 Christopher Montemagno1,2,3*
Christopher Montemagno1,2,3* Gilles Pagès1,2,3*
Gilles Pagès1,2,3*- 1Département de Biologie Médicale, Centre Scientifique de Monaco, Monaco, Monaco
- 2CNRS UMR 7284, Institute for Research on Cancer and Aging of Nice, Université Côte d’Azur, Nice, France
- 3INSERM U1081, Centre Antoine Lacassagne, Nice, France
Angiogenesis, the formation of new blood vessels from preexisting one, represents a critical process for oxygen and nutrient supply to proliferating cells, therefore promoting tumor growth and metastasis. The Vascular Endothelial Growth Factor (VEGF) pathway is one of the key mediators of angiogenesis in cancer. Therefore, several therapies including monoclonal antibodies or tyrosine kinase inhibitors target this axis. Although preclinical studies demonstrated strong antitumor activity, clinical studies were disappointing. Antiangiogenic drugs, used to treat metastatic patients suffering of different types of cancers, prolonged survival to different extents but are not curative. In this review, we focused on different mechanisms involved in resistance to antiangiogenic therapies from early stage resistance involving mainly tumor cells to late stages related to the adaptation of the microenvironment.
Introduction
Angiogenesis is the formation of new blood vessels from pre-existing ones (Hanahan and Folkman, 1996). It is a crucial physiological process that occurs throughout the life time, from the embryo to establish an adequate vasculature for growing and developing organs, to adults during wound healing or ovarian cycle (Folkman and Shing, 1992; Wilting and Christ, 1996; Hazzard and Stouffer, 2000; Tonnesen et al., 2000). Angiogenesis is tightly regulated and disruption of any part of this process induces various disorders, such as psoriasis, diabetic retinopathy, and cancer (Nishida et al., 2006; Walsh, 2007; Crawford et al., 2009; Heidenreich et al., 2009). Angiogenesis involves migration, proliferation and differentiation of endothelial cells (ECs). During the angiogenic cascade, stable vessels undergovascular permeability and a basement membrane degradation by the matrix-metalloproteases (MMPs) liberating extracellular matrix-sequestred growth factors. In response to these growth factors, ECs proliferate and migrate to assemble as lumen-bearing cords with branching structure (Figure 1; Bryan and D’Amore, 2007). Angiogenesis is a tightly balanced mechanism regulated by both pro-angiogenic and anti-angiogenic factors. In tumors, this balance shift toward pro-angiogenic factors sustaining angiogenesis.
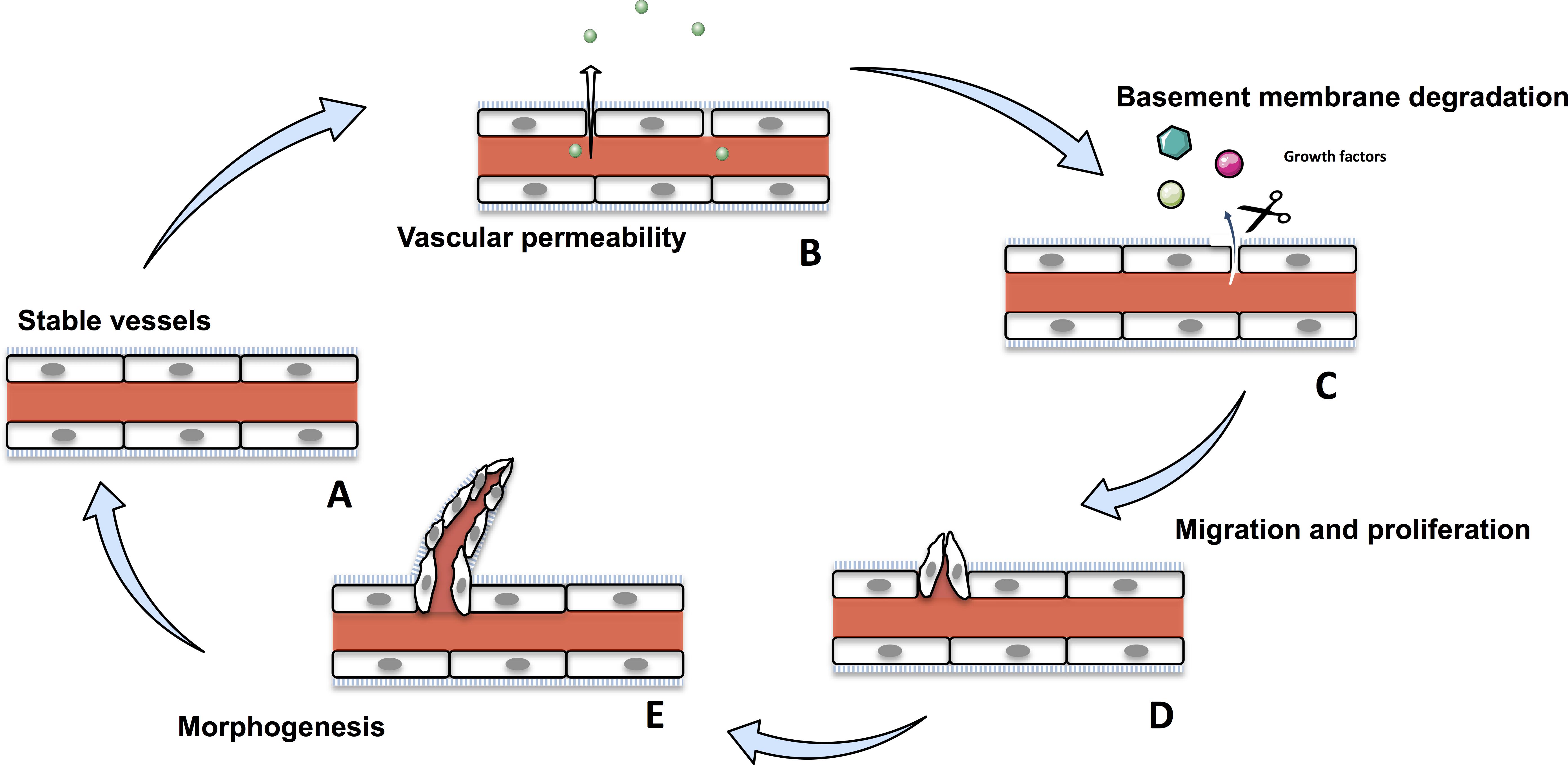
Figure 1. The physiological angiogenic cascade. Stable vessels (A) undergo a vascular permeability allowing extravasation of plasma proteins (B). MMPs degrate the extracellular matrix liberating growth factors (C). ECs proliferate and migrate (D) and undergo morphogenesis and assemble as lumen-bearing cords (E).
One of the first relationships between angiogenesis and cancer was introduced 55 years ago when Ehrmann and Knoth (1968); Greenblatt and Shubi (1968), P highlighted for the first time, that tumors secrete substances targeting ECs that stimulate angiogenesis. Three years later, Judah Folkman observed that the growth of solid tumors relies on this process (Folkman, 1971, 1972). The newly formed vascular network supplies tumor with oxygen, nutrients and growth factors. Based on these observations, Folkman proposed that inhibiting angiogenesis through ECs inhibition should constitute a promising anti-cancer treatment, by preventing nutrients supply and oxygen to tumors. This original concept stipulated that ECs are normal cells incapable of genetic plasticity as compared to tumor cells. Therefore, destruction of the blood vessel should have lead to tumor cell asphyxia and thereafter complete tumor regression. This concept was confirmed by the discovery of several angiogenic factors, such as transforming growth factor-α and β (TGF-α and TGF-β), angiopoietin, epidermal growth factor (EGF), platelet-derived growth factor (PDGF) and vascular endothelial growth factor (VEGF)A (Schreiber et al., 1986; Ferrara and Henzel, 1989; Levéen et al., 1994; van Cruijsen et al., 2005; van Meeteren et al., 2011; Fagiani and Christofori, 2013).
In 1989, the discovery of VEGFA, one of the most important angiogenic factors, by independent teams was a real breakthrough in understanding the mechanisms of angiogenesis (Keck et al., 1989; Leung et al., 1989; Guyot and Pagès, 2015). Four years later, the first monoclonal neutralizing antibody directed against VEGFA was described by the team of N. Ferrara, the winner of the Lasker Award few years laters for the use of these antibodies in eye pathologies especially wet age-related macular degeneration (Kim et al., 1993). This antibody inhibited the growth of experimental models of rhabdomyosarcoma, glioblastoma and colorectal and prostate cancers (Kim et al., 1993; Asano et al., 1995; Warren et al., 1995; Borgström et al., 1998). These promising anti-tumoral effects led to the development of bevacizumab (Avastin®), a humanized anti-VEGFA monoclonal antibody (Presta et al., 1997). Bevacizumab by specifically inhibiting the binding of VEGFA to its receptor VEGFR2 present on ECs, blocks signaling pathways involved in ECs proliferation and subsequently tumor angiogenesis (Shih and Lindley, 2006). In 2004, bevacizumab was approved by the Food and Drug Administration (FDA) as part of combination therapy for metastatic colorectal cancers (Hurwitz et al., 2004). Since 2008, bevacizumab was approved for the treatment of non-small-cell lung, breast, kidney and ovarian cancers in combination with standard chemotherapy (Sandler et al., 2006; Harshman and Srinivas, 2010; Russo et al., 2017). The strong competition in the field led to the development of alternative strategies to inhibit angiogenesis. Since VEGF receptors possess a tyrosine kinase domain, several companies developed small ATP mimetics to inhibit the activity of tyrosine kinases receptors involved in angiogenesis (Qin et al., 2019). Another strategy was designed to inhibit the activity of mTOR, a kinase activated in response to the stimulation of receptors (Faes et al., 2017). The inhibitor molecules sorafenib (Nexavar®), sunitinib (Sutent®), everolimus (Afinitor®), temsirolimus (Torisel®), have for instance been extensively studied in several metastatic cancers (Ebos and Kerbel, 2011; Motzer et al., 2013; Patel et al., 2016; Faes et al., 2017). Sorafenib and sunitinib were the first multikinase inhibitors to be approved, in the therapeutic arsenal for metastatic renal cell carcinoma (RCC) and advanced hepatocellular carcinoma management on the base of increased progression free survival (PFS). However, the impact of these treatment on overall survival (OS) was limited (Yang et al., 2003; Sandler et al., 2006; Miller et al., 2007; Kerbel, 2008; Escudier et al., 2010). Moreover, they induced detrimental side effects such increased blood pressure and hand and foot syndrome (Motzer et al., 2013).
Renal cell carcinoma became a paradigm for the development of more efficient and less toxic agents. Hence, axitinib whose affinity for targets equivalent to those of sunitinib was higher, presented equivalent therapeutic effects with reduced toxicity (Motzer et al., 2013). New drugs were also developed for the treatment of RCC including pazopanib (Votrient®), vandetanib (Caprelsa®) or lenvatinib (Lenvima®) (Llovet et al., 2008; Escudier et al., 2014; Motzer et al., 2015; Rizzo and Porta, 2017). Lenvatinib exploited the inhibition of Fibroblast Growth Factor receptors that are key in endothelial cell proliferation.
Several tumor cells aberrantly expressed VEGFRs and exhibit exacerbated genetic plasticity following anti-angiogenic therapies that is highlighted by several mechanisms of adaptation/resistance. The crosstalk between tumor and stromal cells allows escape mechanisms counteracting the effects of anti-angiogenic therapies.
The objective of this review is to present an overview of the different resistance mechanisms to angiogenic therapies, from the earliest to the late ones, including tumor and stromal cells adaptation. The different mechanisms were divided into “immediate early” resistance mainly reffering to adaptation of tumor cells following exposure to the drug for few minutes/hours; into “early” resistance reffering to days/weeks after treatment exposure and into “late” one occuring several months/years after the treatment and depending on metastasis (Figure 2). Understanding the different spatiotemporal mechanisms leading to such resistance is essential to propose innovative therapeutic strategies for patients presenting innate or acquired resistances.
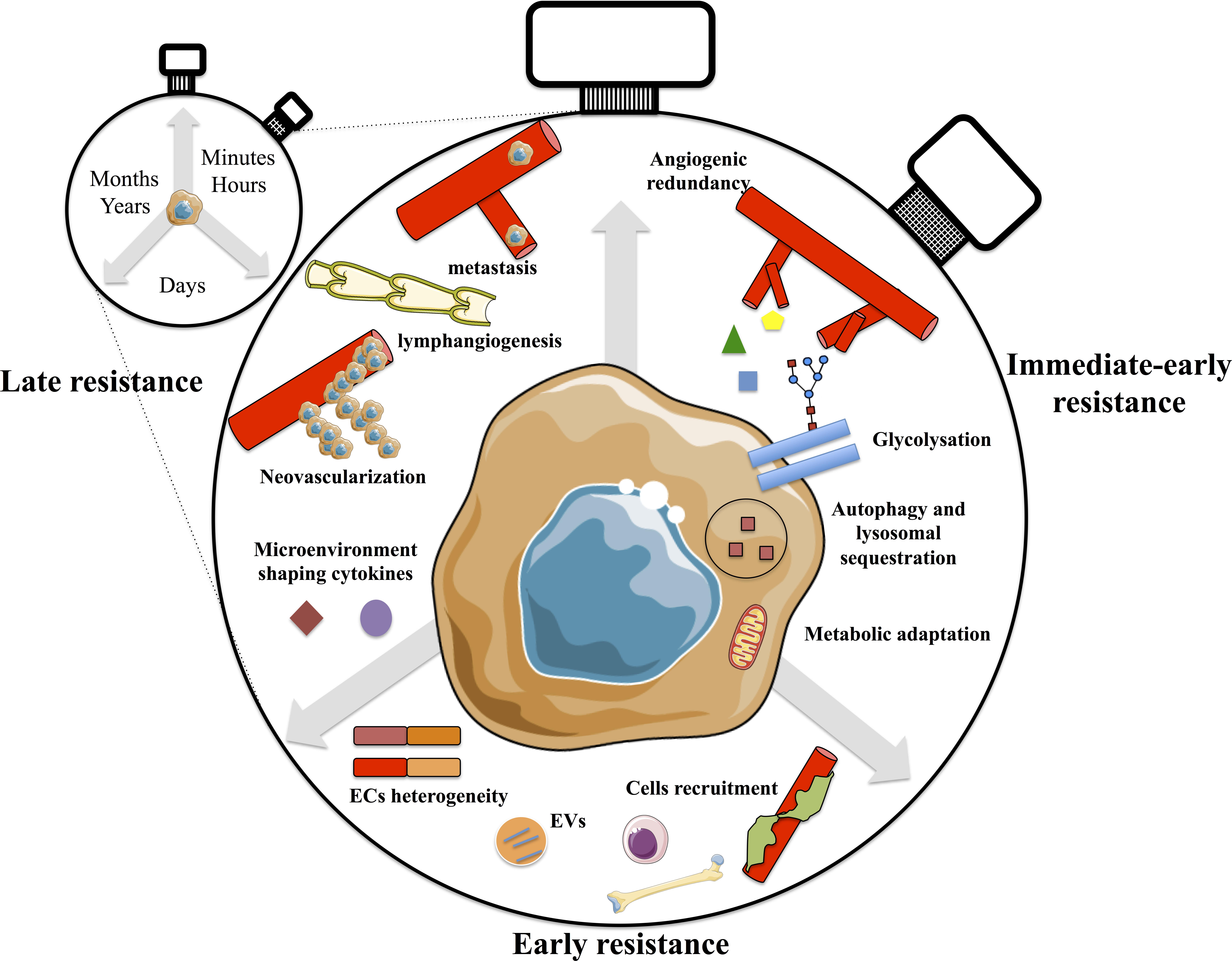
Figure 2. The adaptation to anti-angiogenic therapies; a clock ticking mechanism. Immediate-early resistance occuring within minutes to few hours following anti-angiogenic treatment involved angiogenic redundancy, glycosylation of VEGFR2, metabolic adaptation and sequestration of drugs in lysosomes inducing incomplete autophagy. Early resistance occurs during the following days after treatment and involved BMDCs, stromal cells recruitment into the tumor mass and ECs. The heterogeneity of ECs mediates resistance. Finally, within months following therapy, tumors adopt neovascularization strategies and increased lymphangiogenesis triggering metastasis and poor outcome in patients.
“Immediate-Early” Resistance to Anti-Angiogenic
Redundant Angiogenic Pathways and Hypoxia
Angiogenic redundancy is one of the earliest mechanisms leading to refractoriness or acquired resistance to anti-angiogenic therapies mainly targeting VEGFA and its receptors. Although VEGFA is the best known angiostimulatory protein, angiogenesis can be triggered by several growth factors including angiopoietins (ANGs), epidermal growth factor (EGFs), fibroblast growth factors (FGFs), hepatocyte growth factor (HGF), transforming growth factors (TGFs), placental growth factor (PlGF) or stromal cell-derived factor 1 (SDF1) (Xin et al., 2001; Bergers and Hanahan, 2008; Pardali et al., 2010; Brooks et al., 2012; Fagiani and Christofori, 2013). Except PlGF, which binds to VEGF receptors, all these angiogenic factors signal through different receptors expressed at the membrane of ECs (van Beijnum et al., 2015). This diversity of growth factors extents the toolbox of tumors to create blood vessels. Breast or pancreatic cancers for example rely on these angiogenic factors rather than on VEGFA and are poor responders to bevacizumab (Casanovas et al., 2005). Moreover, preclinical and clinical studies showed that anti-VEGFA antibodies and tyrosine-kinase inhibitors of VEGF receptors stimulate the production of these different growth factors (van Beijnum et al., 2015; Falcon et al., 2016; Haibe et al., 2020).
Preclinical studies for instance demonstrated an increase of SDF1 and PlGF in mice treated with anti-VEGFR2 compounds (Ebos et al., 2007; Fischer et al., 2007). The prolonged use of anti-VEGFR2 antibodies in transgenic mice models of pancreatic cancer stimulated the expression of ANG1 and FGFs. This increase correlated with a shorter survival (Casanovas et al., 2005). Comparable results were reported in head and neck squamous cell carcinoma (HNSCC) xenografts models. Indeed, microarray analysis, showed increased levels of FGF2 and its receptor (FGFR3) in bevacizumab-resistant tumors (Gyanchandani et al., 2013). These results were extended to lung cancer models resistant to angiogenesis-inhibitors, overexpressing EGFRs and FGFRs (Cascone et al., 2011). Clinical studies conducted on bevacizumab-treated colorectal cancer patients evidenced an increase of circulating PlGF, SDF1 and HGF levels (Willett et al., 2005; Kopetz et al., 2010). Equivalent results were obtained for glioblastoma patients treated with cediranib/Recentin®, a tyrosine kinase inhibitor of the VEGFR1, VEGFR2 and VEGFR3 (Batchelor et al., 2007, 2010). FGF and SDF1 increased expression was correlated with tumor relapse in cediranib-treated glioblastoma patients.
Angiopoietin-2
Angiopoietins belong to a family of protein controlling vascular maturation during developmental and pathophysiological angiogenesis (Stratmann et al., 1998; Jeansson et al., 2011; Thurston and Daly, 2012). The predominant angiopoietins are ANG1 and ANG2. ANG1 mediates migration and survival of endothelial cells through binding to Tie2 receptor found on ECs of blood vessels and monocytes, whereas ANG2 promotes cell death and vascular regression (Hanahan, 1997; Maisonpierre et al., 1997). VEGFA and ANG2 promote neovascularization and ANG2 plays a key role in tumor relapse following anti-VEGFA treatment (Asahara et al., 1998). In preclinical models of anti-VEGFR-treated tumors, upregulation of ANG2 stimulates vascular remodeling and sprouting (Crawford and Ferrara, 2009). This observation was supported by clinical studies showing that patients suffering of colorectal cancers who are poor responders to bevacizumab, exhibit high serum levels of ANG2 (Ogawa et al., 2004; Goede et al., 2010). Equivalent results were obtained for melanoma and breast cancer patients treated with anti-angiogenic therapies. Increased serum level of ANG2 correlated with disease progression (Sfiligoi et al., 2003; Helfrich et al., 2009). Preclinical studies recently showed that simultaneous blockade of VEGFA and ANG2 inhibits angiogenesis and tumor growth (Brown et al., 2010; Kienast et al., 2013; Schmittnaegel et al., 2017; Wolf and Langmann, 2019). Clinical trials using such combination are ongoing for the treatment of metastatic colorectal cancers (NCT01688206, NCT02141295). Recently, the vanucizumab, a bispecific anti-ANG2/anti-VEGFA antibody has been evaluated in a phase I study. Vanucizumab displayed acceptable safety profile and encouraging anti-tumor activity (Hidalgo et al., 2018).
Fibroblast-Growth Factors
Fibroblast-growth factors belongs to a family of 22 cell-signaling proteins involved in a broad variety of processes. FGF binds to tyrosine kinase receptors (FGFRs), expressed on tumor and stromal cells including endothelial cells, cancer-associated-fibroblasts or myeloid cells infiltrating tumors (Beenken and Mohammadi, 2009; Ornitz and Itoh, 2015). The FGF pathway promotes cancer progression and angiogenesis by activating RAS/RAF/MEK//ERK and PI3K/AKT/mTOR pathways (LaVallee et al., 1998; Hart et al., 2000). FGFs and FGFRs up-regulation, are involved in mechanisms of resistance to anti-VEGFA therapy (Casanovas et al., 2005; Kopetz et al., 2010). VEGFR2 inhibitors induce FGF2 expression and accelerate the growth murine pancreatic neuroendocrine tumors (Casanovas et al., 2005). Clinical studies on glioblastoma furthers confirmed this observation (Batchelor et al., 2007; Lee et al., 2019).
The proangiogenic role of FGF and its involvement in resistance to VEGFA inhibitors constitute a strong rationale for the development of inhibitors targeting the FGF and VEGFA pathways. The combined inhibition of FGF2 and VEGFA was highly efficient in preclinical models of head and neck carcinoma or pancreatic tumors (Casanovas et al., 2005; Gyanchandani et al., 2013). FGFR inhibitors notably restore the sensibility to bevacizumab in experimental models in mice suggesting a promising therapeutic combination (Gyanchandani et al., 2013). However, clinical investigations failed to demonstrate the relevance of this association (Norden et al., 2015; Semrad et al., 2017). Lenvatinib, a multiple receptor tyrosine kinase inhibiting the VEGFRs, FGFRs, and PDGFRs has shown promising therapeutic effects against various solid tumors and should be considered for counteracting resistance to anti-angiogenic agents (Suyama and Iwase, 2018).
Plateled-Derived-Growth Factor
In the 1970’s several groups demonstrated the existence of growth factors for fibroblasts and smooth muscle cells derived from platelets (Paul et al., 1971; Bowen-Pope and Ross, 1982). These factors were named platelet-derived-growth factors (PDGF) and were one of the first growth factors to be characterized. By binding to their receptors PDGFRs, PDGFs are major mitogens for many cell types and actively participate in angiogenesis (Papadopoulos and Lennartsson, 2018). In cancer, PDGFs exert autocrine loops that stimulate tumor cell proliferation, and paracrine signaling for angiogenesis (Liu et al., 2011; Manzat Saplacan et al., 2017). Upregulation of PDGF was evidenced in glioblastoma patients following anti-angiogenic therapy (Liu T. et al., 2018). The blockade of PDGFR pathway increases the sensibility to VEGFA-neutralizing treatment, giving the rationale for new therapeutic opportunities. However, imatinib, a PDGFR inhibitor, in combination with bevacizumab, failed to demonstrate efficacy in renal cell carcinoma (RCC) patients (Hainsworth et al., 2007; Rock et al., 2007). Despite an increase of PFS, the VEGFRs and PDGFR inhibitor sunitinib is not curative for RCC patients (Motzer et al., 2009).
Based on the redundancy in angiogenic pathways, limited benefits to patients were observed by targeting a single angiogenic growth factor or its receptor. This redundancy is at the origin of innate or acquired resistance, by activation of alternative proliferation/survival pathways. Inhibition of ANG2-, FGF- or PlGF-mediated signaling pathways with those of VEGFA overcomes aspects of resistance to VEGFA blockade, but a sustained inhibition remains to be demonstrated.
Transforming Growth Factor-β
The Transforming Growth factor-β (TGF-β) family regulates cell proliferation, differentiation and apoptosis (Massagué, 2000). In tumors, the role of TGF-β is ambivalent with tumor suppressive effects in early stage, thereafter switching toward tumor progression at later stages (Derynck et al., 2001). TGF-β induces the production of extracellular matrix and stimulates tube formation by ECs therefore inducing angiogenesis (Ferrari et al., 2009). Upregulation of TGF-β expression was reported in mice models of glioma resistant to anti-VEGF therapy (Park et al., 2016). Inhibition of TGF-β in hepatocellular carcinoma (HCC) and glioblastoma revealed anti-angiogenic benefit offering the rational to combine anti-TGF-β agents with anti-VEGF (Fransvea et al., 2009; Comunanza and Bussolino, 2017). The combination of galunisertib, a small inhibitor of TGF-β with sorafinib led to durable response in mice models of breast cancer (Holmgaard et al., 2018). TGF-β is also a major inducer of cancer associated fibroblast (CAF) development and fibrosis that are determinant in tumor aggressiveness. Targeting two hallmarks of cancer with one molecule probably explain the therapeutic response.
Combining anti-VEGF/VEGFR therapies to inhibitors of alternative angiogenic pathways appears relevant. However, the toxicity of such approach is an important issue. Treatment targeting concomitantly VEGFR and receptors involved in relapse is another option. One of the best example was the approval of cabozantinib (Cabometyx®) an inhibitor of VEGFR but also of cMET and AXL to actors involved in relapses after sunitinib treatment. Cabozantinib was approved as a second line treatment for RCC patient experiencing progression on sunitinib (Choueiri et al., 2016). It showed also a better efficacy as compared to sunitinib for RCC patients with poor or intermediate risk (Choueiri et al., 2017).
Hypoxia, a Key Mediator of Angiogenic Redundancy
Hypoxia arises from the combination of high proliferative and metabolic rates with abberant tumor vascularisation with poor oxygen delivery (Semenza, 2014). Beside redundant pro-angiogenic pathways, tumor hypoxia is considered as an “immediate early” response to anti-angiogenic therapy. Although anti-angiogenic therapies reduce and normalize tumor vasculature, limiting tumor hypoxia, alternative theory defends an increased intra-tumor hypoxia (Kerbel and Folkman, 2002; Jain, 2005). Hypoxia plays an important role in resistance to conventional therapies leading to the selection of more aggressive stem cells and a shorter survival (Harris, 2002; Wilson and Hay, 2011; Chen et al., 2018). Indeed, anti-angiogenic agents induce intra-tumoral hypoxia and a concomitant stabilization of the hypoxia-inducible factors 1 and 2 alpha (HIF1/2α). HIF1 is considered as a tumor suppressor whereas HIF2 is considered as an oncogene. HIF1α is a major transcriptional regulator of angiogenic factors. It transactivates hundreds of pro-angiogenic genes, including growth factors (VEGFA, PlFG, FGF-2, PDGF) and their receptors (VEGFRs) (Hirota and Semenza, 2006; Rapisarda and Melillo, 2009). Moreover, HIF1 inhibits the production of anti-angiogenic factors, exacerbating angiogenesis (Hanahan and Folkman, 1996; Laderoute et al., 2000). Hence, HIF1 exerts also potent transcriptional inhibition especially following a long exposure in hypoxic conditions (Hantelys et al., 2019).
Hypoxia and HIF1 activation also trigger EMT and metastasis by regulating the expression of key genes such as c-MET, CXCR4, and lysyl oxidase (LOX), events occurring later as discussed above (Joseph et al., 2018). Moreover, the hypoxic microenvironment generated following anti-angiogenic therapy stimulates β1-integrin expression, a well-known marker of resistance to cancer treatments (Foubert and Varner, 2012) which is consistent with its upregulation in clinical specimens of bevacizumab-resistant glioblastoma (Cordes and Park, 2007). Preclinical studies in mice models of glioblastoma demonstrated also the implication of β1-integrin in resistance to angiogenic therapies (Sidorov et al., 2016). The tumor microenvironment is hypoxic and the active metabolism of tumor cells induces the release of CO2 and lactate (Parks et al., 2013). The effect of hypoxia on tumor metabolism is detailled in the tumor metabolic adaptation part below.
The important role played by HIF in tumor aggressiveness stimulated the development of HIF inhibitors especially HIF2 that has oncogenic properties. Such treatments dissociate the HIF2α HIF1β dimer and consequently inhibit the transcriptional activation of HIF2. This treatment was successfully used in a multi-treated RCC patients (Chen W. et al., 2016). Hence, this treatment combined with classical anti-angiogenic drugs or immunotherapies (see below) is promising and should be further validated (Martínez-Sáez et al., 2017).
Autophagy and Lysosomal Sequestration
Autophagy is a physiological process involving the sequestration of unnecessary or dysfunctional cell components and their degradation in lysosomes (Mizushima, 2007; Janku et al., 2011; David, 2012). In pathophysiological conditions, autophagy is an adaptative response to stress. In cancer, autophagy acts as a double-edged sword by serving as a pro-survival or pro-death process (Mathew et al., 2007). Autophagy plays an important role in enabling tumor cells to overcome harsh conditions arising from the microenvironment following treatment (Chandra et al., 2019). By enhancing the survival of tumor cells, it is indeed now considered as an important mechanism of resistance to cancer drugs (Li et al., 2017; Desantis et al., 2018). Hypoxia-induced autophagy favor the survival of hypoxic tumor cells (Brahimi-Horn et al., 2011). Two mechanisms drive hypoxia-dependent autophagy; the non-selective and the selective autophagy extensively reviewed (Chandra et al., 2019).
A cytoprotective role of autophagy was supported by several preclinical studies using radiation or imatinib as anti-cancer strategies (Miyazawa et al., 2010; Gewirtz, 2014). Resistance to sorafenib in hepatocellular carcinoma was attributed to increased activation of mTOR or Akt pathway triggering autophagy and cell survival (Zhai et al., 2014; Luan et al., 2019). The pro-tumoral role of autophagic processes in mediating resistance to anti-cancer treatments in HCC was highlighted by combining sorafenib to autophagy inhibitors (Shimizu et al., 2012; Lin et al., 2013; Hwang et al., 2015). These preclinical studies gave the proof of concept to initiate clinical trials combining inhibitors of autophagy to sorafenib.
In addition to tumor cells, stromal cells use autophagy as a mechanism of resistance to anti-angiogenic drugs. ECs, the direct targets on anti-angiogenic therapies, are inevitably exposed to drugs via the blood stream. Hence, resistance to sunitinib depends at least, on autophagy processes in ECs (Wu et al., 2020). Sunitinib-resistant RCC display an increased number of lysosomes allowing an enhanced sequestration of the drug which limits its therapeutic activity by isolating the drug from its cytoplasmic targets (Giuliano et al., 2015). The basic pKa of sunitinib induces its lysosomal sequestration., It prevents its accessibility to the tyrosine kinase domains of the receptors targeted by the drug (VEGFR1, 2, 3, PDGFR, CSF1R and cKIT), limiting the efficacy of the treatment.
Tumor Metabolic Adaptation
The updated “Hallmarks of cancer: The Next Generation” includes the deregulation of cellular energetics as a key actor of tumor progression (Hanahan and Weinberg, 2011). Over the last decades, tumor hypoxia, by shaping cell metabolism was demonstrated as a key actor of tumor adaptation to anti-angiogenic therapies. Tumor cell metabolism and angiogenesis are tightly regulated by hypoxia (Semenza, 2014). Several genes involved in glycolysis are under HIF1 control, such as GLUT1, GLUT3, PDK1 or LDHA (Favaro et al., 2011). The more hypoxic the cell, the more glycolysis is used, leading to pyruvate production. Instead of entering the tricarboxylic acid cycle, most of pyruvate is converted to lactate. This excess of lactate diffuses in the extracellular environment and is picked up by oxygenated cells, that revert the lactate to pyruvate and enhance their oxidative phosphorylation (Cassim et al., 2020; Parks et al., 2020). Consequently, their need for glucose decreased, and more glucose is available for the more hypoxic area of tumors (Nakajima and Van Houten, 2013). Following sunitinib treatment, the establishment of this symbiotic loop allows the proliferation of the remaining viable cells despite the dramatic increase of hypoxia following angiogenesis inhibition (Pisarsky et al., 2016).
In addition to low oxygen, increased acidification is also a hallmark of hypoxic tumors. It plays a key role in resistance to anti-cancer therapy (Erra Díaz et al., 2018). While mammalian cells protect their cytosol from acidification through expression of membrane transporters and exchangers such as the Na+/H+ exchanger (L’Allemain et al., 1985) and the monocarboxylate transporter 1 (Halestrap and Price, 1999), hypoxic tumors have developed additional mechanisms to regulate their pH. In solid tumors, the transcription of carbonic anhydrase (CA) IX is controled by HIF1. CAIX catalyzes the hydration of carbon dioxide (CO2) into H+ and bicarbonate (HCO3–) which is rapidly uptaken into cell by Na+-HCO3– transporters sustaining alkaline pHi compatible with cell survival (Parks et al., 2013). In bevacizumab-resistant glioblastomas, increased levels of CAIX and of c-MET were observed (Jahangiri et al., 2013). Analysis of bevacizumab-resistant glioblastoma further revealed modifications in the expression of genes regulating cell metabolism, with (i) an increase of glycolysis-involved genes and (ii) a decrease of genes regulating oxidative phosphorylation (Kumar et al., 2013). Soluble CAIX is also correlated with a poor response to bevacizumab in breast cancers (Janning et al., 2019). Moreover, hypoxia leads to AMPK activation, inducing the metabolic switch from glycolysis to oxidative phosphorylation (McIntyre and Harris, 2015). Following anti-angiogenic therapy, tumor metabolism shifts from glycolysis to lipid consumption allowing tumor relapse (Sounni et al., 2014). Several clinical trials combining metabolism-targeting or hypoxia-targeting drugs with anti-angiogenics are ongoing (McIntyre and Harris, 2015). Recently, exciting novel concepts involving dual blockade of angiogenesis and metabolic adaptation have emerged and could revert the resistance to anti-angiogenic drugs (Jiménez-Valerio and Casanovas, 2017).
Recent findings demonstrated that metabolic reprogramming also occurs in TECs. TECs display upregulation of anabolic pathways in comparison to normal ECs. Unbiased meta-analysis revealed that Aldh18a1 and Sqle were consistently induced in TECs raising the possibility to identify specific targetable TECs markers (Rohlenova et al., 2020).
Glycosylation–Mediated Resistance
Activation of angiogenic receptors also occurs independently of ligand binding, therefore constituting another mechanism of insensitivity to cancer therapies. This process depends in part on galectins. They belong to a family of carbohydrate-binding proteins displaying high affinity for beta-galactoside (Camby et al., 2006). Galectin-1 is overexpressed in tumors and its expression correlated with metastatic dissemination and immune-escape (Hsu et al., 2013). Tumors refractory to anti-VEGFA treatments exhibit enhanced angiogenesis. Anti-VEGFA treatment and hypoxia increased galectin-1 production. Galectin-1 binds to N-glycans glycoproteins on endothelial cells, including VEGFR2. This binding prolongs the presence of VEGFR2 at the cell surface and promotes angiogenesis without VEGFA binding (Croci et al., 2014; Stanley, 2014). Further clinical investigations are needed to consider galectins as relevant targets for anti-angiogenic therapies.
Matrix Metalloproteinases
Matrix metalloproteinases (MMPs) play a key role in angiogenesis and in tumor progression (Deryugina and Quigley, 2010). MMPs can be pro- or anti-angiogenic depending of their categories. On the one hand, MMP-3 and MMP-7 display anti-angiogenic properties (Deryugina and Quigley, 2010). On the other hand, MMP-2 and MMP-9 promote the release of VEGFA from the ECM sustaining angiogenesis (Bergers et al., 2000). MMP-1 induces matrix remodeling and migration of ECs (Chun et al., 2004). Hence, it is now established that MMP inhibitors can induce tumor progression by favoring tumor angiogenesis. Therefore, MMP inhibitors combined to inhibitors of angiogenesis should be considered as a therapeutic option (Winer et al., 2018).
Tumor Stroma
Tumors are a complex association of cancer cells as well as a stromal compartment with cellular and noncellular components. Tumor stroma plays crucial roles in tumor progression and in resistance to treatments. The dense tumor stroma can limit the access of therapeutic agents to their target due to fibrosis, high interstitial pressure and degradation of drugs by stromal enzymes (Valkenburg et al., 2018). The rigid extracellular matrix can reduce blood vessel density, creating a barrier that drugs cannot perfuse (Olive et al., 2009). In parallell, the high interstitial pressure in tumor microenvironment affects drug delivery (Provenzano and Hingorani, 2013). Beside these effects, the cytochrome P450, expressed by fibroblasts, metabolizes toxic molecules including therapeutic drugs contributing to agressive behaviors of tumors (Hirth et al., 2000). In mice models of lung cancer, treatment with bevacizumab led to acquired resistance via upregulation of VEGFA, FGF2 and its receptor FGFR2 and PDGFR in stromal cells (Mitsuhashi et al., 2015). It now becomes evident that cancer therapies should include strategies to target and constrain the tumor stroma. Some agents targeting CXCR4, TGF-β or hyaluronic acid are currently under clinical consideration (Valkenburg et al., 2018).
Early Resistance to Anti-Angiogenic Therapies
Recruitment of Local Stromal Cells
Cells constituting the tumor environment play a key role in the resistance to angiogenesis inhibitors, especially cancer-associated fibroblasts (CAFs) and pericytes.
Cancer-Associated Fibroblasts
Cancer-associated fibroblasts are the principal component of the stroma within the tumor microenvironment. They exhibit diverse functions including matrix remodeling, crosstalk with tumor, endothelial or immune cells, promoting tumorigenesis. CAFs notably allow the recruitment of endothelial progenitors cells (EPCs) and bone-marrow-derived cells (BMDCs) through SDF1 expression and stimulation of its receptor CXCR4 on EPCs (Orimo et al., 2005). The role of EPCs and BMDCs are discussed in the next part. CAFs also promotes angiogenesis through the expression of galectin-1, VEGFA, FGF, HGF or PDGF (Tang et al., 2016; Wang et al., 2019). In tumor cells deficient for VEGFA, CAFs produce VEGFA to sustain angiogenic processes (Dong et al., 2004). CAFs isolated from anti-VEGFA resistant tumors, exhibit high levels of ANG2, and PDGF promoting tumor growth (Crawford and Ferrara, 2009). The pro-angiogenic and pro-invasive role of CAFs in resistance to antiangiogenic drugs can also arise from metalloproteinases (MMPs) production (Sternlicht et al., 1999; Boire et al., 2005).
Blocking the pro-angiogenic role of CAFs with an anti-FGF-2 125I-radiolabeled antibody resulted in the inhibition of HCC tumor growth and decreased angiogenesis (Wang et al., 2012; Hu et al., 2016). Lenvatinib (Lenvima®), which inhibits VEGFRs has a potent anti-angiogenic effect and inhibits also FGF receptors involved in anti-angiogenic resistance. It is now used in the therapeutic arsenal against kidney tumors (Motzer et al., 2016). Brivanib from Bristol Myers Squibb, an anti-VEGFR and FGFR, increased the PFS of 43 patients with recurrent endometrial cancers in a phase II clinical trial (Powell et al., 2014; Hosaka et al., 2018).
Pericytes
Blood vessels are composed of two interacting cell types: the ECs, forming the inner face of vessels, and perivascular cells, called pericytes. Pericytes are peri-endothelial cells that directly interact with ECs, regulating vessel diameter, permeability and therefore the blood flow (Bergers and Song, 2005). Recruitment of pericytes by ECs relies, at least, on the PDGF-PDGFR signaling (Abramsson et al., 2003). Pericytes negatively regulate the proliferation of ECs promoting maturation of neo-vessels (Orlidge and D’Amore, 1987). In preclinical models of glioma or RCC, an increased tumor blood vessel coverage by pericytes following sunitinib or bevacizumab treatments was observed (Norden et al., 2009; Cao et al., 2013; Pinto et al., 2016). Residual tumor vessels, in a preclinical model of colorectal cancer, were heavily covered by pericytes following treatment with Anti-ANG2 antibodies (Thomas et al., 2013). Moreover, the number of vessels covered by pericytes following sunitinib was correlated to aggressiveness of RCC (Cao et al., 2013). Pericyte coverage enhances tumor resistance to these therapies through limited ECs proliferation and through the availability of survival signals (Orlidge and D’Amore, 1987). These different mechanisms highlight the role of pericytes in the resistance to anti-angiogenic treatments observed in the clinic. Therefore, inhibiting blood vessels maturation by targeting blood vessel coverage by pericytes is a relevant strategy to overcome the resistance to anti-angiogenic therapies. Inhibition of PDGFR by imatinib and sunitinib in combination with anti-VEGFR showed anti-tumor effects on experimental tumors in mice (Pietras and Hanahan, 2005). FGF2/FGFR2 signaling and PDGF/PDGFR signaling crosstalk to enhance pericyte proliferation and recruitment (Hosaka et al., 2018). PDGF stimulates the pericytes-fibroblast transition, which contributes to metastatic processes (Hosaka et al., 2016). Therefore, inhibition of PDGF-mediated recruitment of pericytes showed potent anti-tumor effects (Thijssen et al., 2018). Hence, disrupting pericytes support, by using an anti-PDGFR and destabilizing pre-existing tumor vasculature with an anti-VEGFR, is an attractive strategy to overcome tumor refractoriness to conventional anti-angiogenic therapies.
Recruitment of Bone-Marrow Derived-Cells (BMDCS)
Anti-angiogenic therapies normalize vessels but also increase intra-tumoral hypoxia leading to the recruitment of bone marrow-derived cells (BMDCs) (Jain and Duda, 2003). Infiltration of BMDCs in tumors has been linked to tumor progression and angiogenesis for several years (Jain and Duda, 2003). As above-mentioned, anti-angiogenic therapies stimulate the production of pro-angiogenic factors (VEGFA, Angiopoietins, FGFs). However, the stress induced by the treatment stimulates inflammatory pathways involved in the production of cytokines such as SDF1, IL-8 or granulocyte colony-stimulating factor (G-CSF). These cytokines trigger the recruitment of BMDCs that exhibit high plasticity and pro-angiogenic potential limiting the efficacy of anti-angiogenic drugs (van Beijnum et al., 2015).
CD11b+ Gr1+ Myeloid-Derived Suppressor Cells
Myeloid-derived suppressor cells (MDSCs), also known as CD11b+ Gr1+ cells are composed of a mixed population including neutrophils, macrophages and dendritic cells displaying immunosuppressive and pro-tumorigenic capacities (Yang et al., 2004; Marigo et al., 2008; Crawford and Ferrara, 2009). Preclinical and clinical studies evidenced an increased number of MDSCs in cancers, promoting tumorigenesis and angiogenesis (Yang et al., 2004; Serafini et al., 2006; Diaz-Montero et al., 2009). The infiltration of tumors by MDSCs is therefore correlated with a poor outcome in patients. It participates in mechanisms of resistance to anti-angiogenic therapies (Shojaei and Ferrara, 2008). Indeed, tumors resistant to anti-VEGFA-treatments presented increased infiltration of MDSCs in comparison to anti-VEGFA sensitive tumors (Shojaei et al., 2007a). The presence of tumor infiltrating Th-17 cells induces the expression of G-CSF by CAFs and increased production of IL-6 and SDF1 by CAFs and tumor cells, allow the recruitment of MDSCs (Shojaei and Ferrara, 2008; Shojaei et al., 2009). Upregulation of G-CSF by resistant tumors triggers prokinectin-2 (Bv8) overexpression in the bone marrow (BM). Bv8 induces the migration of progenitor cells from the BM to the tumor. Anti-Bv8 antibodies reduce MDSCs recruitment and inhibit tumor growth and angiogenesis, suggesting a role of Bv8 in relapses following anti-VEGFA treatment (Shojaei et al., 2007a, b). Moreover, hypoxia induces resistance to sunitinib in glioblastoma, breast and metastatic RCC by increasing the recruitment of MDSCs to the tumor niche (Finke et al., 2011; Piao et al., 2012). In agreement with these observations, depletion of MDSCs sensitized tumors to anti-angiogenic therapies, highlighting their pivotal role in resistance (Holmgaard et al., 2016).
Among the MDSCs population, increased tumor-infiltration by neutrophils promote resistance to bevacizumab. Neutrophils induce Bv8-dependent tumor angiogenesis independently from the VEGFA signaling. Preclinical studies demonstrated that blockade of Bv8 decreases the recruitment of MDSC and angiogenesis (Shojaei et al., 2007b; Piao et al., 2012).
Macrophages, specialized phagocytic cells, also display plasticity and shape their phenotype in response to environmental conditions, making them a relevant candidate for treatment resistance (Ruffell and Coussens, 2015; Sarode et al., 2020). The first relationship between macrophages and angiogenesis was proposed in Knighton et al. (1983). Depending on their localization and on their polarization profiles, macrophages are pro- or anti-tumoral actors (Cheng et al., 2019). Recruitment of macrophages in tumors is induced by several cytokines including VEGFA or M-CSF. Macrophages secrete growth factors such as VEGFA or EGF triggering angiogenesis (van Beijnum et al., 2015). They also secrete matrix metalloproteases, and physically associate with ECs, promoting angiogenesis. In several preclinical studies, anti-VEGFA therapies reduced macrophage infiltration (Salnikov et al., 2006; Dineen et al., 2008). Nevertheless, specific macrophages with immunoglobin-like and EGF-like domains, the Tie-2-expressing macrophages (TEM), are recruited in hypoxic zones and by ANG2 (Murdoch et al., 2007). TEM also promote angiogenesis and tumor progression in hypoxic environment, through upregulation of HIF1α (De Palma and Naldini, 2011). Therefore, macrophages contribute to anti-angiogenic resistance. Although ANG2 inhibitors do not prevent the recruitment of TEM, they decrease their activity, illustrated by a downregulation of growth factors production and a decrease of their physical association with blood vessels (Mazzieri et al., 2011).
These results suggest that BMDCs are therapeutic targets for counteracting tumor refractoriness to anti-angiogenic therapies. Inhibition of the SDF1 pathway notably prevents BMDCs tumor infiltration and overcomes such resistance (Liu et al., 2010). Equivalent results were obtained with anti-Bv8 antibodies (Hasnis et al., 2014). Clinical studies recently demonstrated that plasma TEM are predictive markers of anti-angiogenic treatment failure in colorectal and ovarian cancers (Jayson et al., 2018). However, clinical investigations consisting in preventing tumor-infiltration of TEM are needed to further consider this therapeutic perspective.
Endothelial Progenitors Cells
The discovery of endothelial progenitor cells (EPCs) in adults and their putative vascular-promoting properties has generated debate in the field of vascular biology (Pasquier and Dias, 2010). EPCs were first isolated in 1997 by Asahara et al. (1997) EPCs are subtypes of stem cells that originate from the bone marrow. A controversy concerning their origin, their isolation and their functioning still exists. EPCs have high proliferative potential, capable of differentiation into mature ECs, therefore contributing to neovascularization and angiogenesis (Asahara et al., 1997; Reale et al., 2016). Several surface markers (CD133, CD34, and VEGFR2) characterize bone marrow derived-EPCs. They acquired CD31 and CD146 expression during their transport to the blood. They become mature ECs in the target tissues where they expressed VEGFR2, CD31, CD136, VE-cadherin, eNOS and von Willebrand factor (Puente et al., 2013). EPCs have a dual role in promoting angiogenesis into the tumor tissue; they regulate the angiogenic process through the production of growth factors and provide structural function in sprouting nascent vessels (Puente et al., 2013). The main chemo-attractants for EPCs in tumor tissue are VEGFA and SDF1, released by ECs, cancer cells and CAFs (Orimo et al., 2005; Grunewald et al., 2006). When recruited, EPCs promote angiogenesis by differentiating in ECs and by incorporating newly formed blood vessels (Puente et al., 2013). Anti-angiogenics, through hypoxia and HIF1α activation lead to the production of VEGFA and SDF1 by tumor cells triggering mobilization and recruitment of EPCs (Ceradini et al., 2004). Activated EPCs secrete pro-angiogenic factors leading to limited effects of anti-angiogenic therapies. Although the precise mechanism of EPCs-induced neovascularization remains poorly understood, recent studies in non-small-cell lung carcinoma (NSCLC) demonstrated a key role of histone deacetylase 7 (HDAC7) in the regulation of angiogenic genes (Wei et al., 2018). Nevertheless, the therapeutic implication of EPCs still remains to be elucitated.
Heterogeneity of Tumor Endothelial Cells
Heterogeneity of tumor endothelial cells (TECs) contributes to resistance to anti-angiogenic therapy (Maishi et al., 2019). TECs cover the inner surfaces of tumor blood vessels and are consequently directly exposed to anti-angiogenic drugs. TECs differ in several points from normal ECs. They display cytogenetic abnormalities, upregulation of pro-angiogenic factors and expression of stemness genes leading to drug resistance (Hida et al., 2004; Maishi et al., 2019). TECs express high levels of VEGFR1, VEGFR2, VEGFR3, and Tie-2 leading to strong responses to their respective angiogenic ligands (Alessandri et al., 1999; Bussolati et al., 2003). Moreover, TECs produce nonconventional growth factors such as biglycan, LOX and pentraxin, sustaining angiogenesis processes (Maishi et al., 2019). These observations led to the development of LOX inhibitors. Inhibition of LOX and biglycan reduces tumor metastasis suggesting the relevance of LOX targeting (Yamamoto et al., 2012; Osawa et al., 2013).
The hypoxic tumor microenvironment stimulates the expression of stemness genes in TECs, such as stem cell antigen 1 (Sca-1), MDR-1 and aldehyde lactate deshydrogenase (ALDH), leading to resistance to paclitaxel and to fluorouracil (5-FU) (Xiong et al., 2009). The vascular stem cells, that constitute a minor population in tumors, were suggested to contribute to tumor resistance to conventional chemotherapy and to anti-angiogenic treatments. Indeed, TECs derived from HCC are also more resistant to sorafenib, in comparison to human umbilical vein endothelial cells (HuVECs) (Xiong et al., 2009).
Transforming growth factors can originate from dedifferentiation of tumor cells, monocytes or from EPCs contributing to high heterogeneity and to resistance to anti-angiogenic treatments. Although cancer cells acquired drug resistance is well documented, the heterogeneity of TECs must be considered as a major actor. Recent single cell RNA-sequencing studies revealed endothelial cell heterogeneity following anti-VEGF therapy (Zhao et al., 2018). TECs can be classified into tip-like, transition and stalk-like cells. The sequencing of 56,771 endothelial cells from human/mouse (peri)-tumoral lung cells revealed different phenotypes following anti-angiogenic treatment. Tip-like signatures correlated with patient survival and tip-like TECs were most sensitive to anti-VEGF therapies (Goveia et al., 2020).
Among TECs-targeting therapies, inhibitors of CXCR4 were scrutinized since TECs are CXCR4-enriched populations is associated with a poor outcome in HCC. Inhibition of CXCR4 induces promising anti-tumor response mainly by preventing recruitment of BMDCs in the tumor mass and must be considered as a future therapeutic option (Kioi et al., 2010).
Extracellular Vesicles
Metastatic dissemination of cancer cells relies on several parameters and notably on the bi-directional communication between primary tumor and future metastatic tissues. This crosstalk essentially involves the production of particles by cancer or stromal cells. These particles are known as Extracellular vesicles (EVs). EVs carry onco peptides, RNA species or lipids from donor to recipient cells, triggering phenotypic changes of the future pre-metastatic niches (Xu et al., 2018). EV stimulate angiogenesis by transporting growth factors (VEGFA, PDGF, FGF-2), transcription factors (STAT3 and STAT5) or micro-RNAs (Todorova et al., 2017).
Recently, the emergence of EVs as a novel player of drug resistance has gained interest. EVs transfer drugs from resistant to sensitive cells triggering cell resistance (Maacha et al., 2019). VEGFA contained in EVs correlates with disease progression in bevacizumab treated patients, raising the possibility that resistance to bevacizumab relies on this process (Ko et al., 2019). Moreover, bevacizumab could be shed and exported by EVs leading to therapeutic escape (Simon et al., 2018).
Late Resistance
The Angiogenic-Dormancy as an Intrinsic Resistance Mechanism
Metastases can remain for months or years in a quiescent, dormant state, in the tissue they colonized. These micro-metastases constitute a residual disease characterized by the persistence of tumor cells, undetectable by conventional diagnostic techniques. The tumor dormancy can be defined as the lag in tumor growth occurring between primary tumor formation and the appearance of clinically detectable metastases (Yadav et al., 2018). The presence of disseminated tumor cells (DTCs) in bone marrow of prostate-cancer and breast-cancer patients have been reported before the development of overt metastases (Banys et al., 2012; Lam et al., 2014). Three molecular mechanisms characterize tumor dormancy: mitotic arrest, immunological and angiogenic dormancy (Senft and Ronai, 2016). The angiogenic dormancy may explain the reasons why angiogenic therapies simply delay tumor progression. More than 20 years ago, pharmacological inhibition of angiogenesis was found to induce dormancy in several mouse models (Holmgren et al., 1995; O’Reilly et al., 1996). The supposed but unproven “angiogenic switch” is supposed to play a key role in the maintenance of the dormancy, since dormant cells upregulate angiogenesis inhibitors such as thrombospondin-1 (TSP-1) (Senft and Ronai, 2016). Despite the lack of clinical evidences, the “angiogenic switch” of dormant cells has to be considered in cancer relapse following treatment arrest.
Induction of Cancer Stem Cells
Cancer stem cells (CSCs) constitute a small population of cells within tumor exhibiting abilities of self-renewal, differentiation and high tumorigenicity potential. They play a key role in the initiation of cancer and in the metastatic cascade. In 2003, CSCs were first identified in human breast and brain cancers (Al-Hajj et al., 2003; Singh et al., 2003). CSCs express CD44, CD24, CD29, CD90, CD133 and aldehyde deshydrogenase (ALDH1) allowing their identification (Yu et al., 2012). CSCs drive angiogenesis in hypoxia and HIF mediates CSCs proliferation and self-renewal (Tong et al., 2018). CSCs was suggested to give rise to endothelial cells and thus neovascularization processes (Fujita and Akita, 2017). Moreover, CSCs can differentiate in pericytes, supporting tumor vessel function (Cheng et al., 2013).
Their tumor initiating properties and their metastatic potential suggest that CSCs are involved in resistance to therapies. Conventional treatments including chemo- and radiation therapies generate the production of CSCs promoting tumor escape (Chen X. et al., 2016; Li et al., 2016; Liu L. et al., 2018). CSCs are actors of anti-angiogenic resistance. Preclinical studies on experimental models of breast cancers showed that sunitinib and bevacizumab increase the CSCs populations through HIF1 activation (Conley et al., 2012). These results indicate that administration of anti-angiogenic agents accelerate tumor growth by increasing CSCs population. Several CSCs-targeting therapies are currently under development. Inhibition of ALDH1 prevents CSCs enrichment and reduces tumor formation of experimental triple-negative breast cancer and NSCLC in mice (Schech et al., 2015; MacDonagh et al., 2017). Evaluation of CD44, CD133 or Hedgehog inhibitors are currently under considerations for further clinical developments (Shibata and Hoque, 2019).
Induction of Epithelial-Mesenchymal Transition and Invasion
Epithelial-mesenchymal transition (EMT) defines the acquisition of characteristics of invasive mesenchymal cells by epithelial cells. EMT is implicated in tumor invasion and metastasis and correlates with poor clinical outcome in several solid tumors (Mittal, 2018). During the EMT process, epithelial cells lose their phenotypes, with a downregulation of E-cadherin and α-catenin and acquire mesenchymal markers (N-cadherin, vimentin, fibronectin) leading to cell mobility and invasiveness (Zeisberg and Neilson, 2009). Several signaling pathways induce EMT (TGF-β, Wnt, Notch), by controlling the transcription factors Snail, Slug, ZEB1/2 and Twist (Garg, 2013). Hypoxia and HIF1α are also well-known drivers of EMT. The expression of Twist and Snail, the downregulation of E-cadherin and the induction of vimentin promoting tumor invasiveness, have been reported following anti-angiogenic treatments (Cooke et al., 2012; Maione et al., 2012). Similarly, enhanced invasiveness and growth capacity of glioblastoma and RCC cells have been demonstrated following VEGFA inhibition (Grepin et al., 2012; Lu et al., 2012). This enhanced invasion abilities led to metastatic dissemination, a later step discussed in part 3.
Several studies highlighted the role of the tyrosine kinase receptor c-MET in promoting tumor invasiveness and metastasis in response to anti-angiogenic therapies (Pàez-Ribes et al., 2009; Lu et al., 2012; Sennino et al., 2012).
Although sunitinib and anti-VEGFA decreased tumor volume, invasiveness, hypoxia and EMT markers are increased (Ebos et al., 2009; Mizumoto et al., 2015). In addition, c-MET and the phosphorylated active forms of c-MET also increased as a consequence of treatment-induced hypoxia. The c-MET pathway is one of the most investigated pathways in the field of resistance to anti-angiogenic therapies. Its stimulation through HGF binding, triggers the activation of the RAS/RAF/MEK/ERK, PI3K/AKT/mTOR, and STAT3 pathways promoting tumor growth and invasiveness (Jeon and Lee, 2017). Bevacizumab-treated glioblastoma patients have increased relapse in comparison to bevacizumab-untreated patients. This clinical observation was recently linked to the upregulation of c-MET and phospho-c-MET (Jahangiri et al., 2013). Hence, c-MET is a robust actor of anti-angiogenic resistance by promoting EMT-like phenotype and invasiveness in glioblastoma. This observation has subsequently led to development of c-MET inhibitors. Cabozantinib, a promising multi-kinase inhibitor of c-MET, VEGFR2, and AXL, improves overall survival of RCC patients with bone metastases (Motzer et al., 2018).
Lymphangiogenesis Induction
Historically, lymphatic vessels were considered as passive participants in metastatic dissemination, only acting as channels for tumor cells transit. Nowadays, it becomes evident that lymphatic vessels have an active role in promoting metastasis. The first pro-lymphangiogenic factors identified more than 20 years ago were the VEGFC and VEGFD that bind to VEGFR3 expressed on lymphatic endothelial cells triggering lymphangiogenesis (Joukov et al., 1996; Yamada et al., 1997). Overexpression of VEGFC and VEGFD increases the number of tumor-associated lymphatic vessels and the incidence of lymph node metastases (Christiansen and Detmar, 2011). Moreover, overexpression of VEGFC and VEGFD is correlated to intra-tumoral lymphatic vessel density, lymph node metastasis and poor outcome in patients with melanoma and breast cancers (Mohammed et al., 2007; van der Schaft et al., 2007). More recently, HGF, c-MET, Tie-2, PDGF and FGF were also identified as pro-lymphangiogenic factors (Christiansen and Detmar, 2011). Immunohistochemical analysis of tumor samples showed that lymphatic vessel invasion (LVI) correlated with lymph node metastasis (Christiansen and Detmar, 2011). Moreover, tumor cells through the expression of chemokine receptors exploit the lymphatic network to form metastases. Indeed, CXCR4 and CCR7 expressed on human breast cancer cells promote metastasis to organs expressing their respective ligands, SDF1 and CCL21 (Müller et al., 2001). CXCR4 is upregulated by hypoxia. Since dissemination to distant organs is governed by the SDF1 gradient, CXCR4/SDF1 antagonists inhibited lymph nodes spreading of cancer cells in experimental tumors in mice (Müller et al., 2001).
Drugs destroying blood vessels stimulate the development of tumor lymphatic vessels contributing to treatment failure. Tumors from sunitinib-treated RCC patients in a neoadjuvant setting exhibit increased lymphatic vessels and increased lymph node invasion. This detrimental effect is explained at least by the stimulation of VEGFC expression following sunitinib administration (Dufies et al., 2017a). Indeed, sunitinib stimulate vegfc gene transcription, mRNA stability and protein production and the subsequent VEGFC-dependent development of lymphatic vessels. Moreover, hypoxia upregulated VEGFC expression (Morfoisse et al., 2014; Ndiaye et al., 2019). Lymphangiogenesis participates in treatment failure and its targeting can be considered in the therapeutic arsenal but only for advanced tumors.
Microenvironment Shaping by Cytokines
The central role played by VEGFA plus ELR+CXCL cytokines and especially CXCXL8/IL-8 was first documented by Sparmann and Bar-Sagi (2004) in colon cancers. The role of ELR+CXCL and their receptors-CXCR1/2 on tumor cell proliferation, angiogenesis and microenvironment adaptation following anti-angiogenic therapies was highly documented (Vandercappellen et al., 2008). The pro-inflammatory interleukin (IL-1 β stimulates CXCL7 production in RCC models resulting in tumor growth (Grepin et al., 2012; Grépin et al., 2014). CXCL7 is a predictive marker of sunitinib efficacy in RCC (Dufies et al., 2017b). CXCL5 in response to lysosomal sequestration of anti-angiogenic drugs plays also a key role in resistance to anti-angiogenic in renal and breast cancers (Giuliano et al., 2019). Inhibitors of CXCR1 and CXCR2 efficiently inhibit the growth of experimental HNSCC and RCC by decreasing tumor cell proliferation, angiogenesis and inflammation (Dufies et al., 2019).
Novel Neovascularization Modalities
Beside angiogenesis, new vascular networks are generated by the attraction of endothelial progenitor cells, intussusseptive angiogenesis, vessel co-option and vasculogenic mimicry.
Vessel Co-option
Tumor can use alternative ways to obtain blood supply, and therefore counteracting the effects of anti-angiogenic therapies. Tumor cells can hijack pre-existing blood vessels of the surrounding non-tumoral tissue and migrate along these vessels. This process, which occurs in the absence of angiogenic growth factors, is called vessel co-option (Kuczynski et al., 2019). Basically, the cancer cells migrate along the surface of pre-existing vessels leading to their incorporation in the tumor mass. Vessel co-option has been extensively reported in histopathological specimens of lung, liver and brain cancers (Nakashima et al., 1995; Pezzella et al., 1997; Offersen et al., 2001; Winkler et al., 2009; Yao et al., 2018). This process sustains the growth of brain metastases emerging from melanomas, liver and breast cancers (Leenders et al., 2004; Kuczynski et al., 2016, 2019).
A major question is whether vascular co-option constitutes an intrinsic resistance or does it occur as an acquired resistance mechanism following therapy. Inhibition of VEGFA promotes cancer invasion, inducing vessel co-option in vivo. Mechanistic studies identified the actin-related protein, Arp2/3, c-MET, ZEB2- and WNT- EMT dependent signaling as promoters of cell motility and vessel co-option (Navis et al., 2013; Depner et al., 2016; Frentzas et al., 2016). Simultaneous blockade of VEGFA and ARP2/3, VEGFA and c-MET or VEGFA and ZEB2 suppresses tumor invasion (Sennino et al., 2012; Depner et al., 2016; Frentzas et al., 2016). Other therapeutic approaches include the blockade of cell-adhesion receptors, since tumor cells adhere to endothelial cells during co-option. Hence, a β1-integrin inhibitor combined with bevacizumab induced sustained anti-tumor response in bevacizumab-resistant glioma xenografts (Carbonell et al., 2013; Jahangiri et al., 2014).
The prognostic value of vessel co-option in cancer patients remains to be elucidated. Bevacizumab-treated colorectal cancer patients with liver metastases demonstrated a limited response due to vessel co-option (Frentzas et al., 2016). Combining cell-motility or cell-adhesion inhibitors with anti-angiogenic compounds deserves to be considered as a therapeutic alternative.
Vasculo Mimicry
The vasculo-mimicry is defined as the formation of vascular-like structures by non-vascular cells. In 1999, it was first reported that tumor can dedifferentiate and form vascular-like structure (Maniotis et al., 1999). Later, vasculo mimicry has been described in several tumor types such as breast, ovarian cancers or Ewing sarcoma (Sood et al., 2001; Shirakawa et al., 2002; van der Schaft et al., 2005). This dedifferentiation is accompanied by the acquisition of endothelial features such as VE-cadherin or Tie-2 expression (Maniotis et al., 1999). In addition, HIF1α is an important regulator in the process of vasculo-mimicry (Delgado-Bellido et al., 2017). Despite this dedifferentiation, tumors remain refractory to anti-angiogenic therapy (van der Schaft et al., 2004). Bevacizumab elicits vasculo-mimicry of tumors leading to tumor escape and metastasis (Xu et al., 2012). Sunitinib stimulates vasculo-mimicry by differentiating tumor cells to endothelial-like cells (Serova et al., 2016; Sun et al., 2017). Nevertheless, further studies are needed to clarify the correlation between vasculo-mimicry and resistance to anti-angiogenic therapies.
Increased Metastasis Rate
The ultimate consequence of the resistance of anti-angiogenic therapies is the increased rate of metastasis. As developed in the previous parts, anti-angiogenesis therapies lead to (i) intrinsic reprogramming of tumor cells with upregulation of alternative pro-angiogenic pathways, increased of lymphangiogenesis-related genes and processes and initiation of EMT (ii) conditioning the microenvironment, with the recruitment of local- and bone marrow-derived cells or used novel neovascularization modalities. All of these mechanisms lead to increased metastatic rate. Ten years ago, Ebos et al. (2009); Pàez-Ribes et al. (2009) were the first to describe the association between anti-angiogenesis drugs and increased distant metastases. Preclinical models of breast cancers showed that sunitinib enhance lung and liver metastasis (Ebos et al., 2009). Anti-angiogenic treatments can make the host more permissive for metastatic seeding. Sunitinib-treated mice exhibit vascular changes such as reduced pericyte coverage and increased leakiness of normal vessels (Chung et al., 2012; Maione et al., 2012; Singh et al., 2012). Therefore, these systemic actions facilitate the creation of a metastatic niche at distance from the primary tumor.
Increased metastasis rate following anti-angiogenic therapies are highly variable and depends on several parameters such as the type of treatment, the dose and the schedule. Singh et al. (2012) showed that sunitinib enhanced the agressiveness of tumor cells whereas the use of an anti-VEGF antibody did not. Chung et al. (2012) further demonstrated that inhibition of VEGF signaling by antibodies does not promote metastasis, in contrast to small molecule RTK inhibitors at elevated-therapeutic drug dosages. Dosing and scheduling of anti-angiogenic administration can also induce resistance. Short-term and high dose of sunitinib increased growth of breast cancer and enhance liver and lung metastasis (Ebos et al., 2009). In contrast, treatment with low dose of sunitinib did not induce metastasis (Welti et al., 2012).
Conclusion and Future of Anti-Angiogenic Therapies
Angiogenesis processes, through the establishment of a new vascular network, are an important contributor to tumor development and metastatic dissemination. Once the tumor has reached 1-2 mm2, the core of tumors become hypoxic and tumor cells counteract hypoxia by the production of angiogenic growth factors. Among them, VEGFA is one of the most important. Targeting the VEGFA/VEGFRs represented a great breakthrough in the therapeutic management of cancer patients. Unfortunately, complete responses are rare, and tumors counteract this inhibition through different processes. The molecular mechanisms of resistance are not fully understood and deciphering them has gained interest. It is now evident that several mechanisms exist. They involve a wide range of processes; (i) the earliest, with the upregulation of genes involved in angiogenic redundancy, EMT or the lysosomal sequestration of drugs, to the latest (ii) with an adaptation of the tumor microenvironment, reflected by the recruitment of progenitors cells, lymphangiogenesis, and adapted neovascularization modalities. All these mechanisms allow tumor metastasis and serve as limitations to anti-angiogenic drug efficacy.
Hence, combining treatments targeting tumor cells and cells of the tumor microenvironment should limit resistance and should improve patients’ survival. One of the first and of the obvious way of resistance involves angiogenic redundancy by multiple growth factors as suggested by the anti-tumoral effects of FGF inhibition and bevacizumab (Casanovas et al., 2005; Gyanchandani et al., 2013). Although combining anti-angiogenic therapies may improve benefit, the other alternative pathways lead to resistance. Moreover, the balance between therapeutic efficacy and toxicity must be evaluated before administration to patients. Another therapeutic strategy consists in targeting BMDCs or pericytes and CAFs in addition to tumor cells. This approach seems relevant since BMDCs and local stromal cells blockade leads to an impairment of tumor growth (Bergers and Hanahan, 2008; Crawford and Ferrara, 2009; Liu et al., 2010). The treatment of patients with diffuse-type giant tumor cells with a CSF-1 antibody elicits objective response (Ries et al., 2014). This result raises the possibility to combine this antibody to anti-angiogenic agents.
Another promising therapeutic strategy consists in targeting lymphangiogenesis and angiogenesis. Lymphangiogenesis induced by anti-angiogenesis dedicated compounds gives rise to node metastasis, leading at term to an increased metastatic rate and poor outcome in patients (Dufies et al., 2017a). Moreover, these lymphatic vessels play a key role in the cancer-induced immune tolerance. Indeed, tumor associated lymphatic vessels upregulated Program-Death Ligand 1 (PDL1) inhibiting T cell activation and therefore anti-tumor response (Dieterich et al., 2017). Recently, the anti-PDL1 antibody, avelumab combined with axitinib was compared to sunitinib for advanced RCC. The progression free survival was 13.8 months and significantly higher than sunitinib alone (8.4 months) (Motzer et al., 2019). A phase III study comparing the anti-PDL1 antibody, atezolizumab, plus bevacizumab versus sunitinib was assessed in metastatic RCC and confirmed these results (Rini et al., 2019b, 3). Among the tested combinations, the anti-PD1, pembrolizumab plus axitinib combo improved the PFS but also the OS of RCC patients (Rini et al., 2019a).
Nevertheless, despite their effects on PFS and OS, these combinations are not curative. The development of animal models mimicking the tumor microenvironment as well as preclinical evaluations of combo therapies are urgently needed to improve patients PFS and OS. To reach the “Golden Age” of tumor treatment as defined by Hsieh et al. (2017) new treatment options are needed either to improve the therapeutic effects of anti-angiogenics and immunotherapies or by inhibiting new relevant pathways involved in innate refractoriness or acquired resistance. The current anti-cancer strategies are based on the inhibition of a specific target playing a key role in tumor development [example: EGFR (lung cancers); HER2 (breast cancers); BRAF (melanoma)]. Because of relapses on these strategies, combinations with conventional chemotherapy [taxanes (breast) platin salts (lung)] or other targeted therapies like anti-angiogenics or immunotherapies have entered in the therapeutic arsenal. However, the second strategy often combined different toxicities and cannot be administered at long terms, limiting the therapeutic index. However, the “magic bullet” does not exist because cancers integrate several mechanisms of evasion to one treatment. Hence, instead of inhibiting several targets with several drugs, the ideal strategy relies on the use of one inhibitor targeting multiple hallmarks of cancers, i.e., tumor cell proliferation/stemness, angiogenesis, chronic inflammation, and immune tolerance.
By destroying the vascular network, antiangiogenic therapies efficacy should have cause vascular network destruction leading to tumor cells asphyxia and nutrient starvation. Moreover, antiangiogenic treatments should have targeted only normal endothelial cells that cannot undergo genetic plasticity, a specific property of tumor cell adaptation to treatments. However, aberrant expression, by tumor cells, of receptors inhibited by antiangiogenic drugs stimulated the genetic adaptation of tumor cells mainly through epigenetic modifications. For example, EZH2 a specific histone methyl transferase is a driver of sunitinib resistance in kidney cancers (Adelaiye-Ogala et al., 2017). In addition to tumor cells, tumor endothelial cells undergo epigenetic modifications crucial for adaptation to the antiangiogenic therapies (Ciesielski et al., 2020).
The correlation between the efficacy of antiangiogenic drugs and tumor grade was also a neglected parameter. Controversial results emerged from their efficacy in non-metastatic versus metastatic kidney cancers. Whereas they are the standard of cancer for metastatic tumors their efficacy as an adjuvant therapy gave conflicting results. The ASSURE trial (NCT00326898) showed no survival benefit relative to placebo whereas the S-TRAC trial showed that sunitinib in an adjuvant setting prolonged the disease free survival for more than 1 year (Haas et al., 2016; Ravaud et al., 2016). These complex features supposed that anti-angiogenic drugs affect other cells than ECs. Hence, the drugs indirectly affect immune cells. Sunitinib for example reverses immune suppression (Finke et al., 2008). In this process, myeloid derived suppressor cells are one of the main targets of sunitinib (Ko et al., 2009). Moreover, inhibition of VEGFA or VEGFR decreased the expression of immune checkpoints involved in immune tolerance, by T cells (Voron et al., 2015). Hence, the crosstalk between angiogenesis and immune cells explain the efficacy of combining antiangiogenic drug to immune checkpoint inhibitors (Motzer et al., 2019; Rini et al., 2019b). Immunetolerance is most of the time encountered in advanced tumors in which angiogenesis is key for metastatic spreading. The relevance of inhibiting angiogenesis was based on these extreme cases. However, blood or lymphatic vessels vehiculate active cytotoxic immune cells to prevent the development of low-grade tumors that did not undergo immune tolerance. Hence, favoring the development of lymphatic vessels through injection of VEGFC decreased the growth of experimental glioblastoma by enabling immunosurveillance (Song et al., 2020). Hence, these experiments completely revisited the notion that angiogenesis is systematically detrimental. The hypothesis that vessels must be normalized in cancer had emerged during the last decade (Goel et al., 2011). This hypothesis stipulates that normalization of tumor vessels will shape the tumor microenvironment leading to the control of tumor progression and to the improvement of the therapeutic response (Martin et al., 2019).
With the advent of the immunotherapy, the blockage of angiogenesis should be reconsidered and the “blasting missil” must be discovered.
It is now evident that targeting only one mechanism involved in cancer development is insufficient. The cancer Hallmarks described by Hanahan and Weimberg probably shape the future treatments to increase the percentage of complete remissions. What is the ideal strategy? Targeting at the same time different Hallmarks with already approved therapies or to find targets that drive concomitantly the different Hallmarks? If these targets exist, a specific inhibitor will serve as a “blasting missile” to destroy the tumor. The reality is probably an intermediate option.
Author Contributions
CM and GP are equally responsible for all parts of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The authors acknowledge funding from the Fondation Xavier-Mora, The Fondation de France and La Ligue contre le Cancer- Equipe labellisée 2019.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abramsson, A., Lindblom, P., and Betsholtz, C. (2003). Endothelial and nonendothelial sources of PDGF-B regulate pericyte recruitment and influence vascular pattern formation in tumors. J. Clin. Invest. 112, 1142–1151. doi: 10.1172/JCI18549
Adelaiye-Ogala, R., Budka, J., Damayanti, N. P., Arrington, J., Ferris, M., Hsu, C.-C., et al. (2017). EZH2 modifies sunitinib resistance in renal cell carcinoma by kinome reprogramming. Cancer Res. 77, 6651–6666. doi: 10.1158/0008-5472.CAN-17-0899
Alessandri, G., Chirivi, R. G., Fiorentini, S., Dossi, R., Bonardelli, S., Giulini, S. M., et al. (1999). Phenotypic and functional characteristics of tumour-derived microvascular endothelial cells. Clin. Exp. Metastasis 17, 655–662. doi: 10.1023/a:1006738901839
Al-Hajj, M., Wicha, M. S., Benito-Hernandez, A., Morrison, S. J., and Clarke, M. F. (2003). Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. U.S.A. 100, 3983–3988. doi: 10.1073/pnas.0530291100
Asahara, T., Chen, D., Takahashi, T., Fujikawa, K., Kearney, M., Magner, M., et al. (1998). Tie2 receptor ligands, Angiopoietin-1 and Angiopoietin-2, modulate VEGF-induced postnatal neovascularization. Circ. Res. 83, 233–240. doi: 10.1161/01.RES.83.3.233
Asahara, T., Murohara, T., Sullivan, A., Silver, M., van der Zee, R., Li, T., et al. (1997). Isolation of putative progenitor endothelial cells for angiogenesis. Science 275, 964–967. doi: 10.1126/science.275.5302.964
Asano, M., Yukita, A., Matsumoto, T., Kondo, S., and Suzuki, H. (1995). Inhibition of tumor growth and metastasis by an immunoneutralizing monoclonal antibody to human vascular endothelial growth factor/vascular permeability factor121. Cancer Res. 55, 5296–5301.
Banys, M., Hartkopf, A. D., Krawczyk, N., Kaiser, T., Meier-Stiegen, F., Fehm, T., et al. (2012). Dormancy in breast cancer. Breast Cancer Targets Ther. 4, 183–191. doi: 10.2147/BCTT.S26431
Batchelor, T. T., Duda, D. G., di Tomaso, E., Ancukiewicz, M., Plotkin, S. R., Gerstner, E., et al. (2010). Phase II study of cediranib, an oral pan-vascular endothelial growth factor receptor tyrosine kinase inhibitor, in patients with recurrent glioblastoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 28, 2817–2823. doi: 10.1200/JCO.2009.26.3988
Batchelor, T. T., Sorensen, A. G., di Tomaso, E., Zhang, W.-T., Duda, D. G., Cohen, K. S., et al. (2007). AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell 11, 83–95. doi: 10.1016/j.ccr.2006.11.021
Beenken, A., and Mohammadi, M. (2009). The FGF family: biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 8, 235–253. doi: 10.1038/nrd2792
Bergers, G., Brekken, R., McMahon, G., Vu, T. H., Itoh, T., Tamaki, K., et al. (2000). Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2, 737–744. doi: 10.1038/35036374
Bergers, G., and Hanahan, D. (2008). Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 8, 592–603. doi: 10.1038/nrc2442
Bergers, G., and Song, S. (2005). The role of pericytes in blood-vessel formation and maintenance. Neuro Oncol. 7, 452–464. doi: 10.1215/S1152851705000232
Boire, A., Covic, L., Agarwal, A., Jacques, S., Sherifi, S., and Kuliopulos, A. (2005). PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell 120, 303–313. doi: 10.1016/j.cell.2004.12.018
Borgström, P., Bourdon, M. A., Hillan, K. J., Sriramarao, P., and Ferrara, N. (1998). Neutralizing anti-vascular endothelial growth factor antibody completely inhibits angiogenesis and growth of human prostate carcinoma micro tumors in vivo. Prostate 35, 1–10. doi: 10.1002/(sici)1097-0045(19980401)35:1<1::aid-pros1>3.0.co;2-o
Bowen-Pope, D. F., and Ross, R. (1982). Platelet-derived growth factor. II. Specific binding to cultured cells. J. Biol. Chem. 257, 5161–5171.
Brahimi-Horn, M. C., Bellot, G., and Pouysségur, J. (2011). Hypoxia and energetic tumour metabolism. Curr. Opin. Genet. Dev. 21, 67–72. doi: 10.1016/j.gde.2010.10.006
Brooks, A. N., Kilgour, E., and Smith, P. D. (2012). Molecular pathways: fibroblast growth factor signaling: a new therapeutic opportunity in cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 18, 1855–1862. doi: 10.1158/1078-0432.CCR-11-0699
Brown, J. L., Cao, Z. A., Pinzon-Ortiz, M., Kendrew, J., Reimer, C., Wen, S., et al. (2010). A human monoclonal anti-ANG2 antibody leads to broad antitumor activity in combination with VEGF inhibitors and chemotherapy agents in preclinical models. Mol. Cancer Ther. 9, 145–156. doi: 10.1158/1535-7163.MCT-09-0554
Bryan, B. A., and D’Amore, P. A. (2007). What tangled webs they weave: Rho-GTPase control of angiogenesis. Cell. Mol. Life Sci. CMLS 64, 2053–2065. doi: 10.1007/s00018-007-7008-z
Bussolati, B., Deambrosis, I., Russo, S., Deregibus, M. C., and Camussi, G. (2003). Altered angiogenesis and survival in human tumor-derived endothelial cells. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 17, 1159–1161. doi: 10.1096/fj.02-0557fje
Camby, I., Le Mercier, M., Lefranc, F., and Kiss, R. (2006). Galectin-1: a small protein with major functions. Glycobiology 16, 137R–157R. doi: 10.1093/glycob/cwl025
Cao, Z., Shang, B., Zhang, G., Miele, L., Sarkar, F. H., Wang, Z., et al. (2013). Tumor cell-mediated neovascularization and lymphangiogenesis contrive tumor progression and cancer metastasis. Biochim. Biophys. Acta 1836, 273–286. doi: 10.1016/j.bbcan.2013.08.001
Carbonell, W. S., DeLay, M., Jahangiri, A., Park, C. C., and Aghi, M. K. (2013). β1 integrin targeting potentiates antiangiogenic therapy and inhibits the growth of bevacizumab-resistant glioblastoma. Cancer Res. 73, 3145–3154. doi: 10.1158/0008-5472.CAN-13-0011
Casanovas, O., Hicklin, D. J., Bergers, G., and Hanahan, D. (2005). Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 8, 299–309. doi: 10.1016/j.ccr.2005.09.005
Cascone, T., Herynk, M. H., Xu, L., Du, Z., Kadara, H., Nilsson, M. B., et al. (2011). Upregulated stromal EGFR and vascular remodeling in mouse xenograft models of angiogenesis inhibitor-resistant human lung adenocarcinoma. J. Clin. Invest. 121, 1313–1328. doi: 10.1172/JCI42405
Cassim, S., Vuèetiæ, M., Ždraleviæ, M., and Pouyssegur, J. (2020). Warburg and beyond: the power of mitochondrial metabolism to collaborate or replace fermentative glycolysis in cancer. Cancers 12:1119. doi: 10.3390/cancers12051119
Ceradini, D. J., Kulkarni, A. R., Callaghan, M. J., Tepper, O. M., Bastidas, N., Kleinman, M. E., et al. (2004). Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat. Med. 10, 858–864. doi: 10.1038/nm1075
Chandra, A., Rick, J., Yagnik, G., and Aghi, M. K. (2019). Autophagy as a mechanism for anti-angiogenic therapy resistance. Semin. Cancer Biol. doi: 10.1016/j.semcancer.2019.08.031
Chen, A., Sceneay, J., Gödde, N., Kinwel, T., Ham, S., Thompson, E. W., et al. (2018). Intermittent hypoxia induces a metastatic phenotype in breast cancer. Oncogene 37, 4214–4225. doi: 10.1038/s41388-018-0259-3
Chen, W., Hill, H., Christie, A., Kim, M. S., Holloman, E., Pavia-Jimenez, A., et al. (2016). Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 539, 112–117. doi: 10.1038/nature19796
Chen, X., Liao, R., Li, D., and Sun, J. (2016). Induced cancer stem cells generated by radiochemotherapy and their therapeutic implications. Oncotarget 8, 17301–17312. doi: 10.18632/oncotarget.14230
Cheng, H., Wang, Z., Fu, L., and Xu, T. (2019). Macrophage polarization in the development and progression of ovarian cancers: an overview. Front. Oncol. 9:421. doi: 10.3389/fonc.2019.00421
Cheng, L., Huang, Z., Zhou, W., Wu, Q., Donnola, S., Liu, J. K., et al. (2013). Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 153, 139–152. doi: 10.1016/j.cell.2013.02.021
Choueiri, T. K., Escudier, B., Powles, T., Tannir, N. M., Mainwaring, P. N., Rini, B. I., et al. (2016). Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR): final results from a randomised, open-label, phase 3 trial. Lancet Oncol. 17, 917–927. doi: 10.1016/S1470-2045(16)30107-3
Choueiri, T. K., Halabi, S., Sanford, B. L., Hahn, O., Michaelson, M. D., Walsh, M. K., et al. (2017). Cabozantinib versus sunitinib as initial targeted therapy for patients with metastatic renal cell carcinoma of poor or intermediate risk: the alliance A031203 CABOSUN trial. J. Clin. Oncol. 35, 591–597. doi: 10.1200/JCO.2016.70.7398
Christiansen, A., and Detmar, M. (2011). Lymphangiogenesis and cancer. Genes Cancer 2, 1146–1158. doi: 10.1177/1947601911423028
Chun, T.-H., Sabeh, F., Ota, I., Murphy, H., McDonagh, K. T., Holmbeck, K., et al. (2004). MT1-MMP-dependent neovessel formation within the confines of the three-dimensional extracellular matrix. J. Cell Biol. 167, 757–767. doi: 10.1083/jcb.200405001
Chung, A. S., Kowanetz, M., Wu, X., Zhuang, G., Ngu, H., Finkle, D., et al. (2012). Differential drug class-specific metastatic effects following treatment with a panel of angiogenesis inhibitors. J. Pathol. 227, 404–416. doi: 10.1002/path.4052
Ciesielski, O., Biesiekierska, M., Panthu, B., Vialichka, V., Pirola, L., and Balcerczyk, A. (2020). The epigenetic profile of tumor endothelial cells. effects of combined therapy with antiangiogenic and epigenetic drugs on cancer progression. Int. J. Mol. Sci. 21:2606. doi: 10.3390/ijms21072606
Comunanza, V., and Bussolino, F. (2017). Therapy for cancer: strategy of combining anti-angiogenic and target therapies. Front. Cell Dev. Biol. 5:101. doi: 10.3389/fcell.2017.00101
Conley, S. J., Gheordunescu, E., Kakarala, P., Newman, B., Korkaya, H., Heath, A. N., et al. (2012). Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc. Natl. Acad. Sci. U.S.A. 109, 2784–2789. doi: 10.1073/pnas.1018866109
Cooke, V. G., LeBleu, V. S., Keskin, D., Khan, Z., O’Connell, J. T., Teng, Y., et al. (2012). Pericyte depletion results in hypoxia-associated epithelial-to-mesenchymal transition and metastasis mediated by met signaling pathway. Cancer Cell 21, 66–81. doi: 10.1016/j.ccr.2011.11.024
Cordes, N., and Park, C. C. (2007). beta1 integrin as a molecular therapeutic target. Int. J. Radiat. Biol. 83, 753–760. doi: 10.1080/09553000701639694
Crawford, T. N., Alfaro, D. V., Kerrison, J. B., and Jablon, E. P. (2009). Diabetic retinopathy and angiogenesis. Curr. Diabetes Rev. 5, 8–13. doi: 10.2174/157339909787314149
Crawford, Y., and Ferrara, N. (2009). Tumor and stromal pathways mediating refractoriness/resistance to anti-angiogenic therapies. Trends Pharmacol. Sci. 30, 624–630. doi: 10.1016/j.tips.2009.09.004
Croci, D. O., Cerliani, J. P., Dalotto-Moreno, T., Méndez-Huergo, S. P., Mascanfroni, I. D., Dergan-Dylon, S., et al. (2014). Glycosylation-dependent lectin-receptor interactions preserve angiogenesis in anti-VEGF refractory tumors. Cell 156, 744–758. doi: 10.1016/j.cell.2014.01.043
David, R. (2012). Metabolism: keeping fit with autophagy. Nat. Rev. Mol. Cell Biol. 13:136. doi: 10.1038/nrm3287
De Palma, M., and Naldini, L. (2011). Angiopoietin-2 TIEs up macrophages in tumor angiogenesis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 17, 5226–5232. doi: 10.1158/1078-0432.CCR-10-0171
Delgado-Bellido, D., Serrano-Saenz, S., Fernández-Cortés, M., and Oliver, F. J. (2017). Vasculogenic mimicry signaling revisited: focus on non-vascular VE-cadherin. Mol. Cancer 16:65. doi: 10.1186/s12943-017-0631-x
Depner, C., Zum Buttel, H., Böğürcü, N., Cuesta, A. M., Aburto, M. R., Seidel, S., et al. (2016). EphrinB2 repression through ZEB2 mediates tumour invasion and anti-angiogenic resistance. Nat. Commun. 7:12329. doi: 10.1038/ncomms12329
Derynck, R., Akhurst, R. J., and Balmain, A. (2001). TGF-beta signaling in tumor suppression and cancer progression. Nat. Genet. 29, 117–129. doi: 10.1038/ng1001-117
Deryugina, E. I., and Quigley, J. P. (2010). Pleiotropic roles of matrix metalloproteinases in tumor angiogenesis: contrasting, overlapping and compensatory functions. Biochim. Biophys. Acta 1803, 103–120. doi: 10.1016/j.bbamcr.2009.09.017
Desantis, V., Saltarella, I., Lamanuzzi, A., Mariggiò, M. A., Racanelli, V., Vacca, A., et al. (2018). Autophagy: a new mechanism of prosurvival and drug resistance in multiple myeloma. Transl. Oncol. 11, 1350–1357. doi: 10.1016/j.tranon.2018.08.014
Diaz-Montero, C. M., Salem, M. L., Nishimura, M. I., Garrett-Mayer, E., Cole, D. J., and Montero, A. J. (2009). Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol. Immunother. CII 58, 49–59. doi: 10.1007/s00262-008-0523-4
Dieterich, L. C., Ikenberg, K., Cetintas, T., Kapaklikaya, K., Hutmacher, C., and Detmar, M. (2017). Tumor-associated lymphatic vessels upregulate PDL1 to inhibit T-Cell activation. Front. Immunol. 8:66. doi: 10.3389/fimmu.2017.00066
Dineen, S. P., Lynn, K. D., Holloway, S. E., Miller, A. F., Sullivan, J. P., Shames, D. S., et al. (2008). Vascular endothelial growth factor receptor 2 mediates macrophage infiltration into orthotopic pancreatic tumors in mice. Cancer Res. 68, 4340–4346. doi: 10.1158/0008-5472.CAN-07-6705
Dong, J., Grunstein, J., Tejada, M., Peale, F., Frantz, G., Liang, W.-C., et al. (2004). VEGF-null cells require PDGFR alpha signaling-mediated stromal fibroblast recruitment for tumorigenesis. EMBO J. 23, 2800–2810. doi: 10.1038/sj.emboj.7600289
Dufies, M., Giuliano, S., Ambrosetti, D., Claren, A., Ndiaye, P. D., Mastri, M., et al. (2017a). Sunitinib Stimulates expression of VEGFC by tumor cells and promotes lymphangiogenesis in clear cell renal cell carcinomas. Cancer Res. 77, 1212–1226. doi: 10.1158/0008-5472.CAN-16-3088
Dufies, M., Giuliano, S., Viotti, J., Borchiellini, D., Cooley, L. S., Ambrosetti, D., et al. (2017b). CXCL7 is a predictive marker of sunitinib efficacy in clear cell renal cell carcinomas. Br. J. Cancer 117, 947–953. doi: 10.1038/bjc.2017.276
Dufies, M., Grytsai, O., Ronco, C., Camara, O., Ambrosetti, D., Hagege, A., et al. (2019). New CXCR1/CXCR2 inhibitors represent an effective treatment for kidney or head and neck cancers sensitive or refractory to reference treatments. Theranostics 9, 5332–5346. doi: 10.7150/thno.34681
Ebos, J. M. L., and Kerbel, R. S. (2011). Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nat. Rev. Clin. Oncol. 8, 210–221. doi: 10.1038/nrclinonc.2011.21
Ebos, J. M. L., Lee, C. R., Christensen, J. G., Mutsaers, A. J., and Kerbel, R. S. (2007). Multiple circulating proangiogenic factors induced by sunitinib malate are tumor-independent and correlate with antitumor efficacy. Proc. Natl. Acad. Sci. U.S.A. 104, 17069–17074. doi: 10.1073/pnas.0708148104
Ebos, J. M. L., Lee, C. R., Cruz-Munoz, W., Bjarnason, G. A., Christensen, J. G., and Kerbel, R. S. (2009). Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 15, 232–239. doi: 10.1016/j.ccr.2009.01.021
Ehrmann, R. L., and Knoth, M. (1968). Choriocarcinoma. Transfilter stimulation of vasoproliferation in the hamster cheek pouch. Studied by light and electron microscopy. J. Natl. Cancer Inst. 41, 1329–1341.
Erra Díaz, F., Dantas, E., and Geffner, J. (2018). Unravelling the interplay between extracellular acidosis and immune cells. Mediators Inflamm. 2018:1218297. doi: 10.1155/2018/1218297
Escudier, B., Bellmunt, J., Négrier, S., Bajetta, E., Melichar, B., Bracarda, S., et al. (2010). Phase III trial of bevacizumab plus interferon alfa-2a in patients with metastatic renal cell carcinoma (AVOREN): final analysis of overall survival. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 28, 2144–2150. doi: 10.1200/JCO.2009.26.7849
Escudier, B., Porta, C., Bono, P., Powles, T., Eisen, T., Sternberg, C. N., et al. (2014). Randomized, controlled, double-blind, cross-over trial assessing treatment preference for pazopanib versus sunitinib in patients with metastatic renal cell carcinoma: PISCES Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 32, 1412–1418. doi: 10.1200/JCO.2013.50.8267
Faes, S., Santoro, T., Demartines, N., and Dormond, O. (2017). Evolving significance and future relevance of anti-angiogenic activity of mTOR inhibitors in cancer therapy. Cancers 9:152. doi: 10.3390/cancers9110152
Fagiani, E., and Christofori, G. (2013). Angiopoietins in angiogenesis. Cancer Lett. 328, 18–26. doi: 10.1016/j.canlet.2012.08.018
Falcon, B. L., Chintharlapalli, S., Uhlik, M. T., and Pytowski, B. (2016). Antagonist antibodies to vascular endothelial growth factor receptor 2 (VEGFR-2) as anti-angiogenic agents. Pharmacol. Ther. 164, 204–225. doi: 10.1016/j.pharmthera.2016.06.001
Favaro, E., Lord, S., Harris, A. L., and Buffa, F. M. (2011). Gene expression and hypoxia in breast cancer. Genome Med. 3:55. doi: 10.1186/gm271
Ferrara, N., and Henzel, W. J. (1989). Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem. Biophys. Res. Commun. 161, 851–858. doi: 10.1016/0006-291x(89)92678-8
Ferrari, G., Cook, B. D., Terushkin, V., Pintucci, G., and Mignatti, P. (2009). Transforming growth factor-beta 1 (TGF-beta1) induces angiogenesis through vascular endothelial growth factor (VEGF)-mediated apoptosis. J. Cell. Physiol. 219, 449–458. doi: 10.1002/jcp.21706
Finke, J., Ko, J., Rini, B., Rayman, P., Ireland, J., and Cohen, P. (2011). MDSC as a mechanism of tumor escape from sunitinib mediated anti-angiogenic therapy. Int. Immunopharmacol. 11, 856–861. doi: 10.1016/j.intimp.2011.01.030
Finke, J. H., Rini, B., Ireland, J., Rayman, P., Richmond, A., Golshayan, A., et al. (2008). Sunitinib reverses type-1 immune suppression and decreases T-regulatory cells in renal cell carcinoma patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 14, 6674–6682. doi: 10.1158/1078-0432.CCR-07-5212
Fischer, C., Jonckx, B., Mazzone, M., Zacchigna, S., Loges, S., Pattarini, L., et al. (2007). Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell 131, 463–475. doi: 10.1016/j.cell.2007.08.038
Folkman, J. (1971). Tumor angiogenesis: therapeutic implications. N. Engl. J. Med. 285, 1182–1186. doi: 10.1056/NEJM197111182852108
Folkman, J. (1972). Anti-angiogenesis: new concept for therapy of solid tumors. Ann. Surg. 175, 409–416. doi: 10.1097/00000658-197203000-00014
Foubert, P., and Varner, J. A. (2012). Integrins in tumor angiogenesis and lymphangiogenesis. Methods Mol. Biol. Clifton. NJ 757, 471–486. doi: 10.1007/978-1-61779-166-6_27
Fransvea, E., Mazzocca, A., Antonaci, S., and Giannelli, G. (2009). Targeting transforming growth factor (TGF)-βRI inhibits activation of β1 integrin and blocks vascular invasion in hepatocellular carcinoma. Hepatology 49, 839–850. doi: 10.1002/hep.22731
Frentzas, S., Simoneau, E., Bridgeman, V. L., Vermeulen, P. B., Foo, S., Kostaras, E., et al. (2016). Vessel co-option mediates resistance to anti-angiogenic therapy in liver metastases. Nat. Med. 22, 1294–1302. doi: 10.1038/nm.4197
Fujita, K., and Akita, M. (2017). Tumor angiogenesis: a focus on the role of cancer stem cells. physiol. pathol. Angiogenesis Signal. Mech. Target. Ther. 2017:215. doi: 10.5772/66402
Garg, M. (2013). Epithelial-mesenchymal transition - activating transcription factors - multifunctional regulators in cancer. World J. Stem Cells 5, 188–195. doi: 10.4252/wjsc.v5.i4.188
Gewirtz, D. A. (2014). The autophagic response to radiation: relevance for radiation sensitization in cancer therapy. Radiat. Res. 182, 363–367. doi: 10.1667/RR13774.1
Giuliano, S., Cormerais, Y., Dufies, M., Grépin, R., Colosetti, P., Belaid, A., et al. (2015). Resistance to sunitinib in renal clear cell carcinoma results from sequestration in lysosomes and inhibition of the autophagic flux. Autophagy 11, 1891–1904. doi: 10.1080/15548627.2015.1085742
Giuliano, S., Dufies, M., Ndiaye, P. D., Viotti, J., Borchiellini, D., Parola, J., et al. (2019). Resistance to lysosomotropic drugs used to treat kidney and breast cancers involves autophagy and inflammation and converges in inducing CXCL5. Theranostics 9, 1181–1199. doi: 10.7150/thno.29093
Goede, V., Coutelle, O., Neuneier, J., Reinacher-Schick, A., Schnell, R., Koslowsky, T. C., et al. (2010). Identification of serum angiopoietin-2 as a biomarker for clinical outcome of colorectal cancer patients treated with bevacizumab-containing therapy. Br. J. Cancer 103, 1407–1414. doi: 10.1038/sj.bjc.6605925
Goel, S., Duda, D. G., Xu, L., Munn, L. L., Boucher, Y., Fukumura, D., et al. (2011). Normalization of the vasculature for treatment of cancer and other diseases. Physiol. Rev. 91, 1071–1121. doi: 10.1152/physrev.00038.2010
Goveia, J., Rholenova, K., Taverna, F., Treps, L., Conradi, L.-C., Pircher, A., et al. (2020). An integrated gene expression landscape profiling approach to identify lung tumor endothelial cell heterogeneity and angiogenic candidates. Cancer Cell 37, 21–36. doi: 10.1016/j.ccell.2019.12.001
Greenblatt, M., and Shubi, P. (1968). Tumor angiogenesis: transfilter diffusion studies in the hamster by the transparent chamber technique. J. Natl. Cancer Inst. 41, 111–124.
Grépin, R., Guyot, M., Giuliano, S., Boncompagni, M., Ambrosetti, D., Chamorey, E., et al. (2014). The CXCL7/CXCR1/2 axis is a key driver in the growth of clear cell renal cell carcinoma. Cancer Res. 74, 873–883. doi: 10.1158/0008-5472.CAN-13-1267
Grepin, R., Guyot, M., Jacquin, M., Durivault, J., Chamorey, E., Sudaka, A., et al. (2012). Acceleration of clear cell renal cell carcinoma growth in mice following bevacizumab/Avastin treatment: the role of CXCL cytokines. Oncogene 31, 1683–1694. doi: 10.1038/onc.2011.360
Grunewald, M., Avraham, I., Dor, Y., Bachar-Lustig, E., Itin, A., Jung, S., et al. (2006). VEGF-induced adult neovascularization: recruitment, retention, and role of accessory cells. Cell 124, 175–189. doi: 10.1016/j.cell.2005.10.036
Guyot, M., and Pagès, G. (2015). “VEGF splicing and the role of VEGF splice variants: from physiological-pathological conditions to specific Pre-mRNA splicing,” in VEGF Signaling Methods in Molecular Biology, ed. L. Fiedler (New York, NY: Springer New York), 3–23. doi: 10.1007/978-1-4939-2917-7_1
Gyanchandani, R., Ortega Alves, M. V., Myers, J. N., and Kim, S. (2013). A proangiogenic signature is revealed in FGF-mediated bevacizumab-resistant head and neck squamous cell carcinoma. Mol. Cancer Res. MCR 11, 1585–1596. doi: 10.1158/1541-7786.MCR-13-0358
Haas, N. B., Manola, J., Uzzo, R. G., Flaherty, K. T., Wood, C. G., Kane, C., et al. (2016). Adjuvant sunitinib or sorafenib for high-risk, non-metastatic renal-cell carcinoma (ECOG-ACRIN E2805): a double-blind, placebo-controlled, randomised, phase 3 trial. Lancet Lond. Engl. 387, 2008–2016. doi: 10.1016/S0140-6736(16)00559-6
Haibe, Y., Kreidieh, M., El Hajj, H., Khalifeh, I., Mukherji, D., Temraz, S., et al. (2020). Resistance mechanisms to anti-angiogenic therapies in cancer. Front. Oncol. 10:221. doi: 10.3389/fonc.2020.00221
Hainsworth, J. D., Spigel, D. R., Sosman, J. A., Burris, H. A., Farley, C., Cucullu, H., et al. (2007). Treatment of advanced renal cell carcinoma with the combination bevacizumab/erlotinib/imatinib: a phase I/II trial. Clin. Genitourin. Cancer 5, 427–432. doi: 10.3816/CGC.2007.n.030
Halestrap, A. P., and Price, N. T. (1999). The proton-linked monocarboxylate transporter (MCT) family: structure, function and regulation. Biochem. J. 343(Pt 2), 281–299.
Hanahan, D. (1997). Signaling vascular morphogenesis and maintenance. Science 277, 48–50. doi: 10.1126/science.277.5322.48
Hanahan, D., and Folkman, J. (1996). Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 86, 353–364. doi: 10.1016/s0092-8674(00)80108-7
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. doi: 10.1016/j.cell.2011.02.013
Hantelys, F., Godet, A.-C., David, F., Tatin, F., Renaud-Gabardos, E., Pujol, F., et al. (2019). Vasohibin1, a new mouse cardiomyocyte IRES trans-acting factor that regulates translation in early hypoxia. eLife 8:e50094. doi: 10.7554/eLife.50094
Harris, A. L. (2002). Hypoxia — a key regulatory factor in tumour growth. Nat. Rev. Cancer 2, 38–47. doi: 10.1038/nrc704
Harshman, L. C., and Srinivas, S. (2010). The bevacizumab experience in advanced renal cell carcinoma. OncoTargets Ther. 3, 179–189.
Hart, K. C., Robertson, S. C., Kanemitsu, M. Y., Meyer, A. N., Tynan, J. A., and Donoghue, D. J. (2000). Transformation and Stat activation by derivatives of FGFR1. FGFR3, and FGFR4. Oncogene 19, 3309–3320. doi: 10.1038/sj.onc.1203650
Hasnis, E., Alishekevitz, D., Gingis-Veltski, S., Bril, R., Fremder, E., Voloshin, T., et al. (2014). Anti-Bv8 antibody and metronomic gemcitabine improve pancreatic adenocarcinoma treatment outcome following weekly gemcitabine therapy. Neoplasia N. Y. N. 16, 501–510. doi: 10.1016/j.neo.2014.05.011
Hazzard, T. M., and Stouffer, R. L. (2000). Angiogenesis in ovarian follicular and luteal development. Best Pract. Res. Clin. Obstet. Gynaecol. 14, 883–900. doi: 10.1053/beog.2000.0133
Heidenreich, R., Röcken, M., and Ghoreschi, K. (2009). Angiogenesis drives psoriasis pathogenesis. Int. J. Exp. Pathol. 90, 232–248. doi: 10.1111/j.1365-2613.2009.00669.x
Helfrich, I., Edler, L., Sucker, A., Thomas, M., Christian, S., Schadendorf, D., et al. (2009). Angiopoietin-2 levels are associated with disease progression in metastatic malignant melanoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 15, 1384–1392. doi: 10.1158/1078-0432.CCR-08-1615
Hida, K., Hida, Y., Amin, D. N., Flint, A. F., Panigrahy, D., Morton, C. C., et al. (2004). Tumor-associated endothelial cells with cytogenetic abnormalities. Cancer Res. 64, 8249–8255. doi: 10.1158/0008-5472.CAN-04-1567
Hidalgo, M., Martinez-Garcia, M., Le Tourneau, C., Massard, C., Garralda, E., Boni, V., et al. (2018). First-in-human phase I study of single-agent vanucizumab, a first-in-class bispecific anti-Angiopoietin-2/Anti-VEGF-A antibody, in adult patients with advanced solid tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 24, 1536–1545. doi: 10.1158/1078-0432.CCR-17-1588
Hirota, K., and Semenza, G. L. (2006). Regulation of angiogenesis by hypoxia-inducible factor 1. Crit. Rev. Oncol. Hematol. 59, 15–26. doi: 10.1016/j.critrevonc.2005.12.003
Hirth, J., Watkins, P. B., Strawderman, M., Schott, A., Bruno, R., and Baker, L. H. (2000). The effect of an individual’s cytochrome CYP3A4 activity on docetaxel clearance. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 6, 1255–1258.
Holmgaard, R. B., Schaer, D. A., Li, Y., Castaneda, S. P., Murphy, M. Y., Xu, X., et al. (2018). Targeting the TGFβ pathway with galunisertib, a TGFβRI small molecule inhibitor, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. J. Immunother. Cancer 6:47. doi: 10.1186/s40425-018-0356-4
Holmgaard, R. B., Zamarin, D., Lesokhin, A., Merghoub, T., and Wolchok, J. D. (2016). Targeting myeloid-derived suppressor cells with colony stimulating factor-1 receptor blockade can reverse immune resistance to immunotherapy in indoleamine 2,3-dioxygenase-expressing tumors. EBioMedicine 6, 50–58. doi: 10.1016/j.ebiom.2016.02.024
Holmgren, L., O’Reilly, M. S., and Folkman, J. (1995). Dormancy of micrometastases: balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat. Med. 1, 149–153. doi: 10.1038/nm0295-149
Hosaka, K., Yang, Y., Nakamura, M., Andersson, P., Yang, X., Zhang, Y., et al. (2018). Dual roles of endothelial FGF-2–FGFR1–PDGF-BB and perivascular FGF-2–FGFR2–PDGFRβ signaling pathways in tumor vascular remodeling. Cell Discov. 4:3. doi: 10.1038/s41421-017-0002-1
Hosaka, K., Yang, Y., Seki, T., Fischer, C., Dubey, O., Fredlund, E., et al. (2016). Pericyte–fibroblast transition promotes tumor growth and metastasis. Proc. Natl. Acad. Sci. U.S.A. 113, E5618–E5627. doi: 10.1073/pnas.1608384113
Hsieh, J. J., Purdue, M. P., Signoretti, S., Swanton, C., Albiges, L., Schmidinger, M., et al. (2017). Renal cell carcinoma. Nat. Rev. Dis. Primer 3:17009. doi: 10.1038/nrdp.2017.9
Hsu, Y.-L., Wu, C.-Y., Hung, J.-Y., Lin, Y.-S., Huang, M.-S., and Kuo, P.-L. (2013). Galectin-1 promotes lung cancer tumor metastasis by potentiating integrin α6β4 and Notch1/Jagged2 signaling pathway. Carcinogenesis 34, 1370–1381. doi: 10.1093/carcin/bgt040
Hu, P.-H., Pan, L.-H., Wong, P. T.-Y., Chen, W.-H., Yang, Y.-Q., Wang, H., et al. (2016). 125I-labeled anti-bFGF monoclonal antibody inhibits growth of hepatocellular carcinoma. World J. Gastroenterol. 22, 5033–5041. doi: 10.3748/wjg.v22.i21.5033
Hurwitz, H., Fehrenbacher, L., Novotny, W., Cartwright, T., Hainsworth, J., Heim, W., et al. (2004). Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 350, 2335–2342. doi: 10.1056/NEJMoa032691
Hwang, K.-E., Kim, Y.-S., Jung, J.-W., Kwon, S.-J., Park, D.-S., Cha, B.-K., et al. (2015). Inhibition of autophagy potentiates pemetrexed and simvastatin-induced apoptotic cell death in malignant mesothelioma and non-small cell lung cancer cells. Oncotarget 6, 29482–29496.
Jahangiri, A., Aghi, M. K., and Carbonell, W. S. (2014). β1 integrin: critical path to antiangiogenic therapy resistance and beyond. Cancer Res. 74, 3–7. doi: 10.1158/0008-5472.CAN-13-1742
Jahangiri, A., De Lay, M., Miller, L. M., Carbonell, W. S., Hu, Y.-L., Lu, K., et al. (2013). Gene expression profile identifies tyrosine kinase c-Met as a targetable mediator of antiangiogenic therapy resistance. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 19, 1773–1783. doi: 10.1158/1078-0432.CCR-12-1281
Jain, R. K. (2005). Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 307, 58–62. doi: 10.1126/science.1104819
Jain, R. K., and Duda, D. G. (2003). Role of bone marrow-derived cells in tumor angiogenesis and treatment. Cancer Cell 3, 515–516. doi: 10.1016/S1535-6108(03)00138-7
Janku, F., McConkey, D. J., Hong, D. S., and Kurzrock, R. (2011). Autophagy as a target for anticancer therapy. Nat. Rev. Clin. Oncol. 8, 528–539. doi: 10.1038/nrclinonc.2011.71
Janning, M., Müller, V., Vettorazzi, E., Cubas-Cordova, M., Gensch, V., Ben-Batalla, I., et al. (2019). Evaluation of soluble carbonic anhydrase IX as predictive marker for efficacy of bevacizumab: a biomarker analysis from the geparquinto phase III neoadjuvant breast cancer trial. Int. J. Cancer 145, 857–868. doi: 10.1002/ijc.32163
Jayson, G. C., Zhou, C., Backen, A., Horsley, L., Marti-Marti, K., Shaw, D., et al. (2018). Plasma Tie2 is a tumor vascular response biomarker for VEGF inhibitors in metastatic colorectal cancer. Nat. Commun. 9:4672. doi: 10.1038/s41467-018-07174-1
Jeansson, M., Gawlik, A., Anderson, G., Li, C., Kerjaschki, D., Henkelman, M., et al. (2011). Angiopoietin-1 is essential in mouse vasculature during development and in response to injury. J. Clin. Invest. 121, 2278–2289. doi: 10.1172/JCI46322
Jeon, H.-M., and Lee, J. (2017). MET: roles in epithelial-mesenchymal transition and cancer stemness. Ann. Transl. Med. 5:5. doi: 10.21037/atm.2016.12.67
Jiménez-Valerio, G., and Casanovas, O. (2017). Angiogenesis and metabolism: entwined for therapy resistance. Trends Cancer 3, 10–18. doi: 10.1016/j.trecan.2016.11.007
Joseph, J. P., Harishankar, M. K., Pillai, A. A., and Devi, A. (2018). Hypoxia induced EMT: a review on the mechanism of tumor progression and metastasis in OSCC. Oral Oncol. 80, 23–32. doi: 10.1016/j.oraloncology.2018.03.004
Joukov, V., Pajusola, K., Kaipainen, A., Chilov, D., Lahtinen, I., Kukk, E., et al. (1996). A novel vascular endothelial growth factor, VEGF-C, is a ligand for the Flt4 (VEGFR-3) and KDR (VEGFR-2) receptor tyrosine kinases. EMBO J. 15, 290–298.
Keck, P. J., Hauser, S. D., Krivi, G., Sanzo, K., Warren, T., Feder, J., et al. (1989). Vascular permeability factor, an endothelial cell mitogen related to PDGF. Science 246, 1309–1312. doi: 10.1126/science.2479987
Kerbel, R., and Folkman, J. (2002). Clinical translation of angiogenesis inhibitors. Nat. Rev. Cancer 2, 727–739. doi: 10.1038/nrc905
Kerbel, R. S. (2008). Tumor angiogenesis. N. Engl. J. Med. 358, 2039–2049. doi: 10.1056/NEJMra0706596
Kienast, Y., Klein, C., Scheuer, W., Raemsch, R., Lorenzon, E., Bernicke, D., et al. (2013). Ang-2-VEGF-A CrossMab, a novel bispecific human IgG1 antibody blocking VEGF-A and Ang-2 functions simultaneously, mediates potent antitumor, antiangiogenic, and antimetastatic efficacy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 19, 6730–6740. doi: 10.1158/1078-0432.CCR-13-0081
Kim, K. J., Li, B., Winer, J., Armanini, M., Gillett, N., Phillips, H. S., et al. (1993). Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature 362, 841–844. doi: 10.1038/362841a0
Kioi, M., Vogel, H., Schultz, G., Hoffman, R. M., Harsh, G. R., and Brown, J. M. (2010). Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J. Clin. Invest. 120, 694–705. doi: 10.1172/JCI40283
Knighton, D. R., Hunt, T. K., Scheuenstuhl, H., Halliday, B. J., Werb, Z., and Banda, M. J. (1983). Oxygen tension regulates the expression of angiogenesis factor by macrophages. Science 221, 1283–1285. doi: 10.1126/science.6612342
Ko, J. S., Zea, A. H., Rini, B. I., Ireland, J. L., Elson, P., Cohen, P., et al. (2009). Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 15, 2148–2157. doi: 10.1158/1078-0432.CCR-08-1332
Ko, S. Y., Lee, W., Kenny, H. A., Dang, L. H., Ellis, L. M., Jonasch, E., et al. (2019). Cancer-derived small extracellular vesicles promote angiogenesis by heparin-bound, bevacizumab-insensitive VEGF, independent of vesicle uptake. Commun. Biol. 2, 1–17. doi: 10.1038/s42003-019-0609-x
Kopetz, S., Hoff, P. M., Morris, J. S., Wolff, R. A., Eng, C., Glover, K. Y., et al. (2010). Phase II trial of infusional fluorouracil, irinotecan, and bevacizumab for metastatic colorectal cancer: efficacy and circulating angiogenic biomarkers associated with therapeutic resistance. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 28, 453–459. doi: 10.1200/JCO.2009.24.8252
Kuczynski, E. A., Vermeulen, P. B., Pezzella, F., Kerbel, R. S., and Reynolds, A. R. (2019). Vessel co-option in cancer. Nat. Rev. Clin. Oncol. 16, 469–493. doi: 10.1038/s41571-019-0181-9
Kuczynski, E. A., Yin, M., Bar-Zion, A., Lee, C. R., Butz, H., Man, S., et al. (2016). Co-option of liver vessels and not sprouting angiogenesis drives acquired sorafenib resistance in hepatocellular carcinoma. JNCI J. Natl. Cancer Inst. 108:djw030. doi: 10.1093/jnci/djw030
Kumar, K., Wigfield, S., Gee, H. E., Devlin, C. M., Singleton, D., Li, J.-L., et al. (2013). Dichloroacetate reverses the hypoxic adaptation to bevacizumab and enhances its antitumor effects in mouse xenografts. J. Mol. Med. Berl. Ger. 91, 749–758. doi: 10.1007/s00109-013-0996-2
Laderoute, K. R., Alarcon, R. M., Brody, M. D., Calaoagan, J. M., Chen, E. Y., Knapp, A. M., et al. (2000). Opposing effects of hypoxia on expression of the angiogenic inhibitor thrombospondin 1 and the angiogenic inducer vascular endothelial growth factor. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 6, 2941–2950.
L’Allemain, G., Paris, S., and Pouysségur, J. (1985). Role of a Na+-dependent Cl-/HCO3- exchange in regulation of intracellular pH in fibroblasts. J. Biol. Chem. 260, 4877–4883.
Lam, H.-M., Vessella, R. L., and Morrissey, C. (2014). The role of the microenvironment – dormant prostate disseminated tumor cells in the bone marrow. Drug Discov. Today Technol. 11, 41–47. doi: 10.1016/j.ddtec.2014.02.002
LaVallee, T. M., Prudovsky, I. A., McMahon, G. A., Hu, X., and Maciag, T. (1998). Activation of the MAP kinase pathway by FGF-1 correlates with cell proliferation induction while activation of the Src pathway correlates with migration. J. Cell Biol. 141, 1647–1658. doi: 10.1083/jcb.141.7.1647
Lee, E. Q., Muzikansky, A., Duda, D. G., Gaffey, S., Dietrich, J., Nayak, L., et al. (2019). Phase II trial of ponatinib in patients with bevacizumab-refractory glioblastoma. Cancer Med. 8, 5988–5994. doi: 10.1002/cam4.2505
Leenders, W. P. J., Küsters, B., Verrijp, K., Maass, C., Wesseling, P., Heerschap, A., et al. (2004). Antiangiogenic therapy of cerebral melanoma metastases results in sustained tumor progression via vessel co-option. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 10, 6222–6230. doi: 10.1158/1078-0432.CCR-04-0823
Leung, D. W., Cachianes, G., Kuang, W. J., Goeddel, D. V., and Ferrara, N. (1989). Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 246, 1306–1309. doi: 10.1126/science.2479986
Levéen, P., Pekny, M., Gebre-Medhin, S., Swolin, B., Larsson, E., and Betsholtz, C. (1994). Mice deficient for PDGF B show renal, cardiovascular, and hematological abnormalities. Genes Dev. 8, 1875–1887. doi: 10.1101/gad.8.16.1875
Li, F., Zhou, K., Gao, L., Zhang, B., Li, W., Yan, W., et al. (2016). Radiation induces the generation of cancer stem cells: a novel mechanism for cancer radioresistance. Oncol. Lett. 12, 3059–3065. doi: 10.3892/ol.2016.5124
Li, Y.-J., Lei, Y.-H., Yao, N., Wang, C.-R., Hu, N., Ye, W.-C., et al. (2017). Autophagy and multidrug resistance in cancer. Chin. J. Cancer 36:52. doi: 10.1186/s40880-017-0219-2
Lin, J., Sampath, D., Nannini, M. A., Lee, B. B., Degtyarev, M., Oeh, J., et al. (2013). Targeting activated Akt with GDC-0068, a novel selective Akt inhibitor that is efficacious in multiple tumor models. Clin. Cancer Res. 19, 1760–1772. doi: 10.1158/1078-0432.CCR-12-3072
Liu, B. Y., Soloviev, I., Chang, P., Lee, J., Huang, X., Zhong, C., et al. (2010). Stromal cell-derived factor-1/CXCL12 contributes to MMTV-Wnt1 tumor growth involving Gr1+CD11b+ cells. PLoS One 5:e8611. doi: 10.1371/journal.pone.0008611
Liu, K.-W., Hu, B., and Cheng, S.-Y. (2011). Platelet-derived growth factor signaling in human malignancies. Chin. J. Cancer 30, 581–584. doi: 10.5732/cjc.011.10300
Liu, L., Yang, L., Yan, W., Zhai, J., Pizzo, D. P., Chu, P., et al. (2018). Chemotherapy induces breast cancer stemness in association with dysregulated monocytosis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 24, 2370–2382. doi: 10.1158/1078-0432.CCR-17-2545
Liu, T., Ma, W., Xu, H., Huang, M., Zhang, D., He, Z., et al. (2018). PDGF-mediated mesenchymal transformation renders endothelial resistance to anti-VEGF treatment in glioblastoma. Nat. Commun. 9, 1–13. doi: 10.1038/s41467-018-05982-z
Llovet, J. M., Ricci, S., Mazzaferro, V., Hilgard, P., Gane, E., Blanc, J.-F., et al. (2008). Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 359, 378–390. doi: 10.1056/NEJMoa0708857
Lu, K. V., Chang, J. P., Parachoniak, C. A., Pandika, M. M., Aghi, M. K., Meyronet, D., et al. (2012). VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell 22, 21–35. doi: 10.1016/j.ccr.2012.05.037
Luan, W., Pang, Y., Li, R., Wei, X., Jiao, X., Shi, J., et al. (2019). Akt/mTOR-mediated autophagy confers resistance to Bet inhibitor JQ1 In ovarian cancer. OncoTargets Ther. 12, 8063–8074. doi: 10.2147/OTT.S220267
Maacha, S., Bhat, A. A., Jimenez, L., Raza, A., Haris, M., Uddin, S., et al. (2019). Extracellular vesicles-mediated intercellular communication: roles in the tumor microenvironment and anti-cancer drug resistance. Mol. Cancer 18:55. doi: 10.1186/s12943-019-0965-7
MacDonagh, L., Gallagher, M. F., Ffrench, B., Gasch, C., Breen, E., Gray, S. G., et al. (2017). Targeting the cancer stem cell marker, aldehyde dehydrogenase 1, to circumvent cisplatin resistance in NSCLC. Oncotarget 8, 72544–72563. doi: 10.18632/oncotarget.19881
Maione, F., Capano, S., Regano, D., Zentilin, L., Giacca, M., Casanovas, O., et al. (2012). Semaphorin 3A overcomes cancer hypoxia and metastatic dissemination induced by antiangiogenic treatment in mice. J. Clin. Invest. 122, 1832–1848. doi: 10.1172/JCI58976
Maishi, N., Annan, D. A., Kikuchi, H., Hida, Y., and Hida, K. (2019). Tumor endothelial heterogeneity in cancer progression. Cancers 11:1511. doi: 10.3390/cancers11101511
Maisonpierre, P. C., Suri, C., Jones, P. F., Bartunkova, S., Wiegand, S. J., Radziejewski, C., et al. (1997). Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 277, 55–60. doi: 10.1126/science.277.5322.55
Maniotis, A. J., Folberg, R., Hess, A., Seftor, E. A., Gardner, L. M., Pe’er, J., et al. (1999). Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am. J. Pathol. 155, 739–752. doi: 10.1016/S0002-9440(10)65173-5
Manzat Saplacan, R. M., Balacescu, L., Gherman, C., Chira, R. I., Craiu, A., Mircea, P. A., et al. (2017). The role of PDGFs and PDGFRs in colorectal cancer. Mediators Inflamm. 2017:e4708076. doi: 10.1155/2017/4708076
Marigo, I., Dolcetti, L., Serafini, P., Zanovello, P., and Bronte, V. (2008). Tumor-induced tolerance and immune suppression by myeloid derived suppressor cells. Immunol. Rev. 222, 162–179. doi: 10.1111/j.1600-065X.2008.00602.x
Martin, J. D., Seano, G., and Jain, R. K. (2019). Normalizing function of tumor vessels: progress. opportunities, and challenges. Annu. Rev. Physiol. 81, 505–534. doi: 10.1146/annurev-physiol-020518-114700
Martínez-Sáez, O., Gajate Borau, P., Alonso-Gordoa, T., Molina-Cerrillo, J., and Grande, E. (2017). Targeting HIF-2 α in clear cell renal cell carcinoma: a promising therapeutic strategy. Crit. Rev. Oncol. Hematol. 111, 117–123. doi: 10.1016/j.critrevonc.2017.01.013
Massagué, J. (2000). How cells read TGF-beta signals. Nat. Rev. Mol. Cell Biol. 1, 169–178. doi: 10.1038/35043051
Mathew, R., Karantza-Wadsworth, V., and White, E. (2007). Role of autophagy in cancer. Nat. Rev. Cancer 7, 961–967. doi: 10.1038/nrc2254
Mazzieri, R., Pucci, F., Moi, D., Zonari, E., Ranghetti, A., Berti, A., et al. (2011). Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell 19, 512–526. doi: 10.1016/j.ccr.2011.02.005
McIntyre, A., and Harris, A. L. (2015). Metabolic and hypoxic adaptation to anti-angiogenic therapy: a target for induced essentiality. EMBO Mol. Med. 7, 368–379. doi: 10.15252/emmm.201404271
Miller, K., Wang, M., Gralow, J., Dickler, M., Cobleigh, M., Perez, E. A., et al. (2007). Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N. Engl. J. Med. 357, 2666–2676. doi: 10.1056/NEJMoa072113
Mitsuhashi, A., Goto, H., Saijo, A., Trung, V. T., Aono, Y., Ogino, H., et al. (2015). Fibrocyte-like cells mediate acquired resistance to anti-angiogenic therapy with bevacizumab. Nat. Commun. 6:8792. doi: 10.1038/ncomms9792
Mittal, V. (2018). Epithelial mesenchymal transition in tumor metastasis. Annu. Rev. Pathol. 13, 395–412. doi: 10.1146/annurev-pathol-020117-043854
Miyazawa, K., Ohtomo, T., Moriya, S., Naito, M., Kuroda, M., Itoh, M., et al. (2010). Cytoprotective autophagy induction by imatinib mesylate in Non-BCR-ABL expressing cells. Blood 116, 4937–4937. doi: 10.1182/blood.V116.21.4937.4937
Mizumoto, A., Yamamoto, K., Nakayama, Y., Takara, K., Nakagawa, T., Hirano, T., et al. (2015). Induction of epithelial-mesenchymal transition via activation of epidermal growth factor receptor contributes to sunitinib resistance in human renal cell carcinoma cell lines. J. Pharmacol. Exp. Ther. 355, 152–158. doi: 10.1124/jpet.115.226639
Mizushima, N. (2007). Autophagy: process and function. Genes Dev. 21, 2861–2873. doi: 10.1101/gad.1599207
Mohammed, R. A. A., Green, A., El-Shikh, S., Paish, E. C., Ellis, I. O., and Martin, S. G. (2007). Prognostic significance of vascular endothelial cell growth factors -A, -C and -D in breast cancer and their relationship with angio- and lymphangiogenesis. Br. J. Cancer 96, 1092–1100. doi: 10.1038/sj.bjc.6603678
Morfoisse, F., Kuchnio, A., Frainay, C., Gomez-Brouchet, A., Delisle, M.-B., Marzi, S., et al. (2014). Hypoxia induces VEGF-C expression in metastatic tumor cells via a HIF-1α-independent translation-mediated mechanism. Cell Rep. 6, 155–167. doi: 10.1016/j.celrep.2013.12.011
Motzer, R. J., Escudier, B., Powles, T., Scheffold, C., and Choueiri, T. K. (2018). Long-term follow-up of overall survival for cabozantinib versus everolimus in advanced renal cell carcinoma. Br. J. Cancer 118, 1176–1178. doi: 10.1038/s41416-018-0061-6
Motzer, R. J., Escudier, B., Tomczak, P., Hutson, T. E., Michaelson, M. D., Negrier, S., et al. (2013). Axitinib versus sorafenib as second-line treatment for advanced renal cell carcinoma: overall survival analysis and updated results from a randomised phase 3 trial. Lancet Oncol. 14, 552–562. doi: 10.1016/S1470-2045(13)70093-7
Motzer, R. J., Hutson, T. E., Glen, H., Michaelson, M. D., Molina, A., Eisen, T., et al. (2015). Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: a randomised, phase 2, open-label, multicentre trial. Lancet Oncol. 16, 1473–1482. doi: 10.1016/S1470-2045(15)00290-9
Motzer, R. J., Hutson, T. E., Ren, M., Dutcus, C., and Larkin, J. (2016). Independent assessment of lenvatinib plus everolimus in patients with metastatic renal cell carcinoma. Lancet Oncol. 17, e4–e5. doi: 10.1016/S1470-2045(15)00543-4
Motzer, R. J., Hutson, T. E., Tomczak, P., Michaelson, M. D., Bukowski, R. M., Oudard, S., et al. (2009). Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 27, 3584–3590. doi: 10.1200/JCO.2008.20.1293
Motzer, R. J., Penkov, K., Haanen, J., Rini, B., Albiges, L., Campbell, M. T., et al. (2019). Avelumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N. Engl. J. Med. 380, 1103–1115. doi: 10.1056/NEJMoa1816047
Müller, A., Homey, B., Soto, H., Ge, N., Catron, D., Buchanan, M. E., et al. (2001). Involvement of chemokine receptors in breast cancer metastasis. Nature 410, 50–56. doi: 10.1038/35065016
Murdoch, C., Tazzyman, S., Webster, S., and Lewis, C. E. (2007). Expression of Tie-2 by human monocytes and their responses to angiopoietin-2. J. Immunol. Baltim. Md 1950, 7405–7411. doi: 10.4049/jimmunol.178.11.7405
Nakajima, E. C., and Van Houten, B. (2013). Metabolic symbiosis in cancer: refocusing the Warburg lens. Mol. Carcinog. 52, 329–337. doi: 10.1002/mc.21863
Nakashima, O., Sugihara, S., Kage, M., and Kojiro, M. (1995). Pathomorphologic characteristics of small hepatocellular carcinoma: a special reference to small hepatocellular carcinoma with indistinct margins. Hepatol. Baltim. Md 22, 101–105.
Navis, A. C., Bourgonje, A., Wesseling, P., Wright, A., Hendriks, W., Verrijp, K., et al. (2013). Effects of dual targeting of tumor cells and stroma in human glioblastoma xenografts with a tyrosine kinase inhibitor against c-MET and VEGFR2. PLoS One 8:e58262. doi: 10.1371/journal.pone.0058262
Ndiaye, P. D., Dufies, M., Giuliano, S., Douguet, L., Grépin, R., Durivault, J., et al. (2019). VEGFC acts as a double-edged sword in renal cell carcinoma aggressiveness. Theranostics 9, 661–675. doi: 10.7150/thno.27794
Nishida, N., Yano, H., Nishida, T., Kamura, T., and Kojiro, M. (2006). Angiogenesis in Cancer. Vasc. Health Risk Manag. 2, 213–219.
Norden, A. D., Drappatz, J., and Wen, P. Y. (2009). Antiangiogenic therapies for high-grade glioma. Nat. Rev. Neurol. 5, 610–620. doi: 10.1038/nrneurol.2009.159
Norden, A. D., Schiff, D., Ahluwalia, M. S., Lesser, G. J., Nayak, L., Lee, E. Q., et al. (2015). Phase II trial of triple tyrosine kinase receptor inhibitor nintedanib in recurrent high-grade gliomas. J. Neurooncol. 121, 297–302. doi: 10.1007/s11060-014-1631-y
Offersen, B. V., Pfeiffer, P., Hamilton-Dutoit, S., and Overgaard, J. (2001). Patterns of angiogenesis in nonsmall-cell lung carcinoma. Cancer 91, 1500–1509.
Ogawa, M., Yamamoto, H., Nagano, H., Miyake, Y., Sugita, Y., Hata, T., et al. (2004). Hepatic expression of ANG2 RNA in metastatic colorectal cancer. Hepatol. Baltim. Md 39, 528–539. doi: 10.1002/hep.20048
Olive, K. P., Jacobetz, M. A., Davidson, C. J., Gopinathan, A., McIntyre, D., Honess, D., et al. (2009). Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324, 1457–1461. doi: 10.1126/science.1171362
O’Reilly, M. S., Holmgren, L., Chen, C., and Folkman, J. (1996). Angiostatin induces and sustains dormancy of human primary tumors in mice. Nat. Med. 2, 689–692. doi: 10.1038/nm0696-689
Orimo, A., Gupta, P. B., Sgroi, D. C., Arenzana-Seisdedos, F., Delaunay, T., Naeem, R., et al. (2005). Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 121, 335–348. doi: 10.1016/j.cell.2005.02.034
Orlidge, A., and D’Amore, P. A. (1987). Inhibition of capillary endothelial cell growth by pericytes and smooth muscle cells. J. Cell Biol. 105, 1455–1462. doi: 10.1083/jcb.105.3.1455
Ornitz, D. M., and Itoh, N. (2015). The fibroblast growth factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 4, 215–266. doi: 10.1002/wdev.176
Osawa, T., Ohga, N., Akiyama, K., Hida, Y., Kitayama, K., Kawamoto, T., et al. (2013). Lysyl oxidase secreted by tumour endothelial cells promotes angiogenesis and metastasis. Br. J. Cancer 109, 2237–2247. doi: 10.1038/bjc.2013.535
Pàez-Ribes, M., Allen, E., Hudock, J., Takeda, T., Okuyama, H., Viñals, F., et al. (2009). Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 15, 220–231. doi: 10.1016/j.ccr.2009.01.027
Papadopoulos, N., and Lennartsson, J. (2018). The PDGF/PDGFR pathway as a drug target. Mol. Aspects Med. 62, 75–88. doi: 10.1016/j.mam.2017.11.007
Pardali, E., Goumans, M.-J., and ten Dijke, P. (2010). Signaling by members of the TGF-beta family in vascular morphogenesis and disease. Trends Cell Biol. 20, 556–567. doi: 10.1016/j.tcb.2010.06.006
Park, S. Y., Piao, Y., Jeong, K. J., Dong, J., and de Groot, J. F. (2016). Periostin (POSTN) Regulates tumor resistance to antiangiogenic therapy in glioma models. Mol. Cancer Ther. 15, 2187–2197. doi: 10.1158/1535-7163.MCT-15-0427
Parks, S. K., Mazure, N. M., Counillon, L., and Pouysségur, J. (2013). Hypoxia promotes tumor cell survival in acidic conditions by preserving ATP levels. J. Cell. Physiol. 228, 1854–1862. doi: 10.1002/jcp.24346
Parks, S. K., Mueller-Klieser, W., and Pouysségur, J. (2020). Lactate and acidity in the cancer microenvironment. Annu. Rev. Cancer Biol. 4, 141–158. doi: 10.1146/annurev-cancerbio-030419-033556
Pasquier, E., and Dias, S. (2010). Endothelial progenitor cells: hope beyond controversy. Curr. Cancer Drug Targets 10, 914–921. doi: 10.2174/156800910793358041
Patel, S. B., Stenehjem, D. D., Gill, D. M., Tantravahi, S. K., Agarwal, A. M., Hsu, J., et al. (2016). Everolimus versus temsirolimus in metastatic renal cell carcinoma after progression with previous systemic therapies. Clin. Genitourin. Cancer 14, 153–159. doi: 10.1016/j.clgc.2015.12.011
Paul, D., Lipton, A., and Klinger, I. (1971). Serum factor requirements of normal and simian virus 40-transformed 3T3 mouse fibroplasts. Proc. Natl. Acad. Sci. U.S.A. 68, 645–652. doi: 10.1073/pnas.68.3.645
Pezzella, F., Pastorino, U., Tagliabue, E., Andreola, S., Sozzi, G., Gasparini, G., et al. (1997). Non-small-cell lung carcinoma tumor growth without morphological evidence of neo-angiogenesis. Am. J. Pathol. 151, 1417–1423.
Piao, Y., Liang, J., Holmes, L., Zurita, A. J., Henry, V., Heymach, J. V., et al. (2012). Glioblastoma resistance to anti-VEGF therapy is associated with myeloid cell infiltration, stem cell accumulation, and a mesenchymal phenotype. Neuro Oncol. 14, 1379–1392. doi: 10.1093/neuonc/nos158
Pietras, K., and Hanahan, D. (2005). A multitargeted, metronomic, and maximum-tolerated dose “chemo-switch” regimen is antiangiogenic, producing objective responses and survival benefit in a mouse model of cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 23, 939–952. doi: 10.1200/JCO.2005.07.093
Pinto, M. P., Sotomayor, P., Carrasco-Avino, G., Corvalan, A. H., and Owen, G. I. (2016). Escaping antiangiogenic therapy: strategies employed by cancer cells. Int. J. Mol. Sci. 17:1489. doi: 10.3390/ijms17091489
Pisarsky, L., Bill, R., Fagiani, E., Dimeloe, S., Goosen, R. W., Hagmann, J., et al. (2016). Targeting Metabolic symbiosis to overcome resistance to anti-angiogenic therapy. Cell Rep. 15, 1161–1174. doi: 10.1016/j.celrep.2016.04.028
Powell, M. A., Sill, M. W., Goodfellow, P. J., Benbrook, D. M., Lankes, H. A., Leslie, K. K., et al. (2014). A phase II trial of brivanib in recurrent or persistent endometrial cancer: an NRG Oncology/Gynecologic oncology group study. Gynecol. Oncol. 135, 38–43. doi: 10.1016/j.ygyno.2014.07.083
Presta, L. G., Chen, H., O’Connor, S. J., Chisholm, V., Meng, Y. G., Krummen, L., et al. (1997). Humanization of an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders. Cancer Res. 57, 4593–4599.
Provenzano, P. P., and Hingorani, S. R. (2013). Hyaluronan, fluid pressure, and stromal resistance in pancreas cancer. Br. J. Cancer 108, 1–8. doi: 10.1038/bjc.2012.569
Puente, P., de la Muz, B., Azab, F., and Azab, A. K. (2013). Cell trafficking of endothelial progenitor cells in tumor progression. Clin. Cancer Res. 19, 3360–3368. doi: 10.1158/1078-0432.CCR-13-0462
Qin, S., Li, A., Yi, M., Yu, S., Zhang, M., and Wu, K. (2019). Recent advances on anti-angiogenesis receptor tyrosine kinase inhibitors in cancer therapy. J. Hematol. Oncol. 12:27. doi: 10.1186/s13045-019-0718-5
Rapisarda, A., and Melillo, G. (2009). Role of the hypoxic tumor microenvironment in the resistance to anti-angiogenic therapies. Drug Resist. Updat. Rev. Comment. Antimicrob. Anticancer Chemother. 12, 74–80. doi: 10.1016/j.drup.2009.03.002
Ravaud, A., Motzer, R. J., Pandha, H. S., George, D. J., Pantuck, A. J., Patel, A., et al. (2016). Adjuvant sunitinib in high-risk renal-cell carcinoma after nephrectomy. N. Engl. J. Med. 375, 2246–2254. doi: 10.1056/NEJMoa1611406
Reale, A., Melaccio, A., Lamanuzzi, A., Saltarella, I., Dammacco, F., Vacca, A., et al. (2016). Functional and biological role of endothelial precursor cells in tumour progression: a new potential therapeutic target in haematological malignancies. Stem Cells Int. 2016:11. doi: 10.1155/2016/7954580
Ries, C. H., Cannarile, M. A., Hoves, S., Benz, J., Wartha, K., Runza, V., et al. (2014). Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 25, 846–859. doi: 10.1016/j.ccr.2014.05.016
Rini, B. I., Plimack, E. R., Stus, V., Gafanov, R., Hawkins, R., Nosov, D., et al. (2019a). Pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N. Engl. J. Med. 380, 1116–1127. doi: 10.1056/NEJMoa1816714
Rini, B. I., Powles, T., Atkins, M. B., Escudier, B., McDermott, D. F., Suarez, C., et al. (2019b). Atezolizumab plus bevacizumab versus sunitinib in patients with previously untreated metastatic renal cell carcinoma (IMmotion151): a multicentre, open-label, phase 3, randomised controlled trial. Lancet 393, 2404–2415. doi: 10.1016/S0140-6736(19)30723-8
Rizzo, M., and Porta, C. (2017). Sunitinib in the treatment of renal cell carcinoma: an update on recent evidence. Ther. Adv. Urol. 9, 195–207. doi: 10.1177/1756287217713902
Rock, E. P., Goodman, V., Jiang, J. X., Mahjoob, K., Verbois, S. L., Morse, D., et al. (2007). Food and drug administration drug approval summary: sunitinib malate for the treatment of gastrointestinal stromal tumor and advanced renal cell carcinoma. Oncologist 12, 107–113. doi: 10.1634/theoncologist.12-1-107
Rohlenova, K., Goveia, J., Garcia-Caballero, M., Subramanian, A., Kalucka, J., Treps, L., et al. (2020). Single-Cell RNA sequencing maps endothelialmetabolic plasticity in pathological angiogenesis. Cell Metab. 31, 862–877. doi: 10.1016/j.cmet.2020.03.009
Ruffell, B., and Coussens, L. M. (2015). Macrophages and therapeutic resistance in cancer. Cancer Cell 27, 462–472. doi: 10.1016/j.ccell.2015.02.015
Russo, A. E., Priolo, D., Antonelli, G., Libra, M., McCubrey, J. A., and Ferraù, F. (2017). Bevacizumab in the treatment of NSCLC: patient selection and perspectives. Lung Cancer Targets Ther. 8, 259–269. doi: 10.2147/LCTT.S110306
Salnikov, A. V., Heldin, N.-E., Stuhr, L. B., Wiig, H., Gerber, H., Reed, R. K., et al. (2006). Inhibition of carcinoma cell-derived VEGF reduces inflammatory characteristics in xenograft carcinoma. Int. J. Cancer 119, 2795–2802. doi: 10.1002/ijc.22217
Sandler, A., Gray, R., Perry, M. C., Brahmer, J., Schiller, J. H., Dowlati, A., et al. (2006). Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N. Engl. J. Med. 355, 2542–2550. doi: 10.1056/NEJMoa061884
Sarode, P., Schaefer, M. B., Grimminger, F., Seeger, W., and Savai, R. (2020). Macrophage and tumor cell cross-talk is fundamental for lung tumor progression: we need to talk. Front. Oncol. 10:324. doi: 10.3389/fonc.2020.00324
Schech, A., Kazi, A., Yu, S., Shah, P., and Sabnis, G. (2015). Histone deacetylase inhibitor entinostat inhibits tumor-initiating cells in triple-negative breast cancer cells. Mol. Cancer Ther. 14, 1848–1857. doi: 10.1158/1535-7163.MCT-14-0778
Schmittnaegel, M., Rigamonti, N., Kadioglu, E., Cassará, A., Wyser Rmili, C., Kiialainen, A., et al. (2017). Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci. Transl. Med. 9:eaak9670. doi: 10.1126/scitranslmed.aak9670
Schreiber, A. B., Winkler, M. E., and Derynck, R. (1986). Transforming growth factor-alpha: a more potent angiogenic mediator than epidermal growth factor. Science 232, 1250–1253. doi: 10.1126/science.2422759
Semenza, G. L. (2014). Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu. Rev. Pathol. 9, 47–71. doi: 10.1146/annurev-pathol-012513-104720
Semrad, T. J., Kim, E. J., Tanaka, M. S., Sands, J., Roberts, C., Burich, R. A., et al. (2017). Phase II study of Dovitinib in patients progressing on anti-vascular endothelial growth factor therapy. Cancer Treat. Res. Commun. 10, 21–26. doi: 10.1016/j.ctarc.2016.12.002
Senft, D., and Ronai, Z. A. (2016). Immunogenic, cellular, and angiogenic drivers of tumor dormancy-a melanoma view. Pigment Cell Melanoma Res. 29, 27–42. doi: 10.1111/pcmr.12432
Sennino, B., Ishiguro-Oonuma, T., Wei, Y., Naylor, R. M., Williamson, C. W., Bhagwandin, V., et al. (2012). Suppression of tumor invasion and metastasis by concurrent inhibition of c-Met and VEGF signaling in pancreatic neuroendocrine tumors. Cancer Discov. 2, 270–287. doi: 10.1158/2159-8290.CD-11-0240
Serafini, P., Borrello, I., and Bronte, V. (2006). Myeloid suppressor cells in cancer: recruitment, phenotype, properties, and mechanisms of immune suppression. Semin. Cancer Biol. 16, 53–65. doi: 10.1016/j.semcancer.2005.07.005
Serova, M., Tijeras-Raballand, A., Santos, C. D., Martinet, M., Neuzillet, C., Lopez, A., et al. (2016). Everolimus affects vasculogenic mimicry in renal carcinoma resistant to sunitinib. Oncotarget 7, 38467–38486. doi: 10.18632/oncotarget.9542
Sfiligoi, C., de Luca, A., Cascone, I., Sorbello, V., Fuso, L., Ponzone, R., et al. (2003). Angiopoietin-2 expression in breast cancer correlates with lymph node invasion and short survival. Int. J. Cancer 103, 466–474. doi: 10.1002/ijc.10851
Shibata, M., and Hoque, M. O. (2019). Targeting cancer stem cells: a strategy for effective eradication of cancer. Cancers 11:732. doi: 10.3390/cancers11050732
Shih, T., and Lindley, C. (2006). Bevacizumab: an angiogenesis inhibitor for the treatment of solid malignancies. Clin. Ther. 28, 1779–1802. doi: 10.1016/j.clinthera.2006.11.015
Shimizu, S., Takehara, T., Hikita, H., Kodama, T., Tsunematsu, H., Miyagi, T., et al. (2012). Inhibition of autophagy potentiates the antitumor effect of the multikinase inhibitor sorafenib in hepatocellular carcinoma. Int. J. Cancer 131, 548–557. doi: 10.1002/ijc.26374
Shirakawa, K., Kobayashi, H., Heike, Y., Kawamoto, S., Brechbiel, M. W., Kasumi, F., et al. (2002). Hemodynamics in vasculogenic mimicry and angiogenesis of inflammatory breast cancer xenograft. Cancer Res. 62, 560–566.
Shojaei, F., and Ferrara, N. (2008). Refractoriness to antivascular endothelial growth factor treatment: role of myeloid cells. Cancer Res. 68, 5501–5504. doi: 10.1158/0008-5472.CAN-08-0925
Shojaei, F., Wu, X., Malik, A. K., Zhong, C., Baldwin, M. E., Schanz, S., et al. (2007a). Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat. Biotechnol. 25, 911–920. doi: 10.1038/nbt1323
Shojaei, F., Wu, X., Qu, X., Kowanetz, M., Yu, L., Tan, M., et al. (2009). G-CSF-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proc. Natl. Acad. Sci. U.S.A. 106, 6742–6747. doi: 10.1073/pnas.0902280106
Shojaei, F., Wu, X., Zhong, C., Yu, L., Liang, X.-H., Yao, J., et al. (2007b). Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature 450, 825–831. doi: 10.1038/nature06348
Sidorov, M., Jahangiri, A., Han, S.-W., De Lay, M., Wagner, J., Castro, B., et al. (2016). 340 c-Met/β1 integrin: a receptor complex driving invasive glioblastoma resistance to antiangiogenic therapy. Neurosurgery 63, 199–200. doi: 10.1227/01.neu.0000489829.38251.85
Simon, T., Pinioti, S., Schellenberger, P., Rajeeve, V., Wendler, F., Cutillas, P. R., et al. (2018). Shedding of bevacizumab in tumour cells-derived extracellular vesicles as a new therapeutic escape mechanism in glioblastoma. Mol. Cancer 17:132. doi: 10.1186/s12943-018-0878-x
Singh, M., Couto, S. S., Forrest, W. F., Lima, A., Cheng, J. H., Molina, R., et al. (2012). Anti-VEGF antibody therapy does not promote metastasis in genetically engineered mouse tumour models. J. Pathol. 227, 417–430. doi: 10.1002/path.4053
Singh, S. K., Clarke, I. D., Terasaki, M., Bonn, V. E., Hawkins, C., Squire, J., et al. (2003). Identification of a cancer stem cell in human brain tumors. Cancer Res. 63, 5821–5828.
Song, E., Mao, T., Dong, H., Boisserand, L. S. B., Antila, S., Bosenberg, M., et al. (2020). VEGF-C-driven lymphatic drainage enables immunosurveillance of brain tumours. Nature 577, 689–694. doi: 10.1038/s41586-019-1912-x
Sood, A. K., Seftor, E. A., Fletcher, M. S., Gardner, L. M., Heidger, P. M., Buller, R. E., et al. (2001). Molecular determinants of ovarian cancer plasticity. Am. J. Pathol. 158, 1279–1288. doi: 10.1016/S0002-9440(10)64079-5
Sounni, N. E., Cimino, J., Blacher, S., Primac, I., Truong, A., Mazzucchelli, G., et al. (2014). Blocking lipid synthesis overcomes tumor regrowth and metastasis after antiangiogenic therapy withdrawal. Cell Metab. 20, 280–294. doi: 10.1016/j.cmet.2014.05.022
Sparmann, A., and Bar-Sagi, D. (2004). Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell 6, 447–458. doi: 10.1016/j.ccr.2004.09.028
Stanley, P. (2014). Galectin-1 pulls the strings on VEGFR2. Cell 156, 625–626. doi: 10.1016/j.cell.2014.01.059
Sternlicht, M. D., Lochter, A., Sympson, C. J., Huey, B., Rougier, J. P., Gray, J. W., et al. (1999). The stromal proteinase MMP3/stromelysin-1 promotes mammary carcinogenesis. Cell 98, 137–146. doi: 10.1016/s0092-8674(00)81009-0
Stratmann, A., Risau, W., and Plate, K. H. (1998). Cell type-specific expression of Angiopoietin-1 and Angiopoietin-2 suggests a role in glioblastoma angiogenesis. Am. J. Pathol. 153, 1459–1466.
Sun, H., Zhang, D., Yao, Z., Lin, X., Liu, J., Gu, Q., et al. (2017). Anti-angiogenic treatment promotes triple-negative breast cancer invasion via vasculogenic mimicry. Cancer Biol. Ther. 18, 205–213. doi: 10.1080/15384047.2017.1294288
Suyama, K., and Iwase, H. (2018). Lenvatinib: a promising molecular targeted agent for multiple cancers. Cancer Control J. Moffitt Cancer Cent. 25:1073274818789361. doi: 10.1177/1073274818789361
Tang, D., Gao, J., Wang, S., Ye, N., Chong, Y., Huang, Y., et al. (2016). Cancer-associated fibroblasts promote angiogenesis in gastric cancer through galectin-1 expression. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 37, 1889–1899. doi: 10.1007/s13277-015-3942-9
Thijssen, V. L., Paulis, Y. W., Nowak-Sliwinska, P., Deumelandt, K. L., Hosaka, K., Soetekouw, P. M., et al. (2018). Targeting PDGF-mediated recruitment of pericytes blocks vascular mimicry and tumor growth. J. Pathol. 246, 447–458. doi: 10.1002/path.5152
Thomas, M., Kienast, Y., Scheuer, W., Bähner, M., Kaluza, K., Gassner, C., et al. (2013). A novel angiopoietin-2 selective fully human antibody with potent anti-tumoral and anti-angiogenic efficacy and superior side effect profile compared to Pan-Angiopoietin-1/-2 inhibitors. PLoS One 8:e54923. doi: 10.1371/journal.pone.0054923
Thurston, G., and Daly, C. (2012). The Complex Role of Angiopoietin-2 in the Angiopoietin–Tie Signaling Pathway. Cold Spring Harb. Perspect. Med. 2:a006550. doi: 10.1101/cshperspect.a006650
Todorova, D., Simoncini, S., Lacroix, R., Sabatier, F., and Dignat-George, F. (2017). Extracellular vesicles in angiogenesis. Circ. Res. 120, 1658–1673. doi: 10.1161/CIRCRESAHA.117.309681
Tong, W.-W., Tong, G.-H., and Liu, Y. (2018). Cancer stem cells and hypoxia-inducible factors (Review). Int. J. Oncol. 53, 469–476. doi: 10.3892/ijo.2018.4417
Tonnesen, M. G., Feng, X., and Clark, R. A. (2000). Angiogenesis in wound healing. J. Investig. Dermatol. Symp. Proc. 5, 40–46. doi: 10.1046/j.1087-0024.2000.00014.x
Valkenburg, K. C., de Groot, A. E., and Pienta, K. J. (2018). Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 15, 366–381. doi: 10.1038/s41571-018-0007-1
van Beijnum, J. R., Nowak-Sliwinska, P., Huijbers, E. J. M., Thijssen, V. L., and Griffioen, A. W. (2015). The great escape; the hallmarks of resistance to antiangiogenic therapy. Pharmacol. Rev. 67, 441–461. doi: 10.1124/pr.114.010215
van Cruijsen, H., Giaccone, G., and Hoekman, K. (2005). Epidermal growth factor receptor and angiogenesis: opportunities for combined anticancer strategies. Int. J. Cancer 117, 883–888. doi: 10.1002/ijc.21479
van der Schaft, D. W. J., Hillen, F., Pauwels, P., Kirschmann, D. A., Castermans, K., Egbrink, M. G. A. O., et al. (2005). Tumor cell plasticity in Ewing sarcoma, an alternative circulatory system stimulated by hypoxia. Cancer Res. 65, 11520–11528. doi: 10.1158/0008-5472.CAN-05-2468
van der Schaft, D. W. J., Pauwels, P., Hulsmans, S., Zimmermann, M., van de Poll-Franse, L. V., and Griffioen, A. W. (2007). Absence of lymphangiogenesis in ductal breast cancer at the primary tumor site. Cancer Lett. 254, 128–136. doi: 10.1016/j.canlet.2007.03.001
van der Schaft, D. W. J., Seftor, R. E. B., Seftor, E. A., Hess, A. R., Gruman, L. M., Kirschmann, D. A., et al. (2004). Effects of angiogenesis inhibitors on vascular network formation by human endothelial and melanoma cells. J. Natl. Cancer Inst. 96, 1473–1477. doi: 10.1093/jnci/djh267
van Meeteren, L. A., Goumans, M.-J., and ten Dijke, P. (2011). TGF-β receptor signaling pathways in angiogenesis; emerging targets for anti-angiogenesis therapy. Curr. Pharm. Biotechnol. 12, 2108–2120. doi: 10.2174/138920111798808338
Vandercappellen, J., Van Damme, J., and Struyf, S. (2008). The role of CXC chemokines and their receptors in cancer. Cancer Lett. 267, 226–244. doi: 10.1016/j.canlet.2008.04.050
Voron, T., Colussi, O., Marcheteau, E., Pernot, S., Nizard, M., Pointet, A.-L., et al. (2015). VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J. Exp. Med. 212, 139–148. doi: 10.1084/jem.20140559
Walsh, D. A. (2007). Pathophysiological mechanisms of angiogenesis. Adv. Clin. Chem. 44, 187–221. doi: 10.1016/s0065-2423(07)44006-9
Wang, F.-T., Sun, W., Zhang, J.-T., and Fan, Y.-Z. (2019). Cancer-associated fibroblast regulation of tumor neo-angiogenesis as a therapeutic target in cancer. Oncol. Lett. 17, 3055–3065. doi: 10.3892/ol.2019.9973
Wang, L., Park, H., Chhim, S., Ding, Y., Jiang, W., Queen, C., et al. (2012). A novel monoclonal antibody to fibroblast growth factor 2 effectively inhibits growth of hepatocellular carcinoma xenografts. Mol. Cancer Ther. 11, 864–872. doi: 10.1158/1535-7163.MCT-11-0813
Warren, R. S., Yuan, H., Matli, M. R., Gillett, N. A., and Ferrara, N. (1995). Regulation by vascular endothelial growth factor of human colon cancer tumorigenesis in a mouse model of experimental liver metastasis. J. Clin. Invest. 95, 1789–1797. doi: 10.1172/JCI117857
Wei, Y., Zhou, F., Zhou, H., Huang, J., Yu, D., and Wu, G. (2018). Endothelial progenitor cells contribute to neovascularization of non-small cell lung cancer via histone deacetylase 7-mediated cytoskeleton regulation and angiogenic genes transcription. Int. J. Cancer 143, 657–667. doi: 10.1002/ijc.31349
Welti, J. C., Powles, T., Foo, S., Gourlaouen, M., Preece, N., and Foster, J. (2012). Contrasting effects of sunitinib within in vivo models of metastasis. Angiogenesis 15, 623–641. doi: 10.1007/s10456-012-9291-z
Willett, C. G., Boucher, Y., Duda, D. G., di Tomaso, E., Munn, L. L., Tong, R. T., et al. (2005). Surrogate markers for antiangiogenic therapy and dose-limiting toxicities for bevacizumab with radiation and chemotherapy: continued experience of a phase I trial in rectal cancer patients. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 23, 8136–8139. doi: 10.1200/JCO.2005.02.5635
Wilson, W. R., and Hay, M. P. (2011). Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 11, 393–410. doi: 10.1038/nrc3064
Wilting, J., and Christ, B. (1996). Embryonic angiogenesis: a review. Naturwissenschaften 83, 153–164. doi: 10.1007/bf01143056
Winer, A., Adams, S., and Mignatti, P. (2018). Matrix metalloproteinase inhibitors in cancer therapy: turning past failures into future successes. Mol. Cancer Ther. 17, 1147–1155. doi: 10.1158/1535-7163.MCT-17-0646
Winkler, F., Kienast, Y., Fuhrmann, M., Von Baumgarten, L., Burgold, S., Mitteregger, G., et al. (2009). Imaging glioma cell invasion in vivo reveals mechanisms of dissemination and peritumoral angiogenesis. Glia 57, 1306–1315. doi: 10.1002/glia.20850
Wolf, A., and Langmann, T. (2019). Anti−VEGF−A/ANG2 combotherapy limits pathological angiogenesis in the eye: a replication study. EMBO Mol. Med. 11:e10362. doi: 10.15252/emmm.201910362
Wu, S., Huang, L., Shen, R., Bernard-Cacciarella, M., Zhou, P., Hu, C., et al. (2020). Drug resistance-related sunitinib sequestration in autophagolysosomes of endothelial cells. Int. J. Oncol. 56, 113–122. doi: 10.3892/ijo.2019.4924
Xin, X., Yang, S., Ingle, G., Zlot, C., Rangell, L., Kowalski, J., et al. (2001). Hepatocyte growth factor enhances vascular endothelial growth factor-induced angiogenesis in vitro and in vivo. Am. J. Pathol. 158, 1111–1120.
Xiong, Y.-Q., Sun, H.-C., Zhang, W., Zhu, X.-D., Zhuang, P.-Y., Zhang, J.-B., et al. (2009). Human hepatocellular carcinoma tumor-derived endothelial cells manifest increased angiogenesis capability and drug resistance compared with normal endothelial cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 15, 4838–4846. doi: 10.1158/1078-0432.CCR-08-2780
Xu, R., Rai, A., Chen, M., Suwakulsiri, W., Greening, D. W., and Simpson, R. J. (2018). Extracellular vesicles in cancer - implications for future improvements in cancer care. Nat. Rev. Clin. Oncol. 15, 617–638. doi: 10.1038/s41571-018-0036-9
Xu, Y., Li, Q., Li, X.-Y., Yang, Q.-Y., Xu, W.-W., and Liu, G.-L. (2012). Short-term anti-vascular endothelial growth factor treatment elicits vasculogenic mimicry formation of tumors to accelerate metastasis. J. Exp. Clin. Cancer Res. CR 31:16. doi: 10.1186/1756-9966-31-16
Yadav, A. S., Pandey, P. R., Butti, R., Radharani, N. N. V., Roy, S., Bhalara, S. R., et al. (2018). The biology and therapeutic implications of tumor dormancy and reactivation. Front. Oncol. 8:72. doi: 10.3389/fonc.2018.00072
Yamada, Y., Nezu, J., Shimane, M., and Hirata, Y. (1997). Molecular cloning of a novel vascular endothelial growth factor, VEGF-D. Genomics 42, 483–488. doi: 10.1006/geno.1997.4774
Yamamoto, K., Ohga, N., Hida, Y., Maishi, N., Kawamoto, T., Kitayama, K., et al. (2012). Biglycan is a specific marker and an autocrine angiogenic factor of tumour endothelial cells. Br. J. Cancer 106, 1214–1223. doi: 10.1038/bjc.2012.59
Yang, J. C., Haworth, L., Sherry, R. M., Hwu, P., Schwartzentruber, D. J., Topalian, S. L., et al. (2003). A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N. Engl. J. Med. 349, 427–434. doi: 10.1056/NEJMoa021491
Yang, L., DeBusk, L. M., Fukuda, K., Fingleton, B., Green-Jarvis, B., Shyr, Y., et al. (2004). Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell 6, 409–421. doi: 10.1016/j.ccr.2004.08.031
Yao, H., Price, T. T., Cantelli, G., Ngo, B., Warner, M. J., Olivere, L., et al. (2018). Leukaemia hijacks a neural mechanism to invade the central nervous system. Nature 560, 55–60. doi: 10.1038/s41586-018-0342-5
Yu, Z., Pestell, T. G., Lisanti, M. P., and Pestell, R. G. (2012). Cancer stem cells. Int. J. Biochem. Cell Biol. 44, 2144–2151. doi: 10.1016/j.biocel.2012.08.022
Zeisberg, M., and Neilson, E. G. (2009). Biomarkers for epithelial-mesenchymal transitions. J. Clin. Invest. 119, 1429–1437. doi: 10.1172/JCI36183
Zhai, B., Hu, F., Jiang, X., Xu, J., Zhao, D., Liu, B., et al. (2014). Inhibition of Akt reverses the acquired resistance to sorafenib by switching protective autophagy to autophagic cell death in hepatocellular carcinoma. Mol. Cancer Ther. 13, 1589–1598. doi: 10.1158/1535-7163.MCT-13-1043
Keywords: VEGFA, anti-angiogenic treatments, resistance, tumor microenvironment, combined therapies
Citation: Montemagno C and Pagès G (2020) Resistance to Anti-angiogenic Therapies: A Mechanism Depending on the Time of Exposure to the Drugs. Front. Cell Dev. Biol. 8:584. doi: 10.3389/fcell.2020.00584
Received: 17 April 2020; Accepted: 16 June 2020;
Published: 07 July 2020.
Edited by:
Lucas Treps, VIB-KU Leuven Center for Cancer Biology, BelgiumReviewed by:
Ali Ibrahim Shamseddine, American University of Beirut, LebanonAndreas Pircher, Innsbruck Medical University, Austria
Copyright © 2020 Montemagno and Pagès. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christopher Montemagno, Y21vbnRlbWFnbm9AY2VudHJlc2NpZW50aWZpcXVlLm1j; Gilles Pagès, Z3BhZ2VzQHVuaWNlLmZy