 Matthew Luzentales-Simpson
Matthew Luzentales-Simpson Yvonne C. F. Pang
Yvonne C. F. Pang Ada Zhang
Ada Zhang James A. Sousa
James A. Sousa Laura M. Sly
Laura M. Sly- Division of Gastroenterology, Department of Pediatrics, BC Children’s Hospital and the University of British Columbia, Vancouver, BC, Canada
Inflammatory bowel diseases (IBD), encompassing ulcerative colitis (UC), and Crohn’s disease (CD), are a group of disorders characterized by chronic, relapsing, and remitting, or progressive inflammation along the gastrointestinal tract. IBD is accompanied by massive infiltration of circulating leukocytes into the intestinal mucosa. Leukocytes such as neutrophils, monocytes, and T-cells are recruited to the affected site, exacerbating inflammation and causing tissue damage. Current treatments used to block inflammation in IBD include aminosalicylates, corticosteroids, immunosuppressants, and biologics. The first successful biologic, which revolutionized IBD treatment, targeted the pro-inflammatory cytokine, tumor necrosis factor alpha (TNFα). Infliximab, adalimumab, and other anti-TNF antibodies neutralize TNFα, preventing interactions with its receptors and reducing the inflammatory response. However, up to 40% of people with IBD become unresponsive to anti-TNFα therapy. Thus, more recent biologics have been designed to block leukocyte trafficking to the inflamed intestine by targeting integrins and adhesins. For example, natalizumab targets the α4 chain of integrin heterodimers, α4β1 and α4β7, on leukocytes. However, binding of α4β1 is associated with increased risk for developing progressive multifocal leukoencephalopathy, an often-fatal disease, and thus, it is not used to treat IBD. To target leukocyte infiltration without this life-threatening complication, vedolizumab was developed. Vedolizumab specifically targets the α4β7 integrin and was approved to treat IBD based on the presumption that it would block T-cell recruitment to the intestine. Though vedolizumab is an effective treatment for IBD, some studies suggest that it may not block T-cell recruitment to the intestine and its mechanism(s) of action remain unclear. Vedolizumab may reduce inflammation by blocking recruitment of T-cells, or pro-inflammatory monocytes and dendritic cells to the intestine, and/or vedolizumab may lead to changes in the programming of innate and acquired immune cells dampening down inflammation.
Introduction
Inflammatory bowel disease (IBD), encompassing ulcerative colitis (UC), and Crohn’s disease (CD) are a group of disorders that are characterized by chronic, relapsing, and remitting, or progressive inflammation along the gastrointestinal tract. UC involves continuous inflammation of the colon and the rectum (Fakhoury et al., 2014). It is limited to the mucosal layer of the intestinal wall. In contrast, CD causes discontinuous, transmural inflammation that can occur anywhere along the digestive tract, though it most commonly affects the distal ileum (Fakhoury et al., 2014). Canada has among the highest prevalence of IBD with 1 in 140 people affected (Kaplan et al., 2019). IBD has traditionally been regarded as a disease of developed and high-income nations, though incidence rates appear to have stabilized with high burden and prevalence. Incidence of IBD is now rapidly rising in newly industrialized countries (GBD 2017 Inflammatory Bowel Disease Collaborators, 2020). There were 6.8 million cases of IBD globally in 2017, with approximately 1.5 million in North America and 2 million in Europe (GBD 2017 Inflammatory Bowel Disease Collaborators, 2020; Jairath and Feagan, 2020). Individuals with IBD suffer from intestinal inflammation that can lead to debilitating symptoms including pain, nausea, and diarrhea (Rosen et al., 2015). In addition to disease burden, the chronic nature of IBD and its requirement for lifelong treatment causes significant economic burden for the individual and society. As analyzed by Park et al. (2020), the annual mean health care cost for people with IBD is over 3-fold higher than for people without IBD (selected from the general population using a health plan member database). Mehta (2016) estimated that extrapolated direct costs of IBD are between $11 to 28 billion in the United States. Including direct and indirect costs associated with loss of productivity and earnings, the total cost of IBD is between $14.6 and 31.6 billion annually (Mehta, 2016).
The etiology of IBD is multifactorial and includes genetic susceptibility, inappropriate immune activity, and environmental triggers. For example, a loss of function gene variant in the NOD2 gene is associated with increased susceptibility to CD due to increased production of pro-inflammatory cytokines (Ogura et al., 2001). Environmental factors affecting incidence of disease include geography, smoking, and pollution (Ananthakrishnan et al., 2018). Genetics, immune responses, and environmental variables also impact the host intestinal microbiota, which is often included as a fourth important factor in the development of IBD (Ananthakrishnan et al., 2018). While the exact cause is unknown, it is widely accepted that IBD occurs in genetically susceptible individuals with environmental influences that result in a dysregulated immune response to commensal intestinal microbiota (Rosen et al., 2015). The wide variation in disease presentation and treatment efficacy reflects the complexity of IBD pathogenesis (Chichlowski and Hale, 2008).
In this review, we will focus on the role of specific immune cells in IBD pathogenesis and how their trafficking and activity may be affected be vedolizumab. IBD is characterized by massive infiltration of circulating leukocytes into the inflamed intestinal mucosa. Diseased sections of the intestines have cytokine profiles that differ from healthy sections, indicating that cytokines play a pivotal role in the incidence and progression of disease (Murch et al., 1993; McAlindon et al., 1998). In particular, immune cells that are isolated from people with IBD display increased expression of pro-inflammatory cytokines and chemokines (Singh et al., 2016). Chemokine production leads to inappropriate recruitment and retention of cells such as T-cells, dendritic cells, and macrophages, which cause inflammation and establish the chronic inflammation that is characteristic of IBD (Singh et al., 2016). Chronic activation and proliferation of these immune cells leads to disruption of healthy tissues and thus further exacerbation of disease (Schippers et al., 2016). Macrophages in particular have a unique role in IBD due to their ability to exhibit pro-inflammatory activity that contributes to disease and Interleukin-10 (IL-10)-mediated anti-inflammatory activity (Kozicky et al., 2015). Anti-inflammatory macrophage activity has recently been described in the resolution of IBD during anti-TNFα therapies in mice and humans (Koelink et al., 2020). Specifically, macrophage IL-10 signaling was described to be the driving force behind the therapeutic effect of these therapies (Koelink et al., 2020). With respect to these studies, IBD therapies can reduce inflammation by targeting leukocyte trafficking to the intestine and pro-inflammatory activity.
There is no standard treatment for IBD. Therapeutic options are non-specific anti-inflammatories that include aminosalicylates, corticosteroids, and other immunosuppressants, and some biologics. New biological therapies have been designed to be specific to the gut to minimize side effects and increase responsiveness (Hazel and O’Connor, 2020). In particular, anti-integrin antibodies target key players in leukocyte trafficking to the gut to reduce immune cell infiltration and the resulting inflammation (Hazel and O’Connor, 2020). Vedolizumab is a therapy that was designed to reduce pathological inflammation in IBD by blocking T-cell recruitment to the intestine (Zeissig et al., 2019). However, some evidence suggests that it does not affect T-cell migration to the intestine (Zeissig et al., 2019). Herein, we describe the potential mechanisms of action by which vedolizumab may reduce pathological inflammation in IBD.
Leukocyte Trafficking and Activation in IBD Pathogenesis
Leukocyte Trafficking
Inflammatory bowel diseases is characterized by immune infiltration from the circulation into the inflamed intestinal mucosa. This migration is facilitated by complex interactions between circulating leukocytes and intestinal endothelial cells. Sialyl LewisX-modified glycoproteins on leukocytes bind selectins on the endothelial cells with low affinity, allowing leukocytes to roll along the endothelium (Wright and Cooper, 2014). Chemokines act as chemoattractants for the rolling leukocytes to promote their infiltration into the mucosa (Wright and Cooper, 2014). Integrins on leukocytes mediate adhesion by binding cellular adhesion molecules (CAMs) that are expressed on the endothelial cells during inflammation (Wright and Cooper, 2014). Integrins are heterodimeric receptors composed of an α and a β subunit, which exist in several forms that can combine to allow tissue-specific adhesion (Arseneau and Cominelli, 2015). For example, the α4β7 receptor is a marker for leukocyte trafficking to the intestine (Clahsen et al., 2015).
Three CAMs have been reported to mediate leukocyte trafficking to the inflamed intestine, contributing to IBD: intracellular adhesion molecule (ICAM)-1, mucosal addressin cellular adhesion molecule (MAdCAM)-1, and vascular cell adhesion molecule (VCAM)-1 (Arseneau and Cominelli, 2015). Pro-inflammatory cytokines that are expressed during inflammation increase ICAM-1 expression, which binds the αLβ2 receptor on leukocytes (Bendjelloul et al., 2000; Arseneau and Cominelli, 2015). MAdCAM-1, which is expressed within Peyer’s patches and intestinal lymphoid tissues, binds the α4β7 receptor on memory/effector T-cells to regulate their homing to the intestine (DeNucci et al., 2010; Arseneau and Cominelli, 2015). It is upregulated at sites of inflammation in people with IBD (Briskin et al., 1997). VCAM-1 binds the α4β1 and α4β7 receptors, which mediate leukocyte trafficking to the central nervous system and intestine, respectively, (Arseneau and Cominelli, 2015; Schippers et al., 2016). Blocking the integrin-CAM interactions in the intestine is used as a strategy for IBD therapy aimed at reducing immune cell infiltration into the inflamed intestine.
T-Cells
T-cells play a prominent role in the regulation of the inflammatory response associated with IBD. In the past, CD was thought to be a Th1-mediated disease characterized by elevated levels of the pro-inflammatory mediators interleukin-2 (IL-2), interferon gamma (IFNγ), and tumor necrosis factor alpha (TNFα; Fais et al., 1991; Breese et al., 1993; Fuss et al., 1996). Since then, additional cytokines have been implicated in the pathogenesis of CD. Neurath et al. (1995) reported that antibodies against the p40 subunit of IL-12, a cytokine which induces Th1 cell differentiation, were able to attenuate 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced colitis (Jacobson et al., 1995; Neurath et al., 1995). Th17 cells were implicated in intestinal inflammation when IL-23, which maintains and expands Th17 populations, was found to share the p40 subunit with IL-12 (Oppmann et al., 2000; Stritesky et al., 2008). Furthermore, IL-23 was also shown to be a key driver of intestinal inflammation in the Helicobacter hepaticus infection and T-cell transfer models of colitis (Hue et al., 2006). UC is characterized by higher expression of IL-5 and IL-13, but not IL-4 (Karttunnen et al., 1994; Fuss et al., 1996). IL-13 is a key effector, synergizing with TNFα to modulate the proteins in tight junction formation, thereby disrupting the epithelial barrier (Heller et al., 2005; Chen and Sundrud, 2016). Recently, Rosen et al. (2017) showed that IL-17A and IL-23 mRNA in pediatric rectal mucosal samples were increased in UC in addition to higher IL-5 and IL-13 mRNA. Thus, targeting T-cell trafficking may reduce the relative concentration of proinflammatory cytokines described to be involved in IBD. Finally, T-regulatory (Treg) cells regulate self-reactive lymphocytes by secreting inhibitory cytokines such as IL-10 and transforming growth factor-β (TGFβ; Taylor et al., 2006). By suppressing immune responses and maintaining tolerance to commensal microbes, Tregs are involved in intestinal homeostasis (Himmel et al., 2012).
Recent developments show multiple IBD susceptibility loci associated with T-cell activation and memory formation (Liu et al., 2015). Genes such as CD28 (T-cell co-stimulation), CCL20 and CCR6 (T-cell migration), NFATC1 (lymphocyte proliferation), NFKBIZ (Th17 development) associated with T-cell function demonstrate the potential for therapeutic strategies, which target different stages of T-cell involvement in IBD, such as recruitment, activation, proliferation, and retention. Genome-Wide Association Studies (GWAS) are crucial to explore potential genes associated with disease susceptibility and are further supported by literature that shows protein-level discrepancies between people with IBD and healthy control study participants. The presence of T-cells in the gut of people with IBD may be mediated via CCR6, CCL20, or the α4β7 integrin (Perez-Jeldres et al., 2019). Thus, blocking the interaction of these molecules with their respective ligands or receptors has not been accepted as the sole mechanism of T-cell trafficking. Drugs such as natalizumab, which binds α4 integrin, and vedolizumab, which targets the α4β7 heterodimer, have been developed specifically to target T-cell trafficking (Hazel and O’Connor, 2020). Therefore, it is crucial that we continue to explore the potential mechanisms of these, and other, therapies in order to improve existing therapies and to develop new ones.
Dendritic Cells
Dendritic cells (DCs) are professional antigen-presenting cells that control the innate and adaptive immune responses. In the intestine, there are two described subsets: conventional (cDCs) and plasmacytoid DCs (pDCs). Depending on their location within the epithelium, cDCs either secrete IL-10 and induce Th2 cells or secrete IL-12 and induce Th1 cells (Guan, 2019). They can be further distinguished by their expression of cell surface receptors, such as the integrin subunit CD103 (αE), which binds β7 to form the αEβ7 complex (Johansson-Lindbom et al., 2005; Clahsen et al., 2015). CD103 facilitates the retention of lymphocytes in the epithelium by binding E-cadherin (Johansson-Lindbom et al., 2005). CD103+ cDCs make up the majority of the DC population in the small intestine (Johansson-Lindbom et al., 2005; Clahsen et al., 2015). They are located in the lamina propria and intraepithelial compartment, but they can migrate to the mesenteric lymph node (MLN) to induce expression of the gut homing receptors CCR9 and α4β7 integrin on B and T-cells (Annacker et al., 2005; Schulz et al., 2009). Additionally, CD103+ cDCs can promote the development of Treg cells (Annacker et al., 2005; Clahsen et al., 2015). In contrast, CD103- (CX3CRI+) cDCs do not migrate (Schulz et al., 2009). Instead, they penetrate the epithelium to sample antigens in the lumen and present antigen to CD4+ T-cells, which differentiate into effector T-cells that secrete pro-inflammatory cytokines (Guan, 2019). Finally, pDCs are rare cells that secrete large quantities of type I interferons (Guan, 2019).
During IBD, DCs are attracted to sites of inflammation in the intestine by chemokines, such as CCL20 and MAdCAM-1 (Guan, 2019). Large numbers of activated DCs are found in the lamina propria and MLN and promote inflammation in murine models of IBD (Berndt et al., 2007; Guan, 2019). Conversely, ablation of DCs can also exacerbate disease (Muzaki et al., 2016). By regulating immune responses and tolerance to the microbiota, DCs play a critical role in IBD pathogenesis.
Macrophages
Macrophages play a role in all stages of inflammation: recognition, response, and resolution. They are highly heterogeneous, demonstrating a wide range of activation states (Kozicky et al., 2015). Cues from the microenvironment polarize macrophages to specific activation states (Kozicky et al., 2015). The three main phenotypes are grouped by function: inflammatory, wound-healing, and regulatory/anti-inflammatory, as reviewed in Steinbach and Plevy (2014). Inflammatory macrophages are promoted by IFNγ from NK and T-helper 1 (Th1) cells and TNFα from antigen presenting cells (Steinbach and Plevy, 2014). TNFα can also result from innate immune stimuli signaling that activates suppressor of cytokine signaling 3 (Steinbach and Plevy, 2014). Inflammatory macrophages produce high levels of the pro-inflammatory cytokines TNFα, IL-12, IL-6, and reactive oxygen and nitrogen species, which promote Th1 and Th17 cell activity and low levels of the anti-inflammatory cytokine, IL-10 (Hausmann et al., 2001; Heinsbroek and Gordon, 2009). Inflammatory macrophages are essential in the response to intracellular infections but can aggravate IBD due to their production of pro-inflammatory cytokines (Heinsbroek and Gordon, 2009). Wound-healing macrophages are induced by IL-4 from granulocytes or Th2 cells in response to tissue injury (Kozicky et al., 2015). They produce relatively lower levels of pro-inflammatory cytokines and higher levels of IL-10, protecting against parasites and promoting wound healing through the suppression of NLRP3 inflammasome activation, angiogenesis, tissue remodeling, and debris scavenging (Steinbach and Plevy, 2014; Yao et al., 2015). Wound-healing macrophages are protective in murine models of intestinal inflammation but may contribute to fibrosis in CD (Steinbach and Plevy, 2014). Regulatory or anti-inflammatory macrophages are a recently described phenotype that require two stimuli, one of which is pro-inflammatory (Mosser and Edwards, 2008; Kozicky et al., 2015). They can be activated by macrophage-derived TGFβ, IL-10, or immune complexes and a pro-inflammatory stimulus (Anderson et al., 2002; Mosser and Edwards, 2008; Kozicky et al., 2015). They produce high levels of IL-10 (Kozicky et al., 2015). In addition, regulatory macrophages express costimulatory molecules that activate T-cells (Steinbach and Plevy, 2014). They further differ from wound-healing macrophages in their lack of extracellular matrix production (Steinbach and Plevy, 2014). Regulatory macrophages play a key role in turning off the inflammatory response by reducing IL-12 synthesis (Kozicky et al., 2019) and are not predicted to promote fibrosis (Steinbach and Plevy, 2014).
In the lamina propria, macrophages control homeostasis by responding to infectious challenges with phagocytic and microbicidal activity while maintaining immune tolerance to commensal microbes. Differentiation into a tolerant phenotype is promoted by the presence of IL-10 and TFGβ in the microenvironment (Smythies et al., 2005). In contrast to the resident macrophages that do not mount an oxidative burst or inflammatory response, circulating blood monocytes are recruited to the sites of inflammation in the intestinal epithelium via chemokines and aggravate disease (Guan, 2019). Infiltration of these blood monocytes to local tissues is facilitated through tight α4β7-MAdCAM-1 interactions among other adhesion molecule and cadherin interactions (Berlin et al., 1993; Gorfu et al., 2009). Acute intestinal inflammation and chronic inflammation cause an influx of pro-inflammatory blood monocytes, which differentiate into inflammatory macrophages and exacerbate disease (Schippers et al., 2016). Macrophages isolated from people with IBD have increased oxidative burst activity and pro-inflammatory cytokine production (Guan, 2019). However, invading monocytes have been shown to dampen the inflammatory response through the release of IL-10 (Koelink et al., 2020). There is growing evidence that intestinal macrophages may play a critical role in the resolution of IBD, especially when activated towards regulatory or wound-healing phenotypes (Koelink et al., 2020). For this reason, macrophages, and subcellular molecules that modulate macrophages like chemokines and cytokines are potential therapeutic targets to ameliorate intestinal inflammation in IBD.
Current Treatments
The increasing disease burden of IBD reflects a need for a greater understanding of the mechanisms of IBD pathogenesis to improve existing therapies and develop new and effective therapies. IBD typically causes significant morbidity and requires lifelong medication in addition to possible dietary and lifestyle changes (Seyedian et al., 2019). There is no standard treatment regimen for individuals with IBD. Therapy relies on non-specific immune suppression to reduce symptoms, maintain remission, and prevent relapse (Hazel and O’Connor, 2020). Therapies include aminosalicylates, non-specific immunosuppressants, steroids, and biologics that target pro-inflammatory cytokines or leukocyte trafficking to the gut (Shi and Ng, 2018; Seyedian et al., 2019; Hazel and O’Connor, 2020). Surgery, which involves removal of the affected region, is an option for some people with acute, severe, refractory UC (Sica and Biancone, 2013), but not CD (Seyedian et al., 2019). Efficacy of therapy can be limited by a lack of primary response, secondary loss of response, and adverse side effects (Shi and Ng, 2018).
Anti-TNFα Biologics
Tumor necrosis factor alpha is an inflammatory cytokine that plays a prominent role in active inflammation associated with IBD (Fakhoury et al., 2014). An increase in TNFα has been shown to induce cell proliferation, differentiation, and upregulation of adhesion molecules on the endothelium to increase cell trafficking to the site of inflammation (Nawroth and Stern, 1986; Fakhoury et al., 2014). Considering its role in IBD, neutralizing TNFα has been used effectively to treat IBD (mechanisms of action reviewed by Park and Jeen (2015) and Adegbola et al. (2018)).
Anti-TNFα biologics, including golimumab, certolizumab, infliximab, and adalimumab, have revolutionized IBD treatment. However, many people with IBD are unresponsive or experience significant adverse effects; up to 40% of people with IBD are predicted to become unresponsive to anti-TNFα antibodies (Shi and Ng, 2018). More specifically, a recent meta-analysis showed secondary loss of response occurs in approximately 33% of people taking infliximab and 41% of people taking adalimumab with a median follow up of 1 year (Qiu et al., 2017). These studies highlight the need for developing new therapeutics to treat IBD.
Anti-interleukin Biologics
Ustekinumab is a monoclonal antibody which binds the p40 subunit of IL-12 and IL-23 and acts as an antagonist (Feagan et al., 2016; Sands et al., 2019). People with CD who received ustekinumab had significantly higher clinical response rates in the UNITI-1 and UNITI-2 clinical trials compared to placebo (Feagan et al., 2016). People with UC who were treated with ustekinumab had significantly higher rates of achieving and maintaining clinical remission compared to placebo (Sands et al., 2019). The FDA approved ustekinumab for treatment of moderate to severe CD in September 2016, and for treatment of moderate to severe UC in October 2019.
Anti-integrin Biologics
Natalizumab
Natalizumab is a humanized monoclonal antibody that targets the α4 chain of the α4β1 and α4β7 integrins expressed on the surface of leukocytes (Leger et al., 1997; Stuve et al., 2006; Haanstra et al., 2013). α4β1 binds VCAM-1 expressed by endothelial cells, which allows leukocytes to firmly adhere to the surface (Alon et al., 1995; Cerutti and Ridley, 2017; Chae et al., 2018). Natalizumab blocks the interaction between α4β1 and VCAM-1, which is required for leukocytes to cross the blood-brain barrier into the central nervous system (CNS; Kumar et al., 1975; Burkly et al., 1991).
Natalizumab was first used successfully for the treatment of relapsing and remitting multiple sclerosis (MS; Polman et al., 2006; Rudick et al., 2006). In January 2008, natalizumab was also approved by the United States Food and Drug Administration (FDA) for treatment of CD (Guagnozzi and Caprilli, 2008). It was the first anti-adhesion biologic used to treat IBD and established evidence for the potential efficacy of blocking leukocyte trafficking to treat IBD. However, clinical trials and market distribution of natalizumab were discontinued due to reports that two people had developed progressive multifocal leukoencephalopathy (PML; Kleinschmidt-DeMasters and Tyler, 2005; Langer-Gould et al., 2005; Van Assche et al., 2005). PML is an aggressive demyelinating disease of the central nervous system (CNS) caused by the opportunistic John Cunningham (JC) virus, for which the majority of the population (75–80%) is seropositive. The recall of natalizumab prompted the retrospective analysis of samples from a deceased person with CD who had been treated with natalizumab in a separate clinical trial (Van Assche et al., 2005). The individual’s serum and brain lesion samples were positive for JC virus, and there was a temporal relationship between natalizumab treatment and increase in viral load (Van Assche et al., 2005). Natalizumab was reapproved by the FDA and European Medicine Agencies (EMA) for the treatment of relapsing-remitting MS but it is not used for CD, due to risk of serious infections (European Medicines Agency, 2007; Ransohoff, 2007; Planas et al., 2014; Avasarala, 2015).
Vedolizumab
Vedolizumab is a humanized monoclonal antibody that was developed to reduce lymphocyte trafficking to the intestine by specifically targeting the α4β7 heterodimer, which is expressed on the surface of gut-specific lymphocytes (Soler et al., 2009; Feagan et al., 2013). In contrast to natalizumab, vedolizumab does not interfere with lymphocyte trafficking to the brain (Feagan et al., 2013).
Three Phase 3 clinical trials evaluated the efficacy and safety of vedolizumab for the induction and maintenance of clinical response and remission in people with moderate to severe UC (GEMINI 1) and CD (GEMINI 2 and GEMINI 3; Feagan et al., 2013; Sandborn et al., 2013; Sands et al., 2014). In the GEMINI 1 trial, 47.1% of people with UC had clinical responses by week 6 compared to 25.5% on placebo (Feagan et al., 2013). For the maintenance arm, 41.8% of people with UC who were treated with vedolizumab every 8 weeks and 44.8% of people with UC who were treated every 4 weeks maintained clinical remission at week 52 of the trial compared to only 15.9% of people with UC on placebo (Feagan et al., 2013). In the GEMINI 2 trial, 14.5% of people with CD who were treated with vedolizumab achieved clinical remission by week 6 compared to 6.8% on placebo (Sandborn et al., 2013). For the maintenance arm, 39.0% of people with CD who received vedolizumab every 8 weeks and 36.4% who received vedolizumab every 4 weeks were in clinical remission at 52 weeks, compared to 21.6% on placebo (Sandborn et al., 2013). In the GEMINI 3 trial, vedolizumab was subsequently shown to be effective for people who have moderate to severe CD and are refractory to TNF antagonists, but induction of remission required 10 weeks of treatment (Sands et al., 2014). Based on this, in May 2014, the FDA and European Medicines Agency (EMA) approved vedolizumab for the treatment of UC and CD (Raine, 2014).
Vedolizumab: Potential Mechanisms of Action
Vedolizumab was designed to reduce intestinal inflammation by interfering with the T-cell trafficking to the intestines (Picarella et al., 1997; Feagan et al., 2013). As mentioned earlier, α4β7 integrin is a receptor expressed on lymphocytes that recognizes MAdCAM-1 (Berlin et al., 1993). MAdCAM-1 is constitutively expressed on venular endothelium and upregulated during inflammation (Briskin et al., 1997). Though already approved by the FDA for treatment of UC and CD, the molecular mechanisms of vedolizumab in humans have not been elucidated and still require further study.
In a study by Zeissig et al. (2019), T-cell trafficking to the intestinal lamina propria in people with CD and UC, who were treated with vedolizumab, was not reduced. Vedolizumab treatment did not affect the intestinal T-cell receptor repertoire or the relative abundance of lamina propria CD4+ T-cells, CD8+ T-cells, central memory T-cells, or effector memory T-cells. However, in vitro and in vivo models of intestinal inflammation have been used to investigate the potential effects of vedolizumab on T-cell migration to the intestine (Fischer et al., 2016; Binder et al., 2018). An inventive in vitro model of blood flow using glass tubes and a peristaltic pump to mimic blood flow through capillaries was highlighted by Binder et al. (2018). The model allowed for researchers to study the effects of vedolizumab treatment on integrin adhesion properties and expression in various T-cell subsets in a distinct, controlled environment (Binder et al., 2018). The group reported that vedolizumab treatment reduced the adhesion of CD4+ and CD8+ T-cells to MAdCAM-1 (Binder et al., 2018). Fischer et al. (2016) injected human T-cells or PBMCs into the ileocolic artery of mice lacking murine lymphocytes and NK cells. They demonstrated that vedolizumab specifically restricts the migration of Tregs from people with UC in this model but does not affect the migration of effector T-cells (Fischer et al., 2016). In contrast, Lord et al. (2018) reported differences in α4β7 expression on circulating lymphocytes, postulating that vedolizumab may preferentially block the recruitment of pro-inflammatory cells to the intestine. Moreover, because Tregs express less α4β7 than effector cells, they may be less affected by vedolizumab and successfully recruited to inflammatory sites where they suppress local inflammation (Lord et al., 2018). Ex vivo work with blood and colonic biopsies from people with IBD, published by Rath et al. (2018), suggests that vedolizumab treatment reduces α4β7 integrin expression on B cells, NK cells, Th1, Th2, and Th17 CD4+ T-cell subsets. In addition, vedolizumab therapy-induced clinical remission was associated with a reduction of α4β7 expression on Th2 and Th17 mucosal CD4+ T-cells, which together could reduce recruitment of pro-inflammatory cells to the gut mucosa. Moreover, Rath et al. (2018) suggested that higher α4β7 expression on T-cells before vedolizumab treatment was associated with clinical remission. Together these studies suggest that vedolizumab may act via selective inhibition of specific T-cell subtypes migrating to the gut.
Clahsen et al. (2015) reported the CD103+ (a subunit of the αEβ7 integrin) subpopulation of cDCs is reduced in MAdCAM-1 deficient mice. CD103+ cDCs play a role in inducing the expression of α4β7 integrin on T-cells and promoting Treg cell development. This may explain the observation made by Fischer et al. (2016) that Treg cell migration was reduced by vedolizumab. If vedolizumab treatment similarly results in fewer CD103+ cDCs, α4β7 expression would be reduced, limiting T-cell recruitment. Fuchs et al. (2017) found an association of αEβ7 expression on effector T-cells with worsened clinical parameters. They propose that αEβ7 upregulation may be an alternative pathway for lymphocytes beyond the α4β7-MAdCAM-1 axis (Fuchs et al., 2017). Furthermore, Zundler et al. (2017) reported that αEβ7 expression was increased on CD8+ T-cells following vedolizumab treatment in people with IBD, suggesting that lymphocytes may use an alternative integrin to ensure their localization in the intestine despite vedolizumab-mediated inhibition of α4β7. Together, this suggests that vedolizumab may indeed cause changes in T-cell recruitment. Though results of these studies suggest that vedolizumab restricts T-cell recruitment via integrin binding, there are limitations in the applicability of in vitro and in vivo animal models when studying vedolizumab, and additional work is required to translate these findings for people with IBD.
Alternative mechanisms of action in blocking monocyte and dendritic cell recruitment have been proposed (Figure 1). Zeissig et al. (2019) investigated the migration of leukocytes to the intestines by staining and labeling peripheral blood leukocytes with Indium-111 and fluorescein for scintigraphy and endomicroscopy. Imaging showed the accumulation of leukocytes in the intestines was not affected by vedolizumab. However, there was a strong association between vedolizumab treatment and the downregulation of genes involved in the innate immune system in the sigmoid colon. Genes that regulate innate effectors, innate immune receptors, chemokines, and chemokine receptors were downregulated in people with UC and CD who achieved clinical remission with vedolizumab. Interestingly, the relative abundance of inflammatory and wound-healing macrophages was also affected by vedolizumab treatment, skewing macrophages toward a healing phenotype (Figure 1A).
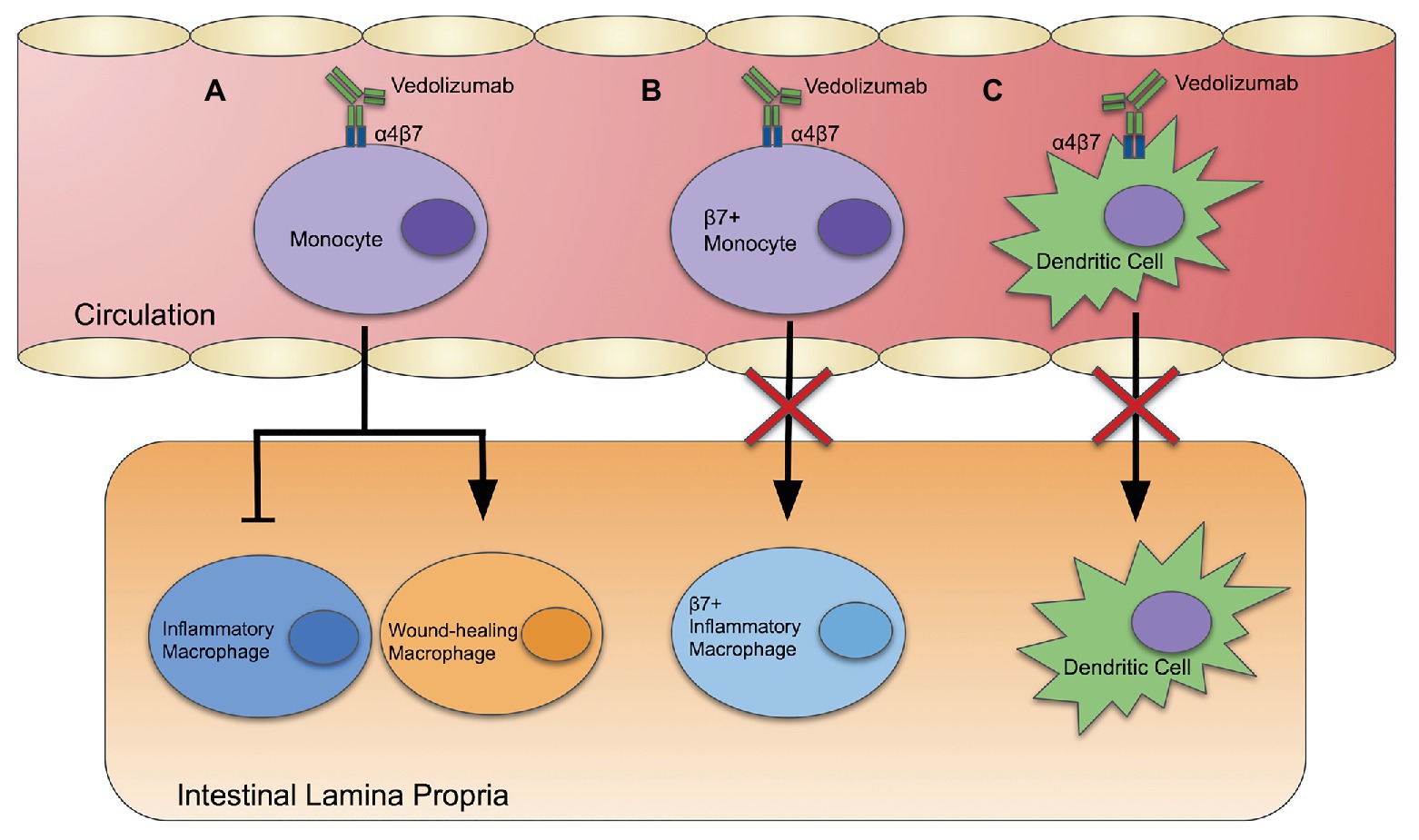
Figure 1. Vedolizumab: three potential mechanisms of action. (A) Vedolizumab binds α4β7 integrin, which alters gene expression of blood monocytes, skewing the population toward a wound-healing phenotype, and away from an inflammatory phenotype. (B) Vedolizumab binds to α4β7 integrin on blood monocytes, thereby inhibiting their ability to enter the intestinal epithelium. (C) Vedolizumab blocks localization of cDC and pDCs in the intestinal epithelium by binding α4β7.
Conversely, vedolizumab treatment has also been reported to interfere with homing of non-classical monocytes, which skew toward wound-healing macrophages (Olingy et al., 2017; Schleier et al., 2020). Approximately 5% of non-classical monocytes express α4β7 integrin, so it has been suggested that vedolizumab may actually disrupt intestinal wound healing and lead to complications (Schleier et al., 2020). Despite that, Danese et al. (2019) found that people with moderate to severe CD who were treated with vedolizumab presented with endoscopic and histological healing at both 26 and 52 weeks. Furthermore, Shen et al. (2019) analyzed data from the GEMINI 1, GEMINI 2, GEMINI Long Term Safety studies, and Vedolizumab Global Safety Database, reporting only a minor difference in the frequency of postoperative complications after intestinal surgery in people treated with vedolizumab compared to placebo. Together, this suggests that the beneficial anti-inflammatory effects of vedolizumab may override concerns about a lack of recruitment of non-classical monocytes, compromised wound healing, and downstream complications.
Schippers et al. (2016) suggested that the therapeutic efficacy of vedolizumab is linked to changes in the innate immune system rather than the adaptive immune system. They reported that the β7-integrin chain leads to recruitment of more inflammatory monocytes to the colon in the dextran sodium sulfate (DSS) model of IBD. In this model, DSS is administered in the drinking water of mice to disrupt the intestinal epithelial layer. The compromised intestinal barrier allows the luminal microbes to interact with underlying immune cells, leading to colitis that models human UC. β7-integrin deficient mice had a lower disease activity index (DAI; an additive score based on weight loss, stool consistency, and fecal blood). Colonic mRNA expression of inducible nitric oxide synthase (iNOS), proinflammatory cytokines, such as IL-6 and TNFα, and the chemokine CCL2 was lower in β7-integrin deficient mice than wild-type mice. RAG2 deficient mice, which lack mature T and B cells (Shinkai et al., 1992), were more susceptible to DSS-induced colitis than their wild-type counterparts. β7-integrin deficiency protected DSS-treated RAG2 deficient mice and mice double deficient in β7 and RAG2 in DAI and histopathology (Schippers et al., 2016). This suggests that an anti-integrin therapy, like vedolizumab, may block the recruitment of proinflammatory monocytes to the site of inflammation (Figure 1B).
Mucosal addressin cellular adhesion molecule primarily interacts with the α4β7 integrin to mediate lymphocyte homing, including effector and memory T-cells. Clahsen et al. (2015) propose a novel role for MAdCAM-1 in mediating intestinal localization of DCs to the intestinal epithelium. They found that MAdCAM-1 deficient mice and β7-integrin deficient mice had lower numbers of cDCs and pDCs in the intestinal epithelium compared to wild-type mice. They propose that MAdCAM-1 may mediate localization of cDCs and pDCs into the gut via the α4β7 integrin. Thus, vedolizumab may work, in part, by blocking recruitment of DCs that promote inflammation (Figure 1C).
Future Directions
A long-term goal including a system wide analysis of the effect of vedolizumab on α4β7- and αEβ7-mediated trafficking, myeloid and T-cell populations in the intestine, and clinical outcomes would be of tremendous value in this field. Additionally, the possibility that selective T-cell trafficking is blocked by vedolizumab should continue to elucidate the effect of less abundant T-cell populations, like Tregs, and the important ratio of Tregs/effector T-cells in the gut after vedolizumab treatment. Vedolizumab may also work in part by blocking pro-inflammatory monocyte and dendritic cell recruitment. Further studies on the properties of leukocyte populations in relation to vedolizumab treatment for people with IBD should be conducted. Additional studies using data derived from people with IBD being treated with vedolizumab are essential to translate potential mechanisms elucidated in vitro and in murine models to people with IBD.
Conclusion
In this paper, we reviewed multiple proposed mechanisms of action for vedolizumab, a relatively new biologic used to treat IBD. It was specifically developed to block T-cell trafficking to the gut, but recent evidence suggests that this may not be its sole mechanism of action. Three alternative mechanisms of action have been reported. Zeissig et al. (2019) examined a mechanism where vedolizumab intervention led to the downregulation of inflammatory gene expression in innate immune cells like monocytes. Clahsen et al. (2015) described a mechanism in which the MAdCAM-1 interaction with α4β7 integrin of pDCs and cDCs is necessary for migration into the intestinal epithelium, where these dendritic cells have been known to exhibit pro-inflammatory phenotypes. Lastly, Schippers et al. (2016) demonstrated a mechanism whereby vedolizumab restricts the recruitment of β7+ effector monocytes to the intestinal epithelium, thereby limiting the inflammatory response. Understanding the mechanism(s) of action of vedolizumab may enable us to improve the efficacy of current treatment and the develop new therapeutic strategies to treat IBD.
Author Contributions
This concept for this work was created by LMS with contributions from ML-S, YP, and AZ. ML-S, YP, and AZ contributed equally to manuscript preparation. Figures were prepared by JS with contributions from ML-S, YP, AZ, and LMS. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by an operating grant from the Canadian Institutes of Health Research (PJT-166100) awarded to LMS.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
LMS is a Biomedical Scholar of the Michael Smith Foundation for Health Research. AZ is supported by a Canada Graduate Scholarship – Master’s program.
References
Adegbola, S. O., Sahnan, K., Warusavitarne, J., Hart, A., and Tozer, P. (2018). Anti-TNF therapy in Crohn’s disease. Int. J. Mol. Sci. 19:2244. doi: 10.3390/ijms19082244
Alon, R., Kassner, P. D., Carr, M. W., Finger, E. B., Hemler, M. E., and Springer, T. A. (1995). The integrin VLA-4 supports tethering and rolling in flow on VCAM-1. J. Cell Biol. 128, 1243–1253. doi: 10.1083/jcb.128.6.1243
Ananthakrishnan, A. N., Bernstein, C. N., Iliopoulos, D., Macpherson, A., Neurath, M. F., Ali, R. A. R., et al. (2018). Environmental triggers in IBD: a review of progress and evidence. Nat. Rev. Gastroenterol. Hepatol. 15, 39–49. doi: 10.1038/nrgastro.2017.136
Anderson, C. F., Gerber, J. S., and Mosser, D. M. (2002). Modulating macrophage function with IgG immune complexes. J. Endotoxin Res. 8, 477–481. doi: 10.1179/096805102125001118
Annacker, O., Coombes, J. L., Malmstrom, V., Uhlig, H. H., Bourne, T., Johansson-Lindbom, B., et al. (2005). Essential role for CD103 in the T cell-mediated regulation of experimental colitis. J. Exp. Med. 202, 1051–1061. doi: 10.1084/jem.20040662
Arseneau, K. O., and Cominelli, F. (2015). Targeting leukocyte trafficking for the treatment of inflammatory bowel disease. Clin. Pharmacol. Ther. 97, 22–28. doi: 10.1002/cpt.6
Avasarala, J. (2015). The TOUCH program and natalizumab: fundamental flaw in patient protection. F1000Res. 4:1450. doi: 10.12688/f1000research.7513.3
Bendjelloul, F., Maly, P., Mandys, V., Jirkovska, M., Prokesova, L., Tuckova, L., et al. (2000). Intercellular adhesion molecule-1 (ICAM-1) deficiency protects mice against severe forms of experimentally induced colitis. Clin. Exp. Immunol. 119, 57–63. doi: 10.1046/j.1365-2249.2000.01090.x
Berlin, C., Berg, E. L., Briskin, M. J., Andrew, D. P., Kilshaw, P. J., Holzmann, B., et al. (1993). Alpha 4 beta 7 integrin mediates lymphocyte binding to the mucosal vascular addressin MAdCAM-1. Cell 74, 185–195. doi: 10.1016/0092-8674(93)90305-a
Berndt, B. E., Zhang, M., Chen, G. H., Huffnagle, G. B., and Kao, J. Y. (2007). The role of dendritic cells in the development of acute dextran sulfate sodium colitis. J. Immunol. 179, 6255–6262. doi: 10.4049/jimmunol.179.9.6255
Binder, M. T., Becker, E., Wiendl, M., Schleier, L., Fuchs, F., Leppkes, M., et al. (2018). Similar inhibition of dynamic adhesion of lymphocytes from IBD patients to MAdCAM-1 by vedolizumab and etrolizumab-s. Inflamm. Bowel Dis. 24, 1237–1250. doi: 10.1093/ibd/izy077
Breese, E., Braegger, C. P., Corrigan, C. J., Walker-Smith, J. A., and MacDonald, T. T. (1993). Interleukin-2- and interferon-gamma-secreting T cells in normal and diseased human intestinal mucosa. Immunology 78, 127–131.
Briskin, M., Winsor-Hines, D., Shyjan, A., Cochran, N., Bloom, S., Wilson, J., et al. (1997). Human mucosal addressin cell adhesion molecule-1 is preferentially expressed in intestinal tract and associated lymphoid tissue. Am. J. Pathol. 151, 97–110.
Burkly, L. C., Jakubowski, A., Newman, B. M., Rosa, M. D., Chi-Rosso, G., and Lobb, R. R. (1991). Signaling by vascular cell adhesion molecule-1 (VCAM-1) through VLA-4 promotes CD3-dependent T cell proliferation. Eur. J. Immunol. 21, 2871–2875. doi: 10.1002/eji.1830211132
Cerutti, C., and Ridley, A. J. (2017). Endothelial cell-cell adhesion and signaling. Exp. Cell Res. 358, 31–38. doi: 10.1016/j.yexcr.2017.06.003
Chae, Y. K., Choi, W. M., Bae, W. H., Anker, J., Davis, A. A., Agte, S., et al. (2018). Overexpression of adhesion molecules and barrier molecules is associated with differential infiltration of immune cells in non-small cell lung cancer. Sci. Rep. 8:1023. doi: 10.1038/s41598-018-19454-3
Chen, M. L., and Sundrud, M. S. (2016). Cytokine networks and T-Cell subsets in inflammatory bowel diseases. Inflamm Bowel Dis. 22, 1157–1167. doi: 10.1097/MIB.0000000000000714
Chichlowski, M., and Hale, L. P. (2008). Bacterial-mucosal interactions in inflammatory bowel disease: an alliance gone bad. Am. J. Physiol. Gastrointest. Liver Physiol. 295, G1139–G1149. doi: 10.1152/ajpgi.90516.2008
Clahsen, T., Pabst, O., Tenbrock, K., Schippers, A., and Wagner, N. (2015). Localization of dendritic cells in the gut epithelium requires MAdCAM-1. Clin. Immunol. 156, 74–84. doi: 10.1016/j.clim.2014.11.005
Danese, S., Sandborn, W. J., Colombel, J. F., Vermeire, S., Glover, S. C., Rimola, J., et al. (2019). Endoscopic, radiologic, and histologic healing with vedolizumab in patients with active Crohn’s disease. Gastroenterology 157, 1007.e1007–1018.e1007. doi: 10.1053/j.gastro.2019.06.038
DeNucci, C. C., Pagan, A. J., Mitchell, J. S., and Shimizu, Y. (2010). Control of alpha4beta7 integrin expression and CD4 T cell homing by the beta1 integrin subunit. J. Immunol. 184, 2458–2467. doi: 10.4049/jimmunol.0902407
European Medicines Agency. (2007). Questions and answers on the recommendation for the refusal of the marketing authorization for Natalizumab Elan Pharma. Available at: https://www.ema.europa.eu/en/documents/smop-initial/questions-answers-recommendation-refusal-marketing-authorization-natalizumab-elan-pharma_en.pdf (Accessed August 15, 2020).
Fais, S., Capobianchi, M. R., Pallone, F., Di Marco, P., Boirivant, M., Dianzani, F., et al. (1991). Spontaneous release of interferon gamma by intestinal lamina propria lymphocytes in Crohn’s disease. Kinetics of in vitro response to interferon gamma inducers. Gut 32, 403–407. doi: 10.1136/gut.32.4.403
Fakhoury, M., Negrulj, R., Mooranian, A., and Al-Salami, H. (2014). Inflammatory bowel disease: clinical aspects and treatments. J. Inflamm. Res. 7, 113–120. doi: 10.2147/JIR.S65979
Feagan, B. G., Rutgeerts, P., Sands, B. E., Hanauer, S., Colombel, J. F., Sandborn, W. J., et al. (2013). Vedolizumab as induction and maintenance therapy for ulcerative colitis. N. Engl. J. Med. 369, 699–710. doi: 10.1056/NEJMoa1215734
Feagan, B. G., Sandborn, W. J., Gasink, C., Jacobstein, D., Lang, Y., Friedman, J. R., et al. (2016). Ustekinumab as induction and maintenance therapy for Crohn’s disease. N. Engl. J. Med. 375, 1946–1960. doi: 10.1056/NEJMoa1602773
Fischer, A., Zundler, S., Atreya, R., Rath, T., Voskens, C., Hirschmann, S., et al. (2016). Differential effects of alpha4beta7 and GPR15 on homing of effector and regulatory T cells from patients with UC to the inflamed gut in vivo. Gut 65, 1642–1664. doi: 10.1136/gutjnl-2015-310022
Fuchs, F., Schillinger, D., Atreya, R., Hirschmann, S., Fischer, S., Neufert, C., et al. (2017). Clinical response to vedolizumab in ulcerative colitis patients is associated with changes in integrin expression profiles. Front. Immunol. 8:764. doi: 10.3389/fimmu.2017.00764
Fuss, I. J., Neurath, M., Boirivant, M., Klein, J. S., de la Motte, C., Strong, S. A., et al. (1996). Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn’s disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J. Immunol. 157, 1261–1270.
GBD 2017 Inflammatory Bowel Disease Collaborators (2020). The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990-2017: a systematic analysis for the global burden of disease study 2017. Lancet Gastroenterol. Hepatol. 5, 17–30. doi: 10.1016/S2468-1253(19)30333-4
Gorfu, G., Rivera-Nieves, J., and Ley, K. (2009). Role of beta7 integrins in intestinal lymphocyte homing and retention. Curr. Mol. Med. 9, 836–850. doi: 10.2174/156652409789105525
Guagnozzi, D., and Caprilli, R. (2008). Natalizumab in the treatment of Crohn’s disease. Biologics 2, 275–284.
Guan, Q. (2019). A comprehensive review and update on the pathogenesis of inflammatory bowel disease. J. Immunol. Res. 2019:7247238. doi: 10.1155/2019/7247238
Haanstra, K. G., Hofman, S. O., Lopes Estevao, D. M., Blezer, E. L., Bauer, J., Yang, L. L., et al. (2013). Antagonizing the alpha4beta1 integrin, but not alpha4beta7, inhibits leukocytic infiltration of the central nervous system in rhesus monkey experimental autoimmune encephalomyelitis. J. Immunol. 190, 1961–1973. doi: 10.4049/jimmunol.1202490
Hausmann, M., Spottl, T., Andus, T., Rothe, G., Falk, W., Scholmerich, J., et al. (2001). Subtractive screening reveals up-regulation of NADPH oxidase expression in Crohn’s disease intestinal macrophages. Clin. Exp. Immunol. 125, 48–55. doi: 10.1046/j.1365-2249.2001.01567.x
Hazel, K., and O’Connor, A. (2020). Emerging treatments for inflammatory bowel disease. Ther. Adv. Chronic Dis. 11:2040622319899297. doi: 10.1177/2040622319899297
Heinsbroek, S. E., and Gordon, S. (2009). The role of macrophages in inflammatory bowel diseases. Expert Rev. Mol. Med. 11:e14. doi: 10.1017/S1462399409001069
Heller, F., Florian, P., Bojarski, C., Richter, J., Christ, M., Hillenbrand, B., et al. (2005). Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology 129, 550–564. doi: 10.1016/j.gastro.2005.05.002
Himmel, M. E., Yao, Y., Orban, P. C., Steiner, T. S., and Levings, M. K. (2012). Regulatory T-cell therapy for inflammatory bowel disease: more questions than answers. Immunology 136, 115–122. doi: 10.1111/j.1365-2567.2012.03572.x
Hue, S., Ahern, P., Buonocore, S., Kullberg, M. C., Cua, D. J., McKenzie, B. S., et al. (2006). Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J. Exp. Med. 203, 2473–2483. doi: 10.1084/jem.20061099
Jacobson, N. G., Szabo, S. J., Weber-Nordt, R. M., Zhong, Z., Schreiber, R. D., Darnell, J. E. Jr., et al. (1995). Interleukin 12 signaling in T helper type 1 (Th1) cells involves tyrosine phosphorylation of signal transducer and activator of transcription (stat)3 and Stat4. J. Exp. Med. 181, 1755–1762. doi: 10.1084/jem.181.5.1755
Jairath, V., and Feagan, B. G. (2020). Global burden of inflammatory bowel disease. Lancet Gastroenterol. Hepatol. 5, 2–3. doi: 10.1016/S2468-1253(19)30358-9
Johansson-Lindbom, B., Svensson, M., Pabst, O., Palmqvist, C., Marquez, G., Forster, R., et al. (2005). Functional specialization of gut CD103+ dendritic cells in the regulation of tissue-selective T cell homing. J. Exp. Med. 202, 1063–1073. doi: 10.1084/jem.20051100
Kaplan, G. G., Bernstein, C. N., Coward, S., Bitton, A., Murthy, S. K., Nguyen, G. C., et al. (2019). The impact of inflammatory bowel disease in Canada 2018: epidemiology. J. Can. Assoc. Gastroenterol. 2(Suppl. 1), S6–S16. doi: 10.1093/jcag/gwy054
Karttunnen, R., Breese, E. J., Walker-Smith, J. A., and MacDonald, T. T. (1994). Decreased mucosal interleukin-4 (IL-4) production in gut inflammation. J. Clin. Pathol. 47, 1015–1018. doi: 10.1136/jcp.47.11.1015
Kleinschmidt-DeMasters, B. K., and Tyler, K. L. (2005). Progressive multifocal leukoencephalopathy complicating treatment with natalizumab and interferon beta-1a for multiple sclerosis. N. Engl. J. Med. 353, 369–374. doi: 10.1056/NEJMoa051782
Koelink, P. J., Bloemendaal, F. M., Li, B., Westera, L., Vogels, E. W. M., van Roest, M., et al. (2020). Anti-TNF therapy in IBD exerts its therapeutic effect through macrophage IL-10 signalling. Gut 69, 1053–1063. doi: 10.1136/gutjnl-2019-318264
Kozicky, L. K., Menzies, S. C., Hotte, N., Madsen, K. L., and Sly, L. M. (2019). Intravenous immunoglobulin (IVIg) or IVIg-treated macrophages reduce DSS-induced colitis by inducing macrophage IL-10 production. Eur. J. Immunol. 49, 1251–1268. doi: 10.1002/eji.201848014
Kozicky, L. K., Zhao, Z. Y., Menzies, S. C., Fidanza, M., Reid, G. S., Wilhelmsen, K., et al. (2015). Intravenous immunoglobulin skews macrophages to an anti-inflammatory, IL-10-producing activation state. J. Leukoc. Biol. 98, 983–994. doi: 10.1189/jlb.3VMA0315-078R
Kumar, A. P., Wrenn, E. L. Jr., Fleming, I. D., Hustu, H. O., Pratt, C. B., and Pinkel, D. (1975). Preoperative therapy for unresectable malignant tumors in children. J. Pediatr. Surg. 10, 657–670. doi: 10.1016/0022-3468(75)90369-3
Langer-Gould, A., Atlas, S. W., Green, A. J., Bollen, A. W., and Pelletier, D. (2005). Progressive multifocal leukoencephalopathy in a patient treated with natalizumab. N. Engl. J. Med. 353, 375–381. doi: 10.1056/NEJMoa051847
Leger, O. J., Yednock, T. A., Tanner, L., Horner, H. C., Hines, D. K., Keen, S., et al. (1997). Humanization of a mouse antibody against human alpha-4 integrin: a potential therapeutic for the treatment of multiple sclerosis. Hum. Antibodies 8, 3–16.
Liu, J. Z., van Sommeren, S., Huang, H., Ng, S. C., Alberts, R., Takahashi, A., et al. (2015). Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 47, 979–986. doi: 10.1038/ng.3359
Lord, J. D., Long, S. A., Shows, D. M., Thorpe, J., Schwedhelm, K., Chen, J., et al. (2018). Circulating integrin alpha4/beta7+ lymphocytes targeted by vedolizumab have a pro-inflammatory phenotype. Clin. Immunol. 193, 24–32. doi: 10.1016/j.clim.2018.05.006
McAlindon, M. E., Hawkey, C. J., and Mahida, Y. R. (1998). Expression of interleukin 1 beta and interleukin 1 beta converting enzyme by intestinal macrophages in health and inflammatory bowel disease. Gut 42, 214–219. doi: 10.1136/gut.42.2.214
Mehta, F. (2016). Report: economic implications of inflammatory bowel disease and its management. Am. J. Manag. Care 22(Suppl. 3), s51–s60.
Mosser, D. M., and Edwards, J. P. (2008). Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969. doi: 10.1038/nri2448
Murch, S. H., Braegger, C. P., Walker-Smith, J. A., and MacDonald, T. T. (1993). Location of tumour necrosis factor alpha by immunohistochemistry in chronic inflammatory bowel disease. Gut 34, 1705–1709. doi: 10.1136/gut.34.12.1705
Muzaki, A. R., Tetlak, P., Sheng, J., Loh, S. C., Setiagani, Y. A., Poidinger, M., et al. (2016). Intestinal CD103(+)CD11b(−) dendritic cells restrain colitis via IFN-gamma-induced anti-inflammatory response in epithelial cells. Mucosal Immunol. 9, 336–351. doi: 10.1038/mi.2015.64
Nawroth, P. P., and Stern, D. M. (1986). Modulation of endothelial cell hemostatic properties by tumor necrosis factor. J. Exp. Med. 163, 740–745. doi: 10.1084/jem.163.3.740
Neurath, M. F., Fuss, I., Kelsall, B. L., Stuber, E., and Strober, W. (1995). Antibodies to interleukin 12 abrogate established experimental colitis in mice. J. Exp. Med. 182, 1281–1290. doi: 10.1084/jem.182.5.1281
Ogura, Y., Bonen, D. K., Inohara, N., Nicolae, D. L., Chen, F. F., Ramos, R., et al. (2001). A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 411, 603–606. doi: 10.1038/35079114
Olingy, C. E., San Emeterio, C. L., Ogle, M. E., Krieger, J. R., Bruce, A. C., Pfau, D. D., et al. (2017). Non-classical monocytes are biased progenitors of wound healing macrophages during soft tissue injury. Sci. Rep. 7:447. doi: 10.1038/s41598-017-00477-1
Oppmann, B., Lesley, R., Blom, B., Timans, J. C., Xu, Y., Hunte, B., et al. (2000). Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 13, 715–725. doi: 10.1016/s1074-7613(00)00070-4
Park, K. T., Ehrlich, O. G., Allen, J. I., Meadows, P., Szigethy, E. M., Henrichsen, K., et al. (2020). The cost of inflammatory bowel disease: an initiative from the Crohn’s & colitis foundation. Inflamm. Bowel Dis. 26, 1–10. doi: 10.1093/ibd/izz104
Park, S. C., and Jeen, Y. T. (2015). Current and emerging biologics for ulcerative colitis. Gut Liver 9, 18–27. doi: 10.5009/gnl14226
Perez-Jeldres, T., Tyler, C. J., Boyer, J. D., Karuppuchamy, T., Yarur, A., Giles, D. A., et al. (2019). Targeting cytokine signaling and lymphocyte traffic via small molecules in inflammatory bowel disease: JAK inhibitors and S1PR agonists. Front. Pharmacol. 10:212. doi: 10.3389/fphar.2019.00212
Picarella, D., Hurlbut, P., Rottman, J., Shi, X., Butcher, E., and Ringler, D. J. (1997). Monoclonal antibodies specific for beta 7 integrin and mucosal addressin cell adhesion molecule-1 (MAdCAM-1) reduce inflammation in the colon of scid mice reconstituted with CD45RBhigh CD4+ T cells. J. Immunol. 158, 2099–2106.
Planas, R., Martin, R., and Sospedra, M. (2014). Long-term safety and efficacy of natalizumab in relapsing-remitting multiple sclerosis: impact on quality of life. Patient Relat. Outcome Meas. 5, 25–33. doi: 10.2147/PROM.S41768
Polman, C. H., O’Connor, P. W., Havrdova, E., Hutchinson, M., Kappos, L., Miller, D. H., et al. (2006). A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med. 354, 899–910. doi: 10.1056/NEJMoa044397
Qiu, Y., Chen, B. L., Mao, R., Zhang, S. H., He, Y., Zeng, Z. R., et al. (2017). Systematic review with meta-analysis: loss of response and requirement of anti-TNFalpha dose intensification in Crohn’s disease. J. Gastroenterol. 52, 535–554. doi: 10.1007/s00535-017-1324-3
Raine, T. (2014). Vedolizumab for inflammatory bowel disease: changing the game, or more of the same? United European Gastroenterol J 2, 333–344. doi: 10.1177/2050640614550672
Ransohoff, R. M. (2007). Natalizumab for multiple sclerosis. N. Engl. J. Med. 356, 2622–2629. doi: 10.1056/NEJMct071462
Rath, T., Billmeier, U., Ferrazzi, F., Vieth, M., Ekici, A., Neurath, M. F., et al. (2018). Effects of anti-integrin treatment with vedolizumab on immune pathways and cytokines in inflammatory bowel diseases. Front. Immunol. 9:1700. doi: 10.3389/fimmu.2018.01700
Rosen, M. J., Dhawan, A., and Saeed, S. A. (2015). Inflammatory bowel disease in children and adolescents. JAMA Pediatr. 169, 1053–1060. doi: 10.1001/jamapediatrics.2015.1982
Rosen, M. J., Karns, R., Vallance, J. E., Bezold, R., Waddell, A., Collins, M. H., et al. (2017). Mucosal expression of type 2 and type 17 immune response genes distinguishes ulcerative colitis from colon-only Crohn’s disease in treatment-naive pediatric patients. Gastroenterology 152, 1345.e1347–1357.e1347. doi: 10.1053/j.gastro.2017.01.016
Rudick, R. A., Stuart, W. H., Calabresi, P. A., Confavreux, C., Galetta, S. L., Radue, E. W., et al. (2006). Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. N. Engl. J. Med. 354, 911–923. doi: 10.1056/NEJMoa044396
Sandborn, W. J., Feagan, B. G., Rutgeerts, P., Hanauer, S., Colombel, J. F., Sands, B. E., et al. (2013). Vedolizumab as induction and maintenance therapy for Crohn’s disease. N. Engl. J. Med. 369, 711–721. doi: 10.1056/NEJMoa1215739
Sands, B. E., Feagan, B. G., Rutgeerts, P., Colombel, J. F., Sandborn, W. J., Sy, R., et al. (2014). Effects of vedolizumab induction therapy for patients with Crohn’s disease in whom tumor necrosis factor antagonist treatment failed. Gastroenterology 147, 618.e613–627.e613. doi: 10.1053/j.gastro.2014.05.008
Sands, B. E., Sandborn, W. J., Panaccione, R., O’Brien, C. D., Zhang, H., Johanns, J., et al. (2019). Ustekinumab as induction and maintenance therapy for ulcerative colitis. N. Engl. J. Med. 381, 1201–1214. doi: 10.1056/NEJMoa1900750
Schippers, A., Muschaweck, M., Clahsen, T., Tautorat, S., Grieb, L., Tenbrock, K., et al. (2016). Beta7-integrin exacerbates experimental DSS-induced colitis in mice by directing inflammatory monocytes into the colon. Mucosal Immunol. 9, 527–538. doi: 10.1038/mi.2015.82
Schleier, L., Wiendl, M., Heidbreder, K., Binder, M. T., Atreya, R., Rath, T., et al. (2020). Non-classical monocyte homing to the gut via alpha4beta7 integrin mediates macrophage-dependent intestinal wound healing. Gut 69, 252–263. doi: 10.1136/gutjnl-2018-316772
Schulz, O., Jaensson, E., Persson, E. K., Liu, X., Worbs, T., Agace, W. W., et al. (2009). Intestinal CD103+, but not CX3CR1+, antigen sampling cells migrate in lymph and serve classical dendritic cell functions. J. Exp. Med. 206, 3101–3114. doi: 10.1084/jem.20091925
Seyedian, S. S., Nokhostin, F., and Malamir, M. D. (2019). A review of the diagnosis, prevention, and treatment methods of inflammatory bowel disease. J. Med. Life 12, 113–122. doi: 10.25122/jml-2018-0075
Shen, B., Blake, A., Lasch, K., Smyth, M., and Bhayat, F. (2019). Vedolizumab use in patients with inflammatory bowel diseases undergoing surgery: clinical trials and post-marketing experience. Gastroenterol. Rep. 7, 322–330. doi: 10.1093/gastro/goz034
Shi, H. Y., and Ng, S. C. (2018). The state of the art on treatment of Crohn’s disease. J. Gastroenterol. 53, 989–998. doi: 10.1007/s00535-018-1479-6
Shinkai, Y., Rathbun, G., Lam, K. P., Oltz, E. M., Stewart, V., Mendelsohn, M., et al. (1992). RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell 68, 855–867. doi: 10.1016/0092-8674(92)90029-c
Sica, G. S., and Biancone, L. (2013). Surgery for inflammatory bowel disease in the era of laparoscopy. World J. Gastroenterol. 19, 2445–2448. doi: 10.3748/wjg.v19.i16.2445
Singh, U. P., Singh, N. P., Murphy, E. A., Price, R. L., Fayad, R., Nagarkatti, M., et al. (2016). Chemokine and cytokine levels in inflammatory bowel disease patients. Cytokine 77, 44–49. doi: 10.1016/j.cyto.2015.10.008
Soler, D., Chapman, T., Yang, L.-L., Wyant, T., Egan, R., and Fedyk, E. R. (2015). The binding specificity and selective antagonism of vedolizumab, an anti-a4ß7 integrin therapeutic antibody in development for inflammatory bowel diseases. J. Pharmacol. Exp. Ther. 330, 864–875. doi: 10.1124/jpet.109.153973
Smythies, L. E., Sellers, M., Clements, R. H., Mosteller-Barnum, M., Meng, G., Benjamin, W. H., et al. (2005). Human intestinal macrophages display profound inflammatory anergy despite avid phagocytic and bacteriocidal activity. J. Clin. Invest. 115, 66–75. doi: 10.1172/JCI19229
Steinbach, E. C., and Plevy, S. E. (2014). The role of macrophages and dendritic cells in the initiation of inflammation in IBD. Inflamm. Bowel Dis. 20, 166–175. doi: 10.1097/MIB.0b013e3182a69dca
Stritesky, G. L., Yeh, N., and Kaplan, M. H. (2008). IL-23 promotes maintenance but not commitment to the Th17 lineage. J. Immunol. 181, 5948–5955. doi: 10.4049/jimmunol.181.9.5948
Stuve, O., Marra, C. M., Jerome, K. R., Cook, L., Cravens, P. D., Cepok, S., et al. (2006). Immune surveillance in multiple sclerosis patients treated with natalizumab. Ann. Neurol. 59, 743–747. doi: 10.1002/ana.20858
Taylor, A., Verhagen, J., Blaser, K., Akdis, M., and Akdis, C. A. (2006). Mechanisms of immune suppression by interleukin-10 and transforming growth factor-beta: the role of T regulatory cells. Immunology 117, 433–442. doi: 10.1111/j.1365-2567.2006.02321.x
Van Assche, G., Van Ranst, M., Sciot, R., Dubois, B., Vermeire, S., Noman, M., et al. (2005). Progressive multifocal leukoencephalopathy after natalizumab therapy for Crohn’s disease. N. Engl. J. Med. 353, 362–368. doi: 10.1056/NEJMoa051586
Wright, R. D., and Cooper, D. (2014). Glycobiology of leukocyte trafficking in inflammation. Glycobiology 24, 1242–1251. doi: 10.1093/glycob/cwu101
Yao, Y., Vent-Schmidt, J., McGeough, M. D., Wong, M., Hoffman, H. M., Steiner, T. S., et al. (2015). Tr1 cells, but not Foxp3+ regulatory T cells, suppress NLRP3 inflammasome activation via an IL-10-dependent mechanism. J. Immunol. 195, 488–497. doi: 10.4049/jimmunol.1403225
Zeissig, S., Rosati, E., Dowds, C. M., Aden, K., Bethge, J., Schulte, B., et al. (2019). Vedolizumab is associated with changes in innate rather than adaptive immunity in patients with inflammatory bowel disease. Gut 68, 25–39. doi: 10.1136/gutjnl-2018-316023
Keywords: vedolizumab, inflammatory bowel disease, cell trafficking, macrophages, innate immunity
Citation: Luzentales-Simpson M, Pang YCF, Zhang A, Sousa JA and Sly LM (2021) Vedolizumab: Potential Mechanisms of Action for Reducing Pathological Inflammation in Inflammatory Bowel Diseases. Front. Cell Dev. Biol. 9:612830. doi: 10.3389/fcell.2021.612830
Edited by:
Zhichao Fan, UCONN Health, United StatesReviewed by:
Rongrong Liu, Northwestern University, United StatesDror S. Shouval, Edmond and Lily Safra Children’s Hospital, Israel
Vito Annese, Fakeeh University Hospital, United Arab Emirates
Copyright © 2021 Luzentales-Simpson, Pang, Zhang, Sousa and Sly. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura M. Sly, bGF1cmFzbHlAbWFpbC51YmMuY2E=