 Christina Z. Chung
Christina Z. Chung Kazuaki Amikura
Kazuaki Amikura Dieter Söll
Dieter Söll- 1Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, CT, United States
- 2Department of Chemistry, Yale University, New Haven, CT, United States
Protein identification has gone beyond simply using protein/peptide tags and labeling canonical amino acids. Genetic code expansion has allowed residue- or site-specific incorporation of non-canonical amino acids into proteins. By taking advantage of the unique properties of non-canonical amino acids, we can identify spatiotemporal-specific protein states within living cells. Insertion of more than one non-canonical amino acid allows for selective labeling that can aid in the identification of weak or transient protein–protein interactions. This review will discuss recent studies applying genetic code expansion for protein labeling and identifying protein–protein interactions and offer considerations for future work in expanding genetic code expansion methods.
Introduction
Heterogeneous environments exist within living cells and this ever-changing environment affects the expression, dynamics, and state of proteins. However, the results and importance of the internal cellular changes are still unclear. Here, we introduce recent progress to observe the internal conditions in living cells using genetic code expansion (GCE).
Proteins are commonly labeled with fluorescent or chemical/light activated molecules in vitro and in vivo for purposes such as protein purification, determining cellular localization, and visualizing and identifying interacting proteins (Jing and Cornish, 2011; Toseland, 2013; Lang and Chin, 2014; Bartoschik et al., 2018; Chaganti et al., 2018). Using GCE to insert non-canonical amino acids (ncAAs) in proteins of interest offers a high degree of specificity for downstream labeling in vitro and in vivo. All ncAAs structures and reactions mentioned in this paper are listed in Scheme 1. The incorporation of ncAAs into cellular proteins can be achieved in either a residue- or site-specific manner through protein translation. Traditionally, the incorporation of ncAAs has been accomplished with structurally similar amino acid analogs via selective pressure incorporation into an auxotrophic host (SPI). This labeling strategy allows easy incorporation of ncAAs into proteins without any genetic manipulation with endogenous tRNAs and aminoacyl-tRNA synthetases (aaRSs) (Budisa, 2004). One canonical amino acid in all cellular proteins is replaced with its derivative, e.g., methionine with selenomethionine (1) or phenylalanine with p-fluorophenylalanine (2) (Cowie and Cohen, 1957; Munier and Cohen, 1959). This strategy even led to the complete proteomic replacement of 20,899 Trp residues with its analog L-β-(thieno[3,2-b]pyrrolyl)alanine (3) in Escherichia coli (Hoesl et al., 2015). SPI has allowed the incorporation of dozens of ncAAs into cellular proteins and can be used for multiple investigative purposes, such as protein purification, determining cellular localization, and identifying interacting proteins (Lang and Chin, 2014). In a recent report, utilizing a photocaged ncAA provided additional control over protein translation (Adelmund et al., 2018) or even optical control of protein bioadhesion properties (Hauf et al., 2017).
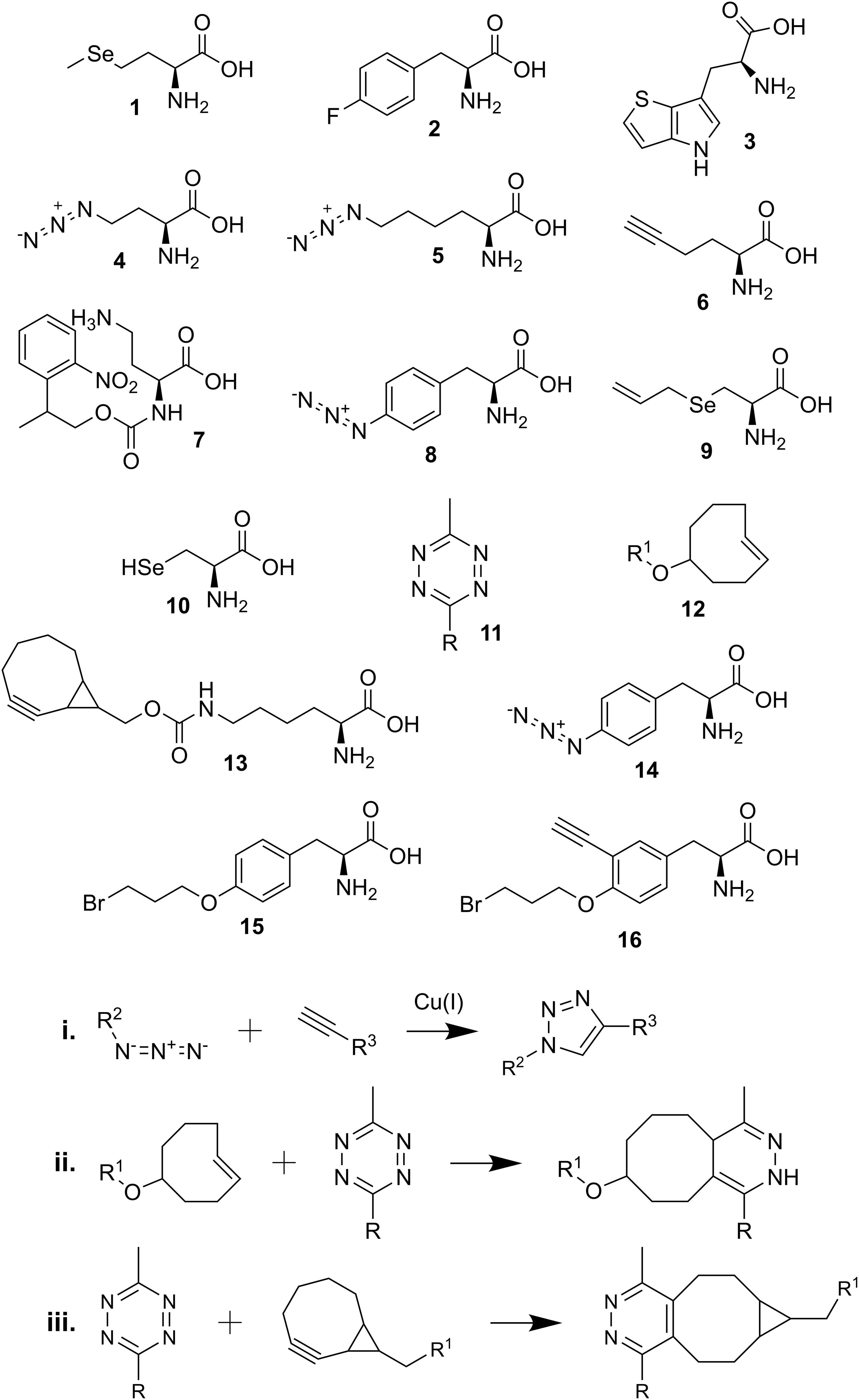
Scheme 1. Chemical structures and reactions described in the review. (1) selenomethionine; (2) p-fluorophenylalanine; (3) L-β-(thieno[3,2-b]pyrrolyl)alanine; (4) azidohomoalanine (Aha); (5) azidonorleucine (Anl); (6) homopropargylglycine (HPG); (7) photocaged Aha (2-(2-nitrophenyl)propyl Aha); (8) p-azido-L-phenylalanine (Azf); (9) Se-allyl selenocysteine (ASec); (10) selenocysteine (Sec); (11) tetrazine-linked dye (R); (12) trans-cyclooct-2-ene (TCO∗)-linked amino acid (R1); (13) bicyclo-non-yne lysine (BCN-Lys); (14) p-azidophenylalanine (Azi); (15) BprY; (16) EB3; (i) copper-catalyzed alkyne-azide reaction; (ii) tetrazine and TCO∗ reaction; (iii) tetrazine and BCN-linked amino acid reaction.
The site-specific incorporation of ncAAs using orthogonal aaRS/tRNA pairs allows observation of a specific protein (Lang and Chin, 2014; Saleh et al., 2019b). However, this method is applicable only to genetically manipulatable organisms where the aaRS/tRNA pairs are orthogonal. In recent years, progress has been made to create aaRS/tRNA pairs that are orthogonal in more organisms (Gohil et al., 2020), including multicellular organisms such as Caenorhabditis elegans, Drosophila melanogaster, Danio rerio, and Mus musculus (Greiss and Chin, 2011; Bianco et al., 2012; Ernst et al., 2016; Chen et al., 2017). Stop and sense codon suppression is commonly used for site-specific incorporation of ncAAs. Although these techniques lead to stochastic incorporation of ncAAs, they are hindered by competition with intrinsic factors such as release factors and native tRNAs. Efforts in the past decade attempt to address this problem by reassigning rare codons to encode ncAAs through genome engineering and synthesis (reviewed in Mukai et al., 2017). Previous studies have accomplished this by eliminating the specific assignment of the amber stop codon to signify translation termination or by reassigning rare arginine codons (AGG, AGA) to encode ncAAs (Mukai et al., 2015a,b; Wang and Tsao, 2016). In a recent study, two sense codons (UCG, UCA) and the amber stop codon in the synthetic E. coli MDS42 strain were completely eliminated for GCE (Fredens et al., 2019). Various reports have utilized cell-free protein synthesis and in vitro transcribed tRNA sets to successfully eliminate or reassign rare codons to encode ncAAs as well as to swap codons for canonical amino acids (Iwane et al., 2016; Cui et al., 2017; Fujino et al., 2020; Hibi et al., 2020). These engineered strains and techniques will contribute to the observation of protein dynamics and the development of protein biochemical studies. This review will present recent studies that develop and utilize various GCE methods to study the cellular dynamics of protein expression and interactions.
Purification of Newly Synthesized Proteins
Metabolic labeling allows cellular protein synthesis to be studied under different environmental or metabolic conditions of cell growth (Beynon and Pratt, 2005). Bioorthogonal ncAA tagging (BONCAT) was developed to label newly synthesized proteins in mammalian cells for purifications and identification (Dieterich et al., 2006, 2007) (Figure 1A). Azidohomoalanine (Aha) (4), a methionine (Met) analog, is charged onto tRNAMet and incorporated into newly synthesized proteins at Met codons. The azide group of Aha is used to selectively purify the proteins produced within a desired timeframe during growth of mammalian cells. Tagged proteins can subsequently be identified through mass spectroscopy (Dieterich et al., 2006). Moreover, azidonorleucine (Anl) (5) has been used in various cell-type specific labeling studies. This azide-containing Met analog has been demonstrated to be charged onto initiator tRNAMeti by a mutant methionyl-tRNA synthetase (MetRS) from E. coli (Ngo et al., 2013) and onto elongator tRNAMet by a mutant murine MetRS (Mahdavi et al., 2016) in mammalian cells for identification of newly synthesized proteins. In both studies, only expression of the mutant MetRS is required to incorporate Anl and cell-specific labeling can be achieved via Cre-induced expression of mutant MetRS (Erdmann et al., 2015; Alvarez-Castelao et al., 2017, 2019; Evans et al., 2020). Alternatively, the alkyne-bearing amino acid homopropargylglycine (HPG) (6) has been suggested as a different Met analog for affinity purification (Landgraf et al., 2015).
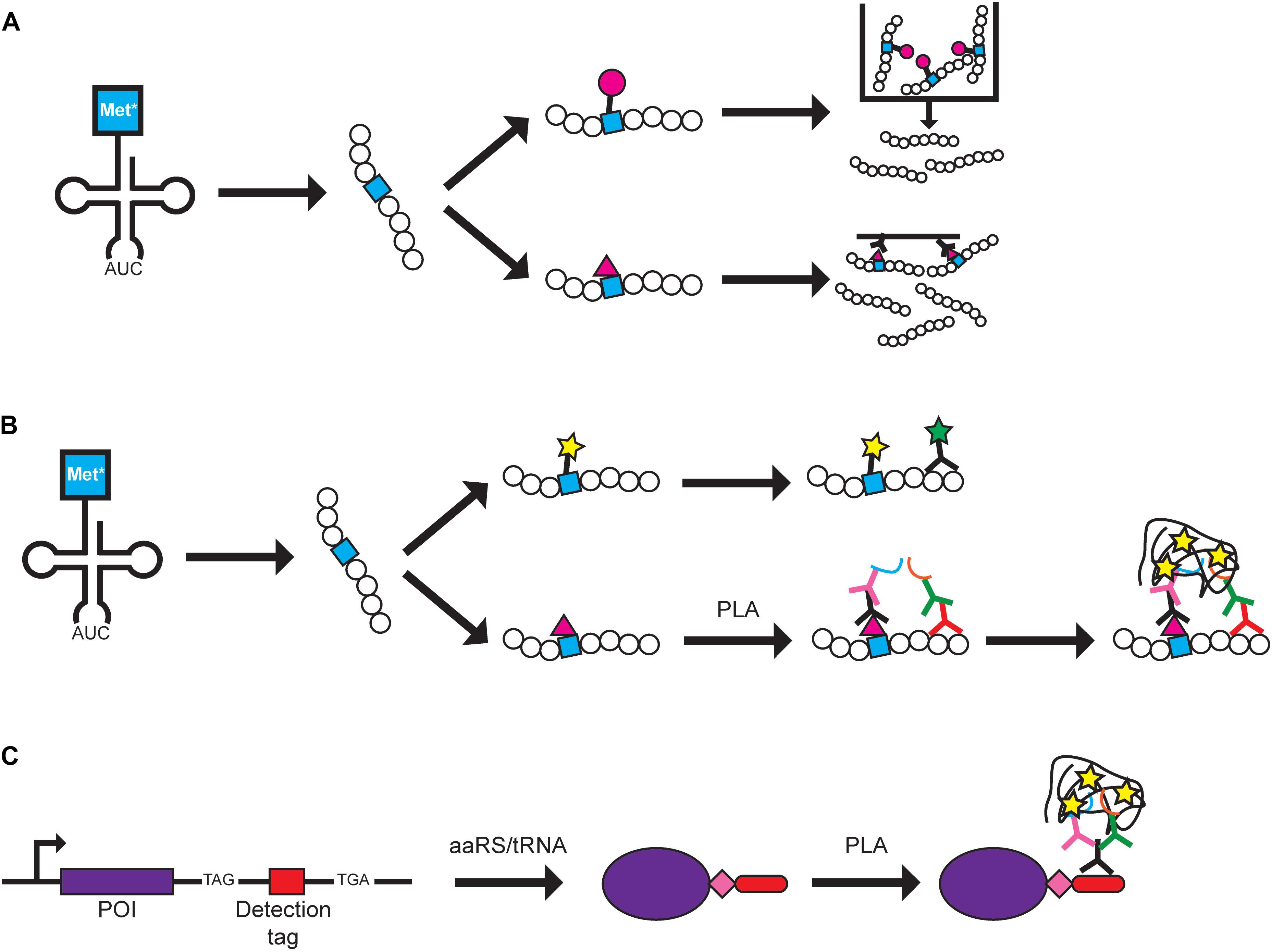
Figure 1. Protein labeling methods using GCE. Examples of how BONCAT and FUNCAT can be used are shown. A Met analog (Met*) is charged onto tRNAMet by MetRS and is incorporated at Met codons. (A) In BONCAT, Met* can be directly conjugated to azide or alkyne-conjugated beads (pink circle) for purification (top pathway) or conjugated to azide or alkyne-conjugated biotin (pink triangle) for affinity purification (bottom pathway). (B) In FUNCAT, Met* can be fluorescently labeled via click chemistry and visualization of a specific protein can be achieved using a fluorescent antibody (top pathway). Alternatively, proximity ligation assay (PLA) can be performed by conjugating Met* to azide- or alkyne-conjugated biotin followed by addition of primary antibodies recognizing biotin and a specific protein. Separate secondary antibodies conjugated to DNA oligonucleotides (blue and orange lines) allow DNA rolling circle amplification that can be targeted by fluorescently labeled complementary probes for signal amplification (bottom pathway). (C) In SCROL, a labeling cassette containing a TAG stop codon, a detection tag (e.g., HA-tag and mCHERRY), and an alternative stop codon (e.g., TGA) are genomically incorporated at the 3′-end of the protein of interest (POI). Transfection of an orthogonal aaRS/tRNA pair and ncAA (pink diamond) produces a tagged protein that can be detected using PLA where a primary antibody targets the tag and two secondary antibodies target different parts of the primary antibody.
While BONCAT was developed in mammalian cell cultures (Dieterich et al., 2006), the method can be adapted for other systems. The labeling method has been combined with Cre-induced MetRS expression for cell-specific labeling in mice (Alvarez-Castelao et al., 2017) and used to study protein synthesis in neurodegenerative disease mouse models (Evans et al., 2019) and during mouse and zebrafish development (Hinz et al., 2012; Saleh et al., 2019a). In each of those studies, the authors were able to identify proteins within specific timeframes and observe changes in the proteome under different conditions. In the plant Arabidopsis thaliana, BONCAT was used to isolate and identify proteins expressed under different conditions of stress via click chemistry to dibenzocyclooctyne beads and mass spectrometry. The authors were able to demonstrate that stress-related proteins were increased upon stress induction (Glenn et al., 2017).
Light-activated BONCAT is a modified version of BONCAT where photocaged Aha (7) can only be incorporated into proteins upon uncaging by light exposure. The amount of protein labeling in HeLa cells could be controlled by varying the light intensity and proteins in specific regions can be labeled by aiming the light source, allowing spatial and temporal control of Aha incorporation (Adelmund et al., 2018). However, proteins that do not encode at least one Met codon within their sequence will not be detected, as the N-terminal Met is often cleaved off (Wingfield, 2017). This can be circumvented by utilizing a different amino acid analog, such as the insertion of p-azido-L-phenylalanine (Azf) (8) at Phe codons in C. elegans by a mutant C. elegans phenylalanyl-tRNA synthetase (Yuet et al., 2015). Nevertheless, BONCAT provides a snapshot of proteins that are produced under various conditions. This can lead to the discovery of proteins that are only produced under specific conditions, and help researchers understand how proteins can play multiple roles.
Protein Visualization Using Fluorescent Molecules
Genetic code expansion has allowed the ability to selectively label proteins in living cells by taking advantage of the unique properties of the encoded ncAA. A protected selenocysteine (Sec) (9) was site-specifically incorporated in the outer membrane protein eCPX (enhanced circularly permuted outer membrane protein OmpX) of E. coli by recoding the UAG stop codon (Liu et al., 2018). By taking advantage of the different pKa values of cysteine (pKa 8.3) and Sec (pKa ∼5.2) (10), selective labeling of the chemically deprotected Sec with a fluorescent dye through thiol chemistry was achieved at pH 5.0 (Liu et al., 2018).
To visualize the spatial localization of newly synthesized proteins, BONCAT was modified to visualize proteins and termed fluorescence non-canonical amino acid tagging (FUNCAT) (Dieterich et al., 2010). Fixed cells from mammalian cell cultures and mouse models with newly synthesized proteins containing Aha or HPG are tagged with fluorescent dyes via click chemistry or can be combined with immunochemistry to visualize specific proteins (Dieterich et al., 2010; Hinz et al., 2012; Evans et al., 2019) (Figure 1B). FUNCAT has also been coupled with the proximity ligation assay (PLA) to visualize the spatial localization of specific proteins in fixed cells (tom Dieck et al., 2015). Primary antibodies recognize either the Aha-tagged protein or a specific protein and each primary antibody is recognized by a secondary antibody that carries different DNA adaptors. The adaptors are used for rolling circle amplification and the resulting DNA product is labeled with complementary fluorescently labeled probes, producing an amplified signal for each target protein molecule (tom Dieck et al., 2015). The authors noted that although it is possible that the PLA signal can result from two closely positioned proteins, it is more likely that both primary antibodies bind to the newly synthesized target protein to produce the PLA signal. Using Met analogs to label newly synthesized proteins has expanded to studying bacteria and microbes in environmental and human samples. The fluorescently labeled Met analog can be combined with other techniques such as FISH or FACS to study the translationally active cell populations (Couradeau et al., 2019; Sebastian and Gasol, 2019; Valentini et al., 2020). Thus, labeling of the Met analog shows promise as a tool that can be widely used in different biological systems in combination with other visualization techniques.
In an effort to further improve upon the specificity of FUNCAT-PLA, SCROL (Stop-Codon-Read-thrOugh-Label) was developed to study expression of a specific protein of interest (Schneider et al., 2018) (Figure 1C). A cassette containing the UAG amber codon, HA-tag, and an alternative stop codon (e.g., UGA opal codon) was genomically inserted at the 3’-end of the target protein using CRISPR/Cas9 in HEK293 cells. It is important to note that, even in the presence of an orthogonal aaRS/tRNA pair, the tagged protein will only be expressed upon addition of the ncAA, and thus, the protein can be labeled at specific timeframes. This would ensure that the cell, and protein of interest, is undisturbed in the absence of the ncAA. In this study, the ncAA was not used for labeling/detecting the proteins but as a method to control expression of the epitope tag. This differs from many other papers where the purpose of incorporating an ncAA is to directly use it in future studies. Thus, additional specialized aaRS/tRNA pairs and ncAAs would not need to be developed for this technique.
An important component of labeling ncAAs in vivo for protein visualization under different cellular conditions is bioorthogonal dye. Tetrazine dyes (11) have been extensively characterized for their use in labeling trans-cyclooct-2-ene (TCO∗) modified ncAAs (12) via click chemistry in a variety of mammalian cells for live cell imaging (Beliu et al., 2019). An attractive trait of the dyes is their small size compared to traditional labeling molecules such as antibodies. By choosing cell membrane-permeable or impermeable dyes based on the target protein, selective labeling can be enhanced. The authors demonstrated selective and efficient labeling of the target proteins that produced superior readings compared to immunolabeling (Beliu et al., 2019). A similar study utilized bicyclo-non-yne lysine (BCN-Lys) (13) to attach a tetrazine dye to the protein of interest via a N-terminal tag in HEK293T and Cos7 cells (Segal et al., 2020). Interestingly, insertion of BCN-Lys in the N-terminal tag was labeled better than BCN insertion within the target protein. In addition, the modified tag was found to be compatible in labeling organelle proteins within their acidic environments. Although the authors demonstrated the ability to switch out the HA-tag with other commonly used tags (Myc and FLAG), the results were not as successful, indicating a need for further optimization (Segal et al., 2020). The ease of simply inserting a tag encoding an ncAA led the authors to state the possibility of genomically inserting the modified tag to monitor endogenously expressed proteins. This would ensure that any effects on protein folding and interactions would be minimized and the use of the tetrazine dyes allows live-cell imaging. Live-cell imaging using fluorescently labeled proteins provides a story of how proteins travel and react within the cell under various conditions, rather than a picture obtained from fixed cells. This could be helpful in studying disease-associated proteins and understanding how cellular and environmental changes affect their spatial distribution.
Insertion of different ncAAs within a single protein allows different molecules to be attached, permitting the application of various techniques. However, the limited number of available codons for reassignment/recoding, orthogonal aaRS/tRNA systems, and orthogonal labeling reactions impairs the actual number of insertable ncAAs. Recently, three different ncAAs were inserted within a single protein using three different orthogonal aaRS/tRNA pairs in E. coli (Italia et al., 2019). The same group had previously engineered an E. coli strain (ATMW1) where the endogenous tryptophanyl–tRNA synthetase/tRNATrp (TrpRS/tRNATrp) pair was replaced by the TrpRS/tRNATrp from Saccharomyces cerevisiae, rendering the E. coli TrpRS/tRNATrp pair available for ncAA insertion (Italia et al., 2017). As three distinct ncAAs were inserted using all three nonsense suppressors, homogenous translation termination was achieved by inserting a TEV cleavage site at the C-terminal end of sfGFP-His followed by TEV protease and 3 consecutive UAA stop codons and all three ncAAs could be labeled in a one-pot reaction (Italia et al., 2019). More recently, two separate classes of pyrrolysyl-tRNA synthetase (PylRS) with the N-terminal domain missing and their corresponding tRNAs were identified to be mutually orthogonal to each other and to a Methanosarcina mazei PylRS/SpetRNAPyl pair in E coli. By co-expressing all three pairs and ribo-Q1 with a GFP reporter, three distinct ncAAs were incorporated by recoding the UAG stop codon and two quadruplet codons (Dunkelmann et al., 2020). The use of quadruplet codons would allow protein termination with a triplet stop codon. Alternatively, competitive labeling of one ncAA reserves a nonsense codon for translation termination while still allowing production of the multi-labeled protein for different studies (Konig et al., 2020). By taking advantage of the ability of BCN-Lys to be labeled by various tetrazine-conjugated fluorescent dyes via click chemistry, two different dyes could be conjugated simultaneously to allow the use of two different techniques that require different fluorescent parameters. This was demonstrated to be feasible in live Cos7 cells, allowing real-time high resolution imaging at a single molecule level (Konig et al., 2020). These studies demonstrate the ability to insert multiple ncAAs within a single protein where each ncAA can take part in a specific chemical reaction. This would greatly increase the yield of samples that require multiple downstream processing steps as constant protein purification for different labeling reactions would not be required.
Identifying Protein–Protein Interactions
Studying the interactions of proteins aids in the understanding of their cellular roles under normal and stress conditions. Co-immunoprecipitation, crosslinking, and BioID are some techniques used to identify interacting proteins (Lin and Lai, 2017; Yu and Huang, 2018; Sears et al., 2019). GCE can be used to insert crosslinking ncAAs at known or potential binding interfaces to increase the chances of identifying interacting proteins as well as minimize interference with protein folding and activity. The light-activated crosslinker p-azidophenylalanine (Azi) (14) was demonstrated in E. coli to be efficiently incorporated and crosslinks with amines of interacting proteins (Chin et al., 2002). Recently, Azi was successfully inserted at recoded UAG stop codons and used to map protein-protein interactions within the inner membrane complex of the protozoan Toxoplasma gondii (Choi et al., 2019). Chemical crosslinkers are an alternative to light-activated crosslinkers. Two chemical crosslinkers have been developed that spontaneously react with cysteine residues that are in close proximity: BprY (15) and EB3 (16). Both crosslinkers are alkyl bromides with EB3 containing an alkyne group for enrichment with biotin. Their applicability in detecting weak and transient interactions was demonstrated in live E. coli cells (Yang et al., 2017). The drawback of these chemical crosslinkers is the assumption that cysteine residues are present at the binding interface. Replicating experiments with different types of crosslinkers could increase the coverage of potential interacting proteins. Using genetically encoded crosslinkers offers a high degree of specificity that can capture strong and transient interactions.
Discussion
The studies discussed in this review demonstrate the potential of utilizing GCE to label proteins in an effort to illuminate the heterogenous intracellular environment. We present the following ideas for consideration to improve the current methods. (1) Although aaRS/tRNA pairs are tested to ensure their orthogonality within a system, cross-reactions may still occur. Developing more specific aaRS/tRNA pairs that minimally cross-react with endogenous aaRSs and tRNAs would lessen the impact on cellular metabolism and allow a more “natural” representation of the protein(s) of interest due to minimal disturbance of the intracellular environment. In addition, there is a need to produce more aaRS/tRNA pairs that are orthogonal in a greater variety of organisms (Greiss and Chin, 2011; Bianco et al., 2012; Ernst et al., 2016; Chen et al., 2017; Gohil et al., 2020) to study proteins in their natural environment with any necessary post-translational modifications that are often missing when produced in model expression organisms such as E. coli. (2) Development of more sensitive detection techniques would enable a greater understanding of localization for more proteins. Although FUNCAT coupled to PLA allows amplification of a signal for a protein of interest (tom Dieck et al., 2015), a high affinity antibody must be available. This is often not the case for many proteins where only low affinity antibodies are available or none have so far been produced. While SCROL offers an alternative where the peptide tag is targeted by an antibody (Schneider et al., 2018), the SCROL cassette must be genomically incorporated to produce a stable cell line. (3) The increasing number of ncAAs allows for the development and optimization of techniques for specific purposes. However, this is often hindered by the limited quantity and price of ncAAs. Many ncAAs are expensive to buy or to chemically synthesize within the lab. It would be beneficial to develop a method to produce greater quantities of ncAAs, biologically or chemically. A related issue is the development of ncAAs for organelle-specific labeling. These ncAAs must be stable in the organellar environment and chemically reactive ncAAs would have to be designed to react specifically within the organellar environment. Light-activated ncAAs would circumvent this issue and would likely offer greater control in activating the ncAA.
Conclusion
In conclusion, GCE has become a versatile tool that can be modified to suit individual needs and incorporated into existing techniques. In the future, it is expected that more tools will be developed for expansion of GCE to other organisms and to improve on existing GCE methods for understanding heterogeneous environments within living cells.
Author Contributions
CZC and KA wrote the manuscript. DS edited it. All authors contributed to the article and approved the submitted version.
Funding
Work in the authors’ laboratory was supported by the National Institute of General Medical Sciences (R35GM122560) and the DOE Office of Basic Energy Sciences (DE-FG0298ER2031).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Oscar Vargas and Natalie Krahn for many discussions.
References
Adelmund, S. M., Ruskowitz, E. R., Farahani, P. E., Wolfe, J. V., and DeForest, C. A. (2018). Light-activated proteomic labeling via photocaged bioorthogonal non-canonical amino acids. ACS Chem. Biol. 13, 573–577. doi: 10.1021/acschembio.7b01023
Alvarez-Castelao, B., Schanzenbacher, C. T., Hanus, C., Glock, C., Tom Dieck, S., Dorrbaum, A. R., et al. (2017). Cell-type-specific metabolic labeling of nascent proteomes in vivo. Nat. Biotechnol. 35, 1196–1201. doi: 10.1038/nbt.4016
Alvarez-Castelao, B., Schanzenbacher, C. T., Langer, J. D., and Schuman, E. M. (2019). Cell-type-specific metabolic labeling, detection and identification of nascent proteomes in vivo. Nat. Protoc. 14, 556–575. doi: 10.1038/s41596-018-0106-6
Bartoschik, T., Galinec, S., Kleusch, C., Walkiewicz, K., Breitsprecher, D., Weigert, S., et al. (2018). Near-native, site-specific and purification-free protein labeling for quantitative protein interaction analysis by microscale thermophoresis. Sci. Rep. 8:4977.
Beliu, G., Kurz, A. J., Kuhlemann, A. C., Behringer-Pliess, L., Meub, M., Wolf, N., et al. (2019). Bioorthogonal labeling with tetrazine-dyes for super-resolution microscopy. Commun. Biol. 2:261.
Beynon, R. J., and Pratt, J. M. (2005). Metabolic labeling of proteins for proteomics. Mol. Cell. Proteomics 4, 857–872. doi: 10.1074/mcp.r400010-mcp200
Bianco, A., Townsley, F. M., Greiss, S., Lang, K., and Chin, J. W. (2012). Expanding the genetic code of Drosophila melanogaster. Nat. Chem. Biol. 8, 748–750. doi: 10.1038/nchembio.1043
Budisa, N. (2004). Prolegomena to future experimental efforts on genetic code engineering by expanding its amino acid repertoire. Angew. Chem. Int. Ed. Engl. 43, 6426–6463. doi: 10.1002/anie.200300646
Chaganti, L. K., Venkatakrishnan, N., and Bose, K. (2018). An efficient method for FITC labelling of proteins using tandem affinity purification. Biosci. Rep. 38:BSR20181764.
Chen, Y., Ma, J., Lu, W., Tian, M., Thauvin, M., Yuan, C., et al. (2017). Heritable expansion of the genetic code in mouse and zebrafish. Cell Res. 27, 294–297. doi: 10.1038/cr.2016.145
Chin, J. W., Santoro, S. W., Martin, A. B., King, D. S., Wang, L., and Schultz, P. G. (2002). Addition of p-azido-L-phenylalanine to the genetic code of Escherichia coli. J. Am. Chem. Soc. 124, 9026–9027. doi: 10.1021/ja027007w
Choi, C. P., Moon, A. S., Back, P. S., Jami-Alahmadi, Y., Vashisht, A. A., Wohlschlegel, J. A., et al. (2019). A photoactivatable crosslinking system reveals protein interactions in the Toxoplasma gondii inner membrane complex. PLoS Biol. 17:e3000475. doi: 10.1371/journal.pbio.3000475
Couradeau, E., Sasse, J., Goudeau, D., Nath, N., Hazen, T. C., Bowen, B. P., et al. (2019). Probing the active fraction of soil microbiomes using BONCAT-FACS. Nat. Commun. 10:2770.
Cowie, D. B., and Cohen, G. N. (1957). Biosynthesis by Escherichia coli of active altered proteins containing selenium instead of sulfur. Biochim. Biophys. Acta 26, 252–261. doi: 10.1016/0006-3002(57)90003-3
Cui, Z., Mureev, S., Polinkovsky, M. E., Tnimov, Z., Guo, Z., Durek, T., et al. (2017). Combining sense and nonsense codon reassignment for site-selective protein modification with unnatural amino acids. ACS Synth. Biol. 6, 535–544. doi: 10.1021/acssynbio.6b00245
Dieterich, D. C., Hodas, J. J., Gouzer, G., Shadrin, I. Y., Ngo, J. T., Triller, A., et al. (2010). In situ visualization and dynamics of newly synthesized proteins in rat hippocampal neurons. Nat. Neurosci. 13, 897–905. doi: 10.1038/nn.2580
Dieterich, D. C., Lee, J. J., Link, A. J., Graumann, J., Tirrell, D. A., and Schuman, E. M. (2007). Labeling, detection and identification of newly synthesized proteomes with bioorthogonal non-canonical amino-acid tagging. Nat. Protoc. 2, 532–540. doi: 10.1038/nprot.2007.52
Dieterich, D. C., Link, A. J., Graumann, J., Tirrell, D. A., and Schuman, E. M. (2006). Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT). Proc. Natl. Acad. Sci. U.S.A. 103, 9482–9487. doi: 10.1073/pnas.0601637103
Dunkelmann, D. L., Willis, J. C. W., Beattie, A. T., and Chin, J. W. (2020). Engineered triply orthogonal pyrrolysyl-tRNA synthetase/tRNA pairs enable the genetic encoding of three distinct non-canonical amino acids. Nat. Chem. 12, 535–544. doi: 10.1038/s41557-020-0472-x
Erdmann, I., Marter, K., Kobler, O., Niehues, S., Abele, J., Muller, A., et al. (2015). Cell-selective labelling of proteomes in Drosophila melanogaster. Nat. Commun. 6:7521.
Ernst, R. J., Krogager, T. P., Maywood, E. S., Zanchi, R., Beranek, V., Elliott, T. S., et al. (2016). Genetic code expansion in the mouse brain. Nat. Chem. Biol. 12, 776–778.
Evans, H. T., Benetatos, J., van Roijen, M., Bodea, L. G., and Gotz, J. (2019). Decreased synthesis of ribosomal proteins in tauopathy revealed by non-canonical amino acid labelling. EMBO J. 38:e101174.
Evans, H. T., Bodea, L. G., and Gotz, J. (2020). Cell-specific non-canonical amino acid labelling identifies changes in the de novo proteome during memory formation. eLife 9:e52990.
Fredens, J., Wang, K., de la Torre, D., Funke, L. F. H., Robertson, W. E., Christova, Y., et al. (2019). Total synthesis of Escherichia coli with a recoded genome. Nature 569, 514–518. doi: 10.1038/s41586-019-1192-5
Fujino, T., Tozaki, M., and Murakami, H. (2020). An amino acid-swapped genetic code. ACS Synth. Biol. doi: 10.1021/acssynbio.0c00196
Glenn, W. S., Stone, S. E., Ho, S. H., Sweredoski, M. J., Moradian, A., Hess, S., et al. (2017). Bioorthogonal noncanonical amino acid tagging (BONCAT) enables time-resolved analysis of protein synthesis in native plant tissue. Plant Physiol. 173, 1543–1553. doi: 10.1104/pp.16.01762
Gohil, N., Bhattacharjee, G., and Singh, V. (2020). “Expansion of the genetic code,” in Advances in Synthetic Biology, ed. V. Singh (Singapore: Springer), 237–249.
Greiss, S., and Chin, J. W. (2011). Expanding the genetic code of an animal. J. Am. Chem. Soc. 133, 14196–14199.
Hauf, M., Richter, F., Schneider, T., Faidt, T., Martins, B. M., Baumann, T., et al. (2017). Photoactivatable mussel-based underwater adhesive proteins by an expanded genetic code. Chembiochem 18, 1819–1823. doi: 10.1002/cbic.201700327
Hibi, K., Amikura, K., Sugiura, N., Masuda, K., Ohno, S., Yokogawa, T., et al. (2020). Reconstituted cell-free protein synthesis using in vitro transcribed tRNAs. Commun. Biol. 3:350.
Hinz, F. I., Dieterich, D. C., Tirrell, D. A., and Schuman, E. M. (2012). Non-canonical amino acid labeling in vivo to visualize and affinity purify newly synthesized proteins in larval zebrafish. ACS Chem. Neurosci. 3, 40–49. doi: 10.1021/cn2000876
Hoesl, M. G., Oehm, S., Durkin, P., Darmon, E., Peil, L., Aerni, H. R., et al. (2015). Chemical evolution of a bacterial proteome. Angew. Chem. Int. Ed. Engl. 54, 10030–10034. doi: 10.1002/anie.201502868
Italia, J. S., Addy, P. S., Erickson, S. B., Peeler, J. C., Weerapana, E., and Chatterjee, A. (2019). Mutually orthogonal nonsense-suppression systems and conjugation chemistries for precise protein labeling at up to three distinct sites. J. Am. Chem. Soc. 141, 6204–6212. doi: 10.1021/jacs.8b12954
Italia, J. S., Addy, P. S., Wrobel, C. J., Crawford, L. A., Lajoie, M. J., Zheng, Y., et al. (2017). An orthogonalized platform for genetic code expansion in both bacteria and eukaryotes. Nat. Chem. Biol. 13, 446–450. doi: 10.1038/nchembio.2312
Iwane, Y., Hitomi, A., Murakami, H., Katoh, T., Goto, Y., and Suga, H. (2016). Expanding the amino acid repertoire of ribosomal polypeptide synthesis via the artificial division of codon boxes. Nat. Chem. 8, 317–325. doi: 10.1038/nchem.2446
Jing, C., and Cornish, V. W. (2011). Chemical tags for labeling proteins inside living cells. Acc. Chem. Res. 44, 784–792. doi: 10.1021/ar200099f
Konig, A. I., Sorkin, R., Alon, A., Nachmias, D., Dhara, K., Brand, G., et al. (2020). Live cell single molecule tracking and localization microscopy of bioorthogonally labeled plasma membrane proteins. Nanoscale 12, 3236–3248. doi: 10.1039/c9nr08594g
Landgraf, P., Antileo, E. R., Schuman, E. M., and Dieterich, D. C. (2015). BONCAT: metabolic labeling, click chemistry, and affinity purification of newly synthesized proteomes. Methods Mol. Biol. 1266, 199–215. doi: 10.1007/978-1-4939-2272-7_14
Lang, K., and Chin, J. W. (2014). Cellular incorporation of unnatural amino acids and bioorthogonal labeling of proteins. Chem. Rev. 114, 4764–4806. doi: 10.1021/cr400355w
Lin, J. S., and Lai, E. M. (2017). Protein-protein interactions: co-immunoprecipitation. Methods Mol. Biol. 1615, 211–219. doi: 10.1007/978-1-4939-7033-9_17
Liu, J., Zheng, F., Cheng, R., Li, S., Rozovsky, S., Wang, Q., et al. (2018). Site-specific incorporation of selenocysteine using an expanded genetic code and palladium-mediated chemical deprotection. J. Am. Chem. Soc. 140, 8807–8816. doi: 10.1021/jacs.8b04603
Mahdavi, A., Hamblin, G. D., Jindal, G. A., Bagert, J. D., Dong, C., Sweredoski, M. J., et al. (2016). Engineered aminoacyl-tRNA synthetase for cell-selective analysis of mammalian protein synthesis. J. Am. Chem. Soc. 138, 4278–4281. doi: 10.1021/jacs.5b08980
Mukai, T., Hoshi, H., Ohtake, K., Takahashi, M., Yamaguchi, A., Hayashi, A., et al. (2015a). Highly reproductive Escherichia coli cells with no specific assignment to the UAG codon. Sci. Rep. 5:9699.
Mukai, T., Yamaguchi, A., Ohtake, K., Takahashi, M., Hayashi, A., Iraha, F., et al. (2015b). Reassignment of a rare sense codon to a non-canonical amino acid in Escherichia coli. Nucleic Acids Res. 43, 8111–8122. doi: 10.1093/nar/gkv787
Mukai, T., Lajoie, M. J., Englert, M., and Söll, D. (2017). Rewriting the genetic code. Annu. Rev. Microbiol. 71, 557–577. doi: 10.1146/annurev-micro-090816-093247
Munier, R., and Cohen, G. N. (1959). [Incorporation of structural analogues of amino acids into bacterial proteins during their synthesis in vivo]. Biochim. Biophys. Acta 31, 378–391.
Ngo, J. T., Schuman, E. M., and Tirrell, D. A. (2013). Mutant methionyl-tRNA synthetase from bacteria enables site-selective N-terminal labeling of proteins expressed in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 110, 4992–4997. doi: 10.1073/pnas.1216375110
Saleh, A. M., Jacobson, K. R., Kinzer-Ursem, T. L., and Calve, S. (2019a). Dynamics of non-canonical amino acid-labeled intra- and extracellular proteins in the developing mouse. Cell. Mol. Bioeng. 12, 495–509. doi: 10.1007/s12195-019-00592-1
Saleh, A. M., Wilding, K. M., Calve, S., Bundy, B. C., and Kinzer-Ursem, T. L. (2019b). Non-canonical amino acid labeling in proteomics and biotechnology. J. Biol. Eng. 13:43.
Schneider, N., Gabelein, C., Wiener, J., Georgiev, T., Gobet, N., Weber, W., et al. (2018). Genetic code expansion method for temporal labeling of endogenously expressed proteins. ACS Chem. Biol. 13, 3049–3053. doi: 10.1021/acschembio.8b00594
Sears, R. M., May, D. G., and Roux, K. J. (2019). BioID as a tool for protein-proximity labeling in living cells. Methods Mol. Biol. 2012, 299–313. doi: 10.1007/978-1-4939-9546-2_15
Sebastian, M., and Gasol, J. M. (2019). Visualization is crucial for understanding microbial processes in the ocean. Philos. Trans. R. Soc. Lond. B Biol. Sci. 374:20190083. doi: 10.1098/rstb.2019.0083
Segal, I., Nachmias, D., Konig, A., Alon, A., Arbely, E., and Elia, N. (2020). A straightforward approach for bioorthogonal labeling of proteins and organelles in live mammalian cells, using a short peptide tag. BMC Biol. 18:5. doi: 10.1186/s12915-019-0708-7
tom Dieck, S., Kochen, L., Hanus, C., Heumuller, M., Bartnik, I., Nassim-Assir, B., et al. (2015). Direct visualization of newly synthesized target proteins in situ. Nat. Methods 12, 411–414. doi: 10.1038/nmeth.3319
Toseland, C. P. (2013). Fluorescent labeling and modification of proteins. J. Chem. Biol. 6, 85–95. doi: 10.1007/s12154-013-0094-5
Valentini, T. D., Lucas, S. K., Binder, K. A., Cameron, L. C., Motl, J. A., Dunitz, J. M., et al. (2020). Bioorthogonal non-canonical amino acid tagging reveals translationally active subpopulations of the cystic fibrosis lung microbiota. Nat. Commun. 11:2287.
Wang, Y., and Tsao, M. L. (2016). Reassigning sense codon AGA to encode noncanonical amino acids in Escherichia coli. Chembiochem 17, 2234–2239. doi: 10.1002/cbic.201600448
Wingfield, P. T. (2017). N-terminal methionine processing. Curr. Protoc. Protein Sci. 88, 6.14.1–6.14.3.
Yang, B., Tang, S., Ma, C., Li, S. T., Shao, G. C., Dang, B., et al. (2017). Spontaneous and specific chemical cross-linking in live cells to capture and identify protein interactions. Nat. Commun. 8:2240.
Yu, C., and Huang, L. (2018). Cross-linking mass spectrometry: an emerging technology for interactomics and structural biology. Anal. Chem. 90, 144–165. doi: 10.1021/acs.analchem.7b04431
Keywords: genetic code expansion, non-canonical amino acids, protein labeling, protein purification, protein–protein interactions
Citation: Chung CZ, Amikura K and Söll D (2020) Using Genetic Code Expansion for Protein Biochemical Studies. Front. Bioeng. Biotechnol. 8:598577. doi: 10.3389/fbioe.2020.598577
Received: 25 August 2020; Accepted: 29 September 2020;
Published: 19 October 2020.
Edited by:
Chenguang Fan, University of Arkansas, United StatesReviewed by:
Nediljko Budisa, University of Manitoba, CanadaWenshe R. Liu, Texas A&M University, United States
Copyright © 2020 Chung, Amikura and Söll. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dieter Söll, ZGlldGVyLnNvbGxAeWFsZS5lZHU=
†These authors have contributed equally to this work