 Hui Lin1,2,3
Hui Lin1,2,3 Kim L. McBride
Kim L. McBride Vidu Garg
Vidu Garg Ming-Tao Zhao
Ming-Tao Zhao- 1Center for Cardiovascular Research, The Abigail Wexner Research Institute, Nationwide Children’s Hospital, Columbus, OH, United States
- 2The Heart Center, Nationwide Children’s Hospital, Columbus, OH, United States
- 3Division of Genetic and Genomic Medicine, Nationwide Children’s Hospital, Columbus, OH, United States
- 4Department of Pediatrics, The Ohio State University College of Medicine, Columbus, OH, United States
- 5Department of Molecular Genetics, The Ohio State University, Columbus, OH, United States
Congenital heart disease (CHD) is the most common cause of infant death associated with birth defects. Recent next-generation genome sequencing has uncovered novel genetic etiologies of CHD, from inherited and de novo variants to non-coding genetic variants. The next phase of understanding the genetic contributors of CHD will be the functional illustration and validation of this genome sequencing data in cellular and animal model systems. Human induced pluripotent stem cells (iPSCs) have opened up new horizons to investigate genetic mechanisms of CHD using clinically relevant and patient-specific cardiac cells such as cardiomyocytes, endothelial/endocardial cells, cardiac fibroblasts and vascular smooth muscle cells. Using cutting-edge CRISPR/Cas9 genome editing tools, a given genetic variant can be corrected in diseased iPSCs and introduced to healthy iPSCs to define the pathogenicity of the variant and molecular basis of CHD. In this review, we discuss the recent progress in genetics of CHD deciphered by large-scale genome sequencing and explore how genome-edited patient iPSCs are poised to decode the genetic etiologies of CHD by coupling with single-cell genomics and organoid technologies.
Introduction
Congenital heart disease (CHD) is a leading cause of birth defect-related death and affects ∼1% of live births in the United States (Hoffman and Kaplan, 2002; Nees and Chung, 2019). CHD is characterized by morphological abnormalities in the cardiac chambers, septa and valves as well as the great vessels arising from the heart. Congenital malformations of all aspects of the heart have been described but the most common types of CHD can be classified into the following categories: (1) cardiac septation defects, (2) conotruncal and aortic arch artery anomalies, (3) right- and left-sided outflow tract obstructive defects, and (4) left-right abnormalities (heterotaxy) (Garg, 2006; Bruneau, 2008). Septation defects consist of atrial septal defects (ASD), ventricular septal defects (VSD) and atrioventricular septal defects (AVSD) while common conotruncal and aortic arch artery anomalies include tetralogy of Fallot (TOF), persistent truncus arteriosus and interrupted aortic arch. Right-sided outflow tract obstructive lesions include pulmonary stenosis and pulmonary valve atresia with intact ventricular septum (PA-IVS), whereas hypoplastic left heart syndrome (HLHS), aortic valve stenosis (AVS) and bicuspid aortic valve (BAV) are common left-sided outflow tract obstructive defects. Abnormalities in left-right signaling in the developing embryos affect cardiac looping, which is critical for proper alignment of the atria chambers to their appropriate-sided ventricles and great vessels. This disruption in proper signaling is associated with complex forms of CHD, such as double outlet right ventricle and double inlet left ventricle, clinically termed as heterotaxy syndrome (Kathiriya and Srivastava, 2000). Other major CHD that does not fit into the abovementioned categories includes isolated valve anomalies (e.g., Ebstein’s anomaly of the tricuspid valve and mitral valve prolapse), total anomalous pulmonary venous connection, anomalous coronary artery and patent ductus arteriosus.
Epidemiologic studies reveal that genetic factors are the predominant cause of CHD whereas environmental factors (exposures, maternal conditions, intrauterine environment, etc.) are also important contributors (Liu et al., 2013; Pierpont et al., 2018). In total, specific genetic and environmental factors can be identified in 20–30% of all CHD cases. Genetic mechanisms underlying the development of CHD are complex and remain elusive using current genetic approaches (Liu et al., 2017; Pierpont et al., 2018). There are limited animal models to study the developmental genetics of CHD, and transgenic mice carrying human variants do not always recapitulate the clinical phenotypes of CHD (Majumdar et al., 2019). Human iPSCs are derived from somatic cells (such as skin fibroblasts or peripheral blood mononuclear cells) and have the potential to generate all cell types in the body originated from the three germ layers (Takahashi et al., 2007; Yu et al., 2007). Compared to animal models, patient iPSCs are clinically relevant and also include the genetic background of the affected individuals in a disease-specific manner, thus providing a powerful tool for studying the contribution of a given genetic variant to CHD. Patient-specific iPSCs can be differentiated into cardiomyocytes, endothelial/endocardial cells, cardiac fibroblasts and smooth muscle cells, which makes it feasible to study complex genetic regulation and gene-environment interactions simultaneously in multiple cell types in the heart (Hu et al., 2016; Zhao et al., 2017a; Gifford et al., 2019). Recent studies demonstrate that genome-edited iPSCs are ideal platforms to elucidate the regulatory roles of non-coding genetic variants in the risk of coronary artery disease and to investigate the contribution of combinatorial interactions of multiple genetic variants to complex cardiovascular disease (Lo Sardo et al., 2018; Deacon et al., 2019).
In this review, we discuss the latest progress on genetic etiologies of CHD uncovered by the state-of-the-art technologies such as whole genome sequencing (WGS) and whole exome sequencing. We explore the fascinating perspectives on using patient-specific iPSCs and CRISPR genome editing to functionally study the genetic and epigenetic (environmental) determinants of CHD.
Genetics of CHD
With the advance of massively parallel sequencing, genetics of CHD have been aggressively explored in the past decade. Large scientific efforts such as NIH-funded Pediatric Cardiac Genomics Consortium (PCGC) have been established to coordinate the investigation of genetic variants present in CHD patient population relevant to clinical outcomes (Pediatric Cardiac Genomics Consortium et al., 2013; Jin et al., 2017). The genetic basis of CHD can be grouped into two categories: syndromic CHD and non-syndromic (isolated) CHD (Pierpont et al., 2018). Syndromic CHD is defined as CHD with other congenital anomalies, neurodevelopmental defects and/or dysmorphic features. Syndromic CHD may be caused by aneuploidy, copy number variants (insertions or deletions > 1,000 nucleotides), or single gene defects. Down syndrome (trisomy 21) is a common chromosome anomaly, and 40–50% of these patients have various types of CHD, with cardiac septation defects being the most common. Turner syndrome is caused by complete or partial loss of an X-chromosome, and left-sided defects (coarctation of the aorta, COA), BAV and HLHS are present in 30% of these patients. 22q11.2 deletion syndrome is one of the most common copy number variants with deletion of more than 40 genes on chromosome 22. Outflow tract defects are present in 75–80% of 22q11.2 patients. Syndromic CHD caused by single-gene defects includes Alagille syndrome (variants in JAG1 and NOTCH2) and Holt-Oram syndrome (variants in TBX5) (Basson et al., 1997; Li et al., 1997b; Table 1). Genetic contributors of isolated CHD have been emerging in the past two decades and most variants are located in genes that are involved in the molecular regulation of cardiac development. Syndromic and isolated CHD display distinct genetic architectures: de novo protein-truncating variants (PTVs) are significantly enriched in syndromic CHD whereas inherited PTVs are mostly derived from unaffected parents in isolated CHD (Sifrim et al., 2016; Jin et al., 2017).
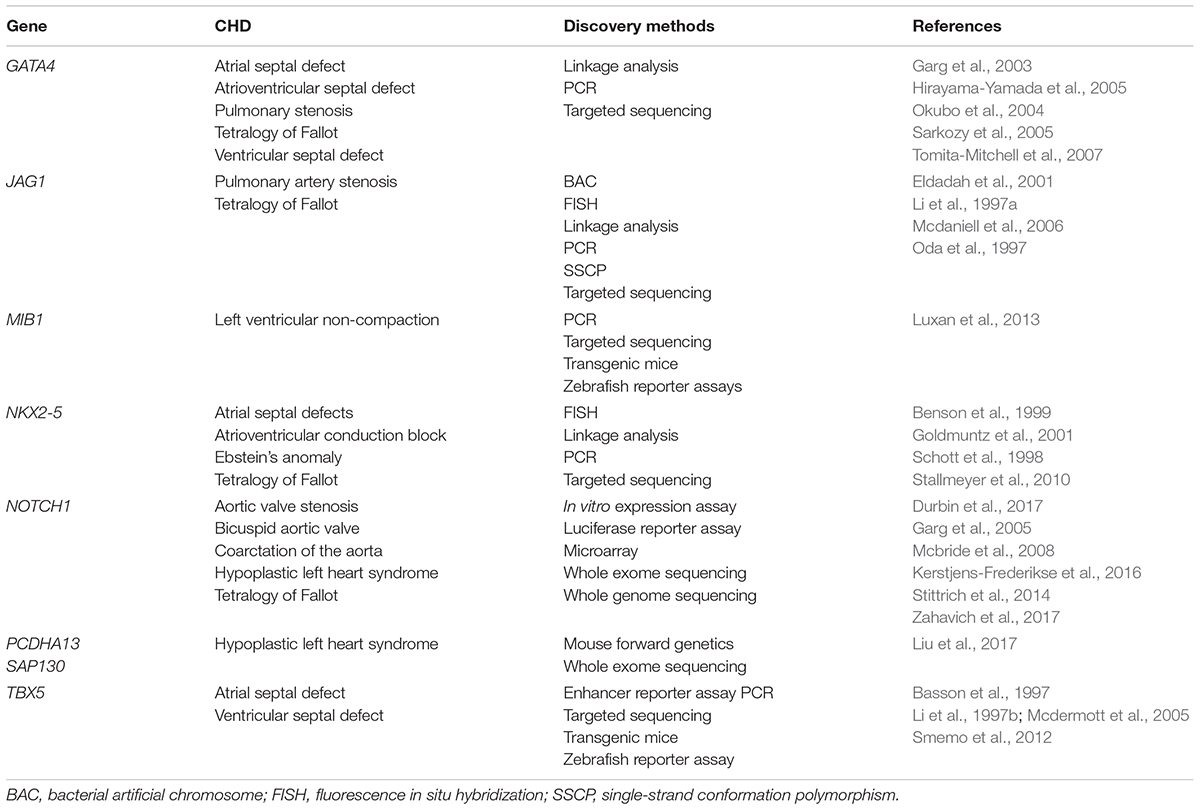
Table 1. A summary of single-gene variants underlying CHD.
Pathogenic variants linked to isolated CHD primarily encode transcription factors, signaling molecules, structural proteins and epigenetic modifiers that are essential for normal cardiac development (Zaidi et al., 2013; Pierpont et al., 2018; Nees and Chung, 2019; Table 1). For instance, genetic variants in highly conserved transcription factors critical for cardiac development are found in both familial and sporadic cases of CHD. NKX2-5 variants are present in patients with TOF and ASD with conduction delay (Schott et al., 1998; Benson et al., 1999; Goldmuntz et al., 2001; Stallmeyer et al., 2010). Pathogenic GATA4 variants are associated with ASD, VSD, AVSD, pulmonary stenosis (PS), and TOF (Garg et al., 2003; Okubo et al., 2004; Hirayama-Yamada et al., 2005; Sarkozy et al., 2005; Tomita-Mitchell et al., 2007). A small subset of GATA4 variant-induced cardiac malformations in humans are recapitulated in transgenic mouse models harboring the mutant human GATA4 variants (Misra et al., 2012; Han et al., 2015).
Components of the NOTCH signaling pathway are linked to both syndromic and isolated CHD. JAG1 variants are observed in ∼90% of Alagille syndrome patients whereas NOTCH2 variants account for additional 1–2% of individuals with Alagille syndrome (Li et al., 1997a; Oda et al., 1997; Mcdaniell et al., 2006; Kamath et al., 2012). Loss-of-function variants in JAG1 cause pulmonary artery stenosis and TOF with or without pulmonary atresia (Eldadah et al., 2001). Heterozygous mutations in DLL4 (ligand) and NOTCH1 (receptor) lead to Adams Oliver syndrome with CHD present in about 25% of these patients (Stittrich et al., 2014; Meester et al., 2015). Variants in RBPJ which interacts with the cleaved NOTCH1 protein to form a transcriptional complex, are also linked to Adams Oliver syndrome (Hassed et al., 2012). Of note, pathogenic NOTCH1 mutations are linked to BAV, HLHS, AVS, COA, and TOF (Garg et al., 2005; Mcbride et al., 2008; Kerstjens-Frederikse et al., 2016; Durbin et al., 2017; Zahavich et al., 2017). Mechanistically, NOTCH1 mutations reduce the ligand binding ability, interrupt the S1 cleavage of NOTCH receptor in the Golgi, and impair the epithelial-to-mesenchymal transition (Riley et al., 2011). In addition, germline mutations in MIB1 which encodes an E3 ubiquitin ligase that promotes endocytosis of NOTCH ligands, lead to left ventricular non-compaction (LVNC) in autosomal-dominant pedigrees (Luxan et al., 2013). Myocardial Mib1 mutations in mice cause the expansion of compact myocardium to proliferative immature trabeculae and interruption of chamber myocardium development.
The encyclopedia of DNA elements (ENCODE) project suggests that more than 80% of human genomic DNA has a biochemical function (Consortium, 2012). The majority of disease-causing variants identified by genome-wide association studies (GWAS) are located in non-coding DNA elements, many of which are embedded in the DNase I hypersensitive (open chromatin) regions (Maurano et al., 2012). GWAS in CHD have similar findings (Cordell et al., 2013; Hu et al., 2013; Mitchell et al., 2015; Hanchard et al., 2016). De novo variants in enhancer elements have been found in several human developmental defects including CHD and neurodevelopmental disorders (Short et al., 2018). For example, sequence variants in a limb-specific enhancer ZRS which is located nearly 1 Mb from its target gene sonic hedgehog (Shh) result in limb malformations such as preaxial polydactyly (Lettice et al., 2003). Copy number variants affecting topological associated domains have also been implicated in disrupting enhancers and causing developmental defects (Lupianez et al., 2015). Distal cis-regulatory elements have been identified in TBX5, of which variants are responsible for Holt-Oram syndrome (Mcdermott et al., 2005; Smemo et al., 2012). Among patients with Holt-Oram syndrome, three quarters have CHD, with ASD and VSD as the most common cardiac defects. A homozygous variant found in an enhancer about 90 kb downstream of TBX5 is associated with isolated ASD and VSD in a cohort of non-syndromic CHD patients. This single-nucleotide variant compromises the enhancer activity driving expression of TBX5 in the heart in both mouse and zebrafish transgenic models (Smemo et al., 2012). Recent WGS and chromatin immunoprecipitation sequencing have enabled researchers to expand the genetic variants in non-coding DNA elements that may have a regulatory role in controlling gene transcription during heart development (Zhao et al., 2017b; Richter et al., 2020). Non-coding de novo variants (DNVs) are significantly enriched in individuals with CHD and potentially exhibit transcriptional and post-transcriptional regulatory effects on genes critical for normal cardiac morphogenesis. Genetic architecture of CHD in cardiac regulatory non-coding DNVs will be further elucidated with the advance of WGS and precise genome editing technologies.
Patient-Specific iPSCs for Modeling Genetics of CHD
Although a genetic etiology is identified in about 1/3 of CHD patients, experimental models to functionally validate genetic variants associated with CHD are far from perfect. Genetically engineered mice have been used for studying fundamental genetics of cardiac development for more than 25 years. Murine models are able to recapitulate some aspects of human cardiac development due to their similar stages of cardiac morphogenesis and adult cardiac structure (Majumdar et al., 2019). However, there are substantial differences in genomic content and physiology between humans and mice. Orthologous heterozygous variants sometimes do not reproduce similar CHD phenotypes when introduced into the mouse genome. Patient-derived iPSCs appear to provide a unique platform to study the genetic mechanisms of CHDs as they retain all the genetic information of the original affected individuals. Combined with CRISPR/Cas9 genome-editing, single-cell genomics, and cardiac organoid engineering technologies, patient-specific iPSCs would greatly complement the murine genetic models of CHD and illustrate novel perspectives on genetic etiologies of CHD for future precision diagnosis and treatment.
Human iPSCs are promising models for studying genetic mechanisms of isolated CHD caused by single-gene defects. In addition to cell-autonomous inherited cardiac disease such as long QT syndrome (Moretti et al., 2010; Itzhaki et al., 2011), ventricular tachycardia (Zhang et al., 2014; Sleiman et al., 2020) and dilated cardiomyopathy (Sun et al., 2012; Hinson et al., 2015), patient iPSCs have been employed to model several types of CHD, including BAV and calcific aortic valve disease (CAVD) (Theodoris et al., 2015), supravalvular aortic stenosis (SVAS) (Ge et al., 2012), cardiac septal defects (Ang et al., 2016), Barth syndrome (Wang et al., 2014), and HLHS (Hrstka et al., 2017; Yang et al., 2017; Miao et al., 2020; Table 2). Human iPSCs can be differentiated to the desired cardiovascular cell types relevant for studying different CHD (Protze et al., 2019), though the immaturity of iPSC-derived cardiomyocytes (iPSC-CMs) continues to be a challenge for recapitulating the physiological scenarios in the heart (Karbassi et al., 2020; Zhao et al., 2020b). Robust cardiac differentiation protocols have been optimized to generate subtype-specific (atrial, ventricular and nodal) cardiomyocytes for precision disease modeling (Zhang et al., 2011; Lee et al., 2017; Protze et al., 2017; Ren et al., 2019; Liang et al., 2020; Zhao et al., 2020a).
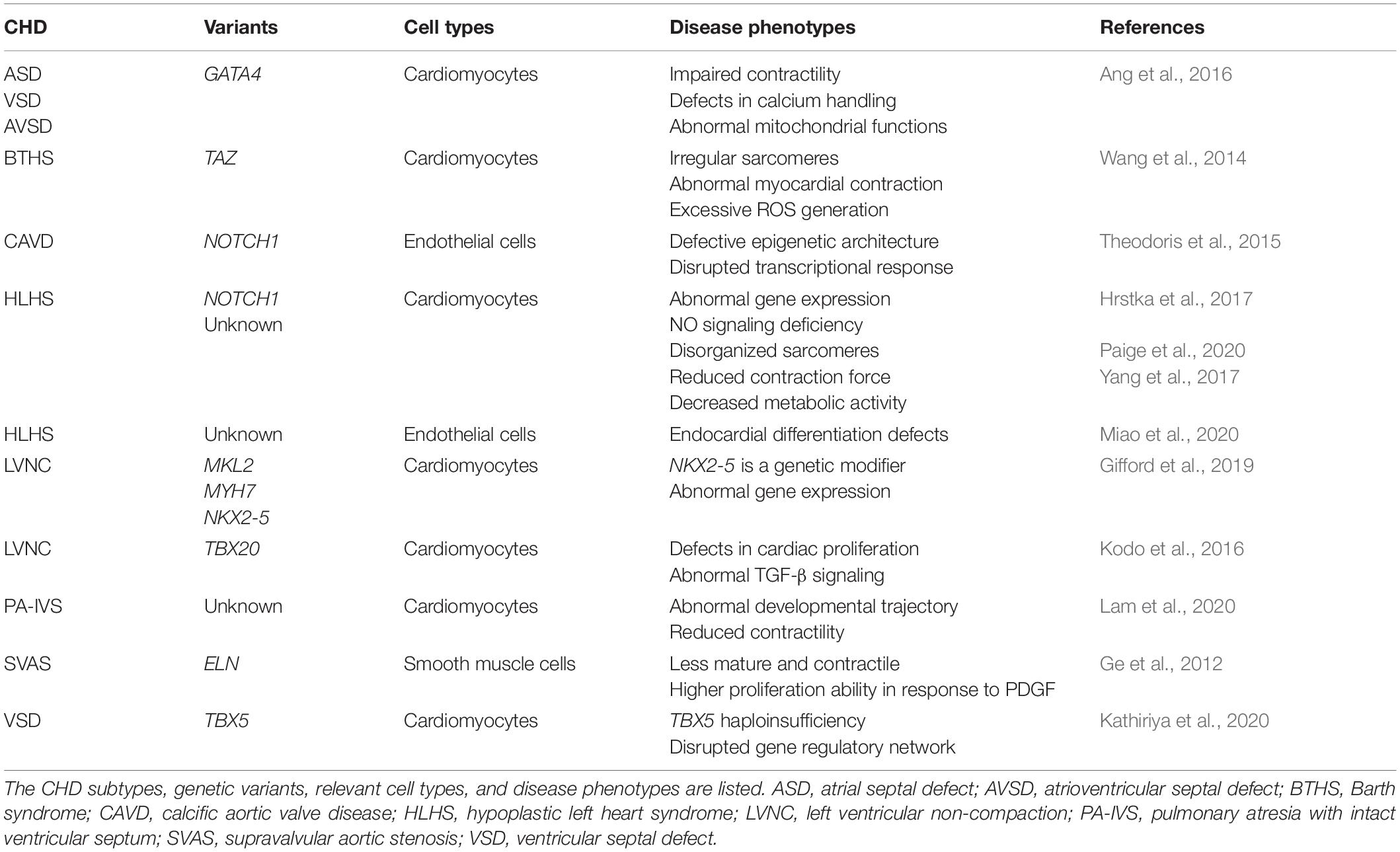
Table 2. Current iPSC models for studying disease mechanisms of CHD.
Human iPSC models of CHD have employed major cardiac cell types such as cardiomyocytes (CMs), vascular smooth muscle cells (SMCs), and endothelial/endocardial cells (ECs) that can be derived from patient-specific iPSCs for laboratory research. These patient-derived cardiac cells carrying genetic variants enable researchers to study the disease mechanisms in a petri dish (Table 2). For example, pathogenic GATA4 variants cause cardiac septal defects and cardiomyopathy. A heterozygous variant in GATA4 (G296S missense) is linked to 100% penetrant ASD, VSD, AVSD or PS (Garg et al., 2003). Human iPSC-CMs from heterozygous GATA4-G296S patients display impaired contractility, defects in calcium handling ability and abnormal mitochondrial functions (Ang et al., 2016). Molecular analysis reveals that mutant GATA4 disrupts the recruitment of TBX5 which binds to cardiac super-enhancers and leads to dysregulation of genes related to cardiac septation. In another study, Theodoris et al. (2015) have derived iPSCs from patients with BAV and CAVD which are linked to NOTCH1 haploinsufficiency. In iPSC-derived endothelial cells (iPSC-ECs), NOTCH1 heterozygous nonsense variants disrupt the epigenetic architecture of NOTCH1-bound enhancers and cause the depression of anti-osteogenic and anti-inflammatory gene regulation networks in response to hemodynamic shear stress (Theodoris et al., 2015). Furthermore, the same group have recently utilized a combination of human iPSC technology, machine learning and network analysis to identify an efficacious therapeutic candidate XCT790 for preventing and treating aortic valve disease in a mouse model, demonstrating the prospective pharmacogenetic applications of CHD patient-specific iPSCs (Theodoris et al., 2020). Ge et al. (2012) have employed iPSC-derived smooth muscle cells (iPSC-SMCs) to investigate how elastin (ELN) gene variants lead to narrowing or blockage of the ascending aorta in SVAS. SVAS iPSC-SMCs harboring ELN variants are less mature and contractile, and show fewer networks of smooth muscle actin filament bundles compared to healthy controls. These SVAS iPSC-SMCs have a higher proliferation ability and migration rate in response to platelet-derived growth factor (PDGF), indicating that SVAS iPSC-SMCs recapitulate the pathological features of SVAS patients and may provide novel insights for future therapies.
Human iPSCs have been utilized to study complex genetics in CHD together with transgenic mouse models and clinical genetics. A recent study reveals that NKX2-5 variants serve as a genetic modifier of a familial LVNC cardiomyopathy with variable age of presentation from childhood to incidental asymptomatic finding in adulthood (Gifford et al., 2019). Human iPSCs were created carrying the inherited compound heterozygous variants in MKL2, MYH7 and NKX2-5 while genetically engineered mice carrying the orthologous variants were also generated. By analyzing the phenotypes from transgenic murine hearts and patient iPSC-CMs, NKX2-5 variants are identified as a genetic modifier for this cardiomyopathy with oligogenic inheritance. In another study, LVNC iPSC lines were generated from patients with TBX20 variants (Kodo et al., 2016). LVNC iPSC-CMs show defects in proliferation which is caused by the abnormal activation of TGF-β signaling. In mice, overexpression of TGF-β1 leads to arrest in cardiac development, disturbed expansion of embryonic cardiomyocytes and trabecular/compact layer ratio in the left ventricle. Mostly recently, Kathiriya and colleagues have generated TBX5 knockout human iPSC lines with different dosages (heterozygous and homozygous) and performed single-cell RNA sequencing and gene regulatory network analysis. TBX5 haploinsufficiency alters the expression of CHD-related genes and reduced TBX5 gene dosage disrupts gene regulatory networks in human iPSC-CMs. Abnormal genetic interaction between Tbx5 and Mef2c leads to ventricular septation defects in transgenic mice with reduced Tbx5 gene dosage (Kathiriya et al., 2020). These studies further highlight the combinatorial advantages of using human iPSCs and transgenic mouse models to reveal genetic mechanisms of CHD pathogenesis (Figure 1).
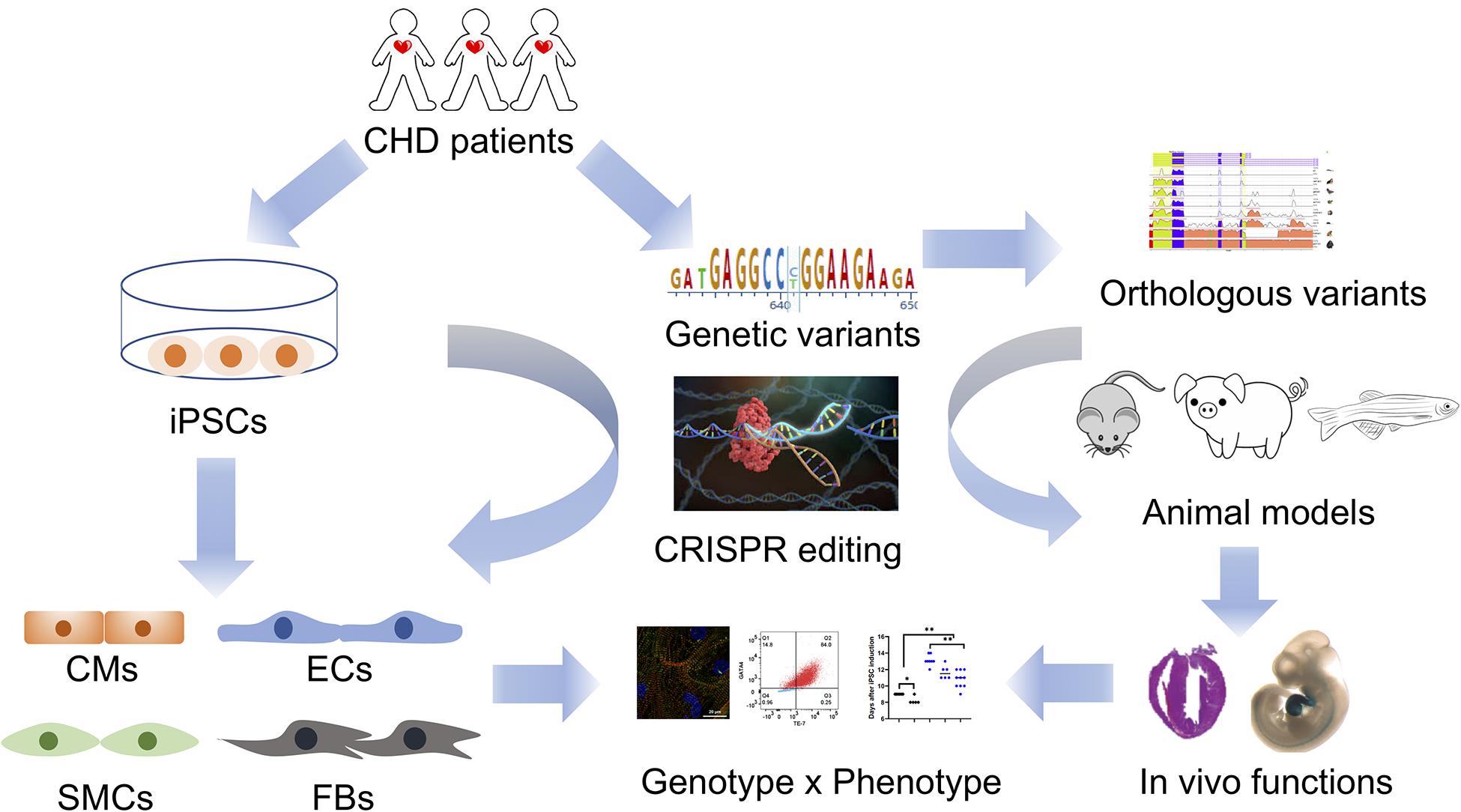
Figure 1. An integrated patient-specific iPSC model for studying genetics of CHD. Whole genome sequencing of CHD patients identifies prospective genetic variants that are retained in patient-specific iPSCs. Cardiovascular defects in CHD are recapitulated in patient iPSC-derived cardiac cells, such as cardiomyocytes (CMs), endocardial/endocardial cells (ECs), vascular smooth muscle cells (SMCs), and fibroblasts (FBs). Genetic variants associated with a CHD phenotype are corrected in diseased iPSCs and introduced to healthy iPSCs using CRISPR/Cas9 genome editing tools. These genome-edited iPSCs are further investigated to validate the cause-effect relationship between a given genetic variant and a CHD phenotype. In parallel, orthologous variants are genetically introduced into animal models (rodents, pigs, zebrafish, etc.) in order to investigate the in vivo effects of a given human variant on heart development. Together, an integrated model including both genome-edited human iPSCs and transgenic animals will yield a more comprehensive illustration of the genetic basis of CHD in the new era of genomic medicine.
Hypoplastic left heart syndrome is a severe form of CHD characterized by aortic and mitral valve atresia or stenosis, leading to a hypoplastic left ventricle and aorta (Saraf et al., 2019). Though HLHS has a strong genetic component, the genetic etiology of HLHS is complex (Mcbride et al., 2005). Further, mouse models are not able to fully recapitulate the clinical phenotype (Liu et al., 2017; Grossfeld et al., 2019). Using HLHS patient-derived iPSC-CMs, multiple studies demonstrate the pathogenic link of NOTCH1 variants to HLHS (Theis et al., 2015; Durbin et al., 2017; Hrstka et al., 2017; Yang et al., 2017). HLHS iPSCs harboring NOTCH1 variants exhibit compromised ability to generate cardiac progenitors and HLHS iPSC-CMs show disorganized sarcomere structures and sarcoplasmic reticulum as well as a blunted drug response (Yang et al., 2017). Another independent study confirms that HLHS iPSCs have a deficiency in cardiomyocyte differentiation and NOTCH signaling pathway (Hrstka et al., 2017). Additionally, abnormalities in the nitric oxide (NO) pathway are found in the cardiac lineage specification of HLHS iPSCs with NOTCH1 mutations. Small molecule supplementation could restore the cardiogenesis, implying a potential therapeutic target for HLHS patients. This study is consistent with the congenital cardiac abnormalities observed in Notch1+/−; Nos3–/– transgenic mice and demonstrates that interaction between NO pathway and NOTCH signaling is required for proper development of the left-sided cardiac structures including the aortic valve (Bosse et al., 2013; Koenig et al., 2016). Recently, Miao et al. (2020) have highlighted the contribution of endocardial defects to the pathogenesis of HLHS using patient iPSCs and single-cell RNA sequencing of human fetuses with under developed left ventricles. Although the genetic causes of these HLHS iPSCs are unclear, endocardial defects lead to abnormal endothelial-to-mesenchymal transition, reduced cardiomyocyte proliferation and maturation, and disrupted fibronectin-integrin signaling. Another study by Mikryukov et al. (2021) has identified a critical role of BMP10 in the specification and maintenance of endocardial cells from human iPSCs. These iPSC-derived endocardial cells can induce trabeculation in iPSC-CMs and generate valvular interstitial-like cells, which are promising in vitro models for studying cardiac valve defects and LVNC. As the intercellular communication between endocardium and myocardium is essential for normal ventricular development (Macgrogan et al., 2018), further investigation would be warranted to illustrate how the abnormal crosstalk signaling leads to hypoplasia of the left ventricle using HLHS iPSC-CMs and iPSC-ECs.
The major challenge for studying genetics of CHD is lack of reliable models to functionally validate genetic variants that are discovered by massive genome sequencing. Although iPSC models are increasingly being used to study the contribution of genetic variation in the development of CHD, limitations should be carefully considered before any translational applications move forward. Human iPSC-CMs are fetal-like cardiomyocytes and show immature structural and physiological characteristics. For example, iPSC-CMs do not have mature structures of myofibrils and T-tubule, and they are misaligned compared to rod-shape adult cardiomyocytes (Karbassi et al., 2020; Zhao et al., 2020b). Enormous efforts have been made to promote the structural and functional maturation of iPSC-CMs, including the addition of thyroid and glucocorticoid hormones (Parikh et al., 2017), physical and electrical conditioning (Ronaldson-Bouchard et al., 2018), and co-culture with stromal cells in 3D cardiac microtissues (Giacomelli et al., 2020). In addition, iPSC-CMs are mostly cultured as a 2D structure which differs from the 3D structure of the human heart. Patient iPSC-derived cardiac organoids may be better models as a 3D substitute for the human heart (Rossi et al., 2018; Richards et al., 2020). However, it is still undetermined whether cardiac organoids can recapitulate the developmental scenarios of CHD pathogenesis. After all, any iPSC-based models are in vitro systems, which are fundamentally distinct from the in vivo environment. Although animal models best represent the in vivo environment, animals are different from humans in terms of physiology and genomics, and may not be clinically relevant. Therefore, we propose an integrated model which incorporates patient-specific iPSCs with transgenic animals (Figure 1). We envision that genetic variants associated with a CHD phenotype are tested in genome-edited iPSCs which are patient-derived and clinically relevant, while orthologous variants are also genetically introduced to animal models (rodents, pigs, zebrafish, etc.) to investigate the in vivo functions. The combination of human iPSCs and transgenic animals will provide us a more comprehensive illustration of pathogenetic mechanisms of CHD.
Outlook
Recent advances in CRISPR/Cas9 genome editing (Adli, 2018), single-cell genomics (Tanay and Regev, 2017) and organoid (Rossi et al., 2018) technologies further propel the discovery of novel mechanisms of CHD development using patient- and disease-specific iPSCs. Precise genome editing technologies can be used to correct a given variant in patient iPSCs and then study whether the disease phenotypes can be rescued in genetically corrected isogenic cardiac cells (Hockemeyer and Jaenisch, 2016; Deacon et al., 2019). Concomitantly, this variant can be introduced to a healthy iPSC line with new genetic background to test whether it is sufficient to cause the disease phenotypes. Moreover, oligogenic inheritance in CHD may be studied in patient iPSCs by simultaneous correction or introduction of a combination of multiple genetic variants (Gifford et al., 2019). Single-cell RNA-seq analysis of human and mouse hearts has provided unprecedented resources on the trajectory of cardiac development in vivo at single-cell resolution and revealed a blueprint on how normal cell fate determination is altered under genetic perturbation and pathological conditions such as CHD (Cui et al., 2019; De Soysa et al., 2019; Litvinukova et al., 2020; Paik et al., 2020). Single-cell transcriptional profiling of healthy and diseased iPSCs during cardiac differentiation would decipher how a given genetic variant affects cardiac differentiation and developmental trajectories, and uncover new molecular insights in the pathogenesis of CHD (Churko et al., 2018; Kathiriya et al., 2020; Lam et al., 2020; Miao et al., 2020; Paige et al., 2020). As heart development is dependent on interaction among multiple cell types in the embryo, cardiac organoids and 3D bio-printing may serve as another tier of disease modeling using patient iPSCs (Lee et al., 2019; Nugraha et al., 2019). Cardiac organoids contain the spatial information of multiple cardiac cell types and lay out a 3-D platform to study the complex interactions between genotypes and phenotypes under normal and diseased conditions using patient-specific iPSCs. Although cardiac organoids have been used for modeling drug-induced toxicity and myocardial infarction (Richards et al., 2020), it is still challenging to generate cardiac structures such as heart valves and septa that can represent the developmental defects in CHD using the current cardiac organoid technologies. Future therapeutic breakthroughs in precision medicine of CHD would require the convergence of precision genome editing, single-cell genomics and cardiac bioengineering, which is built upon clinically relevant and patient-specific iPSC platforms.
Author Contributions
HL and M-TZ: conception and design, figure preparation, manuscript writing, and final approval of manuscript. KM and VG: manuscript writing and final approval of manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the American Heart Association (AHA) Career Development Award 18CDA34110293 (M-TZ), Additional Ventures Innovation Fund (AVIF to M-TZ, KM, and VG), Additional Ventures Single Ventricle Research Fund (SVRF to M-TZ and VG), and National Institute of Health (NIH) grants R01 HL144009 and R01HL132801 (VG). M-TZ was also supported by startup funds from the Abigail Wexner Research Institute at Nationwide Children’s Hospital.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ang, Y.S., Rivas, R.N., Ribeiro, A.J.S., Srivas, R., Rivera, J., Stone, N.R., et al. (2016). Disease model of GATA4 mutation reveals transcription factor cooperativity in human cardiogenesis. Cell 167, 1734–1749. doi: 10.1016/j.cell.2016.11.033
Basson, C.T., Bachinsky, D.R., Lin, R.C., Levi, T., Elkins, J.A., Soults, J., et al. (1997). Mutations in human TBX5 [corrected] cause limb and cardiac malformation in Holt-Oram syndrome. Nat. Genet. 15, 30–35. doi: 10.1038/ng0197-30
Benson, D.W., Silberbach, G.M., Kavanaugh-Mchugh, A., Cottrill, C., Zhang, Y., Riggs, S., et al. (1999). Mutations in the cardiac transcription factor NKX2.5 affect diverse cardiac developmental pathways. J. Clin. Invest. 104, 1567–1573. doi: 10.1172/jci8154
Bosse, K., Hans, C.P., Zhao, N., Koenig, S.N., Huang, N., Guggilam, A., et al. (2013). Endothelial nitric oxide signaling regulates notch1 in aortic valve disease. J. Mol. Cell Cardiol. 60, 27–35. doi: 10.1016/j.yjmcc.2013.04.001
Bruneau, B.G. (2008). The developmental genetics of congenital heart disease. Nature 451, 943–948. doi: 10.1038/nature06801
Churko, J.M., Garg, P., Treutlein, B., Venkatasubramanian, M., Wu, H., Lee, J., et al. (2018). Defining human cardiac transcription factor hierarchies using integrated single-cell heterogeneity analysis. Nat. Commun. 9:4906.
Consortium, E.P. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. doi: 10.1038/nature11247
Cordell, H.J., Topf, A., Mamasoula, C., Postma, A.V., Bentham, J., Zelenika, D., et al. (2013). Genome-wide association study identifies loci on 12q24 and 13q32 associated with tetralogy of fallot. Hum. Mol. Genet. 22, 1473–1481.
Cui, Y., Zheng, Y., Liu, X., Yan, L., Fan, X., Yong, J., et al. (2019). Single-cell transcriptome analysis maps the developmental track of the human heart. Cell Rep. 26, 1934–1950. doi: 10.1016/j.celrep.2019.01.079
De Soysa, T.Y., Ranade, S.S., Okawa, S., Ravichandran, S., Huang, Y., Salunga, H.T., et al. (2019). Single-cell analysis of cardiogenesis reveals basis for organ-level developmental defects. Nature 572, 120–124. doi: 10.1038/s41586-019-1414-x
Deacon, D.C., Happe, C.L., Chen, C., Tedeschi, N., Manso, A.M., Li, T., et al. (2019). Combinatorial interactions of genetic variants in human cardiomyopathy. Nat. Biomed. Eng. 3, 147–157. doi: 10.1038/s41551-019-0348-9
Durbin, M.D., Cadar, A.G., Williams, C.H., Guo, Y., Bichell, D.P., Su, Y.R., et al. (2017). Hypoplastic left heart syndrome sequencing reveals a novel NOTCH1 mutation in a family with single ventricle defects. Pediatr. Cardiol. 38, 1232–1240. doi: 10.1007/s00246-017-1650-5
Eldadah, Z.A., Hamosh, A., Biery, N.J., Montgomery, R.A., Duke, M., Elkins, R., et al. (2001). Familial tetralogy of fallot caused by mutation in the jagged1 gene. Hum. Mol. Genet. 10, 163–169. doi: 10.1093/hmg/10.2.163
Garg, V. (2006). Insights into the genetic basis of congenital heart disease. Cell. Mol. Life Sci. 63, 1141–1148. doi: 10.1007/s00018-005-5532-2
Garg, V., Kathiriya, I.S., Barnes, R., Schluterman, M.K., King, I.N., Butler, C.A., et al. (2003). GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature 424, 443–447. doi: 10.1038/nature01827
Garg, V., Muth, A.N., Ransom, J.F., Schluterman, M.K., Barnes, R., King, I.N., et al. (2005). Mutations in NOTCH1 cause aortic valve disease. Nature 437, 270–274. doi: 10.1038/nature03940
Ge, X., Ren, Y., Bartulos, O., Lee, M.Y., Yue, Z., Kim, K.Y., et al. (2012). Modeling supravalvular aortic stenosis syndrome with human induced pluripotent stem cells. Circulation 126, 1695–1704. doi: 10.1161/circulationaha.112.116996
Giacomelli, E., Meraviglia, V., Campostrini, G., Cochrane, A., Cao, X., Van Helden, R.W.J., et al. (2020). Human-iPSC-derived cardiac stromal cells enhance maturation in 3D cardiac microtissues and reveal non-cardiomyocyte contributions to heart disease. Cell Stem Cell 26, 862–879. doi: 10.1016/j.stem.2020.05.004
Gifford, C.A., Ranade, S.S., Samarakoon, R., Salunga, H.T., De Soysa, T.Y., Huang, Y., et al. (2019). Oligogenic inheritance of a human heart disease involving a genetic modifier. Science 364, 865–870. doi: 10.1126/science.aat5056
Goldmuntz, E., Geiger, E., and Benson, D.W. (2001). NKX2.5 mutations in patients with tetralogy of fallot. Circulation 104, 2565–2568. doi: 10.1161/hc4601.098427
Grossfeld, P., Nie, S., Lin, L., Wang, L., and Anderson, R.H. (2019). Hypoplastic Left Heart Syndrome: A New Paradigm for an Old Disease? J. Cardiovasc. Dev. Dis. 6:10. doi: 10.3390/jcdd6010010
Han, H., Chen, Y., Liu, G., Han, Z., Zhao, Z., Tang, Y., et al. (2015). GATA4 transgenic mice as an in vivo model of congenital heart disease. Int. J. Mol. Med. 35, 1545–1553. doi: 10.3892/ijmm.2015.2178
Hanchard, N.A., Swaminathan, S., Bucasas, K., Furthner, D., Fernbach, S., Azamian, M.S., et al. (2016). A genome-wide association study of congenital cardiovascular left-sided lesions shows association with a locus on chromosome 20. Hum. Mol. Genet. 25, 2331–2341. doi: 10.1093/hmg/ddw071
Hassed, S.J., Wiley, G.B., Wang, S., Lee, J.Y., Li, S., Xu, W., et al. (2012). RBPJ mutations identified in two families affected by adams-oliver syndrome. Am. J. Hum. Genet. 91, 391–395. doi: 10.1016/j.ajhg.2012.07.005
Hinson, J.T., Chopra, A., Nafissi, N., Polacheck, W.J., Benson, C.C., Swist, S., et al. (2015). Heart disease. Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science 349, 982–986. doi: 10.1126/science.aaa5458
Hirayama-Yamada, K., Kamisago, M., Akimoto, K., Aotsuka, H., Nakamura, Y., Tomita, H., et al. (2005). Phenotypes with GATA4 or NKX2.5 mutations in familial atrial septal defect. Am. J. Med. Genet. A 135, 47–52.
Hockemeyer, D., and Jaenisch, R. (2016). Induced pluripotent stem cells meet genome editing. Cell Stem Cell 18, 573–586. doi: 10.1016/j.stem.2016.04.013
Hoffman, J.I., and Kaplan, S. (2002). The incidence of congenital heart disease. J. Am. Coll. Cardiol. 39, 1890–1900.
Hrstka, S.C., Li, X., and Nelson, T.J. Wanek Program Genetics Pipeline Group. (2017). NOTCH1-dependent nitric oxide signaling deficiency in hypoplastic left heart syndrome revealed through patient-specific phenotypes detected in bioengineered cardiogenesis. Stem Cells 35, 1106–1119. doi: 10.1002/stem.2582
Hu, S., Zhao, M.T., Jahanbani, F., Shao, N.Y., Lee, W.H., Chen, H., et al. (2016). Effects of cellular origin on differentiation of human induced pluripotent stem cell-derived endothelial cells. JCI Insight 1:e85558.
Hu, Z., Shi, Y., Mo, X., Xu, J., Zhao, B., Lin, Y., et al. (2013). A genome-wide association study identifies two risk loci for congenital heart malformations in han chinese populations. Nat. Genet. 45, 818–821.
Itzhaki, I., Maizels, L., Huber, I., Zwi-Dantsis, L., Caspi, O., Winterstern, A., et al. (2011). Modelling the long QT syndrome with induced pluripotent stem cells. Nature 471, 225–229.
Jin, S.C., Homsy, J., Zaidi, S., Lu, Q., Morton, S., Depalma, S.R., et al. (2017). Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat. Genet. 49, 1593–1601.
Kamath, B.M., Bauer, R.C., Loomes, K.M., Chao, G., Gerfen, J., Hutchinson, A., et al. (2012). NOTCH2 mutations in Alagille syndrome. J. Med. Genet 49, 138–144.
Karbassi, E., Fenix, A., Marchiano, S., Muraoka, N., Nakamura, K., Yang, X., et al. (2020). Cardiomyocyte maturation: advances in knowledge and implications for regenerative medicine. Nat. Rev. Cardiol. 17, 341–359. doi: 10.1038/s41569-019-0331-x
Kathiriya, I.S., Rao, K.S., Iacono, G., Devine, W.P., Blair, A.P., Hota, S.K., et al. (2020). Modeling human TBX5 haploinsufficiency predicts regulatory networks for congenital heart disease. Dev. Cell 2020:30929–1.
Kathiriya, I.S., and Srivastava, D. (2000). Left-right asymmetry and cardiac looping: implications for cardiac development and congenital heart disease. Am. J. Med. Genet. 97, 271–279. doi: 10.1002/1096-8628(200024)97:4<271::aid-ajmg1277>3.0.co;2-o
Kerstjens-Frederikse, W.S., Van De Laar, I.M., Vos, Y.J., Verhagen, J.M., Berger, R.M., Lichtenbelt, K.D., et al. (2016). Cardiovascular malformations caused by NOTCH1 mutations do not keep left: data on 428 probands with left-sided CHD and their families. Genet. Med. 18, 914–923.
Kodo, K., Ong, S.G., Jahanbani, F., Termglinchan, V., Hirono, K., Inanloorahatloo, K., et al. (2016). iPSC-derived cardiomyocytes reveal abnormal TGF-beta signalling in left ventricular non-compaction cardiomyopathy. Nat. Cell Biol. 18, 1031–1042. doi: 10.1038/ncb3411
Koenig, S.N., Bosse, K., Majumdar, U., Bonachea, E.M., Radtke, F., Garg, V., et al. (2016). Endothelial notch1 is required for proper development of the semilunar valves and cardiac outflow tract. J. Am. Heart. Assoc. 5:e003075.
Lam, Y.Y., Keung, W., Chan, C.H., Geng, L., Wong, N., Breniere-Letuffe, D., et al. (2020). Single-cell transcriptomics of engineered cardiac tissues from patient-specific induced pluripotent stem cell-derived cardiomyocytes reveals abnormal developmental trajectory and intrinsic contractile defects in hypoplastic right heart syndrome. J. Am. Heart Assoc. 9:e016528.
Lee, A., Hudson, A.R., Shiwarski, D.J., Tashman, J.W., Hinton, T.J., Yerneni, S., et al. (2019). 3D bioprinting of collagen to rebuild components of the human heart. Science 365, 482–487. doi: 10.1126/science.aav9051
Lee, J.H., Protze, S.I., Laksman, Z., Backx, P.H., and Keller, G.M. (2017). Human Pluripotent Stem Cell-Derived Atrial and Ventricular Cardiomyocytes Develop from Distinct Mesoderm Populations. Cell Stem Cell 21, 179–194. doi: 10.1016/j.stem.2017.07.003
Lettice, L.A., Heaney, S.J., Purdie, L.A., Li, L., De Beer, P., Oostra, B.A., et al. (2003). A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum. Mol. Genet. 12, 1725–1735. doi: 10.1093/hmg/ddg180
Li, L., Krantz, I.D., Deng, Y., Genin, A., Banta, A.B., Collins, C.C., et al. (1997a). Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat. Genet. 16, 243–251. doi: 10.1038/ng0797-243
Li, Q.Y., Newbury-Ecob, R.A., Terrett, J.A., Wilson, D.I., Curtis, A.R., Yi, C.H., et al. (1997b). Holt-Oram syndrome is caused by mutations in TBX5, a member of the Brachyury (T) gene family. Nat. Genet. 15, 21–29. doi: 10.1038/ng0197-21
Liang, W., Han, P., Kim, E.H., Mak, J., Zhang, R., Torrente, A.G., et al. (2020). Canonical Wnt signaling promotes pacemaker cell specification of cardiac mesodermal cells derived from mouse and human embryonic stem cells. Stem Cells 38, 352–368. doi: 10.1002/stem.3106
Litvinukova, M., Talavera-Lopez, C., Maatz, H., Reichart, D., Worth, C.L., Lindberg, E.L., et al. (2020). Cells of the adult human heart. Nature. 588, 466–472.
Liu, S., Joseph, K.S., Lisonkova, S., Rouleau, J., Van Den Hof, M., Sauve, R., et al. (2013). Association between maternal chronic conditions and congenital heart defects: a population-based cohort study. Circulation 128, 583–589. doi: 10.1161/circulationaha.112.001054
Liu, X., Yagi, H., Saeed, S., Bais, A.S., Gabriel, G.C., Chen, Z., et al. (2017). The complex genetics of hypoplastic left heart syndrome. Nat. Genet. 49, 1152–1159.
Lo Sardo, V., Chubukov, P., Ferguson, W., Kumar, A., Teng, E.L., Duran, M., et al. (2018). Unveiling the role of the most impactful cardiovascular risk locus through haplotype editing. Cell 175, 1796–1810. doi: 10.1016/j.cell.2018.11.014
Lupianez, D.G., Kraft, K., Heinrich, V., Krawitz, P., Brancati, F., Klopocki, E., et al. (2015). Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 161, 1012–1025. doi: 10.1016/j.cell.2015.04.004
Luxan, G., Casanova, J.C., Martinez-Poveda, B., Prados, B., D’amato, G., Macgrogan, D., et al. (2013). Mutations in the NOTCH pathway regulator MIB1 cause left ventricular noncompaction cardiomyopathy. Nat. Med. 19, 193–201. doi: 10.1038/nm.3046
Macgrogan, D., Munch, J., and De La Pompa, J.L. (2018). Notch and interacting signalling pathways in cardiac development, disease, and regeneration. Nat. Rev. Cardiol. 15, 685–704. doi: 10.1038/s41569-018-0100-2
Majumdar, U., Yasuhara, J., and Garg, V. (2019). In Vivo and In Vitro Genetic Models of Congenital Heart Disease. Cold Spring Harb Perspect. Biol. 2019:a036764. doi: 10.1101/cshperspect.a036764
Maurano, M.T., Humbert, R., Rynes, E., Thurman, R.E., Haugen, E., Wang, H., et al. (2012). Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195. doi: 10.1126/science.1222794
Mcbride, K.L., Pignatelli, R., Lewin, M., Ho, T., Fernbach, S., Menesses, A., et al. (2005). Inheritance analysis of congenital left ventricular outflow tract obstruction malformations: Segregation, multiplex relative risk, and heritability. Am. J. Med. Genet. A 134A, 180–186. doi: 10.1002/ajmg.a.30602
Mcbride, K.L., Riley, M.F., Zender, G.A., Fitzgerald-Butt, S.M., Towbin, J.A., Belmont, J.W., et al. (2008). NOTCH1 mutations in individuals with left ventricular outflow tract malformations reduce ligand-induced signaling. Hum. Mol. Genet. 17, 2886–2893. doi: 10.1093/hmg/ddn187
Mcdaniell, R., Warthen, D.M., Sanchez-Lara, P.A., Pai, A., Krantz, I.D., Piccoli, D.A., et al. (2006). NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. Am. J. Hum. Genet. 79, 169–173. doi: 10.1086/505332
Mcdermott, D.A., Bressan, M.C., He, J., Lee, J.S., Aftimos, S., Brueckner, M., et al. (2005). TBX5 genetic testing validates strict clinical criteria for holt-oram syndrome. Pediatr. Res. 58, 981–986. doi: 10.1203/01.pdr.0000182593.95441.64
Meester, J.A., Southgate, L., Stittrich, A.B., Venselaar, H., Beekmans, S.J., Den Hollander, N., et al. (2015). Heterozygous loss-of-function mutations in DLL4 cause adams-oliver syndrome. Am. J. Hum. Genet. 97, 475–482. doi: 10.1016/j.ajhg.2015.07.015
Miao, Y., Tian, L., Martin, M., Paige, S.L., Galdos, F.X., Li, J., et al. (2020). Intrinsic endocardial defects contribute to hypoplastic left heart syndrome. Cell Stem Cell 27, 574–589.
Mikryukov, A.A., Mazine, A., Wei, B., Yang, D., Miao, Y., Gu, M., et al. (2021). BMP10 signaling promotes the development of endocardial cells from human pluripotent stem cell-derived cardiovascular progenitors. Cell Stem Cell 28, 1–16.
Misra, C., Sachan, N., Mcnally, C.R., Koenig, S.N., Nichols, H.A., Guggilam, A., et al. (2012). Congenital heart disease-causing Gata4 mutation displays functional deficits in vivo. PLoS Genet. 8:e1002690. doi: 10.1371/journal.pgen.1002690
Mitchell, L.E., Agopian, A.J., Bhalla, A., Glessner, J.T., Kim, C.E., Swartz, M.D., et al. (2015). Genome-wide association study of maternal and inherited effects on left-sided cardiac malformations. Hum. Mol. Genet. 24, 265–273. doi: 10.1093/hmg/ddu420
Moretti, A., Bellin, M., Welling, A., Jung, C.B., Lam, J.T., Bott-Flugel, L., et al. (2010). Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N. Engl. J. Med. 363, 1397–1409.
Nees, S.N., and Chung, W.K. (2019). Genetic basis of human congenital heart disease. Cold Spring Harb Perspect Biol. 12:a036749.
Nugraha, B., Buono, M.F., Von Boehmer, L., Hoerstrup, S.P., and Emmert, M.Y. (2019). Human cardiac organoids for disease modeling. Clin. Pharmacol. Ther. 105, 79–85. doi: 10.1002/cpt.1286
Oda, T., Elkahloun, A.G., Pike, B.L., Okajima, K., Krantz, I.D., Genin, A., et al. (1997). Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat. Genet. 16, 235–242.
Okubo, A., Miyoshi, O., Baba, K., Takagi, M., Tsukamoto, K., Kinoshita, A., et al. (2004). A novel GATA4 mutation completely segregated with atrial septal defect in a large Japanese family. J. Med. Genet. 41:e97. doi: 10.1136/jmg.2004.018895
Paige, S.L., Galdos, F.X., Lee, S., Chin, E.T., Ranjbarvaziri, S., Feyen, D.A.M., et al. (2020). Patient-specific induced pluripotent stem cells implicate intrinsic impaired contractility in hypoplastic left heart syndrome. Circulation 142, 1605–1608. doi: 10.1161/circulationaha.119.045317
Paik, D.T., Cho, S., Tian, L., Chang, H.Y., and Wu, J.C. (2020). Single-cell RNA sequencing in cardiovascular development, disease and medicine. Nat. Rev. Cardiol. 17, 457–473. doi: 10.1038/s41569-020-0359-y
Parikh, S.S., Blackwell, D.J., Gomez-Hurtado, N., Frisk, M., Wang, L., Kim, K., et al. (2017). Thyroid and glucocorticoid hormones promote functional T-tubule development in human-induced pluripotent stem cell-derived cardiomyocytes. Circ. Res. 121, 1323–1330. doi: 10.1161/circresaha.117.311920
Pediatric Cardiac Genomics Consortium. Gelb, B., Brueckner, M., Chung, W., Goldmuntz, E., Kaltman, J., et al. (2013). The congenital heart disease genetic network study: rationale, design, and early results. Circ. Res. 112, 698–706.
Pierpont, M.E., Brueckner, M., Chung, W.K., Garg, V., Lacro, R.V., Mcguire, A.L., et al. (2018). Genetic basis for congenital heart disease: revisited: a scientific statement from the american heart association. Circulation 138, e653–e711.
Protze, S.I., Lee, J.H., and Keller, G.M. (2019). Human pluripotent stem cell-derived cardiovascular cells: from developmental biology to therapeutic applications. Cell Stem Cell 25, 311–327. doi: 10.1016/j.stem.2019.07.010
Protze, S.I., Liu, J., Nussinovitch, U., Ohana, L., Backx, P.H., Gepstein, L., et al. (2017). Sinoatrial node cardiomyocytes derived from human pluripotent cells function as a biological pacemaker. Nat. Biotechnol. 35, 56–68. doi: 10.1038/nbt.3745
Ren, J., Han, P., Ma, X., Farah, E.N., Bloomekatz, J., Zeng, X.I., et al. (2019). Canonical Wnt5b signaling directs outlying Nkx2.5+ mesoderm into pacemaker cardiomyocytes. Dev Cell 50, 729–743. doi: 10.1016/j.devcel.2019.07.014
Richards, D.J., Li, Y., Kerr, C.M., Yao, J., Beeson, G.C., Coyle, R.C., et al. (2020). Human cardiac organoids for the modelling of myocardial infarction and drug cardiotoxicity. Nat. Biomed. Eng. 4, 446–462. doi: 10.1038/s41551-020-0539-4
Richter, F., Morton, S.U., Kim, S.W., Kitaygorodsky, A., Wasson, L.K., Chen, K.M., et al. (2020). Genomic analyses implicate noncoding de novo variants in congenital heart disease. Nat. Genet. 52, 769–777. doi: 10.1038/s41588-020-0652-z
Riley, M.F., Mcbride, K.L., and Cole, S.E. (2011). NOTCH1 missense alleles associated with left ventricular outflow tract defects exhibit impaired receptor processing and defective EMT. Biochim. Biophys. Acta 1812, 121–129. doi: 10.1016/j.bbadis.2010.10.002
Ronaldson-Bouchard, K., Ma, S.P., Yeager, K., Chen, T., Song, L., Sirabella, D., et al. (2018). Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature 556, 239–243. doi: 10.1038/s41586-018-0016-3
Rossi, G., Manfrin, A., and Lutolf, M.P. (2018). Progress and potential in organoid research. Nat Rev Genet 19, 671–687. doi: 10.1038/s41576-018-0051-9
Saraf, A., Book, W.M., Nelson, T.J., and Xu, C. (2019). Hypoplastic left heart syndrome: From bedside to bench and back. J. Mol. Cell Cardiol. 135, 109–118. doi: 10.1016/j.yjmcc.2019.08.005
Sarkozy, A., Conti, E., Neri, C., D’agostino, R., Digilio, M.C., Esposito, G., et al. (2005). Spectrum of atrial septal defects associated with mutations of NKX2.5 and GATA4 transcription factors. J. Med. Genet. 42:e16. doi: 10.1136/jmg.2004.026740
Schott, J.J., Benson, D.W., Basson, C.T., Pease, W., Silberbach, G.M., Moak, J.P., et al. (1998). Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science 281, 108–111. doi: 10.1126/science.281.5373.108
Short, P.J., Mcrae, J.F., Gallone, G., Sifrim, A., Won, H., Geschwind, D.H., et al. (2018). De novo mutations in regulatory elements in neurodevelopmental disorders. Nature 555, 611–616. doi: 10.1038/nature25983
Sifrim, A., Hitz, M.P., Wilsdon, A., Breckpot, J., Turki, S.H., Thienpont, B., et al. (2016). Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat. Genet. 48, 1060–1065.
Sleiman, Y., Souidi, M., Kumar, R., Yang, E., Jaffre, F., Zhou, T., et al. (2020). Modeling polymorphic ventricular tachycardia at rest using patient-specific induced pluripotent stem cell-derived cardiomyocytes. EBioMedicine 60:103024. doi: 10.1016/j.ebiom.2020.103024
Smemo, S., Campos, L.C., Moskowitz, I.P., Krieger, J.E., Pereira, A.C., Nobrega, M.A., et al. (2012). Regulatory variation in a TBX5 enhancer leads to isolated congenital heart disease. Hum. Mol. Genet. 21, 3255–3263. doi: 10.1093/hmg/dds165
Stallmeyer, B., Fenge, H., Nowak-Gottl, U., and Schulze-Bahr, E. (2010). Mutational spectrum in the cardiac transcription factor gene NKX2.5 (CSX) associated with congenital heart disease. Clin Genet 78, 533–540. doi: 10.1111/j.1399-0004.2010.01422.x
Stittrich, A.B., Lehman, A., Bodian, D.L., Ashworth, J., Zong, Z., Li, H., et al. (2014). Mutations in NOTCH1 cause adams-oliver syndrome. Am. J. Hum. Genet. 95, 275–284. doi: 10.1016/j.ajhg.2014.07.011
Sun, N., Yazawa, M., Liu, J., Han, L., Sanchez-Freire, V., Abilez, O.J., et al. (2012). Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci. Transl. Med. 4:130ra147.
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K., et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872. doi: 10.1016/j.cell.2007.11.019
Tanay, A., and Regev, A. (2017). Scaling single-cell genomics from phenomenology to mechanism. Nature 541, 331–338. doi: 10.1038/nature21350
Theis, J.L., Hrstka, S.C., Evans, J.M., O’byrne, M.M., De Andrade, M., O’leary, P.W., et al. (2015). Compound heterozygous NOTCH1 mutations underlie impaired cardiogenesis in a patient with hypoplastic left heart syndrome. Hum. Genet. 134, 1003–1011. doi: 10.1007/s00439-015-1582-1
Theodoris, C.V., Li, M., White, M.P., Liu, L., He, D., Pollard, K.S., et al. (2015). Human disease modeling reveals integrated transcriptional and epigenetic mechanisms of NOTCH1 haploinsufficiency. Cell 160, 1072–1086. doi: 10.1016/j.cell.2015.02.035
Theodoris, C.V., Zhou, P., Liu, L., Zhang, Y., Nishino, T., Huang, Y., et al. (2020). Network-based screen in iPSC-derived cells reveals therapeutic candidate for heart valve disease. Science 2020:eabd0724. doi: 10.1126/science.abd0724
Tomita-Mitchell, A., Maslen, C.L., Morris, C.D., Garg, V., and Goldmuntz, E. (2007). GATA4 sequence variants in patients with congenital heart disease. J. Med. Genet. 44, 779–783. doi: 10.1136/jmg.2007.052183
Wang, G., Mccain, M.L., Yang, L., He, A., Pasqualini, F.S., Agarwal, A., et al. (2014). Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat. Med. 20, 616–623. doi: 10.1038/nm.3545
Yang, C., Xu, Y., Yu, M., Lee, D., Alharti, S., Hellen, N., et al. (2017). Induced pluripotent stem cell modelling of HLHS underlines the contribution of dysfunctional NOTCH signalling to impaired cardiogenesis. Hum. Mol. Genet. 26, 3031–3045. doi: 10.1093/hmg/ddx140
Yu, J., Vodyanik, M.A., Smuga-Otto, K., Antosiewicz-Bourget, J., Frane, J.L., Tian, S., et al. (2007). Induced pluripotent stem cell lines derived from human somatic cells. Science 318, 1917–1920.
Zahavich, L., Bowdin, S., and Mital, S. (2017). Use of clinical exome sequencing in isolated congenital heart disease. Circ. Cardiovasc. Genet. 10:e001581.
Zaidi, S., Choi, M., Wakimoto, H., Ma, L., Jiang, J., Overton, J.D., et al. (2013). De novo mutations in histone-modifying genes in congenital heart disease. Nature 498, 220–223.
Zhang, M., D’aniello, C., Verkerk, A.O., Wrobel, E., Frank, S., Ward-Van Oostwaard, D., et al. (2014). Recessive cardiac phenotypes in induced pluripotent stem cell models of Jervell and Lange-Nielsen syndrome: disease mechanisms and pharmacological rescue. Proc. Natl. Acad. Sci. U S A. 111, 5383–5392.
Zhang, Q., Jiang, J., Han, P., Yuan, Q., Zhang, J., Zhang, X., et al. (2011). Direct differentiation of atrial and ventricular myocytes from human embryonic stem cells by alternating retinoid signals. Cell Res. 21, 579–587. doi: 10.1038/cr.2010.163
Zhao, M.T., Chen, H., Liu, Q., Shao, N.Y., Sayed, N., Wo, H.T., et al. (2017a). Molecular and functional resemblance of differentiated cells derived from isogenic human iPSCs and SCNT-derived ESCs. Proc. Natl. Acad. Sci. U S A. 114, 11111–11120.
Zhao, M.T., Shao, N.Y., and Garg, V. (2020a). Subtype-specific cardiomyocytes for precision medicine: where are we now? Stem Cells 38, 822–833.
Zhao, M.T., Shao, N.Y., Hu, S., Ma, N., Srinivasan, R., Jahanbani, F., et al. (2017b). Cell Type-Specific Chromatin Signatures Underline Regulatory DNA Elements in Human Induced Pluripotent Stem Cells and Somatic Cells. Circ. Res. 121, 1237–1250. doi: 10.1161/circresaha.117.311367
Keywords: congenital heart disease, human induced pluripotent stem cells, NOTCH signaling, hypoplastic left heart syndrome, genetic models of CHD
Citation: Lin H, McBride KL, Garg V and Zhao M-T (2021) Decoding Genetics of Congenital Heart Disease Using Patient-Derived Induced Pluripotent Stem Cells (iPSCs). Front. Cell Dev. Biol. 9:630069. doi: 10.3389/fcell.2021.630069
Received: 16 November 2020; Accepted: 04 January 2021;
Published: 21 January 2021.
Edited by:
Sveva Bollini, University of Genoa, ItalyReviewed by:
Albano Carlo Meli, INSERM U1046 Physiologie et médecine expérimentale du coeur et des muscles, FranceLuca Sala, Istituto Auxologico Italiano (IRCCS), Italy
Copyright © 2021 Lin, McBride, Garg and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ming-Tao Zhao, TWluZ3Rhby5aaGFvQE5hdGlvbndpZGVDaGlsZHJlbnMuT3Jn