 Oluwaseun B. Ogunbona
Oluwaseun B. Ogunbona Steven M. Claypool
Steven M. Claypool- 1Department of Physiology, School of Medicine, Johns Hopkins University, Baltimore, MD, United States
- 2Department of Pathology & Laboratory Medicine, School of Medicine, Emory University, Atlanta, GA, United States
The mitochondrial carrier family (MCF) is a group of transport proteins that are mostly localized to the inner mitochondrial membrane where they facilitate the movement of various solutes across the membrane. Although these carriers represent potential targets for therapeutic application and are repeatedly associated with human disease, research on the MCF has not progressed commensurate to their physiologic and pathophysiologic importance. Many of the 53 MCF members in humans are orphans and lack known transport substrates. Even for the relatively well-studied members of this family, such as the ADP/ATP carrier and the uncoupling protein, there exist fundamental gaps in our understanding of their biological roles including a clear rationale for the existence of multiple isoforms. Here, we briefly review this important family of mitochondrial carriers, provide a few salient examples of their diverse metabolic roles and disease associations, and then focus on an emerging link between several distinct MCF members, including the ADP/ATP carrier, and cytochrome c oxidase biogenesis. As the ADP/ATP carrier is regarded as the paradigm of the entire MCF, its newly established role in regulating translation of the mitochondrial genome highlights that we still have a lot to learn about these metabolite transporters.
The Solute Carrier (SLC) Family
Transport of substrates across biological membranes between and among organelles is an important feature of eukaryotic cells. The SLC family, the second largest family of membrane proteins, is a large group of membrane transport proteins; in humans, there are 456 known members that are grouped into 65 subfamilies (Höglund et al., 2011; Perland and Fredriksson, 2017). SLCs facilitate the movement of otherwise membrane-impermeable solutes—such as amino acids, ions, nucleotides, sugars and drugs—across biological membranes. The family includes functionally related proteins that mediate the transport and exchange of solutes across cell membranes. Transport can be facilitative by simply allowing solutes to equilibrate across a membrane according to their relative distribution on either side. Additionally, SLCs can mediate secondary active transport by coupling the downhill flow of one substrate, often an ion, to the uphill movement of another substrate against its relative gradient across a membrane. Primary active transporters, ion channels and aquaporins are not included in the SLC family. The criterion for membership in the SLC family is being an integral membrane protein that transports a solute. Not surprisingly, the SLC family is structurally quite diverse. However, within an individual sub-family, members often share more than 20% sequence homology (Hediger et al., 2004). Table 1 describes the current list of SLC family members based on http://slc.bioparadigms.org and provides references that review each subfamily. Families SLC53-65 are newly registered, and are based on a work presented at the BioMedical Transporters 2017 conference in Lausanne, Switzerland.
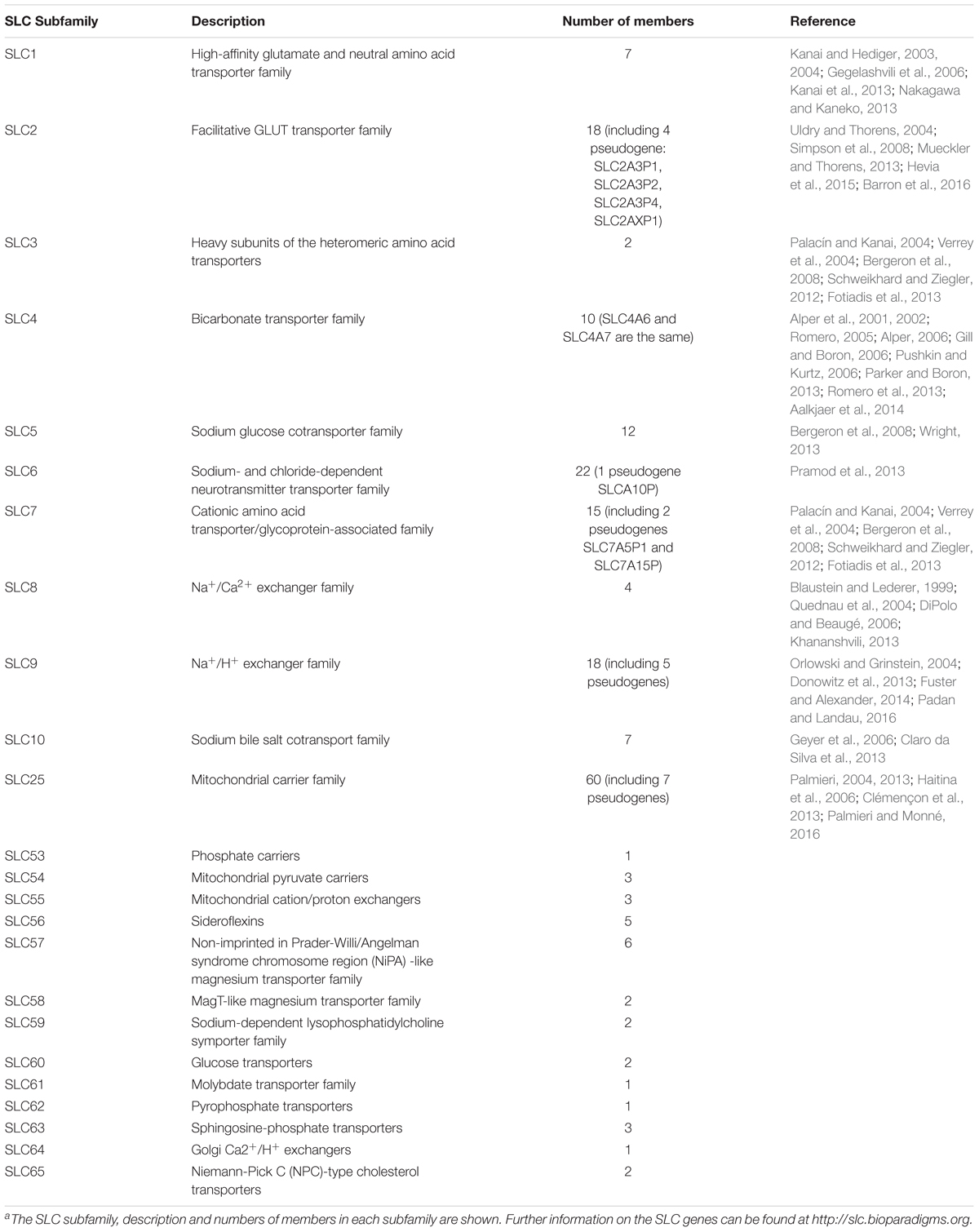
Table 1. Abridged list of current SLC familiesa.
Mitochondrial Carrier Family (SLC25)
The Solute Carrier 25 (SLC25) family transports solutes across the inner mitochondrial membrane (IMM), although several members of this family are localized to other cellular organelles such as chloroplasts and peroxisomes (Visser et al., 2002; Bedhomme et al., 2005). The MCF is the largest SLC subfamily and all members are encoded by the nuclear genome. As such, they are synthesized by cytoplasmic ribosomes and need to be imported from the cytosol to their final location. 35 members have been identified in yeast, 58 members in Arabidopsis thaliana, and 53 members have been identified in humans. As summarized in Table 2 (Palmieri and Pierri, 2010), substrates for approximately one third of the MCF have yet to be identified. Tissue distribution can vary from ubiquitous [e.g., SLC25A6 (Stepien et al., 1992)] to tissue-specific [e.g., SLC25A31 (Dolce et al., 2005; Rodić et al., 2005)].
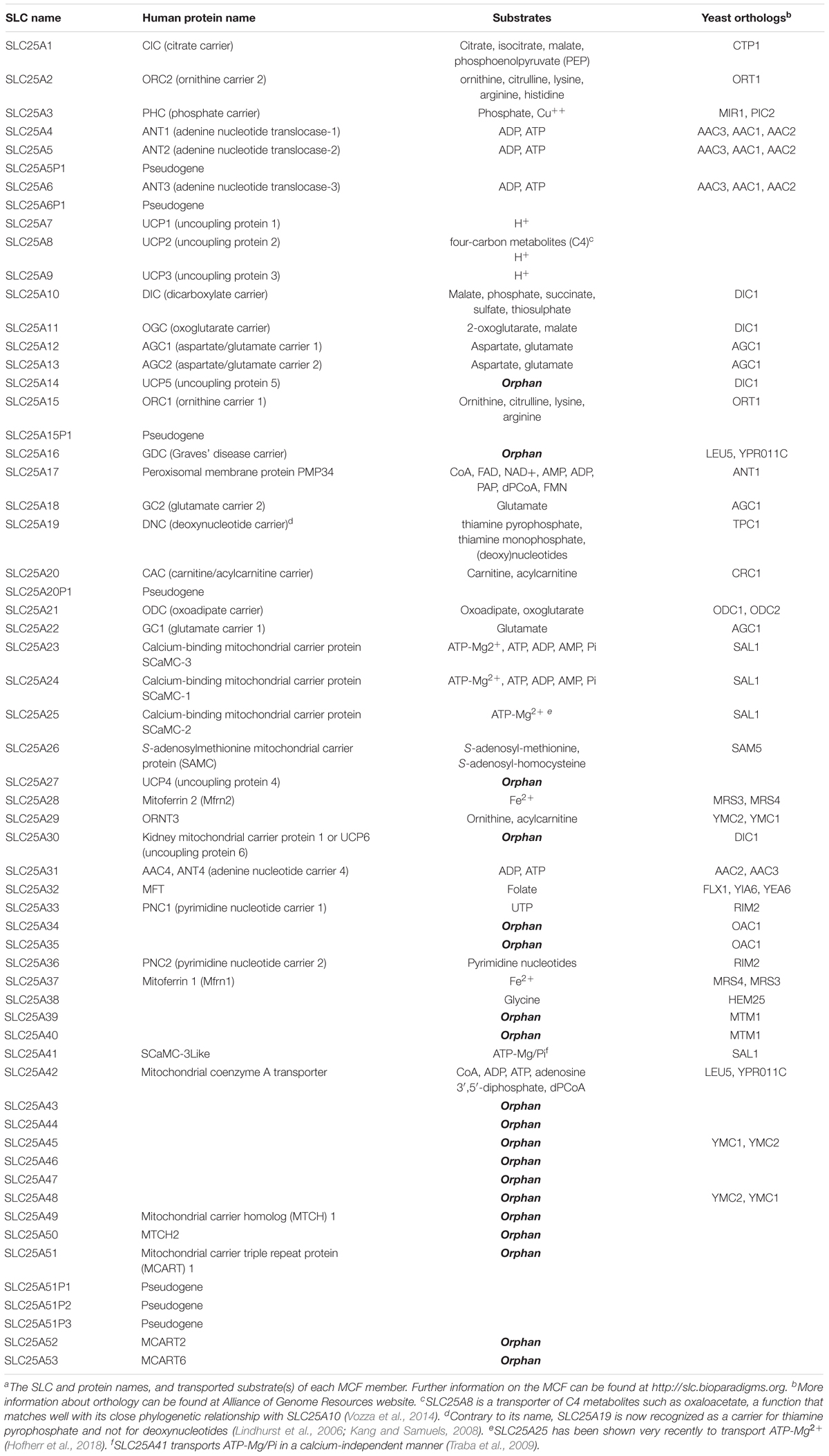
Table 2. Current list of MCF membersa.
SLC25 members are generally characterized by the presence of a tripartite structure of approximately 300 amino acids, six conserved transmembrane regions, and the three-fold repeated MCF signature motif, P-X-[DE]-X-X-[RK] (Figure 1). Although their substrates vary in size and nature, most members catalyze the exchange of one solute for another (antiport), couple the transport of one solute with another (symport), or facilitate the transport of a solute (uniport). Because of their sequence similarity, it is assumed that the transport mechanism is similar for the extended family.
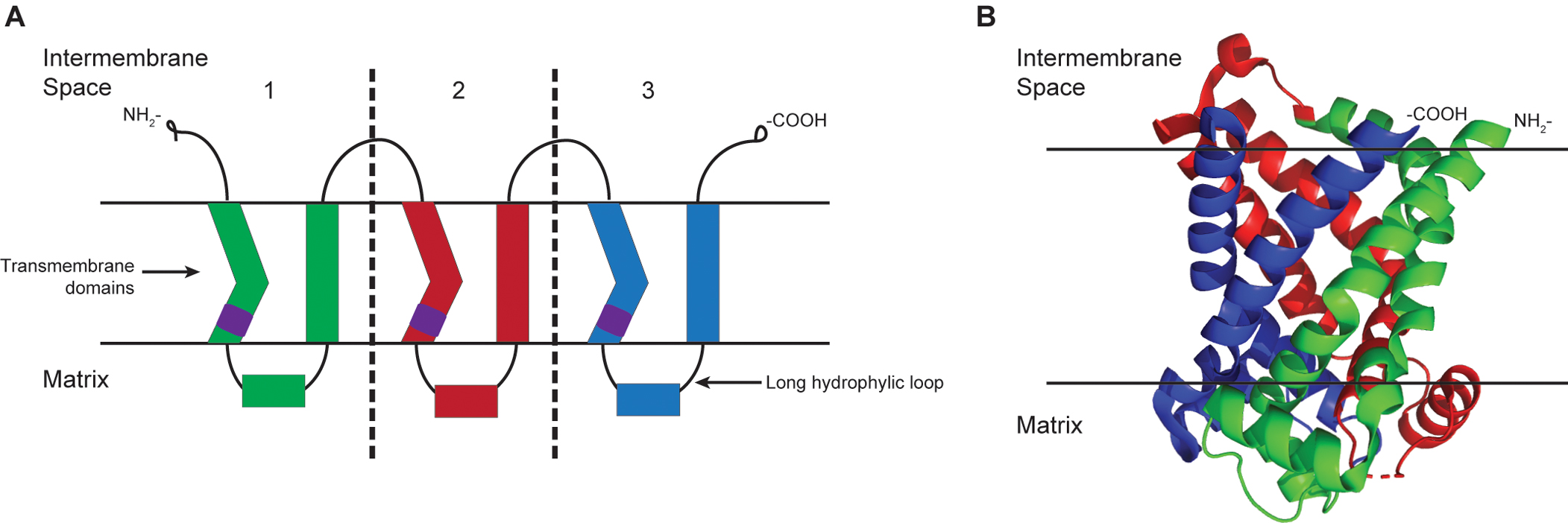
Figure 1. Schematic of the MCF tripartite structure. (A) The structure of members of the mitochondrial carrier family can be seen as three similar parts/domains with approximately 100 amino acids each. In each part, there are two alpha-helix transmembrane segments connected by a long matrix localized hydrophilic loop. The signature motif, PX(D/E)XX(K/R), indicated by purple cylinders, is at the C-terminus of the odd-numbered transmembrane helices. Both the NH2 and COOH termini are on the intermembrane space side of the mitochondrial inner membrane. (B) Structure of the yeast ADP/ATP carrier protein isoform 2 from protein data bank (entry 4C9G) is modified to show how the different domains are folded in a protein.
Physiology of MCF
Collectively, the MCF transports a wide range of solutes across the IMM. In this capacity, they act as important bridges that link many biochemical pathways that are otherwise compartmentalized in either the cytosol or the mitochondrial matrix (Palmieri, 2014). Solutes transported include protons, nucleotides, amino acids, carboxylic acids, inorganic ions, and cofactors. Their fundamental role in enabling metabolic compartmentalization cannot be overemphasized. SLC25 family members are involved in metabolic pathways such as heme synthesis and metal homeostasis (SLC25A28, SLC25A37, and SLC25A38), fatty acid metabolism (SLC23A1 and SLC25A20), amino acid metabolism (SLC25A12, SLC25A13, SLC25A18, and SLC25A22), nucleic acid metabolism (SLC25A26, SLC25A33, and SLC25A36), urea production (SLC25A2, SLC25A13, and SLC25A15) (Shayakul and Hediger, 2004; Shayakul et al., 2013; LeMoine and Walsh, 2015), OXPHOS (SLC25A3, SLC25A4, SLC25A5, SLC25A6, and SLC25A31) and heat generation (SLC25A7 and SLC25A9). As a thorough discussion of the entire SLC family is not the goal of this review, in the next four sections, the physiology of select SLC25 members is briefly discussed to illustrate the diversity of cellular functions in which its members participate.
The ADP/ATP Carrier Protein Is Essential for OXPHOS
Eukaryotic cells make energy in the form of ATP in the mitochondrial matrix and the ATP is translocated through the impermeable IMM to power many processes in the cell. The ADP/ATP carriers (AACs) provide the means of transport of ATP and its precursor ADP, across the IMM. Under physiological conditions, 1 molecule of ADP from the cytosol is exchanged for 1 molecule of matrix-localized ATP by the activity of the ADP/ATP carrier. AAC, referred to as ANT in humans, is a notable MCF member as it was the first to have its amino acid sequenced (Aquila et al., 1982) and its 3D structure solved (Pebay-Peyroula et al., 2003). Similar to all members of the MCF and regarded as a paradigm for this family, AACs are nuclear-encoded, integral membrane proteins with approximately 300 amino acids arranged into three repeats linked by two loops on the cytosolic side. There are two transmembrane α-helices in each repeat connected together by a long loop on the matrix side, giving the carrier a threefold pseudosymmetry (Palmieri, 2013).
One of the most abundant proteins in the IMM, Aacs are encoded by multiple different genes in both unicellular and multicellular eukaryotes. There are three yeast AAC isoforms and four human ANT isoforms. The human ANT isoforms overlap in their expression pattern but exhibit tissue-specificity. ANT1 (SLC25A4) is the most equivalent to yeast Aac2p and the predominant isoform in the heart and skeletal muscle (Stepien et al., 1992). ANT2 (SLC25A5) is mostly expressed in regenerative tissues such as the kidney and liver, ANT3 (SLC25A6) is ubiquitously expressed at low baseline levels, and ANT4 (SLC25A31) is selectively expressed in the testis (Stepien et al., 1992; Doerner et al., 1997; Dolce et al., 2005; Rodić et al., 2005; Kim et al., 2007; Dupont and Stepien, 2011). Aac2p is the most abundant of all three isoforms in yeast and the only one absolutely required for OXPHOS and growth on respiratory carbon sources (Lawson et al., 1990). Aac1p and Aac3p are minor isoforms in yeast that are undetectable at the protein level under normal growth conditions. Aac1p expression is repressed in hypoxic conditions (Gavurníková et al., 1996) and Aac3p expression is induced in anaerobic situations (Sabová et al., 1993).
Originally thought to consist of individual complexes in a functional chain, the advent of Blue-Native Polyacrylamide Gel-Electrophoresis (BN-PAGE) (Schägger and von Jagow, 1991), a gentle electrophoretic technique for the analysis of protein–protein interactions, facilitated the discovery that the respiratory complexes interact to form higher-order supramolecular assemblies of varying stoichiometry termed respiratory supercomplexes (RSCs) (Cruciat et al., 2000; Schägger and Pfeiffer, 2000; Acín-Pérez et al., 2008; Moreno-Lastres et al., 2012; Gu et al., 2016; Letts et al., 2016; Wu et al., 2016). In yeast which lack complex I, RSCs are composed of complexes III and IV whereas in mammals, RSCs consist of complexes I, III, and IV (Schägger and Pfeiffer, 2000). Thus, RSCs are an evolutionarily conserved organizing principle of the electron transport chain. Recently it was demonstrated that RSCs are functional entities (Barrientos and Ugalde, 2013; Lapuente-Brun et al., 2013) whose structures have since provided novel insight into the potential benefits that they may confer (Schäfer et al., 2007; Althoff et al., 2011; Dudkina et al., 2011; Genova and Lenaz, 2014; Gu et al., 2016; Letts et al., 2016; Wu et al., 2016). Functional benefits of RSCs that have been suggested but not yet proven include: improved electron transfer efficiency and reduced ROS generation, each stemming from a substrate channeling based mechanism; increased metabolic flexibility resulting from changes in RSC composition; and finally, enhanced stability and functionality of all participating complexes in the specific context of the protein-dense IMM (Barrientos and Ugalde, 2013; Milenkovic et al., 2017). Nevertheless, there is still some controversy as to the functional and physiological relevance of RSCs.
About 10 years ago, a new functional entity was shown to interact with yeast RSCs: Aac2p (Claypool et al., 2008; Dienhart and Stuart, 2008). More recently, this association was shown to be evolutionarily conserved as two distinct human ANT isoforms also form complexes with RSCs (Lu et al., 2017). Functionally, this conserved interaction could benefit both RSCs and the AACs. Specifically, the electrogenic exchange of ATPin/ADPout by AAC/ANT is positively influenced by the membrane potential (ΔΨ) across the IMM (Krämer and Klingenberg, 1980) which of course is established by the electron transport chain. Similarly, by dissipating the electrical gradient, productive AAC/ANT transport makes it easier for RSCs to pump protons. As such, it is reasonable to hypothesize that this known functional synergy is further enhanced by being physically associated.
The absence of Aac2p in yeast impairs OXPHOS (Lawson et al., 1990; Heidkämper et al., 1996; Muller et al., 1996; Fontanesi et al., 2004; Claypool et al., 2008; Dienhart and Stuart, 2008). Prior mutagenic studies of Aac2p suggested that COX (complex IV) function is dependent on Aac2p function or expression (Muller et al., 1996; Müller et al., 1997). More recently, several groups demonstrated that there is a specific reduction in complex IV activity in yeast strains lacking Aac2p (Heidkämper et al., 1996; Muller et al., 1996; Fontanesi et al., 2004; Claypool et al., 2008; Dienhart and Stuart, 2008). These observations suggest that Aac2p nucleotide transport activity and/or its interaction with the RSCs are critical determinants of optimal COX activity.
ADP/ATP carriers interact with RSCs in the presence of cardiolipin, a unique phospholipid found exclusively in the mitochondrion (Claypool et al., 2008). Available structures of AAC/ANTs depict three tightly bound cardiolipin molecules per monomer (Beyer and Klingenberg, 1985; Pebay-Peyroula et al., 2003; Ruprecht et al., 2014). In the absence of cardiolipin, Aac2p function is impaired and Aac2p assembly is drastically altered (Jiang et al., 2000; Claypool et al., 2008). The absence of cardiolipin also destabilizes the RSCs (Zhang et al., 2002; Pfeiffer et al., 2003), including its association with Aac2p (Claypool et al., 2008). That Aac2p assembly and function is cardiolipin-dependent has led to the hypothesis that the assembly and function of AAC/ANTs may be the “Achilles heel” of a multitude of cardiolipin-based diseases (Klingenberg, 2008; Claypool, 2009). The structural changes in RSCs and Aac2p that occur in the absence of cardiolipin have clear functional consequences (Claypool et al., 2008). However, the relative contribution of each structural change— impaired assembly of RSCs, Aac2p, or RSC-Aac2p— that occurs in the absence of cardiolipin to the associated mitochondrial dysfunction has not been established.
It has also been hotly debated whether the protein exists and/or functions as a monomer or dimer. Mitochondrial carriers were originally accepted to exist and function as homo-dimers (Lin et al., 1980; Klingenberg, 1981; Palmisano et al., 1998; Schroers et al., 1998; Kotaria et al., 1999; Trézéguet et al., 2000; Capobianco et al., 2002; Dyall et al., 2003; Nury et al., 2005; Postis et al., 2005). A number of studies, motivated by the crystal structures (Kunji and Harding, 2003; Pebay-Peyroula et al., 2003; Ruprecht et al., 2014), have challenged this initial view and instead provided evidence that AACs function, and in fact exist in the IMM, as monomers (Kunji and Harding, 2003; Bamber et al., 2006, 2007a,b; Kunji and Crichton, 2010). Obviously, AACs cannot be monomeric in the IMM and interact with RSCs in a cardiolipin-dependent manner. Additional work is needed to reconcile these very different models of AAC connectivity.
Mitoferrins Are Fundamental to Iron Transport
Iron is essential for mitochondrial function (Levi and Rovida, 2009). Mitochondria themselves are intimately involved in the regulation of cellular iron. Iron is important in the heme biosynthetic pathway in the reaction step of ferrous iron incorporation into protoporphyrin IX catalyzed by ferrochelatase (Figure 2) (Ponka, 1997). Heme is needed for synthesis of the mitochondrial cytochromes which are electron carriers critical for OXPHOS. In addition, iron-sulfur cluster biogenesis occurs in the mitochondrial matrix and is tightly linked to many other cellular processes such as heme biosynthesis, ribosome assembly, DNA synthesis, and translation initiation (Lill and Mühlenhoff, 2008; Lill, 2009). SLC25A28 and SLC25A37 encode Mitoferrin 2 (MFRN2) and Mitoferrin 1 (MFRN1), respectively, which are involved in iron import into the mitochondrion. In zebrafish and mammals, MFRN1 is expressed predominantly in hematopoietic tissues whereas MFRN2, with 65% amino acid identity to its paralog, is widely expressed (Shaw et al., 2006; Amigo et al., 2011). MFRN2 has about 38% identity to Mrs3p and Mrs4p (Shaw et al., 2006), two yeast transporters originally identified as suppressors of an intron splicing defect (Waldherr et al., 1993) that have since been associated with iron transport (Foury and Roganti, 2002). Yeast lacking Mrs3p and Mrs4p exhibit poor growth in iron-depleted conditions (Foury and Roganti, 2002). MFRN1 loss-of-function in mice and zebrafish results in reduced iron uptake into mitochondria and defective hemoglobin synthesis (Shaw et al., 2006). In non-erythroid cells, MFRN2 and MFRN1 are both involved in mitochondrial iron uptake (Paradkar et al., 2009). When both transporters are silenced in non-erythroid cells, heme synthesis is severely compromised; further overexpression of one can functionally compensate for the loss of the other (Paradkar et al., 2009). These results establish the fundamental importance of these proteins in mitochondrial iron metabolism in erythroid and non-erythroid cells.
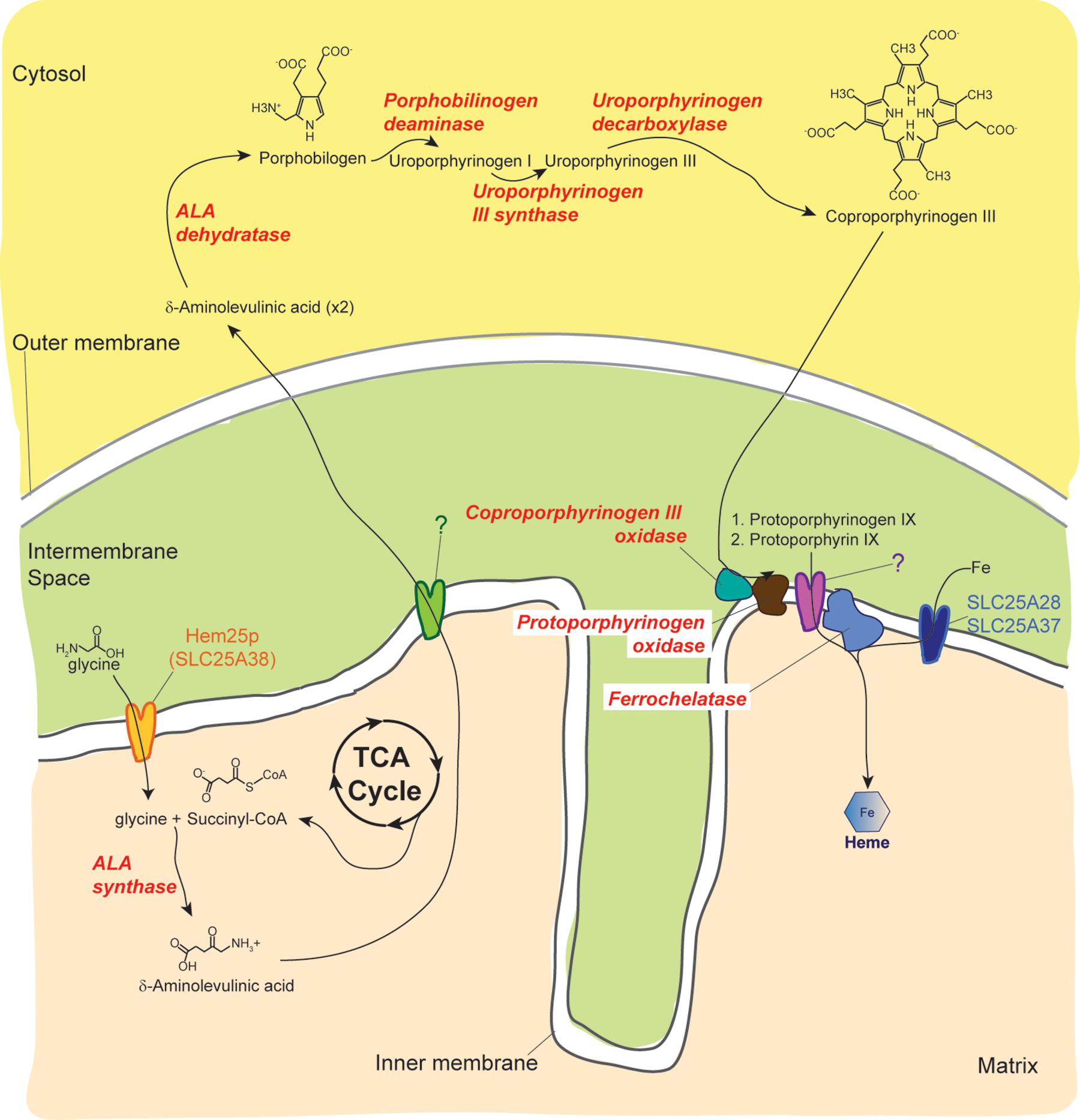
Figure 2. Overview of the heme biosynthetic pathway. Three known MCF members are involved in heme biosynthesis. Following its transport into the matrix by Hem25p/SLC25A38, glycine is condensed with succinyl-CoA by ALA synthase to form δ-aminolevulinic acid. The next four steps of the heme biosynthetic pathway occur in the cytosol. The identity of the protein, which may be a MCF member, that mediates the transport of δ-aminolevulinic acid across the inner membrane has not been determined. The active sites of coproporphyrinogen III oxidase and Protoporphyrinogen oxidase face the intermembrane space. In contrast, the final step in the heme biosynthetic pathway occurs in the matrix and is catalyzed by ferrochelatase. The identity of the protein, which may be a MCF member, that transports protoporphyrin IX to the matrix has not been determined. Ferrochelatase incorporates iron (Fe), transported into the matrix by the mitoferrins SLC25A28 and SLC25A37, into protoporphyrin IX to produce heme.
Uncoupling Proteins Provide a Pathway for Proton Leakage
The UCPs are regulated mitochondrial proteins known to transport protons, anions or other mitochondrial substrates (Jezek et al., 2010; Fedorenko et al., 2012; Porter, 2012; Monné et al., 2018). Six UCP homologs have been discovered in humans—UCP1 or thermogenin (Heaton et al., 1978), UCP2 (Fleury et al., 1997), UCP3 (Boss et al., 1997), UCP4 (Mao et al., 1999), UCP5 or BMCP1 for brain mitochondrial carrier protein 1 (Sanchis et al., 1998), and UCP6 or KMCP1 for kidney mitochondrial carrier protein 1 (Haguenauer et al., 2005). UCPs uncouple OXPHOS from ATP synthesis; they dissipate proton gradients by allowing protons that have been pumped into the intermembrane space by respiratory complexes to flow back into the mitochondrial matrix without being utilized for ATP synthesis (Figure 3). The translocation of hydrogen ion by UCPs requires fatty acids and this activity is inhibited by purine nucleotides such as GDP. Until recently, the role of fatty acids in activating the uncoupling process was hotly debated in relation to the putative UCP transport mechanism. Proton and anion transport by UCP involves fatty acids, as extensively reviewed (Jezek et al., 2018). According to the fatty acid cycling mechanism, protonated free fatty acids spontaneously flip to the matrix IMM leaflet where they release protons based on the proton gradient across the IMM. In turn, UCPs provide a conduit consisting of basic amino acids that facilitates the movement of the FA anion back to the IMS leaflet, a process that is also driven by the electrochemical gradient. In the net, these processes dissipate the proton gradient (Kamp and Hamilton, 1992; Kamp et al., 1993, 1995; Jezek et al., 1997a,b; Jabùrek et al., 2001; Jezek et al., 2010, 2018). This model is validated by nuclear magnetic resonance and functional mutagenesis studies on UCP2 which provided molecular and structural support for this protonophoretic model (Berardi and Chou, 2014).
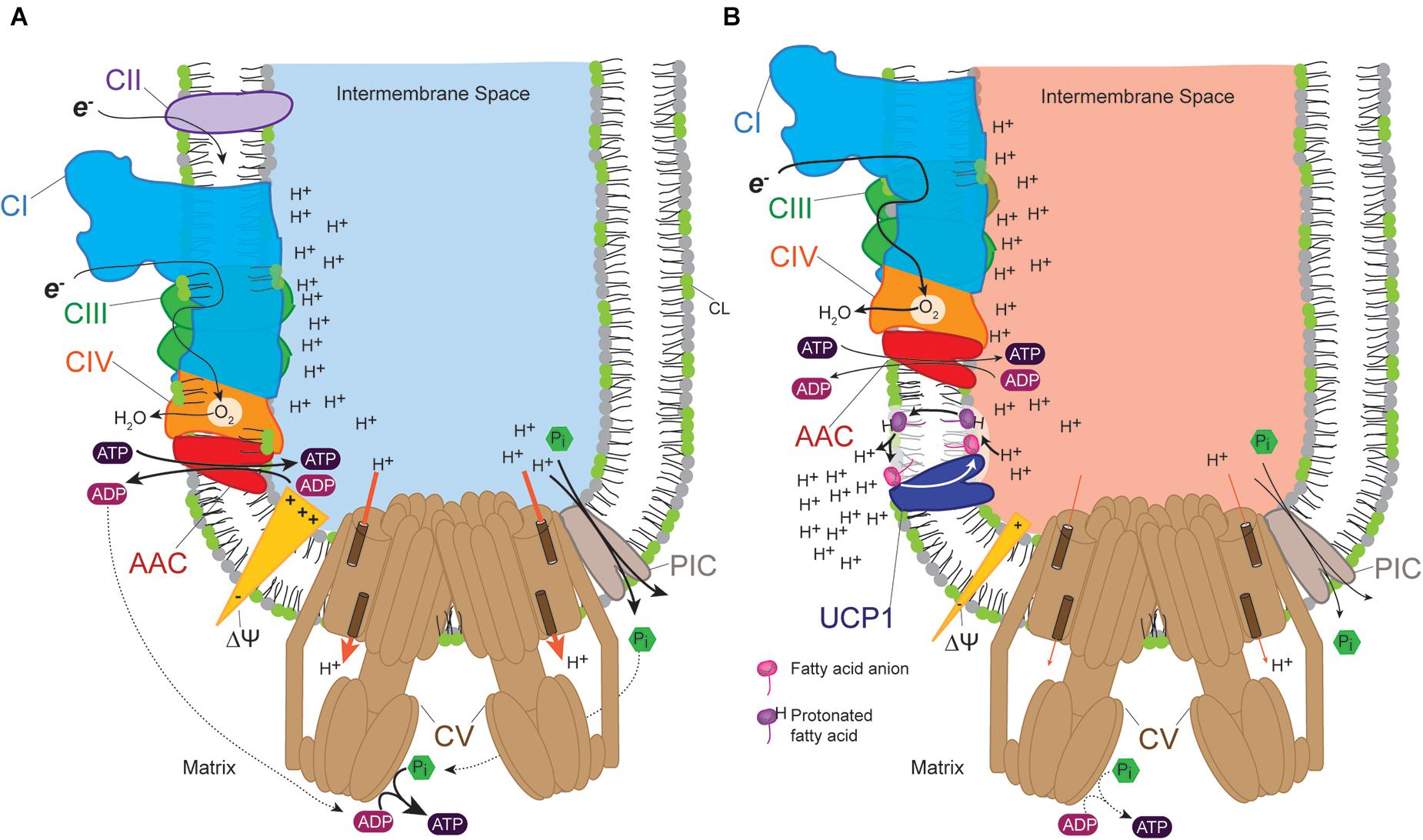
Figure 3. Uncoupling protein (UCP) provides a pathway for proton leak. (A) Proton pumping across the inner membrane into the intermembrane space by complexes I, III, and IV occurs as electrons (e-) flow down the transport chain. This generates a proton-based electrochemical gradient (Δψ) that is consumed by the ATP synthase to produce ATP, thus coupling ATP synthesis to proton pumping. The proton gradient also provides the driving force for the exchange of ADP and ATP by AAC and the uptake of Pi by PIC, transport processes that are essential for OXPHOS. (B) UCPs provide an alternative means to equilibrate the proton gradient which effectively uncouples proton pumping from ATP synthesis. This results in increased flux of electrons through the electron transport chain which generates heat (indicated by red intermembrane space). In (A,B), line thickness reflects relative flux/activity.
The UCP1 is thought to be exclusively found in brown adipose tissue and is the only UCP responsible for adaptive adrenergic non-shivering thermogenesis (Nicholls et al., 1978; Matthias et al., 2000; Golozoubova et al., 2001; Porter, 2008). As such, it is firmly established that UCP1 functions as a true UCP that utilizes the electrochemical gradient generated by the respiratory chain to produce heat instead of ATP. Also, there seems to be a close relationship between mitochondrial ROS and UCP1-dependent thermogenesis although whether or not superoxide modulates UCP1 function is debated (Echtay et al., 2002; Silva et al., 2005; Chouchani et al., 2017). Nonetheless, work with ucp1 knockout (ucp1-/-) mice has established that UCP1 is a target of redox modification in vivo (Chouchani et al., 2016). A recent study showed that brown adipose tissue (BAT) from ucp1-/- mice have reduced respiratory chain proteins and increased host defense signaling following exposure to cold (Kazak et al., 2017). Intriguingly, BAT-derived mitochondria from ucp1-/- mice are more sensitive to calcium overload in a ROS-dependent manner (Kazak et al., 2017). Thus, though UCP1 is traditionally linked to thermogenesis, it is becoming clear that UCP1 function extends beyond thermogenesis.
UCP2-5 are not involved in thermogenesis even though they provide mild uncoupling which may be protective against oxidative stress (Jezek et al., 2018). UCP2 transports C4 metabolites such as oxaloacetate, a function that matches well with its close phylogenetic relationship with the dicarboxylic acid carrier, SLC25A10 (Vozza et al., 2014). It is widely expressed (Fleury et al., 1997; Gimeno et al., 1997) and has numerous pathophysiological roles. For instance, due to its ability to reduce ROS generation, UCP2 participates in both host immunity and the inflammatory response (Nègre-Salvayre et al., 1997; Arsenijevic et al., 2000; Mattiasson and Sullivan, 2006). In addition, UCP2 has been implicated in body mass regulation, glucose metabolism, and carcinogenesis (Zhang et al., 2001; Horimoto et al., 2004; Mattiasson and Sullivan, 2006; Derdak et al., 2008; Li et al., 2013, 2015; Vozza et al., 2014; Sreedhar et al., 2017). UCP3 is expressed mainly in the skeletal muscle and brown adipose tissue, and minimally in the heart (Boss et al., 1997; Vidal-Puig et al., 1997) where it is important for ROS attenuation but not body mass regulation or fatty acid metabolism (Vidal-Puig et al., 2000). Indeed, mitochondria from mice with lower levels of UCP3 have increased ROS production and oxidative damage further suggesting that UCP3 protects against ROS and oxidative damage (Brand et al., 2002). UCP4 is predominantly expressed in the nervous system including different regions of the brain, the spinal cord, hair cells of the inner ear, and Merkel cells in the skin (Liu et al., 2006; Smorodchenko et al., 2009, 2011). UCP4 overexpressing neuronal cell lines have reduced OXPHOS with a corresponding increase in glucose uptake and glycolysis (Liu et al., 2006). These metabolic changes correlate with a drop in ROS production, a reduced tendency for calcium overload and an overall increased resistance to apoptosis (Liu et al., 2006). Overexpression of UCP4 in pre-adipocyte cell lines stimulates proliferation, inhibits differentiation into adipocytes and protects against apoptosis (Zhang et al., 2006). Furthermore, impaired insulin sensitivity and mitochondrial biogenesis, decreased mtDNA level and increased ROS production occurs in adipocyte cell lines overexpressing UCP4 suggesting a global negative impact of UCP4 on mitochondrial function (Gao et al., 2010). However, in UCP4 overexpressing L6 myocytes, insulin sensitivity is improved with no change in intracellular ROS production, mtDNA levels or mitochondrial biogenesis (Gao et al., 2011). Regulated UCP4 expression, therefore, seems to be critical for optimal mitochondrial and cellular function.
UCP5 is expressed principally in the central nervous system and has three different forms (long form, UCP5L with 325 amino acids; short form, UCP5S with 322 amino acids; and short insert form, UCP5S1 with 353 amino acids) (Ramsden et al., 2012). UCP5 overexpression in human SH-SY5Y cells increases proton leak, reduces mitochondrial membrane potential and ATP production, and increases oxygen consumption (Kwok et al., 2010). UCP6 has not been well studied. To date, it is associated with carcinogenesis (Nohara et al., 2012) and its expression in the kidney cortex is increased following pro-oxidant states (Haguenauer et al., 2005).
The Citrate Carrier Has a Pervasive Role in Cellular Metabolism
The ubiquitously expressed mitochondrial CiC, also known as the tricarboxylate carrier, citrate transport protein or dicarboxylic acid transporter, is encoded by SLC25A1 in mammals. It catalyzes the obligate electroneutral exchange of a tricarboxylate such as citrate for another tricarboxylate, a dicarboxylate such as malate, or phosphoenolpyruvate (Klingenberg, 1972; Bisaccia et al., 1989; Gnoni et al., 2009). CiC exports matricial citrate, the first product of the citric acid (Kreb; tricarboxylic acid) cycle, in exchange for cytosolic malate. Citrate is an important regulatory substrate for many metabolic reactions. As such, CiC provides substrates for both fatty acid and cholesterol biosynthesis (Figure 4) and helps in the transfer of reducing equivalents across the IMM (Palmieri et al., 2015). For fatty acid synthesis, citrate is source of carbons and also allosterically regulates the rate-limiting enzyme of this biosynthetic pathway. Cytoplasmic citrate is first cleaved into acetyl-CoA and oxaloacetate by ATP citrate lyase. Acetyl-CoA is then carboxylated into malonyl CoA by acetyl CoA carboxylase, the rate-limiting step in fatty acid synthesis. For cholesterol synthesis, two molecules of acetyl-CoA (also from Citrate by ATP citrate lyase) are condensed to yield acetoacetyl-CoA which is then condensed with another acetyl-CoA to yield 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA). The committed step in cholesterol synthesis is the reduction of cytosolic HMG-CoA to mevalonate. Fatty acid and cholesterol synthesis also requires NADPH and some of these are generated from cytosolic oxaloacetate via malate dehydrogenase and malic enzyme (Palmieri, 2004). Citrate transport from the mitochondrion increases NADPH synthesis and the activity of human CiC is modulated by acetylation in activated immune cells (Palmieri et al., 2015). The orthologous yeast citrate transport protein Ctp1p, preferentially transports tricarboxylates versus other substrates, in contrast to the mammalian transporter which transports tricarboxylates, malate, and phosphoenolpyruvate to similar extents (Kaplan et al., 1995). By supplying citrate-derived acetyl-CoA that is required for normal nucleosome histone acetylation, SLC25A1 and its Drosophila ortholog, scheggia (sea) are also important for the maintenance of genome integrity (Morciano et al., 2009). Recently, it was shown that SLC25A1 is critical for tumor cell metabolism; it enables stem-like properties in cells, contributes to treatment resistance in cancer cells (Fernandez et al., 2018), and has been proposed as a metabolic oncogene (Kolukula et al., 2014). In another recent work, SLC25A1 and its genetic and functional interactions with SLC25A4 is required for optimal neuronal development, function and behavior (Gokhale et al., 2018). Therefore, CiC is critical for intermediary metabolism and has emerging roles in human development and disease including cancer.
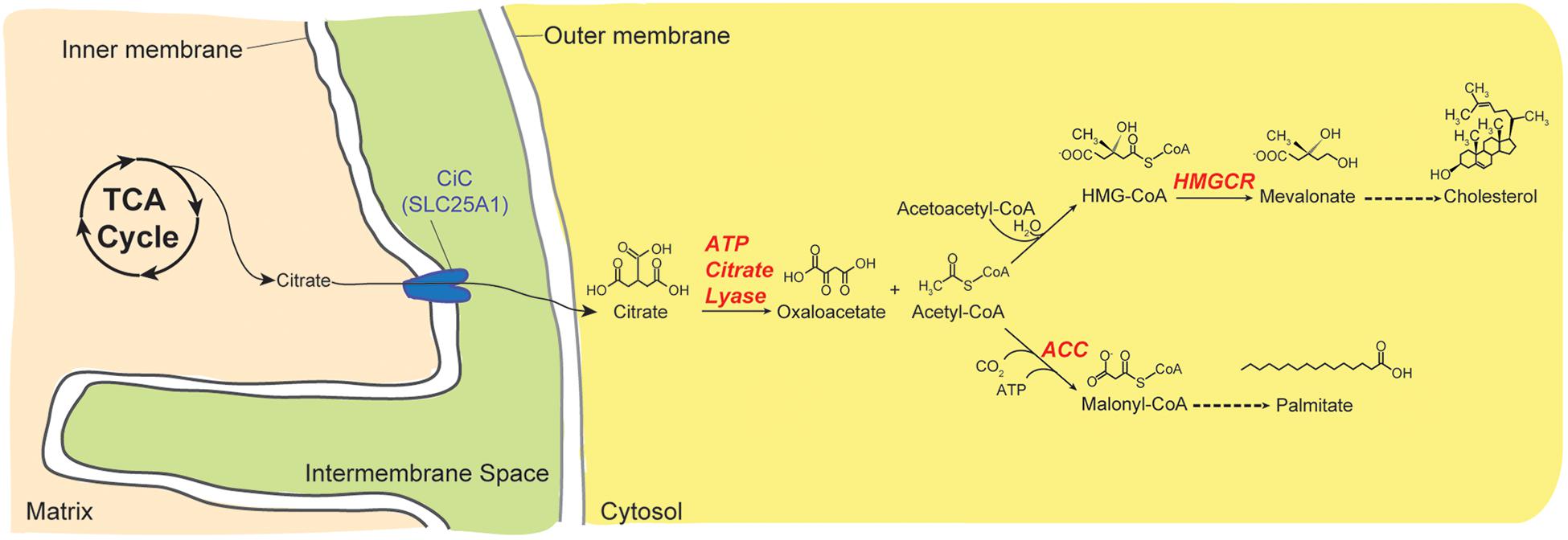
Figure 4. Citrate provides substrates for anabolic pathways. Citrate is a substrate for many biological reactions. Here, the role of citrate in fatty acid and cholesterol synthesis is depicted. Citrate is transported from the mitochondrion into the cytoplasm by CiC/SLC25A1. Once in the cytoplasm, it is broken down into oxaloacetate and acetyl-CoA by the action of ATP Citrate Lyase. For fatty acid synthesis, acetyl-CoA is carboxylated by Acetyl-CoA Carboxylase (ACC) to malonyl-CoA. Alternatively, acetyl-CoA can be condensed with itself to generate acetoacetyl-CoA, which then reacts with another acetyl-CoA and water to form 3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA). Conversion of HMG-CoA to mevalonate, the committed step in cholesterol biosynthesis, is performed by 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase (HMGCR).
Pathology of MCF
The MCF provides substrates for various biochemical processes in the cell. Consistent with their diverse and fundamental roles in metabolism, the absence or dysfunction of assorted MCF members causes a wide variety of disorders including hematologic, neurologic, and cardiac diseases. Underlying many of these disorders is a defect in OXPHOS leading to disturbed mitochondrial energy metabolism that manifests in a wide variety of clinical signs and symptoms. A number of systemic diseases caused by mutations in genes encoding SLC25 members are discussed briefly next and Table 3 displays a summary of MCF-linked diseases clustered by presentation.
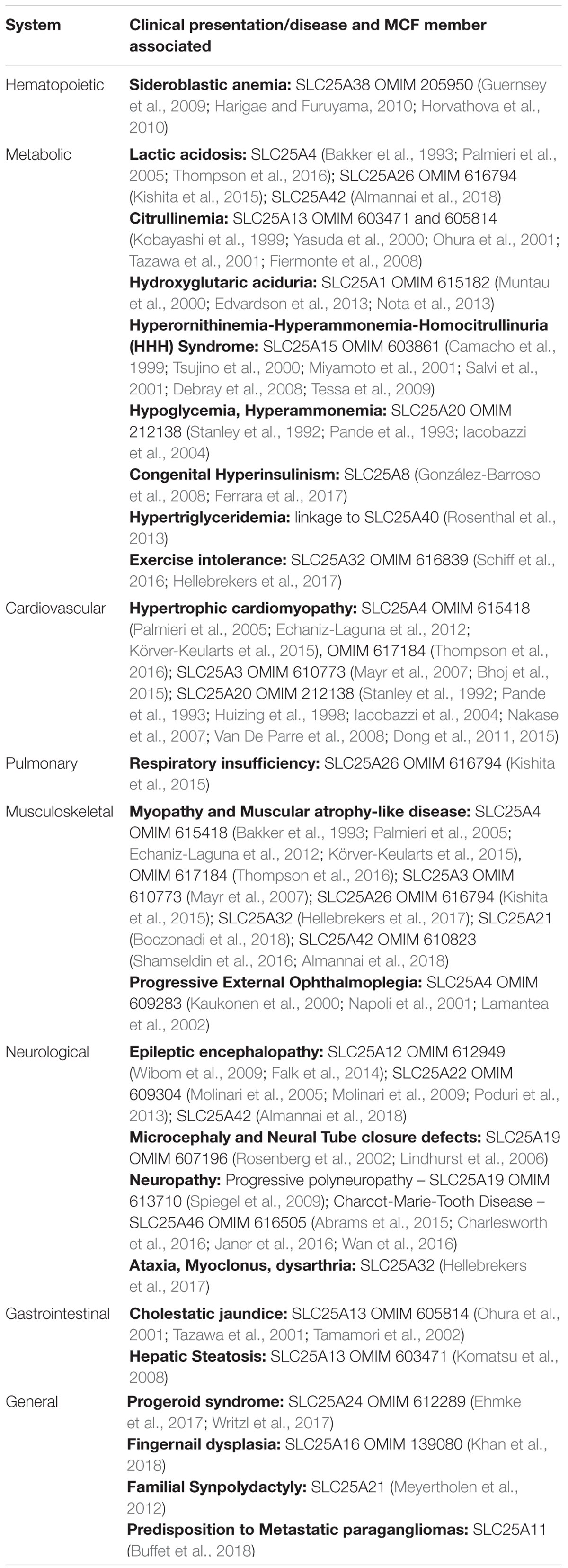
Table 3. Summary of known MCF-involved diseases clustered by systemic presentation.
Mitochondrial carriers encoded by SLC25A28, SLC25A37, and SLC25A38 are important for heme synthesis which requires cellular iron, glycine and succinyl-CoA (Figure 2). Since red blood cells are very sensitive to defects in heme synthesis, dysfunction in any of these mitochondrial carriers causes anemia (Xu et al., 2013). The erythroid specific SLC25A38, which based on its requirement for erythropoiesis was initially predicted to encode an amino acid carrier capable of transporting glycine, is now regarded as a bonafide glycine transporter (Guernsey et al., 2009; Fernández-Murray et al., 2016; Lunetti et al., 2016). Pathogenic mutations in SLC25A38 and knockdown experiments in zebrafish implicate the carrier in the etiology of congenital sideroblastic anemia (Guernsey et al., 2009) (OMIM 610819). Mutations in the carrier are the second most common cause of inherited sideroblastic anemia and may account for about a fifth of all cases (Harigae and Furuyama, 2010; Horvathova et al., 2010). As previously discussed, SLC25A28 and SLC25A37, which encode the mitoferrins, are critical for iron homeostasis. Although no human mutations have been described for these carriers, MFRN1 has been shown to be important for heme synthesis in erythroid cells (Shaw et al., 2006).
SLC25A46 encodes a novel outer mitochondrial membrane protein that is widely expressed in the nervous system (Haitina et al., 2006), and mutated in numerous neurological diseases including optic atrophy spectrum disorder, Charcot-Marie-Tooth type 2, Leigh syndrome, progressive myoclonic ataxia, and lethal congenital pontocerebellar hypoplasia (Abrams et al., 2015; Wan et al., 2016; Terzenidou et al., 2017) (OMIM 610826). Insight into each of these neurological diseases is hampered by the fact that SLC25A46 is an orphan member of the MCF whose substrate(s) has not been defined (Palmieri and Monné, 2016). However, given its unusual localization to the outer mitochondrial membrane, whether SLC25A46 functions as a transporter or instead has novel activities that are unrelated to its SLC25 membership remains an open question. Indeed, SLC25A46 has been implicated in both mitochondrial dynamics and cristae morphology (Abrams et al., 2015; Janer et al., 2016; Steffen et al., 2017).
Mitochondrial energy production is high in cardiac tissue and it is unsurprising that many mitochondrial carriers have been associated with cardiac disease, manifested in most cases as hypertrophic cardiomyopathy. Mutations in SLC25A4, encoding ANT1, are responsible for both the autosomal dominant and recessive cardiomyopathic type mitochondrial DNA depletion syndromes (OMIM 617184 and 615418, respectively) (Palmieri et al., 2005; Echaniz-Laguna et al., 2012; Körver-Keularts et al., 2015; Thompson et al., 2016), in addition to autosomal dominant progressive external ophthalmoplegia (OMIM 609283) (Kaukonen et al., 2000; Napoli et al., 2001; Komaki et al., 2002; Siciliano et al., 2003). Mutations in SLC25A3 that encodes the phosphate carrier cause either hypertrophic cardiomyopathy and impaired function of other organs such as skeletal muscle (Mayr et al., 2007; Mayr et al., 2011) or isolated cardiomyopathy (Bhoj et al., 2015). Interestingly, in the latter case (Bhoj et al., 2015), the mutations discovered in SLC25A3 were a mix of a single nucleotide change and a stretch of indels, both of which could potentially impact the two mammalian isoforms of the protein (Bhoj et al., 2015; Seifert et al., 2015). By disrupting the Urea Cycle, mutations in SLC25A13, whose protein product mediates the electrogenic exchange of aspartate for glutamate (Palmieri et al., 2001), cause adult onset citrulinemia (Kobayashi et al., 1999; Yasuda et al., 2000; Ohura et al., 2001; Tazawa et al., 2001; Fiermonte et al., 2008). Carnitine-acylcarnitine translocase deficiency resulting from many different mutations in SLC25A20 results in a multi-systemic disorder that includes cardiomyopathy as one of its clinical features (OMIM 212138) (Stanley et al., 1992; Pande et al., 1993; Iacobazzi et al., 2004). A genome-wide association study reported an association between UCP5 gene variants and the formation of atherosclerotic plaques suggesting that UCP5 has a protective role against atherosclerosis (Dong et al., 2011). Furthermore, UCP5 expression is increased in embolic stroke and multiple infarction brain lesions probably due to upregulation brought about by chronic ischemic stress (Nakase et al., 2007). However, since UCP5, like SLC25A38 and SLC25A46, is an orphan MCF, the underlying pathogenic mechanism is very much unclear at this time.
Challenges to MCF-Focused Research
Mitochondrial carriers, and SLCs in general, perform a central role in metabolism and their association with a myriad of diseases makes them attractive candidates for basic and translational research. However, it has been noted that this area of research has not grown in commensurate proportion to its size or the gold mine its study could potentially reap (Cesar-Razquin et al., 2015). Most of these carriers are yet to be fully characterized and many of them remain totally uncharacterized. A number of technical factors have hampered growth of research focused on the extended membership of the SLC family that of course includes the MCF. A huge technical hurdle is the systemic absence of validated antibodies specific to most of these proteins. Further, many of the available antibodies are too weak to detect endogenous proteins whose expression is likely low and in general, many of the available reagents have not been rigorously characterized and validated (e.g., absence of signal with appropriate negative controls such as knockout cells). Compounding issues is the fact that many SLC members appear to have low immunogenicity which likely stems from the fact that they are polytopic membrane proteins that often display high interspecies conservation. Finally, transport assays are tedious to perform and limited by the volume of substrates available and/or required to de-orphanize a SLC protein.
Interestingly, a number of carriers have been shown to display substrate promiscuity by transporting more than one type of solute (Fiermonte et al., 2009). Two members in Arabidopsis thaliana, AtUCP1 and AtUCP2, previously thought to be UCPs and therefore named as such were recently assigned the function of transporting amino acids, dicarboxylates, phosphate, sulfate, and thiosulfate (Monné et al., 2018). The Pi carrier in mammals, SLC25A3, was originally described as a phosphate symporter (Wohlrab and Flowers, 1982; Seifert et al., 2015) whose mutation is associated with fatal childhood diseases (Mayr et al., 2007; Bhoj et al., 2015). Pic2p, originally thought to be a second albeit minor Pi carrier in yeast, actually functions as a Cu++ transporter responsible for the import of copper, required for COX assembly, into the mitochondrial matrix (Vest et al., 2013). Similarly, SLC25A3, which shares 65% similarity with Pic2p, was recently shown to have a conserved role for copper transport in vivo and in vitro (Vest et al., 2013; Boulet et al., 2017). Together, these results indicate that even well-established MCF members may have the capacity to transport additional presently unappreciated substrates.
Apparent functional redundancy is another recurring feature of mitochondrial carriers (Taylor, 2017). For example, mitochondrial nucleotide homeostasis not only involves the ADP/ATP carriers (SLC25A4, SLC25A5, SLC25A6, and SLC25A31) but also is influenced by the ATP-Mg/Pi carriers (SLC25A23, SLC25A24, SLC25A25, and SLC25A41). Indeed, in yeast, the calcium-dependent ATP-Mg/Pi carrier, Sal1p, can compensate for the absence of the major isoform of the ADP/ATP carrier, Aac2p, and provide a pool of mitochondrial ATP that is required for yeast viability (Chen, 2004; Cavero et al., 2005; Traba et al., 2008; Laco et al., 2010). Still, there are a lot of unknowns regarding how all of these different transporters maintain the nucleotide pool across the IMM. It has been suggested that different mammalian ADP/ATP carrier isoforms, which do not transport AMP, may have different preferred transport modes, i.e., ATP vs. ADP. For instance, based on its high expression in cancer cells, SLC25A5 (ANT2) was postulated to preferentially import ATP made by glycolysis, an activity that maintains the mitochondrial membrane potential and by extension, other essential mitochondrial functions (Stepien et al., 1992; Giraud et al., 1998; Chevrollier et al., 2005). However, it was recently demonstrated in a range of cancer cells that the uptake of ATP is in fact completely independent of the activity of any ADP/ATP carrier isoform (Maldonado et al., 2016). As such, it remains unresolved whether the different ANT isoforms do or do not have distinct transport activities and/or substrate preferences. Also unclear is how the calcium-dependent ATP-Mg/Pi carriers, which were originally thought to preferentially transport ATP but since demonstrated to also transport other adenine nucleotides (Fiermonte et al., 2004), interface with the ADP/ATP carriers to modulate mitochondrial nucleotide homeostasis. The requirement for calcium provides an opportunity for the flux of adenine nucleotides across the IMM to be regulated. Indeed, the calcium-dependent mitochondrial uptake of adenine nucleotides by SLC25A23 is required for the glucagon-stimulated increase in OXPHOS in liver mitochondria (Amigo et al., 2013). As research progresses in this area, identification of the substrate(s) transported by many of these proteins will provide foundational information as to their physiological roles.
Mitochondrial Translation
The vast majority of mitochondrial proteins are translated in the cytosol and imported thereafter into the mitochondrion. For those proteins encoded by the mitochondrial genome (mtDNA), mitochondria have retained a dedicated, dual-origin translational machinery whose architecture is similar to that of bacteria. Consistent with this bacterial origin, mitochondrial translation is pharmacologically unaffected by cycloheximide, an inhibitor of cytosolic translation, and is instead sensitive to antibiotics such as puromycin and chloramphenicol. Eight polypeptides in yeast and thirteen polypeptides in humans are encoded by mtDNA and thus produced via mitochondrial translation. Apart from genes for these polypeptides, the mtDNA also encodes a set of transfer (t)RNAs, ribosomal (r) RNAs and in yeast, the RNA component of the mitochondrial RNAse P (Towpik, 2005; Walker and Engelke, 2008). The mitochondrial translation cycle is subdivided into four steps— initiation, elongation, termination and recycling—and nuclear encoded polypeptide factors are required at different steps for optimal mitochondrial translation (Towpik, 2005; Smits et al., 2010; Kehrein et al., 2013). An example of a nuclear-encoded factor in yeast is the mitochondrial translation initiation factor 3 (mIF3p), the Saccharomyces cerevisiae homolog of the bacterial translation initiation factor 3 (IF3). Its function is conserved and overlaps with human mIF3p (Atkinson et al., 2012; Kuzmenko et al., 2014), and its absence in yeast disrupts mitochondrial translation (Kuzmenko et al., 2016). In addition, a number of translational activators directly interacting with mRNAs of mitochondrial encoded polypeptides are necessary to optimize the translation of a specific mtDNA-encoded polypeptide, e.g., synthesis of Cox1p is affected when Mss51p is absent or limiting (Siep et al., 2000; Perez-Martinez et al., 2003, 2009; Barrientos et al., 2004; Fontanesi et al., 2011).
Translation of some mitochondrial proteins is tightly coupled to their assembly into respiratory complexes in a manner similar to a mechanism described as “controlled by epistasy of synthesis” (CES) that exists in the biogenesis of photosynthetic protein complexes (Choquet et al., 2001; Towpik, 2005). For example, in yeast, Cox1p synthesis is tightly coupled to the assembly of respiratory complex IV which helps to balance the production of subunits with their assembly into the holoenzyme which in the net preserves mitochondrial proteostasis (Perez-Martinez et al., 2003; Barrientos et al., 2004; Towpik, 2005; Soto et al., 2012).
Emerging Roles in the Biogenesis of Cytochrome C Oxidase
In yeast and in humans, a link between the function of MCF members and COX biogenesis is emerging (Figure 5). For instance, a destabilizing pathogenic mutation in SLC25A46 impairs OXPHOS and ascorbate/TMPD-dependent respiration and reduces steady state levels of complex IV subunits (Janer et al., 2016). These findings are consistent with a complex IV-specific assembly defect in SLC25A46 mutant fibroblasts. At present, the mechanistic basis for the reduced steady state levels of complex IV subunits has not been determined. A role in the assembly of the OXPHOS machinery is perhaps not unexpected given that many SLC members provide substrates that serve as building blocks needed for processes such as mitochondrial DNA replication, transcription, translation, and/or post-translational assembly of protein complexes. However, it is surprising that these defects seem to specifically impact complex IV without significantly affecting the other OXPHOS complexes that are also built from subunits expressed from both the nuclear and mitochondrial genomes.
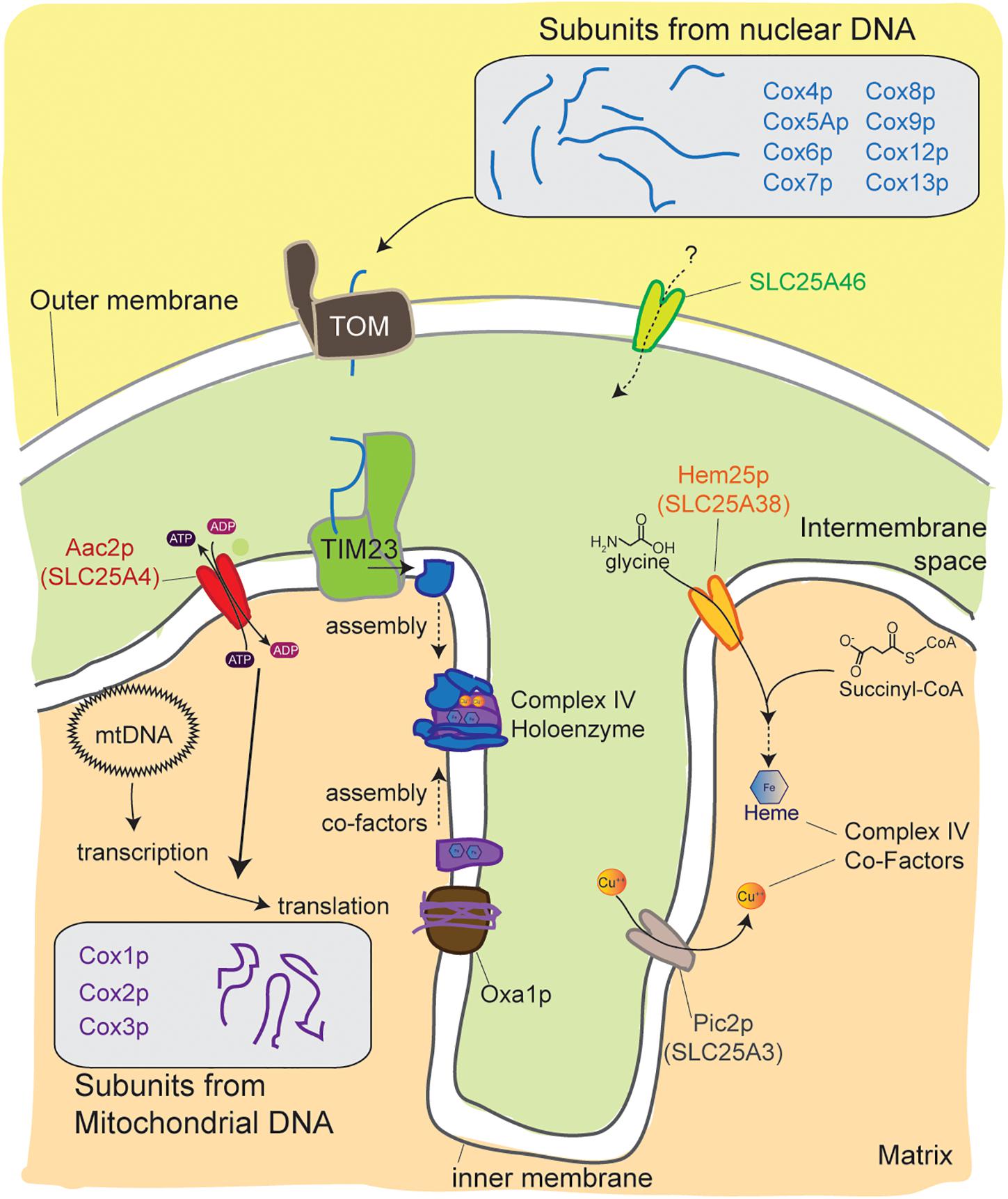
Figure 5. An emerging link between mitochondrial carriers and complex IV biogenesis. Cytochrome c oxidase is assembled from protein subunits that originate from the mitochondrial and nuclear genomes. Mitochondrial carriers support these processes at multiple levels. Mutations in the metazoan-specific SLC25A46, an unusual family member that resides in the outer membrane, decreases complex IV levels by an unclear mechanism. The absence of Aac2p function impairs translation of subunits encoded by mitochondrial DNA. Pic2p and Hem25p help provide co-factors that are essential for complex IV assembly and function. Hem25p transports glycine that is used to make Heme and Pic2p mediates uptake of Cu++. TOM, translocase of the outer membrane; TIM23, translocase of the inner membrane 23; Oxa1p, mitochondrial inner membrane insertase.
Recently, a role for the yeast ortholog of human SLC25A38, Hem25p, important for heme synthesis as a mitochondrial glycine importer, in the stability of respiratory complex proteins was tested (Dufay et al., 2017). While deletion of Hem25p compromises the steady state level of subunits of each respiratory complex, its absence is most detrimental to complex IV (Dufay et al., 2017). Intriguingly, the combined absence of Hem25p and Flx1p, the mitochondrial flavin adenine dinucleotide transporter (ortholog of human SLC25A32), further reduces the steady state level of subunits of the hem25Δ-affected respiratory complexes except complex IV (Dufay et al., 2017). These results are consistent with a model that Hem25p and Flx1p provide heme and FAD, respectively, which are required for the assembly of the respiratory chain complexes.
Knockdown of the two isoforms of SLC25A3, SLC25A3-A, and SLC25A3-B, in many different cell types results in reduced COX holoenzyme levels and activity (Boulet et al., 2017). When SLC25A3 is limiting, the steady state levels of COX4, a nuclear encoded subunit of COX, and mitochondrial copper are reduced (Boulet et al., 2017). Interestingly, COX4 abundance is rescued by copper supplementation (Boulet et al., 2017). Since copper is critical for the assembly of COX (Diaz, 2010; Baile and Claypool, 2013), defects in copper import are likely to impair COX biogenesis. Nevertheless, SLC25A3 function may be modulated and/or linked to the assembly of COX via a pathway that is presently unidentified (Boulet et al., 2017).
The absence of Aac2p in yeast leads to a specific reduction of COX activity while the activity of complex III is unaffected (Dienhart and Stuart, 2008). The reduced complex IV activity likely stems from lower steady state levels of its subunits in the absence of Aac2p. These findings are consistent with the impaired OXPHOS activity that occurs when Aac2p expression is decreased or ablated (Heidkämper et al., 1996; Muller et al., 1996; Müller et al., 1997; Claypool et al., 2008). Intuitively, the mechanistic basis for the reduced steady state levels of complex IV subunits could derive from a defect in any step in its biogenesis that is regulated/modulated by and/or dependent on the nucleotide transport function of Aac2p. Alternatively, the conserved AAC/ANT-RSC interaction may itself be critical for robust COX expression, assembly, and/or function Since the Aac2p and ANT1/ANT2 interactomes all included other SLC25 family members (Claypool et al., 2008; Lu et al., 2017), it is possible that these MCF–MCF interactions are critical for maintaining the abundance of metabolites that are needed for optimal mitochondrial translation and assembly of respiratory complexes.
Our group recently showed that in yeast regulation of complex IV activity by the Aac2p relies solely on the activity of the protein in a way that is mechanistically dependent on mitochondrial translation (Ogunbona et al., 2018). Indeed, similar to the complete absence of Aac2p (Dienhart and Stuart, 2008), an interaction-competent transport-dead mutant of Aac2p exhibits reduced complex IV activity, reduced levels of the complex IV holoenzyme, and reduced steady states levels of complex IV subunits, especially those encoded by mtDNA (Ogunbona et al., 2018). Interestingly, translation of complex IV subunits encoded by mtDNA is specifically decreased in the absence of Aac2p activity, either genetically imposed or due to the presence of AAC-specific inhibitors. In addition, when Aac2p is expressed but non-functional, the turnover of newly synthesized Cox3p is increased (Ogunbona et al., 2018). Together, these results establish a novel link between nucleotide transport and mitochondrial translation of complex IV subunits. A big question moving forward is whether this neoteric mechanism is evolutionarily conserved in mammals. Ongoing and future efforts focused on dissecting the functional significance of distinct aspects of the AAC/ANT interactome are likely to shed further insight into how it supports complex IV biogenesis.
Perspectives
The MCF is an extensive cluster of transport proteins with essential biochemical and physiological functions. Individually and collectively, they have a broad role in intermediary metabolism and are pathophysiologically significant. In spite of this, there remain numerous fundamental gaps in our understanding of their cellular functions and if and how their activities are regulated. At the molecular level, the mechanistic basis for their role(s) in the regulation of many biologic processes, including OXPHOS, is still missing. Even for the best characterized MCF protein, Aac2p, new biology was recently uncovered suggesting that there is likely a ton of new biology to uncover in the biggest subfamily of SLCs. Future research should primarily focus on establishing the biological functions of each and every protein in the family. For proteins with assumed roles in mitochondrial energy production, it will be important to carefully dissect their molecular contributions to OXPHOS. Using the recently uncovered translational regulation of complex IV activity by Aac2p as a paradigm, upcoming research work should remain open-minded to the identification of unanticipated functional relationships that may exist between MCF members and other biological processes. Discovery of new members and novel functions of existing members will give us a better understanding of their pathophysiological roles. Long term, such information may guide development of effective therapeutic strategies to correct or better manage diseases resulting from their dysfunction. With recent advances in biomedical research such as the advent of gene editing technologies (e.g., CRISPR/Cas9), substantial and rapid progress in the field of MCFs is anticipated.
Author Contributions
OO and SC reviewed the literature. OO wrote the initial draft of the review which also served as the Introduction of his Ph.D. thesis (Ogunbona, 2018), submitted to Johns Hopkins University School of Medicine, and generated all of the figures. SC provided guidance and edited the manuscript.
Funding
This work was supported by the National Institutes of Health grant (R01HL108882) to SC and a pre-doctoral fellowship from the American Heart Association (15PRE24480066) to OO.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Gergely Gyimesi (University of Bern, Switzerland) for providing more information on the SLC tables and Dr. Brian O’Rourke (Johns Hopkins University) for critically reading the review.
Abbreviations
AAC, ATP/ATP carrier; ANT, adenine nucleotide translocase; CiC, citrate carrier; COX, cytochrome c oxidase; IMM, inner mitochondrial membrane; MCF, mitochondrial carrier family; OMIM, Online Mendelian Inheritance in Man; OXPHOS, oxidative phosphorylation; ROS, reactive oxygen species; RSC, respiratory supercomplex; SLC, solute carrier; UCP, uncoupling protein.
References
Aalkjaer, C., Boedtkjer, E., Choi, I., and Lee, S. (2014). Cation-coupled bicarbonate transporters. Compr. Physiol. 4, 1605–1637. doi: 10.1002/cphy.c130005
Abrams, A. J., Hufnagel, R. B., Rebelo, A., Zanna, C., Patel, N., Gonzalez, M. A., et al. (2015). Mutations in SLC25A46, encoding a UGO1-like protein, cause an optic atrophy spectrum disorder. Nat. Genet. 47, 926–932. doi: 10.1038/ng.3354
Acín-Pérez, R., Fernández-Silva, P., Peleato, M. L., Pérez-Martos, A., and Enriquez, J. A. (2008). Respiratory active mitochondrial supercomplexes. Mol. Cell 32, 529–539. doi: 10.1016/j.molcel.2008.10.021
Almannai, M., Alasmari, A., Alqasmi, A., Faqeih, E., Al Mutairi, F., Alotaibi, M., et al. (2018). Expanding the phenotype of SLC25A42-associated mitochondrial encephalomyopathy. Clin. Genet. 93, 1097–1102. doi: 10.1111/cge.13210
Alper, S. L. (2006). Molecular physiology of SLC4 anion exchangers. Exp. Physiol. 91, 153–161. doi: 10.1113/expphysiol.2005.031765
Alper, S. L., Chernova, M. N., and Stewart, A. K. (2001). Regulation of Na+-independent Cl-/HCO3- exchangers by pH. JOP 2, 171–175.
Alper, S. L., Darman, R. B., Chernova, M. N., and Dahl, N. K. (2002). The AE gene family of Cl/HCO3- exchangers. J. Nephrol. 15(Suppl. 5), S41–S53.
Althoff, T., Mills, D. J., Popot, J. L., and Kühlbrandt, W. (2011). Arrangement of electron transport chain components in bovine mitochondrial supercomplex I1III2IV1. EMBO J. 30, 4652–4664. doi: 10.1038/emboj.2011.324
Amigo, I., Traba, J., González-Barroso, M. M., Rueda, C. B., Fernández, M., Rial, E., et al. (2013). Glucagon regulation of oxidative phosphorylation requires an increase in matrix adenine nucleotide content through Ca2+ activation of the mitochondrial ATP-Mg/Pi carrier SCaMC-3. J. Biol. Chem. 288, 7791–7802. doi: 10.1074/jbc.M112.409144
Amigo, J. D., Yu, M., Troadec, M. B., Gwynn, B., Cooney, J. D., Lambert, A. J., et al. (2011). Identification of distal cis-regulatory elements at mouse mitoferrin loci using zebrafish transgenesis. Mol. Cell Biol. 31, 1344–1356. doi: 10.1128/MCB.01010-10
Aquila, H., Misra, D., Eulitz, M., and Klingenberg, M. (1982). Complete amino acid sequence of the ADP/ATP carrier from beef heart mitochondria. Hoppe Seylers Z. Physiol. Chem. 363, 345–349.
Arsenijevic, D., Onuma, H., Pecqueur, C., Raimbault, S., Manning, B. S., Miroux, B., et al. (2000). Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat. Genet. 26, 435–439. doi: 10.1038/82565
Atkinson, G. C., Kuzmenko, A., Kamenski, P., Vysokikh, M. Y., Lakunina, V., Tankov, S., et al. (2012). Evolutionary and genetic analyses of mitochondrial translation initiation factors identify the missing mitochondrial IF3 in S. cerevisiae. Nucleic Acids Res. 40, 6122–6134. doi: 10.1093/nar/gks272
Baile, M. G., and Claypool, S. M. (2013). The power of yeast to model diseases of the powerhouse of the cell. Front. Biosci. 18, 241–278.
Bakker, H. D., Scholte, H. R., Van Den Bogert, C., Ruitenbeek, W., Jeneson, J. A., Wanders, R. J., et al. (1993). Deficiency of the adenine nucleotide translocator in muscle of a patient with myopathy and lactic acidosis: a new mitochondrial defect. Pediatr. Res. 33, 412–417. doi: 10.1203/00006450-199304000-00019
Bamber, L., Harding, M., Butler, P. J., and Kunji, E. R. (2006). Yeast mitochondrial ADP/ATP carriers are monomeric in detergents. Proc. Natl. Acad. Sci. U.S.A. 103, 16224–16229. doi: 10.1073/pnas.0607640103
Bamber, L., Harding, M., Monné, M., Slotboom, D. J., and Kunji, E. R. (2007a). The yeast mitochondrial ADP/ATP carrier functions as a monomer in mitochondrial membranes. Proc. Natl. Acad. Sci. U.S.A. 104, 10830–10834. doi: 10.1073/pnas.0703969104
Bamber, L., Slotboom, D. J., and Kunji, E. R. (2007b). Yeast mitochondrial ADP/ATP carriers are monomeric in detergents as demonstrated by differential affinity purification. J. Mol. Biol. 371, 388–395. doi: 10.1016/j.jmb.2007.05.072
Barrientos, A., and Ugalde, C. (2013). I function, therefore I am: overcoming skepticism about mitochondrial supercomplexes. Cell Metab. 18, 147–149. doi: 10.1016/j.cmet.2013.07.010
Barrientos, A., Zambrano, A., and Tzagoloff, A. (2004). Mss51p and Cox14p jointly regulate mitochondrial Cox1p expression in Saccharomyces cerevisiae. EMBO J. 23, 3472–3482. doi: 10.1038/sj.emboj.7600358
Barron, C. C., Bilan, P. J., Tsakiridis, T., and Tsiani, E. (2016). Facilitative glucose transporters: implications for cancer detection, prognosis and treatment. Metabolism 65, 124–139. doi: 10.1016/j.metabol.2015.10.007
Bedhomme, M., Hoffmann, M., Mccarthy, E. A., Gambonnet, B., Moran, R. G., Rébeillé, F., et al. (2005). Folate metabolism in plants: an Arabidopsis homolog of the mammalian mitochondrial folate transporter mediates folate import into chloroplasts. J. Biol. Chem. 280, 34823–34831. doi: 10.1074/jbc.M506045200
Berardi, M. J., and Chou, J. J. (2014). Fatty acid flippase activity of UCP2 is essential for its proton transport in mitochondria. Cell Metab. 20, 541–552. doi: 10.1016/j.cmet.2014.07.004
Bergeron, M. J., Simonin, A., Bürzle, M., and Hediger, M. A. (2008). Inherited epithelial transporter disorders–an overview. J. Inherit. Metab. Dis. 31, 178–187. doi: 10.1007/s10545-008-0861-6
Beyer, K., and Klingenberg, M. (1985). ADP/ATP carrier protein from beef heart mitochondria has high amounts of tightly bound cardiolipin, as revealed by 31P nuclear magnetic resonance. Biochemistry 24, 3821–3826. doi: 10.1021/bi00336a001
Bhoj, E. J., Li, M., Ahrens-Nicklas, R., Pyle, L. C., Wang, J., Zhang, V. W., et al. (2015). Pathologic variants of the mitochondrial phosphate carrier SLC25A3: two new patients and expansion of the cardiomyopathy/skeletal myopathy phenotype with and without lactic acidosis. JIMD Rep. 19, 59–66. doi: 10.1007/8904_2014_364
Bisaccia, F., De Palma, A., and Palmieri, F. (1989). Identification and purification of the tricarboxylate carrier from rat liver mitochondria. Biochim. Biophys. Acta 977, 171–176. doi: 10.1016/S0005-2728(89)80068-4
Blaustein, M. P., and Lederer, W. J. (1999). Sodium/calcium exchange: its physiological implications. Physiol. Rev. 79, 763–854. doi: 10.1152/physrev.1999.79.3.763
Boczonadi, V., King, M. S., Smith, A. C., Olahova, M., Bansagi, B., Roos, A., et al. (2018). Mitochondrial oxodicarboxylate carrier deficiency is associated with mitochondrial DNA depletion and spinal muscular atrophy–like disease. Genet. Med. 20, 1224–1235. doi: 10.1038/gim.2017.251
Boss, O., Samec, S., Paoloni-Giacobino, A., Rossier, C., Dulloo, A., Seydoux, J., et al. (1997). Uncoupling protein-3: a new member of the mitochondrial carrier family with tissue-specific expression. FEBS Lett. 408, 39–42. doi: 10.1016/S0014-5793(97)00384-0
Boulet, A., Vest, K. E., Maynard, M. K., Gammon, M. G., Russell, A. C., Mathews, A. T., et al. (2017). The mammalian phosphate carrier SLC25A3 is a mitochondrial copper transporter required for cytochrome c oxidase biogenesis. J. Biol. Chem. 293, 1887–1896. doi: 10.1074/jbc.RA117.000265
Brand, M. D., Pamplona, R., Portero-Otín, M., Requena, J. R., Roebuck, S. J., Buckingham, J. A., et al. (2002). Oxidative damage and phospholipid fatty acyl composition in skeletal muscle mitochondria from mice underexpressing or overexpressing uncoupling protein 3. Biochem. J. 368, 597–603. doi: 10.1042/BJ20021077
Buffet, A., Morin, A., Castro-Vega, L.-J., Habarou, F., Lussey-Lepoutre, C., Letouzé, E., et al. (2018). Germline mutations in the mitochondrial 2-oxoglutarate/malate carrier SLC25A11 gene confer a predisposition to metastatic paragangliomas. Cancer Res. 78, 1914–1922. doi: 10.1158/0008-5472.Can-17-2463
Camacho, J. A., Obie, C., Biery, B., Goodman, B. K., Hu, C. A., Almashanu, S., et al. (1999). Hyperornithinaemia-hyperammonaemia-homocitrullinuria syndrome is caused by mutations in a gene encoding a mitochondrial ornithine transporter. Nat. Genet. 22, 151–158.
Capobianco, L., Ferramosca, A., and Zara, V. (2002). The mitochondrial tricarboxylate carrier of silver eel: dimeric structure and cytosolic exposure of both N- and C-termini. J. Protein Chem. 21, 515–521. doi: 10.1023/A:1022473504904
Cavero, S., Traba, J., Del arco, A., and Satrústegui, J. (2005). The calcium-dependent ATP-Mg/Pi mitochondrial carrier is a target of glucose-induced calcium signalling in Saccharomyces cerevisiae. Biochem. J. 392, 537–544. doi: 10.1042/bj20050806
Cesar-Razquin, A., Snijder, B., Frappier-Brinton, T., Isserlin, R., Gyimesi, G., Bai, X., et al. (2015). A call for systematic research on solute carriers. Cell 162, 478–487. doi: 10.1016/j.cell.2015.07.022
Charlesworth, G., Balint, B., Mencacci, N. E., Carr, L., Wood, N. W., and Bhatia, K. P. (2016). SLC25A46 mutations underlie progressive myoclonic ataxia with optic atrophy and neuropathy. Mov. Disord. 31, 1249–1251. doi: 10.1002/mds.26716
Chen, X. J. (2004). Sal1p, a calcium-dependent carrier protein that suppresses an essential cellular function associated with the Aac2 isoform of ADP/ATP translocase in Saccharomyces cerevisiae. Genetics 167, 607–617. doi: 10.1534/genetics.103.023655
Chevrollier, A., Loiseau, D., Chabi, B., Renier, G., Douay, O., Malthiery, Y., et al. (2005). ANT2 isoform required for cancer cell glycolysis. J. Bioenerg. Biomembr. 37, 307–316. doi: 10.1007/s10863-005-8642-5
Choquet, Y., Wostrikoff, K., Rimbault, B., Zito, F., Girard-Bascou, J., Drapier, D., et al. (2001). Assembly-controlled regulation of chloroplast gene translation. Biochem. Soc. Trans. 29, 421–426. doi: 10.1042/bst0290421
Chouchani, E. T., Kazak, L., Jedrychowski, M. P., Lu, G. Z., Erickson, B. K., Szpyt, J., et al. (2016). Mitochondrial ROS regulate thermogenic energy expenditure and sulfenylation of UCP1. Nature 532, 112–116. doi: 10.1038/nature17399
Chouchani, E. T., Kazak, L., and Spiegelman, B. M. (2017). Mitochondrial reactive oxygen species and adipose tissue thermogenesis: bridging physiology and mechanisms. J. Biol. Chem. 292, 16810–16816. doi: 10.1074/jbc.R117.789628
Claro da Silva, T., Polli, J. E., and Swaan, P. W. (2013). The solute carrier family 10 (SLC10): beyond bile acid transport. Mol. Aspects Med. 34, 252–269. doi: 10.1016/j.mam.2012.07.004
Claypool, S. M. (2009). Cardiolipin, a critical determinant of mitochondrial carrier protein assembly and function. Biochim. Biophys. Acta 1788, 2059–2068. doi: 10.1016/j.bbamem.2009.04.020
Claypool, S. M., Oktay, Y., Boontheung, P., Loo, J. A., and Koehler, C. M. (2008). Cardiolipin defines the interactome of the major ADP/ATP carrier protein of the mitochondrial inner membrane. J. Cell Biol. 182, 937–950. doi: 10.1083/jcb.200801152
Clémençon, B., Babot, M., and Trézéguet, V. (2013). The mitochondrial ADP/ATP carrier (SLC25 family): pathological implications of its dysfunction. Mol. Aspects Med. 34, 485–493. doi: 10.1016/j.mam.2012.05.006
Cruciat, C. M., Brunner, S., Baumann, F., Neupert, W., and Stuart, R. A. (2000). The cytochrome bc1 and cytochrome c oxidase complexes associate to form a single supracomplex in yeast mitochondria. J. Biol. Chem. 275, 18093–18098. doi: 10.1074/jbc.M001901200
Debray, F. G., Lambert, M., Lemieux, B., Soucy, J. F., Drouin, R., Fenyves, D., et al. (2008). Phenotypic variability among patients with hyperornithinaemia-hyperammonaemia-homocitrullinuria syndrome homozygous for the delF188 mutation in SLC25A15. J. Med. Genet. 45, 759–764. doi: 10.1136/jmg.2008.059097
Derdak, Z., Mark, N. M., Beldi, G., Robson, S. C., Wands, J. R., and Baffy, G. (2008). The mitochondrial uncoupling protein-2 promotes chemoresistance in cancer cells. Cancer Res. 68, 2813–2819. doi: 10.1158/0008-5472.CAN-08-0053
Diaz, F. (2010). Cytochrome c oxidase deficiency: patients and animal models. Biochim. Biophys. Acta 1802, 100–110. doi: 10.1016/j.bbadis.2009.07.013
Dienhart, M. K., and Stuart, R. A. (2008). The yeast Aac2 protein exists in physical association with the cytochrome bc1-COX supercomplex and the TIM23 machinery. Mol. Biol. Cell 19, 3934–3943. doi: 10.1091/mbc.E08-04-0402
DiPolo, R., and Beaugé, L. (2006). Sodium/calcium exchanger: influence of metabolic regulation on ion carrier interactions. Physiol. Rev. 86, 155–203. doi: 10.1152/physrev.00018.2005
Doerner, A., Pauschinger, M., Badorff, A., Noutsias, M., Giessen, S., Schulze, K., et al. (1997). Tissue-specific transcription pattern of the adenine nucleotide translocase isoforms in humans. FEBS Lett. 414, 258–262. doi: 10.1016/S0014-5793(97)01000-4
Dolce, V., Scarcia, P., Iacopetta, D., and Palmieri, F. (2005). A fourth ADP/ATP carrier isoform in man: identification, bacterial expression, functional characterization and tissue distribution. FEBS Lett. 579, 633–637. doi: 10.1016/j.febslet.2004.12.034
Dong, C., Della-Morte, D., Cabral, D., Wang, L., Blanton, S. H., Seemant, C., et al. (2015). Sirtuin/uncoupling protein gene variants and carotid plaque area and morphology. Int. J. Stroke 10, 1247–1252. doi: 10.1111/ijs.12623
Dong, C., Della-Morte, D., Wang, L., Cabral, D., Beecham, A., Mcclendon, M. S., et al. (2011). Association of the sirtuin and mitochondrial uncoupling protein genes with carotid plaque. PLoS One 6:e27157. doi: 10.1371/journal.pone.0027157
Donowitz, M., Ming Tse, C., and Fuster, D. (2013). SLC9/NHE gene family, a plasma membrane and organellar family of Na+/H+ exchangers. Mol. Aspects Med. 34, 236–251. doi: 10.1016/j.mam.2012.05.001
Dudkina, N. V., Kudryashev, M., Stahlberg, H., and Boekema, E. J. (2011). Interaction of complexes I, III, and IV within the bovine respirasome by single particle cryoelectron tomography. Proc. Natl. Acad. Sci. U.S.A. 108, 15196–15200. doi: 10.1073/pnas.1107819108
Dufay, J. N., Fernández-Murray, J. P., and Mcmaster, C. R. (2017). SLC25 family member genetic interactions identify a role for HEM25 in yeast electron transport chain stability. G3 7, 1861–1873. doi: 10.1534/g3.117.041194
Dupont, P. Y., and Stepien, G. (2011). Computational analysis of the transcriptional regulation of the adenine nucleotide translocator isoform 4 gene and its role in spermatozoid glycolytic metabolism. Gene 487, 38–45. doi: 10.1016/j.gene.2011.07.024
Dyall, S. D., Agius, S. C., De Marcos Lousa, C., Trezeguet, V., and Tokatlidis, K. (2003). The dynamic dimerization of the yeast ADP/ATP carrier in the inner mitochondrial membrane is affected by conserved cysteine residues. J. Biol. Chem. 278, 26757–26764. doi: 10.1074/jbc.M302700200
Echaniz-Laguna, A., Chassagne, M., Ceresuela, J., Rouvet, I., Padet, S., Acquaviva, C., et al. (2012). Complete loss of expression of the ANT1 gene causing cardiomyopathy and myopathy. J. Med. Genet. 49, 146–150. doi: 10.1136/jmedgenet-2011-100504
Echtay, K. S., Roussel, D., St-Pierre, J., Jekabsons, M. B., Cadenas, S., Stuart, J. A., et al. (2002). Superoxide activates mitochondrial uncoupling proteins. Nature 415, 96–99. doi: 10.1038/415096a
Edvardson, S., Porcelli, V., Jalas, C., Soiferman, D., Kellner, Y., Shaag, A., et al. (2013). Agenesis of corpus callosum and optic nerve hypoplasia due to mutations in SLC25A1 encoding the mitochondrial citrate transporter. J. Med. Genet. 50, 240–245. doi: 10.1136/jmedgenet-2012-101485
Ehmke, N., Graul-Neumann, L., Smorag, L., Koenig, R., Segebrecht, L., Magoulas, P., et al. (2017). De novo mutations in SLC25A24 cause a craniosynostosis syndrome with hypertrichosis, progeroid appearance, and mitochondrial dysfunction. Am. J. Hum. Genet. 101, 833–843. doi: 10.1016/j.ajhg.2017.09.016
Falk, M. J., Li, D., Gai, X., Mccormick, E., Place, E., Lasorsa, F. M., et al. (2014). AGC1 deficiency causes infantile epilepsy, abnormal myelination, and reduced N-acetylaspartate. JIMD Rep. 14, 77–85. doi: 10.1007/8904_2013_287
Fedorenko, A., Lishko, P. V., and Kirichok, Y. (2012). Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. Cell 151, 400–413. doi: 10.1016/j.cell.2012.09.010
Fernandez, H. R., Gadre, S. M., Tan, M., Graham, G. T., Mosaoa, R., Ongkeko, M. S., et al. (2018). The mitochondrial citrate carrier, SLC25A1, drives stemness and therapy resistance in non-small cell lung cancer. Cell Death Differ. 25, 1239–1258. doi: 10.1038/s41418-018-0101-z
Fernández-Murray, J. P., Prykhozhij, S. V., Dufay, J. N., Steele, S. L., Gaston, D., Nasrallah, G. K., et al. (2016). Glycine and folate ameliorate models of congenital sideroblastic anemia. PLoS Genet. 12:e1005783. doi: 10.1371/journal.pgen.1005783
Ferrara, C. T., Boodhansingh, K. E., Paradies, E., Fiermonte, G., Steinkrauss, L. J., Topor, L. S., et al. (2017). Novel hypoglycemia phenotype in congenital hyperinsulinism due to dominant mutations of uncoupling protein 2. J. Clin. Endocrinol. Metab. 102, 942–949. doi: 10.1210/jc.2016-3164
Fiermonte, G., De Leonardis, F., Todisco, S., Palmieri, L., Lasorsa, F. M., and Palmieri, F. (2004). Identification of the mitochondrial ATP-Mg/Pi transporter. Bacterial expression, reconstitution, functional characterization, and tissue distribution. J. Biol. Chem. 279, 30722–30730. doi: 10.1074/jbc.M400445200
Fiermonte, G., Paradies, E., Todisco, S., Marobbio, C. M., and Palmieri, F. (2009). A novel member of solute carrier family 25 (SLC25A42) is a transporter of coenzyme A and adenosine 3′,5′-diphosphate in human mitochondria. J. Biol. Chem. 284, 18152–18159. doi: 10.1074/jbc.M109.014118
Fiermonte, G., Soon, D., Chaudhuri, A., Paradies, E., Lee, P. J., Krywawych, S., et al. (2008). An adult with type 2 citrullinemia presenting in Europe. N. Engl. J. Med. 358, 1408–1409. doi: 10.1056/NEJMc0707353
Fleury, C., Neverova, M., Collins, S., Raimbault, S., Champigny, O., Levi-Meyrueis, C., et al. (1997). Uncoupling protein-2: a novel gene linked to obesity and hyperinsulinemia. Nat. Genet. 15, 269–272. doi: 10.1038/ng0397-269
Fontanesi, F., Clemente, P., and Barrientos, A. (2011). Cox25 teams up with Mss51, Ssc1, and Cox14 to regulate mitochondrial cytochrome c oxidase subunit 1 expression and assembly in Saccharomyces cerevisiae. J. Biol. Chem. 286, 555–566. doi: 10.1074/jbc.M110.188805
Fontanesi, F., Palmieri, L., Scarcia, P., Lodi, T., Donnini, C., Limongelli, A., et al. (2004). Mutations in AAC2, equivalent to human adPEO-associated ANT1 mutations, lead to defective oxidative phosphorylation in Saccharomyces cerevisiae and affect mitochondrial DNA stability. Hum. Mol. Genet. 13, 923–934. doi: 10.1093/hmg/ddh108
Fotiadis, D., Kanai, Y., and Palacín, M. (2013). The SLC3 and SLC7 families of amino acid transporters. Mol. Aspects Med. 34, 139–158. doi: 10.1016/j.mam.2012.10.007
Foury, F., and Roganti, T. (2002). Deletion of the mitochondrial carrier genes MRS3 and MRS4 suppresses mitochondrial iron accumulation in a yeast frataxin-deficient strain. J. Biol. Chem. 277, 24475–24483. doi: 10.1074/jbc.M111789200
Fuster, D. G., and Alexander, R. T. (2014). Traditional and emerging roles for the SLC9 Na+/H+ exchangers. Pflugers Arch. 466, 61–76. doi: 10.1007/s00424-013-1408-8
Gao, C. L., Ni, Y. H., Liu, G., Chen, X. H., Ji, C. B., Qin, D. N., et al. (2011). UCP4 overexpression improves fatty acid oxidation and insulin sensitivity in L6 myocytes. J. Bioenerg. Biomembr. 43, 109–118. doi: 10.1007/s10863-011-9344-9
Gao, C. L., Zhu, J. G., Zhao, Y. P., Chen, X. H., Ji, C. B., Zhang, C. M., et al. (2010). Mitochondrial dysfunction is induced by the overexpression of UCP4 in 3T3-L1 adipocytes. Int. J. Mol. Med. 25, 71–80.
Gavurníková, G., Sabova, L., Kissová, I., Haviernik, P., and Kolarov, J. (1996). Transcription of the AAC1 gene encoding an isoform of mitochondrial ADP/ATP carrier in Saccharomyces cerevisiae is regulated by oxygen in a heme-independent manner. Eur. J. Biochem. 239, 759–763. doi: 10.1111/j.1432-1033.1996.0759u.x
Gegelashvili, M., Rodriguez-Kern, A., Pirozhkova, I., Zhang, J., Sung, L., and Gegelashvili, G. (2006). High-affinity glutamate transporter GLAST/EAAT1 regulates cell surface expression of glutamine/neutral amino acid transporter ASCT2 in human fetal astrocytes. Neurochem. Int. 48, 611–615. doi: 10.1016/j.neuint.2005.12.033
Genova, M. L., and Lenaz, G. (2014). Functional role of mitochondrial respiratory supercomplexes. Biochim. Biophys. Acta 1837, 427–443. doi: 10.1016/j.bbabio.2013.11.002
Geyer, J., Wilke, T., and Petzinger, E. (2006). The solute carrier family SLC10: more than a family of bile acid transporters regarding function and phylogenetic relationships. Naunyn Schmiedebergs Arch. Pharmacol. 372, 413–431. doi: 10.1007/s00210-006-0043-8
Gill, H. S., and Boron, W. F. (2006). Expression and purification of the cytoplasmic N-terminal domain of the Na/HCO3 cotransporter NBCe1-A: structural insights from a generalized approach. Protein Expr. Purif. 49, 228–234. doi: 10.1016/j.pep.2006.04.001
Gimeno, R. E., Dembski, M., Weng, X., Deng, N., Shyjan, A. W., Gimeno, C. J., et al. (1997). Cloning and characterization of an uncoupling protein homolog: a potential molecular mediator of human thermogenesis. Diabetes 46, 900–906. doi: 10.2337/diab.46.5.900
Giraud, S., Bonod-Bidaud, C., Wesolowski-Louvel, M., and Stepien, G. (1998). Expression of human ANT2 gene in highly proliferative cells: GRBOX, a new transcriptional element, is involved in the regulation of glycolytic ATP import into mitochondria. J. Mol. Biol. 281, 409–418. doi: 10.1006/jmbi.1998.1955
Gnoni, G. V., Priore, P., Geelen, M. J., and Siculella, L. (2009). The mitochondrial citrate carrier: metabolic role and regulation of its activity and expression. IUBMB Life 61, 987–994. doi: 10.1002/iub.249
Gokhale, A., Freeman, A. A. H., Hartwig, C., Bassell, J. L., Zlatic, S. A., Sapp, C., et al. (2018). Systems analysis of the 22q11.2 microdeletion syndrome converges on a mitochondrial interactome necessary for synapse function and behavior. bioRxiv [Preprint]. doi: 10.1101/315143
Golozoubova, V., Hohtola, E., Matthias, A., Jacobsson, A., Cannon, B., and Nedergaard, J. (2001). Only UCP1 can mediate adaptive nonshivering thermogenesis in the cold. FASEB J. 15, 2048–2050. doi: 10.1096/fj.00-0536fje
González-Barroso, M. M., Giurgea, I., Bouillaud, F., Anedda, A., Bellanné-Chantelot, C., Hubert, L., et al. (2008). Mutations in UCP2 in congenital hyperinsulinism reveal a role for regulation of insulin secretion. PLoS One 3:e3850. doi: 10.1371/journal.pone.0003850
Gu, J., Wu, M., Guo, R., Yan, K., Lei, J., Gao, N., et al. (2016). The architecture of the mammalian respirasome. Nature 537, 639–643. doi: 10.1038/nature19359
Guernsey, D. L., Jiang, H., Campagna, D. R., Evans, S. C., Ferguson, M., Kellogg, M. D., et al. (2009). Mutations in mitochondrial carrier family gene SLC25A38 cause nonsyndromic autosomal recessive congenital sideroblastic anemia. Nat. Genet. 41, 651–653. doi: 10.1038/ng.359
Haguenauer, A., Raimbault, S., Masscheleyn, S., Gonzalez-Barroso, M. E. M., Criscuolo, F., Plamondon, J., et al. (2005). A new renal mitochondrial carrier, KMCP1, is up-regulated during tubular cell regeneration and induction of antioxidant enzymes. J. Biol. Chem. 280, 22036–22043. doi: 10.1074/jbc.M412136200
Haitina, T., Lindblom, J., Renström, T., and Fredriksson, R. (2006). Fourteen novel human members of mitochondrial solute carrier family 25 (SLC25) widely expressed in the central nervous system. Genomics 88, 779–790. doi: 10.1016/j.ygeno.2006.06.016
Harigae, H., and Furuyama, K. (2010). Hereditary sideroblastic anemia: pathophysiology and gene mutations. Int. J. Hematol. 92, 425–431. doi: 10.1007/s12185-010-0688-4
Heaton, G. M., Wagenvoord, R. J., Kemp, A., and Nicholls, D. G. (1978). Brown-adipose-tissue mitochondria: photoaffinity labelling of the regulatory site of energy dissipation. Eur. J. Biochem. 82, 515–521. doi: 10.1111/j.1432-1033.1978.tb12045.x
Hediger, M. A., Romero, M. F., Peng, J. B., Rolfs, A., Takanaga, H., and Bruford, E. A. (2004). The ABCs of solute carriers: physiological, pathological and therapeutic implications of human membrane transport proteins: introduction. Pflugers Arch. 447, 465–468. doi: 10.1007/s00424-003-1192-y
Heidkämper, D., Müller, V., Nelson, D. R., and Klingenberg, M. (1996). Probing the role of positive residues in the ADP/ATP carrier from yeast. The effect of six arginine mutations on transport and the four ATP versus ADP exchange modes. Biochemistry 35, 16144–16152. doi: 10.1021/bi960668j
Hellebrekers, D. M. E. I., Sallevelt, S. C. E. H., Theunissen, T. E. J., Hendrickx, A. T. M., Gottschalk, R. W., Hoeijmakers, J. G. J., et al. (2017). Novel SLC25A32 mutation in a patient with a severe neuromuscular phenotype. Eur. J. Hum. Genet. 25, 886–888. doi: 10.1038/ejhg.2017.62
Hevia, D., González-Menéndez, P., Quiros-González, I., Miar, A., Rodríguez-García, A., Tan, D. X., et al. (2015). Melatonin uptake through glucose transporters: a new target for melatonin inhibition of cancer. J. Pineal Res. 58, 234–250. doi: 10.1111/jpi.12210
Hofherr, A., Seger, C., Fitzpatrick, F., Busch, T., Michel, E., Luan, J., et al. (2018). The mitochondrial transporter SLC25A25 links ciliary TRPP2 signaling and cellular metabolism. PLoS Biol. 16:e2005651. doi: 10.1371/journal.pbio.2005651
Höglund, P. J., Nordström, K. J., Schiöth, H. B., and Fredriksson, R. (2011). The solute carrier families have a remarkably long evolutionary history with the majority of the human families present before divergence of Bilaterian species. Mol. Biol. Evol. 28, 1531–1541. doi: 10.1093/molbev/msq350
Horimoto, M., Resnick, M. B., Konkin, T. A., Routhier, J., Wands, J. R., and Baffy, G. (2004). Expression of uncoupling protein-2 in human colon cancer. Clin. Cancer Res. 10, 6203–6207. doi: 10.1158/1078-0432.CCR-04-0419
Horvathova, M., Ponka, P., and Divoky, V. (2010). Molecular basis of hereditary iron homeostasis defects. Hematology 15, 96–111. doi: 10.1179/102453310X12583347009810
Huizing, M., Wendel, U., Ruitenbeek, W., Iacobazzi, V., Ijlst, L., Veenhuizen, P., et al. (1998). Carnitine-acylcarnitine carrier deficiency: identification of the molecular defect in a patient. J. Inherit. Metab. Dis. 21, 262–267. doi: 10.1023/A:1005324323401
Iacobazzi, V., Pasquali, M., Singh, R., Matern, D., Rinaldo, P., Amat Di San Filippo, C., et al. (2004). Response to therapy in carnitine/acylcarnitine translocase (CACT) deficiency due to a novel missense mutation. Am. J. Med. Genet. A 126A, 150–155. doi: 10.1002/ajmg.a.20573
Jabùrek, M., Varecha, M., Jezek, P., and Garlid, K. D. (2001). Alkylsulfonates as probes of uncoupling protein transport mechanism. Ion pair transport demonstrates that direct H+ translocation by UCP1 is not necessary for uncoupling. J. Biol. Chem. 276, 31897–31905. doi: 10.1074/jbc.M103507200
Janer, A., Prudent, J., Paupe, V., Fahiminiya, S., Majewski, J., Sgarioto, N., et al. (2016). SLC25A46 is required for mitochondrial lipid homeostasis and cristae maintenance and is responsible for Leigh syndrome. EMBO Mol. Med. 8, 1019–1038. doi: 10.15252/emmm.201506159
Jezek, P., Holendová, B., Garlid, K. D., and Jaburek, M. (2018). Mitochondrial uncoupling proteins: subtle regulators of cellular redox signaling. Antioxid. Redox Signal. 29, 667–714. doi: 10.1089/ars.2017.7225
Jezek, P., Jabùrek, M., and Garlid, K. D. (2010). Channel character of uncoupling protein-mediated transport. FEBS Lett. 584, 2135–2141. doi: 10.1016/j.febslet.2010.02.068
Jezek, P., Modrianský, M., and Garlid, K. D. (1997a). A structure-activity study of fatty acid interaction with mitochondrial uncoupling protein. FEBS Lett. 408, 166–170.
Jezek, P., Modrianský, M., and Garlid, K. D. (1997b). Inactive fatty acids are unable to flip-flop across the lipid bilayer. FEBS Lett. 408, 161–165.
Jiang, F., Ryan, M. T., Schlame, M., Zhao, M., Gu, Z., Klingenberg, M., et al. (2000). Absence of cardiolipin in the crd1 null mutant results in decreased mitochondrial membrane potential and reduced mitochondrial function. J. Biol. Chem. 275, 22387–22394. doi: 10.1074/jbc.M909868199
Kamp, F., and Hamilton, J. A. (1992). pH gradients across phospholipid membranes caused by fast flip-flop of un-ionized fatty acids. Proc. Natl. Acad. Sci. U.S.A. 89, 11367–11370. doi: 10.1073/pnas.89.23.11367
Kamp, F., Hamilton, J. A., and Westerhoff, H. V. (1993). Movement of fatty acids, fatty acid analogues, and bile acids across phospholipid bilayers. Biochemistry 32, 11074–11086. doi: 10.1021/bi00092a017
Kamp, F., Zakim, D., Zhang, F., Noy, N., and Hamilton, J. A. (1995). Fatty acid flip-flop in phospholipid bilayers is extremely fast. Biochemistry 34, 11928–11937. doi: 10.1021/bi00037a034
Kanai, Y., Clémençon, B., Simonin, A., Leuenberger, M., Lochner, M., Weisstanner, M., et al. (2013). The SLC1 high-affinity glutamate and neutral amino acid transporter family. Mol. Aspects Med. 34, 108–120. doi: 10.1016/j.mam.2013.01.001
Kanai, Y., and Hediger, M. A. (2003). The glutamate and neutral amino acid transporter family: physiological and pharmacological implications. Eur. J. Pharmacol. 479, 237–247. doi: 10.1016/j.ejphar.2003.08.073
Kanai, Y., and Hediger, M. A. (2004). The glutamate/neutral amino acid transporter family SLC1: molecular, physiological and pharmacological aspects. Pflugers Arch. 447, 469–479. doi: 10.1007/s00424-003-1146-4
Kang, J., and Samuels, D. C. (2008). The evidence that the DNC (SLC25A19) is not the mitochondrial deoxyribonucleotide carrier. Mitochondrion 8, 103–108. doi: 10.1016/j.mito.2008.01.001
Kaplan, R. S., Mayor, J. A., Gremse, D. A., and Wood, D. O. (1995). High level expression and characterization of the mitochondrial citrate transport protein from the yeast Saccharomyces cerevisiae. J. Biol. Chem. 270, 4108–4114. doi: 10.1074/jbc.270.8.4108
Kaukonen, J., Juselius, J. K., Tiranti, V., Kyttälä, A., Zeviani, M., Comi, G. P., et al. (2000). Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science 289, 782–785. doi: 10.1126/science.289.5480.782
Kazak, L., Chouchani, E. T., Stavrovskaya, I. G., Lu, G. Z., Jedrychowski, M. P., Egan, D. F., et al. (2017). UCP1 deficiency causes brown fat respiratory chain depletion and sensitizes mitochondria to calcium overload-induced dysfunction. Proc. Natl. Acad. Sci. U.S.A. 114, 7981–7986. doi: 10.1073/pnas.1705406114
Kehrein, K., Bonnefoy, N., and Ott, M. (2013). Mitochondrial protein synthesis: efficiency and accuracy. Antioxid. Redox Signal. 19, 1928–1939. doi: 10.1089/ars.2012.4896
Khan, S., Ansar, M., Khan, A. K., Shah, K., Muhammad, N., Shahzad, S., et al. (2018). A homozygous missense mutation in SLC25A16 associated with autosomal recessive isolated fingernail dysplasia in a Pakistani family. Br. J. Dermatol. 178, 556–558. doi: 10.1111/bjd.15661
Khananshvili, D. (2013). The SLC8 gene family of sodium-calcium exchangers (NCX) - structure, function, and regulation in health and disease. Mol. Aspects Med. 34, 220–235. doi: 10.1016/j.mam.2012.07.003
Kim, Y. H., Haidl, G., Schaefer, M., Egner, U., Mandal, A., and Herr, J. C. (2007). Compartmentalization of a unique ADP/ATP carrier protein SFEC (Sperm Flagellar Energy Carrier, AAC4) with glycolytic enzymes in the fibrous sheath of the human sperm flagellar principal piece. Dev. Biol. 302, 463–476. doi: 10.1016/j.ydbio.2006.10.004
Kishita, Y., Pajak, A., Bolar, N. A., Marobbio, C. M., Maffezzini, C., Miniero, D. V., et al. (2015). Intra-mitochondrial methylation deficiency due to mutations in SLC25A26. Am. J. Hum. Genet. 97, 761–768. doi: 10.1016/j.ajhg.2015.09.013
Klingenberg, M. (1972). Kinetic study of the tricarboxylate carrier in rat liver mitochondria. Eur. J. Biochem. 26, 587–594. doi: 10.1111/j.1432-1033.1972.tb01801.x
Klingenberg, M. (1981). Membrane protein oligomeric structure and transport function. Nature 290, 449–454. doi: 10.1038/290449a0
Klingenberg, M. (2008). The ADP and ATP transport in mitochondria and its carrier. Biochim. Biophys. Acta 1778, 1978–2021. doi: 10.1016/j.bbamem.2008.04.011
Kobayashi, K., Sinasac, D. S., Iijima, M., Boright, A. P., Begum, L., Lee, J. R., et al. (1999). The gene mutated in adult-onset type II citrullinemia encodes a putative mitochondrial carrier protein. Nat. Genet. 22, 159–163.
Kolukula, V. K., Sahu, G., Wellstein, A., Rodriguez, O. C., Preet, A., Iacobazzi, V., et al. (2014). SLC25A1, or CIC, is a novel transcriptional target of mutant p53 and a negative tumor prognostic marker. Oncotarget 5, 1212–1225. doi: 10.18632/oncotarget.1831
Komaki, H., Fukazawa, T., Houzen, H., Yoshida, K., Nonaka, I., and Goto, Y. (2002). A novel D104G mutation in the adenine nucleotide translocator 1 gene in autosomal dominant progressive external ophthalmoplegia patients with mitochondrial DNA with multiple deletions. Ann. Neurol. 51, 645–648. doi: 10.1002/ana.10172
Komatsu, M., Yazaki, M., Tanaka, N., Sano, K., Hashimoto, E., Takei, Y., et al. (2008). Citrin deficiency as a cause of chronic liver disorder mimicking non-alcoholic fatty liver disease. J. Hepatol. 49, 810–820. doi: 10.1016/j.jhep.2008.05.016
Körver-Keularts, I. M., De Visser, M., Bakker, H. D., Wanders, R. J., Vansenne, F., Scholte, H. R., et al. (2015). Two novel mutations in the SLC25A4 gene in a patient with mitochondrial myopathy. JIMD Rep. 22, 39–45. doi: 10.1007/8904_2015_409
Kotaria, R., Mayor, J. A., Walters, D. E., and Kaplan, R. S. (1999). Oligomeric state of wild-type and cysteine-less yeast mitochondrial citrate transport proteins. J. Bioenerg. Biomembr. 31, 543–549. doi: 10.1023/A:1005460810527
Krämer, R., and Klingenberg, M. (1980). Modulation of the reconstituted adenine nucleotide exchange by membrane potential. Biochemistry 19, 556–560. doi: 10.1021/bi00544a025
Kunji, E. R., and Crichton, P. G. (2010). Mitochondrial carriers function as monomers. Biochim. Biophys. Acta 1797, 817–831. doi: 10.1016/j.bbabio.2010.03.023
Kunji, E. R., and Harding, M. (2003). Projection structure of the atractyloside-inhibited mitochondrial ADP/ATP carrier of Saccharomyces cerevisiae. J. Biol. Chem. 278, 36985–36988. doi: 10.1074/jbc.C300304200
Kuzmenko, A., Atkinson, G. C., Levitskii, S., Zenkin, N., Tenson, T., Hauryliuk, V., et al. (2014). Mitochondrial translation initiation machinery: conservation and diversification. Biochimie 100, 132–140. doi: 10.1016/j.biochi.2013.07.024
Kuzmenko, A., Derbikova, K., Salvatori, R., Tankov, S., Atkinson, G. C., Tenson, T., et al. (2016). Aim-less translation: loss of Saccharomyces cerevisiae mitochondrial translation initiation factor mIF3/Aim23 leads to unbalanced protein synthesis. Sci. Rep. 6:18749. doi: 10.1038/srep18749
Kwok, K. H., Ho, P. W., Chu, A. C., Ho, J. W., Liu, H. F., Yiu, D. C., et al. (2010). Mitochondrial UCP5 is neuroprotective by preserving mitochondrial membrane potential, ATP levels, and reducing oxidative stress in MPP+ and dopamine toxicity. Free Radic. Biol. Med. 49, 1023–1035. doi: 10.1016/j.freeradbiomed.2010.06.017
Laco, J., Zeman, I., Pevala, V., Polèic, P., and Kolarov, J. (2010). Adenine nucleotide transport via Sal1 carrier compensates for the essential function of the mitochondrial ADP/ATP carrier. FEMS Yeast Res. 10, 290–296. doi: 10.1111/j.1567-1364.2010.00606.x
Lamantea, E., Tiranti, V., Bordoni, A., Toscano, A., Bono, F., Servidei, S., et al. (2002). Mutations of mitochondrial DNA polymerase gammaA are a frequent cause of autosomal dominant or recessive progressive external ophthalmoplegia. Ann. Neurol. 52, 211–219. doi: 10.1002/ana.10278
Lapuente-Brun, E., Moreno-Loshuertos, R., Acín-Pérez, R., Latorre-Pellicer, A., Colás, C., Balsa, E., et al. (2013). Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science 340, 1567–1570. doi: 10.1126/science.1230381
Lawson, J. E., Gawaz, M., Klingenberg, M., and Douglas, M. G. (1990). Structure-function studies of adenine nucleotide transport in mitochondria. I. Construction and genetic analysis of yeast mutants encoding the ADP/ATP carrier protein of mitochondria. J. Biol. Chem. 265, 14195–14201.
LeMoine, C. M., and Walsh, P. J. (2015). Evolution of urea transporters in vertebrates: adaptation to urea’s multiple roles and metabolic sources. J. Exp. Biol. 218, 1936–1945. doi: 10.1242/jeb.114223
Letts, J. A., Fiedorczuk, K., and Sazanov, L. A. (2016). The architecture of respiratory supercomplexes. Nature 537, 644–648. doi: 10.1038/nature19774
Levi, S., and Rovida, E. (2009). The role of iron in mitochondrial function. Biochim. Biophys. Acta 1790, 629–636. doi: 10.1016/j.bbagen.2008.09.008
Li, W., Nichols, K., Nathan, C. A., and Zhao, Y. (2013). Mitochondrial uncoupling protein 2 is up-regulated in human head and neck, skin, pancreatic, and prostate tumors. Cancer Biomark. 13, 377–383. doi: 10.3233/CBM-130369
Li, W., Zhang, C., Jackson, K., Shen, X., Jin, R., Li, G., et al. (2015). UCP2 knockout suppresses mouse skin carcinogenesis. Cancer Prev. Res. 8, 487–491. doi: 10.1158/1940-6207.CAPR-14-0297-T
Lill, R. (2009). Function and biogenesis of iron-sulphur proteins. Nature 460, 831–838. doi: 10.1038/nature08301
Lill, R., and Mühlenhoff, U. (2008). Maturation of iron-sulfur proteins in eukaryotes: mechanisms, connected processes, and diseases. Annu. Rev. Biochem. 77, 669–700. doi: 10.1146/annurev.biochem.76.052705.162653
Lin, C. S., Hackenberg, H., and Klingenberg, E. M. (1980). The uncoupling protein from brown adipose tissue mitochondria is a dimer. A hydrodynamic study. FEBS Lett. 113, 304–306. doi: 10.1016/0014-5793(80)80614-4
Lindhurst, M. J., Fiermonte, G., Song, S., Struys, E., De Leonardis, F., Schwartzberg, P. L., et al. (2006). Knockout of SLC25A19 causes mitochondrial thiamine pyrophosphate depletion, embryonic lethality, CNS malformations, and anemia. Proc. Natl. Acad. Sci. U.S.A. 103, 15927–15932. doi: 10.1073/pnas.0607661103
Liu, D., Chan, S. L., De Souza-Pinto, N. C., Slevin, J. R., Wersto, R. P., Zhan, M., et al. (2006). Mitochondrial UCP4 mediates an adaptive shift in energy metabolism and increases the resistance of neurons to metabolic and oxidative stress. Neuromolecular Med. 8, 389–414. doi: 10.1385/NMM:8:3:389
Lu, Y. W., Acoba, M. G., Selvaraju, K., Huang, T. C., Nirujogi, R. S., Sathe, G., et al. (2017). Human adenine nucleotide translocases physically and functionally interact with respirasomes. Mol. Biol. Cell 28, 1489–1506. doi: 10.1091/mbc.E17-03-0195
Lunetti, P., Damiano, F., De Benedetto, G., Siculella, L., Pennetta, A., Muto, L., et al. (2016). Characterization of human and yeast mitochondrial glycine carriers with implications for heme biosynthesis and anemia. J. Biol. Chem. 291, 19746–19759. doi: 10.1074/jbc.M116.736876
Maldonado, E. N., Dehart, D. N., Patnaik, J., Klatt, S. C., Gooz, M. B., and Lemasters, J. J. (2016). ATP/ADP turnover and import of glycolytic ATP into mitochondria in cancer cells is independent of the adenine nucleotide translocator. J. Biol. Chem. 291, 19642–19650. doi: 10.1074/jbc.M116.734814
Mao, W., Yu, X. X., Zhong, A., Li, W., Brush, J., Sherwood, S. W., et al. (1999). UCP4, a novel brain-specific mitochondrial protein that reduces membrane potential in mammalian cells. FEBS Lett. 443, 326–330. doi: 10.1016/S0014-5793(98)01713-X
Matthias, A., Ohlson, K. B., Fredriksson, J. M., Jacobsson, A., Nedergaard, J., and Cannon, B. (2000). Thermogenic responses in brown fat cells are fully UCP1-dependent. UCP2 or UCP3 do not substitute for UCP1 in adrenergically or fatty scid-induced thermogenesis. J. Biol. Chem. 275, 25073–25081. doi: 10.1074/jbc.M000547200
Mattiasson, G., and Sullivan, P. G. (2006). The emerging functions of UCP2 in health, disease, and therapeutics. Antioxid. Redox Signal. 8, 1–38. doi: 10.1089/ars.2006.8.1
Mayr, J. A., Merkel, O., Kohlwein, S. D., Gebhardt, B. R., Böhles, H., Fötschl, U., et al. (2007). Mitochondrial phosphate-carrier deficiency: a novel disorder of oxidative phosphorylation. Am. J. Hum. Genet. 80, 478–484. doi: 10.1086/511788
Mayr, J. A., Zimmermann, F. A., Horváth, R., Schneider, H. C., Schoser, B., Holinski-Feder, E., et al. (2011). Deficiency of the mitochondrial phosphate carrier presenting as myopathy and cardiomyopathy in a family with three affected children. Neuromuscul. Disord. 21, 803–808. doi: 10.1016/j.nmd.2011.06.005
Meyertholen, K., Ravnan, J. B., and Matalon, R. (2012). Identification of a novel 14q13.3 deletion involving the SLC25A21 gene associated with familial synpolydactyly. Mol. Syndromol. 3, 25–29. doi: 10.1159/000339177
Milenkovic, D., Blaza, J. N., Larsson, N. G., and Hirst, J. (2017). The enigma of the respiratory chain supercomplex. Cell Metab. 25, 765–776. doi: 10.1016/j.cmet.2017.03.009
Miyamoto, T., Kanazawa, N., Kato, S., Kawakami, M., Inoue, Y., Kuhara, T., et al. (2001). Diagnosis of Japanese patients with HHH syndrome by molecular genetic analysis: a common mutation, R179X. J. Hum. Genet. 46, 260–262. doi: 10.1007/s100380170075
Molinari, F., Kaminska, A., Fiermonte, G., Boddaert, N., Raas-Rothschild, A., Plouin, P., et al. (2009). Mutations in the mitochondrial glutamate carrier SLC25A22 in neonatal epileptic encephalopathy with suppression bursts. Clin. Genet. 76, 188–194. doi: 10.1111/j.1399-0004.2009.01236.x
Molinari, F., Raas-Rothschild, A., Rio, M., Fiermonte, G., Encha-Razavi, F., Palmieri, L., et al. (2005). Impaired mitochondrial glutamate transport in autosomal recessive neonatal myoclonic epilepsy. Am. J. Hum. Genet. 76, 334–339. doi: 10.1086/427564
Monné, M., Daddabbo, L., Gagneul, D., Obata, T., Hielscher, B., Palmieri, L., et al. (2018). Uncoupling proteins 1 and 2 (UCP1 and UCP2) from Arabidopsis thaliana are mitochondrial transporters of aspartate, glutamate and dicarboxylates. J. Biol. Chem. 293, 4213–4227. doi: 10.1074/jbc.RA117.000771
Morciano, P., Carrisi, C., Capobianco, L., Mannini, L., Burgio, G., Cestra, G., et al. (2009). A conserved role for the mitochondrial citrate transporter Sea/SLC25A1 in the maintenance of chromosome integrity. Hum. Mol. Genet. 18, 4180–4188. doi: 10.1093/hmg/ddp370
Moreno-Lastres, D., Fontanesi, F., García-Consuegra, I., Martín, M. A., Arenas, J., Barrientos, A., et al. (2012). Mitochondrial complex I plays an essential role in human respirasome assembly. Cell Metab. 15, 324–335. doi: 10.1016/j.cmet.2012.01.015
Mueckler, M., and Thorens, B. (2013). The SLC2 (GLUT) family of membrane transporters. Mol. Aspects Med. 34, 121–138. doi: 10.1016/j.mam.2012.07.001
Muller, V., Basset, G., Nelson, D. R., and Klingenberg, M. (1996). Probing the role of positive residues in the ADP/ATP carrier from yeast. The effect of six arginine mutations of oxidative phosphorylation and AAC expression. Biochemistry 35, 16132–16143. doi: 10.1021/bi960667r
Müller, V., Heidkämper, D., Nelson, D. R., and Klingenberg, M. (1997). Mutagenesis of some positive and negative residues occurring in repeat triad residues in the ADP/ATP carrier from yeast. Biochemistry 36, 16008–16018. doi: 10.1021/bi971867l
Muntau, A. C., Röschinger, W., Merkenschlager, A., Van Der Knaap, M. S., Jakobs, C., Duran, M., et al. (2000). Combined D-2- and L-2-hydroxyglutaric aciduria with neonatal onset encephalopathy: a third biochemical variant of 2-hydroxyglutaric aciduria? Neuropediatrics 31, 137–140. doi: 10.1055/s-2000-7497
Nakagawa, T., and Kaneko, S. (2013). SLC1 glutamate transporters and diseases: psychiatric diseases and pathological pain. Curr. Mol. Pharmacol. 6, 66–73. doi: 10.2174/18744672113069990033
Nakase, T., Yoshida, Y., and Nagata, K. (2007). Amplified expression of uncoupling proteins in human brain ischemic lesions. Neuropathology 27, 442–447. doi: 10.1111/j.1440-1789.2007.00815.x
Napoli, L., Bordoni, A., Zeviani, M., Hadjigeorgiou, G. M., Sciacco, M., Tiranti, V., et al. (2001). A novel missense adenine nucleotide translocator-1 gene mutation in a Greek adPEO family. Neurology 57, 2295–2298. doi: 10.1212/WNL.57.12.2295
Nègre-Salvayre, A., Hirtz, C., Carrera, G., Cazenave, R., Troly, M., Salvayre, R., et al. (1997). A role for uncoupling protein-2 as a regulator of mitochondrial hydrogen peroxide generation. FASEB J. 11, 809–815. doi: 10.1096/fasebj.11.10.9271366
Nicholls, D. G., Bernson, V. S., and Heaton, G. M. (1978). The identification of the component in the inner membrane of brown adipose tissue mitochondria responsible for regulating energy dissipation. Exp. Suppl. 32, 89–93. doi: 10.1007/978-3-0348-5559-4_9
Nohara, K., Tateishi, Y., Suzuki, T., Okamura, K., Murai, H., Takumi, S., et al. (2012). Late-onset increases in oxidative stress and other tumorigenic activities and tumors with a Ha-ras mutation in the liver of adult male C3H mice gestationally exposed to arsenic. Toxicol. Sci. 129, 293–304. doi: 10.1093/toxsci/kfs203
Nota, B., Struys, E. A., Pop, A., Jansen, E. E., Fernandez Ojeda, M. R., Kanhai, W. A., et al. (2013). Deficiency in SLC25A1, encoding the mitochondrial citrate carrier, causes combined D-2- and L-2-hydroxyglutaric aciduria. Am. J. Hum. Genet. 92, 627–631. doi: 10.1016/j.ajhg.2013.03.009
Nury, H., Dahout-Gonzalez, C., Trézéguet, V., Lauquin, G., Brandolin, G., and Pebay-Peyroula, E. (2005). Structural basis for lipid-mediated interactions between mitochondrial ADP/ATP carrier monomers. FEBS Lett. 579, 6031–6036. doi: 10.1016/j.febslet.2005.09.061
Ogunbona, O. B. (2018). ADP/ATP Carrier Activity and Mitochondrial Translation-Dependent Regulation of Oxidative Phosphorylation in Saccharomyces cerevisiae. Doctor of Philosophy, Johns Hopkins University, Baltimore, MD.
Ogunbona, O. B., Baile, M. G., and Claypool, S. M. (2018). Cardiomyopathy-associated mutation in the ADP/ATP carrier reveals translation-dependent regulation of cytochrome c oxidase activity. Mol. Biol. Cell 29, 1449–1464. doi: 10.1091/mbc.E17-12-0700
Ohura, T., Kobayashi, K., Tazawa, Y., Nishi, I., Abukawa, D., Sakamoto, O., et al. (2001). Neonatal presentation of adult-onset type II citrullinemia. Hum. Genet. 108, 87–90. doi: 10.1007/s004390000448
Orlowski, J., and Grinstein, S. (2004). Diversity of the mammalian sodium/proton exchanger SLC9 gene family. Pflugers Arch. 447, 549–565. doi: 10.1007/s00424-003-1110-3
Padan, E., and Landau, M. (2016). Sodium-proton (Na+/H+) antiporters: properties and roles in health and disease. Met. Ions Life Sci. 16, 391–458. doi: 10.1007/978-3-319-21756-7_12
Palacín, M., and Kanai, Y. (2004). The ancillary proteins of HATs: SLC3 family of amino acid transporters. Pflugers Arch. 447, 490–494. doi: 10.1007/s00424-003-1062-7
Palmieri, E. M., Spera, I., Menga, A., Infantino, V., Porcelli, V., Iacobazzi, V., et al. (2015). Acetylation of human mitochondrial citrate carrier modulates mitochondrial citrate/malate exchange activity to sustain NADPH production during macrophage activation. Biochim. Biophys. Acta 1847, 729–738. doi: 10.1016/j.bbabio.2015.04.009
Palmieri, F. (2004). The mitochondrial transporter family (SLC25): physiological and pathological implications. Pflugers Arch. 447, 689–709. doi: 10.1007/s00424-003-1099-7
Palmieri, F. (2013). The mitochondrial transporter family SLC25: identification, properties and physiopathology. Mol. Aspects Med. 34, 465–484. doi: 10.1016/j.mam.2012.05.005
Palmieri, F. (2014). Mitochondrial transporters of the SLC25 family and associated diseases: a review. J. Inherit. Metab. Dis. 37, 565–575. doi: 10.1007/s10545-014-9708-5
Palmieri, F., and Monné, M. (2016). Discoveries, metabolic roles and diseases of mitochondrial carriers: a review. Biochim. Biophys. Acta 1863, 2362–2378. doi: 10.1016/j.bbamcr.2016.03.007
Palmieri, F., and Pierri, C. L. (2010). Mitochondrial metabolite transport. Essays Biochem. 47, 37–52. doi: 10.1042/bse0470037
Palmieri, L., Alberio, S., Pisano, I., Lodi, T., Meznaric-Petrusa, M., Zidar, J., et al. (2005). Complete loss-of-function of the heart/muscle-specific adenine nucleotide translocator is associated with mitochondrial myopathy and cardiomyopathy. Hum. Mol. Genet. 14, 3079–3088. doi: 10.1093/hmg/ddi341
Palmieri, L., Pardo, B., Lasorsa, F. M., Del Arco, A., Kobayashi, K., Iijima, M., et al. (2001). Citrin and Aralar1 are Ca2+-stimulated aspartate/glutamate transporters in mitochondria. EMBO J. 20, 5060–5069. doi: 10.1093/emboj/20.18.5060
Palmisano, A., Zara, V., Hönlinger, A., Vozza, A., Dekker, P. J., Pfanner, N., et al. (1998). Targeting and assembly of the oxoglutarate carrier: general principles for biogenesis of carrier proteins of the mitochondrial inner membrane. Biochem. J. 333(Pt 1), 151–158. doi: 10.1042/bj3330151
Pande, S. V., Brivet, M., Slama, A., Demaugre, F., Aufrant, C., and Saudubray, J. M. (1993). Carnitine-acylcarnitine translocase deficiency with severe hypoglycemia and auriculo ventricular block. Translocase assay in permeabilized fibroblasts. J. Clin. Invest. 91, 1247–1252. doi: 10.1172/JCI116288
Paradkar, P. N., Zumbrennen, K. B., Paw, B. H., Ward, D. M., and Kaplan, J. (2009). Regulation of mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin 2. Mol. Cell Biol. 29, 1007–1016. doi: 10.1128/MCB.01685-08
Parker, M. D., and Boron, W. F. (2013). The divergence, actions, roles, and relatives of sodium-coupled bicarbonate transporters. Physiol. Rev. 93, 803–959. doi: 10.1152/physrev.00023.2012
Pebay-Peyroula, E., Dahout-Gonzalez, C., Kahn, R., Trézéguet, V., Lauquin, G. J., and Brandolin, G. (2003). Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature 426, 39–44. doi: 10.1038/nature02056
Perez-Martinez, X., Broadley, S. A., and Fox, T. D. (2003). Mss51p promotes mitochondrial Cox1p synthesis and interacts with newly synthesized Cox1p. EMBO J. 22, 5951–5961. doi: 10.1093/emboj/cdg566
Perez-Martinez, X., Butler, C. A., Shingu-Vazquez, M., and Fox, T. D. (2009). Dual functions of Mss51 couple synthesis of Cox1 to assembly of cytochrome c oxidase in Saccharomyces cerevisiae mitochondria. Mol. Biol. Cell 20, 4371–4380. doi: 10.1091/mbc.E09-06-0522
Perland, E., and Fredriksson, R. (2017). Classification systems of secondary active transporters. Trends Pharmacol. Sci. 38, 305–315. doi: 10.1016/j.tips.2016.11.008
Pfeiffer, K., Gohil, V., Stuart, R. A., Hunte, C., Brandt, U., Greenberg, M. L., et al. (2003). Cardiolipin stabilizes respiratory chain supercomplexes. J. Biol. Chem. 278, 52873–52880. doi: 10.1074/jbc.M308366200
Poduri, A., Heinzen, E. L., Chitsazzadeh, V., Lasorsa, F. M., Elhosary, P. C., Lacoursiere, C. M., et al. (2013). SLC25A22 is a novel gene for migrating partial seizures in infancy. Ann. Neurol. 74, 873–882. doi: 10.1002/ana.23998
Ponka, P. (1997). Tissue-specific regulation of iron metabolism and heme synthesis: distinct control mechanisms in erythroid cells. Blood 89, 1–25.
Porter, R. K. (2008). Uncoupling protein 1: a short-circuit in the chemiosmotic process. J. Bioenerg. Biomembr. 40, 457–461. doi: 10.1007/s10863-008-9172-8
Porter, R. K. (2012). Studies on the function and regulation of mitochondrial uncoupling proteins. Adv. Exp. Med. Biol. 748, 171–184. doi: 10.1007/978-1-4614-3573-0_7
Postis, V., De Marcos Lousa, C., Arnou, B., Lauquin, G. J., and Trézéguet, V. (2005). Subunits of the yeast mitochondrial ADP/ATP carrier: cooperation within the dimer. Biochemistry 44, 14732–14740. doi: 10.1021/bi051648x
Pramod, A. B., Foster, J., Carvelli, L., and Henry, L. K. (2013). SLC6 transporters: structure, function, regulation, disease association and therapeutics. Mol. Aspects Med. 34, 197–219. doi: 10.1016/j.mam.2012.07.002
Pushkin, A., and Kurtz, I. (2006). SLC4 base (HCO3-, CO32-) transporters: classification, function, structure, genetic diseases, and knockout models. Am. J. Physiol. Renal Physiol. 290, F580–F599. doi: 10.1152/ajprenal.00252.2005
Quednau, B. D., Nicoll, D. A., and Philipson, K. D. (2004). The sodium/calcium exchanger family-SLC8. Pflugers Arch. 447, 543–548. doi: 10.1007/s00424-003-1065-4
Ramsden, D. B., Ho, P. W., Ho, J. W., Liu, H. F., So, D. H., Tse, H. M., et al. (2012). Human neuronal uncoupling proteins 4 and 5 (UCP4 and UCP5): structural properties, regulation, and physiological role in protection against oxidative stress and mitochondrial dysfunction. Brain Behav. 2, 468–478. doi: 10.1002/brb3.55
Rodić, N., Oka, M., Hamazaki, T., Murawski, M. R., Jorgensen, M., Maatouk, D. M., et al. (2005). DNA methylation is required for silencing of ANt4, an adenine nucleotide translocase selectively expressed in mouse embryonic stem cells and germ cells. Stem Cells 23, 1314–1323. doi: 10.1634/stemcells.2005-0119
Romero, M. F. (2005). Molecular pathophysiology of SLC4 bicarbonate transporters. Curr. Opin. Nephrol. Hypertens 14, 495–501. doi: 10.1097/01.mnh.0000168333.01831.2c
Romero, M. F., Chen, A. P., Parker, M. D., and Boron, W. F. (2013). The SLC4 family of bicarbonate (HCO3-) transporters. Mol. Aspects Med. 34, 159–182. doi: 10.1016/j.mam.2012.10.008
Rosenberg, M. J., Agarwala, R., Bouffard, G., Davis, J., Fiermonte, G., Hilliard, M. S., et al. (2002). Mutant deoxynucleotide carrier is associated with congenital microcephaly. Nat. Genet. 32, 175–179. doi: 10.1038/ng948
Rosenthal, E. A., Ranchalis, J., Crosslin, D. R., Burt, A., Brunzell, J. D., Motulsky, A. G., et al. (2013). Joint linkage and association analysis with exome sequence data implicates SLC25A40 in hypertriglyceridemia. Am. J. Hum. Genet. 93, 1035–1045. doi: 10.1016/j.ajhg.2013.10.019
Ruprecht, J. J., Hellawell, A. M., Harding, M., Crichton, P. G., Mccoy, A. J., and Kunji, E. R. (2014). Structures of yeast mitochondrial ADP/ATP carriers support a domain-based alternating-access transport mechanism. Proc. Natl. Acad. Sci. U.S.A. 111, E426–E434. doi: 10.1073/pnas.1320692111
Sabová, L., Zeman, I., Supek, F., and Kolarov, J. (1993). Transcriptional control of AAC3 gene encoding mitochondrial ADP/ATP translocator in Saccharomyces cerevisiae by oxygen, heme and ROX1 factor. Eur. J. Biochem. 213, 547–553. doi: 10.1111/j.1432-1033.1993.tb17793.x
Salvi, S., Santorelli, F. M., Bertini, E., Boldrini, R., Meli, C., Donati, A., et al. (2001). Clinical and molecular findings in hyperornithinemia-hyperammonemia-homocitrullinuria syndrome. Neurology 57, 911–914. doi: 10.1212/WNL.57.5.911
Sanchis, D., Fleury, C., Chomiki, N., Goubern, M., Huang, Q., Neverova, M., et al. (1998). BMCP1, a novel mitochondrial carrier with high expression in the central nervous system of humans and rodents, and respiration uncoupling activity in recombinant yeast. J. Biol. Chem. 273, 34611–34615. doi: 10.1074/jbc.273.51.34611
Schäfer, E., Dencher, N. A., Vonck, J., and Parcej, D. N. (2007). Three-dimensional structure of the respiratory chain supercomplex I1III2IV1 from bovine heart mitochondria. Biochemistry 46, 12579–12585. doi: 10.1021/bi700983h
Schägger, H., and Pfeiffer, K. (2000). Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 19, 1777–1783. doi: 10.1093/emboj/19.8.1777
Schägger, H., and von Jagow, G. (1991). Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal. Biochem. 199, 223–231. doi: 10.1016/0003-2697(91)90094-A
Schiff, M., Veauville-Merllié, A., Su, C. H., Tzagoloff, A., Rak, M., Ogier De Baulny, H., et al. (2016). SLC25A32 mutations and riboflavin-responsive exercise intolerance. N. Engl. J. Med. 374, 795–797. doi: 10.1056/NEJMc1513610
Schroers, A., Burkovski, A., Wohlrab, H., and Krämer, R. (1998). The phosphate carrier from yeast mitochondria. Dimerization is a prerequisite for function. J. Biol. Chem. 273, 14269–14276. doi: 10.1074/jbc.273.23.14269
Schweikhard, E. S., and Ziegler, C. M. (2012). Amino acid secondary transporters: toward a common transport mechanism. Curr. Top. Membr. 70, 1–28. doi: 10.1016/B978-0-12-394316-3.00001-6
Seifert, E. L., Ligeti, E., Mayr, J. A., Sondheimer, N., and Hajnóczky, G. (2015). The mitochondrial phosphate carrier: role in oxidative metabolism, calcium handling and mitochondrial disease. Biochem. Biophys. Res. Commun. 464, 369–375. doi: 10.1016/j.bbrc.2015.06.031
Shamseldin, H. E., Smith, L. L., Kentab, A., Alkhalidi, H., Summers, B., Alsedairy, H., et al. (2016). Mutation of the mitochondrial carrier SLC25A42 causes a novel form of mitochondrial myopathy in humans. Hum. Genet. 135, 21–30. doi: 10.1007/s00439-015-1608-8
Shaw, G. C., Cope, J. J., Li, L., Corson, K., Hersey, C., Ackermann, G. E., et al. (2006). Mitoferrin is essential for erythroid iron assimilation. Nature 440, 96–100. doi: 10.1038/nature04512
Shayakul, C., Clémençon, B., and Hediger, M. A. (2013). The urea transporter family (SLC14): physiological, pathological and structural aspects. Mol. Aspects Med. 34, 313–322. doi: 10.1016/j.mam.2012.12.003
Shayakul, C., and Hediger, M. A. (2004). The SLC14 gene family of urea transporters. Pflugers Arch. 447, 603–609. doi: 10.1007/s00424-003-1124-x
Siciliano, G., Tessa, A., Petrini, S., Mancuso, M., Bruno, C., Grieco, G. S., et al. (2003). Autosomal dominant external ophthalmoplegia and bipolar affective disorder associated with a mutation in the ANT1 gene. Neuromuscul. Disord. 13, 162–165. doi: 10.1016/S0960-8966(02)00221-3
Siep, M., Van Oosterum, K., Neufeglise, H., Van Der Spek, H., and Grivell, L. A. (2000). Mss51p, a putative translational activator of cytochrome c oxidase subunit-1 (COX1) mRNA, is required for synthesis of Cox1p in Saccharomyces cerevisiae. Curr. Genet. 37, 213–220. doi: 10.1007/s002940050522
Silva, J. P., Shabalina, I. G., Dufour, E., Petrovic, N., Backlund, E. C., Hultenby, K., et al. (2005). SOD2 overexpression: enhanced mitochondrial tolerance but absence of effect on UCP activity. EMBO J. 24, 4061–4070. doi: 10.1038/sj.emboj.7600866
Simpson, I. A., Dwyer, D., Malide, D., Moley, K. H., Travis, A., and Vannucci, S. J. (2008). The facilitative glucose transporter GLUT3: 20 years of distinction. Am. J. Physiol. Endocrinol. Metab. 295, E242–E253. doi: 10.1152/ajpendo.90388.2008
Smits, P., Smeitink, J., and Van Den Heuvel, L. (2010). Mitochondrial translation and beyond: processes implicated in combined oxidative phosphorylation deficiencies. J. Biomed. Biotechnol. 2010:737385. doi: 10.1155/2010/737385
Smorodchenko, A., Rupprecht, A., Fuchs, J., Gross, J., and Pohl, E. E. (2011). Role of mitochondrial uncoupling protein 4 in rat inner ear. Mol. Cell. Neurosci. 47, 244–253. doi: 10.1016/j.mcn.2011.03.002
Smorodchenko, A., Rupprecht, A., Sarilova, I., Ninnemann, O., Bräuer, A. U., Franke, K., et al. (2009). Comparative analysis of uncoupling protein 4 distribution in various tissues under physiological conditions and during development. Biochim. Biophys. Acta 1788, 2309–2319. doi: 10.1016/j.bbamem.2009.07.018
Soto, I. C., Fontanesi, F., Liu, J., and Barrientos, A. (2012). Biogenesis and assembly of eukaryotic cytochrome c oxidase catalytic core. Biochim. Biophys. Acta 1817, 883–897. doi: 10.1016/j.bbabio.2011.09.005
Spiegel, R., Shaag, A., Edvardson, S., Mandel, H., Stepensky, P., Shalev, S. A., et al. (2009). SLC25A19 mutation as a cause of neuropathy and bilateral striatal necrosis. Ann. Neurol. 66, 419–424. doi: 10.1002/ana.21752
Sreedhar, A., Petruska, P., Miriyala, S., Panchatcharam, M., and Zhao, Y. (2017). UCP2 overexpression enhanced glycolysis via activation of PFKFB2 during skin cell transformation. Oncotarget 8, 95504–95515. doi: 10.18632/oncotarget.20762
Stanley, C. A., Hale, D. E., Berry, G. T., Deleeuw, S., Boxer, J., and Bonnefont, J. P. (1992). Brief report: a deficiency of carnitine-acylcarnitine translocase in the inner mitochondrial membrane. N. Engl. J. Med. 327, 19–23. doi: 10.1056/NEJM199207023270104
Steffen, J., Vashisht, A. A., Wan, J., Jen, J. C., Claypool, S. M., Wohlschlegel, J. A., et al. (2017). Rapid degradation of mutant SLC25A46 by the ubiquitin-proteasome system results in MFN1/2-mediated hyperfusion of mitochondria. Mol. Biol. Cell 28, 600–612. doi: 10.1091/mbc.e16-07-0545
Stepien, G., Torroni, A., Chung, A. B., Hodge, J. A., and Wallace, D. C. (1992). Differential expression of adenine nucleotide translocator isoforms in mammalian tissues and during muscle cell differentiation. J. Biol. Chem. 267, 14592–14597.
Tamamori, A., Okano, Y., Ozaki, H., Fujimoto, A., Kajiwara, M., Fukuda, K., et al. (2002). Neonatal intrahepatic cholestasis caused by citrin deficiency: severe hepatic dysfunction in an infant requiring liver transplantation. Eur. J. Pediatr. 161, 609–613. doi: 10.1007/s00431-002-1045-2
Taylor, E. B. (2017). Functional properties of the mitochondrial carrier system. Trends Cell Biol. 27, 633–644. doi: 10.1016/j.tcb.2017.04.004
Tazawa, Y., Kobayashi, K., Ohura, T., Abukawa, D., Nishinomiya, F., Hosoda, Y., et al. (2001). Infantile cholestatic jaundice associated with adult-onset type II citrullinemia. J. Pediatr. 138, 735–740. doi: 10.1067/mpd.2001.113264
Terzenidou, M. E., Segklia, A., Kano, T., Papastefanaki, F., Karakostas, A., Charalambous, M., et al. (2017). Novel insights into SLC25A46-related pathologies in a genetic mouse model. PLoS Genet. 13:e1006656. doi: 10.1371/journal.pgen.1006656
Tessa, A., Fiermonte, G., Dionisi-Vici, C., Paradies, E., Baumgartner, M. R., Chien, Y. H., et al. (2009). Identification of novel mutations in the SLC25A15 gene in hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome: a clinical, molecular, and functional study. Hum. Mutat. 30, 741–748. doi: 10.1002/humu.20930
Thompson, K., Majd, H., Dallabona, C., Reinson, K., King, M. S., Alston, C. L., et al. (2016). Recurrent de novo dominant mutations in SLC25A4 cause severe early-onset mitochondrial disease and loss of mitochondrial DNA copy number. Am. J. Hum. Genet. 99, 860–876. doi: 10.1016/j.ajhg.2016.11.001
Towpik, J. (2005). Regulation of mitochondrial translation in yeast. Cell. Mol. Biol. Lett. 10, 571–594.
Traba, J., Froschauer, E. M., Wiesenberger, G., Satrústegui, J., and Del Arco, A. (2008). Yeast mitochondria import ATP through the calcium-dependent ATP-Mg/Pi carrier Sal1p, and are ATP consumers during aerobic growth in glucose. Mol. Microbiol. 69, 570–585. doi: 10.1111/j.1365-2958.2008.06300.x
Traba, J., Satrústegui, J., and Del Arco, A. (2009). Characterization of SCaMC-3-like/SLC25A41, a novel calcium-independent mitochondrial ATP-Mg/Pi carrier. Biochem. J. 418, 125–133. doi: 10.1042/bj20081262
Trézéguet, V., Le Saux, A., David, C., Gourdet, C., Fiore, C., Dianoux, A., et al. (2000). A covalent tandem dimer of the mitochondrial ADP/ATP carrier is functional in vivo. Biochim. Biophys. Acta 1457, 81–93. doi: 10.1016/S0005-2728(99)00115-2
Tsujino, S., Kanazawa, N., Ohashi, T., Eto, Y., Saito, T., Kira, J., et al. (2000). Three novel mutations (G27E, insAAC, R179X) in the ORNT1 gene of Japanese patients with hyperornithinemia, hyperammonemia, and homocitrullinuria syndrome. Ann. Neurol. 47, 625–631. doi: 10.1002/1531-8249(200005)47:5<625::AID-ANA10>3.0.CO;2-Q
Uldry, M., and Thorens, B. (2004). The SLC2 family of facilitated hexose and polyol transporters. Pflugers Arch. 447, 480–489. doi: 10.1007/s00424-003-1085-0
Van De Parre, T. J., Martinet, W., Verheye, S., Kockx, M. M., Van Langenhove, G., Herman, A. G., et al. (2008). Mitochondrial uncoupling protein 2 mediates temperature heterogeneity in atherosclerotic plaques. Cardiovasc. Res. 77, 425–431. doi: 10.1093/cvr/cvm003
Verrey, F., Closs, E. I., Wagner, C. A., Palacin, M., Endou, H., and Kanai, Y. (2004). CATs and HATs: the SLC7 family of amino acid transporters. Pflugers Arch. 447, 532–542. doi: 10.1007/s00424-003-1086-z
Vest, K. E., Leary, S. C., Winge, D. R., and Cobine, P. A. (2013). Copper import into the mitochondrial matrix in Saccharomyces cerevisiae is mediated by Pic2, a mitochondrial carrier family protein. J. Biol. Chem. 288, 23884–23892. doi: 10.1074/jbc.M113.470674
Vidal-Puig, A., Solanes, G., Grujic, D., Flier, J. S., and Lowell, B. B. (1997). UCP3: an uncoupling protein homologue expressed preferentially and abundantly in skeletal muscle and brown adipose tissue. Biochem. Biophys. Res. Commun. 235, 79–82. doi: 10.1006/bbrc.1997.6740
Vidal-Puig, A. J., Grujic, D., Zhang, C. Y., Hagen, T., Boss, O., Ido, Y., et al. (2000). Energy metabolism in uncoupling protein 3 gene knockout mice. J. Biol. Chem. 275, 16258–16266. doi: 10.1074/jbc.M910179199
Visser, W. F., Van Roermund, C. W., Waterham, H. R., and Wanders, R. J. (2002). Identification of human PMP34 as a peroxisomal ATP transporter. Biochem. Biophys. Res. Commun. 299, 494–497. doi: 10.1016/S0006-291X(02)02663-3
Vozza, A., Parisi, G., De Leonardis, F., Lasorsa, F. M., Castegna, A., Amorese, D., et al. (2014). UCP2 transports C4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc. Natl. Acad. Sci. U.S.A. 111, 960–965. doi: 10.1073/pnas.1317400111
Waldherr, M., Ragnini, A., Jank, B., Teply, R., Wiesenberger, G., and Schweyen, R. J. (1993). A multitude of suppressors of group II intron-splicing defects in yeast. Curr. Genet. 24, 301–306. doi: 10.1007/BF00336780
Walker, S. C., and Engelke, D. R. (2008). A protein-only RNase P in human mitochondria. Cell 135, 412–414. doi: 10.1016/j.cell.2008.10.010
Wan, J., Steffen, J., Yourshaw, M., Mamsa, H., Andersen, E., Rudnik-Schöneborn, S., et al. (2016). Loss of function of SLC25A46 causes lethal congenital pontocerebellar hypoplasia. Brain 139, 2877–2890. doi: 10.1093/brain/aww212
Wibom, R., Lasorsa, F. M., Töhönen, V., Barbaro, M., Sterky, F. H., Kucinski, T., et al. (2009). AGC1 deficiency associated with global cerebral hypomyelination. N. Engl. J. Med. 361, 489–495. doi: 10.1056/NEJMoa0900591
Wohlrab, H., and Flowers, N. (1982). pH gradient-dependent phosphate transport catalyzed by the purified mitochondrial phosphate transport protein. J. Biol. Chem. 257, 28–31.
Wright, E. M. (2013). Glucose transport families SLC5 and SLC50. Mol. Aspects Med. 34, 183–196. doi: 10.1016/j.mam.2012.11.002
Writzl, K., Maver, A., Kovačič, L., Martinez-Valero, P., Contreras, L., Satrustegui, J., et al. (2017). De novo mutations in SLC25A24 cause a disorder characterized by early aging, bone dysplasia, characteristic face, and early demise. Am. J. Hum. Genet. 101, 844–855. doi: 10.1016/j.ajhg.2017.09.017
Wu, M., Gu, J., Guo, R., Huang, Y., and Yang, M. (2016). Structure of mammalian respiratory supercomplex I1III2IV1. Cell 167, 1598.e–1609.e. doi: 10.1016/j.cell.2016.11.012
Xu, W., Barrientos, T., and Andrews, N. C. (2013). Iron and copper in mitochondrial diseases. Cell Metab. 17, 319–328. doi: 10.1016/j.cmet.2013.02.004
Yasuda, T., Yamaguchi, N., Kobayashi, K., Nishi, I., Horinouchi, H., Jalil, M. A., et al. (2000). Identification of two novel mutations in the SLC25A13 gene and detection of seven mutations in 102 patients with adult-onset type II citrullinemia. Hum. Genet. 107, 537–545. doi: 10.1007/s004390000430
Zhang, C. Y., Baffy, G., Perret, P., Krauss, S., Peroni, O., Grujic, D., et al. (2001). Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, beta cell dysfunction, and type 2 diabetes. Cell 105, 745–755. doi: 10.1016/S0092-8674(01)00378-6
Zhang, M., Mileykovskaya, E., and Dowhan, W. (2002). Gluing the respiratory chain together. Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J. Biol. Chem. 277, 43553–43556. doi: 10.1074/jbc.C200551200
Keywords: ADP/ATP carrier, cytochrome c oxidase, mitochondrial carrier family, mitochondrial translation, respiratory supercomplexes, solute carrier family
Citation: Ogunbona OB and Claypool SM (2019) Emerging Roles in the Biogenesis of Cytochrome c Oxidase for Members of the Mitochondrial Carrier Family. Front. Cell Dev. Biol. 7:3. doi: 10.3389/fcell.2019.00003
Received: 01 November 2018; Accepted: 10 January 2019;
Published: 31 January 2019.
Edited by:
Atan Gross, Weizmann Institute of Science, IsraelReviewed by:
Flavia Fontanesi, University of Miami, United StatesAraceli Del Arco, University of Castilla–La Mancha, Spain
Copyright © 2019 Ogunbona and Claypool. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Steven M. Claypool, c2NsYXlwbzFAamhtaS5lZHU=