 Nicolò Ilacqua1†
Nicolò Ilacqua1† Miguel Sánchez-Álvarez2†
Miguel Sánchez-Álvarez2† Magdalena Bachmann1
Magdalena Bachmann1 Veronica Costiniti1
Veronica Costiniti1 Miguel A. Del Pozo2
Miguel A. Del Pozo2 Marta Giacomello1*
Marta Giacomello1*- 1Department of Biology, University of Padova, Padova, Italy
- 2Mechanoadaptation and Caveolae Biology Lab, Cell and Developmental Biology Area, Centro Nacional de Investigaciones Cardiovasculares Carlos III, Madrid, Spain
Mitochondria-endoplasmic reticulum (ER) contacts (MERCs) are sites at which the outer mitochondria membrane and the Endoplasmic Reticulum surface run in parallel at a constant distance. The juxtaposition between these organelles determines several intracellular processes such as to name a few, Ca2+ and lipid homeostasis or autophagy. These specific tasks can be exploited thanks to the enrichment (or re-localization) of dedicated proteins at these interfaces. Recent proteomic studies highlight the tissue specific composition of MERCs, but the overall mechanisms that control MERCs plasticity remains unclear. Understanding how proteins are targeted at these sites seems pivotal to clarify such contextual function of MERCs. This review aims to summarize the current knowledge on protein localization at MERCs and the possible contribution of the mislocalization of MERCs components to human disorders.
Introduction
The term “synapse” refers to a site at which two neurons are close enough to communicate to each other. Here, electrical or chemical signals are integrated to determine specific responses, such as the generation of action potentials. The concept of synapse and synaptic integration can be extended to other cells. For example, immunological synapses have been defined for T cells (Norcross, 1984; Paul and Seder, 1994; Grakoui et al., 1999; Bromley et al., 2001; Viola et al., 2010): these are the sites at which signaling cascades originating from T cell Receptors are ultimately “decoded and integrated” to achieve either activation or tolerance.
This concept can be further extended to contact sites between intracellular compartments such as those amongst mitochondria and Endoplasmic Reticulum (ER). Mitochondria-ER contacts (MERCs) are sites in which the surfaces of the two organelles juxtapose at a constant distance, for several nm in length. These contacts can be isolated through subcellular fractionation procedures and the membrane fraction corresponding to the MERCs is known as MAMs (mitochondria associated membranes; Vance, 1990; Rusiñol et al., 1994). Thus, MAMs are the biochemical counterpart of MERCs (Giacomello and Pellegrini, 2016).
Cues comprising information from cell growth signaling, metabolic, and stress-responsive programmes are integrated at MERCs, determining cell wellbeing/homeostasis. Therefore, it is not surprising that the disruption of MERCs has been associated with an ever-growing number of pathologies, as an element contributing to the propagation of functional imbalances across cellular systems—such as lipid imbalance and insulin resistant states (Arruda et al., 2014).
MERCs-associated functions, composition, and extension seem to be tailored to specialized tissues- further stressing their relevance for the fine tuning and integration of multiple functions. But how are MERCs defined and how is their specific composition dictated? Viola et al. (2010) proposed that integration of different signaling steps may promote the rearrangement of lipids within membranes, thus providing specialized platforms at which signals can be generated, amplified, or even blunted. Whether this principle applies to MERCs is a standing question to explore. The latter have been already shown to have “lipid raft”-like properties (Hayashi and Fujimoto, 2010; Area-Gomez et al., 2012), although this aspect needs further clarification. Besides “raft”-like domains, MERCs are characterized by the presence of proteins that either tether the two organelles together or dictate their biological function. These “molecular bridges” appear as electron dense rods in EM images (Csordás et al., 2006). Among the potential tethers identified to date, the most studied in higher eukaryotes is Mitofusin2 (Mfn2), first discovered as a key factor for mitochondrial fusion (Chen et al., 2003). Its presence at the surface of the ER and the evidence that ER-located Mfn2 binds to the OMM located Mfn2 and Mfn1 (de Brito and Scorrano, 2008; Naon et al., 2016) strongly suggested its involvement in the control of MERCs. Another key protein required for MERCs assembly and activity is PhosphoAcidic Cluster Sorting protein 2 (PACS2; Simmen et al., 2005): its ablation decreases the interaction between the two organelles and the activities of the MAMs resident proteins phosphatidyl serine synthase 1 (PSS1) and long-chain fatty acid acetyl-CoA synthase (ACSL4; Piccini et al., 1998; Simmen et al., 2005). A third tethering complex proposed for higher eukaryotes includes the integral ER protein Vesicle-Associated membrane Protein associated protein B (VAPB) and the OMM Protein Tyrosine Phosphatase Interacting Protein 51 (PTPIP51). Interestingly, the VAPB-PTPIP51 tethering complex negatively controls autophagy and is dysregulated in frontotemporal dementia (De Vos et al., 2012; Stoica et al., 2014; Gomez-Suaga et al., 2017).
Although these (and others, for a detailed list please refer to Table 1, and to: Area-Gomez et al., 2012; De Mario et al., 2016) structural components of MERCs have been uncovered, it is nowadays clear that MERCs display cell-specific tissue composition, as highlighted by a number of proteomic analyses (Poston et al., 2011; Horner et al., 2015; Liu et al., 2015; Sala-Vila et al., 2016). While the interest on the biology of MERCs has recently soared, further systematic studies are required to get a complete view of the MERCs toolkit.
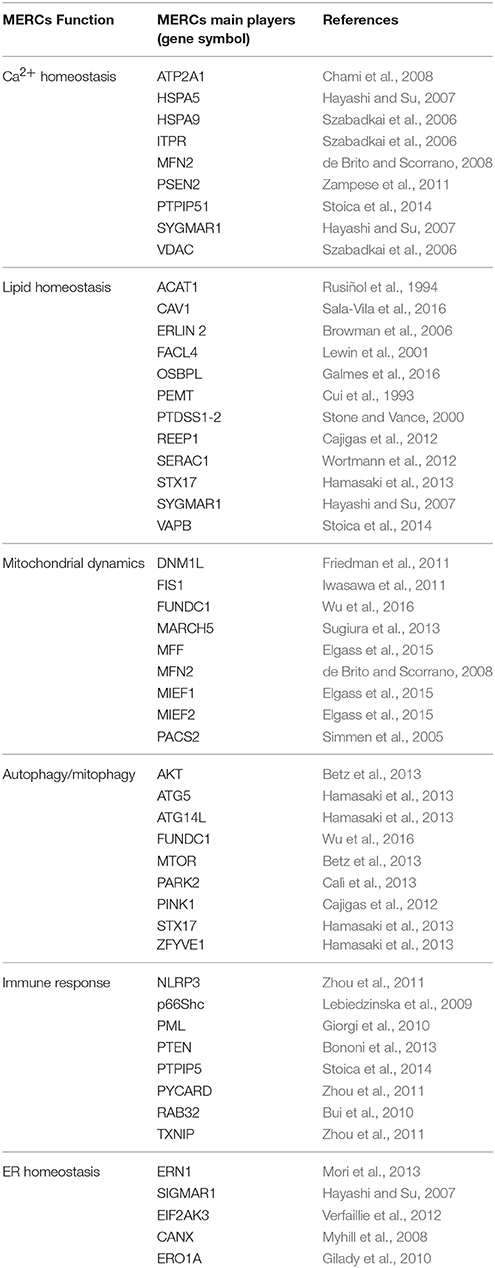
Table 1. Overview of the main MERCs functions and actors.
Notably, while proteins involved in the maintenance of lipid and Ca2+ homeostasis can be retrieved at MERCs in basal conditions, some proteins enrich in these sub-compartments only upon stimulation. Thus, MERCs, similarly to membrane rafts, function as platforms for composite signal transduction complexes. How proteins are recruited to these “biological interfaces” and retained there still needs to be clarified and is fundamental to understand MERCs physiological role. This review focuses on this aspect, and aims to highlight the principles determining protein enrichment/translocation at MERCs.
MERCs Functions at a Glance
As stated above, mitochondria-ER contact sites (MERCs) appear in electron microscopy (EM) as the parallel juxtaposition of the ER surface to the Outer Mitochondrial Membrane (OMM), at a distance ranging from 10 to 80 nm (Giacomello and Pellegrini, 2016). The length and width of the cleft separating both organelles and the protein composition of the communicating membranes are strictly bound to the processes in which MERCs are involved (summarized in Table 1). A number of recent reviews have already summarized in detail the role of MERCs in different subcellular pathways (Rowland and Voeltz, 2012; De Mario et al., 2016; Eisenberg-Bord et al., 2016; Prudent and McBride, 2017). Here, we will just provide a quick overview of the main MERCs functions.
The most established roles of MERCs pertain to their contribution to lipid and Ca2+ handling. Almost three decades ago, (Vance, 1990) highlighted the importance of MERCs for lipid homeostasis. Indeed these contact sites shape the specific route for phospholipid interconversion, allowing for the synthesis of phosphatidylethanolamine and phosphatidylcholine from serine and contributing to the composition of mitochondrial membranes (Rusiñol et al., 1994; Vance, 2014). These MERCs-associated routes may turn essential under restrictive conditions such as ethanolamine deficiency (Flis and Daum, 2013). Notably while the synthesis of cholesterol and its precursors, minoritary components of the OMM, is located at the ER, they can be converted into other molecules such as steroid hormones in MAMs (Bosch et al., 2011b; Sala-Vila et al., 2016). Thus, mitochondria-ER contacts appear as the sites at which coordination among lipid homeostasis and other cell functions occurs.
MERCs are also the site of Ca2+ exchange between the two organelles: they host a protein complex composed of the inositol triphosphate receptor (IP3R), the voltage-dependent anion channel (VDAC) and the chaperone grp75 (Szabadkai et al., 2006), which allows for rapid mitochondrial Ca2+ uptake through the Ca2+ Uniporter. The efficient shuttling of Ca2+ between both organelles depends on the width of the cleft that separates them: an optimal length of 15–25 nm allows both the assembly of the IP3R-grp75-VDAC complex and a fast Ca2+ exchange; on the contrary, a distance below 10 nm impedes the formation of the complex due to steric hindrance (Csordás et al., 2006). On the other hand, a distance above 25 nm would decrease the Ca2+ diffusion rate and hence blunt mitochondria Ca2+ uptake (Giacomello and Pellegrini, 2016). Mitochondral Ca2+ levels impinge on the activity of pyruvate, isocitrate, and α-ketoglutarate dehydrogenases (Denton et al., 1972, 1978), and hence on cell bioenergetics (Cárdenas et al., 2010). On the other hand, if pronounced, a sustained mitochondrial Ca2+ uptake can be read as a cell death signal able to trigger permeability of the mitochondrial membranes and opening of the permeability transition pore (PTP) (Bernardi et al., 2001; Hurst et al., 2017). Thus, MERCs also contribute to determine the cell fate (Simmen et al., 2005; Bui et al., 2010; Iwasawa et al., 2011; Prudent and McBride, 2017).
More recently, a bunch of additional functions have been ascribed to MERCs. For example they have been proposed as the site of autophagosome formation, thus playing a key role in autophagy (Hamasaki et al., 2013; Martínez-Pizarro et al., 2016). MERCs appear to couple mtDNA synthesis with mitochondrial division, that is also regulated by the interaction between mitochondria and ER (Friedman et al., 2011; Elgass et al., 2015; Lewis et al., 2016), and can behave as a scaffold that ultimately coordinates immune signaling and inflammasome formation (Lerner et al., 2012; Horner et al., 2015).
Finally, in yeast, mitochondria-ER interaction appears also fundamental for appropriate maintenance of cellular iron homeostasis and mitochondrial biogenesis (Wu et al., 2016; Ellenrieder et al., 2017; Xue et al., 2017).
Subcellular Localization: Targeting Sequences and More
Protein subcellular distribution relies on several mechanisms (Figure 1). The most common is the presence of a targeting peptide within the protein, which determines its sorting to specific sites. Two questions must be considered in this case: whether specialized or “consensus”-based mechanisms exist, and whether they are subjected to regulation.
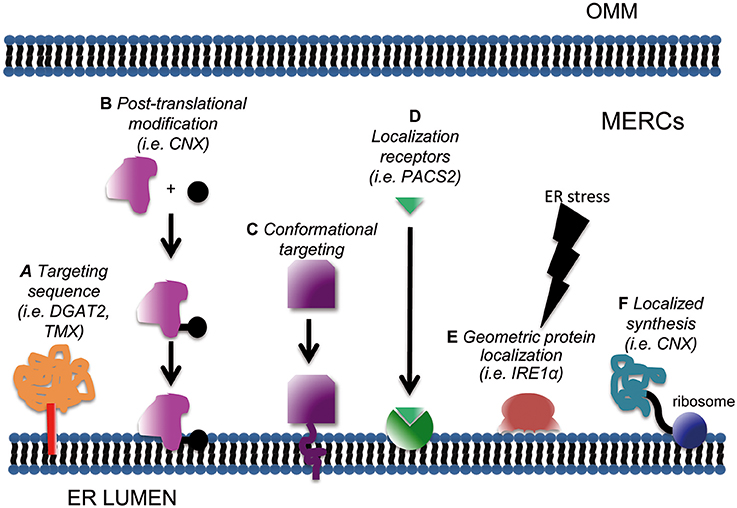
Figure 1. Schematic of subcellular mechanisms for protein targeting at MERCs. Enrichment at mitochondria-ER interface can be achieved through classical targeting sequences (A); post-translational modifications such as phosphorylation, acetylation or sumoylation (B); conformational targeting (C), localization signal receptors (D), geometric protein localization (E) and localized synthesis (F).
So far a robust consensus motif targeting proteins to MERCs has not been defined. Localization at the OMM or ER surface appears sufficient for a protein to be retrieved at MAMs, in a proportion that varies depending on the cell type, culture conditions, oxidation state, or specific metabolic status of the cell. For example, ~90% of the chaperone calnexin (CNX) is homogenously spread in the ER in basal conditions, concentrating at MERCs up to 70% under specific (stress) stimuli (Lynes et al., 2012). To date, only a few putative MERCs-targeting signals have been identified: this is the case for example of a stretch of 67 aa in the cytoplasmic N-terminus of DGAT2 (Stone et al., 2009). Another peculiar motif resides in the transmembrane and cytosolic domains of the transmembrane thioredoxin protein TMX, which are “necessary and sufficient” to ensure TMX accumulation at MAMs (Lynes et al., 2012). As stated above, a unique MERCs-targeting motif has not been identified yet. This could depend on the requirement of post-translational modifications (PTMs) and/or conformational determinants. PTMs as mechanisms for the regulated targeting of proteins at MERCs will be discussed separately in the following section, since a number of evidence substantiating this possibility have been already reported. Another mechanism both hampering the identification of “consensus target motifs” and rendering “alternative compartmentalization” possible might reside in mRNA processing. This remains still a hypothesis, since evidence for the existence of alternative splicing programmes dictating protein localization at MERCs is still lacking.
Another hypothetical MERCs-targeting mechanism would be the existence of “conformational” motifs. That is, a domain composed not by a linear stretch of amino acid, but by a functional interaction surface determined by the rearrangement of the 3D structure of the protein. The possibility of a “conformational” domain appears particularly intriguing for MERCs, since it would provide a mean for switchable recruitment of proteins. If this possibility holds true, it would perfectly match with the highly plastic lipid environment of MERCs. In fact, one of the means for PTMs-regulated targeting to MERCs might rely on such conditional conformational state (see below). Molecular threading for “tridimensional alignment” has been classically very challenging in terms of computational power requirements, but recent advances may ease these approaches to study conformational MERCs-targeting domains.
Another widespread mechanism for regulated protein compartmentalization relies on masking of localization domains: interaction with other partner molecules may confine them into the cytosol or other subdomains. This mechanism usually keeps “silent” (i.e., inactive) a certain protein until its release, which induces its re-localization at sites where specific interacting partners and/or target functions are (Bauer et al., 2015). Theoretically the concept of domain masking could be extended also to MERCs: targeting motifs would be exposed only if the specific function of the protein of interest is needed at these sites.
Subcellular targeting relies also on appropriate “localization signal receptors” (Bauer et al., 2015). This term denotes the presence of a sequestering/scaffolding protein able to bind its ligand and restrict its diffusion. Sequestration of a protein implies that the density of binding sites within a subcellular domain is high enough to significantly limit its mobility toward other locations. Interestingly, the affinity of a localization signal for its receptor can be modulated, especially through PTMs such as phosphorylation, lysine acetylation, or SUMOylation of either the receptor or of the ligand. Changes in the affinity of this interaction can either increase or decrease the compartmentalization of the ligand, by unveiling or, alternatively by masking, any signaling peptide. Thus, PTMs would exert their function not only by modulating the activity of proteins, but also by controlling their localization. As a consequence, they can shape the composition (and hence the function) of subcellular compartments. As to MERCs, an example of localization signal receptor is PACS2, which mediates the localization and enrichment of the CNX at these sites (Myhill et al., 2008). It is interesting to note that not only proteins, but also lipids and phospholipids could behave as localization receptors. For example, it has been shown that phosphatidylinositol (3,5) diphosphate can act as a membrane-targeting molecule, mediating the binding of different proteins to biological membranes (Ferguson et al., 2009; Salminen et al., 2013). Due to the special lipid composition of MERCs, we further elaborate in a separate section on this topic (see below).
Another common protein targeting strategy is “localized synthesis” (Kejiou and Palazzo, 2017). mRNAs encoding for a given polypeptide can be localized to specific subcellular domains where they are either kept silent, waiting for specific stimuli to trigger translation, or efficiently translated if necessary in basal conditions (Kejiou and Palazzo, 2017). Classical examples of spatial protein segregation by localized synthesis can be found in neurons (Rangaraju et al., 2017). Here, localized production is required to quickly shape the response of neurites to the signals coming from synapses. Interestingly, a study conducted to describe the local transcriptome in the synaptic neuropil of CAI Hippocampus (i.e., the mRNA enriched in this specific cell subdomain) highlighted the presence of MERCs resident or regulatory proteins, among which also calnexin, mTOR, Pink1, presenilin2, REEP1, Sigma-1 Receptor, α-synuclein, VAPB (Table S10 of Cajigas et al., 2012). These data support the hypothesis that local translation also contributes to the plasticity of MERCs, that with their activity could in turn match the needs of specialized cell structures.
Finally it's worth mentioning two additional mechanisms, although yet unproven for protein targeting at MERCs. A first one has been termed “geometric protein localization” (Ramamurthi et al., 2009; Updegrove and Ramamurthi, 2017). Mostly studied in bacteria, it relies on the ability of some proteins to “sense” membrane curvature and bind to specific geometric cues. In this case protein localization will be dictated by the shape of the membrane, independently of its composition (Hatzakis et al., 2009; Bhatia et al., 2010). These proteins are often characterized by membrane-binding amphipathic helices (Updegrove and Ramamurthi, 2017) that recognize even small changes in the curvature of membranes and enrich at these special sites. Interestingly, some curvature-sensing proteins have been reported in eukaryotes: this is the case of dynamins (Ramachandran and Schmid, 2008), cytochrome b5 (Taylor and Roseman, 1995), and interestingly, the ER stress-transducer IRE1 (Halbleib et al., 2017; see below). Recently, it was also reported that membrane-anchored proteins can efficiently sense membrane curvature, the latter being an additional mechanism for their efficient clustering (Hatzakis et al., 2009). As to MERCs, so far they have been characterized and described according to specific parameters: the relative length of the ER surface portion that run in parallel to the OMM and the width of the cleft that separates the two organelles (Giacomello and Pellegrini, 2016). Whether they are characterized by a particular membrane curvature range remains, at least to our knowledge, to be defined. The study of “geometric protein localization” at MERCs remains also challenged by the lack of appropriate readouts, compatible with the manipulation of such properties specifically at those sites.
The second mechanism, already shown to operate in the context of ER segregation and partitioning in yeast, pertains to the establishment of “protein boundaries” able to restrict the lateral diffusion of other membrane components (Chao et al., 2014). In this case, cytoskeleton-associated proteins (septins) are the effectors of such compartmentalization. Intriguingly, MERCs constitute in some experimental models points for actin- and microtubule-assisted mitochondrial fission (Ji et al., 2015; Prudent and McBride, 2016). So far, no clear connection has been established between MERC composition and cytoskeletal components, nor septins.
Post-Translational Modification: “Localization” or “Relocation” at MERCs?
PTMs are normally used by the cell to modulate the activity, stability, interaction profile and/or subcellular segregation of proteins. MERCs components are no exception: substantial evidence exists for multiple PTMs fine-tuning their properties, including their localization. As such, most MERCs proteins subjected to PTMs are characterized by (at least) a dual localization: either they have a broad subcellular distribution (for example, at the cytosol, or at the ER, or at the OMM), and then they enrich at MAMs, or the reverse (i.e., they appear located at MERCs, and upon PTMs, they redistribute to other subcellular compartments).
The first possibility applies to CNX. Its binding to PACS2 appears not sufficient for its enrichment at MERCs (Myhill et al., 2008), palmitoylation being the additional trigger necessary for its complete relocation at these interfaces (Lynes and Simmen, 2011). Examples have also been described for the second scenario. Impairment of ER oxidizing conditions causes Ero1α to lose its MAMs localization (Gilady et al., 2010), while heme oxygenase-1 relocates from MAMs to rough ER in the absence of palmitoylation (Lynes and Simmen, 2011). Another example is N-myristoylation: when the carbohydrate-binding protein starch binding domain-containing protein 1 (Stbd1) undergoes this modification, it is mostly retrieved on the ER, its wild type form being on the contrary enriched at MAMs “by default” (Demetriadou et al., 2017).
Generally speaking, PTMs also represent a switch for the activity of several enzymes. Thus, PTMs might control MERCs activity in a dual fashion: on one side, through a modulatory function, by directly enhancing or decreasing the activity of a MERCs-resident enzyme; on the other, by enhancing the contribution of MERCs to a subcellular process through re- or delocalization of specific proteins. Transient PTMs-dependent recruitment at MERCs may explain, at least in part, why changes in the expression levels of some proteins have a profound effect on ER-mitochondria apposition, despite they are not enriched in purified MAM fractions (Wieckowski et al., 2009; Eisenberg-Bord et al., 2016; Naon et al., 2016). Such an example has recently been reported for a novel OMM-ER tethering complex composed by two proteins broadly distributed to the OMM and ER surface: SYNJ2BP and RRBP1 (Hung et al., 2017). These data support the hypothesis of “auxiliary tethers,” according to which some proteins would not be strictly necessary to form a (functional) contact site, but they would be able to do so whenever their MERCs-related function was needed (Eisenberg-Bord et al., 2016).
“Lipid Raft”-Like Behavior as a Mechanism for Compartmentalization at MERCs
Another layer for regulating MERCs compartmentalization comes from the distinct lipid composition of the membrane domains delimiting MERCs (Figure 2). The concept of dynamic membrane nanodomains or “lipid rafts” initially introduced by Simons and van Meer (1988) almost 30 years ago states that one of the key properties of such membrane patches is the efficient accruing and stabilization of transmembrane proteins and membrane-associated activities (Simons and Sampaio, 2011). Correspondences between mitochondria-ER contacts and generic lipid rafts have been highlighted (Area-Gomez et al., 2012; Annunziata et al., 2013): MAMs have a significantly higher content in cholesterol and sphingolipids as compared with bulk ER membranes (Annunziata et al., 2013; Vance, 2014; Sala-Vila et al., 2016); they seem to have a lower degree on curvature as compared with surrounding ER regions (Rowland and Voeltz, 2012).
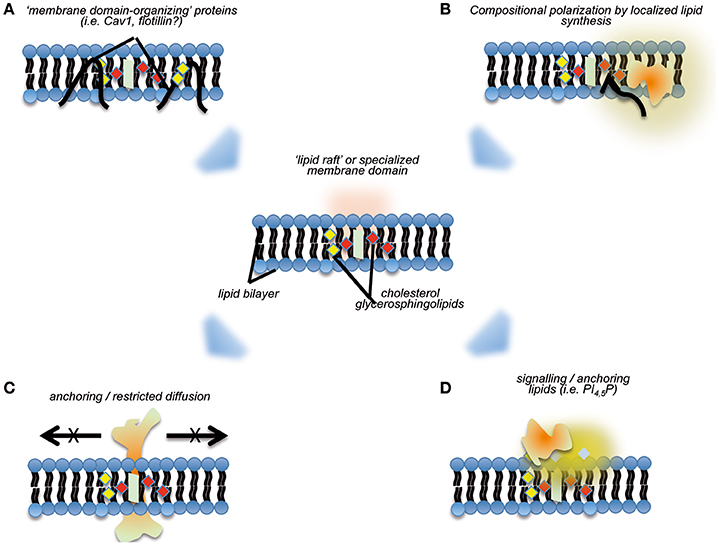
Figure 2. Cartoon summarizing aspects pertaining to the organization of “nanodomains” on membranes, and their potential effects regarding recruitment of specific activities. “Lipid raft”-like domains may be stabilized by specialized membrane-binding proteins, such as caveolins (A), and/or through directional local synthesis and accumulation of specific lipid species (B). The subsequent definition of such membrane nanodomains may preclude lateral diffusion of specific transmembrane proteins (C), and or act as “molecular beacons” for the specific recruitment of proteins (D).
The “lipid raft”-like organization of MERCs could help to explain some of their properties and functions. As mentioned above, it would induce the stabilization and limitation of lateral diffusion of ensembles of membrane proteins, or “polarization”—a property that would benefit the initial localization and subsequent sequential assembly of tethering complexes. The full collection of MERCs components in higher eukaryotes remains to be listed, but certain properties of the better-defined tethering complexes in yeast (termed ERMES) support this concept. First, ERMES components exhibit aberrant distribution if expressed in the absence of their partners (Kornmann et al., 2009, 2011; Stroud et al., 2011). Second, analysis of their structure revealed that their membrane-binding domains are fully functional only if they form complexes with appropriate stoichiometry, and only in this case they can recognize organelle contact sites. Thus, it seems that (a) MERCs lipid composition is an essential, but not a sufficient, feature to ensure MERCs targeting; and (b) cooperative recruitment may ensure the regulated, reversible assembly of tethering complexes. The differential lipid composition of MERCs appears critical for the targeting of specific proteins to this subcellular compartment in higher eukaryotes, a major example being constituted by the Prion protein (PrPC). While in basal conditions PrPC localizes mostly at the plasma membrane, pro-apoptotic stimuli can induce its relocalization to the MAMs, transducing a pro-death signal from the surface into the cell (Mattei et al., 2011).
Local synthesis and modification of lipids are hallmarks of lipid rafts. These have been proposed as potential mechanisms for the formation and stabilization of these nanodomains: a directional flux of specific lipid building blocks might favor per se the establishment of regions characterized by differential composition (Simons and Sampaio, 2011). A major subset of processes enabled by MERCs comprises lipid anabolism and trans-organelle transport of lipids (a classical example being phospholipids, see above). Furthermore, recent surveys highlight the enrichment of lipid metabolism functions at MERCs, including cholesterol synthesis and modification and fatty acid catabolism (Sala-Vila et al., 2016). A still open question is whether such local lipid metabolism sustains the differential composition of MERCs or not. Intriguingly, and further related to these observations, membrane proteins typically ascribed as lipid raft scaffolds/organizers, such as caveolin-1 (Cav1) are specifically enriched at MERCs. Cav1 is a cholesterol-binding protein famous for its role as an essential scaffold of plasma membrane nanodomains named caveolae (Parton and del Pozo, 2013). However, Cav1 also assembles in oligomers in the ER and determines cholesterol trafficking across subcellular compartments-including MERCs (reviewed in Bosch et al., 2011a). Cav1 genetic ablation is associated with increased cholesterol content and altered composition of MAMs derived from hepatocytes: Cav1 absence preferentially affects MAMs components involved in cholesterol and fatty acid metabolism, thus stressing the importance of lipid precursor fluxes for the organization and stabilization of these organelle contacts (Sala-Vila et al., 2016).
Altogether, these evidences favor another emerging function of MERCs, acting as “gauges” for lipid homeostasis in the cell. Similarly to plasma membrane lipid rafts, MERCs are sensitive to conditions disrupting or causing imbalances in lipid metabolism and membrane composition (Zhuang et al., 2005; Vance, 2014). Since MERCs are a platform in which many cell pathways converge, their “design” as elements highly sensitive to changes in lipid homeostasis allows for the integration of virtually all those signaling networks with lipid homeostasis. Thus, their dynamics might contribute substantially to phenomena such as dyslipidemia-associated modulation of proteostasis (see below) or insulin resistance (Arruda et al., 2014; Tubbs et al., 2014). Indeed, key regulators of the Pi3K/AKT/mTOR pathway have been reported to specifically localize at MERCs (Betz et al., 2013; Bononi et al., 2013). It is likely that the lipid raft-like properties of MERCs drive such recruitment and that of additional molecular beacons like phosphatidylinositol phosphate species (Hill et al., 2002; Goswami et al., 2005; Simons and Sampaio, 2011).
UPR and ER Stress Signaling at MERCs
Besides being the site for the synthesis, folding, and maturation of secreted and organelle-targeted proteins (Braakman and Bulleid, 2011), the ER also allocates other essential tasks, including lipid homeostasis and mobilization, red/ox control and Ca2+ flux regulation. Therefore, the ER constitutes a “hub” through which specific imbalances (i.e., dyslipidemia) can be easily propagated to other cellular systems, underpinning complex pathogenic processes such as obesity-related diseases and cancer. Eukaryotes have evolved a complex surveillance system to cope with functional imbalances in the ER, generally termed ER stress: the Unfolded Protein Response (Ellgaard and Helenius, 2003; Chakrabarti et al., 2011; UPR). UPR regulates either pro-survival programmes, aimed at enhancing ER capacity and/or lowering its functional demand; or pro-death pathways, in case of sustained unresolved ER stress (Ron, 2002; Rutkowski and Kaufman, 2004; Naidoo, 2009).
The UPR includes three signaling branches, associated with three ER-resident transmembrane transducers. The first is the Activation Transcription Factor-6 (ATF6). ATF6 is translocated to Golgi membranes, where it is sequentially cleaved by the S1P and S2P proteases (Haze et al., 1999). This yields an N-terminal fragment which acts as a leucine-zipper transcription factor and drives the expression of adaptive programmes. ATF6 signaling leads to the induction of the turnover system “ER Associated Degradation” (ERAD) and of ER chaperones (for a more detailed overview of UPR and ERAD, please refer to Yoshida et al., 2000; Okada et al., 2003; Galehdar et al., 2010; Tsai and Weissmann, 2010; Smith et al., 2011; Hetz, 2012; Arensdorf et al., 2013). The second branch relies on PRKR-like endoplasmic reticulum kinase (PERK), one of the four eIF2alpha-kinases expressed in higher eukaryotes. Upon “sensing” alterations in ER function or integrity through its luminal domain and its transmembrane segment, PERK oligomerizes and becomes catalytically active, repressing mRNA pools and thus reducing the ER load (Harding et al., 1999). PERK activation also favors translation of the activation transcription factor 4 (ATF4), which controls the expression of master regulators of cell survival and apoptosis (Lu et al., 2004). The third and most conserved UPR transducer is the inositol-requiring enzyme 1 (IRE1, of which two isoforms exist, being IRE1α the essential and most ubiquitous one; Tirasophon et al., 1998). IRE1 catalyzes the unconventional splicing of the X-box binding protein 1(XBP1) mRNA, yielding to a potent transcriptional activator that orchestrates adaptive programmes like the physical expansion of the ER itself (Harding et al., 1999; Yoshida et al., 2001; Calfon et al., 2002; Hetz et al., 2006).
As an integral part of ER, it is not surprising that MERCs function and structure are linked to UPR signaling. IRE1 has been found in MAMs, and in turn the ER-mitochondrial interaction significantly impacts on its activation (Mori et al., 2013). In particular, upon acute ER stress, the MERCs-resident chaperone Sigma-1 receptor (SigR1) stabilizes IRE1α, thus favoring its activation and UPR initiation (Hayashi and Su, 2007; Mori et al., 2013). PERK has also been retrieved in MAMs (Verfaillie et al., 2012), where it directly associates with Mfn2. This interaction seems to increase the activation threshold of PERK (Muñoz et al., 2013). Further, conditions disrupting MERCs, such as Mfn2 knockdown, are associated with a sustained activation of some UPR/ER stress response transducers even in basal conditions (Ngoh et al., 2012; Sebastián et al., 2012; Schneeberger et al., 2013). Recent evidence suggests that changes of MERCs dynamics can influence not only UPR triggering thresholds and amplitude, but also UPR shutdown dynamics. For example, the MERCs stabilization appears as an essential component to induce IRE1 shutdown during ER stress recovery (Sanchez-Alvarez et al., 2017). Conversely, MERCs also coordinate cell functioning and UPR activation: increased coupling of ER and mitochondria accompanies early phases of ER stress and sustains the metabolic adaptations necessary for the cell to cope with non-physiological conditions (Bravo et al., 2011). Further potential ties between MERCs and adaptive UPR signaling pertain to activities determining red/ox potential in the ER. For example, protein disulfide isomerases such as PDIA6 accrue at MERCs (Vance and Vance, 2009) and regulate IRE1 activation (Eletto et al., 2014, 2016). ER stress associated with ROS dysregulation is likely transduced by the PERK-dependent branch at MERCs too (Verfaillie et al., 2012). Hence, the confinement of UPR transduction at MERCs appears fundamental for the integration of the UPR response with multiple signaling pathways.
Does ER homeostasis surveillance influence MERCs composition? Specific adaptations at MERCs take place during ER stress. In these circumstances expression levels of CNX at the plasma membrane decrease (Wiest et al., 1995; Okazaki et al., 2000), increasing in parallel at MAMs (Myhill et al., 2008; Lynes et al., 2012). Another prominent example are programmes favoring cell apoptosis in the face of unresolved or excessive ER stress. Sustained PERK activation contributes to stabilize MERCs, enabling lipid peroxidation at the mitochondrial membrane- which in turn enhances expression and/or mitochondrial recruitment of proapoptotic regulators such as Bax and Ca2+ uptake (McCullough et al., 2001; Puthalakath et al., 2007; Galehdar et al., 2010; Verfaillie et al., 2012). A peculiar case of MERCs targeting regulated by UPR is embodied by IRE1 and PERK themselves. As stated above, they continuously monitor misfolded protein levels in the ER lumen through specialized domains. Deletion mutants of yeast IRE1 and of vertebrate IRE1 and PERK lacking luminal domains appear insensitive to acute protein misfolding, but retain sensitivity to conditions altering ER composition or physical properties, such as increased global acyl chain saturation or cholesterol content (Brodsky and Skach, 2011; Volmer et al., 2013; Volmer and Ron, 2015). At least in the case of IRE1, such “membrane monitoring” relies on the features of its transmembrane domain (Halbleib et al., 2017; Kono et al., 2017). The latter mechanism is likely to determine the segregation of UPR transducers to MERCs, possibly in combination with other regulatory layers such as transient dimerization or conformational changes. Recruitment of these UPR sensors at MERCs further contributes to the integration between UPR signaling and cell metabolism (Walter and Ron, 2011).
Disrupted MERCs Localization: Potential Impact in Human Disorders
In general, it is well-established that defective subcellular localization can either alter the activity of a protein and/or the subcellular processes in which it is involved. If we extend this concept to MERCs and we take into account that they participate in a myriad of essential process (see above), it seems obvious that their alteration or adaptation to stress conditions could both worsen and propagate imbalances across cellular systems—a phenomenon that appears to be common-place for complex diseases. For instance, the disruption of general mechanisms impacting on MERCs, such as protein palmitoylation, could simultaneously affect their integrity and that of other cellular functions. This could be the basis of phenotypic variability and epistatic effects across many different disorders, ranging from schizophrenia and other neurodegenerative disorders to tumor development (Giorgi et al., 2010; Mórotz et al., 2012; Sander et al., 2015).
MERCs dysfunctions could be caused not only by mutations of proteins that exert their function at these sites, but also by impaired targeting of MERCs-resident proteins. The subsequent pathological conditions associated to MERCs defects will be more evident in tissues where that specific MERCs protein is mostly expressed/active. This is the case, for example, of CNX. CNX acts mostly as a chaperone for glycoproteins, which are key molecules for the development and maintenance of myelin structure (Denzel et al., 2002; Quarles, 2002). Hence, it is predictable that defective CNX would cause myelinopathy: this is actually the case, as confirmed in CNX knockout mice (Kraus et al., 2010). Demyelination has diverse causes, such as for example mutations in myelin basic proteins or altered activity of enzymes responsible for the production of cholesteryl esters: defective MERCs activity should be added to the list of possible mechanisms underlying it.
As reported above, MERCs have been implied in metabolic diseases, like obesity and diabetes (Tubbs and Rieusset, 2017). Hepatocytes from obese mice are characterized by increased coupling between mitochondria and ER, and the consequent mitochondrial Ca2+ overload is paralleled by higher mitochondrial reactive oxygen species (ROS) production and abnormal glucose metabolism (Arruda et al., 2014). This phenotype can be ameliorated upon silencing of PACS-2 and IP3R1, leading to lower cell stress and increased glucose tolerance (Arruda et al., 2014).
Another MERCs protein, Mfn2, besides being the genetic cause of an inherited peripheral neuropathy (Charcot Marie Tooth 2a), has also been associated with metabolic dysfunctions (Sebastián et al., 2012; Boutant et al., 2017). A recent study highlighted that metabolic transitions in liver are accompanied by changes in the MERCs structure (Sood et al., 2014), further suggesting that MERCs play an active role in metabolic processes: hence, even mild dysfunction of MERCs could exacerbate a given pathological condition.
Downregulation or mutations of a protein that regulates broad physicochemical properties of MERCs may alter the recruitment or stability of defined subsets of MERCs components, thus preferentially impacting specific functions or metabolic routes. An example of such scenario is showcased by models of genetic deficiency in Cav1 protein. Quantitative proteomic profiling of MAM fractions purified from livers of Cav1KO mice shows a depletion of steroid metabolism and fatty acid catabolism regulators (Sala-Vila et al., 2016). It remains to be elucidated whether these changes are due to aberrant membrane composition (i.e., high free cholesterol), and which is their contribution to the metabolic phenotypes associated with Cav1 deficiency (i.e., lipodystrophy and metabolic inflexibility; Bosch et al., 2011b; Fernández-Rojo et al., 2013; Parton and del Pozo, 2013). MERCs-associated lipid metabolism might be of relevance not only for metabolic phenotypes: it could add to the pathogenesis of neurodegenerative disorders like Alzheimer's disease (AD). One of the main gene products associated to familial cases of AD (presenilins, PSs) is enriched in MAMS, although the link between MAMs and sporadic AD is less obvious (Zampese et al., 2011). Importantly, a genetic connection exists between phospholipid/cholesterol dyshomeostasis and AD (Mapstone et al., 2014; Chang et al., 2017). Further, inhibition of cholesterol transport impairs PSs localization at the ER, inducing their accumulation in vesicles and enhancing the production of the main component of AD neurofibrillary tangles, Aβ (Runz et al., 2002). Finally, an apolipoprotein E variant associated with higher risk of lipid metabolism-associated disorders (ApoE4) specifically alters MERCs lipid metabolism and favor AD-like changes in vitro (Area-Gomez et al., 2012).
Another interesting example of a potentially pathological mislocalization of a MERCs component is C19orf12. Mutations in this protein, whose physiological function is yet unknown, are the genetic cause of Mitochondrial Membrane Protein Associated Neurodegeneration (MPAN, Hartig et al., 2013). This severe, early-onset pathological condition is characterized by optic atrophy, generalized dystonia, neuropathy, and psychiatric symptoms. Interestingly, C19orf12 has been retrieved at mitochondria, ER and MAMs, and its mutated forms appear to mislocalize. The evidence that fibroblasts from MPAN patients are characterized by higher mitochondria Ca2+ uptake suggest that this protein somehow regulates MERCs function, its localization likely causing enhanced ER-mitochondria Ca2+ transfer and hence increased sensitivity to apoptosis (Venco et al., 2015). Notably, C19orf12 mutations have been linked to Parkinson's Disease (PD), strengthening the possibility that PD is linked to defective MERCs function. This hypothesis is substantiated by a number of additional findings. For example α-synuclein (α-syn), a protein whose mutations are linked to PD, has been also retrieved in MAMs (Eliezer et al., 2001; Jao et al., 2008; Guardia-Laguarta et al., 2014). The group of E. Schon demonstrated that mutated α-syn has lower affinity for MERCs, thus challenging the theory that mutant α-syn are “gain of function”, and favoring a “loss of MAMs function” hypothesis (Guardia-Laguarta et al., 2014, 2015). Additional data support defective MERCs contribution to the etiology of PD: mutants for parkin (PARK2), DJ-1 (PARK7), and PINK1 (PARK6), all causing recessive early-onset PD cases, can impact on ER-mitochondria tethering, mitochondrial quality control, and Ca2+ transfer between the two organelles (Li et al., 2005; Narendra et al., 2008; Davison et al., 2009; Ziviani et al., 2010; Calì et al., 2013).
Mutated MERCs proteins are not the only reason for the development of MERCs linked diseases or symptoms: being “raft”-like domains, changes in lipid homeostasis could exert deleterious effects on their structure/composition. One of such example could be atherosclerosis. It has been shown that an ER overload of cholesterol in murine macrophages causes prolonged ER stress and UPR activation culminating in apoptosis, substantially contributing to the progression of this disease (Tabas, 2002; Feng et al., 2003). An intriguing question is whether MERCs are modulated in arterial wall cell populations at different stages of atherosclerosis progression, and what their pathogenic impact may be. Notably, MERCs could also take part to the inflammatory response involved in such pathologies, as they have also been involved in the activation of the inflammasome complex and ILβ production, although this needs to be more deeply investigated (Zhou et al., 2011; Lerner et al., 2012; Marchi et al., 2014).
Altogether these findings suggest that defective MERCs localization is likely not only to constitute discrete, primary elements of pathogenesis, but also to be a source of epistatic effects underlying the impact of additive risk factors.
Conclusions
In the last years the interest on MERCs biology has exponentially grown, due to the evidence that at these interfaces many biological processes integrate and that MERCs defects underlie several pathological conditions. Many proteins have been retrieved in the biochemical counterparts of MERCs (that are, MAM fractions, see above) but so far the mechanisms responsible for targeting at MERCs have yet not been fully elucidated. Interestingly, post-translational modifications such as palmitoylation, miristoylation, and oxidation seem to gain the upper hand over a more canonical targeting signal. Another standing question pertains as to how special conditions, such as ER stress, specifically contribute to determine MERCs composition and hence functional state. It is likely that several independent features of an ER-stressed cell take part to such remodeling. Acute ER stress is frequently associated with alteration (mostly attenuation) of signaling pathways which are considered to stabilize MERCs, namely AKT-mTOR signaling (Betz et al., 2013), or with increased ER Ca2+ levels, that could in turn enhance (at least in early stress phases) mitochondria-ER proximity as an adaptive response for the maintenance of intracellular Ca2+ homeostasis (Bravo et al., 2011). Chronic ER stress as well can induce MERCs remodeling: for example, lipid imbalance associated with obesity might promote connectivity between the two organelles, in an attempt to restore equilibrium among different lipid species and to exert a tighter control on Ca2+ homeostasis, which is significantly perturbed in such dyslipidemic states (Fu et al., 2011; Arruda et al., 2014).
Overall, despite a small number of elegant studies on MERCs targeting mechanisms and protein relocation at MAM fractions have been published (such as for example, Myhill et al., 2008; Lynes et al., 2012), this aspect in the field of MERCs biology appears to be just at its infancy. Exciting findings lie ahead, and their discovery will certainly represent another step forward into the complexity of cellular signal transduction, as well as in the understanding of pathological processes.
Author Contributions
MG, MS-Á, and MD: conceived, designed, and wrote the manuscript; NI, MB, and VC: drafted and revised the article; MS-Á and VC prepared figures.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer IB and handling Editor declared their shared affiliation.
Acknowledgments
This work was supported by CARIPARO Starting Grant 2016 AIFbiol (to MG) and DiBio Departmental Research Project PRID Seed 2017 (to MG); MD received support from grants SAF2014-51876-R from MINECO (Spanish Ministry of Economy and Competitiveness) and 15-0404 from the Worldwide Cancer Research Foundation. MS-Á is recipient of an IPP-CNIC postdoctoral fellow award. The CNIC is supported by the Spanish Ministry of Economy and Competitiveness (MINECO) and the Pro-CNIC Foundation, and is a Severo Ochoa Center of Excellence (SEV-2015-0505). We thank I. Navarro-Lérida and Aleix Sala-Vila for generation of data critical for this review.
Abbreviations
AD, Alzheimer's disease; ASC, apoptosis-associated speck-like protein containing a CARD; ATF4/6, activating transcription factor 4/6; BIM, Bcl2-interacting mediator of cell death; CHOP, C/EBP-homologous protein; CNX, calnexin; DGAT2, diacylglycerol O-acyltransferase 2; eIF2, eukaryotic translation initiation factor 2; ER, endoplasmic reticulum; ERAD, ER-associated degradation; ERMES, endoplasmic reticulum (ER)-mitochondria encounter structure; FACL4, fatty acid-CoA ligase 4; GRP75, 75 kDa glucose-regulated protein; IMM, inner mitochondrial membrane; IP3R, inositol triphosphate receptor; IRE1, inositol requiring enzyme 1; MAM, mitochondria-associated membrane; MERC, mitochondria-ER contact site; MFN1/2, Mitofusin 1/2; MPAN, mitochondrial membrane protein associated neurodegeneration; mTOR, mammalian target of rapamycin; OMM, outer mitochondrial membrane; PACS2, phosphofurin acidic cluster sorting protein 2; PC, phosphatidylcholine; PD, Parkinson's disease; PE, phosphatidylethanolamine; PERK, protein kinase R-like endoplasmic reticulum kinase; PI(3,5)P2, phosphatidylinositol 3,5-bisphosphate; PI3K, phosphoinositide 3-kinase; PrP, prion protein; PS, phosphatidylserine; PSD, phosphatidylserine decarboxylase; PSS1/2, phosphatidylserine synthase 1/2; PTEN, phosphatase and tensin homolog; PTM, post-translational modifications; PTP, permeability transition pore; PTPIP51, protein tyrosine phosphatase interacting protein 51; PUMA, p53 up-regulated modulator of apoptosis; REEP1, receptor expression-enhancing protein 1; ROS, reactive oxygen species; RRBP1, ribosome-binding protein 1; S1/2P, site 1/2 protease; SMP, synaptotagmin-like mitochondrial-lipid binding protein; STBD1, starch binding domain-containing protein 1; SYNJ2BP, synaptojanin-2-binding protein; TMX, thioredoxin-related transmembrane protein; TXNIP, thioredoxin-interacting protein; UPR, unfolded protein response; VAPB, vesicle-associated membrane protein associated protein B; VDAC, voltage-gated anion channel; XBP1, X-box binding protein 1.
References
Annunziata, I., Patterson, A., and d'Azzo, A. (2013). Mitochondria-associated ER membranes (MAMs) and glycosphingolipid enriched microdomains (GEMs): isolation from mouse brain. J. Vis. Exp. 4:e50215. doi: 10.3791/50215
Area-Gomez, E., del Carmen Lara Castillo, M., Tambini, M. D., Guardia-Laguarta, C., de Groof, A. J. C., Madra, M., et al. (2012). Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 31, 4106–4123. doi: 10.1038/emboj.2012.202
Arensdorf, A. M., Diedrichs, D., and Rutkowski, D. T. (2013). Regulation of the transcriptome by ER stress: non-canonical mechanisms and physiological consequences. Front. Genet. 4:256. doi: 10.3389/fgene.2013.00256
Arruda, A. P., Pers, B. M., Parlakgül, G., Güney, E., Inouye, K., and Hotamisligil, G. S. (2014). Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat. Med. 20, 1427–1435. doi: 10.1038/nm.3735
Bauer, N. C., Doetsch, P. W., and Corbett, A. H. (2015). Mechanisms regulating protein localization. Traffic 16, 1039–1061. doi: 10.1111/tra.12310
Bernardi, P., Petronilli, V., Di Lisa, F., and Forte, M. (2001). A mitochondrial perspective on cell death. Trends Biochem. Sci. 26, 112–117. doi: 10.1016/S0968-0004(00)01745-X
Betz, C., Stracka, D., Prescianotto-Baschong, C., Frieden, M., Demaurex, N., and Hall, M. N. (2013). Feature Article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc. Natl. Acad. Sci. U.S.A. 110, 12526–12534. doi: 10.1073/pnas.1302455110
Bhatia, V. K., Hatzakis, N. S., and Stamou, D. (2010). A unifying mechanism accounts for sensing of membrane curvature by BAR domains, amphipathic helices and membrane-anchored proteins. Semin. Cell Dev. Biol. 21, 381–390. doi: 10.1016/j.semcdb.2009.12.004
Bononi, A., Bonora, M., Marchi, S., Missiroli, S., Poletti, F., Giorgi, C., et al. (2013). Identification of PTEN at the ER and MAMs and its regulation of Ca(2+) signaling and apoptosis in a protein phosphatase-dependent manner. Cell Death Differ. 20, 1631–1643. doi: 10.1038/cdd.2013.77
Bosch, M., Marí, M., Gross, S. P., Fernández-Checa, J. C., and Pol, A. (2011a). Mitochondrial cholesterol: a connection between caveolin, metabolism, and disease. Traffic 12, 1483–1489. doi: 10.1111/j.1600-0854.2011.01259.x
Bosch, M., Marí, M., Herms, A., Fernández, A., Fajardo, A., Kassan, A., et al. (2011b). Caveolin-1 deficiency causes cholesterol-dependent mitochondrial dysfunction and apoptotic susceptibility. Curr. Biol. 21, 681–686. doi: 10.1016/j.cub.2011.03.030
Boutant, M., Kulkarni, S. S., Joffraud, M., Ratajczak, J., Valera-Alberni, M., Combe, R., et al. (2017). Mfn2 is critical for brown adipose tissue thermogenic function. EMBO J. 36, 1543–1558. doi: 10.15252/embj.201694914
Braakman, I., and Bulleid, N. J. (2011). Protein folding and modification in the mammalian endoplasmic reticulum. Annu. Rev. Biochem. 80, 71–99. doi: 10.1146/annurev-biochem-062209-093836
Bravo, R., Vicencio, J. M., Parra, V., Troncoso, R., Munoz, J. P., Bui, M., et al. (2011). Increased ER–mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J. Cell Sci. 124, 2143–2152. doi: 10.1242/jcs.080762
Brodsky, J. L., and Skach, W. R. (2011). Protein folding and quality control in the endoplasmic reticulum: recent lessons from yeast and mammalian cell systems. Curr. Opin. Cell Biol. 23, 464–475. doi: 10.1016/j.ceb.2011.05.004
Bromley, S.K., Burack, W.R., Johnson, K.G., Somersalo, K., Sims, T.N., Sumen, C., et al. (2001). The immunological synapse. Annu. Rev. Immunol. 19, 375–396. doi: 10.1158/2326-6066.CIR-14-0161
Browman, D.T., Resek, M.E., Zajchowski, L.D., and Robbins, S.M. (2006). Erlin-1 and erlin-2 are novel members of the prohibitin family of proteins that define lipid-raft-like domains of the ER. J. Cell Sci. 119(Pt 15), 3149–3160. doi: 10.1242/jcs.03060
Bui, M., Gilady, S. Y., Fitzsimmons, R. E. B., Benson, M. D., Lynes, E. M., Gesson, K., et al. (2010). Rab32 modulates apoptosis onset and mitochondria-associated membrane (MAM) properties. J. Biol. Chem. 285, 31590–31602. doi: 10.1074/jbc.M110.101584
Cajigas, I. J., Tushev, G., Will, T. J., tom Dieck, S., Fuerst, N., and Schuman, E. M. (2012). The local transcriptome in the synaptic neuropil revealed by deep sequencing and high-resolution imaging. Neuron 74, 453–466. doi: 10.1016/j.neuron.2012.02.036
Calì, T., Ottolini, D., Negro, A., and Brini, M. (2013). Enhanced parkin levels favor ER-mitochondria crosstalk and guarantee Ca(2+) transfer to sustain cell bioenergetics. Biochim. Biophys. Acta 1832, 495–508. doi: 10.1016/j.bbadis.2013.01.004
Calfon, M., Zeng, H., Urano, F., Till, J. H., Hubbard, S. R., Harding, H. P., et al. (2002). IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415, 92–96. doi: 10.1038/415092a
Cárdenas, C., Miller, R. A., Smith, I., Bui, T., Molgó, J., Müller, M., et al. (2010). Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 142, 270–283. doi: 10.1016/j.cell.2010.06.007
Chakrabarti, A., Chen, A. W., and Varner, J. D. (2011). A review of the mammalian unfolded protein response. Biotechnol. Bioeng. 108, 2777–2793. doi: 10.1002/bit.23282
Chami, M., Oulès, B., Szabadkai, G., Tacine, R., Rizzuto, R., and Paterlini-Bréchot, P. (2008). Role of SERCA1 truncated isoform in the proapoptotic calcium transfer from ER to mitochondria during ER stress. Mol. Cell 32, 641–651. doi: 10.1016/j.molcel.2008.11.014
Chang, T. Y., Yamauchi, Y., Hasan, M. T., and Chang, C. C. (2017). Cellular cholesterol homeostasis in Alzheimer's disease. J. Lipid Res. 58, 2239–2254. doi: 10.1194/jlr.R075630
Chao, J. T., Wong, A. K. O., Tavassoli, S., Young, B. P., Chruscicki, A., Fang, N. N., et al. (2014). Polarization of the endoplasmic reticulum by ER-septin tethering. Cell 158, 620–632. doi: 10.1016/j.cell.2014.06.033
Chen, H., Detmer, S. A., Ewald, A. J., Griffin, E. E., Fraser, S. E., and Chan, D. C. (2003). Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell. Biol. 160, 189–200. doi: 10.1083/jcb.200211046
Csordás, G., Renken, C., Várnai, P., Walter, L., Weaver, D., Buttle, K. F., et al. (2006). Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 174, 915–921. doi: 10.1083/jcb.200604016
Cui, Z., Vance, J. E., Chen, M. H., Voelker, D. R., and Vance, D. E. (1993). Cloning and expression of a novel phosphatidylethanolamine N-methyltransferase. A specific biochemical and cytological marker for a unique membrane fraction in rat liver. J. Biol. Chem. 268, 16655–16663.
Davison, E. J., Pennington, K., Hung, C. C., Peng, J., Rafiq, R., Ostareck-Lederer, A., et al. (2009). Proteomic analysis of increased Parkin expression and its interactants provides evidence for a role in modulation of mitochondrial function. Proteomics 9, 4284–4297. doi: 10.1002/pmic.200900126
de Brito, O. M., and Scorrano, L. (2008). Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610. doi: 10.1038/nature07534
De Mario, A., Quintana-Cabrera, R., Martinvalet, D., and Giacomello, M. (2016). (Neuro)degenerated mitochondria-ER contacts. Biochem. Biophys. Res. Commun. 483, 1096–1109. doi: 10.1016/j.bbrc.2016.07.056
De Vos, K. J., Mórotz, G. M., Stoica, R., Tudor, E. L., Lau, K.-F., Ackerley, S., et al. (2012). VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum. Mol. Genet. 21, 1299–1311. doi: 10.1093/hmg/ddr559
Demetriadou, A., Morales-Sanfrutos, J., Nearchou, M., Baba, O., Kyriacou, K., Tate, E. W., et al. (2017). Mouse Stbd1 is N-myristoylated and affects ER-mitochondria association and mitochondrial morphology. J. Cell Sci. 130, 903–915. doi: 10.1242/jcs.195263
Denton, R. M., Randle, P. J., and Martin, B. R. (1972). Stimulation by calcium ions of pyruvate dehydrogenase phosphate phosphatase. Biochem. J. 128, 161–163. doi: 10.1042/bj1280161
Denton, R. M., Richards, D. A., and Chin, J. G. (1978). Calcium ions and the regulation of NAD+-linked isocitrate dehydrogenase from the mitochondria of rat heart and other tissues. Biochem. J. 176, 899–906. doi: 10.1042/bj1760899
Denzel, A., Molinari, M., Trigueros, C., Martin, J. E., Velmurgan, S., Brown, S., et al. (2002). Early postnatal death and motor disorders in mice congenitally deficient in calnexin expression. Mol. Cell. Biol. 22, 7398–7404. doi: 10.1128/MCB.22.21.7398-7404.2002
Eisenberg-Bord, M., Shai, N., Schuldiner, M., and Bohnert, M. (2016). A tether is a tether is a tether: tethering at membrane contact sites. Dev. Cell 39, 395–409. doi: 10.1016/j.devcel.2016.10.022
Eletto, D., Eletto, D., Boyle, S., and Argon, Y. (2016). PDIA6 regulates insulin secretion by selectively inhibiting the RIDD activity of IRE1. FASEB J. 30, 653–665. doi: 10.1096/fj.15-275883
Eletto, D., Eletto, D., Dersh, D., Gidalevitz, T., and Argon, Y. (2014). Protein disulfide Isomerase A6 controls the decay of IRE1α signaling via disulfide-dependent association. Mol. Cell. 53, 562–576. doi: 10.1016/j.molcel.2014.01.004
Elgass, K. D., Smith, E. A., LeGros, M. A., Larabell, C. A., and Ryan, M. T. (2015). Analysis of ER-mitochondria contacts using correlative fluorescence microscopy and soft X-ray tomography of mammalian cells. J. Cell Sci. 128, 2795–2804. doi: 10.1242/jcs.169136
Eliezer, D., Kutluay, E., Bussell, R., and Browne, G. (2001). Conformational properties of alpha-synuclein in its free and lipid-associated states. J. Mol. Biol. 307, 1061–1073. doi: 10.1006/jmbi.2001.4538
Ellenrieder, L., Rampelt, H., and Becker, T. (2017). Connection of protein transport and organelle contact sites in mitochondria. J. Mol. Biol. 429, 2148–2160. doi: 10.1016/j.jmb.2017.05.023
Ellgaard, L., and Helenius, A. (2003). Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 4, 181–191. doi: 10.1038/nrm1052
Feng, B., Yao, P. M., Li, Y., Devlin, C. M., Zhang, D., Harding, H. P., et al. (2003). The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat. Cell Biol. 5, 781–792. doi: 10.1038/ncb1035
Ferguson, M. A., Kinoshita, T., and Hart, G. W. (2009). “Glycosylphosphatidylinositol anchors,” in Essentials of Glycobiology, eds A. Varki, R. D. Cummings, J. D. Esko, H. H. Freeze, P. Stanley, C. R. Bertozzi, et al. (Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press). Available online at: http://www.ncbi.nlm.nih.gov/books/NBK1966/
Fernández-Rojo, M. A., Gongora, M., Fitzsimmons, R. L., Martel, N., Martin, S. D., Nixon, S. J., et al. (2013). Caveolin-1 is necessary for hepatic oxidative lipid metabolism: evidence for crosstalk between Caveolin-1 and bile acid signaling. Cell Rep. 4, 238–247. doi: 10.1016/j.celrep.2013.06.017
Flis, V. V., and Daum, G. (2013). Lipid transport between the endoplasmic reticulum and mitochondria. Cold Spring Harb. Perspect. Biol. 5:a013235. doi: 10.1101/cshperspect.a013235
Friedman, J. R., Lackner, L. L., West, M., DiBenedetto, J. R., Nunnari, J., and Voeltz, G. K. (2011). ER tubules mark sites of mitochondrial division. Science 334, 358–362. doi: 10.1126/science.1207385
Fu, S., Yang, L., Li, P., Hofmann, O., Dicker, L., Hide, W., et al. (2011). Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 473, 528–531. doi: 10.1038/nature09968
Galehdar, Z., Swan, P., Fuerth, B., Callaghan, S. M., Park, D. S., and Cregan, S. P. (2010). Neuronal apoptosis induced by endoplasmic reticulum stress is regulated by ATF4-CHOP-mediated induction of the Bcl-2 homology 3-only member PUMA. J. Neurosci. 30, 16938–16948. doi: 10.1523/JNEUROSCI.1598-10.2010
Galmes, R., Houcine, A., van Vliet, A. R., Agostinis, P., Jackson, C. L., and Giordano, F. (2016). ORP5/ORP8 localize to endoplasmic reticulum-mitochondria contacts and are involved in mitochondrial function. EMBO Rep. 17, 800–810. doi: 10.15252/embr.201541108
Giacomello, M., and Pellegrini, L. (2016). The coming of age of the mitochondria-ER contact: a matter of thickness. Cell Death Differ. 23, 1417–1427. doi: 10.1038/cdd.2016.52
Gilady, S. Y., Bui, M., Lynes, E. M., Benson, M. D., Watts, R., Vance, J. E., et al. (2010). Ero1alpha requires oxidizing and normoxic conditions to localize to the mitochondria-associated membrane (MAM). Cell Stress Chaperones 15, 619–629. doi: 10.1007/s12192-010-0174-1
Giorgi, C., Ito, K., Lin, H. K., Santangelo, C., Wieckowski, M. R., Lebiedzinska, M., et al. (2010). PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science 330, 1247–1251. doi: 10.1126/science.1189157
Gomez-Suaga, P., Paillusson, S., Stoica, R., Noble, W., Hanger, D. P., and Miller, C. C. J. (2017). The ER-mitochondria tethering complex VAPB-PTPIP51 regulates autophagy. Curr. Biol. 27, 371–385. doi: 10.1016/j.cub.2016.12.038
Goswami, R., Singh, D., Phillips, G., Kilkus, J., and Dawson, G. (2005). Ceramide regulation of the tumor suppressor phosphatase PTEN in rafts isolated from neurotumor cell lines. J. Neurosci. Res. 81, 541–550. doi: 10.1002/jnr.20550
Grakoui, A., Bromley, S. K., Sumen, C., Davis, M. M., Shaw, A. S., Allen, P. M., et al. (1999). The immunological synapse: a molecular machine controlling T cell activation. Science 285, 221–227.
Guardia-Laguarta, C., Area-Gomez, E., Schon, E. A., and Przedborski, S. (2015). Novel subcellular localization for α-synuclein: possible functional consequences. Front. Neuroanat. 9:17. doi: 10.3389/fnana.2015.00017
Guardia-Laguarta, C., Area-Gomez, E., Rüb, C., Liu, Y., Magrané, J., Becker, D., et al. (2014). α-Synuclein is localized to mitochondria-associated ER membranes. J. Neurosci. 34, 249–259. doi: 10.1523/JNEUROSCI.2507-13.2014
Halbleib, K., Pesek, K., Covino, R., Hofbauer, H. F., Wunnicke, D., Hänelt, I., et al. (2017). Activation of the unfolded protein response by lipid bilayer stress. Mol. Cell 67, 673.e8–684.e8. doi: 10.1016/j.molcel.2017.06.012
Hamasaki, M., Furuta, N., Matsuda, A., Nezu, A., Yamamoto, A., Fujita, N., et al. (2013). Autophagosomes form at ER-mitochondria contact sites. Nature 495, 389–393. doi: 10.1038/nature11910
Harding, H. P., Zhang, Y., and Ron, D. (1999). Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397, 271–274. doi: 10.1038/16729
Hartig, M., Prokisch, H., Meitinger, T., and Klopstock, T. (2013). Mitochondrial membrane protein-associated neurodegeneration (MPAN). Int. Rev. Neurobiol. 110, 73–84. doi: 10.1016/B978-0-12-410502-7.00004-1
Hatzakis, N. S., Bhatia, V. K., Larsen, J., Madsen, K. L., Bolinger, P.Y., Kunding, A. H., et al. (2009). How curved membranes recruit amphipathic helices and protein anchoring motifs. Nat. Chem. Biol. 5, 835–841. doi: 10.1038/nchembio.213
Hayashi, T., and Fujimoto, M. (2010). Detergent-resistant microdomains determine the localization of sigma-1 receptors to the endoplasmic reticulum-mitochondria junction. Mol. Pharmacol. 77, 517–528. doi: 10.1124/mol.109.062539
Hayashi, T., and Su, T. P. (2007). Sigma-1 receptor chaperones at the ER- mitochondrion interface regulate Ca2+ signalling and cell survival. Cell 131, 596–610. doi: 10.1016/j.cell.2007.08.036
Haze, K., Yoshida, H., Yanagi, H., Yura, T., and Mori, K. (1999). Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 10, 3787–3799. doi: 10.1091/mbc.10.11.3787
Hetz, C. (2012). The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 13, 89–102. doi: 10.1038/nrm3270
Hetz, C., Bernasconi, P., Fisher, J., Lee, A.H., Bassik, M.C., Antonsson, B., et al. (2006). Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1alpha. Science 312, 572–576. doi: 10.1126/science.1123480
Hill, M. M., Feng, J., and Hemmings, B. A. (2002). Identification of a plasma membrane Raft-associated PKB Ser473 kinase activity that is distinct from ILK and PDK1. Curr. Biol. 12, 1251–1255. doi: 10.1016/S0960-9822(02)00973-9
Horner, S. M., Wilkins, C., Badil, S., Iskarpatyoti, J., and Gale, M. (2015). Proteomic analysis of mitochondrial-associated ER Membranes (MAM) during RNA virus infection reveals dynamic changes in protein and organelle trafficking. PLoS ONE 10:e0117963. doi: 10.1371/journal.pone.0117963
Hung, V., Lam, S. S., Udeshi, N. D., Svinkina, T., Guzman, G., Mootha, V. K., et al. (2017). Proteomic mapping of cytosol-facing outer mitochondrial and ER membranes in living human cells by proximity biotinylation. Elife 6:e24463. doi: 10.7554/eLife.24463
Hurst, S., Hoek, J., and Sheu, S.-S. (2017). Mitochondrial Ca2+ and regulation of the permeability transition pore. J. Bioenerg. Biomembr. 49, 27–47. doi: 10.1007/s10863-016-9672-x
Iwasawa, R., Mahul-Mellier, A. L., Datler, C., Pazarentzos, E., and Grimm, S. (2011). Fis1 and Bap31 bridge the mitochondria-ER interface to establish a platform for apoptosis induction. EMBO J. 30, 556–568. doi: 10.1038/emboj.2010.346
Jao, C. C., Hegde, B. G., Chen, J., Haworth, I. S., and Langen, R. (2008). Structure of membrane-bound alpha-synuclein from site-directed spin labeling and computational refinement. Proc. Natl. Acad. Sci. U.S.A. 105, 19666–19671. doi: 10.1073/pnas.0807826105
Ji, W. K., Hatch, A. L., Merrill, R. A., Strack, S., and Higgs, H. N. (2015). Actin filaments target the oligomeric maturation of the dynamin GTPase Drp1 to mitochondrial fission sites. Elife 4:e11553. doi: 10.7554/eLife.11553
Kejiou, N. S., and Palazzo, A. F. (2017). mRNA localization as a rheostat to regulate subcellular gene expression. Wiley Interdiscipl. Rev. RNA 8:e1416. doi: 10.1002/wrna.1416
Kono, N., Amin-Wetzel, N., and Ron, D. (2017). Generic membrane spanning features endow IRE1α with responsiveness to membrane aberrancy. Mol. Biol. Cell 28, 2318–2332. doi: 10.1091/mbc.E17-03-0144
Kornmann, B., Currie, E., Collins, S. R., Schuldiner, M., Nunnari, J., Weissman, J. S., et al. (2009). An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science 325, 477–481. doi: 10.1126/science.1175088
Kornmann, B., Osman, C., and Walter, P. (2011). The conserved GTPase Gem1 regulates endoplasmic reticulum-mitochondria connections. Proc. Natl. Acad. Sci. U.S.A. 108, 14151–14156. doi: 10.1073/pnas.1111314108
Kraus, A., Groenendyk, J., Bedard, K., Baldwin, T. A., Krause, K. H., Dubois-Dauphin, M., et al. (2010). Calnexin deficiency leads to dysmyelination. J. Biol. Chem. 285, 18928–18938. doi: 10.1074/jbc.M110.107201
Lebiedzinska, M., Duszynski, J., Rizzuto, R., Pinton, P., and Wieckowski, M. R. (2009). Age-related changes in levels of p66Shc and serine 36-phosphorylated p66Shc in organs and mouse tissues. Arch. Biochem. Biophys. 486, 73–80. doi: 10.1016/j.abb.2009.03.007
Lerner, A. G., Upton, J. P., Praveen, P. V., Ghosh, R., Nakagawa, Y., Igbaria, A., et al. (2012). IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 16, 250–264. doi: 10.1016/j.cmet.2012.07.007
Lewin, T. M., Kim, J. H., Granger, D. A., Vance, J. E., and Coleman, R. A. (2001). Acyl-CoA synthetase isoforms 1, 4, and 5 are present in different subcellular membranes in rat liver and can be inhibited independently. J. Biol. Chem. 276, 24674–24679. doi: 10.1074/jbc.M102036200
Lewis, S. C., Uchiyama, L. F., and Nunnari, J. (2016). ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 353:aaf5549. doi: 10.1126/science.aaf5549
Li, Y., Tomiyama, H., Sato, K., Hatano, Y., Yoshino, H., Atsumi, M., et al. (2005). Clinicogenetic study of PINK1 mutations in autosomal recessive early-onset parkinsonism. Neurology 64, 1955–1957. doi: 10.1212/01.WNL.0000164009.36740.4E
Liu, Z., Du, X., Deng, J., Gu, M., Hu, H., Gui, M., et al. (2015). The interactions between mitochondria and sarcoplasmic reticulum and the proteome characterization of mitochondrion-associated membrane from rabbit skeletal muscle. Proteomics 15, 2701–2704. doi: 10.1002/pmic.201400493
Lu, P. D., Harding, H. P., and Ron, D. (2004). Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J. Cell Biol. 167, 27–33. doi: 10.1083/jcb.200408003
Lynes, E. M., and Simmen, T. (2011). Urban planning of the endoplasmic reticulum (ER): how diverse mechanisms segregate the many functions of the ER. Biochim. Biophys. Acta 1813, 1893–1905. doi: 10.1016/j.bbamcr.2011.06.011
Lynes, E. M., Bui, M., Yap, M. C., Benson, M. D., Schneider, B., Ellgaard, L., et al. (2012). Palmitoylated TMX and calnexin target to the mitochondria-associated membrane. EMBO J. 31, 457–470. doi: 10.1038/emboj.2011.384
Mapstone, M., Cheema, A. K., Fiandaca, M. S., Zhong, X., Mhyre, T. R., MacArthur, L. H., et al. (2014). Plasma phospholipids identify antecedent memory impairment in older adults. Nat. Med. 20, 415–418. doi: 10.1038/nm.3466
Marchi, S., Giorgi, C., Oparka, M., Duszynski, J., Wieckowski, M. R., and Pinton, P. (2014). Oncogenic and oncosuppressive signal transduction at mitochondria-associated endoplasmic reticulum membranes. Mol. Cell Oncol. 1:e956469. doi: 10.4161/23723548.2014.956469
Martínez-Pizarro, A., Desviat, L. R., Ugarte, M., Pérez, B., and Richard, E. (2016). Endoplasmic reticulum stress and autophagy in homocystinuria patients with remethylation defects. PLoS ONE 11:e0150357. doi: 10.1371/journal.pone.0150357
Mattei, V., Matarrese, P., Garofalo, T., Tinari, A., Gambardella, L., Ciarlo, L., et al. (2011). Recruitment of cellular prion protein to mitochondrial raft-like microdomains contributes to apoptosis execution. Mol. Biol. Cell 22, 4842–4853. doi: 10.1091/mbc.E11-04-0348
McCullough, K. D., Martindale, J. L., Klotz, L. O., Aw, T. Y., and Holbrook, N. J. (2001). Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell. Biol. 21, 1249–1259. doi: 10.1128/MCB.21.4.1249-1259.2001
Mori, T., Hayashi, T., Hayashi, E., and Su, T. P. (2013). Sigma-1 receptor chaperone at the ER-mitochondrion interface mediates the mitochondrion-ER-nucleus signaling for cellular survival. PLoS ONE 8:e76941. doi: 10.1371/journal.pone.0076941
Mórotz, G. M., De Vos, K. J., Vagnoni, A., Ackerley, S., Shaw, C. E., and Miller, C. C. (2012). Amyotrophic lateral sclerosis-associated mutant VAPBP56S perturbs calcium homeostasis to disrupt axonal transport of mitochondria. Hum. Mol. Genet. 21, 1979–1988. doi: 10.1093/hmg/dds011
Muñoz, J. P., Ivanova, S., Sánchez-Wandelmer, J., Martínez-Cristóbal, P., Noguera, E., Sancho, A., et al. (2013). Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J. 32, 2348–2361. doi: 10.1038/emboj.2013.168
Myhill, N., Lynes, E. M., Nanji, J. A., Blagoveshchenskaya, A. D., Fei, H., Carmine Simmen, K., et al. (2008). The subcellular distribution of calnexin is mediated by PACS-2. Mol. Biol. Cell 19, 2777–2788. doi: 10.1091/mbc.E07-10-0995
Naidoo, N. (2009). ER and aging-Protein folding and the ER stress response. Ageing Res. Rev. 8, 150–159. doi: 10.1016/j.arr.2009.03.001
Naon, D., Zaninello, M., Giacomello, M., Varanita, T., Grespi, F., Lakshminaranayan, S., et al. (2016). Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum–mitochondria tether. Proc. Natl. Acad. Sci. U.S.A. 113, 11249–11254. doi: 10.1073/pnas.1606786113
Narendra, D., Tanaka, A., Suen, D. F., and Youle, R. J. (2008). Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183, 795–803. doi: 10.1083/jcb.200809125
Ngoh, G. A., Papanicolaou, K. N., and Walsh, K. (2012). Loss of mitofusin 2 promotes endoplasmic reticulum stress. J. Biol. Chem. 287, 20321–20332. doi: 10.1074/jbc.M112.359174
Okada, T., Haze, K., Nadanaka, S., Yoshida, H., Seidah, N. G., Hirano, Y., et al. (2003). A serine protease inhibitor prevents endoplasmic reticulum stress-induced cleavage but not transport of the membrane-bound transcription factor ATF6. J. Biol. Chem. 278, 31024–31032. doi: 10.1074/jbc.M300923200
Okazaki, Y., Ohno, H., Takase, K., Ochiai, T., and Saito, T. (2000). Cell surface expression of calnexin, a molecular chaperone in the endoplasmic reticulum. J. Biol. Chem. 275, 35751–35758. doi: 10.1074/jbc.M007476200
Parton, R. G., and del Pozo, M. A. (2013). Caveolae as plasma membrane sensors, protectors and organizers. Nat. Rev. Mol. Cell Biol. 14, 98–112. doi: 10.1038/nrm3512
Piccini, M., Vitelli, F., Bruttini, M., Pober, B. R., Jonsson, J. J., Villanova, M., et al. (1998). FACL4, a new gene encoding long-chain acyl-CoA synthetase 4, is deleted in a family with Alport syndrome, elliptocytosis, and mental retardation. Genomics 47, 350–358.
Poston, C. N., Duong, E., Cao, Y., and Bazemore-Walker, C. R. (2011). Proteomic analysis of lipid raft-enriched membranes isolated from internal organelles. Biochem. Biophys. Res. Commun. 415, 355–360. doi: 10.1016/j.bbrc.2011.10.072
Prudent, J., and McBride, H. M. (2017). The mitochondria-endoplasmic reticulum contact sites: a signalling platform for cell death. Curr. Opin. Cell Biol. 47, 52–63. doi: 10.1016/j.ceb.2017.03.007
Prudent, J., and McBride, H. M. (2016). Mitochondrial dynamics: ER actin tightens the Drp1 noose. Curr. Biol. 26, R207–R209. doi: 10.1016/j.cub.2016.01.009
Puthalakath, H., O'Reilly, L. A., Gunn, P., Lee, L., Kelly, P. N., Huntington, N. D., et al. (2007). ER stress triggers apoptosis by activating BH3-only protein bim. Cell 129, 1337–1349. doi: 10.1016/j.cell.2007.04.027
Quarles, R. H. (2002). Myelin sheaths: glycoproteins involved in their formation, maintenance and degeneration. Cell. Mol. Life Sci. 59, 1851–1871. doi: 10.1007/PL00012510
Ramachandran, R., and Schmid, S. L. (2008). Real-time detection reveals that effectors couple dynamin's GTP-dependent conformational changes to the membrane. EMBO J. 27, 27–37. doi: 10.1038/sj.emboj.7601961
Ramamurthi, K. S., Lecuyer, S., Stone, H. A., and Losick, R. (2009). Geometric cue for protein localization in a bacterium. Science 323, 1354–1357. doi: 10.1126/science.1169218
Rangaraju, V., tom Dieck, S., and Schuman, E. M. (2017). Local translation in neuronal compartments: how local is local? EMBO Rep. 18, 693–711. doi: 10.15252/embr.201744045
Ron, D. (2002). Translational control in the endoplasmic reticulum stress response. J. Clin. Invest. 110, 1383–1388. doi: 10.1172/JCI0216784
Rowland, A. A., and Voeltz, G. K. (2012). Endoplasmic reticulum-mitochondria contacts: function of the junction. Nat. Rev. Mol. Cell Biol. 13, 607–625. doi: 10.1038/nrm3440
Runz, H., Rietdorf, J., Tomic, I., de Bernard, M., Beyreuther, K., Pepperkok, R., et al. (2002). Inhibition of intracellular cholesterol transport alters presenilin localization and amyloid precursor protein processing in neuronal cells. J. Neurosci. 22, 1679–1689.
Rusiñol, A. E., Cui, Z., Chen, M. H., and Vance, J. E. (1994). A unique mitochondria-associated membrane fraction from rat liver has a high capacity for lipid synthesis and contains pre-Golgi secretory proteins including nascent lipoproteins. J. Biol. Chem. 269, 27494–27502.
Rutkowski, D. T., and Kaufman, R. J. (2004). A trip to the ER: coping with stress. Trends Cell Biol. 14, 20–28. doi: 10.1016/j.tcb.2003.11.001
Sala-Vila, A., Navarro-Lérida, I., Sánchez-Alvarez, M., Bosch, M., Calvo, C., López, J. A., et al. (2016). Interplay between hepatic mitochondria-associated membranes, lipid metabolism and caveolin-1 in mice. Sci. Rep. 6:27351. doi: 10.1038/srep27351
Salminen, A., Kaarniranta, K., Kauppinen, A., Ojala, J., Haapasalo, A., Soininen, H., et al. (2013). Impaired autophagy and APP processing in Alzheimer's disease: the potential role of Beclin 1 interactome. Prog. Neurobiol. 106–107, 33–54. doi: 10.1016/j.pneurobio.2013.06.002
Sanchez-Alvarez, M., del Pozo, M. A., and Bakal, C. (2017). AKT-mTOR signaling modulates the dynamics of IRE1 RNAse activity by regulating ER-mitochondria contacts. Sci. Rep. 7:16497. doi: 10.1038/s41598-017-16662-1
Sander, E. L., Wootton, J. T., and Allesina, S. (2015). What can interaction webs tell us about species roles? PLoS Comput. Biol. 11:e1004330. doi: 10.1371/journal.pcbi.1004330
Schneeberger, M., Dietrich, M. O., Sebastián, D., Imbernón, M., Casta-o, C., Garcia, A., et al. (2013). Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell 155, 172–187. doi: 10.1016/j.cell.2013.09.003
Sebastián, D., Hernández-Alvarez, M. I., Segalés, J., Sorianello, E., Mu-oz, J. P., Sala, D., et al. (2012). Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc. Natl. Acad. Sci. U.S.A. 109, 5523–5528. doi: 10.1073/pnas.1108220109
Simmen, T., Aslan, J. E., Blagoveshchenskaya, A. D., Thomas, L., Wan, L., Xiang, Y., et al. (2005). PACS-2 controls endoplasmic reticulum–mitochondria communication and Bid-mediated apoptosis. EMBO J. 24, 717–729. doi: 10.1038/sj.emboj.7600559
Simons, K., and Sampaio, J. L. (2011). Membrane organization and lipid rafts. Cold Spring Harb. Perspect. Biol. 3:a004697. doi: 10.1101/cshperspect.a004697
Simons, K., and van Meer, G. (1988). Lipid sorting in epithelial cells. Biochemistry 27, 6197–6202. doi: 10.1021/bi00417a001
Smith, M. H., Ploegh, H. L., and Weissman, J. S. (2011). Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science 334, 1086–1090. doi: 10.1126/science.1209235
Sood, A., Jeyaraju, D. V., Prudent, J., Caron, A., Lemieux, P., McBride, H. M., et al. (2014). A Mitofusin-2-dependent inactivating cleavage of Opa1 links changes in mitochondria cristae and ER contacts in the postprandial liver. Proc. Natl. Acad. Sci. U.S.A. 111, 16017–16022. doi: 10.1073/pnas.1408061111
Stoica, R., Vos, K. J. D., Paillusson, S., Mueller, S., Sancho, R. M., Lau, K.-F., et al. (2014). ER–mitochondria associations are regulated by the VAPB–PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 5:3996. doi: 10.1038/ncomms4996
Stone, S. J., Levin, M. C., Zhou, P., Han, J., Walther, T. C., and Farese, R. V. (2009). The endoplasmic reticulum enzyme DGAT2 is found in mitochondria-associated membranes and has a mitochondrial targeting signal that promotes its association with mitochondria. J. Biol. Chem. 284, 5352–5361. doi: 10.1074/jbc.M805768200
Stone, S. J., and Vance, J. E. (2000). Phosphatidylserine synthase-1 and -2 are localized to mitochondria-associated membranes. J. Biol. Chem. 275, 34534–34540. doi: 10.1074/jbc.M002865200
Stroud, D. A., Oeljeklaus, S., Wiese, S., Bohnert, M., Lewandrowski, U., Sickmann, A., et al. (2011). Composition and topology of the endoplasmic reticulum-mitochondria encounter structure. J. Mol. Biol. 413, 743–750. doi: 10.1016/j.jmb.2011.09.012
Sugiura, A., Nagashima, S., Tokuyama, T., Amo, T., Matsuki, Y., Ishido, S., et al. (2013). MITOL regulates endoplasmic reticulum-mitochondria contacts via Mitofusin2. Mol. Cell. 51, 20–34. doi: 10.1016/j.molcel.2013.04.023
Szabadkai, G., Bianchi, K., Várnai, P., De Stefani, D., Wieckowski, M. R., Cavagna, D., et al. (2006). Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 175, 901–911. doi: 10.1083/jcb.200608073
Tabas, I. (2002). Consequences of cellular cholesterol accumulation: basic concepts and physiological implications. J. Clin. Invest. 110, 905–911. doi: 10.1172/JCI0216452
Taylor, K. M., and Roseman, M. A. (1995). Effect of cholesterol, fatty acyl chain composition, and bilayer curvature on the interaction of cytochrome b5 with liposomes of phosphatidylcholines. Biochemistry 34, 3841–3850. doi: 10.1021/bi00011a042
Tirasophon, W., Welihinda, A. A., and Kaufman, R. J. (1998). A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 12, 1812–1824. doi: 10.1101/gad.12.12.1812
Tsai, Y. C., and Weissmann, A. M. (2010). The unfolded protein response, degradation from the endoplasmic reticulum, and cancer. Genes Cancer 1, 764–778. doi: 10.1177/1947601910383011
Tubbs, E., and Rieusset, J. (2017). Metabolic signaling functions of ER-mitochondria contact sites: role in metabolic diseases. J. Mol. Endocrinol. 58, R87–R106. doi: 10.1530/JME-16-0189
Tubbs, E., Theurey, P., Vial, G., Bendridi, N., Bravard, A., Chauvin, M.-A., et al. (2014). Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 63, 3279–3294. doi: 10.2337/db13-1751
Updegrove, T. B., and Ramamurthi, K. S. (2017). Geometric protein localization cues in bacterial cells. Curr. Opin. Microbiol. 36, 7–13. doi: 10.1016/j.mib.2016.12.001
Vance, D. E., and Vance, J. E. (2009). Physiological consequences of disruption of mammalian phospholipid biosynthetic genes. J. Lipid Res. 50(Suppl.), S132–S137. doi: 10.1194/jlr.R800048-JLR200
Vance, J. E. (1990). Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 265, 7248–7256.
Vance, J. E. (2014). MAM (mitochondria-associated membranes) in mammalian cells: lipids and beyond. Biochim. Biophys. Acta 1841, 595–609. doi: 10.1016/j.bbalip.2013.11.014
Venco, P., Bonora, M., Giorgi, C., Papaleo, E., Iuso, A., Prokisch, H., et al. (2015). Mutations of C19orf12, coding for a transmembrane glycine zipper containing mitochondrial protein, cause mis-localization of the protein, inability to respond to oxidative stress and increased mitochondrial Ca2+. Front. Genet. 6:185. doi: 10.3389/fgene.2015.00185
Verfaillie, T., Rubio, N., Garg, A. D., Bultynck, G., Rizzuto, R., Decuypere, J.-P., et al. (2012). PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 19, 1880–1891. doi: 10.1038/cdd.2012.74
Viola, A., Contento, R. L., and Molon, B. (2010). Signaling amplification at the immunological synapse. Curr. Top. Microbiol. Immunol. 340, 109–122. doi: 10.1007/978-3-642-03858-7_6
Volmer, R., and Ron, D. (2015). Lipid-dependent regulation of the unfolded protein response. Curr. Opin. Cell Biol. 33, 67–73. doi: 10.1016/j.ceb.2014.12.002
Volmer, R., van der Ploeg, K., and Ron, D. (2013). Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc. Natl. Acad. Sci. U.S.A. 110, 4628–4633. doi: 10.1073/pnas.1217611110
Walter, P., and Ron, D. (2011). The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086. doi: 10.1126/science.1209038
Wieckowski, M. R., Giorgi, C., Lebiedzinska, M., Duszynski, J., and Pinton, P. (2009). Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat. Protoc. 4, 1582–1590. doi: 10.1038/nprot.2009.151
Wiest, D. L., Burgess, W. H., McKean, D., Kearse, K. P., and Singer, A. (1995). The molecular chaperone calnexin is expressed on the surface of immature thymocytes in association with clonotype-independent CD3 complexes. EMBO J. 14, 3425–3433.
Wortmann, S. B., Vaz, F. M., Gardeitchik, T., Vissers, L. E., Renkema, G. H., Schuurs-Hoeijmakers, J. H., et al. (2012). Mutations in the phospholipid remodeling gene SERAC1 impair mitochondrial function and intracellular cholesterol trafficking and cause dystonia and deafness. Nat. Genet. 44, 797–802. doi: 10.1038/ng.2325
Wu, W., Li, W., Chen, H., Jiang, L., Zhu, R., and Feng, D. (2016). FUNDC1 is a novel mitochondrial-associated-membrane (MAM) protein required for hypoxia-induced mitochondrial fission and mitophagy. Autophagy 12, 1675–1676. doi: 10.1080/15548627.2016.1193656
Xue, Y., Schmollinger, S., Attar, N., Campos, O. A., Vogelauer, M., Carey, M. F., et al. (2017). Endoplasmic reticulum-mitochondria junction is required for iron homeostasis. J. Biol. Chem. 292, 13197–13204. doi: 10.1074/jbc.M117.784249
Yoshida, H., Matsui, T., Yamamoto, A., Okada, T., and Mori, K. (2001). XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891. doi: 10.1016/S0092-8674(01)00611-0
Yoshida, H., Okada, T., Haze, K., Yanagi, H., Yura, T., Negishi, M., et al. (2000). ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol. Cell Biol. 20, 6755–6767. doi: 10.1128/MCB.20.18.6755-6767.2000
Zampese, E., Fasolato, C., Kipanyula, M. J., Bortolozzi, M., Pozzan, T., and Pizzo, P. (2011). Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc. Natl. Acad. Sci. U.S.A. 108, 2777–2782. doi: 10.1073/pnas.1100735108
Zhou, R., Yazdi, A. S., Menu, P., and Tschopp, J. (2011). A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225. doi: 10.1038/nature09663
Zhuang, L., Kim, J., Adam, R. M., Solomon, K. R., and Freeman, M. R. (2005). Cholesterol targeting alters lipid raft composition and cell survival in prostate cancer cells and xenografts. J. Clin. Invest. 115, 959–968. doi: 10.1172/JCI200519935
Keywords: mitochondria-ER contact sites, protein targeting, post-translational modifications, lipid rafts, ER stress
Citation: Ilacqua N, Sánchez-Álvarez M, Bachmann M, Costiniti V, Del Pozo MA and Giacomello M (2017) Protein Localization at Mitochondria-ER Contact Sites in Basal and Stress Conditions. Front. Cell Dev. Biol. 5:107. doi: 10.3389/fcell.2017.00107
Received: 12 September 2017; Accepted: 24 November 2017;
Published: 12 December 2017.
Edited by:
Nuno Raimundo, Universitätsmedizin Göttingen, GermanyReviewed by:
Ivan Bogeski, University of Göttingen, GermanyXin Qi, Case Western Reserve University School of Medicine, United States
Ove Eriksson, University of Helsinki, Finland
Copyright © 2017 Ilacqua, Sánchez-Álvarez, Bachmann, Costiniti, Del Pozo and Giacomello. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marta Giacomello, bWFydGEuZ2lhY29tZWxsb0B1bmlwZC5pdA==
†These authors have contributed equally to this work.