 Suresh Kumar1*
Suresh Kumar1* Trilochan Mohapatra2
Trilochan Mohapatra2- 1Division of Biochemistry, ICAR-Indian Agricultural Research Institute, New Delhi, India
- 2Indian Council of Agricultural Research, New Delhi, India
Gene regulation depends on dynamic and reversibly modifiable biological and chemical information in the epigenome/epitranscriptome. Accumulating evidence suggests that messenger RNAs (mRNAs) are generated in flashing bursts in the cells in a precisely regulated manner. However, the different aspects of the underlying mechanisms are not fully understood. Cellular RNAs are post-transcriptionally modified at the base level, which alters the metabolism of mRNA. The current understanding of epitranscriptome in the animal system is far ahead of that in plants. The accumulating evidence indicates that the epitranscriptomic changes play vital roles in developmental processes and stress responses. Besides being non-genetically encoded, they can be of reversible nature and involved in fine-tuning the expression of gene. However, different aspects of base modifications in mRNAs are far from adequate to assign the molecular basis/functions to the epitranscriptomic changes. Advances in the chemogenetic RNA-labeling and high-throughput next-generation sequencing techniques are enabling functional analysis of the epitranscriptomic modifications to reveal their roles in mRNA biology. Mapping of the common mRNA modifications, including N6-methyladenosine (m6A), and 5-methylcytidine (m5C), have enabled the identification of other types of modifications, such as N1-methyladenosine. Methylation of bases in a transcript dynamically regulates the processing, cellular export, translation, and stability of the mRNA; thereby influence the important biological and physiological processes. Here, we summarize the findings in the field of mRNA base modifications with special emphasis on m6A, m5C, and their roles in growth, development, and stress tolerance, which provide a new perspective for the regulation of gene expression through post-transcriptional modification. This review also addresses some of the scientific and technical issues in epitranscriptomic study, put forward the viewpoints to resolve the issues, and discusses the future perspectives of the research in this area.
Introduction
From the genome to proteome, several proficient biological processes regulate cellular growth and functions. Transcription of a gene is a truthful process, as the timing and rate of transcription are subjected to strict regulation, and its accuracy is vital for the vigor and development of the cell (Wang et al., 2018). Because translation of mRNA is a vital process in all living organisms, and assembly of the translational machinery followed by movement along the mRNA consumes ∼40% of cellular energy, the process needs to be precisely regulated to conserve energy. The ‘Central Dogma of life’ describes that genetic information is transformed from DNA to protein through RNA. Both DNA and histone proteins are reversibly modified (epigenetic modifications) to fine-tune the expression of genes/phenotypes (Fu Y. et al., 2014). An analogous process for RNA (epitranscriptomic modification) has been a missing component of the central dogma (Figure 1). Reversible biochemical modifications are known now to occur in most of the constituent processes of the central dogma, which dynamically control gene expression. The spectrum of epigenetic base modifications detected so far in DNA is relatively limited (six), about 170 distinct modifications have been identified in RNAs (Boccaletto et al., 2018; Kadumuri and Janga, 2018; Shen et al., 2019; Boo and Kim, 2020; Selmi et al., 2021). RNAs play vital roles in biological systems, not only as structural components [i.e., ribosomal RNAs (rRNAs)], translators [i.e., transfer RNAs (tRNAs)], and messengers (i.e., mRNAs, conveying genetic information to the protein) but also as regulators [i.e., small interfering RNAs (siRNAs), enhancer RNAs (eRNAs)] of several biological processes. The functions of rRNAs, tRNAs, and mRNAs are regulated through co- or post-transcriptional chemical modifications (Boccaletto et al., 2018; Boo and Kim, 2020), the exact role of many of these base modifications remain enigmatic. Although extensive base modifications in rRNAs and tRNAs in terms of the variety/abundance of modifications are well known and have remained undisputed for many decades (Jackman and Alfonzo, 2013), all other classes of RNA are subjected to enzymatic modifications (Xu L. et al., 2017). Several post-transcriptional base modifications in messenger RNA (mRNA) have only recently been identified. Such mRNA base modifications affect different cellular processes like pre-mRNA splicing, mRNA export, translation, and degradation, which shape the cellular transcriptome and proteome. Recent findings indicate that the level of proteins in a cell does not necessarily correspond with the mRNA level (Khan et al., 2013; Wu et al., 2013), which might vary because of various post-transcriptional regulation, including epitranscriptomic modifications affecting mRNA biology. The recent advances in experimental techniques have facilitated the identification of different epitranscriptomic modifications in the coding and untranslated regions (UTRs) of mRNAs (Zhao et al., 2020). While the functions of some of the epitranscriptomic modifications are known, occurrence and function of many other diverse epitranscriptomic modifications are still to be established.
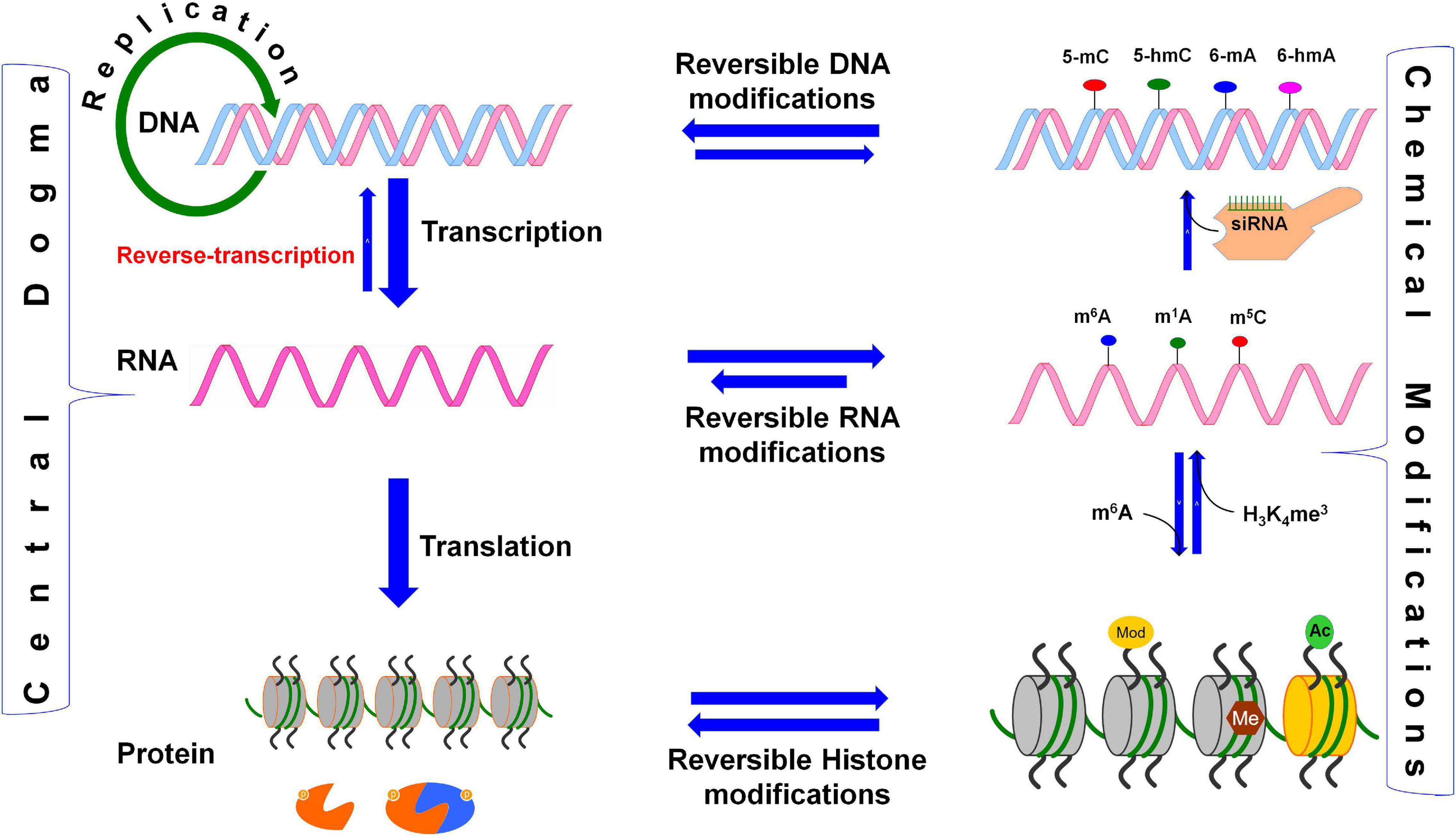
Figure 1. Reversible biochemical modifications affect the transfer of genetic information (the Central Dogma). As per the central dogma, the genetic information passes from DNA, through RNA, to protein. However, epigenetic DNA base modifications [e.g., 5-methylcytosine (5-mC), 5-hydroxymethylcytosine (5-hmC), N6-methyladenine (6-mA), and N6-hydroxymethyladenine (6-hmA)] and histone protein modifications [e.g., methylation (me) and acetylation (ac) of amino acids] affects RNA metabolism (including splicing, export, stability, and translation efficiency) and play crucial roles in the regulation of cellular growth, development, and protection from environmental stress. Similarly, the dynamic RNA modifications [e.g., N6-methyladenosine (m6A), N1-methyladenosine (m1A), and N6-hydroxymethyladenosine (hm6A)] encrypt an additional layer of information and dynamically regulate the biological processes. Small-interfering RNA (siRNA) plays important role in recruitment of DNA methyltranferase for DNA base modification, methylated mRNA bases (e.g., m6A) play role in protein synthesis, the histone 3 (H3) protein trimethylated (me3) at 4th lysine of (H3K4me3) affects the transcription process.
The dynamic and reversible RNA base modifications are catalyzed by distinct enzymes like methyltransferases (writers), and removed by demethylases (erasers). These modifications are interpreted by a modification-specific binding proteins known as readers. Characterization of writers, readers, and erasers is further advancing our epitranscriptomic understanding of functional genomics. Similar to the epigenetic modifications of DNA bases (Kumar et al., 2018), mRNA base modifications provide another layer of information created by the writers/erasers and interpreted by the readers. Like the reversible nature of DNA base modifications (Wang et al., 2016), some of the mRNA base modifications are known to be reversed by their respective eraser. Although translation process is typically controlled by translation factors and certain non-coding RNAs (ncRNAs), base modifications play equally important role in mRNA metabolism and translation process. Thus, the mRNA base modifications create the epitranscriptomic regulatory machinery that is being elucidated in the animal as well as the plant systems. It is now apparent that mRNA is a dynamic and reversibly modifiable biomolecule (Figure 2) that play crucial roles in post-transcriptional regulation of gene expression (Zhao et al., 2017a).
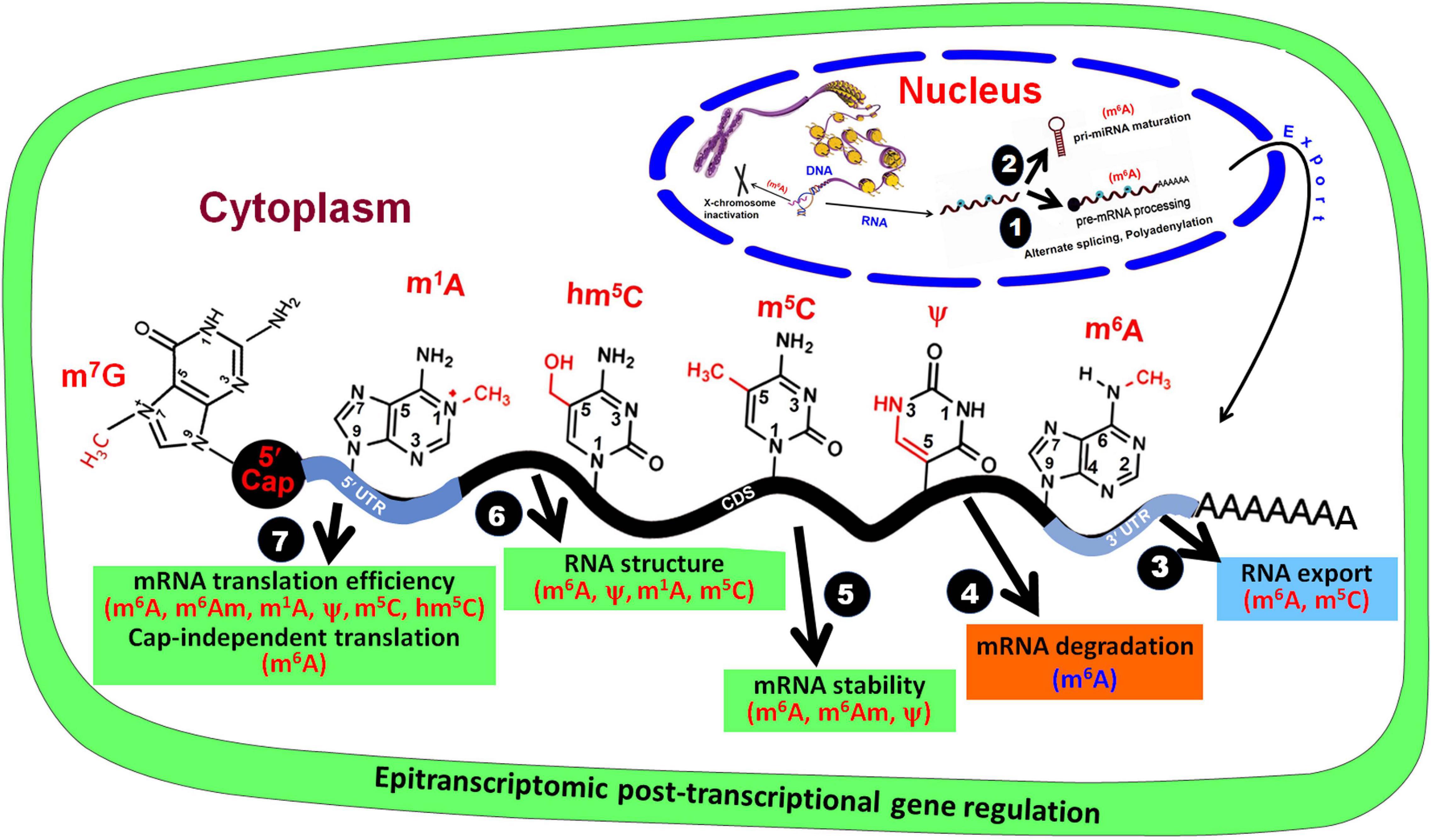
Figure 2. Base modifications in mRNA affect post-transcriptional gene regulation. In the nucleus, RNA base modifications affect (1) pre-mRNA processing and (2) pri-miRNA maturation, and (3) their export from the nucleus. In the cytoplasm, RNA base modifications regulate (4) mRNA degradation, (5) mRNA stability, (6) RNA structure, and (7) mRNA translation efficiency.
Many of the mRNA base modifications involve attachment of a methyl (CH3) group at a particular position either on the base [e.g., N6-methyladenosine (m6A), N1-methyladenosine (m1A), 5-methylcytidine (m5C), 3-methylcytidine (m3C), N7-methylguanosine (m7G), and 1-methylguanosine (m1G)], ribose sugar (e.g., 2′-O-methyladenosine), or on both base and sugar [e.g., N6,2′-O-dimethyladenosine (m6Am)] (Dominissini et al., 2012; Linder et al., 2015; Dominissini et al., 2016; Molinie et al., 2016; Mauer et al., 2017). Thus, methylation of bases at different position has distinct impact on RNA biology by affecting folding, stability, cellular localization, and/or interaction with other RNAs/proteins (Wu et al., 2016). The m6A is one of the most common, reversible epitranscriptomic marks, functionally pertinent in both animal and plant mRNAs (Batista et al., 2014; Shen et al., 2016). Moreover, writers, readers, and erasers for m6A are known in animals as well as plants (Bokar et al., 1997; Zhong et al., 2008; Liu et al., 2014; Du et al., 2016; Liu and Pan, 2016; Patil et al., 2016; Roundtree and He, 2016; Martinez-Perez et al., 2017; Zhao et al., 2017b; Arribas-Hernandez et al., 2018; Scutenaire et al., 2018; Wei et al., 2018; Liang et al., 2020). The m6A destabilizes A = U pairing due to altered energetics/steric hindrance; however, the donor and acceptor in the hydrogen bond remain the same (Roost et al., 2015).
On the other hand, CH3 of m1A in RNA provides a positive charge (which interacts with negatively charged phosphate in the backbone) and it bulges out of the Watson–Crick hydrogen bond resulting in a strong electrostatic interaction (Helm, 2006). Moreover, 2′-O-methylation confers hydrophobicity, which protects the RNA from nucleolytic attack and stabilizes RNA coiling (Kumar et al., 2014). Thus, structure and functions of dynamic RNA modifications during the developmental process and environmental stress, and their effects on gene expression have emerged as a new branch of functional genomics known as ‘epitranscriptomics.’
To decipher the biological functions of a modified RNA base, it is vital to identify the writer/reader/eraser that modulates the modification. However, high-throughput detection methods for many of these modifications are still lacking. The recent advances in high-throughput next-generation sequencing (NGS) together with the novel chemogenetic RNA-labelng techniques have provided unprecedented opportunities to understand the RNA structure and functions. Such advances provide a better understanding of the presence and dynamics of base modifications like m6A (Zhao et al., 2017b), m5C (Cui Q. et al., 2017; David et al., 2017; Fang et al., 2020), 5-hydroxymethylcytidine (hm5C) (Huber et al., 2015; Delatte et al., 2016; Zhang et al., 2016), and m1A (Dominissini et al., 2016; Li et al., 2016a; Shen et al., 2016; Xiong et al., 2018) in RNAs. Base modifications, such as addition of 5′ cap (e.g., N7-methylguanosine, m7G-cap), and RNA editing are vital for mRNA stability (Kiledjian, 2018), translation (Topisirovic et al., 2011; Holstein et al., 2016) and functional diversity (Peng et al., 2018). More importantly, NAD+ has been reported to be a new/alternative RNA cap in diverse organisms including bacteria, yeast, human (Cahová et al., 2015; Jiao et al., 2017; Walters et al., 2017; Frindert et al., 2018), and plant (Wang et al., 2019). Thousands of transcripts for the protein-coding genes from nuclear and mitochondrial genomes in Arabidopsis were observed to contain NAD+ cap (Wang et al., 2019). These clearly indicate that NAD+ cap is one of the evolutionarily conserved caps that affects mRNA metabolic processes. A comprehensive understanding of the distribution, function, and regulation of RNA base modification will further increase the available knowledge on epitranscriptomic regulation of gene expression.
Epitranscriptomic base modifications have become an interesting topic of research and review, particularly in the animal system (Meyer et al., 2012; Carlile et al., 2014; Fu Y. et al., 2014; Dominissini et al., 2016; Peer et al., 2017; Angelova et al., 2018; Arribas-Hernandez et al., 2018; Khoddami et al., 2019; Leonardi et al., 2020). Now, the epitranscriptomic modifications in plants like Arabidopsis (Luo et al., 2014; Wan et al., 2015; Shen et al., 2016; Zuber et al., 2016; Cui Q. et al., 2017; David et al., 2017; Duan et al., 2017), rice (Li Y. et al., 2014), maize (Luo et al., 2019; Miao et al., 2019), and tomato (Zhou et al., 2019) are also being studied. However, our knowledge of plant epitranscriptomic modifications, except for the 5′-cap and poly-A-tail, is limited to uridylation (de Almeida et al., 2018), m6A (Li et al., 2018), and m5C (Cui X. et al., 2017; David et al., 2017). Other types of modifications can also be expected to occur in plant mRNAs but their existence/detection and roles/functions remain to be explored. Considering the crucial and dynamic roles of epitranscriptomic modifications in many biological processes like embryo development, leaf morphogenesis, root development, floral transition, fruit ripening, and stress tolerance, the importance and future perspectives of epitranscriptomic research in plants are being discussed (Hu et al., 2019; Shen et al., 2019; Liang et al., 2020). The present review focuses on recent developments in base modifications in RNAs, particularly m6A and m5C in plant mRNAs, their biochemical properties, and functions. Moreover, the review discusses technological advances in high-throughput detection methods to elucidate epitranscriptomic modifications, as well as the technological limitations. Further advances in the next-generation detection techniques and functional analysis of RNA base modifications might facilitate epitranscriptomic manipulation of the traits of interest.
Biochemistry of Adenosine Methylation in mRNA
Methylation of adenosine (A) at N6 position [in both syn- (energetically favored) and anti-conformation] results in the formation of m6A (Zou et al., 2016). The methyltransferase-like 14 (METTL14) complex and Wilm’s tumor-associated protein (WTAP) work in cooperation with METTL3, and cofactors KIAA1429, RBM15/RBM15B which constitute a functional methyltransferase to create m6A in mammalian mRNAs at a consensus sequence of R−−m6A−C−H (where R = A/G, and H = A/C/U) (Patil et al., 2016). Emerging evidence suggests that VIRMA/KIAA1429 recruits the catalytic core (METTL3/WTAP/METTL14) for a sequence-specific methylation of A to m6A (Yue et al., 2018). Recent studies suggest that ZC3H13 is another component of the m6A writer-complex, and it regulates the methylation of A (Knuckles et al., 2018; Wen et al., 2018). Moreover, m6A mark gets erased by the enzymes like fat mass and obesity-associated protein (FTO) and alkylation repair homolog protein 5 (ALKBH5), which convert it back to A (Jia et al., 2011). FTO oxidatively removes m6A through N6-hydroxymethyladenosine (hm6A) and N6-formyladenosine (f6A) intermediates (Fu Y. et al., 2014). Thus, m6A is a reversible epitranscriptomic modification, which functions to regulate gene expression.
m6A Writer
An RNA methyltransferase complex is comprised of methyltransferase-like 3 (METTL3) (Bokar et al., 1997), METTL14 (Liu et al., 2014), KIAA1429/VIRMA (Schwartz et al., 2014b; Yue et al., 2018), HAKAI (Ruzicka et al., 2017), RNA binding motif protein 15 (RBM15) (Patil et al., 2016), Wilm’s tumor 1-associating protein (WTAP) (Ping et al., 2014), and a zinc finger CCCH domain-containing protein 13 (ZC3H13) (Frye et al., 2018; Wen et al., 2018; Yang et al., 2018). It is involved in methylation/modification of adenosine to m6A in mammals. While METTL3 is known to methylate single-stranded RNAs (ssRNAs) in a sequence-specific (RRACH) manner, METTL16 methylates structured RNAs having a nonamer sequence (UACAGAGAA; the targeted adenosine for methylation is marked with bold face) (Pendleton et al., 2017). Thus, METTL16 is another m6A-specific methyltransferase which targets U6 snRNA and human MAT2A mRNA encoding for S-adenosylmethionine (SAM) synthetase (Pendleton et al., 2017). Interestingly, SAM is the methyl group donor for methylation of DNA, RNA, and proteins.
In Arabidopsis, the m6A writer complex is composed of adenosine methyltransferase (MTA) (METTL3 ortholog), its homolog MTB (METTL14 ortholog), FKBP12 interacting protein 37 (FIP37) (WTAP ortholog), VIRLIZER/KIAA1229 (VIR), and HAKAI (Ruzicka et al., 2017) (Table 1). Although the components of plant writer complex were observed to be distributed in the nucleoplasm, but FIP37 and VIR do not affect alternative splicing of transcripts (Shen et al., 2016; Ruzicka et al., 2017). While WTAP interacts with METLL3, METTL14, VIRMA, and HAKAI in mammals (Yue et al., 2018), Arabidopsis FIP37 (a WTAP ortholog in mammals) interacts directly with MTA only (Ruzicka et al., 2017). This clearly indicates that the mechanism of adenine methylation (m6A) is conserved among the eukaryotes; however, some unique features of m6A modification might have been evolved in plants. Most of the constituents of m6A writer complex, excluding HAKAI, are needed for the embryonic development. Moreover, m6A plays diverse roles in various other developmental processes in plants. Hence even after conserved m6A modification machinery in eukaryotes, it appears that individual members of m6A writer complex has achieved functional divergence in plants.
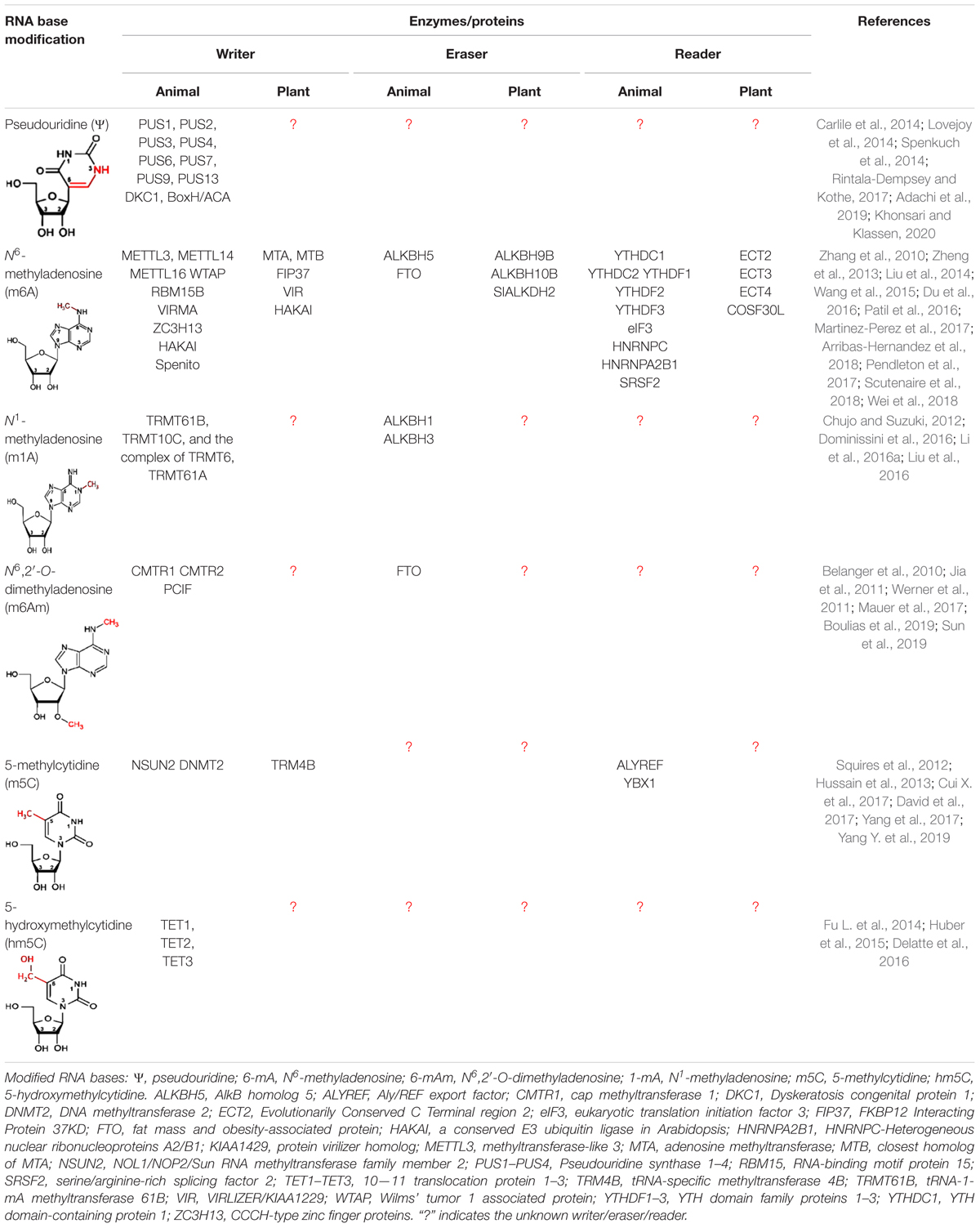
Table 1. Modified RNA bases, their modulators, and interpreters.
m6A Reader
Methyladenosine (m6A) affects several mRNA metabolic processes in both nucleus and cytoplasm through the recruitment of m6A-binding protein (RBP), also known as m6A reader (Wang et al., 2015; Yue et al., 2015; Zhou et al., 2015; Xiao et al., 2016; Li A. et al., 2017; Yang et al., 2017; Zhao et al., 2017b; Scutenaire et al., 2018; Wei et al., 2018). Two important classes of m6A readers known so far include the YTH domain-containing protein (Zhang et al., 2010) and the heterogeneous nuclear ribonucleo-protein (HNRNP) (Alarcon et al., 2015a). Arabidopsis and rice genomes contain several genes (13 and 12, respectively) for the YTH homolog known as ‘evolutionarily conserved C-terminal region’ (ECT) (Li D. et al., 2014); however, their role as an m6A-reader has only recently been recognized (Arribas-Hernandez et al., 2018; Scutenaire et al., 2018; Wei et al., 2018). These genes exhibit distinct/diverse expression pattern in different organs at different developmental stages, and under different stress conditions. A plant-specific motif URUAW (R = G or A; W = U or A) was reported in the ECT2-binding sites, which is different from the YTH-binding motif observed in human (Xiao et al., 2016; Shen et al., 2019). The binding of ECT2 at m6A increases the stability of the transcript responsible for trichome morphogenesis/development in Arabidopsis. ECT2 functions with ECT3 and ECT4 to regulate leaf formation/morphogenesis (Arribas-Hernandez et al., 2018). Structural analysis of the m6A binding domain in yeast and mammalian YTH revealed that the it recognizes m6A in mRNA through a hydrophobic aromatic cage containing three conserved tryptophan residues (Luo and Tong, 2014; Theler et al., 2014; Xu et al., 2014). Mutation in the hydrophobic cage of ECT2 and ECT3 was reported to abolish the function of m6A recognition (Arribas-Hernandez et al., 2018; Wei et al., 2018; Scutenaire et al., 2018), which suggest that specific-binding of ECT to m6A is essential for their functional activity in leaf and trichome development. Occurrence of a number of YTH proteins in Arabidopsis and rice, having very high sequence similarity (Li D. et al., 2014), might help elucidating their roles in interpreting m6A epitranscriptome in plants by creating/using multiple knockout mutants.
m6A Eraser
Since the formation of m6A is a reversible process, it is dynamically removed from the mRNA by two ALKBH family m6A demethylases namely ‘Fat mass and obesity-associated protein’ (FTO) (Jia et al., 2011) and α-ketoglutarate-dependent dioxygenase homolog 5 (ALKBH5) (Zheng et al., 2013) in mammals. In Arabidopsis (13) and rice (9) a number of ALKBH family proteins have been reported (Mielecki et al., 2012; Liang et al., 2020). Phylogenetic analysis showed no orthologs of FTO to be present in plants (Liang et al., 2020). But, the existence of multiple copies of ALKBH5 orthologs [six orthologs (ALKBH9A/B/C and ALKBH10A/B/C) in Arabidopsis] suggests redundant functions of these proteins in m6A demethylation. They are differentially expressed in different tissues (Duan et al., 2017) with their diverse subcellular localization (Mielecki et al., 2012). This again suggests their role in functional divergence in m6A dynamics in plants (Burgess et al., 2016). ALKBH9B, ALKBH10B, and SLALKBH2 (Zhou et al., 2019) remove m6A from mRNA in Arabidopsis (Duan et al., 2017; Martinez-Perez et al., 2017). ALKBH10B removes m6A from mRNAs for several regulators, which enhances the stability of the transcripts and promotes floral transition. Thus, m6A promotes degradation of mRNAs for developmental regulators in Arabidopsis (Duan et al., 2017). This indicates that it might potentially be used as an epitranscriptomic mark for modulating flowering time in crop plants.
Occurrence of m6A
Occurrence of m6A has been observed across the animals, plants, single-cell organisms (archaea, bacteria, and yeast), and viruses (Zhao et al., 2017a). Three independent studies showed consensus on adenosine methylation (m6A) motif RRACH in yeast, mammals, and plants (Dominissini et al., 2012; Schwartz et al., 2013; Luo et al., 2014). It has been detected in mRNAs of many plant species, including Arabidopsis, maize, wheat, oat, and rice (Zhong et al., 2008). In Arabidopsis, m6A content varies in different tissues, ranging from 0.4% in seeds to 1.5% in young seedlings (Zhong et al., 2008). Three independent studies reported m6A mapping in different ecotypes and tissues of Arabidopsis (Luo et al., 2014; Wan et al., 2015; Shen et al., 2016). m6A was reported to be widely distributed in >5,000 transcripts, and accumulated near the start and the stop codons, as well as in the 3′ UTR (Luo et al., 2014). However, occurrence of m6A near the start codon was not detected in the methylome of leaf, flower, and root of Arabidopsis (Wan et al., 2015), probably because of the dynamic nature of the modified m6A. Differentially methylated mRNAs were observed in leaf, flower, and root of Arabidopsis (Wan et al., 2015), indicating the role of m6A in tissue/organ differentiation. The m6A writers MTA/MTB, FIP37/VIRILIZER/HAKAI were reported to be involved in embryo and plant development (Shen et al., 2016; Ruzicka et al., 2017; Hu et al., 2019). YTH/ECT and ALKBH, reader and eraser, respectively, play important role in growth, development and flowering in Arabidopsis (Duan et al., 2017; Arribas-Hernandez et al., 2018; Scutenaire et al., 2018; Wei et al., 2018). Differential methylation of several transcripts in root, leaf, and flower of Arabidopsis (Wan et al., 2015), suggests that m6A dynamics of specific transcripts might be an integral part of tissue/organ differentiation in plants (Shen et al., 2019). Another recent work on epitranscriptomic profiling of salt-treated Arabidopsis leaf reported m6A enrichment in the transcripts for salt- and osmotic-stress responses (Anderson et al., 2018).
In addition to Arabidopsis, the enzymes associated with epitranscriptomic modifications have been reported in some of the agronomically important plants like Nicotiana sylvestris, maize, rice, and tomato. The methylases and demethylases have also been reported in plants, and they are evolutionarily conserved. Any change in their expression shows a significant alteration in the m6A content in polyadenylated transcriptome, and drastic physiological impacts. Analysis of m6A landscape in rice (Li D. et al., 2014) exhibited a similar pattern that was observed in Arabidopsis, which indicates a conserved m6A distribution in plants. Accumulating evidences also indicate that writers/readers and erasers play important roles in abiotic stress responses in plants (Hu et al., 2019). Zhang F. et al. (2019) identified a panicle-specific m6A motif UGWAMH (W = U/A; M = C/A; H = U/A/C) in rice. Despite the progress being made in understanding m6A landscape in crop plants, the writers, readers, erasers for m6A and its functions in plant growth, development, and survival under the stress are yet to be elucidated. However, the position, pattern, and motif of m6A suggest that the writers, readers, and erasers might be conserved across the kingdoms.
Methylation at Other Positions in Adenosine
In addition to the m6A, the human epitranscriptome is known to contain other modified/methylated forms of adenosine like m1A and m6Am (Hauenschild et al., 2015; Molinie et al., 2016). Methylation at the N1 position of adenosine creates N1-methyladenosine (m1A), and it has been prevalent in rRNA and tRNA. However, the occurrence of m1A has also been reported in the human transcriptome (Li X. et al., 2017), which can be erased by ALKBH3 (Li et al., 2016b). The CH3 group at N1 position of m1A interferes with standard base pairing (Hauenschild et al., 2015), which affects mRNA folding around the transcription start site (TSS) and facilitates initiation of translation. Despite the progress in the detection of modified nucleosides, transcriptome-wide distributions of m1A in plants remain unknown. When adenosine is methylated at the C2 position of ribose sugar [by 2′-O-methyltransferase (CMTR: Cap methyltransferase) to form 2′-O-methyladenosine (Am) (Werner et al., 2011) and then it is methylated at the N6 position of adenosine [by an unidentified nucleo-cytoplasmic methyltransferase], it forms N6, 2′-O-dimethyladenosine (m6Am). The m6Am modification is exclusively distributed at the TSS (generally after the m7G cap) in certain mRNAs (Linder et al., 2015) at a frequency of 0.003% (Molinie et al., 2016). m6Am was reported to be mediated by phosphorylated CTD interacting factor 1 (PCIF1) which catalyzes methylation of m6A to m6Am at the 5′ end of mRNA (Sendinc et al., 2019). Although such epitranscriptomic modifications play important roles in mammals, they are remained to be identified/characterized in plant.
Modification of Other Bases in mRNA
Besides the modifications of adenosine, epitranscriptome is known to contain methylation/modification at other bases, for example, m5C, hm5C, m3C, ac4C, m1G, m7G, 8-oxo-G, Uridylation, Pseudouridine (ψ), and Inosine (I), particulately in animal systems (reviewed by Shen et al., 2019; Boo and Kim, 2020). While the occurrence of some of the modified bases (e.g., m5C, hm5C, m7G, and ψ) have been confirmed (Huber et al., 2015; Vandivier et al., 2015; Burgess et al., 2016; Zuber et al., 2016; Cui Q. et al., 2017; Martinez-Perez et al., 2017; Malbec et al., 2019), presence of m1G has been predicted in Arabidopsis epitranscriptome. Many of these epitranscriptomic modifications like m3C, m7G, 8-oxoG, and I play important roles in animals (Palladino et al., 2000; Torres et al., 2014; Arimbasseri et al., 2016; Xu L. et al., 2017; Malbec et al., 2019), but their existence/identification and functional characterization remains to be confirmed in plants.
Cytosine Modifications in mRNA
Occurrence of methylcytidine (m5C) is common in tRNAs and rRNAs (Squires and Preiss, 2010), but it has also been identified in mRNAs and ncRNAs (Squires et al., 2012). Since m5C is less abundant (0.4% of total cytosine, compared to ∼1.5% of m6A in human transcripts), much less has been researched on its occurrence and functions (Squires et al., 2012; Ke et al., 2015). Detection of m5C in mRNAs of different plant species, including Arabidopsis, Medicago, rice, maize, and foxtail millet, has been reported (Cui Q. et al., 2017). Change in m5C level across the tissues in Arabidopsis, with a gradual increase during vegetative growth, suggest a dynamic change in m5C content during plant growth and development. More than one thousand m5C were detected on transcriptome-wide analysis of shoot, root, and siliques of Arabidopsis, but only a few dozen of them were commonly present among these tissues (David et al., 2017). m5C is generally accumulated in the coding sequence (CDS) of the mRNA in HACCR (where H = A, U or C; R = A or G) and CTYCTYC (Y = U or C) motifs in Arabidopsis (Cui Q. et al., 2017). A marginal increase in expression of TRM4B (an m5C writer) was observed under cold stress in Arabidopsis, but it showed decreased expression under heat stress. However, the expression level of TRM4B was not altered in rice under abiotic stresses (Zou et al., 2016). TRM4B has been further characterized in plants (David et al., 2017; Cui Q. et al., 2017), and m5C was observed to be required for root development and oxidative stress responses (David et al., 2017). TRM4B loss-of-function mutants of Arabidopsis exhibited down-regulated expression of short hypocotyl 2 (SHY2) and indoleacetic acid-induced protein 16 (IAA16) genes involved in root development. Stability of the transcripts of such genes was observed to be positively correlated with the m5C modification/content (Cui Q. et al., 2017).
Writer, Reader, and Eraser of m5C
Formation of m5C in human mRNA is catalyzed by methyltransferases such as DNMT2 and NSUN2 (Squires et al., 2012; Bohnsack et al., 2019). NSUN6, a Type II m5C site-specific methyltransferase, was reported to negatively correlate m5C methylation with translation efficiency (Liu et al., 2020). Recently, Selmi et al. (2021) mapped NSUN6-dependent m5C sites in human transcripts, which were located in protein coding RNAs at 3′-UTR within a consensus sequence (CTCCA) motif, and mark translation termination. Eight m5C methyltransferases are encoded by Arabidopsis genome, two of them are tRNA-specific methyltransferase 4A (TRM4A) and TRM4B (Chen et al., 2010; Cui Q. et al., 2017). While TRM4A is responsible for m5C in tRNA, TRM4B targets mRNA for the modification. A recent study demonstrated that an RRM motif-containing ALY protein binds to m5C-containing mRNAs in Arabidopsis (Pfaff et al., 2018). The aly mutants showed shorter primary roots, defective reproductive development including abnormal flowers and reduced seed production (Pfaff et al., 2018). Thus, m5C is another important epitranscriptomic mark that affects plant growth, development and adaptive responses in plants. Although m5C is reported to be further oxidized to hm5C by a family of Ten-eleven translocation (TET) enzymes (Huber et al., 2015; Delatte et al., 2016), varying hm5C content in different Arabidopsis tissues indicate that it is a dynamic epitranscriptomic mark in plants (Shen et al., 2019). Despite the progress in detecting/distribution of hm5C, its oxidation to m5C in mRNA is still not fully demonstrated. However, further research would be required to identify m5C readers/erasers, and elucidate the mechanisms/functions of m5C-mediated regulation of gene expression.
Methylation at Other Positions in Cytosine
Cytosine can also be acetylated at the N4 position by an N-acetyltransferase (NAT10) to form ac4C. Such modification is commonly found in tRNA, rRNA, but it has also been observed in mRNA (Dong et al., 2016; Arango et al., 2018). ac4C was observed distributed in coding and non-coding RNAs in human, abundant near the TSS (Arango et al., 2018). The occurrence of ac4C increases mRNA half-life and promotes translation efficiency. NAT10 acts as the primary ac4C writer, and NAT10 knocking out reduces ac4C content in RNA. In yeast, orphan box C/D snoRNAs complex guides Kre33 (a yeast homolog of human NAT10) to the target sites for ac4C modification (Sharma et al., 2017). However, it is still not known whether ac4C is a reversible or not, as neither an ac4C reader nor its deacetylation process is known. Moreover, its occurrence in plant and role/function in gene regulation is not yet known.
Modification of Other Bases in mRNA
Uridylation (addition of uridines at the 3′ without any template) of mRNA, targeted for degradation, has been reported in both mice and Arabidopsis (Shen and Goodman, 2004; Zhang et al., 2017). Uridylation of mRNAs in plants is catalyzed by UTP: RNA uridylytransferase1 (URT1) and terminal uridylyltransferase (TUTase) (Sement et al., 2013; Lim et al., 2014). Pseudouridine (Ψ), also known as the 5th base of RNA and the first modified RNA base (Davis and Allen, 1957), is a C−glycosidic rotational isomeric form of uridine (U), wherein U is attached to a ribose sugar through a carbon–carbon (instead of a nitrogen-carbon) glycosidic bond. Formation of Ψ in eukaryotes involves an RNA-dependent pseudouridine synthase (PUS) such as Cbf5 which uses a cofactor box (H/ACA ribonucleo-proteins) as a guide. Ψ formation may also occur through an RNA-independent PUS that does not require any cofactor (Carlile et al., 2014; Spenkuch et al., 2014). Ψ may further get methylated by EMG1 at the N1 position to generate 1-methylpseudouridine (m1Ψ) (Wurm et al., 2010). Although Ψ is mainly distributed around the CDS and the 3′ UTR of the gene (Carlile et al., 2014; Li et al., 2015), its occurrence is yet to be mapped in plant mRNAs.
Oxidation of RNA bases due to excessive reactive oxygen species (ROS, e.g., superoxide, hydroxyl radicals, and hydrogen peroxide) generates different oxidized RNA bases like 8-oxoG, 8-oxo-7,8-dihydroadenosine, 5-hydroxycytidine, cytosine glycol, and 5-hydroxyuridine (Yan and Zaher, 2019). 8-Oxo-7,8-dihydroguanosine (8-oxoG, an oxidized form of guanine base) is one of the most abundant variants of guanosine found in mammalian cells associated with neurodegenerative diseases (Nunomura et al., 2017). This determines the fate of mRNA, including stability and translation (Yan et al., 2019). AU-rich element RBP 1 (AUF1) and Y-box binding protein 1 (YBX1) preferentially bind to 8-oxoG to trigger rapid degradation of 8-oxoG–containing mRNAs (Ishii et al., 2015). Recently poly(C)-binding protein 1 (PCBP1) was identified as an 8-oxoG reader protein. However, the binding of PCBP1 requires two 8-oxoGs located nearby in the RNA, and this is associated with cellular apoptosis under oxidative stress (Ishii et al., 2018). Reversal of 8-oxoG to a normal guanosine base, as observed in the case of many other RNA base modifications, is not yet known. Moreover, the occurrence of such modified RNA base(s) in plant can be expected, particularly under environmental stresses when ROS production increases significantly, but their existence has not yet been reported.
Effect of Modified Base on mRNA Metabolism
Modified bases influence mRNA metabolism, including splicing, export, translation, and degradation of the transcript. Many of the functions of m6A in mRNA metabolism in animal system are well-known (Wang et al., 2014; Liu et al., 2015; Haussmann et al., 2016; Shi et al., 2017; Kasowitz et al., 2018). However, only some of the functions of m6A and its readers like ECT2 are known in plants, including the regulation of 3′ UTR processing and improving mRNA stability (Wei et al., 2018). Moreover, some other functions of the core components of the methyltransferase complex (MTA, MTB, and FIP37) in plant development and survival under abiotic stresses were deciphered by mutation/knock-out studies in Arabidopsis (Tzafrir et al., 2003; Vespa et al., 2004; Zhong et al., 2008; Bodi et al., 2012). The function of another modified adenosine base, m6Am (a close homolog of m6A), is yet poorly understood, but it has been reported to improve translation efficiency and mRNA stability in mice by protecting the mRNA from decapping enzymes like DCP2 (Mauer et al., 2017).
m5C facilitates binding of ALYREF (an mRNA export adaptor) and removal of NSUN2 (an m5C writer), which disrupt mRNA transport from the nucleus (Yang et al., 2017). In Arabidopsis, a reduced ribosomal occupancy was observed in the m5C–marked mRNAs, indicating interfering role of m5C in binding of translational machinery (Cui Q. et al., 2017). A decreased m5C content accelerates mRNA decay, which indicates that it is another important epitranscriptomic mark affecting mRNA stability and translation efficiency in plants (Cui Q. et al., 2017).
Other modified bases, such as ψ, have been depicted to be involved in splicing and undisrupted translation of mRNA in yeast and mammals (Carlile et al., 2014; Karijolich et al., 2015). URT1-dependent uridylation and poly-A binding protein (PABP) in plants was reported to prevent excessive deadenylation, and thus, protects mRNA from degradation in Arabidopsis (Zuber et al., 2016). The occurrence of 8-oxoG considerably inhibits the efficiency of peptide bond formation, which restricts translation and triggers mRNA degradation (Boo and Kim, 2020). Transcription factors (TFs; e.g., ZFP217-dependent METTL3 and HIF-dependent ALKBH5), and miRNAs (e.g., miRNA responsible for RNA-dependent METTL3 activity) may also trigger the expression of writers and erasers of modified bases, demonstrating the feedback activation. This suggests a complex interplay between the modified bases and regulatory pathways. Thus, the stimuli and signaling/regulatory processes that fine-tune the transcription and translation processes of a gene might also affect the activity of writers, readers, and erasers through various RNA modifications. The same signaling pathway may also activate or inactivate the synthesis of readers and erasers through post-translational modifications.
Role of Modified Base on mRNA Translation
Translation process is regulated by the binding of ribosome and initiation factor activities, including phosphorylation of the ‘eukaryotic initiation factor 2’ (eIF2) (Pavitt, 2018). Translation efficiency was reported to be moderately increased in the METTL3-knockout mutants of mouse embryonic stem cells and embryoid bodies, which suggest a negative regulatory role of m6A on translation efficacy (Liu et al., 2015). However, the binding of YTHDF1 (a cytoplasmic m6A reader) cooperates with ribosomes and initiation factors to increase translation efficiency (Wang et al., 2015). Recent studies demonstrate that m6A promotes translation efficacy of mRNAs (Li A. et al., 2017; Shi et al., 2017; Weng et al., 2018). Similarly, IGF2BPs (m6A-binding proteins) help reinforcing the stability and increase the translation efficiency of m6A-containg mRNAs (Huang et al., 2018). Studies also suggested that the presence of m6A in 5′ UTR of an mRNA promotes initiation of cap-independent translation (Meyer et al., 2015) and the IGF2BPs-mediated translation (Huang et al., 2018). It has also been reported that eIF3 directly binds to m6A−harboring 5′ UTR and engages the 43S ribosomal complex to begin the translation process, even in the absence of eIF4E (a cap-binding factor) (Meyer et al., 2015). The presence of m6A in the coding region of mRNA has been reported to disrupt tRNA boarding and elongation of the translation process in vitro (Choi et al., 2016). m6A has also been reported to negatively regulate the translation process by serving as a link between transcription and translation processes (Slobodin et al., 2017). All of these findings support the regulatory functions of m6A in mRNA translation.
Recent mapping studies indicate that m1A is abundant in the 5′ UTR of mRNA (Dominissini et al., 2016; Li X. et al., 2017), which is associated with higher translational efficiency; however, the underlying mechanism is yet to be discovered. In addition to this, the presence of m6Am creates hindrance in the binding of mRNA-decapping enzyme DCP2, which improves the stability of the transcript (Mauer et al., 2017). Moreover, m6Am also makes mRNA resistant to microRNA-mediated degradation (Mauer et al., 2017). Similarly, m5C has been reported to stabilize RNA secondary structure; hence, it influences translational fidelity (Helm, 2006; Squires and Preiss, 2010). While the presence of m5C at the first position in the CCC codon was reported to reduce translational product by ∼40% using bacterial whole-cell extract, its presence at the 2nd position of the codon was reported to suppress translation termination (Hoernes et al., 2016). In contrast, hm5C has been reported to activate translation in Drosophila melanogaster (Delatte et al., 2016). The effects of Ψ on translation efficiency depend on its position in a codon.
Although m6A has been known to promote translation efficiency in the animal system (Meyer et al., 2015; Wang et al., 2015; Slobodin et al., 2017), a little is known about its functions in plants where it works differently. In maize, m6A was found to be negatively correlated with translation efficiency; however, this depends on the location and content of m6A in the gene (Luo et al., 2019). Similarly, m5C was also reported to be associated with reduced efficiency of translation in Arabidopsis (Cui Q. et al., 2017). A recent study reports m5C to play important role in mRNA stability (Yang L. et al., 2019), which in turn improves translation efficiency. Thus, the role of different methylated bases in mRNA translation needs to be further explored to better understand the epitranscriptomic regulation of gene expression in plants.
Role of Modified Base on mRNA Splicing, Export, and Decay
Transcripts with modified bases get easily exported, translated, and degraded, probably due to the binding of the reader at the modified base. Studies provide convincing evidence for the regulatory function of m6A on processing of pre-mRNA and pri-miRNA (Alarcon et al., 2015b). A family of nuclear hnRNPs, an m6A-binding protein accelerate processing of pri-miRNAs through interaction with DGCR8 (Alarcon et al., 2015a). An hnRNPA2B1 modulate alternative splicing of transcripts (Alarcon et al., 2015a). Moreover, hnRNPC plays an important role in the pre-mRNAs processing (Rajagopalan et al., 2012). Pfaff et al. (2018) reported that an RRM motif-containing ALY protein binds to m5C-containing mRNAs and helps in mRNA export in Arabidopsis. Reports suggest that controlling RNA modification regulates mRNA stability which ultimately fine tunes the gene expression. Research demonstrates that alternatively spliced mRNAs in animals retain more m6A sites and the binding sites for METTL3. Geula et al. (2015) reported that a METTL3-deficient mouse embryonic stem cell retains intron and shows exon skipping. Thus, m6A exerts its effect through binding of the reader proteins, particularly a family of proteins containing YTH domain (Xu et al., 2014). YTHDF2 (a well-established m6A reader) specifically binds to m6A−containing mRNA to deploy CCR4–NOT deadenylase complex (Du et al., 2016) for mRNA transport to the processing bodies (Wang et al., 2014), which promotes degradation of mRNA through translocation of the transcript (Sheth and Parker, 2003). This indicates a linkage between m6A and mRNA degradation. m6A modification and binding of readers also affect mRNA splicing and alternative polyadenylation (Xiao et al., 2016; Kasowitz et al., 2018). An alternative to 3′–5′ exoribonucleolytic cleavage on mRNA, endoribonucleolytic cleavage of the m6A-containing mRNAs is mediated by interaction among the YTHDF2, heat-responsive protein 12 (HRSP12), and P/MRP (an endoribonuclease RNase) complex (Park et al., 2019). Presence of the 8-oxoG in mRNA causes ribosome stalling followed by no-go decay (Ikeuchi et al., 2018). The roles of modified RNA base in regulation of mRNA stability/decay have recently been reviewed by Boo and Kim (2020).
Effects of Methylated Base on Biological Processes
Complex cellular processes are intricately regulated by mRNA methylation. According to the cellular needs, mRNA export/localization is altered by RNA base methylation (Roundtree et al., 2017; Yang et al., 2017; Chen et al., 2019). The presence of m6A in transcripts of pluripotent TFs prompts transcriptomic flexibility in embryonic stem cells of mouse and human (Batista et al., 2014; Geula et al., 2015). Sequestration of METTL3 by ZFP217 indicates a complex interplay between epitranscriptome and TFs (Aguilo et al., 2015). The depletion of m6A from glioblastoma stem cells due to METTL3/14 knockdown was reported to promote self-regeneration and tumorigenesis (Cui X. et al., 2017). In Zebrafish (Danio rerio), m6A coordinates the elimination of maternal mRNAs with the help of Ythdf2 which is essential for maternal-to-zygotic transition (Cui X. et al., 2017). Heat-shock stress suppresses cap-dependent translation and induces adenine methylation (formation of m6A) at 5′ UTR of the transcripts (Meyer et al., 2015; Zhou et al., 2015). Although cells can discriminate between self (modified) and non-self (unmodified) RNAs, epitranscriptome plays an important role in immune responses also (Kariko et al., 2012; Hull and Bevilacqua, 2016). A study on the precursor cells of neurons revealed that m5C regulates differentiation and motility of neural stem cells in mice and humans (Flores et al., 2017). Mutants for FIP37 displayed about 85% reduction in m6A content and a massive proliferation of apical meristem in the shoot (Shen et al., 2016). Loss of m6A in FIP37 mutants of Arabidopsis was reported to be a key regulator of transcripts like WUSCHEL and SHOOTMERISTEMLESS, which results in the accumulation of transcripts due to their decreased decay (Shen et al., 2016).
Advances in epitranscriptomics have revealed several potential biological roles of post-transcriptional mRNA modifications (Zhao et al., 2017a). Reports demonstrate that methylated transcripts have shorter 3′ UTRs and lesser stability than its unmethylated counterpart (Molinie et al., 2016). Thus, methylation of mRNA base, and synthesis/binding of TFs/regulatory proteins get synchronized in response to the development processes and environmental stimuli (Zhao et al., 2017b). In mouse brain, the m6A level was reported to increase throughout the lifespan (Meyer et al., 2012). Studies have shown the role of m6A accumulation in learning and memory in mouse mediated by Ythdf1 binding in response to stimuli (Shi et al., 2018). Moreover, a recent study suggests the stress-mediated regulation of m6A accumulation in patients with depression, indicating that the dysregulation of m6A is associated with the development of mental disorders (Engel et al., 2017). An impaired build-up of m6A disrupts sex determination in Drosophila, and it causes embryonic-lethality in plants (Haussmann et al., 2016). Moreover, reduced accumulation of m6A inhibits the differentiation of embryonic stem cells in mammals (Batista et al., 2014; Geula et al., 2015). Some of the studies also suggest that the presence of m6A in mRNA plays a crucial role in spermatogenesis in mice (Mus musculus L.) (Hsu et al., 2017; Xu K. et al., 2017).
In Arabidopsis, the deficiency of mRNA adenosine methylase enzyme (a homolog of METTL3) has serious effects on plant growth and development (Bodi et al., 2012). Mutation studies on m6A methyltransferase core components (MTA, MTB, and FIP37) in Arabidopsis suggest that m6A is essential for the survival of the plant (Zhong et al., 2008). Arabidopsis mutants for FIP37 displayed an 85% reduction in m6A content and massive proliferation of apical meristem in the shoot (Shen et al., 2016). Knockdown of MTB in Arabidopsis was reported to cause a considerable reduction in height of the plant, while hypomorphic vir allele produced defective roots and the VIR null mutants were observed to be embryo-lethal (Ruzicka et al., 2017). A distinct pattern of m6A accumulation was observed in different organs of Arabidopsis, which suggests that m6A plays a role in organogenesis and it has tissue-specific functions (Wan et al., 2015). The content of m6A in Arabidopsis transcripts is controlled by 13 different ALKBHs (Mielecki et al., 2012) which indicate dynamic expression and diverse subcellular localization of ALKBH in plant. The alkbh10b mutants of Arabidopsis showed elevated m6A content in >1,000 transcripts and delayed floral transition, indicating that it mediates demethylation (removal of m6A) of regulatory transcripts (Duan et al., 2017). A wild-type ALKBH10B could restore the alkbh10 mutant phenotype, suggesting that m6A is an important regulator of flowering time in plants. The regulatory function of ECTs in leaf morphogenesis and trichome development has recently been demonstrated (Arribas-Hernandez et al., 2018; Scutenaire et al., 2018; Wei et al., 2018). ECT2 binds at m6A sites in the trichome development-related genes and improves the mRNA stability. Transcripts of the genes in ect2 mutant get degraded at an accelerated rate and affect trichome branching (Wei et al., 2018), which suggests that m6A mediates trichome and leaf development by the recruitment of reader proteins.
A large number of differentially methylated transcripts were observed in leaf, flower, and root of Arabidopsis, while >14,000 transcripts were found to contain m6A in rice leaf (Wan et al., 2015). Findings suggest that m6A might be involved in tissue-differentiation in plants. OsMTA2 and OsFIP were identified to be the important components of RNA m6A methyltransferase complex, and m6A is involved in the regulation of sporogenesis, particularly male gametogenesis in rice (Zhang F. et al., 2019). A loss-of-function mutation in OsFIP resulted in the early degeneration of microspores, irregular meiosis in prophase I. Tomato slalkbh2 mutants showed delayed fruit ripening phenotypes and increased m6A content compared with the wild-type plants (Zhou et al., 2019). SlALKBH2 is involved in the demethylation of the SlDML2 mRNA and regulates its degradation. SlDML2 encodes a DNA demethylase that regulates the expression of SlALKBH2 through DNA (5-mC) methylation. This suggests a novel mechanism of gene regulation connecting epigenetics (DNA methylation, 5-mC) and epitranscriptomics (mRNA modification, m6A) (Zhou et al., 2019). Loss of function mutation in Arabidopsis for TRM4B (an m5C writer) resulted in defective root phenotype because of the decreased content of m5C in the genes involved in root development (Cui Q. et al., 2017).
mRNA base modifications (m6A and m5C) are sensitive to environmental changes in plants. The m5C content was reported to decrease under drought and heat stress in Arabidopsis (Cui Q. et al., 2017). Similarly, m6A content was reported to decrease in drought stress (Zhou et al., 2019), suggesting the epitranscriptomic regulation of stress responses in plants. The findings indicate that m6A and m5C play important roles in post-transcriptional regulation of gene expression in plants. Several studies on the writers, readers, and erasers in plants demonstrate that mRNA modification is an important molecular mechanism for regulating plant development and environmental responses (Shen et al., 2016; Cui Q. et al., 2017; Duan et al., 2017; Martinez-Perez et al., 2017; Scutenaire et al., 2018). More importantly, most of the above-mentioned functions result due to silencing/over-expressing of gene or due to the combined action of reader/eraser but not only due to the removal/accumulation of any RNA base modification. Thus, the authors agree with the limitations of the studies/reports, and realize the importance of the factor(s) involved. Moreover, the mechanism for synchronized response of writers, readers, and erasers to internal/external stimuli is still elusive. Despite the progress in understanding the functions of m6A and m5C in plants, the mode of action of their writers, readers, and erasers are yet to be discovered.
Detection of Modified Base in RNA
Post-transcriptional modifications in RNA bases have been reported to play essential roles in various functional RNAs. These modifications alter the structure, processing, and functions of RNAs. A comprehensive understanding of the biochemical modifications in mRNA bases and the changes in accompanying non-covalent interactions is required to gain insights into the functional diversity. m6A is the most abundant modified mRNA base and the first epitranscriptomic modification mapped (Dominissini et al., 2012; Meyer et al., 2012). Although marvelous progress has been made in understanding the modified mRNA bases (Liang et al., 2020), in-depth insights into the dynamics, structure, and functions of such epitranscriptomic modification in this fascinating messenger biomolecule are essential. Detection of the modified RNA base helps understanding its dynamics and biological functions. Modern high-throughput technologies together with the conventional methods (Table 2) are expected to advance the field of epitranscriptomics by generating data and discoveries. However, most of the current methods of detecting modified base are specific for a particular modification but recently Khoddami et al. (2019) reported a method (RNA bisulfite sequencing, RBS-seq) for transcriptome-wide detection of multiple base modifications (m5C, Ψ, and m1A) simultaneously at single-base resolution. In this section, we present an overview of the technological advancements in the detection methods, their applications, and their limitations.
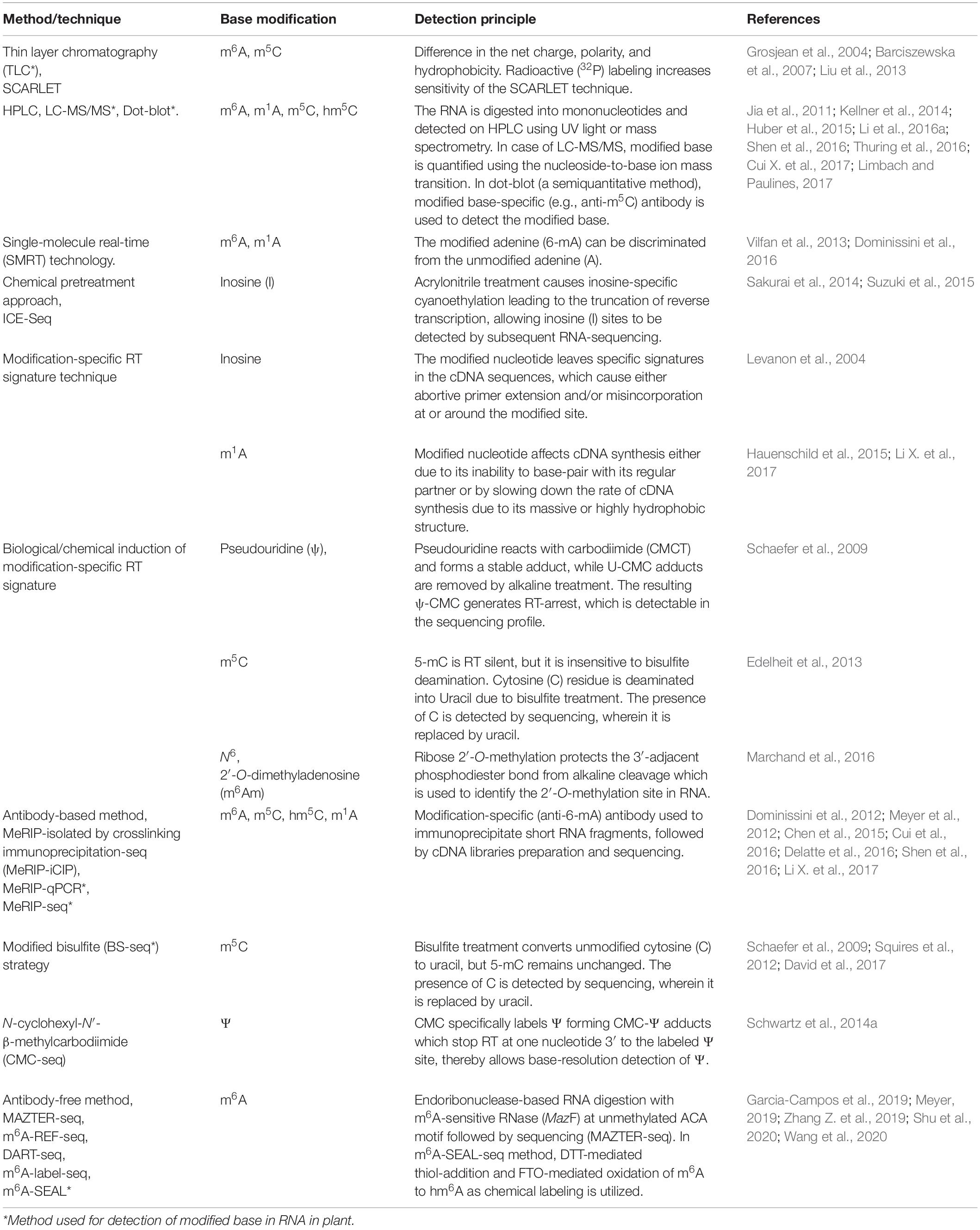
Table 2. Techniques for detection of modified RNA base.
Thin-Layer Chromatography
Thin-layer chromatography (TLC) has been one of the conventional methods for revealing RNA nucleobase modification (Keith, 1995). Generally, a modified base differs from its unmodified counterpart in terms of the net charge, polarity, and/or hydrophobicity, which allow their separation through chromatography. TLC separation of bases can be performed in one-dimension (1D) or two-dimensions (2D) using microcrystalline cellulose as a stationary phase. Using this method 2D-TLC maps for several modified RNA bases have been prepared (Grosjean et al., 2004; Barciszewska et al., 2007; Zhong et al., 2008). The sensitivity of the method can be increased by using radioactive (32P) labeling [site-specific cleavage and radioactive labeling, ligation-assisted extraction, and thin layer chromatography (SCARLET)] to detect the modification within an individual transcript (Liu et al., 2013). However, the TLC-based method fails to provide information about the location/context of the modified base.
High-Performance Liquid Chromatography and MS
The content of modified RNA nucleobases can also be determined in digested mononucleosides by using high/ultra-performance liquid chromatography (UPLC) followed by mass spectrometry (MS) (Thuring et al., 2016). The method has been extensively used earlier for the detection and quantification of modified RNA bases. MS coupled to a nano-chromatography system reduces the amount (to picomole) of the sample required. Detection of a modified RNA base through ‘matrix-assisted laser desorption/ionization−time-of-flight’ (MALDI-TOF) is still being optimized (Schwartz and Motorin, 2017). Detection of the modified RNA bases can also be performed using methods like dot-blot and LC-MS/MS (Jia et al., 2011; Delatte et al., 2016; Shen et al., 2016; Cui Q. et al., 2017). Ross et al. (2016) reported LC-MS/MS based, sequence specific detection of modified nucleosides in tRNAs from bacteria and human.
Reverse Transcription-Based Techniques
Reverse transcription (RT)-based techniques use the primer-extension method to reveal the modified RNA bases. The presence of the modified base in mRNA interrupts/inhibits primer extension, which facilitates its context-specific positioning. However, comparative sequence analysis with unmodified RNA transcripts is necessary to eliminate structural RT-stops. The advantage of the RT-based technique includes its applicability and sensitivity to a complex mixture of mRNAs, but it requires pure and concentrated mRNA molecules. Li X. et al. (2017) used a technique named m1A-MAP to detect the modified base at single-nucleotide resolution to profile m1A in the human transcriptome. Since m1A causes truncation and/or misincorporation during cDNA synthesis from the transcript (Hauenschild et al., 2015), a more precise method in detecting the position of m1A at single-base resolution. Nevertheless, inosine cannot be detected directly by the RT-based technique, as it can base-pair with cytosine. However, inosine-specific cyanoethylation treatment, using acrylonitrile, converts inosine into N1-cyanoethylinosine (ce1I) which disable base pairing of inosine with cytosine. This allows inosine sites to be detected by subsequent ‘inosine chemical erasing’ (ICE)-sequencing (ICE-Seq) in mRNA (Suzuki et al., 2015).
NGS Technologies-Based Method for Detection of RNA Modification
The advances in next-generation high-throughput sequencing technologies for the detection of RNA base modifications have considerably improved the epitranscriptomic studies (Li et al., 2016b). Currently, the sequencing technologies employ an amplification step to generate clusters, which provides exceptionally high sequencing output with <0.1% error. However, the read length remains shorter (500–600 nt) in most of the cases. The BS-seq method combines bisulfite conversion followed by NGS to map m5C, which has been successfully used for epitranscriptomic analyses in several animals and plants (Squires et al., 2012; David et al., 2017). Although the BS-seq method precisely identifies the site of m5C at single-base resolution (Figure 3A), it possesses two technical disadvantages. First, the BS-seq fails to distinguish between m5C and other modified cytosine bases (e.g., hm5C) in mRNA (Nestor et al., 2010). Second, bisulfite treatment during sample preparation causes degradation of mRNA and thus impedes amplification of m5C–containing mRNA which limits the applicability of this method.
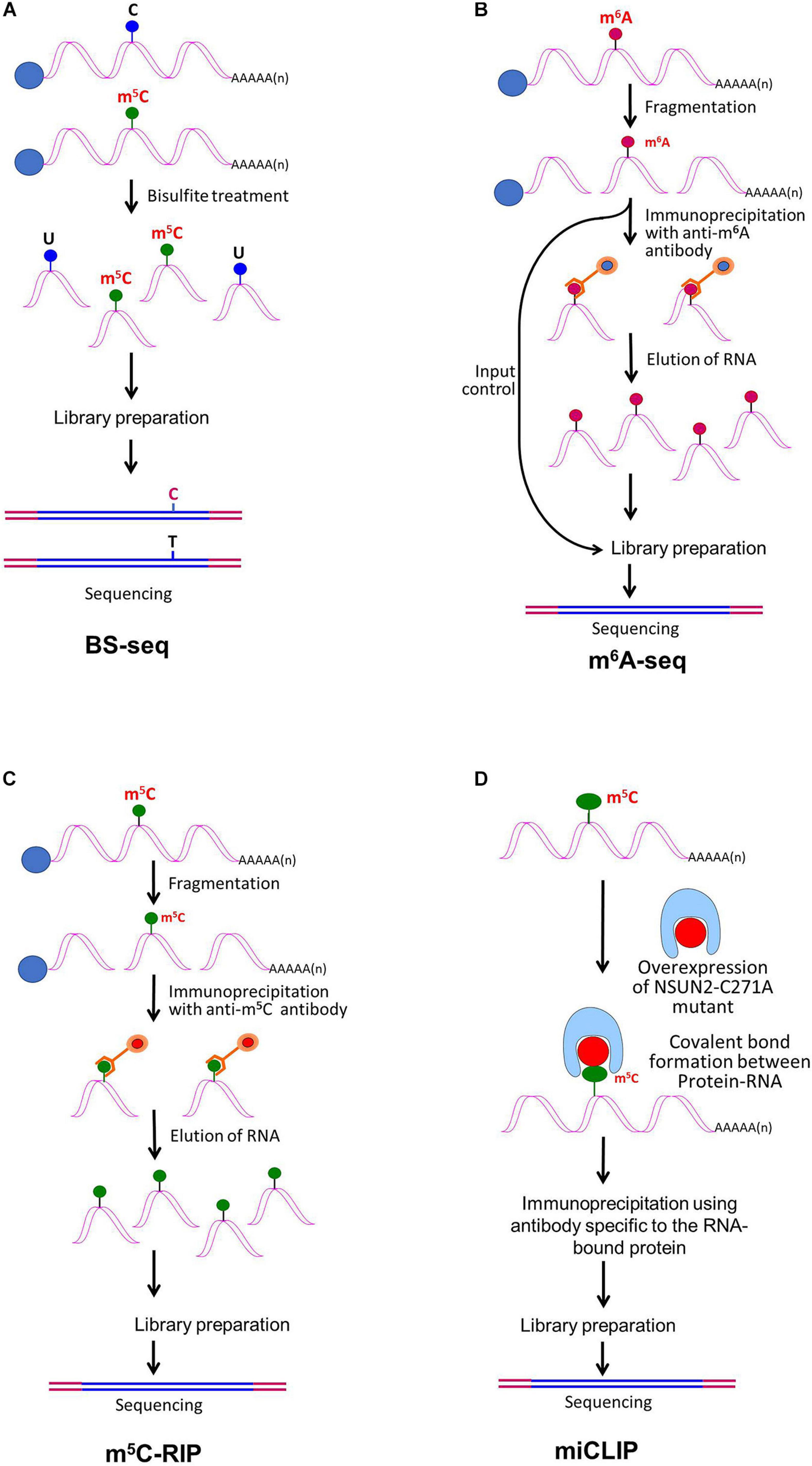
Figure 3. Detection of modified bases in mRNA. (A) Bisulfite sequencing (BS-seq) for the detection of 5-methylcytosine (m5C). Purified mRNA is fragmented into small (100–200 nt) fragments, and subjected to bisulfite treatment. Bisulfite treatment causes converts cytosine (C) to uracil (U), but m5C remains unchanged. Presence of C is detected by sequencing, wherein it is replaced by T. (B) Purified mRNAs are fragmented into 100–200 nt, followed by immunoprecipitation using anti-m6A antibody to enrich the sample with fragments containing the modified base, library preparation, and high-throughput deep-sequencing for detection of m6A. (C) Purified mRNAs are fragmented followed by immunoprecipitation using anti-m5C antibody of the fragments containing the modified base, library preparation, and sequencing. (D) m5C individual-nucleotide-resolution crosslinking and immunoprecipitation (m5C-miCLIP) exploites catalytic activity of cysteine-to-alanine mutation (C271A) mutant of NSUN2 (methyltransferase) which inhibits release of the enzyme from the protein–RNA complex making stable covalent bond between NSun2 and its RNA targets. Antibody specific to the RNA bound protein is used for immunoprecipitation, followed by library preparation and sequencing. This allows detection of low-abundance methylated RNAs without the need of deep sequencing.
The single-molecule sequencing approach uses either of the two NGS principles. While single-molecule real-time (SMRT) technology uses nanowell (zero-mode waveguide, ZMW; Pacific BioSciences) (Vilfan et al., 2013), Nanopore sequencing (Min-ION, Oxford Nanopore Technologies) uses the change in electrical charge to detect the modified base in mRNA passing through nanopore-forming proteins (Liu et al., 2019). One of the advantages of single-molecule sequencing approaches is very long (>10,000 nt) read length, but the accuracy of the sequence is compromised. Such technologies are useful for analyzing modified bases in mRNA, particularly in a context-specific manner, as these methods allow direct sequencing of mRNA without converting it into DNA (such conversion causes the loss of modified base). However, these technologies require extensive optimization for their use in the detection of modified bases in RNA. The first experimentation on using reverse transcriptase (instead of a DNA polymerase) as the enzyme in ZMW of SMRT for direct sequencing of modified bases in mRNA was carried out by Vilfan et al. (2013). The entry of a modified base (m6A), present in the template mRNA, into the nanowell (ZMW) causes increased ‘inter-pulse duration’ (IPD) compared to its unmodified counterpart (adenosine). The potential of SMRT sequencing to detect m6A was demonstrated by Vilfan et al. (2013). Similarly, Nanopore sequencing has successfully been used to detect m6A in native RNA (Liu et al., 2019). Further optimization of the single-molecule sequencing technologies will revolutionize epitranscriptomic research on modified bases in mRNA.
Recently, Fang et al. (2020) reported a new method CRISPR integrated gRNA and reporter sequencing (CIGAR -seq) by combining pooled CRISPR screen and the reporters associated with RNA modification. Using the CIGAR-seq method, they could discover NSUN6 as a novel m5C methyltransferase in mRNA. Subsequently, they could demonstrate that this method can be successfully used to identify the regulators of other mRNA modifications such as m1A.
Antibody-Based Methods for Detection of Modified Bases
RNA base modification, particularly m6A, is a widespread epitranscriptomic change that influences nearly every aspect of mRNA biology. Our understanding of the RNA base modification has been facilitated by the recent developments in the use of an antibody to immunoprecipitate RNAs containing modified base, and the high-throughput sequencing technologies. Methyl-RNA-immunoprecipitation-sequencing (MeRIP-seq) (Meyer et al., 2012) and m6A-seq (Dominissini et al., 2012) use immunoprecipitation with the help of modification (m6A or m5C)−specific antibody followed by sequencing (Figure 3B) (Yang Y. et al., 2019). Similarly, hMeRIP-seq relies on the anti-hm5C antibody to detect hm5C in Drosophila mRNA (Delatte et al., 2016). acRIP-Seq uses an ac4C-specific antibody to identify 4,000 ac4C in the human transcriptome (Arango et al., 2018). However, these antibody-based detection methods cannot detect hm5C and ac4C at single-base resolution, but the success in the single-base resolution of m6A by sequencing might help to optimize the method (Yuan et al., 2019). This enables studying the dynamics of epitranscriptome, a post-transcriptional regulatory mechanism for gene expression. The modification-specific antibody is used for enrichment/collection of the sample with the fragments containing the modified base. To detect m5C in mRNA, bisulfite-based technique cannot be used with much success. Hence m5C RNA immunoprecipitation (m5C-RIP) was used (Figure 3C) by Edelheit et al. (2013). In this method, an anti-m5C antibody is used to immunoprecipitate and enrich the modified base containing mRNA fragments, followed by library preparation and sequencing. Methylation individual-nucleotide-resolution crosslinking and immunoprecipitation’ (miCLIP) was used (Figure 3D) to identify m5C in RNA (Hussain et al., 2013; Khoddami and Cairns, 2013). This approach exploits the enzymatic activity of m5C methyltransferase containing a cysteine-to-alanine mutation (C271A) in NSUN2 which inhibits release of the enzyme from protein–RNA complex. This results in a covalent bond between the enzyme and its RNA targets. Antibody specific to the RNA-bound protein is used to immunoprecipitate the fragments containing the modified base, followed by library preparation and sequencing. The immunoprecipitation allows detection of methylated bases/RNAs in low-abundance without the need of deep sequencing. Subsequently, miCLIP was used to map m6A at single-base resolution (Linder et al., 2015).
Antibody-Free Sequencing Methods
Many of the RNA base modification detection methods rely on the use of antibodies for immunoprecipitation. However, an antibody may fail to distinguish between two different modified forms of a nucleobase, such as m6A and m6Am. Moreover, the methods are dependent on the specificity of the antibody, which emphasizes the desire for the antibody-free method to draw transcriptome-wide atlas of the modified base. Hong et al. (2018) developed an antibody-independent method to detect m6A at single-nucleotide resolution via 4SedTTP incorporation and FTO demethylation. Since the 4SedTTP stably base pair with A but cause truncation on m6A-T pairing during reverse transcription. The RT stop signals of RNA with/without FTO treatment is then compared to determine the exact sites of m6A. Recently, endoribonuclease-based RNA digestion with m6A-sensitive RNase (MazF to cleave RNA at unmethylated ACA motifs) followed by sequencing (MAZTER-seq) (Garcia-Campos et al., 2019), and m6A-sensitive RNA-endoribonuclease-facilitated sequencing (m6A-REF-seq) (Zhang Z. et al., 2019) methods were used as antibody-independent methods. Another antibody-free m6A sequencing (deamination adjacent to RNA modification targets, DART-seq) method was devised, using APOBEC1-YTH (cytidine deaminase fused with m6A-binding YTH domain) protein which deaminates C to U at the site adjacent to m6A. This helps to identify m6A sites in mRNA (Meyer, 2019). Moreover, two chemical labeling methods viz. m6A-label-seq (Shu et al., 2020), and m6A-SEAL (Wang et al., 2020) have also been developed. Wang et al. (2020) combined dithiothreitol (DTT)-mediated thiol-addition reaction [that converts the unstable hm6A to stable N6-dithiolsitolmethyladenosine (dm6A)] with FTO-mediated enzymatic oxidation of m6A to hm6A to develop FTO-assisted m6A selective chemical labeling (m6A-SEAL) method for detection of m6A in mRNA. In a transcriptome-wide m6A-SEAL-seq analysis, they could identify 8,605 m6A in human embryonic kidney and 12,297 m6A in rice leaf. Currently, most of the epitranscriptomic studies employ a detection method with NGS technology for context-specific mapping of the modified base at single-base resolution.
Challenges in the Detection of Modified Bases
A major challenge in detection of the modified mRNA base has been the relatively low count of the modified base within the vast mRNA repertoire Another challenge is the precise quantification and mapping of modified RNA residues at a single-nucleotide level. Additional challenge stems from substantial background signals often present in the maps prepared. The inability of the technique to discriminate between misincorporation/RT-stop due to the modified base and background-pause/misincorporation either due to RNA structure, RT-error, or technical errors of sequencing platform (Schwartz and Motorin, 2017). Since the same antibody can recognize both m6A (in RNA) and 6-mA (in DNA), the contaminating DNA must be removed to get the real level of the modified base (Liang et al., 2020). Besides, there are many other limitations including intrinsic bias on secondary structures. For example, the m6A specific-antibody fails to distinguish between m6A and m6Am (Schwartz et al., 2013; Linder et al., 2015). Although CMC-based Ψ sequencing has been successful in identifying Ψ at the single-base resolution, it has been associated with the problem of RNA degradation because of the alkaline treatment step (see Zhao et al., 2020). Moreover, current sequencing technologies have not been able to detect hm5C and m1A, particularly at the single-base resolution, which limits the functional characterization of these modified bases. Some of the challenges in the detection of modified RNA bases at technological, experimental, and analytical levels are described here.
Sequencing by synthesis approach has many restrictions in detecting the base modification. The specific antibody or chemical required for the detection of a modified base (indirect detection of the modified base) is known for only a limited number of modifications, which may show cross-reactivity. The antibody-based immunoprecipitation (IP) sequencing method (e.g., m6A-seq or MeRIP) uses a 100–200 nt mapping window which fails to precisely identify m6A sites (Molinie et al., 2016). The photocrosslinking-assisted m6A sequencing (PA-m6A-seq), m6A individual-nucleotide-resolution crosslinking and immunoprecipitation (miCLIP), and UV-CLIP techniques suffer from low crosslinking yield and use an indirect method to infer m6A sites. The location of m6A is inferred near the antibody crosslinking point (the tyrosine residue of antibody and RNA base), but the crosslinking point might be at varying distance from the m6A-binding sites, which creates difficulty in precise identification of the m6A site, particularly when m6A occurs in a cluster (Meyer et al., 2012; Linder et al., 2015). Even the direct detection methods like SMRT face certain challenges such as the ZMW stumbles when a stretch m6A gets incorporated; hence, the current throughput level is too low for transcriptome-wide analysis.
Careful selection/inclusion of input controls for base modification mapping is crucial. Mostly, well-known modified bases in rRNA/tRNA serve as intrinsic controls for evaluating the sensitivity and specificity of analytical methods. The conventional transcriptome analysis uses millions of cells from a tissue, epitranscriptome being highly dynamic, the cell-specific analysis would be necessary for the detection/quantification of a particular base modification and its functional characterization (Helm and Motorin, 2017). Furthermore, biological and/or technical replicates (at least 2–3) are very important to filter out falsely-detected sites, as well as to assess the robustness/reproducibility of the detection and quantification method.
Future Perspectives
Recent studies have provided unprecedented mechanistic insights into RNA base modifications, and NGS-based technologies for detecting RNA base modifications are further improving the scenario. Modern chemical biology tools would be applied to expedite the epitranscriptomic studies. High-throughput technologies to simultaneously identify different modified bases in the same RNA molecule will have considerable applicability, as modified bases may have cumulative effects on regulating biological functions. Plants provide a unique system to elucidate the biological functions of modified RNA bases and their regulatory aspects through investigating epitranscriptomic alterations in higher eukaryotes, which are otherwise difficult to be elucidated using an animal system (Shen et al., 2019). Using a combination of techniques including genetic ablation and NGS-based mapping, the regulatory roles of the epitranscriptome in several developmental processes in plants have been demonstrated. Compared with the writers and erasers, readers for the modified base play a more significant role in responses to environmental stresses. This suggests that deciphering the location/context of epitranscriptomic marks is more important than merely detecting the changes (writing/erasing) in the marks for improved stress adaptation in plants. Therefore, it is important to characterize the role of reader proteins in the epitranscriptomic regulation of gene expression under environmental stresses (Hu et al., 2019). Association between epitranscriptome and stress responses in plants indicates that such epitranscriptomic marks might be utilized in the future as important epimarks for the development of stress-tolerant crop plants (Vandivier and Gregory, 2018). In the line of the success in the detection of m6A at single-base resolution, a similar sequencing method would be optimized for hm5C and m1A mapping. Nevertheless, for functional characterization of epitranscriptomic modifications, quantification of the absolute stoichiometry of RNA modifications is crucial.
However, several questions need to be answered before we can devise appropriate strategies to better utilize the epitranscriptomeic information. Some of these include, why only selected mRNAs get modified? Why are only certain adenosine/m6A or cytosine/m5C at selected sites gets methylated/demethylated? How does the modified base affect downstream mRNA processing? How do different readers recognize their targets? How are the writer, reader, and eraser for a nucleobase get co-ordinately regulated by the developmental/environmental signal? Even if we get answers to some of these questions, several other questions would require to be answered. For example, how do the different mRNA base modifications influence the dynamics/function of each other? Experiments designed to answer some of these questions are underway in laboratories worldwide, and we expect that the next 5 years of research in epitranscriptomics would be more exciting than the past!.
Conclusion
During the past few years, many RNA modifications and their functional versatility could be discovered due to the advances in chemogenetic RNA labeling techniques, high-throughput NGS, and functional validation. Several other dynamic base modifications in mRNA are also being identified, which would require functional characterization for advances in epitranscriptomics. Numerous other epitranscriptomic modifications may be identified in the future which may show interaction with other modified bases in modulating metabolic pathways. The biological functions of several mRNA base modifications are still poorly understood, their detection at single-base resolution using technological advancements such as nanowell (SMRT) and nanopore (Oxford Nanopore) sequencing is very much promising. However, proper experimental design with a sufficient number of replications, and inclusion of controls would be very important to rule out false-positive results and for the highest confidence level. Moreover, identifying the enzyme(s) involved in modification of RNA base (reader), and replacing it with an unmodified base (eraser) is necessary for devising strategies to manipulate the expression of a gene. However, several fundamental questions remain to be answered, including whether modified bases are conserved among plant species. Answering these questions would substantially improve our knowledge of epitranscriptomics and its effects on plant growth, fitness, and survival under environmental stress. Such investigations, particularly comprehensive studies to demonstrate a linkage between epigenetic and epitranscriptomic regulations, would offer potential new strategies for the manipulation of crop plants with better plasticity/adaptability to the changing climatic conditions. Comprehensive studies on the correlation between epigenetic and epitranscriptomic regulation of gene expression might provide some newer aspects (Song and Yi, 2017) for the manipulation of a trait through epigenome/epitranscriptome editing to develop climate-smart crop plants for the 21st century (Kumar, 2019).
Author Contributions
SK and TM conceived the review. SK prepared the manuscript. SK and TM revised the manuscript and approved the final draft. Both authors contributed to the article and approved the submitted version.
Funding
The epigenomics and epitranscriptomics research are being carried out with financial supports from National Agricultural Science Fund (NASF/ABP-70161/2018-19), and an Extramural Research grant [18(3)/2018-O&P] from the Indian Council of Agricultural Research, Government of India, New Delhi.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adachi, H., De Zoysa, M. D., and Yu, Y. T. (2019). Post-transcriptional pseudouridylation in mRNA as well as in some major types of noncoding RNAs. Biochem. Biophys. Acta Gene Regul. Mech. 1862, 230–239. doi: 10.1016/j.bbagrm.2018.11.002
Aguilo, F., Zhang, F., Sancho, A., Fidalgo, M., Cecilia, S. D., Vashisht, A., et al. (2015). Coordination of m6A mRNA methylation and gene transcription by ZFP217 regulates pluripotency and reprogramming. Cell Stem Cell 17, 689–704. doi: 10.1016/j.stem.2015.09.005
Alarcon, C. R., Goodarzi, H., Lee, H., Liu, X., Tavazoie, S., and Tavazoie, S. F. (2015a). HNRNPA2B1 is a mediator of m6A-dependent nuclear RNA processing events. Cell 162, 1299–1308. doi: 10.1016/j.cell.2015.08.011
Alarcon, C. R., Lee, H., Goodarzi, H., Halberg, N., and Tavazoie, S. F. (2015b). N6-methyladenosine marks primary microRNAs for processing. Nature 519, 482–485. doi: 10.1038/nature14281
Anderson, S. J., Kramer, M. C., Gosai, S. J., Yu, X., Vandivier, L. E., and Nelson, A. D. L. (2018). N6-methyladenosine inhibits local ribonucleolytic cleavage to stabilize mRNAs in Arabidopsis. Cell Rep. 25, 1146–1157. doi: 10.1016/j.celrep.2018.10.020
Angelova, M. T., Dimitrova, D. G., Dinges, N., Lence, T., Worpenberg, L., and Carre, C. (2018). The emerging field of epitranscriptomics in neurodevelopmental and neuronal disorders. Front. Bioeng. Biotechnol. 6:46. doi: 10.3389/fbioe.2018.00046
Arango, D., Sturgill, D., Alhusaini, N., Dillman, A. A., Sweet, T. J., and Hanson, G. (2018). Acetylation of cytidine in mRNA promotes translation efficiency. Cell 175, 1872–1886. doi: 10.1016/j.cell.2018.10.030
Arimbasseri, A. G., Iben, J., Wei, F. Y., Rijal, K., Tomizawa, K., Hafner, M., et al. (2016). Evolving specificity of tRNA 3-methyl-cytidine-32 (m3C32) modification: a subset of tRNAsSer requires N6-isopentenylation of A37. RNA 22, 1400–1410. doi: 10.1261/rna.056259.116
Arribas-Hernandez, L., Bressendorff, S., Hansen, M. H., Poulsen, C., Erdmann, S., and Brodersen, P. (2018). An m6A-YTH module controls developmental timing and morphogenesis in Arabidopsis. Plant Cell 30, 952–967. doi: 10.1105/tpc.17.00833
Barciszewska, M. Z., Barciszewska, A. M., and Rattan, S. I. S. (2007). TLC-based detection of methylated cytosine: application to aging epigenetics. Biogerontology 8, 673–678. doi: 10.1007/s10522-007-9109-3
Batista, P. J., Molinie, B., Wang, J., Qu, K., Zhang, J., Li, L., et al. (2014). m6A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 15, 707–719. doi: 10.1016/j.stem.2014.09.019
Belanger, F., Stepinski, J., Darzynkiewicz, E., and Pelletier, J. (2010). Characterization of hMTr1, a human Cap1 2′-O-ribose methyltransferase. J. Biol. Chem. 285, 33037–33044. doi: 10.1074/jbc.M110.155283
Boccaletto, P., Machnicka, M. A., Purta, E., Pia̧tkowski, P., Bagiński, B., and Wirecki, T. K. (2018). MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 46, 303–307. doi: 10.1093/nar/gkx1030
Bodi, Z., Zhong, S., Mehra, S., Song, J., Li, H., Graham, N., et al. (2012). Adenosine methylation in Arabidopsis mRNA is associated with the 3′ end and reduced levels cause developmental defects. Front. Plant Sci. 3:48. doi: 10.3389/fpls.2012.00048
Bohnsack, K. E., Höbartner, C., and Bohnsack, M. T. (2019). Eukaryotic 5-methylcytosine (m5C) RNA methyltransferases: mechanisms, cellular functions, and links to disease. Genes 10:102. doi: 10.3390/genes10020102
Bokar, J. A., Shambaugh, M. E., Polayes, D., Matera, A. G., and Rottman, F. M. (1997). Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA 3, 1233–1247.
Boo, S. H., and Kim, Y. K. (2020). The emerging role of RNA modifications in the regulation of mRNA stability. Expt. Mol. Med. 52, 400–408. doi: 10.1038/s12276-020-0407-z
Boulias, K., Toczydłowska-Socha, D., Hawley, B. R., Liberman, N., Takashima, K., and Zaccara, S. (2019). Identification of the m(6)Am methyltransferase PCIF1 reveals the location and functions of m(6)Am in the transcriptome. Mol. Cell 75, 631–643.e638. doi: 10.1016/j.molcel.2019.06.006
Burgess, A., David, R., and Searle, I. R. (2016). Deciphering the epitranscriptome: a green perspective. J. Integr. Plant Biol. 58, 822–835. doi: 10.1111/jipb.12483
Cahová, H., Winz, M. L., Höfer, K., Nübel, G., and Jäschke, A. (2015). NAD captureSeq indicates NAD as a bacterial cap for a subset of regulatory RNAs. Nature 519, 374–377. doi: 10.1038/nature14020
Carlile, T. M., Rojas-Duran, M. F., Zinshteyn, B., Shin, H., Bartoli, K. M., and Gilbert, W. V. (2014). Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 515, 143–146. doi: 10.1038/nature13802
Chen, K., Lu, Z., Wang, X., Fu, Y., and Luo, G. Z. (2015). High-resolution N6-methyladenosine (m6A) map using photo-crosslinking-assisted m6A sequencing. Angew. Chem. Int. Ed. Engl. 54, 1587–1590. doi: 10.1002/anie.201410647
Chen, P., Jäger, G., and Zheng, B. (2010). Transfer RNA modifications and genes for modifying enzymes in Arabidopsis thaliana. BMC Plant Biol. 10:201. doi: 10.1186/1471-2229-10-201
Chen, R. X., Chen, X., Xia, L. P., Zhang, J. X., Pan, Z. Z., Ma, X. D., et al. (2019). N6-methyladenosine modification of circNSUN2 facilitates cytoplasmic export and stabilizes HMGA2 to promote colorectal liver metastasis. Nat. Commun. 10:4695. doi: 10.1038/s41467-019-12651-2
Choi, J., Ieong, K. W., Demirci, H., Chen, J., Petrov, A., Prabhakar, A., et al. (2016). N6-methyladenosine in mRNA disrupts tRNA selection and translation-elongation dynamics. Nat. Str. Mol. Biol. 23, 110–115. doi: 10.1038/nsmb.3148
Chujo, T., and Suzuki, T. (2012). Trmt61B is a methyltransferase responsible for 1-methyladenosine at position 58 of human mitochondrial tRNAs. RNA 18, 2269–2276. doi: 10.1261/rna.035600.112
Cui, Q., Shi, H., Ye, P., Li, L., Qu, Q., Sun, G., et al. (2017). m6A RNA methylation regulates the self-renewal and tumorigenesis of glioblastma stem cells. Cell Rep. 18, 2622–2634. doi: 10.1016/j.celrep.2017.02.059
Cui, X., Liang, Z., Shen, L., Zhang, Q., Bao, S., and Geng, Y. (2017). 5-Methylcytosine RNA methylation in Arabidopsis thaliana. Mol. Plant 10, 1387–1399. doi: 10.1016/j.molp.2017.09.013
Cui, X., Meng, J., Zhang, S., Chen, Y., and Huang, Y. (2016). A novel algorithm for calling mRNA m6A peaks by modeling biological variances in MeRIP-seq data. Bioinformatics 32, 378–385. doi: 10.1093/bioinformatics/btw281
David, R., Burgess, A., Parker, B., Li, J., Pulsford, K., and Sibbritt, T. (2017). Transcriptome-wide mapping of RNA 5-methylcytosine in Arabidopsis mRNAs and noncoding RNAs. Plant Cell 29, 445–460. doi: 10.1105/tpc.16.00751
Davis, F. F., and Allen, F. W. (1957). Ribonucleic acids from yeast which contain a fifth nucleotide. J. Biol. Chem. 227, 907–915. doi: 10.1016/S0021-9258(18)70770-9
de Almeida, C., Scheer, H., Zuber, H., and Gagliardi, D. (2018). RNA uridylation: a key posttranscriptional modification shaping the coding and noncoding transcriptome. Wiley Interdiscip. Rev. RNA 9:e1440. doi: 10.1002/wrna.1440
Delatte, B., Wang, F., Ngoc, L. V., Collignon, E., Bonvin, E., Deplus, R., et al. (2016). Transcriptome-wide distribution and function of RNA hydroxymethylcytosine. Science 351, 282–285. doi: 10.1126/science.aac5253
Dominissini, D., Moshitch-Moshkovitz, S., Schwartz, S., Salmon-Divon, M., Ungar, L., and Osenberg, S. (2012). Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206. doi: 10.1038/nature11112
Dominissini, D., Nachtergaele, S., Moshitch-Moshkovitz, S., Peer, E., Kol, N., Ben-Haim, M. S., et al. (2016). The dynamic N1-methyladenosine methylome in eukaryotic messenger RNA. Nature 530, 441–446. doi: 10.1038/nature16998
Dong, C., Niu, L., Song, W., Xiong, X., Zhang, X., and Zhang, Z. (2016). tRNA modification profiles of the fast-proliferating cancer cells. Biochem. Biophys. Res. Commun. 476, 340–345. doi: 10.1016/j.bbrc.2016.05.124
Du, H., Zhao, Y., He, J., Zhang, Y., Xi, H., Liu, M., et al. (2016). YTHDF2 destabilizes m6A-containing RNA through direct recruitment of the CCR4–NOT deadenylase complex. Nat. Commun. 7:12626. doi: 10.1038/ncomms12626
Duan, H. C., Wei, L. H., Zjang, C., Wang, Y., Chen, L., and Lu, Z. (2017). ALKBH10B is an RNA N6-methyladenosine demethylase affecting Arabidopsis floral transition. Plant Cell 29, 2995–3011. doi: 10.1105/tpc.16.00912
Edelheit, S., Schwartz, S., Mumbach, M. R., Wurtzel, O., and Sorek, R. (2013). Transcriptome-wide mapping of 5-methylcytidine RNA modifications in bacteria. archaea, and yeast reveals m5C within archaeal mRNAs. PLoS Genet. 9:e1003602. doi: 10.1371/journal.pgen.1003602
Engel, M., Roeh, S., Eggert, C., Kaplick, P. M., Tietze, L., and Namendorf, C. (2017). The role of m6A-RNA methylation in stress response regulation. BioRxiv [Preprint] doi: 10.1101/200402
Fang, L., Wang, W., Li, G., Zhang, L., Li, J., Gan, D., et al. (2020). CIGAR-seq, a CRISPR/Cas-based method for unbiased screening of novel mRNA modification regulators. Mol. Syst. Biol. 16:e10025. doi: 10.15252/msb.202010025
Flores, J. V., Cordero-Espinoza, L., Oeztuerk-Winder, F., Andersson-Rolf, A., Selmi, T., Blanco, S., et al. (2017). Cytosine-5 RNA methylation regulates neural stem cell differentiation and motility. Stem Cell Rep. 8, 112–124. doi: 10.1016/j.stemcr.2016.11.014
Frindert, J., Zhang, Y., Nübel, G., Kahloon, M., Kolmar, L., Hotz-Wagenblatt, A., et al. (2018). Identification, biosynthesis, and decapping of NAD-capped RNAs in B. subtilis. Cell Rep. 24, 1890–1901. doi: 10.1016/j.celrep.2018.07.047
Frye, M., Harada, B., Behm, M., and He, C. (2018). RNA modifications modulate gene expression during development. Science 361, 1346–1349. doi: 10.1126/science.aau1646
Fu, L., Guerrero, C. R., Zhong, N., Amato, N. J., Liu, Y., and Liu, S. (2014). Tet-mediated formation of 5-hydroxymethylcytosine in RNA. J. Am. Chem. Soc. 136, 11582–11585. doi: 10.1021/ja505305z
Fu, Y., Dominissini, D., Rechavi, G., and He, C. (2014). Gene expression regulation mediated through reversible m6A RNA methylation. Nat. Rev. Genet. 15, 293–306. doi: 10.1038/nrg3724
Garcia-Campos, M. A., Edelheit, S., Toth, U., Safra, M., Shachar, R., and Viukov, S. (2019). Deciphering the “m6A code” via antibody-independent quantitative profiling. Cell 3, 731–747. doi: 10.1016/j.cell.2019.06.013
Geula, S., Moshitch-Moshkovitz, S., Dominissini, D., Mansour, A. A., Kol, N., Salmon-Divon, M., et al. (2015). m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science 347, 1002–1006. doi: 10.1126/science.1261417
Grosjean, H., Keith, G., and Droogmans, L. (2004). Detection and quantification of modified nucleotides in RNA using thin-layer chromatography. Methods Mol. Biol. 265, 357–391. doi: 10.1385/1-59259-775-0:357
Hauenschild, R., Tserovski, L., Schmid, K., Th€uring, K., Winz, M. L., Sharma, S., et al. (2015). The reverse transcription signature of N-1-methyladenosine in RNA-Seq is sequence dependent. Nucleic Acids Res. 43, 9950–9964. doi: 10.1093/nar/gkv895
Haussmann, I. U., Bodi, Z., Sanchez-Moran, E., Mongan, N. P., Archer, N., Fray, R. G., et al. (2016). m6A potentiates Sxl alternative pre-mRNA splicing for robust Drosophila sex determination. Nature 540, 301–304. doi: 10.1038/nature20577
Helm, M. (2006). Post-transcriptional nucleotide modification and alternative folding of RNA. Nucleic Acids Res. 34, 721–733. doi: 10.1093/nar/gkj471
Helm, M., and Motorin, Y. (2017). Detecting RNA modifications in the epitranscriptome: predict and validate. Nat. Rev. Genet. 18, 275–291. doi: 10.1038/nrg.2016.169
Hoernes, T. P., Clementi, N., Faserl, K., Glasner, H., Breuker, K., and Lindner, H. (2016). Nucleotide modifications within bacterial messenger RNAs regulate their translation and are able to rewire the genetic code. Nucleic Acids Res. 44, 852–862. doi: 10.1093/nar/gkv1182
Holstein, J. M., Anhäuser, L., and Rentmeister, A. (2016). Modifying the 5′ cap for click reactions of eukaryotic mRNA and to tune translation efficiency in living cells. Angew. Chem. Int. Ed. 55, 10899–10903. doi: 10.1002/anie.201604107
Hong, T., Yuan, Y., Chen, Z., Xi, K., Wang, T., Xie, Y., et al. (2018). Precise antibody-independent m6A identification via 4SedTTP-involved and FTO-assisted strategy at single-nucleotide resolution. J. Am. Chem. Soc. 140, 5886–5889. doi: 10.1021/jacs.7b13633
Hsu, P. J., Zhu, Y., Ma, H., Gho, Y., Shi, X., and Liu, Y. (2017). Ythdc2 is an N6-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. 27, 1115–1127. doi: 10.1038/cr.2017.99
Hu, J., Manduzio, S., and Kang, H. (2019). Epitranscriptomic RNA methylation in plant development and abiotic stress responses. Front. Plant Sci. 10:500. doi: 10.3389/fpls.2019.00500
Huang, H., Weng, H., Sun, W., Qin, X., Shi, H., and Wu, H. (2018). Recognition of RNA N6-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 20, 285–295. doi: 10.1038/s41556-018-0045-z
Huber, S. M., Van Delet, P., Mendil, L., Bachman, M., Smollett, K., and Werner, F. (2015). Formation and abundance of 5-hydroxymethylcytosine in RNA. Chembiochem 16, 752–755. doi: 10.1002/cbic.201500013
Hull, C. M., and Bevilacqua, P. C. (2016). Discriminating self and non-self by RNA: roles for RNA structure, misfolding, and modification in regulating the innate immune sensor PKR. Acc. Chem. Res. 49, 1242–1249. doi: 10.1021/acs.accounts.6b00151
Hussain, S., Sajini, A. A., Blanco, S., Dietmann, S., Lombard, P., Sugimoto, Y., et al. (2013). NSun2-mediated cytosine-5 methylation of vault noncoding RNA determines its processing into regulatory small RNAs. Cell Rep. 4, 255–261. doi: 10.1016/j.celrep.2013.06.029
Ikeuchi, K., Izawa, T., and Inada, T. (2018). Recent progress on the molecular mechanism of quality controls induced by ribosome stalling. Front. Genet. 9:743. doi: 10.3389/fgene.2018.00743
Ishii, T., Hayakawa, H., Igawa, T., Sekiguchi, T., and Sekiguchi, M. (2018). Specific binding of PCBP1 to heavily oxidized RNA to induce cell death. Proc. Natl Acad. Sci. USA 115, 6715–6720. doi: 10.1073/pnas.1806912115
Ishii, T., Hayakawa, H., Sekiguchi, T., Adachi, N., and Sekiguchi, M. (2015). Role of Auf1 in elimination of oxidatively damaged messenger RNA in human cells. Free Radic. Biol. Med. 79, 109–116. doi: 10.1016/j.freeradbiomed.2014.11.018
Jackman, J. E., and Alfonzo, J. D. (2013). Transfer RNA modifications: nature’s combinatorial chemistry playground. Wiley Interdiscip. Rev. RNA. 4, 35–48. doi: 10.1002/wrna.1144
Jia, G. F., Fu, Y., Zhao, X., Dai, Q., Zheng, G., and Yang, Y. (2011). N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 7, 885–887. doi: 10.1038/nchembio.687
Jiao, X., Doamekpor, S. K., Bird, J. G., Nickels, B. E., Tong, L., Hart, R. P., et al. (2017). 5’ End nicotinamide adenine dinucleotide cap in human cells promotes RNA decay through DXO-mediated deNADding. Cell 168, 1015–1027. doi: 10.1016/j.cell.2017.02.019
Kadumuri, R. V., and Janga, S. C. (2018). Epitranscriptomic code and its alterations in human disease. Trends Mol. Med. 24, 886–903. doi: 10.1016/j.molmed.2018.07.010
Karijolich, J., Yi, C., and Yu, Y. T. (2015). Transcriptome-wide dynamics of RNA pseudouridylation. Nat. Rev. Mol. Cell Biol. 16, 581–585. doi: 10.1038/nrm4040
Kariko, K., Muramatsu, H., Keller, J. M., and Weissman, D. (2012). Increased erythropoiesis in mice injected with submicrogram quantities of pseudouridine-containing mRNA encoding erythropoietin. Mol. Ther. 20, 948–953. doi: 10.1038/mt.2012.7
Kasowitz, S. D., Ma, J., Anderson, S. J., Leu, N. A., Xu, Y., Gregory, B. D., et al. (2018). Nuclear m6A reader YTHDC1 regulates alternative polyadenylation and splicing during mouse oocyte development. PLoS Genet. 14:e1007412. doi: 10.1371/journal.pgen.1007412
Ke, S., Alemu, E. A., Mertens, C., Gantman, E. C., Fak, J. J., and Mele, A. (2015). A majority of m6A residues are in the last exons, allowing the potential for 30 UTR regulation. Genes Dev. 29, 2037–2053. doi: 10.1101/gad.269415.115
Keith, G. (1995). Mobilities of modified ribonucleotides on two-dimensional cellulose thin-layer chromatography. Biochimie 77, 142–144. doi: 10.1016/0300-9084(96)88118-1
Kellner, S., Ochel, A., Thüring, K., Spenkuch, F., Neumann, J., Sharma, S., et al. (2014). Absolute and relative quantification of RNA modifications via biosynthetic isotopomers. Nucleic Acids Res. 42:e142. doi: 10.1093/nar/gku733
Khan, Z., Ford, M. J., Cusanovich, D. A., Mitrano, A., Pritchard, J. K., and Gilad, Y. (2013). Primate transcript and protein expression levels evolve under compensatory selection pressures. Science 342, 1100–1104. doi: 10.1126/science.1242379
Khoddami, V., and Cairns, B. R. (2013). Identification of direct targets and modified bases of RNA cytosine methyltransferases. Nat. Biotechnol. 31, 458–464. doi: 10.1038/nbt.2566
Khoddami, V., Yerra, A., Mosbruger, T. L., Fleming, A. M., Burrows, C. J., and Cairns, B. R. (2019). Transcriptome-wide profiling of multiple RNA modifications simultaneously at single-base resolution. Proc. Natl. Acad. Sci. USA 116, 6784–6789. doi: 10.1073/pnas.1817334116
Khonsari, B., and Klassen, R. (2020). Impact of Pus1 pseudouridine synthase on specific decoding events in Saccharomyces cerevisiae. Biomolecules 10:729.
Kiledjian, M. (2018). Eukaryotic RNA 5′-end NAD+ capping and DeNADding. Trends Cell Biol. 28, 454–464. doi: 10.1016/j.tcb.2018.02.005
Knuckles, P., Lence, T., Haussmann, I. U., Jacob, D., Kreim, N., Carl, S. H., et al. (2018). Zc3h13/Flacc is required for adenosine methylation by bridging the mRNA-binding factor Rbm15/Spenito to the m6A machinery component Wtap/Fl(2)d. Genes Dev. 32, 415–429. doi: 10.1101/gad.309146.117
Kumar, S. (2019). Epigenomics for crop improvement: current status and future perspectives. J. Genet. Cell Biol. 3, 128–134.
Kumar, S., Chinnusamy, V., and Mohapatra, T. (2018). Epigenetics of modified DNA bases: 5-methylcytosine and beyond. Front. Genet. 9:640. doi: 10.3389/fgene.2018.00640
Kumar, S., Mapa, K., and Maiti, S. (2014). Understanding the effect of locked nucleic acid and 2′-O-methyl modification on the hybridization thermodynamics of a miRNA-mRNA pair in the presence and absence of AfPiwi protein. Biochemistry 53, 1607–1615. doi: 10.1021/bi401677d
Leonardi, A., Kovalchuk, N., Yin, L., Endres, L., Evke, S., Nevins, S., et al. (2020). The epitranscriptomic writer ALKBH8 drives tolerance and protects mouse lungs from the environmental pollutant naphthalene. Epigenetics 15, 1121–1138. doi: 10.1080/15592294.2020.1750213
Levanon, E. Y., Eisenberg, E., Yelin, R., Nemzer, S., Hallegger, M., and Shemesh, R. (2004). Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat. Biotechnol. 22, 1001–1005. doi: 10.1038/nbt996
Li, A., Chen, Y. S., Ping, X. L., Yang, X., Xiao, W., and Yang, Y. (2017). Cytoplasmic m6A reader YTHDF3 promotes mRNA translation. Cell Res. 27, 444–447. doi: 10.1038/cr.2017.10
Li, D., Zhang, H., Hong, Y., Huamg, L., Li, X., Zhang, Y., et al. (2014). Genome-wide identification, biochemical characterization, and expression analyses of the YTH domain-containing RNA-binding protein family in Arabidopsis and rice. Plant Mol. Biol. Rep. 32, 1169–1186. doi: 10.1007/s11105-014-0724-2
Li, X., Xiong, X., and Yi, C. (2016b). Epitranscriptome sequencing technologies: decoding RNA modifications. Nat. Methods 14, 23–31. doi: 10.1038/nmeth.4110
Li, X., Xiong, X., Wang, K., Wang, L., Shu, X., Ma, S., et al. (2016a). Transcriptome-wide mapping reveals reversible and dynamic N(1)-methyladenosine methylome. Nat. Chem. Biol. 12, 311–316. doi: 10.1038/nchembio.2040
Li, X., Xiong, X., Zhang, M., Wang, K., Chen, Y., Zhou, J., et al. (2017). Base-resolution mapping reveals distinct m1A methylome in nuclear- and mitochondrial-encoded transcripts. Mol. Cell 68, 993–1005. doi: 10.1016/j.molcel.2017.10.019
Li, X., Zhu, P., Ma, S., Song, J., Bai, J., Sun, F., et al. (2015). Chemical pulldown reveals dynamic pseudouridylation of the mammalian transcriptome. Nat. Chem. Biol. 11, 592–597. doi: 10.1038/nchembio.1836
Li, Y., Wang, X., Li, C., Hu, S., Yu, J., and Song, S. (2014). Transcriptome-wide N6-methyladenosine profiling of rice callus and leaf reveals the presence of tissue-specific competitors involved in selective mRNA modification. RNA Biol. 11, 1180–1188. doi: 10.4161/rna.36281
Li, Z., Shi, J., Yu, L., Zhao, X., Ran, L., and Hu, D. (2018). N6-methyl-adenosine level in Nicotiana tabacum is associated with tobacco mosaic virus. Virol. J. 15:87. doi: 10.1186/s12985-018-0997-4
Liang, Z., Riaz, A., Chachar, S., Ding, Y., Du, H., and Gu, X. (2020). Epigenetic modifications of mRNA and DNA in plants. Mol. Plant 13, 14–30. doi: 10.1016/j.molp.2019.12.007
Lim, J., Ha, M., Chang, H., Kwon, S. C., Simanshu, D. K., Patel, D. J., et al. (2014). Uridylation by TUT4 and TUT7 marks mRNA for degradation. Cell 159, 1365–1376. doi: 10.1016/j.cell.2014.10.055
Limbach, P. A., and Paulines, M. J. (2017). Going global: the new era of mapping modifications in RNA. Wiley Interdiscip. Rev. RNA 8:e1367. doi: 10.1002/wrna.1367
Linder, B., Grozhik, A. V., Olarerin-George, A. O., Meydan, C., Mason, C. E., and Jaffrey, S. R. (2015). Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 12, 767–772. doi: 10.1038/nmeth.3453
Liu, F., Clark, W., Luo, G., Wang, X., Fu, Y., and Wei, J. (2016). ALKBH1-mediated tRNA demethylation regulates translation. Cell 167, 816–828. doi: 10.1016/j.cell.2016.09.038
Liu, H., Begik, O., Lucas, M. C., Ramirez, J. M., Mason, C. E., and Wiener, D. (2019). Accurate detection of m6A RNA modifications in native RNA sequences. Nat. Commun. 10:4079. doi: 10.1038/s41467-019-11713-9
Liu, J., Huang, T., Zhang, Y., Zhao, T., Zhao, X., Chen, W., et al. (2020). Sequence- and structure-selective mRNA m5C methylation by NSUN6 in animals. Natl. Sci. Rev. nwaa273. doi: 10.1093/nsr/nwaa273
Liu, J., Yue, Y., Han, D., Wang, X., Fu, Y., and Zhang, L. (2014). A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 10, 93–95. doi: 10.1038/nchembio.1432
Liu, N., and Pan, T. (2016). N6-methyladenosine–encoded epitranscriptomics. Nat. Struc. Mol. Biol. 23, 98–102. doi: 10.1038/nsmb.3162
Liu, N., Dai, Q., Zheng, G., He, C., Parisien, M., and Pan, T. (2015). N6-methyladenosine-dependent RNA structural switches regulate RNA–protein interactions. Nature 518, 560–564. doi: 10.1038/nature14234
Liu, N., Parisien, M., Dai, Q., Zheng, G., He, C., and Pan, T. (2013). Probing N6-methyladenosine RNA modification status at single nucleotide resolution in mRNA and long noncoding RNA. RNA 19, 1848–1856. doi: 10.1261/rna.041178.113
Lovejoy, A. F., Riordan, D. P., and Brown, P. O. (2014). Transcriptome-wide mapping of pseudouridines: pseudouridine synthases modify specific mRNAs in S. cerevisiae. PLoS One 9:e110799. doi: 10.1371/journal.pone.0110799
Luo, G. Z., Macqueen, A., Zheng, G., Duan, H., Dore, L. C., and Lu, Z. (2014). Unique features of the m6A methylome in Arabidopsis thaliana. Nat. Commun. 5:5630. doi: 10.1038/ncomms6630
Luo, J., Wang, Y., Wang, M., Zhang, L., Peng, H., and Zhou, Y. Y. (2019). Natural variation in RNA m6A methylation and its relationship with translational status. Plant Physiol. 182, 332–344. doi: 10.1104/pp.19.00987
Luo, S., and Tong, L. (2014). Molecular basis for the recognition of methylated adenines in RNA by the eukaryotic YTH domain. Proc. Natl. Acad. Sci. USA 111, 13834–13839. doi: 10.1073/pnas.1412742111
Malbec, L., Zhang, T., Chen, Y. S., Zhang, Y., Sun, B. F., Shi, B.-Y., et al. (2019). Dynamic methylome of internal mRNA N7-methylguanosine and its regulatory role in translation. Cell Res. 29, 927–941. doi: 10.1038/s41422-019-0230-z
Marchand, V., Blanloeil-Oillo, F., Helm, M., and Motorin, Y. (2016). Illumina-based RiboMethSeq approach for mapping of 2′-O-Me residues in RNA. Nucleic Acids Res. 44:e135. doi: 10.1093/nar/gkw547
Martinez-Perez, M., Aparicio, F., López-Gresa, M. P., Bellés, J. M., Sánchez-Navarro, J. A., and Pallás, V. (2017). Arabidopsis m6A demethylase activity modulates viral infection of a plant virus and the m6A abundance in its genomic RNAs. Proc. Natl. Acad. Sci. USA 14, 10755–10760. doi: 10.1073/pnas.1703139114
Mauer, J., Luo, X., Blanjoie, A., Jiao, X., Grozhik, A. V., Patil, D. P., et al. (2017). Reversible methylation of m6Am in the 5′ cap controls mRNA stability. Nature 541, 371–375. doi: 10.1038/nature21022
Meyer, K. D. (2019). DART-seq: an antibody-free method for global m(6)A detection. Nat. Methods 16, 1275–1280. doi: 10.1038/s41592-019-0570-0
Meyer, K. D., Patil, D. P., Zhou, J., Zinoviev, A., Skabkin, M. A., Elemento, O., et al. (2015). 5′ UTR m6A promotes cap-independent translation. Cell 163, 999–1010. doi: 10.1016/j.cell.2015.10.012
Meyer, K. D., Saletore, Y., Zumbo, P., Elemento, O., Mason, C. E., and Jaffrey, S. R. (2012). Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 149, 1635–1646. doi: 10.1016/j.cell.2012.05.003
Miao, Z., Zhang, T., Qi, Y., Song, J., Han, Z., and Ma, C. (2019). Evolution of the RNA N6-methyladenosine methylome mediated by genomic duplication. Plant Physiol. 182, 345–360. doi: 10.1104/pp.19.00323
Mielecki, D., Zugaj, D. Ł, Muszewska, A., Piwowarski, J., Chojnacka, A., Mielecki, M., et al. (2012). Novel AlkB dioxygenases–alternative models for in silico and in vivo studies. PLoS One 7:e30588. doi: 10.1371/journal.pone.0030588
Molinie, B., Wang, J., Lim, K. S., Hillebrand, R., Lu, Z. X., Van Wittenberghe, N., et al. (2016). m6A-LAIC-seq reveals the census and complexity of the m6A epitranscriptome. Nat. Methods 13, 692–698. doi: 10.1038/nmeth.3898
Nestor, C., Ruzov, A., Meehan, R., and Dunican, D. (2010). Enzymatic approaches and bisulfite sequencing cannot distinguish between 5-methylcytosine and 5-hydroxymethylcytosine in DNA. Biotechniques 48, 317–319. doi: 10.2144/000113403
Nunomura, A., Lee, H. G., Zhu, X., and Perry, G. (2017). Consequences of RNA oxidation on protein synthesis rate and fidelity: implications for the pathophysiology of neuropsychiatric disorders. Biochem Soc. Trans. 45, 1053–1066. doi: 10.1042/BST20160433
Palladino, M. J., Keegan, L. P., O’Connell, M. A., and Reenan, R. A. (2000). A-to-I pre-mRNA editing in drosophila is primarily involved in adult nervous system function and integrity. Cell 102, 437–449. doi: 10.1016/S0092-8674(00)00049-0
Park, O. H., Ha, H., Lee, Y., Boo, S. H., Kwon, D. H., and Song, H. K. (2019). Endoribonucleolytic cleavage of m(6)A-containing RNAs by RNase P/MRP complex. Mol. Cell 74, 494–507 e498. doi: 10.1016/j.molcel.2019.02.034
Patil, D. P., Chen, C. K., Pickering, B. F., Chow, A., Jackson, C., Guttman, M., et al. (2016). m6A RNA methylation promotes XIST-mediated transcriptional repression. Nature 537, 369–373. doi: 10.1038/nature19342
Pavitt, G. D. (2018). Regulation of translation initiation factor eIF2B at the hub of the integrated stress response. Wiley Interdiscip. Rev. RNA 9:e1491. doi: 10.1002/wrna.1491
Peer, E., Rechavi, G., and Dominissini, D. (2017). Epitranscriptomics: regulation of mRNA metabolism through modifications. Curr. Opin. Chem. Biol. 41, 93–98. doi: 10.1016/j.cbpa.2017.10.008
Pendleton, K. E., Chen, B., Liu, K., Hunter, O. V., Xie, Y., Tu, B. P., et al. (2017). The U6 snRNA m(6)A methyltransferase METTL16 regulates SAM synthetase intron retention. Cell 169, 824–835. doi: 10.1016/j.cell.2017.05.003
Peng, X., Xu, X., Wang, Y., Hawke, D. H., Yu, S., Han, L., et al. (2018). A-to-I RNA editing contributes to proteomic diversity in cancer. Cancer Cell 33, 817–828. doi: 10.1016/j.ccell.2018.03.026
Pfaff, C., Ehrnsberger, H. F., Flores-Tornero, M., Sørensen, B. B., Schubert, T., Längst, G., et al. (2018). ALY RNA-binding proteins are required for nucleocytosolic mRNA transport and modulate plant growth and development. Plant Physiol. 177, 226–240. doi: 10.1104/pp.18.00173
Ping, X. L., Sun, B. F., Wang, L., Xiao, W., Yang, X., Wang, W. J., et al. (2014). Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 24, 177–189. doi: 10.1038/cr.2014.3
Rajagopalan, L. E., Westmark, C. J., Jarzembowski, J. A., and Malter, J. S. (2012). hnRNP C tetramer measures RNA length to classify RNA polymerase II transcripts for export. Science 335, 1643–1646. doi: 10.1126/science.1218469
Rintala-Dempsey, A. C., and Kothe, U. (2017). Eukaryotic stand-alone pseudouridine synthases- RNA modifying enzymes and emerging regulators of gene expression? RNA Biol. 14, 1185–1196. doi: 10.1080/15476286.2016.1276150
Roost, C., Lynch, S. R., Batista, P. J., Qu, K., Chang, H. Y., and Kool, E. T. (2015). Structure and thermodynamics of N6-methyladenosine in RNA: a spring-loaded base modification. J. Am. Chem. Soc. 137, 2107–2115. doi: 10.1021/ja513080v
Ross, R., Cao, X., Yu, N., and Limbach, P. A. (2016). Sequence mapping of transfer RNA chemical modifications by liquid chromatography tandem mass spectrometry. Methods 107, 73–78. doi: 10.1016/j.ymeth.2016.03.016
Roundtree, I. A., and He, C. (2016). RNA epigenetics–chemical messages for posttranscriptional gene regulation. Curr. Opin. Chem. Biol. 30, 46–51. doi: 10.1016/j.cbpa.2015.10.024
Roundtree, I. A., Luo, G. Z., Zhang, Z., Wang, X., Zhou, T., Cui, Y., et al. (2017). YTHDC1 mediates nuclear export of N6-methyladenosine methylated mRNAs. Elife 6:e31311. doi: 10.7554/eLife.31311.040
Ruzicka, K., Zhang, M., Campilho, A., Bodi, Z., Kashif, M., Saleh, M., et al. (2017). Identification of factors required for m6A mRNA methylation in Arabidopsis reveals a role for the conserved E3 ubiquitin ligase HAKAI. New Phytol. 215, 157–172. doi: 10.1111/nph.14586
Sakurai, M., Ueda, H., Yano, T., Okada, S., Terajima, H., Mitsuyama, T., et al. (2014). A biochemical landscape of A-to-I RNA editing in the human brain transcriptome. Genome Res. 24, 522–534. doi: 10.1101/gr.162537.113
Schaefer, M., Pollex, T., Hanna, K., and Lyko, F. (2009). RNA cytosine methylation analysis by bisulfite sequencing. Nucleic Acids Res. 37:e12. doi: 10.1093/nar/gkn954
Schwartz, S., Agarwala, S. D., Mumbach, M. R., Jovanovic, M., Mertins, P., Shishkin, A., et al. (2013). High-resolution mapping reveals a conserved, widespread, dynamic mRNA methylation program in yeast meiosis. Cell 155, 1409–1421. doi: 10.1016/j.cell.2013.10.047
Schwartz, S., and Motorin, Y. (2017). Next-generation sequencing technologies for detection of modified nucleotides in RNAs. RNA Biol. 14, 1124–1137. doi: 10.1080/15476286.2016.1251543
Schwartz, S., Bernstein, D. A., Mumbach, M. R., Jovanovic, M., Herbst, R. H., and León-Ricardo, B. X. (2014a). Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell 159, 148–162. doi: 10.1016/j.cell.2014.08.028
Schwartz, S., Mumbach, M. R., Jovanovic, M., Wang, T., Maciag, K., and Bushkin, G. G. (2014b). Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep. 8, 284–296. doi: 10.1016/j.celrep.2014.05.048
Scutenaire, J., Deragon, J.-M., Jean, V., Benhamed, M., Raynaud, C., Favory, J. J., et al. (2018). The YTH domain protein ECT2 is an m6A reader required for normal trichome branching in Arabidopsis. Plant Cell 30, 986–1005. doi: 10.1105/tpc.17.00854
Selmi, T., Hussain, S., Dietmann, S., Heiß, M., Borland, K., Flad, S., et al. (2021). Sequence- and structure-specific cytosine-5 mRNA methylation by NSUN6. Nucleic. Acids Res. 49, 1006–1022. doi: 10.1093/nar/gkaa1193
Sement, F. M., Ferrier, E., Zuber, H., Merret, R., Alioua, M., Deragon, J. M., et al. (2013). Uridylation prevents 3′ trimming of oligoadenylated mRNAs. Nucleic Acids Res. 41, 7115–7127. doi: 10.1093/nar/gkt465
Sendinc, E., Valle-Garcia, D., Dhall, A., Chen, H., Henriques, T., Navarrete-Perea, J., et al. (2019). PCIF1 catalyzes m6Am mRNA methylation to regulate gene expression. Mol. Cell 75, 631–643. doi: 10.1016/j.molcel.2019.05.030
Sharma, S., Yang, J., van Nues, R., Watzinger, P., Kötter, P., and Lafontaine, D. L. J. (2017). Specialized box C/D snoRNPs act as antisense guides to target RNA base acetylation. PLoS Genet. 13:e1006804. doi: 10.1371/journal.pgen.1006804
Shen, B., and Goodman, H. M. (2004). Uridine addition after microRNA-directed cleavage. Science 306, 997–997. doi: 10.1126/science.1103521
Shen, L., Liang, Z., Gu, X., Chen, Y., Teo, Z. W., Hou, X., et al. (2016). N(6)-methyladenosine RNA modification regulates shoot stem cell fate in Arabidopsis. Dev. Cell 38, 186–200. doi: 10.1016/j.devcel.2016.06.008
Shen, L., Liang, Z., Wong, C. E., and Yu, H. (2019). Messenger RNA modifications in plants. Trends Plant Sci. 24, 328–341. doi: 10.1016/j.tplants.2019.01.005
Sheth, U., and Parker, R. (2003). Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science 300, 805–808. doi: 10.1126/science.1082320
Shi, H., Wang, X., Lu, Z., Zhao, B. S., Ma, H., Hsu, P. J., et al. (2017). YTHDF3 facilitates translation and decay of N6-methyladenosine-modified RNA. Cell Res. 27, 315–328. doi: 10.1038/cr.2017.15
Shi, H., Zhang, X., Weng, Y. L., Lu, Z., Liu, Y., Lu, Z., et al. (2018). m6A facilitates hippocampus-dependent learning and memory through YTHDF1. Nature 563, 249–253. doi: 10.1038/s41586-018-0666-1
Shu, X., Cao, J., Cheng, M., Xiang, S., Gao, M., Li, T., et al. (2020). A metabolic labeling method detects m6A transcriptome-wide at single base resolution. Nat. Chem. Biol. 16, 887–895. doi: 10.1038/s41589-020-0526-9
Slobodin, B., Han, R., Calderone, V., Vrielink, J. A. O., Loayza-Puch, F., Elkon, R., et al. (2017). Transcription impacts the efficiency of mRNA translation via co-transcriptional N6-adenosine methylation. Cell 169, 326–337. doi: 10.1016/j.cell.2017.03.031
Song, J., and Yi, C. (2017). Chemical modifications to RNA: a new layer of gene expression regulation. ACS Chem. Biol. 12, 316–325. doi: 10.1021/acschembio.6b00960
Spenkuch, F., Motorin, Y., and Helm, M. (2014). Pseudouridine: still mysterious, but never a fake (uridine)! RNA Biol. 11, 1540–1554. doi: 10.4161/15476286.2014.992278
Squires, J. E., and Preiss, T. (2010). Function and detection of 5-methylcytosine in eukaryotic RNA. Epigenomics 2, 709–715. doi: 10.2217/epi.10.47
Squires, J. E., Patel, H. R., Nousch, M., Sibbritt, T., Humphreys, D. T., Parker, B. J., et al. (2012). Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 40, 5023–5033. doi: 10.1093/nar/gks144
Sun, H., Zhang, M., Li, K., Bai, D., and Yi, C. (2019). Cap-specific, terminal N(6)-methylation by a mammalian m(6)Am methyltransferase. Cell Res. 29, 80–82. doi: 10.1038/s41422-018-0117-4
Suzuki, T., Ueda, H., Okada, S., and Sakurai, M. (2015). Transcriptome-wide identification of adenosine-to-inosine editing using the ICE-seq method. Nat. Protoc. 10, 715–732. doi: 10.1038/nprot.2015.037
Theler, D., Dominguez, C., Blatter, M., Boudet, J., and Allain, F. H. (2014). Solution structure of the YTH domain in complex with N6-methyladenosine RNA: a reader of methylated RNA. Nucleic Acids Res. 42, 13911–13919. doi: 10.1093/nar/gku1116
Thuring, K., Schmid, K., Keller, P., and Helm, M. (2016). Analysis of RNA modifications by liquid chromatography-tandem mass spectrometry. Methods 107, 48–56. doi: 10.1016/j.ymeth.2016.03.019
Topisirovic, I., Svitkin, Y. V., Sonenberg, N., and Shatkin, A. J. (2011). Cap and cap-binding proteins in the control of gene expression. RNA 2, 277–298. doi: 10.1002/wrna.52
Torres, A. G., Pineyro, D., Filonava, L., Stracker, T. H., Batlle, E., and Ribas, et al. (2014). A-to-I editing on tRNAs: biochemical, biological and evolutionary implications. FEBS Lett. 588, 4279–4286. doi: 10.1016/j.febslet.2014.09.025
Tzafrir, I., Dickerman, A., Brazhnik, O., Nguyen, Q., McElver, J., Frye, C., et al. (2003). The Arabidopsis seedgenes project. Nucleic Acids Res. 31, 90–93. doi: 10.1093/nar/gkg028
Vandivier, L. E., and Gregory, B. D. (2018). New insights into the plant epitranscriptome. J. Exp. Bot. 69, 4659–4665. doi: 10.1093/jxb/ery262
Vandivier, L. E., Campos, R., Kuksa, P. P., Silverman, I. M., Wang, L.-S., and Gregory, B. D. (2015). Chemical modifications mark alternatively spliced and uncapped messenger RNAs in Arabidopsis. Plant Cell 27, 3024–3037. doi: 10.1105/tpc.15.00591
Vespa, L., Vachon, G., Berger, F., Perazza, D., Faure, J.-D., and Herzog, M. (2004). The immunophilin-interacting protein AtFIP37 from Arabidopsis is essential for plant development and is involved in trichome endoreduplication. Plant Physiol. 134, 1283–1292. doi: 10.1104/pp.103.028050
Vilfan, I. D., Tsai, Y. C., Clark, T. A., Wegener, J., Dai, Q., Yi, C., et al. (2013). Analysis of RNA base modification and structural rearrangement by single-molecule real-time detection of reverse transcription. J. Nanobiotechnol. 11:8. doi: 10.1186/1477-3155-11-8
Walters, R. W., Matheny, T., Mizoue, L. S., Rao, B. S., Muhlrad, D., and Parker, R. (2017). Identification of NAD+ capped mRNAs in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA. 114, 480–485. doi: 10.1073/pnas.1619369114
Wan, Y., Tang, K., Zhang, D., Xie, S., Zhu, X., Wang, Z., et al. (2015). Transcriptome-wide high-throughput deep m6A-seq reveals unique differential m6A methylation patterns between three organs in Arabidopsis thaliana. Genome Biol. 16:272. doi: 10.1186/s13059-015-0839-2
Wang, X., Li, Q., Yuan, W., Kumar, S., and Qian, W. (2016). The cytosolic Fe-S cluster assembly component MET18 is required for the full enzymatic activity of ROS1 in active DNA demethylation. Sci Rep. 6:26443. doi: 10.1038/srep26443
Wang, X., Lu, Z., Gomez, A., Hon, G. C., Yue, Y., Han, D., et al. (2014). N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120. doi: 10.1038/nature12730
Wang, X., Zhao, B. S., Roundtree, I. A., Lu, Z., Han, D., Ma, H., et al. (2015). N6-methyladenosine modulates messenger RNA translation efficiency. Cell 161, 1388–1399. doi: 10.1016/j.cell.2015.05.014
Wang, Y., Ni, T., Wang, W., and Liu, F. (2018). Gene transcription in bursting: a unified mode for realizing accuracy and stochasticity. Biol. Rev. 94, 248–258. doi: 10.1111/brv.12452
Wang, Y., Xiao, Y., Dong, S., Yu, Q., and Jia, G. (2020). Antibody-free enzyme assisted chemical approach for detection of N6-methyladenosine. Nat. Chem. Biol. 16, 896–903. doi: 10.1038/s41589-020-0525-x
Wang, Y., Li, S., Zhao, Y., You, C., Le, B., Gong, Z., etal. (2019). NAD+-capped RRNAs are widespread in the Arabidopsis transcriptome and can probably be translated. Proc. Natl. Acad. Sci. USA. 116, 12094–12102. doi: 10.1073/pnas.1903682116
Wei, L. H., Song, P., Wang, Y., Lu, Z., Tang, Q., Yu, Q., et al. (2018). The m6A reader ECT2 controls trichome morphology by affecting mRNA stability in Arabidopsis. Plant Cell 30, 968–985. doi: 10.1105/tpc.17.00934
Wen, J., Lv, R., Ma, H., Shen, H., He, C., Wang, J., et al. (2018). Zc3h13 regulates nuclear RNA m6A methylation and mouse embryonic stem cell self-renewal. Mol. Cell 69, 1028–1038. doi: 10.1016/j.molcel.2018.02.015
Weng, Y. L., Wang, X., An, R., Cassin, J., Vissers, C., Liu, Y., et al. (2018). Epitranscriptomic m6A regulation of axon regeneration in the adult mammalian nervous system. Neuron 97, 313–325. doi: 10.1016/j.neuron.2017.12.036
Werner, M., Purta, E., Kaminska, K. H., Cymerman, I. A., Campbell, D. A., Mittra, B., et al. (2011). 2′-O-ribose methylation of cap2 in human: function and evolution in a horizontally mobile family. Nucleic Acids Res. 39, 4756–4768. doi: 10.1093/nar/gkr038
Wu, L., Candille, S. I., Choi, Y., Xie, D., Jiang, L., Li-Pook-Than, J., et al. (2013). Variation and genetic control of protein abundance in humans. Nature 499, 79–82. doi: 10.1038/nature12223
Wu, R., Jiang, D., Wang, Y., and Wang, X. (2016). N(6)-methyladenosine (m(6)A) methylation in mRNA with a dynamic and reversible epigenetic modification. Mol. Biotechnol. 58, 450–459. doi: 10.1007/s12033-016-9947-9
Wurm, J. P., Meyer, B., Bahr, U., Held, M., Frolow, O., Kotter, B., et al. (2010). The ribosome assembly factor Nep1 responsible for bowen-conradi syndrome is a pseudouridine-N1-specific methyltransferase. Nucleic Acids Res. 38, 2387–2398. doi: 10.1093/nar/gkp1189
Xiao, W., Adhikari, S., Dahal, U., Chen, Y. S., Hao, Y. J., Sun, B. F., et al. (2016). Nuclear m6A reader YTHDC1 regulates mRNA splicing. Mol. Cell 61, 507–519. doi: 10.1016/j.molcel.2016.01.012
Xiong, X., Li, X., and Yi, C. (2018). N1-methyladenosine methylome in messenger RNA and non-coding RNA. Curr. Opin. Chem. Biol. 45, 179–186. doi: 10.1016/j.cbpa.2018.06.017
Xu, C., Wang, X., Liu, K., Roundtree, I. A., Tempel, W., Li, Y., et al. (2014). Structural basis for selective binding of m6A RNA by the YTHDC1 YTH domain. Nat. Chem. Biol. 10, 927–929. doi: 10.1038/nchembio.1654
Xu, K., Yang, Y., Feng, G. H., Sun, B. F., Chen, J. Q., Li, Y. F., et al. (2017). Mettl3-mediated m6A regulates spermatogonial differentiation and meiosis initiation. Cell Res. 27, 1100–1114. doi: 10.1038/cr.2017.100
Xu, L., Liu, X., Sheng, N., Oo, K. S., Liang, J., Chionh, Y. H., et al. (2017). Three distinct 3-methylcytidine (m3C) methyltransferases modify tRNA and mRNA in mice and humans. J. Biol. Chem. 292, 14695–14703. doi: 10.1074/jbc.M117.798298
Yan, L. L., and Zaher, H. S. (2019). How do cells cope with RNA damage and its consequences? J. Biol. Chem. 294, 15158–15171. doi: 10.1074/jbc.REV119.006513
Yan, L. L., Simms, C. L., McLoughlin, F., Vierstra, R. D., and Zaher, H. S. (2019). Oxidation and alkylation stresses activate ribosome-quality control. Nat. Commun. 10:5611. doi: 10.1038/s41467-019-13579-3
Yang, L., Perrera, V., Saplaoura, E., Apelt, F., Bahin, M., Kramdi, A., et al. (2019). m5C Methylation guides systemic transport of messenger RNA over graft junctions in plants. Curr. Biol. 29, 2465–2476. doi: 10.1016/j.cub.2019.06.042
Yang, X., Yang, Y., Sun, B. F., Chen, Y. S., Xu, J. W., Lai, W. Y., et al. (2017). 5-methylcytosine promotes mRNA export—NSUN2 as the methyltransferase and ALYREF as an m5C reader. Cell Res. 27, 606–625. doi: 10.1038/cr.2017.55
Yang, Y. Y., Hsu, P. J., Chen, Y. S., and Yang, Y. G. (2018). Dynamic transcriptomic m6A decoration: writers, erasers, readers and functions in RNA metabolism. Cell Res. 28, 616–624. doi: 10.1038/s41422-018-0040-8
Yang, Y., Wang, L., Han, X., Yang, W.-L., Zhang, M., Ma, H.-L., et al. (2019). RNA 5-methylcytosine facilitates the maternal-to-zygotic transition by preventing maternal mRNA decay. Mol. Cell 75, 1188–1202. doi: 10.1016/j.molcel.2019.06.033
Zhou, Y. L., Zhang, X. X., and Song, C. X. (2019). Bisulfite-free and base-resolution analysis of 5-methylcytidine and 5-hydroxymethylcytidine in RNA with peroxotungstate. Chem. Commun. 55, 2328–2331. doi: 10.1039/C9CC00274J
Yue, Y., Hu, J., and He, C. (2015). RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev. 29, 1343–1355. doi: 10.1101/gad.262766.115
Yue, Y., Liu, J., Cui, X., Cao, J., Luo, G., Zhang, Z., et al. (2018). VIRMA mediates preferential m6A mRNA methylation in 3′UTR and near stop codon and associates with alternative polyadenylation. Cell Discov. 4:10. doi: 10.1038/s41421-018-0019-0
Zhang, F., Zhang, Y.-C., Lio, J.-Y., Yu, Y., Zhou, Y.-F., Feng, Y.-Z., et al. (2019). The subunit of RNA N6-methyladenosine methyltransferase OsFIP regulates early degeneration of microspores in rice. PLoS Genet. 15:e1008120. doi: 10.1371/journal.pgen.1008120
Zhang, H. Y., Xiong, J., Qi, B. L., Feng, Y. Q., and Yuan, B. F. (2016). The existence of 5-hydroxymethylcytosine and 5-formylcytosine in both DNA and RNA in mammals. Chem. Commun. 52, 737–740. doi: 10.1039/C5CC07354E
Zhang, Z., Chen, L. Q., Zhao, Y. L., Yang, C. G., Roundtree, I. A., Zhang, Z., et al. (2019). Single-base mapping of m6A by an antibody-independent method. Sci. Adv. 5:eaax0250. doi: 10.1126/sciadv.aax0250
Zhang, Z., Hu, F., Sung, M. W., Shu, C., Castillo-González, C., Koiwa, H., et al. (2017). RISC-interacting clearing 3′- 5′ exoribonucleases (RICEs) degrade uridylated cleavage fragments to maintain functional RISC in Arabidopsis thaliana. eLife 6:e24466. doi: 10.7554/eLife.24466
Zhang, Z., Theler, D., Kaminska, K. H., Hiller, M., and De La Grange, P. (2010). The YTH domain is a novel RNA binding domain. J. Biol. Chem. 285, 14701–14710. doi: 10.1074/jbc.M110.104711
Zhao, B. S., Roundtree, I. A., and He, C. (2017a). Post-transcriptional gene regulation by mRNA modification. Nat. Rev. Mol. Cell Biol. 18, 31–42. doi: 10.1038/nrm.2016.132
Zhao, B. S., Wang, X., Beadell, A. V., Lu, Z., Shi, H., Kuuspalu, A., et al. (2017b). m6A-dependent maternal mRNA clearance facilitates zebrafish maternal-to-zygotic transition. Nature 542, 475–478. doi: 10.1038/nature21355
Zhao, L. Y., Song, J., Liu, Y., Song, C.-X., and Yi, C. (2020). Mapping the epigenetic modifications of DNA and RNA. Protein Cell 11, 792–808. doi: 10.1007/s13238-020-00733-7
Zheng, G., Dahl, J. A., Niu, Y., Fedorcsak, P., Huang, C.-M., Li, C. J., et al. (2013). ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell. 49, 18–29. doi: 10.1016/j.molcel.2012.10.015
Zhong, S., Li, H., Bodi, Z., Button, J., Vespa, L., Herzog, M., et al. (2008). MTA is an Arabidopsis messenger RNA adenosine methylase and interacts with a homolog of a sex-specific splicing factor. Plant Cell 20, 1278–1288. doi: 10.1105/tpc.108.058883
Zhou, J., Wan, J., Gao, X., Zhang, X., Jaffrey, S., and Qian, S. B. (2015). Dynamic m6A mRNA methylation directs translational control of heat shock response. Nature 526, 591–594. doi: 10.1038/nature15377
Zhou, L., Tian, S., and Qin, G. (2019). RNA methylomes reveal the m6A mediated regulation of DNA demethylase gene SlDML2 in tomato fruit ripening. Genome Biol. 20:156. doi: 10.1186/s13059-019-1771-7
Zou, S., Toh, J. D., Wong, K. H., Gao, Y. G., Hong, W., and Woon, E. C. (2016). N6-Methyladenosine: a conformational marker that regulates the substrate specificity of human demethylases FTO and ALKBH5. Sci. Rep. 6:25677. doi: 10.1038/srep25677
Keywords: epitranscriptomics, RNA modification, post-transcriptional regulation, 5-methylcytidine, N6-methyladenosine, RNA metabolism, mRNA methylation, central dogma
Citation: Kumar S and Mohapatra T (2021) Deciphering Epitranscriptome: Modification of mRNA Bases Provides a New Perspective for Post-transcriptional Regulation of Gene Expression. Front. Cell Dev. Biol. 9:628415. doi: 10.3389/fcell.2021.628415
Received: 11 November 2020; Accepted: 22 February 2021;
Published: 16 March 2021.
Edited by:
Jia Meng, Xi’an Jiaotong-Liverpool University, ChinaReviewed by:
Xudong Zhang, University of California, Riverside, United StatesSandra Blanco, University of Salamanca, Spain
Copyright © 2021 Kumar and Mohapatra. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Suresh Kumar, c3VyZXNoa3VtYXJAaWFyaS5yZXMuaW4=; c3VyZXNoa3VtYXIzX2luQHlhaG9vLmNvLnVr; orcid.org/0000-0002-7127-3079