 Yibo Luo
Yibo Luo Ejaz Ahmad
Ejaz Ahmad Song-Tao Liu
Song-Tao Liu- Department of Biological Sciences, University of Toledo, Toledo, OH, United States
The mitotic checkpoint monitors kinetochore-microtubule attachment, delays anaphase onset and prevents aneuploidy when unattached or tensionless kinetochores are present in cells. Mitotic arrest deficiency 1 (MAD1) is one of the evolutionarily conserved core mitotic checkpoint proteins. MAD1 forms a cell cycle independent complex with MAD2 through its MAD2 interaction motif (MIM) in the middle region. Such a complex is enriched at unattached kinetochores and functions as an unusual catalyst to promote conformational change of additional MAD2 molecules, constituting a crucial signal amplifying mechanism for the mitotic checkpoint. Only MAD2 in its active conformation can be assembled with BUBR1 and CDC20 to form the Mitotic Checkpoint Complex (MCC), which is a potent inhibitor of anaphase onset. Recent research has shed light on how MAD1 is recruited to unattached kinetochores, and how it carries out its catalytic activity. Here we review these advances and discuss their implications for future research.
Introduction
The mitotic checkpoint (or spindle checkpoint or spindle assembly checkpoint) is a crucial mechanism to maintain chromosomal stability. It functions during every prometaphase, detecting the lack of microtubule occupancy or tension at kinetochores and delaying anaphase onset for error correction and faithful chromosome segregation (Lara-Gonzalez et al., 2012; Foley and Kapoor, 2013; Jia et al., 2013; London and Biggins, 2014b; Musacchio, 2015; Liu and Zhang, 2016) (Figure 1). Defects in the mitotic checkpoint may lead to cancer (Weaver and Cleveland, 2007). Sustained mitotic checkpoint triggered by microtubule-targeted cancer drugs or other small molecule inhibitors in clinical trials, on the other hand, causes prolonged mitotic arrest and cancer cell death (Liu and Yen, 2008). Understanding how the mitotic checkpoint works is of significance not only for the fundamental biological question of cell division, but also for improving cancer therapy.
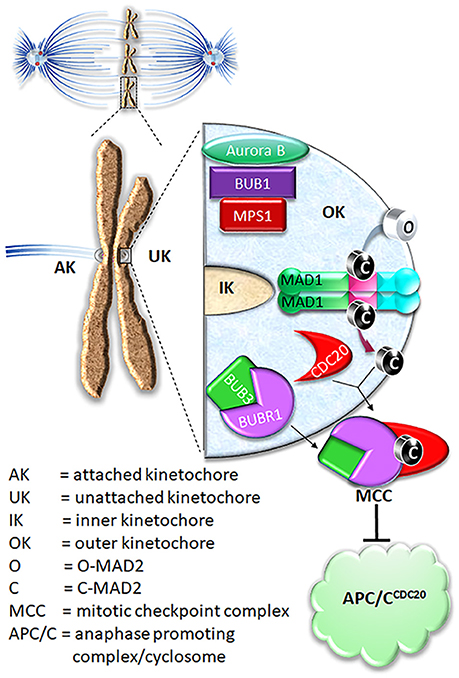
Figure 1. Current model on MAD2 O-C conversion and MCC assembly. In prometaphase, unattached kinetochores initiate mitotic checkpoint signaling, leading to assembly of the mitotic checkpoint complex (MCC) consisting of BUBR1, CDC20, BUB3, and MAD2. The MCC inhibits APC/C ubiquitination activity until all chromosomes are correctly attached to microtubules. For MCC assembly, MAD2 needs to be converted from O conformation into C conformation.
Mitotic arrest deficiency 1 (MAD1) is one of the core mitotic checkpoint genes first identified in S. cerevisiae and is evolutionarily conserved among most eukaryotic cells (Li and Murray, 1991; Vleugel et al., 2012). Insight into the mechanistic underpinnings of MAD1 function in the mitotic checkpoint has accumulated steadily, with several exciting results obtained in the past few years, yet there are still significant gaps on its working mechanisms. In this review we discuss recent results and potential future directions on MAD1 research. We mostly focus on research in human cells and on two questions: what is the kinetochore receptor for MAD1 and how MAD1 helps MAD2 conformational change.
Current Model on MAD1 Functioning Mechanism
Shortly after identification of the core mitotic checkpoint genes, their protein products were found to be concentrated at the unattached or tensionless kinetochores, connecting the genetic and cellular aspects of the mitotic checkpoint control mechanisms (Chen et al., 1996; Waters et al., 1998). MAD1, MAD2, and MPS1 levels drop below detection at attached kinetochores, indicating their sensitivity to the kinetochore-microtubule attachment status. It was later established that the major function of MAD1 is to recruit MAD2 to unattached kinetochores, where MAD2 molecules are converted from open (O-MAD2) to closed (C-MAD2) conformation. The conformational change of MAD2 is a critical signal amplification mechanism for the mitotic checkpoint (Mapelli and Musacchio, 2007; Luo and Yu, 2008).
O-MAD2 is the predominant conformer in interphase cells (Luo et al., 2004; Fava et al., 2011). In the current model, a 2:2 MAD1:C-MAD2 tetramer localized at unattached kinetochores functions as the catalyst for MAD2 O-C conversion, initiating the mitotic checkpoint response when cells enter mitosis (De Antoni et al., 2005). It was suggested that O-MAD2 hetero-dimerizes with the C-MAD2 subunit in the MAD1:C-MAD2 tetramer and converts into C-MAD2 (Mapelli and Musacchio, 2007; Luo and Yu, 2008). The mechanism of conversion is unknown but possibly involves one or more intermediate MAD2 conformations (I-MAD2) (Mapelli and Musacchio, 2007; Luo and Yu, 2008; Hara et al., 2015). C-MAD2 conformation is required to interact with BUBR1 and CDC20 to form the Mitotic Checkpoint Complex (MCC), which binds and inhibits the anaphase promoting complex/cyclosome (APC/CCDC20) (Lara-Gonzalez et al., 2012; London and Biggins, 2014b; Musacchio, 2015; Liu and Zhang, 2016) (Figure 1). As the E3 ubiquitin ligase activity of the APC/CCDC20 is inhibited by the MCC, cells are arrested at the metaphase. It is important to note that the CDC20 molecule associated with the APC/C is a substrate-binding and activator subunit for the APC/C, and is separate from the second CDC20 molecule as a subunit of the MCC (Izawa and Pines, 2015; Alfieri et al., 2016; Yamaguchi et al., 2016). C-MAD2 in cells may also directly bind to free CDC20, blocking its association with the APC/C core thus keeping APC/C activity low (Fang et al., 1998; Izawa and Pines, 2012).
Structure of MAD1
Human MAD1 is a protein of 718 amino acids (Figure 2). The structure of full length MAD1 has not been solved yet, mainly due to low solubility of recombinant MAD1, although Faesen et al. recently reported purification of full-length MAD1 (co-expressed with MAD2) from Tnao38 insect cells (Faesen et al., 2017). MAD1 is often simplified as a coiled coil molecule along its entire length, but structural analyses and predictions indicated several non-coiled coil segments interrupting the coiled coil regions (Lupas et al., 1991) (Figure 2A). The N-terminal domain of MAD1 (1–485 residues, MAD1NTD) is thought to target the protein to nuclear envelope or kinetochores, is required for MAD1 dimerization and interacts with many other proteins (Chen et al., 1998; Martin-Lluesma et al., 2002; Rodriguez-Bravo et al., 2014; Akera et al., 2015; Ji et al., 2018). A predicted structure of MAD1NTD is presented in Figure 2C. The best characterized MAD1 domain contains its MAD2 interaction motif (MIM, 485–584 residues). The solved crystal structure of MAD1MIM in complex with C-MAD2 is the cornerstone for the now classical model of the MAD1:C-MAD2 catalyst (Sironi et al., 2002). In the structure, MAD1MIM forms a dimer, with each monomer utilizing the disordered loop spanning 530–550 residues to trap one molecule of C-MAD2 and assemble a 2:2 heterotetramer (Figure 2D) (Sironi et al., 2002). Although the MIM motif was suggested to be absent in MAD1 homologs in Salpingoeca rosetta (a choanoflagellate), Micromonas pusilla (a green algae), and Naegleria gruberi (an amoeboflagellate) (Vleugel et al., 2012), the particular sequence alignments need to be treated more cautiously as the annotated MAD1s might only be partial sequences from genome projects. The C-terminal domain of MAD1 (585–718 residues, MAD1CTD) is also evolutionarily conserved and many results have confirmed its functional importance in maintaining the mitotic checkpoint (Chen et al., 1999; De Antoni et al., 2005; Yang et al., 2008; Ballister et al., 2014; Heinrich et al., 2014; Kruse et al., 2014; Kuijt et al., 2014; Ji et al., 2018). The N-terminal portion of MAD1CTD adopts an α-helical structure and forms a coiled coil stem (597–637 residues) while its C-terminal (638–718) residues fold into a globular domain (Figure 2E). The overall MAD1CTD dimer structure resembles the kinetochore-binding domains of Spc25 and Csm1 or the RWD domain (Kim et al., 2012).
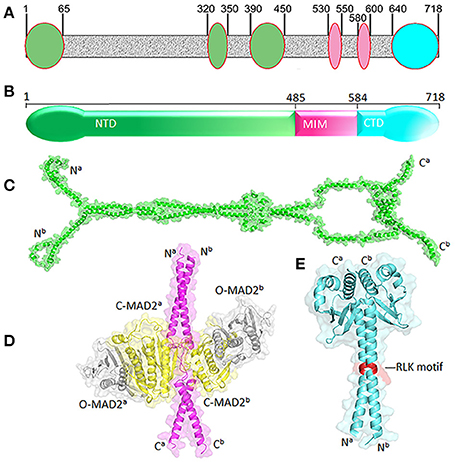
Figure 2. Structural features of MAD1. (A) Non-coiled coil segments (ovals) are scatted along likely coiled-coil regions of MAD1 (in gray) as predicted by COILS program (Lupas et al., 1991). (B) Diagram of MAD1 showing its N-terminal domain (NTD), the domain containing the MAD2 interaction motif (MIM) and C-terminal domain (CTD). (C) Predicted model of MAD1NTD dimer by Galaxy Homomer (Baek et al., 2017). (D) Crystal structure of MAD1MIM (pink) dimer bound with two molecules of C-MAD2 (yellow) as in PDB 1GO4 (Sironi et al., 2002). Two O-MAD2 molecules (gray) from 2V64 (Mapelli et al., 2007) are also fitted. (E) Crystal structure of MAD1CTD dimer from 4DZO (Kim et al., 2012). The RLK motifs are shown in red. Superscripts a&b denote two different chains of the same molecule. PyMol was used for structure visualization and model generation.
Kinetochore Receptors of MAD1
MAD1 forms a cell cycle independent complex with MAD2. The yeast MAD1:MAD2 complex is stable enough to tolerate harsh conditions such as 5M NaCl and 1M Urea (Chen et al., 1999). The observed stable complex most likely reflects C-MAD2 bound to MAD1MIM, as point mutations at the MAD1MIM abolished most of MAD2 signals in both imaging and immunoprecipitation experiments (Sironi et al., 2002; Maldonado and Kapoor, 2011; Ji et al., 2018). Such MAD1:C-MAD2 complexes are localized to the nuclear envelope during interphase, coinciding with a fraction of MPS1, and have been shown to generate a basal level of C-MAD2 in cells that contributes to the mitotic checkpoint (Chen et al., 1998; Campbell et al., 2001; Liu et al., 2003a; Lee et al., 2008; Rodriguez-Bravo et al., 2014). However, it is generally accepted that C-MAD2 production peaks during prometaphase, when the MAD1:C-MAD2 complex is localized to the unattached kinetochores (Chen et al., 1998; Tipton et al., 2011a). Therefore, although the interaction between MAD1MIM and C-MAD2 does not change in interphase and prometaphase cells, some mitosis-specific modifications or interactions must have occurred to the MAD1:C-MAD2 tetramer, the catalyst for the MAD2 O to C conformation conversion reaction, to make it more efficient in generating C-MAD2. To better understand the enhanced catalytic activity of the MAD1:C-MAD2 complex in prometaphase cells, identifying its kinetochore receptor(s) is certainly an important question.
In earlier efforts to characterize kinetochore receptor(s) for MAD1, people have tried to refine the region in MAD1 for kinetochore targeting and determine the dependency relationship of MAD1 to various proteins for kinetochore localization. Biochemical assays have also uncovered multiple MAD1 interacting proteins (Figure 3). MAD1NTD was long thought to be responsible for its kinetochore localization (Chung and Chen, 2002). However, MAD1CTD also plays a role in MAD1 kinetochore localization (Kim et al., 2012). Based on epistatic analysis using siRNAs, MAD1 kinetochore localization was mapped into a CENP-I NDC80MPS1MAD1MAD2 dependency hierarchy (Martin-Lluesma et al., 2002; Liu et al., 2003b, 2006; Matson and Stukenberg, 2014). Other results concluded that ROD and ZW10 are also required for MAD1 localization (Kops et al., 2005). Additional knockdown and knockout based analyses, together with genetic mutants in model organisms, have provided clues to the requirement of other proteins (including BUB1) for MAD1 kinetochore localization (Johnson et al., 2004; Meraldi and Sorger, 2005; Liu et al., 2006; Qian et al., 2017). Biochemically, MAD1NTD interacts with NDC80 (Martin-Lluesma et al., 2002), PLK1 (Chi et al., 2008), NEK2 (Lou et al., 2004), TPR (Lince-Faria et al., 2009), CEP57 (Zhou et al., 2016), and CENP-E (Akera et al., 2015), while MAD1CTD binds directly to BUB1 as well as CDC20 (Ji et al., 2017) (Figure 3). Our recent work has shown that both the NTD and CTD of MAD1 interact with MPS1 and that MAD1 has additional binding sites for MAD2 outside the MIM region (Ji et al., 2018). We next discuss MAD1 interacting proteins as potential kinetochore receptors for the MAD1:C-MAD2 complex.
NDC80MPS1MAD1MAD2 dependency hierarchy (Martin-Lluesma et al., 2002; Liu et al., 2003b, 2006; Matson and Stukenberg, 2014). Other results concluded that ROD and ZW10 are also required for MAD1 localization (Kops et al., 2005). Additional knockdown and knockout based analyses, together with genetic mutants in model organisms, have provided clues to the requirement of other proteins (including BUB1) for MAD1 kinetochore localization (Johnson et al., 2004; Meraldi and Sorger, 2005; Liu et al., 2006; Qian et al., 2017). Biochemically, MAD1NTD interacts with NDC80 (Martin-Lluesma et al., 2002), PLK1 (Chi et al., 2008), NEK2 (Lou et al., 2004), TPR (Lince-Faria et al., 2009), CEP57 (Zhou et al., 2016), and CENP-E (Akera et al., 2015), while MAD1CTD binds directly to BUB1 as well as CDC20 (Ji et al., 2017) (Figure 3). Our recent work has shown that both the NTD and CTD of MAD1 interact with MPS1 and that MAD1 has additional binding sites for MAD2 outside the MIM region (Ji et al., 2018). We next discuss MAD1 interacting proteins as potential kinetochore receptors for the MAD1:C-MAD2 complex.
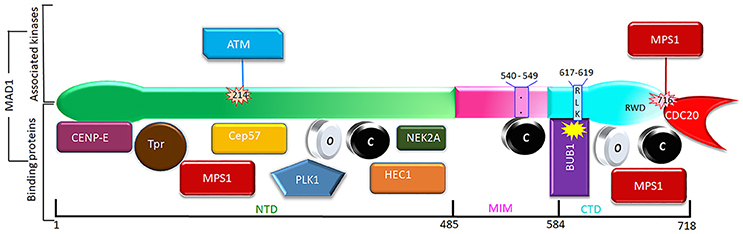
Figure 3. Phosphorylation (above) and interacting partners (below) of MAD1. Only the phosphorylation events and interactions with better-defined functional implications are shown. The interacting proteins are roughly grouped based on their binding to the NTD, MIM, or CTD of MAD1.
BUB1 as a Kinetochore Receptor of MAD1
In budding yeast, an RLK motif in the MAD1CTD was long known to direct interaction with BUB1 when the mitotic checkpoint is activated (Brady and Hardwick, 2000). London et al. demonstrated that BUB1 is phosphorylated in the middle region by MPS1 kinase, and that the phosphorylated region then binds to MAD1 and recruits it to kinetochores (London and Biggins, 2014a). Except in C. elegans (Moyle et al., 2014), the RLK motif in MAD1 and the corresponding region in BUB1 (conserved domain 1 or CD1) are evolutionarily conserved, and the RLK motif is essential for MAD1 kinetochore targeting in fission yeast and human cells (Klebig et al., 2009; Kim et al., 2012; Heinrich et al., 2014; Mora-Santos et al., 2016). Puzzlingly, direct interaction between BUB1 and MAD1 was not observed in human cell lysates (Kim et al., 2012). However, recently several labs have confirmed that human BUB1 is phosphorylated at S459 by CDK1, and then further phosphorylated by MPS1 at T461. The doubly phosphorylated BUB1 directly binds to the RLK motif in MAD1 and recruits MAD1 to kinetochores (Faesen et al., 2017; Ji et al., 2017; Zhang et al., 2017). It should be noted that targeting of BUB1 to the kinetochore also requires MPS1 activity. Specifically, MPS1 phosphorylates the outer kinetochore protein KNL1 at threonine (T) residues of its “MELT” repeat motifs. The phosphorylated MELT motifs are recognized by BUB3, which in turn recruits its binding partner BUB1 to kinetochores (London et al., 2012; Shepperd et al., 2012; Yamagishi et al., 2012; Primorac et al., 2013; Vleugel et al., 2013; Krenn et al., 2014; Zhang et al., 2014).
Intriguingly, Qian et al recently reported that the BUB1:MAD1 interaction only occurs transiently in early prometaphase in unperturbed mitosis (Qian et al., 2017). During prolonged prometaphase after cells were challenged by microtubule drugs nocodazole and taxol or the Eg5 kinesin inhibitor STLC, MAD1 levels at unattached kinetochores remained high, even in the absence of BUB1 (Qian et al., 2017). These results agreed with earlier observations implying BUB1-independent mechanisms for MAD1 kinetochore localization. For example, in both fission yeast and Drosophila, MELT-phosphomimetic KNL1 mutants are sufficient to retain BUB1 but not MAD1 at kinetochores (Shepperd et al., 2012; Vleugel et al., 2012).
The ROD/ZW10/ZWILCH Complex as a Kinetochore Receptor for MAD1
The ROD/ZW10/ZWILCH or RZZ complex are kinetochore proteins found only in metazoans (Karess, 2005). Multiple studies showed that the RZZ complex is required for MAD1 and MAD2 localization at kinetochores and mitotic checkpoint signaling under all treatment conditions (Basto et al., 2000; Chan et al., 2000; Buffin et al., 2005; Kops et al., 2005; Silió et al., 2015; Qian et al., 2017). More recent data supported that a pool of RZZ requires the N-terminal region of KNL1 for kinetochore localization in BUB1-dependent manner (Caldas et al., 2015; Zhang et al., 2015). Interestingly, the same CD1 domain of BUB1 is required for this pool of RZZ and MAD1 kinetochore localization (Zhang et al., 2015, 2017). As mentioned above, BUB1 might not be the only kinetochore receptor for MAD1, but MAD1 kinetochore localization always requires RZZ (Qian et al., 2017). There is another pool of RZZ whose localization is dependent neither on KNL1 nor on BUB1, but possibly on NDC80 that is associated with CENP-T (Caldas et al., 2015; Samejima et al., 2015). It awaits to be further studied whether this pool of RZZ retains MAD1 at kinetochores during nocodazole treatment when the BUB1:MAD1 interaction is lost (see BUB1 as a Kinetochore Receptor of MAD1). While MAD1 is found in RZZ immunoprecipitates, direct interaction between MAD1 and the RZZ complex subunits remains to be established (Défachelles et al., 2015).
NDC80 as a Kinetochore Receptor for MAD1
NDC80 is a subunit of the NDC80/NUF2/SPC24/SPC25 complex, whose microtubule binding activity is primarily responsible for end-on attachment of microtubules to kinetochores (Santaguida and Musacchio, 2009). Depletion of NDC80 and NUF2 caused mis-localization of MAD1:MAD2 complex and defective checkpoint activation (Meraldi et al., 2004). Depletion of SPC25 also resulted in loss of NDC80 and MAD1 from kinetochores, but had no impact on BUB1 levels at kinetochores (Bharadwaj et al., 2004). These results lent support to the notion that MAD1 has one or more kinetochore receptors that involve the NDC80 complex. Yeast two-hybrid assay showed NDC80 interacts with MAD1NTD (Martin-Lluesma et al., 2002), but the interaction has not been observed by immunoprecipitation in cell lysates. However, immunoprecipitation in cell lysates might not be a gold standard in assessing protein-protein interactions at kinetochores, where proteins are normally enriched to much higher concentrations than in the cytosol, are spatially proximal to multiple proteins, and may undergo particular posttranslational modifications such as phosphorylation. In addition, the concentration of any particular endogenous protein in a cell lysate at 1 mg/ml total protein level can be 10~1,000 times lower than its intracellular level. These factors might cause failure in detecting a physiologically relevant protein-protein interaction such as the MPS1:NDC80 interaction by immunoprecipitation using cell lysates (Aravamudhan et al., 2015; Hiruma et al., 2015; Ji et al., 2015). Testing protein-protein interactions using recombinant proteins in vitro and immunofluorescence of protein mutants in cells could complement the results from yeast two-hybrid and immunoprecipitation using cell lysates. It remains to be characterized whether and how direct MAD1 interaction with NDC80 could occur at kinetochores, and whether MAD1-NDC80 interaction could serve as a microtubule attachment sensitive checkpoint activation mechanism.
CEP57 as a Kinetochore Receptor for MAD1
Cep57 is a microtubule binding protein that localizes at centrosomes and kinetochores in Xenopus and human cells (Emanuele and Stukenberg, 2007; Zhou et al., 2016). Zhou et al. showed that Cep57 kinetochore localization depends on its interaction with Mis12 of the KMN network through its N-terminal region (Zhou et al., 2016). Cep57 also helps recruit MAD1 and MAD2 to kinetochores. The recruitment is mediated by direct interaction between MAD1 and the Cep57 C-terminal region which overlaps with its microtubule binding domain. Interestingly, the microtubule binding of Cep57 at kinetochores competitively displaces MAD1, suggesting a mechanism to couple MAD1 localization with microtubule attachment status at kinetochores (Zhou et al., 2016). However, the mitotic checkpoint defects in Cep57 depleted cells are much weaker compared to MAD1 or MAD2 depletion, again suggesting the presence of other proteins in recruiting MAD1 to unattached kinetochores.
Consideration of Other Proteins as Potential Kinetochore Receptor for MAD1
Kinetochores as a platform may provide a MAD1 recruitment interface that is contributed by multiple distinct proteins, and the interface may dynamically change at different cell cycle stages or when cells are exposed to different drugs. Individual interactions might be weak, but the collective force can be both strong and highly specific (Kim et al., 2012). In the literature, there are many other proteins shown to affect MAD1 kinetochore localization. Most of them may work indirectly through affecting the MAD1 receptors discussed above, but it is also possible some might directly contribute to kinetochore localization of MAD1. We briefly comment on some of these proteins (Figure 3).
Nek2 kinase interacts with MAD1, but it is degraded during prometaphase, so the significance of its interaction with MAD1 in terms of mitotic checkpoint signaling is unclear (Lou et al., 2004; Hayes et al., 2006; Sedgwick et al., 2013). Similarly, inhibition or knockdown of Plk1 kinase causes strong prometaphase arrest, arguing against the possibility Plk1 is a major kinetochore receptor for the MAD1:C-MAD2 complex, although recent reports suggested that Plk1 might contribute to the mitotic checkpoint by potentiating MPS1 or phosphorylating CDC20 (Sumara et al., 2004; Lénárt et al., 2007; Chi et al., 2008; Liu et al., 2012; Espeut et al., 2015; O'Connor et al., 2015; von Schubert et al., 2015; Jia et al., 2016; Ikeda and Tanaka, 2017). Plk1 and MPS1 share many substrates, and Plk1 even phosphorylates many MPS1 autophosphorylation sites, so Plk1 functions in the mitotic checkpoint in C. elegans, which naturally lack a MPS1 homolog (Dou et al., 2011; Espeut et al., 2015; von Schubert et al., 2015). As mentioned, MPS1 was long known to be essential for MAD1 kinetochore localization (Martin-Lluesma et al., 2002; Liu et al., 2003a). In light of recent results, MPS1 may phosphorylate BUB1 to create MAD1 binding sites at kinetochores (Ji et al., 2017; Zhang et al., 2017). We showed that MPS1 binds and phosphorylates both MAD1NTD and MAD1CTD at least in vitro (Ji et al., 2018). MAD1 phosphorylation by MPS1 has also been shown by other groups (Faesen et al., 2017; Ji et al., 2017). However the MAD1CTD:MPS1 interaction was weakened when MAD1CTD was phosphorylated by MPS1, arguing against the role of MPS1 as the kinetochore receptor for MAD1 (Ji et al., 2018). TPR interacts with MAD1 and MAD2 in both interphase and mitosis, but it is largely dispensable for MAD1 kinetochore localization (Lee et al., 2008; Rodriguez-Bravo et al., 2014). CENP-E interaction with MAD1 was suggested to be involved in chromosome alignment. Whether it helps initiate MAD1 activity for the mitotic checkpoint is unclear (Akera et al., 2015).
MAD1 in the Catalysis of MAD2 O-C Conversion and MCC Assembly
Although kinetochore localization of MAD1 is important for its regulation and activity, the localization itself is not sufficient for MAD2 O-C conversion and the mitotic checkpoint response (i.e., maintaining metaphase arrest). This principle was first demonstrated in experiments by Maldonado and Kapoor (2011). When MAD1 was fused with centromere protein Mis12 to be constitutively targeted to kinetochores, cells were arrested at metaphase even after all kinetochores were attached by microtubules. However, the arrest was abrogated by inhibitors of MPS1 or Aurora B kinases, suggesting that these conserved kinases affect catalytic functions of MAD1 or other aspects of the mitotic checkpoint signaling (Maldonado and Kapoor, 2011). Using a similar strategy, we and others have shown that removing either the NTD or CTD regions from MAD1 negatively impacted the mitotic checkpoint, most likely due to reduced catalysis of MAD2 O-C conversion and/or MCC assembly (Tipton et al., 2013; Ballister et al., 2014; Heinrich et al., 2014; Kruse et al., 2014; Kuijt et al., 2014; Ji et al., 2018). Further understanding the MAD2 O-C conversion mechanisms requires dissection of contribution of MAD1 dimerization, interactions between different MAD1 domains, and the interplay of MAD1 with different kinases (Figure 3).
MAD1 Dimerization
MAD1 forms a dimer mostly through its coiled coil segments (Figure 1), but whether the dimerization is regulated or is functionally important is largely unknown. Kim et al. identified several mutations in the CTD globular domain that disrupted both homodimerization and kinetochore localization of MAD1 (Kim et al., 2012). ATM, a canonical DNA damage checkpoint protein, mediates S214 phosphorylation that regulates MAD1 dimerization as well as heterodimerization with MAD2, and contributes to mitotic checkpoint function and chromosomal stability (Yang et al., 2014). A recent report suggested that PTEN affects MAD1 dimerization in interphase (Liu et al., 2017). When tested as fusions with Mis12, both MAD1NTD and MAD1CTD are required for mitotic checkpoint signaling (Ji et al., 2018). We showed that both MAD1CTD and MAD1NTD are required for MAD1 dimerization, and MPS1 kinase might regulate MAD1 dimerization (Ji et al., 2018). However, as MAD1NTD and MAD1CTD both have important binding partners, we cannot conclusively ascribe the checkpoint defects caused by MAD1 truncations to defective MAD1 dimerization.
MAD1 Inter-domain Interactions
Previously it was reported that MAD1CTD can possibly fold back to be close to the MAD1MIM:C-MAD2 catalytic center and the recruited MAD2 molecules undergoing conformational conversion (Sironi et al., 2002). We did not detect direct interaction of recombinant MAD1MIM with either MAD1NTD or MAD1CTD (Ji et al., 2018), arguing against the possibility that the α-helical stem in the CTD forms an anti-parallel coiled coil with a helical portion of MIM (Sironi et al., 2002; Heinrich et al., 2014). This agrees with the current MAD1CTD dimer structure, in which the α-helical portions in two CTD monomers form a coiled coil (Kim et al., 2012). However, studies on the phosphorylation of Thr716 which is two residues away from the C-terminal end, on mutations of several charged residues at the MAD1CTD globular domain, and on mutations along the MAD1CTD α-helical portion (Ser610, or RLK617−619, or Tyr634), revealed that MAD1CTD positively contributes to mitotic checkpoint signaling, most likely by promoting MAD2 O-C conversion and/or MCC assembly (see below) (Heinrich et al., 2014; Kruse et al., 2014; Faesen et al., 2017; Ji et al., 2017, 2018). How this regulation occurs is still unclear, but a posttranslational modification-driven CTD fold-back is a possible mechanism.
Interestingly we found that MAD1NTD and MAD1CTD directly interact with each other, and that the interaction is reduced after phosphorylation by MPS1 (Ji et al., 2018). We speculate that relaxing the interaction between MAD1NTD and MAD1CTD elevates the efficiency of the MAD1:C-MAD2 catalyst, and that interphase MAD1 might be in a ground-state conformation. We are still uncertain whether the interaction between MAD1NTD and MAD1CTD occurs within a MAD1 dimer or between MAD1 dimers. Nevertheless, cellular experiments suggested that both NTD and CTD contribute positively to the mitotic checkpoint (Ji et al., 2018).
MAD1 in MAD2 O-C Conversion
As mentioned, the major activity of MAD1 is to catalyze MAD2 O-C conversion during mitotic checkpoint signaling. However, how the MAD1:C-MAD2 heterotetramer catalyzes the O-MAD2 converting into C-MAD2 is largely unknown. In the past two years, several publications have advanced our understanding of the underlying mechanisms. Ji et al. in the Yu lab reported that MAD1 Thr716 phosphorylation by MPS1 helps generate the APC/C inhibitory activity in a series of in vitro reconstitution experiments (Ji et al., 2017). Faesen et al. established a FRET assay to detect MAD2 conformational conversion by measuring its interaction with BUBR1 and formation of the MCC, both of which require C-MAD2 (Faesen et al., 2017). They found that MPS1 phosphorylates MAD1 directly, after which MAD1:MAD2 complexes accelerate MAD2 O-C conversion. Moreover, phosphorylation of the MAD1CTD at S699, S713, and T716 by MPS1 is essential for this acceleration. How MAD1CTD and its phosphorylation facilitate the MAD2 O-C conversion was unclear from these studies. Our recently published results suggested at least one mechanism how MAD1CTD and MAD1NTD can assist MAD2 O-C conversion. We found that both MAD1NTD and MAD1CTD directly bind to both O and C conformers of MAD2. We also showed that mutating T716 into alanine significantly reduced the MAD1CTD binding to both C- and O-MAD2 (Ji et al., 2018).
Expanding the MAD1 functional domains beyond its classical MIM region to NTD and CTD helps address one major puzzle in mitotic checkpoint studies. It is well known that more C-MAD2 is produced when cells enter mitosis (Luo et al., 2004; Fava et al., 2011), but how could this happen if the levels of MAD2 and the assumed catalyst for MAD2 O-C conversion, the stable MAD1MIM:C-MAD2 heterotetramer, do not change throughout the cell cycle? We proposed a model that during interphase MAD1NTD and MAD1CTD interact with each other, limiting their binding to O- or C-MAD2 (Figure 4). Despite MAD1MIM binding to the C-MAD2, the MAD1 configuration during interphase is at a ground state that shows only low level catalytic activity for MAD2 O-C conversion. When cells enter mitosis, MPS1 activity relaxes the MAD1NTD and MAD1CTD interaction, and more O-MAD2 is recruited to MAD1, through either the MAD1NTD or MAD1CTD. The law of mass action, together with modification at the MAD1MIM:C-MAD2 catalytic center and probably also in the MAD1CTD, promote more production of C-MAD2 (Figure 4) (Ji et al., 2018). I-MAD2 has been surmised to exist during MAD2 O-C conversion and was shown to share structure similarity to C-MAD2 (Mapelli et al., 2007; Hara et al., 2015). We showed that both MAD1NTD or MAD1CTD bind to at least one form of I-MAD2 (MAD2ΔN10) (Ji et al., 2018). Such binding may also facilitate MAD2 O-C conversion.
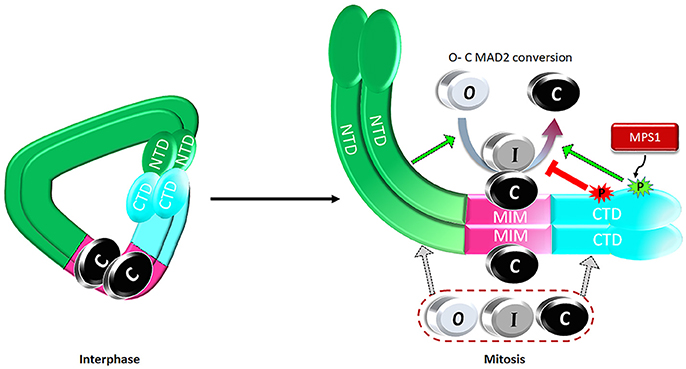
Figure 4. A proposed model on regulation of the catalytic activity of the MAD1:C-MAD2 heterotetramer to drive MAD2 O-C conversion. See text for details.
MAD1 in MCC Assembly
Studies on MAD1NTD and MAD1CTD also indicated direct involvement of MAD1 in facilitating MCC assembly, an activity intricately linked with but possibly separated from catalyzing MAD2 O-C conversion. The MCC is assembled from CDC20, C-MAD2 and the cell cycle independent BUBR1:BUB3 subcomplex (Sudakin et al., 2001; Liu and Zhang, 2016). MAD1 at kinetochores may help bring the MCC subunits in spatial proximity to promote MCC assembly. MAD1 phosphorylation at Thr716 by MPS1 kinase was shown to increase its interaction with CDC20 through a basic motif 27RWQRK31 in CDC20 (Ji et al., 2017). MAD1 at least in prometaphase is recruited to kinetochores by BUB1, which in turn is localized through KNL1 (Caldas et al., 2015; Zhang et al., 2015, 2017; Faesen et al., 2017; Ji et al., 2017). The BUBR1:BUB3 subcomplex can be recruited to KNL1 directly or through interaction with BUB1 (Overlack et al., 2015; Zhang et al., 2016). In addition, both BUB1 and BUBR1 directly interact with CDC20 at kinetochores (Lischetti et al., 2014; Diaz-Martinez et al., 2015; Di Fiore et al., 2015). Together with C-MAD2 produced by the MAD1:C-MAD2 heterotetramer, the spatial proximity of the MCC subunits, guided by protein-protein interactions, presumably increases the efficiency of MCC assembly. Such explanation links MCC production to the unattached kinetochores as the assembly platform. The energy favorable MCC assembly may also be coupled with MAD2 O-C conversion to help overcome the activation energy barrier for MAD2 conformational change (Faesen et al., 2017; Ji et al., 2017, 2018). During MCC assembly, MAD2 uses its “safety belt” structural motif to interact with CDC20, and its “dimerization domain” to interact with BUBR1 (Tipton et al., 2011b; Chao et al., 2012; Alfieri et al., 2016; Yamaguchi et al., 2016). We found that the MAD1NTD or MAD1CTD association with C-MAD2 or I-MAD2 does not require these two known MAD2 motifs (Ji et al., 2018). This may create some advantage to relay newly converted C-MAD2 from MAD1 to the MCC, as those C-MAD2 molecules still have the “safety belt” and the “dimerization domain” readily accessible for MCC assembly.
Conclusions
As one of the most important checkpoint proteins, MAD1 forms a cell cycle independent MAD1:C-MAD2 complex to catalyze the MAD2 O-C conversion at unattached kinetochores, thus amplifying the signals for the mitotic checkpoint and promoting the formation of the MCC, the potent inhibitor of anaphase onset. Previous research on MAD1 has focused on its MIM domain. The line of work provided tremendous insights into the MAD1 working mechanism but still left many questions, the predominant one being how to reconcile the cell cycle independent MAD1MIM:C-MAD2 catalyst formation with cell cycle dependent C-MAD2 production. Recent results suggested that MAD1NTD and MAD1CTD domains may be an integral part of the regulatory mechanisms to control MAD1 activity in promoting MAD2 O-C conversion. In addition, MAD1 may also be part of the scaffold to streamline MCC assembly at kinetochores. In the near future, it is crucial to clarify the kinetochore receptor(s) for MAD1 and to establish how microtubule attachment disrupts the interaction between MAD1 and its kinetochore receptors. It will also be intriguing to further elucidate how different domains of MAD1 respond to modifications by protein kinases such as MPS1, BUB1 and Aurora B, and their counter-acting phosphatases to control MAD2 O-C conversion and MCC assembly. MAD1 demands and deserves more attention.
Author Contributions
YL and EA contribute equally to the manuscript. Both YL and EA drafted the manuscript and made the figures. S-TL revised the whole manuscript critically for important intellectual content. All authors give final approval of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The work in the authors' lab is supported by National Institutes of Health (R01CA169500) and Elsa U. Pardee Foundation. We thank Dr. William Taylor for critical reading of the manuscript.
References
Akera, T., Goto, Y., Sato, M., Yamamoto, M., and Watanabe, Y. (2015). Mad1 promotes chromosome congression by anchoring a kinesin motor to the kinetochore. Nat. Cell Biol. 17, 1124–1133. doi: 10.1038/ncb3219
Alfieri, C., Chang, L., Zhang, Z., Yang, J., Maslen, S., Skehel, M., et al. (2016). Molecular basis of APC/C regulation by the spindle assembly checkpoint. Nature 536, 431–436. doi: 10.1038/nature19083
Aravamudhan, P., Goldfarb, A. A., and Joglekar, A. P. (2015). The kinetochore encodes a mechanical switch to disrupt spindle assembly checkpoint signalling. Nat. Cell Biol. 17, 868–879. doi: 10.1038/ncb3179
Baek, M., Park, T., Heo, L., Park, C., and Seok, C. (2017). GalaxyHomomer: a web server for protein homo-oligomer structure prediction from a monomer sequence or structure. Nucleic Acids Res. 45, W320–W324. doi: 10.1093/nar/gkx246
Ballister, E. R., Riegman, M., and Lampson, M. A. (2014). Recruitment of Mad1 to metaphase kinetochores is sufficient to reactivate the mitotic checkpoint. J. Cell Biol. 204, 901–908. doi: 10.1083/jcb.201311113
Basto, R., Gomes, R., and Karess, R. E. (2000). Rough deal and Zw10 are required for the metaphase checkpoint in Drosophila. Nat. Cell Biol. 2, 939–943. doi: 10.1038/35046592
Bharadwaj, R., Qi, W., and Yu, H. (2004). Identification of two novel components of the human NDC80 kinetochore complex. J. Biol. Chem. 279, 13076–13085. doi: 10.1074/jbc.M310224200
Brady, D. M., and Hardwick, K. G. (2000). Complex formation between Mad1p, Bub1p and Bub3p is crucial for spindle checkpoint function. Curr. Biol. 10, 675–678. doi: 10.1016/S0960-9822(00)00515-7
Buffin, E., Lefebvre, C., Huang, J., Gagou, M. E., and Karess, R. E. (2005). Recruitment of Mad2 to the kinetochore requires the Rod/Zw10 complex. Curr. Biol. 15, 856–861. doi: 10.1016/j.cub.2005.03.052
Caldas, G. V., Lynch, T. R., Anderson, R., Afreen, S., Varma, D., and DeLuca, J. G. (2015). The RZZ complex requires the N-terminus of KNL1 to mediate optimal Mad1 kinetochore localization in human cells. Open Biol. 5:150160. doi: 10.1098/rsob.150160
Campbell, M. S., Chan, G. K., and Yen, T. J. (2001). Mitotic checkpoint proteins HsMAD1 and HsMAD2 are associated with nuclear pore complexes in interphase. J. Cell Sci. 114(Pt 5), 953–963.
Chan, G. K., Jablonski, S. A., Starr, D. A., Goldberg, M. L., and Yen, T. J. (2000). Human Zw10 and ROD are mitotic checkpoint proteins that bind to kinetochores. Nat. Cell Biol. 2, 944–947. doi: 10.1038/35046598
Chao, W. C., Kulkarni, K., Zhang, Z., Kong, E. H., and Barford, D. (2012). Structure of the mitotic checkpoint complex. Nature 484, 208–213. doi: 10.1038/nature10896
Chen, R. H., Brady, D. M., Smith, D., Murray, A. W., and Hardwick, K. G. (1999). The spindle checkpoint of budding yeast depends on a tight complex between the Mad1 and Mad2 proteins. Mol. Biol. Cell 10, 2607–2618. doi: 10.1091/mbc.10.8.2607
Chen, R. H., Shevchenko, A., Mann, M., and Murray, A. W. (1998). Spindle checkpoint protein Xmad1 recruits Xmad2 to unattached kinetochores. J. Cell Biol. 143, 283–295. doi: 10.1083/jcb.143.2.283
Chen, R. H., Waters, J. C., Salmon, E. D., and Murray, A. W. (1996). Association of spindle assembly checkpoint component XMAD2 with unattached kinetochores. Science 274, 242–246. doi: 10.1126/science.274.5285.242
Chi, Y. H., Haller, K., Ward, M. D., Semmes, O. J., Li, Y., and Jeang, K. T. (2008). Requirements for protein phosphorylation and the kinase activity of polo-like kinase 1 (Plk1) for the kinetochore function of mitotic arrest deficiency protein 1 (Mad1). J. Biol. Chem. 283, 35834–35844. doi: 10.1074/jbc.M804967200
Chung, E., and Chen, R. H. (2002). Spindle checkpoint requires Mad1-bound and Mad1-free Mad2. Mol. Biol. Cell 13, 1501–1511. doi: 10.1091/mbc.02-01-0003
De Antoni, A., Pearson, C. G., Cimini, D., Canman, J. C., Sala, V., Nezi, L., et al. (2005). The Mad1/Mad2 complex as a template for Mad2 activation in the spindle assembly checkpoint. Curr. Biol. 15, 214–225. doi: 10.1016/j.cub.2005.01.038
Défachelles, L., Raich, N., Terracol, R., Baudin, X., Williams, B., Goldberg, M. E., et al. (2015). RZZ and Mad1 dynamics in Drosophila mitosis. Chromosome Res. 23, 333–342. doi: 10.1007/s10577-015-9472-x
Diaz-Martinez, L. A., Tian, W., Li, B., Warrington, R., Jia, L., Brautigam, C. A., et al. (2015). The Cdc20-binding Phe box of the spindle checkpoint protein BubR1 maintains the mitotic checkpoint complex during mitosis. J. Biol. Chem. 290, 2431–2443. doi: 10.1074/jbc.M114.616490
Di Fiore, B., Davey, N. E., Hagting, A., Izawa, D., Mansfeld, J., Gibson, T. J., et al. (2015). The ABBA motif binds APC/C activators and is shared by APC/C substrates and regulators. Dev. Cell 32, 358–372. doi: 10.1016/j.devcel.2015.01.003
Dou, Z., von Schubert, C., Körner, R., Santamaria, A., Elowe, S., and Nigg, E. A. (2011). Quantitative mass spectrometry analysis reveals similar substrate consensus motif for human Mps1 kinase and Plk1. PLoS ONE 6:e18793. doi: 10.1371/journal.pone.0018793
Emanuele, M. J., and Stukenberg, P. T. (2007). Xenopus Cep57 is a novel kinetochore component involved in microtubule attachment. Cell 130, 893–905. doi: 10.1016/j.cell.2007.07.023
Espeut, J., Lara-Gonzalez, P., Sassine, M., Shiau, A. K., Desai, A., and Abrieu, A. (2015). Natural loss of Mps1 kinase in nematodes uncovers a role for polo-like Kinase 1 in spindle checkpoint initiation. Cell Rep. 12, 58–65. doi: 10.1016/j.celrep.2015.05.039
Faesen, A. C., Thanasoula, M., Maffini, S., Breit, C., Müller, F., van Gerwen, S., et al. (2017). Basis of catalytic assembly of the mitotic checkpoint complex. Nature 542, 498–502. doi: 10.1038/nature21384
Fang, G., Yu, H., and Kirschner, M. W. (1998). The checkpoint protein MAD2 and the mitotic regulator CDC20 form a ternary complex with the anaphase-promoting complex to control anaphase initiation. Genes Dev. 12, 1871–1883. doi: 10.1101/gad.12.12.1871
Fava, L. L., Kaulich, M., Nigg, E. A., and Santamaria, A. (2011). Probing the in vivo function of Mad1:C-Mad2 in the spindle assembly checkpoint. EMBO J. 30, 3322–3336. doi: 10.1038/emboj.2011.239
Foley, E. A., and Kapoor, T. M. (2013). Microtubule attachment and spindle assembly checkpoint signalling at the kinetochore. Nat. Rev. Mol. Cell Biol. 14, 25–37. doi: 10.1038/nrm3494
Hara, M., Özkan, E., Sun, H., Yu, H., and Luo, X. (2015). Structure of an intermediate conformer of the spindle checkpoint protein Mad2. Proc. Natl. Acad. Sci. U.S.A. 112, 11252–11257. doi: 10.1073/pnas.1512197112
Hayes, M. J., Kimata, Y., Wattam, S. L., Lindon, C., Mao, G., Yamano, H., et al. (2006). Early mitotic degradation of Nek2A depends on Cdc20-independent interaction with the APC/C. Nat. Cell Biol. 8, 607–614. doi: 10.1038/ncb1410
Heinrich, S., Sewart, K., Windecker, H., Langegger, M., Schmidt, N., Hustedt, N., et al. (2014). Mad1 contribution to spindle assembly checkpoint signalling goes beyond presenting Mad2 at kinetochores. EMBO Rep. 15, 291–298. doi: 10.1002/embr.201338114
Hiruma, Y., Sacristan, C., Pachis, S. T., Adamopoulos, A., Kuijt, T., Ubbink, M., et al. (2015). CELL DIVISION CYCLE. Competition between MPS1 and microtubules at kinetochores regulates spindle checkpoint signaling. Science 348, 1264–1267. doi: 10.1126/science.aaa4055
Ikeda, M., and Tanaka, K. (2017). Plk1 bound to Bub1 contributes to spindle assembly checkpoint activity during mitosis. Sci. Rep. 7:8794. doi: 10.1038/s41598-017-09114-3
Izawa, D., and Pines, J. (2012). Mad2 and the APC/C compete for the same site on Cdc20 to ensure proper chromosome segregation. J. Cell Biol. 199, 27–37. doi: 10.1083/jcb.201205170
Izawa, D., and Pines, J. (2015). The mitotic checkpoint complex binds a second CDC20 to inhibit active APC/C. Nature 517, 631–634. doi: 10.1038/nature13911
Ji, W., Luo, Y., Ahmad, E., and Liu, S. T. (2018). Direct interactions of mitotic arrest deficient 1 (MAD1) domains with each other and MAD2 conformers are required for mitotic checkpoint signaling. J. Biol. Chem. 293, 484–496. doi: 10.1074/jbc.RA117.000555
Ji, Z., Gao, H., Jia, L., Li, B., and Yu, H. (2017). A sequential multi-target Mps1 phosphorylation cascade promotes spindle checkpoint signaling. Elife 6:e22513. doi: 10.7554/eLife.22513
Ji, Z., Gao, H., and Yu, H. (2015). CELL DIVISION CYCLE. Kinetochore attachment sensed by competitive Mps1 and microtubule binding to Ndc80C. Science 348, 1260–1264. doi: 10.1126/science.aaa4029
Jia, L., Kim, S., and Yu, H. (2013). Tracking spindle checkpoint signals from kinetochores to APC/C. Trends Biochem. Sci. 38, 302–311. doi: 10.1016/j.tibs.2013.03.004
Jia, L., Li, B., and Yu, H. (2016). The Bub1-Plk1 kinase complex promotes spindle checkpoint signalling through Cdc20 phosphorylation. Nat. Commun. 7:10818. doi: 10.1038/ncomms10818
Johnson, V. L., Scott, M. I., Holt, S. V., Hussein, D., and Taylor, S. S. (2004). Bub1 is required for kinetochore localization of BubR1, Cenp-E, Cenp-F and Mad2, and chromosome congression. J. Cell Sci. 117(Pt 8), 1577–1589. doi: 10.1242/jcs.01006
Karess, R. (2005). Rod-Zw10-Zwilch: a key player in the spindle checkpoint. Trends Cell Biol. 15, 386–392. doi: 10.1016/j.tcb.2005.05.003
Kim, S., Sun, H., Tomchick, D. R., Yu, H., and Luo, X. (2012). Structure of human Mad1 C-terminal domain reveals its involvement in kinetochore targeting. Proc. Natl. Acad. Sci. U.S.A. 109, 6549–6554. doi: 10.1073/pnas.1118210109
Klebig, C., Korinth, D., and Meraldi, P. (2009). Bub1 regulates chromosome segregation in a kinetochore-independent manner. J. Cell Biol. 185, 841–858. doi: 10.1083/jcb.200902128
Kops, G. J., Kim, Y., Weaver, B. A., Mao, Y., McLeod, I., Yates, J. R., et al. (2005). ZW10 links mitotic checkpoint signaling to the structural kinetochore. J. Cell Biol. 169, 49–60. doi: 10.1083/jcb.200411118
Krenn, V., Overlack, K., Primorac, I., van Gerwen, S., and Musacchio, A. (2014). KI motifs of human Knl1 enhance assembly of comprehensive spindle checkpoint complexes around MELT repeats. Curr. Biol. 24, 29–39. doi: 10.1016/j.cub.2013.11.046
Kruse, T., Larsen, M. S., Sedgwick, G. G., Sigurdsson, J. O., Streicher, W., Olsen, J. V., et al. (2014). A direct role of Mad1 in the spindle assembly checkpoint beyond Mad2 kinetochore recruitment. EMBO Rep. 15, 282–290. doi: 10.1002/embr.201338101
Kuijt, T. E., Omerzu, M., Saurin, A. T., and Kops, G. J. (2014). Conditional targeting of MAD1 to kinetochores is sufficient to reactivate the spindle assembly checkpoint in metaphase. Chromosoma 123, 471–480. doi: 10.1007/s00412-014-0458-9
Lara-Gonzalez, P., Westhorpe, F. G., and Taylor, S. S. (2012). The spindle assembly checkpoint. Curr. Biol. 22, R966–R980. doi: 10.1016/j.cub.2012.10.006
Lee, S. H., Sterling, H., Burlingame, A., and McCormick, F. (2008). Tpr directly binds to Mad1 and Mad2 and is important for the Mad1-Mad2-mediated mitotic spindle checkpoint. Genes Dev. 22, 2926–2931. doi: 10.1101/gad.1677208
Lénárt, P., Petronczki, M., Steegmaier, M., Di Fiore, B., Lipp, J. J., Hoffmann, M., et al. (2007). The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr. Biol. 17, 304–315. doi: 10.1016/j.cub.2006.12.046
Li, R., and Murray, A. W. (1991). Feedback control of mitosis in budding yeast. Cell 66, 519–531. doi: 10.1016/0092-8674(81)90015-5
Lince-Faria, M., Maffini, S., Orr, B., Ding, Y., Florindo, C., Sunkel, C. E., et al. (2009). Spatiotemporal control of mitosis by the conserved spindle matrix protein Megator. J. Cell Biol. 184, 647–657. doi: 10.1083/jcb.200811012
Lischetti, T., Zhang, G., Sedgwick, G. G., Bolanos-Garcia, V. M., and Nilsson, J. (2014). The internal Cdc20 binding site in BubR1 facilitates both spindle assembly checkpoint signalling and silencing. Nat. Commun. 5:5563. doi: 10.1038/ncomms6563
Liu, D., Davydenko, O., and Lampson, M. A. (2012). Polo-like kinase-1 regulates kinetochore-microtubule dynamics and spindle checkpoint silencing. J. Cell Biol. 198, 491–499. doi: 10.1083/jcb.201205090
Liu, S. T., Chan, G. K., Hittle, J. C., Fujii, G., Lees, E., and Yen, T. J. (2003a). Human MPS1 kinase is required for mitotic arrest induced by the loss of CENP-E from kinetochores. Mol. Biol. Cell 14, 1638–1651. doi: 10.1091/mbc.02-05-0074
Liu, S. T., Hittle, J. C., Jablonski, S. A., Campbell, M. S., Yoda, K., and Yen, T. J. (2003b). Human CENP-I specifies localization of CENP-F, MAD1 and MAD2 to kinetochores and is essential for mitosis. Nat. Cell Biol. 5, 341–345. doi: 10.1038/ncb953
Liu, S. T., Rattner, J. B., Jablonski, S. A., and Yen, T. J. (2006). Mapping the assembly pathways that specify formation of the trilaminar kinetochore plates in human cells. J. Cell Biol. 175, 41–53. doi: 10.1083/jcb.200606020
Liu, S.-T., and Yen, T. J. (2008). “The kinetochore as target for cancer drug development,” in The Kinetochore: From Molecular Discoveries to Cancer Therapy, eds P. De Wulf and W. C. Earnshaw (New York, NY: Springer), 455–479.
Liu, S. T., and Zhang, H. (2016). The mitotic checkpoint complex (MCC): looking back and forth after 15 years. AIMS Mol. Sci. 3, 597–634. doi: 10.3934/molsci.2016.4.597
Liu, Y., Du, X., Zhang, S., Liu, Y., Zhang, Q., Yin, Q., et al. (2017). PTEN regulates spindle assembly checkpoint timing through MAD1 in interphase. Oncotarget 8, 98040–98050. doi: 10.18632/oncotarget.20532
London, N., and Biggins, S. (2014a). Mad1 kinetochore recruitment by Mps1-mediated phosphorylation of Bub1 signals the spindle checkpoint. Genes Dev. 28, 140–152. doi: 10.1101/gad.233700.113
London, N., and Biggins, S. (2014b). Signalling dynamics in the spindle checkpoint response. Nat. Rev. Mol. Cell Biol. 15, 736–747. doi: 10.1038/nrm3888
London, N., Ceto, S., Ranish, J. A., and Biggins, S. (2012). Phosphoregulation of Spc105 by Mps1 and PP1 regulates Bub1 localization to kinetochores. Curr. Biol. 22, 900–906. doi: 10.1016/j.cub.2012.03.052
Lou, Y., Yao, J., Zereshki, A., Dou, Z., Ahmed, K., Wang, H., et al. (2004). NEK2A interacts with MAD1 and possibly functions as a novel integrator of the spindle checkpoint signaling. J. Biol. Chem. 279, 20049–20057. doi: 10.1074/jbc.M314205200
Luo, X., Tang, Z., Xia, G., Wassmann, K., Matsumoto, T., Rizo, J., et al. (2004). The Mad2 spindle checkpoint protein has two distinct natively folded states. Nat. Struct. Mol. Biol. 11, 338–345. doi: 10.1038/nsmb748
Luo, X., and Yu, H. (2008). Protein metamorphosis: the two-state behavior of Mad2. Structure 16, 1616–1625. doi: 10.1016/j.str.2008.10.002
Lupas, A., Van Dyke, M., and Stock, J. (1991). Predicting coiled coils from protein sequences. Science 252, 1162–1164. doi: 10.1126/science.252.5009.1162
Maldonado, M., and Kapoor, T. M. (2011). Constitutive Mad1 targeting to kinetochores uncouples checkpoint signalling from chromosome biorientation. Nat. Cell Biol. 13, 475–482. doi: 10.1038/ncb2223
Mapelli, M., Massimiliano, L., Santaguida, S., and Musacchio, A. (2007). The Mad2 conformational dimer: structure and implications for the spindle assembly checkpoint. Cell 131, 730–743. doi: 10.1016/j.cell.2007.08.049
Mapelli, M., and Musacchio, A. (2007). MAD contortions: conformational dimerization boosts spindle checkpoint signaling. Curr. Opin. Struct. Biol. 17, 716–725. doi: 10.1016/j.sbi.2007.08.011
Martin-Lluesma, S., Stucke, V. M., and Nigg, E. A. (2002). Role of hec1 in spindle checkpoint signaling and kinetochore recruitment of mad1/mad2. Science 297, 2267–2270. doi: 10.1126/science.1075596
Matson, D. R., and Stukenberg, P. T. (2014). CENP-I and Aurora B act as a molecular switch that ties RZZ/Mad1 recruitment to kinetochore attachment status. J. Cell Biol. 205, 541–554. doi: 10.1083/jcb.201307137
Meraldi, P., Draviam, V. M., and Sorger, P. K. (2004). Timing and checkpoints in the regulation of mitotic progression. Dev. Cell 7, 45–60. doi: 10.1016/j.devcel.2004.06.006
Meraldi, P., and Sorger, P. K. (2005). A dual role for Bub1 in the spindle checkpoint and chromosome congression. EMBO J. 24, 1621–1633. doi: 10.1038/sj.emboj.7600641
Mora-Santos, M. D., Hervas-Aguilar, A., Sewart, K., Lancaster, T. C., Meadows, J. C., and Millar, J. B. (2016). Bub3-Bub1 binding to Spc7/KNL1 toggles the spindle checkpoint switch by licensing the interaction of Bub1 with Mad1-Mad2. Curr. Biol. 26, 2642–2650. doi: 10.1016/j.cub.2016.07.040
Moyle, M. W., Kim, T., Hattersley, N., Espeut, J., Cheerambathur, D. K., Oegema, K., et al. (2014). A Bub1-Mad1 interaction targets the Mad1-Mad2 complex to unattached kinetochores to initiate the spindle checkpoint. J. Cell Biol. 204, 647–657. doi: 10.1083/jcb.201311015
Musacchio, A. (2015). The molecular biology of spindle assembly checkpoint signaling dynamics. Curr. Biol. 25, R1002–R1018. doi: 10.1016/j.cub.2015.08.051
O'Connor, A., Maffini, S., Rainey, M. D., Kaczmarczyk, A., Gaboriau, D., Musacchio, A., et al. (2015). Requirement for PLK1 kinase activity in the maintenance of a robust spindle assembly checkpoint. Biol. Open 5, 11–19. doi: 10.1242/bio.014969
Overlack, K., Primorac, I., Vleugel, M., Krenn, V., Maffini, S., Hoffmann, I., et al. (2015). A molecular basis for the differential roles of Bub1 and BubR1 in the spindle assembly checkpoint. Elife 4:e05269. doi: 10.7554/eLife.05269
Primorac, I., Weir, J. R., Chiroli, E., Gross, F., Hoffmann, I., van Gerwen, S., et al. (2013). Bub3 reads phosphorylated MELT repeats to promote spindle assembly checkpoint signaling. Elife 2:e01030. doi: 10.7554/eLife.01030
Qian, J., García-Gimeno, M. A., Beullens, M., Manzione, M. G., Van der Hoeven, G., Igual, J. C., et al. (2017). An attachment-independent biochemical timer of the spindle assembly checkpoint. Mol. Cell. 68, 715.e5–730.e5. doi: 10.1016/j.molcel.2017.10.011
Rodriguez-Bravo, V., Maciejowski, J., Corona, J., Buch, H. K., Collin, P., Kanemaki, M. T., et al. (2014). Nuclear pores protect genome integrity by assembling a premitotic and mad1-dependent anaphase inhibitor. Cell 156, 1017–1031. doi: 10.1016/j.cell.2014.01.010
Samejima, I., Spanos, C., Alves Fde, L., Hori, T., Perpelescu, M., Zou, J. C., et al. (2015). Whole-proteome genetic analysis of dependencies in assembly of a vertebrate kinetochore. J. Cell Biol. 211, 1141–1156. doi: 10.1083/jcb.201508072
Santaguida, S., and Musacchio, A. (2009). The life and miracles of kinetochores. EMBO J. 28, 2511–2531. doi: 10.1038/emboj.2009.173
Sedgwick, G. G., Hayward, D. G., Di Fiore, B., Pardo, M., Yu, L., Pines, J., et al. (2013). Mechanisms controlling the temporal degradation of Nek2A and Kif18A by the APC/C-Cdc20 complex. EMBO J. 32, 303–314. doi: 10.1038/emboj.2012.335
Shepperd, L. A., Meadows, J. C., Sochaj, A. M., Lancaster, T. C., Zou, J., Buttrick, G. J., et al. (2012). Phosphodependent recruitment of Bub1 and Bub3 to Spc7/KNL1 by Mph1 kinase maintains the spindle checkpoint. Curr. Biol. 22, 891–899. doi: 10.1016/j.cub.2012.03.051
Silió, V., McAinsh, A. D., and Millar, J. B. (2015). KNL1-Bubs and RZZ provide two separable pathways for checkpoint activation at human kinetochores. Dev. Cell 35, 600–613. doi: 10.1016/j.devcel.2015.11.012
Sironi, L., Mapelli, M., Knapp, S., De Antoni, A., Jeang, K. T., and Musacchio, A. (2002). Crystal structure of the tetrameric Mad1-Mad2 core complex: implications of a “safety belt” binding mechanism for the spindle checkpoint. EMBO J. 21, 2496–2506. doi: 10.1093/emboj/21.10.2496
Sudakin, V., Chan, G. K., and Yen, T. J. (2001). Checkpoint inhibition of the APC/C in HeLa cells is mediated by a complex of BUBR1, BUB3, CDC20, and MAD2. J. Cell Biol. 154, 925–936. doi: 10.1083/jcb.200102093
Sumara, I., Giménez-Abián, J. F., Gerlich, D., Hirota, T., Kraft, C., de la Torre, C., et al. (2004). Roles of polo-like kinase 1 in the assembly of functional mitotic spindles. Curr. Biol. 14, 1712–1722. doi: 10.1016/j.cub.2004.09.049
Tipton, A. R., Ji, W., Sturt-Gillespie, B., Bekier, M. E., Wang, K., Taylor, W. R., et al. (2013). Monopolar spindle 1 (MPS1) kinase promotes production of closed MAD2 (C-MAD2) conformer and assembly of the mitotic checkpoint complex. J. Biol. Chem. 288, 35149–35158. doi: 10.1074/jbc.M113.522375
Tipton, A. R., Tipton, M., Yen, T., and Liu, S. T. (2011a). Closed MAD2 (C-MAD2) is selectively incorporated into the mitotic checkpoint complex (MCC). Cell Cycle 10, 3740–3750. doi: 10.4161/cc.10.21.17919
Tipton, A. R., Wang, K., Link, L., Bellizzi, J. J., Huang, H., Liu, S. T., et al. (2011b). BUBR1 and closed MAD2 (C-MAD2) interact directly to assemble a functional mitotic checkpoint complex. J. Biol. Chem. 286, 21173–21179. doi: 10.1074/jbc.M111.238543
Vleugel, M., Hoogendoorn, E., Snel, B., and Kops, G. J. (2012). Evolution and function of the mitotic checkpoint. Dev. Cell 23, 239–250. doi: 10.1016/j.devcel.2012.06.013
Vleugel, M., Tromer, E., Omerzu, M., Groenewold, V., Nijenhuis, W., Snel, B., et al. (2013). Arrayed BUB recruitment modules in the kinetochore scaffold KNL1 promote accurate chromosome segregation. J. Cell Biol. 203, 943–955. doi: 10.1083/jcb.201307016
von Schubert, C., Cubizolles, F., Bracher, J. M., Sliedrecht, T., Kops, G. J. P. L., and Nigg, E. A. (2015). Plk1 and Mps1 cooperatively regulate the spindle assembly checkpoint in human cells. Cell Rep. 12, 66–78. doi: 10.1016/j.celrep.2015.06.007
Waters, J. C., Chen, R. H., Murray, A. W., and Salmon, E. D. (1998). Localization of Mad2 to kinetochores depends on microtubule attachment, not tension. J. Cell Biol. 141, 1181–1191. doi: 10.1083/jcb.141.5.1181
Weaver, B. A., and Cleveland, D. W. (2007). Aneuploidy: instigator and inhibitor of tumorigenesis. Cancer Res. 67, 10103–10105. doi: 10.1158/0008-5472.CAN-07-2266
Yamagishi, Y., Yang, C. H., Tanno, Y., and Watanabe, Y. (2012). MPS1/Mph1 phosphorylates the kinetochore protein KNL1/Spc7 to recruit SAC components. Nat. Cell Biol. 14, 746–752. doi: 10.1038/ncb2515
Yamaguchi, M., VanderLinden, R., Weissmann, F., Qiao, R., Dube, P., Brown, N. G., et al. (2016). Cryo-EM of mitotic checkpoint complex-bound APC/C reveals reciprocal and conformational regulation of ubiquitin ligation. Mol. Cell 63, 593–607. doi: 10.1016/j.molcel.2016.07.003
Yang, C., Hao, J., Kong, D., Cui, X., Zhang, W., Wang, H., et al. (2014). ATM-mediated Mad1 Serine 214 phosphorylation regulates Mad1 dimerization and the spindle assembly checkpoint. Carcinogenesis 35, 2007–2013. doi: 10.1093/carcin/bgu087
Yang, M., Li, B., Liu, C. J., Tomchick, D. R., Machius, M., Rizo, J., et al. (2008). Insights into mad2 regulation in the spindle checkpoint revealed by the crystal structure of the symmetric mad2 dimer. PLoS Biol. 6:e50. doi: 10.1371/journal.pbio.0060050
Zhang, G., Kruse, T., López-Méndez, B., Sylvestersen, K. B., Garvanska, D. H., Schopper, S., et al. (2017). Bub1 positions Mad1 close to KNL1 MELT repeats to promote checkpoint signalling. Nat. Commun. 8:15822. doi: 10.1038/ncomms15822
Zhang, G., Lischetti, T., Hayward, D. G., and Nilsson, J. (2015). Distinct domains in Bub1 localize RZZ and BubR1 to kinetochores to regulate the checkpoint. Nat. Commun. 6:7162. doi: 10.1038/ncomms8162
Zhang, G., Lischetti, T., and Nilsson, J. (2014). A minimal number of MELT repeats supports all the functions of KNL1 in chromosome segregation. J. Cell Sci. 127 (Pt 4), 871–884. doi: 10.1242/jcs.139725
Zhang, G., Mendez, B. L., Sedgwick, G. G., and Nilsson, J. (2016). Two functionally distinct kinetochore pools of BubR1 ensure accurate chromosome segregation. Nat. Commun. 7:12256. doi: 10.1038/ncomms12256
Keywords: mitosis, mitotic checkpoint, kinetochore, MAD1, MAD2, protein conformation
Citation: Luo Y, Ahmad E and Liu S-T (2018) MAD1: Kinetochore Receptors and Catalytic Mechanisms. Front. Cell Dev. Biol. 6:51. doi: 10.3389/fcell.2018.00051
Received: 07 March 2018; Accepted: 18 April 2018;
Published: 07 May 2018.
Edited by:
Uttam Surana, Institute of Molecular and Cell Biology (A*STAR), SingaporeReviewed by:
Andrew Burgess, Anzac Research Institute, AustraliaBrian Gabrielli, The University of Queensland, Australia
Copyright © 2018 Luo, Ahmad and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Song-Tao Liu, c2xpdUB1dG5ldC51dG9sZWRvLmVkdQ==
†These authors have contributed equally to this work.