 Jingxin Yang†
Jingxin Yang† Yiming Pan
Yiming Pan Juan Kong
Juan Kong- Department of Clinical Nutrition, Shengjing Hospital of China Medical University, Shenyang, China
Energy substrate imbalance is a major cause of cardiac dysfunction. Vitamin D/vitamin D receptor (VD/VDR) deficiency is involved in the pathogenesis of various cardiac diseases; however, the exact underlying mechanism remains unclear. The aim of this study was to investigate whether vitamin D modulates mitochondrial fatty acid oxidase via sirtuin 3 signaling to protect the myocardium. 1-Alpha-hydroxylase-defficient mice exhibited a high metabolic rate and lower myocardial contractility than wild-type mice. Sirtuin 3 upregulation was detected in high-fat diet-fed mice receiving vitamin D3 compared with that in high-fat diet-fed mice. Both sirtuin 3 blockade and knockout inhibited the VD/VDR-induced downregulation of fatty acid oxidase in myocardial mitochondria. VD/VDR suppressed fatty acid metabolism by upregulating sirtuin 3 and lowering mitochondrial fat uptake, thereby improving myocardial function and balancing energy substrates, rather than by altering fat endocytosis and exocytosis.
Introduction
Cardiomyocyte energy metabolism encompasses the processes of substrate absorption, distribution, and utilization. There are three main energy substrate sources in heart cells, namely glycolysis (10–20%), lactic acid (10–20%), and fatty acid oxidation (60–90%) (van Bilsen, 2004). Together, these maintain a relatively constant substrate availability in order to meet the organ’s energy demands. Heart cells adjust carbohydrate and fatty acid metabolism to adapt to various pathophysiological conditions, switching between glucose and fatty acids as the main energy source. The imbalance of energy substrates, especially the excess of fatty acids, leads to the initiation of the pathological processes in cardiac tissue, including myocardial hypertrophy and myocardial necrosis (Fillmore et al., 2014; Fukushima and Lopaschuk, 2016). Although myocardial function is known to be affected by hyperlipidemia, cardiac hypertrophy, coronary atherosclerosis, and other conditions, the detailed mechanisms underlying their effects remain unclear.
Vitamin D (VD) plays an important role in the pathogenesis of cardiovascular disease (Ku et al., 2013; Brondum-Jacobsen et al., 2012). As a liposoluble secosteroid and prohormone (Kong et al., 2013), it binds to the vitamin D receptor (VDR) (Kong et al., 2010) to regulate gene expression. VDR, a member of the nuclear receptor superfamily, plays an essential role in modulating lipid metabolism (Moya et al., 2010; Vluggens and Reddy, 2012). VD exerts its physiological effects as its bioactive form [1, 25(OH)2D3] hydroxylated by 1-alpha-hydroxylase [1α(OH)ase], which encoded by the Cyp27b1 gene. 1,25(OH)2D3 activates the VDR to form a heterodimer complex with the retinoid X receptor, which then interacts vitamin D response elements (VDREs) in the promoter region of target genes, thus, driving the stimulation of fatty acid oxidation and the inhibition of lipogenesis by activating endogenous and exogenous SIRT1 (Sabir et al., 2017). Cyp27b1–/– mice have vitamin D metabolism disorders and developed hypertension, cardiac hypertrophy, and cardiac contractile dysfunction (Zhang et al., 2015). Cholecalciferol cholesterol emulsion (CCE), which is composed of tea oil and VD3(93.75 μg/ml), is a safe and reliable vitamin D analog that is used in clinical practice to cure infant rickets caused by vitamin D deficiency. VDR deficiency leads to protein succinylation in white adipose tissue, providing a crucial clue regarding the underlying mechanisms of VDR in energy metabolism (Su et al., 2017).
Sirtuins are a family of nicotinamide acetylated (NAD)-dependent histone deacetylases, which are particularly sensitive to metabolic fluctuations (Lombard et al., 2007; Sadhukhan et al., 2016). Among mitochondrial sirtuins, SIRT3 appears to have the broadest deacetylase activity and is highly expressed in mitochondria-rich tissues, including muscle, liver, kidney, and heart (Lombard et al., 2007; Fukushima and Lopaschuk, 2016). In recent years, SIRT3 has been found to regulate the deacetylation of fatty acid oxidase [long-chain fatty acyl-CoA dehydrogenase (LCAD)](Hirschey et al., 2010) and electron transfer chain complexes in the mitochondria (Vassilopoulos et al., 2014). SIRT3 significantly reduces fatty acyl-coenzyme A in the tricarboxylic acid pathway as well as oxygen demand, indirectly decreasing oxygen consumption and fatty acid β-oxidation (Sack, 2012). In addition, fatty acid oxidation is upregulated in Sirt3 knockout mice, which clearly indicates the involvement of SIRT3 in the regulation of this process (Kendrick et al., 2011). Under hyperlipidemia, the inhibition of SIRT3 and the acetylation of mitochondrial protein leads to the excessive utilization of fatty acids as energy substrates in addition to excess oxygen consumption, in turn adversely affecting cardiac cells (Lawrence et al., 1998; Sack, 2012; Fukushima and Lopaschuk, 2016). More importantly, cardiomyocyte SIRT3 expression can be inhibited by a high-fat diet (Fukushima and Lopaschuk, 2016). The deficiency of SIRT3, an anti-hypertrophic molecule, is associated with the development of cardiac hypertrophy (Sundaresan et al., 2009). However, the ability of VD to regulate SIRT3 and the exact role of the latter in VD’s effects on myocardial energy metabolism remain elusive.
To investigate the possible mechanisms underlying the cardioprotective effects of VD/VDR, we sought to link the VD/VDR signaling system with fatty acid metabolism via myocardial SIRT3.
Materials and Methods
Our experimental process is shown in Figure 1.
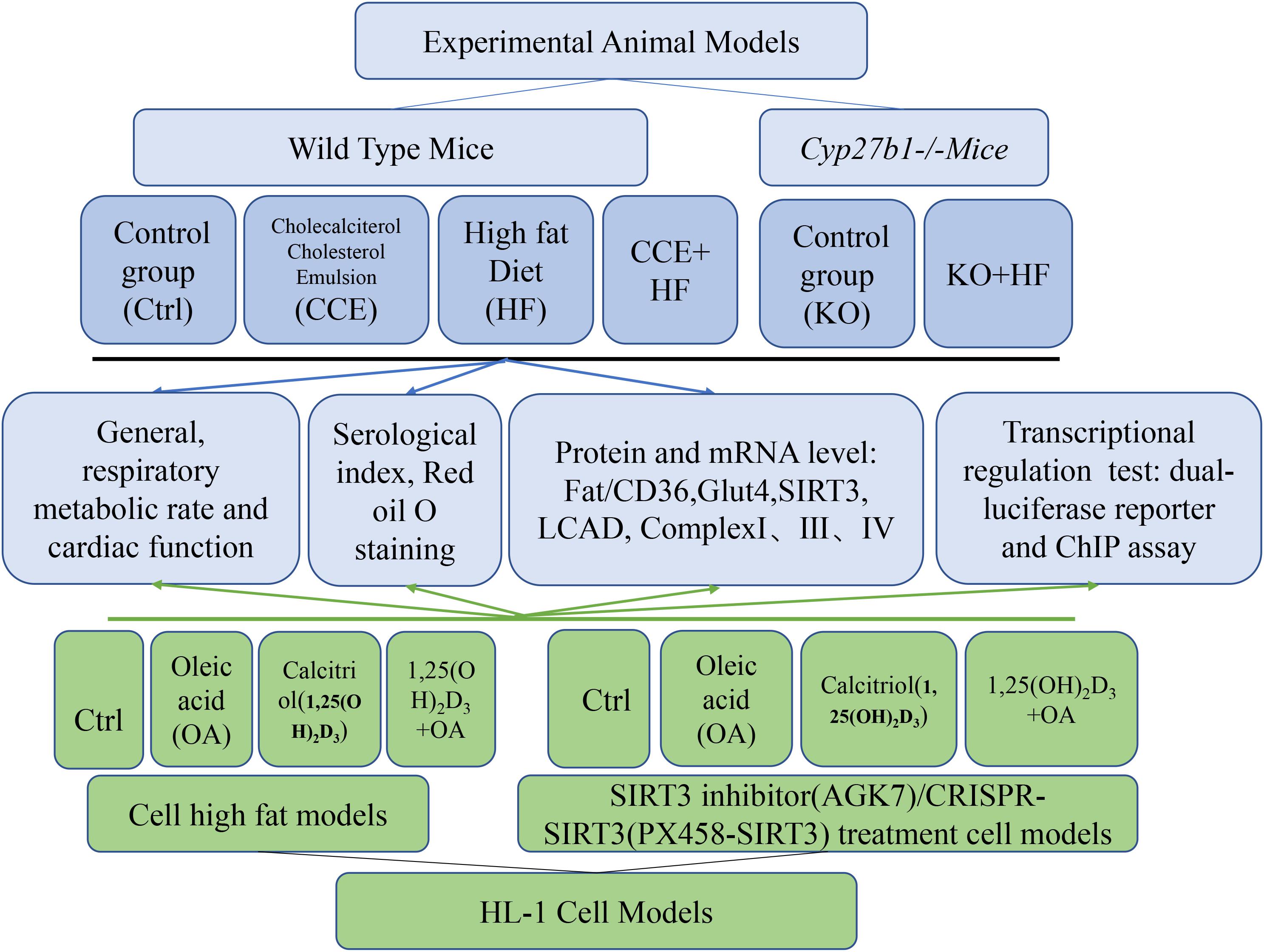
Figure 1. General graphical outline of the experimental design.
Animal Studies
ICR (Institute of Cancer Research) mice, aged 4 weeks, were purchased from Changsheng Biotechnology Co., Ltd. (Liaoning, China) and housed in the Animal Center of the Experimental Department at the Shengjing Hospital of China Medical University. Mice were maintained on a 12-h light/dark cycle and chow diet with free access to water and food. Cyp27b1–/– mice of an ICR background were used in the current study. 1α(OH)ase is encoded by Cyp27b1, and its absence is characterized by VD deficiency. The knockout mice were given a high-calcium diet to maintain normal blood calcium levels. All experimental protocols in our study were approved by the Animal Experimental Committee under No. 2018PS347K. All animal procedures were carried out to minimize suffering in accordance with the guidelines established by the Animal Experimental Committee.
Cyp27b1–/–mice, originally on a BALB/c background, were repeatedly backcrossed with wild-type mice on an ICR background for 12 generations to obtain Cyp27b1–/–mice on an ICR background. Cyp27b1–/– mice were generated through the breeding of heterozygous (Cyp27b1±) mice and identified via PCR-based genotyping.
WT mice were identified using the following Cyp27b1 primer pairs:
5′-GAAGTCCCTCCTGACACAGAAACCT-3′ (forward)
and 5′-CTCATAGAGTGTCCAGGAGAGCGTA-3′ (rev erse).
The neomycin resistance gene primer pair used was 5′-ACAACAGACAATCGGCTGCTC-3′ (forward) and 5′-CTC ATAGAGTGTCCAGGAGAGCGTA-3′ (reverse).
Humans and mice are converted into body surface area equivalent doses. For people aged 14–18, the recommended dietary allowance (RDA) of VD from Institute of Medicine is 600IU/d, the daily requirement of VD given by Endocrine Society is 600-1000IU/d, and the recommended nutrient intake (RNI) of VD from Chinese Nutrition Society is 400IU/d. To sum up, take an adult who needs to intake 1000IU (25 μg) of VD per day as an example. The average body weight of adult human and mice is 70 kg and 20 g, respectively. The conversion coefficient between humans and mice is 0.026, so mice consumed dose of VD3 per day per kg weight is 25 μg/70 kg ∗ 70 kg ∗ 0.0026/20 g = 3.25 μg/kg/d.
The wild-type mice fed a normal diet were randomly divided into a control group and a group receiving CCE via the drinking water (CCE: water = 10 μl:100 ml) for 14 weeks (n = 15 in each group). In addition, wild-type mice were fed a high-fat diet (HF, 45% Cal/fat) and randomized into an HF group and a CCE + HF group (n = 15). TheCyp27b1–/– mice were randomly divided into a KO mice group fed a normal diet and a KO + HF group (n = 10) fed the high-fat diet for 14 weeks (n = 10). Body weight as well as food and water intake were monitored weekly. Body composition was determined using the Minispec (Bruker, LF-50). Rectal temperatures were measured at 4 p.m. using a rectal probe attached to a digital thermometer (Runjing Inc., TES-1310). All experimental mice were sacrificed under anesthesia. Blood and heart tissues were harvested and frozen for further analysis.
Cell Culture and Treatment
The HL-1 cardiac cell line purchased from BeNa Culture Collection (BNCC, 288890) and the Cos-7 fibroblast-like cell line were cultured in DMEM with 5% glucose, 10% fetal bovine serum (FBS), and 1% penicillin/streptomycin. The cells were treated with 2 × 10–8 mol/l calcitriol for 2 h and subsequently challenged with 0.25 mmol/L oleic acid (OA) (Sigma-Aldrich) for 24 h.
Biochemical Measurements
Serum triglyceride, total cholesterol, glucose, free fatty acids, calcium, and potassium were determined using commercial kits (Nanjing Jiancheng Bioengineering Institute).
RNA Extraction and Real-Time RT-PCR
Total RNA isolated using TRIzol (Invitrogen, 15596026) was reverse-transcribed using the RT reagent kit (Takara, RR047A). Real-time PCR was conducted using SYBR Premix Ex Taq II (Takara, RR8320A). The specific primers used were synthesized by Life Co., and are listed below:
Vdr Forward CTGGTGACTTTGACCGGAAT;
Vdr Reverse CTGCACCTCCTCATCTGTGA;
Glut4 Forward CTCTCGTCACTGACTGCACA;
Glu4 Reverse CTCTCGTCACTGACTGCACA;
Sirt1 Forward CGGCTACCGAGGTCCATATAC;
Sirt1 Reverse ACAATCTGCCACAGCGTCAT;
Sirt2 Forward AGCCGGACCGATTCAGA;
Sirt2 Reverse TCGAGGGTCAGCTCGTCTA;
Sirt3 Forward TTTCATGTTGGCCAAGGAGC;
Sirt3 Reverse GCTGTTACAAAGGTCCCGTG;
Sirt4 Forward GGGTCCTGAGCTCTTCTTGG;
Sirt4 Reverse CCGCTCATTCTTATTCTGTCTGG;
Sirt5 Forward CTGATGCGACCTCTCCTGAT;
Sirt5 Reverse CCCCGAGATGATGGCTATGT;
Sirt6 Forward ACGCAGTACGTCAGAGACAC;
Sirt6 Reverse TCTCTCAGCTCCCCTCTACA;
Sirt7 Forward CTACAACCGGTGGCAGGAT;
Sirt7 Reverse AGTGACTTCCTACTGTGGCT.
Transmission Electron Microscopy
Heart tissue samples were fixed in 2.5% glutaraldehyde with 1% paraformaldehyde, according to standard protocols. Then, 80–100 nm-thick sections were prepared.
Western Blotting
Protein samples were isolated from tissue and cells using the Minute total protein extraction kit (Invent, SD001). Separated proteins were transferred to polyvinylidene difluoride membranes (Millipore Sigma Co., Ltd., Burlington, MA, United States). The membranes were probed with antibodies against SIRT3 (10099-1-AP, 1:500), LCAD (17526-1-AP, 1:300), Complex I (12444-1-AP, 1:1,000), Complex III (14865-1-AP, 1:1,000), Complex IV (18443-1-AP, 1:2,000), FAT/CD36 (18836-1-AP, 1:500), CPT-1β (22170-1-AP, 1:500), and GADPH (60004-1-Ig, 1:5,000) purchased from Proteintech as well as with an antibody against MTTP from Absin (abs136111a, 1:1,000) and antibodies against VDR (sc-1008, 1:1,000) and GLUT4 (sc-53566, 1:500) purchased from Santa Cruz. Following overnight primary antibody incubation, membranes were incubated with a secondary antibody (ZSGB-Bio, Beijing, China) for 1 h. The protein bands of interest were detected using Amersham ECL reagents (Cytiva, Little Chalfont, United Kingdom).
Oil Red O Staining
Lipid accumulation in the cytoplasm was evaluated using Oil Red O staining. Myocardial tissue was formalin-fixed and frozen in Tissue Tek OCT compound (Sakura Finetek, 4,583). Then, 5 μm-thick cryosections were stained via Oil Red O with hematoxylin (Beyotime, C0107) used as a counterstain. Cells were fixed in 4% buffered paraformaldehyde and stained with Oil Red O and hematoxylin.
Measurement of Cellular Oxygen Consumption Rate and Extracellular Acidification Rate
HL-1 cells were seeded in XF8 plates at a density of 60,000 cells per well to form an even monolayer, followed by treatment with OA, calcitriol, or AGK7 (SIRT3 inhibitor, Santa Cruz Biotechnology, sc-203281) for 24 h. Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured in XF media under basal conditions following mitochondrial inhibitor treatment.
Indirect Calorimetry
Mice were maintained in a comprehensive lab animal OxyletPro system (Panlab, Metabolism 3.0) through which oxygen consumption and carbon dioxide production were continuously measured for 48 h according to the instructions of the manufacturer.
Luciferase Reporter Assays
VDREs located at nt-776 to 780 of the Sirt3 promoter region were predicted using PROMO web. A PCR fragment with VDREs was inserted into a pGL3-basic vector confirmed by sequencing. The primers used were as follows: forward, 5′-TTCTGGTGAGAAAATTCCTGGG-3′; reverse, 5′-GACTAA CCTGGGTTACACAGCA-3′. The plasmids with VDRE and the Renilla luciferase control vector (PRL-TK) were then co-transfected into Cos-7 cells using Lipofectamine 3000 (Invitrogen, L3000-015). The luciferase activity was determined using a Dual Luciferase Assay System (Promega, e2920) with a Modulus Microplate Reader (BioTek, Synergy H1).
Chromatin Immunoprecipitation Assay
The chromatin immunoprecipitation assay was performed using a commercial kit (Cell Signaling, 56383). Briefly, chromatin was digested and sonicated into 200–500 bp DNA fragments. The precipitated DNA was subjected to PCR analysis. The VDREs were identified using the following primer pairs: forward, 5′-CTGACAAAGCATGGGAAAGAGTG-3′ and reverse, 5′-TTGAAGGTGAGAGGGTTGAGG-3′.
CRISPR/Cas9 System Construction and Transfection
Genome engineering was performed using a CRISPR/Cas9 system according to published protocols (Ran et al., 2013). A single guide RNA that targets the region within exon 2 of the mouse Sirt3 was selected from genome-wide mouse sgRNA libraries. The sgRNA-optimized CRISPR design was inserted into the pSpCas9(BB)-2A-GFP vector. The primers targeting Sirt3 were as follows: sgRNA top, 5′-TCTATACACAGAACATCGAC-3′, sgRNA bottom, 5′-GTCGATGTTCTGTGTATAGA-3′. The plasmids were then amplified, purified, sequenced, and transfected into cardiomyocytes using a jetPRIME transfection reagent (Polyplus-transfection). Western blotting was performed to confirm transfection.
Assessment of Myocardial Function
The tracheal intubation was connected to a small animal ventilator (DW-3000B, Beijing Zhongshidichuang Co., Ltd.) under intraperitoneal anesthesia. A pressure-volume catheter was then placed in the left ventricle via an apical stab approach. Electrocardiogram, the maximal contractile force (Fc), and heart rate were measured using a MedLab biological signal acquisition and analysis system (JH-2, Beijing Zhongshidichuang Co., Ltd.).
Malondialdehyde Production and Superoxide Dismutase Activity
The mice were anesthetized with ether and orbital blood was collected. The blood sample was stored at 4°C for 1 h, and then centrifuged at 3,500 g for 10 min, and then the serum supernatant was taken out for determination. All test kits were purchased from Nanjing Jiancheng Biological Engineering Co., Ltd., and operated in accordance with the manufacturer’s instructions. The thiobarbituric acid colorimetric method was used to determine the concentration of Malondialdehyde (MDA), and the xanthine oxidase method was used to determine the activity of superoxide dismutase (SOD).
Statistical Analysis
Statistical analysis was performed with the Prism 8.0 software (GraphPad). An unpaired Student’s t-test or one-way analysis of variance followed by a Bonferroni test were used for statistical analysis after equal variance was confirmed with an F test. A p-value less than 0.05 was considered to indicate statistical significance.
Results
VD Regulated Energy Metabolism in High-Fat Diet-Fed Mice
To investigate the effects of VD on energy metabolism, we examined physiological changes in each group of mice. No significant difference was observed in food intake, water intake, and body temperature among all groups (Figures 2A–C). The body weight of Cyp27b1–/– mice was lower than that of wild-type control mice. Similarly, KO mice showed a resistance to body weight gain in response to the high-fat diet compared with that in wild-type mice. Furthermore, the highest body weight gain was observed in CCE-HF mice (Figure 2D). HF mice exhibited higher O2 consumption and serum free fatty acid levels (Figures 2E–G) than CCE-HF or control mice. There were no differences in serum calcium and potassium levels between all groups (Figures 2H,I), suggesting that CCE is a safe VD analog. These data indicated that VD plays a role in O2 consumption and lipid metabolism in vivo.
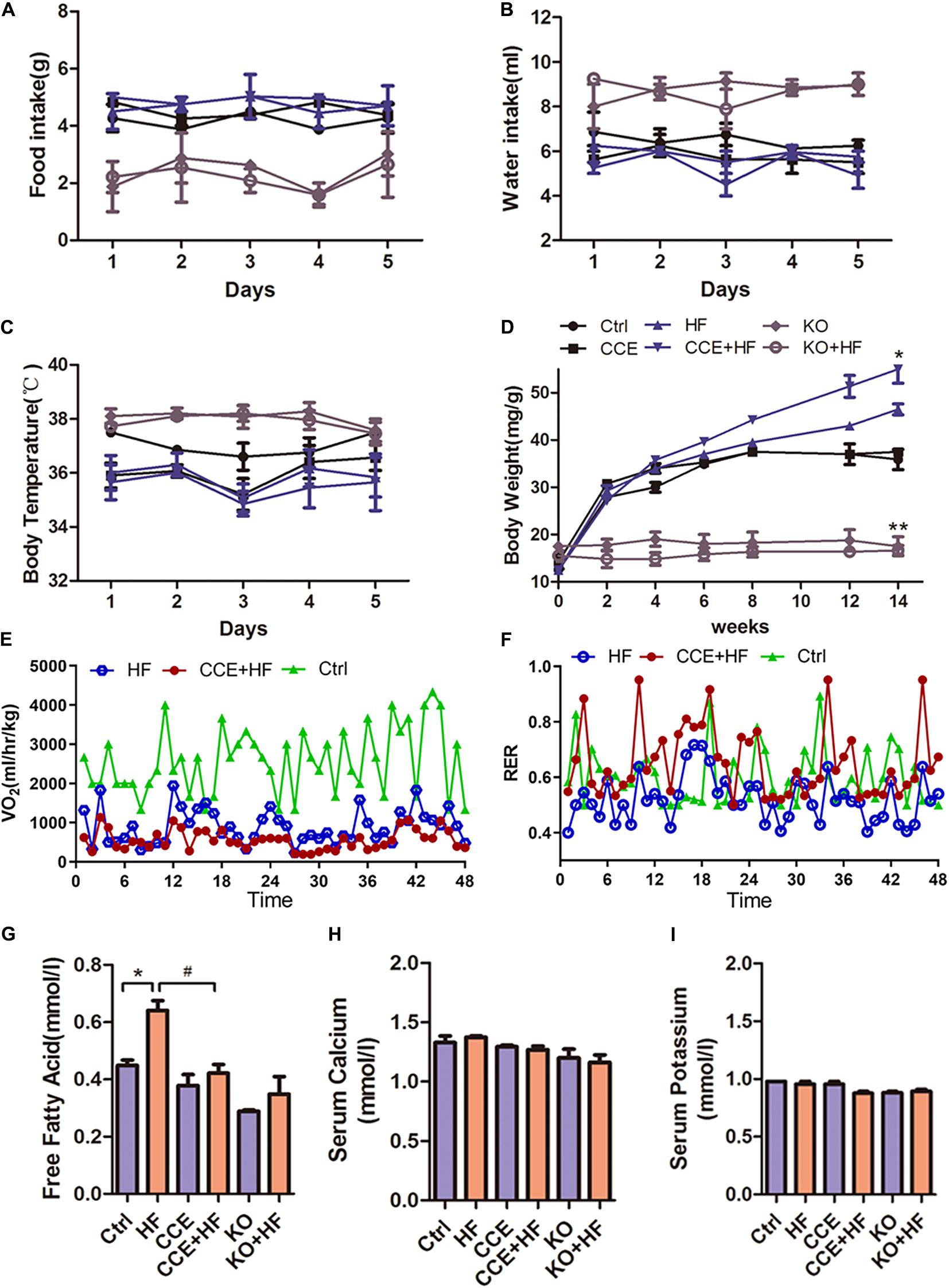
Figure 2. Effect of vitamin D on energy metabolism in mice fed a high-fat diet. (A) Body weight changes in control or KO mice receiving CCE or a high-fat diet for 14 weeks (*p < 0.05 or **p < 0.01). (B) Food intake, (C) water intake, and (D) body temperature in each group were monitored for 5 days, and no changes were observed in all groups (p > 0.05). (E) O2 consumption and (F) respiratory exchange rate indicated a lower metabolic rate in the CCE + HF group than in the HF group. Serum lipid levels analysis: (G) free fatty acids, (H) serum calcium, and (I) serum potassium, with relative changes shown for all groups (*p < 0.05 vs. Ctrl, #p < 0.05 vs. HF). Values are expressed as the mean ± SEM. Ctrl, control group. CCE, Fed cholecalciferol cholesterol emulsion [a precursor of 1, 25(OH)2D3] group. HF, Fed high fat die group. CCE + HF, Fed cholecalciferol cholesterol emulsion [a precursor of 1, 25(OH)2D3] and high fat die group. KO, Cyp27b1–/– mice, KO + HF, Cyp27b1–/– mice fed with high fat die group.
VD Regulated Cardiomyocyte Fatty Acid Metabolism and Protected Against Hyperlipidemia in vivo
There were no obvious changes in the levels of endocytosis marker FAT/CD36 and exocytosis marker MTTP between HF and CCE-HF mice (Figure 3A). However, the expression of CPT-1β, a marker of mitochondrial fatty acid uptake, was significantly lower in CCE + HF mice than in HF mice, suggesting that CCE could suppress mitochondrial fatty acid utilization instead of excessive endocytosis. It is possible that VD regulates fatty acid metabolism by reducing mitochondrial fatty acid uptake and fat burning, rather than by altering fatty acid endocytosis and exocytosis. In addition, our data indicated that glucose transporter GLUT4 was upregulated in the CCE + HF group compared with that in the HF group (Figure 3A), indicating that to balance the fatty acid consumption, the body regulates glucose metabolism in the presence of VD to maintain the ratio of energy substrates. The loops of pressure-volume in Cyp27b1–/–mice hearts were much smaller than those in wild-type mouse hearts, indicative of the markedly reduced cardiac contractility in the absence of 1α(OH)ase (Figure 3B). The blood pressure and the ratio of heart weight to body weight of KO mice were higher than those of control group mice. Furthermore, the blood pressure of HF group animals was increased compared with that in control and CCE + HF mice (Figures 3C,D). Electrocardiogram and cardiac contractility analysis revealed higher, sharper R waves and worsened myocardial contractile rhythm in Cyp27b1–/–mice and HF mice than in control animals, suggesting that VD could alleviate cardiac hypertrophy and improve cardiac function (Figure 3E). Taken together, VD regulated cardiomyocyte fatty acid metabolism and protected cardiac function.
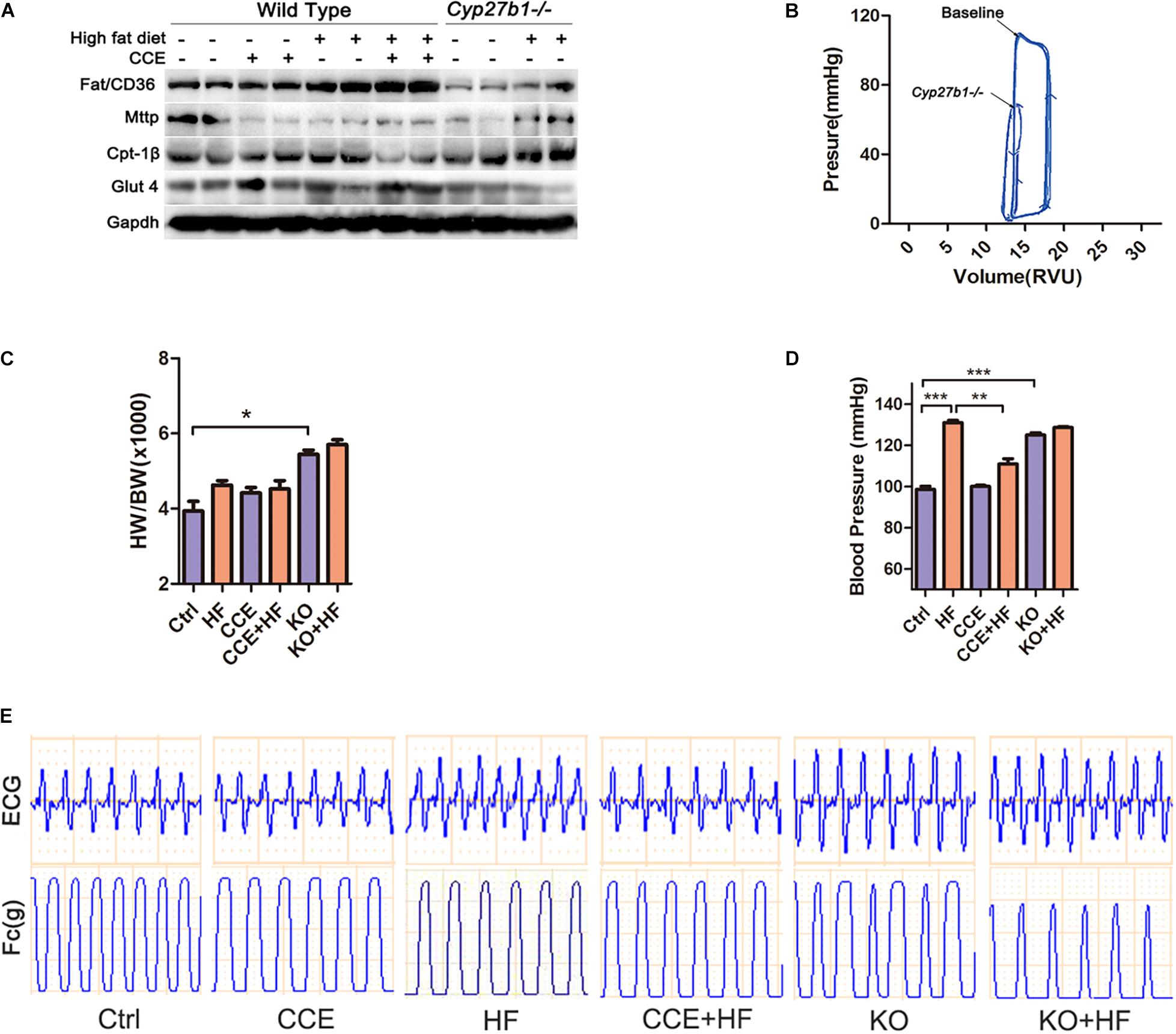
Figure 3. Effect of vitamin D on lipid metabolism and cardiac function in vivo. (A) Western blot of lipid metabolism-associated proteins. (B) The left ventricular pressure-volume loop analysis revealed that heart contractility of Cyp27b1–/– mice was lower than the baseline. (C) Heart weight normalized to mouse body weight in animals fed a high-fat diet with or without CCE. (D) Invasive blood pressure measured via the carotid artery was lower in the CCE + HF group than the HF group (*p < 0.05, **p < 0.01, ***p < 0.001). (E) The curves of ECG and myocardial contractility were recorded for each group. Ctrl, control group. CCE, Fed cholecalciferol cholesterol emulsion [a precursor of 1, 25(OH)2D3] group. HF, Fed high fat die group. CCE + HF, Fed cholecalciferol cholesterol emulsion [a precursor of 1, 25(OH)2D3] and high fat die group. KO, Cyp27b1–/– mice. KO + HF, Cyp27b1–/– mice fed with high fat die group.
VD/VDR Promoted the Expression of SIRT3 and Mitochondrial Respiration in the Mouse Myocardium
To explore the mechanism through which VD reduced mitochondrial fatty acid uptake, we focused on the expression of various enzymes associated with oxidative metabolism in the mitochondria. SIRT3, VDR, and 1α(OH)ase were considerably upregulated at both the mRNA and protein levels in response to CCE treatment (Figures 4A,B) and were significantly reduced in the heart tissue of KO mice (Figure 4C) when compared with those in the heart tissue of control group animals. Consistent with previous studies (Bharathi et al., 2013), we found that a high-fat diet inhibited SIRT3 activity and protein expression, an effect that was reversed by CCE treatment. Similarly, CCE blocked the upregulated expression of LCAD caused by hyperlipidemia (Figure 4C). Transmission electron microscopy revealed severe damage of mitochondria in HF and KO mice, which CCE treatment rescued (Figure 4D). Compared with the HF group, in VD-HF animals, the protein expression levels of mitochondrial respiratory chain complexes I, III, and IV were increased (Figure 4E). Figures 4F,G indicate that VD significantly alleviated malondialdehyde (MDA) production and promoted superoxide dismutase (SOD) activity to protect cardiomyocytes against oxidative damage. Taken together, VD/VDR upregulated SIRT3 and mitochondrial respiratory chain complexes, while decreasing hyperlipidemia-induced oxidative damage.
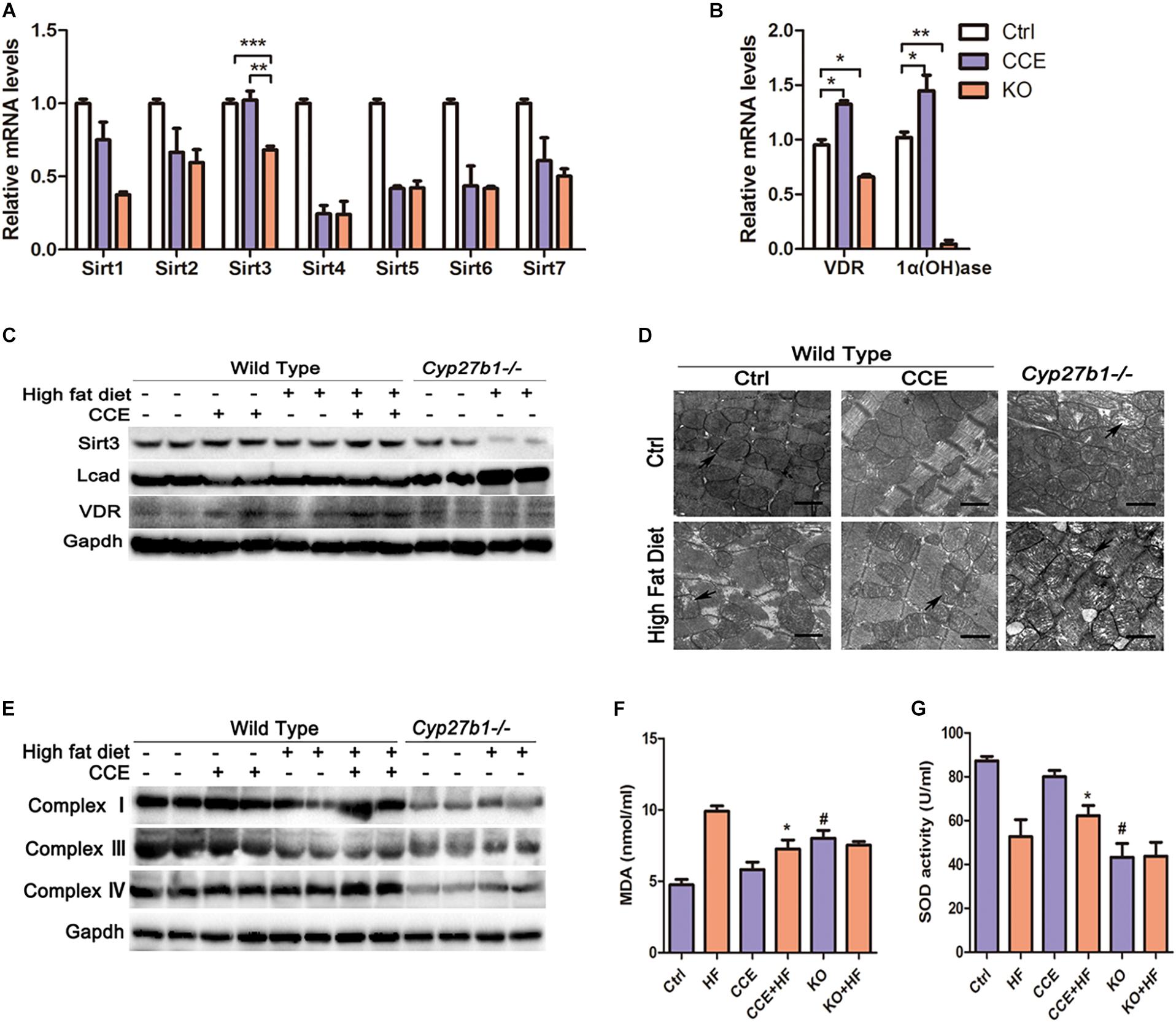
Figure 4. Vitamin D increased the expression of SIRT3 and mitochondrial complexes in high-fat diet-fed mice. (A,B) RT-qPCR analysis of sirtuins, VDR, and 1α(OH)ase revealed that the expression levels of Sirt3, VDR, and 1α(OH)ase were decreased in the KO group and increased following CCE treatment. Values are expressed as the mean ± SEM, *p < 0.05, **p ≤ 0.01, and ***p ≤ 0.001. (C) Western blotting analysis of SIRT3, LCAD, and VDR protein levels. (D) Electron microscopy images indicated mitochondrial arrangement and morphological characteristics (12,000×). Aberrant mitochondrial structures are indicated by a black arrow. (E) Western blotting indicated that CCE upregulated mitochondrial complex I, III, and IV protein expression. Serum levels of (F) MDA and (G) SOD with relative changes shown for the CCE + HF group compared with the HF group (#p < 0.05 vs. Ctrl, *p < 0.05 vs. HF). Ctrl, control group. CCE, Fed cholecalciferol cholesterol emulsion [a precursor of 1, 25(OH)2D3] group. HF, Fed high fat die group. CCE + HF, Fed cholecalciferol cholesterol emulsion [a precursor of 1, 25(OH)2D3] and high fat die group. KO, Cyp27b1–/– mice. KO + HF, Cyp27b1–/– mice fed with high fat die group.
VD/VDR-Mediated Mitochondrial Respiration Required SIRT3 in vitro
As a SIRT3 antagonist, AGK7 cleared accumulated oil droplets in VD-HF cells, which suggested that VD might increase intracellular lipid droplets through SIRT3 (Figures 5A,B). In addition, AGK7 blocked the decrease of LCAD protein expression and the increase of mitochondrial complex expression following treatment with VD in OA (Figure 5C). However, VD was unable to reduce the expression of LCAD and upregulated mitochondrial complexes in the absence of SIRT3. We then examined the degree of oxidative damage in cells, and found that SOD and MDA levels did not change significantly after blocking SIRT3 (Figures 5D,E). Similarly, LCAD protein expression was markedly upregulated in the Sirt3–/– cardiomyocytes following 1,25(OH)2D3-OA treatment relative to SIRT3-competent cells. The opposite trend was observed with regard to complex I, III and IV protein expression (Figures 5F,G). These results indicated that VD/VDR regulated mitochondrial respiratory metabolism in a SIRT3-dependent manner.
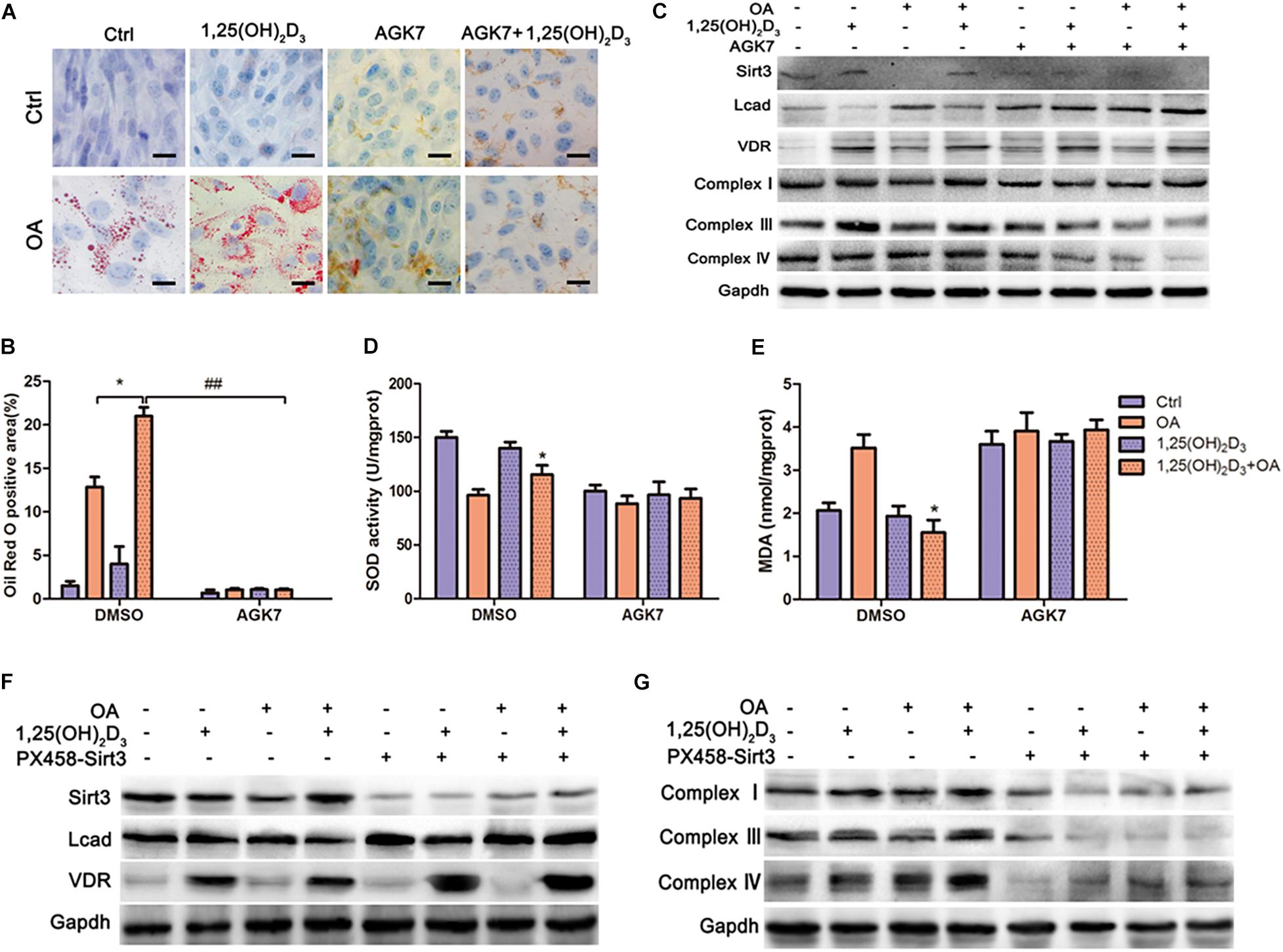
Figure 5. VD/VDR regulated lipid metabolism and mitochondrial respiratory metabolism via SIRT3 to protect HL-1 cells against oxidative damage. (A,B) representative Oil Red O staining in cells treated with 1,25(OH)2D3, oleic acid (OA), and AGK7 (SIRT3 inhibitor) (original magnification: 400×). *p < 0.05, ##p < 0.01. (C) Protein levels of SIRT3, LCAD, VDR, complexes I, III, and IV in HL-1 cells treated with 1,25(OH)2D3, OA, and AGK7 were analyzed via western blot. Analysis of (D) MDA and (E) SOD with relative changes shown for the 1,25(OH)2D3 + OA group compared with the OA group (*p < 0.05 vs. OA). (F,G) Western blot analysis of SIRT3, LCAD, complexes I, III and IV in cells with and/or without 1,25(OH)2D3, knockout of Sirt3 via CRISPR/Cas9, and OA, indicated that VD/VDR did not regulate fatty acid metabolism in the absence SIRT3.OA, oleic acid. PX458-Sirt3, knockout of Sirt3 via CRISPR/Cas9. AGK7, SIRT3 inhibitor.
AGK7 Partly Abolished the Effect of VD/VDR on Energy Substrate Homeostasis in HL-1 Cells
The oxygen consumption of mitochondria in HL-1 cells treated with 1,25(OH)2D3-OA was considerably lower than that of OA-treated cells, as determined via the Seahorse analyzer (Figure 6A,B). However, there was no difference in OCR and ATP production between 1,25(OH)2D3-OA- and OA-treated cells following AGK7 addition, supporting the crucial role of VD in mitochondrial metabolism via SIRT3. Furthermore, AGK7 stimulated high OCRs (Figures 6C,D). Interestingly, blocking SIRT3 suppressed glycolysis (Figures 6E,F), indicating that changes in a single substrate can affect overall energy homeostasis and that the maintenance of cell glycolysis by VD stabilized the proportion of energy substrates.
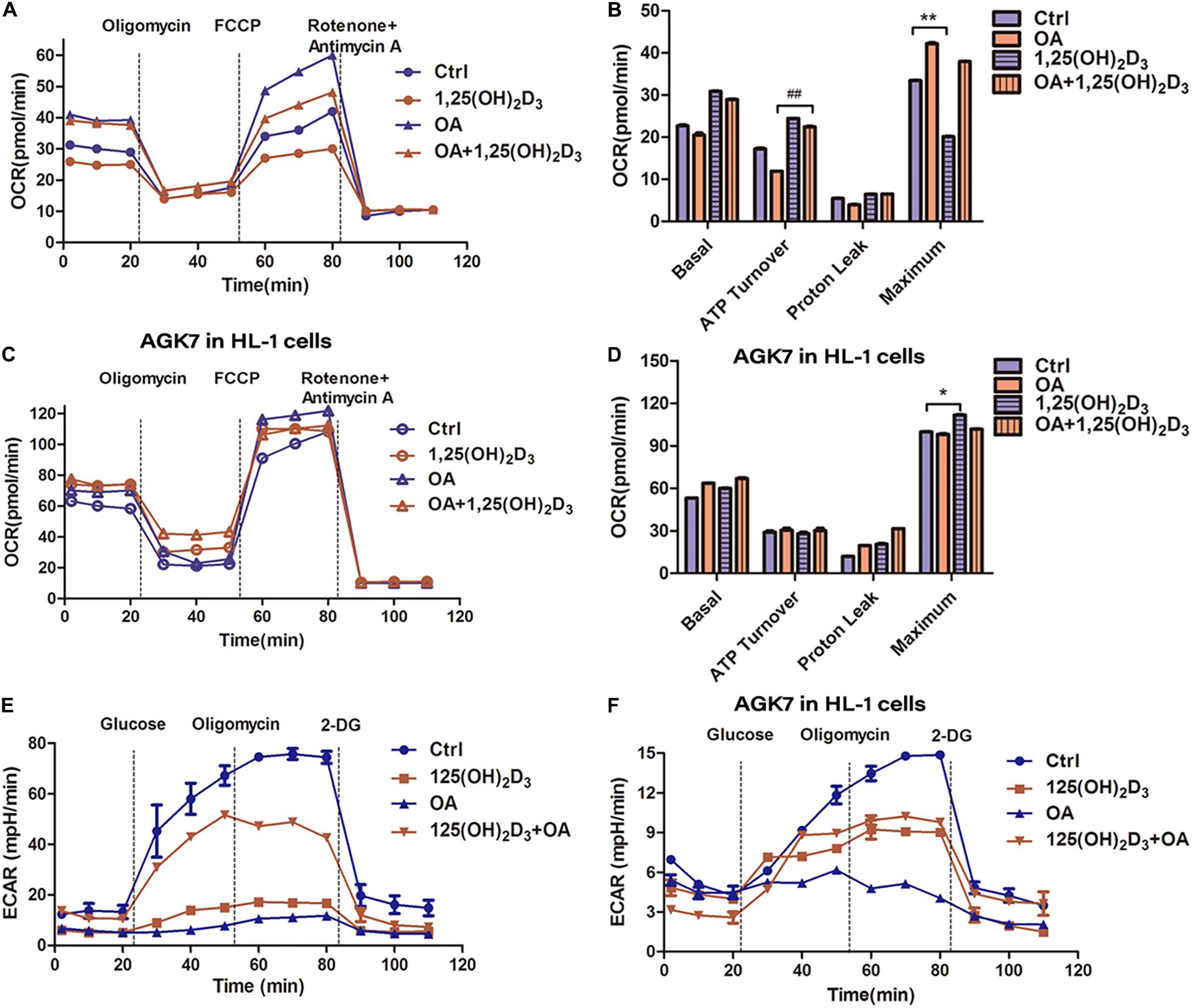
Figure 6. VD/VDR regulated energy substrate homeostasis in HL-1 cells via SIRT3. (A) Analysis of oxygen consumption rate (OCR) in the mitochondria of HL-1 cells treated with 1,25(OH)2D3 and OA. (B,D) The analysis of OCR in HL-1 cells treated with 1,25(OH)2D3, OA, and AGK7, indicating that VD treatment had no effect in the absence of SIRT3 *p < 0.05, **p < 0.01, ##p ≤ 0.01. (C) ATP turnover was determined based on the OCR prior to oligomycin minus the OCR after oligomycin, indicating that mitochondrial respiration was decreased, and ATP was increased in the 1,25(OH)2D3 + OA-treated cells compared with the OA-treated cells. (E,F) The analysis of extracellular acidification rate (ECAR) in HL-1 cells treated with 1,25(OH)2D3, OA, and AGK7. Ctrl, control group. OA, oleic acid. AGK7, SIRT3 inhibitor.
VDR Binds to the VDRE to Promote Sirt3 Expression
The transcriptional binding sites for VDR in the Sirt3 promoter were predicted using PROMO virtual laboratory. A twofold ratio of Firefly to Renilla was induced by calcitriol (Figure 7A). Moreover, chromatin immunoprecipitation indicated that VD promoted the binding of VDR to the SIRT3 VDRE in HL-1 cardiac myocyte cells (Figures 7B,C). These results suggested that VD increased the expression of Sirt3 through VDR binding at the VDRE within the Sirt3 promoter.
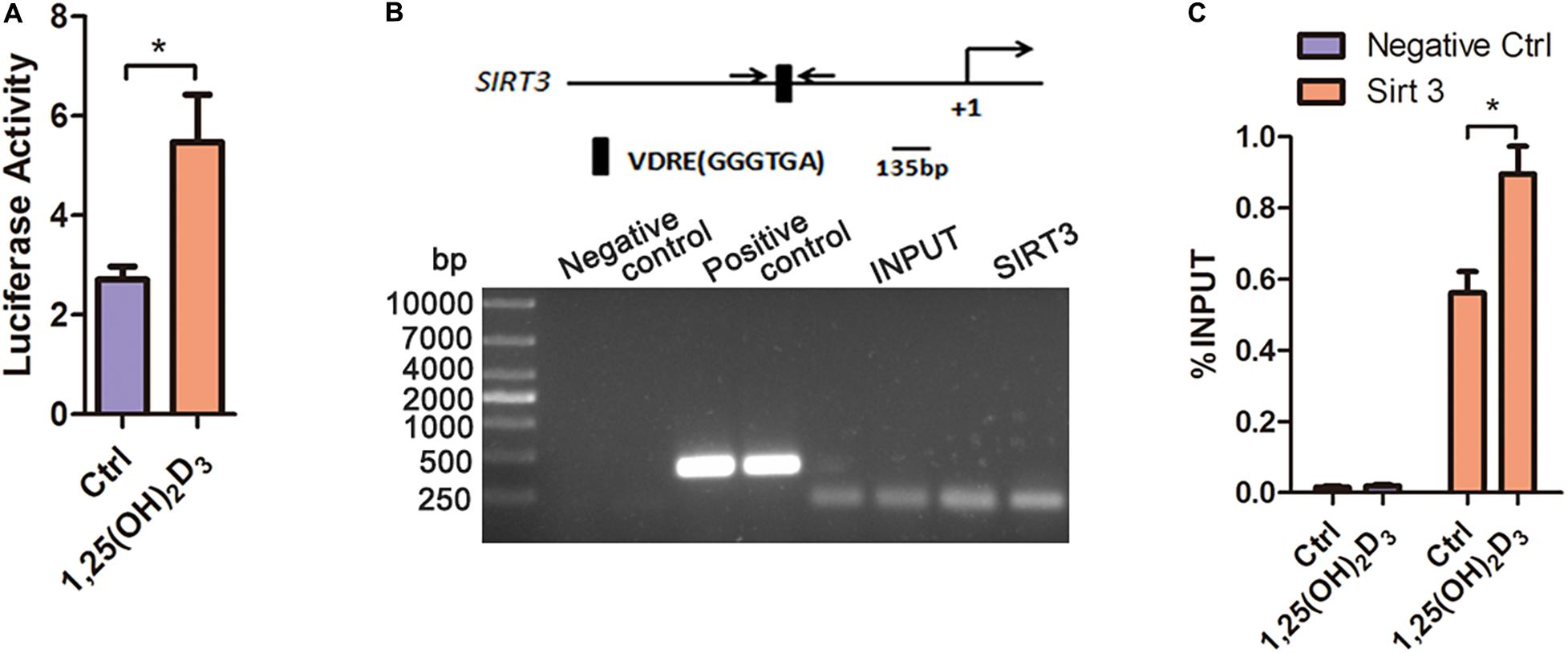
Figure 7. The binding of VDR to the VDRE in the promoter region of Sirt3 increased Sirt3 transcriptional activity. (A) Luciferase assay analysis indicated that 1,25(OH)2D3 increased Sirt3 transcriptional activity, *p < 0.05. (B,C) PCR and RT-qPCR analysis of chromatin immunoprecipitation confirmed that VD promoted the binding of VDR to the VDRE in the Sirt3 promoter, *p < 0.05.
Discussion
A high-fat diet intake can cause undesirable changes in cardiac structure and function. Mice on a high-fat diet will develop obesity, cardiac remodeling, contractile dysfunction, mitochondrial abnormalities, an abnormal ultrastructure, inflammation, cardiomyocyte apoptosis, etc (Ren et al., 2020; Gong et al., 2020; Zhang and Ren, 2016). Our experimental results were in agreement with some of the above (Figures 2D, 3D,E, 4D). CCE significantly improved cardiac function and myocardial contractility in mice with hyperlipidemia (Figure 3H). Interestingly, CCE markedly reduced the metabolic rate of hyperlipidemic mice, with no obvious changes in diet, water intake, and body temperature (Figure 2). Previous works have suggested a role for VDR in the regulation of energy metabolism in vivo (Cianferotti and Demay, 2007; Nimitphong et al., 2012). In addition, VDR–/– and Cyp27b1–/–mice were reported to exhibit increased energy expenditure (Bouillon et al., 2014). Because maintaining a certain energy substrate ratio is a prerequisite for normal cardiac function, it is speculated that VD/VDR can protect cardiomyocytes via regulation of fatty acid utilization.
Hyperlipidemia is known to compromise cardiac function (Zittermann, 2006; Forman et al., 2007; Lee et al., 2008). However, the exact underlying mechanism remains obscure. Cardiac energy metabolite dysregulation seems to be an important factor contributing to cardiovascular disease (Fillmore et al., 2014). One of our major hypotheses is that VD can regulate the unbalanced utilization of energy substrates, thereby improving myocardial function. As expected, we observed that CCE (vitamin D3) significantly reduced lipid utilization and increased glucose uptake to balance substrates in hyperlipidemic mice (Figures 3–5). The current work provided substantial evidence regarding the VD-mediated reduction of fatty acid utilization. VD deficiency caused excessive fatty acid consumption in the heart. In contrast, VD induced an increase of free lipid droplets in cardiomyocytes in vivo (Figure 3A) as well as in vitro (Figure 5A). Owing to the increase of lipid droplets in the cytoplasm, it was presumed that VD either increased cellular fat uptake or reduced fat utilization by cells. However, unlike in the control group, VD did not change the expression of CD36 (endocytosis marker) and MTTP (exocytosis marker), suggesting that the amount of lipid inflow and outflow from cells was not changed by VD (Figure 3C). In contrast, the expression of CPT-1β, a marker of mitochondrial fatty acid uptake (Zhang et al., 2016), was significantly decreased by VD, indicating that VD regulated lipid metabolism by reducing mitochondrial fatty acid utilization rather than by regulating fat turnover via endo/exocytosis.
To balance lipid metabolism, VD enhanced glucose uptake in myocardial cells via upregulating of GLUT4 (Figure 3C). GLUT4 has a protective effect against hyperglycemia in diet-induced obese mice (Atkinson et al., 2013). Therefore, we suggest that VD induced GLUT4 to maintain the ratio between fat and glucose. Mitochondria have been established as essential organelles for the oxidation of fatty acids, with mitochondrial damage potentially leading to cardiac insufficiency (Drose et al., 2014; Fillmore et al., 2014). As expected, VD considerably decreased the mitochondrial and oxidative damage induced by hyperlipidemia (Figure 4).
SIRT3 is a well-established regulatory factor of lipid metabolism, which participates in mitochondrial deacetylation (Sundaresan et al., 2009; Sol et al., 2012). Cardiac dysfunction, interstitial fibrosis, myocardial cell apoptosis, and mitochondrial damage, accompanied by autophagy and mitochondrial inhibition in streptozotocin-induced diabetes models were exacerbated by knocking out Sirt3. In contrast, SIRT3 overexpression activated autophagy and the mitochondria, inhibiting mitochondrial damage as well as cardiomyocyte apoptosis, thereby protecting cardiomyocytes in vitro (Yu et al., 2017). However, as observed in previous studies, a high-fat diet induced an upregulated fatty acid oxidation rate and decreased SIRT3 expression. We also confirmed that VD improved cardiac function by upregulating SIRT3 (Figures 4, 5). SIRT3 regulates mitochondrial fatty acid oxidation through LCAD, a downstream target of SIRT3 (Hirschey et al., 2010; Zhao et al., 2010; Hirschey et al., 2011; Rardin et al., 2013). Furthermore, a high-fat diet or Sirt3 knockout effectively enhanced LCAD acetylation, resulting in increased fatty acid peroxidation (Alrob et al., 2014). In particular, SIRT3 regulated mitochondrial fatty acid oxidation by inhibiting lysine 318/322 acetylation near the LCAD active site (Bharathi et al., 2013). As expected, our results revealed that VD considerably suppressed LCAD expression, presumably reducing the effects of mitochondrial lipid peroxidation (Figures 5D,E). These observations suggested that VD regulates fatty acid metabolism through the upregulation of SIRT3 to suppress LCAD in cardiomyocytes. To further confirm that VD regulated cardiac fatty acid metabolism through SIRT3, AGK7 and Sirt3-Cas9 plasmids were used to block SIRT3 activity and expression, respectively, in HL-1 cardiomyocytes. The Oil Red O staining and Seahorse analysis indicated that the regulation of fatty acid metabolism by VD was dependent on SIRT3 (Figure 6). Previously carried out acetylation proteomics revealed that the hyperacetylation of mitochondrial proteins was closely related to lower levels of NAD+, the co-substrate of mitochondrial complexes and SIRT3 (Hirschey et al., 2011). The production of ATP required mitochondrial complexes as well as cofactors, the availability of which was directly determined by intracellular NAD+/NADH levels (Drose et al., 2014). A defect of complex I led to a decrease in the NAD+/NADH ratio, which caused the hyperacetylation of mitochondrial protein in cardiac tissue (Karamanlidis et al., 2013). As CCE significantly induced complex I expression, we speculated that another mechanism through which VD regulated fatty acid metabolism may be the indirect enhancement of SIRT3 via complex I upregulation. This may represent another cardioprotective mechanism of VD.
The hydroxylation of vitamin D3 by 25-hydroxylase (CYP2R1 or CYP27A1) generates 25(OH)D3, which is further hydroxylated by CYP27B1 to produce biologically active 1,25 (OH)2D3. Under the action of CYP11A1, VD is converted into mono-di- and trihydroxy-D3 products, which are then further modified by CYP27B1, CYP27A1 and CYP24A1 (Slominski et al., 2012, 2014a, 2015a,b, 2017). Knockout of Cyp27b1 can prevent mice from synthesizing active VD and lead to VD deficiency. The active form of VD3 exerts pleiotropic activity, and these different effects are mediated through interaction with VDR and/or with the retinoic acid orphan receptors (ROR)α, RORγ and aryl hydrocarbon receptor (Slominski et al., 2014b, 2018, 2020). VD regulates gene transcription through VDR activation, which then binds to the VDRE in target gene promoter regions as a heterodimer along with the retinoid X receptor (Haussler et al., 1997; Barrett et al., 2016). Therefore, we predicted and confirmed that VDR modulated Sirt3 expression by binding to the VDRE within the Sirt3 promoter region (Figure 7). These results further confirmed that VD regulated SIRT3 under hyperlipidemia.
In summary, we identified that the VD/VDR signaling system balanced the utilization of energy substrates to protect the heart against hyperlipidemia (Figure 8). The novel mechanisms underlying these cardioprotective effects included the following: VD/VDR (1) regulated the expression of Sirt3 to suppress LCAD, (2) indirectly induced mitochondria complex expression via SIRT3 to prevent oxidative damage, (3) regulated transcriptional activity by binding to the VDRE within the Sirt3 promoter, (4) upregulated glucose transport, and (5) suppressed mitochondrial fatty acid utilization to balance energy substrates.
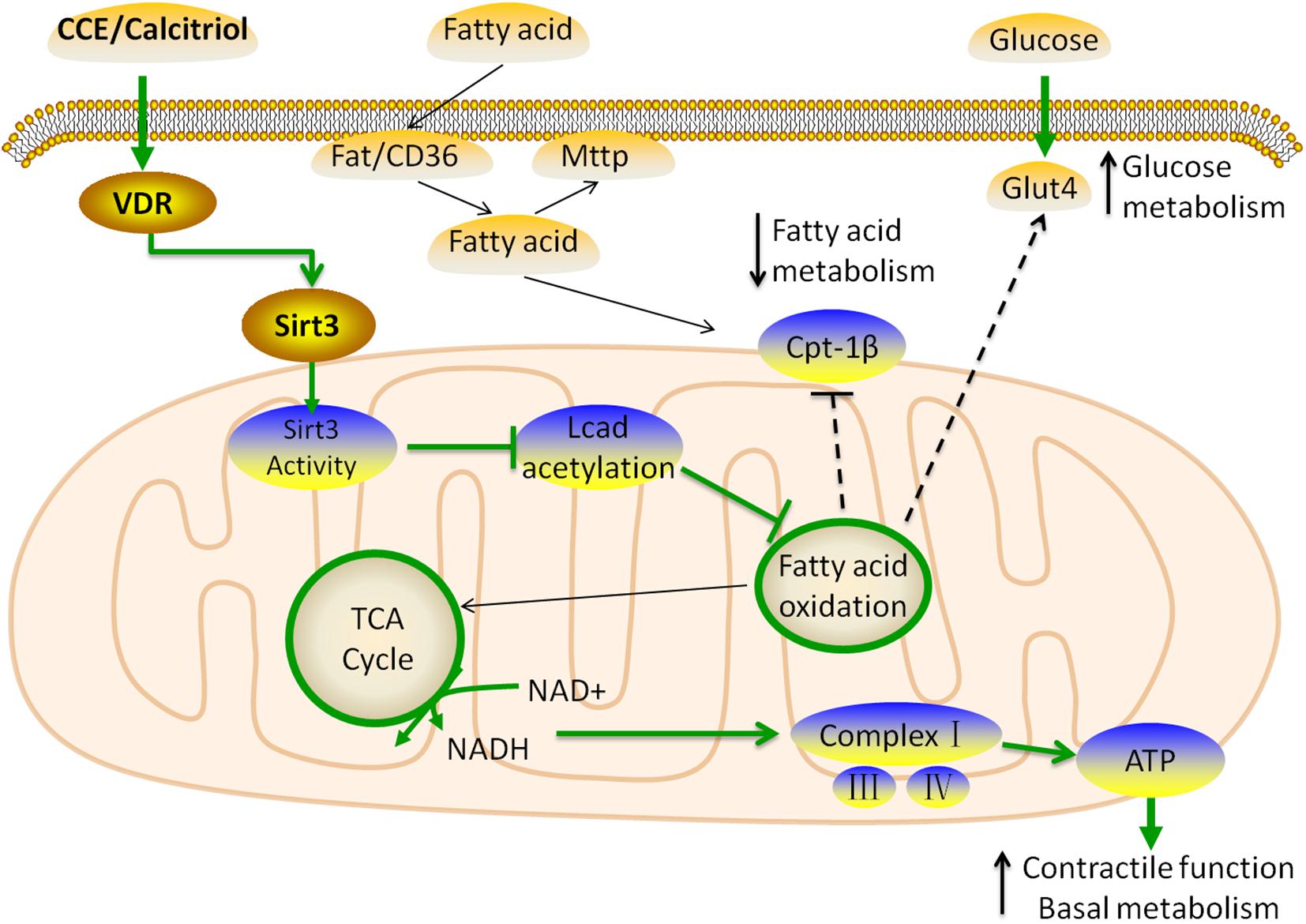
Figure 8. Schematic model for the role of vitamin D in the regulation of cardiac fatty acid metabolism. The VDR activated by CCE and calcitriol upregulated the expression of Sirt3 through transcriptional regulation. SIRT3 inhibits the expression of LCAD and stimulates that of mitochondrial complexes through the use of common substrate NAD+, resulting in decreased fatty acid β-oxidation. VDR does not affect the inflow (Fat/CD36) and outflow (MTTP) of fatty acids, and ultimately suppresses CPT-1β to reduce the uptake of fatty acids in the mitochondria. In addition, to balance energy substrate use, VDR upregulates GLUT4, thus, promoting glucose metabolism. These changes lead to a balance of glucose and fatty acid metabolism in myocardial cells, which is beneficial to myocardial function.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The animal study was reviewed and approved by Animal Experimental Committee of China Medical University.
Author Contributions
JK, JY, and YZ: conceptualization. JY and YZ: methodology. CS and YP: software. JY, YZ, NL, ZL, XL, and YL: investigation. JK: resources. JY and YZ: writing—original draft preparation. JY, YZ, and JK: writing—review and editing. JK: funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the National Natural Science Foundation of China, protocol no. (82073545,81570811); Clinical projects in ShengJing Hospital in 2019–Clinical study on homocysteine and vitamin/trace elements in blood; the Newly Established Nationwide Medical Experts Committee Funded Program in 2018; and ShengJing Hospital 345 Talent Project and CNS-ZD Tizhi and Health Fund, protocol no.CNS-ZD2019059.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank ZhaoYue for the COS-7 cell line. We would like to thank Dengshun Miao from Nanjing Medical University for the Cyp27b1-/- mice. We would like to thank the Experimental and Animal Center in Shengjing Hospital of China Medical University for their support with the experiment.
Abbreviations
VD, vitamin D; VDR, vitamin D receptor; 1 α (OH)ase, 1-alpha-hydroxylase; CCE, cholecalciterol cholesterol emulsion; LCAD, long-chain fatty acyl-CoA dehydrogenase; OA, oleic acid; MDA, malondialdehyde; SOD, superoxide dismutase; OCR, oxygen consumption rate; ECAR, extracellular acidification rate
References
Alrob, O. A., Sankaralingam, S., Ma, C., Wagg, C. S., Fillmore, N., Jaswal, J. S., et al. (2014). ‘Obesity-induced lysine acetylation increases cardiac fatty acid oxidation and impairs insulin signalling’. Cardiovasc. Res. 103, 485–497. doi: 10.1093/cvr/cvu156
Atkinson, B. J., Griesel, B. A., King, C. D., Josey, M. A., and Olson, A. L. (2013). ‘Moderate GLUT4 overexpression improves insulin sensitivity and fasting triglyceridemia in high-fat diet-fed transgenic mice’. Diabetes 62, 2249–2258. doi: 10.2337/db12-1146
Barrett, K. G., Fang, H., Kocarek, T. A., and Runge-Morris, M. (2016). ‘Transcriptional regulation of cytosolic sulfotransferase 1C2 by vitamin D receptor in LS180 human colorectal adenocarcinoma cells’. Drug Metab. Dispos. 44, 1431–1434. doi: 10.1124/dmd.116.070300
Bharathi, S. S., Zhang, Y., Mohsen, A. W., Uppala, R., Balasubramani, M., Schreiber, E., et al. (2013). ‘Sirtuin 3 (SIRT3) protein regulates long-chain acyl-CoA dehydrogenase by deacetylating conserved lysines near the active site’. J. Biol. Chem. 288, 33837–33847. doi: 10.1074/jbc.M113.510354
Bouillon, R., Carmeliet, G., Lieben, L., Watanabe, M., Perino, A., Auwerx, J., et al. (2014). Vitamin D and energy homeostasis: of mice and men. Nat. Rev. Endocrinol. 10, 79–87. doi: 10.1038/nrendo.2013.226
Brondum-Jacobsen, P., Benn, M., Jensen, G. B., and Nordestgaard, B. G. (2012). 25-hydroxyvitamin d levels and risk of ischemic heart disease, myocardial infarction, and early death: population-based study and meta-analyses of 18 and 17 studies. Arterioscler. Thromb. Vasc. Biol. 32, 2794–2802. doi: 10.1161/ATVBAHA.112.248039
Cianferotti, L., and Demay, M. B. (2007). ‘VDR-mediated inhibition of DKK1 and SFRP2 suppresses adipogenic differentiation of murine bone marrow stromal cells’. J. Cell Biochem. 101, 80–88. doi: 10.1002/jcb.21151
Drose, S., Brandt, U., and Wittig, I. (2014). ‘Mitochondrial respiratory chain complexes as sources and targets of thiol-based redox-regulation’. Biochim. Biophys. Acta 1844, 1344–1354. doi: 10.1016/j.bbapap.2014.02.006
Fillmore, N., Mori, J., and Lopaschuk, G. D. (2014). ‘Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy’. Br. J. Pharmacol. 171, 2080–2090. doi: 10.1111/bph.12475
Forman, J. P., Giovannucci, E., Holmes, M. D., Bischoff-Ferrari, H. A., Tworoger, S. S., Willett, W. C., et al. (2007). ‘Plasma 25-hydroxyvitamin D levels and risk of incident hypertension’. Hypertension 49, 1063–1069. doi: 10.1161/HYPERTENSIONAHA.107.087288
Fukushima, A., and Lopaschuk, G. D. (2016). ‘Acetylation control of cardiac fatty acid beta-oxidation and energy metabolism in obesity, diabetes, and heart failure’. Biochim. Biophys. Acta 1862, 2211–2220. doi: 10.1016/j.bbadis.2016.07.020
Gong, Y., Li, G., Tao, J., Wu, N. N., Kandadi, M. R., Bi, Y., et al. (2020). ‘Double knockout of Akt2 and AMPK accentuates high fat diet-induced cardiac anomalies through a cGAS-STING-mediated mechanism’. Biochim. Biophys. Acta Mol. Basis Dis. 1866:165855. doi: 10.1016/j.bbadis.2020.165855
Haussler, M. R., Haussler, C. A., Jurutka, P. W., Thompson, P. D., Hsieh, J. C., Remus, L. S., et al. (1997). ‘The vitamin D hormone and its nuclear receptor: molecular actions and disease states’. J. Endocrinol. 154(Suppl.), S57–S73.
Hirschey, M. D., Shimazu, T., Goetzman, E., Jing, E., Schwer, B., Lombard, D. B., et al. (2010). ‘SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation’. Nature 464, 121–125. doi: 10.1038/nature08778
Hirschey, M. D., Shimazu, T., Jing, E., Grueter, C. A., Collins, A. M., Aouizerat, B., et al. (2011). ‘SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome’. Mol. Cell 44, 177–190. doi: 10.1016/j.molcel.2011.07.019
Karamanlidis, G., Lee, C. F., Garcia-Menendez, L., Kolwicz, S. C., Suthammarak, W., Gong, G., et al. (2013). Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab. 18, 239–250. doi: 10.1016/j.cmet.2013.07.002
Kendrick, A. A., Choudhury, M., Rahman, S. M., McCurdy, C. E., Friederich, M., Van Hove, J. L. K., et al. (2011). ‘Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation’. Biochem. J. 433, 505–514. doi: 10.1042/BJ20100791
Kong, J., Kim, G. H., Wei, M., Sun, T., Li, G., Liu, S. Q., et al. (2010). ‘Therapeutic effects of vitamin D analogs on cardiac hypertrophy in spontaneously hypertensive rats’. Am. J. Pathol. 177, 622–631. doi: 10.2353/ajpath.2010.091292
Kong, J., Zhu, X., Shi, Y., Liu, T., Chen, Y., Bhan, I., et al. (2013). ‘VDR attenuates acute lung injury by blocking Ang-2-Tie-2 pathway and renin-angiotensin system’. Mol. Endocrinol. 27, 2116–2125. doi: 10.1210/me.2013-1146
Ku, Y. C., Liu, M. E., Ku, C. S., Liu, T. Y., and Lin, S. L. (2013). ‘Relationship between vitamin D deficiency and cardiovascular disease’. World J. Cardiol. 5, 337–346. doi: 10.4330/wjc.v5.i9.337
Lawrence, R. C., Helmick, C. G., Arnett, F. C., Deyo, R. A., Felson, D. T., Giannini, E. H., et al. (1998). ‘Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States’. Arthritis Rheum. 41, 778–799. doi: 10.1002/1529-0131(199805)41:5<778::AID-ART4>3.0.CO;2-V
Lee, J. H., O’Keefe, J. H., Bell, D., Hensrud, D. D., and Holick, M. F. (2008). ‘Vitamin D deficiency an important, common, and easily treatable cardiovascular risk factor?’. J. Am. Coll. Cardiol. 52, 1949–1956. doi: 10.1016/j.jacc.2008.08.050
Lombard, D. B., Alt, F. W., Cheng, H. L., Bunkenborg, J., Streeper, R. S., Mostoslavsky, R., et al. (2007). ‘Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation’. Mol. Cell. Biol. 27, 8807–8814. doi: 10.1128/MCB.01636-07
Moya, M., Gomez-Lechon, M. J., Castell, J. V., and Jover, R. (2010). ‘Enhanced steatosis by nuclear receptor ligands: a study in cultured human hepatocytes and hepatoma cells with a characterized nuclear receptor expression profile’. Chem. Biol. Interact. 184, 376–387. doi: 10.1016/j.cbi.2010.01.008
Nimitphong, H., Holick, M. F., Fried, S. K., and Lee, M. J. (2012). ‘25-hydroxyvitamin D(3) and 1,25-dihydroxyvitamin D(3) promote the differentiation of human subcutaneous preadipocytes’. PLoS One 7:e52171. doi: 10.1371/journal.pone.0052171
Ran, F. A., Hsu, P. D., Wright, J., Agarwala, V., Scott, D. A., and Zhang, F. (2013). ‘Genome engineering using the CRISPR-Cas9 system’. Nat. Protoc. 8, 2281–2308. doi: 10.1038/nprot.2013.143
Rardin, M. J., Newman, J. C., Held, J. M., Cusack, M. P., Sorensen, D. J., Li, B., et al. (2013). ‘Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways’. Proc. Natl. Acad. Sci. U. S. A. 110, 6601–6606. doi: 10.1073/pnas.1302961110
Ren, J., Sun, M., Zhou, H., Ajoolabady, A., Zhou, Y., Tao, J., et al. (2020). FUNDC1 interacts with FBXL2 to govern mitochondrial integrity and cardiac function through an IP3R3-dependent manner in obesity. Sci. Adv. 6:eabc8561. doi: 10.1126/sciadv.abc8561
Sabir, M. S., Khan, Z., Hu, C., Galligan, M. A., Dussik, C. M., Mallick, S., et al. (2017). ‘SIRT1 enzymatically potentiates 1,25-dihydroxyvitamin D3 signaling via vitamin D receptor deacetylation’. J. Steroid Biochem. Mol. Biol. 172, 117–129. doi: 10.1016/j.jsbmb.2017.06.010
Sack, M. N. (2012). ‘The role of SIRT3 in mitochondrial homeostasis and cardiac adaptation to hypertrophy and aging’. J. Mol. Cell Cardiol. 52, 520–525. doi: 10.1016/j.yjmcc.2011.11.004
Sadhukhan, S., Liu, X. J., Ryu, D., Nelson, O. D., Stupinski, J. A., Li, Z., et al. (2016). ‘Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function’. Proc. Natl. Acad. Sci. U. S. A. 113, 4320–4325. doi: 10.1073/pnas.1519858113
Slominski, A. T., Brożyna, A. A., Zmijewski, M. A., Jóżwicki, W., Jetten, A. M., Mason, R. S., et al. (2017). ‘Vitamin D signaling and melanoma: role of vitamin D and its receptors in melanoma progression and management’. Lab. Invest. 97, 706–724. doi: 10.1038/labinvest.2017.3
Slominski, A. T., Chaiprasongsuk, A., Janjetovic, Z., Kim, T. K., Stefan, J., Slominski, R. M., et al. (2020). ‘Photoprotective properties of vitamin D and lumisterol hydroxyderivatives’. Cell Biochem. Biophys. 78, 165–180. doi: 10.1007/s12013-020-00913-6
Slominski, A. T., Kim, T. K., Janjetovic, Z., Brożyna, A. A., Żmijewski, M. A., Xu, H., et al. (2018). Differential and overlapping effects of 20,23(OH)2D3 and 1,25(OH)2D3 on gene expression in human epidermal keratinocytes: identification of AhR as an alternative receptor for 20,23(OH)2D3. Int. J. Mol. Sci. 19:3072. doi: 10.3390/ijms19103072
Slominski, A. T., Kim, T. K., Li, W., Postlethwaite, A., Tieu, E. W., Tang, E. K. Y., et al. (2015a). ‘Detection of novel CYP11A1-derived secosteroids in the human epidermis and serum and pig adrenal gland’. Sci. Rep. 5:14875. doi: 10.1038/srep14875
Slominski, A. T., Kim, T. K., Shehabi, H. Z., Semak, I., Tang, E. K., Nguyen, M. N., et al. (2012). ‘In vivo evidence for a novel pathway of vitamin D3 metabolism initiated by P450scc and modified by CYP27B1’. FASEB J. 26, 3901–3915. doi: 10.1096/fj.12-208975
Slominski, A. T., Kim, T. K., Shehabi, H. Z., Tang, E. K., Benson, H. A., Semak, I., et al. (2014a). ‘In vivo production of novel vitamin D2 hydroxy-derivatives by human placentas, epidermal keratinocytes, Caco-2 colon cells and the adrenal gland’. Mol. Cell. Endocrinol. 383, 181–192. doi: 10.1016/j.mce.2013.12.012
Slominski, A. T., Kim, T. K., Takeda, Y., Janjetovic, Z., Brozyna, A. A., Skobowiat, C., et al. (2014b). ‘RORα and ROR γ are expressed in human skin and serve as receptors for endogenously produced noncalcemic 20-hydroxy- and 20,23-dihydroxyvitamin D’. FASEB J. 28, 2775–2789. doi: 10.1096/fj.13-242040
Slominski, A. T., Li, W., Kim, T. K., Semak, I., Wang, J., Zjawiony, J. K., et al. (2015b). ‘Novel activities of CYP11A1 and their potential physiological significance’. J. Steroid Biochem. Mol. Biol. 151, 25–37. doi: 10.1016/j.jsbmb.2014.11.010
Sol, E. M., Wagner, S. A., Weinert, B. T., Kumar, A., Kim, H. S., Deng, C. X., et al. (2012). ‘Proteomic investigations of lysine acetylation identify diverse substrates of mitochondrial deacetylase sirt3’. PLoS One 7:e50545. doi: 10.1371/journal.pone.0050545
Su, H., Lou, Y., Fu, Y., Zhang, Y., Liu, N., Liu, Z., et al. (2017). ‘Involvement of the vitamin D receptor in energy metabolism revealed by profiling of lysine succinylome of white adipose tissue’. Sci. Rep. 7:14132. doi: 10.1038/s41598-017-14477-8
Sundaresan, N. R., Gupta, M., Kim, G., Rajamohan, S. B., Isbatan, A., and Gupta, M. P. (2009). ‘Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice’. J. Clin. Invest. 119, 2758–2771. doi: 10.1172/JCI39162
van Bilsen, M. (2004). “‘Energenetics” of heart failure’. Ann. N. Y. Acad. Sci. 1015, 238–249. doi: 10.1196/annals.1302.020
Vassilopoulos, A., Pennington, J. D., Andresson, T., Rees, D. M., Bosley, A. D. I, Fearnley, M., et al. (2014). ‘SIRT3 deacetylates ATP synthase F1 complex proteins in response to nutrient- and exercise-induced stress’. Antioxid. Redox. Signal 21, 551–564. doi: 10.1089/ars.2013.5420
Vluggens, A., and Reddy, J. K. (2012). ‘Nuclear receptors and transcription factors in the development of fatty liver disease’. Curr. Drug Metab. 13, 1422–1435. doi: 10.2174/138920012803762710
Yu, W., Gao, B., Li, N., Wang, J., Qiu, C., Zhang, G., et al. (2017). ‘Sirt3 deficiency exacerbates diabetic cardiac dysfunction: role of Foxo3A-Parkin-mediated mitophagy’. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 1973–1983. doi: 10.1016/j.bbadis.2016.10.021
Zhang, W., Chen, L., Zhang, L., Xiao, M., Ding, J., Goltzman, D., et al. (2015). ‘Administration of exogenous 1,25(OH)2D3 normalizes overactivation of the central renin-angiotensin system in 1α(OH)ase knockout mice’. Neurosci. Lett. 588, 184–189. doi: 10.1016/j.neulet.2015.01.013
Zhang, Y., Fang, X., Dai, M., Cao, Q., Tan, T., He, W., et al. (2016). ‘Cardiac-specific down-regulation of carnitine palmitoyltransferase-1b (CPT-1b) prevents cardiac remodeling in obese mice’. Obesity (Silver Spring) 24, 2533–2543. doi: 10.1002/oby.21665
Zhang, Y., and Ren, J. (2016). ‘Epigenetics and obesity cardiomyopathy: from pathophysiology to prevention and management’. Pharmacol. Ther. 161, 52–66. doi: 10.1016/j.pharmthera.2016.03.005
Zhao, S., Xu, W., Jiang, W., Yu, W., Lin, Y., Zhang, T., et al. (2010). ‘Regulation of cellular metabolism by protein lysine acetylation’. Science 327, 1000–1004. doi: 10.1126/science.1179689
Keywords: Vitamin D, fatty acid metabolism, Sirtuin 3, mitochondria, cardiac function
Citation: Yang J, Zhang Y, Pan Y, Sun C, Liu Z, Liu N, Fu Y, Li X, Li Y and Kong J (2021) The Protective Effect of 1,25(OH)2D3 on Myocardial Function is Mediated via Sirtuin 3-Regulated Fatty Acid Metabolism. Front. Cell Dev. Biol. 9:627135. doi: 10.3389/fcell.2021.627135
Received: 08 November 2020; Accepted: 30 March 2021;
Published: 26 April 2021.
Edited by:
Maria Barile, University of Bari Aldo Moro, ItalyReviewed by:
Andrzej T. Slominski, University of Alabama at Birmingham, United StatesJun Ren, University of Washington, United States
Copyright © 2021 Yang, Zhang, Pan, Sun, Liu, Liu, Fu, Li, Li and Kong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan Kong, a29uZ2pAY211LmVkdS5jbg==
†These authors have contributed equally to this work