 Nihan Ozturk
Nihan Ozturk Indrabahadur Singh
Indrabahadur Singh Aditi Mehta
Aditi Mehta Thomas Braun
Thomas Braun Guillermo Barreto
Guillermo Barreto- 1LOEWE Research Group Lung Cancer Epigenetic, Max-Planck-Institute for Heart and Lung Research, Bad Nauheim, Germany
- 2Department of Cardiac Development and Remodeling, Max-Planck-Institute for Heart and Lung Research, Bad Nauheim, Germany
High mobility group (HMG) proteins are the most abundant non-histone chromatin associated proteins. HMG proteins bind to DNA and nucleosome and alter the structure of chromatin locally and globally. Accessibility to DNA within chromatin is a central factor that affects DNA-dependent nuclear processes, such as transcription, replication, recombination, and repair. HMG proteins associate with different multi-protein complexes to regulate these processes by mediating accessibility to DNA. HMG proteins can be subdivided into three families: HMGA, HMGB, and HMGN. In this review, we will focus on recent advances in understanding the function of HMGA family members, specifically their role in gene transcription regulation during development and cancer.
Introduction
Chromatin constitutes the physiological template for transcription, thereby increasing the complexity of gene transcription regulation. Nucleosomes are the structural and functional units of chromatin. A nucleosome is built of DNA surrounding a histone octamer, which consists of two H2A–H2B dimers and one (H3–H4)2 tetramer. In addition to nucleosomes, chromatin consists of non-histone chromatin-associated proteins. It is well known that chromatin mediated regulation of transcription involves DNA methylation and histone modifications (Zhang et al., 2005; Karlic et al., 2010; Ha et al., 2011; Gohlke et al., 2013). However the precise biological function of non-histone chromatin-associated proteins is not yet clear. High mobility group (HMG) proteins are the most abundant non-histone chromatin associated proteins. Based on their DNA binding domains, the HMG proteins are subdivided into three families (Figure 1A): HMGA (containing AT-hooks), HMGB (containing HMG-boxes) and HMGN (containing nucleosomal binding domains) (Bustin, 2001; Catez and Hock, 2010). HMG proteins can recognize structure rather than a particular nucleotide sequence and are able to bind to specific structures in DNA or chromatin in a sequence-independent fashion via their respective functional motifs (Reeves, 2001). HMG proteins do not possess intrinsic transcriptional activity while having the ability to modulate transcription of their target genes by altering the chromatin structure at the promoter and/or enhancers (Reeves, 2010). Thus, they are called architectural transcription factors. In this review, we will focus on recent advances in understanding the function of HMGA family members. HMGA family is comprised of HMGA1a, HMGA1b, HMGA1c, and HMGA2. They are expressed highly during embryonic development and in transformed cells. They are encoded by two distinct genes. The HMGA1 gene gives rise to three proteins (HMGA1a, HMGA1b, and HMGA1c) by alternative splicing of a common transcript. With the exception of HMGA1c, the HMGA proteins contain three short basic repeats called AT-hook motif and a C-terminal acidic tail (Figure 1B) (Sgarra et al., 2004; Fusco and Fedele, 2007; Hammond and Sharpless, 2008; Pfannkuche et al., 2009). The amino acid sequence of the AT-hook motif is K/RXRGRP (X = glycine or proline) and is flanked on either side by positively charged residues. These characteristics determine the binding preference of HMGA proteins to both the minor groove of AT-rich DNA stretches (Cui and Leng, 2007; Winter et al., 2011) and to nucleosomes in a cooperative manner (Li et al., 2006). HMGA proteins, when free in solution, possess very little, if any, secondary structure as shown by biophysical techniques such as circular dichroism (CD) (Lehn et al., 1988) and nuclear magnetic resonance (NMR) spectroscopy (Evans et al., 1995). However, when bound to DNA or proteins specifically, the AT-hooks undergo from a disordered to ordered conformational change and influence the conformation of bound DNA substrates in different ways including bending, straightening, unwinding, and inducing looping in linear DNA molecules. The function of the C-terminal acidic region is poorly understood. However, there are speculations that C-terminal acidic tail is involved in protein-protein interaction and recruitment of factors during regulation of gene transcription (Xu et al., 2011).
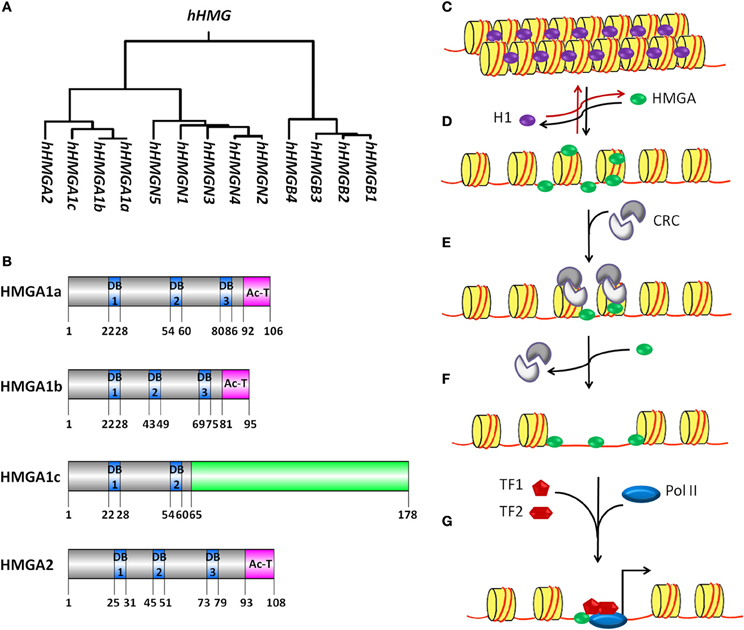
Figure 1. The HMG proteins superfamily and model for HMGA proteins mediated transcription activation. (A) Genealogical tree of human HMG proteins. HMG proteins are divided into three families depending on their DNA binding domains: HMGA (containing AT-hooks), HMGB (containing HMG-boxes) and HMGN (containing nucleosomal binding domains). CLUSTALW software was used to build rooted genealogical tree with branch length (UPGMA) of human HMG group family members. (B) Schematic representation of human HMGA proteins. HMGA1a, HMGA1b, HMGA1c, and HMGA2 proteins contain unique DNA binding domains (DB, blue boxes) with a characteristic AT-hook motif and a C terminal acidic domain (pink boxes). HMGA1a and HMGA1b proteins show high sequence similarity. HMGA1b lacks eleven amino acids before the second DB when compared to HMGA1a and 1c. The amino acid sequence of HMGA1c differs from the one from HGMA1a and 1b starting from amino acid 65 (green box). GPS DOG 2.0 software was used to illustrate protein structure domains (Ren et al., 2009). (C–G) A model for the putative mechanism of HMGA proteins as factors promoting nucleosome mobility and accessibility to specific DNA sites for transcription activation is depicted. (C) The nucleosome is built of 146 bp DNA (red line) surrounding the histone octamer (yellow cylinder), which consist of two H2A-H2B dimers and one (H3–H4)2 tetramer. The linker histone H1 (purple oval) binds to linker DNA outside the histone octamer at the position where the DNA enters and exits the nucleosome core particle. (D) HMGA proteins (green oval) can compete and displace histone H1 from the chromatin. HMGA binding to linker DNA in the chromatin and the subsequent displacement of histone H1 leads to decompactness of the chromatin. (E) The relaxed chromatin structure acts as an anchoring site for the recruitment of chromatin remodeling complexes (CRC, gray). Binding of this complex is enhanced by HMGA proteins and induces eviction of core histones and/or mobilization of complete histone octamers. (F) The chromatin remodeling events make sequence-specific sites on the DNA accessible for transcription factor (TF, red pentagon and hexagon) binding. (G) HMGA proteins might facilitate the formation of transcription factor complexes binding to these sequence-specific sites. Later, RNA polymerase II (Pol II, blue oval) is recruited for transcription initiation (black arrow).
HMGA family members show high sequence homology; the only difference between HMGA1a and HMGA1b is an internal deletion of eleven amino acids before the second AT-hook domain. Although the sequence similarity may suggest that these two isoforms could be biologically interchangeable, there are reports that show the opposite. For example, it was shown that tetracycline-regulated induction of HMGA1b in the human breast epithelial cell line MCF7 caused them to progress much more rapidly to a metastatic and highly malignant phenotype than did induced overexpression of HMGA1a (Reeves et al., 2001). The differences in their posttranslational modifications may explain the different cellular processes they are involved in. Also the different spacing of AT-hook domains between the different HMGA proteins could have a role in their different target gene selection and/or binding affinity (Cleynen and Van De Ven, 2008).
HMGA and Chromatin
In the nucleus of eukaryotic cells, genomic DNA is highly organized and packed into chromatin. Chromatin serves as substrate for all kinds of DNA dependent processes and represents a strong barrier to sequence specific recognition sites on the DNA. Thus, to overcome this DNA sequence accessibility barrier, it is a prerequisite to open the higher order chromatin structure so that transcription regulators can access to specific DNA sequences and execute their function. Histone H1 binds to linker DNA and increases the compactness of higher order chromatin (Figures 1C,D). HMGA proteins compete with histone H1 for binding to linker DNA thereby inducing a loosening of the chromatin structure, which has been demonstrated by different techniques including fluorescence recovery after photobleaching (FRAP), nuclear fractionation analysis and micrococcal nuclease digestion (MNase) assays (Zhao et al., 1993; Catez et al., 2004; Kishi et al., 2012). However, the molecular mechanism underlying the replacement of histone H1 by HMGA proteins which results in chromatin opening is not well understood. Several post-translational modifications have been reported for histone H1, such as phosphorylation, methylation, acetylation and poly-ADP-ribosylation (PARylation) (Snijders et al., 2008). Similar to the core histones, these post-translational modifications of the linker histone H1 play a role in regulation of chromatin structure. PARP-1 mediated PARylation of histone H1 leads to nucleosome-specific exchange of histone H1 by HMGB proteins inducing local changes of chromatin structure (Ju et al., 2006). In a similar manner, histone H1 post-translational modifications could facilitate replacement of histone H1 by HMGA proteins thereby inducing chromatin decompaction. It is well known that the globular domain of histone H1 interacts and binds with linker DNA. Several motifs for kinases are located inside the globular domain or flanking it. Phosphorylation of these sites modulate the binding affinity of histone H1 to linker DNA (Contreras et al., 2003) and might play a role during replacement of histone H1 by HMGA proteins.
Histone H1 eviction from the chromatin is not enough to facilitate the access of regulatory elements on target genes because the DNA is still wrapped around the core histones hindering the accessibility of transcription factors to their binding elements. Hence, there is further need of either eviction or mobilization of core histones (Figures 1E,F). Several chromatin remodelers might be involved in these processes. For example, the FACT complex has been reported to participate in H2A/H2B histone eviction/deposition (Belotserkovskaya et al., 2003), whereas ASF1 is involved in H3/H4 histone eviction/deposition (Schwabish and Struhl, 2006). HMGA proteins bind to both nucleosomes (Li et al., 2006) and chromatin remodelers (Malini et al., 2011) suggesting a role of these proteins in eviction and/or mobilization of core histones during transcriptional regulation.
HMGA and Transcription
In eukaryotes, two types of core promoters are used for initiation of gene transcription: promoters that are enriched with the di-nucleotide sequence CpG and promoters that are CpG poor (Antequera, 2003; Ramirez-Carrozzi et al., 2009; Deaton and Bird, 2011). CpG poor core promoters usually contain a TATA box, initiator sequences (INR), a TFIIB recognition element (BRE) and downstream promoter elements (DPE) (Butler and Kadonaga, 2002; Levine and Tjian, 2003). These promoters have specific single transcription start sites (TSS). In addition, a strong stimulation of RNA polymerase II (Pol II) dependent transcription initiation has been reported for these promoters by the synergistic interplay of the TATA box and INR core promoter elements. Recently, HMGA1 was identified as one of the factors required for the synergy between the TATA box and INR elements (Figure 1G) (Xu et al., 2011). However, there is still need of further investigation to test the relevance of this finding as a general mechanism of transcription initiation. Gene transcription can also be regulated by the interplay of core promoter elements along with regulatory DNA elements, such as enhancers and silencers, which might be located several kilo base pairs (kbp) upstream or downstream of the promoter. In response to defined signals, specific proteins bind to the enhancer and form a complex called enhanceosome (Munshi et al., 1999; Yie et al., 1999; Panne et al., 2007; Panne, 2008). Looping of the DNA brings the enhaceosome and the core promoter in close proximity resulting in enhanced gene transcription. HMGA proteins are involved in enhanceosome formation (Bouallaga et al., 2000, 2003). In addition, it has been reported that HMGA proteins have the ability to bend DNA (Chen et al., 2010). Thus, a model can be suggested in which HMGA proteins participate not only in the formation of the enhanceosome but also in DNA looping and chromatin rearrangements that occur to bring enhanceosomes and core promoter in close proximity so that a coordinated assembly of the transcription initiation complex at core promoter can take place.
It has been reported that the C-terminal domain of HMGA proteins (Figure 1B) is required for the interaction of HMGA1 with TFIID (Xu et al., 2011). This interaction seems to mediate core promoter specific functions since a C-terminal deletion mutant of HMGA1 fails to initiate transcription (Figure 1G). In addition, this domain contains several conserved phosphorylation sites, for example SQ sites, which are substrates for the kinases DNA-PK, ATM and ATR. These kinases have been involved in different signal transduction pathways. In addition, ATM and ATR interact with and phosphorylate HMGA group proteins (Pentimalli et al., 2008; Palmieri et al., 2011; Natarajan et al., 2013). It would be interesting to elucidate the biological relevance of this finding within the context of different signal transduction pathways to understand the signaling events responsible for fine-tuning the HMGA mediated cell specific gene transcription.
The second type of promoters, which contain CpG islands, lack TATA boxes and display multiple heterogeneous TSSs. These promoters tend to be enriched with binding elements for SP1, NRF-1, E2F, and ETS transcription factors (Landolin et al., 2010; Deaton and Bird, 2011). It was shown that HMGA1 interacts with SP1 and facilitates its binding to both the human insulin receptor (INSR) gene promoter and the herpes simplex virus latency-active promoter 2 (Figure 1G) (French et al., 1996; Foti et al., 2003). This suggests that HMGA family members may be involved in the transcriptional activation of CpG rich promoters. Although previous reports support the specific binding of HMGA to AT-rich sequences on the DNA (Cleynen et al., 2007; Cui and Leng, 2007; Winter et al., 2011), it would be interesting to compare transcriptome data after HMGA gain-of-function with whole-genome chromatin immunoprecipitation sequencing (ChIP-seq) data after HMGA-ChIP to find out whether HMGA mediated regulation of CpG rich promoter is a general mechanism or it is only restricted to specific genes.
HMGA Role During Development
HMGA proteins are present at high levels in various undifferentiated tissues during embryonic development and their levels are strongly reduced, or almost absent in case of HMGA2, in the corresponding adult tissues (Pfannkuche et al., 2009). Hmga1 and Hmga2 are highly and ubiquitously expressed during early embryonic development (Chiappetta et al., 1996; Hirning-Folz et al., 1998). At later stages, Hmga1 expression becomes restricted to specific organs of all three germ layers, the ectoderm, mesoderm and endoderm. Although Hmga1 can be detected in most adult tissues, its expression is higher in testis, skeletal muscle, and thymus (Chiappetta et al., 1996). Hmga2 expression is mainly limited to the mesenchyme and is hardly detectable in healthy adult tissues, except testis, skeletal muscle and adipose tissues (Anand and Chada, 2000; Chieffi et al., 2002; Caron et al., 2005). HMGA proteins in adult organs have been implicated in maintaining and activating stem/progenitor cells in different tissues (Figure 2A) (Nishino et al., 2008; Li et al., 2012). When compared to wild-type embryonic stem (ES) cells, Hmga1−/− ES cells form fewer and smaller embryoid bodies (EBs) and show reduced capacity to differentiate to T lymphocytes (Caron et al., 2005). Hmga1−/− mice suffer from hematologic malignancies and cardiac hypertrophy resulting from a direct regulation of cardiomyocyte growth (Figure 2A) (Fedele et al., 2006a). In addition, Hmga1−/− mice show reduced insulin receptor expression leading to type 2 diabetes (Foti et al., 2005; Fedele et al., 2006a). The interaction of HMGA1 with retinoblastoma protein (RB1, also known as pRB) and CCAAT/enhancer-binding protein β (C/EBPβ) was shown to be important for proper regulation of genes required for adipocyte differentiation (Figure 2A) (Esposito et al., 2009). Transgenic mice carrying an Hmga1b truncated gene exhibit gigantism with an increased amount of the retroperitoneal and subcutaneous white adipose tissue, possibly due to activation of E2F transcription factor 1 (E2F1) pathway. These mice also develop B-cell lymphomas similar to that occurring in Hmga1−/− mice (Fedele et al., 2011).
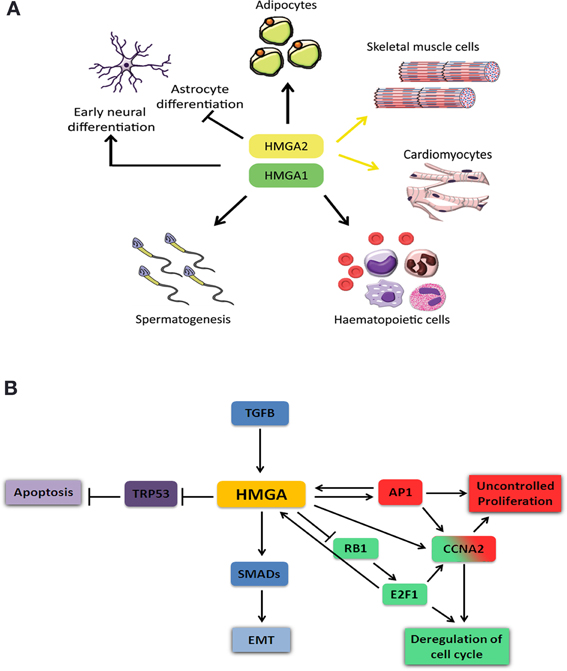
Figure 2. Schematic representation of HMGA role during cell differentiation and cancer. (A) HMGA proteins are required for proper development and progenitor cell differentiation to adipocyte, skeletal muscle, cardiac muscle, spermatozoids, and hematopoietic cells. They have a dual role in neuronal differentiation, where they are required during early neural progenitor differentiation but inhibit astrocyte differentiation at later stages. (B) HMGA proteins are involved in cellular processes that induce tumor formation and metastasis. Abnormal high expression of HMGA proteins results in tumor formation and metastasis. HMGA proteins deregulate cell cycle and thereby increase cell proliferation through either inhibition of RB1 and induction of E2F1 (green) or activation of AP1 and increased Cyclin A2 (Ccna2) expression (red). HMGA interaction with TRP53 blocks its activity and results in inhibition of apoptosis (purple). TGFB positively regulates HMGA expression (blue). HMGA interaction with SMADs leads to epithelial mesenchymal transition (EMT).
Hmga2−/− mice show a pygmy phenotype due to reduced expression of the insulin-like growth factor 2 mRNA binding protein 2 (Igf2bp2) gene (Zhou et al., 1995; Brants et al., 2004; Cleynen et al., 2007; Li et al., 2012). The Hmga2 deficient mice also show reduced fat tissue which is linked to a function of Hmga2 in pre-adipocyte precursor cell proliferation (Figure 2A) (Anand and Chada, 2000). In addition, Hmga2−/− embryonic fibroblasts have a proliferative defect. A recent study shows that Hmga2 is extremely important for the self-renewal potential of hematopoietic stem cells (HSCs, Figure 2A) (Copley et al., 2013). On the other hand, transgenic mice overexpressing a carboxyl-terminally truncated version of Hmga2 show a giant phenotype, are obese and develop lymphomas (Baldassarre et al., 2001; Fedele et al., 2001). It was shown that HMGA2 is necessary for the commitment of mouse embryonic stem cells to the skeletal muscle (Figure 2A) (Caron et al., 2005; Li et al., 2012) and cardiac muscle lineages (Figure 2A) (Monzen et al., 2008). HMGA proteins are also important for the proliferation of early stage neural precursor cells (NPCs) and for their neurogenic potential and overexpression of these genes can reprogram late stage NPCs into cells with early stage-specific capacities suppressing astrogenesis (Figure 2A) (Kishi et al., 2012). HMGA proteins are required for normal sperm development; both Hmga1−/− and Hmga2−/− mice show impaired spermatogenesis and the latter are sterile (Figure 2A) (Chieffi et al., 2002; Liu et al., 2003).
HMGA Proteins and Cancer
Hmga1 and Hmga2 are highly expressed in transformed cells in a variety of malignant and benign tumors of different origins (Fusco and Fedele, 2007; Hock et al., 2007). Transformation of a rat thyroid cell line (FRTL5) by the Kirsten murine sarcoma virus (KiMSV) increased sharply the level of phosphorylated HMGA proteins along with phosphorylated histone proteins H1 and H2A, and ubiquitinated H2A. Thus, the association of HMGA proteins with the neoplastic phenotype was demonstrated for the first time (Giancotti et al., 1985). Subsequently, a direct role of HMGA proteins in tumorigenesis was proven with a loss of function of HMGA2 in normal rat thyroid cells which prevented retrovirally induced neoplastic transformation (Berlingieri et al., 1995). More direct evidence of the tumorigenic potential of HMGA proteins came when normal cells in culture overexpressing these proteins underwent oncogenic transformation (Wood et al., 2000). For example, the human breast epithelial cell line MCF7 acquired the ability to form both primary and metastatic tumors in nude mice after overexpression of HMGA1 (Reeves et al., 2001). In general, there are accumulating reports supporting that HMGA proteins are required and sufficient for neoplastic transformation (Di Cello et al., 2008; Winslow et al., 2011; Morishita et al., 2013; Sun et al., 2013).
HMGA proteins contribute to tumor formation by inhibiting the apoptotic function of the transformation related protein 53 (TRP53, also known as P53) (Figure 2B). Two mechanisms have been reported for this inhibition. HMGA1 directly binds to and inactivates TRP53 thereby modulating the transcription of its target genes Cdkn1a (cyclin-dependent kinase inhibitor 1A, also known as p21), Bcl2 (B cell leukemia/lymphoma 2) and Bax (Bcl2-associated X protein and cyclin-dependent kinase inhibitor 1A), while enabling the active transcription of the TRP53 inhibitor Mdm2 (Frasca et al., 2006; Pierantoni et al., 2006; Esposito et al., 2010, 2012). The second mechanism is indirect and involves translocation of HIPK2 (homeodomain-interacting protein kinase 2) to the cytoplasm. HIPK2 phosphorylates p53 at S46 in the nucleus to induce apoptosis after non-repairable DNA damage has taken place (Pierantoni et al., 2007). The cytoplasmic translocation of HIPK2 abolishes the apoptotic function of TRP53.
Another mechanism by which HMGA proteins exert their tumorigenic activity is by interfering with cell cycle (Figure 2B). RB1 is one of the main cell cycle regulator proteins and keeps the E2F1 inactive in a complex with HDAC1 resulting in transcriptional repression. HMGA2 is able to bind RB1 and displace HDAC1 from the complex. Subsequent recruitment of the histone acetyl transferase EP300 (E1A binding protein p300, also known as P300) to the complex results in acetylation of DNA associated histones. E2F1 leads to activation of the protein complex and target gene transcription (Fedele et al., 2006b). The activation of E2F1 mediated by HMGA2 was shown to induce pituitary adenomas. Interestingly, a recent study has shown that human HMGA1 promoter is target of E2F1 and the deregulated RB1/E2F1 pathway might contribute to deregulation of HMGA1 in cancer (Figure 2B) (Massimi et al., 2013).
HMGA proteins interfere with cell cycle also by regulating the expression of Cyclin A2 (Ccna2) gene through the E2F1 (Schulze et al., 1995) and AP1 complexes (Casalino et al., 2007) (Figure 2B). AP1 complex can be composed of three Jun proteins (JUN, JUNB and JUND) and four Fos proteins (FOS, FOSB, FRA1, and FRA2). It was reported that HMGA protein levels increased in RAS-transformed rat thyroid cells, which in return increased the transcription of Junb and Fra1 (Vallone et al., 1997). In these transformed cells, AP1 complex is mainly composed of JUNB-FRA1 heterodimers, and this compositional change leads to increased transcriptional activity of the AP1 complex explaining the high levels of Ccna2.
During epithelial-mesenchymal transition (EMT), polarized epithelial cells undergo several biochemical processes that result in a transition to a mesenchymal cell phenotype (Thiery and Sleeman, 2006; Kalluri and Weinberg, 2009). These processes include altered cell-cell and cell-extracellular matrix (ECM) interactions and reorganization of the cytoskeleton, both leading to an increased ability of the cells to migrate. Although EMT occurs during normal embryonic development, it is also a key process during tumorigenesis and metastasis. The transforming growth factor beta (TGFB) signaling pathway is able to induce EMT and has been involved in cancer initiation and metastasis. TGFB increases the expression of Hmga2 by SMAD3 mediated inhibition of let-7, a microRNA that targets Hmga2 transcript (Lee and Dutta, 2007). HMGA2 was found to be required for TGFB signaling and to interact with several SMAD proteins which are mediators of the TGFB signaling (Figure 2B) (Thuault et al., 2006, 2008; Watanabe et al., 2009; Wu et al., 2011; Zha et al., 2012). Moreover, HMGA and SMADs have been shown to coregulate the expression of SNAIL1 (Protein snail homolog 1), which is the master effector for the induction of EMT in mammary epithelial cells (Thuault et al., 2008). Taking all these together, it will be of interest from the therapeutic point of view to investigate whether HMGA proteins are required for TGFB induced EMT during tumorigenesis and metastasis. Indeed, the multiple roles of TGFB signaling in tumor progression have promoted the development of therapeutic agents based on the inhibition of this signaling pathway (Golestaneh and Mishra, 2005; Ikushima et al., 2009). Interestingly, it has been recently shown that HMGA2 increased the expression of the TGFB type II receptor thereby enhancing TGFB signaling in epithelial carcinomas (Morishita et al., 2013).
LIN-28, a developmentally regulated RNA binding protein which is expressed at high levels in undifferentiated ES cells, has been shown to inhibit let-7 microRNA biogenesis (Thornton and Gregory, 2012).The balance between LIN-28 and let-7 affects the expression of Hmga2 and therefore is important for ES cell self-renewal and differentiation (Li et al., 2012; Copley et al., 2013) and maintenance of an undifferentiated state in cancer cells (Shell et al., 2007). Moreover, a recent study has shown that Hmga2 could promote cancer progression by acting as a competing endogeneous RNA (ceRNA) for let-7, adding another complexity to the mechanism by which Hmga2 can contribute to tumorigenesis (Kumar et al., 2013). LIN-28/let-7 axis also regulates glucose uptake (Zhu et al., 2011) and has the ability to reprogram cells toward glycolytic metabolism (Thornton and Gregory, 2012). This different metabolism, which is a hallmark of cancer, is known as the “Warburg Effect.”
Chromosomal rearrangements have also been reported in a variety of common benign tumors. Among these, rearrangements of the HMGA2 gene at 12q15 seem to be the most common rearrangements in human tumors of mesenchymal origin. The role of HMGA translocations in the formation of various human tumors has been previously reviewed (Fusco and Fedele, 2007; Wu and Wei, 2013).
Conclusions and Perspective
High mobility group (HMG) proteins are the most abundant non-histone chromatin associated proteins. Members of the HMGA subfamily contain a unique DNA-binding domain, the AT-hook, which can bind AT-rich DNA sequences with high affinity. HMGA proteins act as architectural transcription factors, which means that although they do not possess intrinsic transcriptional activity, they have the ability to modulate transcription of their target genes by altering the chromatin structure in different ways: displacement of histone H1 leading to chromatin decompaction, high-order chromatin rearrangements resulting in DNA looping that brings enhancers and promoter in close proximity, promoting assembly of regulatory nucleoprotein complexes, such as enhanceosomes and transcription initiation complexes, facilitating nucleosome remodeling and thereby accessibility of DNA to transcription factors and recruiting transcription factors to specific promoters.
HMGA proteins undergo several post-translational modifications, such as phosphorylation (Nissen et al., 1991; Wang et al., 1995; Schwanbeck et al., 2000; Pierantoni et al., 2001; Di Agostino et al., 2004; Zhang and Wang, 2007), methylation (Sgarra et al., 2003; Edberg et al., 2004, 2005; Sgarra et al., 2006), acetylation (Munshi et al., 1998, 2001; Zhang et al., 2007), sumoylation (Cao et al., 2008) and ribosylation (Elton and Reeves, 1986). Here, we have discussed only the role of phosphorylation of HMGA proteins mediated by the kinases DNA-PK, ATM and ATR at the SQ sites during transcriptional regulation. The biological relevance of the other modifications is not well understood and requires more attention. It would be interesting to analyze also the other post-translational modifications to understand the molecular mechanisms underlying their role in fine-tuning the interaction of HMGA proteins with other transcription factors, with the chromatin and with the DNA.
HMGA proteins are present at high levels in various undifferentiated tissues and several studies confirmed their central role during normal embryonic development. They contribute to the plasticity of ES cells by maintaining an open chromatin state evicting linker histone H1 which facilitates formation of nucleoprotein structures at specific promoters (Pfannkuche et al., 2009; Kishi et al., 2012). Hence, it is not surprising that HMGA protein levels are developmentally regulated; they are strongly reduced or almost absent in adult organs, where they have been implicated in maintaining and activating stem/progenitor cells in different tissues. Regeneration of adult organs during normal homeostatic turnover or after injury requires a proper balance between self-renewal and differentiation of tissue-specific progenitor cells. On the other hand, re-expression of both Hmga1 and Hmga2 at high levels in the adult stage is seen in a variety of malignant and benign tumors, which might be in part due to their role in transcription activation of certain cell proliferation genes. Characterization of the role of HMGA proteins in the regulatory mechanisms controlling the proper balance between expansion and differentiation of progenitor cells will have a profound impact on our understanding and treatment of diseases originated from improper regulation of HMGA proteins, such as cancer, diabetes and idiopathic pulmonary fibrosis (Pandit et al., 2010).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Adriana Contreras and Julio Cordero for helpful discussion. Guillermo Barreto is funded by the “LOEWE-Initiative der Landesförderung” (Wiesbaden, Germany) (III L 4—518/15.004 2009) and the “Deutsche Forschungsgemeinschaft” (DFG, Bonn, Germany) (BA 4036/1-1).
References
Anand, A., and Chada, K. (2000). In vivo modulation of Hmgic reduces obesity. Nat. Genet. 24, 377–380. doi: 10.1038/74207
Antequera, F. (2003). Structure, function and evolution of CpG island promoters. Cell. Mol. Life Sci. 60, 1647–1658. doi: 10.1007/s00018-003-2185-x
Baldassarre, G., Fedele, M., Battista, S., Vecchione, A., Klein-Szanto, A. J. P., Santoro, M., et al. (2001). Onset of natural killer cell lymphomas in transgenic mice carrying a truncated HMGI-C gene by the chronic stimulation of the IL-2 and IL-15 pathway. Proc. Natl. Acad. Sci. U.S.A. 98, 7970–7975. doi: 10.1073/pnas.141224998
Belotserkovskaya, R., Oh, S., Bondarenko, V. A., Orphanides, G., Studitsky, V. M., and Reinberg, D. (2003). FACT facilitates transcription-dependent nucleosome alteration. Science 301, 1090–1093. doi: 10.1126/science.1085703
Berlingieri, M. T., Manfioletti, G., Santoro, M., Bandiera, A., Visconti, R., Giancotti, V., et al. (1995). Inhibition of Hmgi-C protein-synthesis suppresses retrovirally induced neoplastic transformation of rat-thyroid cells. Mol. Cell. Biol. 15, 1545–1553.
Bouallaga, I., Massicard, S., Yaniv, M., and Thierry, F. (2000). An enhanceosome containing the Jun B/Fra-2 heterodimer and the HMG-I(Y) architectural protein controls HPV 18 transcription. EMBO Rep. 1, 422–427. doi: 10.1093/embo-reports/kvd091
Bouallaga, I., Teissier, S., Yaniv, M., and Thierry, F. (2003). HMG-I(Y) and the CBP/p300 coactivator are essential for human papillomavirus type 18 enhanceosome transcriptional activity. Mol. Cell. Biol. 23, 2329–2340. doi: 10.1128/Mcb.23.7.2329-2340.2003
Brants, J. R., Ayoubi, T. A. Y., Chada, K., Marchal, K., Van De Ven, W. J. M., and Petit, M. M. R. (2004). Differential regulation of the insulin-like growth factor II mRNA-binding protein genes by architectural transcription factor HMGA2. FEBS Lett. 569, 277–283. doi: 10.1016/j.febslet.2004.05.075
Bustin, M. (2001). Revised nomenclature for high mobility group (HMG) chromosomal proteins. Trends Biochem. Sci. 26, 152–153. doi: 10.1016/S0968-0004(00)01777-1
Butler, J. E. F., and Kadonaga, J. T. (2002). The RNA polymerase II core promoter: a key component in the regulation of gene expression. Genes Dev. 16, 2583–2592. doi: 10.1101/Gad.1026202
Cao, X., Clavijo, C., Li, X., Lin, H. H., Chen, Y., Shih, H. M., et al. (2008). SUMOylation of HMGA2: selective destabilization of promyelocytic leukemia protein via proteasome. Mol. Cancer Ther. 7, 923–934. doi: 10.1158/1535-7163.mct-07-0540
Caron, L., Bost, F., Prot, M., Hofman, P., and Binetruy, B. (2005). A new role for the oncogenic high-mobility group A2 transcription factor in myogenesis of embryonic stem cells. Oncogene 24, 6281–6291. doi: 10.1038/sj.onc.1208781
Casalino, L., Bakiri, L., Talotta, F., Weitzman, J. B., Fusco, A., Yaniv, M., et al. (2007). Fra-1 promotes growth and survival in RAS-transformed thyroid cells by controlling cyclin A transcription. EMBO J. 26, 1878–1890. doi: 10.1038/sj.emboj.7601617
Catez, F., and Hock, R. (2010). Binding and interplay of HMG proteins on chromatin: Lessons from live cell imaging. Biochim. Biophys. Acta Gene Regul. Mech. 1799, 15–27. doi: 10.1016/j.bbagrm.2009.11.001
Catez, F., Yang, H., Tracey, K. J., Reeves, R., Misteli, T., and Bustin, M. (2004). Network of dynamic interactions between histone H1 and high-mobility-group proteins in chromatin. Mol. Cell. Biol. 24, 4321–4328. doi: 10.1128/MCB.24.10.4321-4328.2004
Chen, B., Young, J., and Leng, F. F. (2010). DNA bending by the mammalian high-mobility group protein AT Hook 2. Biochemistry 49, 1590–1595. doi: 10.1021/Bi901881c
Chiappetta, G., Avantaggiato, V., Visconti, R., Fedele, M., Battista, S., Trapasso, F., et al. (1996). High level expression of the HMGI (Y) gene during embryonic development. Oncogene 13, 2439–2446.
Chieffi, P., Battista, S., Barchi, M., Di Agostino, S., Pierantoni, G. M., Fedele, M., et al. (2002). HMGA1 and HMGA2 protein expression in mouse spermatogenesis. Oncogene 21, 3644–3650. doi: 10.1038/sj.onc.1205501
Cleynen, I., Brants, J. R., Peeters, K., Deckers, R., Debiec-Rychter, M., Sciot, R., et al. (2007). HMGA2 regulates transcription of the Imp2 gene via an intronic regulatory element in cooperation with nuclear factor-kappaB. Mol. Cancer Res. 5, 363–372. doi: 10.1158/1541-7786.MCR-06-0331
Cleynen, I., and Van De Ven, W. J. (2008). The HMGA proteins: a myriad of functions (Review). Int. J. Oncol. 32, 289–305. Available online at: http://www.spandidos-publications.com/ijo/32/2/289
Contreras, A., Hale, T. K., Stenoien, D. L., Rosen, J. M., Mancini, M. A., and Herrera, R. E. (2003). The dynamic mobility of histone H1 is regulated by cyclin/CDK phosphorylation. Mol. Cell. Biol. 23, 8626–8636. doi: 10.1128/Mcb.23.23.8626-8636.2003
Copley, M. R., Babovic, S., Benz, C., Knapp, D. J., Beer, P. A., Kent, D. G., et al. (2013). The Lin28b-let-7-Hmga2 axis determines the higher self-renewal potential of fetal haematopoietic stem cells. Nat. Cell Biol. 15, 916–925. doi: 10.1038/ncb2783
Cui, T. J., and Leng, F. F. (2007). Specific recognition of AT-Rich DNA sequences by the mammalian high mobility group-protein AT-hook 2: a SELEX study. Biochemistry 46, 13059–13066. doi: 10.1021/Bi701269s
Deaton, A. M., and Bird, A. (2011). CpG islands and the regulation of transcription. Genes Dev. 25, 1010–1022. doi: 10.1101/Gad.2037511
Di Agostino, S., Fedele, M., Chieffi, P., Fusco, A., Rossi, P., Geremia, R., et al. (2004). Phosphorylation of high-mobility group protein A2 by Nek2 kinase during the first meiotic division in mouse spermatocytes. Mol. Biol. Cell 15, 1224–1232. doi: 10.1091/mbc.E03-09-0638
Di Cello, F., Hillion, J., Hristov, A., Wood, L. J., Mukherjee, M., Schuldenfrei, A., et al. (2008). HMGA2 participates in transformation in human lung cancer. Mol. Cancer Res. 6, 743–750. doi: 10.1158/1541-7786.Mcr-07-0095
Edberg, D. D., Adkins, J. N., Springer, D. L., and Reeves, R. (2005). Dynamic and differential in vivo modifications of the isoform HMGA1a and HMGA1b chromatin proteins. J. Biol. Chem. 280, 8961–8973. doi: 10.1074/jbc.M407348200
Edberg, D. D., Bruce, J. E., Siems, W. F., and Reeves, R. (2004). In vivo posttranslational modifications of the high mobility group A1a proteins in breast cancer cells of differing metastatic potential. Biochemistry 43, 11500–11515. doi: 10.1021/bi049833i
Elton, T. S., and Reeves, R. (1986). Purification and postsynthetic modifications of Friend erythroleukemic cell high mobility group protein HMG-I. Anal. Biochem. 157, 53–62.
Esposito, F., Pierantoni, G. M., Battista, S., Melillo, R. M., Scala, S., Chieffi, P., et al. (2009). Interaction between HMGA1 and retinoblastoma protein is required for adipocyte differentiation. J. Biol. Chem. 284, 25993–26004. doi: 10.1074/jbc.M109.034280
Esposito, F., Tornincasa, M., Chieffi, P., De Martino, I., Pierantoni, G. M., and Fusco, A. (2010). High-Mobility Group A1 proteins regulate p53-mediated transcription of Bcl-2 Gene. Cancer Res. 70, 5379–5388. doi: 10.1158/0008-5472.Can-09-4199
Esposito, F., Tornincasa, M., Federico, A., Chiappetta, G., Pierantoni, G. M., and Fusco, A. (2012). High-mobility group A1 protein inhibits p53-mediated intrinsic apoptosis by interacting with Bcl-2 at mitochondria. Cell Death Dis. 3:e383. doi: 10.1038/cddis.2012.126
Evans, J. N., Zajicek, J., Nissen, M. S., Munske, G., Smith, V., and Reeves, R. (1995). 1H and 13C NMR assignments and molecular modelling of a minor groove DNA-binding peptide from the HMG-I protein. Int. J. Pept. Protein Res. 45, 554–560.
Fedele, M., Battista, S., Manfioletti, G., Croce, C. M., Giancotti, V., and Fusco, A. (2001). Role of the high mobility group A proteins in human lipomas. Carcinogenesis 22, 1583–1591. doi: 10.1093/carcin/22.10.1583
Fedele, M., Fidanza, V., Battista, S., Pentimalli, F., Klein-Szanto, A. J. P., Visone, R., et al. (2006a). Haploinsufficiency of the Hmga1 gene causes cardiac hypertrophy and myelo-lymphoproliferative disorders in mice. Cancer Res. 66, 2536–2543. doi: 10.1158/0008-5472.Can-05-1889
Fedele, M., Visone, R., De Martino, I., Palmieri, D., Valentino, T., Esposito, F., et al. (2011). Expression of a truncated Hmga1b gene induces gigantism, lipomatosis and B-cell lymphomas in mice. Eur. J. Cancer 47, 470–478. doi: 10.1016/j.ejca.2010.09.045
Fedele, M., Visone, R., De Martino, I., Troncone, G., Palmieri, D., Battista, S., et al. (2006b). HMGA2 induces pituitary tumorigenesis by enhancing E2F1 activity. Cancer Cell 9, 459–471. doi: 10.1016/j.ccr.2006.04.024
Foti, D., Chiefari, E., Fedele, M., Iuliano, R., Brunetti, L., Paonessa, F., et al. (2005). Lack of the architectural factor HMGA1 causes insulin resistance and diabetes in humans and mice. Nat. Med. 11, 765–773. doi: 10.1038/nm1254
Foti, D., Iuliano, R., Chiefari, E., and Brunetti, A. (2003). A nucleoprotein complex containing Sp1, C/EBP beta, and HMGI-Y controls human insulin receptor gene transcription. Mol. Cell. Biol. 23, 2720–2732. doi: 10.1128/MCB.23.8.2720-2732.2003
Frasca, F., Rustighi, A., Malaguarnera, R., Altamura, S., Vigneri, P., Del Sal, G., et al. (2006). HMGA1 inhibits the function of p53 family members in thyroid cancer cells. Cancer Res. 66, 2980–2989. doi: 10.1158/0008-5472.Can-05-2637
French, S. W., Schmidt, M. C., and Glorioso, J. C. (1996). Involvement of a high-mobility-group protein in the transcriptional activity of herpes simplex virus latency-active promoter 2. Mol. Cell. Biol. 16, 5393–5399.
Fusco, A., and Fedele, M. (2007). Roles of HMGA proteins in cancer. Nat. Rev. Cancer 7, 899–910. doi: 10.1038/Nrc2271
Giancotti, V., Berlingieri, M. T., Difiore, P. P., Fusco, A., Vecchio, G., and Crane-Robinson, C. (1985). Changes in nuclear proteins on transformation of rat epithelial thyroid cells by a murine sarcoma retrovirus. Cancer Res. 45, 6051–6057.
Gohlke, J., Scholz, C. J., Kneitz, S., Weber, D., Fuchs, J., Hedrich, R., et al. (2013). DNA methylation mediated control of gene expression is critical for development of crown gall tumors. PLoS Genet. 9:e1003267. doi: 10.1371/journal.pgen.1003267
Golestaneh, N., and Mishra, B. (2005). TGF-beta, neuronal stem cells and glioblastoma. Oncogene 24, 5722–5730. doi: 10.1038/sj.onc.1208925
Ha, M., Ng, D. W. K., Li, W. H., and Chen, Z. J. (2011). Coordinated histone modifications are associated with gene expression variation within and between species. Genome Res. 21, 590–598. doi: 10.1101/gr.116467.110
Hammond, S. M., and Sharpless, N. E. (2008). HMGA2, MicroRNAs, and Stem Cell Aging. Cell 135, 1013–1016. doi: 10.1016/j.cell.2008.11.026
Hirning-Folz, U., Wilda, M., Rippe, V., Bullerdiek, J., and Hameister, H. (1998). The expression pattern of the Hmgic gene during development. Genes Chromosomes Cancer 23, 350–357. doi: 10.1002/(Sici)1098-2264(199812)23:4<350::Aid-Gcc10>3.0.Co;2-E
Hock, R., Furusawa, T., Ueda, T., and Bustin, M. (2007). HMG chromosomal proteins in development and disease. Trends Cell Biol. 17, 72–79. doi: 10.1016/j.tcb.2006.12.001
Ikushima, H., Todo, T., Ino, Y., Takahashi, M., Miyazawa, K., and Miyazono, K. (2009). Autocrine TGF-beta signaling maintains tumorigenicity of glioma-initiating cells through Sry-related HMG-box factors. Cell Stem Cell 5, 504–514. doi: 10.1016/j.stem.2009.08.018
Ju, B. G., Lunyak, V. V., Perissi, V., Garcia-Bassets, I., Rose, D. W., Glass, C. K., et al. (2006). A topoisomerase II beta-mediated dsDNA break required for regulated transcription. Science 312, 1798–1802. doi: 10.1126/science.1127196
Kalluri, R., and Weinberg, R. A. (2009). The basics of epithelial-mesenchymal transition. J. Clin. Invest. 119, 1420–1428. doi: 10.1172/Jci39104
Karlic, R., Chung, H. R., Lasserre, J., Vlahovicek, K., and Vingron, M. (2010). Histone modification levels are predictive for gene expression. Proc. Natl. Acad. Sci. U.S.A. 107, 2926–2931. doi: 10.1073/pnas.0909344107
Kishi, Y., Fujii, Y., Hirabayashi, Y., and Gotoh, Y. (2012). HMGA regulates the global chromatin state and neurogenic potential in neocortical precursor cells. Nat. Neurosci. 15, 1127–1133. doi: 10.1038/Nn.3165
Kumar, M. S., Armenteros-Monterroso, E., East, P., Chakravorty, P., Matthews, N., Winslow, M. M., et al. (2013). HMGA2 functions as a competing endogenous RNA to promote lung cancer progression. Nature 505, 212–217. doi: 10.1038/nature12785
Landolin, J. M., Johnson, D. S., Trinklein, N. D., Aldred, S. F., Medina, C., Shulha, H., et al. (2010). Sequence features that drive human promoter function and tissue specificity. Genome Res. 20, 890–898. doi: 10.1101/gr.100370.109
Lee, Y. S., and Dutta, A. (2007). The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev. 21, 1025–1030. doi: 10.1101/Gad.1540407
Lehn, D. A., Elton, T. S., Johnson, K. R., and Reeves, R. (1988). A conformational study of the sequence specific binding of Hmg-I (Y) with the Bovine Interleukin-2 Cdna. Biochem. Int. 16, 963–971.
Levine, M., and Tjian, R. (2003). Transcription regulation and animal diversity. Nature 424, 147–151. doi: 10.1038/Nature01763
Li, O., Vasudevan, D., Davey, C. A., and Droge, P. (2006). High-level expression of DNA architectural factor HMGA2 and its association with nucleosomes in human embryonic stem cells. Genesis 44, 523–529. doi: 10.1002/dvg.20242
Li, Z. Z., Gilbert, J. A., Zhang, Y. Y., Zhang, M. S., Qiu, Q., Ramanujan, K., et al. (2012). An HMGA2-IGF2BP2 Axis regulates myoblast proliferation and myogenesis. Dev. Cell 23, 1176–1188. doi: 10.1016/j.devcel.2012.10.019
Liu, J., Schiltz, J. F., Ashar, H. R., and Chada, K. K. (2003). Hmga1 is required for normal sperm development. Mol. Reprod. Dev. 66, 81–89. doi: 10.1002/mrd.10323
Malini, E., Maurizio, E., Bembich, S., Sgarra, R., Edomi, P., and Manfioletti, G. (2011). HMGA Interactome: new insights from phage display technology. Biochemistry 50, 3462–3468. doi: 10.1021/Bi200101f
Massimi, I., Guerrieri, F., Petroni, M., Veschi, V., Truffa, S., Screpanti, I., et al. (2013). The HMGA1 protoncogene frequently deregulated in cancer is a transcriptional target of E2F1. Mol. Carcinog. 52, 526–534. doi: 10.1002/Mc.21887
Monzen, K., Ito, Y., Naito, A. T., Kasai, H., Hiroi, Y., Hayashi, D., et al. (2008). A crucial role of a high mobility group protein HMGA2 in cardiogenesis. Nat. Cell Biol. 10, 567–574. doi: 10.1038/Ncb1719
Morishita, A., Zaidi, M. R., Mitoro, A., Sankarasharma, D., Szabolcs, M., Okada, Y., et al. (2013). HMGA2 is a driver of tumor metastasis. Cancer Res. 73, 4289–4299. doi: 10.1158/0008-5472.CAN-12-3848
Munshi, N., Agalioti, T., Lomvardas, S., Merika, M., Chen, G. Y., and Thanos, D. (2001). Coordination of a transcriptional switch by HMGI(Y) acetylation. Science 293, 1133–1136. doi: 10.1126/science.293.5532.1133
Munshi, N., Merika, M., Yie, J., Senger, K., Chen, G., and Thanos, D. (1998). Acetylation of HMG I(Y) by CBP turns off IFN beta expression by disrupting the enhanceosome. Mol. Cell 2, 457–467
Munshi, N., Yie, Y., Merika, M., Senger, K., Lomvardas, S., Agalioti, T., et al. (1999). The IFN-beta enhancer: a paradigm for understanding activation and repression of inducible gene expression. Cold Spring Harb. Symp. Quant. Biol. 64, 149–159.
Natarajan, S., Hombach-Klonisch, S., Droge, P., and Klonisch, T. (2013). HMGA2 Inhibits Apoptosis through Interaction with ATR-CHK1 Signaling Complex in Human Cancer Cells. Neoplasia 15, 263–280. doi: 10.1593/Neo.121988.
Nishino, J., Kim, I., Chada, K., and Morrison, S. J. (2008). Hmga2 promotes neural stem cell self-renewal in young but not old mice by reducing p16Ink4a and p19Arf Expression. Cell 135, 227–239. doi: 10.1016/j.cell.2008.09.017
Nissen, M. S., Langan, T. A., and Reeves, R. (1991). Phosphorylation by cdc2 kinase modulates DNA binding activity of high mobility group I nonhistone chromatin protein. J. Biol. Chem. 266, 19945–19952.
Palmieri, D., Valentino, T., D'angelo, D., De Martino, I., Postiglione, I., Pacelli, R., et al. (2011). HMGA proteins promote ATM expression and enhance cancer cell resistance to genotoxic agents. Oncogene 30, 3024–3035. doi: 10.1038/Onc.2011.21
Pandit, K. V., Corcoran, D., Yousef, H., Yarlagadda, M., Tzouvelekis, A., Gibson, K. F., et al. (2010). Inhibition and Role of let-7d in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 182, 220–229. doi: 10.1164/rccm.200911-1698OC
Panne, D. (2008). The enhanceosome. Curr. Opin. Struct. Biol. 18, 236–242. doi: 10.1016/j.sbi.2007.12.002
Panne, D., Maniatis, T., and Harrison, S. C. (2007). An atomic model of the interferon-beta enhanceosome. Cell 129, 1111–1123. doi: 10.1016/j.cell.2007.05.019
Pentimalli, F., Palmieri, D., Pacelli, R., Garbi, C., Cesari, R., Martin, E., et al. (2008). HMGA1 protein is a novel target of the ATM kinase. Eur. J. Cancer 44, 2668–2679. doi: 10.1016/j.ejca.2008.07.033
Pfannkuche, K., Summer, H., Li, O., Hescheler, J., and Droge, P. (2009). The High Mobility Group Protein HMGA2: a co-regulator of chromatin structure and pluripotency in stem cells? Stem Cell Rev. Rep. 5, 224–230. doi: 10.1007/s12015-009-9078-9
Pierantoni, G. M., Fedele, M., Pentimalli, F., Benvenuto, G., Pero, R., Viglietto, G., et al. (2001). High mobility group I (Y) proteins bind HIPK2, a serine-threonine kinase protein which inhibits cell growth. Oncogene 20, 6132–6141. doi: 10.1038/sj.onc.1204635
Pierantoni, G. M., Rinaldo, C., Esposito, F., Mottolese, M., Soddu, S., and Fusco, A. (2006). High Mobility Group A1 (HMGA1) proteins interact with p53 and inhibit its apoptotic activity. Cell Death Differ. 13, 1554–1563. doi: 10.1038/sj.cdd.4401839
Pierantoni, G. M., Rinaldo, C., Mottolese, M., Di Benedetto, A., Esposito, F., Soddu, S., et al. (2007). High-mobility group A1 inhibits p53 by cytoplasmic relocalization of its proapoptotic activator HIPK2. J. Clin. Invest. 117, 693–702. doi: 10.1172/Jc129852
Ramirez-Carrozzi, V. R., Braas, D., Bhatt, D. M., Cheng, C. S., Hong, C., Doty, K. R., et al. (2009). A unifying model for the selective regulation of inducible transcription by CpG Islands and nucleosome remodeling. Cell 138, 114–128. doi: 10.1016/j.cell.2009.04.020
Reeves, R. (2001). Molecular biology of HMGA proteins: hubs of nuclear function. Gene 277, 63–81. doi: 10.1016/S0378-1119(01)00689-8
Reeves, R. (2010). Nuclear functions of the HMG proteins. Biochim. Biophys. Acta Gene Regul. Mech. 1799, 3–14. doi: 10.1016/j.bbagrm.2009.09.001
Reeves, R., Edberg, D. D., and Li, Y. (2001). Architectural transcription factor HMGI(Y) promotes tumor progression and mesenchymal transition of human epithelial cells. Mol. Cell. Biol. 21, 575–594. doi: 10.1128/Mcb.21.2.575-594.2001
Ren, J., Wen, L., Gao, X., Jin, C., Xue, Y., and Yao, X. (2009). DOG 1.0: illustrator of protein domain structures. Cell Res. 19, 271–273. doi: 10.1038/cr.2009.6
Schulze, A., Zerfass, K., Spitkovsky, D., Middendorp, S., Berges, J., Helin, K., et al. (1995). Cell-cycle regulation of the cyclin-a gene promoter is mediated by a variant E2f site. Proc. Natl. Acad. Sci. U.S.A. 92, 11264–11268. doi: 10.1073/pnas.92.24.11264
Schwabish, M. A., and Struhl, K. (2006). Asf1 mediates histone eviction and deposition during elongation by RNA polymerase II. Mol. Cell 22, 415–422. doi: 10.1016/j.molcel.2006.03.014
Schwanbeck, R., Manfioletti, G., and Wisniewski, J. R. (2000). Architecture of high mobility group protein I-C.DNA complex and its perturbation upon phosphorylation by Cdc2 kinase. J. Biol. Chem. 275, 1793–1801. doi: 10.1074/jbc.275.3.1793
Sgarra, R., Diana, F., Bellarosa, C., Dekleva, V., Rustighi, A., Toller, M., et al. (2003). During apoptosis of tumor cells HMGA1a protein undergoes methylation: identification of the modification site by mass spectrometry. Biochemistry 42, 3575–3585. doi: 10.1021/Bi0273381
Sgarra, R., Lee, J., Tessari, M. A., Altamura, S., Spolaore, B., Giancotti, V., et al. (2006). The AT-hook of the chromatin architectural transcription factor high mobility group A1a is arginine-methylated by protein arginine methyltransferase 6. J. Biol. Chem. 281, 3764–3772. doi: 10.1074/jbc.M510231200
Sgarra, R., Rustighi, A., Tessari, M. A., Di Bernardo, J., Altamura, S., Fusco, A., et al. (2004). Nuclear phosphoproteins HMGA and their relationship with chromatin structure and cancer. FEBS Lett. 574, 1–8. doi: 10.1016/j.febslet.2004.08.013
Shell, S., Park, S. M., Radjabi, A. R., Schickel, R., Kistner, E. O., Jewell, D. A., et al. (2007). Let-7 expression defines two differentiation stages of cancer. Proc. Natl. Acad. Sci. U.S.A. 104, 11400–11405. doi: 10.1073/pnas.0704372104
Snijders, A. P., Pongdam, S., Lambert, S. J., Wood, C. M., Baldwin, J. P., and Dickman, M. J. (2008). Characterization of post-translational modifications of the linker histones H1 and H5 from chicken erythrocytes using mass spectrometry. J. Proteome Res. 7, 4326–4335. doi: 10.1021/pr800260a
Sun, M., Song, C. X., Huang, H., Frankenberger, C. A., Sankarasharma, D., Gomes, S., et al. (2013). HMGA2/TET1/HOXA9 signaling pathway regulates breast cancer growth and metastasis. Proc. Natl. Acad. Sci. U.S.A. 110, 9920–9925. doi: 10.1073/pnas.1305172110
Thiery, J. P., and Sleeman, J. P. (2006). Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 7, 131–142. doi: 10.1038/nrm1835
Thornton, J. E., and Gregory, R. I. (2012). How does Lin28 let-7 control development and disease? Trends Cell Biol. 22, 474–482. doi: 10.1016/j.tcb.2012.06.001
Thuault, S., Tan, E. J., Peinado, H., Cano, A., Heldin, C. H., and Moustakas, A. (2008). HMGA2 and Smads Co-regulate SNAIL1 expression during induction of epithelial-to-mesenchymal transition. J. Biol. Chem. 283, 33437–33446. doi: 10.1074/jbc.M802016200
Thuault, S., Valcourt, U., Petersen, M., Manfioletti, G., Heldin, C. H., and Moustakas, A. (2006). Transforming growth factor-beta employs HMGA2 to elicit epithelial-mesenchymal transition. J. Cell Biol. 174, 175–183. doi: 10.1083/jcb.200512110
Vallone, D., Battista, S., Pierantoni, G. M., Fedele, M., Casalino, L., Santoro, M., et al. (1997). Neoplastic transformation of rat thyroid cells requires the junB and fra-1 gene induction which is dependent on the HMGI-C gene product. EMBO J. 16, 5310–5321. doi: 10.1093/emboj/16.17.5310
Wang, D. Z., Ray, P., and Boothby, M. (1995). Interleukin 4-inducible phosphorylation of HMG-I(Y) is inhibited by rapamycin. J. Biol. Chem. 270, 22924–22932.
Watanabe, S., Ueda, Y., Akaboshi, S., Hino, Y., Sekita, Y., and Nakao, M. (2009). HMGA2 Maintains Oncogenic RAS-Induced epithelial-mesenchymal transition in human pancreatic cancer cells. Am. J. Pathol. 174, 854–868. doi: 10.2353/ajpath.2009.080523
Winslow, M. M., Dayton, T. L., Verhaak, R. G. W., Kim-Kiselak, C., Snyder, E. L., Feldser, D. M., et al. (2011). Suppression of lung adenocarcinoma progression by Nkx2-1. Nature 473, U101–U120. doi: 10.1038/Nature09881
Winter, N., Nimzyk, R., Bosche, C., Meyer, A., and Bullerdiek, J. (2011). Chromatin Immunoprecipitation to Analyze DNA Binding Sites of HMGA2. PLoS ONE 6:e18837. doi: 10.1371/journal.pone.0018837
Wood, L. J., Maher, J. F., Bunton, T. E., and Resar, L. M. S. (2000). The oncogenic properties of the HMG-I gene family. Cancer Res. 60, 4256–4261.
Wu, J. J., and Wei, J. J. (2013). HMGA2 and high-grade serous ovarian carcinoma. J. Mol. Med. 91, 1155–1165. doi: 10.1007/s00109-013-1055-8
Wu, J. J., Liu, Z. J., Shao, C. S., Gong, Y. Q., Hernando, E., Lee, P., et al. (2011). HMGA2 Overexpression-induced ovarian surface epithelial transformation is mediated through regulation of EMT Genes. Cancer Res. 71, 349–359. doi: 10.1158/0008-5472.Can-10-2550
Xu, M. Y., Sharma, P., Pan, S. Q., Malik, S., Roeder, R. G., and Martinez, E. (2011). Core promoter-selective function of HMGA1 and mediator in initiator-dependent transcription. Genes Dev. 25, 2513–2524. doi: 10.1101/gad.177360.111
Yie, J., Merika, M., Munshi, N., Chen, G., and Thanos, D. (1999). The role of HMG I(Y) in the assembly and function of the IFN-beta enhanceosome. EMBO J. 18, 3074–3089. doi: 10.1093/emboj/18.11.3074
Zha, L., Wang, Z. W., Tang, W. X., Zhang, N., Liao, G., and Huang, Z. (2012). Genome-wide analysis of HMGA2 transcription factor binding sites by ChIP on chip in gastric carcinoma cells. Mol. Cell. Biochem. 364, 243–251. doi: 10.1007/s11010-012-1224-z
Zhang, Q., and Wang, Y. (2007). Homeodomain-interacting protein kinase-2 (HIPK2) phosphorylates HMGA1a at Ser-35, Thr-52, and Thr-77 and modulates its DNA binding affinity. J. Proteome Res. 6, 4711–4719. doi: 10.1021/pr700571d
Zhang, Q., Zhang, K., Zou, Y., Perna, A., and Wang, Y. (2007). A quantitative study on the in vitro and in vivo acetylation of high mobility group A1 proteins. J. Am. Soc. Mass Spectrom. 18, 1569–1578. doi: 10.1016/j.jasms.2007.05.020
Zhang, Y., Fatima, N., and Dufau, M. L. (2005). Coordinated changes in DNA methylation and histone modifications regulate silencing/derepression of luteinizing hormone receptor gene transcription. Mol. Cell. Biol. 25, 7929–7939. doi: 10.1128/Mcb.25.18.7929-7939.2005
Zhao, K., Kas, E., Gonzalez, E., and Laemmli, U. K. (1993). Sar-dependent mobilization of histone H1 by Hmg-I/Y in-Vitro - Hmg-I/Y Is Enriched in H1-depleted chromatin. EMBO J. 12, 3237–3247.
Zhou, X. J., Benson, K. F., Ashar, H. R., and Chada, K. (1995). Mutation Responsible for the Mouse Pygmy Phenotype in the Developmentally-Regulated Factor Hmgi-C. Nature 376, 771–774. doi: 10.1038/376771a0
Keywords: HMGA, chromatin structure, transcription, development and cancer
Citation: Ozturk N, Singh I, Mehta A, Braun T and Barreto G (2014) HMGA proteins as modulators of chromatin structure during transcriptional activation. Front. Cell Dev. Biol. 2:5. doi: 10.3389/fcell.2014.00005
Received: 02 December 2013; Paper pending published: 08 December 2013;
Accepted: 07 February 2014; Published online: 06 March 2014.
Edited by:
Michael E. Symonds, The University of Nottingham, UKReviewed by:
Huabing Li, Yale University School of Medicine, USAPeter Droge, Nanyang Technological University, Singapore
Kailin Yang, Cleveland Clinic, USA
Copyright © 2014 Ozturk, Singh, Mehta, Braun and Barreto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guillermo Barreto, Lung Cancer Epigenetics Group, Max-Planck Institute of Heart and Lung Research, Parkstrasse 1, Bad Nauheim 61231, Germany e-mail:Z3VpbGxlcm1vLmJhcnJldG9AbXBpLWJuLm1wZy5kZQ==
†Member of the Universities of Giessen and Marburg Lung Center (UGMLC) and the German Center of Lung Research (DZL).