 Pasquale Cocchiaro1,2,3,4,5†
Pasquale Cocchiaro1,2,3,4,5† Valeria De Pasquale1†
Valeria De Pasquale1† Rossella Della Morte6
Rossella Della Morte6 Simona Tafuri6
Simona Tafuri6 Luigi Avallone6Anne Pizard2,3,4,5
Luigi Avallone6Anne Pizard2,3,4,5 Anna Moles7*
Anna Moles7* Luigi Michele Pavone1*
Luigi Michele Pavone1*- 1Department of Molecular Medicine and Medical Biotechnology, University of Naples Federico II, Naples, Italy
- 2Faculty of Medicine, Institut National de la Santé Et de la Recherche Médicale, “Défaillance Cardiaque Aigüe et Chronique”, Nancy, France
- 3Université de Lorraine, Nancy, France
- 4Institut Lorrain du Coeur et des Vaisseaux, Center for Clinical Investigation 1433, Nancy, France
- 5CHRU de Nancy, Hôpitaux de Brabois, Nancy, France
- 6Department of Veterinary Medicine and Animal Productions, University of Naples Federico II, Naples, Italy
- 7Institute of Cellular Medicine, Newcastle University, Newcastle upon Tyne, United Kingdom
Kidney disease is worldwide the 12th leading cause of death affecting 8–16% of the entire population. Kidney disease encompasses acute (short-lasting episode) and chronic (developing over years) pathologies both leading to renal failure. Since specific treatments for acute or chronic kidney disease are limited, more than 2 million people a year require dialysis or kidney transplantation. Several recent evidences identified lysosomal proteases cathepsins as key players in kidney pathophysiology. Cathepsins, originally found in the lysosomes, exert important functions also in the cytosol and nucleus of cells as well as in the extracellular space, thus participating in a wide range of physiological and pathological processes. Based on their catalytic active site residue, the 15 human cathepsins identified up to now are classified in three different families: serine (cathepsins A and G), aspartate (cathepsins D and E), or cysteine (cathepsins B, C, F, H, K, L, O, S, V, X, and W) proteases. Specifically in the kidney, cathepsins B, D, L and S have been shown to regulate extracellular matrix homeostasis, autophagy, apoptosis, glomerular permeability, endothelial function, and inflammation. Dysregulation of their expression/activity has been associated to the onset and progression of kidney disease. This review summarizes most of the recent findings that highlight the critical role of cathepsins in kidney disease development and progression. A better understanding of the signaling pathways governed by cathepsins in kidney physiopathology may yield novel selective biomarkers or therapeutic targets for developing specific treatments against kidney disease.
Introduction
Kidneys are complex organs whose excretory, biosynthetic and metabolic activities are essential for healthy living. They regulate body fluid balance, blood pressure, waste removal, and red blood cells production (Preuss, 1993; Adamson, 1996). Kidney functions take place through mechanisms of filtration, reabsorption and secretion occurring in the nephrons, the basic structural and functional units of the kidney (Figure 1). Nephron components filter the blood free of cells and large proteins, producing an ultrafiltrate composed of the other smaller circulating elements. The ultrafiltrate enters tubule segments to produce the final urine by removing (reabsorption) or adding (secretion) substances from or to the tubular fluid (Gueutin et al., 2012; Mount, 2014). Indeed, by adapting the quality composition of urine to the needs of the body, kidneys keep the organism in balance of water, hydrogen ion concentration, electrolytes, and minerals, and eliminate the toxic substances produced in the body. Deregulation of kidney functions may lead to severe pathological conditions affecting different tissues and organs.
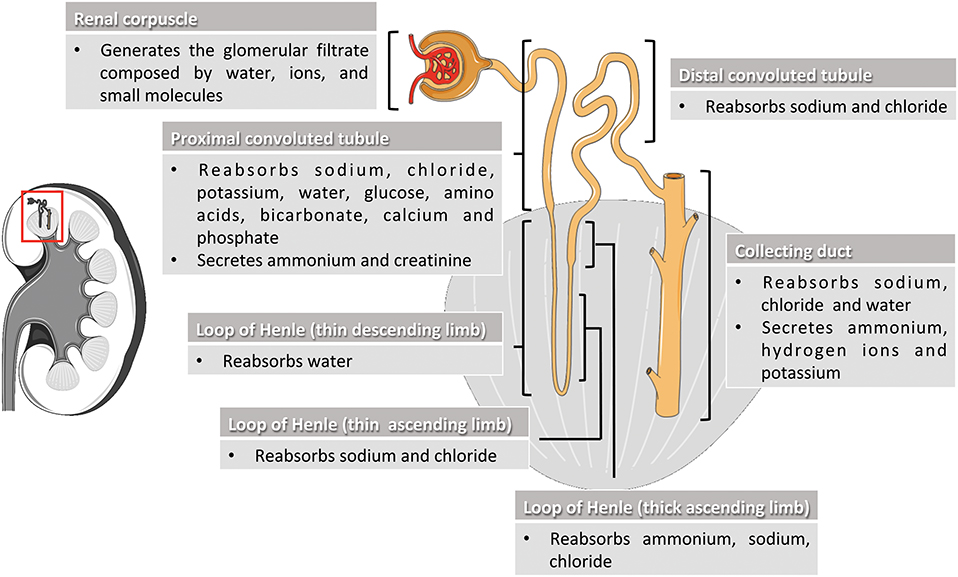
Figure 1. Nephron segments and their main physiological function. The nephron is the functional unit of the kidney and is composed by the renal corpuscle and the renal tubule. In the renal corpuscle, the glomerular filtrate is generated by filtration of water, ions, and small molecules from the bloodstream. The glomerular filtrate is transformed into urine by reabsorption and secretion of different molecules through the different sections of the renal tubule (proximal convoluted tubule, loop of Henle, and distal convoluted tubule) and the collecting duct system. Kidney and nephron image adapted from Smart Servier Medical Art under Creative Commons Attribution 3.0 Unported License.
Kidney diseases are worldwide the 12th leading cause of death and the 17th cause for loss of healthy life years (Glassock et al., 2017). They are classified into two major groups of pathologies depending on the length of the disease encompassing acute kidney injury (AKI), which is an abrupt reduction of kidney functions within 48 hours (Mehta et al., 2007; Rewa and Bagshaw, 2014), and chronic kidney disease (CKD) that is a gradual loss of renal function over years (Jha et al., 2013; Hill et al., 2016). AKI is associated with an high mortality rate (30–70%) and can have long-term consequences predisposing to CKD development (Coca et al., 2009). Due to the lack of adequate specific treatments, many patients (>2 millions worldwide) progress from CKD to end-stage renal disease and organ failure, requiring dialysis, or kidney transplantation (Hu and Coresh, 2017). Management of AKI and CKD represents a massive burden for the health care systems (Kerr et al., 2014), and CKD is on the rise due to the aging of the population and the clinical complications associated with diabetes and hypertension (Jobs et al., 2011; Tonelli and Riella, 2014). Therefore, there is an urgent need to increase our understanding on kidney disease pathogenesis to find new selective biomarkers or therapeutic candidates for drug development.
In this scenario, emerging evidence demonstrate the important role for lysosomal proteases cathepsins (Cts) in the onset and progression of kidney disease (Svara et al., 2010; Moallem et al., 2011; Ozkayar et al., 2015; Cocchiaro et al., 2016; Fox et al., 2016; Yamamoto-Nonaka et al., 2016; Conley et al., 2017). Lysosomes are ubiquitous organelles responsible for the catabolism and recycling of different types of macromolecules and constitute the major degradative compartment of the cell (Cuervo and Dice, 1998). They are involved in the renal epithelial molecular machinery underlying kidney physiology (Surendran et al., 2014). Two classes of proteins mediate lysosomal activity: integral lysosomal membrane proteins and soluble lysosomal hydrolases.
Among hydrolases, Cts are implicated in multiple cellular processes ranging from the processing of proteins and hormones to the regulation of cell cycle, autophagy, cell death, and immune response (Ciechanover, 2012). Altered expression and/or activity of Cts have been associated with a variety of human diseases (Reiser et al., 2010; Pišlar and Kos, 2014; Stoka et al., 2016). Since a growing number of studies deals with the involvement of Cts in kidney physiopathology, this review aims to highlight the most recent advances in our understanding of the molecular mechanisms by which lysosomal Cts promote kidney disease.
Proteases Cathepsins
To date more than 20 types of Cts have been identified in animals, plants, and microorganisms. In humans, 15 types of Cts have been reported, which can be classified into 3 distinct groups based on the amino acid that comprises the active site residue: serine (Cts A and G), cysteine (Cts B, C, H, F, L, K, O, S, V, X, W), and aspartate proteases (Cts D and E) (Table 1).
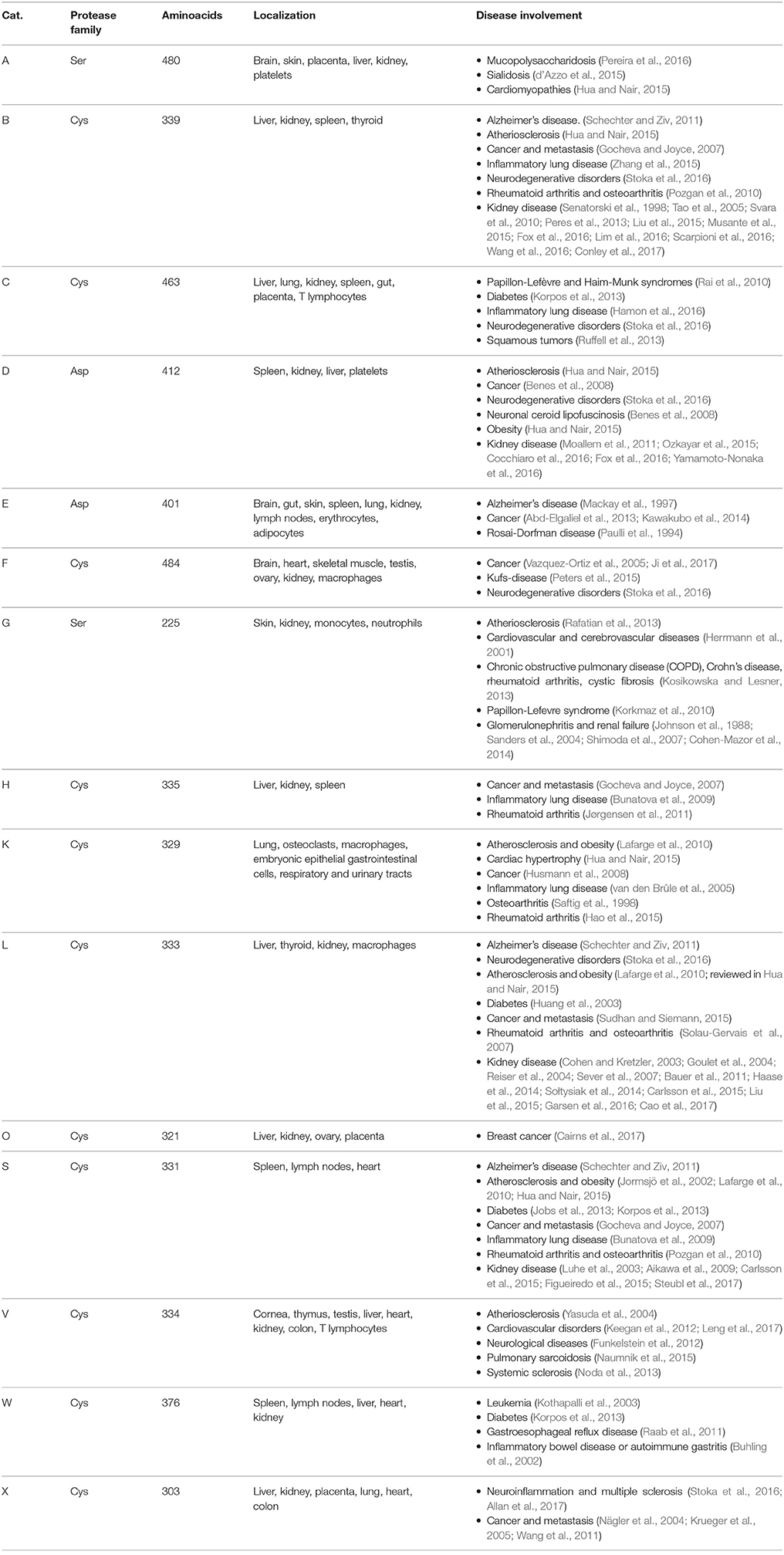
Table 1. Classification, tissue localization and disease involvement of human cathepsins.
Mainly localized in the lysosomes where their activity is facilitated by the lysosomal acidic environment, under certain circumstances, Cts can also be found in the intracellular and extracellular spaces (Stoka et al., 2001, 2005, 2016; Jordans et al., 2009). Indeed, leakage of CtD from the lysosome into the cytosol induces apoptosis (Liaudet-Coopman et al., 2006). In addition, Cts B, D, G, K, L, S, and X participate in the degradation of the major extracellular matrix components in various pathophysiological processes (Brix et al., 2008).
Almost all types of Cts share a common synthetic pathway (Ishidoh and Kominami, 2002). They are synthesized as inactive preproenzyme, and following translocation into the endoplasmic reticulum (ER), the N-terminal signal peptide of the precursor protein is cleaved with simultaneous N-linked glycosylation of the proenzyme (zymogen) (Erickson, 1989; Wiederanders et al., 2003). The propeptide is transported to the Golgi apparatus where it is further glycosylated and phosphorylated to form a mannose-6-phosphate protein that is recognized by the mannose-6-phosphate receptor and carried toward the lysosome where it is hydrolyzed to the active form. This general mechanism of Ct biosynthesis and transport may vary in some cases. The proteolytic cleavage of the zymogen may occur either through an autocatalytic process which is facilitated by the binding of the zymogen to glycosaminoglycans (GAGs) or through the action of other proteases (Dahl et al., 2001; Vasiljeva et al., 2005; Caglic et al., 2007).
Although Cts show similarities in their cellular localization and biosynthesis, they are expressed at different levels in tissues and organs (Table 1). While some Cts such as B, H, L, C, and O are ubiquitously expressed, other Cts such as F, K, S, V, X, and W show a more limited cell and tissue distribution and expression. The differences in tissue localization and expression levels suggest specific cellular functions for different Cts (Brix et al., 2008; Reiser et al., 2010; Stoka et al., 2016). The relevance of the Cts physiological roles in different organs and tissues is supported by multiple evidence demonstrating that abnormal levels or activity of Cts correlate with numerous human diseases, including inflammatory and cardiovascular diseases, neurodegenerative disorders, diabetes, obesity, cancer, kidney dysfunction, and others (Table 1). In particular, depending on the cell type localization, Cts B, D, L, and S regulate in the kidney different physiopathological processes, by activating signaling pathways that ultimately may result in kidney disease (Figure 2).
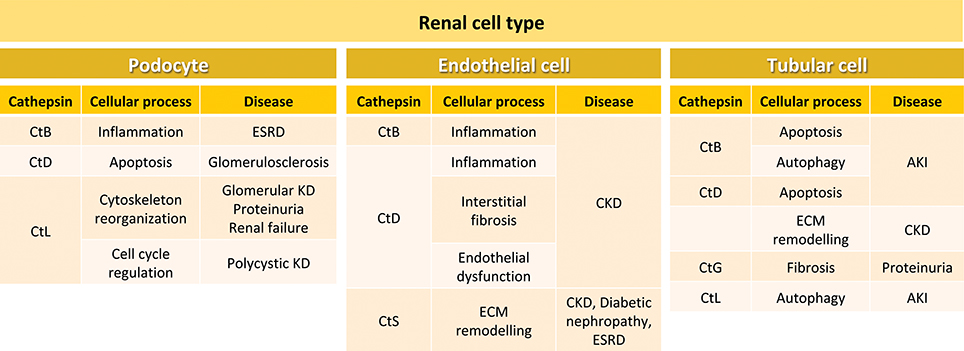
Figure 2. Cellular processes and kidney diseases involving cathepsins in different renal cell types. In podocytes, CtB participates in inflammation during ESRD, CtD is involved in apoptosis in glomerulosclerosis, and CtL plays a role in cytoskeleton reorganization and cell cycle regulation during glomerular kidney disease, proteinuria, renal failure and polycystic kidney disease. In endothelial cells, CtB and CtD are involved in inflammation. In addition, CtD participates in interstitial fibrosis and endothelial dysfunction during CKD. CtS is important in CKD, diabetic nephropathy and ESRD. In tubular cells, CtsB is involved in apoptosis and autophagy during AKI, and CtD in apoptosis and ECM remodeling during CKD. CtG participates in fibrosis during proteinuria, and CtL in autophagy in AKI. ESRD, end-stage renal disease; KD, kidney disease; CKD, chronic kidney disease; ECM, extracellular matrix; AKI, acute kidney injury.
Cathepsins in Acute Kidney Injury (AKI)
AKI is characterized by a relatively sudden reduction, within 48 hours, in kidney function or production, processing, and excretion of ultrafiltrate by the kidney (decreased glomerular filtration rate, GFR) (Mehta et al., 2007). Permanent damage to the microvasculature with subsequent abnormalities in kidney structure and function are caused by AKI. Incomplete recovery from AKI leads to the development of CKD (Venkatachalam et al., 2015; Sud et al., 2016). To date, no effective treatments for AKI are available.
A variety of insults may promote the onset of AKI, leading all of them to epithelial tubular cell death. Increasing evidence demonstrates that ER dysfunction and mitochondrial stress causing tubular damage are important factors in the pathogenesis of AKI (Tábara et al., 2014; Ishimoto and Inagi, 2016; Duann and Lin, 2017; Galvan et al., 2017). Cts play important roles in the signaling pathways driving apoptotic and necrotic cell death, by degrading different substrates and/or contributing to mitochondrial destabilization (Turk et al., 2002; Stoka et al., 2005; Turk and Stoka, 2007). Increased expression levels and activation of CtB have been observed in the human proximal tubular epithelial cell line HK-2 undergoing to apoptosis (Wang et al., 2008). Autophagy induction in proximal tubular cells occurs during AKI (Livingston and Dong, 2014). The activity of CtB and CtL decreases when autophagy-lysosome pathway in HK-2 is disrupted by advanced glycation end products in diabetic nephropathy (Liu et al., 2015). Decreased activity of CtB correlates with an impairment of the autophagic flux and worsening of the renal function in a tubular epithelial cell model (Herzog et al., 2012). In a rat model of AKI, a significant decrease of CtB was detected in the affected proximal tubules, which correlated with increased severity of the histopathological lesions of the tubules (Svara et al., 2010). However, although autophagy causes cell death under certain conditions, a renoprotective role for autophagy in AKI has been established (Jiang et al., 2012). Urinary CtB levels have shown a strong inverse correlation with surrogate markers of nephron number in intrauterine growth-restricted neonates and pre-term infants, suggesting that urinary CtB activity may represent an useful tool for early predicting renal susceptibility to damage in low birth weight neonates (Aisa et al., 2016). Serum CtB concentration directly correlates with the loss of renal function in healthy individuals and the aging-related decrease of kidney function in the normal population (Wang et al., 2016).
The protease CtD is highly expressed in damaged tubular cells suggesting a possible contribution of CtD to cell death in AKI (Cocchiaro et al., 2016). During apoptosis, lysosomal membrane permeabilization allows translocation of CtD from the lysosome into the cytosol where it can exert its pro-apoptotic function. Cytosolic CtD cleaves Bid protein into tBid triggering the insertion of Bax protein into the mitochondrial membrane. This leads to cytochrome c release from the mitochondria into the cytosol, and the activation of pro-caspases 9 and 3 (Stoka et al., 2001). Enhanced CtD expression has been found in murine models of AKI (Kimura et al., 2012; Cocchiaro et al., 2016). CtD has been recently identified as a possible novel prognostic marker for AKI, as it is differentially regulated in urine from late/non recovered vs. early/recovered AKI patients (Aregger et al., 2014).
Translocation of CtL from the lysosome into the cytoplasm is a key event in the induction of glomerular kidney disease (Sever et al., 2007). The onset of proteinuria in kidney dysfunctions reflects a migratory event in the foot processes of the podocytes that correlates with the activation of CtL (Reiser et al., 2004; Cao et al., 2017). Three substrates have been described for cytosolic CtL in podocytes: CD2-associated protein, synaptopodin and dynamin (Sever et al., 2007; Mundel and Reiser, 2010; Yaddanapudi et al., 2011). These proteins are crucial for maintaining the normal cytoskeleton architecture of podocytes, and their degradation by CtL results in the reorganization of the actin cytoskeleton, proteinuria and renal failure (Reiser et al., 2010; Garsen et al., 2016). An emerging role of nuclear CtL in polycystic kidney disease comes out from the evidence that a CtL isoform lacking of a signal peptide localizes to the nucleus in S phase and processes the CDP/Cux transcription factor, thus regulating cell cycle progression (Goulet et al., 2004). The quantification of CtL has been demonstrated to provide a better predictive value for AKI than creatinine, urea and urine output (Haase et al., 2014). Genome expression studies performed with RNA from kidneys of 7-week-old male and female double transgenic rats (dTGRs), harboring human renin and angiotensinogen genes, showed that CtL was differentially expressed between the sexes and was strongly associated with the degree of renal injury (Bauer et al., 2011).
Finally, CtG has been identified as a critical component sustaining neutrophil-mediated acute tissue pathology and subsequent fibrosis after renal ischemia/reperfusion injury (Shimoda et al., 2007). It has been shown that CtG mediates marked changes in glomerular permeability in vivo, contributing to proteinuria (Johnson et al., 1988).
Cathepsins in Chronic Kidney Disease (CKD)
In spite of advance in the development of treatment approaches to improve outcomes, CKD is still associated with a high morbidity and mortality rate for patients affected by kidney dysfunctions (Hill et al., 2016; Glassock et al., 2017). AKI can contribute or worse the progression of CKD because of an abnormal or incomplete repair response (Chawla et al., 2014). The primary glomerular injury leads to a decreased post-glomerular flow, which finally results into peri-tubular capillary loss. Alternatively, renal injury can trigger an inflammatory response that recruits profibrotic cytokines such as transforming growth factor-β, and further induces the transformation of renal epithelial and endothelial cells to myofibroblasts (De Chiara and Crean, 2016; Cruz-Solbes and Youker, 2017). The histopathological hallmark of CKD is tubulointerstitial fibrosis, which is currently thought to be the best predictor to assess progression toward end-stage renal disease (Liu, 2006).
During CKD, CtD plays critical roles in inflammation and endothelial dysfunction (Erdmann et al., 2008; Ozkayar et al., 2015; Fox et al., 2016). Elevated expression levels of CtD have been found in human and murine damaged kidneys. Inhibition of CtD by Pepstatin A in murine models of progressive CKD resulted in a reduction of interstitial fibrosis (Fox et al., 2016). CtD inhibition led to an increase in extracellular protease activity of urokinase-type plasminogen activator (uPA) due to altered lysosomal recycling; UPA processes plasminogen into plasmin, which can degrade extracellular matrix proteins (Eddy, 2009). A role for CtD in podocytes, responsible for maintaining the ultrafiltration barrier thus preventing urinary protein loss, has also been reported (Yamamoto-Nonaka et al., 2016). In a podocyte-specific knock-out mouse model, the absence of CtD resulted in podocyte apoptotic cell death, and in age–dependent, late–onset glomerulosclerosis (Alghamdi et al., 2017). Therefore, CtD activity in kidney could be different depending on the cell type, and further studies will be required to clarify this issue. In CKD, CtD serum levels were significantly higher and correlated with endothelial dysfunction in patients (Ozkayar et al., 2015). However, no correlation was found between serum CtD levels and traditional cardiovascular risk factors, indicating that enhanced CtD could be a selective risk factor for endothelial dysfunction in kidney disease.
Altered levels of CtB activity have been detected under pathological processes in kidney (Ling et al., 1998; Senatorski et al., 1998; Svara et al., 2010). Toll-like receptor 3 (TLR3), which activates both the innate and adaptive immune systems, is cleaved and activated by CtB (Garcia-Cattaneo et al., 2012). CtB-dependent activation of TLR3 leads to the activation of the transcription factors NF-κB and interferon regulatory factor 3, resulting into the production of type I interferons and pro-inflammatory cytokines such as IL-6 and IL-8 (Kawasaki and Kawai, 2014). In the kidney, inflammation promotes the progression of glomerular sclerotic pathologies resulting in end-stage renal disease (Anders and Muruve, 2011; Lim et al., 2016). It has been demonstrated that CtB mediates the signaling pathway activating the inflammasome, a large multiprotein complex containing NOD-like receptor with pyrin domain 3 (NLRP3) which triggers the production of proinflammatory cytokines in response to infection and tissue injury (Conley et al., 2017). NLRP3 inflammasome activation by CtB may promote glomerular inflammation and other cell damages resulting into glomerular injury and end-stage renal disease. Inflammasome activation may occur not only in immune cells but also in residential cells such as endothelial cells and podocytes in the glomeruli (Conley et al., 2017). Thus, NLRP3 inflammasome has been suggested as a potential target for the treatment of progressive CKD (Scarpioni et al., 2016). A correlation between serum CtB concentration and the age-related decline in renal function has been described in healthy individuals (Wang et al., 2016). CtB has also been shown to be involved in diabetic nephropathy (Musante et al., 2015). Other reports demonstrate a reduction of CtB activity during polycystic kidney disease (Schaefer et al., 1996; Hartz and Wilson, 1997; Tao et al., 2005), puromycin induced nephrosis (Huang et al., 1999), and rat and human diabetic nephropathy (Shechter et al., 1994; Grzebyk et al., 2013; Peres et al., 2013). Conversely, CtB expression increased in unilateral ureteric obstruction mouse model, however, its inhibition led to no reduction in kidney fibrosis (Fox et al., 2016).
The expression of CtL results to be enhanced in various glomerular diseases such as focal segmental glomerulosclerosis, membranous glomerulonephritis, and diabetic nephropathy (Baricos et al., 1991; Sever et al., 2007). Induction of CtL expression in podocytes has been associated with the development of proteinuria in puromycin aminonucleoside induced-kidney failure (Reiser et al., 2004), and streptozotocin-induced diabetic nephropathy (Garsen et al., 2016). CtL can contribute to the development of kidney disease by different mechanisms. Cytoplasmic CtL cleaves the GTPase dynamin resulting in podocyte failure and proteinuria (Sever et al., 2007). In addition, CtL activates proteins such as heparanase that are involved in the pathogenesis of diabetic nephropathy (Garsen et al., 2016). Interestingly, CtL expression levels resulted to be lower in males than in females, but the increase in CtL detected with disease progression was greater in males. This evidence strongly suggests that estrogens regulate CtL expression and activity (Bauer et al., 2011). In CKD patients, serum CtL activity is markedly elevated and its levels positively correlate with the severity of proteinuria (Cohen and Kretzler, 2003; Sever et al., 2007; Cao et al., 2017). The presence and severity of proteinuria in patients with CKD is associated with higher mortality and morbidity (Hemmelgarn et al., 2010; Garsen et al., 2016). Elevated CtL activity correlates with higher hospital admission rates in CKD patients (Cao et al., 2017). Urinary excretion of CtL was higher in children with type 1 diabetes mellitus with respect to healthy patients (Sołtysiak et al., 2014).
In contrast with other Ct members, CtS remains catalytically active under neutral pH (optimum pH values, 6.0–7.5) and its main physiological role is outside the lysosome. Intracellularly, CtS has an important role in the intrinsic apoptotic pathways inducing cleavage of both caspase-3 and poly ADP ribose polymerase (Wang et al., 2015). CtS can translocate to the cell surface and be secreted into the extracellular milieu, participating in the degradation of extracellular matrix proteins (Jordans et al., 2009; Wilkinson et al., 2015). Beside its ability to degrade fibers, CtS may activate the protease-activated receptor-2 (PAR2) in endothelial cells (Elmariah et al., 2014). Indeed, in vitro studies demonstrated that CtS may damage the integrity and barrier function of glomerular endothelial cells (Aikawa et al., 2009; Lafarge et al., 2010). In human and mouse type 2 diabetic nephropathy, CtS mRNA resulted to be expressed only in CD68(+) intrarenal monocytes, while the protein was found along endothelial cells and inside proximal tubular epithelial cells (Kumar et al., 2016). High circulating levels of CtS have been correlated with increased mortality risk in the human population (Jobs et al., 2011) because of its involvement in the complex pathways leading to cardiovascular disease, cancer and impaired kidney function (Feldreich et al., 2016). In vivo studies demonstrated that CtS-induced elastolysis stimulates arterial and aortic valve calcification in CKD, suggesting that CtS might be a therapeutic target to prevent cardiovascular complications in CKD (Aikawa et al., 2009). Up-regulation of CtS has been detected in ochratoxin A-induced nephropathy (Luhe et al., 2003). Furthermore, selective CtS inhibition attenuates atherogenesis in hypercholesterolemic mice with CKD (Figueiredo et al., 2015). In mice, serum levels of CtS and markers of inflammation-related endothelial dysfunction, such as soluble tumor-necrosis-factor receptors (sTNFR) 1 and 2, increase with the decline of estimated GFR, while in human cohortes an increase of GFR was associated with a decrease of CtS (Steubl et al., 2017). However, in patients with end-stage renal disease, high levels of CtS were associated with sTNFR1/2 activation (Carlsson et al., 2015). These findings indicate that CtS activity increases with CKD progression, thus representing a potential marker of disease progression.
Conclusions and Perspective
Kidney disease, characterized by the progressive loss of kidney functions, occurs through different steps of damage leading to organ failure and end-stage renal disease. Due to the lack of specific treatments to stop disease progression (Mehta et al., 2007; Black et al., 2010), kidney disease remains an important clinical problem affecting millions of people worldwide (Jha et al., 2013; Hu and Coresh, 2017). In addition, the traditional clinical markers used to assess and monitor kidney function such as serum creatinine, GFR, and the presence of proteinuria often miss the early stages of the disease delaying essential treatment (Mårtensson et al., 2012; Haase et al., 2014; Wasung et al., 2015). Indeed, both of the two major groups of kidney disease, AKI and CKD, are still associated with increasing morbidity and mortality (Coca et al., 2009; Kerr et al., 2014; Hill et al., 2016; Glassock et al., 2017). Therefore, there is an urgent need to better understand the biological events driving AKI and CKD in order to either find more accurate and sensitive biomarkers of cell injury that may predict disease progression or identify critical cellular and molecular mediators that may provide novel therapeutic targets. Lysosomal Cts have emerged in the recent years as important players in kidney disease, thus suggesting their detection as early diagnostic approach. Moreover, targeting Cts or their downstream signaling seems a promising treatment strategy to slow down kidney disease progression. Nevertheless, further studies are required to assess the suitability, specificity and drugability of Cts in human kidney disease.
Author Contributions
PC, VDP, LMP, and AM has conceived, designed the work, written and revised the manuscript. RDM, ST, LA, and AP have collaborated to design, to write and revise the manuscript.
Funding
The work has been supported by a grant from University of Naples Federico II to LMP, and by Northern Countries Kidney Research Fund (NCKRF), Wellcome Trust Institutional Strategic Support and MRC Confidence in Concept funds granted to AM. AM salary is funded through Newcastle University Research Fellowship (NURF) scheme.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abd-Elgaliel, W. R., Cruz-Monserrate, Z., Wang, H., Logsdon, C. D., and Tung, C. H. (2013). Pancreatic cancer-associated Cathepsin E as a drug activator. J. Control Release 167, 221–227. doi: 10.1016/j.jconrel.2013.02.007
Adamson, J. W. (1996). Regulation of red blood cell production. Am. J. Med. 101, 4S−6S. doi: 10.1016/S0002-9343(96)00160-X
Aikawa, E., Aikawa, M., Libby, P., Figueiredo, J. L., Rusanescu, G., Iwamoto, Y., et al. (2009). Arterial and aortic valve calcification abolished by elastolytic cathepsin S deficiency in chronic renal disease. Circulation 119, 1785–1794. doi: 10.1161/CIRCULATIONAHA.108.827972
Aisa, M. C., Cappuccini, B., Barbati, A., Orlacchio, A., Baglioni, M., and Di Renzo, G. C. (2016). Biochemical parameters of renal impairment/injury and surrogate markers of nephron number in intrauterine growth-restricted and preterm neonates at 30-40 days of postnatal corrected age. Pediatr. Nephrol. 31, 2277–2287. doi: 10.1007/s00467-016-3484-4
Alghamdi, T. A., Majumder, S., Thieme, K., Batchu, S. N., White, K. E., Liu, Y., et al. (2017). Janus Kinase 2 regulates transcription factor EB expression and autophagy completion in glomerular podocytes. J. Am. Soc. Nephrol. 28, 2641–2653. doi: 10.1681/ASN.2016111208
Allan, E. R. O., Campden, R. I., Ewanchuk, B. W., Tailor, P., Balce, D. R., McKenna, N. T., et al. (2017). A role for cathepsin Z in neuroinflammation provides mechanistic support for an epigenetic risk factor in multiple sclerosis. J. Neuroinflammation 14:103. doi: 10.1186/s12974-017-0874-x
Anders, H. J., and Muruve, D. A. (2011). The inflammasomes in kidney disease. J. Am. Soc. Nephrol. 22, 1007–1018. doi: 10.1681/ASN.2010080798
Aregger, F., Uehlinger, D. E., Witowski, J., Brunisholz, R. A., Hunziker, P., Frey, F. J., et al. (2014). Identification of IGFBP-7 by urinary proteomics as a novel prognostic marker in early acute kidney injury. Kidney Int. 85, 909–919. doi: 10.1038/ki.2013.363
Baricos, W. H., Cortez, S. L., Le, Q. C., Wu, L. T., Shaw, E., Hanada, K., et al. (1991). Evidence suggesting a role for cathepsin L in an experimental model of glomerulonephritis. Arch. Biochem. Biophys. 288, 468–472. doi: 10.1016/0003-9861(91)90222-5
Bauer, Y., Hess, P., Qiu, C., Klenk, A., Renault, B., Wanner, D., et al. (2011). Identification of cathepsin L as a potential sex-specific biomarker for renal damage. Hypertension 57, 795–801. doi: 10.1161/HYPERTENSIONAHA.110.157206
Benes, P., Vetvicka, V., and Fusek, M. (2008). Cathepsin D–many functions of one aspartic protease. Crit. Rev. Oncol. Hematol. 68, 12–28. doi: 10.1016/j.critrevonc.2008.02.008
Black, C., Sharma, P., Scotland, G., McCullough, K., McGurn, D., Robertson, L., et al. (2010). Early referral strategies for management of people with markers of renal disease: a systematic review of the evidence of clinical effectiveness, cost-effectiveness and economic analysis. Health Technol. Assess. 14, 1–184. doi: 10.3310/hta14210
Brix, K., Dunkhorst, A., Mayer, K., and Jordans, S. (2008). Cysteine cathepsins: cellular roadmap to different functions. Biochimie 90, 194–207. doi: 10.1016/j.biochi.2007.07.024
Buhling, F., Kellner, U., Guenther, D., Kahl, S., Brömme, D., Weber, E., et al. (2002). Characterization of novel anti-cathepsin W antibodies and cellular distribution of cathepsin W in the gastrointestinal tract. Biol. Chem. 383, 1285–1289. doi: 10.1515/BC.2002.144
Bunatova, K., Obermajer, N., Kotyza, J., Pesek, M., and Kos, J. (2009). Levels of cathepsins S and H in pleural fluids of inflammatory and neoplastic origin. Int. J. Biol. Markers 24, 47–51. doi: 10.5301/JBM.2009.1216
Caglic, D., Pungercar, J. R., Pejler, G., Turk, V., and Turk, B. (2007). Glycosaminoglycans facilitate procathepsin B activation through disruption of propeptide-mature enzyme interactions. J. Biol. Chem. 282, 33076–33085. doi: 10.1074/jbc.M705761200
Cairns, J., Ingle, J. N., Wickerham, L. D., Weinshilboum, R., Liu, M., and Wang, L. (2017). SNPs near the cysteine proteinase cathepsin O gene (CTSO) determine tamoxifen sensitivity in ERα-positive breast cancer through regulation of BRCA1. PLoS Genet. 13:e1007031. doi: 10.1371/journal.pgen.1007031
Cao, Y., Liu, X., Li, Y., Lu, Y., Zhong, H., Jiang, W., et al. (2017). Cathepsin L activity correlates with proteinuria in chronic kidney disease in humans. Int. Urol. Nephrol. 49, 1409–1417. doi: 10.1007/s11255-017-1626-7
Carlsson, A. C., Carrero, J. J., Stenvinkel, P., Bottai, M., Barany, P., Larsson, A., et al. (2015). Endostatin, cathepsin S, and cathepsin L, and their association with inflammatory markers and mortality in patients undergoing hemodialysis. Blood Purif. 39, 259–265. doi: 10.1159/000381664
Chawla, L. S., Eggers, P. W., Star, R. A., and Kimmel, P. L. (2014). Acute kidney injury and chronic kidney disease as interconnected syndromes. N. Engl. J. Med. 371, 58–66. doi: 10.1056/NEJMra1214243
Ciechanover, A. (2012). Intracellular protein degradation: from a vague idea thru the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Biochim. Biophys. Acta 1824, 3–13. doi: 10.1016/j.bbapap.2011.03.007
Coca, S. G., Yusuf, B., Shlipak, M. G., Garg, A. X., and Parikh, C. R. (2009). Long-term risk of mortality and other adverse outcomes after acute kidney injury: a systematic review and meta-analysis. Am. J. Kidney Dis. 53, 961–973. doi: 10.1053/j.ajkd.2008.11.034
Cocchiaro, P., Fox, C., Tregidgo, N. W., Howarth, R., Wood, K. M., Situmorang, G. R., et al. (2016). Lysosomal protease cathepsin D; a new driver of apoptosis during acute kidney injury. Sci. Rep. 6:27112. doi: 10.1038/srep27112
Cohen, C. D., and Kretzler, M. (2003). Gene-expression analysis of microdissected renal biopsies. Methods Mol. Med. 86, 285–293. doi: 10.1385/1-59259-392-5:285
Cohen-Mazor, M., Mazor, R., Kristal, B., and Sela, S. (2014). Elastase and cathepsin G from primed leukocytes cleave vascular endothelial cadherin in hemodialysis patients. Biomed. Res. Int. 2014:459640. doi: 10.1155/2014/459640
Conley, S. M., Abais, J. M., Boini, K. M., and Li, P. L. (2017). Inflammasome activation in chronic glomerular diseases. Curr. Drug Targets 18, 1019–1029. doi: 10.2174/1389450117666160817103435
Cruz-Solbes, A. S., and Youker, K. (2017). Epithelial to mesenchymal transition (EMT) and endothelial to mesenchymal transition (EndMT): role and implications in kidney fibrosis. Results Probl. Cell Differ. 60, 345–372. doi: 10.1007/978-3-319-51436-9_13
Cuervo, A. M., and Dice, J. F. (1998). Lysosomes, a meeting point of proteins, chaperones, and proteases. J. Mol. Med. 76, 6–12. doi: 10.1007/s109-1998-8099-y
Dahl, S. W., Halkier, T., Lauritzen, C., Dolenc, I., Pedersen, J., Turk, V., et al. (2001). Human recombinant pro-dipeptidyl peptidase I (cathepsin C) can be activated by cathepsins L and S but not by autocatalytic processing. Biochemistry 40, 1671–1678. doi: 10.1021/bi001693z
d'Azzo, A., Machado, E., and Annunziata, I. (2015). Pathogenesis, emerging therapeutic targets and treatment in sialidosis. Expert Opin. Orphan Drugs 3, 491–504. doi: 10.1517/21678707.2015.1025746
De Chiara, L., and Crean, J. (2016). Emerging transcriptional mechanisms in the regulation of epithelial to mesenchymal transition and cellular plasticity in the kidney. J. Clin. Med. 5:E6. doi: 10.3390/jcm5010006
Duann, P., and Lin, P. H. (2017). Mitochondria damage and kidney disease. Adv. Exp. Med. Biol. 982, 529–551. doi: 10.1007/978-3-319-55330-6_27
Eddy, A. A. (2009). Serine proteases, inhibitors and receptors in renal fibrosis. Thromb. Haemost. 101, 656–664. doi: 10.1160/TH08-12-0779
Elmariah, S. B., Reddy, V. B., and Lerner, E. A. (2014). Cathepsin S signals via PAR2 and generates a novel tethered ligand receptor agonist. PLoS ONE 9:e99702. doi: 10.1371/journal.pone.0099702
Erdmann, S., Ricken, A., Hummitzsch, K., Merkwitz, C., Schliebe, N., Gaunitz, F., et al. (2008). Inflammatory cytokines increase extracellular procathepsin D in permanent and primary endothelial cell cultures. Eur. J. Cell. Biol. 87, 311–323. doi: 10.1016/j.ejcb.2008.01.005
Erickson, A. H. (1989). Biosynthesis of lysosomal endopeptidases. J. Cell. Biochem. 40, 31–41. doi: 10.1002/jcb.240400104
Feldreich, T., Carlsson, A. C., Risérus, U., Larsson, A., Lind, L., and Ärnlöv, J. (2016). The association between serum cathepsin L and mortality in older adults. Atherosclerosis 254, 109–116. doi: 10.1016/j.atherosclerosis.2016.09.062
Figueiredo, J. L., Aikawa, M., Zheng, C., Aaron, J., Lax, L., Libby, P., et al. (2015). Selective cathepsin S inhibition attenuates atherosclerosis in apolipoprotein E–deficient mice with chronic renal disease. Am. J. Pathol. 185, 1156–1166. doi: 10.1016/j.ajpath.2014.11.026
Fox, C., Cocchiaro, P., Oakley, F., Howarth, R., Callaghan, K., Leslie, J., et al. (2016). Inhibition of lysosomal protease cathepsin D reduces renal fibrosis in murine chronic kidney disease. Sci. Rep. 6:20101. doi: 10.1038/srep20101
Funkelstein, L., Lu, W. D., Koch, B., Mosier, C., Toneff, T., Taupenot, L., et al. (2012). Human cathepsin V protease participates in production of enkephalin and NPY neuropeptide neurotransmitters. Biol. Chem. 287, 15232–15241. doi: 10.1074/jbc.M111.310607
Galvan, D. L., Green, N. H., and Danesh, F. R. (2017). The hallmarks of mitochondrial dysfunction in chronic kidney disease. Kidney Int. 92, 1051–1057. doi: 10.1016/j.kint.2017.05.034
Garcia-Cattaneo, A., Gobert, F. X., Müller, M., Toscano, F., Flores, M., Lescure, A., et al. (2012). Cleavage of Toll-like receptor 3 by cathepsins B and H is essential for signaling. Proc. Natl. Acad. Sci. U.S.A. 109, 9053–9058. doi: 10.1073/pnas.1115091109
Garsen, M., Rops, A. L., Dijkman, H., Willemsen, B., van Kuppevelt, T. H., Russel, F. G., et al. (2016). Cathepsin L is crucial for the development of early experimental diabetic nephropathy. Kidney Int. 90, 1012–1022. doi: 10.1016/j.kint.2016.06.035
Glassock, R. J., Warnock, D. G., and Delanaye, P. (2017). The global burden of chronic kidney disease: estimates, variability and pitfalls. Nat. Rev. Nephrol. 13, 104–114. doi: 10.1038/nrneph.2016.163
Gocheva, V., and Joyce, J. A. (2007). Cysteine cathepsins and the cutting edge of cancer invasion. Cell Cycle 6, 60–64. doi: 10.4161/cc.6.1.3669
Goulet, B., Baruch, A., Moon, N. S., Poirier, M., Sansregret, L. L., Erickson, A., et al. (2004). A cathepsin L isoform that is devoid of a signal peptide localizes to the nucleus in S phase and processes the CDP/Cux transcription factor. Mol. Cell. 14, 207–219. doi: 10.1016/S1097-2765(04)00209-6
Grzebyk, E., Knapik-Kordecka, M., and Piwowar, A. (2013). Advanced glycation end-products and cathepsin cysteine protease in type 2 diabetic patients. Pol. Arch. Med. Wewn. 123, 364–370. doi: 10.20452/pamw.1821
Gueutin, V., Deray, G., and Isnard-Bagnis, C. (2012). Renal physiology. Bull. Cancer 99, 237–249. doi: 10.1152/ajprenal.00239.2017
Haase, M., Bellomo, R., Albert, C., Vanpoucke, G., Thomas, G., Laroy, W., et al. (2014). The identification of three novel biomarkers of major adverse kidney events. Biomark. Med. 8, 1207–1217. doi: 10.2217/bmm.14.90
Hamon, Y., Legowska, M., Hervé, V., Dallet-Choisy, S., Marchand-Adam, S., Vanderlynden, L., et al. (2016). Neutrophilic cathepsin C is maturated by a multistep proteolytic process and secreted by activated cells during inflammatory lung diseases. J. Biol. Chem. 291, 8486–8499. doi: 10.1074/jbc.M115.707109
Hao, L., Zhu, G., Lu, Y., Wang, M., Jules, J., Zhou, X., et al. (2015). Deficiency of cathepsin K prevents inflammation and bone erosion in rheumatoid arthritis and periodontitis and reveals its shared osteoimmune role. FEBS Lett. 589, 1331–1339. doi: 10.1016/j.febslet.2015.04.008
Hartz, P. A., and Wilson, P. D. (1997). Functional defects in lysosomal enzymes in autosomal dominant polycystic kidney disease (ADPKD): abnormalities in synthesis, molecular processing, polarity, and secretion. Biochem. Mol. Med. 60, 8–26. doi: 10.1006/bmme.1996.2542
Hemmelgarn, B. R., Manns, B. J., Lloyd, A., James, M. T., Klarenbach, S., Quinn, R. R., et al. (2010). Relation between kidney function, proteinuria, and adverse outcomes. JAMA 303, 423–429. doi: 10.1001/jama.2010.39
Herrmann, S. M., Funke-Kaiser, H., Schmidt-Petersen, K., Nicaud, V., Gautier-Bertrand, M., Evans, A., et al. (2001). Characterization of polymorphic structure of cathepsin G gene: role in cardiovascular and cerebrovascular diseases. Arterioscler. Thromb. Vasc. Biol. 21, 1538–1543. doi: 10.1161/hq0901.095555
Herzog, C., Yang, C., Holmes, A., and Kaushal, G. P. (2012). zVAD-fmk prevents cisplatin-induced cleavage of autophagy proteins but impairs autophagic flux and worsens renal function. Am. J. Physiol. Renal Physiol. 303, F1239–F1250. doi: 10.1152/ajprenal.00659.2011
Hill, N. R., Fatoba, S. T., Oke, J. L., Hirst, J. A., O'Callaghan, C. A., Lasserson, D. S., et al. (2016). Global prevalence of chronic kidney disease – a systematic review and meta-analysis. PLoS ONE 11:e0158765. doi: 10.1371/journal.pone.0158765
Hu, J. R., and Coresh, J. (2017). The public health dimension of chronic kidney disease: what we have learnt over the past decade. Nephrol. Dial. Transplant. 32, ii113–ii120. doi: 10.1093/ndt/gfw416
Hua, Y., and Nair, S. (2015). Proteases in cardiometabolic diseases: pathophysiology, molecular mechanisms and clinical applications. Biochim. Biophys. Acta 1852, 195–208. doi: 10.1016/j.bbadis.2014.04.032
Huang, S., Schaefer, R. M., Reisch, S., Paczek, L., Schaefer, L., Teschner, M., et al. (1999). Suppressed activities of cathepsins and metalloproteinases in the chronic model of puromycin aminonucleoside nephrosis. Kidney Blood Press Res. 22, 121–127. doi: 10.1159/000025917
Huang, X., Vaag, A., Carlsson, E., Hansson, M., Ahrén, B., and Groop, L. (2003). Impaired cathepsin L gene expression in skeletal muscle is associated with type 2 diabetes. Diabetes 52, 2411–2418. doi: 10.2337/diabetes.52.9.2411
Husmann, K., Muff, R., Bolander, M. E., Sarkar, G., Born, W., and Fuchs, B. (2008). Cathepsins and osteosarcoma: expression analysis identifies cathepsin K as an indicator of metastasis. Mol. Carcinog. 47, 66–73. doi: 10.1002/mc.20362
Ishidoh, K., and Kominami, E. (2002). Processing and activation of lysosomal proteinases. Biol. Chem. 383, 1827–1831. doi: 10.1515/BC.2002.206
Ishimoto, Y., and Inagi, R. (2016). Mitochondria: a therapeutic target in acute kidney injury. Nephrol. Dial. Transplant. 31, 1062–1069. doi: 10.1093/ndt/gfv317
Jha, V., Garcia-Garcia, G., Iseki, K., Li, Z., Naicker, S., Plattner, B., et al. (2013). Chronic kidney disease: global dimension and perspectives. Lancet 382, 260–272. doi: 10.1016/S0140-6736(13)60687-X
Ji, C., Zhao, Y., Kou, Y. W., Shao, H., Guo, L., Bao, C. H., et al. (2017). Cathepsin F knockdown induces proliferation and inhibits apoptosis in gastric cancer cells. Oncol. Res. doi: 10.3727/096504017X14928634401204. [Epub ahead of print].
Jiang, M., Wei, Q., Dong, G., Komatsu, M., Su, Y., and Dong, Z. (2012). Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 82, 1271–1283. doi: 10.1038/ki.2012.261
Jobs, E., Ingelsson, E., Risérus, U., Nerpin, E., Jobs, M., Sundström, J., et al. (2011). Association between serum cathepsin S and mortality in older adults. JAMA 306, 1113–1121. doi: 10.1001/jama.2011.1246
Jobs, E., Risérus, U., Ingelsson, E., Sundström, J., Jobs, M., Nerpin, E., et al. (2013). Serum cathepsin S is associated with decreased insulin sensitivity and the development of type 2 diabetes in a community-based cohort of elderly men. Diabetes Care 36, 163–165. doi: 10.2337/dc12-0494
Johnson, R. J., Couser, W. G., Alpers, C. E., Vissers, M., Schulze, M., and Klebanoff, S. J. (1988). The human neutrophil serine proteinases, elastase and cathepsin G, can mediate glomerular injury in vivo. J. Exp. Med. 168, 1169–1174. doi: 10.1084/jem.168.3.1169
Jordans, S., Jenko-Kokalj, S., Kühl, N. M., Tedelind, S., Sendt, W., Brömme, D., et al. (2009). Monitoring compartment-specific substrate cleavage by cathepsins B, K, L and S at physiological pH and redox conditions. BMC Biochem. 10:23. doi: 10.1186/1471-2091-10-23
Jørgensen, I., Kos, J., Krašovec, M., Troelsen, L., Klarlund, M., Jensen, T. W., et al. (2011). Serum cysteine proteases and their inhibitors in rheumatoid arthritis: relation to disease activity and radiographic progression. Clin. Rheumatol. 30, 633–638. doi: 10.1007/s10067-010-1585-1
Jormsjö, S., Wuttge, D. M., Sirsjö, A., Whatling, C., Hamsten, A., Stemme, S., et al. (2002). Differential expression of cysteine and aspartic proteases during progression of atherosclerosis in apolipoprotein E-deficient mice. Am. J. Pathol. 161, 939–945. doi: 10.1016/S0002-9440(10)64254-X
Kawakubo, T., Yasukochi, A., Toyama, T., Takahashi, S., Okamoto, K., Tsukuba, T., et al. (2014). Repression of cathepsin E expression increases the risk of mammary carcinogenesis and links to poor prognosis in breast cancer. Carcinogenesis 35, 714–726. doi: 10.1093/carcin/bgt373
Kawasaki, T., and Kawai, T. (2014). Toll-Like receptor signaling pathways. Front. Immunol. 5:461. doi: 10.3389/fimmu.2014.00461
Keegan, P. M., Surapaneni, S., and Platt, M. O. (2012). Sickle cell disease activates peripheral blood mononuclear cells to induce cathepsins K and V activity in endothelial cells. Anemia 2012:201781. doi: 10.1155/2012/201781
Kerr, M., Bedford, M., Matthews, B., and O'Donoghue, D. (2014). The economic impact of acute kidney injury in England. Nephrol. Dial Transplant. 29, 1362–1368. doi: 10.1093/ndt/gfu016
Kimura, A., Ishida, Y., Inagaki, M., Nakamura, Y., Sanke, T., Mukaida, N., et al. (2012). Interferon-γ is protective in cisplatin-induced renal injury by enhancing autophagic flux. Kidney Int. 82, 1093–1104. doi: 10.1038/ki.2012.240
Korkmaz, B., Horwitz, M. S., Jenne, D. E., and Gauthier, F. (2010). Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol. Rev. 62, 726–759. doi: 10.1124/pr.110.002733
Korpos, É., Kadri, N., Kappelhoff, R., Wegner, J., Overall, C. M., Weber, E., et al. (2013). The peri-islet basement membrane, a barrier to infiltrating leukocytes in type 1 diabetes in mouse and human. Diabetes 62, 531–542. doi: 10.2337/db12-0432
Kosikowska, P., and Lesner, A. (2013). Inhibitors of cathepsin G: a patent review (2005 to present). Expert Opin. Ther. Pat. 23, 1611–1624. doi: 10.1517/13543776.2013.835397
Kothapalli, R., Bailey, R. D., Kusmartseva, I., Mane, S., Epling-Burnette, P. K., Loughran, T. P., et al. (2003). Constitutive expression of cytotoxic proteases and down-regulation of protease inhibitors in LGL leukemia. Int. J. Oncol. 22, 33–39. doi: 10.3892/ijo.22.1.33
Krueger, S., Kalinski, T., Hundertmark, T., Wex, T., Küster, D., Peitz, U., et al. (2005). Up-regulation of cathepsin X in Helicobacter pylori gastritis and gastric cancer. J. Pathol. 207, 32–42. doi: 10.1002/path.1820
Kumar, Vr. S., Darisipudi, M. N., Steiger, S., Devarapu, S. K., Tato, M., Kukarni, O. P., et al. (2016). Cathepsin S cleavage of protease-activated receptor-2 on endothelial cells promotes microvascular diabetes complications. J. Am. Soc. Nephrol. 27, 1635–1649. doi: 10.1681/ASN.2015020208
Lafarge, J. C., Naour, N., Clément, K., and Guerre-Millo, M. (2010). Cathepsins and cystatin C in atherosclerosis and obesity. Biochimie 92, 1580–1586. doi: 10.1016/j.biochi.2010.04.011
Leng, Y. P., Ma, Y. S., Li, X. G., Chen, R. F., Zeng, P. Y., Li, X. H., et al. (2017). l-Homocysteine-induced cathepsin V mediates the vascular endothelial inflammation in hyperhomocysteinaemia. Br. J. Pharmacol. doi: 10.1111/bph.13920. [Epub ahead of print].
Liaudet-Coopman, E., Beaujouin, M., Derocq, D., Garcia, M., Glondu-Lassis, M., Laurent-Matha, V., et al. (2006). Cathepsin D newly discovered functions of a long-standing aspartic protease in cancer and apoptosis. Cancer Lett. 237, 167–179. doi: 10.1016/j.canlet.2005.06.007
Lim, B. J., Yang, J. W., Do, W. S., and Fogo, A. B. (2016). Pathogenesis of focal segmental glomerulosclerosis. J. Pathol. Transl. Med. 50, 405–410. doi: 10.4132/jptm.2016.09.21
Ling, H., Ardjomand, P., Samvakas, S., Simm, A., Busch, G. L., Lang, F., et al. (1998). Mesangial cell hypertrophy induced by NH4Cl: role of depressed activities of cathepsins due to elevated lysosomal pH. Kidney Int. 53, 1706–1712. doi: 10.1046/j.1523-1755.1998.00952.x
Liu, W. J., Shen, T. T., Chen, R. H., Wu, H. L., Wang, Y. J., Deng, J. K., et al. (2015). Autophagy-lysosome pathway in renal tubular epithelial cells is disrupted by advanced glycation end products in diabetic nephropathy. J. Biol. Chem. 290, 20499–20510. doi: 10.1074/jbc.M115.666354
Liu, Y. (2006). Renal fibrosis: new insights into the pathogenesis and therapeutics. Kidney Int. 69, 213–217. doi: 10.1038/sj.ki.5000054
Livingston, M. J., and Dong, Z. (2014). Autophagy in acute kidney injury. Semin. Nephrol. 34, 17–26. doi: 10.1016/j.semnephrol.2013.11.004
Luhe, A., Hildebrand, H., Bach, U., Dingermann, T., and Ahr, H. J. (2003). A new approach to studying ochratoxin A (OTA)-induced nephrotoxicity: expression profiling in vivo and in vitro employing cDNA microarrays. Toxicol. Sci. 73, 315–328. doi: 10.1093/toxsci/kfg073
Mackay, E. A., Ehrhard, A., Moniatte, M., Guenet, C., Tardif, C., Tarnus, C., et al. (1997). A possible role for cathepsins D, E, and B in the processing of beta-amyloid precursor protein in Alzheimer's disease. Eur. J. Biochem. 244, 414–425. doi: 10.1111/j.1432-1033.1997.00414.x
Mårtensson, J., Martling, C. R., and Bell, M. (2012). Novel biomarkers of acute kidney injury and failure: clinical applicability. Br. J. Anaesth. 109, 843–850. doi: 10.1093/bja/aes357
Mehta, R. L., Kellum, J. A., Shah, S. V., Molitoris, B. A., Ronco, C., Warnock, D. G., et al. (2007). Acute kidney injury network: report of an initiative to improve outcomes in acute kidney injury. Crit. Care 11:31. doi: 10.1186/cc5713
Moallem, S. A., Nazemian, F., Eliasi, S., Alamdaran, S. A., Shamsara, J., and Mohammadpour, A. H. (2011). Correlation between cathepsin D serum concentration and carotid intima-media thickness in hemodialysis patients. Int. Urol. Nephrol. 43, 841–848. doi: 10.1007/s11255-010-9729-4
Mount, D. B. (2014). Thick ascending limb of the loop of Henle. Clin. J. Am. Soc. Nephrol. 9, 1974–1986. doi: 10.2215/CJN.04480413
Mundel, P., and Reiser, J. (2010). Proteinuria: an enzymatic disease of the podocyte? Kidney Int. 77, 571–580. doi: 10.1038/ki.2009.424
Musante, L., Tataruch, D., Gu, D., Liu, X., Forsblom, C., Groop, P. H., et al. (2015). Proteases and protease inhibitors of urinary extracellular vesicles in diabetic nephropathy. J. Diabetes Res. 2015:289734. doi: 10.1155/2015/289734
Nägler, D. K., Krüger, S., Kellner, A., Ziomek, E., Menard, R., Buhtz, P., et al. (2004). Up-regulation of cathepsin X in prostate cancer and prostatic intraepithelial neoplasia. Prostate 60, 109–119. doi: 10.1002/pros.20046
Naumnik, W., Ossolinska, M., Płonska, I., Chyczewska, E., and Niklinski, J. (2015). Endostatin and cathepsin-V in bronchoalveolar lavage fluid of patients with pulmonary sarcoidosis. Adv. Exp. Med. Biol. 833, 55–61. doi: 10.1007/5584_2014_26
Noda, S., Asano, Y., Takahashi, T., Akamata, K., Aozasa, N., Taniguchi, T., et al. (2013). Decreased cathepsin V expression due to Fli1 deficiency contributes to the development of dermal fibrosis and proliferative vasculopathy in systemic sclerosis. Rheumatology 52, 790–799. doi: 10.1093/rheumatology/kes379
Ozkayar, N., Piskinpasa, S., Akyel, F., Turgut, D., Bulut, M., Turhan, T., et al. (2015). Relation between serum cathepsin D levels and endothelial dysfunction in patients with chronic kidney disease. Nefrologia 35, 72–79. doi: 10.3265/Nefrologia.pre2014
Paulli, M., Feller, A. C., Boveri, E., Kindl, S., Berti, E., Rosso, R., et al. (1994). Cathepsin D and E co-expression in sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease) and Langerhans' cell histiocytosis: further evidences of a phenotypic overlap between these histiocytic disorders. Virchows Arch. 424, 601–606. doi: 10.1007/BF00195773
Pereira, V. G., Queiroz, M. T., and D'Almeida, V. (2016). Differential expression of microRNAs from miR-17 family in the cerebellum of mucopolysaccharidosis type I mice. Gene 595, 207–211. doi: 10.1016/j.gene.2016.10.007
Peres, G. B., Juliano, M. A., Simões, M. J., and Michelacci, Y. M. (2013). Lysosomal enzymes are decreased in the kidney of diabetic rats. Biochim. Biophys. Acta 1832, 85–95. doi: 10.1016/j.bbadis.2012.09.011
Peters, J., Rittger, A., Weisner, R., Knabbe, J., Zunke, F., Rothaug, M., et al. (2015). Lysosomal integral membrane protein type-2 (LIMP-2/SCARB2) is a substrate of cathepsin-F, a cysteine protease mutated in type-B-Kufs-disease. Biochem. Biophys. Res. Commun. 457, 334–340. doi: 10.1016/j.bbrc.2014.12.111
Pišlar, A., and Kos, J. (2014). Cysteine cathepsins in neurological disorders. Mol. Neurobiol. 49, 1017–1030. doi: 10.1007/s12035-013-8576-6
Pozgan, U., Caglic, D., Rozman, B., Nagase, H., Turk, V., and Turk, B. (2010). Expression and activity profiling of selected cysteine cathepsins and matrix metalloproteinases in synovial fluids from patients with rheumatoid arthritis and osteoarthritis. Biol. Chem. 391, 571–579. doi: 10.1515/bc.2010.035
Raab, A. K., Mönkemüller, K., Kandulski, A., Weber, E., Malfertheiner, P., and Wex, T. (2011). Expression pattern of cathepsin W isoforms in peripheral blood and gastroesophageal mucosa of patients with gastroesophageal reflux disease. Biol. Chem. 392, 1167–1172. doi: 10.1515/BC.2011.192
Rafatian, N., Karunakaran, D., Rayner, K. J., Leenen, F. H., Milne, R. W., and Whitman, S. C. (2013). Cathepsin G deficiency decreases complexity of atherosclerotic lesions in apolipoprotein E-deficient mice. Am. J. Physiol. Heart Circ. Physiol. 305, H1141–H1148. doi: 10.1152/ajpheart.00618.2012
Rai, R., Thiagarajan, S., Mohandas, S., Natarajan, K., Shanmuga Sekar, C., and Ramalingam, S. (2010). Haim Munk syndrome and Papillon Lefevre syndrome–allelic mutations in cathepsin C with variation in phenotype. Int. J. Dermatol. 49, 541–543. doi: 10.1111/j.1365-4632.2010.04300.x
Reiser, J., Adair, B., and Reinheckel, T. (2010). Specialized roles for cysteine cathepsins in health and disease. J. Clin. Invest. 120, 3421–3431. doi: 10.1172/JCI42918
Reiser, J., Oh, J., Shirato, I., Asanuma, K., Hug, A., Mundel, T. M., et al. (2004). Podocyte migration during nephrotic syndrome requires a coordinated interplay between cathepsin L and α 3 integrin. J. Biol. Chem. 279, 34827–34832. doi: 10.1074/jbc.M401973200
Rewa, O., and Bagshaw, S. M. (2014). Acute kidney injury-epidemiology, outcomes and economics. Nat. Rev. Nephrol. 10, 193–207. doi: 10.1038/nrneph.2013.282
Ruffell, B., Affara, N. I., Cottone, L., Junankar, S., Johansson, M., DeNardo, D. G., et al. (2013). Cathepsin C is a tissue-specific regulator of squamous carcinogenesis. Genes Dev. 27, 2086–2098. doi: 10.1101/gad.224899.113
Saftig, P., Hunziker, E., Wehmeyer, O., Jones, S., Boyde, A., Rommerskirch, W., et al. (1998). Impaired osteoclastic bone resorption leads to osteopetrosis in cathepsin-K-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 95, 13453–13458. doi: 10.1073/pnas.95.23.13453
Sanders, J. S., van Goor, H., Hanemaaijer, R., Kallenberg, C. G., and Stegeman, C. A. (2004). Renal expression of matrix metalloproteinases in human ANCA-associated glomerulonephritis. Nephrol. Dial. Transplant. 19, 1412–1419. doi: 10.1093/ndt/gfh186
Scarpioni, R., Ricardi, M., and Albertazzi, V. (2016). Secondary amyloidosis in autoinflammatory diseases and the role of inflammation in renal damage. World J. Nephrol. 5, 66–75. doi: 10.5527/wjn.v5.i1.66
Schaefer, L., Han, X., Gretz, N., and Schaefer, R. M. (1996). Alterations of cathepsins B, H and L in proximal tubules from polycystic kidneys of the Han:SPRD rat. Kidney Int. 50, 424–431. doi: 10.1038/ki.1996.332
Schechter, I., and Ziv, E. (2011). Cathepsins S, B and L with aminopeptidases display β-secretase activity associated with the pathogenesis of Alzheimer's disease. Biol. Chem. 392, 555–569. doi: 10.1515/bc.2011.054
Senatorski, G., Paczek, L., Sułowicz, W., Gradowska, L., and Bartłomiejczyk, I. (1998). Urine activity of cathepsin B, collagenase and urine excretion of TGF-β1 and fibronectin in membranous glomerulonephritis. Res. Exp. Med. 198, 199–206. doi: 10.1007/s004330050103
Sever, S., Altintas, M. M., Nankoe, S. R., Möller, C. C., Ko, D., Wei, C., et al. (2007). Proteolytic processing of dynamin by cytoplasmic cathepsin L is a mechanism for proteinuric kidney disease. J. Clin. Invest. 117, 2095–2104. doi: 10.1172/JCI32022
Shechter, P., Boner, G., and Rabkin, R. (1994). Tubular cell protein degradation in early diabetic renal hypertrophy. J. Am. Soc. Nephrol. 4, 1582–1587.
Shimoda, N., Fukazawa, N., Nonomura, K., and Fairchild, R. L. (2007). Cathepsin G is required for sustained inflammation and tissue injury after reperfusion of ischemic kidneys. Am. J. Pathol. 170, 930–940. doi: 10.2353/ajpath.2007.060486
Solau-Gervais, E., Zerimech, F., Lemaire, R., Fontaine, C., Huet, G., and Flipo, R. M. (2007). Cysteine and serine proteases of synovial tissue in rheumatoid arthritis and osteoarthritis. Scand. J. Rheumatol. 36, 373–377. doi: 10.1080/03009740701340172
Sołtysiak, J., Skowronska, B., Fichna, P., Stankiewicz, W., Lewandowska-Stachowiak, M., Ostalska-Nowicka, D., et al. (2014). Neutrophil gelatinase-associated lipocalin and cathepsin L as early predictors of kidney dysfunction in children with type 1 diabetes. Endokrynol. Pol. 65, 479–484. doi: 10.5603/EP.2014.0067
Steubl, D., Kumar, S. V., Tato, M., Mulay, S. R., Larsson, A., Lind, L., et al. (2017). Circulating cathepsin-S levels correlate with GFR decline and sTNFR1 and sTNFR2 levels in mice and humans. Sci. Rep. 7:43538. doi: 10.1038/srep43538
Stoka, V., Turk, B., and Turk, V. (2005). Lysosomal cysteine proteases: structural features and their role in apoptosis. IUBMB Life 57, 347–353. doi: 10.1080/15216540500154920
Stoka, V., Turk, B., Schendel, S. L., Kim, T. H., Cirman, T., and Snipas, S. J. (2001). Lysosomal protease pathways to apoptosis. cleavage of bid, not pro-caspases, is the most likely route. J. Biol. Chem. 276, 3149–3157. doi: 10.1074/jbc.M008944200
Stoka, V., Turk, V., and Turk, B. (2016). Lysosomal cathepsins and their regulation in aging and neurodegeneration. Ageing Res. Rev. 32, 22–37. doi: 10.1016/j.arr.2016.04.010
Sud, M., Tangri, N., Pintilie, M., Levey, A. S., and Naimark, D. M. (2016). Progression to stage 4 chronic kidney disease and death, acute kidney injury and hospitalization risk: a retrospective cohort study. Nephrol. Dial. Transplant. 31, 1122–1130. doi: 10.1093/ndt/gfv389
Sudhan, D. R., and Siemann, D. W. (2015). Cathepsin L targeting in cancer treatment. Pharmacol. Ther. 155, 105–116. doi: 10.1016/j.pharmthera.2015.08.007
Surendran, K., Vitiello, S. P., and Pearce, D. A. (2014). Lysosome dysfunction in the pathogenesis of kidney diseases. Pediatr. Nephrol. 29, 2253–2261. doi: 10.1007/s00467-013-2652-z
Svara, T., Pogacnik, M., and Juntes, P. (2010). Distribution and amount of cathepsin B in gentamicin-induced acute kidney injury in rats. Pol. J. Vet. Sci. 13, 75–82.
Tábara, L. C., Poveda, J., Martin-Cleary, C., Selgas, R., Ortiz, A., and Sanchez-Niño, M. D. (2014). Mitochondria-targeted therapies for acute kidney injury. Expert Rev. Mol. Med. 16:e13. doi: 10.1017/erm.2014.14
Tao, Y., Kim, J., Faubel, S., Wu, J. C., Falk, S. A., Schrier, R. W., et al. (2005). Caspase inhibition reduces tubular apoptosis and proliferation and slows disease progression in polycystic kidney disease. Proc. Natl. Acad. Sci. U.S.A. 102, 6954–6959. doi: 10.1073/pnas.0408518102
Tonelli, M., and Riella, M. (2014). Chronic kidney disease and the ageing population. Nephron Clin. Pract. 128, 319–322. doi: 10.1159/000362458
Turk, B., and Stoka, V. (2007). Protease signalling in cell death: caspases versus cysteine cathepsins. FEBS Lett. 581, 2761–2767. doi: 10.1016/j.febslet.2007.05.038
Turk, B., Stoka, V., Rozman-Pungercar, J., Cirman, T., Droga-Mazovec, G., Oreic, K., et al. (2002). Apoptotic pathways: involvement of lysosomal proteases. Biol. Chem. 383, 1035–1044. doi: 10.1515/BC.2002.112
van den Brûle, S., Misson, P., Bühling, F., Lison, D., and Huaux, F. (2005). Overexpression of cathepsin K during silica-induced lung fibrosis and control by TGF-β. Respir. Res. 6:84. doi: 10.1186/1465-9921-6-84
Vasiljeva, O., Dolinar, M., Rozman-Pungerčar, J., Turk, V., and Turk, B. (2005). Recombinant human procathepsin S is capable of autocatalytic processing at neutral pH in the presence of glycosaminoglycans, FEBS Lett. 579, 1285–1290. doi: 10.1016/j.febslet.2004.12.093
Vazquez-Ortiz, G., Pina-Sanchez, P., Vazquez, K., Duenas, A., Taja, L., Mendoza, P., et al. (2005). Overexpression of cathepsin F, matrix metalloproteinases 11 and 12 in cervical cancer. BMC Cancer 5:68. doi: 10.1186/1471-2407-5-68
Venkatachalam, M. A., Weinberg, J. M., Kriz, W., and Bidani, A. K. (2015). Failed tubule recovery, AKI-CKD transition, and kidney disease progression. J. Am. Soc. Nephrol. 26, 1765–1776. doi: 10.1681/ASN.2015010006
Wang, C., Jiang, Z., Yao, J., Wu, X., Sun, L., Liu, C., et al. (2008). Participation of cathepsin B in emodin-induced apoptosis in HK-2 Cells. Toxicol. Lett. 181, 196–204. doi: 10.1016/j.toxlet.2008.05.013
Wang, J., Chen, L., Li, Y., and Guan, X. Y. (2011). Overexpression of cathepsin Z contributes to tumor metastasis by inducing epithelial-mesenchymal transition in hepatocellular carcinoma. PLoS ONE 6:e24967. doi: 10.1371/journal.pone.0024967
Wang, N., Bai, X., Jin, B., Han, W., Sun, X., and Chen, X. (2016). The association of serum cathepsin B concentration with age-related cardiovascular-renal subclinical state in a healthy Chinese population. Arch. Gerontol. Geriatr. 65, 146–155. doi: 10.1016/j.archger.2016.03.015
Wang, X., Xiong, L., Yu, G., Li, D., Peng, T., Luo, D., et al. (2015). Cathepsin S silencing induces apoptosis of human hepatocellular carcinoma cells. Am. J. Transl. Res. 7, 100–110.
Wasung, M. E., Chawla, L. S., and Madero, M. (2015). Biomarkers of renal function, which and when? Clin. Chim. Acta 438, 350–357. doi: 10.1016/j.cca.2014.08.039
Wiederanders, B., Kaulmann, G., and Schilling, K. (2003). Functions of propeptide parts in cysteine proteases. Curr. Protein Pept. Sci. 4, 309–326. doi: 10.2174/1389203033487081
Wilkinson, R. D., Williams, R., Scott, C. J., and Burden, R. E. (2015). Cathepsin S: therapeutic, diagnostic, and prognostic potential. Biol. Chem. 396, 867–882. doi: 10.1515/hsz-2015-0114
Yaddanapudi, S., Altintas, M. M., Kistler, A. D., Fernandez, I., Möller, C. C., Wei, C., et al. (2011). CD2AP in mouse and human podocytes controls a proteolytic program that regulates cytoskeletal structure and cellular survival. J. Clin. Invest. 121, 3965–3980. doi: 10.1172/JCI58552
Yamamoto-Nonaka, K., Koike, M., Asanuma, K., Takagi, M., Oliva Trejo, J. A., Seki, T., et al. (2016). Cathepsin D in podocytes is important in the pathogenesis of proteinuria and CKD. J. Am. Soc. Nephrol. 27, 2685–2700. doi: 10.1681/ASN.2015040366
Yasuda, Y., Li, Z., Greenbaum, D., Bogyo, M., Weber, E., and Brömme, D. (2004). Cathepsin V, a novel and potent elastolytic activity expressed in activated macrophages. J. Biol. Chem. 279, 36761–36770. doi: 10.1074/jbc.M403986200
Keywords: cathepsins, acute kidney injury, chronic kidney disease, lysosomal proteases, signaling pathways
Citation: Cocchiaro P, De Pasquale V, Della Morte R, Tafuri S, Avallone L, Pizard A, Moles A and Pavone LM (2017) The Multifaceted Role of the Lysosomal Protease Cathepsins in Kidney Disease. Front. Cell Dev. Biol. 5:114. doi: 10.3389/fcell.2017.00114
Received: 16 October 2017; Accepted: 07 December 2017;
Published: 19 December 2017.
Edited by:
Andrei Surguchov, University of Kansas Medical Center Research Institute, United StatesReviewed by:
Kamel Laghmani, Centre de Recherche des Cordeliers, INSERM/UPMC/CNRS - U1138, ERL8228, FranceMaurizio Renna, University of Cambridge, United Kingdom
Copyright © 2017 Cocchiaro, De Pasquale, Della Morte, Tafuri, Avallone, Pizard, Moles and Pavone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Moles, YW5uYS5tb2xlcy5mZXJuYW5kZXpAZ21haWwuY29t
Luigi Michele Pavone, bHVpZ2ltaWNoZWxlLnBhdm9uZUB1bmluYS5pdA==
†These authors have contributed equally to the work.