E., Hoff, G., et al. (2011). SPG20, a novel biomarker for early detection of colorectal cancer, encodes a regulator of cytokinesis. Oncogene 30, 3967–3978. doi: 10.1038/onc.2011.109
Liu, H. A., and Setiono, R. (1998). Incremental feature selection. Appl. Intell. 9, 217–230. doi: 10.1023/A:1008363719778
Mandon, M., and Cyr, D. G. (2015). Tricellulin and its role in the epididymal epithelium of the rat. Biol. Reprod. 92:66. doi: 10.1095/biolreprod.114.120824
Marzese, D. M., Scolyer, R. A., Roqué, M., Vargas-Roig, L. M., Huynh, J. L., Wilmott, J. S., et al. (2014). DNA methylation and gene deletion analysis of brain metastases in melanoma patients identifies mutually exclusive molecular alterations. Neuro Oncol. 16, 1499–1509. doi: 10.1093/neuonc/nou107
Matthews, B. W. (1975). Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta 405, 442–451. doi: 10.1016/0005-2795(75)90109-9
Mcevoy, J., Nagahawatte, P., Finkelstein, D., Richards-Yutz, J., Valentine, M., Ma, J., et al. (2014). RB1 gene inactivation by chromothripsis in human retinoblastoma. Oncotarget 5, 438–450. doi: 10.18632/oncotarget.1686
Michaelsen, S. R., Aslan, D., Urup, T., Poulsen, H. S., Gronbaek, K., Broholm, H., et al. (2018). DNA methylation levels of the ELMO gene promoter CpG Islands in human glioblastomas. Int. J. Mol. Sci. 19:679. doi: 10.3390/ijms19030679
Moore, L. D., Le, T., and Fan, G. (2013). DNA methylation and its basic function. Neuropsychopharmacology 38, 23–38. doi: 10.1038/npp.2012.112
Pelaseyed, T., Bergström, J. H., Gustafsson, J. K., Ermund, A., Birchenough, G. M., Schütte, A., et al. (2014). The mucus and mucins of the goblet cells and enterocytes provide the first defense line of the gastrointestinal tract and interact with the immune system. Immunol. Rev. 160, 8–20. doi: 10.1111/imr.12182
Peng, H. C., Long, F. H., and Ding, C. (2005). Feature selection based on mutual information: criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans. Pattern Anal. Mach. Intell. 27, 1226–1238. doi: 10.1109/TPAMI.2005.159
Petell, C. J., Alabdi, L., He, M., San Miguel, P., Rose, R., and Gowher, H. (2016). An epigenetic switch regulates de novo DNA methylation at a subset of pluripotency gene enhancers during embryonic stem cell differentiation. Nucleic Acids Res. 44, 7605–7617. doi: 10.1093/nar/gkw426
Rani, V., Mccullough, M., and Chandu, A. (2013). Assessment of laminin-5 in oral dysplasia and squamous cell carcinoma. J. Oral. Maxillofac. Surg. 71, 1873–1879. doi: 10.1016/j.joms.2013.04.032
Razandi, M., Pedram, A., and Levin, E. R. (2000). Plasma membrane estrogen receptors signal to antiapoptosis in breast cancer. Mol. Endocrinol. 14, 1434–1447. doi: 10.1210/mend.14.9.0526
Renaud, F., Mariette, C., Vincent, A., Wacrenier, A., Maunoury, V., Leclerc, J., et al. (2016). The serrated neoplasia pathway of colorectal tumors: identification of MUC5AC hypomethylation as an early marker of polyps with malignant potential. Int. J. Cancer 138, 1472–1481. doi: 10.1002/ijc.29891
Renaud, F., Vincent, A., Mariette, C., Crepin, M., Stechly, L., Truant, S., et al. (2015). MUC5AC hypomethylation is a predictor of microsatellite instability independently of clinical factors associated with colorectal cancer. Int. J. Cancer 136, 2811–2821. doi: 10.1002/ijc.29342
Rezvani, N., Alibakhshi, R., Vaisi-Raygani, A., Bashiri, H., and Saidijam, M. (2017). Detection of SPG20 gene promoter-methylated DNA, as a novel epigenetic biomarker, in plasma for colorectal cancer diagnosis using the methylight method. Oncol. Lett. 13, 3277–3284. doi: 10.3892/ol.2017.5815
Rivkin, N., Chapnik, E., Mildner, A., Barshtein, G., Porat, Z., Kartvelishvily, E., et al. (2017). Erythrocyte survival is controlled by microRNA-142. Haematologica 102, 676–685. doi: 10.3324/haematol.2016.156109
Robbins, H. L., and Hague, A. (2016). The PI3K/Akt pathway in tumors of endocrine tissues. Front. Endocrinol. 6:188. doi: 10.3389/fendo.2015.00188
Sahm, F., Schrimpf, D., Stichel, D., Jones, D. T. W., Hielscher, T., Schefzyk, S., et al. (2017). DNA methylation-based classification and grading system for meningioma: a multicentre, retrospective analysis. Lancet Oncol. 18, 682–694. doi: 10.1016/S1470-2045(17)30155-9
Salari, N., Shohaimi, S., Najafi, F., Nallappan, M., and Karishnarajah, I. (2014). A novel hybrid classification model of genetic algorithms, modified k-nearest neighbor and developed backpropagation neural network. PLoS ONE 9:e112987. doi: 10.1371/journal.pone.0112987
Sandoval, J., Diaz-Lagares, A., Salgado, R., Servitje, O., Climent, F., Ortiz-Romero, P. L., et al. (2015). MicroRNA expression profiling and DNA methylation signature for deregulated microRNA in cutaneous T-cell lymphoma. J. Invest. Dermatol. 135, 1128–1137. doi: 10.1038/jid.2014.487
Santamato, A., Fransvea, E., Dituri, F., Caligiuri, A., Quaranta, M., Niimi, T., et al. (2011). Hepatic stellate cells stimulate HCC cell migration via laminin-5 production. Clin. Sci. 121, 159–168. doi: 10.1042/CS20110002
Schmuker, M., Pfeil, T., and Nawrot, M. P. (2014). A neuromorphic network for generic multivariate data classification. Proc. Natl. Acad. Sci. U. S. A. 111, 2081–2086. doi: 10.1073/pnas.1303053111
Sethakorn, N., and Dulin, N. O. (2013). RGS expression in cancer: oncomining the cancer microarray data. J. Recept. Sig. Transd. 33, 166–171. doi: 10.3109/10799893.2013.773450
Somoracz, A., Korompay, A., Torzsok, P., Patonai, A., Erdelyi-Belle, B., Lotz, G., et al. (2014). Tricellulin expression and its prognostic significance in primary liver carcinomas. Pathol. Oncol. Res. 20, 755–764. doi: 10.1007/s12253-014-9758-x
Uchil, P. D., Pawliczek, T., Reynolds, T. D., Ding, S., Hinz, A., Munro, J. B., et al. (2014). TRIM15 is a focal adhesion protein that regulates focal adhesion disassembly. J. Cell. Sci. 127, 3928–3942. doi: 10.1242/jcs.143537
Voisin, S., Almen, M. S., Zheleznyakova, G. Y., Lundberg, L., Zarei, S., Castillo, S., et al. (2015). Many obesity-associated SNPs strongly associate with DNA methylation changes at proximal promoters and enhancers. Genome Med. 7:103. doi: 10.1186/s13073-015-0225-4
Wang, L., Yang, L., Luo, Y., and Zheng, Y. (2003). A novel strategy for specifically down-regulating individual Rho GTPase activity in tumor cells. J. Biol. Chem. 278, 44617–44625. doi: 10.1074/jbc.M308929200
Wang, S., Li, Y., Han, F., Hu, J., Yue, L., Yu, Y., et al. (2009). Identification and characterization of MARVELD1, a novel nuclear protein that is down-regulated in multiple cancers and silenced by DNA methylation. Cancer Lett. 282, 77–86. doi: 10.1016/j.canlet.2009.03.008
Xia, L. H., Yan, Q. H., Sun, Q. D., and Gao, Y. P. (2018). MiR-411-5p acts as a tumor suppressor in non-small cell lung cancer through targeting PUM1. Eur. Rev. Med. Pharmacol. Sci. 22, 5546–5553.
Yasui, N., Kajimoto, K., Sumiya, T., Okuda, T., and Iwai, N. (2007). The monocyte chemotactic protein-1 gene may contribute to hypertension in Dahl salt-sensitive rats. Hypertension Res. 30, 185–193. doi: 10.1291/hypres.30.185
Yu, Q., Xiang, L., Yin, L., Liu, X., Yang, D., and Zhou, J. (2017). Loss-of-function of miR-142 by hypermethylation promotes TGF-beta-mediated tumour growth and metastasis in hepatocellular carcinoma. Cell. Prolif. 50:e12384. doi: 10.1111/cpr.12384
Zhang, D., Wu, B., Wang, P., Wang, Y., Lu, P., Nechiporuk, T., et al. (2017). Non-CpG methylation by DNMT3B facilitates REST binding and gene silencing in developing mouse hearts. Nucleic Acids Res. 45, 3102–3115. doi: 10.1093/nar/gkw1258
Zhang, X., Chen, L., Guo, Z.-H., and Liang, H. (2019). Identification of human membrane protein types by incorporating network embedding methods. IEEE Access 7, 140794–140805. doi: 10.1109/ACCESS.2019.2944177
Zhao, X., Chen, L., Guo, Z.-H., and Liu, T. (2019). Predicting drug side effects with compact integration of heterogeneous networks. Curr. Bioinform. 14, 709–720. doi: 10.2174/1574893614666190220114644
Zhao, X., Chen, L., and Lu, J. (2018). A similarity-based method for prediction of drug side effects with heterogeneous information. Math. Biosci. 306, 136–144. doi: 10.1016/j.mbs.2018.09.010
Zhi, X., Tao, J., Zhang, L., Tao, R., Ma, L., and Qin, J. (2016). Silencing speckle-type POZ protein by promoter hypermethylation decreases cell apoptosis through upregulating hedgehog signaling pathway in colorectal cancer. Cell. Death Dis. 7:e2569. doi: 10.1038/cddis.2016.435
Zhou, J.-P., Chen, L., and Guo, Z.-H. (2020a). iATC-NRAKEL: an efficient multi-label classifier for recognizing anatomical therapeutic chemical classes of drugs. Bioinformatics 36, 1391–1396. doi: 10.1093/bioinformatics/btz757
Zhou, J.-P., Chen, L., Wang, T., and Liu, M. (2020b). iATC-FRAKEL: a simple multi-label web-server for recognizing anatomical therapeutic chemical classes of drugs with their fingerprints only. Bioinformatics doi: 10.1093/bioinformatics/btaa166. [Epub ahead of print].
Zhou, S., Treloar, A. E., and Lupien, M. (2016). Emergence of the noncoding cancer genome: a target of genetic and epigenetic alterations. Cancer Discov. 6, 1215–1229. doi: 10.1158/2159-8290.CD-16-0745
Zuo, P., Fu, Z., Tao, T., Ye, F., Chen, L., Wang, X., et al. (2013). The expression of glia maturation factors and the effect of glia maturation factor-gamma on angiogenic sprouting in zebrafish. Exp. Cell. Res. 319, 707–717. doi: 10.1016/j.yexcr.2013.01.004
Keywords: methylation signature, dys-methylated pattern, cell line, rule, classification
Citation: Zhang S, Zeng T, Hu B, Zhang Y-H, Feng K, Chen L, Niu Z, Li J, Huang T and Cai Y-D (2020) Discriminating Origin Tissues of Tumor Cell Lines by Methylation Signatures and Dys-Methylated Rules. Front. Bioeng. Biotechnol. 8:507. doi: 10.3389/fbioe.2020.00507
Received: 07 March 2020; Accepted: 30 April 2020;
Published: 26 May 2020.
Edited by:
Yang Yang, Shanghai Jiao Tong University, ChinaCopyright © 2020 Zhang, Zeng, Hu, Zhang, Feng, Chen, Niu, Li, Huang and Cai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tao Huang, dG9odWFuZ3Rhb0AxMjYuY29t; Yu-Dong Cai, Y2FpX3l1ZEAxMjYuY29t
†These authors have contributed equally to this work