Abstract
Macro (Autophagy) is a catabolic process that relies on the cooperative function of two organelles: the lysosome and the autophagosome. The recent discovery of a transcriptional gene network that co-regulates the biogenesis and function of these two organelles, and the identification of transcription factors, miRNAs and epigenetic regulators of autophagy, demonstrated that this catabolic process is controlled by both transcriptional and post-transcriptional mechanisms. In this review article, we discuss the nuclear events that control autophagy, focusing particularly on the role of the MiT/TFE transcription factor family. In addition, we will discuss evidence suggesting that the transcriptional regulation of autophagy could be targeted for the treatment of human genetic diseases, such as lysosomal storage disorders (LSDs) and neurodegeneration.
Introduction
Autophagy is an evolutionary conserved catabolic process devoted to the degradation of intracellular components. Three main types of autophagy have been described to date: macroautophagy, microautophagy, and chaperon-mediated autophagy. Macroautophagy involves the formation of a double-membrane vesicle, the autophagosome, which captures cytoplasmic contents and then fuses with lysosomes to generate autophagolysosomes, structures in which cargo substrates are degraded by lysosomal enzymes (Mizushima et al., 2008; He and Klionsky, 2009; Hurley and Schulman, 2014). In microautophagy, cytoplasmic constituents are directly imported into the lysosome and degraded (Ahlberg et al., 1982; Mijaljica et al., 2011; Sahu et al., 2011), while chaperon-mediated autophagy is characterized by the translocation of cytosolic proteins harboring the pentapeptide KFERQ sequence across the lysosomal membrane for degradation (Kaushik and Cuervo, 2012). Thus, the three types of autophagy rely on functional lysosomes to digest intracellular cargos.
Macroautophagy (herein referred to as autophagy) is constitutively active, albeit at low levels, in most cells of our body as part of the constitutive turnover of cytosolic components (Mizushima and Komatsu, 2011). This is generally referred as “basal autophagy.” In addition, different cellular stimuli, in particular nutrient starvation, can potently stimulate autophagy to enhance the degradation of cytosolic components to generate energy (Kaur and Debnath, 2015). Two nutrient-responsive kinases, mTORC1 and AMPK, rapidly respond to nutrient fluctuations and phosphorylate critical regulators of autophagosome biogenesis and maturation (e.g., fusion with lysosomes) (Egan et al., 2011). In particular, in the presence of nutrients, mTORC1 phosphorylates two fundamental autophagy initiation proteins, unc-51-like autophagy activating kinase (ULK)1 and ATG13, inhibiting their pro-autophagic activity (Hosokawa et al., 2009). Conversely, nutrient depletion inactivates mTORC1 and concomitantly activates AMPK, which phosphorylates ULK1 and ATG13 on specific amino acid residues promoting ULK1/ATG13 complex activity and autophagy initiation (Shang et al., 2011). In addition, several other mechanisms of post-translational regulation of autophagy in response to nutrient fluctuations have been described and reviewed elsewhere [see for example reviews (He and Klionsky, 2009; Kuma and Mizushima, 2010; Rabinowitz and White, 2010; Mizushima et al., 2011)].
The modulation of autophagy in the maintenance of cellular homeostasis goes far beyond the response to nutrient fluctuation, as cells exploit autophagy to eliminate damaged organelles, misfolded proteins, and invading organisms (Deretic et al., 2006; Mizushima et al., 2008). Deregulation of these autophagy-dependent cytoprotective functions has been associated to different pathologies, including immune disorders, neurodegenerative diseases, cancer and aging (Deretic et al., 2006; Hara et al., 2006; Komatsu et al., 2006; Harris and Rubinsztein, 2011; Mizushima and Komatsu, 2011; White, 2015).
For a long time, autophagy was considered as a pathway exclusively regulated by cytosolic processes. This concept was supported by the observation that enucleated cells still form autophagosomes (Morselli et al., 2011). However, increasing amounts of evidence collected in the last decade clearly indicate that nuclear transcriptional and epigenetic events play a major role in autophagy regulation. This review aims to summarize the “nuclear” control of autophagy, focusing in particular on the co-regulation of autophagy and lysosome biogenesis by the transcription factor EB (TFEB).
Transcriptional Regulation of Autophagy
The first observation that autophagy can be induced at the transcriptional level was made in yeast in Kirisako et al. (1999), who reported that nitrogen starvation induced the upregulation of the essential autophagy gene Apg8p, the homologous of mammalian LC3. In the last 10 years several laboratories demonstrated that transcription factors that enhance the expression of autophagy genes (even few of them) increase autophagy and the degradation of unwanted substrates [see below and (Lapierre et al., 2015; Füllgrabe et al., 2016)]. These observations opened a new, unexpected, scenario indicating that autophagy activity could in fact be modulated from the nucleus.
TFEB and MiT Factors
Transcription factor EB is a member of the microphthalmia/ transcription factor E (MiT/TFE) family of transcription factors (TFs) that also includes MITF, TFE3, and TFEC proteins (Hemesath et al., 1994). They belong to the larger family of basic helix-loop-helix leucine zipper (bHLH-Zip) transcription factors, such as MYC, MAD, and MAX, and share a basic DNA-binding domain, and an HLH plus a leucine zipper domain important for dimerization (Beckmann et al., 1990; Sato et al., 1997; Steingrimsson et al., 2002). The homo- or hetero- dimerization is necessary to activate transcription. MiT/TFE members can only form heterodimers among each other due to structural constraints in their leucine zipper domain (Hemesath et al., 1994; Pogenberg et al., 2012). Binding to DNA is mediated by the recognition of a common DNA hexanucleotide sequence (CACGTG) known as the E-box (Hemesath et al., 1994). This sequence conforms to the canonical CANNTG motif, recognized by other bHLH-Zip transcription factors, however, specific nucleotide residues that flank this motif characterize the coordinated lysosomal expression and regulation (CLEAR) motif (GTCACGTGAC) that is preferentially recognized by MiT/TFE members (Sardiello et al., 2009; Palmieri et al., 2011; Martina et al., 2014). Bioinformatic analysis identified one or more CLEAR motifs in the promoter region of many lysosomal genes. Notably, these genes belong to different functional lysosomal categories, (ion channels, hydrolases, and transmembrane proteins, etc.) so that TFEB activation leads to a global enhancement of lysosomal catabolic efficiency (Sardiello et al., 2009).
In addition, TFEB also regulates the expression of genes involved in different steps of the autophagy process, such as genes important for autophagy initiation (BECN1, WIPI1, ATG9B, and NRBF2) autophagosome membrane elongation (GABARAP, MAP1LC3B, and ATG5), but also genes important for substrate capture (SQSTM1) and for autophagosomes trafficking and fusion with lysosomes (UVRAG, RAB7) (Palmieri et al., 2011; Settembre et al., 2011). As a result, TFEB activation induces a striking increase in autophagy flux. Similarly, TFE3 and MITF were successively identified as regulators of autophagy and lysosomal biogenesis (Martina et al., 2014; Ploper et al., 2015).
Transcription factor EB activity is largely controlled by its subcellular localization, which is mainly regulated by phosphorylation (Puertollano et al., 2018). Phosphorylated TFEB is sequestered into the cytosol, hence the transcriptional induction of its target genes is inhibited. Conversely, upon nutrient starvation, TFEB is dephosphorylated and rapidly translocates into the nucleus where it binds to the promoter of target genes (Settembre et al., 2011). To date, different kinases that phosphorylate TFEB have been identified. mTOR, as part of the protein complex mTORC1, represents the main kinase responsible for TFEB phosphorylation in presence of amino acids (Peña-Llopis et al., 2011; Martina et al., 2012; Roczniak-Ferguson et al., 2012; Settembre et al., 2012). Inhibition of TFEB activity via phosphorylation of conserved amino acid residues (Ser 142, Ser 211, Ser122, and Ser138) is part of a larger metabolic response mediated by mTORC1 aimed to shut-off catabolic pathways while turning on anabolic ones when nutrients are available (Martina et al., 2012; Roczniak-Ferguson et al., 2012; Settembre et al., 2012; Vega-Rubin-de-Celis et al., 2017; Napolitano G. et al., 2018). Similarly, mTORC1 also regulates the nuclear localization of TFE3 and some isoforms of MITF, thus efficiently inhibiting transcriptional induction of lysosome biogenesis and autophagy (Martina et al., 2014).
In addition, mTORC1 can inhibit TFEB transcriptional activity by modulating the zinc finger transcription factors harboring Kruppel-associated box (KRAB) and SCAN domain (ZKSCAN3) activity (Chauhan et al., 2013). ZKSCAN3 represses a large group of lysosomal and autophagy genes when nutrients, in particular amino acids, are present in the cell. Conversely, treatment with the mTOR inhibitor Torin1 induced ZKSCAN3 nuclear exclusion. Silencing of ZKSCAN3 augmented TFEB-mediated lysosomal and autophagic activation suggesting that these two transcription factors act in opposite ways to regulate autophagy in response to nutrient fluctuations (Figure 1A). While this mechanism appears to be relevant in cell culture experiments, its relevance in vivo is unclear (Pan et al., 2017).
FIGURE 1
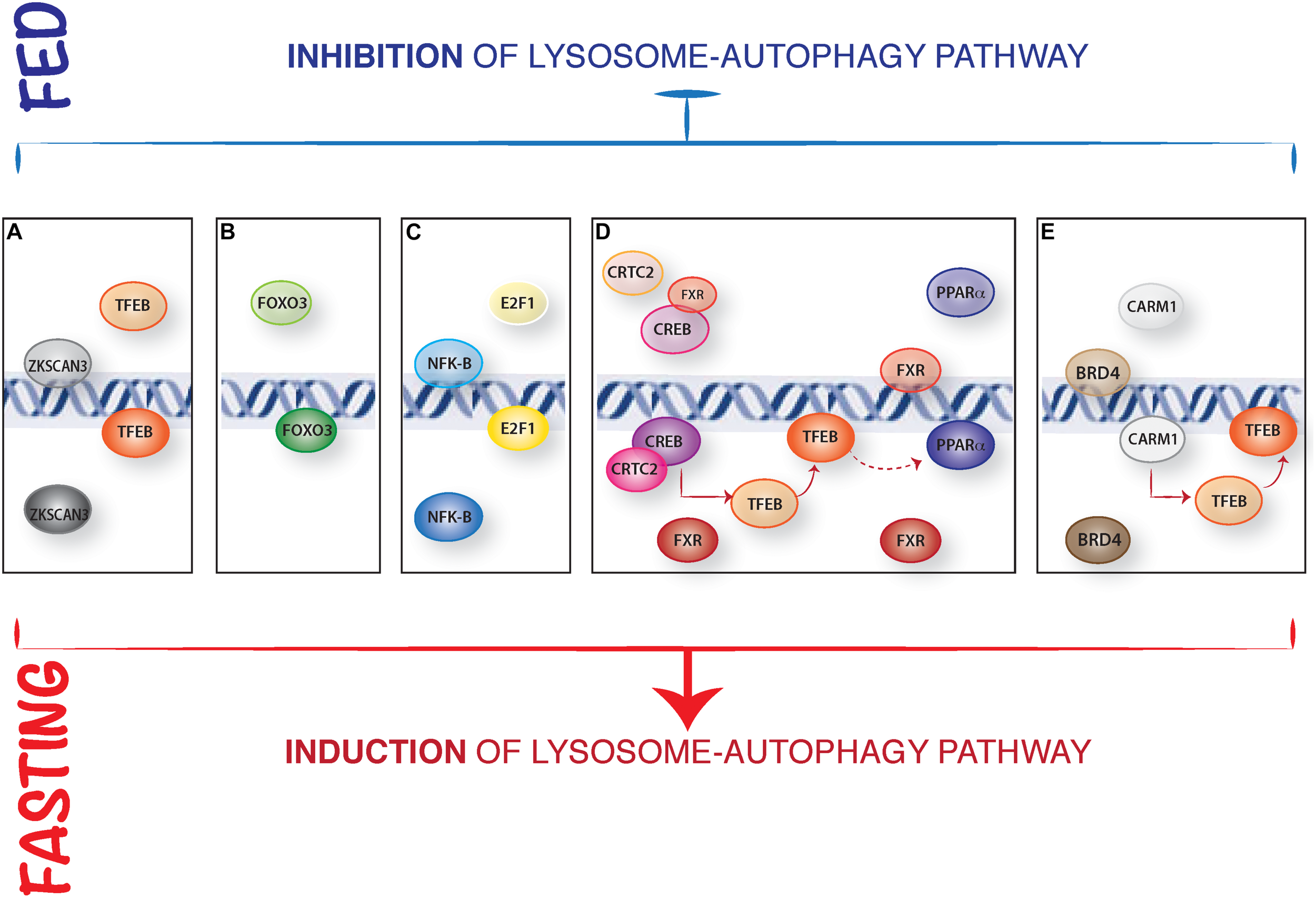
Representative model of the nuclear control of lysosome-autophagy pathway. (A) Opposed regulation of ZKSCAN3 and TFEB. In presence of nutrients, TFEB is cytosolic, and the transcription factor ZKSCAN3 localizes in the nucleus, inhibiting lysosome gene expression. During starvation, ZKSCAN3 translocates into the cytosol and TFEB translocates into the nucleus where activates lysosome-autophagy gene expression. (B) The nuclear translocation and activation of FOXOs transcription factors is induced IN serum starved condition. (C) NFKB binds to the promoter and represses Bnip3 expression in fed conditions, while during starvation Bnip3 expression is promoted by E2F1. (D) In presence of nutrients, FXR inhibits autophagy by preventing the binding of PPARα to DNA and by inhibiting CREB interaction with its coactivator CRTC2. Conversely, during starvation FXR activity is inhibited, and CREB-CRTC2 complex is formed and binds to the promoters of lysosomal autophagy genes and of TFEB; similarly, starvation-mediated inhibition of FXR allows PPARα binding to the DR1 elements in the promoters of autophagy genes. (E) Epigenetic regulation of autophagy: in fed status, CARM1 is inactive and BRD4 represses the expression of autophagic and lysosomal genes regulating Histone3 lysine9 methylation. In fast state, BRD4 is inactive and CARM1 translocates into the nucleus promoting lysosomal-autophagy gene expression via a positive Histone3 Arginine17 methylation and inducing TFEB transcriptional activity.
In addition to mTORC1, other growth-regulating kinases control TFEB nuclear localization. ERK2 was the first kinase to be associated with TFEB phosphorylation in response to nutrients availability (Settembre et al., 2011). In particular, ERK2 mediated phosphorylation of TFEB at Ser142 inhibited TFEB nuclear translocation thus limiting transcriptional activation of its downstream target genes (Settembre et al., 2011; Li et al., 2018, 2019). Subsequently, the glycogen synthase kinase 3 beta (GSK3B) was identified as the kinase responsible for TFEB phosphorylation at Ser134 and 138 (Li et al., 2016). This event, coupled to phosphorylation at Ser142 by ERK2 and mTORC1, unmasks a nuclear export localization signal required for TFEB cytosolic accumulation (Li et al., 2018). Moreover, the Akt and the PKCβ kinases phosphorylate TFEB at c-terminal critical serines, but this phosphorylation seems to control TFEB stability rather than its nuclear localization (Ferron et al., 2013; Palmieri et al., 2017).
Transcription factor EB nuclear translocation can also be triggered by activation of the calcium and calmodulin dependent serine/threonine phosphatase calcineurin (Medina et al., 2015). Notably, the calcium efflux through the lysosomal cation channel Mucolipin1 triggers calcineurin-mediated TFEB dephosphorylation and activation, hence providing a mechanistic explanation of autophagy regulation by calcium signaling.
More recently, the protein phosphatase 2A (PP2A) has been shown to dephosphorylate TFEB upon induction of acute oxidative stress by sodium arsenite (Martina and Puertollano, 2018).
To date, the mechanisms controlling TFEB nuclear export are less characterized but seem to be dependent on the CRM1 exportin and on the presence of a TFEB nuclear export sequence (Napolitano G. et al., 2018). Intriguingly, mTOR-dependent TFEB re-phosphorylation in the nucleus seems to play a major role in TFEB nuclear export.
These studies indicate that several signaling events regulate TFEB subcellular localization, thus placing the transcriptional activation of the lysosomal-autophagy pathway as a general response to cope with different types of cellular stresses.
FOXO Factors
The class O of forkhead box transcription factors (FOXO) family has an established role in autophagy regulation (Webb and Brunet, 2014). In mammals, this family includes four members: FOXO1, FOXO3, FOXO4, and FOXO6. The activity of three out of four members (FOXO1, FOXO3, and FOXO4) is mainly regulated by AKT phosphorylation in response to growth factors and insulin stimulation. FOXO3 was the first FOXO member identified as a transcriptional regulator of several autophagy genes (ATG4, ATG12, BECN1, BNIP3, LC3, ULK1, ULK2, and VPS34) in muscle (Mammucari et al., 2007; Zhao et al., 2007; Sanchez et al., 2012). Similar to what reported for MiT/TFE family of transcription factors, FOXO3 transcriptional activity is mostly regulated by a nuclear/cytosolic shuttling. Once activated by growth factors, AKT phosphorylates FOXO3 and this results in its cytoplasmic retention, thus inhibiting transcriptional activation of its target genes. Later on, another member of this family, FOXO1, was also described as a transcriptional regulator of different autophagy genes (Liu et al., 2009; Xu et al., 2011; Xiong et al., 2012). However, FOXO1 also induces autophagy in a transcriptional-independent way: in response to oxidative stress or serum starvation, FOXO1 is acetylated in the cytosol and binds to Atg7 thus favoring autophagy induction by direct interaction with key regulators of autophagosome biogenesis (Zhao et al., 2010; Figure 1B). More recently FOXO transcription factors have been shown to cooperatively control autophagy in cartilage and protect against osteoarthritis (Matsuzaki et al., 2018).
Most notably, a study using Caenorhabditis elegans demonstrated that DAF16 (FOXO in mammals) physically and functionally cooperates with HLH30 (TFEB in mammals) to ensure appropriate expression of target genes during organismal responses to stressors (Lin et al., 2018). It will be important to understand whether a FOXO-TFEB cooperation occurs also in mammals.
P53
Different studies suggest that P53, the most studied tumor suppressor protein, is an inducer of the autophagy pathway. P53 was initially described to promote autophagy by inhibiting the mTORC1 pathway, through transcriptional induction of Sestrin proteins, which activate AMPK while inhibiting mTORC1 lysosomal recruitment (Budanov and Karin, 2008; Chantranupong et al., 2014), and by inducing the expression of the Damaged-regulated- modulator DRAM, a lysosomal protein, which induces autophagy through a yet not identified mechanism (Crighton et al., 2006). Subsequently, a combined CHIP-SEQ and RNA-SEQ analysis performed on mouse embryonic fibroblasts (MEFs) upon DNA-damage, revealed that P53 controls the expression of several genes essential for autophagy induction (LKB1, ULK1/2), and autophagosome maturation (ATG4, ATG7, and ATG10) (Kenzelmann Broz et al., 2013). Moreover, P53 regulates both FOXO3a expression and activity (You et al., 2004; Fu et al., 2009; Miyaguchi et al., 2009; Renault et al., 2011), and promotes TFEB/TFE3 nuclear translocation upon DNA damage (Jeong et al., 2018), thus controlling key upstream modulators of the autophagy pathway.
However, cytoplasmatic P53 may also act as a negative regulator of autophagy, although the mechanisms underlying this inhibitory regulation are still elusive (Green and Kroemer, 2009; Comel et al., 2014). Further studies are needed to fully define the role of P53 in the regulation of autophagy pathway.
E2F1/NF-kB Axis
The transcription factors E2F1 and NF-kB regulate autophagy through the regulation of BNIP3 expression (Tracy et al., 2007; Gang et al., 2011). BNIP3 is a hypoxia-induced activator of autophagy that disrupts the inhibitory binding of B-cell lymphoma 2 (BCL-2) to Beclin1, a component of the class III phosphatidylinositol-3-OH kinase (PI3K) complex, that promotes autophagosome biogenesis. During normoxia, NF-kB constitutively binds to the promoter of BNIP3 repressing its expression (Shaw et al., 2008). Hypoxia reduces the occupancy of NF-kB on the BNIP3 promoter thus allowing E2F1 to induce its expression and activate autophagy (Figure 1C). In addition, E2F1 can also promote the expression of other autophagy genes, such as ULK1, LC3, and ATG5 (Polager et al., 2008).
CREB-FXR and PPARα-FXR Circuits
The farnesoid X receptor (FXR) represses liver autophagy during feeding conditions (Thomas et al., 2008; Calkin and Tontonoz, 2012). FXR is activated by increased bile acid levels after feeding and transcriptionally represses several autophagy genes through two apparently independent mechanisms. Seok et al. (2014) proposed that FXR inhibits the transcriptional activity of the fasting-activated cAMP response element-binding protein (CREB) by impeding the interaction between CREB and its coactivator CRTC2. Upon fasting, FXR inhibition is relieved thus allowing the CREB-CRTC2 complex to form and induce the expression of many autophagy genes, including ATG7, ULK1, and TFEB (Figure 1D). Interestingly, TFEB also regulates the expression of genes important for lipid metabolism in the liver, suggesting that its role in the FXR-CREB axis might be not limited to autophagy regulation (Settembre et al., 2013). In addition, Lee et al. (2014) identified the nuclear receptor Peroxisome Proliferator Activated Receptor alpha (PPARα) as the transcriptional activator that opposes FXR in response to nutrient availability. FXR and PPARα share the ability to bind to specific DNA sites (DR1 elements) in the promoter regions of many autophagy-related genes, so that these two nuclear receptors compete for the binding to the same target genes. Fasting activates PPARα while inhibiting FXR, thus inducing transcriptional activation of autophagy genes in liver (Figure 1D). Notably, TFEB transcriptionally enhances the expression of PPARα and its coactivator peroxisome proliferator activated receptor gamma 1 alpha (PGC1α) (Settembre et al., 2013), suggesting that the induction of TFEB expression by CREB could in turn potentiate PPARα activity. Thus, it is possible that both the FXR-CREB and FXR-PPARα circuits coexist and participate to the coordination of autophagy with other metabolic processes (e.g., lipid degradation) occurring in the liver.
Epigenetic Regulation of Autophagy
Histone post-translational modifications, such as methylation, acetylation, and deacetylation, influence the overall chromatin structure, thus affecting the accessibility of transcription factors to chromatin (Lawrence et al., 2016). To date, several examples of epigenetic regulations of the autophagy pathway have been described.
Histone Methylation
The epigenetic reader Bromodomain-containing protein 4 (BRD4) has been identified as a repressor of a transcriptional program that promotes autophagy and lysosome biogenesis (Sakamaki et al., 2017). In presence of nutrients, BRD4 represses the expression of several autophagic and lysosomal genes by recruiting the histone lysine methyltransferase G9a, which deposits a repressive H3K9diMe in the promoters of lysosomal and autophagy genes. Conversely, nutrient depletion promotes AMPK-mediated BRD4 inhibition and the expression of lysosomal and autophagic genes through a yet-to be characterized transcriptional regulator.
The co-activator-associated arginine-methyltransferase 1 (CARM1) was recently identified as a key autophagy regulator (Shin et al., 2016). Glucose (but also amino acid) starvation leads to a CARM1-dependent increase in histone H3 Arg17 dimethylation levels at the promoters of autophagy and lysosomal genes and this is critical for proper autophagy activation. Mechanistically, upon starvation CARM1 translocates into the nucleus where binds TFEB and promotes the transcriptional activation of its target genes. CARM1 seems to be essential for TFEB-mediated autophagy activation since TFEB overexpression fails to increase autophagy in cells lacking CARM1 (Figure 1E).
Histone Acetylation
Recently, a global decrease in acetylation levels of H4K16 was described upon nutrient starvation and/or mTOR inhibition (Füllgrabe et al., 2013). This downregulation translates into a transcriptional repression of key autophagy genes in order to prevent a chronic autophagy induction, which could be lethal. These responses are dependent on the histone acetyltransferase hMOF/KAT8/MYST1.
The NAD+-dependent deacetylase Sirt1 regulates autophagy through its deacetylase activity on non-histone cytosolic targets (Lee et al., 2008; Bao and Sack, 2010). Sirt1 may induce autophagy directly by deacetylating autophagy proteins such as ATG5, ATG7 and LC3. Sirt1 might also control the stability of mRNAs encoding for lysosomal enzymes (Latifkar et al., 2019). Moreover, Sirt1 deacetylates the transcriptional regulators of autophagy FOXO1 and FOXO3, enhancing their transcriptional activity (Brunet et al., 2004). Finally, Sirt1 promotes autophagy by activating AMPK, via deacetylation of LKB1 (Lan et al., 2008), while inhibiting mTORC1 signaling favoring its interaction with the TSC1/TSC2 complex (Ghosh et al., 2010).
Additional epigenetic modifications related to autophagy induction are H3K9 methylation (Artal-Martinez de Narvajas et al., 2013), H3K56 acetylation (Chen et al., 2012) and H4K20 methylation (Kourmouli et al., 2004). These are associated with suppression of autophagy, even if further studies are required to clarify their regulation.
MITF Factors and Human Diseases
The autophagy pathway is important in several processes required to maintain cellular homeostasis, including adaptation to metabolic stress, removal of dangerous cargo, and prevention of DNA damage. If any of these protective functions are impaired, onset and progression of several diseases, such as infection, cancer, neurodegeneration, cardiovascular diseases, and aging may be favored (Mizushima et al., 2008; Harris and Rubinsztein, 2011; Mizushima and Komatsu, 2011; White, 2015). Therefore, it is not surprising that a long list of diseases is associated to mutations in autophagy-related genes [recently reviewed in Levine and Kroemer (2019)]. However, it is important to note that several autophagy proteins participate to other cellular processes, such as vesicular trafficking, phagocytosis, exocytosis, and even cell cycle regulation and immunity, thus the link between disease manifestation and autophagy dysfunction might be difficult to establish (Levine and Kroemer, 2019). This is particularly true for transcription factors, that control the expression of target genes implicated in a number of diverse cellular functions. The activity and/or the localization of TFEB has been reported to be deregulated in several neurodegenerative diseases, such as X-linked spinal and bulbar muscular atrophy (Cortes et al., 2014), Parkinson disease (Decressac et al., 2013), Huntington disease (Tsunemi et al., 2012), and Alzheimer disease (Reddy et al., 2016). These neurodegenerative disorders are characterized by intracellular protein aggregation and autophagy dysfunction, which is predicted to contribute to disease establishment (Menzies et al., 2015). Notably, forced overexpression of TFEB in cellular and murine models of these disorders significantly reduced protein aggregation attenuating pathological manifestation, suggesting that TFEB represents an appealing target for therapy (Sardiello et al., 2009; Dehay et al., 2010; Tsunemi et al., 2012; Decressac et al., 2013; Polito et al., 2014; Xiao et al., 2014, 2015; Chauhan et al., 2015; Kilpatrick et al., 2015).
Lysosomal storage disorders (LSDs) are a class of rare diseases due to mutations in genes encoding for lysosomal proteins (Ballabio and Gieselmann, 2009; Cox and Cachón-González, 2012; Platt et al., 2018). As a consequence, cells show progressive accumulation of indigested material within lysosomes and, eventually, impaired autophagy flux. Interestingly, TFEB was found to be predominantly nuclear in several LSD cellular models (Sardiello et al., 2009; Bartolomeo et al., 2017). The increased nuclear localization of TFEB may be interpreted as an attempt to compensate for the decreased autophagy flux and lysosomal degradative function. While in this context the physiological induction of the TFEB seems to be unable to fully counteract disease progression, TFEB overexpression in different LSDs, such as multiple sulfatase deficiency and mucopolysaccharidosis IIIA (Medina et al., 2011), Pompe disease (Spampanato et al., 2013), Batten disease (Palmieri et al., 2017), Gaucher and Tay Sachs disease (Song et al., 2013), and cystinosis (Rega et al., 2016) resulted effective in reducing lysosomal storage. This effect is most likely the consequence of TFEB’s ability to concomitantly induce lysosomal exocytosis, autophagy and lysosome biogenesis. Similarly, TFEB overexpression in liver had beneficial effects in mouse models of alpha1-antitrypsin deficiency and hepatic hyperammonemia (Pastore et al., 2013; Soria et al., 2018). Notably, by increasing the autophagic degradation of intracellular lipid droplets, TFEB also represents a potential therapeutic target to fight metabolic syndrome associated with obesity (Settembre et al., 2013). Despite the induction of TFEB activity looks as a promising therapeutic tool for several diseases, the side effects of its long-term overexpression must be considered. The over-activation of MiT family of transcription factors is associated with different types of cancer. MITF genomic amplification is frequently found in melanoma, while chromosomal translocations and rearrangements of TFE3 and TFEB are associated with pediatric renal cell carcinomas and alveolar soft part sarcoma (Argani et al., 2001; Haq and Fisher, 2011; Kauffman et al., 2014). Moreover, upregulation of MiT/TFE members has also been observed in pancreatic ductal adenocarcinoma (Perera et al., 2015).
How over-activation of these TFs may favor pro-tumorigenic processes is not completely clear, but recent data indicate that hyper-activation of mTORC1 signaling is a common feature of MiT/TFE associated malignancies (Di Malta et al., 2017). This signaling deregulation depends on the constitutive induction of the essential components of the mTORC1 amino acid sensing machinery RagD and RagC GTPases, direct downstream targets of MiT/TFE TFs. Interestingly, at least in pancreatic ductal adenocarcinoma, the upregulation of MiT/TFE factors leads to simultaneous mTORC1 hyperactivation and autophagy induction and presumably both pathways are exploited by tumor cells to efficiently compete with non-transformed cells (Perera et al., 2015, 2019; Di Malta et al., 2017). In light of the pathological consequences of the constitutive activation of MiT/TFE factors, a pulsatile approach aimed at enhancing TFEB activity only for a certain time-frame could represent a therapeutic strategy for diseases that might benefit of the stimulation of the lysosomal/autophagy pathway.
Conclusion
In the last years, several studies provided conclusive evidence that autophagy is a transcriptionally regulated process. However, despite different transcriptional modulators of autophagy have been identified, we still know very little about the physiological relevance of this nuclear regulation. The most likely hypothesis is that transcriptional regulation of autophagy cooperates with the post-translational regulation to achieve a fine tuning of autophagy flux particularly in conditions of prolonged starvation or chronic stress. Indeed, the degradation of autophagy proteins, in particular those serving as cargo receptors, is enhanced during autophagy, and similarly lysosomes are utilized during the formation of autolysosomes. Hence, the transcriptional induction of lysosomal and autophagy genes might counteract the depletion of the correspondent proteins during autophagy. Consistently, the translation of mRNAs encoding for proteins with catabolic roles is spared from the general inhibition of protein synthesis during nutrient starvation (Saikia et al., 2016). Additionally, the transcriptional regulation of autophagy might participate to biological processes that are regulated independently of the nutrient status of the cells, such as cellular differentiation and tissue development (Cinque et al., 2015). It will be important in the next years to understand whether different transcription factors regulate selective types of autophagy in a tissue and time specific fashion and if their modulation can be exploited for therapeutic purposes.
A selective modulation of autophagy might be beneficial for the treatment of several diseases for which there are no currently available therapies. Notably, several therapeutic benefits associated to administration of widely used drugs, such as aspirin and metformin, and food compounds, such as resveratrol and curcumin, might be due their ability to induce TFEB nuclear translocation and autophagy (Bao et al., 2016; Zhang et al., 2016; Wang et al., 2017; Yan et al., 2017; Chandra et al., 2018). Currently, whether these molecules can be repositioned for the treatment of genetic diseases is largely unexplored. Lastly, the use of computational approaches combined to an integrated analysis of omics data represents an invaluable tool to identify novel transcriptional modulators of autophagy (Napolitano F. et al., 2018).
Statements
Author contributions
CDM and CS wrote the manuscript. LC prepared the figure and wrote the figure legend.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
Ahlberg J. Marzella L. Glaumann H. (1982). Uptake and degradation of proteins by isolated rat liver lysosomes. suggestion of a microautophagic pathway of proteolysis.Lab. Invest.47523–532.
2
Argani P. Antonescu C. R. Illei P. B. Lui M. Y. Timmons C. F. Newbury R. et al (2001). Primary renal neoplasms with the ASPL-TFE3 gene fusion of alveolar soft part sarcoma.Am. J. Pathol.159179–192.
3
Artal-Martinez de Narvajas A. Gomez T. S. Zhang J.-S. Mann A. O. Taoda Y. Gorman J. A. et al (2013). Epigenetic regulation of autophagy by the methyltransferase G9a.Mol. Cell. Biol.333983–3993. 10.1128/MCB.00813-13
4
Ballabio A. Gieselmann V. (2009). Lysosomal disorders: from storage to cellular damage.Biochim. Et Biophy. Acta1793684–696. 10.1016/j.bbamcr.2008.12.001
5
Bao J. Sack M. N. (2010). Protein deacetylation by sirtuins: delineating a post-translational regulatory program responsive to nutrient and redox stressors.Cell. Mol. Life Sci.673073–3087. 10.1007/s00018-010-0402-y
6
Bao J. Zheng L. Zhang Q. Li X. Zhang X. Li Z. et al (2016). Deacetylation of TFEB promotes fibrillar Aβ degradation by upregulating lysosomal biogenesis in microglia.Protein Cell7417–433. 10.1007/s13238-016-0269-2
7
Bartolomeo R. Cinque L. De Leonibus C. Forrester A. Salzano A. C. Monfregola J. et al (2017). MTORC1 hyperactivation arrests bone growth in lysosomal storage disorders by suppressing autophagy.J. Clin. Invest.1273717–3729. 10.1172/JCI94130
8
Beckmann H. Su L. K. Kadesch T. (1990). TFE3: a helix-loop-helix protein that activates transcription through the immunoglobulin enhancer MuE3 motif.Genes Dev.4167–179.
9
Brunet A. Sweeney L. B. Sturgill J. F. Chua K. F. Greer P. L. Lin Y. et al (2004). Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase.Science3032011–2015. 10.1126/science.1094637
10
Budanov A. V. Karin M. (2008). P53 target genes sestrin1 and sestrin2 connect genotoxic stress and MTOR signaling.Cell134451–460. 10.1016/j.cell.2008.06.028
11
Calkin A. C. Tontonoz P. (2012). Transcriptional integration of metabolism by the nuclear sterol-activated receptors LXR and FXR.Nat. Rev. Mol. Cell Biol.13213–224. 10.1038/nrm3312
12
Chandra S. Jana M. Pahan K. (2018). Aspirin induces lysosomal biogenesis and attenuates amyloid plaque pathology in a mouse model of alzheimer’s disease via PPARα.J. Neurosci.386682–6699. 10.1523/JNEUROSCI.0054-18.2018
13
Chantranupong L. Wolfson R. L. Orozco J. M. Saxton R. A. Scaria S. M. Bar-Peled L. et al (2014). The sestrins interact with GATOR2 to negatively regulate the amino-acid-sensing pathway upstream of MTORC1.Cell Rep.91–8. 10.1016/j.celrep.2014.09.014
14
Chauhan S. Ahmed Z. Bradfute S. B. Arko-Mensah J. Mandell M. A. Choi S. W. et al (2015). Pharmaceutical screen identifies novel target processes for activation of autophagy with a broad translational potential.Nat. Comm.6:8620. 10.1038/ncomms9620
15
Chauhan S. Goodwin J. G. Chauhan S. Manyam G. Wang J. Kamat A. M. et al (2013). ZKSCAN3 is a master transcriptional repressor of autophagy.Mol. Cell5016–28. 10.1016/j.molcel.2013.01.024
16
Chen H. Fan M. Pfeffer L. M. Laribee R. N. (2012). The Histone H3 lysine 56 acetylation pathway is regulated by target of rapamycin (TOR) signaling and functions directly in ribosomal RNA biogenesis.Nucleic Acids Res.406534–6546. 10.1093/nar/gks345
17
Cinque L. Forrester A. Bartolomeo R. Svelto M. Venditti R. Montefusco S. et al (2015). FGF signalling regulates bone growth through autophagy.Nature528272–275. 10.1038/nature16063
18
Comel A. Sorrentino G. Capaci V. Del Sal G. (2014). The cytoplasmic side of P53’s oncosuppressive activities.FEBS Lett.5882600–2609. 10.1016/j.febslet.2014.04.015
19
Cortes C. J. Miranda H. C. Frankowski H. Batlevi Y. Young J. E. Le A. et al (2014). Polyglutamine-expanded androgen receptor interferes with TFEB to elicit autophagy defects in SBMA.Nat. Neurosci.171180–1189. 10.1038/nn.3787
20
Cox T. M. Cachón-González M. B. (2012). The cellular pathology of lysosomal diseases.J. Pathol.226241–254. 10.1002/path.3021
21
Crighton D. Wilkinson S. O’Prey J. Syed N. Smith P. Harrison P. R. et al (2006). DRAM, a P53-induced modulator of autophagy, is critical for apoptosis.Cell126121–134. 10.1016/j.cell.2006.05.034
22
Decressac M. Mattsson B. Weikop P. Lundblad M. Jakobsson J. Björklund A. (2013). TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity.Proc. Natl. Acad. Sci. U.S.A.110E1817–E1826. 10.1073/pnas.1305623110
23
Dehay B. Bové J. Rodríguez-Muela N. Perier C. Recasens A. Boya P. et al (2010). Pathogenic lysosomal depletion in parkinson’s disease.J. Neurosci.3012535–12544. 10.1523/JNEUROSCI.1920-10.2010
24
Deretic V. Singh S. Master S. Harris J. Roberts E. Kyei G. et al (2006). Mycobacterium tuberculosis inhibition of phagolysosome biogenesis and autophagy as a host defence mechanism.Cell. Microbiol.8719–727. 10.1111/j.1462-5822.2006.00705.x
25
Di Malta C. Siciliano D. Calcagni A. Monfregola J. Punzi S. Pastore N. et al (2017). Transcriptional activation of RagD GTPase controls MTORC1 and promotes cancer growth.Science3561188–1192. 10.1126/science.aag2553
26
Egan D. Kim J. Shaw R. J. Guan K. L. (2011). The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and MTOR.Autophagy7643–644.
27
Ferron M. Settembre C. Shimazu J. Lacombe J. Kato S. Rawlings D. J. et al (2013). A RANKL-PKCβ-TFEB signaling cascade is necessary for lysosomal biogenesis in osteoclasts.Genes Dev.27955–969. 10.1101/gad.213827.113
28
Fu W. Ma Q. Chen L. Li P. Zhang M. Ramamoorthy S. et al (2009). MDM2 acts downstream of P53 as an E3 ligase to promote FOXO ubiquitination and degradation.J. Biol. Chem.28413987–14000. 10.1074/jbc.M901758200
29
Füllgrabe J. Ghislat G. Cho D. Rubinsztein D. C. (2016). Transcriptional regulation of mammalian autophagy at a glance.J Cell Sci.1293059–3066. 10.1242/jcs.188920
30
Füllgrabe J. Lynch-Day M. A. Heldring N. Li W. Struijk R. B. Ma Q. et al (2013). The histone H4 lysine 16 acetyltransferase HMOF regulates the outcome of autophagy.Nature500468–471. 10.1038/nature12313
31
Gang H. Dhingra R. Wang Y. Mughal W. Gordon J. W. Kirshenbaum L. A. (2011). Epigenetic regulation of E2F-1-dependent Bnip3 transcription and cell death by nuclear factor-KB and histone deacetylase-1.Pediatr. Cardiol.32263–266. 10.1007/s00246-011-9893-z
32
Ghosh H. S. McBurney M. Robbins P. D. (2010). SIRT1 negatively regulates the mammalian target of rapamycin.PLoS One5:e9199. 10.1371/journal.pone.0009199
33
Green D. R. Kroemer G. (2009). Cytoplasmic functions of the tumour suppressor P53.Nature4581127–1130. 10.1038/nature07986
34
Haq R. Fisher D. E. (2011). Biology and clinical relevance of the micropthalmia family of transcription factors in human cancer.J. Clin. Oncol.293474–3482. 10.1200/JCO.2010.32.6223
35
Hara T. Nakamura K. Matsui M. Yamamoto A. Nakahara Y. Suzuki-Migishima R. et al (2006). Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice.Nature441885–889. 10.1038/nature04724
36
Harris H. Rubinsztein D. C. (2011). Control of autophagy as a therapy for neurodegenerative disease.Nat. Rev. Neurol.8108–117. 10.1038/nrneurol.2011.200
37
He C. Klionsky D. J. (2009). Regulation mechanisms and signaling pathways of autophagy.Ann. Rev. Genet.4367–93. 10.1146/annurev-genet-102808-114910
38
Hemesath T. J. Steingrímsson E. McGill G. Hansen M. J. Vaught J. Hodgkinson C. A. et al (1994). Microphthalmia, a critical factor in melanocyte development, defines a discrete transcription factor family.Genes Dev.82770–2780. 10.1101/gad.8.22.2770
39
Hosokawa N. Hara T. Kaizuka T. Kishi C. Takamura A. Miura Y. et al (2009). Nutrient-dependent MTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy.Mol. Biol. Cell201981–1991. 10.1091/mbc.e08-12-1248
40
Hurley J. H. Schulman B. A. (2014). Atomistic autophagy: the structures of cellular self-digestion.Cell157300–311. 10.1016/j.cell.2014.01.070
41
Jeong E. Brady O. A. Martina J. A. Pirooznia M. Tunc I. Puertollano R. (2018). The transcription factors TFE3 and TFEB amplify P53 dependent transcriptional programs in response to DNA damage.ELife7:e40856. 10.7554/eLife.40856
42
Kauffman E. C. Ricketts C. J. Rais-Bahrami S. Yang Y. Merino M. J. Bottaro D. P. et al (2014). Molecular genetics and cellular features of TFE3 and TFEB fusion kidney cancers.Nat. Rev. Urol.11465–475. 10.1038/nrurol.2014.162
43
Kaur J. Debnath J. (2015). Autophagy at the crossroads of catabolism and anabolism.Nat. Rev. Mol Cell Biol.16461–472. 10.1038/nrm4024
44
Kaushik S. Cuervo A. M. (2012). Chaperone-mediated autophagy: a unique way to enter the lysosome world.Trends Cell Biol.22407–417. 10.1016/j.tcb.2012.05.006
45
Kenzelmann Broz D. Spano Mello S. Bieging K. T. Jiang D. Dusek R. L. Brady C. A. et al (2013). Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key P53 responses.Genes Dev.271016–1031. 10.1101/gad.212282.112
46
Kilpatrick K. Zeng Y. Hancock T. Segatori L. (2015). Genetic and chemical activation of TFEB mediates clearance of aggregated α-Synuclein.PLoS One10:e0120819. 10.1371/journal.pone.0120819
47
Kirisako T. Baba M. Ishihara N. Miyazawa K. Ohsumi M. Yoshimori T. et al (1999). Formation process of autophagosome is traced with Apg8/Aut7p in yeast.J. Cell Biol.147435–446. 10.1083/jcb.147.2.435
48
Komatsu M. Waguri S. Chiba T. Murata S. Iwata J. Tanida I. et al (2006). Loss of autophagy in the central nervous system causes neurodegeneration in mice.Nature441880–884. 10.1038/nature04723
49
Kourmouli N. Jeppesen P. Mahadevhaiah S. Burgoyne P. Wu R. Gilbert D. M. et al (2004). Heterochromatin and tri-methylated lysine 20 of histone H4 in animals.J. Cell Sci.117(Pt 12), 2491–2501. 10.1242/jcs.01238
50
Kuma A. Mizushima N. (2010). Physiological role of autophagy as an intracellular recycling system: with an emphasis on nutrient metabolism.Semin. Cell Dev. Biol.21683–690. 10.1016/j.semcdb.2010.03.002
51
Lan F. Cacicedo J. M. Ruderman N. Ido Y. (2008). SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. possible role in AMP-activated protein kinase activation.J. Biol. Chem.28327628–27635. 10.1074/jbc.M805711200
52
Lapierre L. R. Kumsta C. Sandri M. Ballabio A. Hansen M. (2015). Transcriptional and epigenetic regulation of autophagy in aging.Autophagy11867–880. 10.1080/15548627.2015.1034410
53
Latifkar A. Ling L. Hingorani A. Johansen E. Clement A. Zhang X. et al (2019). Loss of sirtuin 1 alters the secretome of breast cancer cells by impairing lysosomal integrity.Dev. Cell49393.e–408.e. 10.1016/j.devcel.2019.03.011
54
Lawrence M. Daujat S. Schneider R. (2016). Lateral thinking: how histone modifications regulate gene expression.Trends Genet.3242–56. 10.1016/j.tig.2015.10.007
55
Lee I. H. Cao L. Mostoslavsky R. Lombard D. B. Liu J. Bruns N. E. et al (2008). A role for the NAD-dependent deacetylase sirt1 in the regulation of autophagy.Proc. Natl. Acad. Sci. U.S.A.1053374–3379. 10.1073/pnas.0712145105
56
Lee J. M. Wagner M. Xiao R. Kim K. Feng D. Lazar M. A. et al (2014). Nutrient-sensing nuclear receptors coordinate autophagy.Nature516112–115. 10.1038/nature13961
57
Levine B. Kroemer G. (2019). Biological functions of autophagy genes: a disease perspective.Cell17611–42. 10.1016/j.cell.2018.09.048
58
Li L. Friedrichsen H. J. Andrews S. Picaud S. Volpon L. Ngeow K. et al (2018). A TFEB nuclear export signal integrates amino acid supply and glucose availability.Nat. Comm.9:2685. 10.1038/s41467-018-04849-7
59
Li S. Song Y. Quach C. Guo H. Jang G.-B. Maazi H. et al (2019). Transcriptional regulation of autophagy-lysosomal function in BRAF-driven melanoma progression and chemoresistance.Nat. Comm.10:1693. 10.1038/s41467-019-09634-8
60
Li Y. Xu M. Ding X. Yan C. Song Z. Chen L. et al (2016). Protein kinase C controls lysosome biogenesis independently of MTORC1.Nat. Cell Biol.181065–1077. 10.1038/ncb3407
61
Lin X.-X. Sen I. Janssens G. E. Zhou X. Fonslow B. R. Edgar D. et al (2018). DAF-16/FOXO and HLH-30/TFEB Function as combinatorial transcription factors to promote stress resistance and longevity.Nat. Comm.9:4400. 10.1038/s41467-018-06624-0
62
Liu H.-Y. Han J. Cao S. Y. Hong T. Zhuo D. Shi J. et al (2009). Hepatic autophagy is suppressed in the presence of insulin resistance and hyperinsulinemia: inhibition of Foxo1-dependent expression of key autophagy genes by insulin.J. Biol. Chem.28431484–31492. 10.1074/jbc.M109.033936
63
Mammucari C. Milan G. Romanello V. Masiero E. Rudolf R. Del Piccolo P. et al (2007). FoxO3 controls autophagy in skeletal muscle in vivo.Cell Metab.6458–471. 10.1016/j.cmet.2007.11.001
64
Martina J. A. Chen Y. Gucek M. Puertollano R. (2012). MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB.Autophagy8903–914. 10.4161/auto.19653
65
Martina J. A. Diab H. I. Lishu L. Jeong-A L. Patange S. Raben N. et al (2014). The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris.Sci. Signal.7:ra9. 10.1126/scisignal.2004754
66
Martina J. A. Puertollano R. (2018). Protein phosphatase 2A stimulates activation of TFEB and TFE3 transcription factors in response to oxidative stress.J. Biol. Chem.29312525–12534. 10.1074/jbc.RA118.003471
67
Matsuzaki T. Alvarez-Garcia O. Mokuda S. Nagira K. Olmer M. Gamini R. et al (2018). FoxO transcription factors modulate autophagy and proteoglycan 4 in cartilage homeostasis and osteoarthritis.Sci. Transl. Med.10:eaan0746. 10.1126/scitranslmed.aan0746
68
Medina D. L. Di Paola S. Peluso I. Armani A. De Stefani D. Venditti R. et al (2015). Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB.Nat. Cell Biol.17288–299. 10.1038/ncb3114
69
Medina D. L. Fraldi A. Bouche V. Annunziata F. Mansueto G. Spampanato C. et al (2011). Transcriptional activation of lysosomal exocytosis promotes cellular clearance.Dev. Cell21421–430. 10.1016/j.devcel.2011.07.016
70
Menzies F. M. Fleming A. Rubinsztein D. C. (2015). Compromised autophagy and neurodegenerative diseases.Nat. Rev. Neurosci.16345–357. 10.1038/nrn3961
71
Mijaljica D. Prescott M. Devenish R. J. (2011). Microautophagy in mammalian cells: revisiting a 40-year-old conundrum.Autophagy7673–682.
72
Miyaguchi Y. Tsuchiya K. Sakamoto K. (2009). P53 negatively regulates the transcriptional activity of FOXO3a under oxidative stress.Cell Biol. Int.33853–860. 10.1016/j.cellbi.2009.04.017
73
Mizushima N. Komatsu M. (2011). Autophagy: renovation of cells and tissues.Cell147728–741. 10.1016/j.cell.2011.10.026
74
Mizushima N. Levine B. Cuervo A. M. Klionsky D. J. (2008). Autophagy fights disease through cellular self-digestion.Nature4511069–1075. 10.1038/nature06639
75
Mizushima N. Yoshimori T. Ohsumi Y. (2011). The role of atg proteins in autophagosome formation.Ann. Rev. Cell Dev. Biol.27107–132. 10.1146/annurev-cellbio-092910-154005
76
Morselli E. Mariño G. Bennetzen M. V. Eisenberg T. Megalou E. Schroeder S. et al (2011). Spermidine and resveratrol induce autophagy by distinct pathways converging on the acetylproteome.J. Cell Biol.192615–629. 10.1083/jcb.201008167
77
Napolitano F. Carrella D. Mandriani B. Pisonero-Vaquero S. Sirci F. Medina D. L. et al (2018). Gene2drug: a computational tool for pathway-based rational drug repositioning.Bioinformatics341498–1505. 10.1093/bioinformatics/btx800
78
Napolitano G. Esposito A. Choi H. Matarese M. Benedetti V. Di Malta C. et al (2018). mTOR-dependent phosphorylation controls TFEB nuclear export.Nat. Comm.9:3312. 10.1038/s41467-018-05862-6
79
Palmieri M. Impey S. Kang H. di Ronza A. Pelz C. Sardiello M. et al (2011). Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways.Hum. Mol. Genet.203852–3866. 10.1093/hmg/ddr306
80
Palmieri M. Pal R. Nelvagal H. R. Lotfi P. Stinnett G. R. Seymour M. L. et al (2017). MTORC1-independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases.Nat. Comm.8:14338. 10.1038/ncomms14338
81
Pan H. Yan Y. Liu C. Finkel T. (2017). The role of ZKSCAN3 in the transcriptional regulation of autophagy.Autophagy131235–1238. 10.1080/15548627.2017.1320635
82
Pastore N. Blomenkamp K. Annunziata F. Piccolo P. Mithbaokar P. Maria Sepe R. et al (2013). Gene transfer of master autophagy regulator tfeb results in clearance of toxic protein and correction of hepatic disease in alpha-1-anti-trypsin deficiency.EMBO Mol. Med.5397–412. 10.1002/emmm.201202046
83
Peña-Llopis S. Vega-Rubin-de-Celis S. Schwartz J. C. Wolff N. C. Tran T. A. Zou L. et al (2011). Regulation of TFEB and V-ATPases by MTORC1.EMBO J.303242–3258. 10.1038/emboj.2011.257
84
Perera R. M. Di Malta C. Ballabio A. (2019). MiT/TFE family of transcription factors, lysosomes, and cancer.Ann. Rev. Cancer Biol.3203–222. 10.1146/annurev-cancerbio-030518-055835
85
Perera R. M. Stoykova S. Nicolay B. N. Ross K. N. Fitamant J. Boukhali M. et al (2015). Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism.Nature524361–365. 10.1038/nature14587
86
Platt F. M. d’Azzo A. Davidson B. L. Neufeld E. F. Tifft C. J. (2018). Lysosomal storage diseases.Nat. Rev. Dis. Primers4:27. 10.1038/s41572-018-0025-24
87
Ploper D. Taelman V. F. Robert L. Perez B. S. Titz B. Chen H.-W. et al (2015). MITF drives endolysosomal biogenesis and potentiates Wnt signaling in melanoma cells.Proc. Natl. Acad. Sci. U.S.A.112E420–E429. 10.1073/pnas.1424576112
88
Pogenberg V. Ogmundsdóttir M. H. Bergsteinsdóttir K. Schepsky A. Phung B. Deineko V. et al (2012). Restricted leucine zipper dimerization and specificity of DNA recognition of the melanocyte master regulator MITF.Genes Dev.262647–2658. 10.1101/gad.198192.112
89
Polager S. Ofir M. Ginsberg D. (2008). E2F1 regulates autophagy and the transcription of autophagy genes.Oncogene274860–4864. 10.1038/onc.2008.117
90
Polito V. A. Li H. Martini-Stoica H. Wang B. Yang L. Xu Y. et al (2014). Selective clearance of aberrant tau proteins and rescue of neurotoxicity by transcription factor EB.EMBO Mol. Med.61142–1160. 10.15252/emmm.201303671
91
Puertollano R. Ferguson S. M. Brugarolas J. Ballabio A. (2018). The complex relationship between TFEB transcription factor phosphorylation and subcellular localization.EMBO J.37:e98804. 10.15252/embj.201798804
92
Rabinowitz J. D. White E. (2010). Autophagy and metabolism.Science3301344–1348. 10.1126/science.1193497
93
Reddy K. Cusack C. L. Nnah I. C. Khayati K. Saqcena C. Huynh T. B. et al (2016). Dysregulation of nutrient sensing and clearance in presenilin deficiency.Cell Rep.142166–2179. 10.1016/j.celrep.2016.02.006
94
Rega L. R. Polishchuk E. Montefusco S. Napolitano G. Tozzi G. Zhang J. et al (2016). Activation of the transcription factor EB rescues lysosomal abnormalities in cystinotic kidney cells.Kidney Int.89862–873. 10.1016/j.kint.2015.12.045
95
Renault V. M. Thekkat P. U. Hoang K. L. White J. L. Brady C. A. Kenzelmann Broz D. et al (2011). The pro-longevity gene foxo3 is a direct target of the P53 tumor suppressor.Oncogene303207–3221. 10.1038/onc.2011.35
96
Roczniak-Ferguson A. Petit C. S. Froehlich F. Qian S. Ky J. Angarola B. et al (2012). The transcription factor TFEB links MTORC1 signaling to transcriptional control of lysosome homeostasis.Sci. Signal.5ra42–ra42. 10.1126/scisignal.2002790
97
Sahu R. Kaushik S. Clement C. C. Cannizzo E. S. Scharf B. Follenzi A. et al (2011). Microautophagy of cytosolic proteins by late endosomes.Dev. Cell20131–139. 10.1016/j.devcel.2010.12.003
98
Saikia M. Wang X. Mao Y. Wan J. Pan T. Qian S.-B. (2016). Codon optimality controls differential MRNA translation during amino acid starvation.RNA221719–1727. 10.1261/rna.058180.116
99
Sakamaki J.-I. Wilkinson S. Hahn M. Tasdemir N. O’Prey J. Clark W. et al (2017). Bromodomain protein BRD4 is a transcriptional repressor of autophagy and lysosomal function.Mol. Cell66517.e–532.e. 10.1016/j.molcel.2017.04.027
100
Sanchez A. M. Csibi A. Raibon A. Cornille K. Gay S. Bernardi H. et al (2012). AMPK promotes skeletal muscle autophagy through activation of forkhead FoxO3a and interaction with Ulk1.J. Cell. Biochem.113695–710. 10.1002/jcb.23399
101
Sardiello M. Palmieri M. di Ronza A. Medina D. L. Valenza M. Gennarino V. A. et al (2009). A gene network regulating lysosomal biogenesis and function.Science325473–477. 10.1126/science.1174447
102
Sato S. Roberts K. Gambino G. Cook A. Kouzarides T. Goding C. R. (1997). CBP/P300 as a co-factor for the microphthalmia transcription factor.Oncogene143083–3092. 10.1038/sj.onc.1201298
103
Seok S. Fu T. Choi S.-E. Li Y. Zhu R. Kumar S. et al (2014). Transcriptional regulation of autophagy by an FXR-CREB axis.Nature516108–111. 10.1038/nature13949
104
Settembre C. De Cegli R. Mansueto G. Saha P. K. Vetrini F. Visvikis O. et al (2013). TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop.Nat. Cell Biol.15647–658. 10.1038/ncb2718
105
Settembre C. Di Malta C. Polito V. A. Garcia Arencibia M. Vetrini F. Erdin S. et al (2011). TFEB links autophagy to lysosomal biogenesis.Science3321429–1433. 10.1126/science.1204592
106
Settembre C. Zoncu R. Medina D. L. Vetrini F. Erdin S. Erdin S. et al (2012). A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via MTOR and TFEB: self-regulation of the lysosome via MTOR and TFEB.EMBO J.311095–1108. 10.1038/emboj.2012.32
107
Shang L. Chen S. Du F. Li S. Zhao L. Wang X. (2011). Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK.Proc. Natl. Acad. Sci. U.S.A.1084788–4793. 10.1073/pnas.1100844108
108
Shaw J. Yurkova N. Zhang T. Gang H. Aguilar F. Weidman D. et al (2008). Antagonism of E2F-1 regulated Bnip3 transcription by NF-KappaB is essential for basal cell survival.Proc. Natl. Acad. Sci. U.S.A.10520734–20739. 10.1073/pnas.0807735105
109
Shin H.-J. Kim H. Oh S. Lee J.-G. Kee M. Ko H.-J. et al (2016). AMPK-SKP2-CARM1 signalling cascade in transcriptional regulation of autophagy.Nature534553–557. 10.1038/nature18014
110
Song W. Wang F. Savini M. Ake A. di Ronza A. Sardiello M. et al (2013). TFEB regulates lysosomal proteostasis.Hum. Mol. Genet.221994–2009. 10.1093/hmg/ddt052
111
Soria L. R. Allegri G. Melck D. Pastore N. Annunziata P. Paris D. et al (2018). Enhancement of hepatic autophagy increases ureagenesis and protects against hyperammonemia.Proc. Natl. Acad. Sci. U.S.A.115391–396. 10.1073/pnas.1714670115
112
Spampanato C. Feeney E. Li L. Cardone M. Lim J.-A. Annunziata F. et al (2013). Transcription factor EB (TFEB) is a new therapeutic target for pompe disease.EMBO Mol. Med.5691–706. 10.1002/emmm.201202176
113
Steingrimsson E. Tessarollo L. Pathak B. Hou L. Arnheiter H. Copeland N. G. et al (2002). Mitf and Tfe3, two members of the Mitf-Tfe family of BHLH-Zip transcription factors, have important but functionally redundant roles in osteoclast development.Proc. Natl. Acad. Sci. U.S.A.994477–4482. 10.1073/pnas.072071099
114
Thomas C. Pellicciari R. Pruzanski M. Auwerx J. Schoonjans K. (2008). Targeting bile-acid signalling for metabolic diseases.Nat. Rev. Drug Dis.7678–693. 10.1038/nrd2619
115
Tracy K. Dibling B. C. Spike B. T. Knabb J. R. Schumacker P. Macleod K. F. (2007). BNIP3 is an RB/E2F target gene required for hypoxia-induced autophagy.Mol. Cell. Biol.276229–6242. 10.1128/MCB.02246-2246
116
Tsunemi T. Ashe T. D. Morrison B. E. Soriano K. R. Au J. Vázquez Roque R. A. et al (2012). PGC-1α rescues huntington’s disease proteotoxicity by preventing oxidative stress and promoting TFEB function.Sci. Transl. Med.4:142ra97. 10.1126/scitranslmed.3003799
117
Vega-Rubin-de-Celis S. Peña-Llopis S. Konda M. Brugarolas J. (2017). Multistep regulation of TFEB by MTORC1.Autophagy13464–472. 10.1080/15548627.2016.1271514
118
Wang C. Niederstrasser H. Douglas P. M. Lin R. Jaramillo J. Li Y. et al (2017). Small-molecule TFEB pathway agonists that ameliorate metabolic syndrome in mice and extend C. elegans lifespan.Nat. Comm.8:2270. 10.1038/s41467-017-02332-3
119
Webb A. E. Brunet A. (2014). FOXO transcription factors: key regulators of cellular quality control.Trends Biochem. Sci.39159–169. 10.1016/j.tibs.2014.02.003
120
White E. (2015). The role for autophagy in cancer.J. Clin. Invest.12542–46. 10.1172/JCI73941
121
Xiao Q. Yan P. Ma X. Liu H. Perez R. Zhu A. et al (2014). Enhancing astrocytic lysosome biogenesis facilitates Aβ clearance and attenuates amyloid plaque pathogenesis.J. Neurosci.349607–9620. 10.1523/JNEUROSCI.3788-13.2014
122
Xiao Q. Yan P. Ma X. Liu H. Perez R. Zhu A. et al (2015). Neuronal-targeted TFEB accelerates lysosomal degradation of APP, Reducing Aβ generation and amyloid plaque pathogenesis.J. Neurosci.3512137–12151. 10.1523/JNEUROSCI.0705-15.2015
123
Xiong X. Tao R. DePinho R. A. Dong X. C. (2012). The autophagy-related gene 14 (Atg14) is regulated by forkhead box O transcription factors and circadian rhythms and plays a critical role in hepatic autophagy and lipid metabolism.J. Biol. Chem.28739107–39114. 10.1074/jbc.M112.412569
124
Xu P. Das M. Reilly J. Davis R. J. (2011). JNK regulates FoxO-dependent autophagy in neurons.Genes Dev.25310–322. 10.1101/gad.1984311
125
Yan Q. Han C. Wang G. Waddington J. L. Zheng L. Zhen X. (2017). Activation of AMPK/MTORC1-mediated autophagy by metformin reverses Clk1 deficiency-sensitized dopaminergic neuronal death.Mol. Pharmacol.92640–652. 10.1124/mol.117.109512
126
You H. Jang Y. You-Ten A. I. Okada H. Liepa J. Wakeham A. et al (2004). P53-dependent inhibition of FKHRL1 in response to DNA damage through protein kinase SGK1.Proc. Natl. Acad. Sci. U.S.A.10114057–14062. 10.1073/pnas.0406286101
127
Zhang J. Wang J. Xu J. Lu Y. Jiang J. Wang L. et al (2016). Curcumin targets the TFEB-lysosome pathway for induction of autophagy.Oncotarget775659–75671. 10.18632/oncotarget.12318
128
Zhao J. Brault J. J. Schild A. Cao P. Sandri M. Schiaffino S. et al (2007). FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells.Cell Metab.6472–483. 10.1016/j.cmet.2007.11.004
129
Zhao Y. Yang J. Liao W. Liu X. Zhang H. Wang S. et al (2010). Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity.Nat. Cell Biol.12665–675. 10.1038/ncb2069
Summary
Keywords
autophagy, TFEB, genetic diseases, nucleus, transcription, lysosomal storage disease
Citation
Di Malta C, Cinque L and Settembre C (2019) Transcriptional Regulation of Autophagy: Mechanisms and Diseases. Front. Cell Dev. Biol. 7:114. doi: 10.3389/fcell.2019.00114
Received
26 February 2019
Accepted
05 June 2019
Published
02 July 2019
Volume
7 - 2019
Edited by
Sovan Sarkar, University of Birmingham, United Kingdom
Reviewed by
Marco Sandri, University of Padua, Italy; Cecilia Bucci, University of Salento, Italy
Updates
Copyright
© 2019 Di Malta, Cinque and Settembre.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chiara Di Malta, dimalta@tigem.it; settembre@tigem.it
This article was submitted to Membrane Traffic, a section of the journal Frontiers in Cell and Developmental Biology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.