 Alice Turdo1
Alice Turdo1 Veronica Veschi1
Veronica Veschi1 Miriam Gaggianesi1
Miriam Gaggianesi1 Aurora Chinnici1
Aurora Chinnici1 Paola Bianca1
Paola Bianca1 Matilde Todaro2
Matilde Todaro2 Giorgio Stassi1*
Giorgio Stassi1*- 1Department of Surgical, Oncological and Stomatological Sciences, University of Palermo, Palermo, Italy
- 2Department of PROMISE, University of Palermo, Palermo, Italy
Notwithstanding cancer patients benefit from a plethora of therapeutic alternatives, drug resistance remains a critical hurdle. Indeed, the high mortality rate is associated with metastatic disease, which is mostly incurable due to the refractoriness of metastatic cells to current treatments. Increasing data demonstrate that tumors contain a small subpopulation of cancer stem cells (CSCs) able to establish primary tumor and metastasis. CSCs are endowed with multiple treatment resistance capabilities comprising a highly efficient DNA damage repair machinery, the activation of survival pathways, enhanced cellular plasticity, immune evasion and the adaptation to a hostile microenvironment. Due to the presence of distinct cell populations within a tumor, cancer research has to face the major challenge of targeting the intra-tumoral as well as inter-tumoral heterogeneity. Thus, targeting molecular drivers operating in CSCs, in combination with standard treatments, may improve cancer patients’ outcomes, yielding long-lasting responses. Here, we report a comprehensive overview on the most significant therapeutic advances that have changed the known paradigms of cancer treatment with a particular emphasis on newly developed compounds that selectively affect the CSC population. Specifically, we are focusing on innovative therapeutic approaches including differentiation therapy, anti-angiogenic compounds, immunotherapy and inhibition of epigenetic enzymes and microenvironmental cues.
Cancer Stem Cells as a Main Determinant of Therapy Refractoriness
Cancer stem cells (CSCs) are defined as being a subpopulation of cells within the heterogeneous tumor mass. This subset of cells is endowed with the ability to self-renew and differentiate into non-CSCs, indicating their capability of reproducing the tumor of origin when transplanted into immunocompromised mice. CSCs are also considered responsible for the metastatic spreading and chemoresistance. Strong evidence suggests that conventional treatments, including radio- and chemotherapy, spare the CSC subset, which is responsible for minimal residual disease (MRD) and cancer relapse (Valent et al., 2012). Indeed, CSCs are characterized by more pronounced levels of drug transporters, enhanced DNA-damage repair mechanisms and the ability to escape the cytotoxic chemotherapy by maintaining a quiescent state. New emerging therapeutic approaches using immunotherapy, anti-angiogenic compounds and/or epigenetic probes aim to overcome the CSC resistance to treatments. CSCs have been thoroughly investigated in the past decades, starting in 1971 when they were observed by Perce and Wallance, who described aggressive undifferentiated cells that are able to generate squamous cell carcinoma in vivo (Lobo et al., 2007). CSCs were first identified in Myeloid Leukemia in 1997 and since then they have been proposed to be the tumor initiating cells responsible for disease recurrence and metastasis formation. Bonnet and Dick identified a subpopulation of tumor initiating cells with marked stem-like properties in acute myeloid leukemia (AML). Later, several groups also identified CSCs in solid tumors, including breast, brain, thyroid, melanoma, colon, pancreatic, liver, prostate, lung, head and neck, ovarian, and stomach cancers (Lapidot et al., 1994; Bonnet and Dick, 1997; Al-Hajj et al., 2003; Hemmati et al., 2003; Singh et al., 2004; Collins et al., 2005; Ma et al., 2007; Fukuda et al., 2009; Boiko et al., 2010; Todaro et al., 2010). Based on these studies, a large number of biomarkers can be adopted to identify CSCs (Table 1).
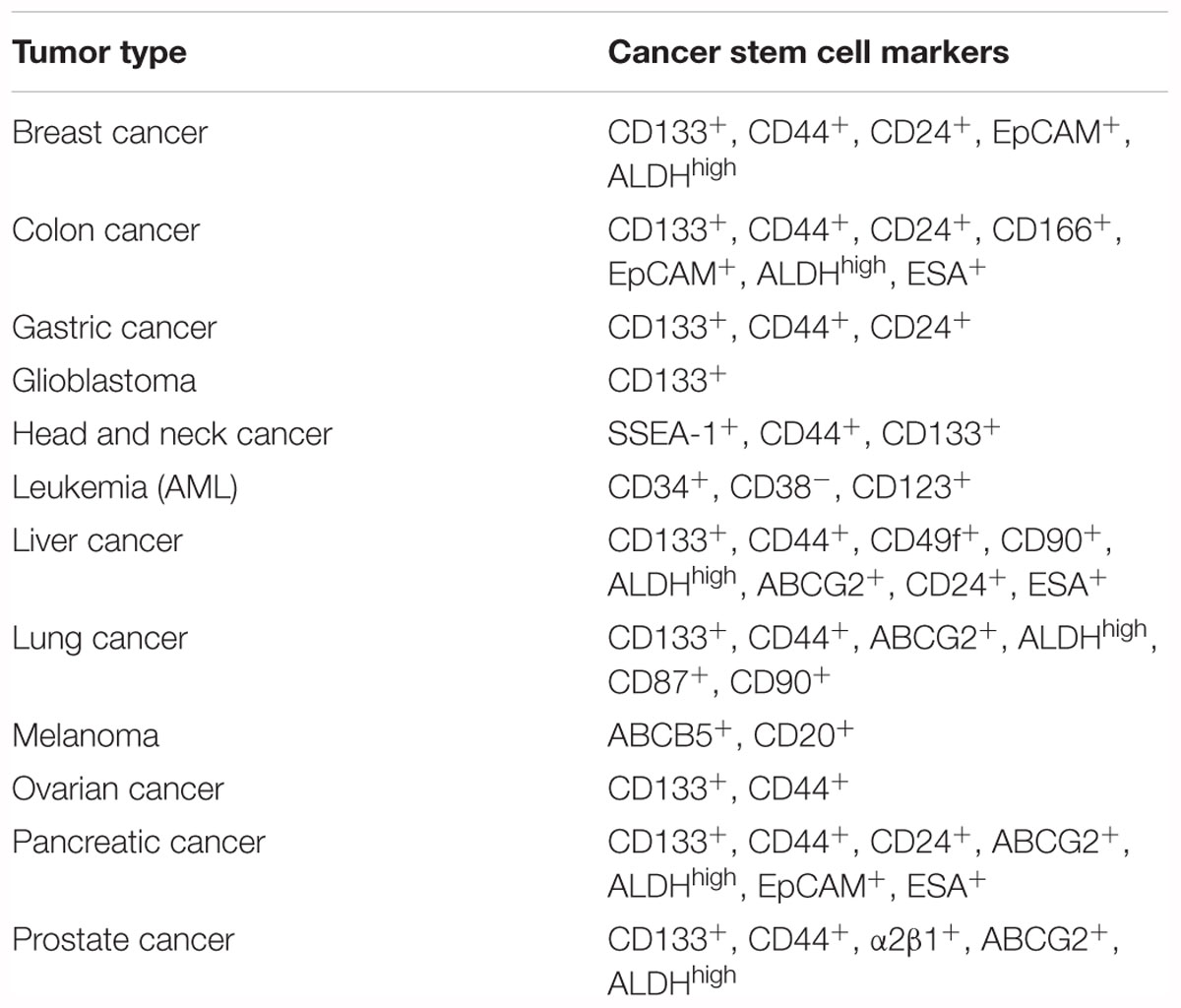
Table 1. Expression of CSCs markers according to tumor types.
Interfering With the Intrinsic Mechanisms of Therapy Resistance in CSCs
Cancer stem cells own a superior capability to survive current therapeutic regimens, meaning that chemo- and radiotherapy are not sufficient to successfully eradicate cancer and are inadequate, especially when the diagnosis occurs at a later stage (Valent et al., 2012; Ajani et al., 2015). Recent evidence showed that the CSC subpopulation is enriched after chemotherapy, suggesting that this subset is responsible for the majority of treatment failure (Visvader and Lindeman, 2008; Alison et al., 2011). Chemoresistance is favored by several mechanisms, among which cellular plasticity. Indeed, Liu et al. (2014) and Luo and Wicha (2019) demonstrated that breast CSCs can switch from proliferating epithelial characteristics to a mesenchymal state which contributes to metastatic dissemination and resistance to therapies.
Nevertheless, the resistance of CSCs to therapy is usually not limited to one drug and this phenomenon referred to as multidrug resistance (MDR) (Efferth et al., 2008). MDR is the result of the endogenous expression of detoxifying enzymes, increased drug efflux pump levels, enhanced DNA repair activity, reduced drug response and activated survival pathways (Singh and Settleman, 2010). These features, combined with the capability of CSCs to evade the immune system, to activate an epithelial to mesenchymal transition (EMT) program and to adapt their metabolism under scarce nutrient conditions, render CSCs almost an imperishable cancer population (Figure 1).
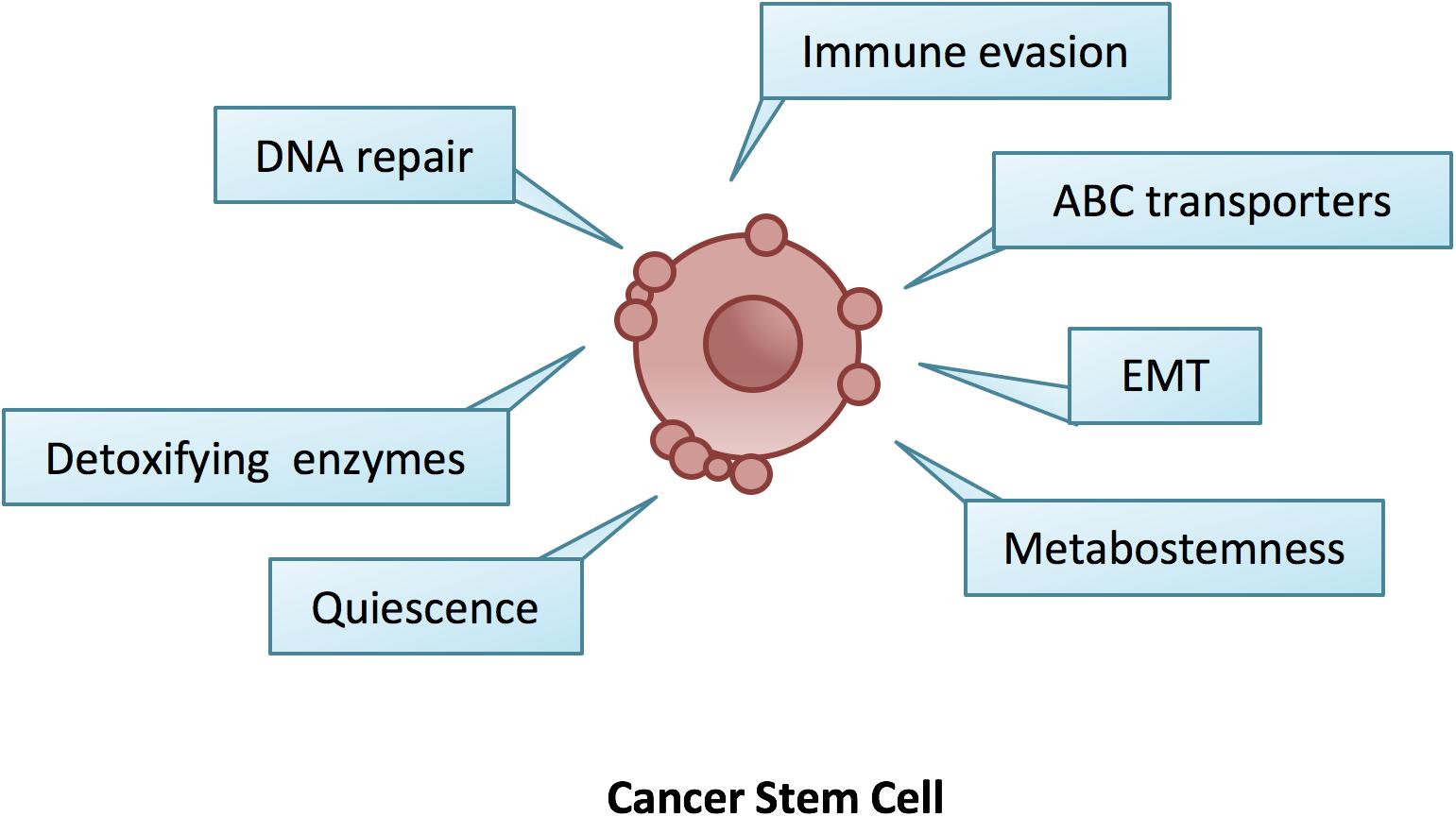
Figure 1. The hallmarks of cancer stem cells. CSCs are endowed with a number of innate ad adaptive responses such as quiescence, EMT, increased DNA repair and detoxifying enzymes, metabostemness, immune evasion and over-expression of ABC transporters, which gave them the ability to survive changes in the microenvironment and anti-cancer therapies.
The aldehyde dehydrogenase (ALDH) 1 belongs to the ALDH superfamily, which is composed by 19 enzymes (Hsu et al., 1999). ALDH1 is the main isoform that by oxidizing aldehydes to carboxylic acids and retinol to retinoic acid, allows the detoxification from drugs and reactive oxygen species (ROS) (Singh et al., 2013). ALDH is known to be expressed by normal stem cells, for this reason its activity may be an intrinsic characteristic of CSCs as well. As a result, high levels of ALDH1 activity were found in CSCs, thus representing a reliable marker for the identification of this subset (Charafe-Jauffret et al., 2010). ALDH1 positive cells showed an increased potential of forming xenograft tumors in AML and breast cancer (Cheung et al., 2007; Ginestier et al., 2007). Thereafter, ALDH1+ cells from stomach, lung, liver, head and neck, pancreas, cervix, thyroid, prostate, colon, bladder, and ovary tumors were successfully transplanted into mice (Ma et al., 2008a). The implication of the ALDH superfamily in detoxification suggests that these enzymes may have a key role in CSCs’ chemoresistance. Indeed, it has been demonstrated that ALDH expression confers resistance to several chemotherapeutic agents, such as cyclophosphamide, cisplatin, paclitaxel, docetaxel, doxorubicin, and gemcitabine in leukemia, medulloblastoma, adenocarcinoma, colon and breast cancer (Hilton, 1984; Friedman et al., 1992; Tanei et al., 2009; Duong et al., 2012). Moreover, the inhibition of ALDH activity with disulfiram, sorafenib, and sulforaphane can sensitize CSCs to therapy, providing further confirmation of ALDH role in chemoresistance (Rausch et al., 2010).
A large number of studies have demonstrated that the reduction of chemotherapy efficiency is related to an increased drug efflux from cancer cells. This is caused by the aberrant expression of a family of proteins known as ATP-binding cassette (ABC) transporters, which belong to a family of 49 molecules, usually implicated in membrane trafficking using ATP as a source of energy. Among these proteins, ABCB1 (also known as MDR1 or P-gp), ABCG2 (also known as BCRP1), ABCB5 and ABCC1 were largely studied and characterized (Leonard et al., 2003; Lobo et al., 2007). Starting from these premises, it was hypothesized that CSCs may over-express ABC transporters as compared to non-CSCs. Indeed, several groups independently demonstrated that CSCs share features with the Hoechst dye excluding side population (SP), which highly expresses efflux pumps able to induce resistance to harmful toxins and chemotherapeutic compounds (Hirschmann-Jax et al., 2004; Ho et al., 2007). ABC transporters are involved in the resistance to a wide array of drugs. In particular, it was demonstrated that ABCB1 is over-expressed in breast CSCs, causing their resistance to doxorubicin and paclitaxel, and in multiple myeloma stem cells, refractory to carfilzomib (Wright et al., 2008; Hawley et al., 2013). On the other hand, ABCG2 is responsible for the resistance of hepatocellular CSCs to 5-fluorouracile, mephedrone, and cisplatin, whereas ABCB5 was found on circulating melanoma cells resistant to doxorubicin (Frank et al., 2005; Shi et al., 2008). The inhibition of these transporters represents a useful tool to overcome CSCs’ chemoresistance. This was demonstrated by Frank et al., who targeted ABCB5 by way of a specific blocking monoclonal antibody to restore melanoma cells’ sensitivity to doxorubicin, and by Lancet group who demonstrated the sensitizing effect of zosuquidar, a P-gp inhibitor (Frank et al., 2005; Lancet et al., 2009).
The B-cell lymphoma-2 (BCL-2) family plays a pivotal role in regulating cell fate. The pro-survival proteins belonging to this family are BCL-2 itself, B-cell lymphoma extra large (BCL-xL), BCL-2-like-2 (BCL-W), BCL-2-related protein A1A (BCL-A1A), and myeloid cell leukemia sequence-1 (MCL1), whereas the pro-apoptotic molecules include BCL2-associated-X-protein (BAX) and BCL-2 homologous antagonist killer (BAK) (Kelly and Strasser, 2011). Among these molecules, BCL-2 was found over-expressed in breast CSCs, while both BCL-2 and BCL-xL were found up-regulated in leukemia CSCs (Konopleva et al., 2002; Madjd et al., 2009). The role of the BCL-2 family has been further elucidated by Strasser et al. (1990) who demonstrated that BCL-2 over-expression promotes tumorigenesis. Consequently, the inhibition of BCL-2 downstream pathways caused an increased sensitization to chemotherapy in colon and hepatocellular CSCs (Todaro et al., 2007; Ma et al., 2008b).
In vitro evidence suggests that CSCs are slow-cycling if compared to non-CSCs (Viale et al., 2009). Interestingly, quiescence makes CSCs less sensitive to cell-cycle directed therapies such as vinca alkaloids, which prevents the polarization of microtubules and taxanes, known to stabilize existing microtubules (Gascoigne and Taylor, 2009).
Chemotherapeutic agents and radiotherapy are used in clinical setting to induce DNA damage. Of note, CSCs do not respond to therapy due to increased activity of DNA repair machinery (Bao et al., 2006; Eyler et al., 2008; McCord et al., 2009; Ropolo et al., 2009). In fact, in glioma and breast CSCs, a higher phosphorylation of DNA repair proteins was observed, in particular in ATM, CHK1, and CHK2 (Eyler and Rich, 2008; Gallmeier et al., 2011; Maugeri-Sacca et al., 2011). Moreover, ovarian and lung CSCs are enriched after cisplatin treatment, a further indication that chemotherapy is limited to kill the proliferating fraction of the tumor bulk (Levina et al., 2008; Rizzo et al., 2011).
Furthermore, it has been demonstrated that chemotherapy induced damage stimulates glioblastoma multiforme and bladder CSCs to divide and thus to repopulate tumor bulk (Chen et al., 2012; Kurtova et al., 2015). On the other hand, this induced proliferation may be exploited to increase the efficacy of therapeutic regimens (Saito et al., 2010). Interestingly, the induction of CSC differentiation by using the bone morphogenic protein 4 (BMP4) renders these cells more susceptible to standard and targeted anti-cancer therapies (Lombardo et al., 2011). Furthermore, the all-trans retinoic acid is among the most common drugs used to cause differentiation of stem cells particularly in acute promyelocytic leukemia (Nowak et al., 2009). Inhibitors of epigenetic modulators such as DNA methyltransferase 1 (DNMT1), histone deacetylases (HDACs) and bromodomain and extra-terminal (BET) inhibitors have shown capabilities to function as differentiation therapies for CSCs in various tumor types (Toh et al., 2017).
Additionally, one cancer hallmark is the activation of angiogenesis, which concurs with the nurture of the tumor mass by stimulating de novo vessels formation (Hanahan and Weinberg, 2011).
Targeting the ‘Metabostemness’
Compelling evidence suggests that stem-like features can be acquired as a result of metabolic shifts, which are able to render normal stem cells or differentiated cancer cells more susceptible to epigenetic reprogramming. These cells are thus more likely to move up the cancer cell hierarchy by their expression of pluripotent genes. The metabolic insults, able to induce this reprogramming into CSCs in the context of a pre-malignant tumor, are collectively termed ‘metabostemness’ (Menendez and Alarcon, 2014). Consistently, some of the intermediates deriving from mutated metabolic enzymes, involved in glycolysis, tricarboxylic acid cycle, oxidative phosphorylation (OXPHOS) and mitochondrial fatty acid oxidation, act as oncometabolites for DNA and histones epigenetic modifications by driving tumorigenesis (Menendez and Alarcon, 2014). For this reason, targeting metabolic processes may represent a successful strategy. In particular, in most cases OXPHOS is the preferential source of energy rather than glycolysis, probably because of the low levels of glucose in tumors. Moreover, increased OXPHOS is a hallmark of resistance to chemotherapy (Lee et al., 2017). Therefore, it is not surprising that the targeting of OXPHOS via the BCL-2 inhibitor venetoclax in combination with the hypomethylating agent azacitidine was able to impair leukemia stem cells (LSCs) proliferation and metabolic activity (Jones et al., 2018; Pollyea et al., 2018). Accordingly, the OXPHOS inhibitor salinomycin was able to kill breast CSCs (Gupta et al., 2009). Interestingly, it has been shown that, during relapse, LSCs are able to rescue OXPHOS levels after amino acid depletion thanks to increased mitochondrial fatty acid oxidation (FAO) (Jones et al., 2018). FAO can also be promoted by the crosstalk with adipose tissue, which fuels LSCs metabolism by acting as a niche and promoting LSCs chemoresistance (Ye et al., 2016). In addition, the targeting of lipolysis, and in particular of COPI-Arf1 complex, was shown to be a promising tool for the eradication of CSCs in adult Drosophila (Singh et al., 2016). The crucial role of mitochondria in CSCs impelled several groups to develop therapeutic strategies aimed at their targeting (Skoda et al., 2018). Notably, mitochondrial biogenesis can be abrogated through the estrogen-related receptor α inhibitor XCT790 (Deblois and Giguere, 2011; Deblois et al., 2013), whereas their fission can be impaired thanks to the dynamin-related protein 1 (DRP1) inhibitors Mdivi-1 and P110 (Xie et al., 2015). In addition, it has been demonstrated that DRP1 activation may be promoted by the interaction between cyclooxygenase-2 (COX-2) and mitochondria. For this reason COX-2 inhibitors, resveratrol and celecoxib, were repositioned as mitochondrial fission inhibitors (Guo et al., 2015; Cilibrasi et al., 2017). Inhibitors of mitochondrial respiration were used to target pancreatic CSC subset (Sancho et al., 2016). Likewise, the inhibition of the mitochondrial complex I through the repositioning of the antidiabetic drug Metformin was recently proposed with encouraging results (Wheaton et al., 2014).
Cancer Stem Cells, Tumor Microenvironment, Angiogenesis and Metastasis: How to Disrupt This Intricate Network?
Angiogenesis is a multistep physiological process, characterized by the formation of new vessels from preexisting ones, which governs many biological activities, such as development and tissue repair. In order to maintain tissue homeostasis, angiogenesis is tightly regulated by a balance between pro- and anti-angiogenic factors (Hanahan and Folkman, 1996). In pathological conditions, such as cancer, this balance is destroyed favoring the secretion of pro-angiogenic factors. The term “tumor angiogenesis” was used for the first time by Folkman (1971) to point out the sprouting of cancer-associated neo-vessels from existing vessels that are in close proximity. Proliferating cancer cells require oxygen and high amount of nutrients, leading to the formation of hypoxic areas in the innermost part of the tumor. Under hypoxic condition, CSCs increase hypoxia-inducible factor-1 (HIF-1) expression and activate the HIF-1 pathway, enhancing the secretion of many angiogenic growth factors (Pugh and Ratcliffe, 2003; Gilbertson and Rich, 2007). In particular, high levels of vascular endothelial growth factor-A (VEGF-A) recruit VEGF receptors (VEGFRs)-expressing endothelial cells (ECs), named tip cells. After VEGF-A binding, tip cells up-regulate cell proliferation, cytoskeleton remodeling and migration pathways (MAPK, PI3K/AKT, RhoA), sprout toward tumor cells and activate the adjacent ECs (stalk cells) to form new tumor vessels (Ricciuti et al., 2017). In addition to ECs, CSCs’ secreted cytokines prime the microenvironment (tumor microenvironment, TME) and recruit myeloid cells to fuel cancer progression. In particular, cancer-associated fibroblasts (CAFs) and activated tumor-associated macrophages (TAMs) secrete high levels of metalloproteases (MMPs), growth factors and interleukins to sustain angiogenesis and to promote CSC invasion (Bhowmick et al., 2004; Crawford and Ferrara, 2009; Owen and Mohamadzadeh, 2013). Furthermore, it has been reported that de novo vessel formation may be boosted by CSCs from different tumor types; this process is termed vascular mimicry (Weis and Cheresh, 2011). It has been described that breast and glioblastoma CSCs could give rise to both ECs and pericytes supporting tumor growth and progression (Bussolati et al., 2009; Ricci-Vitiani et al., 2010; Wang et al., 2010; Cheng et al., 2013). Unlike normal vasculature, tumor vessels are tortuous and more permeable due to the lower presence of pericytes (Jain, 2005; Sawada et al., 2012). This “leakiness” reduces the capacity of chemotherapeutic agents to target cancer cells and facilitate the intravasation of metastatic cancer cells. These circulating tumor cells (CTCs) possess a CSC-like phenotype, characterized by a high expression of EMT-related genes (Burgess, 2013; Grillet et al., 2017). Although many cancer cells are able to intravasate, only few cells survive in the bloodstream and extravasate, activating a mesenchymal-epithelial transition program (Tam and Weinberg, 2013). The persistence of extravasated cells requires the presence of a favorable host microenvironment (metastatic niche) and the escape from immune cell surveillance. For these reasons, tumor cells remain in a dormant state, which can last many years, and, after the release of molecules and growth factors by the metastatic niche, they restart to proliferate and disseminate (Gao et al., 2012; Giancotti, 2013) (Figure 2).
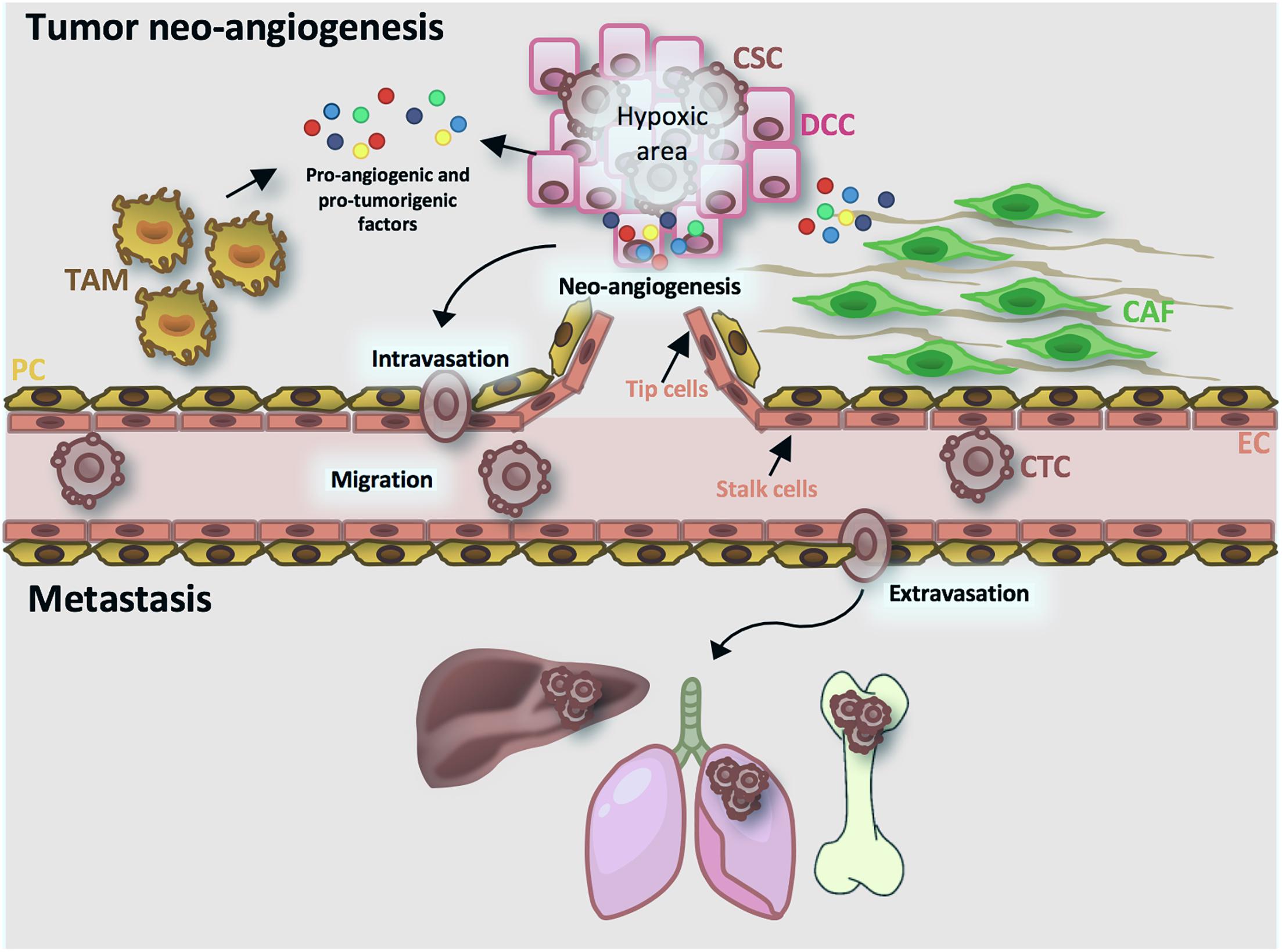
Figure 2. Tumor angiogenesis and metastatic process. Cancer cells secrete pro-angiogenic and pro-tumorigenic factors (MMPs, VEGF-A, HIF-1A, cytokines, chemokines, growth factors). VEGF-A activates endothelial cells (ECs) in tip cells, that direct the sprouting vessels, and stalk cells, implicated in vessel stability. Moreover, cancer cells-released cytokines activate cancer-associated fibroblasts (CAFs) and activated tumor-associated macrophages (TAMs), that in turn favor the intravasation of cancer stem cells (CSCs). Circulating cancer cells (CTCs) through the bloodstream reach target organ, extravasate and start to proliferate and disseminate. CSC, cancer stem cell; CTC, tumor circulating cells; DCC, differentiated cancer cell; CAF, cancer associated fibroblast; TAM, tumor associated macrophage; EC, endothelial cell; PC, pericyte cell.
Targeting Tumor Angiogenesis and Metastasis
The possibility of specifically blocking tumor angiogenesis and the metastatic process could have a clinical impact on cancer patients’ outcome. Bevacizumab is a humanized monoclonal antibody that binds to VEGF, impairing VEGF/VEGFR interaction, approved in 2004 by the Food and Drug Administration (FDA) for the treatment of metastatic colorectal cancer (CRC) in combination with standard therapy (Ferrara et al., 2004; Hurwitz et al., 2004). Then, it was approved for the treatment of other metastatic cancers, among which non-squamous NSCLC and cervical cancer (Tewari et al., 2014; Lin et al., 2016). In 2008, bevacizumab was approved for the treatment of metastatic Her2 negative breast cancer in combination with paclitaxel. However, other studies did not show a significant overall survival (OS) and the FDA withdrew the approval in 2011 for breast cancer treatment (Miller et al., 2007; Aalders et al., 2017). Conversely, the European Medicines Agency maintains bevacizumab approval in combination with chemotherapy. Another strategy to inhibit tumor angiogenesis is the use of tyrosine kinase inhibitors, such as sorafenib and sunitinib. Sorafenib is an inhibitor of VEGFR-1,-2,-3 and PDGFR-β, approved for the treatment of metastatic renal cell carcinoma and unresectable hepatocellular carcinoma (Wilhelm et al., 2006; Escudier et al., 2007), whereas sunitinib blocks VEGFR-2 and PDGFR phosphorylation and is used for gastrointestinal tumor and metastatic renal cell carcinoma (Sun et al., 2003; Motzer et al., 2007). Although anti-angiogenic therapy may potentially have clinical implication, the increase of OS is insufficient. This is probably due to (i) acquired resistance (Lu et al., 2012); (ii) the increment of tumor hypoxia (Erler et al., 2009) and (iii) the diminished delivery of chemotherapeutic agents (Jain, 2005).
Metalloproteases are crucial mediators of tumor angiogenesis and cell migration (Kessenbrock et al., 2010). Although many MMP inhibitors have been developed and many clinical trials have been conducted, none of these have increased patients’ OS (Coussens et al., 2002; Winer et al., 2018). On the contrary, MMP inhibitors have numerous side effects, due to the MMP’s role in numerous physiological processes. In order to obtain clinical benefits, inhibitors should be highly selective for MMPs that drive tumor progression.
The dysregulation of stem cell-specific signaling pathways, such as Notch, Wnt and Hedgehog, could reduce metastatic progression. In glioma patients, the use of a gamma secretase inhibitor (RO4929097) reduced CSC number; unfortunately the prolonged use of this inhibitor led to the acquisition of angiogenesis-mediated resistance (Pan et al., 2016; Xu et al., 2016). Vismodegib, an inhibitor of a component of the Hedgehog pathway, Smoothened, was used in combination with gemcitabine in pancreatic cancer, without affecting CSC number (Catenacci et al., 2015). In order to increase the efficacy of these inhibitors that target CSCs and block metastasis development, further studies must be carried out especially to reduce the side effects.
Cytokines, chemokines and growth factors secreted by TME cells enhance the migration capacity of cancer cells and promote angiogenesis (Wakefield and Hill, 2013; Scala, 2015; Gaggianesi et al., 2017). Therefore the inhibition of their receptors could have clinical benefits. In fact, reparixin, an inhibitor of IL-8 receptor CXCR1, reduced the breast CSC population and lung metastases (Ginestier et al., 2010) and is used in combination with paclitaxel in an ongoing clinical trial in triple negative breast cancer patients (Marcucci et al., 2016).
Harnessing the Immune System to Flush Out and Eradicate Cancer Stem Cells
The new frontier of cancer treatment is aimed at strengthening the immune system’s defenses against cancer cells. In the last decade the remarkable progress made on immunotherapy heralded an impressive novelty in the management of patients affected by a variety of cancers. The results obtained by immune-based therapies in terms of durable objective response rate exceeded expectations and it is no wonder that the scientists James Allison and Tasuku Honjo were recently awarded the 2018 Nobel prize in medicine for their pioneering discoveries in immunotherapy. Their studies were different, although based on the same principle: to fight cancer by harnessing the immune system.
Several compounds based on the inhibition of immune checkpoints have been approved by the FDA since 2011. Ever since, the most promising of these therapies have been antibodies targeting the cytotoxic T lymphocyte-associated protein 4 (CTLA-4) or the programmed cell death 1 (PD-1) pathway, administered as single therapy or in combination.
The role of CTLA-4 as a negative regulator of T cell activation was discovered in the laboratory of James Allison and Jeffrey Bluestone. To induce antitumor responses, T cells are initially activated in the lymph node in two subsequent steps (i) engagement of T cell receptor (TCR) with a tumor antigen MHC complex on antigen presenting cells (APCs) and (ii) binding of CD28 to the costimulatory molecule B7. Following T cell activation, CTLA-4 translocates from the intracellular compartment to the cells’ surface to compete with the costimulatory molecules, causing the inhibition of T cell proliferation. The blockade of this essential immune checkpoint with monoclonal antibodies enables T cells to active, expand and reach the tumor burden, where they can find the cognate antigen presented by cancer cells (Ribas and Wolchok, 2018).
Otherwise, Tasuku Honjo demonstrated that TCR engagement at the tumor site causes the expression of the PD-1 receptor that binds the PD-1 ligand (PD-L1) on cancer cells, causing the exhaustion of T cells and hampering the antitumor cytotoxic T cell responses (Okazaki et al., 2013).
These two mechanisms are generally implemented to impede the overstimulation of the immune system but in the context of cancer, they become detrimental for cancer cell elimination. Nevertheless, an immune checkpoint blockade could be exploited to potentiate the antitumor immune response.
Ipilimumab was the first CTLA-4 inhibitor that entered the clinic and was approved by the FDA in 2011. A substantial portion of advanced melanoma patients treated with ipilimumab had a durable response that was unluckily accompanied by toxicity, such as colitis and the inflammation of endocrine glands. Nivolumab and pembrolizumab were the first anti-PD-1 compounds approved by the FDA for melanoma (2014) and NSCLC (2015) followed by the approval of anti-PD-L1 antibodies, atezolizumab, avelumab, and durvalumab. Interestingly, the anti-PD-1 pathway inhibitors were approved for the first time based on their genetic background as for example, the presence of unstable microsatellite rather than the cancer type. Objective response rate was high varying from 15% for head and neck, gastroesophageal, bladder and urinary tract cancers and reaching almost 90% for Hodgkin’s disease. Of interest, an ongoing phase 2 clinical trial is assessing the optimal adaptive dosage of an ipilimumab and nivolumab combination in metastatic melanoma patients (NCT03122522). Moreover, other promising results have been achieved by preclinical studies that show a synergistic effect of anti-HER2 antibodies and immune checkpoint inhibitors in breast cancer (Su et al., 2018).
Albeit immune checkpoint inhibitors are considered the spearhead of immunotherapy against cancers that show an high mutation burden, the expected accumulation of neoantigen and the high PD-1/PD-L1 expression (Bailey et al., 2018) may not produce a greater antitumor response (Gide et al., 2018).
According to the new iRECIST criteria of tumor response following the administration of immunotherapy (Seymour et al., 2017), patients undergoing treatment resistance may experience pseudoprogression or hyperprogression, which consists in the initial increase of tumor volume followed by its decrease or in a faster progression of the disease as compared to the predicted rate, respectively (Champiat et al., 2017; Siefker-Radtke and Curti, 2018). The mechanisms of resistance to immune checkpoint inhibitors may be caused by the persistence of a subpopulation of CSCs. Indeed, the activation of transcriptomic profiles characterized by genes involved in EMT, angiogenesis and stemness causes the lack of T cell recognition and immunotherapy refractoriness (Spranger et al., 2015; Hugo et al., 2016). Indeed, CSCs can evade the immune system (Lee et al., 2016; Hsu et al., 2018) mainly due to the high expression levels of PD-L1 (Wu et al., 2017), the down-regulation of molecules involved in the presentation of the antigen to T cells (Bruttel and Wischhusen, 2014) and their capacity to promote the formation of an immune suppressive microenvironment (Jachetti et al., 2015; Sorrentino et al., 2018; Szarynska et al., 2018). On the other hand, the high levels of PD-L1 expressed by the CSCs render them potentially susceptible to treatments with checkpoint inhibitors, which can be combined with other immune-based therapies for an effective response (Figure 3).
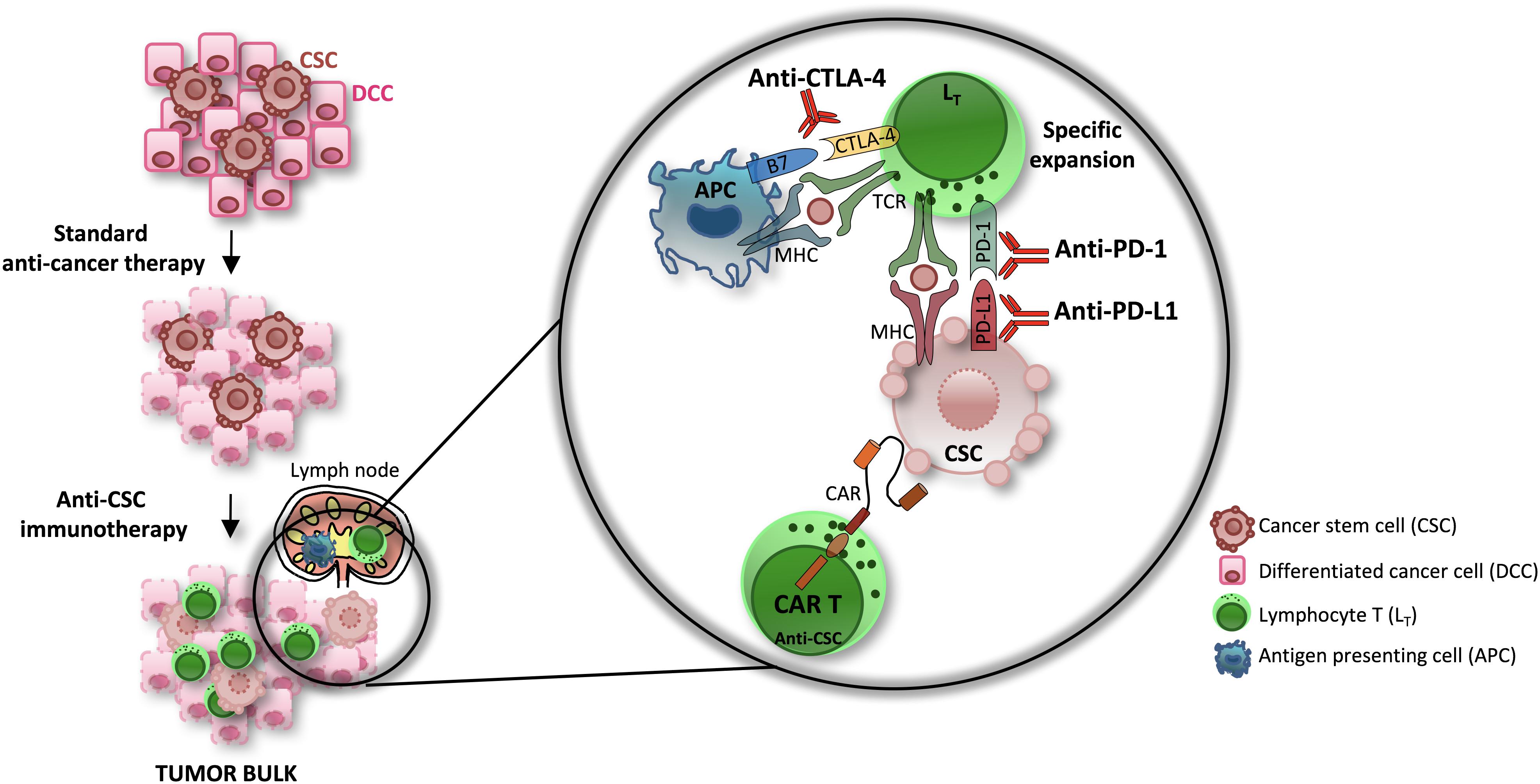
Figure 3. The efficacy of immune-based therapies in the eradication of CSCs. The standard anti-cancer therapies are able to affect differentiated cancer cells (DCCs) while sparing CSCs. Novel immunotherapy approaches have shown promising therapeutic efficacy in several type of cancers. Combinations of checkpoint inhibitors (CTLA-4 and PD-1/PD-L1 pathway inhibitors) and CAR T cell transfer, in particular, efficiently eliminate the CSCs subpopulation.
For instance, in a syngeneic melanoma mouse model, the combination of immune checkpoint inhibitors (PD-L1 inhibitors and CTLA-4 inhibitors) with CSC lysate-pulsed dendritic cells (DCs) vaccine augmented T cell antitumor response and led to tumor regression (Zheng et al., 2018).
The chimeric antigen receptor (CAR) T cell transfer is currently being investigated (Gargett et al., 2016) and holds great promise in the treatment of liquid and solid malignancies (Grupp et al., 2013; Mount et al., 2018). CAR T cells are constituted by an antigen receptor linked by a single chain fragment to an intracellular domain, usually supplemented with a co-stimulatory molecule. CAR T cells offer an exceptional substrate for the development of selective CSCs therapies, being potentially able to recognize any antigen exposed on the surface of CSCs (Guo et al., 2018). A case report has recently been described by Feng et al. (2017), which shows the efficacy of the subsequent infusion of CAR T anti-EGFR and anti-CD133, a well known marker that identifies CSCs, in a patient affected by cholangiocarcinoma. The CAR T-EGFR and CAR T-CD133 are currently under clinical evaluation (NCT01869166 and NCT02541370). Moreover, CAR T cells, which target the CSC marker EpCAM, reduced prostate cancer progression in preclinical models (Deng et al., 2015). Thus, the CAR T cell-based therapies offer the opportunity to specifically eliminate the CSC subpopulation and are a valid alternative to checkpoint inhibitors, in a subset of cancer with paucity of neoantigens expression. Additionally, CAR T cells transfer could strengthen the efficacy of CTLA-4/PD-1 pathway inhibitors and targeted therapies.
Hence, contrary to the targeted therapies, which are almost mutation-related and could induce the reactivation of alternative survival pathways, immunotherapy offers the opportunity to achieve long-lasting responses in a broad range of tumor types, by overcoming the highly adaptive behavior of CSCs.
Epigenetic Reprogramming and Cancer Stem Cells
Dynamic epigenetic reprogramming of the CSC subpopulation adds a further layer of inter- and intra-tumor heterogeneity to the complexity of tumors, which represents a hurdle for successful therapies. Epigenetics is the study of heritable changes and phenotypes not encoded in DNA (Dawson, 2017). The epigenetic enzymes responsible for histone modifications (writers, erasers, and readers) and DNA methylation (DNMT) have been extensively described (Arrowsmith et al., 2012). The histone methylation and acetylation are catalyzed by histone methyltransferases (HMTs) and histone acetyltransferases (HATs), while the histone demethylation and deacetylation are catalyzed by the histone demethylases (HDMs) and HDACs, respectively. Acetylated histones tend to be less compact and more accessible to RNA polymerase and transcriptional machinery, thereby enabling the transcription of nearby genes. Methylated histones can be either repressive or activating, depending on the site and degree of methylation. In particular, histone H3/H4 acetylation (H3Ac, H4Ac) and H3 lysine 4 methylation (H3K4me) are generally associated with active transcription, while histone H3 lysine 9 and 27 methylation (H3K9me, H3K27me) are commonly linked to gene repression (Bird, 1986; Jones and Takai, 2001; Venters and Pugh, 2007; McCabe et al., 2009). The well-known “histone code” hypothesis is based on the knowledge that different patterns of histone modifications on each histone determine the ultimate transcriptional event, either gene expression or silencing (Strahl and Allis, 2000). Several interrelated molecular mechanisms contribute to epigenetic gene regulation, such as chromatin remodeling via ATP-dependent processes and exchange of histone variants, regulation by non-coding RNAs, methylation and related modifications of cytosines on DNA, as well as covalent modification of histones. Local chromatin state at gene promoter is governed by DNMT and posttranslational histone modifications, thus playing an essential role in transcription regulation. DNMT1, DNMT3A, and DNMT3B are responsible for the methylation of the CpG islands, CpG-dense regions that are included in the majority of human gene promoters. While the unmethylated status of CpG islands is aimed to maintain promoter chromatin in a transcriptionally permissive state, their methylation is linked to gene silencing (e.g., X-chromosome inactivation, tumor suppressor gene silencing in cancers). The chromatin remodeling complexes, including the SWI/SNF complex, are at least five families that use ATP-hydrolysis to modify chromatin structure and remodel nucleosomes. Polycomb repressive complexes (PRC1 and PRC2) are epigenetic repressors of transcriptional programs fundamental for the cell’s identity, development, differentiation and lineage specification, by catalyzing the trimethylation of histone 3 lysine 27 (H3K27me3) (Di Croce and Helin, 2013). Recently, it has been demonstrated that EZH2, the functional enzymatic component of the PRC2, is required for stable self-renewal and differentiation not only in mouse but also in human embryonic stem cells (Collinson et al., 2016).
Epigenetic alterations, including DNMT and histone modifications, are a key manifestation of the stem cells’ differentiation into various tissue subtypes. The increasing number of recently discovered mutations in epigenetic regulators has shed new light on the importance of epigenetic dysregulation in tumor initiation and in the biology of CSCs. These may originate from a deregulated epigenetic reprogramming, which leads to the loss of differentiation genes and to the reestablishment of stem cell-specific characteristics. Epigenetic mechanisms play an important role in endowing stem cell characteristics to cancer cells. This is well established in many types of cancer, as: (1) CSC markers are directly regulated by epigenetic modifications (i.e., CD133 and DCLK1) (Yi et al., 2008; Vedeld et al., 2014); (2) CSCs exhibit mutations in chromatin remodeler components (loss of function mutations of PRC2 complex and deregulation of EZH2) (van Vlerken et al., 2013); (3) EMT, which confers cells with tumor-initiating capabilities and CSC properties (Mani et al., 2008), is finely controlled by epigenetic mechanisms (Kanwar et al., 2010; Beck et al., 2015; Avgustinova and Benitah, 2016).
This link between epigenetics and CSCs suggests that epigenetic alterations may be key therapeutic targets in this abnormal subpopulation. Furthermore, the development of specific epigenetic enzymes inhibitors has been a promising area of drug discovery, due in part to the “druggability” of these critical regulators. Therefore, an extensive investigation of the epigenetic enzymatic activities that are critical for the reprogramming of CSCs toward differentiation may be crucial for the tailoring and designing of new therapeutic strategies against a variety of deadly tumors. Hence, epigenetics enzymes are fundamental in regulating survival pathways, EMT, metastatic phenotype and chemoresistance in CSCs (Figure 4).
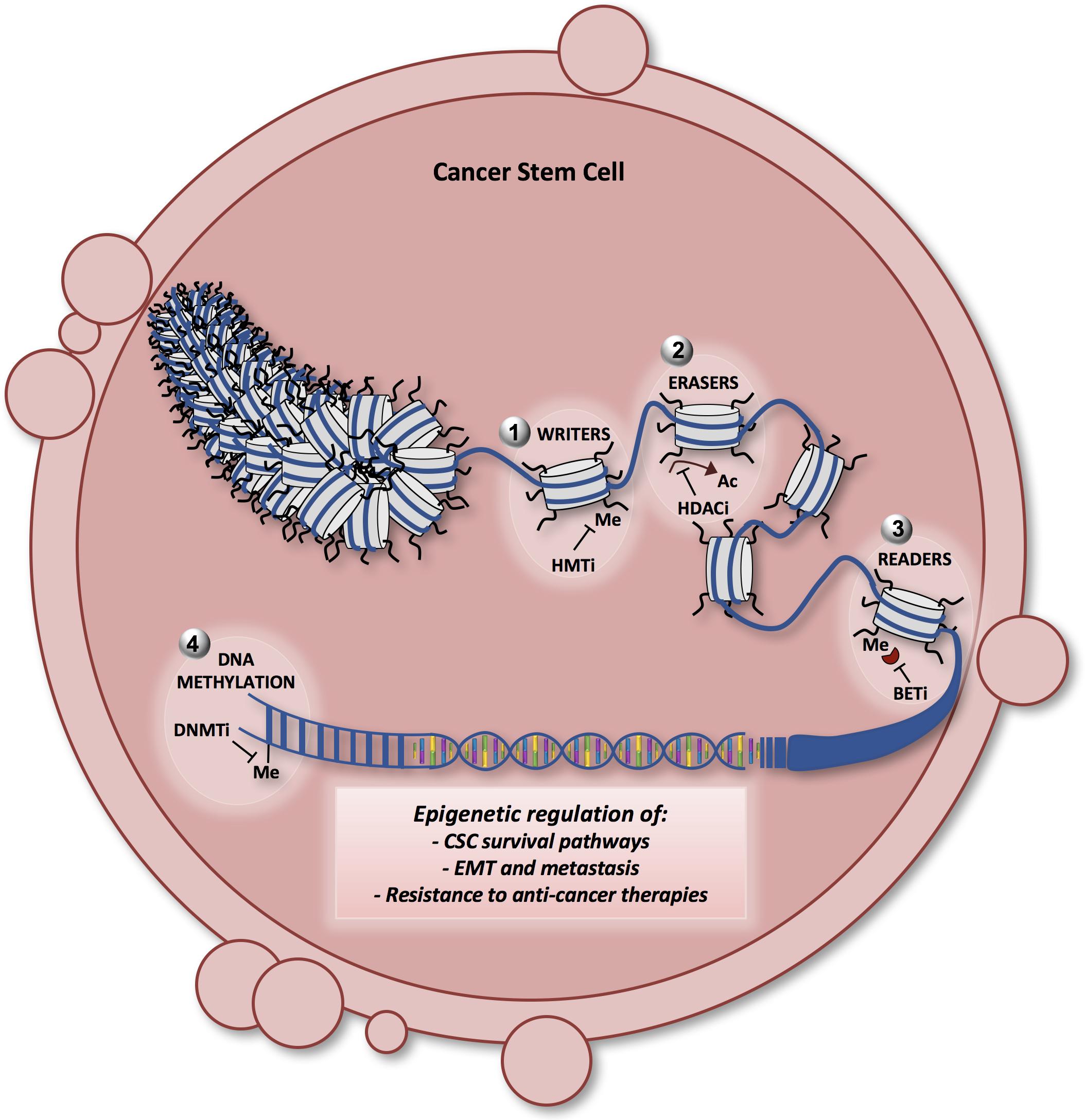
Figure 4. Model showing the different layers of epigenetic regulation in CSCs and the potential therapeutic approaches. The chromatin fiber and the nucleosome are represented in the nucleus of a cancer stem cell. Epigenetic enzymes [writers (1), erasers (2), readers (3), and DNA methyltransferases (4)] are the principal actors in regulating the key survival pathways in CSCs, such as the Notch, Wnt and Hedgehog signaling. Moreover, epigenetic alterations guide the epithelial-mesenchymal transition (EMT) and the aberrant process of metastatization in CSCs, contributing to CSC resistance to therapy. Many of the latest generation compounds (epigenetic probes) have been designed to target the epigenetic enzymes involved in the CSC survival, maintenance, EMT and metastasis. DNMTi (DNA methyltransferase inhibitors): decitabine, azacitidine; HMTi (histone methyltransferase inhibitors) such as EZH2, DOT1L, and SETD8 inhibitors; HDACi (histone deacetylase inhibitors): vorinostat, romidepsin; BETi (bromodomain inhibitors): JQ1, I-BET762. Me, methylation; Ac, acetylation.
CSC Formation and Maintenance
Of note, many epigenetic mechanisms that promote the acquisition of uncontrolled self-renewal and CSC formation are based on driver mutations that have been found in principal epigenetic regulators, in both chromatin-related driver genes and DNA-methylation-related genes (Wainwright and Scaffidi, 2017).
LSCs bear the fusion protein product of the KMT2/MLL gene. This gene encodes for a HMT involved in many biological processes. Importantly, the MLL fusion proteins have been associated with an oncogenic role due to their ability to initiate the tumorigenic process in both AML and acute lymphoblastic leukemia (ALL) cells (Cozzio et al., 2003; Krivtsov et al., 2006; Somervaille et al., 2009).
In about 33% of pediatric glioblastoma patients, gain-of-function mutations have been identified in the gene encoding for histone H3. The most represented alteration is a K27M substitution, which leads to the impaired functions of the PRC2 complex and a lack of gene repression, which in turn leads to the aberrant activation of oncogenic programs and self-renewal ability (Lewis et al., 2013). DNMTs are mutated in 25% of AML patients. These mutations hamper the enzymatic activity and leads to the propagation of pre-LSCs.
Importantly, the dynamic cooperation between the genetic and epigenetic alterations in cancer initiation and promotion has been supported by recent evidence, especially in CRC model.
DNA methyltransferases have been shown to play a key role in the initiation and progression of CRC. Many tumor suppressor gene promoters are hypermethylated in CRC (MLH1, RB, P16, RARB, SFRP). The expression and the activity of the DNMTs seem to be controlled by APC mutation, a driver event in CRC (Hammoud et al., 2013), confirming once again that genetic and epigenetic interactions may cooperate to induce tumor initiation and progression. Specifically, it has been shown that the expression levels of DNMT1 are higher in CRCs compared to normal controls, suggesting that the elevated levels of this DNA methyltransferase may determine a dysregulation in the methylome by suppressing the transcription of the tumor suppressor genes. Moreover, this supports the hypothesis that deregulation of DNMT in CR-CSCs could be a crucial event during cancer progression.
Crucial pathways involved in CSC maintenance, such as Wnt/β-catenin, Notch and Hedgehog signaling pathways are finely regulated by epigenetic mechanisms. These pathways in physiological conditions control self-renewal and development in embryonic and adult stem cells. DNMT, aberrant histone modification and also non-coding RNA have been identified as epigenetic aberrations in the main regulators of these pathways in CSCs. For instance, aberrant DNMT silences Wnt inhibitory factor genes with a tumor suppressor role, such as WIF-1, AXIN-2, SFRP-1, and DKK1 (Suzuki et al., 2004). The promoter of DKK1 is also silenced by decreased acetylation of H3K16 and increased H3K27 trimethylation (Hussain et al., 2009). In multiple myeloma cells, an enhanced histone acetylation has been found at the promoter region of JAGGED2, a Notch receptor ligand, leading to the activation of Notch signaling by overexpression of its ligand (Ghoshal et al., 2009). The histone methylation of H3K27 is inhibited on the promoters of two Notch signaling target genes, HES1 and HES5. This is accomplished by the serine-threonine kinase receptor-associated protein (STRAP), which interacts with PRC2 complex components, thus leading to gene activation in CRC. SNF5, a member of a chromatin remodeler complex SWI/SNF, binds directly Gli1, which is the down-stream effector of the Hedgehog signaling pathway, leading to a repression of the target genes transcription (42). Indeed, in human malignant rhabdoid tumors inactivation of SNF5 results in an aberrant activation of Hedgehog signaling. Moreover, HDAC1 is required to transcriptionally activate Gli1 and Gli2. However, this inhibitory mechanism is hampered by the frequent somatic mutations in REN gene, which encodes for the E3-ubiquitin ligase complex that mediates the degradation of HDAC1 (Di Marcotullio et al., 2004; Canettieri et al., 2010). Aberrant DNA hypomethylation of the Sonic Hedgehog ligand promoter is responsible for the pathway activation.
Therefore, the integration of genetic and epigenetic mechanisms disrupts the balance between self-renewal and pro-differentiation stimuli thus generating an aberrant program that sustains CSC survival.
EMT, Metastasis, and Resistance to Therapies in CSCs
The concept of CSCs in the maintenance and progression of many types of cancer is now well accepted and continues to evolve (Todaro et al., 2007, 2014; Kemper et al., 2010). This cell status is dynamic during cancer progression as it is mainly affected by genetic and epigenetic changes and influenced by the TME. Another characteristic of CSCs is their ability to invade and metastasize by acquiring the EMT phenotype that can be determined by examining the expression of E-cadherin (CDH1) and vimentin, which represent the effectors for Wnt and Notch signaling. It has been reported that Wnt/β-catenin signaling plays a critical role in regulating growth and maintenance of CR-CSCs (Kanwar et al., 2010). In particular, our group identified CD44v6 as a marker of metastatic potential that defines the CR-CSC subpopulation (Todaro et al., 2014). In CR-CSCs the up-regulation of Wnt signaling is correlated with a higher CD44v6 expression, suggesting that this population may retain metastatic traits and chemoresistance.
Many different epigenetic mechanisms have been linked to the activation of an uncontrolled EMT process. The loss of E-cadherin can be defined as a hallmark of EMT given the lack of the cell–cell adhesion. Of note, DNMT of the CDH1 promoter by recruiting HDACs to the promoter site results in histone deacetylation and transcriptional silencing. Furthermore, EZH2 and the PRC2 complex mediate the histone methylation of the CDH1 promoter, repressing its expression (Cao et al., 2008).
MiR-200 family members have been associated with a role in repressing EMT and invasion through a direct binding to ZEB1 and ZEB2 (zing finger E-box-binding homeobox 1 and 2), which are two transcription factors. Epigenetic silencing of these miRNAs by DNMT and H3K27 tri-methylation induces the acquisition of both an EMT-like and CSC phenotype (Tellez et al., 2011).
One of the most common mechanisms of drug resistance, subjected to epigenetic regulation in CSCs, is mediated by a pronounced expression of the drug efflux transporters, such as the ATP-binding cassette family (ABCG2, MDR1, MRP1). Decreased HDAC1 levels and increased histone acetylation and phosphorylation are responsible of an enhanced expression of ABCG2 (To et al., 2008).
Alternative Epigenetic Mechanisms of CSCs Regulation
On one hand, it is well known that the addition or removal of epigenetic marks on the histones of the nucleosomes play a crucial role in regulating the gene expression of oncogenic drivers or oncosuppressors (Louis and Shohet, 2015). On the other hand, oncogenes and tumor suppressors can themselves be activators of epigenetic mechanisms fundamental in CSCs by the induction of a “non-canonical” epigenetic program. Indeed, recent data have demonstrated that MYC favors a stem cell-like phenotype in mammary epithelial cells and induces an alternative epigenetic program, supported by the activation of de novo enhancers and repression of lineage-specifying transcription factors, which causes loss of cell identity and the activation of oncogenic pathways (Poli et al., 2018). Moreover, HMTs can methylate non-histone proteins such as the pivotal tumor suppressor gene TP53. It has been demonstrated that the tumor suppressor function of WT p53 is inhibited by repressive epigenetic pathways. p53 and “stemness” may be considered as conceptual antagonists. p53 suppresses self-renewal and promotes differentiation of adult stem cells. Inactivation of p53, by deletion, mutation, or expression of dominant-negative isoforms of p53 family members, enriches stem cell populations including CSCs (Molchadsky and Rotter, 2017). Some HMTs (SETD8 and SMYD2) have been found to regulate the methylation of non-histone proteins in particular p53 in lysine residues. These modifications such as the monomethylation on lysine 370 and lysine 382 of p53 (p53K370me1 and p53K382me1) have been associated with a pro-tumorigenic function (Zhu et al., 2016; Veschi et al., 2017). Further studies are needed to better elucidate these mechanisms and their targeting as a therapeutic approach in CSCs.
Treatments That Target Epigenetic Modifications in CSCs
The dynamic nature of epigenetics indicates that it may be possible to alter cancer-associated epigenetic states through direct manipulation of the molecular factors involved in this process. Currently, the major challenge in epigenetic drug discovery is to identify selective compounds with significant in vitro cellular activity at nM concentrations and well tolerated in vivo. Recently, mostly by using high throughput screening approaches, many studies identified and characterized new epigenetic regulators and their roles in various cancers. These findings represent the translational basis for the initiation of clinical trials in the area of specific epigenetic target classes. HDACs and DNMTs were the first epigenetic targets to be approved for cancer application by the FDA, but more recently additional families of epigenetic regulators have been the subject of intense studies, such as, methyltransferases (EZH2, SETD8, DOT1L, PRMT5), demethylase (LSD1, KDM4B), and BET proteins. Some potent inhibitors are now being studied in a clinical setting, more specifically in hematological and solid tumors. Early results are encouraging, despite relevant toxicity.
Histone deacetylases are key regulators of histone acetylation levels and are mostly associated with enhanced gene transcription. HDACs remove acetyl groups on histones’ lysine residues and maintain cell balance by opposing the function of the HATs. Despite promising anti-cancer data from clinical trials, HDAC inhibitors need to be considered as pan-inhibitors with associated side effects, although increasing efforts have been made to develop selective HDAC inhibitors. Vorinostat (SAHA) represents the first FDA approved pan-HDAC inhibitor that targets HDAC1-3 and 6. Currently, there are 6 clinical trials using Vorinostat targeting refractory or recurrent pediatric cancers and adult tumors. Romidepsin is an FDA approved selective HDAC1/2 inhibitor that is well tolerated in clinical trials for advanced pediatric and adult tumors (Children’s Oncology et al., 2006; Amiri-Kordestani et al., 2013). DNA demethylating or hypomethylating agents, such as DNMTs inhibitors (DNMTi, azacitidine and decitabine), are currently in clinical phases I and II for a variety of tumors, including CRC.
The chromatin readers (BET family) recruit additional chromatin modifiers and remodeling enzymes, which serve as the effectors of the modification. For instance, acetylated histones serve as docking sites for bromodomain containing proteins (Dhalluin et al., 1999; Dey et al., 2003). Thus, the histone code imparts a tertiary level of genomic control beyond the DNA sequence and corresponding transcription factors (He et al., 2013). BET inhibitors have been demonstrated to successfully target CSCs in MLL-driven ALL and in other cancers. Among the first selective and more efficacious BET inhibitors, JQ1 is able to target c-MYC in many different cancers and I-BET762 is in Phase I-II clinical trial for NUT midline carcinoma, neuroblastoma and other tumors1 (Filippakopoulos et al., 2010; Filippakopoulos and Knapp, 2014). However, nowadays many deleterious effects on healthy cells and resistance mechanisms to the BET inhibitors have been elucidated.
One of the major areas of interest regarding drug discovery is the great potential of combination therapies, especially in the case of resistance to existing standard therapy and or refractory states. Combination strategies, including pan-HDAC inhibitors in association with other agents and/or small molecules (chemotherapy, anti-GD2 antibody, retinoic acid, DNMTi, JQ1), are under evaluation in many pediatric and adult cancers. Specifically, the addition of JQ1 or EZH2 inhibitors to panobinostat (HDAC inhibitor) showed synergistic effects in vitro and in vivo (Shahbazi et al., 2016; Chen et al., 2018).
A detailed overview of the synergistic therapies with BET inhibitors and other epigenetic drugs or targeted agents can be found in Ramadoss and Mahadevan (2018). Aside from the above mentioned combination of HDAC and BET inhibitors, synergistic effects have also been demonstrated in combinatorial treatments using HDACs and DNMTs inhibitors, or DOT1L and DNMTs inhibitors in MLL-arranged leukemia cells (Klaus et al., 2014).
Concluding Remarks
In the past few years, an improved survival rates in cancer patients has been witnessed, due to early diagnosis and the advent of new targeted therapies. However, there are still millions of patients who die every year. Tumor recurrence and relapse may be driven by a variety of molecular events that are modulated according to different treatment pressure. It is now clear that within the tumor bulk there is a subpopulation of cancer cells, named CSCs, which are mainly responsible for the anti-cancer drug refractoriness. Thus, the novel frontiers of cancer treatment are aimed at defeating CSCs by using newly discovered drug delivery methods. For instance, one appealing approach is represented by the use of nanotechnology as an efficient tool for detection and elimination of CSCs (Qin et al., 2017). Nanomaterials including gold particles, origami and tetrahedron DNA nanostructures, liposomes, graphene and nanodiamond have been loaded with chemotherapeutics compounds or agents effective against CSCs, such as Salinomycin and Hedgehog pathway inhibitors (Xu et al., 2012; Yao et al., 2014). The enzymatic functionalization of nanomaterials with ligands of cell surface markers of CSCs, such as CD44 and CD133, is crucial to confer specificity in CSCs binding and targeting (Yao et al., 2014; Ni et al., 2015; Al Faraj et al., 2016). The potential clinical application of these carriers rely also on their high solubility, fast internalization and photothermal features.
The urgent need of successful cancer cures is due to the mechanisms of CSC resistance, which are disparate and act at different levels, including activation of survival pathways, metabolic adaptation, epigenetic modifications and immune escape. All these aspects have been thoroughly investigated in this review with the aim of offering an overview and food for thought on the novel developed therapeutic strategies to improve cancer patients management.
Author Contributions
AT, VV, MG, AC, and PB drafted the manuscript. MT and GS critically reviewed this manuscript.
Funding
This work was supported by grants from Associazione Italiana per la Ricerca sul Cancro (AIRC) (14415) to MT and AIRC (16746) and AIRC 5X1000 (9979) to GS. AT is a recipient of an AIRC fellowship (18000). AC is a Ph.D. student of Molecular and Experimental Medicine Ph.D. Program.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Francesco Calò for the editing of figures and Tatiana Terranova for editing and proofreading the manuscript.
Footnotes
- ^ http://www.cancer.gov/clinicaltrials NCT01587703
References
Aalders, K. C., Tryfonidis, K., Senkus, E., and Cardoso, F. (2017). Anti-angiogenic treatment in breast cancer: facts, successes, failures and future perspectives. Cancer Treat. Rev. 53, 98–110. doi: 10.1016/j.ctrv.2016.12.009
Ajani, J. A., Song, S., Hochster, H. S., and Steinberg, I. B. (2015). Cancer stem cells: the promise and the potential. Semin. Oncol. 42 (Suppl. 1), S3–S17. doi: 10.1053/j.seminoncol.2015.01.001
Al Faraj, A., Shaik, A. S., Al Sayed, B., Halwani, R., and Al Jammaz, I. (2016). Specific targeting and noninvasive imaging of breast cancer stem cells using single-walled carbon nanotubes as novel multimodality nanoprobes. Nanomedicine 11, 31–46. doi: 10.2217/nnm.15.182
Al-Hajj, M., Wicha, M. S., Benito-Hernandez, A., Morrison, S. J., and Clarke, M. F. (2003). Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. U.S.A. 100, 3983–3988. doi: 10.1073/pnas.0530291100
Alison, M. R., Lim, S. M., and Nicholson, L. J. (2011). Cancer stem cells: problems for therapy? J. Pathol. 223, 147–161. doi: 10.1002/path.2793
Amiri-Kordestani, L., Luchenko, V., Peer, C. J., Ghafourian, K., Reynolds, J., Draper, D., et al. (2013). Phase I trial of a new schedule of romidepsin in patients with advanced cancers. Clin. Cancer Res. 19, 4499–4507. doi: 10.1158/1078-0432.CCR-13-0095
Arrowsmith, C. H., Bountra, C., Fish, P. V., Lee, K., and Schapira, M. (2012). Epigenetic protein families: a new frontier for drug discovery. Nat Rev. Drug Discov. 11, 384–400. doi: 10.1038/nrd3674
Avgustinova, A., and Benitah, S. A. (2016). The epigenetics of tumour initiation: cancer stem cells and their chromatin. Curr. Opin. Genet. Dev. 36, 8–15. doi: 10.1016/j.gde.2016.01.003
Bailey, M. H., Tokheim, C., Porta-Pardo, E., Sengupta, S., Bertrand, D., Weerasinghe, A., et al. (2018). Comprehensive characterization of cancer driver genes and mutations. Cell 173, 371–385.e18. doi: 10.1016/j.cell.2018.02.060
Bao, S., Wu, Q., McLendon, R. E., Hao, Y., Shi, Q., Hjelmeland, A. B., et al. (2006). Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444, 756–760. doi: 10.1038/nature05236
Beck, B., Lapouge, G., Rorive, S., Drogat, B., Desaedelaere, K., Delafaille, S., et al. (2015). Different levels of twist1 regulate skin tumor initiation, stemness, and progression. Cell Stem Cell 16, 67–79. doi: 10.1016/j.stem.2014.12.002
Bhowmick, N. A., Neilson, E. G., and Moses, H. L. (2004). Stromal fibroblasts in cancer initiation and progression. Nature 432, 332–337. doi: 10.1038/nature03096
Bird, A. P. (1986). CpG-rich islands and the function of DNA methylation. Nature 321, 209–213. doi: 10.1038/321209a0
Boiko, A. D., Razorenova, O. V., van de Rijn, M., Swetter, S. M., Johnson, D. L., Ly, D. P., et al. (2010). Human melanoma-initiating cells express neural crest nerve growth factor receptor CD271. Nature 466, 133–137. doi: 10.1038/nature09161
Bonnet, D., and Dick, J. E. (1997). Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 3, 730–737.
Bruttel, V. S., and Wischhusen, J. (2014). Cancer stem cell immunology: key to understanding tumorigenesis and tumor immune escape? Front. Immunol. 5:360. doi: 10.3389/fimmu.2014.00360
Burgess, D. J. (2013). Breast cancer: circulating and dynamic EMT. Nat. Rev. Cancer 13:148. doi: 10.1038/nrc3475
Bussolati, B., Grange, C., Sapino, A., and Camussi, G. (2009). Endothelial cell differentiation of human breast tumour stem/progenitor cells. J. Cell Mol. Med. 13, 309–319. doi: 10.1111/j.1582-4934.2008.00338.x
Canettieri, G., Di Marcotullio, L., Greco, A., Coni, S., Antonucci, L., Infante, P., et al. (2010). Histone deacetylase and Cullin3-REN(KCTD11) ubiquitin ligase interplay regulates Hedgehog signalling through gli acetylation. Nat. Cell Biol. 12, 132–142. doi: 10.1038/ncb2013
Cao, Q., Yu, J., Dhanasekaran, S. M., Kim, J. H., Mani, R. S., Tomlins, S. A., et al. (2008). Repression of E-cadherin by the polycomb group protein EZH2 in cancer. Oncogene 27, 7274–7284. doi: 10.1038/onc.2008.333
Catenacci, D. V., Junttila, M. R., Karrison, T., Bahary, N., Horiba, M. N., Nattam, S. R., et al. (2015). Randomized phase Ib/II study of gemcitabine plus placebo or vismodegib, a hedgehog pathway inhibitor, in patients with metastatic pancreatic cancer. J. Clin. Oncol. 33, 4284–4292. doi: 10.1200/JCO.2015.62.8719
Champiat, S., Dercle, L., Ammari, S., Massard, C., Hollebecque, A., Postel-Vinay, S., et al. (2017). Hyperprogressive Disease is a new pattern of progression in cancer patients treated by anti-PD-1/PD-L1. Clin. Cancer Res. 23, 1920–1928. doi: 10.1158/1078-0432.CCR-16-1741
Charafe-Jauffret, E., Ginestier, C., Iovino, F., Tarpin, C., Diebel, M., Esterni, B., et al. (2010). Aldehyde dehydrogenase 1-positive cancer stem cells mediate metastasis and poor clinical outcome in inflammatory breast cancer. Clin. Cancer Res. 16, 45–55. doi: 10.1158/1078-0432.CCR-09-1630
Chen, J., Li, Y., Yu, T. S., McKay, R. M., Burns, D. K., Kernie, S. G., et al. (2012). A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 488, 522–526. doi: 10.1038/nature11287
Chen, L., Alexe, G., Dharia, N. V., Ross, L., Iniguez, A. B., Conway, A. S., et al. (2018). CRISPR-Cas9 screen reveals a MYCN-amplified neuroblastoma dependency on EZH2. J. Clin. Invest. 128, 446–462. doi: 10.1172/JCI90793
Cheng, L., Huang, Z., Zhou, W., Wu, Q., Donnola, S., Liu, J. K., et al. (2013). Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 153, 139–152. doi: 10.1016/j.cell.2013.02.021
Cheung, A. M., Wan, T. S., Leung, J. C., Chan, L. Y., Huang, H., Kwong, Y. L., et al. (2007). Aldehyde dehydrogenase activity in leukemic blasts defines a subgroup of acute myeloid leukemia with adverse prognosis and superior NOD/SCID engrafting potential. Leukemia 21, 1423–1430. doi: 10.1038/sj.leu.2404721
Children’s Oncology, G., Fouladi, M., Furman, W. L., Chin, T., Freeman, B. B., III, Dudkin, L., et al. (2006). Phase I study of depsipeptide in pediatric patients with refractory solid tumors: a children’s oncology group report. J. Clin. Oncol. 24, 3678–3685. doi: 10.1200/JCO.2006.06.4964
Cilibrasi, C., Riva, G., Romano, G., Cadamuro, M., Bazzoni, R., Butta, V., et al. (2017). Resveratrol Impairs glioma stem cells proliferation and motility by modulating the wnt signaling pathway. PLoS One 12:e0169854. doi: 10.1371/journal.pone.0169854
Collins, A. T., Berry, P. A., Hyde, C., Stower, M. J., and Maitland, N. J. (2005). Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 65, 10946–10951. doi: 10.1158/0008-5472.CAN-05-2018
Collinson, A., Collier, A. J., Morgan, N. P., Sienerth, A. R., Chandra, T., Andrews, S., et al. (2016). Deletion of the polycomb-group protein EZH2 leads to compromised self-renewal and differentiation defects in human embryonic stem cells. Cell Rep. 17, 2700–2714. doi: 10.1016/j.celrep.2016.11.032
Coussens, L. M., Fingleton, B., and Matrisian, L. M. (2002). Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science 295, 2387–2392. doi: 10.1126/science.1067100
Cozzio, A., Passegue, E., Ayton, P. M., Karsunky, H., Cleary, M. L., and Weissman, I. L. (2003). Similar MLL-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes Dev. 17, 3029–3035. doi: 10.1101/gad.1143403
Crawford, Y., and Ferrara, N. (2009). Tumor and stromal pathways mediating refractoriness/resistance to anti-angiogenic therapies. Trends Pharmacol. Sci. 30, 624–630. doi: 10.1016/j.tips.2009.09.004
Dawson, M. A. (2017). The cancer epigenome: concepts, challenges, and therapeutic opportunities. Science 355, 1147–1152. doi: 10.1126/science.aam7304
Deblois, G., and Giguere, V. (2011). Functional and physiological genomics of estrogen-related receptors (ERRs) in health and disease. Biochim. Biophys. Acta 1812, 1032–1040. doi: 10.1016/j.bbadis.2010.12.009
Deblois, G., St-Pierre, J., and Giguere, V. (2013). The PGC-1/ERR signaling axis in cancer. Oncogene 32, 3483–3490. doi: 10.1038/onc.2012.529
Deng, Z., Wu, Y., Ma, W., Zhang, S., and Zhang, Y. Q. (2015). Adoptive T-cell therapy of prostate cancer targeting the cancer stem cell antigen EpCAM. BMC Immunol. 16:1. doi: 10.1186/s12865-014-0064-x
Dey, A., Chitsaz, F., Abbasi, A., Misteli, T., and Ozato, K. (2003). The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc. Natl. Acad. Sci. U.S.A. 100, 8758–8763. doi: 10.1073/pnas.1433065100
Dhalluin, C., Carlson, J. E., Zeng, L., He, C., Aggarwal, A. K., and Zhou, M. M. (1999). Structure and ligand of a histone acetyltransferase bromodomain. Nature 399, 491–496. doi: 10.1038/20974
Di Croce, L., and Helin, K. (2013). Transcriptional regulation by polycomb group proteins. Nat. Struct. Mol. Biol. 20, 1147–1155. doi: 10.1038/nsmb.2669
Di Marcotullio, L., Ferretti, E., De Smaele, E., Argenti, B., Mincione, C., Zazzeroni, F., et al. (2004). REN(KCTD11) is a suppressor of Hedgehog signaling and is deleted in human medulloblastoma. Proc. Natl. Acad. Sci. U.S.A. 101, 10833–10838. doi: 10.1073/pnas.0400690101
Duong, H. Q., Hwang, J. S., Kim, H. J., Kang, H. J., Seong, Y. S., and Bae, I. (2012). Aldehyde dehydrogenase 1A1 confers intrinsic and acquired resistance to gemcitabine in human pancreatic adenocarcinoma MIA PaCa-2 cells. Int. J. Oncol. 41, 855–861. doi: 10.3892/ijo.2012.1516
Efferth, T., Konkimalla, V. B., Wang, Y. F., Sauerbrey, A., Meinhardt, S., Zintl, F., et al. (2008). Prediction of broad spectrum resistance of tumors towards anticancer drugs. Clin. Cancer Res. 14, 2405–2412. doi: 10.1158/1078-0432.CCR-07-4525
Erler, J. T., Bennewith, K. L., Cox, T. R., Lang, G., Bird, D., Koong, A., et al. (2009). Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 15, 35–44. doi: 10.1016/j.ccr.2008.11.012
Escudier, B., Eisen, T., Stadler, W. M., Szczylik, C., Oudard, S., Siebels, M., et al. (2007). Sorafenib in advanced clear-cell renal-cell carcinoma. N. Engl. J. Med. 356, 125–134. doi: 10.1056/NEJMoa060655
Eyler, C. E., Foo, W. C., LaFiura, K. M., McLendon, R. E., Hjelmeland, A. B., and Rich, J. N. (2008). Brain cancer stem cells display preferential sensitivity to Akt inhibition. Stem Cells 26, 3027–3036. doi: 10.1634/stemcells.2007-1073
Eyler, C. E., and Rich, J. N. (2008). Survival of the fittest: cancer stem cells in therapeutic resistance and angiogenesis. J. Clin. Oncol. 26, 2839–2845. doi: 10.1200/JCO.2007.15.1829
Feng, K. C., Guo, Y. L., Liu, Y., Dai, H. R., Wang, Y., Lv, H. Y., et al. (2017). Cocktail treatment with EGFR-specific and CD133-specific chimeric antigen receptor-modified T cells in a patient with advanced cholangiocarcinoma. J. Hematol. Oncol. 10:4. doi: 10.1186/s13045-016-0378-7
Ferrara, N., Hillan, K. J., Gerber, H. P., and Novotny, W. (2004). Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat. Rev. Drug Discov. 3, 391–400. doi: 10.1038/nrd1381
Filippakopoulos, P., and Knapp, S. (2014). Targeting bromodomains: epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 13, 337–356. doi: 10.1038/nrd4286
Filippakopoulos, P., Qi, J., Picaud, S., Shen, Y., Smith, W. B., Fedorov, O., et al. (2010). Selective inhibition of BET bromodomains. Nature 468, 1067–1073. doi: 10.1038/nature09504
Folkman, J. (1971). Tumor angiogenesis: therapeutic implications. N. Engl. J. Med. 285, 1182–1186. doi: 10.1056/NEJM197111182852108
Frank, N. Y., Margaryan, A., Huang, Y., Schatton, T., Waaga-Gasser, A. M., Gasser, M., et al. (2005). ABCB5-mediated doxorubicin transport and chemoresistance in human malignant melanoma. Cancer Res. 65, 4320–4333. doi: 10.1158/0008-5472.CAN-04-3327
Friedman, H. S., Colvin, O. M., Kaufmann, S. H., Ludeman, S. M., Bullock, N., Bigner, D. D., et al. (1992). Cyclophosphamide resistance in medulloblastoma. Cancer Res. 52, 5373–5378.
Fukuda, K., Saikawa, Y., Ohashi, M., Kumagai, K., Kitajima, M., Okano, H., et al. (2009). Tumor initiating potential of side population cells in human gastric cancer. Int. J. Oncol. 34, 1201–1207.
Gaggianesi, M., Turdo, A., Chinnici, A., Lipari, E., Apuzzo, T., Benfante, A., et al. (2017). IL4 Primes the dynamics of breast cancer progression via DUSP4 inhibition. Cancer Res. 77, 3268–3279. doi: 10.1158/0008-5472.CAN-16-3126
Gallmeier, E., Hermann, P. C., Mueller, M. T., Machado, J. G., Ziesch, A., De Toni, E. N., et al. (2011). Inhibition of ataxia telangiectasia- and Rad3-related function abrogates the in vitro and in vivo tumorigenicity of human colon cancer cells through depletion of the CD133(+) tumor-initiating cell fraction. Stem Cells 29, 418–429. doi: 10.1002/stem.595
Gao, H., Chakraborty, G., Lee-Lim, A. P., Mo, Q., Decker, M., Vonica, A., et al. (2012). The BMP inhibitor Coco reactivates breast cancer cells at lung metastatic sites. Cell 150, 764–779. doi: 10.1016/j.cell.2012.06.035
Gargett, T., Yu, W., Dotti, G., Yvon, E. S., Christo, S. N., Hayball, J. D., et al. (2016). GD2-specific CAR T cells undergo potent activation and deletion following antigen encounter but can be protected from activation-induced cell death by PD-1 blockade. Mol. Ther. 24, 1135–1149. doi: 10.1038/mt.2016.63
Gascoigne, K. E., and Taylor, S. S. (2009). How do anti-mitotic drugs kill cancer cells? J. Cell Sci. 122(Pt 15), 2579–2585. doi: 10.1242/jcs.039719
Ghoshal, P., Nganga, A. J., Moran-Giuati, J., Szafranek, A., Johnson, T. R., Bigelow, A. J., et al. (2009). Loss of the SMRT/NCoR2 corepressor correlates with JAG2 overexpression in multiple myeloma. Cancer Res. 69, 4380–4387. doi: 10.1158/0008-5472.CAN-08-3467
Giancotti, F. G. (2013). Mechanisms governing metastatic dormancy and reactivation. Cell 155, 750–764. doi: 10.1016/j.cell.2013.10.029
Gide, T. N., Wilmott, J. S., Scolyer, R. A., and Long, G. V. (2018). Primary and Acquired resistance to immune checkpoint inhibitors in metastatic melanoma. Clin. Cancer Res. 24, 1260–1270. doi: 10.1158/1078-0432.CCR-17-2267
Gilbertson, R. J., and Rich, J. N. (2007). Making a tumour’s bed: glioblastoma stem cells and the vascular niche. Nat. Rev. Cancer 7, 733–736. doi: 10.1038/nrc2246
Ginestier, C., Hur, M. H., Charafe-Jauffret, E., Monville, F., Dutcher, J., Brown, M., et al. (2007). ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 1, 555–567. doi: 10.1016/j.stem.2007.08.014
Ginestier, C., Liu, S., Diebel, M. E., Korkaya, H., Luo, M., Brown, M., et al. (2010). CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J. Clin. Invest. 120, 485–497. doi: 10.1172/JCI39397
Grillet, F., Bayet, E., Villeronce, O., Zappia, L., Lagerqvist, E. L., Lunke, S., et al. (2017). Circulating tumour cells from patients with colorectal cancer have cancer stem cell hallmarks in ex vivo culture. Gut 66, 1802–1810. doi: 10.1136/gutjnl-2016-311447
Grupp, S. A., Kalos, M., Barrett, D., Aplenc, R., Porter, D. L., Rheingold, S. R., et al. (2013). Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 368, 1509–1518. doi: 10.1056/NEJMoa1215134
Guo, Y., Feng, K., Wang, Y., and Han, W. (2018). Targeting cancer stem cells by using chimeric antigen receptor-modified T cells: a potential and curable approach for cancer treatment. Protein Cell 9, 516–526. doi: 10.1007/s13238-017-0394-6
Guo, Z., Jiang, J. H., Zhang, J., Yang, H. J., Yang, F. Q., Qi, Y. P., et al. (2015). COX-2 Promotes migration and invasion by the side population of cancer stem cell-like hepatocellular carcinoma cells. Medicine 94:e1806. doi: 10.1097/MD.0000000000001806
Gupta, P. B., Onder, T. T., Jiang, G., Tao, K., Kuperwasser, C., Weinberg, R. A., et al. (2009). Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 138, 645–659. doi: 10.1016/j.cell.2009.06.034
Hammoud, S. S., Cairns, B. R., and Jones, D. A. (2013). Epigenetic regulation of colon cancer and intestinal stem cells. Curr. Opin. Cell Biol. 25, 177–183. doi: 10.1016/j.ceb.2013.01.007
Hanahan, D., and Folkman, J. (1996). Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 86, 353–364.
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. doi: 10.1016/j.cell.2011.02.013
Hawley, T. S., Riz, I., Yang, W., Wakabayashi, Y., Depalma, L., Chang, Y. T., et al. (2013). Identification of an ABCB1 (P-glycoprotein)-positive carfilzomib-resistant myeloma subpopulation by the pluripotent stem cell fluorescent dye CDy1. Am. J. Hematol. 88, 265–272. doi: 10.1002/ajh.23387
He, S., Liu, Z., Oh, D. Y., and Thiele, C. J. (2013). MYCN and the epigenome. Front. Oncol. 3:1. doi: 10.3389/fonc.2013.00001
Hemmati, H. D., Nakano, I., Lazareff, J. A., Masterman-Smith, M., Geschwind, D. H., Bronner-Fraser, M., et al. (2003). Cancerous stem cells can arise from pediatric brain tumors. Proc. Natl. Acad. Sci. U.S.A. 100, 15178–15183. doi: 10.1073/pnas.2036535100
Hilton, J. (1984). Role of aldehyde dehydrogenase in cyclophosphamide-resistant L1210 leukemia. Cancer Res. 44, 5156–5160.
Hirschmann-Jax, C., Foster, A. E., Wulf, G. G., Nuchtern, J. G., Jax, T. W., Gobel, U., et al. (2004). A distinct ”side population” of cells with high drug efflux capacity in human tumor cells. Proc. Natl. Acad. Sci. U.S.A. 101, 14228–14233. doi: 10.1073/pnas.0400067101
Ho, M. M., Ng, A. V., Lam, S., and Hung, J. Y. (2007). Side population in human lung cancer cell lines and tumors is enriched with stem-like cancer cells. Cancer Res. 67, 4827–4833. doi: 10.1158/0008-5472.CAN-06-3557
Hsu, J. M., Xia, W., Hsu, Y. H., Chan, L. C., Yu, W. H., Cha, J. H., et al. (2018). STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat. Commun. 9:1908. doi: 10.1038/s41467-018-04313-6
Hsu, L. C., Chang, W. C., Hoffmann, I., and Duester, G. (1999). Molecular analysis of two closely related mouse aldehyde dehydrogenase genes: identification of a role for Aldh1, but not Aldh-pb, in the biosynthesis of retinoic acid. Biochem. J. 339(Pt 2), 387–395.
Hugo, W., Zaretsky, J. M., Sun, L., Song, C., Moreno, B. H., Hu-Lieskovan, S., et al. (2016). Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell 165, 35–44. doi: 10.1016/j.cell.2016.02.065
Hurwitz, H., Fehrenbacher, L., Novotny, W., Cartwright, T., Hainsworth, J., Heim, W., et al. (2004). Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl. J. Med. 350, 2335–2342. doi: 10.1056/NEJMoa032691
Hussain, M., Rao, M., Humphries, A. E., Hong, J. A., Liu, F., Yang, M., et al. (2009). Tobacco smoke induces polycomb-mediated repression of Dickkopf-1 in lung cancer cells. Cancer Res. 69, 3570–3578. doi: 10.1158/0008-5472.CAN-08-2807
Jachetti, E., Caputo, S., Mazzoleni, S., Brambillasca, C. S., Parigi, S. M., Grioni, M., et al. (2015). Tenascin-C Protects cancer stem-like cells from immune surveillance by arresting T-cell activation. Cancer Res. 75, 2095–2108. doi: 10.1158/0008-5472.CAN-14-2346
Jain, R. K. (2005). Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 307, 58–62. doi: 10.1126/science.1104819
Jones, C. L., Stevens, B. M., D’Alessandro, A., Reisz, J. A., Culp-Hill, R., Nemkov, T., et al. (2018). Inhibition of Amino acid metabolism selectively targets human leukemia stem cells. Cancer Cell 34, 724–740.e4. doi: 10.1016/j.ccell.2018.10.005
Jones, P. A., and Takai, D. (2001). The role of DNA methylation in mammalian epigenetics. Science 293, 1068–1070. doi: 10.1126/science.1063852
Kanwar, S. S., Yu, Y., Nautiyal, J., Patel, B. B., and Majumdar, A. P. (2010). The Wnt/beta-catenin pathway regulates growth and maintenance of colonospheres. Mol. Cancer 9:212. doi: 10.1186/1476-4598-9-212
Kelly, P. N., and Strasser, A. (2011). The role of Bcl-2 and its pro-survival relatives in tumourigenesis and cancer therapy. Cell Death Differ. 18, 1414–1424. doi: 10.1038/cdd.2011.17
Kemper, K., Grandela, C., and Medema, J. P. (2010). Molecular identification and targeting of colorectal cancer stem cells. Oncotarget 1, 387–395. doi: 10.18632/oncotarget.101003
Kessenbrock, K., Plaks, V., and Werb, Z. (2010). Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 141, 52–67. doi: 10.1016/j.cell.2010.03.015
Klaus, C. R., Iwanowicz, D., Johnston, D., Campbell, C. A., Smith, J. J., Moyer, M. P., et al. (2014). DOT1L inhibitor EPZ-5676 displays synergistic antiproliferative activity in combination with standard of care drugs and hypomethylating agents in MLL-rearranged leukemia cells. J. Pharmacol. Exp. Ther. 350, 646–656. doi: 10.1124/jpet.114.214577
Konopleva, M., Zhao, S., Hu, W., Jiang, S., Snell, V., Weidner, D., et al. (2002). The anti-apoptotic genes Bcl-X(L) and Bcl-2 are over-expressed and contribute to chemoresistance of non-proliferating leukaemic CD34+ cells. Br. J. Haematol. 118, 521–534.
Krivtsov, A. V., Twomey, D., Feng, Z., Stubbs, M. C., Wang, Y., Faber, J., et al. (2006). Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 442, 818–822. doi: 10.1038/nature04980
Kurtova, A. V., Xiao, J., Mo, Q., Pazhanisamy, S., Krasnow, R., Lerner, S. P., et al. (2015). Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature 517, 209–213. doi: 10.1038/nature14034
Lancet, J. E., Baer, M. R., Duran, G. E., List, A. F., Fielding, R., Marcelletti, J. F., et al. (2009). A phase I trial of continuous infusion of the multidrug resistance inhibitor zosuquidar with daunorubicin and cytarabine in acute myeloid leukemia. Leuk Res. 33, 1055–1061. doi: 10.1016/j.leukres.2008.09.015
Lapidot, T., Sirard, C., Vormoor, J., Murdoch, B., Hoang, T., Caceres-Cortes, J., et al. (1994). A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 367, 645–648. doi: 10.1038/367645a0
Lee, K. M., Giltnane, J. M., Balko, J. M., Schwarz, L. J., Guerrero-Zotano, A. L., Hutchinson, K. E., et al. (2017). MYC and MCL1 cooperatively promote chemotherapy-resistant breast cancer stem cells via regulation of mitochondrial oxidative phosphorylation. Cell Metab. 26, 633–647.e7. doi: 10.1016/j.cmet.2017.09.009
Lee, Y., Shin, J. H., Longmire, M., Wang, H., Kohrt, H. E., Chang, H. Y., et al. (2016). CD44+ cells in head and neck squamous cell carcinoma suppress T-cell-mediated immunity by selective constitutive and inducible expression of PD-L1. Clin. Cancer Res. 22, 3571–3581. doi: 10.1158/1078-0432.CCR-15-2665
Leonard, G. D., Fojo, T., and Bates, S. E. (2003). The role of ABC transporters in clinical practice. Oncologist 8, 411–424.
Levina, V., Marrangoni, A. M., DeMarco, R., Gorelik, E., and Lokshin, A. E. (2008). Drug-selected human lung cancer stem cells: cytokine network, tumorigenic and metastatic properties. PLoS One 3:e3077. doi: 10.1371/journal.pone.0003077
Lewis, P. W., Muller, M. M., Koletsky, M. S., Cordero, F., Lin, S., Banaszynski, L. A., et al. (2013). Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340, 857–861. doi: 10.1126/science.1232245
Lin, Z., Zhang, Q., and Luo, W. (2016). Angiogenesis inhibitors as therapeutic agents in cancer: challenges and future directions. Eur. J. Pharmacol. 793, 76–81. doi: 10.1016/j.ejphar.2016.10.039
Liu, S., Cong, Y., Wang, D., Sun, Y., Deng, L., Liu, Y., et al. (2014). Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. 2, 78–91. doi: 10.1016/j.stemcr.2013.11.009
Lobo, N. A., Shimono, Y., Qian, D., and Clarke, M. F. (2007). The biology of cancer stem cells. Annu. Rev. Cell Dev. Biol. 23, 675–699. doi: 10.1146/annurev.cellbio.22.010305.104154
Lombardo, Y., Scopelliti, A., Cammareri, P., Todaro, M., Iovino, F., Ricci-Vitiani, L., et al. (2011). Bone morphogenetic protein 4 induces differentiation of colorectal cancer stem cells and increases their response to chemotherapy in mice. Gastroenterology 140, 297–309. doi: 10.1053/j.gastro.2010.10.005
Louis, C. U., and Shohet, J. M. (2015). Neuroblastoma: molecular pathogenesis and therapy. Annu. Rev. Med. 66, 49–63. doi: 10.1146/annurev-med-011514-023121
Lu, K. V., Chang, J. P., Parachoniak, C. A., Pandika, M. M., Aghi, M. K., Meyronet, D., et al. (2012). VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell 22, 21–35. doi: 10.1016/j.ccr.2012.05.037
Luo, M., and Wicha, M. S. (2019). Targeting cancer stem cell redox metabolism to enhance therapy responses. Semin. Radiat. Oncol. 29, 42–54. doi: 10.1016/j.semradonc.2018.10.003
Ma, S., Chan, K. W., Hu, L., Lee, T. K., Wo, J. Y., Ng, I. O., et al. (2007). Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology 132, 2542–2556. doi: 10.1053/j.gastro.2007.04.025
Ma, S., Chan, K. W., Lee, T. K., Tang, K. H., Wo, J. Y., Zheng, B. J., et al. (2008a). Aldehyde dehydrogenase discriminates the CD133 liver cancer stem cell populations. Mol. Cancer Res. 6, 1146–1153. doi: 10.1158/1541-7786.MCR-08-0035
Ma, S., Lee, T. K., Zheng, B. J., Chan, K. W., and Guan, X. Y. (2008b). CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene 27, 1749–1758. doi: 10.1038/sj.onc.1210811
Madjd, Z., Mehrjerdi, A. Z., Sharifi, A. M., Molanaei, S., Shahzadi, S. Z., and Asadi-Lari, M. (2009). CD44+ cancer cells express higher levels of the anti-apoptotic protein Bcl-2 in breast tumours. Cancer Immun. 9:4.
Mani, S. A., Guo, W., Liao, M. J., Eaton, E. N., Ayyanan, A., Zhou, A. Y., et al. (2008). The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133, 704–715. doi: 10.1016/j.cell.2008.03.027
Marcucci, F., Rumio, C., and Lefoulon, F. (2016). Anti-cancer stem-like cell compounds in clinical development - an overview and critical appraisal. Front. Oncol. 6:115. doi: 10.3389/fonc.2016.00115
Maugeri-Sacca, M., Vigneri, P., and De Maria, R. (2011). Cancer stem cells and chemosensitivity. Clin. Cancer Res. 17, 4942–4947. doi: 10.1158/1078-0432.CCR-10-2538
McCabe, M. T., Brandes, J. C., and Vertino, P. M. (2009). Cancer DNA methylation: molecular mechanisms and clinical implications. Clin. Cancer Res. 15, 3927–3937. doi: 10.1158/1078-0432.CCR-08-2784
McCord, A. M., Jamal, M., Williams, E. S., Camphausen, K., and Tofilon, P. J. (2009). CD133+ glioblastoma stem-like cells are radiosensitive with a defective DNA damage response compared with established cell lines. Clin. Cancer Res. 15, 5145–5153. doi: 10.1158/1078-0432.CCR-09-0263
Menendez, J. A., and Alarcon, T. (2014). Metabostemness: a new cancer hallmark. Front. Oncol. 4:262. doi: 10.3389/fonc.2014.00262
Miller, K., Wang, M., Gralow, J., Dickler, M., Cobleigh, M., Perez, E. A., et al. (2007). Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N. Engl. J. Med. 357, 2666–2676. doi: 10.1056/NEJMoa072113
Molchadsky, A., and Rotter, V. (2017). p53 and its mutants on the slippery road from stemness to carcinogenesis. Carcinogenesis 38, 347–358. doi: 10.1093/carcin/bgw092
Motzer, R. J., Hutson, T. E., Tomczak, P., Michaelson, M. D., Bukowski, R. M., Rixe, O., et al. (2007). Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N. Engl. J. Med. 356, 115–124. doi: 10.1056/NEJMoa065044
Mount, C. W., Majzner, R. G., Sundaresh, S., Arnold, E. P., Kadapakkam, M., Haile, S., et al. (2018). Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M(+) diffuse midline gliomas. Nat. Med. 24, 572–579. doi: 10.1038/s41591-018-0006-x
Ni, M., Xiong, M., Zhang, X., Cai, G., Chen, H., Zeng, Q., et al. (2015). Poly(lactic-co-glycolic acid) nanoparticles conjugated with CD133 aptamers for targeted salinomycin delivery to CD133+ osteosarcoma cancer stem cells. Int. J. Nanomed. 10, 2537–2554. doi: 10.2147/IJN.S78498
Nowak, D., Stewart, D., and Koeffler, H. P. (2009). Differentiation therapy of leukemia: 3 decades of development. Blood 113, 3655–3665. doi: 10.1182/blood-2009-01-198911
Okazaki, T., Chikuma, S., Iwai, Y., Fagarasan, S., and Honjo, T. (2013). A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nat. Immunol. 14, 1212–1218. doi: 10.1038/ni.2762
Owen, J. L., and Mohamadzadeh, M. (2013). Macrophages and chemokines as mediators of angiogenesis. Front. Physiol. 4:159. doi: 10.3389/fphys.2013.00159
Pan, E., Supko, J. G., Kaley, T. J., Butowski, N. A., Cloughesy, T., Jung, J., et al. (2016). Phase I study of RO4929097 with bevacizumab in patients with recurrent malignant glioma. J. Neurooncol. 130, 571–579. doi: 10.1007/s11060-016-2263-1
Poli, V., Fagnocchi, L., Fasciani, A., Cherubini, A., Mazzoleni, S., Ferrillo, S., et al. (2018). MYC-driven epigenetic reprogramming favors the onset of tumorigenesis by inducing a stem cell-like state. Nat. Commun. 9:1024. doi: 10.1038/s41467-018-03264-2
Pollyea, D. A., Stevens, B. M., Jones, C. L., Winters, A., Pei, S., Minhajuddin, M., et al. (2018). Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat. Med. 24, 1859–1866. doi: 10.1038/s41591-018-0233-1
Pugh, C. W., and Ratcliffe, P. J. (2003). Regulation of angiogenesis by hypoxia: role of the HIF system. Nat. Med. 9, 677–684. doi: 10.1038/nm0603-677
Qin, W., Huang, G., Chen, Z., and Zhang, Y. (2017). Nanomaterials in targeting cancer stem cells for cancer therapy. Front Pharmacol. 8:1. doi: 10.3389/fphar.2017.00001
Ramadoss, M., and Mahadevan, V. (2018). Targeting the cancer epigenome: synergistic therapy with bromodomain inhibitors. Drug Discov. Today 23, 76–89. doi: 10.1016/j.drudis.2017.09.011
Rausch, V., Liu, L., Kallifatidis, G., Baumann, B., Mattern, J., Gladkich, J., et al. (2010). Synergistic activity of sorafenib and sulforaphane abolishes pancreatic cancer stem cell characteristics. Cancer Res. 70, 5004–5013. doi: 10.1158/0008-5472.CAN-10-0066
Ribas, A., and Wolchok, J. D. (2018). Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355. doi: 10.1126/science.aar4060
Ricciuti, B., Foglietta, J., Bianconi, V., Sahebkar, A., and Pirro, M. (2017). Enzymes involved in tumor-driven angiogenesis: a valuable target for anticancer therapy. Semin. Cancer Biol. doi: 10.1016/j.semcancer.2017.11.005 [Epub ahead of print].
Ricci-Vitiani, L., Pallini, R., Biffoni, M., Todaro, M., Invernici, G., Cenci, T., et al. (2010). Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 468, 824–828. doi: 10.1038/nature09557
Rizzo, S., Hersey, J. M., Mellor, P., Dai, W., Santos-Silva, A., Liber, D., et al. (2011). Ovarian cancer stem cell-like side populations are enriched following chemotherapy and overexpress EZH2. Mol. Cancer Ther. 10, 325–335. doi: 10.1158/1535-7163.MCT-10-0788
Ropolo, M., Daga, A., Griffero, F., Foresta, M., Casartelli, G., Zunino, A., et al. (2009). Comparative analysis of DNA repair in stem and nonstem glioma cell cultures. Mol. Cancer Res. 7, 383–392. doi: 10.1158/1541-7786.MCR-08-0409
Saito, Y., Uchida, N., Tanaka, S., Suzuki, N., Tomizawa-Murasawa, M., Sone, A., et al. (2010). Induction of cell cycle entry eliminates human leukemia stem cells in a mouse model of AML. Nat. Biotechnol. 28, 275–280. doi: 10.1038/nbt.1607
Sancho, P., Barneda, D., and Heeschen, C. (2016). Hallmarks of cancer stem cell metabolism. Br. J. Cancer 114, 1305–1312. doi: 10.1038/bjc.2016.152
Sawada, J., Urakami, T., Li, F., Urakami, A., Zhu, W., Fukuda, M., et al. (2012). Small GTPase R-Ras regulates integrity and functionality of tumor blood vessels. Cancer Cell 22, 235–249. doi: 10.1016/j.ccr.2012.06.013
Scala, S. (2015). Molecular pathways: targeting the CXCR4-CXCL12 Axis–untapped potential in the tumor microenvironment. Clin. Cancer Res. 21, 4278–4285. doi: 10.1158/1078-0432.CCR-14-0914
Seymour, L., Bogaerts, J., Perrone, A., Ford, R., Schwartz, L. H., Mandrekar, S., et al. (2017). iRECIST: guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol. 18, e143–e152. doi: 10.1016/S1470-2045(17)30074-8
Shahbazi, J., Liu, P. Y., Atmadibrata, B., Bradner, J. E., Marshall, G. M., Lock, R. B., et al. (2016). The bromodomain inhibitor JQ1 and the histone deacetylase inhibitor panobinostat synergistically reduce N-Myc expression and induce anticancer effects. Clin. Cancer Res. 22, 2534–2544. doi: 10.1158/1078-0432.CCR-15-1666
Shi, G. M., Xu, Y., Fan, J., Zhou, J., Yang, X. R., Qiu, S. J., et al. (2008). Identification of side population cells in human hepatocellular carcinoma cell lines with stepwise metastatic potentials. J. Cancer Res. Clin. Oncol. 134, 1155–1163. doi: 10.1007/s00432-008-0407-1
Siefker-Radtke, A., and Curti, B. (2018). Immunotherapy in metastatic urothelial carcinoma: focus on immune checkpoint inhibition. Nat. Rev. Urol. 15, 112–124. doi: 10.1038/nrurol.2017.190
Singh, A., and Settleman, J. (2010). EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 29, 4741–4751. doi: 10.1038/onc.2010.215
Singh, S., Brocker, C., Koppaka, V., Chen, Y., Jackson, B.C., Matsumoto, A., et al. (2013). Aldehyde dehydrogenases in cellular responses to oxidative/electrophilic stress. Free Radic. Biol. Med. 56, 89–101. doi: 10.1016/j.freeradbiomed.2012.11.010
Singh, S. K., Hawkins, C., Clarke, I. D., Squire, J. A., Bayani, J., Hide, T., et al. (2004). Identification of human brain tumour initiating cells. Nature 432, 396–401. doi: 10.1038/nature03128
Singh, S. R., Zeng, X., Zhao, J., Liu, Y., Hou, G., Liu, H., et al. (2016). The lipolysis pathway sustains normal and transformed stem cells in adult Drosophila. Nature 538, 109–113. doi: 10.1038/nature19788
Skoda, J., Borankova, K., Jansson, P. J., Huang, M. L., Veselska, R., and Richardson, D. R. (2018). Pharmacological targeting of mitochondria in cancer stem cells: an ancient organelle at the crossroad of novel anti-cancer therapies. Pharmacol. Res. 139, 298–313. doi: 10.1016/j.phrs.2018.11.020
Somervaille, T. C., Matheny, C. J., Spencer, G. J., Iwasaki, M., Rinn, J. L., Witten, D. M., et al. (2009). Hierarchical maintenance of MLL myeloid leukemia stem cells employs a transcriptional program shared with embryonic rather than adult stem cells. Cell Stem Cell 4, 129–140. doi: 10.1016/j.stem.2008.11.015
Sorrentino, C., Ciummo, S. L., Cipollone, G., Caputo, S., Bellone, M., and Di Carlo, E. (2018). Interleukin-30/IL27p28 shapes prostate cancer stem-like cell behavior and is critical for tumor onset and metastasization. Cancer Res. 78, 2654–2668. doi: 10.1158/0008-5472.CAN-17-3117
Spranger, S., Bao, R., and Gajewski, T. F. (2015). Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 523, 231–235. doi: 10.1038/nature14404
Strahl, B. D., and Allis, C. D. (2000). The language of covalent histone modifications. Nature 403, 41–45. doi: 10.1038/47412
Strasser, A., Harris, A. W., Bath, M. L., and Cory, S. (1990). Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature 348, 331–333. doi: 10.1038/348331a0
Su, S., Zhao, J., Xing, Y., Zhang, X., Liu, J., Ouyang, Q., et al. (2018). Immune checkpoint inhibition overcomes ADCP-induced immunosuppression by macrophages. Cell 175, 442–457.e3. doi: 10.1016/j.cell.2018.09.007
Sun, L., Liang, C., Shirazian, S., Zhou, Y., Miller, T., Cui, J., et al. (2003). Discovery of 5-[5-fluoro-2-oxo-1,2- dihydroindol-(3Z)-ylidenemethyl]-2,4- dimethyl-1H-pyrrole-3-carboxylic acid (2-diethylaminoethyl)amide, a novel tyrosine kinase inhibitor targeting vascular endothelial and platelet-derived growth factor receptor tyrosine kinase. J. Med. Chem. 46, 1116–1119. doi: 10.1021/jm0204183
Suzuki, H., Watkins, D. N., Jair, K. W., Schuebel, K. E., Markowitz, S. D., Chen, W. D., et al. (2004). Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat. Genet. 36, 417–422. doi: 10.1038/ng1330
Szarynska, M., Olejniczak, A., Kobiela, J., Laski, D., Sledzinski, Z., and Kmiec, Z. (2018). Cancer stem cells as targets for DC-based immunotherapy of colorectal cancer. Sci. Rep. 8:12042. doi: 10.1038/s41598-018-30525-3
Tam, W. L., and Weinberg, R. A. (2013). The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat. Med. 19, 1438–1449. doi: 10.1038/nm.3336
Tanei, T., Morimoto, K., Shimazu, K., Kim, S. J., Tanji, Y., Taguchi, T., et al. (2009). Association of breast cancer stem cells identified by aldehyde dehydrogenase 1 expression with resistance to sequential paclitaxel and epirubicin-based chemotherapy for breast cancers. Clin. Cancer Res. 15, 4234–4241. doi: 10.1158/1078-0432.CCR-08-1479
Tellez, C. S., Juri, D. E., Do, K., Bernauer, A. M., Thomas, C. L., Damiani, L. A., et al. (2011). EMT and stem cell-like properties associated with miR-205 and miR-200 epigenetic silencing are early manifestations during carcinogen-induced transformation of human lung epithelial cells. Cancer Res. 71, 3087–3097. doi: 10.1158/0008-5472.CAN-10-3035
Tewari, K. S., Sill, M. W., Long, H. J., III, Penson, R. T., Huang, H., Ramondetta, L. M., et al. (2014). Improved survival with bevacizumab in advanced cervical cancer. N. Engl. J. Med. 370, 734–743. doi: 10.1056/NEJMoa1309748
To, K. K., Polgar, O., Huff, L. M., Morisaki, K., and Bates, S. E. (2008). Histone modifications at the ABCG2 promoter following treatment with histone deacetylase inhibitor mirror those in multidrug-resistant cells. Mol. Cancer Res. 6, 151–164. doi: 10.1158/1541-7786.MCR-07-0175
Todaro, M., Alea, M. P., Di Stefano, A. B., Cammareri, P., Vermeulen, L., Iovino, F., et al. (2007). Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell 1, 389–402. doi: 10.1016/j.stem.2007.08.001
Todaro, M., Gaggianesi, M., Catalano, V., Benfante, A., Iovino, F., Biffoni, M., et al. (2014). CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell Stem Cell 14, 342–356. doi: 10.1016/j.stem.2014.01.009
Todaro, M., Iovino, F., Eterno, V., Cammareri, P., Gambara, G., Espina, V., et al. (2010). Tumorigenic and metastatic activity of human thyroid cancer stem cells. Cancer Res. 70, 8874–8885. doi: 10.1158/0008-5472.CAN-10-1994
Toh, T. B., Lim, J. J., and Chow, E. K. (2017). Epigenetics in cancer stem cells. Mol. Cancer 16:29. doi: 10.1186/s12943-017-0596-9
Valent, P., Bonnet, D., De Maria, R., Lapidot, T., Copland, M., Melo, J. V., et al. (2012). Cancer stem cell definitions and terminology: the devil is in the details. Nat. Rev. Cancer 12, 767–775. doi: 10.1038/nrc3368
van Vlerken, L. E., Kiefer, C. M., Morehouse, C., Li, Y., Groves, C., Wilson, S. D., et al. (2013). EZH2 is required for breast and pancreatic cancer stem cell maintenance and can be used as a functional cancer stem cell reporter. Stem Cells Transl. Med. 2, 43–52. doi: 10.5966/sctm.2012-0036
Vedeld, H. M., Skotheim, R. I., Lothe, R. A., and Lind, G. E. (2014). The recently suggested intestinal cancer stem cell marker DCLK1 is an epigenetic biomarker for colorectal cancer. Epigenetics 9, 346–350. doi: 10.4161/epi.27582
Venters, B. J., and Pugh, B. F. (2007). Chromatin meets RNA polymerase II. Genome Biol. 8:319. doi: 10.1186/gb-2007-8-11-319
Veschi, V., Liu, Z., Voss, T. C., Ozbun, L., Gryder, B., Yan, C., et al. (2017). Epigenetic siRNA and chemical screens identify SETD8 inhibition as a therapeutic strategy for p53 activation in high-risk neuroblastoma. Cancer Cell 31, 50–63. doi: 10.1016/j.ccell.2016.12.002
Viale, A., De Franco, F., Orleth, A., Cambiaghi, V., Giuliani, V., Bossi, D., et al. (2009). Cell-cycle restriction limits DNA damage and maintains self-renewal of leukaemia stem cells. Nature 457, 51–56. doi: 10.1038/nature07618
Visvader, J. E., and Lindeman, G. J. (2008). Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat. Rev. Cancer 8, 755–768. doi: 10.1038/nrc2499
Wainwright, E. N., and Scaffidi, P. (2017). Epigenetics and cancer stem cells: unleashing, hijacking, and restricting cellular plasticity. Trends Cancer 3, 372–386. doi: 10.1016/j.trecan.2017.04.004
Wakefield, L. M., and Hill, C. S. (2013). Beyond TGFbeta: roles of other TGFbeta superfamily members in cancer. Nat. Rev. Cancer 13, 328–341. doi: 10.1038/nrc3500
Wang, R., Chadalavada, K., Wilshire, J., Kowalik, U., Hovinga, K. E., Geber, A., et al. (2010). Glioblastoma stem-like cells give rise to tumour endothelium. Nature 468, 829–833. doi: 10.1038/nature09624
Weis, S. M., and Cheresh, D. A. (2011). Tumor angiogenesis: molecular pathways and therapeutic targets. Nat. Med. 17, 1359–1370. doi: 10.1038/nm.2537
Wheaton, W. W., Weinberg, S. E., Hamanaka, R. B., Soberanes, S., Sullivan, L. B., Anso, E., et al. (2014). Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 3:e02242. doi: 10.7554/eLife.02242
Wilhelm, S., Carter, C., Lynch, M., Lowinger, T., Dumas, J., Smith, R. A., et al. (2006). Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat. Rev. Drug Discov. 5, 835–844. doi: 10.1038/nrd2130
Winer, A., Adams, S., and Mignatti, P. (2018). Matrix metalloproteinase inhibitors in cancer therapy: turning past failures into future successes. Mol. Cancer Ther. 17, 1147–1155. doi: 10.1158/1535-7163.MCT-17-0646
Wright, M. H., Calcagno, A. M., Salcido, C. D., Carlson, M. D., Ambudkar, S. V., and Varticovski, L. (2008). Brca1 breast tumors contain distinct CD44+/CD24- and CD133+ cells with cancer stem cell characteristics. Breast Cancer Res. 10:R10. doi: 10.1186/bcr1855
Wu, Y., Chen, M., Wu, P., Chen, C., Xu, Z. P., and Gu, W. (2017). Increased PD-L1 expression in breast and colon cancer stem cells. Clin. Exp. Pharmacol. Physiol. 44, 602–604. doi: 10.1111/1440-1681.12732
Xie, Q., Wu, Q., Horbinski, C. M., Flavahan, W. A., Yang, K., Zhou, W., et al. (2015). Mitochondrial control by DRP1 in brain tumor initiating cells. Nat. Neurosci. 18, 501–510. doi: 10.1038/nn.3960
Xu, R., Shimizu, F., Hovinga, K., Beal, K., Karimi, S., Droms, L., et al. (2016). Molecular and clinical effects of notch inhibition in glioma patients: a phase 0/I trial. Clin. Cancer Res. 22, 4786–4796. doi: 10.1158/1078-0432.CCR-16-0048
Xu, Y., Chenna, V., Hu, C., Sun, H. X., Khan, M., Bai, H., et al. (2012). Polymeric nanoparticle-encapsulated hedgehog pathway inhibitor HPI-1 (NanoHHI) inhibits systemic metastases in an orthotopic model of human hepatocellular carcinoma. Clin. Cancer Res. 18, 1291–1302. doi: 10.1158/1078-0432.CCR-11-0950
Yao, H. J., Zhang, Y. G., Sun, L., and Liu, Y. (2014). The effect of hyaluronic acid functionalized carbon nanotubes loaded with salinomycin on gastric cancer stem cells. Biomaterials 35, 9208–9223. doi: 10.1016/j.biomaterials.2014.07.033
Ye, H., Adane, B., Khan, N., Sullivan, T., Minhajuddin, M., Gasparetto, M., et al. (2016). Leukemic stem cells evade chemotherapy by metabolic adaptation to an adipose tissue niche. Cell Stem Cell 19, 23–37. doi: 10.1016/j.stem.2016.06.001
Yi, J. M., Tsai, H. C., Glockner, S. C., Lin, S., Ohm, J. E., Easwaran, H., et al. (2008). Abnormal DNA methylation of CD133 in colorectal and glioblastoma tumors. Cancer Res. 68, 8094–8103. doi: 10.1158/0008-5472.CAN-07-6208
Zheng, F., Dang, J., Zhang, H., Xu, F., Ba, D., Zhang, B., et al. (2018). Cancer stem cell vaccination with PD-L1 and CTLA-4 blockades enhances the eradication of melanoma stem cells in a mouse tumor model. J. Immunother. 41, 361–368. doi: 10.1097/CJI.0000000000000242
Keywords: cancer stem cells, metastasis, anti-cancer therapies, immunotherapy, epigenetic inhibitors
Citation: Turdo A, Veschi V, Gaggianesi M, Chinnici A, Bianca P, Todaro M and Stassi G (2019) Meeting the Challenge of Targeting Cancer Stem Cells. Front. Cell Dev. Biol. 7:16. doi: 10.3389/fcell.2019.00016
Received: 14 November 2018; Accepted: 01 February 2019;
Published: 18 February 2019.
Edited by:
Hasan Korkaya, Augusta University, United StatesReviewed by:
Bradley Doble, McMaster University, CanadaShree Ram Singh, National Cancer Institute at Frederick, United States
Copyright © 2019 Turdo, Veschi, Gaggianesi, Chinnici, Bianca, Todaro and Stassi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giorgio Stassi, Z2lvcmdpby5zdGFzc2lAdW5pcGEuaXQ=