 Sayako Hirose1
Sayako Hirose1 Takeru Makiyama1*
Takeru Makiyama1* Dario Melgari2,3
Dario Melgari2,3 Yuta Yamamoto1,4
Yuta Yamamoto1,4 Yimin Wuriyanghai1Fumika Yokoi1Suguru Nishiuchi1Takeshi Harita1Mamoru Hayano1
Yimin Wuriyanghai1Fumika Yokoi1Suguru Nishiuchi1Takeshi Harita1Mamoru Hayano1 Hirohiko Kohjitani1Jingshan Gao1Asami Kashiwa1Misato Nishikawa5
Hirohiko Kohjitani1Jingshan Gao1Asami Kashiwa1Misato Nishikawa5 Jie Wu2,6Jun Yoshimoto7Kazuhisa Chonabayashi5
Jie Wu2,6Jun Yoshimoto7Kazuhisa Chonabayashi5 Seiko Ohno4
Seiko Ohno4 Yoshinori Yoshida5
Yoshinori Yoshida5 Minoru Horie2,8Takeshi Kimura1
Minoru Horie2,8Takeshi Kimura1- 1Department of Cardiovascular Medicine, Kyoto University Graduate School of Medicine, Kyoto, Japan
- 2Department of Cardiovascular and Respiratory Medicine, Shiga University of Medical Science, Otsu, Japan
- 3Institute of Cardiometabolism and Nutrition, Sorbonne University, Paris, France
- 4Department of Bioscience and Genetics, National Cerebral and Cardiovascular Center, Suita, Japan
- 5Center for iPS Cell Research and Application (CiRA), Institute for Integrated Cell-Material Sciences, Kyoto University, Kyoto, Japan
- 6Department of Pharmacology, Medical School of Xi’an Jiaotong University, Xi’an, China
- 7Department of Cardiology, Shizuoka Children’s Hospital, Shizuoka, Japan
- 8Center for Epidemiologic Research in Asia, Shiga University of Medical Science, Otsu, Japan
Background: Long QT syndrome type 3 (LQT3) is caused by gain-of-function mutations in the SCN5A gene, which encodes the α subunit of the cardiac voltage-gated sodium channel. LQT3 patients present bradycardia and lethal arrhythmias during rest or sleep. Further, the efficacy of β-blockers, the drug used for their treatment, is uncertain. Recently, a large multicenter LQT3 cohort study demonstrated that β-blocker therapy reduced the risk of life-threatening cardiac events in female patients; however, the detailed mechanism of action remains unclear.
Objectives: This study aimed to establish LQT3-human induced pluripotent stem cells (hiPSCs) and to investigate the effect of propranolol in this model.
Method: An hiPSCs cell line was established from peripheral blood mononuclear cells of a boy with LQT3 carrying the SCN5A-N1774D mutation. He had suffered from repetitive torsades de pointes (TdPs) with QT prolongation since birth (QTc 680 ms), which were effectively treated with propranolol, as it suppressed lethal arrhythmias. Furthermore, hiPSCs were differentiated into cardiomyocytes (CMs), on which electrophysiological functional assays were performed using the patch-clamp method.
Results: N1774D-hiPSC-CMs exhibited significantly prolonged action potential durations (APDs) in comparison to those of the control cells (N1774D: 440 ± 37 ms vs. control: 272 ± 22 ms; at 1 Hz pacing; p < 0.01). Furthermore, N1774D-hiPSC-CMs presented gain-of-function features: a hyperpolarized shift of steady-state activation and increased late sodium current compared to those of the control cells. 5 μM propranolol shortened APDs and inhibited late sodium current in N1774D-hiPSC-CMs, but did not significantly affect in the control cells. In addition, even in the presence of intrapipette guanosine diphosphate βs (GDPβs), an inhibitor of G proteins, propranolol reduced late sodium current in N1774D cells. Therefore, these results suggested a unique inhibitory effect of propranolol on late sodium current unrelated to β-adrenergic receptor block in N1774D-hiPSC-CMs.
Conclusion: We successfully recapitulated the clinical phenotype of LQT3 using patient-derived hiPSC-CMs and determined that the mechanism, by which propranolol inhibited the late sodium current, was independent of β-adrenergic receptor signaling pathway.
Introduction
Congenital long QT syndrome (LQT) is an inherited arrhythmogenic disease, associated with lethal arrhythmic events and sudden cardiac death. Patients with LQT are currently classified into over 15 genetic subtypes, and LQT1-3 accounts for approximately 90% of the genotyped patients, specifically: 40–55%, 30–45%, and 5–10% are LQT1, LQT2, and LQT3, respectively (Schwartz et al., 2012; Mizusawa et al., 2014). LQT1 and LQT2 are caused by loss-of-function mutations in KCNQ1 and KCNH2 genes, which encode cardiac slowly (IKs) and rapidly (IKr) activating delayed rectifier potassium channels, respectively (Moss et al., 2007; Shimizu et al., 2009). On the other hand, LQT3 is caused by gain-of function mutations in the SCN5A gene, which encodes cardiac voltage-gated sodium channels (Bennett et al., 1995).
A number of studies reported genotype-phenotype correlations among those three major genotypes (Moss et al., 2000; Schwartz et al., 2001; Priori et al., 2004; Hobbs et al., 2006), and genetic testing for patients with LQT is highly recommended for identifying carriers in the families and determining the appropriate choice of gene-specific treatment (Ackerman et al., 2011; Skinner et al., 2019). Patients with LQT1 and LQT2 suffer from cardiac events that occur during exercise or emotional stress (Schwartz et al., 2001, 2012). They are usually treated with β-blockers, as they are highly effective in reducing cardiac event and mortality rates (Schwartz et al., 2001; Priori et al., 2004). By contrast, patients with LQT3 experience cardiac events during rest or sleep (Schwartz et al., 2001, 2012), and β-blocker therapy has resulted less effective or even harmful in preventing those cardiac events, according to small cohort studies and the clinical features of LQT3 (Moss et al., 2000; Schwartz et al., 2001; Priori et al., 2004). Recently, a large clinical cohort study, which consisted of 406 patients with LQT3, demonstrated that β-blocker therapy reduced the number of cardiac events in female patients >1 year old (Wilde et al., 2016), thus β-blocker therapy has regained importance as an optimal medicine for LQT3. However, the pharmacological mechanism by which β-blockers benefit LQT3 patients is still unclear.
In the present study, we investigated the cellular mechanism by which β-blockers affected late sodium currents in human LQT3 cardiomyocytes (CMs). To this end, we established a human induced pluripotent stem cell line (hiPSCs) from a young male carrying SCN5A-N1774D genotype and whose repetitive torsades de pointes (TdPs) were effectively treated with propranolol. In N1774D-hiPSC-derived CMs, prolonged action potential durations (APDs) and increased late sodium current at baseline were attenuated after propranolol administration. More importantly, we determined that late sodium current inhibition by propranolol was unrelated to β-adrenergic receptor signaling pathway in the LQT3 hiPSC-CM model.
Materials and Methods
Generation and Characterization of LQT3-hiPSCs
We collected peripheral blood mononuclear cells from a patient carrying SCN5A-N1774D after obtaining the written informed consent, and employed an integration-free method using episomal vectors to generate hiPSCs (Okita et al., 2013). We used three different patient-derived iPSCs clones in this study. As control hiPSCs lines, we used two different lines (201B7 and 253G1) generated from a healthy donor (Takahashi et al., 2007; Nakagawa et al., 2008). This study was approved by the Kyoto University ethics review broad and conformed to the principles of the Declaration of Helsinki.
The pluripotency of established LQT3-hiPSCs were assessed using immunostaining and teratoma assay (Yamamoto et al., 2017; Wuriyanghai et al., 2018). Briefly, the hiPSC colonies were fixed with 4% paraformaldehyde for 20 min at 4°C. The cells were permeabilized in 0.2% Triton X-100 (Nacalai Tesque, Kyoto, Japan) and blocked with 5% FBS. The samples were stained overnight at 4°C with the following primary antibodies: mouse monoclonal anti-OCT3/4 (1:50; Santa Cruz Biotechnology, Delaware, CA, United States), mouse monoclonal anti-SSEA4 (1:200; Santa Cruz Biotechnology), and mouse monoclonal anti-TRA 1–60 (1:200; Santa Cruz Biotechnology). The secondary antibody was donkey anti-mouse Alexa fluor 488 (1:1000, Invitrogen, Carlsbad, CA, United States). The nuclei were stained with DAPI (1:2000, Wako Pure Chemical Industries, Osaka, Japan). The specimens were observed under a fluorescence microscope (Biozero BZ-9000; KEYENCE, Osaka, Japan). For teratoma assay, the hiPSCs were injected into severe combined immunodeficiency disease scid/scid mice under the testis capsule. Tumor samples were surgically dissected at 8 weeks, fixed in 10% formalin and stained with hematoxylin and eosin. All animal experiments were performed in accordance with the “Guide for the Care and Use of Laboratory Animals” (2011) of the National Institutes of Health and the Regulation on Animal Experimentation at Kyoto University, and approved by Ethics Committee of Kyoto University.
Cardiomyocyte Differentiation
Cardiomyocyte differentiation was induced with an embryoid body (EB) differentiation system as previously described (Yang et al., 2008; Wuriyanghai et al., 2018). The hiPSCs aggregated to form EBs and cultured in suspension for 20 days. On the 20th day of culture, EBs were dispersed into small clusters using collagenase B (Roche Diagnostics GmbH, Mannheim, Germany) and trypsin EDTA (Nacalai Tesque) and were plated onto 0.1% gelatin coated dishes. After day 20, hiPSC-CMs were maintained in DMEM/F12 with 2% fetal bovine serum, 2 mmol/L L-glutamine, 0.1 mmol/L non-essential amino acids, 0.1 mmol/L β-mercaptoethanol, 50 U/ml penicillin, and 50 μg/ml streptomycin. The medium was renewed every 2–3 days. Differentiated 6–8-week-old hiPSC-CMs were enzymatically dispersed into single cells using collagenase B and trypsin EDTA. The cells were plated on 0.1% gelatin coated glass cover slips. Patch-clamp experiments were performed 4–7 days after the procedures. Cardiac differentiation was performed more than five times with each hiPSCs clone and the data pooled from different lines or clones among the control and N1774D group were analyzed.
Electrophysiological Assay
Electrophysiological assays were performed using the whole-cell patch-clamp technique with the Multiclamp 700B microelectrode amplifier and Axon Digidata 1440 digitizer hardware (Molecular Devices, San Jose, CA, United States), as described previously (Ma et al., 2011; Hayano et al., 2017; Wuriyanghai et al., 2018).
Action potentials (APs) were recorded with perforated patch-clamp in the current mode at a bath temperature of 36 ± 1°C. APs were evoked at a constant pacing rate of 1 Hz, with 5 ms depolarizing current injections of 50–200 pA. The external solution contained (in mM): 140 NaCl, 5.4 KCl, 1.8 CaCl2, 1.0 MgCl2, 10 HEPES, 10 Glucose, pH 7.40 (adjusted with NaOH) and pipette solution contained (in mM): 150 KCl, 5 EGTA, 5 MgATP, 10 HEPES, 5 NaCl, 2 CaCl2 with pH of 7.2 and 300–500 μg/ml amphotericin B. The patch pipettes had a resistance of 3–6 MΩ. Ventricular-type action potentials were defined by the morphology of APs and the classification based on previous report (Matsa et al., 2011): a deep diastolic membrane potential (<−50 mV), a sharp systolic depolarization, a long plateau phase, and AP duration at 90% repolarization (action potential duration: APD90)/APD at 50% repolarization (APD50) ratio <1.4.
Sodium current was recorded at the bath temperature of 22–23°C using whole-cell voltage-clamp technique. Pipette resistances were between 0.8 and 1.8 MΩ, with access resistances of <5 MΩ. The extracellular bath solution contained (in mM): 137 NaCl, 100 TEA-Cl, 1.8 CaCl2, 2 MgCl2, 10 HEPES, 10 Glucose, 0.2 NiCl2, 0.005 Nifedipine (pH was adjusted to 7.4 with CsOH). The intrapipette solution was contained (in mM): 5 NaCl, 70 CsCl, 40 CSAs, 2.0 CaCl2, 10 EGTA, 10 HEPES, 5 MgATP, pH 7.3 (adjusted with CsOH). The standard holding potential was −100 mV. Each protocol in detail is illustrated in the inset.
The current-voltage relationships were fit with the Boltzmann equation: I = (V−Vrev) × Gmax × [1 + exp (V−V1/2)/k]–1, where I is the peak sodium current during the test pulse potential V. The parameters estimated by the fitting are reversal potential (Vrev), maximum conductance (Gmax), and slop factor (k). Steady-state availability was fit to the Boltzmann equation: I/I max = [1 + exp (V−V1/2)/k]–1 to determine the membrane potential for V1/2 (half-maximal inactivation) and k (slop factor). I max is the maximum peak sodium current. Data for the time course of recovery from inactivation were fitted by a biexponential equation: I(t)/I max = Af [1−exp(−t/τf)] + As [1−exp(−t/τs)], where Af and As represent fractions of each components, respectively. Time course of entry into the slow inactivation state and development of closed-state inactivation were fitted with a single exponential equation: I/I max = y0 + A[1−exp(−t/τ)], where the transfer rate of sodium channels from closed-state to inactivated closed-state without an intervening opening state was measured by a double pulse protocol. Time course for development of closed-state inactivation was fit with a single exponential equation, I/I max = y0 + A [1−exp(−t/τ)].
Late sodium currents were recorded for 800 ms at −10 mV before and after 20 μM tetrodotoxin (TTX; Nacalai Tesque) application. The pipette solution contained (in mM): 5 NaCl, 70 CsCl, 40 CSAsp, 10 EGTA, 10 HEPES, 5 MgATP, pH 7.3 (adjusted with CsOH).
To assess the effect of propranolol for APD and sodium current, recordings were performed before and after the administration of 5 μM propranolol hydrochloride (FUJIFILM Wako, Osaka, Japan).
Statistics
Data are expressed as mean ± the standard error of measurement. Statistical comparisons were analyzed using appropriate student’s t-test and paired t-tests. A p value of <0.05 was considered statistically significant.
Results
Clinical Patient Profile
The proband was a 1-day-old infant who exhibited severe bradycardia at 27 weeks of gestation (Kato et al., 2014). His surface electrocardiogram showed an extremely prolonged QT interval (QTc 680 ms) with functional atrio-ventricular block and repeated TdPs immediately after his birth. He was suspected of long QT syndrome and thus propranolol was administered to suppress TdPs. But the treatment was not enough to suppress the electrical storm. Therefore, mexiletine was added and, as a result, TdPs disappeared.
Additionally, genetic testing identified a heterozygous SCN5A mutation, named p. N1774D, confirming the diagnosis of LQT3. Therefore, treatment of the patient with propranolol was discontinued. However, TdPs recurred after its interruption, and thus propranolol administration was resumed, and as a consequence, TdPs immediately disappeared.
Generation and Characterization of SCN5A-N1774D-hiPSCs
Patient-specific LQT3 hiPSCs were generated from the peripheral blood mononuclear cells of the pediatric patient carrying the SCN5A-N1774D mutation. SCN5A-N1774D-hiPSCs displayed characteristics of human embryonic stem cell morphology and expressed human pluripotency makers: OCT3/4, SSEA4, and TRA-1-60 (Figure 1A). To evaluate the pluripotency of the generated hiPSC line, teratoma formation assays were performed using scid/scid mice. The teratomas contained tissues derivatives of three germ layers: ectoderm, endoderm, and mesoderm (Figure 1B). Furthermore, the SCN5A-N1774D mutation (c.5319A > G) was confirmed in the patient-derived hiPSCs, but not identified in the control samples (Figure 1C).
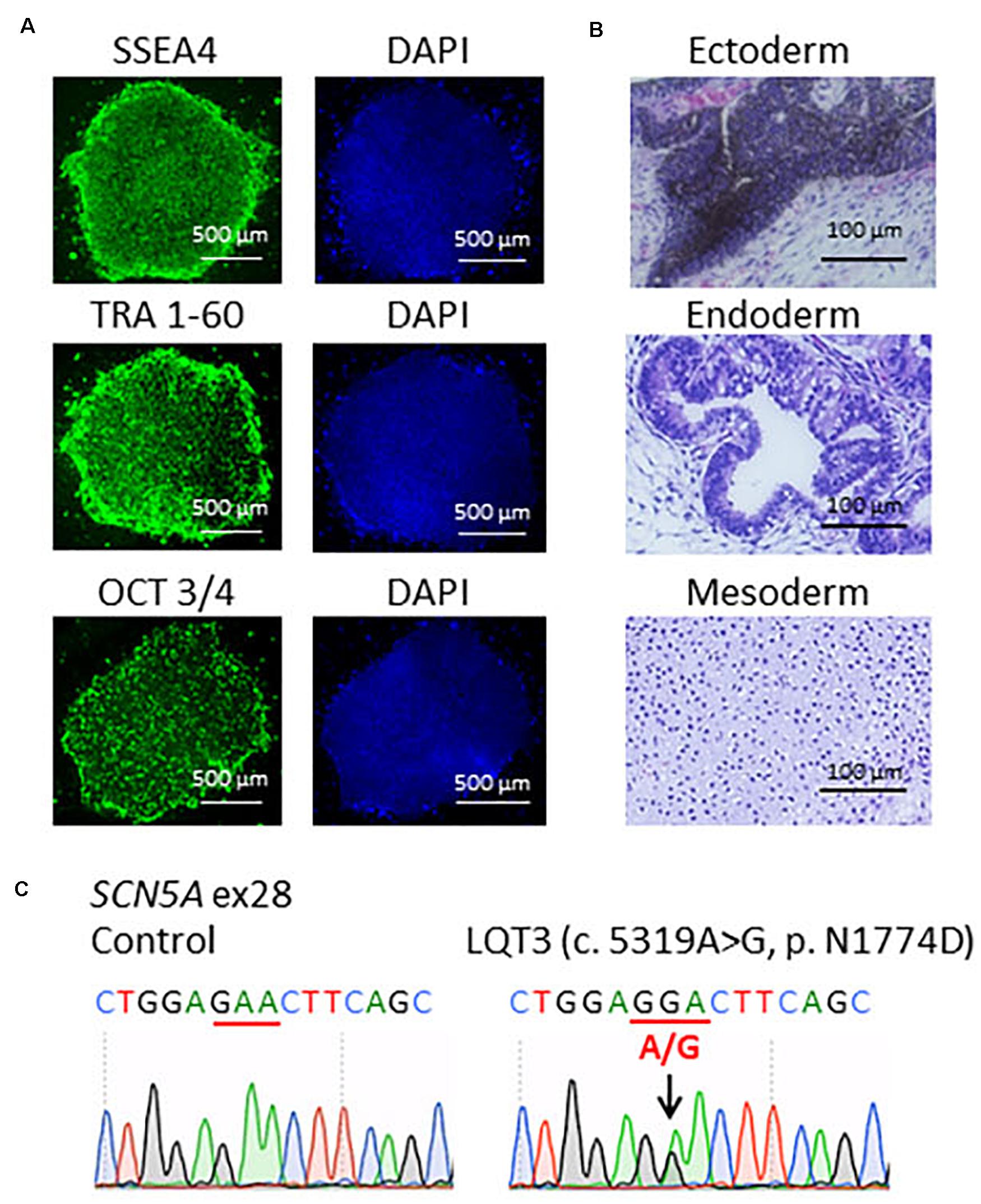
Figure 1. Characterization of LQT3-hiPSCs. (A) Immunofluorescence staining for stem cells makers. LQT3-hiPSC colonies derived from the peripheral blood mononuclear cells of a patient with SCN5A-N1774D expressed pluripotency markers; SSEA4, TRA 1-60, and OCT 3/4. Blue (right) showed 40-6-diamidino-2-phenylindole (DAPI) staining of nuclei. Scale bars = 500 μm. (B) Hematoxylin-eosin staining of teratomas formed from LQT3-hiPSC showed differentiation of the cells into various tissue derived from all three germ layers: melanocytes (ectoderm), gut-like structures (endoderm), and cartilage tissue (mesoderm). (C) DNA sequences of the SCN5A gene identified in the control hiPSCs and LQT3 cells carrying N1774D heterozygous mutation in LQT3-hiPSCs, not control hiPSCs. Scale bars: 100 μm.
Prolonged APDs in N1774D-hiPSC-CMs
Action potentials of hiPSC-CMs were recorded using the current-clamp technique. Figure 2A shows typical ventricular-type APs. For this study, we measured APDs at 50% and 90% repolarization, APD50 and APD90, respectively, maximum diastolic potential, and AP amplitude.
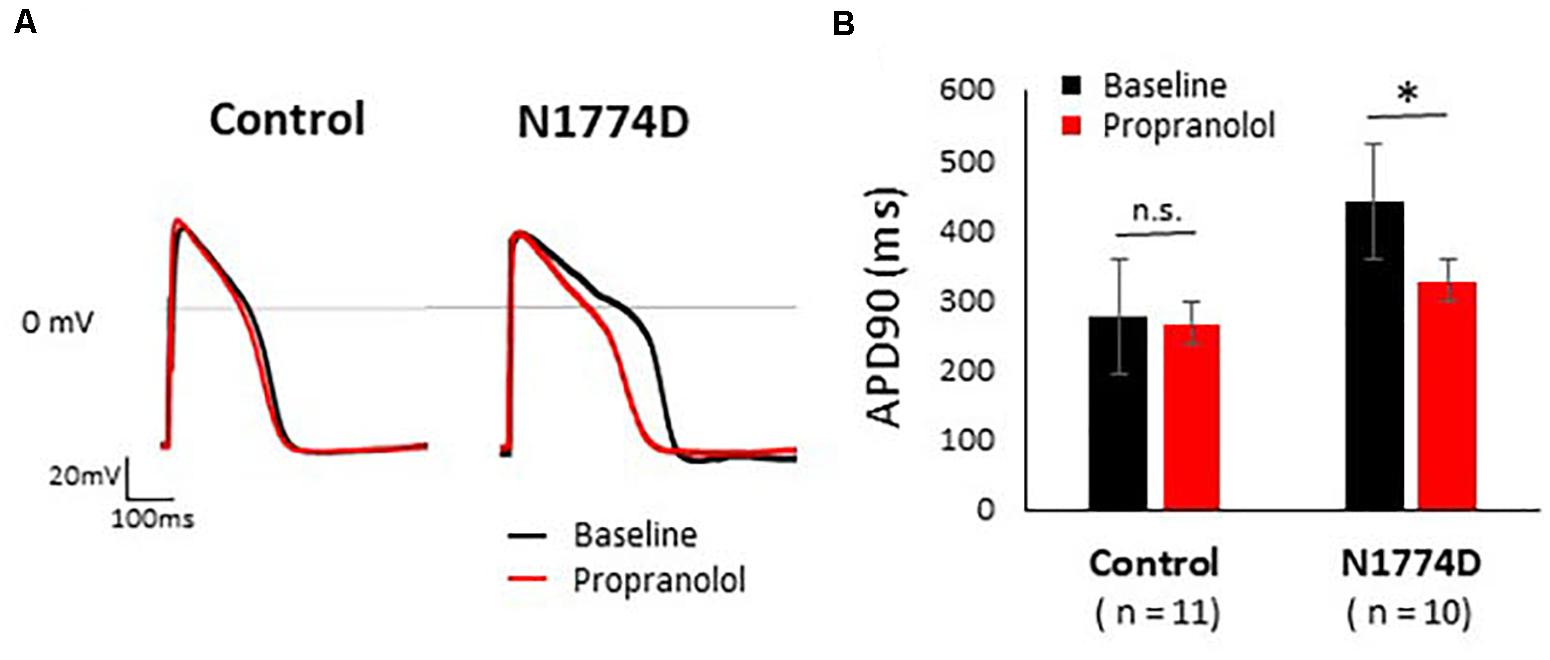
Figure 2. Effect of propranolol on action potential recording in control and N1774D-hiPSC-CMs. (A) Representative traces of paced ventricular-type action potential (AP) at 1 Hz pacing at baseline (black line) and after the administration 5 μM propranolol (red line) in control (left) and N1774D-hiPSC-CMs (right). (B) Summarized data in effects of propranolol on AP duration of control and N1774D-hiPSC-CMs. The data pooled from different lines or clones among the control and N1774D group were analyzed. ∗ p < 0.001, vs. control. APD90 was measured at 90% repolarization (APD90). APD90 values in N1774D-hiPSC-CMs were significantly prolonged compared with those in control. Propranolol significantly shortened the values of APD90 in N1774D-hiPSC-CMs. ∗ p < 0.001, vs. baseline.
Action potential durations in N1774D-hiPSC-CMs were significantly prolonged compared to those in control hiPSC-CMs at 1 Hz pacing (APD50 and APD90: N1774D-hiPSC-CMs, 440 ± 37 ms and 377 ± 33 ms, respectively, n = 11, vs. control, 272 ± 22 ms and 192 ± 18 ms, respectively, n = 10; p < 0.01; Figure 2 and Table 1). However, no significant difference in other AP parameters was observed (Table 1).
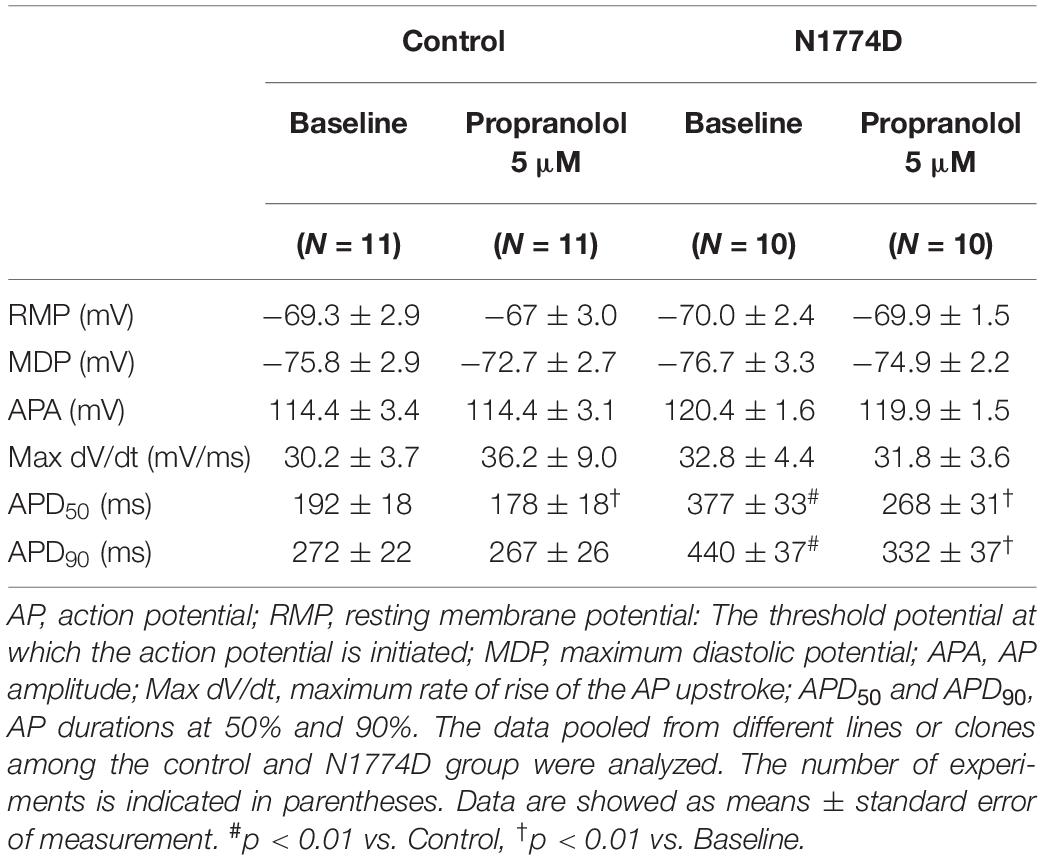
Table 1. AP parameters at baseline and after administration of propranolol in control and N1774D-hiPSC-CMs at 1 Hz pacing.
N1774D-hiPSC-CMs Exhibited Increased Late Sodium Current
The sodium channel currents in N1774D-hiPSC-CMs and control cells were recorded by using the voltage-clamp technique (Figure 3 and Table 2). Figure 3A illustrates representative whole-cell current traces of N1774D-hiPSC-CMs and control cells. As shown in Figure 3B, the peak sodium current densities of N1774D-hiPSC-CMs were significantly increased, compared to those of the control cells (N1774D: −333 ± 62 pA/pF, n = 15 vs. control: −175 ± 33 pA/pF, n = 15; p < 0.005; Table 2). Moreover, the steady-state activation of sodium channels, shifted to more negative potentials by 13 mV in N1774D-hiPSC-CMs, compared to that of the control cells (Figure 3C and Table 2). No significant differences in the voltage dependence of inactivation and other kinetic properties were noted between control cells and N1774D-hiPSC-CMs.
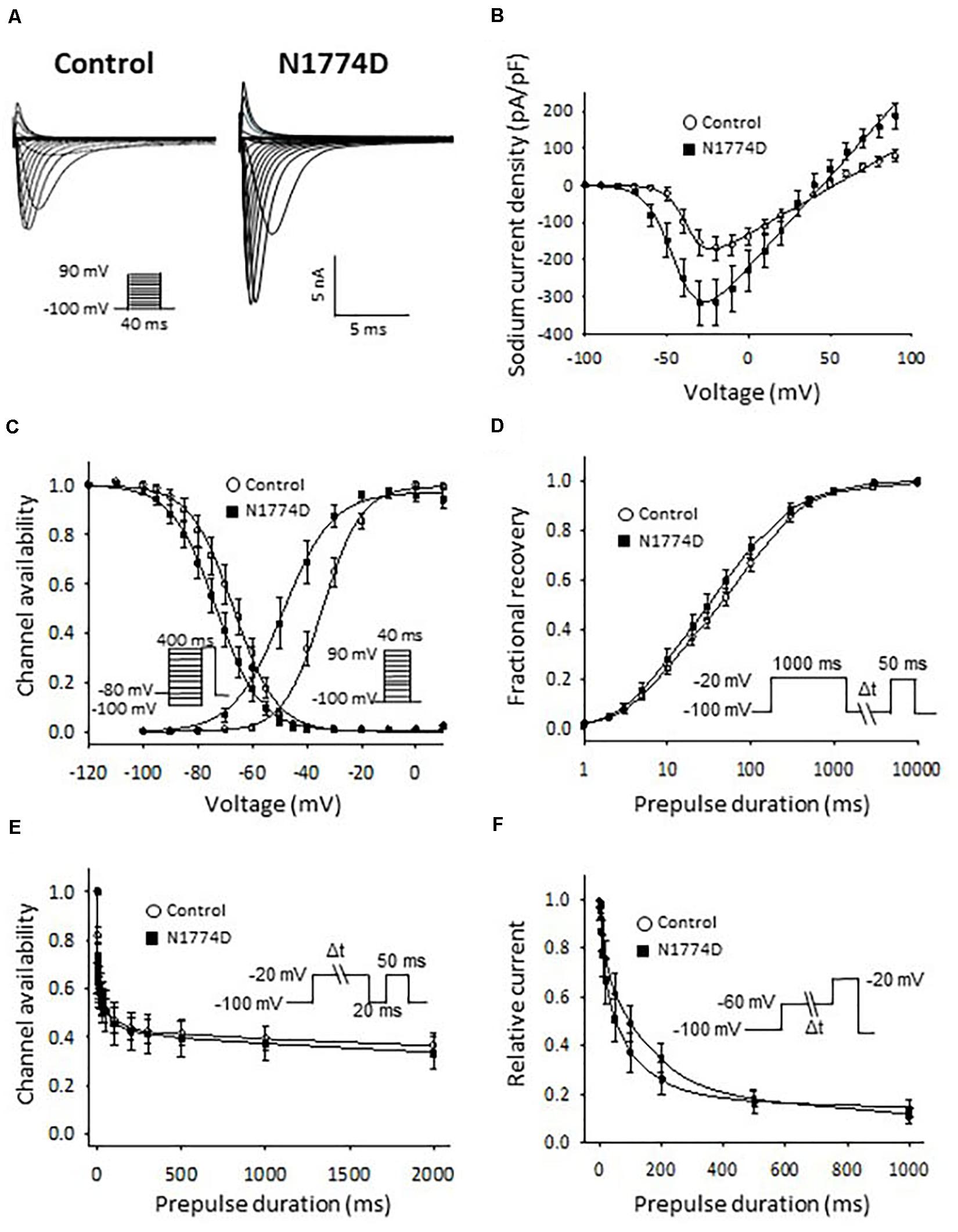
Figure 3. Sodium current recordings and gating properties of sodium channels in hiPSC-CMs. (A) Representative traces of sodium currents in control and N1774D-hiPSC-CMs. The pulse protocol is shown in the inset. (B) Average current-voltage relationship for peak sodium current in control (n = 15, open circles) and N1774D channels (n = 15, closed squares). Data were fitted with the Boltzmann equation (see “Materials and Methods”). The currents were normalized to the cell capacitance to give a measure of peak current densities. The peak current densities were significantly lager in N1774D. (C) Voltage dependence of steady-state inactivation and activation in control and N1774D-hiPSC-CMs. Curves were fit using the Boltzmann equation. The activation curve negatively shifted by 13 mV. (D) Time course of recovery from inactivation was obtained by a double pulse potential shown in inset. Experimental data were fit to a biexponential. (E) Onset of slow inactivation. Time course of entry into the slow inactivation state was measured by a double pulse protocol shown in inset. Curves were fit with a shingle exponential equation. (F) Closed-state inactivation. The transfer rate of sodium channels from closed-state to inactivated closed-state without an intervening opening state was elicited with a double pulse protocol shown in inset. The data pooled from different lines or clones among the control and N1774D group were analyzed.
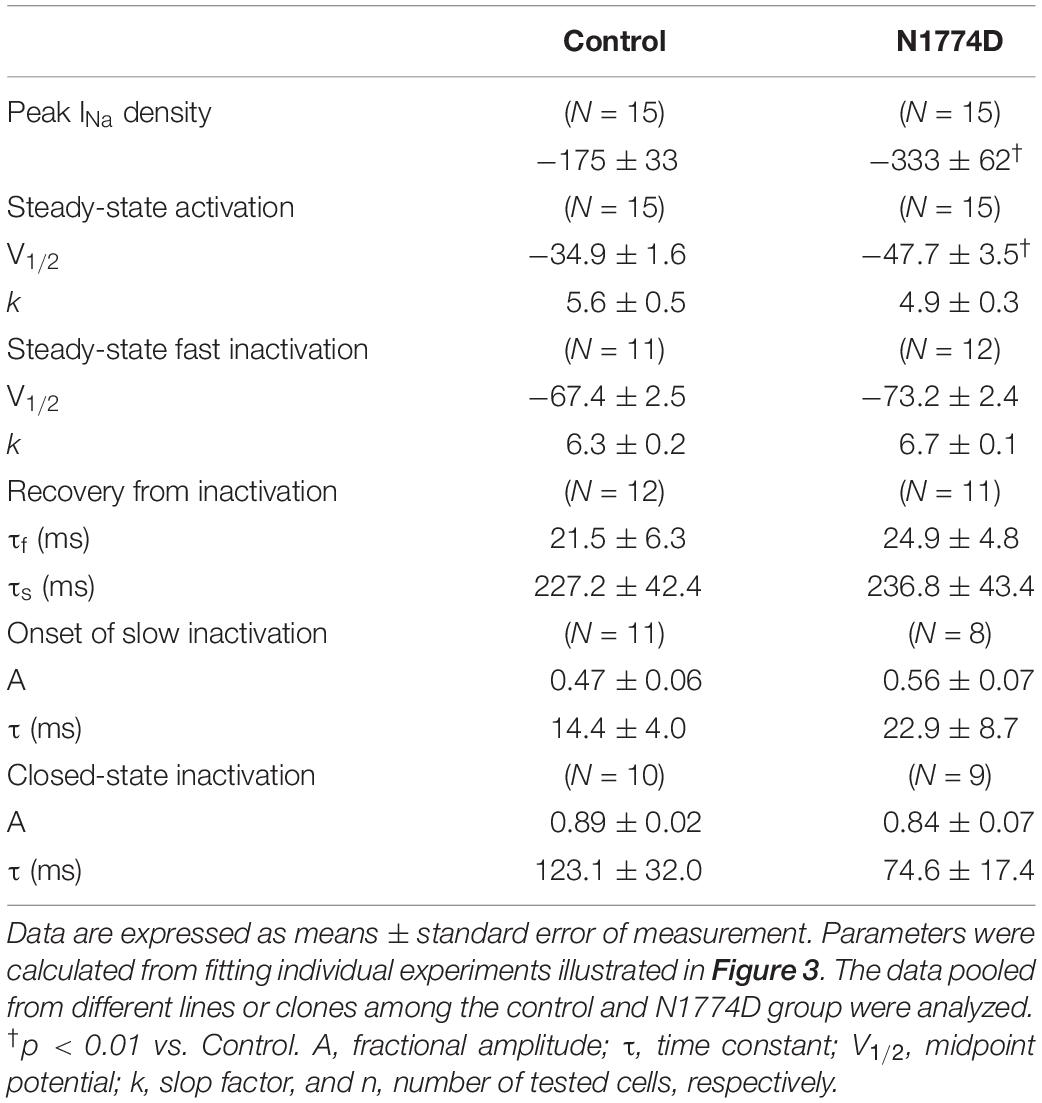
Table 2. Biophysical properties in control and N1774D-hiPSC-CMs.
Next, we measured the late sodium current for both cell lines. Figure 4A shows representative whole-cell sodium current traces from control and N1774D-hiPSC-CMs. The ratio of late/peak sodium current in N1774D-hiPSC-CMs was significantly increased compared with that in control cells (N1774D: 0.47 ± 0.04%, n = 11, vs. control, 0.04 ± 0.01%, n = 5; p < 0.0001; Figure 4B).
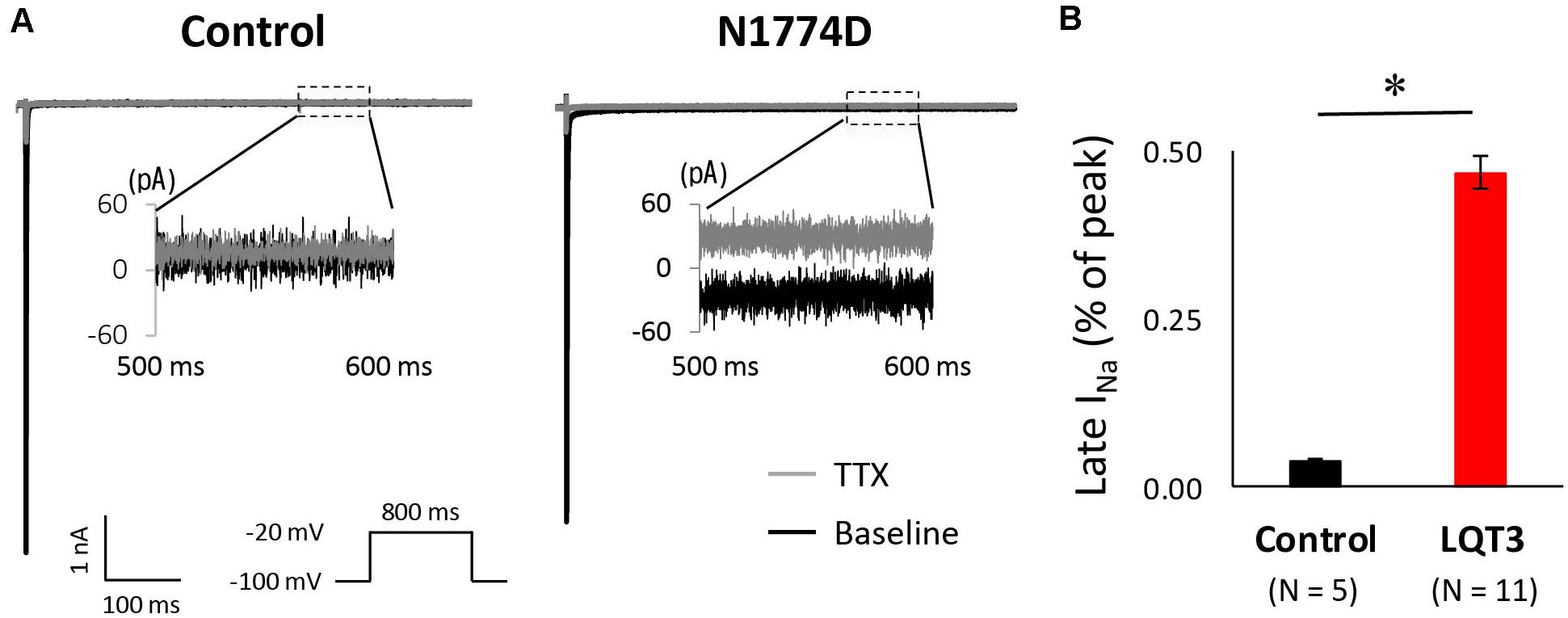
Figure 4. Late sodium current in control and N1774D-hiPSC-CMs. (A) Representative traces of sodium currents in the absence (black line) and presence (gray line) of 20 μM tetrodotoxin (TTX). The used protocol is shown in the lower panel. Inset shows late sodium current between 500 and 600 ms. Tetrodotoxin-sensitive current was calculated by subtraction. (B) Mean late sodium current of control and N1774D channels. Late sodium current is presented as the percentage of late sodium current to peak sodium current. The data pooled from different lines or clones among the control and N1774D group were analyzed. The late sodium current was significantly increased. ∗p < 0.001, vs. control.
Propranolol Shortened APDs and Reduced Late Sodium Current in N1774D-hiPSC-CMs
We assessed the effect of propranolol on APDs and late sodium current using hiPSC-CMs. Propranolol shortened APDs in N1774D-hiPSC-CMs (APD90: 440 ± 37 ms at baseline vs. 332 ± 37 ms after propranolol treatment, respectively, n = 10; at 1 Hz pacing; p < 0.01; Figure 2 and Table 1), but propranolol did not significantly affect APDs in the control cells (APD90: 272 ± 22 ms at baseline vs. 267 ± 26 ms after propranolol treatment, respectively, n = 11; at 1 Hz pacing; p = 0.15; Figure 2 and Table 1).
Figure 5A shows the representative traces of late sodium current at baseline and after administration of 5 μM propranolol to N1774D-hiPSC-CMs. Propranolol treatment decreased the ratio of late/peak sodium current by approximately 25%, from 0.53 ± 0.05 to 0.40 ± 0.06% (n = 7; p < 0.001; Figure 5B). These results suggested that propranolol attenuated late sodium current and thus shortened APDs in N1774D-hiPSC-CMs.
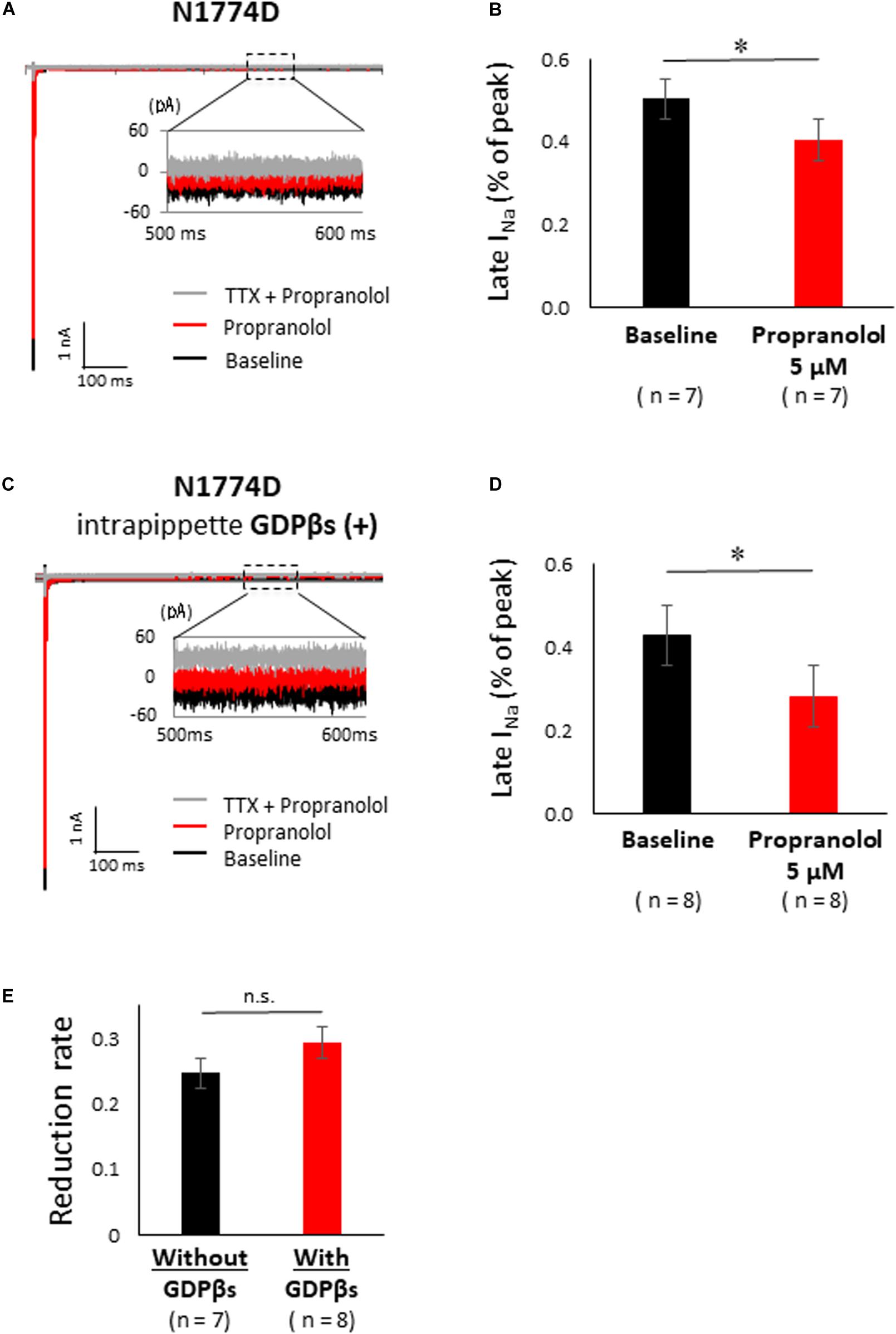
Figure 5. Effect of propranolol on late sodium current in N1774-hiPSC-CMs. (A) Typical late sodium current traces recorded at baseline (black line), after 5 μM propranolol application (red line), and after the additional treatment with 20 μM tetrodotoxin (TTX) (gray line). (B) Statistical analysis of the effect of propranolol on late sodium current. The data pooled from different clones in N1774D-hiPSC-CMs were analyzed. Late sodium current was normalized to peak sodium current. Propranolol significantly reduced the ratio of late/peak sodium current. ∗p < 0.001, vs. baseline. (C) Representative trace of late sodium current in the presence of intrapipette GDPβs. Black line is at baseline, red line is in the presence of 5 μM propranolol, and gray line is in the presence of 5 μM propranolol and 20 μM TTX. We recorded sodium currents in the presence of 5 μM propranolol following addition of 20 μM TTX after recording at baseline. (D) Summary of efficacy of propranolol on late sodium current after intrapipette GDPβs treatment. Propranolol significantly reduced the ratio of late/peak sodium current in the presence of intrapipette GDPβs. ∗p < 0.001, vs. baseline. (E) Reduction rate of late sodium current with and without GDPβs. There is no significant difference between in the absence and the presence of intrapipette GDPβs. GDPβs, guanosine diphosphate βs; TTX, tetrodotoxin.
Propranolol Directly Inhibited the Late Sodium Current in N1774D-hiPSC-CMs
Finally, to examine whether propranolol affected the sodium channels through G protein cascade or not, we recorded late sodium currents by using intrapipette guanosine diphosphate βs (GDPβs), a G protein inhibitor (Figure 5C). The late sodium current was significantly reduced by treatment with propranolol and 1 mM intrapipette GDPβs (baseline, 0.43 ± 0.07% vs. propranolol 0.28 ± 0.07%, n = 8; p = 0.01; Figure 5D). However, the reduction rate of late sodium current by propranolol treatment was not statistically different after addition of intrapipette GDPβs (without GDPβs: 24.7 ± 4.8% vs. with GDPβs: 30.9 ± 6.9%; p = 0.48; Figure 5E). Thus, propranolol directly blocked sodium channels and did not affect them via G protein pathway through β-adrenergic receptors.
Additionally, the suppression of late sodium current by propranolol was larger than that of the peak current whether in absence or presence of intrapipette GDPβs (Figure 6). No significant difference in the suppression rate of the peak and the late sodium currents were observed whether in absence or presence of GDPβs (Figure 6).
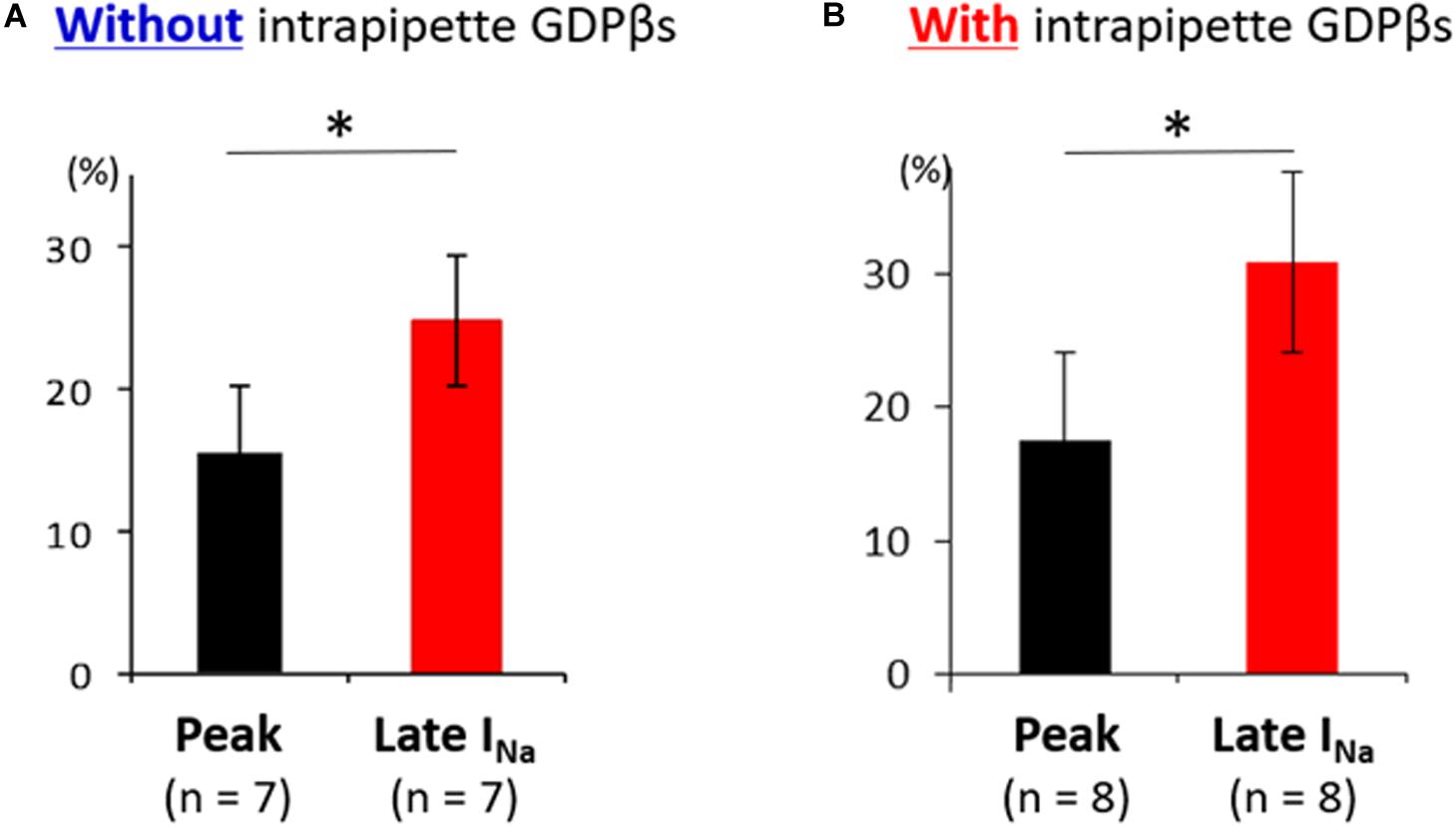
Figure 6. Effect of propranolol on peak and late sodium current in N1774D-hiPSC-CMs. Effect of propranolol on peak and late sodium current in the absence (A) and the presence of intrapipette GDPβs (B). The data pooled from different clones in N1774D-hiPSC-CMs were analyzed. (A) Without intrapipette GDPβs, the rate of reduction was larger in late sodium current compared to in peak sodium current. (B) In the presence of intrapipette GDPβs, the rate of reduction was also larger in late sodium current compared to in peak sodium current. ∗p < 0.001, vs. peak sodium current.
Discussion
Patient-Specific iPSC-CMs as a Cell Model for Studying SCN5A-N1774D Associated LQT3
The efficacy of β-blockers has been controversial in patients with LQT3 (Moss et al., 2000; Schwartz et al., 2001; Priori et al., 2004). Recently, a large clinical study demonstrated the efficacy of β-blockers on patients with LQT3 (Wilde et al., 2016); however, the pharmacological mechanism of β-blockers in LQT3 remains unclear.
Recently, the iPSC technology enables us to analyze the disease-causing mechanism of inherited arrhythmia disorder using self-beating cardiomyocytes with the same genetic background as the patient, and several studies of hiPSC-based disease modeling of LQT3 have been reported (Davis et al., 2012; Fatima et al., 2013; Ma et al., 2013; Terrenoire et al., 2013; Spencer et al., 2014; Yoshinaga et al., 2019). To elucidate the mechanism, we established the LQT3-hiPSC-CM cell model from a patient carrying the SCN5A-N1774D mutation who was effectively treated with propranolol. In accordance with his clinical course, propranolol attenuated the increased of late sodium current and prolonged APDs in N1774D-hiPSC-CMs, compared to the control cells. In addition, by using GDPβs, an inhibitor of G proteins, we demonstrated that propranolol inhibited the late sodium current through a pathway other than β-adrenergic receptor signaling pathway. Thus, the efficacy and pharmacological mechanism of propranolol have never, to our knowledge, been hitherto elucidated in an LQT3-hiPSC-CM model.
In our previous report (Kato et al., 2014), we analyzed SCN5A-N1774D channels using a heterologous expression system in HEK 293 cells and determined that N1774D channels displayed increased peak sodium current densities (2.2 times), a hyperpolarized shift of steady-state activation curve by 7.9 mV, and increased late sodium current densities by 1.7 times, compared to those of wild-type channels. In our LQT3-hiPSC model, the steady-state activation curve was negatively shifted by 12.8 mV and the peak and late sodium current densities were 1.9 and 9.3 times greater, respectively, when compared to those of the control hiPSC-CMs (Figures 2, 3). Considering that our hiPSCs carried the heterozygous SCN5A-N1774D mutation, the electrophysiological properties of hiPSC-CMs showed similar trends but different degrees of change in sodium channel kinetics to those of the heterologous expression system, possibly it might result from the differences between the two cell types.
β-Blocker Therapy for Patients With LQT3
β-blocker therapy is effective for treatment of patients especially with LQT1 and LQT2, who experience cardiac events after adrenergic stimulation, such as exercise or emotional stress. In LQT1 and LQT2, β-blockers clinically reduce the rate of cardiac events and mortality (Moss et al., 2000; Schwartz et al., 2001; Priori et al., 2004). Therefore, based on the clinical evidence, β-blocker therapy is recommended as the first-line treatment for LQT1 and LQT2 (Priori et al., 2013).
In contrast, Schwartz et al. (2001) reported that the recurrence of syncope and death rates in patients with LQT3 were higher (50% and 17%, respectively, n = 18) than those with LQT1 and 2. Additionally, Priori et al. (2004) reported that 9 out of 28 (32%) LQT3 patients experienced a cardiac event while on β-blocker therapy at a rate four-fold higher than that of LQT1 patients. Even though the number of LQT3 patients enrolled in the study was small, β-blocker therapy in LQT3 was suggested to be ineffective in preventing fatal arrhythmias and even contraindicated or harmful due to the clinical feature of LQT3, that cardiac events mainly occur at rest, during sleep or bradycardia.
However, recently, the efficacy of β-blocker therapy was analyzed in a large multicenter LQT3 cohort study (Wilde et al., 2016). Wilde et al. (2016) investigated 406 LQT3 patients and demonstrated that β-blocker therapy reduced the risk of life-threatening cardiac events in female patients >1 year of age. In the present study, our patient suffered from repetitive TdPs since birth and propranolol was definitely effective in suppressing those events. Despite accumulating clinical evidence that β-blocker therapy might be beneficial to LQT3 patients, the underlying mechanism by which β-blockers suppress malignant ventricular arrhythmias remains mostly unclear. In our LQT3-hiPSC model, prolonged APDs at baseline were attenuated after propranolol administration (Figure 5), which was consistent with the clinical efficacy of β-blocker therapy in LQT3.
Why Are β-Blockers Effective in Patients With LQT3?
Cardiac Sodium Channel and β-Adrenergic Stimulation
Cardiac sodium channels are known to be modulated by β-adrenergic stimulation via phosphorylation sites on protein kinases A and C, PKA and PKC, respectively (Grant, 2009). In cardiac sodium channels, PKA activation increased the peak sodium current density and shifted both steady-state activation and inactivation to the hyperpolarized direction (Matsuda et al., 1992; Ono et al., 1993). On the contrary, PKC activation reduced peak sodium current densities and caused a negative shift of the steady-state inactivation curve (Valdivia et al., 2009).
PKA activation did not affect the late sodium current in wild-type sodium channels. On the other hand, in LQT3-related mutant sodium channels, PKA activation had little or no effect on Y1795C and Y1795H channels, but it enhanced the late sodium current in ΔKPQ, D1790G, R1623G, and V2016M channels (Chandra et al., 1999; Tateyama et al., 2003; Tsurugi et al., 2009; Chen et al., 2016).
In LQT3 mouse models, propranolol efficacy to prevent lethal arrhythmias has been contradictory. Fabritz et al. (2010) showed that chronic propranolol therapy did not suppress the carbachol-mediated ventricular arrhythmias, such as TdPs, in a heterozygous knock-in SCN5A-ΔKPQ LQT3 mice. On the other hand, Calvillo et al. (2014) reported that propranolol pretreatment prevented carbachol-mediated malignant ventricular tachyarrhythmias in the SCN5A-ΔKPQ LQT3 knock-in transgenic mice. Thus, the pharmacological mechanism of β-blockers in LQT3 mice and human cardiomyocytes derived from patients with LQT3 has remained uncertain until now. In the present study, we demonstrated that propranolol attenuated the prolonged APD, by reducing the late sodium current in the patient-derived LQT3-hiPSC-CMs, and provided new supportive evidence of its efficacy on LQT3 by using a human CM model.
Propranolol Has an Inhibitory Effect on Sodium Channels but Not Through β-Adrenergic Receptor Blockade in an LQT3-hiPSC Model
Propranolol was previously reported to have a unique blocking effect on sodium channels, which was not identified in other β-blockers (Wang et al., 2010). In a heterologous expression system using HEK293 cells, only propranolol, not metoprolol or nadolol, blocked the cardiac sodium channels similar to local anesthetics, and not by acting as β-blockers (Wang et al., 2010). They also demonstrated that the block depends on a critical D4/S6 residue, F1760, involved in local anesthetic effects. In addition, Bankston and Kass (2010) demonstrated that propranolol decreased late sodium current in F1473C and ΔKPQ mutant channels. In this study, we showed no significant difference in the suppression rate of late sodium current by propranolol in presence, or not, of intrapipette GDPβs, an inhibitor of G proteins (Figure 5E). This was found to be consistent with the previous reports that propranolol has a sodium channel blocking effect through interfering with the cascade other than G protein cascades (Bankston and Kass, 2010; Wang et al., 2010).
Interestingly, Bankston and Kass (2010) also reported that propranolol within 40 nM to 40 μM range preferential inhibited late but not peak sodium currents. In this study, administration of 5 μM propranolol reduced late sodium current, but it slightly decreased peak sodium current (Figure 6 and Table 3). Regarding the plasma concentration of propranolol, it was reported that there was wide inter-individual variation from 0.04 to 4 μM in peak plasma concentrations, and an increase in the dose up to plasma levels of approximately 0.4 μM was suggested when sufficient therapeutic effects were not achieved (Woosley et al., 1979). Unfortunately, the plasma concentration of propranolol in the patient in this study was not available, and we recorded APs and late sodium current with only 5 μM propranolol using hiPSC-CM model. In addition, we did not examine the effect of propranolol on the kinetics of sodium channel current, which was demonstrated in a heterologous expression system (Wang et al., 2010). Therefore, further examination is needed to reveal the detailed relationship between propranolol dose and the effects on electrophysiological properties in hiPSC-CMs.
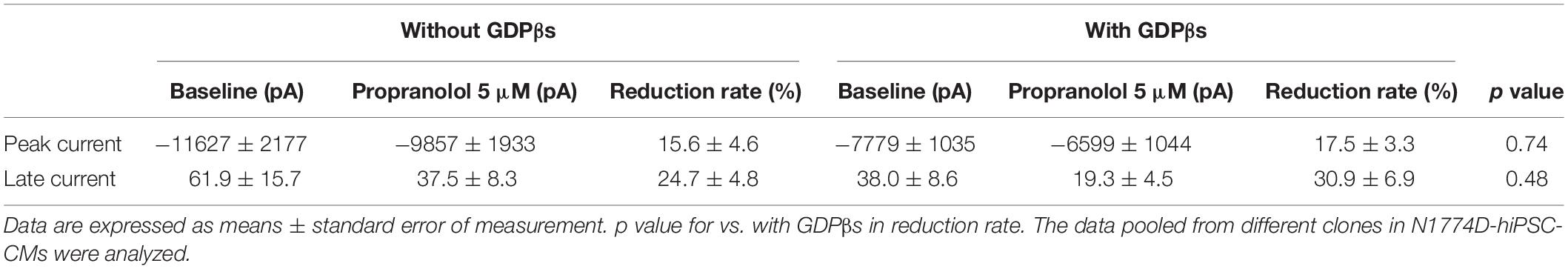
Table 3. The change of peak and late sodium current after the administration of propranolol in N1774D-hiPSC-CMs.
Study Limitations
In the present study, we did not employ the isogenic control line which will prove the phenotype is directly imputable to the mutation. In addition, we did not examine the efficacy of other β-blockers. Another limitation is that hiPSC-CM is an in vitro cellular model; therefore, it is impossible to assess the role of sympathetic nervous system. Clinically, left cardiac sympathetic denervation was reported to inhibit cardiac events in LQT3 patients (Schwartz et al., 2004), which indicated that β-blockers would have an antiarrhythmic effect by suppressing sympathetic nervous system in LQT3.
Conclusion
We successfully recapitulated the LQT3 disease phenotype in the SCN5A-N1774D-hiPSC-CMs. Additionally, the propranolol efficacy and mode of action were demonstrated in this cell model. Specifically, propranolol blocked sodium channels by means of a unique mechanism unrelated to β-adrenergic signaling pathway, by preferentially inhibiting late rather than peak sodium current.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of Kyoto University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. The animal study was reviewed and approved by Ethics Committee of Kyoto University.
Author Contributions
SH, TM, and MHo: conceptualization. SH and TM: methodology and writing – original draft. SH, TM, DM, JW, YYa, YW, FY, SN, TH, MHa, HK, JG, AK, MN, JY, and KC: investigation. TM, DM, MHa, and MHo: writing – review and editing. TM and MHo: funding acquisition. TM, SO, YYo, and MHo: resources. DM, SO, YYo, MHo, and TK: supervision. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the JSPS KAKENHI (Grant Numbers JP16K09499 and JP19K08538 to TM), Suzuken Memorial Foundation (TK and TM), research grant from the Japan Agency for Medical Research and Development, AMED (Grant Number 19ek0109219h0003 to MHo and TM).
Conflict of Interest
YYo owns stock in iPS Portal.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank the proband and his family for their participation in this study. We are also grateful for Kyoko Yoshida for her technical assistance.
References
Ackerman, M. J., Priori, S. G., Willems, S., Berul, C., Brugada, R., Calkins, H., et al. (2011). HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm. 8, 1308–1339. doi: 10.1016/j.hrthm.2011.05.020
Bankston, J. R., and Kass, R. S. (2010). Molecular determinants of local anesthetic action of beta-blocking drugs: implications for therapeutic management of long QT syndrome variant 3. J. Mol. Cell Cardiol. 48, 246–253. doi: 10.1016/j.yjmcc.2009.05.012
Bennett, P. B., Yazawa, K., Makita, N., and George, A. L. Jr. (1995). Molecular mechanism for an inherited cardiac arrhythmia. Nature 376, 683–685. doi: 10.1038/376683a0
Calvillo, L., Spazzolini, C., Vullo, E., Insolia, R., Crotti, L., and Schwartz, P. J. (2014). Propranolol prevents life-threatening arrhythmias in LQT3 transgenic mice: implications for the clinical management of LQT3 patients. Heart Rhythm. 11, 126–132. doi: 10.1016/j.hrthm.2013.10.029
Chandra, R., Chauhan, V. S., Starmer, C. F., and Grant, A. O. (1999). beta-Adrenergic action on wild-type and KPQ mutant human cardiac Na+ channels: shift in gating but no change in Ca2+:Na+ selectivity. Cardiovasc. Res. 42, 490–502. doi: 10.1016/s0008-6363(99)00042-5
Chen, J., Makiyama, T., Wuriyanghai, Y., Ohno, S., Sasaki, K., Hayano, M., et al. (2016). Cardiac sodium channel mutation associated with epinephrine-induced QT prolongation and sinus node dysfunction. Heart Rhythm. 13, 289–298. doi: 10.1016/j.hrthm.2015.08.021
Davis, R. P., Casini, S., van den Berg, C. W., Hoekstra, M., Remme, C. A., Dambrot, C., et al. (2012). Cardiomyocytes derived from pluripotent stem cells recapitulate electrophysiological characteristics of an overlap syndrome of cardiac sodium channel disease. Circulation 125, 3079–3091. doi: 10.1161/CIRCULATIONAHA.111.066092
Fabritz, L., Damke, D., Emmerich, M., Kaufmann, S. G., Theis, K., Blana, A., et al. (2010). Autonomic modulation and antiarrhythmic therapy in a model of long QT syndrome type 3. Cardiovasc. Res. 87, 60–72. doi: 10.1093/cvr/cvq029
Fatima, A., Kaifeng, S., Dittmann, S., Xu, G., Gupta, M. K., Linke, M., et al. (2013). The disease-specific phenotype in cardiomyocytes derived from induced pluripotent stem cells of two long QT syndrome type 3 patients. PLoS One 8:e83005. doi: 10.1371/journal.pone.0083005
Grant, A. O. (2009). Cardiac ion channels. Circ. Arrhythm. Electrophysiol. 2, 185–194. doi: 10.1161/CIRCEP.108.789081
Hayano, M., Makiyama, T., Kamakura, T., Watanabe, H., Sasaki, K., Funakoshi, S., et al. (2017). Development of a patient-derived induced pluripotent stem cell model for the investigation of SCN5A-D1275N-related cardiac sodium channelopathy. Circ. J. 81, 1783–1791. doi: 10.1253/circj.CJ-17-0064
Hobbs, J. B., Peterson, D. R., Moss, A. J., McNitt, S., Zareba, W., Goldenberg, I., et al. (2006). Risk of aborted cardiac arrest or sudden cardiac death during adolescence in the long-QT syndrome. JAMA 296, 1249–1254. doi: 10.1001/jama.296.10.1249
Kato, K., Makiyama, T., Wu, J., Ding, W. G., Kimura, H., Naiki, N., et al. (2014). Cardiac channelopathies associated with infantile fatal ventricular arrhythmias: from the cradle to the bench. J. Cardiovasc. Electrophysiol. 25, 66–73. doi: 10.1111/jce.12270
Ma, D., Wei, H., Zhao, Y., Lu, J., Li, G., Sahib, N. B., et al. (2013). Modeling type 3 long QT syndrome with cardiomyocytes derived from patient-specific induced pluripotent stem cells. Int. J. Cardiol. 168, 5277–5286. doi: 10.1016/j.ijcard.2013.08.015
Ma, J., Guo, L., Fiene, S. J., Anson, B. D., Thomson, J. A., Kamp, T. J., et al. (2011). High purity human-induced pluripotent stem cell-derived cardiomyocytes: electrophysiological properties of action potentials and ionic currents. Am. J. Physiol. Heart Circ. Physiol. 301, H2006–H2017. doi: 10.1152/ajpheart.00694.2011
Matsa, E., Rajamohan, D., Dick, E., Young, L., Mellor, I., Staniforth, A., et al. (2011). Drug evaluation in cardiomyocytes derived from human induced pluripotent stem cells carrying a long QT syndrome type 2 mutation. Eur. Heart J. 32, 952–962. doi: 10.1093/eurheartj/ehr073
Matsuda, J. J., Lee, H., and Shibata, E. F. (1992). Enhancement of rabbit cardiac sodium channels by beta-adrenergic stimulation. Circ. Res. 70, 199–207. doi: 10.1161/01.res.70.1.199
Mizusawa, Y., Horie, M., and Wilde, A. A. (2014). Genetic and clinical advances in congenital long QT syndrome. Circ. J. 78, 2827–2833. doi: 10.1253/circj.cj-14-0905
Moss, A. J., Shimizu, W., Wilde, A. A., Towbin, J. A., Zareba, W., Robinson, J. L., et al. (2007). Clinical aspects of type-1 long-QT syndrome by location, coding type, and biophysical function of mutations involving the KCNQ1 gene. Circulation 115, 2481–2489. doi: 10.1161/CIRCULATIONAHA.106.665406
Moss, A. J., Zareba, W., Hall, W. J., Schwartz, P. J., Crampton, R. S., Benhorin, J., et al. (2000). Effectiveness and limitations of beta-blocker therapy in congenital long-QT syndrome. Circulation 101, 616–623. doi: 10.1161/01.cir.101.6.616
Nakagawa, M., Koyanagi, M., Tanabe, K., Takahashi, K., Ichisaka, T., Aoi, T., et al. (2008). Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat. Biotechnol. 26, 101–106. doi: 10.1038/nbt1374
Okita, K., Yamakawa, T., Matsumura, Y., Sato, Y., Amano, N., Watanabe, A., et al. (2013). An efficient nonviral method to generate integration-free human-induced pluripotent stem cells from cord blood and peripheral blood cells. Stem Cells 31, 458–466. doi: 10.1002/stem.1293
Ono, K., Fozzard, H. A., and Hanck, D. A. (1993). Mechanism of cAMP-dependent modulation of cardiac sodium channel current kinetics. Circ. Res. 72, 807–815. doi: 10.1161/01.res.72.4.807
Priori, S. G., Napolitano, C., Schwartz, P. J., Grillo, M., Bloise, R., Ronchetti, E., et al. (2004). Association of long QT syndrome loci and cardiac events among patients treated with beta-blockers. JAMA 292, 1341–1344. doi: 10.1001/jama.292.11.1341
Priori, S. G., Wilde, A. A., Horie, M., Cho, Y., Behr, E. R., Berul, C., et al. (2013). HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm. 10, 1932–1963. doi: 10.1016/j.hrthm.2013.05.014
Schwartz, P. J., Crotti, L., and Insolia, R. (2012). Long-QT syndrome: from genetics to management. Circ. Arrhythm. Electrophysiol. 5, 868–877. doi: 10.1161/CIRCEP.111.962019
Schwartz, P. J., Priori, S. G., Cerrone, M., Spazzolini, C., Odero, A., Napolitano, C., et al. (2004). Left cardiac sympathetic denervation in the management of high-risk patients affected by the long-QT syndrome. Circulation 109, 1826–1833. doi: 10.1161/01.CIR.0000125523.14403.1E
Schwartz, P. J., Priori, S. G., Spazzolini, C., Moss, A. J., Vincent, G. M., Napolitano, C., et al. (2001). Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation 103, 89–95. doi: 10.1161/01.cir.103.1.89
Shimizu, W., Moss, A. J., Wilde, A. A., Towbin, J. A., Ackerman, M. J., January, C. T., et al. (2009). Genotype-phenotype aspects of type 2 long QT syndrome. J. Am. Coll. Cardiol. 54, 2052–2062. doi: 10.1016/j.jacc.2009.08.028
Skinner, J. R., Winbo, A., Abrams, D., Vohra, J., and Wilde, A. A. (2019). Channelopathies that lead to sudden cardiac death: clinical and genetic aspects. Heart Lung Circ. 28, 22–30. doi: 10.1016/j.hlc.2018.09.007
Spencer, C. I., Baba, S., Nakamura, K., Hua, E. A., Sears, M. A., Fu, C. C., et al. (2014). Calcium transients closely reflect prolonged action potentials in iPSC models of inherited cardiac arrhythmia. Stem Cell Rep. 3, 269–281. doi: 10.1016/j.stemcr.2014.06.003
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K., et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872. doi: 10.1016/j.cell.2007.11.019
Tateyama, M., Rivolta, I., Clancy, C. E., and Kass, R. S. (2003). Modulation of cardiac sodium channel gating by protein kinase A can be altered by disease-linked mutation. J. Biol. Chem. 278, 46718–46726. doi: 10.1074/jbc.M308977200
Terrenoire, C., Wang, K., Tung, K. W., Chung, W. K., Pass, R. H., Lu, J. T., et al. (2013). Induced pluripotent stem cells used to reveal drug actions in a long QT syndrome family with complex genetics. J. Gen. Physiol. 141, 61–72. doi: 10.1085/jgp.201210899
Tsurugi, T., Nagatomo, T., Abe, H., Oginosawa, Y., Takemasa, H., Kohno, R., et al. (2009). Differential modulation of late sodium current by protein kinase A in R1623Q mutant of LQT3. Life Sci. 84, 380–387. doi: 10.1016/j.lfs.2009.01.001
Valdivia, C. R., Ueda, K., Ackerman, M. J., and Makielski, J. C. (2009). GPD1L links redox state to cardiac excitability by PKC-dependent phosphorylation of the sodium channel SCN5A. Am. J. Physiol. Heart Circ. Physiol. 297, H1446–H1452. doi: 10.1152/ajpheart.00513.2009
Wang, D. W., Mistry, A. M., Kahlig, K. M., Kearney, J. A., and Xiang, J. (2010). Propranolol blocks cardiac and neuronal voltage-gated sodium channels. Front. Pharmacol. 1:144. doi: 10.3389/fphar.2010.00144
Wilde, A. A., Moss, A. J., Kaufman, E. S., Shimizu, W., Peterson, D. R., Benhorin, J., et al. (2016). Clinical aspects of type 3 long-QT syndrome: an international multicenter study. Circulation 134, 872–882. doi: 10.1161/CIRCULATIONAHA.116.021823
Woosley, R. L., Kornhauser, D., Smith, R., Reele, S., Higgins, S. B., Nies, A. S., et al. (1979). Suppression of chronic ventricular arrhythmias with propranolol. Circulation 60, 819–827. doi: 10.1161/01.cir.60.4.819
Wuriyanghai, Y., Makiyama, T., Sasaki, K., Kamakura, T., Yamamoto, Y., Hayano, M., et al. (2018). Complex aberrant splicing in the induced pluripotent stem cell-derived cardiomyocytes from a patient with long QT syndrome carrying KCNQ1-A344Aspl mutation. Heart Rhythm. 15, 1566–1574. doi: 10.1016/j.hrthm.2018.05.028
Yamamoto, Y., Makiyama, T., Harita, T., Sasaki, K., Wuriyanghai, Y., Hayano, M., et al. (2017). Allele-specific ablation rescues electrophysiological abnormalities in a human iPS cell model of long-QT syndrome with a CALM2 mutation. Hum. Mol. Genet. 26, 1670–1677. doi: 10.1093/hmg/ddx073
Yang, L., Soonpaa, M. H., Adler, E. D., Roepke, T. K., Kattman, S. J., Kennedy, M., et al. (2008). Human cardiovascular progenitor cells develop from a KDR+ embryonic-stem-cell-derived population. Nature 453, 524–528. doi: 10.1038/nature06894
Keywords: long QT syndrome type 3, induced pluripotent stem cell, sodium channel, β blocker, arrhythmia
Citation: Hirose S, Makiyama T, Melgari D, Yamamoto Y, Wuriyanghai Y, Yokoi F, Nishiuchi S, Harita T, Hayano M, Kohjitani H, Gao J, Kashiwa A, Nishikawa M, Wu J, Yoshimoto J, Chonabayashi K, Ohno S, Yoshida Y, Horie M and Kimura T (2020) Propranolol Attenuates Late Sodium Current in a Long QT Syndrome Type 3-Human Induced Pluripotent Stem Cell Model. Front. Cell Dev. Biol. 8:761. doi: 10.3389/fcell.2020.00761
Received: 12 February 2020; Accepted: 20 July 2020;
Published: 13 August 2020.
Edited by:
Masayuki Yazawa, Columbia University, United StatesReviewed by:
Yukiomi Tsuji, Nagasaki University, JapanMilena Bellin, Leiden University Medical Center, Netherlands
Copyright © 2020 Hirose, Makiyama, Melgari, Yamamoto, Wuriyanghai, Yokoi, Nishiuchi, Harita, Hayano, Kohjitani, Gao, Kashiwa, Nishikawa, Wu, Yoshimoto, Chonabayashi, Ohno, Yoshida, Horie and Kimura. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takeru Makiyama, bWFraXlhbWFAa3VocC5reW90by11LmFjLmpw