Abstract
Autosomal dominant osteopetrosis type II (ADO II), characterized by increased bone mass and density, is caused by mutations in the chloride channel 7 (CLCN7) gene. In this study, a novel missense variant in CLCN7 (c.1678A > G; p.Met560Val) was identified in three symptomatic subjects and one carrier of a Chinese family with ADO II. Notably, bone formation markers, including osteocalcin and total procollagen type N-terminal propeptide, have increased or presented at the upper limit of the normal range in the three patients. Serum factors secreted by osteoclast lineage cells and affecting the CD31hiEMCNhi vessel formation, such as tartrate-resistant acid phosphatase 5b, platelet-derived growth factor-BB, vascular endothelial growth factor, and SLIT3, had a higher expression in three ADO II subjects than in 15 healthy age-matched and sex-matched controls. Moreover, the conditioned medium was obtained from preosteoclast induced from the ADO II patients’ peripheral blood mononuclear cells. It was found to promote the CD31hiEMCNhi vessel formation of human microvascular endothelial cells and osteogenic differentiation of bone marrow-derived stem cells. Taken together, our finding revealed a novel CLCN7 variant associated with ADO II and suggested that the sclerotic bone was potentially associated with the increase of the CD31hiEMCNhi vessel formation and bone formation.
Introduction
Autosomal dominant osteopetrosis type II (ADO II; OPTA2, Online Mendelian Inheritance in Man: 166600), also known as “Albers Schönberg disease,” is the most common type of osteopetrosis with an estimated prevalence of 2.5/100,000 (Bénichou et al., 2000; Del Fattore et al., 2008). It is characterized by segmentary osteosclerosis, predominantly involving in the vertebral endplates (“rugger-jersey spine”), iliac wings (“bone within bone” sign), and the skull base. Previous studies revealed that ADO II was a phenotypically heterogeneous disease whose clinical spectrums ranged from unaffected radiography mutation carriers to affected skeletal patients yet asymptomatic to severe manifestations including fracture, osteomyelitis, visual loss, and bone marrow failure (Bénichou et al., 2000; Waguespack et al., 2007).
The bone homeostasis was maintained by balancing osteoclast-mediated resorption and osteoblast-mediated formation (Wang et al., 2018; Hayashi et al., 2019; Rodriguez-Laguna et al., 2019). The disrupted bone mass and structure of osteopetrosis resulted from the impaired osteoclast-mediated bone resorption caused by genetic factors (Tolar et al., 2004; Sobacchi et al., 2013). Bénichou et al. (2001) previously considered that ADO II was genetically homogeneous with a single disease-causing gene on 16p13.3. Accumulated studies showed that roughly 70% of patients harbored heterozygous missense mutations of the CLCN7 gene in a dominant-negative form (Cleiren et al., 2001). To date, more than 40 mutations in CLCN7 has been identified in different osteopetrosis families (Leisle et al., 2011; Pang et al., 2016). CLCN7 (Online Mendelian Inheritance in Man: 602727) is a multi-pass membrane protein that acts as a Cl-/H + exchanger by voltage gate (Leisle et al., 2011). It mainly resides in the late endosomes, lysosomes, and the ruffled membrane of osteoclasts. In osteoclasts, the ruffled border contains Cl-channels that are inserted by exocytotic fusion together with the H + -ATPase to ensure the acidification of extracellular resorption lacuna. Pathogenic mutations in the CLCN7 gene impairing acidification of extracellular resorption lacuna were assumed to prevent degradation of the calcified and organic matrix of bone (Weinert et al., 2010). Furthermore, some studies suggested that chloride channels could be a drug target for osteopetrosis (Capulli et al., 2015). Mattia et al. demonstrated that CLCN7 mutant-specific small interfering RNAs silenced dominant-negative CLCN7 transcripts and subsequently ameliorated the bone phenotype in ADO II (Verkman and Galietta, 2009). Recent research also demonstrated that the specific small interfering RNA therapy normalized extra-skeletal manifestations on the ADO II mouse (Maurizi et al., 2019).
Some studies suggested that CLCN7 mutations regulate bone formation in patients with osteopetrosis (Barvencik et al., 2014; Coudert et al., 2014). The vessels in the bone niche were reported to play great roles in bone formation (Abarrategi et al., 2018; Hao et al., 2019). A specific endothelium in the murine skeletal system, termed type H, strongly positive for CD31 and endomucin, is crucial in bone formation and regeneration (Kusumbe et al., 2014; Ramasamy et al., 2014; Langen et al., 2017). Accumulated evidence suggests that the type H vessels exert on bone formation in many aspects, ranging from mediating local growth of the vasculature, to providing a necessary microenvironment for perivascular osteoprogenitors, and coupling angiogenesis to osteogenesis (Yang et al., 2017). Previous findings revealed that platelet-derived growth factor-BB (PDGF-BB) secreted by preosteoclast could induce CD31hiEmcnhi vessels subtype subsequently prevented bone loss in osteoporosis and bone-loss diseases (Xie et al., 2014). Furthermore, we recently found a mutation in Reg1cp involved in high bone mass syndrome through promoting angiogenesis (Yang et al., 2019). However, whether the bone accrual is associated with the coupling between angiogenesis and osteogenesis or not remains unclear.
Here, we ascertained a missense variant in CLCN7 in a Chinese family. This is the first description genetically confirmed with this heterozygous missense mutation (c.1678A > G; p. Met560Val) responsible for ADO II. Additionally, these results indicated that the mutation might be associated with the dysfunction of the angiogenesis and osteogenesis, which further explained the elevated bone formation markers in this ADO II family.
Materials and Methods
Family Collection
The Chinese family who suffered from ADO II was recruited from Xiangya Hospital Central South University. There were three affected individuals (II3, II7, and III2 in Figure 1A) in this family, and all of the affected subjects were not born of consanguineous parents. The patients underwent a series of clinical evaluation including laboratory and radiography examination in Xiangya Hospital, such as blood route, liver function test, renal function test, serum calcium (Ca), phosphate (P), alkaline phosphatase (ALP), β-Cross Laps of type I collagen containing cross-linked C-telopeptide, osteocalcin (OC), total procollagen type N-terminal propeptide (TPINP), and intact parathyroid hormone (PTH). Additionally, the patients were carried out with bone mineral density of the total body measured by a dual-energy X-ray absorptiometry densitometer and X-ray of bilateral hip joints and lumbar. The study was approved by the Ethics Committee of Xiangya Hospital Central South University. All subjects involved in this study signed informed consent documents before participating in the project.
FIGURE 1
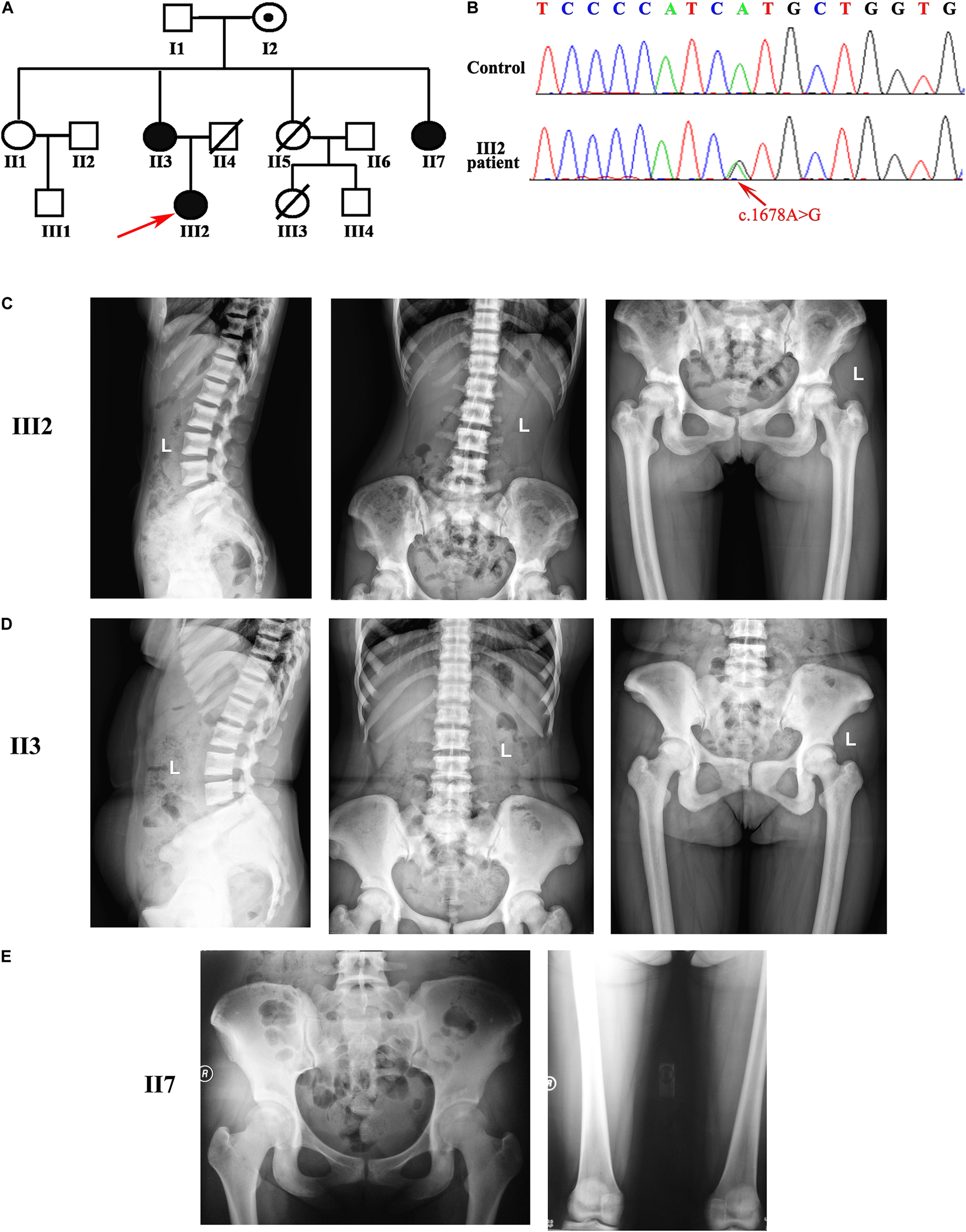
Pedigree of the osteopetrosis family was associated with the mutation in CLCN7. (A) Pedigree of a Chinese family with osteopetrosis. Squares represent males, and circles represent females. Red arrow indicates the proband in this family. White symbols represent unaffected subjects, and black symbols indicate affected subjects. Symbols with an embedded black dot represent asymptomatic carriers. Symbols with a diagonal line represent deceased subjects. (B) Sanger sequence chromatograms from the proband and unaffected II1. c.1678A > G transition is indicated by the red arrow. (C–E) Radiographic image reveals osteosclerosis in three affected patients (radiographic images of III2 in the top row, images of II3 in the second row, and images of II7 in the bottom row). Images showed vertebral endplates (“rugger-jersey spine”; III2 and II3) and osteosclerosis of the pelvis and femoral (III2, II3, and II7).
Whole-Genome Sequence
Genomic DNA was collected and extracted from peripheral whole blood samples using Qiagen DNA kits. The genomic DNA met the sequencing requirements of the purity (optical density 260/280 > 1.8) and concentration (50 ng/ml) of each sample. Whole-genome sequencing was conducted by Annoroad Gene Technology by using Illumina/TruSeq Nano DNA HT Library Preparation Kit for capture and Illumina sequencing platforms for data generation (150-bp paired-end, 300 cycles). Sequencing was performed with 150-bp paired-end reads on the Illumina NovaSeq 6000 sequencing platform. The mean coverage was 40×.
Variant Filtering and Analysis
Sequence alignment was performed with the Burrows–Wheeler Aligner mem using human genome assembly hg19 as the reference, then produce called variants with RTG tools Hyplotype Caller. Annotation of gene context was carried out with annovar using Single Nucleotide Polymorphism Database, Refseq gene as the reference.
Sanger Sequence
The variant was amplified by using DNA polymerase [MIX (GREEN), TSINGKE, China], and the former and reverse primers are CTTGAGGAGTCCACACCCAC and CTGGGAGCAGGTGGATGG, respectively. Forward and reverse sequencing reactions were performed with the Big Dye Terminator Cycle Sequencing Ready Reaction Kit (PE Applied Biosystems), and the products were analyzed on an ABI 3730XL automated sequencer (PE Applied Biosystems).
Enzyme-Linked Immunosorbent Assay
Serum was obtained from peripheral blood using 4,000 rpm for 5 min. Then, we used the ELISA kits to test the expression of tartrate-resistant acid phosphatase 5b (TRAP-5b), PDGF-BB, vascular endothelial growth factor (VEGF; R&D Systems), and SLIT3 (SED353Mi) in serum according to the manufacturers’ instructions. The ELISA was replicated three times for the affected patients, and the mean values were used.
Cell Culture
A total of 15 ml of heparin anti-coagulated peripheral blood was obtained from every subject, including all members of this family and healthy age- and sex-matched controls. Then, peripheral blood mononuclear cells (PBMCs) were washed using phosphate-buffered saline twice and cultured in minimum essential medium Eagle-α containing 10% fetal bovine serum, 100 U ml–1 penicillin, 100 μg ml–1 streptomycin sulfate, and 50 ng ml–1 macrophage colony-stimulating factor (M-CSF; R&D Systems Inc.). Non-adherent cells were incubated with M-CSF (50 ng ml–1) to obtain pure monocytes and macrophages. Then, PBMCs were cultured in 24-well plates at 1 × 105 cells per well with 50 ng ml–1 of M-CSF and 100 ng ml–1 receptor activator of nuclear factor-kappa-B ligand. Cells became preosteoclasts after a 3-day culture. We collected serum-containing conditioned medium from the preosteoclasts. After centrifugation (2,500 rpm for 10 min at 4°C), we aliquoted conditioned media and stored them at −80°C. Human BMSCs (HUXMA-0100; Cyagen Biosciences) were cultured in a human mesenchymal stem cell growth medium (HUXMA-90011; Cyagen Biosciences). To induce osteogenic differentiation of BMSCs, BMSCs were cultured in 48-well using the serum-containing conditioned medium. Then, we collected protein to test ALP activity on day 2, RNA on the 3rd day, and performed the ALP staining on day 7. Human microvascular endothelial cells (HMECs) were cultured with MCDB131 medium (Gibco) supplemented 10% fetal bovine serum, 100 U ml–1 penicillin, and 100 μg ml–1 streptomycin.
Real-Time Quantitative Polymerase Chain Reaction
Quantitative PCR analysis was performed as described previously (Xiao et al., 2020). Briefly, we extracted RNA from cells using Trizol reagent (Takara). Then, 1,000-ng RNA was reverse-transcribed into first-strand complementary DNA using the Reverse Transcription Kit (Takara). Quantitative PCR was performed using SYBR Green PCR Master Mix (Takara), and messenger RNA expression was normalized to reference genes GAPDH.
Tube Formation Assay
The Matrigel (BD Biosciences) was plated in 48-well plates and then incubated at 37°C for 45 min. HMECs were seeded in 48-well plates at 5 × 104 cells per well on polymerized Matrigel. The cells were cultured in the preosteoclast-conditioned medium collected from patients or control individuals for 4 h incubation at 37°C. Then, the tube formation was observed through microscopy, and the number of tube branches was quantified in four random microscope visual fields of each well.
Alkaline Phosphatase Activity
On the second day of osteogenic differentiation, the cell lysates were homogenized for ALP activity assay by spectrophotometric measurement of p-nitrophenol release using an ALP Assay Kit (P0321S, Beyotime) according to the manufacturer’s instructions.
Alkaline Phosphatase Staining
The cells were washed using phosphate-buffered saline for three times, followed by 10% paraformaldehyde for 5 min. Then, cells were incubated in ALP incubation buffer (0.2-g barbital sodium, 0.4-g magnesium sulfate, 0.2-g calcium chloride, and 0.3-g beta-glycerophosphate) at 37°C for 2 h. Next, cells were washed with 2% calcium chloride and incubated with 2% cobaltous nitrate for 5 min. Finally, cells were incubated in 1:80 ammonium sulfate for 10 s. The ALP staining was observed through a microscope.
Statistical Analysis
Data were imported into Excel and scaled and normalized to appropriate controls. Unpaired, two-tailed Student’s t-tests were performed for the comparisons of two groups. Critical P-values were Bonferroni corrected and expressed as follows: *P < 0.05. **P < 0.01.
Results
Clinical Characteristics of Patients
A Chinese patient, aged 22 years, was admitted to the Department of Endocrinology with a history of bone pain for 6 years. There was a family history of a similar disease of her mother (II3) and aunt (II7) shown in Figure 1A. All these three patients had a mild phenotype of osteopetrosis only with bone pain on hip joints and back. The proband also had a history of fracture in the hip. On examination, we did not find poor nutritional status, short stature, malformed craniofacial appearance, hepatosplenomegaly, and intellectual disability in the proband and other affected patients in this family.
Laboratory findings of the three patients showed that the hematopoietic function, liver function, kidney function, total serum calcium, and total serum phosphorus were within the normal ranges except for a slight increase of aspartate transaminase of II3 (Table 1). For bone turnover markers, β cross-linked C-telopeptide of type 1 collagen and ALP have not detected any abnormalities in this family (Table 2). However, markers of bone formation in the proband, such as OC and TPINP, were both found at the upper limit of the normal range (Table 2). Moreover, the levels of OC, TPINP, and PTH (parathormone) were found to be elevated in her mother (II3). The level of OC in her aunt (II7) also slightly increased. Thus, we hypothesized that bone formation in these patients had been elevated (Table 2).
TABLE 1
| Parameters | III2 | II3 | II7 | Normal range |
| WBC (109⋅L–1) | 4.2 | 5.3 | 5.46 | 3.5–9.5 |
| Hb (g⋅L–1) | 124 | 126 | 126 | 115–150 |
| RBC (1012⋅L–1) | 4.52 | 4.34 | 4.19 | 3.80–5.10 |
| PLT (109⋅L–1) | 295 | 240 | 146 | 125–350 |
| ALT (μ/L) | 9.3 | 21.6 | 20.4 | 7.0–40.0 |
| AST (μ/L) | 32.4 | 46.5 | 22.5 | 13.0–35.0 |
| BUN (mmol/L) | 4.58 | 5.56 | 6.95 | 2.60–7.50 |
| Cre (μmol/L) | 55 | 69 | 53 | 41.0–111.0 |
| UA (μmol/L) | 287.5 | 289.5 | 246 | 155.0–357.0 |
| Na (mmol/L) | 140 | 142.8 | 142 | 137.0–147.0 |
| K (mmol/L) | 4.54 | 4.22 | 3.6 | 3.50–5.30 |
| Cl (mmol/L) | 103.4 | 107.8 | 105.6 | 99.0–110.0 |
| Ca (mmol/L) | 2.45 | 2.28 | 2.22 | 2.00–2.60 |
| Mg (mmol/L) | 0.91 | 0.84 | 0.98 | 0.66–1.07 |
| P (mmol/L) | 1.41 | 1.45 | 1.25 | 0.86–1.78 |
Hematopoietic, hepatorenal, and serum electrolytic data of ADO II subjects.
WBC, white blood cells; RBC, red blood cells; Hb, hemoglobin; PLT, platelet; ALT, alanine transaminase; AST, aspartate transaminase; BUN, blood urea nitrogen; Cre, creatinine; and UA, uric acid.
TABLE 2
| III2 | II3 | II7 | Normal range | |
| ALP (μ/L) | 55.3 | 64.5 | 46.8 | 45.0–125.0 |
| OC (ng/ml) | 20.44 | 26.79 | 45 | 4.11–21.87 |
| TPINP (ng/ml) | 58.37 | 131.4 | NA | 8.53–64.32 |
| PTH (pg/ml) | 44.8 | 83.8 | 59.85 | 15.0–68.3 |
| β-CTX (pg/ml) | 290.5 | 243.5 | 572 | 68–680 |
Bone biomarker of ADO II subjects.
ALP, alkaline phosphatase; OC, osteocalcin; TPINP, total procollagen type N-terminal propeptide; PTH, parathormone; β-CTX, β cross-linked C-telopeptide of type 1 collagen; and NA, not available.
The total bone mineral density values of the three patients at lumbar and hip sites, measured by dual-energy X-ray absorptiometry, are shown in Table 3. The three affected patients were all with regular menstruation. The Z values at different sites of the three patients were significantly higher than the those of the sex-matched and age-matched controls (Table 3). Radiological features of the three patients revealed diffused osteosclerosis, predominantly at the vertebral endplates (“rugger-jersey spine”). The superior ramus of the pubis, sacral vertebrae, femoral head, and greater trochanter of femur also presented diffuse osteosclerosis. The boundaries among cancellous bone, cortical bone, and bone medullary disappeared (Figures 1C–E). Based on their clinical and radiological assessment, the family was diagnosed with ADO II.
TABLE 3
| Subjects | Age | Total | Lumbar | Left hip | |||
| Total BMD (g/cm2) | Z value | Total BMD (g/cm2) | Z value | Total BMD (g/cm2) | Z value | ||
| III2 | 21 | 1.611 | 5.8 | 2.221 | 12.1 | 1.591 | 6.9 |
| II 3 | 42 | 2.063 | 8.0 | 2.304 | 12.7 | 1.918 | 9.6 |
| II7 | 29 | 1.973 | 7.6 | 2.245 | 11.2 | 1.855 | 8.3 |
Bone mass density of the ADO II subjects.
BMD, Bone mineral density.
Genetic Analysis With Whole-Genome Sequencing and Identification of the Pathogenic Variant
We performed the whole-genome sequence in the three affected patients. Firstly, we filtered the common variants, which were unlikely to cause the rare heritable disorder (allele frequency greater than 0.1% in the 1000 Genomes database and Exome Variant Server). Then, we excluded the variants with low confidence and the putative impact. Next, we tried to find the shared variant in the three patients. Interestingly, a heterozygous missense variant (c.1678A > G, p.Met560Val) in CLCN7 segregated with osteopetrosis among three affected patients. We then performed the Sanger sequence in all members of this family and found the variant occurred in three patients and a non-penetrant carrier (I2 in Figure 1A was asymptomatic and did not present abnormal bone phenotype as well as laboratory findings; Figure 1B).
Various mutations in CLCN7 have been reported to be involved in the pathogenesis of osteopetrosis (Figure 2). In gnomAD database, the PopMax Allele Frequency and allele count of this variant were 0.00000900058 and 1, respectively, (the total Allele number was 245442). Moreover, this variant was not found in East Asian with 17228 Allele number in total. Pro560 in CLCN7, which is substituted to a Valine in three patients, is phylogenetically highly conserved among both vertebrates and invertebrates (Figure 3) [Phylop] score of 6.111 and [PhastCons] score of 1. CLCN7 gene was also predicted to be intolerant whose Residual Variation Intolerance Score is 7.0381% (Table 4). Furthermore, the effect of missense mutation Met560Val in CLCN7 was predicted by different variant effect prediction software (PolyPhen-2, MutationTaster, CADD, MPC). It suggested that the Met560Val substitution is likely to be deleterious (PolyPhen score of 0.991, probably damaging; MutationTaster score of 0.999, disease-causing; CADD score of 24.3, damaging; MPC score of 1.0788, possibly damaging; Table 4). Based on the genetic analysis, this study revealed that the heterozygous missense mutation was possibly responsible for the ADO II.
FIGURE 2
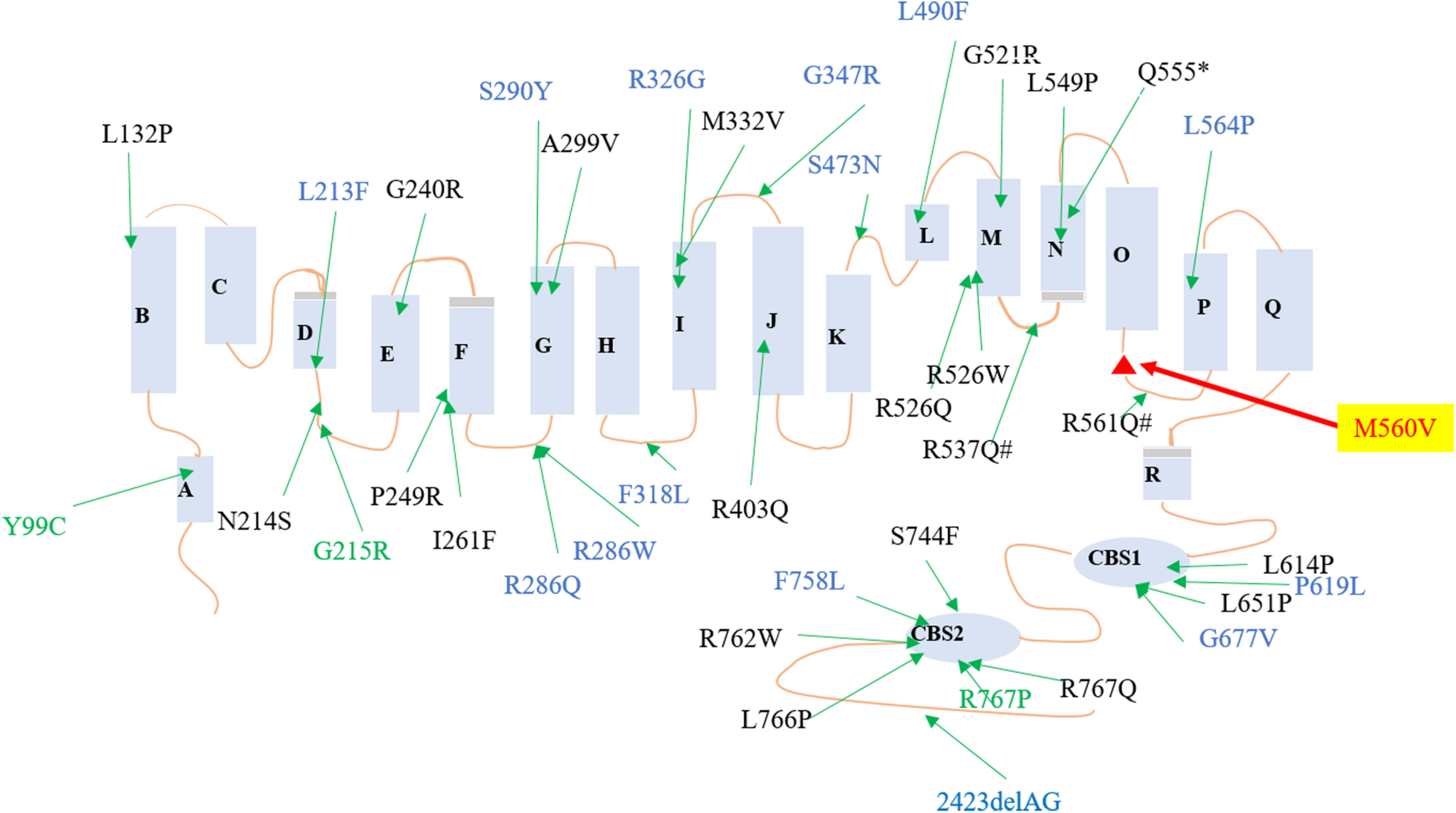
Reported mutations of CLCN7 in osteopetrosis family. Red mutation was found in this family. Black mutations were reported to cause osteopetrosis autosomal recessive 4, blue mutations were reported to cause ADO II, and green mutations were reported to cause different types of osteopetrosis.
FIGURE 3
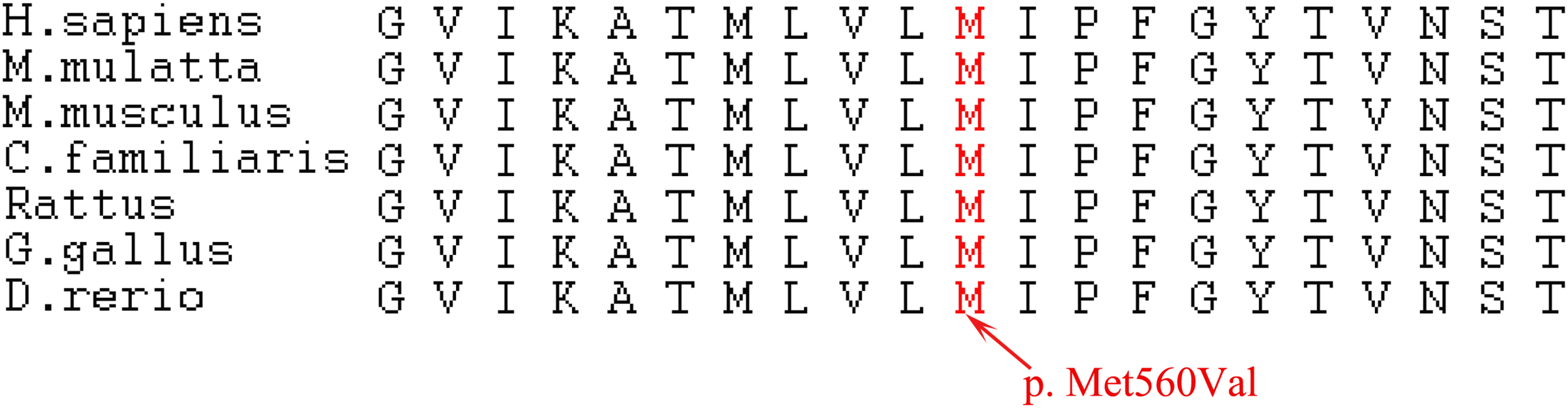
Conservation of the mutated amino acids. Alignment of the CLCN7 proteins, showing the conservation of the mutated residues among different species. p. Met560Val transition is indicated by the red arrow.
TABLE 4
| Variant (Missense) | Numbers | Rarity in gnomAD | Conservation (Extremely conserved) | Pathogenic prediction (Damaging) | ||||||
| Patients | Carrier | PopMax AF | Allele count | Phylop score | PhastCon score | MutationT-aster score | MPC | CADD score Phred | RVIS% | |
| c.1678A > G p.Met560Val | 3 | 1 | 9.00058E-06 | 1 | 6.111 | 1 | 0.999 | 1.0788 | 24.3 | 7.04% |
Summary of the CLCN7 mutation in this family with ADO II.
PopMax AF, Maximum allele frequency in any one population; MPC, mutant prevention concentration; CADD, Combined Annotation Dependent Depletion; and RVIS, Residual Variation Intolerance Score.
CLCN7 Mutation Induces the Type H Vessel Formation and Osteogenic Differentiation in vitro
The cross-talks between osteoclasts and other cells, including osteoblasts, endothelium cells, have been well established (Phan et al., 2004; Ramasamy et al., 2014; Xie et al., 2014; Yang et al., 2017, 2019; Chen et al., 2018; Li et al., 2019). Additionally, PDGF-BB secreted by preosteoclasts could result in elevated bone mass through inducing angiogenesis during coupling with osteogenesis (Xie et al., 2014). To investigate if the osteosclerotic bone of these patients was associated with the coupling between angiogenesis and osteogenesis, we collected the serum from ADO II individuals and 15 normal controls for further studies. TRAP-5b is well-established to be secreted by osteoclasts, and PDGF-BB, VEGF, and SLIT3 can also be secreted by osteoclasts/preosteoclasts, which were reported to be involved in the type H formation (Bollerslev et al., 2013; Sivaraj and Adams, 2016; Kim et al., 2018; Peng et al., 2020). Thus, we selected them to see if they have been altered in ADO II patients. Surprisingly, the level of TRAP-5b, PDGF-BB, VEGF, and SLIT3 showed a remarkable increase in ADO II patients compared with the 15 control groups with matched age and sex (Figures 4A–D). To further confirm the possible effect of type H vessels in this kindred, we purified the PBMCs from the patients and control normal subjects. Then, the collected preosteoclast condition medium from osteoclastic-induced PBMCs has been used to culture the HMECs. As expected, the expression of CD31, EMCN, and vessel growth factors, including VEGFA, VEGFB, PDGFA, and PDGFB, has increased in the cells treated with preosteoclast-conditioned medium from ADO II cells compared with the cells from control individuals (Figures 5A–F). Moreover, the tube formation also has been promoted by the ADO II-conditioned medium (Figures 5G,H). Even the expression of RUNX2 was not altered by the conditioned medium from patients; other osteogenic markers in hBMSCs, such as ALPL and SP7, elevated (Figures 6A–C). Consistent with these results, both ALP activity and ALP staining showed a significant difference between the two groups (Figures 6D,E). Together, these data suggested that the CLCN7 mutation might cause the bone accrual partially because of the elevated coupling of bone formation with type H formation.
FIGURE 4
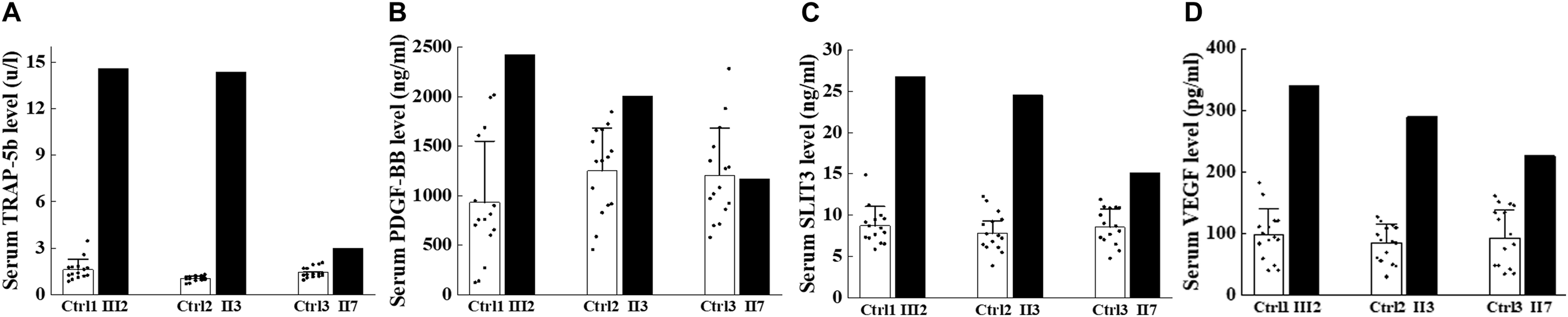
Expression of factors secreted by osteoclast lineage cells elevated in the affected patients. Serum analysis of ADO II subjects and age-matched and sex-matched. (A) Expression of serum TRAP-5b. (B) Expression of serum PDGF-BB. (C) Expression of serum SLIT3. (D) Expression of serum VEGF. Number of controls = 15. Data are shown as mean ± SD.
FIGURE 5
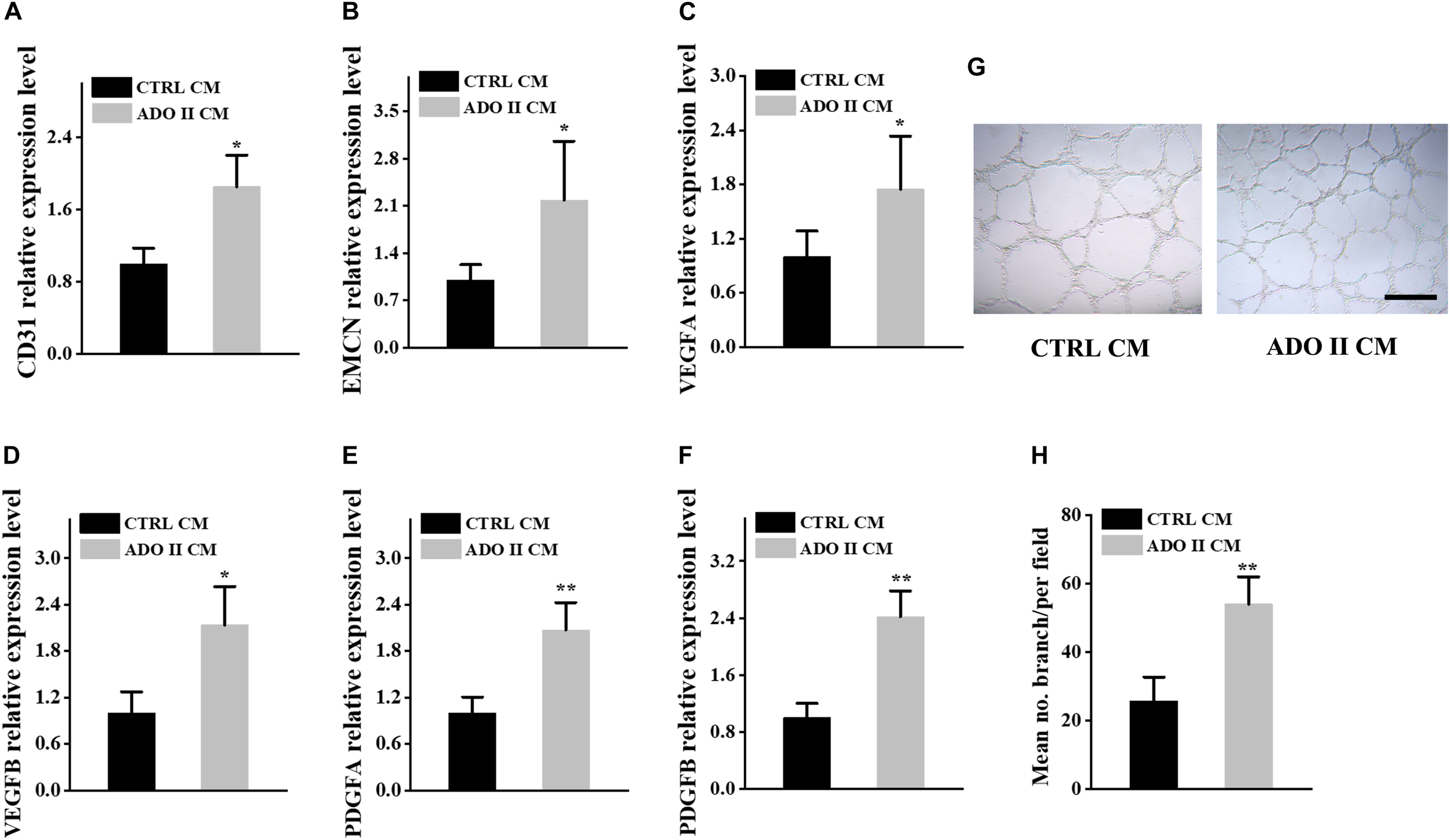
Preosteoclast-conditioned medium from ADO II patients promotes vessel formation in vitro. HMECs were treated with a preosteoclast-conditioned medium from ADO II patients or control groups. (A,B) qRT–PCR analysis of the relative levels of CD31(A), EMCN(B), VEGFA(C), VEGFB(D), PDGFA(E), and PDGFB(F) in HMECs. (G) Representative images of tube formation. (H) Quantification of tube branch numbers per field. CTRL CM, control conditioned medium; ADO II CM, ADO II conditioned medium. These experiments were replicated three times. Data are shown as mean ± SD. *P < 0.05 and **P < 0.01.
FIGURE 6
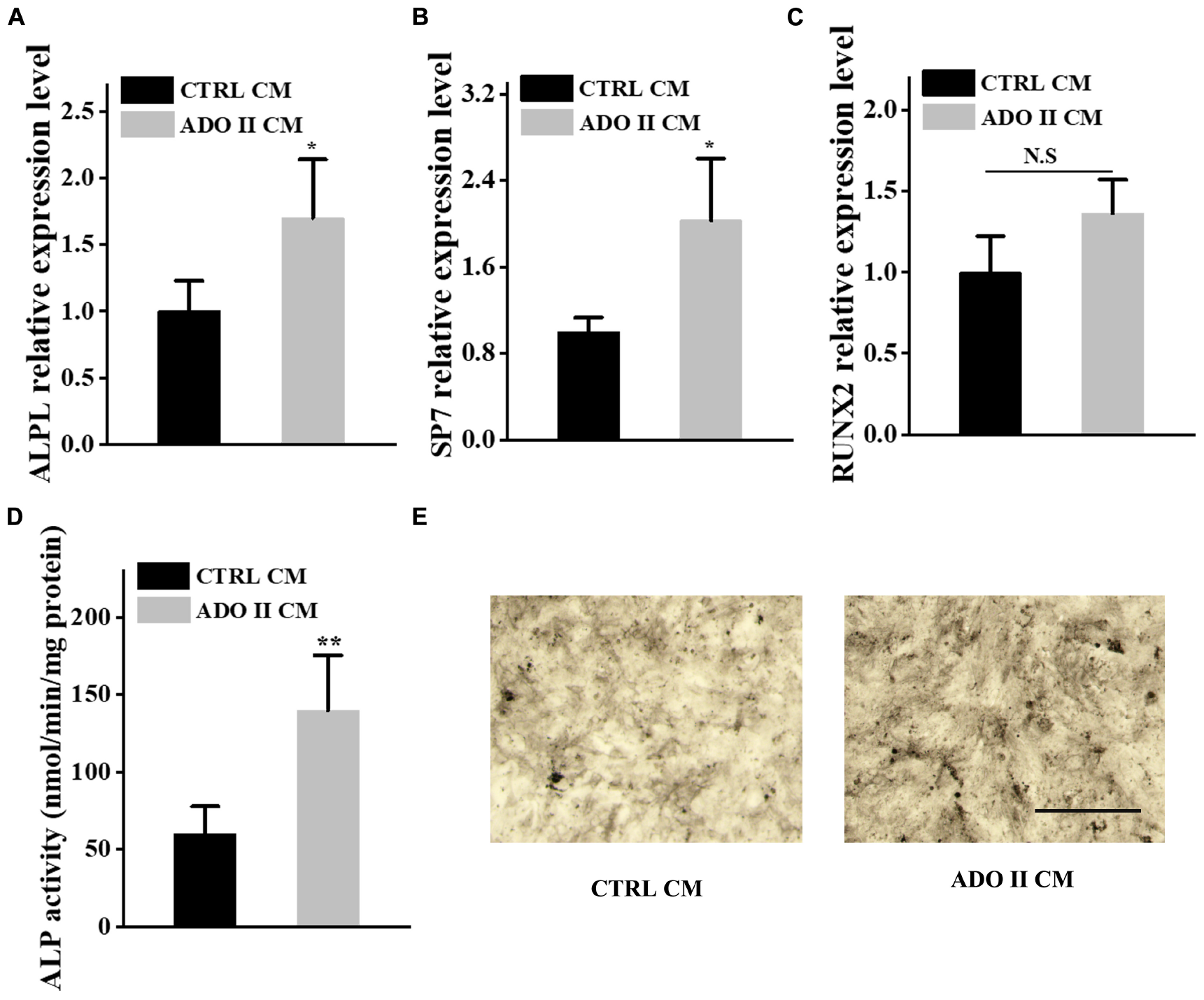
Preosteoclast-conditioned medium from ADO II patients enhances osteoblastic differentiation in vitro. BMSCs were treated with a preosteoclast-conditioned medium from ADO II patients or control groups. (A–C) qRT–PCR analysis of the relative levels of ALPL(A), SP7(B), and RUNX2(C) in BMSCs. (D) ALP activity in BMSCs. (E) Representative images of ALP staining in BMSCs. Scale bar = 300 μm. These experiments were replicated three times. Data are shown as mean ± SD. *P < 0.05 and **P < 0.01.
Discussion
Osteopetrosis is a rare genetic condition of increased bone mass and density generally due to a defect of bone resorption. The common complications are confined to the skeleton, including bone fracture, bone pain, osteomyelitis, and rare bone marrow failure (Bénichou et al., 2000). Bone pain on the back was described in three affected patients and fracture on the hip of the proband. Their prominent radiographic signatures of vertebral endplates (“rugger-jersey spine”) and diffused bone sclerosis were found in three affected patients.
The mutation analysis in this family was in agreement with the clinical diagnosis of ADO-II. Previous studies that reported several mutations in CLCN7 could result in severe recessive, dominant, and intermediate osteopetrosis (Frattini et al., 2003). Currently, genetic and molecular diagnoses were available for patients with ADO II, especially to the missense mutations in the CLCN7 gene (Bollerslev et al., 2013). Above 70% of patients harbored heterozygous dominant-negative mutations of the CLCN7 gene (Cleiren et al., 2001). These CLCN7-related mutations are usually located in different structures of its protein, such as intramembrane α helices, extracellular loops, Cl-binding sites, and CBS domains (Pang et al., 2016; Jentsch and Pusch, 2018). We identified a heterozygous variant p.Met560Val located in the loop between helix N and helix O in three patients and an unaffected carrier. There was only one allele of the identified mutation in gnomAD and was not presented in East Asian populations. Additionally, Steven et al. have estimated the penetrance of ADO II to be approximately 66% (62 clinically affected individuals/94 subjects with the gene mutation), which means among the individuals with CLCN7 mutations, one third were asymptomatic gene carriers without osteopetrosis-related signs in clinical, biochemical, and radiological results (Waguespack et al., 2003). Thus, the carrier did not present the related osteosclerotic bone associated with the incomplete penetrance. The phenotypical and genetic heterogeneity and incomplete penetrance remain unclear in skeletal dysplasia (Alam et al., 2017; Chen et al., 2020). It is likely to be related to the environmental factors, methylated gene modification, the complex interaction between other genes and CLCN7, and even some unidentified loci (Zhang et al., 2009; Leisle et al., 2011; Boudin et al., 2016), which need more further studies to elucidate.
The previous investigation indicated that ADO was one of the osteoclast-rich forms of osteopetrosis (Bollerslev et al., 2013). The bone biopsies from patients with ADO showed increased size and number of osteoclasts (Bollerslev et al., 1993). Chen et al. revealed that c.1856C > T mutation in CLCN7 identified in an ADO II case resulted in enhanced but hypofunctional osteoclastogenesis (Chen et al., 2016). Besides, previous studies showed that the serum TRAP-5b was significantly elevated in osteoclast-rich ADO II mice and patients (Chen et al., 2016; Alam et al., 2017). Consistent with these studies, the elevated serum TRAP-5b was also found in these patients with ADO II (Alatalo et al., 2004). The prominently elevated biochemical measurements of TRAP-5b suggested that the variant affected the function of osteoclast and indicated the molecular feature to be osteoclast-rich osteopetrosis in this family.
Osteopetrosis is a clinically heterogenetic disorder. In this Chinese family with the CLCN7 variant, the serum bone formation markers (OC, TPINP, and PTH) were at the upper limit range in the proband. They also elevated in the proband’s mother (II3), and the level of OC increased remarkably in the aunt (II7). The variable level of bone formation markers in the three affected patients might be associated with the characterization of clinical heterogeneity or some factors, such as age, lifestyle, and other environmental stimulus (Lundberg et al., 2018; Chevalier et al., 2020). Taken together, the upregulated or upper limit range of serum bone formation markers implied that the bone formation increased. It is extensively reported that the coupling of angiogenesis with osteogenesis plays a great role in bone modeling and remodeling (Phan et al., 2004; Ramasamy et al., 2014; Xie et al., 2014; Yang et al., 2017, 2019; Chen et al., 2018; Li et al., 2018). Previous findings revealed that PDGF-BB secreted by preosteoclasts could stimulate angiogenesis during coupling with osteogenesis (Xie et al., 2014). Furthermore, we demonstrated that Reg1cp mutation potentially caused high bone mass syndrome by regulating the coupling of angiogenesis with osteogenesis (Yang et al., 2019). Other studies revealed that the secret factors, including VEGF and SLIT3 by osteoclast lineage cells, could regulate type H formation during bone regeneration (Xu et al., 2018; Peng et al., 2020). Based on these findings, we investigated if this CLCN7 mutation contributed to the increased bone mass by regulating the process of angiogenesis and osteogenesis. Intriguingly, we found that the levels of PDGF-BB, VEGF, and SLIT3 were increased in ADO II individuals. Of note, the conditioned medium from PBMCs of ADO II patients promoted vessel formation of HMECs as well as osteogenic differentiation of hBMSCs. These results gave some new insights into the pathogenesis in this family.
In this study, we identified a novel missense variant (p.Met560Val) in CLCN7 through whole-genome sequencing and detected it in all affected family members by Sanger sequence. The rarity, conservation, and pathogenic predication all supported that this CLCN7 mutation might be a disease-causing one. Furthermore, the in vitro experiments showed that the variant promoted the vessel formation and osteogenic differentiation in this family. To conclude, we presented a novel heterozygous mutation (p.Met560Val) in CLCN7 segregated with ADO II in this Chinese family and revealed that it might be associated with the increased type H vessel formation and bone formation.
Statements
Data availability statement
The whole-genome sequencing data were deposited on the Genbank (Sequence Read Archive) database with the accession no. PRJNA669897.
Ethics statement
The studies involving human participants were reviewed and approved by Ethics Committee of the Xiangya Hospital of Central South University. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
HP designed the experiments, performed whole-genome sequencing analysis, carried out most of the experiments, and drafted the manuscript. TW and H-BH recruited the patients. TW supervised the experiments, analyzed results, and proofread the manuscript. All authors were involved in the final approval of the submitted version.
Acknowledgments
The authors would like to thank the family for their participation in this study. The authors gratefully acknowledge Prof. Stephen P. Robertson at the University of Otago, providing the platform and the method of genetic analysis.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
AbarrategiA.MianS. A.PassaroD.Rouault-PierreK.GreyW.BonnetD. (2018). Modeling the human bone marrow niche in mice: from host bone marrow engraftment to bioengineering approaches.J. Exp. Med.215729–743. 10.1084/jem.20172139
2
AlamI.McQueenA. K.ActonD.ReillyA. M.Gerard-O’RileyR. L.OakesD. K.et al (2017). Phenotypic severity of autosomal dominant osteopetrosis type II (ADO2) mice on different genetic backgrounds recapitulates the features of human disease.Bone9434–41. 10.1016/j.bone.2016.10.016
3
AlataloS. L.IvaskaK. K.WaguespackS. G.EconsM. J.VäänänenK.HalleenJ. M. (2004). Osteoclast-derived serum tartrate-resistant acid phosphatase 5b in Albers-Schonberg disease (type II autosomal dominant osteopetrosis).Clin. Chem.50883–890. 10.1373/clinchem.2003.029355
4
BarvencikF.KurthI.KoehneT.StauberT.ZustinJ.TsiakasK.et al (2014). CLCN7 and TCIRG1 mutations differentially affect bone matrix mineralization in osteopetrotic individuals.J. Bone Miner. Res.29982–991. 10.1002/jbmr.2100
5
BénichouO.CleirenE.GramJ.BollerslevJ.de VernejoulM.-C.Van HulW. (2001). Mapping of autosomal dominant osteopetrosis type II (Albers-Schönberg disease) to chromosome 16p13.3.Am. J. Hum. Genet.69647–654. 10.1086/323132
6
BénichouO. D.LaredoJ. D.de VernejoulM. C. (2000). Type II autosomal dominant osteopetrosis (Albers-Schönberg disease): clinical and radiological manifestations in 42 patients.Bone2687–93. 10.1016/S8756-3282(99)00244-6
7
BollerslevJ.HenriksenK.Frost NielsenM.BrixenK.Van HulW. (2013). Autosomal dominant osteopetrosis revisited: lessons from recent studies.Eur. J. Endocrinol.169R39–R57. 10.1530/EJE-13-0136
8
BollerslevJ.MarksS. C.Jr.PockwinseS.KassemM.BrixenK. (1993). Ultrastructural investigations of bone resorptive cells in two types of autosomal dominant osteopetrosis.Bone14865–869. 10.1016/8756-3282(93)90316-3
9
BoudinE.FijalkowskiI.HendrickxG.Van HulW. (2016). Genetic control of bone mass.Mol. Cell. Endocrinol.4323–13. 10.1016/j.mce.2015.12.021
10
CapulliM.MauriziA.VenturaL.RucciN.TetiA. (2015). Effective small interfering RNA therapy to treat CLCN7-dependent autosomal dominant osteopetrosis type 2.Mol. Ther. Nucleic Acids4:e248. 10.1038/mtna.2015.21
11
ChenX.WangZ.DuanN.ZhuG.SchwarzE. M.XieC. (2018). Osteoblast–osteoclast interactions.Connect. Tissue Res.5999–107. 10.1080/03008207.2017.1290085
12
ChenX.ZhangK.HockJ.WangC.YuX. (2016). Enhanced but hypofunctional osteoclastogenesis in an autosomal dominant osteopetrosis type II case carrying a c.1856C>T mutation in CLCN7.Bone Res.4:16035. 10.1038/boneres.2016.35
13
ChenY.-H.GrigelionieneG.NewtonP. T.GullanderJ.ElfvingM.HammarsjöA.et al (2020). Absence of GP130 cytokine receptor signaling causes extended Stüve-Wiedemann syndrome.J. Exp. Med.217:e20191306. 10.1084/jem.20191306
14
ChevalierC.KieserS.ÇolakoğluM.HadadiN.BrunJ.RigoD.et al (2020). Warmth prevents bone loss through the gut microbiota.Cell Metab.32575.e7–590.e7. 10.1016/j.cmet.2020.08.012
15
CleirenE.BénichouO.Van HulE.GramJ.BollerslevJ.SingerF. R.et al (2001). Albers-Schonberg disease (autosomal dominant osteopetrosis, type II) results from mutations in the ClCN7 chloride channel gene.Hum. Mol. Genet.102861–2867. 10.1093/hmg/10.25.2861
16
CoudertA. E.Del FattoreA.BaulardC.OlasoR.SchiltzC.ColletC.et al (2014). Differentially expressed genes in autosomal dominant osteopetrosis type II osteoclasts reveal known and novel pathways for osteoclast biology.Lab. Investig.94275–285. 10.1038/labinvest.2013.140
17
Del FattoreA.CapparielloA.TetiA. (2008). Genetics, pathogenesis and complications of osteopetrosis.Bone4219–29. 10.1016/j.bone.2007.08.029
18
FrattiniA.PangrazioA.SusaniL.SobacchiC.MiroloM.AbinunM.et al (2003). Chloride channel ClCN7 mutations are responsible for severe recessive, dominant, and intermediate osteopetrosis.J. Bone Miner. Res.181740–1747. 10.1359/jbmr.2003.18.10.1740
19
HaoX.GuH.ChenC.HuangD.ZhaoY.XieL.et al (2019). Metabolic imaging reveals a unique preference of symmetric cell division and homing of leukemia-initiating cells in an endosteal niche.Cell Metab.29950.e6–965.e6. 10.1016/j.cmet.2018.11.013
20
HayashiM.NakashimaT.YoshimuraN.OkamotoK.TanakaS.TakayanagiH. (2019). Autoregulation of osteocyte sema3a orchestrates estrogen action and counteracts bone aging.Cell Metab.29627.e5–637.e5. 10.1016/j.cmet.2018.12.021
21
JentschT. J.PuschM. (2018). CLC chloride channels and transporters: structure, function, physiology, and disease.Physiol. Rev.981493–1590. 10.1152/physrev.00047.2017
22
KimB. J.LeeY. S.LeeS. Y.BaekW. Y.ChoiY. J.MoonS. A.et al (2018). Osteoclast-secreted SLIT3 coordinates bone resorption and formation.J. Clin. Invest.1281429–1441. 10.1172/JCI91086
23
KusumbeA. P.RamasamyS. K.AdamsR. H. (2014). Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone.Nature507323–328. 10.1038/nature13145
24
LangenU. H.PitulescuM. E.KimJ. M.Enriquez-GascaR.SivarajK. K.KusumbeA. P.et al (2017). Cell-matrix signals specify bone endothelial cells during developmental osteogenesis.Nat. Cell Biol.19189–201. 10.1038/ncb3476
25
LeisleL.LudwigC. F.WagnerF. A.JentschT. J.StauberT. (2011). ClC-7 is a slowly voltage-gated 2Cl−/1H+-exchanger and requires Ostm1 for transport activity.EMBO J.302140–2152. 10.1038/emboj.2011.137
26
LiC.XiaoY.YangM.SuT.SunX.GuoQ.et al (2018). Long noncoding RNA Bmncr regulates mesenchymal stem cell fate during skeletal aging.J. Clin. Invest.1285251–5266. 10.1172/JCI99044
27
LiX.SunX.CarmelietP. (2019). Hallmarks of endothelial cell metabolism in health and disease.Cell Metab.30414–433. 10.1016/j.cmet.2019.08.011
28
LundbergJ. O.CarlströmM.WeitzbergE. (2018). Metabolic effects of dietary nitrate in health and disease.Cell Metab.289–22. 10.1016/j.cmet.2018.06.007
29
MauriziA.CapulliM.CurleA.PatelR.UcciA.CôrtesJ. A.et al (2019). Extra-skeletal manifestations in mice affected by Clcn7-dependent autosomal dominant osteopetrosis type 2 clinical and therapeutic implications.Bone Res.7:17. 10.1038/s41413-019-0055-x
30
PangQ.ChiY.ZhaoZ.XingX.LiM.WangO.et al (2016). Novel mutations of CLCN7 cause autosomal dominant osteopetrosis type II (ADO-II) and intermediate autosomal recessive osteopetrosis (IARO) in Chinese patients.Osteoporos. Int.271047–1055. 10.1007/s00198-015-3320-x
31
PengY.WuS.LiY.CraneJ. L. (2020). Type H blood vessels in bone modeling and remodeling.Theranostics10426–436. 10.7150/thno.34126
32
PhanT. C. A.XuJ.ZhengM. H. (2004). Interaction between osteoblast and osteoclast: impact in bone disease.Histol. Histopathol.191325–1344. 10.14670/HH-19.1325
33
RamasamyS. K.KusumbeA. P.WangL.AdamsR. H. (2014). Endothelial Notch activity promotes angiogenesis and osteogenesis in bone.Nature507376–380. 10.1038/nature13146
34
Rodriguez-LagunaL.AgraN.IbañezK.Oliva-MolinaG.GordoG.KhuranaN.et al (2019). Somatic activating mutations in PIK3CA cause generalized lymphatic anomaly.J. Exp. Med.216407–418. 10.1084/jem.20181353
35
SivarajK. K.AdamsR. H. (2016). Blood vessel formation and function in bone.Development1432706–2715. 10.1242/dev.136861
36
SobacchiC.SchulzA.CoxonF. P.VillaA.HelfrichM. H. (2013). Osteopetrosis: genetics, treatment and new insights into osteoclast function.Nat. Rev. Endocrinol.9522–536. 10.1038/nrendo.2013.137
37
TolarJ.TeitelbaumS. L.OrchardP. J. (2004). Osteopetrosis.N. Engl. J. Med.3512839–2849. 10.1056/NEJMra040952
38
VerkmanA. S.GaliettaL. J. V. (2009). Chloride channels as drug targets.Nat. Rev. Drug Discov.8153–171. 10.1038/nrd2780
39
WaguespackS. G.HuiS. L.DiMeglioL. A.EconsM. J. (2007). Autosomal dominant osteopetrosis: clinical severity and natural history of 94 subjects with a chloride channel 7 gene mutation.J. Clin. Endocrinol. Metab.92771–778. 10.1210/jc.2006-1986
40
WaguespackS. G.KollerD. L.WhiteK. E.FishburnT.CarnG.BuckwalterK. A.et al (2003). Chloride channel 7 (ClCN7) gene mutations and autosomal dominant osteopetrosis, type II.J. Bone Miner. Res.181513–1518. 10.1359/jbmr.2003.18.8.1513
41
WangC.HockermanS.JacobsenE. J.AlippeY.SelnessS. R.HopeH. R.et al (2018). Selective inhibition of the p38α MAPK–MK2 axis inhibits inflammatory cues including inflammasome priming signals.J. Exp. Med.2151315–1325. 10.1084/jem.20172063
42
WeinertS.JabsS.SupanchartC.SchweizerM.GimberN.RichterM.et al (2010). Lysosomal pathology and osteopetrosis upon loss of H+-driven lysosomal Cl-accumulation.Science3281401–1403. 10.1126/science.1188072
43
XiaoY.-Z.YangM.XiaoY.GuoQ.HuangY.LiC.-J.et al (2020). Reducing hypothalamic stem cell senescence protects against aging-associated physiological decline.Cell Metab.31534.e5–548.e5. 10.1016/j.cmet.2020.01.002
44
XieH.CuiZ.WangL.XiaZ.HuY.XianL.et al (2014). PDGF-BB secreted by preosteoclasts induces angiogenesis during coupling with osteogenesis.Nat. Med.201270–1278. 10.1038/nm.3668
45
XuR.YallowitzA.QinA.WuZ.ShinD. Y.KimJ.-M.et al (2018). Targeting skeletal endothelium to ameliorate bone loss.Nat. Med.24823–833. 10.1038/s41591-018-0020-z
46
YangM.GuoQ.PengH.XiaoY.-Z.XiaoY.HuangY.et al (2019). Krüppel-like factor 3 inhibition by mutated lncRNA Reg1cp results in human high bone mass syndrome.J. Exp. Med.2161944–1964. 10.1084/jem.20181554
47
YangM.LiC.-J.SunX.GuoQ.XiaoY.SuT.et al (2017). MiR-497 195 cluster regulates angiogenesis during coupling with osteogenesis by maintaining endothelial Notch and HIF-1α activity.Nat. Commun.81–11. 10.1038/ncomms16003
48
ZhangZ.HeJ.ZhangH.HuW.FuW.GuJ.et al (2009). Identification of the CLCN7 gene mutations in two Chinese families with autosomal dominant osteopetrosis (type II).J. Bone Miner. Metab.27444–451. 10.1007/s00774-009-0051-0
Summary
Keywords
autosomal dominant osteopetrosis type II, CLCN7, variant, CD31hiEmcnhi vessel formation, bone formation
Citation
Peng H, He H-B and Wen T (2020) A Novel Variant in CLCN7 Regulates the Coupling of Angiogenesis and Osteogenesis. Front. Cell Dev. Biol. 8:599826. doi: 10.3389/fcell.2020.599826
Received
28 August 2020
Accepted
20 October 2020
Published
16 November 2020
Volume
8 - 2020
Edited by
Chao Liang, Hong Kong Baptist University, Hong Kong
Reviewed by
Li Hui, Shaanxi Provincial People’s Hospital, China; Peng Cheng, Nanjing Medical University, China
Updates
Copyright
© 2020 Peng, He and Wen.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ting Wen, tingwen163@126.com
This article was submitted to Molecular Medicine, a section of the journal Frontiers in Cell and Developmental Biology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.