 Matthias Gaestel
Matthias Gaestel- Department of Biochemistry, Hannover Medical University, Hannover, Germany
Downstream of MAPKs, such as classical/atypical ERKs and p38 MAPKs, but not of JNKs, signaling is often mediated by protein kinases which are phosphorylated and activated by MAPKs and, therefore, designated MAPK-activated protein kinases (MAPKAPKs). Recently, novel insights into the specificity of the assembly of MAPK/MAPKAPK hetero-dimeric protein kinase signaling complexes have been gained. In addition, new functional aspects of MKs have been described and established functions have been challenged. This short review will summarize recent developments including the linear motif (LM) in MKs, the ERK-independent activation of RSK, the RSK-independent effects of some RSK-inhibitors and the challenged role of MK5/PRAK in tumor suppression.
Introduction
Besides phosphorylation of other substrates, ERKs and p38 MAPKs are able to signal further downstream by the activation of so called MAPK-activated protein kinases (MAPKAPKs) (reviewed in Cargnello and Roux, 2011). These downstream kinases are the p90 ribosomal-S6-kinases (RSK1-3), the mitogen- and stress-activated protein kinases MSK1/2, the MAPK-interacting kinases MNK1/2 and the MAPKAP kinases MK2, MK3 and MK5/PRAK (Gaestel, 2006). Specific signaling complexes between MAPK and their target MAPKAPKs exist and are the structural basis for the functional downstream-extension of MAPK cascades. Canonical activation pathways have been defined for the exclusive activation of RSKs by ERK1/2, the exclusive activation of MK2/3 by p38α/β as well as the more promiscuous activation of MNKs and MSKs by both ERKs and p38 and of MK5/PRAK by p38β/ERK3/4. Here, I will discuss novel findings regarding the molecular basis of specific and productive signaling complexes between MAPKs and MAPKAPKs, the non-canonical activation of RSKs and recent challenges arising from off target effects of the widely used RSK inhibitors SL0101 and BI-D1870. Furthermore, the challenge of the anticipated tumor-suppressive function of MK5/PRAK is discussed.
Novel Insights
The Molecular Basis for MAPKAPK's Specific Interaction with MAPKs: Classical D Motifs and Reverse D-Motifs Constitute the Linear Motif (LM)
Specific interactions of MAPKs with their activators and substrates are established via the common docking (CD) motif of MAPKs (D-X2-D/E) and the docking (D) motif (R/K-R/K-X2−6-Ø-X-Ø) or kinase-interacting motif (KIM) (L/V-X2-R/K-R/K-X5-L) of the substrate or activator (Tanoue et al., 2000; reviewed in Gaestel, 2008). However, while these interactions fully govern the recognition and phosphorylation of unstructured regions in substrates such as transcription factors, the CD-D-interaction is not completely sufficient for establishing the specificity of binding of MAPKs to important activators and other substrates. The isolated D-motifs of MKK3/6 (p38 specific MAPKK) or MKK1/2 (ERK specific MAPKK) are, for instance, not able to discriminate between p38α and ERK2, but bind to both kinases with comparable affinity (Garai et al., 2012). Further structural analyses have revealed that the CD motif of MAPKs can be divided into the negatively charged CD groove and various further hydrophobic pockets or grooves which are able to interact with the basic core of the D-motif and a pattern of further hydrophobic residues located N terminal to the D-motif designated reverse D (revD) motif (Ø-X-Ø-X2-Ø-X4−6-Ø-X2-R/K-R/K) (Garai et al., 2012). Interestingly, this revD motif allows clear discrimination in binding affinity between p38α and ERK2. While the revD motif of RSK1 displays high affinity to ERK2, its binding affinity to p38α is 20-fold lower. Vice versa, the revD motif of MK2 shows strong affinity to p38α but only weak interaction with ERK2 (Garai et al., 2012).
Only the revD of MNK1, which is activated by both ERK2 and p38α, displays similar affinity to both kinases. Hence, a linear motif (LM) formed by the overlapping D and revD motifs is necessary and sufficient to guarantee specific interaction in the binary MAPK/MAPKAPK complexes such as ERK2/RSK1 and p38α/MK2. The following alignment shows the D-, KIM-, and revD motifs identified in MAPK substrates and activators. Together, these overlapping motifs should be regarded as the linear motif (LM). Ø stands for a hydrophobic amino acid, Xn for the number n of variable amino acids:
D: R/K-R/K-X2−6-Ø-X-Ø
KIM: L/V-X2-R/K-R/K-X5-L
revD: Ø-X-Ø-X2-Ø-X4−6-Ø-X2-R/K-R/K
LM: Ø-X-Ø-X2-Ø-X4-6-Ø-X2-R/K-R/K-X2-6-Ø-X-Ø
Although the CD motif-LM-interaction is essential for various MAPK/MAPKAPK complexes, the CD-motif of the atypical MAPKs ERK 3 and ERK4 is not sufficient for the activation of MK5/PRAK. Instead, a novel FRIEDE interaction motif in loop L16 C-terminal to the CD-motif is necessary for MK5/PRAK binding of ERK3/4 (Aberg et al., 2009). Interestingly, the L16 FRIEDE motif in ERK3/4 is activated by phosphorylation of the atypical activation loop SEG in an allosteric manner. The FRIEDE motif interacts with the C-terminus of MK5/PRAK and a mutant lacking 50 C-terminal amino acids but still containing the D-domain of MK5/PRAK is unable to bind to ERK3/4 (Aberg et al., 2006). Hence, this interaction is clearly different from the CD-LM-module.
Primary MAPK/MAPKAPK Complexes Formed by LM-CD Motif Interaction
The binding of the LM of a MAPKAPK and the CD grooves of MAPKs (Figures 1A,B) is the first step of formation of specific signaling complexes but does not necessarily lead to the formation of a signaling competent complex with both MAPK and MAPKAPK activity. However, the primary “encounter” complex formation (Figure 1B) is already able to cause mutual stabilization of the MAPKs/MAPKAPKs in the specific complex. In vivo, this stabilization is reflected by the findings that in non-stimulated MK2-deficient cells the p38α level is significantly reduced (Kotlyarov et al., 2002) and that p38α-deficient resting cells display reduced MK2 levels (Sudo et al., 2005). Furthermore, the formation of primary, non-productive kinase complexes is able to prevent binding to (and activation by) non-specific MAPKs and crosstalk with other signaling pathways. This is demonstrated in vitro by the fact that addition of inactive p38α strongly increases the specificity of ppERK2 toward RSK1 and blocks ppERK2's activity against MK2 (Alexa et al., 2015). This finding implies that the stoichiometry between specific MAPKs and MAPKAPKs is an important determinant to maintain the specificity of signaling also in vivo. Taking into account that other MAPK substrates and activators compete with MAPKAPKs in binding to the CD motif, signaling complex formation in vivo is likely highly sensitive to the local concentrations of these competing interactors. In this regard a high complexity of regulation will also arise due to the fact that local sub-cellular concentrations of many signaling molecules are also signal-regulated. Equal importantly, this result also indicates that artificial overexpression of a specific MAPK or MAPKAPK, which can lead to significant stoichiometric alterations between specific MAPKs and/or MAPKAPKs in the cell, could also lead to artificial activation of non-specific signaling pathways. This would explain the initial observation that MK3, a kinase downstream to p38α/β, is activated by ERKs, JNKs, and p38 in cells overexpressing these MAPKs (Ludwig et al., 1996) or that MK5/PRAK, a kinase activated by the atypical ERK3/4 (see below), also displays docking to p38α when both kinases are overexpressed (New et al., 2003).
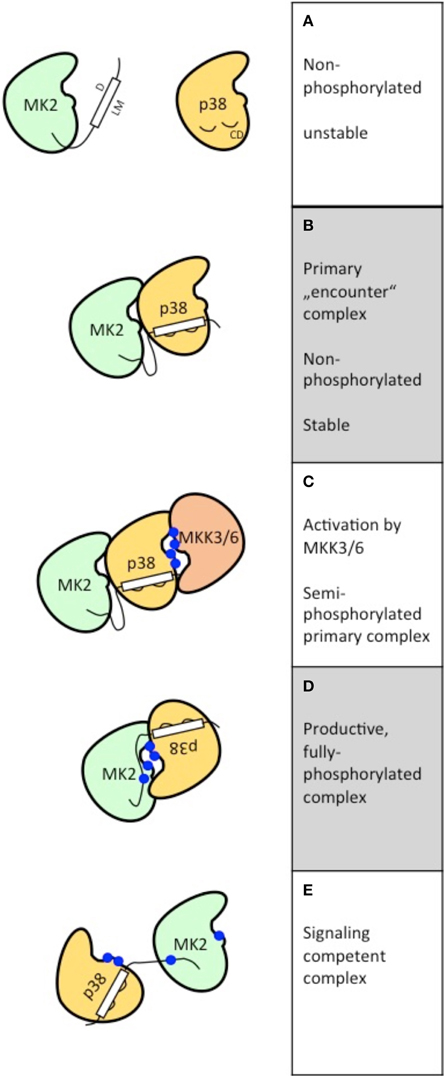
Figure 1. Schematic representation of the postulated steps to reach signaling competent, fully active binary kinase complexes between MAPKs (here p38α) and MAPKAPKs (here MK2). The features of the five different states postulated (A–E) are depicted at the right.
The three-dimensional structure of a primary MAPK-MAPKAPK-complex between non-phosphorylated p38α/MK2 has been established (White et al., 2007). In this complex the LM of MK2 is bound to the CD motif of p38α. Both kinases bind in a parallel “head to head” orientation (Figure 1B), but catalytic and substrate regions are distantly located at different sides of the kinase heterodimer making it unlikely that this is a signaling competent complex. However, this orientation would enable upstream activators, such as MKK3 or MKK6, to phosphorylate the activation loop of p38α leading to a semi-phosphorylated primary complex (Figure 1C).
Productive Dimerization Leading to Active Signaling Complexes
The three-dimensional structure of another non-phosphorylated MAPK/MAPKAPK complex consisting of ERK2 and RSK1 has recently been determined revealing a structure for a pre-catalytic state of anti-parallel “head to tail” orientation where both kinases face each other and the activation loop of RSK2 is located close to the catalytic center of ERK2 (Alexa et al., 2015). After phosphorylation of ERK2 by the upstream activator MEK1/2 only minor readjustments of the orientation of the binary complex seem necessary to activate RSK1 by phosphorylation of the CTD leading to a productive signaling module (Alexa et al., 2015).
In the case of p38α/MK2 more complex changes in orientation of the molecules in the complex seem necessary to enable p38 to phosphorylate the regulatory sites of MK2 (Figure 1D). It could be assumed that these changes are allosterically induced by phosphorylation of p38α at the activation loop. After phosphorylation of the regulatory sites of MK2 at the activation loop and in the hinge region between catalytic core and C-terminal extension, MK2 itself undergoes a structural transition involving a major conformational change of the atypically structured APE motif of MK2 (Alexa et al., 2015). As a result of this process a fully active signaling complex is formed (Figure 1E). The transition from the primary “encounter” complex to the fully active p38α/MK2 signaling complex is accompanied by a reduction of the affinity of interaction reflected by a increase of the Kd-value from 2.5 nM for non-phosphorylated MK2 and p38α to about 60 nM for phosphorylated MK2 and p38α (Lukas et al., 2004). Interestingly, a number of proteins and cellular structures, such as LIMK1 (Kobayashi et al., 2006), keratin K8/K20, or K8/K18 complexes (Menon et al., 2010) and the neighboring immediate early promoter binding factors CREB/SRF (Heidenreich et al., 1999; Ronkina et al., 2011) are substrates for both p38α and MK2 indicating that the fully active p38α/MK2 complex might act cooperatively to phosphorylate these proteins and structures.
Non-Canonical Activation of RSK in Dendritic Cells
Although there is a specific interaction between ERKs and RSKs via the CD-LM-interaction in many cell types, an ERK-independent but p38α-dependent activation of RSK by MK2 and MK3 has been described in dendritic cells. In these cells MK2/3 bypass phosphorylation of the C-terminal kinase domain (KD) by ERKs by directly phosphorylating the auto-phosphorylation site S386 between N- and C-terminal KD, a prerequisite for the activation of the N-terminal KD by PDK1 (Zaru et al., 2007). Recently, the structural and functional basis for the cell type-specific operation of this alternative activation mechanism of RSKs has been characterized further (Zaru et al., 2014). It has turned out that the non-canonical activation of RSKs is specific for hematopoietic cells, such as dendritic cells and macrophages, and that the C-terminal KD of RSK is dispensable for this activation. Furthermore, the existence of the non-canonical activation mechanism is accompanied by an increased constitutive cytoplasmic localization of p38α/MK2/3 in these cells and a very low activation of ERKs by inflammatory stimuli, such as LPS. Hence, in these cells a certain plasticity of MAPK signaling guarantees the LPS-induced TLR-mediated interferon-β induction via the p38α/MK2/3-RSK-pathway. The interaction between MK2/3 and RSK in these cells seems rather transient (Zaru et al., 2014) and it is not clear whether further cell type-specific protein partners facilitate this interaction in macrophages and dendritic cells.
Established Functions Challenged
Challenged Specificity of the Compounds BI-D1870 and SL0101 and mTORC1-Related Function of RSKs
In tests against a panel of recombinant protein kinases the compounds BI-D1870 and SL0101 appeared as relatively specific inhibitors for RSK1 and RSK2 (Bain et al., 2007). However, said panel did not contain mTOR or mTORC1 and a recent study demonstrated that BI-D1870 and SL0101 also modulate mTORC1-p70S6K signaling in different directions (Roffé et al., 2015). Since SL0101 clearly also inhibits mTORC1-p70S6K signaling, the demonstration that RSK phosphorylates ribosomal protein S6, a substrate of p70S6K, using this inhibitor is challenged. Interestingly, BI-D1870 increased p70S6K activation in an ERK1/2- and RSK-independent manner by a mechanism unknown to date. In the light of these findings, the interpretation of the results presented in nearly 100 publications describing effects of these inhibitors without confirming these effects by further experiments, such as knockdown or overexpression of active kinase, should be reassessed. Meanwhile, novel and more specific RSK-inhibitors have also been identified (Jain et al., 2015) enabling us to better define the in vivo function of these kinases.
Challenged Function of MK5/PRAK as Tumor Suppressor
Controversial discussions regarding the activation mechanism and function of MK5/PRAK have been published. As seen from the LM alignment below, the sequence of the LM present in this protein kinase bears similarity to both the LM of RSK and MK2, indicating possible interaction with ERKs or p38 MAPKs:
RSK1: 721-PQLKPIESSILAQRRVRKLPS-741
:::. .. .: .
MK5/PRAK: 348-VSLKPLHSVNNPILRKRKLLGTK-364
. .: . . ::.: ::. .
MK2: 372-IKIKKIEDASNPLLLKRRKKARA-392
LM: ØXØXXØXXXXXXØXXRRXXXØXØ
KK
In line with this similarity, activation of MK5/PRAK has been observed by p38 MAPKs and by ERKs when these kinases were overexpressed in mammalian cells (New et al., 1998; Ni et al., 1998). Furthermore, there is the FRIEDE-binding region (see above) in the C-terminal stretch of 50 amino acids, which enables interaction of this kinase with ERK3/4 (Aberg et al., 2009). Overexpression of both p38α and ERK3/4 leads to phosphorylation of MK5/PRAK at its regulatory site T182 and its activation as measured by phosphorylation of the peptide PRAKtide. While several publications describe a p38-dependent activation of MK5/PRAK (New et al., 1998), others could not detect activation of MK5/PRAK by stimuli, which activate p38 MAPKs, such as arsenite or high osmolarity (sorbitol) treatment (Shi et al., 2003). ERK3/4 activity and binding of the FRIEDE motif to MK5/PRAK can be stimulated by phosphorylation by p21-activated kinase 1 (PAK1) in the SEG motif in the activation loop (De la Mota-Peynado et al., 2011; Déléris et al., 2011) connecting MK5 to signaling of the small GTP-ase Rac. Furthermore, acetylation of MK5/PRAK at lysine K364 in the putative LM has also been described to increase its activity, although it should interfere with binding of the appropriate MAPK (Zheng et al., 2013), and various substrates of MK5/PRAK, such as p53 (Sun et al., 2007), HSP27 (Kostenko et al., 2009), FoxO3a (Kress et al., 2011), Foxo1 (Chow et al., 2013), and Rheb (Zheng et al., 2011) have been proposed.
The function of MK5/PRAK has been mainly characterized by two different mouse knockout approaches targeting exon 6 and exon 8, respectively (Shi et al., 2003; Sun et al., 2007). Surprisingly, it has recently turned out that both knockout approaches for MK5/PRAK failed to delete the entire protein (Ronkina et al., 2015). Instead, two different truncated MK5/PRAK forms are still present in the knockout mice (Table 1). Since these mutants display different biochemical and cellular properties (Table 1) it is not surprising that the effects of expression of these mutants in cellular systems differ: While MEFs from the Δex6-targeted mice did not show altered p21WAF level in response to H-Ras-G12V expression and did not grow in soft agar, Δex8-targeted MEFs displayed reduced levels of the key marker of tumor suppression p21WAF and growth in soft agar (Sun et al., 2007; Ronkina et al., 2015). It is highly probable that the properties of the different MK5/PRAK deletion mutants also contribute to the phenotype of the targeted mice in the established DMBA one-step skin tumor model. Hence, it is not surprising that Δex6-targeted mice did not display increased skin tumor formation in this model, while Δex8-targeted did (Sun et al., 2007; Ronkina et al., 2015). Since the results obtained using the Δex8-targeted mice formed the basis for the formulation of the tumor suppressive role of MK5/PRAK (Sun et al., 2007) as well as for the identification of MK5/PRAK as a tumor-promoting angiogenic factor (Yoshizuka et al., 2012), these roles are challenged and should be revisited by MK5 targeting approaches deleting the entire protein kinase. Once the phenotype of the real MK5/PRAK knockout mouse has been described, these results should serve to clarify the physiological function of MK5/PRAK.
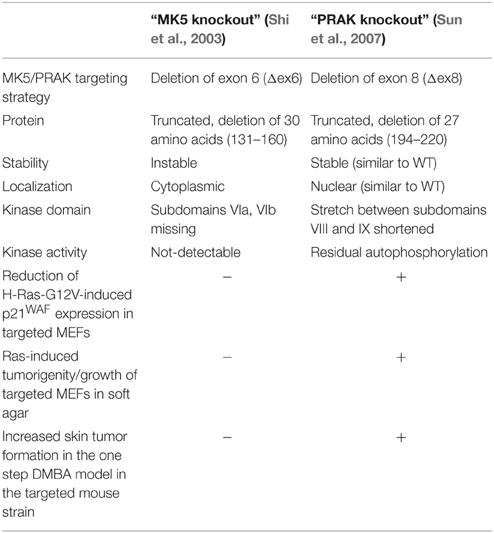
Table 1. Comparison of the results of the targeting approaches for MK5/PRAK.
Funding
The work of MG was funded by Deutsche Forschungsgemeinschaft.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author would like to thank Drs. Achim Gossler and Alexey Kotlyarov for discussion of the manuscript, the reviewers for very helpful comments and Stefanie Hall for proofreading.
References
Aberg, E., Perander, M., Johansen, B., Julien, C., Meloche, S., Keyse, S. M., et al. (2006). Regulation of MAPK-activated protein kinase 5 activity and subcellular localization by the atypical MAPK ERK4/MAPK4. J. Biol. Chem. 281, 35499–35510. doi: 10.1074/jbc.M606225200
Aberg, E., Torgersen, K. M., Johansen, B., Keyse, S. M., Perander, M., and Seternes, O.-M. (2009). Docking of PRAK/MK5 to the atypical MAPKs ERK3 and ERK4 defines a novel MAPK interaction motif. J. Biol. Chem. 284, 19392–19401. doi: 10.1074/jbc.M109.023283
Alexa, A., Gógl, G., Glatz, G., Garai, Á., Zeke, A., Varga, J., et al. (2015). Structural assembly of the signaling competent ERK2–RSK1 heterodimeric protein kinase complex. Proc. Natl. Acad. Sci. U.S.A. 112, 2711–2716. doi: 10.1073/pnas.1417571112
Bain, J., Plater, L., Elliott, M., Shpiro, N., Hastie, C. J., McLauchlan, H., et al. (2007). The selectivity of protein kinase inhibitors: a further update. Biochem. J. 408, 297–315. doi: 10.1042/BJ20070797
Cargnello, M., and Roux, P. P. (2011). Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 75, 50–83. doi: 10.1128/MMBR.00031-10
Chow, K. T., Timblin, G. A., McWhirter, S. M., and Schlissel, M. S. (2013). MK5 activates Rag transcription via Foxo1 in developing B cells. J. Exp. Med. 210, 1621–1634. doi: 10.1084/jem.20130498
De la Mota-Peynado, A., Chernoff, J., and Beeser, A. (2011). Identification of the atypical MAPK Erk3 as a novel substrate for p21-activated kinase (Pak) activity. J. Biol. Chem. 286, 13603–13611. doi: 10.1074/jbc.M110.181743
Déléris, P., Trost, M., Topisirovic, I., Tanguay, P.-L., Borden, K. L. B., Thibault, P., et al. (2011). Activation loop phosphorylation of ERK3/ERK4 by group I p21-activated kinases (PAKs) defines a novel PAK-ERK3/4-MAPK-activated protein kinase 5 signaling pathway. J. Biol. Chem. 286, 6470–6478. doi: 10.1074/jbc.M110.181529
Gaestel, M. (2006). MAPKAP kinases - MKs - two“s company, three”s a crowd. Nat. Rev. Mol. Cell Biol. 7, 120–130. doi: 10.1038/nrm1834
Gaestel, M. (2008). Specificity of signaling from MAPKs to MAPKAPKs: kinases' tango nuevo. Front. Biosci. 13, 6050–6059. doi: 10.2741/3136
Garai, Á., Zeke, A., Gógl, G., Töro, I., Fördos, F., Blankenburg, H., et al. (2012). Specificity of linear motifs that bind to a common mitogen-activated protein kinase docking groove. Sci. Signal. 5, ra74. doi: 10.1126/scisignal.2003004
Heidenreich, O., Neininger, A., Schratt, G., Zinck, R., Cahill, M. A., Engel, K., et al. (1999). MAPKAP kinase 2 phosphorylates serum response factor in vitro and in vivo. J. Biol. Chem. 274, 14434–14443.
Jain, R., Mathur, M., Lan, J., Costales, A., Atallah, G., Ramurthy, S., et al. (2015). Discovery of potent and selective rsk inhibitors as biological probes. J. Med. Chem. 58, 6766–6783. doi: 10.1021/acs.jmedchem.5b00450
Kobayashi, M., Nishita, M., Mishima, T., Ohashi, K., and Mizuno, K. (2006). MAPKAPK-2-mediated LIM-kinase activation is critical for VEGF-induced actin remodeling and cell migration. EMBO J. 25, 713–726. doi: 10.1038/sj.emboj.7600973
Kostenko, S., Johannessen, M., and Moens, U. (2009). PKA-induced F-actin rearrangement requires phosphorylation of Hsp27 by the MAPKAP kinase MK5. Cell. Signal. 21, 712–718. doi: 10.1016/j.cellsig.2009.01.009
Kotlyarov, A., Yannoni, Y., Fritz, S., Laass, K., Telliez, J.-B., Pitman, D., et al. (2002). Distinct cellular functions of MK2. Mol. Cell Biol. 22, 4827–4835. doi: 10.1128/MCB.22.13.4827-4835.2002
Kress, T. R., Cannell, I. G., Brenkman, A. B., Samans, B., Gaestel, M., Roepman, P., et al. (2011). The MK5/PRAK kinase and Myc form a negative feedback loop that is disrupted during colorectal tumorigenesis. Mol. Cell 41, 445–457. doi: 10.1016/j.molcel.2011.01.023
Ludwig, S., Engel, K., Hoffmeyer, A., Sithanandam, G., Neufeld, B., Palm, D., et al. (1996). 3pK, a novel mitogen-activated protein (MAP) kinase-activated protein kinase, is targeted by three MAP kinase pathways. Mol. Cell Biol. 16, 6687–6697.
Lukas, S. M., Kroe, R. R., Wildeson, J., Peet, G. W., Frego, L., Davidson, W., et al. (2004). Catalysis and function of the p38 alpha. MK2a signaling complex. Biochemistry 43, 9950–9960. doi: 10.1021/bi049508v
Menon, M. B., Schwermann, J., Singh, A. K., Franz-Wachtel, M., Pabst, O., Seidler, U., et al. (2010). p38 MAP kinase and MAPKAP kinases MK2/3 cooperatively phosphorylate epithelial keratins. J. Biol. Chem. 285, 33242–33251. doi: 10.1074/jbc.M110.132357
New, L., Jiang, Y., and Han, J. (2003). Regulation of PRAK subcellular location by p38 MAP kinases. Mol. Biol. Cell 14, 2603–2616. doi: 10.1091/mbc.E02-08-0538
New, L., Jiang, Y., Zhao, M., Liu, K., Zhu, W., Flood, L. J., et al. (1998). PRAK, a novel protein kinase regulated by the p38 MAP kinase. EMBO J. 17, 3372–3384. doi: 10.1093/emboj/17.12.3372
Ni, H., Wang, X. S., Diener, K., and Yao, Z. (1998). MAPKAPK5, a novel mitogen-activated protein kinase (MAPK)-activated protein kinase, is a substrate of the extracellular-regulated kinase (ERK) and p38 kinase. Biochem. Biophys. Res. Commun. 243, 492–496. doi: 10.1006/bbrc.1998.8135
Roffé, M., Lupinacci, F. C., Soares, L. C., Hajj, G. N., and Martins, V. R. (2015). Cellular signalling. Cell. Signal. 27, 1630–1642. doi: 10.1016/j.cellsig.2015.04.004
Ronkina, N., Johansen, C., Bohlmann, L., Lafera, J., Menon, M. B., Tiedje, C., et al. (2015). Comparative analysis of two gene-targeting approaches challenges the tumor-suppressive role of the protein kinase MK5/PRAK. PLoS ONE 10:e0136138. doi: 10.1371/journal.pone.0136138.s006
Ronkina, N., Menon, M. B., Schwermann, J., Arthur, J. S. C., Legault, H., Telliez, J.-B., et al. (2011). Stress induced gene expression: a direct role for MAPKAP kinases in transcriptional activation of immediate early genes. Nucleic Acids Res. 39, 2503–2518. doi: 10.1093/nar/gkq1178
Shi, Y., Kotlyarov, A., Laabeta, K., Gruber, A. D., Butt, E., Marcus, K., et al. (2003). Elimination of protein kinase MK5/PRAK activity by targeted homologous recombination. Mol. Cell Biol. 23, 7732–7741. doi: 10.1128/MCB.23.21.7732-7741.2003
Sudo, T., Kawai, K., Matsuzaki, H., and Osada, H. (2005). p38 mitogen-activated protein kinase plays a key role in regulating MAPKAPK2 expression. Biochem. Biophys. Res. Commun. 337, 415–421. doi: 10.1016/j.bbrc.2005.09.063
Sun, P., Yoshizuka, N., New, L., Moser, B. A., Li, Y., Liao, R., et al. (2007). PRAK is essential for ras-induced senescence and tumor suppression. Cell 128, 295–308. doi: 10.1016/j.cell.2006.11.050
Tanoue, T., Adachi, M., Moriguchi, T., and Nishida, E. (2000). A conserved docking motif in MAP kinases common to substrates, activators and regulators. Nat. Cell Biol. 2, 110–116. doi: 10.1038/35000065
White, A., Pargellis, C. A., Studts, J. M., Werneburg, B. G., and Farmer, B. T. (2007). Molecular basis of MAPK-activated protein kinase 2:p38 assembly. Proc. Natl. Acad. Sci. U.S.A. 104, 6353–6358. doi: 10.1073/pnas.0701679104
Yoshizuka, N., Chen, R. M., Xu, Z., Liao, R., Hong, L., Hu, W.-Y., et al. (2012). A novel function of p38-regulated/activated kinase in endothelial cell migration and tumor angiogenesis. Mol. Cell Biol. 32, 606–618. doi: 10.1128/MCB.06301-11
Zaru, R., Edgar, A. J., Hanauer, A., and Watts, C. (2014). Structural and functional basis for p38-MK2-activated Rsk signaling in toll-like receptor-stimulated dendritic cells. Mol. Cell Biol. 35, 132–140. doi: 10.1128/MCB.00773-14
Zaru, R., Ronkina, N., Gaestel, M., Arthur, J. S. C., and Watts, C. (2007). The MAPK-activated kinase Rsk controls an acute Toll-like receptor signaling response in dendritic cells and is activated through two distinct pathways. Nat. Immunol. 8, 1227–1235. doi: 10.1038/ni1517
Zheng, H., Seit-Nebi, A., Han, X., Aslanian, A., Tat, J., Liao, R., et al. (2013). A posttranslational modification cascade involving p38, Tip60, and PRAK mediates oncogene-induced senescence. Mol. Cell 50, 699–710. doi: 10.1016/j.molcel.2013.04.013
Keywords: p38 MAPK, ERK3/4, common docking motif, macrophage-specific activation, dendritic cells, mouse gene-targeting strategy, Ras-induced senescence, DMBA-induced skin tumors
Citation: Gaestel M (2016) MAPK-Activated Protein Kinases (MKs): Novel Insights and Challenges. Front. Cell Dev. Biol. 3:88. doi: 10.3389/fcell.2015.00088
Received: 16 October 2015; Accepted: 18 December 2015;
Published: 08 January 2016.
Edited by:
Ana Cuenda, Spanish National Research Council, SpainReviewed by:
Ole-Morten Seternes, UiT The Arctic University of Norway, NorwayChristopher James Caunt, University of Bath, UK
Copyright © 2016 Gaestel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthias Gaestel, Z2Flc3RlbC5tYXR0aGlhc0BtaC1oYW5ub3Zlci5kZQ==