 Sebastian Köbbing
Sebastian Köbbing Lars M. Blank1
Lars M. Blank1 Nick Wierckx
Nick Wierckx- 1Institute of Applied Microbiology - iAMB, Aachen Biology and Biotechnology – ABBt, RWTH Aachen University, Aachen, Germany
- 2Institute of Bio- and Geosciences (IBG-1: Biotechnology), Forschungszentrum Jülich GmbH, Jülich, Germany
Understanding the composability of genetic elements is central to synthetic biology. Even for seemingly well-known elements such as a sigma 70 promoter the genetic context-dependent variability of promoter activity remains poorly understood. The lack of understanding of sequence to function results in highly limited de novo design of novel genetic element combinations. To address this issue, we characterized in detail concatenated “stacked” synthetic promoters including varying spacer sequence lengths and compared the transcription strength to the output of the individual promoters. The proxy for promoter activity, the msfGFP synthesis from stacked promoters was consistently lower than expected from the sum of the activities of the single promoters. While the spacer sequence itself had no activity, it drastically affected promoter activities when placed up- or downstream of a promoter. Single promoter-spacer combinations revealed a bivalent effect on msfGFP synthesis. By systematic analysis of promoter and spacer combinations, a semi-empirical correlation was developed to determine the combined activity of stacked promoters.
Introduction
The Pseudomonads are a promising group of bacteria for industrial applications (Wierckx et al., 2005; Tiso et al., 2014; Aparicio et al., 2018). A versatile metabolism enables them to grow on several carbon sources like glucose and glycerol, but also on a wide range of aliphatics and aromatics (Jiménez et al., 2002; Nikel et al., 2014; Köhler et al., 2015). Different Pseudomonas strains have been engineered for the production of chemicals with industrial importance from different renewable carbon sources, like furandicarboxylic acid, rhamnolipids, and aromatics (Wierckx et al., 2005; Sun et al., 2007; Blank et al., 2008; Koopman et al., 2010; Meijnen et al., 2011; Wynands et al., 2018). Pseudomonas is highly tolerant to chemical stresses and can survive harmful conditions caused by oxidative stress (Isken and de Bont, 1998; Ramos et al., 2002; Wierckx et al., 2005; Wynands et al., 2018). Some strains can thrive under a second phase of toxic hydrophobic solvents such as toluene or styrene (Heipieper et al., 2007; Kusumawardhani et al., 2018). P. putida KT2440 is a non-pathogenic representative of this versatile group of bacteria (Nelson et al., 2002). The strain is able to produce and accumulate polyhydroxyalkanoates (PHA) as a storage polymer in granules under nitrogen depletion from different carbon sources like glycerol, glucose, ethylene glycol, 1,4-butanediol, or fatty acids (Sun et al., 2007; Wang and Nomura, 2010; Franden et al., 2018; Li et al., 2019, 2020).
Parallel to the increasing industrial interest in Pseudomonads, an ever-increasing set of synthetic biology tools is developed for this genus. These include genomic integration tools like transposon Tn5 (de Lorenzo et al., 1990; Herrero et al., 1990; Nikel and de Lorenzo, 2013) or Tn7 (Lambertsen et al., 2004; Damron et al., 2013; Silva-Rocha and de Lorenzo, 2014), as well as a suite of tools for targeted and marker-less integration (Martínez-García and de Lorenzo, 2011). To obtain gene replacements, counter-selection procedures were established, like sacB originating from Bacillus subtilis (Schweizer, 1992). New tools are based on CRISPR/Cas9 showing high potential for whole-genome engineering approaches (Jiang et al., 2013; Aparicio et al., 2018). These tools enable a deep genetic and metabolic re-factoring of different Pseudomonads as exemplified in the engineering of streamlined chassis strains (Shen et al., 2017; Wynands et al., 2018; Sánchez-Pascuala et al., 2019).
Especially when such deep engineering entails the (over-) expression of many homologous or heterologous genes, balanced and reliable gene expression is required, which doesn't unnecessarily burden the cell. In this context, calibrated synthetic promoter libraries enable modulation of enzyme expression in metabolic pathways and protein production (Rud et al., 2006; Solem et al., 2007). Two major ways to generate a promoter library are prominently used. A low degeneracy approach, where only a few random nucleotides are introduced, reduces the number of possible generated promoter sequences and thus decreases the number of sequences, which have to be tested (Mutalik et al., 2013). This allows a deeper insight into promoter sequence-activity relationships. On the other hand, high degeneracy promoter libraries based on a degenerated core promoter sequence lead to billions of different possibilities (Zobel et al., 2015; Gilman and Love, 2016; Elmore et al., 2017). While the sequence space clearly outnumbers the experimental space possible to address, a high resolution of different promoter activities is possible. The use of calibrated and standardized synthetic promoters covering a range of activities are commonly used (Zobel et al., 2015). Constitutive synthetic promoters are generally based on sigma-70 (σ70) factor core promoters (Gruber and Gross, 2003). The σ70 factor encoded by rpoD guides the RNA polymerase to many promoters active during growth including the expression of housekeeping genes (Kang et al., 1997; Potvin et al., 2008). Varying the consensus sequences of the −10 and −35 elements, which are recognized by the holoenzyme as part of the core promotor (Lodge et al., 1990; McLean et al., 1997), leads to weaker expression strength in E. coli and P. aeruginosa (McLean et al., 1997). The σ70 factors consensus sequence of P. putida KT2440 and P. aeruginosa are identical (McLean et al., 1997; Zobel et al., 2015).
Characterization of synthetic promoters has been performed using plasmid-based expression systems or genomically integrated probes (Jensen and Hammer, 1998; Hammer et al., 2006; Zobel et al., 2015). However, varying plasmid copy numbers and high fitness costs for the host makes plasmid-based expression systems less suitable for promoter characterization in particular, and for metabolic engineering in general (Gao et al., 2014; Jahn et al., 2014; Lindmeyer et al., 2015; San Millan and MacLean, 2017). Genomic integration of the probe is preferred for characterization procedures (Zobel et al., 2015). The major difference is the fact that many of the plasmids used are multicopy, which increases the variability of the reporter output by copy number variations. In addition, an often-overlooked disadvantage of using multicopy plasmids for synthetic promoter screening is that they favor the selection of relatively weak promoters, as the combined effect of a strong constitutive promoter at high copy number may pose a too high burden. Genomic integration abolishes these copy number effects, as well as clonal variations, which have also been observed for different Pseudomonas strains (Friehs, 2004; Gao et al., 2014; Zobel et al., 2015). Nevertheless, the integration site in the genome must be chosen wisely and must be the same for all promoters. The expression activity differs not only for single genes, but also in larger regions on the genome (“hot” and “cold” spots). Therefore, we used a mini Tn7 transposon, which integrates in a targeted manner downstream of the glmS gene in the attTn7 site of a broad range of bacteria including P. putida KT2440, thereby enabling reliable and stable expression (Lambertsen et al., 2004; Choi et al., 2005; Zobel et al., 2015).
Synthetic promoter libraries are described for P. putida KT2440 (Zobel et al., 2015; Elmore et al., 2017). However, the predictability and composability of these promoters in different genetic contexts is poorly understood. Li et al. (2012) has shown that different numbers of promoters in tandem direction result in increased activities. Several other publications feature tandem promoters, but so far without a characterization of these promoter combinations that focusses on composability and predictability of the activity of these genetic elements (Dixon, 1984; Martens et al., 2004; Tamsir et al., 2011). The combination of promoters in different contexts is also a key element in logic gate construction in synthetic biology, and sensitivity to genetic context is considered a challenge there (Stanton et al., 2013).
In this work, we stacked (concatenated) promoters of a previously published promoter library from Zobel et al. (2015) in series and analyzed the resulting activities as single genomically integrated probes by measuring msfGFP expression (Landgraf, 2012). The obvious assumption that the combination of two promoters would yield their summed activity proved to be false. The reasons for this are investigated and a semi-empirical correlation was developed to reliably predict stacked synthetic promoter activities. This provided insights into the context-dependent activity of promoters that may foster a better predictability and composability of this key element of synthetic biology.
Materials and Methods
Bacterial Strains, Plasmids, and Cultivation Conditions
Strains and plasmids used and generated in this study are listed in Table 1. For cloning chemically competent E. coli PIR2 (Life Technologies, Carlsbad, USA) were used (Hanahan, 1983). Cultivation of E. coli was performed in lysogeny broth (LB) with 5 g L−1 NaCl (Sambrook et al., 1989). For solid media 15 g L−1 agar was added to the medium before autoclaving. To maintain the mini Tn7 plasmid in E. coli, 50 mg L−1 kanamycin was added to either liquid or solid medium. For pRK600 (Keen et al., 1988) bearing strains chloramphenicol (10 mg L−1) and for pTnS-1 (Choi et al., 2005) ampicillin (100 mg L−1) was used. Cultivation of E. coli strains was carried out at 37°C. Integration of the mini Tn7 transposon was performed by patch mating on LB agar plates and subsequent cultivation overnight at 30°C (Zobel et al., 2015). This was done with a mini Tn7 suicide plasmid-bearing donor strain, acceptor strain P. putida KT2440, and two helper-strains. E. coli HB101 (Boyer and Roulland-Dussoix, 1969) bearing plasmid pRK600 (Keen et al., 1988) with mobilization genes. E. coli DH5αλpir bearing pTnS-1 (Choi et al., 2005) encodes a transposase for transposition of the mini Tn7 transposon. For selection and counter-selection of Tn7-bearing P. putida KT2440, cetrimide agar plates containing 30 mg L−1 gentamycin and 1 % glycerol were used.

Table 1. Strains and plasmids used and generated in this study.
Cultivations of P. putida KT2440 derivatives for promoter characterization were performed in minimal medium containing 3.88 g L−1 K2HPO4 and 1.63 g L−1 NaH2PO4 with 20 mM glucose as the sole carbon source (Hartmans et al., 1989).
DNA Techniques
All oligonucleotides used in this study are listed in Supplementary Table 1. For the generation of different length spacer sequences PCR was used (Supplementary Table 2). Up to 40 bp length a forward oligonucleotide (SK11, SK34, SK36, or SK38) and reverse oligonucleotide SK2 were used with plasmid pBG14g as template (Supplementary Table 3). Q5 polymerase (New England Biolabs) with proofreading activity was used for amplification and reactions were prepared as described by the manufacturer. The resulting fragments were cut out from agarose gels and purified with the DNA Gel Extraction kit from New England Biolabs. Longer spacer sequences from 50 to 100 bp were assembled by PCR with two long oligonucleotides with complementary 3′-ends and an initial annealing step in the PCR (Figure 1, Supplementary Table 4). Dimer formation of 3′-ends was checked in silico to ensure that stacked promoter constructs can be formed by annealing of two oligonucleotides (https://www.thermofisher.com/de/de/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/thermo-scientific-web-tools/multiple-primer-analyzer.html). A detailed approach is described in the Supporting Information. PCR fragments were purified with PCR & DNA Cleanup Kit (New England Biolabs). Promoter-less plasmid pBG was used as backbone. Generated spacer fragments were digested by PacI and AvrII (New England Biolabs) at 37°C. Cloning procedures are following the rules of SEVA and are thus compatible with other constructs (Silva-Rocha et al., 2013). Plasmid pBG was additionally treated with alkaline phosphatase (Fermentas) to circumvent self-ligation. Digested fragments were purified using PCR & DNA Cleanup Kit (New England Biolabs). DNA concentrations of eluted DNA were measured with a NanoDrop One (Thermo Scientific). Fragments and backbone with adjusted concentrations were ligated using T4 ligase (New England Biolabs) at room temperature for 30 min. Transformation of ligated plasmids was performed by heat shock into chemically competent E. coli PIR2 cells (Hanahan, 1983). Plasmid isolation was done with Plasmid Miniprep Kit (New England Biolabs) and sequences were confirmed by Sanger sequencing (Eurofins Genomics). Oligonucleotide combinations and detailed PCR protocols are described in Supplementary Table 5.
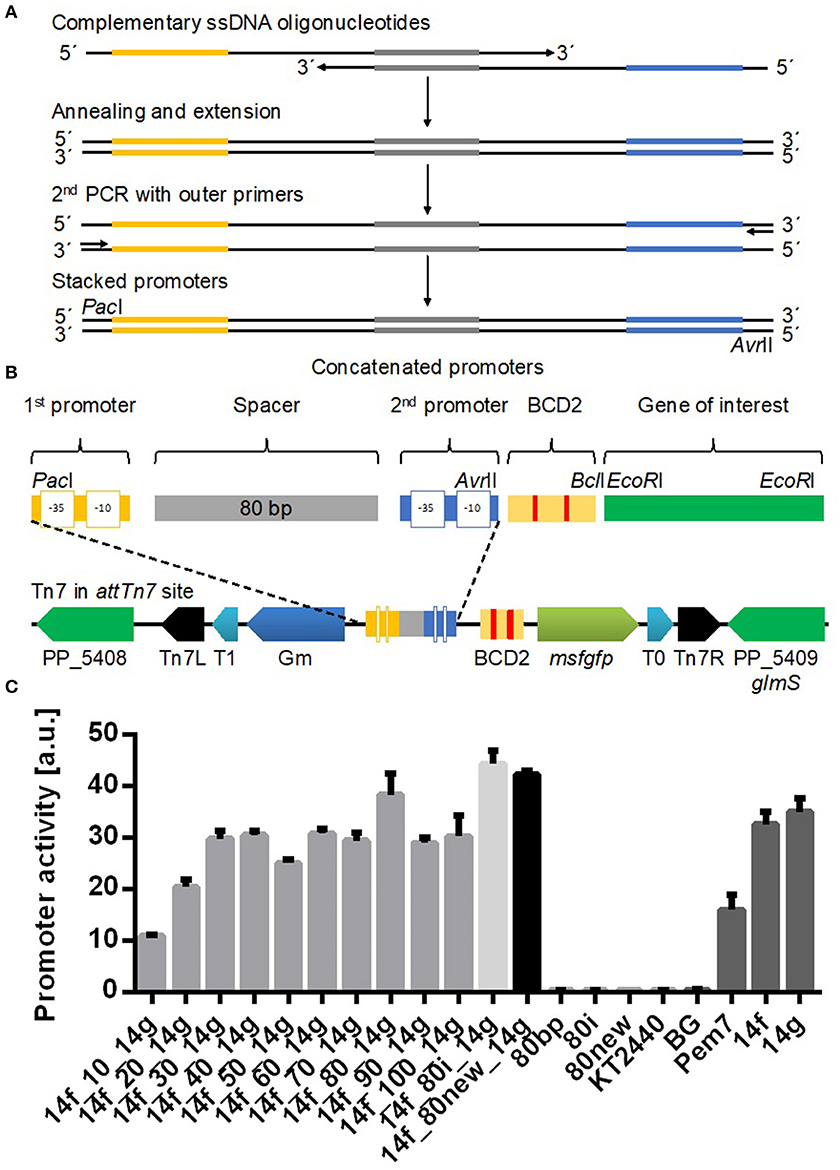
Figure 1. Identification of the optimal spacer length between the two promoters 14f and 14g using a mini Tn7 vector (Zobel et al., 2015). (A) Two PCR reactions were performed to generate stacked promoters with longer (>45 bp) spacer sequences and promoter combinations with the 80i spacer sequence. In a first PCR reaction long single stranded DNA oligonucleotides containing one promoter sequence (yellow or blue) were annealed via complementary sequences in the spacer (gray) and extended by Q5 polymerase to double stranded DNA. The resulting dsDNA fragment was amplified in a second PCR. (B) Structural organization of stacked promoters and of the mini Tn7 used in this study after genomic integration. Stacked promoters consisting of promoter sequences at two positions separated by a spacer were inserted via restriction sites PacI and AvrII. BCD2 element for translational coupling and msfGFP as reporter gene. Tn7 module contains a GmR marker for selection and two terminators (T0 and T1) for insulation of the probe. Tn7R and Tn7L are recognized by a transposase. (C) Tested spacers contained between 10 and 100 bp. P. putida KT2440 attTn7::BGf##g-mfsGFP, where ## refers to the number of nucleotides in the spacer sequence (gray bars), were cultured in a BioLector in minimal medium with 20 mM glucose in a 96 well plate. The control strains BG13 with the Pem7 promoter of average strength, the individual promoters 14f and 14g, and promoterless BG and wild type P. putida KT2440, as well as additional controls with two 80 bp spacers 80i and 80new are also shown. Identical strains from at least two different transformations were tested, with three biological replicates each. Error bars indicate the standard error of the mean (n > 6).
For the construction of promoter positions and spacer controls, previously cloned plasmids containing stacked promoters were used as template (Supplementary Tables 6, 7).
Construction of the single nucleotide polymorphism (SNP) library was done by PCR with plasmid pBG14g as template and primers SK63-SK92 containing single degenerate nucleotides.
We used colony PCR with oligonucleotides SK4 and SK5 to verify the correct and full-length genomic integration of Tn7 at the attTn7 site in P. putida KT2440. Single colonies were picked with a pipette tip or toothpick and lysed in 30 μL lysis buffer containing 60 % alkaline PEG 200 (pH adjusted to 13–13.5 with 2M KOH) for 15 min at room temperature (Wynands et al., 2018). As template one microliter was used for the PCR reaction (Taq 2X Master Mix, New England BioLabs).
Measuring Fluorescence and Determination of Promoter Activity
For the identification of SNPs in the obtained promoter library for the individual positions we measured GFP fluorescence. We cultivated E. coli PIR2 mini Tn7 plasmid-bearing strains in 0.5 mL LB medium containing 50 mg L−1 kanamycin at 30°C in 96 well System Duetz plates (Enzyscreen, The Netherlands). Fluorescence of msfGFP was measured in a synergyMX plate reader (Biotek, Bad Friedrichshall, Germany). Samples were measured in black bottom 96 well plates at an excitation wavelength of 488 nm and emission wavelength of 520 nm. Absorption was measured at 600 nm in clear bottom 96 well plates. From strains showing different intensities for GFP fluorescence the plasmid was isolated and sequenced. Followed by genomic integration of desired plasmids in P. putida KT2440 by triparental mating.
Growth and fluorescence measurements of integrated promoter constructs in P. putida KT2440 were performed with a Biolector (M2P Labs, Baesweiler, Germany) in 96 well plates (Greiner Bio-One) with a filling volume of 200 μL. Cultures were inoculated to an optical density at 600 nm of 0.1 for each strain from precultures cultivated at 30 °C at 300 rpm in 24 well System Duetz plates (Enzyscreen, Heemstede, The Netherlands) containing 1.5 mL of previously described minimal medium. The Biolector was set to 30°C, 900 rpm and humidity control of 85 %. Two internal filter modules of the device were used for online measurement. Fluorescence of GFP was measured at excitation wavelength at 488 nm and emission wavelength of 520 nm with gain 50. Biomass was determined at 620 nm with gain 40 as scattered light. Scattered light was correlated to OD600 with a dilution series of a stationary phase culture. Determination of promoter activity was done with Microsoft Excel by calculating the slope of GFP fluorescence to optical density during the exponential phase.
Determination of Transcript Levels by Quantitative Real Time PCR
Transcription levels of msfGFP was determined by quantitative real time PCR. RNA was isolated from chosen strains grown on minimal medium containing 20 mM glucose as sole carbon source in 24 well System Duetz plates at 30°C and 300 rpm (Hartmans et al., 1989). Biological duplicates of each strain were cultivated until an optical density of 1.0 was reached. One milliliter of cell cultures were harvested, supernatant discarded and the resulting pellet resuspended with 1 mL RNAlaterTM Stabilization Solution (ThermoFisher Scientific). Afterwards the cells were resuspended in 700 μL lyse solution (New England Biolabs, Monarch™ Total RNA Miniprep Kit) and transferred to bead beating tubes containing glass bead with a size of 0.5 mm (Zymo Research, Irvine, USA). Tubes were beaten for 1 min to destroy the cells (Mini-Beadbeater-16, Biospec Products, Bartlesville, USA). Cells debris were removed by centrifugation at 13.000 rpm for 2 min. The supernatant was transferred to an RNase-free tube and used for further works. RNA isolation from lysed samples followed the manual from the kit Monarch Total RNA Miniprep Kit (New England Biolabs). Elution of RNA was done with 50 μL RNase free water. RNA concentration was measured with a NanoDrop One (Thermo Scientific) at 260 nm. Samples were adjusted to a final RNA concentration of 280 ng in a total volume of 40 μL, dilution was done with RNase-free water. An additional DNase treatment was done by adding 5 μL DNaseI and 5 μL DNase I reaction buffer (New England Biolabs) to the RNA isolates. Digestion was done at 37°C for 10 min and DNase inactivation at 75°C for 10 min. For cDNA synthesis LunaScript RT SuperMix Kit (New England Biolabs) was performed as describe in the manual.
Determination of primer efficiencies was done with diluted cDNA from BG14f_80. cDNA was diluted 1:10, 1:20, 1:40, 1:80, 1:160, 1:320, and 1:640. 1.25 μL of each used for the qRT-PCR reaction. A total volume of 10 μL containing 5 μL Universal qPCR Master Mix (New England Biolabs), 0.25 μL of each oligonucleotide, 1.25 μL sample and 3.25 μL RNase-free water were used. We tested oligonucleotide combinations for the target gene msfGFP and housekeeping gene rpoD in a CFX Connect Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, USA) using a protocol described in the manual of Universal qPCR Master Mix (New England Biolabs). CFX Manager software (Bio-Rad Laboratories, Hercules, USA) was used for the calculation of resulting primer efficiencies and an online tool was used to calculated the amplification factor (https://www.thermofisher.com/de/de/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/thermo-scientific-web-tools/qpcr-efficiency-calculator.html). Tested oligonucleotide combinations for msfGFP achieved a value of 1.97 and rpoD of 2.02 (Udvardi et al., 2008).
Each cDNA sample was diluted 1:10 and analyzed as technical duplicate. Volumes for each reaction are described above. As negative control the same amount of water was added to the reaction instead of cDNA. Examination from resulting Ct values was done with Microsoft Excel and a ΔCt method was applied (Pfaffl, 2001). To exclude genomic DNA in the samples, isolated RNA was used in separate reactions with oligonucleotides for rpoD.
Statistics
Each promoter construct was characterized in 2–3 independent transformations performed on different days. Three clones from each transformant were tested in a Biolector to determine promoter activities, yielding a total of 6–9 biological replicates. For each construct the mean and standard error of the mean was calculated from these combined biological replicates. Significance of difference of the activity of constructs with different spacer lengths was analyzed by one-way ANOVA with Turkey's post-hoc comparison. Coefficient of variation, determined by dividing the absolute difference of the predicted and experimental value by the experimental value, was used to compare the accuracy of prediction of stacked promoter activities.
Results and Discussion
Identification of the Optimal Distance Between Two Promoters
For the characterization of stacked promoters, we used a mini Tn7 transposon, which integrates as single copy into the attTn7 site downstream of the glmS gene in the genome of P. putida KT2440 (Bagdasarian et al., 1981; Choi et al., 2005; Zobel et al., 2015). The transposon is designed to characterize promoters in a reliable and reproducible manner, featuring a BCD2 element to reduce GOI-based expression variability (Mutalik et al., 2013), an msfGFP (Landgraf, 2012) gene as reporter, and two flanking terminators to minimize genomic read-through (Figure 1; Zobel et al., 2015).
In order to determine the optimal distance between two promoters, we stacked the 14f and 14g promoters from a previously published synthetic promoter library (Zobel et al., 2015) with spacer sequences with increasing length from 10 to 100 bp by extension at the 3'-end. The promoters are referred to solely by their SEVA code (Zobel et al., 2015; 14a-g, with a being the weakest and g being the strongest) for ease of reference. The spacer was randomly generated (http://www.faculty.ucr.edu/~mmaduro/random.htm) and manually curated for unwanted restriction sites as well as putative ribosome binding sites, −35, and −10 like sequences, which could disturb the analysis due to intrinsic activity. The spacer was created with a GC content of 62%, similar to the genomic average of P. putida KT2440 (Nelson et al., 2002).
A promoterless construct BG and wildtype P. putida KT2440 were used as negative controls. As positive controls we used the single calibrated promoters described in Zobel et al. (2015), including Pem7 (Martínez-García et al., 2015) which reaches half of the activity of promoter 14g. With short spacer sequences of <35 bp, the activity of the stacked promoter is lower than that of either of the single promoters (Figure 1). This result is likely caused by steric hindrance of the RNA polymerase holoenzyme, since the combined sigma factor and RNA polymerase cover around 80 bp upstream of the promoter (Schmitz and Galas, 1979). A spacer length of 40–70 and 90–100 bp resulted in activities of comparable strength. A significantly higher activity was observed for 14f_80_14g with an 80 bp spacer compared to all other spacer lengths (one-way ANOVA with Turkey's post-hoc comparison).
To exclude that this 80 bp spacer is an outlier due to possible activating sequences, the experiment was repeated with a reverse complement version of the spacer (80i) and a new, independently generated spacer sequence (80new, Supplementary Table 2). All three 80 bp control spacers led to comparable activities, indicating that this distance between two promoters is promoting additive activity of the two promoters. It is interesting to note that the spacer length of 80 bp matches the sequence covered by the RNA polymerase holoenzyme (Schmitz and Galas, 1979), although this correlation should not be confused with causation. Promoterless controls with only 80i and 80new also show no activity, excluding any intrinsic activity from the spacers themselves (Figure 1).
With the 80i spacer a cumulative effect occurred, with the total output of the stacked promoters being higher than the individual activities. However, the output was much lower than the sum of the two individual promoters for each tested spacer length. For further characterization, we used the 80i spacer since it enabled the highest promoter activity.
Characterization of Context Effects on Stacked Promoters
We hypothesized two possible ways how these stacked promoters are affected. The primary hypothesis is that of an effect of the spacer on the promoter. The alternative hypothesis is a mutual influence of one promoter on the other (Callen et al., 2004; Shearwin et al., 2005). To test these hypotheses, we constructed 14 different stacked promoter combinations and 12 controls to determine the influence of the 80i spacer on single promoter activities. Following the rules provided by SEVA (Martínez-García et al., 2015), the promoter is integrated between restriction sites PacI and AvrII. The spacer is an additional sequence in the probe vector published by Zobel et al. (2015) and is not replacing any sequences from the original construct. The constructs are named according to their composition, i.e., in 14f_80i promoter 14f is cloned upstream of the 80i spacer. After genomic integration of the Tn7 transposon all strains were characterized in a BioLector system (Figure 2).
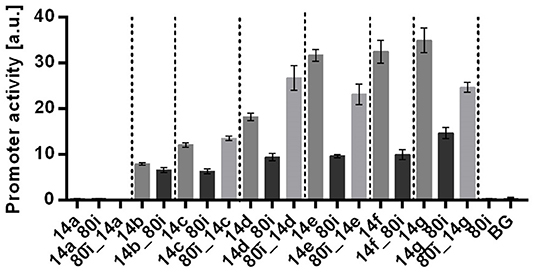
Figure 2. Characterization of context-depended promoter activities using the 80i spacer. Promoters with both up- and downstream spacer were tested. Shown are promoter activities for the original promoters (dark bars) from Zobel et al. (2015) and promoter-spacer (14x_80i) as well as spacer-promoter (80i_14x) combinations (gray bars), where x stands for promoter 14a to 14g. All constructs were genomically integrated in P. putida KT2440. Strains were cultured in a BioLector in minimal medium with 20 mM glucose in a 96 well plate Identical strains from at least two different transformations were tested, with three biological replicates each. Vertical dotted lines are separating individual sets. Error bars indicate the standard error of the mean (n > 6).
The single promoter controls without spacer reached activities that are comparable to those initially described by Zobel et al. (2015). In contrast, single promoter combined with the 80i spacer, either upstream or downstream, were strongly affected in their activity (Figure 2). With downstream placement of the spacer, all promoters were negatively affected, with decreases up to 70% for 14f_80i. In contrast, no clear trend could be discerned with upstream placement of the spacer, with most combinations having decreased activities up to 28%, but 80i_14c gained 12 % and 80i_14d even 50% activity.
These results show that the spacer has a drastic effect on all tested promoters. This is in spite of the fact that the spacer itself doesn't display any promoter activity (Figure 1), nor does it contain any discernible sequences that might affect promoter activity, such as AT-rich UP elements (Estrem et al., 1998). In addition, up- and downstream effects of the spacer are unpredictable. Most of the single promoters show a decreased activity when combined with the spacer, which could be explained by missing upstream activating elements potentially present in the original construct such as the AT-rich PacI restriction site. While no consistent correlation between spacer position and promoter activity is discernible, the results do confirm the primary hypothesis that the activity of the promoters is affected by the spacer.
To further test if, besides the effect of the spacer, the stacked promoters also affect each other, all seven calibrated promoters were stacked with 14g at the second position. As additional control, the reverse-order combination 14g_80i_14a was also included. As expected from the abovementioned results, all of these combinations led to much lower activities than expected from the sum of the individual promoters disregarding context-effects (Figure 3). Interestingly, the combinations 14a_80i_14g and 14g_80i_14a, which only differ in the order of the promoters, reached completely different activities. During the cloning of these stacks, a triple ‘ffg’ promoter consisting of two 14f and one 14g sequences separated by two spacers was accidentally created, in which the second 14f is shorter by two nucleotides between the −35 and −10 elements. Deriving sequence-function relationships from this promoter would be too complex, but the fact that it is around 45% stronger than the strongest promoter combination 14g_80i_14g makes it useful in applications where very high expression is needed (Lenzen et al., 2019; Bator et al., 2020) (Supplementary Figure 1).
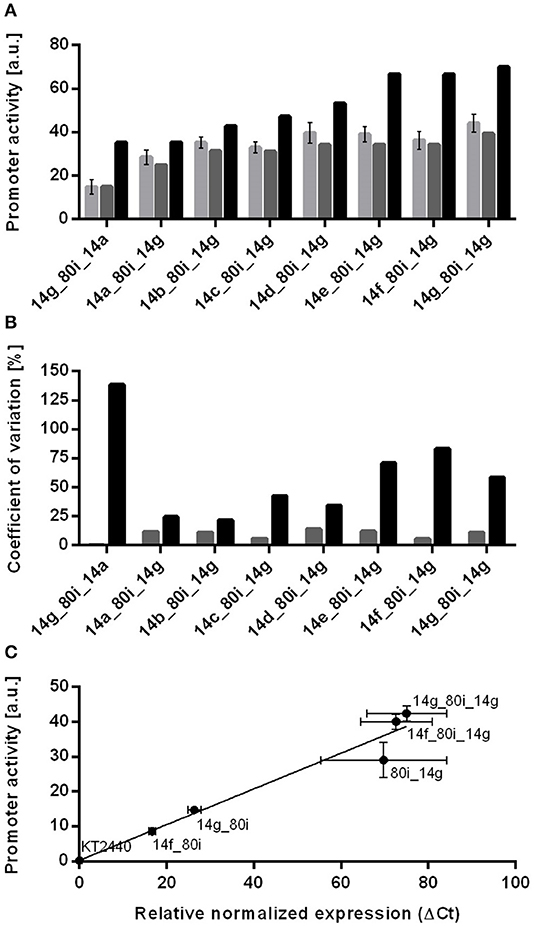
Figure 3. Comparison of experimental values, context-specific and context-unspecific prediction of promoter activities for stacked promoter with the 80i spacer separating promoters 14a to 14g. (A) Shown are determined promoter activities for stacked promoters (light gray bars), context-specific activities calculated with context-depended values (gray bars) and context-unspecific activities using original promoter activities (black bars). All constructs were genomically integrated into P. putida KT2440. Cultivation was done in a BioLector in minimal medium with 20 mM glucose in a 96 well plate. Identical strains from at least three different transformations were tested, with three biological replicates each. Error bars indicate the standard error of the mean (n > 9). (B) Coefficient of variation (CV) of context-specific (light gray bars) and context-unspecific (black bars) prediction of resulting promoter activities. (C) Plot of msfGFP transcription levels normalized to rpoD determined by quantitative real time PCR (qRT-PCR) and promoter activities from single promoter controls and stacked promoters. Wild type P. putida KT2440 was used as negative control. All constructs are genomically integrated into the genome of P. putida KT2440. Cultivations to determine promoter activities were done in a BioLector in minimal medium with 20 mM glucose in a 96 well plate. Cultivation to determine transcription levels was done in 24 well System Duetz plates containing minimal medium with 20 mM glucose. Identical strains from at least two different transformations were tested, with three biological replicates each. Error bars indicate the standard error of the mean (n > 6).
When comparing the activities of these stacks to the single promoter-spacer controls above, it becomes apparent that the immediate context of single promoters is the major determinant for the prediction of promoter activity. The sum of the single promoter activities greatly overestimates the activities of stacked promoters by as much as 140% for the 14g_80i_14a combination (Figure 3). In contrast, the sum of the context-specific controls provides a much more accurate prediction, i.e., 14g_80i + 80i_14a = 14g_80i_14a. In this case, the coefficient of variance between context-specific prediction and experimental values is lower than 15% for all tested combinations. This strongly suggests that, once the direct context of the individual promoters is sufficiently taken into account, the stacked promoters don't affect each other's activity.
Beyond having different promoter contexts, the abovementioned constructs also generate different 5′-terminal mRNA ends, which may cause differences in mRNA stability or translation initiation rates. In order to minimize the effect of these differences, a bicistronic design (Mutalik et al., 2013) was included in the reporter construct. To verify whether the altered expression of context-affected constructs is caused by increased transcription, we performed quantitative real time PCR (qRT-PCR) on selected constructs. Determined transcript levels correlate well with promoter activities derived from fluorescence measurements (r2 = 0.95, Figure 3), confirming that the spacer influences transcription, rather than translation. Attempts to determine the relative contributions of the first or second promoter by qRT-PCR were inconclusive. In principle, stacked promoters generate two overlapping transcripts of different length, which might be distinguished with different primers pairs. However, longer transcripts show a shift in Ct value compared to shorter amplicons, and suitable primer pairs for similar lengths could not be found (Debode et al., 2017).
Using an SNP Promoter Library for Stacked Promoters
A change in context greatly affects promoter activitiy, and there are large quantitative differences for each tested promoter-spacer combination. Given that the main variable between these constructs is the promoter sequence, this might be due to specific DNA-DNA interactions between promoter and spacer, which influence promoter activity, or RNA-RNA interactions, which affect RNA stability. To further investigate the sequence-activity relationship, we generated a single nucleotide polymorphism (SNP) library based on promoter 14g (Figure 4). Such a library yields promoters with very similar sequences, but large differences in activity. If the variability of the impact of the spacer on the activity is indeed caused by DNA-DNA or RNA-RNA interactions, using promoters with more similar sequences can be expected to reduce this variability. The library contained 90 different promoter sequences, which were generated in 30 PCR reaction with one degenerate nucleotide in the sequence. Changes of the core promoter sequence were inserted within the −35 element (position 1–6), in the interspaced region (position 7–23, 30), and in the −10 element (position 24–29). For each position four different promoters can occur, of which one will correspond to the original 14g sequence. Initial screening of mini libraries (14 clones each) of these low degeneracy promoters was performed in plasmid-bearing E. coli PIR2 strains by analyzing msf GFP fluorescence. Aberrations compared to original pBG14g-bearing E. coli PIR2 strains were recognized (Figure 4). For nearly each position clones were found with either a higher or lower fluorescence signal than the original pBG14g plasmid. Variations in the interspaced region generally had a lower effect on expression strength, while changing single nucleotides in the −35 and −10 consensus sequences yielded more clones with decreased promoter activity, as expected (Lodge et al., 1990; McLean et al., 1997). We therefore focused further characterization on these elements in order to obtain a set of promoters with a range of activity that is comparable to the previously described calibrated promoter library from Zobel et al. (2015) (Figure 4).
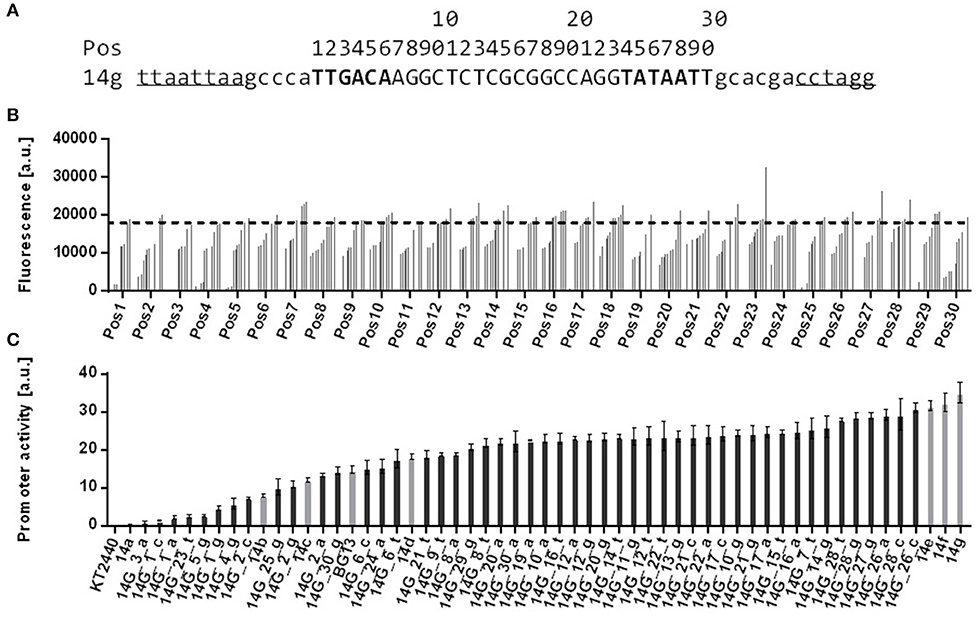
Figure 4. Comparison of screened and characterized promoter sequences based on 14g with single nucleotide polymorphisms. (A) Original promoter sequence of 14g with highlighted−35 and−10 elements (in bold). Restriction sites PacI and AvrII are underlined and positions of the modified core promoter sequence are given with numbers above the sequence. (B) msfGFP fluorescence of E. coli PIR2 bearing plasmid pBG14g with degenerate bases at 30 positions along the core promoter sequence. Changed position is shown on the x axis. Determined values are ranked by fluorescence intensity of 14 strains tested for each position. Strains were cultivated in 96 well System Duetz plates with LB medium supplemented with 50 mg L−1 kanamycin. Fluorescence and optical density were measured with a plate reader. The dotted line indicates the promoter activity of the 14g control. (C) Chosen SNP promoter constructs were genomically integrated into P. putida KT2440. Strains are named 14G_##n, whereas ## stands for the position in the promoter sequence and n for a nucleotide (A, C, G or T). Cultivation was done in a BioLector in minimal medium with 20 mM glucose in 96 well plates. Identical strains from at least two different transformations were tested, with three biological replicates each. Error bars indicate the standard error of the mean (n > 6).
After initial screening of positions in the SNP promoter sequences, we selected three such positions within the −35 or −10 elements for further characterization. Introducing a degenerate base in these three positions yields nine different promoters with a good spread of activity, and these promoters were combined with the 80i spacer in the downstream position (Figure 5). Changing position 26 in the −10 sequence resulted in a slightly decreased activity, while changes in positions 1 and 2 in the −35 sequence yielded larger decreases, which is in accordance with Lodge et al. (1990).
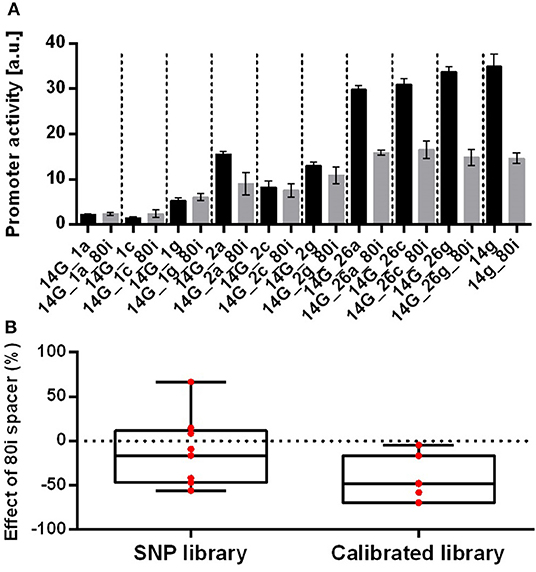
Figure 5. Characterization of the effect of the 80i spacer on a single nucleotide exchange promoter library based on promoter 14g genomically integrated in P. putida KT2440. (A) Promoter activities derived from GFP fluorescence analysis of SNP promoters with (gray bars) and without (black bars) downstream 80i spacer. Cultivation was done in a BioLector in minimal medium with 20 mM glucose in 96 well plates. Identical strains from at least two different transformations were tested, with three biological replicates each. Vertical dotted lines are separating individual sets. Error bars indicate the standard error of the mean (n > 6). (B) Box and whiskers plot of the relative effect of the downstream 80i spacer on promoter activities of the SNP library and the original calibrated promoter library from Zobel et al. (2015). The effect of the spacer is calculated as the % change of the promoter with downstream 80i sequence compared to the original promoter. Data points are indicated in red.
We have seen that small changes in the 14g promoter sequence can strategically affect activity in a mini-promoter-library (Supplementary Table 10). In spite of the relative uniformity of the promoter sequences in this library, combination of these promoters with the 80i spacer again strongly affected the activities with both in- and decreases up to 66 %. The reduced sequence variability did not reduce the quantitative variability of the spacer effect compared to the CalPro library from Zobel et al. (2015). Both have a high coefficient of variation of 40% for the SNP library and 25% for the CalPro library (Figure 5). This strongly suggests that the promoter sequence per se does not cause the context-dependent effect, suggesting that other factors such as the varying transcription-initiation rates are in play.
Conclusion
In this work, we aimed to increase the composability and predictability of synthetic promoters by investigating the effects of differing contexts on their activity. In the combination of two promoters, the length of the spacer region is crucial for reaching higher and cumulative effects, with 80 bp being the optimal. Even though the spacer sequence has no intrinsic activity, it strongly and unpredictably influences the activity of promoters by up- and downstream effects. By accounting for this influence, the activity of two stacked promoters can be accurately predicted with coefficients of variance below 15%. A strong reduction of sequence variability was achieved using an SNP library, but this reduction did not reduce the quantitative variability of the spacer effect. This strongly indicates that nucleotide-nucleotide interactions between promoter and spacer do not play a prominent role. Clearly, context-specific effects of synthetic promoters are not yet fully understood in Pseudomonas. Although the semi-empirical approach for prediction of stacked promoter activities provides an accurate workaround to this, further work is needed to understand the fundamental interaction of genetic elements and their surroundings.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Author Contributions
NW and SK designed the experiments. SK performed all molecular engineering and experiments, prepared figures and wrote the manuscript. NW supervised the study and edited the manuscript. LB advised on all experiments, analyzed and discussed data, and edited the manuscript.
Funding
We gratefully acknowledge funding for SK from the European Union's Horizon 2020 research and innovation program under grant agreement no. 633962 for the project P4SB. NW was supported by the German Research Foundation (DFG) through the Emmy Noether program (WI 4255/1-1). The laboratory of LB was partially funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbioe.2020.00551/full#supplementary-material
References
Aparicio, T., de Lorenzo, V., and Martinez-Garcia, E. (2018). CRISPR/Cas9-based counterselection boosts recombineering efficiency in Pseudomonas putida. Biotechnol. J. 13:e1700161. doi: 10.1002/biot.201700161
Bagdasarian, M., Lurz, R., Ruckert, B., Franklin, F., Bagdasarian, M., Frey, J., et al. (1981). Specific-purpose plasmid cloning vectors. II. broad host range, high copy number, RSF1010-derived vectors, and a host-vector system for gene cloning in Pseudomonas. Gene 16, 237–247. doi: 10.1016/0378-1119(81)90080-9
Bator, I., Wittgens, A., Rosenau, F., Tiso, T., and Blank, L. M. (2020). Comparison of three xylose pathways in Pseudomonas putida KT2440 for the synthesis of valuable products. Front. Bioeng. Biotechnol. 7:480. doi: 10.3389/fbioe.2019.00480
Blank, L. M., Ionidis, G., Ebert, B. E., Bühler, B., and Schmid, A. (2008). Metabolic response of Pseudomonas putida during redox biocatalysis in the presence of a second octanol phase. FEBS J. 275, 5173–5190. doi: 10.1111/j.1742-4658.2008.06648.x
Boyer, H. W., and Roulland-Dussoix, D. (1969). A complementation analysis of the restriction and modification of DNA in Escherichia coli. J. Mol. Biol. 41, 459–472. doi: 10.1016/0022-2836(69)90288-5
Callen, B. P., Shearwin, K. E., and Egan, J. B. (2004). Transcriptional interference between convergent promoters caused by elongation over the promoter. Mol. Cell. 14, 647–656. doi: 10.1016/j.molcel.2004.05.010
Choi, K. H., Gaynor, J. B., White, K. G., Lopez, C., Bosio, C. M., Karkhoff-Schweizer, R. R., et al. (2005). A Tn7-based broad-range bacterial cloning and expression system. Nat. Methods 2, 443–448. doi: 10.1038/nmeth765
Damron, F. H., McKenney, E. S., Barbier, M., Liechti, G. W., Schweizer, H. P., and Goldberg, J. B. (2013). Construction of mobilizable mini-Tn7 vectors for bioluminescent detection of gram-negative bacteria and single-copy promoter lux reporter analysis. Appl. Environ. Microb. 79, 4149–4153. doi: 10.1128/AEM.00640-13
de Lorenzo, V., Herrero, M., Jakubzik, U., and Timmis, K. N. (1990). Mini-Tn5 transposon derivatives for insertion mutagenesis, promoter probing, and chromosomal insertion of cloned DNA in gram-negative bacteria. J. Bacteriol. 172, 6568–6572. doi: 10.1128/JB.172.11.6568-6572.1990
Debode, F., Marien, A., Janssen, É., Bragard, C., and Berben, G. (2017). Influence of the amplicon length on real-time PCR results. Biotechnol. Agron. Soc. 21, 3–11. doi: 10.25518/1780-4507.13461
Dixon, R. (1984). Tandem promoters determine regulation of the Klebsiella pneumonia glutamine synthetase (gln) gene. Nucleic Acids Res. 12, 7811–7830. doi: 10.1093/nar/12.20.7811
Elmore, J. R., Furches, A., Wolff, G. N., Gorday, K., and Guss, A. M. (2017). Development of a high efficiency integration system and promoter library for rapid modification of Pseudomonas putida KT2440. Metab. Eng. Commun. 5, 1–8. doi: 10.1016/j.meteno.2017.04.001
Estrem, S. T., Gaal, T., Ross, W., and Gourse, R. L. (1998). Identification of an UP element consensus sequence for bacterial promoters. Proc. Natl. Acad. Sci. U.S.A. 95, 9761–9766. doi: 10.1073/pnas.95.17.9761
Franden, M. A., Jayakody, L. N., Li, W. J., Wagner, N. J., Cleveland, N. S., Michener, W. E., et al. (2018). Engineering Pseudomonas putida KT2440 for efficient ethylene glycol utilization. Metab. Eng. 48, 197–207. doi: 10.1016/j.ymben.2018.06.003
Friehs, K. (2004). Plasmid copy number and plasmid stability. Adv. Biochem. Eng. Biotechnol. 86, 47–82. doi: 10.1007/b12440
Gao, Y., Liu, C., Ding, Y., Sun, C., Zhang, R., Xian, M., et al. (2014). Development of genetically stable Escherichia coli strains for poly(3-hydroxypropionate) production. PLoS ONE 9:e97845. doi: 10.1371/journal.pone.0097845
Gilman, J., and Love, J. (2016). Synthetic promoter design for new microbial chassis. Biochem. Soc. Trans. 44, 731–737. doi: 10.1042/BST20160042
Gruber, T. M., and Gross, C. A. (2003). Multiple sigma subunits and the partitioning of bacterial transcription space. Annu. Rev. Microbiol. 57, 441–466. doi: 10.1146/annurev.micro.57.030502.090913
Hammer, K., Mijakovic, I., and Jensen, P. R. (2006). Synthetic promoter libraries-tuning of gene expression. Trends Biotechnol. 24, 53–55. doi: 10.1016/j.tibtech.2005.12.003
Hanahan, D. (1983). Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166, 557–580. doi: 10.1016/S0022-2836(83)80284-8
Hartmans, S., Smits, J. P., van der Werf, M. J., Volkering, F., and de Bont, J. A. M. (1989). Metabolism of styrene oxide and 2-phenylethanol in the styrene-degrading Xanthobacter strain 124X. Appl. Environ. Microb. 55, 2850–2855. doi: 10.1128/AEM.55.11.2850-2855.1989
Heipieper, H. J., Neumann, G., Cornelissen, S., and Meinhardt, F. (2007). Solvent-tolerant bacteria for biotransformations in two-phase fermentation systems. Appl. Microbiol. Biotechnol. 74, 961–973. doi: 10.1007/s00253-006-0833-4
Herrero, M., de Lorenzo, V., and Timmis, K. N. (1990). Transposon vectors containing non-antibiotic resistance selection markers for cloning and stable chromosomal insertion of foreign genes in gram-negative bacteria. J. Bacteriol. 172, 6557–6567. doi: 10.1128/JB.172.11.6557-6567.1990
Isken, S., and de Bont, J. A. (1998). Bacteria tolerant to organic solvents. Extremophiles 2, 229–238. doi: 10.1007/s007920050065
Jahn, M., Vorpahl, C., Turkowsky, D., Lindmeyer, M., Buhler, B., Harms, H., et al. (2014). Accurate determination of plasmid copy number of flow-sorted cells using droplet digital PCR. Anal. Chem. 86, 5969–5976. doi: 10.1021/ac501118v
Jensen, P. R., and Hammer, K. (1998). Artificial promoters for metabolic optimization. Biotechnol. Bioeng. 58, 191–195. doi: 10.1002/(SICI)1097-0290(19980420)58:2/3<191::AID-BIT11>3.0.CO;2-G
Jiang, W., Bikard, D., Cox, D., Zhang, F., and Marraffini, L. A. (2013). RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 31, 233–239. doi: 10.1038/nbt.2508
Jiménez, J. I., Miñambres, B., García, J. L., and Díaz, E. (2002). Genomic analysis of the aromatic catabolic pathways from Pseudomonas putida KT2440. Environ. Microbiol. 4, 824–841. doi: 10.1046/j.1462-2920.2002.00370.x
Kang, J-G., Hahn, M-Y., Roe, J-H., and Ishihama, A. (1997). Identification of sigma factors for growth phase-related promoter selectivity of RNA polymerases from Streptomyces coelicolor A3(2). Nucleic Acids Res. 25, 2566–2573. doi: 10.1093/nar/25.13.2566
Keen, N. T., Tamaki, S., Kobayashi, D., and Trollinger, D. (1988). Improved broad-host-range plasmids for DNA cloning in gram-negative bacteria. Gene 70, 191–197. doi: 10.1016/0378-1119(88)90117-5
Köhler, K. A. K., Blank, L. M., Frick, O., and Schmid, A. (2015). D-Xylose assimilation via the weimberg pathway by solvent tolerant Pseudomonas taiwanensis VLB120. Environ. Microbiol. 17, 156–170. doi: 10.1111/1462-2920.12537
Koopman, F., Wierckx, N., de Winde, J. H., and Ruijssenaars, H. J. (2010). Efficient whole-cell biotransformation of 5-(hydroxymethyl)furfural into FDCA, 2,5-furandicarboxylic acid. Bioresour. Technol. 101, 6291–6296. doi: 10.1016/j.biortech.2010.03.050
Kusumawardhani, H., Hosseini, R., and de Winde, J. H. (2018). Solvent tolerance in bacteria: fulfilling the promise of the biotech era? Trends Biotechnol. 36, 1025–1039. doi: 10.1016/j.tibtech.2018.04.007
Lambertsen, L., Sternberg, C., and Molin, S. (2004). Mini-Tn7 transposons for site-specific tagging of bacteria with fluorescent proteins. Environ. Microbiol. 6, 726–732. doi: 10.1111/j.1462-2920.2004.00605.x
Landgraf, D. (2012). Quantifying Localizations and Dynamics in Single Bacterial Cells. Doctoral Dissertation, Harvard University, Cambridge, MA, USA.
Lenzen, C., Wynands, B., Otto, M., Bolzenius, J., Mennicken, P., Blank, L. M., et al. (2019). High-yield production of 4-hydroxybenzoate from glucose or glycerol by an engineered Pseudomonas taiwanensis VLB120. Front. Bioeng. Biotechnol. 7:130. doi: 10.3389/fbioe.2019.00130
Li, M., Wang, J., Geng, Y., Li, Y., Wang, Q., Liang, Q., et al. (2012). A strategy of gene overexpression based on tandem repetitive promoters in Escherichia coli. Microb. Cell Fact. 11:19. doi: 10.1186/1475-2859-11-19
Li, W. J., Jayakody, L. N., Franden, M. A., Wehrmann, M., Daun, T., Hauer, B., et al. (2019). Laboratory evolution reveals the metabolic and regulatory basis of ethylene glycol metabolism by Pseudomonas putida KT2440. Environ. Microbiol. 21, 3669–3682. doi: 10.1111/1462-2920.14703
Li, W. J., Narancic, T., Kenny, S. T., Niehoff, P. J., O'Connor, K. E., Blank, L. M., et al. (2020). Unraveling 1,4-butanediol metabolism in Pseudomonas putida KT2440. Front. Microbiol. 11:382. doi: 10.3389/fmicb.2020.00382
Lindmeyer, M., Jahn, M., Vorpahl, C., Müller, S., Schmid, A., and Bühler, B. (2015). Variability in subpopulation formation propagates into biocatalytic variability of engineered Pseudomonas putida strains. Front. Microbiol. 6:1042. doi: 10.3389/fmicb.2015.01042
Lodge, J., Williams, R., Bell, A., Chan, B., and Busby, S. (1990). Comparison of promoter activities in Escherichia coli and Pseudomonas aeruginosa: use of a new broad-host-range promoter-probe plasmid. FEMS Microbiol. Lett. 67, 221–225. doi: 10.1111/j.1574-6968.1990.tb13867.x
Martens, J. A., Laprade, L., and Winston, F. (2004). Intergenic transcription is required to repress the Saccharomyces cerevisiae SER3 gene. Nature 429, 571–574. doi: 10.1038/nature02538
Martínez-García, E., Aparicio, T., Goñi-Moreno, A., Fraile, S., and de Lorenzo, V. (2015). SEVA 2.0: an update of the standard European vector architecture for de-/re-construction of bacterial functionalities. Nucleic Acids Res. 43, D1183–D1189. doi: 10.1093/nar/gku1114
Martínez-García, E., and de Lorenzo, V. (2011). Engineering multiple genomic deletions in gram-negative bacteria: analysis of the multi-resistant antibiotic profile of Pseudomonas putida KT2440. Environ. Microbiol. 13, 2702–2716. doi: 10.1111/j.1462-2920.2011.02538.x
McLean, B. W., Wiseman, S. L., and Kropinski, A. M. (1997). Functional analysis of sigma-70 consensus promoters in Pseudomonas aeruginosa and Escherichia coli. Can. J. Microbiol. 43, 981–985. doi: 10.1139/m97-141
Meijnen, J. P., de Winde, J. H., and Ruijssenaars, H. J. (2011). Sustainable production of fine chemicals by the solvent-tolerant Pseudomonas putida S12 using lignocellulosic feedstock. Int. Sugar J. 113, 24–30.
Mutalik, V. K., Guimaraes, J. C., Cambray, G., Lam, C., Christoffersen, M. J., Mai, Q. A., et al. (2013). Precise and reliable gene expression via standard transcription and translation initiation elements. Nat. Methods 10, 354–360. doi: 10.1038/nmeth.2404
Nelson, K. E., Weinel, C., Paulsen, I. T., Dodson, R. J., Hilbert, H., Martins dos Santos, V. A., et al. (2002). Complete genome sequence and comparative analysis of the metabolically versatile Pseudomonas putida KT2440. Environ. Microbiol. 4, 799–808. doi: 10.1046/j.1462-2920.2002.00366.x
Nikel, P. I., and de Lorenzo, V. (2013). Implantation of unmarked regulatory and metabolic modules in gram-negative bacteria with specialised mini-transposon delivery vectors. J. Biotechnol. 163, 143–154. doi: 10.1016/j.jbiotec.2012.05.002
Nikel, P. I., Kim, J., and de Lorenzo, V. (2014). Metabolic and regulatory rearrangements underlying glycerol metabolism in Pseudomonas putida KT2440. Environ. Microbiol. 16, 239–254. doi: 10.1111/1462-2920.12224
Pfaffl, M. W. (2001). A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res. 29:e45. doi: 10.1093/nar/29.9.e45
Potvin, E., Sanschagrin, F., and Levesque, R. C. (2008). Sigma factors in Pseudomonas aeruginosa. FEMS Microbiol. Rev. 32, 38–55. doi: 10.1111/j.1574-6976.2007.00092.x
Ramos, J. L., Duque, E., Gallegos, M. T., Godoy, P., Ramos-Gonzalez, M. I., Rojas, A., et al. (2002). Mechanisms of solvent tolerance in gram-negative bacteria. Annu. Rev. Microbiol. 56, 743–768. doi: 10.1146/annurev.micro.56.012302.161038
Rud, I., Jensen, P. R., Naterstad, K., and Axelsson, L. (2006). A synthetic promoter library for constitutive gene expression in Lactobacillus plantarum. Microbiology 152, 1011–1019. doi: 10.1099/mic.0.28599-0
Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989). Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
San Millan, A., and MacLean, R. C. (2017). Fitness costs of plasmids: a limit to plasmid transmission. Microbiol. Spectr. 5, 1–12. doi: 10.1128/microbiolspec.MTBP-0016-2017
Sánchez-Pascuala, A., Fernández-Cabezón, L., de Lorenzo, V., and Nikel, P. I. (2019). Functional implementation of a linear glycolysis for sugar catabolism in Pseudomonas putida. Metab. Eng. 54, 200–211. doi: 10.1016/j.ymben.2019.04.005
Schmitz, A., and Galas, D. J. (1979). The interaction of RNA polymerase and lac repressor with the lac control region. Nucleic Acids Res. 6, 111–137. doi: 10.1093/nar/6.1.111
Schweizer, H. P. (1992). Allelic exchange in Pseudomonas aeruginosa using novel ColE1-type vectors and a family of cassettes containing a portable oriT and the counter-selectable Bacillus subtilis sacB marker. Mol. Microbiol. 6, 1195–1204. doi: 10.1111/j.1365-2958.1992.tb01558.x
Shearwin, K. E., Callen, B. P., and Egan, J. B. (2005). Transcriptional interference-a crash course. Trends Genet. 21, 339–345. doi: 10.1016/j.tig.2005.04.009
Shen, X., Wang, Z., Huang, X., Hu, H., Wang, W., and Zhang, X. (2017). Developing genome-reduced Pseudomonas chlororaphis strains for the production of secondary metabolites. BMC Genomics 18:715. doi: 10.1186/s12864-017-4127-2
Silva-Rocha, R., and de Lorenzo, V. (2014). Chromosomal integration of transcriptional fusions. Methods Mol. Biol. 1149, 479–489. doi: 10.1007/978-1-4939-0473-0_37
Silva-Rocha, R., Martínez-García, E., Calles, B., Chavarría, M., Arce-Rodríguez, A., de Las Heras, A., et al. (2013). The Standard European Vector Architecture (SEVA): a coherent platform for the analysis and deployment of complex prokaryotic phenotypes. Nucleic Acids Res. 41, D666–D675. doi: 10.1093/nar/gks1119
Solem, C., Koebmann, B., Yang, F., and Jensen, P. R. (2007). The las enzymes control pyruvate metabolism in Lactococcus lactis during growth on maltose. J. Bacteriol. 189, 6727–6730. doi: 10.1128/JB.00902-07
Stanton, B. C., Nielsen, A. A. K., Tamsir, A., Clancy, K., Peterson, T., and Voigt, C. A. (2013). Genomic mining of prokaryotic repressors for orthogonal logic gates. Nat. Chem. Biol. 10, 99–105. doi: 10.1038/nchembio.1411
Sun, Z., Ramsay, J. A., Guay, M., and Ramsay, B. A. (2007). Carbon-limited fed-batch production of medium-chain-length polyhydroxyalkanoates from nonanoic acid by Pseudomonas putida KT2440. Appl. Microbiol. Biot. 74, 69–77. doi: 10.1007/s00253-006-0655-4
Tamsir, A., Tabor, J. J., and Voigt, C. A. (2011). Robust multicellular computing using genetically encoded NOR gates and chemical ‘wires’. Nature 469, 212–215. doi: 10.1038/nature09565
Tiso, T., Wierckx, N., Blank, L., and Grunwald, P. (2014). Non-pathogenic Pseudomonas as Platform for Industrial Biocatalysis. Singapore: Pan Stanford Publishing.
Udvardi, M. K., Czechowski, T., and Scheible, W-R. (2008). Eleven golden rules of quantitative RT-PCR. Plant Cell 20, 1736–1737. doi: 10.1105/tpc.108.061143
Wang, Q., and Nomura, C. T. (2010). Monitoring differences in gene expression levels and polyhydroxyalkanoate (PHA) production in Pseudomonas putida KT2440 grown on different carbon sources. J. Biosci. Bioeng. 110, 653–659. doi: 10.1016/j.jbiosc.2010.08.001
Wierckx, N. J. P., Ballerstedt, H., De Bont, J. A. M., and Wery, J. (2005). Engineering of solvent-tolerant Pseudomonas putida S12 for bioproduction of phenol from glucose. Appl. Environ. Microb. 71, 8221–8227. doi: 10.1128/AEM.71.12.8221-8227.2005
Wynands, B., Lenzen, C., Otto, M., Koch, F., Blank, L. M., and Wierckx, N. (2018). Metabolic engineering of Pseudomonas taiwanensis VLB120 with minimal genomic modifications for high-yield phenol production. Metab. Eng. 47, 121–133. doi: 10.1016/j.ymben.2018.03.011
Keywords: Pseudomonas putida, synthetic biology, synthetic promoter libraries, Tn7 transposon, tandem promoter, heterologous expression
Citation: Köbbing S, Blank LM and Wierckx N (2020) Characterization of Context-Dependent Effects on Synthetic Promoters. Front. Bioeng. Biotechnol. 8:551. doi: 10.3389/fbioe.2020.00551
Received: 14 March 2020; Accepted: 07 May 2020;
Published: 12 June 2020.
Edited by:
Yunzi Luo, Sichuan University, ChinaReviewed by:
Wenjing Cui, Jiangnan University, ChinaMario Andrea Marchisio, Tianjin University, China
Copyright © 2020 Köbbing, Blank and Wierckx. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nick Wierckx, bi53aWVyY2t4QGZ6LWp1ZWxpY2guZGU=