 Jaehyun Cha
Jaehyun Cha Jinhee Lee
Jinhee Lee Byoung Wook Jeon
Byoung Wook Jeon Yong Hwan Kim
Yong Hwan Kim Inchan Kwon
Inchan Kwon- 1School of Materials Science and Engineering, Gwangju Institute of Science and Technology (GIST), Gwangju, Republic of Korea
- 2School of Energy and Chemical Engineering, Ulsan National Institute of Science and Technology (UNIST), Ulsan, Republic of Korea
- 3Research Center for Innovative Energy and Carbon Optimized Synthesis for Chemicals (Inn-ECOSysChem), Gwangju Institute of Science and Technology (GIST), Gwangju, Republic of Korea
It is challenging to capture carbon dioxide (CO2), a major greenhouse gas in the atmosphere, due to its high chemical stability. One potential practical solution to eliminate CO2 is to convert CO2 into formate using hydrogen (H2) (CO2 hydrogenation), which can be accomplished with inexpensive hydrogen from sustainable sources. While industrial flue gas could provide an adequate source of hydrogen, a suitable catalyst is needed that can tolerate other gas components, such as carbon monoxide (CO) and oxygen (O2), potential inhibitors. Our proposed CO2 hydrogenation system uses the hydrogenase derived from Ralstonia eutropha H16 (ReSH) and formate dehydrogenase derived from Methylobacterium extorquens AM1 (MeFDH1). Both enzymes are tolerant to CO and O2, which are typical inhibitors of metalloenzymes found in flue gas. We have successfully demonstrated that combining ReSH- and MeFDH1-immobilized resins can convert H2 and CO2 in real flue gas to formate via a nicotinamide adenine dinucleotide-dependent cascade reaction. We anticipated that this enzyme system would enable the utilization of diverse H2 and CO2 sources, including waste gases, biomass, and gasified plastics.
1 Introduction
Global warming is an issue that has garnered significant attention in recent years due to its potentially catastrophic impacts on the environment and human societies (Frölicher et al., 2018; Hughes et al., 2018). One of the primary causes of global warming is the release of carbon dioxide (CO2) into the atmosphere, mainly due to human activities such as burning fossil fuels (Salam and Noguchi, 2005; Armaroli and Balzani, 2011; Olah et al., 2011; Al-Ghussain, 2019). To mitigate the effects of global warming, it is crucial to develop effective strategies for reducing CO2 emissions (Takemura and Suzuki, 2019). Due to its stability as a fully oxidized gaseous molecule at room temperature and pressure, capturing CO2 from the atmosphere presents a formidable challenge (Rochelle, 2009; Ghalei et al., 2017; Ding and Jiang, 2018). Moreover, as large-scale captured CO2 holds little industrial value, the most promising approach to regenerate combusted CO2 is through CO2 hydrogenation (Li et al., 2018; Ye et al., 2019).
One potential approach to CO2 hydrogenation is the conversion of CO2 and hydrogen (H2) into formate, using hydrogen as a renewable energy source (Wang et al., 2015; Wang et al., 2019). Formate, one of the most basic C1 compounds, exists as a non-flammable liquid at ambient temperature and pressure, serving as both a precursor for various syntheses and a primary storage medium for captured CO2 (Kothandaraman et al., 2015). Further hydrogenation of formate can lead to the production of formaldehyde or methanol (Wang et al., 2015), while microorganisms can utilize it to generate biopolymers (Hwang et al., 2020). However, to enable the hydrogenation of a massive amount of CO2, cheap H2 derived from sustainable sources is required.
Industrial flue gas can address this issue as a cheap H2 source. According to an International Energy Agency report, around 30% of industrial CO2 emissions came from the steel industry (Kim et al., 2019). The steel mill generates various flue gases such as coke oven gas (COG), blast furnace gas (BFG), and Lintz-Donawitz gas (LDG) (Lee et al., 2020). In particular, COG has a high H2 content of nearly 60% (Moral et al., 2022) (Table 1). These gases are promising cheap H2 resources but are used inefficiently for auxiliary facilities in steel mills as heat or power sources for boilers (Xiang et al., 2016). While it is unsurprising that efforts have been made to harness this H2 for CO2 hydrogenation, the presence of multiple gases in the industrial flue gas necessitates the use of a catalyst that is practically free of side reactions and tolerant to other gas components like carbon monoxide (CO) and oxygen (O2). To address this issue, we propose a CO2 hydrogenation system that utilizes multiple enzymes, taking advantage of their high reactivity and substrate specificity under ambient temperature and pressure conditions.
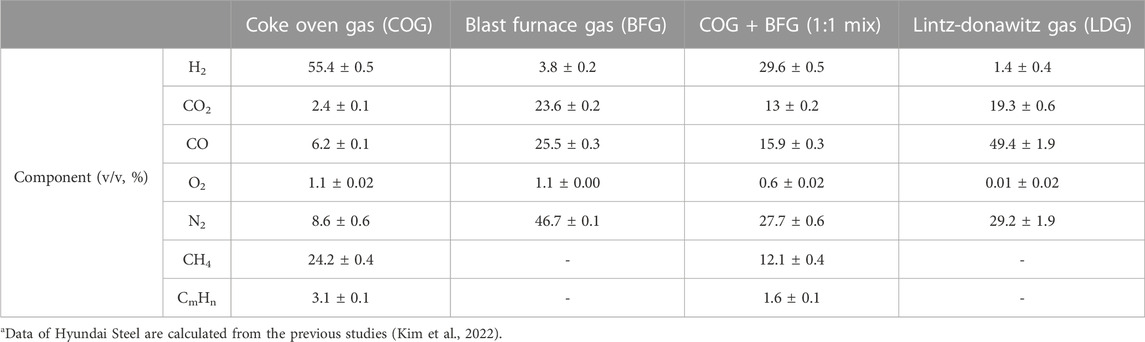
TABLE 1. Composition of flue gases from Hyundai Steela.
The CO2 hydrogenation reaction occurs as two half-reactions: H2 oxidation and CO2 reduction (equation (1)-3) (Loges et al., 2008; Reda et al., 2008). The enzymes responsible for each reaction are hydrogenase (H2ase) for H2 oxidation (Lubitz et al., 2014) and formate dehydrogenase (FDH) for CO2 reduction (Appel et al., 2013; Amao, 2018; Moon et al., 2020). The conversion of H2 and CO2 to formate was reported through a cascade reaction involving the reaction of H2ase and FDH and electron transfer (Sokol et al., 2019). Flue gas contains CO, O2, nitrogen (N2), etc., in addition to the substrate H2 and CO2. The H2ase and FDH used in the previous study as well as most other H2ases and FDHs were derived from anaerobic microorganisms, and so their metal-active sites are often attacked and inactivated even in the presence of trace amounts of O2 (Fontecilla-Camps et al., 2007; Niks and Hille, 2018). CO also acts as an irreversible inhibitor of most H2ases (Bagley et al., 1994; Vincent et al., 2007).
We previously demonstrated the feasibility of the combined use of O2-tolerant H2ase and FDH using pure gases (Cha et al., 2023a). However, for CO2 hydrogenation using the real flue gas, several issues should be solved. First, with the ultimate goal of long-term repeated or continuous reactions, enzymes should be immobilized on solid supports. We developed the suitable pair of immobilization tag and affinity resin for each enzyme. Second, the enzymes need to be tolerant to both CO and O2. Therefore, we investigated the impact of CO or O2 on the kinetic parameters of both H2ase and FDH. Third, the H2ase and FDH, capable of electron transfer between the two reactions, must satisfy catalytic bias. Therefore, we selected H2nase and FDH, which exhibit a catalytic bias towards a desired direction to the greatest extent possible.
First, we chose the hydrogenase from Ralstonia eutropha H16 (ReSH) consisting of heterodimeric [NiFe] hydrogenase (HoxHY) subunits and diaphorase (HoxFU) subunits, stably oxidized H2 and reduced NAD+ to NADH under oxic conditions (Lauterbach and Lenz, 2013; Wulff et al., 2014; Horch et al., 2015). In addition, it has also been reported that ReSH is not sensitive to CO, unlike other hydrogenases (Burgdorf et al., 2005). Resistant to O2 and CO, ReSH is a promising H2ase candidate that can reduce NAD+ to NADH by consuming hydrogen in real flue gas. FDHs are classified as metal-independent or metal-dependent based on the active site (molybdenum or tungsten) (Ünlü et al., 2021; Alpdağtaş et al., 2022). Although metal-independent FDH exhibits O2-independent activity, the strategies like mutation and immobilization have been reported to enhance its activity and stability due to its inherently low activity (Çakar et al., 2020; Tülek et al., 2023). FDHs from Clostridium carboxidovorans strain P7T (Alissandratos et al., 2013), Chaetomium thermophilum (Çakar et al., 2020), Rhodobacter capsulatus (Hartmann and Leimkühler, 2013), and Methylobacterium extorquens AM1 (Laukel et al., 2003) have maintained high CO2 reduction activity under oxic conditions. Second, we chose tungsten-containing FDH1 from M. extorquens AM1 (MeFDH1), consisting of alpha and beta subunits, as FDH to produce formate via enzymatic NAD+ regeneration stably (Baccour et al., 2020). It was reported that MeFDH1 has the great catalytic bias towards CO2 reduction. The resistance of MeFDH1 to CO has not been reported.
In this study, we constructed an enzymatic reactor with ReSH-immobilized resins, MeFDH1-immobilized resins, and NAD+, which showed CO2 hydrogenation from real flue gas (Figure 1). The kinetic parameters were determined to ensure each enzyme could withstand the potential critical inhibitors O2 and CO in the flue gas environment. The gaseous substrate needs to be bubbled to supply the enzymatic reactor continuously. To reduce aeration damage, the reactor was constructed with the enzymes immobilized on an agarose based affinity resin, respectively.
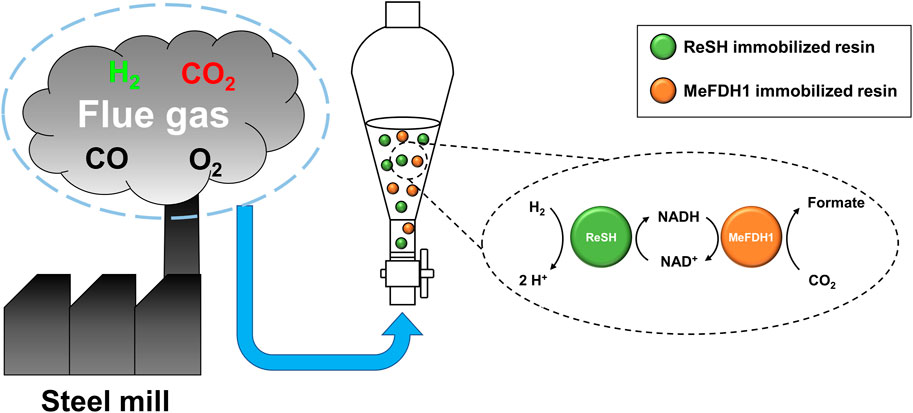
FIGURE 1. Schematic showing an enzymatic reactor for flue gas conversion. The reactor converts H2 and CO2 to formate via an NAD+-dependent cascade reaction of immobilized ReSH and MeFDH1.
2 Materials and methods
2.1 Materials
COG and BFG were supplied by Hyundai steel (Ulsan, South Korea). Ni-NTA agarose and a gravity-flow polypropylene column (PP column, 10 mL) were purchased from Qiagen (Hilden, Germany). Strep-Tactin XT 4 Flow high-capacity resin was obtained from IBA Life Sciences (Göttingen, Germany). Disposable PD-10 desalting columns were purchased from Cytiva (Marlborough, MA, United States). Unless otherwise stated, all other chemical reagents were purchased from Sigma-Aldrich (St. Louis, MO, United States).
2.2 Preparation of immobilized and purified ReSH and MeFDH1
Strains and expression conditions of ReSH and MeFDH1 were described in previous studies (Jang et al., 2018; Cha et al., 2023a). To purify ReSH, cell pellets were resuspended in 50 mM potassium phosphate (Kpi) buffer (pH 7.0) containing 1 mg/mL lysozyme to a concentration of 1 g/10 mL. The resuspended cells were lysed by sonication (amplitude 28%, on/off 2 s/4 s) for 1 h. Insoluble cell debris was removed by centrifugation at 13,000 × g for 30 min. Strep-Tactin XT 4Flow high-capacity resin was mixed with the clear supernatants incubated at 4 °C for 30 min. The resin was washed with 50 mM Kpi buffer (pH 7.0) containing 300 mM potassium chloride on a PP column to remove impurities. After running off the wash solution, plug the end of the pp column, add 200 mM Kpi (pH 6.5), and resuspended the resin to obtain the ReSH immobilized resin for cascade reactions. The ReSH-immobilized resin was stored at 4°C until use. In order to obtain purified ReSH, the resin was eluted with 3 mL of 200 mM Kpi (pH 6.5) containing 50 mM biotin and buffer-exchanged with 200 mM Kpi buffer (pH 6.5) using a PD-10 column.
The purification of MeFDH1 was started with the cell lysis by sonication in an anaerobic chamber. The wet cell pellet was suspended into the buffer A (50 mM (3-(N-morpholino) propanesulfonic acid (MOPS), 200 mM NaCl, 20 mM imidazole, 2 mM dithioerythritol (DTE) and 2 µM resazurin, pH 7.0) with 1 g wet cell/10 mL buffer concentration and sonicated (amplitude 35%, on/off 2 s/2 s). The lysate was centrifuged at 11,000 rpm at 4 °C for 20 min. The supernatant and Ni-NTA agarose bead were mixed together and incubated for 15 min to bind protein. The protein-bound Ni-NTA agarose bead was separated from the mixture using a gravity flow column. The collected protein-bound Ni-NTA agarose bead was washed with buffer A. The MeFDH1-bound Ni-NTA agarose bead was stored at 4°C until use. For the activity and kinetic property assay, the purified MeFDH1 was eluted by buffer B (50 mM MOPS, 200 mM NaCl, 300 mM imidazole, 2 mM DTE and 2 µM resazurin, pH 7.0).
Protein purity was verified by SDS-PAGE (Figs. Sa and b). The concentrations of purified ReSH was determined by measuring their absorbance at 280 nm using a microplate reader (Synergy, BioTek, Winooski, VT, United States), as previously reported for other proteins (Bak et al., 2020; Kim et al., 2021). The concentrations of purified MeFDH1 was determined by measuring their absorbance at 280 nm using a NanoDrop 1 C (Thermo fisher scientific, Waltham, Massachusetts, United States). The extinction coefficients of ReSH and MeFDH1 were calculated to be 165,710 and 153,735 M−1⋅cm−1, respectively, based on their amino acid sequences.
2.3 Enzyme kinetics
The enzyme reaction kinetic parameters of ReSH were measured for the NAD+-dependent oxidation of H2 to H+ in the presence or absence of O2 and CO. The sealing cuvette was filled with 900 μL of 200 mM Kpi buffer (pH 6.5) containing NAD+ and sealed; then, 100% H2 and mix gas containing 1) 100% N2, 2) 96% N2 and 4% O2 3) 60% N2 and 40% CO, and 4) 56% N2, 4% O2 and 40% CO were injected simultaneously for 30 min at 10 mL/min. ReSH (6 mL, 80 nM) was purged with 10 mL/min N2 gas bubbling in a 25 mL sealing vial for 30 min to remove O2 from the air. The reaction was initiated by mixing 100 μL of 80 nM ReSH with a gas-saturated solution in a sealed cuvette. The final concentration of NAD+ was varied from 0 to 4 mM.
The enzyme reaction kinetic parameters of MeFDH1 were measured for the NADH-dependent reduction of CO2 to formate in the presence or absence of O2 and CO. The 200 mM Kpi buffer (pH 6.5) was purged with gases containing 1) 100% N2, 2) 100% O2 and 3) 100% CO for 1 h in closed serum bottles. Each gas-purged buffer was mixed to make 1) 100% N2, 2) 98% N2 and 2% O2, 3) 80% N2 and 20% CO and 4) 78% N2, 20% CO and 2% O2 dissolved buffers. The powder of sodium bicarbonate and NADH was dissolved into each buffers (100 mM of sodium bicarbonate and 6.25—100 µM of NADH). The 980 μL of buffers are finally aliquoted into the cuvettes and sealed. By injecting 10 ug of MeFDH1 in 20 uL of buffer B, the reaction was started.
All measurements were performed in triplicate based on the change in the absorbance at 340 nm in the cuvette. The absorbane changes for ReSH and MeFDHS1 were measured using the T60 UV-Vis spectrophotometer (PG Instruments Ltd., Lutterworth, UK)and the UV-1650 PC (Shimadzu, Kyoto, Japan), respectively. The inverse of initial reaction rates was plotted versus the substrate concentration according to the Hanes-Woolf plot to calculate the kinetic parameters.
2.4 Formate production and quantification
For the cascade reaction, the gas content was controlled in a gas flow reactor (Figure 1). The reactor was filled with 6 mL of reactor containing 20 U of immobilized ReSH, 1 U of immobilized MeFDH1, 1 mM NAD+, and 1% (v/v) antifoam A. All components were resuspended or dissolved in 200 mM Kpi buffer (pH 6.5). The reaction was initiated by a 30 mL/min substrate gas injection. Formate production was sampled every 30 min during incubation for 1.5 h, and 20 μL of 6 N H2SO4 was added to the 200 μL sample to inactivate the enzymes immediately. Additionally, 180 μL of distilled water was mixed with the sample, and the aggregated enzymes were removed by syringe filter. Formate production was quantified by HPLC (1,260, Agilent, CA, United States) equipped with a diode-array detector and an Aminex HPX-87H column (BIO-RAD, CA, United States) with a mobile phase of 5 μM H2SO4 at a flow rate of 0.6 mL/min.
3 Results and discussion
3.1 Preparation of ReSH and MeFDH1
Recombinant ReSH and MeFDH1 were expressed in R. eutropha and M. extorquens, respectively. The ReSH and MeFDH1 were immobilized on the strep tactin XT 4flow resin and Ni-NTA agarose affinity resins, respectively, as described in the Materials and Methods section. A fraction of these was eluted separately to obtain purified proteins. The SDS-PAGE analysis was performed to verify the existence of subunits and purity of each. Five bands of purified ReSH subunits were observed, which matched their expected molecular weights (HoxF, 68,110 Da; HoxH, 54,863 Da; HoxU, 26,173 Da; HoxY, 22,881 Da; HoxI, 18,567 Da) (Supplementary Figure S1a). Two bands of purified MeFDH1 subunits were also observed, consistent with their expected molecular weights (FdhA1: 107,341 Da, FdhA2: 62,321 Da) (Supplementary Figure S1b). The purity of both enzymes was high. To calculate the enzyme units of ReSH- or MeFDH1 immobilized resin, we measured the concentration and enzymatic activity of purified ReSH and MeFDH1, respectively. The ReSH-immobilized resin had an NAD+-dependent H2 oxidation activity of 140.9 U/mL. The MeFDH1-immobilized resin had an NADH-dependent CO2 reduction activity of 0.53 U/mL. These results showed that the immobilized ReSH and MeFDH1 were successfully prepared.
3.2 pH-dependency of enzymes
We investigated the pH-dependent enzyme activities of NAD+-dependent H2 oxidation of ReSH and NADH-dependent CO2 reduction of MeFDH1 to determine the buffer conditions in the reactor for the cascade reaction. The specific activity of ReSH tended to increase with increasing pH from 6 to 8 (Figure 2A). In contrast, the specific activity of MeFDH1 tended to decrease with increasing pH, with the maximum activity at pH 6.5 (Figure 2B). Since the specific activity of the two enzymes is maximized at different points, we decided to use pH 6.5 as the cascade reaction condition where the activity of MeFDH1, which produces formate, is maximal.
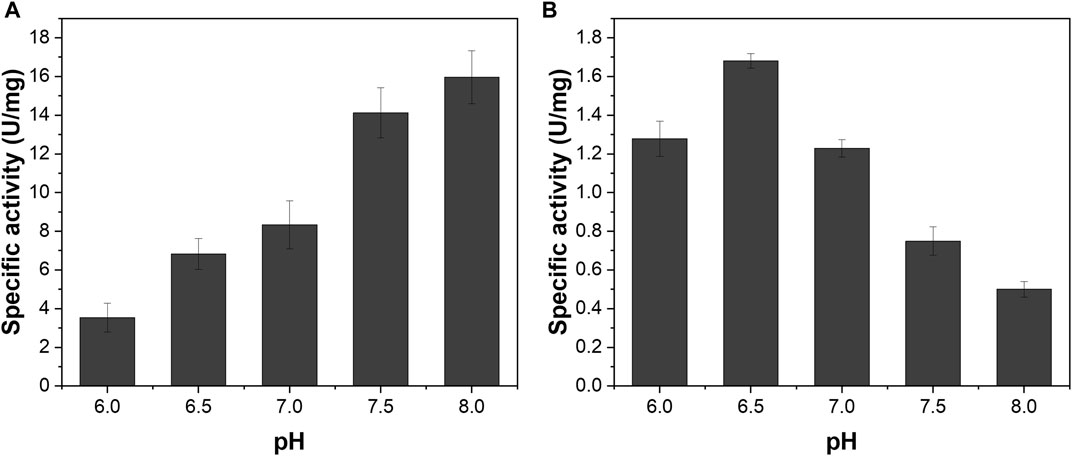
FIGURE 2. pH-dependency of (A) ReSH and (B) MeFDH1 activity. The enzyme activities were determined in the pH range of 6.0–8.0 in 200 mM Kpi buffer.
3.3 Enzyme kinetics in the flue gas mimic conditions
We investigated whether both ReSH and MeFDH1 can tolerate the potential inhibitors (O2 and CO) present in the flue gas at pH 6.5. The final concentrations of O2 and CO were 2% and 20%, respectively, which is higher than those inside the COG, to evaluate enzyme activity inhibition. The NAD+-dependent H2 oxidation reaction rate by ReSH was measured, and the Hanes-Woolf plot was fitted to calculate the kinetic parameters using Origin 2022 program (Supplementary Figure S2). The kcat and Km values of ReSH were not substantially different in four conditions: control without any O2 and CO, 2% O2, 20% CO, and both 2% O2 and 20% CO (Table 2). Similarly, The NADH-dependent CO2 reduction reaction rate by MeFDH1 was measured, and the Hanes-Woolf plot was fitted to calculate the kinetic parameters (Supplementary Figure S3). The kcat and Km of MeFDH1 were not substantially different in four conditions: control without any O2 and CO, 2% O2, 20% CO, and both 2% O2 and 20% CO (Table 3). These results show that at concentrations below 2% O2 and 20% CO, the enzymatic activities of ReSH and MeFDH1 are not substantially inhibited, indicating their potential for use in flue gas conversion.
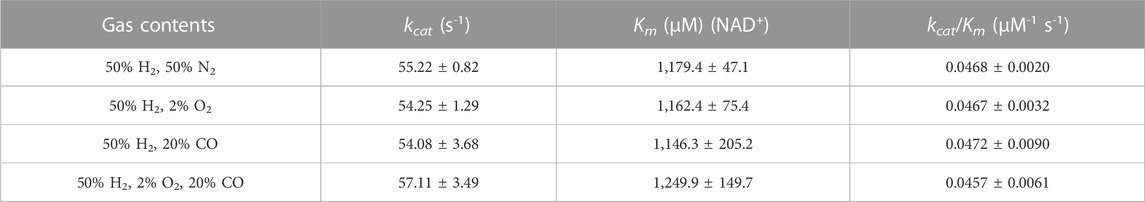
TABLE 2. Kinetic parameters of ReSH in the presence of O2 or CO.
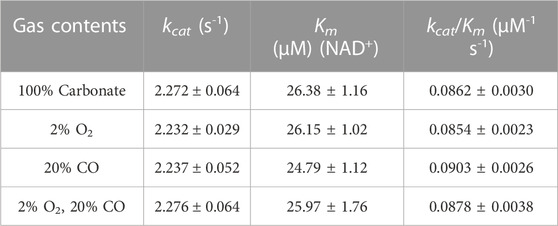
TABLE 3. Kinetic parameters of MeFDH1 in the presence of O2 or CO.
3.4 Enzymatic reactor optimization
We optimized the components of the enzymatic reactor for a smooth cascade reaction. The reaction mechanism is that ReSH oxidizes H2, and NAD+ is reduced to NADH, which is then oxidized back to NAD+ by MeFDH1, which reduces CO2 to formate. Since MeFDH1 has a Km value of about 25 uM, we used 1 mM of NAD+ as a sufficient initial input to produce the formate at Vmax. For formate production to continue at the maximum rate with the minimal reverse reaction of MeFDH1, NAD+ must be reduced immediately so that NADH is dominant during the reaction. To achieve this, the enzyme unit of ReSH must be higher than that of MeFDH1. We fixed the MeFDH1 immobilized resin at 1 U and varied the ReSH immobilized resin from 0 to 20 U (Unit ratio ReSH: MeFDH1 = 0:1 to 20:1) and measured the formate produced when supplied with gas substrate H2 and CO2 for 1 h (Figure 3). Formate production was quantified via HPLC. The retention time of formate was 13.010 min (Cha et al., 2023a). No formate was produced in the reactor without ReSH. Compared to the 1:1 ReSH to MeFDH1 unit ratio, all experiments with an excess of ReSH produced about 2 mM of formate, and experiments with an excess of ReSH over 5:1 tended to saturate formate production. Based on these results, we determined that the unit ratio of ReSH to MeFDH1 was 20:1 for sufficient formate productivity.
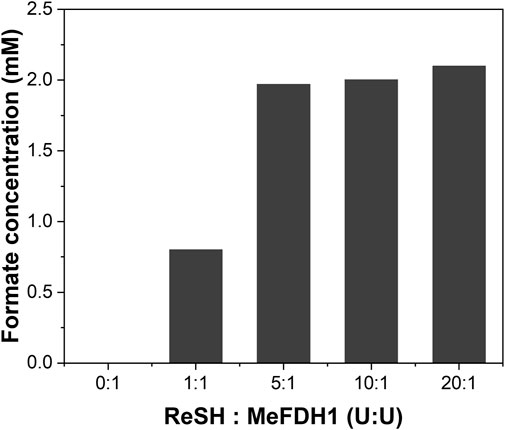
FIGURE 3. Formate produced during 1 h at different ratios of ReSH to MeFDH1. The activity of ReSH was varied with 0, 1, 5, 10, and 20 U (ReSH:MeFDH1 = 0:1, 1:1, 5:1, 10:1, 20:1) in the presence of 1 U MeFDH1 and 1 mM NAD+.
3.5 Formate production from flue gas mimic condition
We demonstrated CO2 hydration using a flue gas mimic. We decided to implement a specific flue gas mimic condition by mixing different syngas, but most flue gases have a high percentage of either H2 or CO 2. For example, COG had more than 50% H2 but less than 3% CO2, and BFG had 20% CO2 but less than 4% H2. So the flue gas mimic realized them in a ratio similar to the gas contents after mixing them 1:1 (Table 1). 20 U of the ReSH immobilized resin and 1 U of the MeFDH1 immobilized resin and 1 mM NAD+ were mixed and placed in a gas flow reactor. Formate production was monitored for 2 h at 30 min intervals with a continuous flow of flue gas mimic to the enzymatic reactor containing a mixture of all reaction components (Figure 4). No formate was detected in the negative control group, missing one of ReSH, MeFDH1, and NAD+. Following the trend of previous kinetics, we found that formate was produced in 2% O2 and 20% CO. The formate tended to reach about 2 mM around 2 h and then saturate without further increase. We speculated that the elevated formate concentration increased the reverse reaction rate, causing it to reach equilibrium.
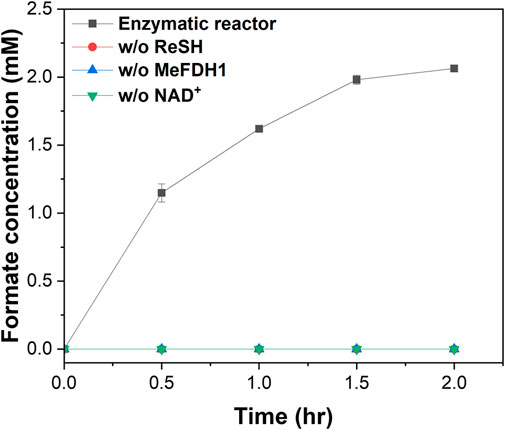
FIGURE 4. Formate production by enzymatic reactors in the presence of O2 and CO. As negative controls, enzymatic reactors were constructed lacking ReSH, MeFDH, and NAD+, respectively. The flue gas mimic containing 30% H2, 15% CO2, 15% CO, 2% O2, and 38% N2 were fed to the enzymatic reactor at 30 mL/min for 2 h. All measurements were performed in triplicate.
3.6 Formate production using real flue gases
We investigated formate production by feeding real flue gases to the enzymatic reactor. We provided the COG, BFG, and a 1:1 mix of COG and BFG to the enzymatic reactor and monitored the formate production (Figure 5). Contrary to our expectations, both COG, which is rich in H2 and relatively low in CO2, and BFG, which is low in H2 and high in CO2, saturated in formate production around 2 mM at 2 h, just like the flue gas mimic, and we saw the same trend in formate production when we fed a 1:1 mixture of the two gases. We speculated that even at relatively low concentrations of H2 in BFG or CO2 in COG, the reaction rates of ReSH and MeFDH1 were high enough to have a Vmax due to the continuous gas flow. In practice, however, CO2 and H2 react in a 1:1 mole ratio, so mixing the flue gases to produce formate, as we have shown, is a significant step toward industrial-scale CO2 hydration. However, to solve the current situation where formate production is saturated, it is necessary to configure a repeated batch or continuous flow reactor to separate the formate produced from the reactor continuously. As described in the Introduction section, FDHs for enzymatic CO2 reduction are categorized into metal-independent and metal-dependent types. Metal-independent FDHs are not affected by O2 but generally exhibit relatively low catalytic activities (with a range of kcat/Km 0.0004–0.08 s-1 mM-1) (Alpdağtaş et al., 2022). On the other hand, metal-dependent FDHs display high catalytic activity (with a range of kcat/Km 100∼103 s-1 mM-1), but most of them face difficulties in utilizing real CO2 sources due to susceptibility to inactivation by O2 (Moon et al., 2020; Calzadiaz-Ramirez and Meyer, 2022). The MeFDH1 enzyme used in this study is expected to serve as a compromise, as it demonstrates catalytic activity falling between these two ranges and displays resistance to both O2 and CO.
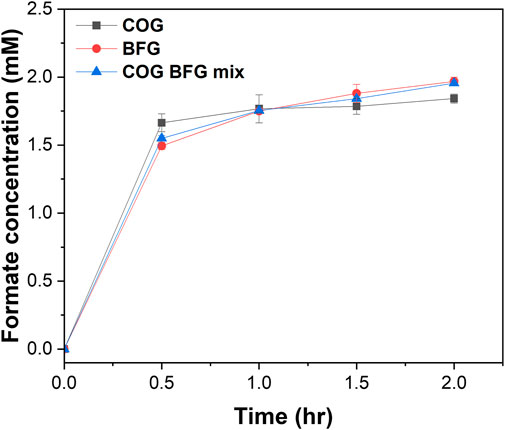
FIGURE 5. Formate production by the enzymatic reactors using real flue gas. COG, BFG, and 1:1 mixture of COG and BFG were fed to the enzymatic reactor at 30 mL/min for 2 h. All measurements were performed in triplicate.
4 Conclusion
We have successfully demonstrated CO2 hydrogenation using hydrogen in the real flue gas from the steel industry via an NAD+-dependent cascade reaction in an enzymatic reactor that combines a specific H2ase and FDH. We confirmed that ReSH and MeFDH1, previously reported as O2-tolerant, are also resistant to CO, a potential inhibitor of metalloenzymes. We used both H2 and CO2 from the flue gas in this study. However, the enzymatic reactor system developed in this study is expected to enable the utilization of CO2 and H2 obtained from other sources, such as industrial waste gases, biomass and gasified plastics. We plan to improve the stability of the enzymes for the long-term operation of the reactor and improve the cofactor co-immobilization system (Cha et al., 2023b) for implementation as a continuous reactor.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author contributions
JC: Conceptualization, Data curation, Formal Analysis, Writing–original draft. JL: Data curation, Methodology, Writing–original draft. BJ: Formal Analysis, Investigation, Writing–original draft. YK: Funding acquisition, Project administration, Supervision, Writing–review and editing. IK: Conceptualization, Funding acquisition, Project administration, Supervision, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Research Foundation of Korea (NRF), the Ministry of Science, and ICT (grant numbers 2021R1A5A1028138, 2020R1A5A1019631, and 2015M3D3A1A01064923).
Acknowledgments
The authors are grateful to Professor Oliver Lenz (Technische Universität Berlin) for providing cells expressing ReSH.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbioe.2023.1265272/full#supplementary-material
References
Al-Ghussain, L. (2019). Global warming: review on driving forces and mitigation. Environ. Prog. Sustain. Energy 38, 13–21. doi:10.1002/ep.13041
Alissandratos, A., Kim, H.-K., Matthews, H., Hennessy, J. E., Philbrook, A., and Easton, C. J. (2013). Clostridium carboxidivorans strain P7T recombinant formate dehydrogenase catalyzes reduction of CO(2) to formate. Appl. Environ. Microbiol. 79, 741–744. doi:10.1128/AEM.02886-12
Amao, Y. (2018). Formate dehydrogenase for CO2 utilization and its application. J. CO2 Util. 26, 623–641. doi:10.1016/j.jcou.2018.06.022
Appel, A. M., Bercaw, J. E., Bocarsly, A. B., Dobbek, H., DuBois, D. L., Dupuis, M., et al. (2013). Frontiers, opportunities, and challenges in biochemical and chemical catalysis of CO2 fixation. Chem. Rev. 113, 6621–6658. doi:10.1021/cr300463y
Armaroli, N., and Balzani, V. (2011). The legacy of fossil fuels. Chem. – Asian J. 6, 768–784. doi:10.1002/asia.201000797
Baccour, M., Lamotte, A., Sakai, K., Dubreucq, E., Mehdi, A., Kano, K., et al. (2020). Production of formate from CO2 gas under ambient conditions: towards flow-through enzyme reactors. Green Chem. 22, 3727–3733. doi:10.1039/D0GC00952K
Bagley, K. A., Van Garderen, C. J., Chen, M., Duin, E. C., Albracht, S. P., and Woodruff, W. H. (1994). Infrared studies on the interaction of carbon monoxide with divalent nickel in hydrogenase from Chromatium vinosum. Biochemistry 33, 9229–9236. doi:10.1021/bi00197a026
Bak, M., Park, J., Min, K., Cho, J., Seong, J., Hahn, Y. S., et al. (2020). Recombinant peptide production platform coupled with site-specific albumin conjugation enables a convenient production of long-acting therapeutic peptide. Pharmaceutics 12, 364. doi:10.3390/pharmaceutics12040364
Burgdorf, T., Linden, E., Bernhard, M., Yin, Q., Back, J., Hartog, A., et al. (2005). The soluble NAD+-Reducing [NiFe]-Hydrogenase from Ralstonia eutropha H16 consists of six subunits and can Be specifically activated by NADPH. J. Bacteriol. 187, 3122–3132. doi:10.1128/JB.187.9.3122-3132.2005
Çakar, M. M., Ruupunen, J., Mangas-Sanchez, J., Birmingham, W. R., Yildirim, D., Turunen, O., et al. (2020). Engineered formate dehydrogenase from Chaetomium thermophilum, a promising enzymatic solution for biotechnical CO2 fixation. Biotechnol. Lett. 42, 2251–2262. doi:10.1007/s10529-020-02937-7
Cha, J., Bak, H., and Kwon, I. (2023a). Hydrogen-fueled CO2 reduction using oxygen-tolerant oxidoreductases. Front. Bioeng. Biotechnol. 10, 1078164. doi:10.3389/fbioe.2022.1078164
Cha, J., Cho, J., and Kwon, I. (2023b). Self-sufficient reusable biocatalytic system outfitted with multiple oxidoreductases and flexible polypeptide-based cofactor swing arms. ACS Sustain. Chem. Eng. 11, 3710–3719. doi:10.1021/acssuschemeng.2c06676
Ding, M., and Jiang, H. (2018). Incorporation of imidazolium-based poly(ionic liquid)s into a metal–organic framework for CO2 capture and conversion. ACS Catal. 8, 3194–3201. doi:10.1021/acscatal.7b03404
Fontecilla-Camps, J. C., Volbeda, A., Cavazza, C., and Nicolet, Y. (2007). Structure/function relationships of [NiFe]- and [FeFe]-Hydrogenases. Chem. Rev. 107, 4273–4303. doi:10.1021/cr050195z
Frölicher, T. L., Fischer, E. M., and Gruber, N. (2018). Marine heatwaves under global warming. Nature 560, 360–364. doi:10.1038/s41586-018-0383-9
Ghalei, B., Sakurai, K., Kinoshita, Y., Wakimoto, K., Isfahani, A. P., Song, Q., et al. (2017). Enhanced selectivity in mixed matrix membranes for CO2 capture through efficient dispersion of amine-functionalized MOF nanoparticles. Nat. Energy 2, 17086. doi:10.1038/nenergy.2017.86
Hartmann, T., and Leimkühler, S. (2013). The oxygen-tolerant and NAD+-dependent formate dehydrogenase from Rhodobacter capsulatus is able to catalyze the reduction of CO2 to formate. FEBS J. 280, 6083–6096. doi:10.1111/febs.12528
Horch, M., Lauterbach, L., Mroginski, M. A., Hildebrandt, P., Lenz, O., and Zebger, I. (2015). Reversible active site sulfoxygenation can explain the oxygen tolerance of a NAD+-Reducing [NiFe] hydrogenase and its unusual infrared spectroscopic properties. J. Am. Chem. Soc. 137, 2555–2564. doi:10.1021/ja511154y
Hughes, T. P., Kerry, J. T., Baird, A. H., Connolly, S. R., Dietzel, A., Eakin, C. M., et al. (2018). Global warming transforms coral reef assemblages. Nature 556, 492–496. doi:10.1038/s41586-018-0041-2
Hwang, H. W., Yoon, J., Min, K., Kim, M.-S., Kim, S.-J., Cho, D. H., et al. (2020). Two-stage bioconversion of carbon monoxide to biopolymers via formate as an intermediate. Chem. Eng. J. 389, 124394. doi:10.1016/j.cej.2020.124394
Jang, J., Jeon, B. W., and Kim, Y. H. (2018). Bioelectrochemical conversion of CO2 to value added product formate using engineered Methylobacterium extorquens. Sci. Rep. 8, 7211. doi:10.1038/s41598-018-23924-z
Kim, S., Kim, M., Kim, Y. T., Kwak, G., and Kim, J. (2019). Techno-economic evaluation of the integrated polygeneration system of methanol, power and heat production from coke oven gas. Energy Convers. Manag. 182, 240–250. doi:10.1016/j.enconman.2018.12.037
Kim, S., Kwon, K., Tae, G., and Kwon, I. (2021). Nano-entrapping multiple oxidoreductases and cofactor for all-in-one nanoreactors. ACS Sustain. Chem. Eng. 9, 6741–6747. doi:10.1021/acssuschemeng.1c00843
Kim, S. M., Lee, J., Kang, S. H., Heo, Y., Yoon, H.-J., Hahn, J.-S., et al. (2022). O2-tolerant CO dehydrogenase via tunnel redesign for the removal of CO from industrial flue gas. Nat. Catal. 5, 807–817. doi:10.1038/s41929-022-00834-y
Kothandaraman, J., Czaun, M., Goeppert, A., Haiges, R., Jones, J.-P., May, R. B., et al. (2015). Amine-free reversible hydrogen storage in formate salts catalyzed by ruthenium pincer complex without pH control or solvent change. ChemSusChem 8, 1442–1451. doi:10.1002/cssc.201403458
Laukel, M., Chistoserdova, L., Lidstrom, M. E., and Vorholt, J. A. (2003). The tungsten-containing formate dehydrogenase from Methylobacterium extorquens AM1: purification and properties. Eur. J. Biochem. 270, 325–333. doi:10.1046/j.1432-1033.2003.03391.x
Lauterbach, L., and Lenz, O. (2013). Catalytic production of hydrogen peroxide and water by oxygen-tolerant [NiFe]-Hydrogenase during H2 cycling in the presence of O2. J. Am. Chem. Soc. 135, 17897–17905. doi:10.1021/ja408420d
Lee, J.-K., Shin, S., Kwak, G.-J., Lee, M.-K., Lee, I.-B., and Yoon, Y.-S. (2020). Techno-economic evaluation of polygeneration system for olefins and power by using steel-mill off-gases. Energy Convers. Manag. 224, 113316. doi:10.1016/j.enconman.2020.113316
Li, W., Wang, H., Jiang, X., Zhu, J., Liu, Z., Guo, X., et al. (2018). A short review of recent advances in CO2 hydrogenation to hydrocarbons over heterogeneous catalysts. RSC Adv. 8, 7651–7669. doi:10.1039/c7ra13546g
Loges, B., Boddien, A., Junge, H., and Beller, M. (2008). Controlled generation of hydrogen from formic acid amine adducts at room temperature and application in H 2/O 2 fuel cells. Angew. Chem. Int. Ed. 4, 3962–3965. doi:10.1002/anie.200705972
Moon, M., Park, G. W., Lee, J., Lee, J.-S., and Min, K. (2020). Recent progress in formate dehydrogenase (FDH) as a non-photosynthetic CO2 utilizing enzyme: A short review. J. CO2 Util. 42, 101353. doi:10.1016/j.jcou.2020.101353
Moral, G., Ortiz-Imedio, R., Ortiz, A., Gorri, D., and Ortiz, I. (2022). Hydrogen recovery from coke oven gas. Comparative analysis of technical alternatives. Ind. Eng. Chem. Res. 61, 6106–6124. doi:10.1021/acs.iecr.1c04668
Niks, D., and Hille, R. (2018). “Chapter Eleven - reductive activation of CO2 by formate dehydrogenases,” in Enzymes of energy technology methods in enzymology. Editor F. Armstrong (China: Academic Press), 277–295. doi:10.1016/bs.mie.2018.10.013
Olah, G. A., Prakash, G. K. S., and Goeppert, A. (2011). Anthropogenic chemical carbon cycle for a sustainable future. J. Am. Chem. Soc. 133, 12881–12898. doi:10.1021/ja202642y
Reda, T., Plugge, C. M., Abram, N. J., and Hirst, J. (2008). Reversible interconversion of carbon dioxide and formate by an electroactive enzyme. Proc. Natl. Acad. Sci. U. S. A. 105, 10654–10658. doi:10.1073/pnas.0801290105
Rochelle, G. T. (2009). Amine scrubbing for CO2 capture. Sci. (80-. ) 325, 1652–1654. doi:10.1126/science.1176731
Salam, M. A., and Noguchi, T. (2005). Impact of human activities on carbon dioxide (CO2) emissions: A statistical analysis. Environmentalist 25, 19–30. doi:10.1007/s10669-005-3093-4
Sokol, K. P., Robinson, W. E., Oliveira, A. R., Zacarias, S., Lee, C.-Y., Madden, C., et al. (2019). Reversible and selective interconversion of hydrogen and carbon dioxide into formate by a semiartificial formate hydrogenlyase mimic. J. Am. Chem. Soc. 141, 17498–17502. doi:10.1021/jacs.9b09575
Takemura, T., and Suzuki, K. (2019). Weak global warming mitigation by reducing black carbon emissions. Sci. Rep. 9, 4419. doi:10.1038/s41598-019-41181-6
Tülek, A., Günay, E., Servili, B., Essiz, S., Binay, B., and Yildirim, D. (2023). Sustainable production of formic acid from CO2 by a novel immobilized mutant formate dehydrogenase. Sep. Purif. Technol. 309, 123090. doi:10.1016/j.seppur.2022.123090
Vincent, K. A., Parkin, A., and Armstrong, F. A. (2007). Investigating and exploiting the electrocatalytic properties of hydrogenases. Chem. Rev. 107, 4366–4413. doi:10.1021/cr050191u
Wang, W.-H., Himeda, Y., Muckerman, J. T., Manbeck, G. F., and Fujita, E. (2015). CO2 hydrogenation to formate and methanol as an alternative to photo- and electrochemical CO2 reduction. Chem. Rev. 115, 12936–12973. doi:10.1021/acs.chemrev.5b00197
Wang, Y., Tan, L., Tan, M., Zhang, P., Fang, Y., Yoneyama, Y., et al. (2019). Rationally designing bifunctional catalysts as an efficient strategy to boost CO2 hydrogenation producing value-added aromatics. ACS Catal. 9, 895–901. doi:10.1021/acscatal.8b01344
Wulff, P., Day, C. C., Sargent, F., and Armstrong, F. A. (2014). How oxygen reacts with oxygen-tolerant respiratory [NiFe]-hydrogenases. Proc. Natl. Acad. Sci. 111, 6606–6611. doi:10.1073/pnas.1322393111
Xiang, D., Yang, S., and Qian, Y. (2016). Techno-economic analysis and comparison of coal based olefins processes. Energy Convers. Manag. 110, 33–41. doi:10.1016/j.enconman.2015.12.011
Keywords: carbon dioxide, hydrogen, formate, flue gas, hydrogenase, formate dehydrogenase
Citation: Cha J, Lee J, Jeon BW, Kim YH and Kwon I (2023) Real flue gas CO2 hydrogenation to formate by an enzymatic reactor using O2- and CO-tolerant hydrogenase and formate dehydrogenase. Front. Bioeng. Biotechnol. 11:1265272. doi: 10.3389/fbioe.2023.1265272
Received: 22 July 2023; Accepted: 22 September 2023;
Published: 03 October 2023.
Edited by:
Heiko Lange, University of Milano-Bicocca, ItalyReviewed by:
Baris Binay, Gebze Technical University, TürkiyeYoung Joo Yeon, Gangneung–Wonju National University, Republic of Korea
Copyright © 2023 Cha, Lee, Jeon, Kim and Kwon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yong Hwan Kim, bWV0YWxraW1AdW5pc3QuYWMua3I=; Inchan Kwon, aW5jaGFuQGdpc3QuYWMua3I=
†These authors have contributed equally to this work